
Inhibition of Tumor Necrosis Factor Alpha (TNFα) Activity in Rat Brain Is Associated with...
7
Journal of Cerebral Blood Flow and Metaboli.,m 16:378-384 © 1996 The International Society of Cerebral Blood Flow and Metabolism Published by Lippincott-Raven Publishers, Philadelphia Rapid Communication Inhibition of Tumor Necrosis Factor Alpha (TNFa) Activity in Rat Brain Is Associated with Cerebