
TNFα acting on TNFR1 promotes breast cancer growth via p42/P44 MAPK, JNK, Akt and NF-κB-dependent...
21
Research Article TNFα acting on TNFR1 promotes breast cancer growth via p42/P44 MAPK, JNK, Akt and NF-κB-dependent pathways Martín A. Rivas a , Romina P. Carnevale a , Cecilia J. Proietti a , Cinthia Rosemblit a , Wendy Beguelin a , Mariana Salatino a , Eduardo H. Charreau a , Isabel Frahm b , Sandra Sapia c , Peter Brouckaert d , Patricia V. Elizalde a , Roxana Schillaci a, ⁎ a Laboratory of Molecular Mechanisms of Carcinogenesis, Instituto de Biología y Medicina Experimental, CONICET, Vuelta de Obligado 2490, Buenos Aires, C1428ADN, Argentina b Servicio de Patología, Sanatorio Mater Dei, Buenos Aires, Argentina c Laboratorio de Patología, Fundaleu, Buenos Aires, Argentina d Department of Molecular Biomedical Research, VIB and Ghent University, Ghent, Belgium ARTICLE INFORMATION ABSTRACT Article Chronology: Received 11 May 2007 Revised version received 8 October 2007 Accepted 10 October 2007 Available online 13 October 2007 Tumor necrosis factor α (TNFα) enhances proliferation of chemically-induced mammary tumors and of T47D human cell line through not fully understood pathways. Here, we explored the intracellular signaling pathways triggered by TNFα, the participation of TNFα receptor (TNFR) 1 and TNFR2 and the molecular mechanism leading to breast cancer growth. We demonstrate that TNFα induced proliferation of C4HD murine mammary tumor cells and of T47D cells through the activation of p42/p44 MAPK, JNK, PI3-K/Akt pathways and nuclear factor-kappaB (NF-κB) transcriptional activation. A TNFα-specific mutein selectively binding to TNFR1 induced p42/p44 MAPK, JNK, Akt activation, NF-κB transcriptional activation and cell proliferation, just like wild-type TNFα, while a mutein selective for TNFR2 induced only p42/p44 MAPK activation. Interestingly, blockage of TNFR1 or TNFR2 with specific antibodies was enough to impair TNFα signaling and biological effect. Moreover, in vivo TNFα administration supported C4HD tumor growth. We also demonstrated, for the first time, that injection of a selective inhibitor of NF-κB activity, Bay 11-7082, resulted in regression of TNFα-promoted tumor. Bay 11-7082 blocked TNFα capacity to induce cell proliferation and up-regulation of cyclin D1 and of Bcl-x L in vivo and in vitro. Our results reveal evidence for TNFα as a breast tumor promoter, and provide novel data for a future therapeutic approach using TNFα antagonists and NF-κB pharmacological inhibitors in established breast cancer treatment. © 2007 Elsevier Inc. All rights reserved. Keywords: Breast cancer TNFα NF-κB TNFα receptors Introduction Tumor necrosis factor alpha (TNFα) is a pleiotropic cytokine that can regulate a wide variety of cellular responses including pro- liferation, differentiation, inflammation, and cell death. Al- though TNFα was originally characterized to cause hemorrhagic tumor necrosis at high concentrations in many types of cancer, low concentrations of TNFα seem to increase tumor growth and progression [1]. TNFα binds to TNFα receptors (TNFRs) that recruit several proteins which function as a platform adapter EXPERIMENTAL CELL RESEARCH 314 (2008) 509 – 529 * Corresponding author. Fax: +54 11 4786 2564. E-mail address: [email protected] (R. Schillaci). 0014-4827/$ – see front matter © 2007 Elsevier Inc. All rights reserved. doi:10.1016/j.yexcr.2007.10.005 available at www.sciencedirect.com www.elsevier.com/locate/yexcr
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of TNFα acting on TNFR1 promotes breast cancer growth via p42/P44 MAPK, JNK, Akt and NF-κB-dependent...

E X P E R I M E N T A L C E L L R E S E A R C H 3 1 4 ( 2 0 0 8 ) 5 0 9 – 5 2 9
ava i l ab l e a t www.sc i enced i rec t . com
www.e l sev i e r. com/ loca te /yexc r
Research Article
TNFα acting on TNFR1 promotes breast cancer growth viap42/P44 MAPK, JNK, Akt and NF-κB-dependent pathways
Martín A. Rivasa, Romina P. Carnevalea, Cecilia J. Proiettia, Cinthia Rosemblita,Wendy Beguelina, Mariana Salatinoa, Eduardo H. Charreaua, Isabel Frahmb,Sandra Sapiac, Peter Brouckaertd, Patricia V. Elizaldea, Roxana Schillacia,⁎aLaboratory of Molecular Mechanisms of Carcinogenesis, Instituto de Biología y Medicina Experimental,CONICET, Vuelta de Obligado 2490, Buenos Aires, C1428ADN, ArgentinabServicio de Patología, Sanatorio Mater Dei, Buenos Aires, ArgentinacLaboratorio de Patología, Fundaleu, Buenos Aires, ArgentinadDepartment of Molecular Biomedical Research, VIB and Ghent University, Ghent, Belgium
A R T I C L E I N F O R M A T I O N
* Corresponding author. Fax: +54 11 4786 2564E-mail address: [email protected] (R. Sc
0014-4827/$ – see front matter © 2007 Elsevidoi:10.1016/j.yexcr.2007.10.005
A B S T R A C T
Article Chronology:Received 11 May 2007Revised version received8 October 2007Accepted 10 October 2007Available online 13 October 2007
Tumor necrosis factor α (TNFα) enhances proliferation of chemically-induced mammarytumors and of T47D human cell line through not fully understood pathways. Here, weexplored the intracellular signaling pathways triggered by TNFα, the participation of TNFαreceptor (TNFR) 1 and TNFR2 and themolecularmechanism leading to breast cancer growth.We demonstrate that TNFα induced proliferation of C4HD murine mammary tumor cellsand of T47D cells through the activation of p42/p44 MAPK, JNK, PI3-K/Akt pathways andnuclear factor-kappaB (NF-κB) transcriptional activation. A TNFα-specific mutein selectivelybinding to TNFR1 induced p42/p44 MAPK, JNK, Akt activation, NF-κB transcriptionalactivation and cell proliferation, just like wild-type TNFα, while a mutein selective forTNFR2 induced only p42/p44MAPKactivation. Interestingly, blockage of TNFR1 or TNFR2withspecific antibodies was enough to impair TNFα signaling and biological effect. Moreover, invivo TNFα administration supported C4HD tumor growth.We also demonstrated, for the firsttime, that injectionof a selective inhibitor ofNF-κBactivity, Bay11-7082, resulted in regressionof TNFα-promoted tumor. Bay 11-7082 blocked TNFα capacity to induce cell proliferation andup-regulationof cyclinD1andof Bcl-xL in vivo and in vitro. Our results reveal evidence for TNFαas a breast tumor promoter, and provide novel data for a future therapeutic approach usingTNFα antagonists and NF-κB pharmacological inhibitors in established breast cancertreatment.
© 2007 Elsevier Inc. All rights reserved.
Keywords:Breast cancerTNFαNF-κBTNFα receptors
Introduction
Tumornecrosis factor alpha (TNFα) is a pleiotropic cytokine thatcan regulate a wide variety of cellular responses including pro-liferation, differentiation, inflammation, and cell death. Al-
.hillaci).
er Inc. All rights reserved
thoughTNFαwasoriginally characterized to cause hemorrhagictumor necrosis at high concentrations inmany types of cancer,low concentrations of TNFα seem to increase tumor growth andprogression [1]. TNFα binds to TNFα receptors (TNFRs) thatrecruit several proteins which function as a platform adapter
.

510 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 4 ( 2 0 0 8 ) 5 0 9 – 5 2 9
and trigger different signaling pathways depending on cell type.These receptors, 55-kDa type1 receptor (TNFR1) and75-kDa type2 receptor (TNFR2), are glycoproteins with a single transmem-brane domain [2]. Each receptor shares only 28% homology inthe ligand-binding extracellular domain with its counterpart,and is devoid of enzymatic activity. Through its cytoplasmicdeath domain, TNFR1 recruits, among others, TNFR1-associateddeath domain protein (TRADD) and TNF receptor-associatedfactor (TRAF)-2, which is known to be required for activation ofc-Jun N-terminal kinase (JNK), of p42/p44 mitogen-activatedprotein kinase (MAPK) and of p38 MAPK [3]. On the other hand,although lacking the death domain, TNFR2 is known to bind toTRAF2 through another adapter protein, TRAF1 [4], thus pro-viding amechanismfor someshared activity betweenTNFRs. Inaddition, TNFR1 also activatesmitogen-activatedproteinkinasekinase kinase 3 which directly phosphorylates IκBβ kinase(IKKβ) [5]. Moreover, TRAF-2 can also associate with the nuclearfactor-kappaB (NF-κB) inducing kinase (NIK), necessary tophosphorylate IKKα kinase together with Akt [6,7]. Then, theactivated IKKs cause phosphorylation of IκBs, cytoplasmic inhi-bitors of the transcription factor NF-κB, triggering their ubiqui-tination and subsequent degradation,which in turn leads to therelease and nuclear translocation of p65 and p50 NF-κB familymembers [8]. This pathway, knownas canonical, has so far beenthe best described and is probably themost importantmediatorof NF-κB activation in response to TNFα. Another form of NF-κBactivation, the non-canonical pathway, has been describedparticularly in B cells, and is IκB-independent. It is induced e.g.by lymphotoxin β, and leads to NIK- and IKKα-dependentprocessing of the p100 NF-κB family member precursor protein,which results in release of p52 [9].
In the normal mammary gland, TNFα is known to play aphysiological role in development, proliferation and branch-ing morphogenesis [10]. TNFR1 expression has been reportedto mediate TNFα-induced proliferation of normal mammaryepithelial cells, while TNFR2 activation induces casein accu-mulation [11]. The fact that in TNFα-deficient mice UV doesnot evoke skin cancer, provides evidence that TNFα is a keycytokine for tumor promotion [12]. Particularly in breast can-cer, TNFα has been reported to induce apoptosis or to inhibit invitro proliferation in MCF-7 cells [13], while other breast cancercell lines have been shown to be resistant to TNFα-inducedapoptosis [14]. Differences in susceptibility to TNFα-inducedapoptosis may be explained, among other factors, by differ-ences in TNFR expression and p38 MAPK activation [13,15]. Onthe other hand, resistance to the TNFα cytotoxic effect iscaused by constitutive ErbB-2 activation of Akt/NF-κB pathwayin breast cancer cell lines [14]. Furthermore, TNFα can also bemitogenic in some breast cancer cells, as in the 1-methyl-1-nitrosourea-induced rat mammary tumor model [16] and inthe human breast cancer cell line T47D [17]. Intracellular sig-naling pathways triggered by TNFα leading to breast cancerproliferation have only partially been delineated, with NF-κBplaying an important role [16]. However, the participation ofTNFR1 and TNFR2, the molecular mechanisms by which TNFαcontrols breast cancer growth, and the involvement of up-stream kinases in the regulation of this process, remain poorlyexplored.
In the present study, we have described TNFα proliferativeeffect on C4HD murine mammary tumor cells. C4HD cells
belong to an experimentalmodel of hormonal carcinogenesis inwhich the synthetic progestin medroxyprogesterone acetateinduced mammary adenocarcinomas in female Balb/c mice[18,19]. Taking advantage of the mitogenic effect of TNFα onT47D human breast cancer cell line and on C4HD cells, weelucidated the signal transduction pathways involved in TNFα-mediated breast cancer proliferation. We demonstrated adifferential activation of p42/p44 MAPK, JNK and phosphatidy-linositol 3-kinase (PI3-K)/Akt pathways, NF-κB transcriptionalactivation and cell proliferation after independent ligandengagementofTNFR1orTNFR2 inbreast cancer cells.Moreover,we found that blockage of each receptor with their specificantibodies inhibited these effects. Finally, this is the first reportto demonstrate that in vivo administration of TNFα promotesgrowth of a primary breast tumor, and that injection of theselective pharmacological inhibitor of NF-κB, Bay 11-7082,results in tumor regression. Our results add further evidenceon TNFα as a tumor promoter, and provide novel data for therational use of TNFα antagonists and of NF-κB pharmacologicalinhibitors as therapies for breast cancer treatment.
Materials and methods
Animals and tumors
Experiments were carried out in virgin female Balb/c mice,raised at the Instituto de Biología y Medicina Experimental(IBYME) of Buenos Aires. All animal studies were conductedin accordance with the highest standards of animal care asoutlined in the NIH guide for the Care and Use of LaboratoryAnimals, and were approved by the IBYME Animal ResearchCommittee. Hormone-dependent mammary ductal tumorC4HD was originated in mice treated with 40mg medroxypro-gesterone acetate (MPA, Medrosterona, Laboratorios Gador,Buenos Aires, Argentina) every 3months for 1year, and hasbeen maintained by serial transplantation in animals treatedwith 40mg s.c. MPA depot in the opposite flank to tumorinoculum [18–20]. C4HD tumor expresses progesterone andestrogen receptors [18–20].
Cell cultures and proliferation assays
Primary cultures of epithelial cells from C4HD tumors, growingin MPA-treated mice, were performed as previously described[18–21]. Inbrief, C4HD tumorswereaseptically removed,mincedand washed with Dulbecco's Modified Eagle's Medium /F12without phenol red (Sigma, St. Louis, MO) with 100U/ml peni-cillin, and with 100μg/ml streptomycin (DMEM). Tissue wassuspended in 5ml of enzymatic solution [(trypsin 2.5mg/ml,albumin 5mg/ml and collagenase type II (Gibco BRL, Gaither-burg, MD) 239U/ml] in PBS and incubated at 37 °C for 40min,under continuous stirring. Enzyme action was stopped byadding DMEM + 5% heat-inactivated fetal calf serum (FCS, GenS.A., Buenos Aires, Argentina). Epithelial and fibroblastic cellswere separated by suspension in 15ml DMEM+2% FCS andallowed to sediment for 20min. The upper 15ml was discardedand sedimented cells, corresponding to the epithelial-enrichedfraction,were resuspended in15mlDMEM+2%FCSandallowedto sediment for 20min. This procedure was repeated until no

Fig. 1 – TNFα promotes proliferation of primary cultures of C4HD cells andT47D cell line. (A) Primary cultures of epithelial C4HDcells or (B) T47D cell line were incubated for 48 h in DMEM+0.1% ChFCS supplemented with mouse TNFα (mTNFα) or humanTNFα (hTNFα) at various concentrations. (C) and (D) Twenty ng/ml mTNFα or hTNFα was preincubated for 30 min in thepresence or absence of different concentrations of Etanercept and then added to C4HD or T47D cells respectively. Incorporationof [3H]-thymidine was used to measure DNA synthesis. Data are presented as mean±SE of octuplicate samples. Theexperiments shown are representative of a total of four. b vs. a, P<0.001.
511E X P E R I M E N T A L C E L L R E S E A R C H 3 1 4 ( 2 0 0 8 ) 5 0 9 – 5 2 9
fibroblasts were observed in the supernatant. Epithelial cellswere plated in culture flaskswithDMEM+ 10% FCS, and allowedto attach for 24–48h. Purity of epithelial cultures was evaluatedby cytokeratin staining. C4HD cells were trypsinized and platedat 104 cells/well in a P96 plate. As C4HD cells grow in thepresence of progestins, all the experiments were performed inDMEM + 0.1% charcoal-stripped FCS (ChFCS).
Human breast cancer cell line T47D was obtained from theAmerican Type Culture Collection. This cell line expressesboth TNFRs [22]. In experiments assessing TNFα activity, T47Dcells were cultured and subjected to the treatments describedfor C4HD cells.
In all cases, cells were starved for 72h inDMEM + 0.1%ChFCSand then subjected to the different treatments. Cells weretreated for 48h with different concentrations of murine orhuman TNFα (Cell Sciences, Canton, MA) or genetically engi-neered murine TNFα muteins, that bind selectively to either of
the TNFR subtypes [23]. Muteins were constructed by replacingspecific amino acids from the region which contains the dif-ferences between mTNFα and hTNFα [23]. The following mu-teins were used: mTNF-D71S/Y72T/Δ73H/E89T (R1-TNF) whichselectivelyactivatesmouseTNFR1,hTNF-S71D/T72Y/H73Δ/T89E(R1R2-TNF), a human TNFα with the ability to bind to TNFR1 aswell as toTNRF2 [23], andmTNF-R32Y/A145R (R2-TNF), amurineTNFα which selectively activates mouse TNFR2. The latter hasno affinity to mouse TNFR1 and its dissociation from TNFR2 issimilar to that of wild-type mTNFα, determined by the directreceptor binding assay with Surface Plasmon Resonance tech-nology provided by BIAcore (Biacore, Uppsala, Sweden). R2-TNFwasnot able to induceNF-κBactivity in L929 cells evenwhen it isused at 1μg/ml but retained strong proliferating activity on CT6thymoma cells, a TNFR2-mediated effect. It is cleared from cir-culation just like wild-type mTNFα (Dr. P. Brouckaert, unpub-lished). In selected experiments, rabbit antibodies blocking

512 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 4 ( 2 0 0 8 ) 5 0 9 – 5 2 9
mouse TNFR1 or TNFR2 kindly provided by Dr. W.A. Buurman(Maastricht University, Maastricht, TheNetherlands), were used[24]. The following inhibitors were added to cells 30min beforeincubation with TNFα: SP600125 (Calbiochem, La Jolla, CA),LY294002 and Bay 11-7082 (all from Sigma) for blocking JNK, PI3-K/Akt and NF-κB respectively, and PD98059 and U0126 forblocking p42/p44 MAPK. Controls were performed in order toverify that DMSO (1:2000) did not modify TNFα-induced proli-feration. To perform certain experiments, the anti-TNFα Eta-nercept (p75 fusionprotein) from Immunex (Enbrelk; ThousandOaks, CA) was purchased from a pharmaceutical supplier.
After 24h incubation, 50% of media was replaced by freshmedia, and cells were incubated for another 24h in thepresence of 1μCi [3H]-thymidine (NEN, Dupont, Boston, MA;specific activity: 20Ci/mmol). Cells were then trypsinized andharvested with a Nunc harvester that aspirates and lyses cellsand transfers DNA onto glass filters, while allowing unincor-porated [3H]-thymidine to wash out. The filters were countedusing standard scintillation procedures. Assays were per-formed in octuplicate. In earlier experiments we had demon-strated that [3H]-thymidine uptake correlatedwith the numberof cells/well [25].

513E X P E R I M E N T A L C E L L R E S E A R C H 3 1 4 ( 2 0 0 8 ) 5 0 9 – 5 2 9
Flow cytometry analysis and cell counting
C4HD cells were harvested with PBS + EDTA 1mM. Cells wereincubated with rabbit anti-mouse TNFR1 or TNFR2, washedand then incubated with an isothiocianate fluorescein (FITC)-conjugated F(ab)2 fragment of anti-rabbit IgG (Dako Corpora-tion, Carpinteria, CA). Cells were washed and analyzed usingFACScalibur cytometer (Becton Dickinson, La Jolla, CA). A totalof 104 cells/sample was analyzed. Background staining wasevaluated in cells incubated with a rabbit IgG isotype controlfollowed by secondary antibody stain. Data analysis wasperformed using Cell Quest software (Becton Dickinson).
For cell cycle analysis, primary cultures of C4HD cells werestarved for 72h in DMEM + 0.1% ChFCS and then subjected tothe different treatments. Cells were harvested at 24, 36 and48h, and fixed in 70% ethanol for 24h at 4 °C, as we previouslydescribed [21]. They were washed twice with PBS, followed byRNA digestion (RNAse A 50U/ml) and propidium iodide (20μg/ml) staining for 30min at room temperature in the dark. Cellcycle analysis was performed using a FACScalibur flowcytometer (Becton Dickinson) and Modfit LT software.
For apoptosis analysis and cell counting, primary culturesof C4HD cells were starved for 72h in DMEM + 0.1% ChFCS andthen subjected to the different treatments for 96h. Apoptosiswas investigated through cell surface binding of fluorescentAnnexin V by using FITC–Annexin V binding assay (Immuno-tech) [19]. Cell counting was used to further corroborate that[3H]-thymidine uptake correlated with cell growth. Viable cellswere counted with Trypan Blue dye.
Western blot analysis
Lysates were prepared from C4HD or T47D cells subjected tothe different treatments as described in each experiment. Cellswere lysed in a buffer containing 50mM Tris (pH 7.4), 150mMNaCl, 1mM EDTA, 1mM EGTA, 10% glycerol, 1% Nonidet P-40,0.1% SDS, 1mM Mg2Cl, 1mM phenylmethylsulfonylfluoride(PMSF), 10μg/ml leupeptin, 5μg/ml pepstatin, 5μg/ml aprotinin,1mM sodium orthovanadate and 25mM NaF. Lysates werecentrifuged at 12,000×g for 30min at 4 °C and protein content inthe supernatant was determined using a Bio-Rad kit (Rich-mond, CA). Protein lysates from C4HD tumors growing inmicesubjected to different treatments were prepared as previouslydescribed [18]. Proteins were solubilized in sample buffer(60mM Tris–HCl, pH 6.8, 2% SDS, 10% glycerol, 0.7mM, 2β-
Fig. 2 – Proliferative effect of TNFα requires p42/p44 MAPK, JNKtreated between 0 and 60 min in DMEM+0.1% ChFCS supplemenfive μg protein (for p42/p44 MAPK) and 50 μg protein (for p38 MAdescribed in Materials andmethods, were electrophoresed and im(first panels), anti-phospho-p38 MAPK antibody (third panels), anserine 473-Akt antibody (seventh panels). Membranes were thenkinase (second, forth, sixth and eighth panels). The relative activgraphically represented in a bar plot. Values are expressed as thkinases vs. total kinases (AU). Data are presented as mean±SE ocells were pretreated for 30min eitherwith 10μMPD98059, 10μMeither left untreated or treatedwith 20 ng/mlmTNFα or hTNFα reFig. 1. The right panels of (B) and (C) show representative Westeexperiment shown is representative of a total of three with simi
Mercaptoethanol and 0.01% bromophenol blue) and subjectedto SDS-PAGE. Proteins were electroblotted onto nitrocellulose.Membranes were immunoblotted with the following antibo-dies: p42/p44 MAPK (C-14), phospho-p42/p44 MAPK (E-4), totalp38 (A-12), phospho p38 (D-8), JNK (N-18), phospho JNK (G-7),cyclin D1(72-13G), and Bcl-xL (S-18), all from Santa Cruz Bio-technology (Santa Cruz, CA), Akt, and phospho Akt (Ser 473),phospho IκBα (Ser32/36), IκBα from Cell Signaling (Beverly,MA), actin (Clone ACTN05) from Neomarkers (Freemont, CA)and TNFR1 or TNFR2 [24]. After washing, membranes wereincubated with HRP-conjugated secondary antibody (VectorLaboratories, Burlingame, CA). Enhanced chemiluminescence(ECL) was performed according to the manufacturer's instruc-tions (Amersham, Buckinghamshire, England).
Transient transfections
In experiments assessing TNFα-induced transcriptional acti-vation of NF-κB, we used a κB-Luc vector (κB sites from HIVpromoter), kindly provided by Dr. M. Bell (Mayo Clinic, Ro-chester, MN). C4HD cells were transfected for 24h in DMEMsupplemented with 10nM MPA and 2.5% ChFCS, and T47D inDMEM+10% FCS without antibiotics. FuGENE 6 transfectionreagent technique (Roche Biochemicals, IN) was used in accor-dance to the manufacturer's instructions. C4HD and T47Dcells were transiently co-transfectedwith 1μg of κB-Luc vector,and 10ng renilla luciferase expression vector CMV-pRL (Pro-mega, Madison, WI) to correct variations in transfectionefficiency. As control, cells were also transfected with apGL3-basic reporter lacking κB. Cells were starved for 24h inmedium supplemented with 0.1% ChFCS and pretreated for30min with SP600125, PD98059, LY294002, Bay 11-7082 orcontrol DMSO and were then either left untreated or treatedwith TNFα or muteins for 18h. We also assessed the roles ofp42/p44MAPK, JNK, Akt and IκBα on NF-κB activation usinginterfering mutants. For that purpose we co-transfected C4HDand T47D with 0.5μg pCDNA Raf 301, to block Raf/MEK/p42/p44MAPK pathway and with 1μg of the pEBG SEK kr (contain-ing a Lys 129 to Arg mutation of SEK) to block JNK, kindlyprovided by Dr. O. Coso (University of Buenos Aires, Argen-tina), together with κB-Luc vector and CMV-pRL. We also used0.5μg green fluorescent protein dominant negative of p85subunit of PI-3K (Δp85), kindly provided by Dr. Y. Shimizu(University of Minnesota, MN) and 0.5μg super-repressor IκB(ssIκB) that contains mutated sites at Ser32 and Ser36 which
and PI3-K/Akt activation. (A) C4HD and T47D cells wereted with 20 ng/ml of mTNFα or hTNFα respectively. TwentyPK, JNK and Akt) from whole-cell lysates, obtained asmunoblotted with an anti-phospho-p42/p44MAPK antibodyti-phospho JNK antibody (fifth panels) and anti-phosphostripped and hybridized with antibodies against each totalation of each kinase was assessed from each blot and ise ratio of arbitrary densitometric units of phosphorylatedf three experiments. In addition, (B) C4HD cells and (C) T47D(C4HD) or 5μM (T74D) SP600125 or 2μMLY294002, and then
spectively for 48 h. Proliferationwasmeasured as described inrn blots of inhibitor effectiveness of each pathway. Thelar results. b vs. a, P<0.001.
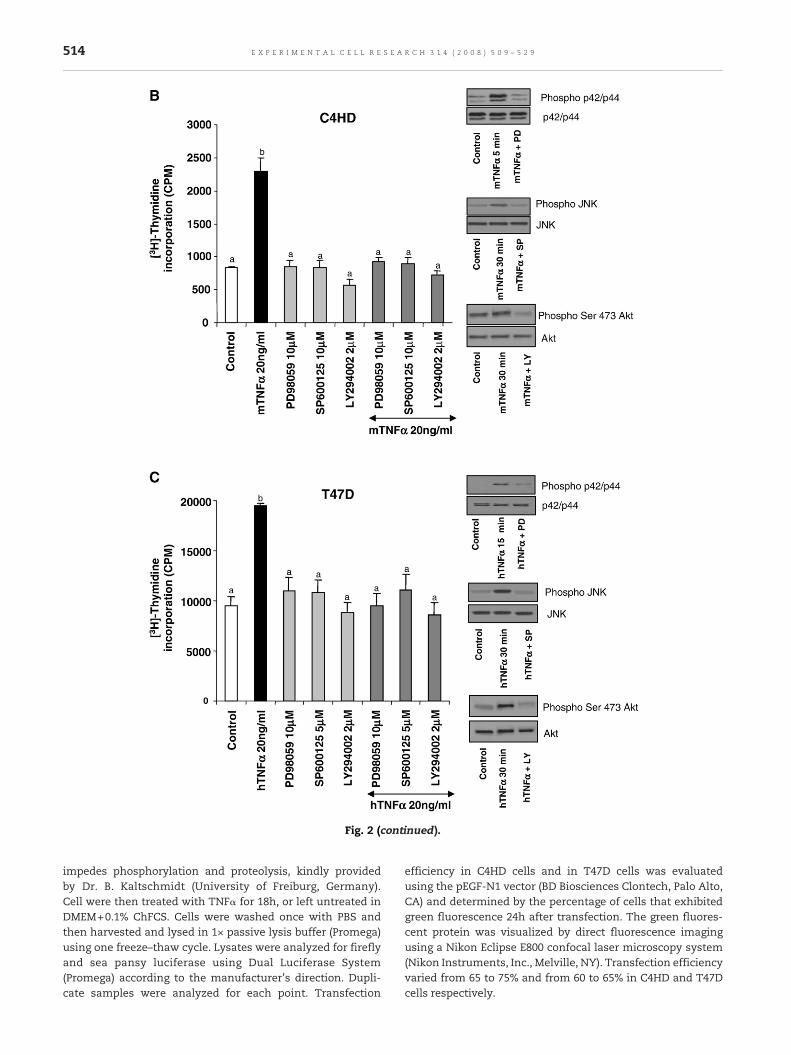
Fig. 2 (continued).
514 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 4 ( 2 0 0 8 ) 5 0 9 – 5 2 9
impedes phosphorylation and proteolysis, kindly providedby Dr. B. Kaltschmidt (University of Freiburg, Germany).Cell were then treated with TNFα for 18h, or left untreated inDMEM+0.1% ChFCS. Cells were washed once with PBS andthen harvested and lysed in 1× passive lysis buffer (Promega)using one freeze–thaw cycle. Lysates were analyzed for fireflyand sea pansy luciferase using Dual Luciferase System(Promega) according to the manufacturer's direction. Dupli-cate samples were analyzed for each point. Transfection
efficiency in C4HD cells and in T47D cells was evaluatedusing the pEGF-N1 vector (BD Biosciences Clontech, Palo Alto,CA) and determined by the percentage of cells that exhibitedgreen fluorescence 24h after transfection. The green fluores-cent protein was visualized by direct fluorescence imagingusing a Nikon Eclipse E800 confocal laser microscopy system(Nikon Instruments, Inc., Melville, NY). Transfection efficiencyvaried from 65 to 75% and from 60 to 65% in C4HD and T47Dcells respectively.

Fig. 3 – TNFR1 and TNFR2 differentially induce p42/p44 MAPK, JNK, Akt activation and proliferation in C4HD cells. (A) and (B)Expression of TNFR1 and TNFR2 in C4HD cells. (A) A total of 50 μg protein from C4HD cells or mouse thymocytes lysateswas electrophoresed and immunoblottedwith TNFR1 or TNFR2 antibodies. Membraneswere stripped and immunoblottedwithanti-actin antibody as loading control. (B) C4HD cells were stained by indirect immunofluorescence using anti-TNFR1 oranti-TNFR2 as first antibodies, and FITC-conjugated antibodies as second ones, and fluorescence was analyzed by flowcytometry. Representative histograms of TNFR1 and TNFR2 expression are shown. Gray areas define the TNFR expressionsuperimposed over histograms of cells stained with an irrelevant isotype-matched antibody (white area). The experimentsshown in (A) and (B) are representative of a total of three. (C) C4HD cells were preincubated for 30min in the absence or presenceof 5 μg/ml blocking antibodies anti-TNFR1 or anti-TNFR2 or control isotype antibody, and then treated for 5 min or 15 min withTNFα to perform Western blots for p42/p44 MAPK or JNK and Akt respectively or (D) cells were also treated with murine orhuman TNFα or with muteins specific to TNFR1 (R1-TNF, 20 ng/ml), to TNFR2 (R2-TNF, 200 ng/ml) or to both receptors(R1R2-TNF, 20 ng/ml). Proteins from the cell lysates were electrophoresed and immunoblotted with an anti-phospho-p42/p44MAPK antibody (first panel), an anti-phospho JNK antibody (third panel) and an anti-phospho-Ser 473-Akt antibody (fifth panel)as described in Fig. 2A. Experiments shown are representative of four performed. To determine proliferation, cells werepreincubated with 5 μg/ml blocking antibodies anti-TNFR1 or anti-TNFR2 for 30 min prior to TNFα treatment, after which astandard 3H-thymidine incorporation assay was performed. (E) Control isotype antibodies were also assessed under the samecondition. Data shown are mean±SE of three experiments. Significance for b vs. a, P<0.001. (F) Cells were also treated withdifferent concentrations of TNFαmuteins R1-TNF, R2-TNF or R1R2-TNF, 20 ng/ml wild-type mTNFα or hTNF. Proliferation wasmeasured as described in Fig. 1. The experiment shown is representative of the three performed. Significance for b vs. a P<0.01;c vs. a P<0.05; d vs. a P<0.001.
515E X P E R I M E N T A L C E L L R E S E A R C H 3 1 4 ( 2 0 0 8 ) 5 0 9 – 5 2 9

Fig. 3 (continued).
516 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 4 ( 2 0 0 8 ) 5 0 9 – 5 2 9
In vivo C4HD tumor growth
Micewere inoculated subcutaneously (s.c.) into the left flankwitha fragment of C4HD tumor (1mm3) and were injected either s.c.with 10ng mTNFα/day/mouse in 50μl of PBS, or with 50μl of PBS/day/mouse, in all cases in the absence of MPA depot. Animalswere carefully monitored, and body weight was measuredweekly. Tumor width (W) and length (L) were measured with aVernier caliper three times aweek and tumor volume (mm3)wascalculated as (L × W2)/2. At day 16, animals treated with TNFαwere separated at random in four experimental groups whentumors reached a mean volume of 60–70mm3, and treated asfollows: 1) 10ngmTNFα/day/mouse, 2) 10ngmTNFα/day/mouse+5mg/kg Bay 11-7082 on days 16, 19 and 21, 3) 10ng mTNFα/day/mouse + vehicle on days 16, 19 and 21, or 4) PBS 50μl/day/mouse.Bay 11-7082 was injected intraperitoneally and dissolved in 0.5%methyl cellulose (200μl/mouse). Tumor growth rates weredetermined as the slopes of growth curves. At day 26, animalswere euthanized and tumors were removed. Tissues to be usedformolecular studieswerestoredat−80 °Candtissues for routineH&E histopathological analysis were fixed in 10% bufferedformalin. Samples of liver, lung, spleen and kidney were alsotaken for histological examination. All samples were examinedby an experienced pathologist who was blinded to therapy.Several slides were examined from each tumor and organs.
Immunohistochemistry
Formalin-fixedparaffin-embedded tissueswere cut to 4μm,andsections were deparaffinized in xylene and graded alcohols.Stainingwas performedmanually, endogenous peroxidase wasblocked with 0.03% hydrogen peroxide for 5min, and antigenretrievalwas carried outwith 10mMsodiumcitrate buffer pH6.0following the manufacturer's instructions. Expression of p65NF-κB and phospho Akt was studied on consecutive sections ofsamples using antibodies to p65 (C22B4) and Akt (Ser 473), bothfrom Cell Signalling, or a control isotype antibody andwas thendeveloped using the Envision signal detection system (DakoCy-tomation Corp). Sections were then treated with 3,3-diamino-benzidine as chromogen for 1min, and counterstained withhematoxylin. The expression was analyzed blindly and inde-pendently by two pathologists.
Statistical analysis
The differences between control and experimental groups wereanalyzed by ANOVA followed by Tukey t test among groups.Linear regression analysis was performed on tumor growthcurves, and the slopeswere compared usingANOVA to evaluatethe statistical significance of the differences.
Results
TNFα stimulates proliferation of murine C4HD and humanT47D cells
We first examined whether TNFα regulates proliferation ofC4HD murine mammary tumor cells. Primary cultures of C4HDcells were treated with several concentrations of murine TNFα(mTNFα), from 1 to 200ng/ml, after which an increase in cellproliferation that reached a plateau at 10ng/ml TNFα was ob-served (Fig. 1A). As previously reported, TNFα also had proli-ferative activity on thehumancell lineT47D [17]. A similar dose–response curve of human TNFα (hTNFα) on T47D proliferation,with amaximumeffect at 10ng/ml of hTNFα, was also observed(Fig. 1B).
We neutralized TNFα action with Etanercept, a fusion pro-tein between TNFR2 and the Fc fraction of human IgG used toblock TNFα inflammatory activity in humans [26]. Severalconcentrations of Etanercept (0.1–2μg/ml) were preincubatedwith mTNFα or hTNFα prior to addition to C4HD or T47D cellsrespectively. Proliferation assays showed that 0.1μg/ml of Eta-nercept was enough to abolish TNFα-induced proliferation inC4HD and in T47D cells (Figs. 1C and D), suggesting that theeffect was TNFα-specific.
TNFα induces breast cancer cell proliferation throughstimulation of p42/p44 MAPK, JNK and PI3-K/Akt pathways
In order to investigate the contribution of kinase activation inTNFα-induced breast cancer proliferation, we explored themain cellular signaling pathways activated byTNFα. C4HDandT47D cells were incubated along 0–60min with TNFα and ac-tivation of p42/p44 MAPK, p38 MAPK, JNK and Akt wasdetermined by Western blot using antibodies against the

517E X P E R I M E N T A L C E L L R E S E A R C H 3 1 4 ( 2 0 0 8 ) 5 0 9 – 5 2 9
phosphorylated form of these kinases. Fig. 2 shows thatphospho-p42/p44 MAPK were activated within 5min of TNFαtreatment, with activation diminishing at 10min and reachingbasal levels at 30min in C4HD cells (Fig. 2A, first and second leftpanels). TNFα treatment of T47D cells for 15–30min inducedstrong p42/p44MAPKphosphorylationwhich returned to basallevels at 60min (Fig. 2A first and second right panels).
TNFα is known to usually induce activation of the stress-activated protein kinases p38 MAPK and JNK. In our case, asshown in Fig. 2A (third and fourth panels), TNFα did not inducep38 MAPK activation either in C4HD or in T47D cells under thecondition assessed. As control, we stimulated MCF-7 cell linewith hTNFα, where p38 MAPK activation was evident at 15min(14 ± 4 fold of basal levels, not shown). On the other hand, inC4HD and T47D cells, TNFα-induced phosphorylation of JNK-1
Fig. 4 – Blockage of JNK and PI3-K/Akt activities inhibits NF-κB tracells were treated between 0 and 180 min in DMEM+0.1% ChFCSrespectively. Twenty five μg protein from whole-cell lysates, obtelectrophoresed and immunoblotted with an anti-phospho IκBαand anti-actin antibody (third panels). (B) C4HD and T47D cells wLY294002 or Bay 11-7082 before TNFα stimulation during 10 or 3lysates, obtained as described in Materials and methods, was eleIκBα antibody (upper panels) and an anti-IκBα antibody (lowertransfected with 1 μg/well of κB-luciferase construct and with 10control, and then pretreated for 30 min with either PD98059, SP6treated with TNFα during 18 h. Cells were also transfected withwere harvested and lysed. Luciferase and renilla activities werem(G) T47D cells were transiently transfected with 0.5 μg/well of Iκ(a dominant negative of upstream kinase of p42/p44MAPK or JNKwith 1 μg/well κB-luciferase construct and 10 ng/well of CMV-pRstimulated with TNFα for 18 h and underwent the procedures deffectiveness of each construct in C4HD cells. (H) C4HD cells transmuteins R1-TNF, R1R2-TNF (20 ng/ml) or R2-TNF (200 ng/ml) or wpresented as n-fold induction of luciferase activity with respect tData shown represent the mean of six independent experiments
at 15–30minwith residual activity retainedat60min (Fig. 2A fifthand sixth panels). These results demonstrate that in breastcancer cells, TNFα is able to stimulate the stress-activated pro-tein kinase JNK, but not p38 MAPK.
In several models, Akt phosphorylation can be induced byTNFα, suggesting that its activation can account for at leastsome antiapoptotic effects of TNFα [7]. These results promptedus to determine whether Akt was activated by TNFα in C4HDand T47D cells. As shown in Fig. 2A (seventh and eighth leftpanels) a significant increase in Akt (Ser 473) phosphorylationwas detected in C4HD cells between 5 and 30min of TNFαstimulation that declinedat 60min. To ensure that inductionofAkt activity was not unique to C4HD, Akt activity was alsodetermined in T47D cells. Indeed, addition of TNFα stimulatedAkt phosphorylation in T47D, reaching its maximum within
nscriptional activity in breast cancer cells. (A) C4HD and T47Dsupplemented with 20 ng/ml of mTNFα or hTNFαained as described in Materials and methods, wasantibody (first panels), anti-IκBα antibody (second panels)ere pretreated for 30 min either with PD98059, SP600125,0 min, respectively. Twenty five μg protein from whole-cellctrophoresed and immunoblotted with an anti-phosphopanels). (C) C4HD and (D) T47D cells were transientlyng/well of renilla expression vector (CMV-pRL) as internal
00125, LY294002 or Bay 11-7082, and then left untreated ora p-GL3-basic reporter lacking κB insertion. In all cases, cellseasured as described inMaterials andmethods. (E) C4HD andBss (an inhibitor of IκB phosphorylation), Raf301, SEK krrespectively), or Δp85 (a dominant negative of PI3-K) togetherL expression vector as internal control. Cells wereescribed in (C). (F) Representative Western blot of inhibitorfected with the κB-luc construct were stimulated with TNFαith 20 ng/ml of wild-type murine or human TNFα. Results areo control cells growing in the absence of TNFα stimulation.±SE. b vs. a, P<0.001.

Fig. 4 (continued).
518 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 4 ( 2 0 0 8 ) 5 0 9 – 5 2 9
10–15min and returning to basal levels after 60min (Fig. 2Aseventh and eighth right panels). Densitometric quantificationof kinases activation is shown in Fig. 2A, lower panels.
To dissect individual kinase contribution to the TNFα-mediated proliferative effect, we used specific pharmacologicalinhibitors. Pretreatment of C4HD and T47D cells with PD98059(MEK inhibitor), LY294002 (PI3-K inhibitor) or SP600125 (JNKinhibitor), suppressed proliferation induced by TNFα (Figs. 2BandC respectively) revealing themasessential pathways in saideffect. The right panels of these figures show representativeWestern blots of the effectiveness of the inhibitors.
TNFR1 and TNFR2 differentially induce p42/p44 MAPK, JNKand Akt pathway activation
Biological effects of TNFα are exerted through binding to bothTNFR1 and TNFR2 [2]. To explore the presence of TNFRs in
C4HD cells, we performed Western blot analysis in whole-cellextracts. Murine thymocytes extracts were included as po-sitive controls, since they are known to express both recep-tors, being TNFR2 present in larger amounts. As shown inFig. 3A, C4HD cells had similar levels of TNFR1 to thoseobserved in thymocytes, while TNFR2 expression was lowerthan in thymocytes. TNFR2 revealed two components of thereceptor of 70kDa and 60kDa, similar to those described inL929 cells [27]. Western blot data were supplemented withflow cytometry analysis of cell surface expression of bothreceptors after staining with anti-TNFR1 or anti-TNFR2 anti-bodies. Fig. 3B shows approximately equal expression levels ofboth TNFα receptors in C4HD cells.
To gain further insight into the contribution of TNFRs in theactivation of p42/p44 MAPK, JNK and Akt pathways in C4HDcells, we used blocking antibodies to TNFRs as well as TNFαmuteins. These tools were specifically designed for murine
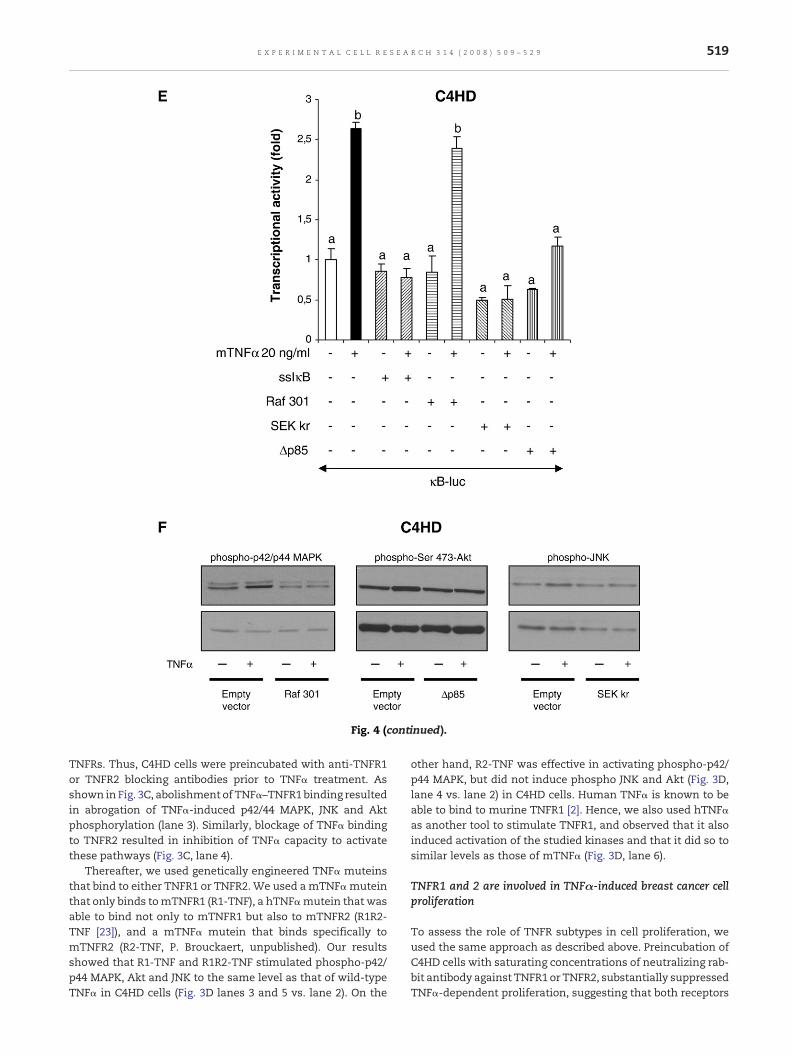
Fig. 4 (continued).
519E X P E R I M E N T A L C E L L R E S E A R C H 3 1 4 ( 2 0 0 8 ) 5 0 9 – 5 2 9
TNFRs. Thus, C4HD cells were preincubated with anti-TNFR1or TNFR2 blocking antibodies prior to TNFα treatment. Asshown in Fig. 3C, abolishment of TNFα–TNFR1binding resultedin abrogation of TNFα-induced p42/44 MAPK, JNK and Aktphosphorylation (lane 3). Similarly, blockage of TNFα bindingto TNFR2 resulted in inhibition of TNFα capacity to activatethese pathways (Fig. 3C, lane 4).
Thereafter, we used genetically engineered TNFα muteinsthat bind to either TNFR1 or TNFR2. We used amTNFαmuteinthat only binds tomTNFR1 (R1-TNF), a hTNFαmutein that wasable to bind not only to mTNFR1 but also to mTNFR2 (R1R2-TNF [23]), and a mTNFα mutein that binds specifically tomTNFR2 (R2-TNF, P. Brouckaert, unpublished). Our resultsshowed that R1-TNF and R1R2-TNF stimulated phospho-p42/p44 MAPK, Akt and JNK to the same level as that of wild-typeTNFα in C4HD cells (Fig. 3D lanes 3 and 5 vs. lane 2). On the
other hand, R2-TNF was effective in activating phospho-p42/p44 MAPK, but did not induce phospho JNK and Akt (Fig. 3D,lane 4 vs. lane 2) in C4HD cells. Human TNFα is known to beable to bind to murine TNFR1 [2]. Hence, we also used hTNFαas another tool to stimulate TNFR1, and observed that it alsoinduced activation of the studied kinases and that it did so tosimilar levels as those of mTNFα (Fig. 3D, lane 6).
TNFR1 and 2 are involved in TNFα-induced breast cancer cellproliferation
To assess the role of TNFR subtypes in cell proliferation, weused the same approach as described above. Preincubation ofC4HD cells with saturating concentrations of neutralizing rab-bit antibody against TNFR1 or TNFR2, substantially suppressedTNFα-dependent proliferation, suggesting that both receptors

Fig. 4 (continued).
520 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 4 ( 2 0 0 8 ) 5 0 9 – 5 2 9
play an important role in C4HD proliferation (Fig. 3E). TNFαmuteins at concentrations ranging from 2–200ng/ml, wereadded to C4HD cells and a [3H]-thymidine incorporation assaywasperformed. Proliferation inducedbyR1-TNF resembled theone obtained with wild-type TNFα (Fig. 1A), reaching itsmaximum at 20ng/ml (Fig. 3F). Interestingly, addition of R2-TNF induced C4HD proliferation only at 200ng/ml, though lessthan its wild-type counterpart at 20ng/ml. Finally, R1R2-TNFproved to be the most active in inducing proliferation, with200ng/ml increasing proliferation 50% above that of wild-type
TNFα at the same dose (Fig. 3F). Addition of hTNFα-inducedproliferation of C4HD cells to the same level as that of mTNFα.
TNFα induces NF-κB transcriptional activation mainlythrough TNFR1, JNK and Akt-dependent pathways
NF-κB is a transcription factor usually stimulated by TNFα inseveral cell types. To accomplish NF-κB transcriptional acti-vation through the canonical pathway, cytoplasmic inhibitorIκBαmust be phosphorylated, ubiquitinated anddegradated. To

521E X P E R I M E N T A L C E L L R E S E A R C H 3 1 4 ( 2 0 0 8 ) 5 0 9 – 5 2 9
determine the time course of IκBα phosphorylation and/ordegradation, we treated C4HD and T47D cells along 0–180minwith TNFα. The data revealed that themaximum level of TNFα-induced IκBα phosphorylation occurred at 10min in C4HD, andat 30min in T47D cells (Fig. 4A). Degradation was detected at60min of TNFα stimulation in both cell types (Fig. 4A). Todetermine whether p42/p44 MAPK, JNK and Akt pathways may
Fig. 5 – NF-κB activation is essential for TNFα-induced Bcl-xL ancells. C4HD and T47D cells were pretreated for 30 min with eithetreated with 20 ng/ml mTNFα or hTNFα respectively for 48 h. (Aimmunoblotted with anti-Bcl-xL or anti-cyclin D1 antibodies. Memantibody. The experiment shown is representative of the three pand analyzed for cell cycle distribution by flow cytometry after 3three experiments performed. Mean percentages of total cells inindicated. b vs. a and e vs. d P<0.05, b vs. c P<0.01. Bay concentrcells cultured for 96 h and cell surface Annexin V–FITC bindingwaThe percentage of apoptotic cells (FITC–Annexin V positive cells)the three experiments performed. Bay concentration was 1 μM.exclusion and the number of cells at day 1 was 2×105 cells. Thesimilar results. b vs. a P<0.01, b vs. c P<0.001.(E) Proliferation asshown is representative of the three performed. b vs. a P<0.001.
affect TNFα-inducible phosphorylation, we examined the levelsof phospho IκBα in C4HD and T47D cells pretreated with therespective pharmacological inhibitors and then stimulatedwithTNFα for 10min or 30min respectively. LY294002 and SP600125were able to effectively prevent IκBα phosphorylation, whilePD98059 did not have any effect in any of the cell types (Fig. 4B).For further confirmation, p42/p44 MAPK blockage was also
d cyclin D1 expression and proliferation in breast cancerr Bay 11-7082 or vehicle (DMSO) and then left untreated or) Fifty μg protein from cell lysates was electrophoresed andbranes were then stripped and hybridizedwith an anti-actinerformed. (B) C4HD cells were stained with propidium iodide6 or 48 h treatment. Representative histograms are shown ofthe cell cycle phases±SD of the three experiments areation was 1 μM. (C) Apoptosis assay was performed in C4HDsmeasured by flow cytometry. b vs. a P<0.05, b vs. c P<0.001.is indicated in each histogram as mean percentage±SD of(D) Cell count assay was performed with Trypan blue dyeassay was run in octuplicates and repeated four times withsay was performed as described in Fig. 1. The experiment

Fig. 5 (continued).
522 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 4 ( 2 0 0 8 ) 5 0 9 – 5 2 9
performedwith2μMU0126, aMEK1/MEK2 inhibitor,with similarresults (data not shown). Bay 11-7082, an IκBα phosphorylationinhibitor [28], was included as control.
To investigate TNFα capacity to induce NF-κB transcrip-tional activation in C4HD cells and to further confirm thiseffect in T47D cells, cells were transiently transfected with a
Fig. 6 – TNFα promotes in vivo C4HD growth and can be preventinoculated subcutaneously (s.c) into the left flank with a fragmen10 ngmTNFα/day/mouse, dissolved in PBS (●), or with PBS 50 μlanimals were separated into three experimental groups (n=5/gro10 ng mTNFα/day/mouse+5 mg/kg Bay 11-7082 on days 16, 19 acellulose) on days 16, 19 and 21 (□) or 10 ng mTNFα/day/mouseday 16 was submitted to TNFα withdrawal (▲), is shown in theweek in order to calculate volume as described in Materials andtumors±SD. The experiment was repeated twice with similar reimmunohistochemistry (IHC). Tissue sections of C4HD tumor obdifferent treatments as described in (A). TNFα: Ductal mammaryglandular cells, separated by scanty vascular stroma (H&E ×100).arrowheads at highermagnification (inset, H&E ×400). TNFα+BayIn a non-necrotic area the tumor shows absence of mitosis (insespecimen shows nuclear p65 immunoreactivity in tumor cells. Tp65 staining in tumor cells. IHC for phospho-Ser 473-Akt. TNFα anand cytoplasmic localization of phospho Akt in tumor cells. Tissisotype antibodies (inset), and bound antibodies were visualizedcounterstained with hematoxylin, magnification ×400. (C) Effectdegradation and (D) on Bcl-xL, cyclin D1 expression in C4HD tumofrom tumor lysates was electrophoresed and immunoblotted witand anti-cyclin D1 antibodies. Two representative samples of tuTNFα+Bay11-7082 (lanes 3 and 4) are shown. Membranes wereThe experiment shown is representative of the three performed
κB-luciferase reporter construct and a renilla expression vec-tor as internal control. Treatment of C4HD and T47D cells withTNFα induced a 3- and a 4-fold increase in κB transcriptionalactivation, respectively (Figs. 4C and D). Addition of Bay 11-7082 completely inhibited TNFα-induced activation of the κB-luc reporter plasmid in C4HD and T47D cells (Figs. 4C and D).
ed through blockage of NF-κB activation. (A) Mice weret of C4HD tumor (1 mm3) and were injected either with/day/mouse (○) for the first 16 days. On day 16, TNFα-treatedup) and received respectively the following treatments:nd 21 (■), 10 ng mTNFα/day/mouse+vehicle (200 μl methyl(●). An additional group of TNFα-treated animals, which oninset. Width and length were measured three times amethods. Each point represents the mean volume of 5sults. (B) Histopathological analysis andtained after TNFα administration and then submitted tocarcinoma composed of pseudolobules of highly cohesiveAtypical features and several mitotic figures are indicated by11-7082: Tumor showsnecrosis, apoptosis andhyalinization.t, H&E ×400). IHC analysis for p65. TNFα: C4HD breast cancerNFα+Bay 11-7082: Tumor shows cytoplasmic but no nucleard TNFα+Bay 11-7082: Breast cancer specimens shownuclearue sections were probed with specific antibodies or controlusing immunoperoxidase detection. Sections wereof Bay 11-7082 treatment on IκBα phosphorylation andr obtained after TNFα administration. A total of 50 μg proteinh (C) anti-phospho IκBα and anti-IκBα, or with (D) anti-Bcl-xL
mor from mice treated with TNFα (lanes 1 and 2) andthen stripped and hybridized with an anti-actin antibody..

523E X P E R I M E N T A L C E L L R E S E A R C H 3 1 4 ( 2 0 0 8 ) 5 0 9 – 5 2 9
It is well known that TNFα triggers signaling pathways suchas PI3-K/Akt, that converge on the activation of transcriptionfactorNF-κB [7].However, theparticipationofp42/p44MAPK, andJNKonNF-κB transcriptional regulation remainspoorlyexplored.To investigate signaling pathways that contribute to TNFα-induced transcriptional activation of NF-κB and, correlatingwiththe previous data on IκBα phosphorylation, κB-luc-transfectedcells were pretreated with the specific pharmacological inhibi-tors of p42/p44 MAPK, PI3-K/Akt and JNK pathways, and then
stimulated with TNFα. Addition of SP600125 and LY294002 toC4HD and T47D cells abolished TNFα capacity to activate theκB-luc reporter construct, while PD98059 or U0126 (not shown)partially inhibited TNFα-induced NF-κB transcriptional activa-tion (Figs. 4C and D).
To further confirm the role of p42/p44 MAPK, PI3-K/Akt andJNK pathways on TNFα-induced NF-κB transcriptional activa-tion, we used dominant-negative mutants that interfere withthose pathways. Results show that transfection of C4HD and
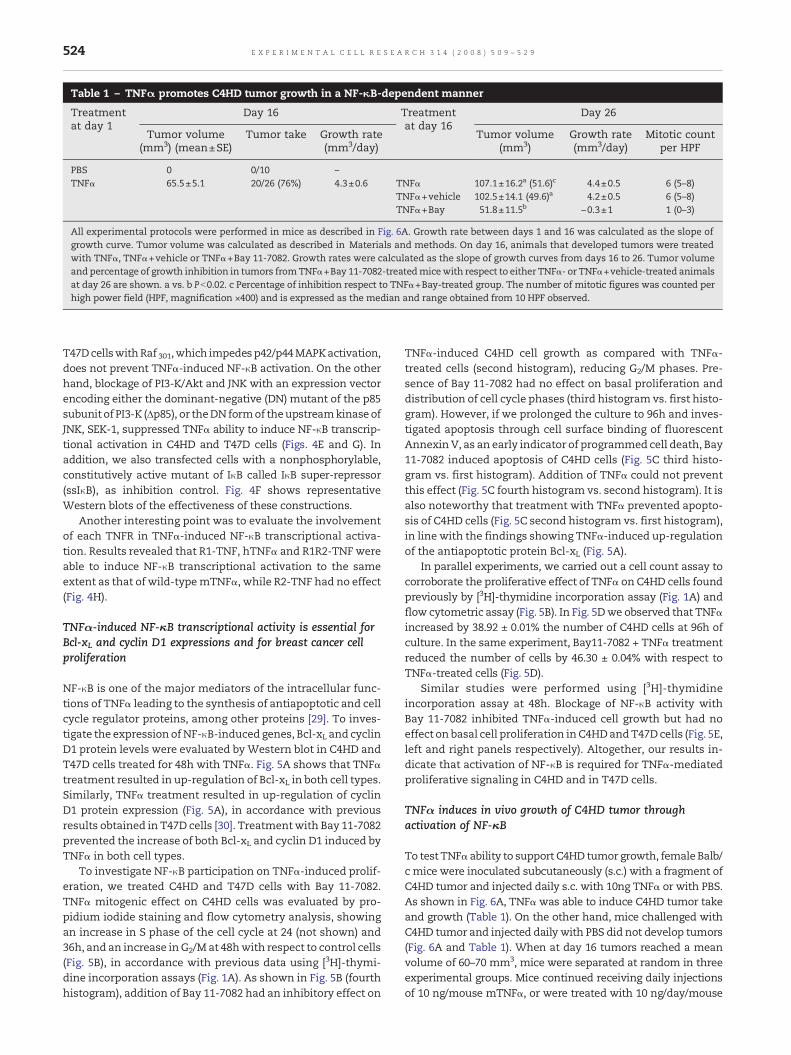
Table 1 – TNFα promotes C4HD tumor growth in a NF-κB-dependent manner
Treatmentat day 1
Day 16 Treatmentat day 16
Day 26
Tumor volume(mm3) (mean±SE)
Tumor take Growth rate(mm3/day)
Tumor volume(mm3)
Growth rate(mm3/day)
Mitotic countper HPF
PBS 0 0/10 –TNFα 65.5±5.1 20/26 (76%) 4.3±0.6 TNFα 107.1±16.2a (51.6)c 4.4±0.5 6 (5–8)
TNFα+vehicle 102.5±14.1 (49.6)a 4.2±0.5 6 (5–8)TNFα+Bay 51.8±11.5b −0.3±1 1 (0–3)
All experimental protocols were performed in mice as described in Fig. 6A. Growth rate between days 1 and 16 was calculated as the slope ofgrowth curve. Tumor volume was calculated as described in Materials and methods. On day 16, animals that developed tumors were treatedwith TNFα, TNFα+vehicle or TNFα+Bay 11-7082. Growth rates were calculated as the slope of growth curves from days 16 to 26. Tumor volumeand percentage of growth inhibition in tumors fromTNFα+Bay 11-7082-treatedmicewith respect to either TNFα- or TNFα+vehicle-treated animalsat day 26 are shown. a vs. b Pb0.02. c Percentage of inhibition respect to TNFα+Bay-treated group. The number of mitotic figures was counted perhigh power field (HPF, magnification ×400) and is expressed as the median and range obtained from 10 HPF observed.
524 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 4 ( 2 0 0 8 ) 5 0 9 – 5 2 9
T47DcellswithRaf 301,which impedesp42/p44MAPKactivation,does not prevent TNFα-induced NF-κB activation. On the otherhand, blockage of PI3-K/Akt and JNK with an expression vectorencoding either the dominant-negative (DN) mutant of the p85subunit of PI3-K (Δp85), or theDNformof theupstreamkinase ofJNK, SEK-1, suppressed TNFα ability to induce NF-κB transcrip-tional activation in C4HD and T47D cells (Figs. 4E and G). Inaddition, we also transfected cells with a nonphosphorylable,constitutively active mutant of IκB called IκB super-repressor(ssIκB), as inhibition control. Fig. 4F shows representativeWestern blots of the effectiveness of these constructions.
Another interesting point was to evaluate the involvementof each TNFR in TNFα-induced NF-κB transcriptional activa-tion. Results revealed that R1-TNF, hTNFα and R1R2-TNF wereable to induce NF-κB transcriptional activation to the sameextent as that of wild-typemTNFα, while R2-TNF had no effect(Fig. 4H).
TNFα-induced NF-κB transcriptional activity is essential forBcl-xL and cyclin D1 expressions and for breast cancer cellproliferation
NF-κB is one of the major mediators of the intracellular func-tions of TNFα leading to the synthesis of antiapoptotic and cellcycle regulator proteins, among other proteins [29]. To inves-tigate the expression ofNF-κB-induced genes, Bcl-xL and cyclinD1 protein levels were evaluated byWestern blot in C4HD andT47D cells treated for 48h with TNFα. Fig. 5A shows that TNFαtreatment resulted in up-regulation of Bcl-xL in both cell types.Similarly, TNFα treatment resulted in up-regulation of cyclinD1 protein expression (Fig. 5A), in accordance with previousresults obtained in T47D cells [30]. Treatmentwith Bay 11-7082prevented the increase of both Bcl-xL and cyclin D1 induced byTNFα in both cell types.
To investigate NF-κB participation on TNFα-induced prolif-eration, we treated C4HD and T47D cells with Bay 11-7082.TNFα mitogenic effect on C4HD cells was evaluated by pro-pidium iodide staining and flow cytometry analysis, showingan increase in S phase of the cell cycle at 24 (not shown) and36h, and an increase inG2/M at 48hwith respect to control cells(Fig. 5B), in accordance with previous data using [3H]-thymi-dine incorporation assays (Fig. 1A). As shown in Fig. 5B (fourthhistogram), addition of Bay 11-7082 had an inhibitory effect on
TNFα-induced C4HD cell growth as compared with TNFα-treated cells (second histogram), reducing G2/M phases. Pre-sence of Bay 11-7082 had no effect on basal proliferation anddistribution of cell cycle phases (third histogram vs. first histo-gram). However, if we prolonged the culture to 96h and inves-tigated apoptosis through cell surface binding of fluorescentAnnexin V, as an early indicator of programmed cell death, Bay11-7082 induced apoptosis of C4HD cells (Fig. 5C third histo-gram vs. first histogram). Addition of TNFα could not preventthis effect (Fig. 5C fourth histogram vs. second histogram). It isalso noteworthy that treatment with TNFα prevented apopto-sis of C4HD cells (Fig. 5C second histogram vs. first histogram),in line with the findings showing TNFα-induced up-regulationof the antiapoptotic protein Bcl-xL (Fig. 5A).
In parallel experiments, we carried out a cell count assay tocorroborate the proliferative effect of TNFα on C4HD cells foundpreviously by [3H]-thymidine incorporation assay (Fig. 1A) andflow cytometric assay (Fig. 5B). In Fig. 5Dwe observed that TNFαincreased by 38.92 ± 0.01% the number of C4HD cells at 96h ofculture. In the same experiment, Bay11-7082 + TNFα treatmentreduced the number of cells by 46.30 ± 0.04% with respect toTNFα-treated cells (Fig. 5D).
Similar studies were performed using [3H]-thymidineincorporation assay at 48h. Blockage of NF-κB activity withBay 11-7082 inhibited TNFα-induced cell growth but had noeffect on basal cell proliferation inC4HDandT47Dcells (Fig. 5E,left and right panels respectively). Altogether, our results in-dicate that activation of NF-κB is required for TNFα-mediatedproliferative signaling in C4HD and in T47D cells.
TNFα induces in vivo growth of C4HD tumor throughactivation of NF-κB
To test TNFα ability to support C4HD tumor growth, femaleBalb/c mice were inoculated subcutaneously (s.c.) with a fragment ofC4HD tumor and injected daily s.c. with 10ng TNFα or with PBS.As shown in Fig. 6A, TNFαwas able to induce C4HD tumor takeand growth (Table 1). On the other hand, mice challenged withC4HD tumor and injected daily with PBS did not develop tumors(Fig. 6A and Table 1). When at day 16 tumors reached a meanvolume of 60–70 mm3, mice were separated at random in threeexperimental groups. Mice continued receiving daily injectionsof 10 ng/mouse mTNFα, or were treated with 10 ng/day/mouse

525E X P E R I M E N T A L C E L L R E S E A R C H 3 1 4 ( 2 0 0 8 ) 5 0 9 – 5 2 9
mTNFα plus 5 mg/kg Bay 11-7082 on days 16, 19 and 21, or with10ng/day/mousemTNFαplus vehicle. The dose of 5mg/kg Bay11-7082 was selected for the experiments because 1 mg/kg ofBay had hardly reduced TNFα-promoted tumor growth (notshown). Growth curves of tumor in the presence of TNFα plusvehicle were not distinguishable from the curve obtainedwith TNFα alone (Fig. 6A). Administration of Bay 11-7082 toC4HD growing with TNFα proved effective to inhibit bothtumor growth rate and tumor volume relative to TNFα orTNFα plus vehicle-treated animals (Fig. 6A and Table 1). Inorder to elucidate the behavior of this tumor promoted byTNFα, on day 16we interrupted TNFα treatment.We observeda progressive and significant regression of the tumor afterTNFαwithdrawal (Fig. 6A, inset). Altogether these data revealthat TNFα allowed C4HD tumor take and that its furtherprogressive growth was TNFα-dependent.
Tumors were excised at day 26 of each protocol and theirhistopathological features were evaluated by hematoxylin–eosin (H&E) staining of histological sections. Fig. 6B showsrepresentative sections of tumors from TNFα and from TNFαplus Bay 11-7082 experimental groups. C4HD tumors grown inmice receiving TNFα were ductal mammary carcinomacomposed of solid pseudolobules of highly cohesive glandularcells that seldom show tubular differentiation, separated byscanty fibroblastic stroma (Fig. 6B). On the other hand, about40–50%of tumormass from tumors grown inmice treatedwithTNFα plus Bay 11-7082 showed necrosis, apoptosis and hya-linization. Moreover, these tumors also showed a significantlylower mitotic index as compared to tumors of TNFα-treatedanimals (Table 1). Histological examination of liver, lung,kidney and spleen did not reveal any pathological change inTNFα treated animals. Only moderate hepatocyte dysplasiawas found in the Bay 11-7082-treated group. No body weightloss was observed in any mice group along the experiment.
As Bay 11-7082 is an inhibitor of IκBα degradation andblocks NF-κB translocation from cytoplasm to nucleus, weinvestigated the subcellular localization of the p65 familymember of NF-κB in breast tumor tissue sections by immu-nohistochemistry (IHC). In C4HD tumors grown in mice re-ceiving TNFα, we observed p65 nuclear staining in nearly allbreast cancer cells, a protein that was not detectable in thestromal cells (Fig. 6B). Tumors grown in mice treated withTNFα plus Bay 11-7082 exhibited only positive cytoplasmicstaining for p65 (Fig. 6B). These data suggest that TNFα wasable in vivo to induce the translocation of p65 to the nucleus ofthe tumoral cells, whereas Bay 11-7082 inhibited this effect.We then explored the phospho-Ser 473-Akt localization inthese tumor specimens. We observed a similar pattern ofphospho Akt revealed by nuclear and cytoplasmic staining intumoral cells from mice treated with TNFα or with TNFα+Bay11-7082, which suggests that phospho Akt was not affected byaddition of the IκB inhibitor Bay 11-7082.
To further establish whether in vivo administration of Bay11-7082 was effective in blocking IκBα degradation in C4HDtumors growing with TNFα, IκBα expression was evaluated inprotein extracts from tumors excised at day 26. Fig. 6C showsthat TNFα ability to induce IκBα phosphorylation and degra-dation in C4HD tumors was inhibited by the administration ofBay 11-7082 (Fig. 6C). Finally, we explored the expression ofNF-κB-regulated proteins Bcl-xL and cyclin D1, in C4HD tumors
growing with TNFα and then treated as described above. Asshown in Fig. 6C, Western blot analysis revealed that Bay 11-7082 administration inhibited the expression of Bcl-xL andcyclin D1 in tumors from mice receiving TNFα (Fig. 6D).
Discussion
In the present study, we analyzedmultiple signal transductionevents generated by TNFα in breast cancer cells, and definedtheir role in cell proliferation. Using mouse C4HD and humanT47D breast cancer cells, we showed that TNFα induces cellcycle progression throughactivation of p42/p44MAPK, JNKandAkt pathways, but not of p38 MAPK. The response of T47D andC4HD cells to TNFα involves transient although strong p42/p44MAPK activation. This pattern of p42/p44MAPK activationwasalso described in mouse fibroblasts, macrophages and intes-tinal epithelial cells [31–33] where its induction is essential forcell growth. In C4HD and T47D cells, p42/p44MAPK activationwas strongerwhen compared to that of JNK, and different fromthe pattern reported in cells where TNFα induces cell death[34]. On the contrary, in cells where TNFα induces apoptosis(i.e. KYM-1, HeLa-TNFR2 and MCF-7 cells) activation of JNK isusually observed to be stronger, sustained in time and to beaccompanied by weak p42/p44 MAPK activation [35,36].Participation of JNK in determining the biological response toTNFα has been elusive. Experiments performed in cells inwhich NF-κB activation was genetically impaired demonstrat-ed that JNK activation promotes TNFα-induced cell death [34].Nonetheless, JNK activity per se is not a simple on–off switchand its level and duration determine its biological function. InNF-κB competent cells, TNFα leads to rapid but transient JNKactivation due to the action of JNK phosphatases, and does notelicit cell death [3]. On the other hand, PI3-K and its target Akthave emerged as critical signaling molecules that regulatemultiple cellular processes, including survival and prolifera-tion in numerous systems [37]. In the course of our experi-ments, TNFα was able to induce JNK and Akt activation onlytransiently. These findings are consistent with results ob-tained in cardiomyocytes and in gastric cancer in which TNFαpromotes growth through the activation of Akt and JNK [38,39].Our results indicate that TNFα activates three signal transduc-tion pathways in breast cancer cells, p42/p44 MAPK, Akt andJNK, eachwith a specific profile, all three of themnecessary forbreast cancer proliferation.
Although there are several reports on TNFR1 and TNFR2signaling by means of various approaches, our report is thefirst to investigate the role of each TNFR in primary cultures ofbreast cancer cells that endogenously express TNFRs subtypeswithout interference of artificial over-expression or knock-outstrategies. Stimulation of TNFR1 with the specific mutein R1-TNF activates p42/p44MAPK, JNK, Akt and NF-κB and inducesproliferation to the same level as that of wild-type TNFα. Onthe other hand, R2-TNF induces p42/p44MAPK activation as itswild-type counterpart, but has no effect on JNK, Akt and NF-κBactivation. Only a high concentration of R2-TNF (200 ng/ml)hardly induced cell proliferation. These differences in kinaseresponses may in part be due to the different adaptor proteinsthat TNFR1 and TNFR2 recruit after TNFα binding. It is knownthat the first protein recruited to TNFR1 is TRADD, which

526 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 4 ( 2 0 0 8 ) 5 0 9 – 5 2 9
recruits TRAF2 with very high affinity [40]. On the other hand,TNFR2 recruits TRAF2 directly. Thismight explainwhy TNFα isa better activator of TNFR1, which uses TRADD as an adaptor,than TNFR2, which does not interact with TRADD. Anotherpossible explanation of the different signaling outcome ofTNFR2, which only stimulates p42/p44 MAPK, would be theevidence produced by Tartaglia et al., who demonstrated thatthe half-life time of TNFα binding to TNFR1 is >3 h, and toTNFR2 is 10 min, measured by exchange rates at cell surface[41]. The results obtained in our work suggest that kinetics ofmutein binding to TNFRs is parallel to kinase activation. Itmaybe argued that themore rapid onset of p42/p44MAPK, reachingits maximum activation at 5 min of TNFα stimulation, allowsmutein R2-TNF to stimulate this pathway. This could provethat prolonged interaction with TNFR with its ligand is neces-sary to fully activate PI3-K/Akt and JNK pathways, as in thecase of mutein R1-TNF. Our present findings demonstrate thatTNFR1 as well as TNFR2 independent ligation with TNFαmuteins results in C4HD cell signaling and proliferation, al-though to a different degree. However, the exact ratio betweenTNFR1 and TNFR2 needed for the same magnitude of signaltransduction is not clear. This may be because differentialreceptor clustering is needed to obtain a downstream signal.
In this studywehaveuseddifferent strategies to blockTNFαaction in breast cancer cell proliferation. The use of blockingantibodies against TNFR1 or TNFR2 in murine C4HD cellseliminated TNFα-induced activation of p42/p44 MAPK, PI3-K/Akt and JNK and cell proliferation. These results indicate thatfor effective TNFα signal transduction and biological effect,both receptors need to remain intact. Moreover, addition ofEtanercept, a TNFR2 fusion protein with human Fc IgG, wasable to neutralize TNFα effect not only inC4HDcells, but also inhuman cell line T47D. Altogether, these data reveal that tar-geting only one of the receptors or the cytokine itself would beuseful to prevent TNFα-mediated breast cancer proliferation.The fact that TNFR2 is only present in certain cells, and thatparticularly high levels were found in in situ breast carcinomas[42], unlike the constitutive expression of TNFR1 in almost anycell type, would have to be taken into account for therapeuticapproaches using antibodies to TNFRs.
NF-κB is one of the major transcription factors stimulatedby TNFα and has a pivotal role in controlling the initiation andprogression of cancer [43]. However, the mechanism by whichTNFRs can activate NF-κB is not fully understood. In ourexperimental model of mouse breast cancer, we observed thatTNFα mutein R1-TNF can fully activate NF-κB transcriptionalactivation, while R2-TNF only activates it poorly. Similarly, McFarlane described that stimulation of NF-κB is mainly throughTNFR1 in human cervical carcinoma cells [44]. Many reportsindicate that TNFR2 alone is insufficient to activate NF-κB[31,45] and that only its over-expression can mediate ligand-induced NF-κB transcriptional activation [46]. We also as-sessed the role of kinase activation in TNFα-induced NF-κBactivation. Blockage of JNK or PI3-K/Akt signaling pathwayseither with pharmacological inhibitors, SP600125 or LY294002,or with dominant-negative vectors, SEK1 kr or Δp85 respec-tively, abolished NF-κB transcriptional activation in TNFα-treated C4HD and T47D cells. Recently, two reports haveaddressed this issue. One demonstrated that TNFα-inducedIL-6 production is regulated by JNK activation of NF-κB in renal
epithelial cells [47]. The other demonstrated that JNK activa-tion is essential for IL-1β-induced ICAM-1 gene expressionthrough the NF-κB pathway in a human pulmonary epithelialcell carcinoma [48]. In both cases, the JNK activation profileresembled the one obtained in T47D and C4HD cells. On theother hand, Akt plays a central role in NF-κB activationbecause it interacts with IKKα upon TNFα stimulation andinduces phosphorylation of threonine 23 [7], and because it isalso involved in phosphorylation of p65 transactivationdomain by the IKK complex [49,50]. In line with our findings,Condorelli et al. demonstrated that Akt and JNK pathways areinvolved in regulating NF-κB and that both pathways arecritical for the hypertrophic effects of TNFα in cardiomyocytes[38]. Furthermore, our data showed that p42/p44 MAPK acti-vation was minimally involved in NF-κB transcriptionalactivation, as blockage with PD98059 or U0126, or with thedominant-negative Raf 301 had little or no effect respectivelyon TNFα-induced NF-κB activity in C4HD and T47D cells. Ourfindings suggest that p42/p44 MAPK activation is essential forTNFα-induced proliferation, although another transcriptionfactor, different from NF-κB, would be involved.
TNFα activation of NF-κB has been associated with at leasttwo aspects of tumorigenesis: the promotion of cancer cellproliferation and the prevention of apoptosis [43]. These in-clude regulation of genes such as cyclin D1 and Bcl-xL, amongothers [8]. It has recently been demonstrated that cyclin D1 is arequirement for breast carcinogenesis [51], and that NF-κBpromotes G1-to-S-phase transition in T47D human mammarycarcinoma cells through regulation of cyclin D1 expression[30,52]. On the other hand, NF-κB is also an inhibitor of pro-grammed cell death. Karin and Lin described that the exactmode by which NF-κB inhibited apoptosis may vary from onecell type to another [29]. In the present work, we found thatTNFα-induced up-regulation of both cyclin D1 and Bcl-xLprotein expression in C4HD and T47D cells was inhibited byBay 11-7082, an IκB inhibitor, demonstrating the involvementof NF-κB in this effect. We also observed that TNFαwas able toinhibit apoptosis of C4HDand to increase cell number, and thatBay 11-7082 blocked both TNF's effects. These findings areconsistent with a mechanism of tumor progression by whichTNFα-induced NF-κB activation contributes to both prolifera-tion and prevention of apoptosis in breast cancer cells. Takenas a whole, our results provided a time course between TNFRsactivation and breast cancer proliferation. The rapid onset ofJNK and PI3-K/Akt activation is necessary for IκB phosphory-lation and degradation (1 h), which results in NF-κB transcrip-tional activation andup-regulation of cyclinD1 andBcl-xL geneexpression (detectable as soon as 4 h and 8 h respectively, datanot shown) and leading to breast cancer proliferation.
One of themost exciting findings of our present work is thatwe were able to demonstrate in vivowhat we had found in vitro:TNFα ability to support breast cancer growth. In fact, weextended our results to show that TNFα is able to induce C4HDtumor take and to support its in vivo growth, events which donot occur when TNFα is absent. Although there are manyreports illustratingTNFα in vivo capacity to promote skin tumors[12,53], few describe this activity in breast cancer. In this regard,Scott et al. described that administration of a neutralizingantibody to TNFα the day before inoculation of 410.4 mammarytumor cells was able to inhibit their in vivo growth [53]. Our

527E X P E R I M E N T A L C E L L R E S E A R C H 3 1 4 ( 2 0 0 8 ) 5 0 9 – 5 2 9
findings indicate that blockage of NF-κB through in vivo ad-ministration of Bay 11-7082 inhibits TNFα-induced C4HD tumorgrowth and prevents up-regulation of Bcl-xL and cyclin D1. Invitro treatment of C4HD cells with Bay 11-7082 and TNFαarrested cells on G0/1 phase at 48 h and induced apoptosis at96 h of in vitro culture. Histological evaluation of tumors fromBay 11-7082 plus TNFα-treated animals revealed a low mitoticindex and a high percentage of necrosis and apoptosis. Ef-fectiveness of Bay 11-7082 inhibition in this group of animalswas confirmed by immunohistochemistry showing that p65could not translocate to the nucleus of the tumoral cells. Thisexperiment indicates that NF-κB blockage in breast cancer cellsin vivo leads to tumor cell arrest and then to death. Althoughtumor regressionwasnot complete after Bay 11-7082 treatment,most of tumor tissue at the end of the experiment showednecrosis, apoptosis and hyalinization and the remaining tu-moral cells were arrested. In vitro data showed that p42/p44MAPK activation is essential for TNFα-induced proliferation butcould involve another transcription factor different fromNF-κB.Therefore p42/p44 MAPK pathway may account for marginaltumor cell survival.
Interestingly, in the present work we demonstrated thatTNFα is able to support growth of C4HD, a tumor which waspreviously reported to be progestin-dependent [25]. Progestinsare known to be involved in controllingmammary tumorigen-esis [54]. Our data could be useful to shed light into possiblecross-talks between progestins and TNFα. In experiments cur-rently being performed in our lab, we studied progesteronereceptor binding by using the single-saturating-dose 3H R5020binding assay, and observed that TNFαwas able to up-regulateprogesterone-binding sites in C4HD cells at 48 h of treatment(data not shown). This suggests that TNFα could sensitizebreast cancer tumor to progestin effect by increasing proges-terone receptor.
In conclusion, to the best of our knowledge, we have for thefirst time demonstrated that an established solid tumor can beinhibited by blockage of NF-κB through administration of aspecific pharmacological inhibitor. Previous studies on theinhibition of NF-κB activity in vivo have used transfected cellswith a non-degradable IκB mutant, which prevents nucleartranslocation of NF-κB [55]. Another widespread strategy hasbeen to administrate selective inhibitors of NF-κB at the sametime as tumor challenge or a few days afterwards [56,57]. It isworth mentioning that in the model here described, we treatedan established breast tumor resembling the clinical condition,and that administration of a NF-κB pharmacological inhibitor iseasier and more efficient than gene transfer in vivo. Taking intoaccount that constitutively expressed NF-κB has been found tocorrelatewith a higher likelihood ofmetastasis andworse prog-nosis for patients with breast cancer [58], small molecule inhib-itors of NF-κB would be useful as adjuvant for existing therapy,to more efficiently and selectively destroy malignant cells.
Acknowledgments
The authors wish to thank Dr. Alfredo A. Molinolo (NIH,Bethesda, MD) for his constant help and support, Dr. Elisa Balde Kier Joffé, Dr. Eduardo Arzt and Dr. Omar Coso (Universityof Buenos Aires, Buenos Aires, Argentina), and Dr. W. A.
Buurman (Maastricht University, Maastricht, The Nether-lands), for their helpful discussions and critical reading ofthe manuscript. This work was supported by grants IDB 201/OC-AR PICT 2002 05-11055 and IDB 1728/OC-AR PICT 2004 05-25301, both from the National Agency of Scientific Promotionof Argentina, by grant PIP 5391 from the Argentine NationalCouncil of Scientific Research (CONICET) and by Oncomed-Reno CONICET 1819/03, from the Henry Moore Institute ofArgentina. PB is supported by the FWO-Vlaanderen.
R E F E R E N C E S
[1] S. Wu, C.M. Boyer, R.S. Whitaker, A. Berchuck, J.R. Wiener, J.B.Weinberg, R.C. Bast Jr., Tumor necrosis factor alpha asan autocrine and paracrine growth factor for ovarian cancer:monokine induction of tumor cell proliferation and tumornecrosis factor alpha expression, Cancer Res. 53 (1993)1939–1944.
[2] J. Rothe, G. Gehr, H. Loetscher, W. Lesslauer, Tumor necrosisfactor receptors — structure and function, Immunol. Res. 11(1992) 81–90.
[3] Z.G. Liu, H. Hsu, D.V. Goeddel, M. Karin, Dissection of TNFreceptor 1 effector functions: JNK activation is not linked toapoptosis while NF-kappaB activation prevents cell death,Cell 87 (1996) 565–576.
[4] M. Rothe, S.C.Wong,W.J. Henzel, D.V. Goeddel, A novel familyof putative signal transducers associated with thecytoplasmic domain of the 75 kDa tumor necrosis factorreceptor, Cell 78 (1994) 681–692.
[5] J. Yang, Y. Lin, Z. Guo, J. Cheng, J. Huang, L. Deng, W. Liao, Z.Chen, Z. Liu, B. Su, The essential role ofMEKK3 inTNF-inducedNF-kappaB activation, Nat. Immunol. 2 (2001) 620–624.
[6] H. Hsu, J. Xiong, D.V. Goeddel, The TNF receptor 1-associatedprotein TRADD signals cell death and NF-kappa Bactivation, Cell 81 (1995) 495–504.
[7] O.N. Ozes, L.D. Mayo, J.A. Gustin, S.R. Pfeffer, L.M. Pfeffer, D.B.Donner, NF-kappaB activation by tumour necrosis factorrequires the Akt serine–threonine kinase, Nature 401 (1999)82–85.
[8] M. Karin, Nuclear factor-kappaB in cancer development andprogression, Nature 441 (2006) 431–436.
[9] J.L. Pomerantz, D. Baltimore, Two pathways to NF-kappaB,Mol. Cell 10 (2002) 693–695.
[10] P.P. Lee, J.J. Hwang, G. Murphy, M.M. Ip, Functionalsignificance of MMP-9 in tumor necrosis factor-inducedproliferation and branching morphogenesis of mammaryepithelial cells, Endocrinology 141 (2000) 3764–3773.
[11] L.M. Varela, M.M. Ip, Tumor necrosis factor-alpha: amultifunctional regulator of mammary gland development,Endocrinology 137 (1996) 4915–4924.
[12] M. Suganuma, S. Okabe, M.W. Marino, A. Sakai, E. Sueoka, H.Fujiki, Essential role of tumor necrosis factor alpha(TNF-alpha) in tumor promotion as revealed byTNF-alpha-deficient mice, Cancer Res. 59 (1999) 4516–4518.
[13] M.E. Burow, C.B.Weldon, Y. Tang, G.L. Navar, S. Krajewski, J.C.Reed, T.G. Hammond, S. Clejan, B.S. Beckman, Differences insusceptibility to tumor necrosis factor alpha-inducedapoptosis among MCF-7 breast cancer cell variants, CancerRes. 58 (1998) 4940–4946.
[14] B.P. Zhou, M.C. Hu, S.A. Miller, Z. Yu, W. Xia, S.Y. Lin, M.C.Hung, HER-2/neu blocks tumor necrosis factor-inducedapoptosis via the Akt/NF-kappaB pathway, J. Biol. Chem. 275(2000) 8027–8031.
[15] C.B. Weldon, A.P. Parker, D. Patten, S. Elliott, Y. Tang, D.E.Frigo, C.M. Dugan, E.L. Coakley, N.N. Butler, J.L. Clayton, J.

528 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 4 ( 2 0 0 8 ) 5 0 9 – 5 2 9
Alam, T.J. Curiel, B.S. Beckman, B.M. Jaffe, M.E. Burow,Sensitization of apoptotically-resistant breast carcinomacells to TNF and TRAIL by inhibition of p38 mitogen-activatedprotein kinase signaling, Int. J. Oncol. 24 (2004) 1473–1480.
[16] L.M. Varela, N.C. Stangle-Castor, S.F. Shoemaker, W.K.Shea-Eaton, M.M. Ip, TNFalpha induces NFkappaB/p50 inassociation with the growth and morphogenesis of normaland transformed ratmammary epithelial cells, J. Cell. Physiol.188 (2001) 120–131.
[17] G. Pirianov, K.W. Colston, Interactions of vitamin D analogueCB1093, TNFalpha and ceramide on breast cancer cellapoptosis, Mol. Cell. Endocrinol. 172 (2001) 69–78.
[18] M. Salatino, R. Schillaci, C.J. Proietti, R. Carnevale, I. Frahm,A.A.Molinolo, A. Iribarren, E.H. Charreau, P.V. Elizalde,Inhibition of in vivo breast cancer growth by antisenseoligodeoxynucleotides to type I insulin-like growth factorreceptor mRNA involves inactivation of ErbBs, PI-3K/Akt andp42/p44 MAPK signaling pathways but not modulation ofprogesterone receptor activity, Oncogene 23 (2004) 5161–5174.
[19] C. Proietti, M. Salatino, C. Rosemblit, R. Carnevale, A. Pecci, A.R.Kornblihtt, A.A. Molinolo, I. Frahm, E.H. Charreau, R.Schillaci, P.V. Elizalde, Progestins induce transcriptionalactivation of signal transducer and activator of transcription 3(Stat3) via a Jak- and Src-dependent mechanism in breastcancer cells, Mol. Cell. Biol. 25 (2005) 4826–4840.
[20] C. Lanari, A.A. Molinolo, C.D. Pasqualini, Induction ofmammary adenocarcinomas by medroxyprogesteroneacetate in BALB/c female mice, Cancer Lett. 33 (1986) 215–223.
[21] M. Salatino, L. Labriola, R. Schillaci, E.H. Charreau, P.V.Elizalde, Mechanisms of cell cycle arrest in response toTGF-beta in progestin-dependent and -independent growthof mammary tumors, Exp. Cell Res. 265 (2001) 152–166.
[22] I.S. Mathiasen, C.M. Hansen, L. Foghsgaard, M. Jaattela,Sensitization to TNF-induced apoptosis by 1,25-dihydroxyvitamin D(3) involves up-regulation of the TNF receptor 1 andcathepsin B, Int. J. Cancer 93 (2001) 224–231.
[23] P. Ameloot, W. Fiers, P. De Bleser, C.F. Ware, P. Vandenabeele,P. Brouckaert, Identification of tumor necrosis factor (TNF)amino acids crucial for binding to the murine p75 TNFreceptor and construction of receptor-selective mutants,J. Biol. Chem. 276 (2001) 37426–37430.
[24] M.A. Dentener, F.T. Smit, G.J. Francot, W.A. Buurman,Characterization of two monoclonal antibodies directedagainst bactericidal/permeability-increasing protein, J. Infect.Dis. 170 (1994) 1483–1489.
[25] G. Dran, I.A. Luthy, A.A. Molinolo, F. Montecchia, E.H.Charreau, C.D. Pasqualini, C. Lanari, Effect ofmedroxyprogesterone acetate (MPA) and serum factors oncell proliferation in primary cultures of an MPA-inducedmammary adenocarcinoma, Breast Cancer Res. Treat.35 (1995) 173–186.
[26] P.T. van der, S.M. Coyle, M. Levi, P.M. Jansen, M. Dentener, K.Barbosa, W.A. Buurman, C.E. Hack, J.W. ten Cate, J.M.Agosti, S.F. Lowry, Effect of a recombinant dimeric tumornecrosis factor receptor on inflammatory responses tointravenous endotoxin in normal humans, Blood 89 (1997)3727–3734.
[27] K.C. Sheehan, J.K. Pinckard, C.D. Arthur, L.P. Dehner, D.V.Goeddel, R.D. Schreiber, Monoclonal antibodies specific formurine p55 and p75 tumor necrosis factor receptors:identification of a novel in vivo role for p75, J. Exp. Med.181 (1995) 607–617.
[28] J.W. Pierce, R. Schoenleber, G. Jesmok, J. Best, S.A. Moore, T.Collins, M.E. Gerritsen, Novel inhibitors of cytokine-inducedIkappaBalpha phosphorylation and endothelial celladhesion molecule expression show anti-inflammatoryeffects in vivo, J. Biol. Chem. 272 (1997) 21096–21103.
[29] M. Karin, A. Lin, NF-kappaB at the crossroads of life anddeath, Nat. Immunol. 3 (2002) 221–227.
[30] M.F. Rubio, S. Werbajh, E.G. Cafferata, A. Quaglino, G.P.Colo, I.M. Nojek, E.C. Kordon, V.E. Nahmod, M.A. Costas,TNF-alpha enhances estrogen-induced cell proliferation ofestrogen-dependent breast tumor cells through a complexcontaining nuclear factor-kappa B, Oncogene 25 (2006)1367–1377.
[31] A. Kalb, H. Bluethmann, M.W. Moore, W. Lesslauer, Tumornecrosis factor receptors (Tnfr) in mouse fibroblasts deficientin Tnfr1 or Tnfr2 are signaling competent and activate themitogen-activated protein kinase pathway with differentialkinetics, J. Biol. Chem. 271 (1996) 28097–28104.
[32] A. Mukhopadhyay, J. Suttles, R.D. Stout, B.B. Aggarwal,Genetic deletion of the tumor necrosis factor receptor p60 orp80 abrogates ligand-mediated activation of nuclearfactor-kappa B and of mitogen-activated protein kinases inmacrophages, J. Biol. Chem. 276 (2001) 31906–31912.
[33] G.C. Kaiser, F. Yan, D.B. Polk, Conversion of TNF alpha fromantiproliferative to proliferative ligand in mouse intestinalepithelial cells by regulating mitogen-activated proteinkinase, Exp. Cell Res. 249 (1999) 349–358.
[34] G. Tang, Y. Minemoto, B. Dibling, N.H. Purcell, Z. Li, M. Karin,A. Lin, Inhibition of JNK activation through NF-kappaB targetgenes, Nature 414 (2001) 313–317.
[35] O.J. Jupp, S.M. McFarlane, H.M. Anderson, A.F. Littlejohn, A.A.Mohamed, R.H. MacKay, P. Vandenabeele, D.J. MacEwan, TypeII tumour necrosis factor-alpha receptor (TNFR2) activatesc-Jun N-terminal kinase (JNK) but not mitogen-activatedprotein kinase (MAPK) or p38 MAPK pathways, Biochem. J. 359(2001) 525–535.
[36] F. Tang, G. Tang, J. Xiang, Q. Dai, M.R. Rosner, A. Lin, Theabsence of NF-kappaB-mediated inhibition of c-JunN-terminal kinase activation contributes to tumor necrosisfactor alpha-induced apoptosis, Mol. Cell. Biol. 22 (2002)8571–8579.
[37] A. Kauffmann-Zeh, P. Rodriguez-Viciana, E. Ulrich, C. Gilbert,P. Coffer, J. Downward, G. Evan, Suppression ofc-Myc-induced apoptosis by Ras signalling through PI(3)K andPKB, Nature 385 (1997) 544–548.
[38] G. Condorelli, C. Morisco, M.V. Latronico, P.P. Claudio, P. Dent,P. Tsichlis, G. Condorelli, G. Frati, A. Drusco, C.M. Croce, C.Napoli, TNF-alpha signal transduction in rat neonatal cardiacmyocytes: definition of pathways generating from theTNF-alpha receptor, FASEB J. 16 (2002) 1732–1737.
[39] S.J. Park, Y.Y. Kim, J.W. Ju, B.G. Han, S.I. Park, B.J. Park,Alternative splicing variants of c-FLIP transduce thedifferential signal through the Raf or TRAF2 in TNF-inducedcell proliferation, Biochem. Biophys. Res. Commun. 289 (2001)1205–1210.
[40] Y.C. Park, H. Ye, C. Hsia, D. Segal, R.L. Rich, H.C. Liou, D.G.Myszka, H. Wu, A novel mechanism of TRAF signalingrevealed by structural and functional analyses of theTRADD–TRAF2 interaction, Cell 101 (2000) 777–787.
[41] L.A. Tartaglia, D. Pennica, D.V. Goeddel, Ligand passing: the75-kDa tumor necrosis factor (TNF) receptor recruits TNF forsignaling by the 55-kDa TNF receptor, J. Biol. Chem. 268 (1993)18542–18548.
[42] I. Garcia-Tunon, M. Ricote, A. Ruiz, B. Fraile, R. Paniagua, M.Royuela, Role of tumor necrosis factor-alpha and its receptorsin human benign breast lesions and tumors (in situ andinfiltrative), Cancer Sci. 97 (2006) 1044–1049.
[43] D.S. Basseres, A.S. Baldwin, Nuclear factor-kappaB andinhibitor of kappaB kinase pathways in oncogenic initiationand progression, Oncogene 25 (2006) 6817–6830.
[44] S.M. McFarlane, G. Pashmi, M.C. Connell, A.F. Littlejohn, S.J.Tucker, P. Vandenabeele, D.J. MacEwan, Differentialactivation of nuclear factor-kappaB by tumour necrosis factor receptor subtypes. TNFR1 predominates whereasTNFR2 activates transcription poorly, FEBS Lett. 515 (2002)119–126.

529E X P E R I M E N T A L C E L L R E S E A R C H 3 1 4 ( 2 0 0 8 ) 5 0 9 – 5 2 9
[45] G.B. Chainy, S. Singh, U. Raju, B.B. Aggarwal, Differentialactivation of the nuclear factor-kappa B by TNF muteinsspecific for the p60 and p80 TNF receptors, J. Immunol. 157(1996) 2410–2417.
[46] V. Haridas, B.G. Darnay, K. Natarajan, R. Heller, B.B. Aggarwal,Overexpressionof thep80TNF receptor leads toTNF-dependentapoptosis, nuclear factor-kappa B activation, and c-Jun kinaseactivation, J. Immunol. 160 (1998) 3152–3162.
[47] S. de Haij, A.C. Bakker, R.N. van der Geest, G. Haegeman, W.Vanden Berghe, J. Aarbiou, M.R. Daha, C. van Kooten,NF-kappaB mediated IL-6 production by renal epithelial cellsis regulated by c-jun NH2-terminal kinase, J. Am. Soc.Nephrol. 16 (2005) 1603–1611.
[48] F.S. Lin, C.C. Lin, C.S. Chien, S.F. Luo, C.M. Yang, Involvementof p42/p44 MAPK, JNK, and NF-kappaB in IL-1beta-inducedICAM-1 expression in human pulmonary epithelial cells,J. Cell Physiol 202 (2005) 464–473.
[49] L.V. Madrid, M.W. Mayo, J.Y. Reuther, A.S. Baldwin Jr., Aktstimulates the transactivation potential of the RelA/p65Subunit of NF-kappa B through utilization of the Ikappa Bkinase and activation of themitogen-activated protein kinasep38, J. Biol. Chem. 276 (2001) 18934–18940.
[50] N. Sizemore, N. Lerner, N. Dombrowski, H. Sakurai, G.R. Stark,Distinct roles of the Ikappa B kinase alpha and betasubunits in liberating nuclear factor kappa B (NF-kappa B)from Ikappa B and in phosphorylating the p65 subunit ofNF-kappa B, J. Biol. Chem. 277 (2002) 3863–3869.
[51] Q. Yu, Y. Geng, P. Sicinski, Specific protection against breastcancers by cyclin D1 ablation, Nature 411 (2001) 1017–1021.
[52] M. Hinz, D. Krappmann, A. Eichten, A. Heder, C. Scheidereit,M. Strauss, NF-kappaB function in growth control:regulation of cyclin D1 expression and G0/G1-to-S-phasetransition, Mol. Cell. Biol. 19 (1999) 2690–2698.
[53] K.A. Scott, R.J. Moore, C.H. Arnott, N. East, R.G. Thompson, B.J.Scallon, D.J. Shealy, F.R. Balkwill, An anti-tumor necrosisfactor-alpha antibody inhibits the development ofexperimental skin tumors, Mol. Cancer Ther. 2 (2003) 445–451.
[54] C.L. Clarke, R.L. Sutherland, Progestin regulation of cellularproliferation, Endocr. Rev. 11 (1990) 266–301.
[55] J.L. Luo, S. Maeda, L.C. Hsu, H. Yagita, M. Karin, Inhibition ofNF-kappaB in cancer cells converts inflammation-inducedtumor growth mediated by TNFalpha to TRAIL-mediatedtumor regression, Cancer Cells 6 (2004) 297–305.
[56] B.K. Park, H. Zhang, Q. Zeng, J. Dai, E.T. Keller, T. Giordano, K.Gu, V. Shah, L. Pei, R.J. Zarbo, L. McCauley, S. Shi, S.Chen, C.Y. Wang, NF-kappaB in breast cancer cellspromotes osteolytic bone metastasis by inducingosteoclastogenesis via GM-CSF, Nat. Med. 13 (2007) 62–69.
[57] G.Matsumoto, J. Namekawa,M.Muta, T. Nakamura, H. Bando,K. Tohyama, M. Toi, K. Umezawa, Targeting of nuclear factorkappaB pathways by dehydroxymethylepoxyquinomicin, anovel inhibitor of breast carcinomas: antitumor andantiangiogenic potential in vivo, Clin. Cancer Res. 11 (2005)1287–1293.
[58] D.K. Biswas, Q. Shi, S. Baily, I. Strickland, S. Ghosh, A.B.Pardee, J.D. Iglehart, NF-kappa B activation in human breastcancer specimens and its role in cell proliferation andapoptosis, Proc. Natl. Acad. Sci. U. S. A 101 (2004) 10137–10142.




















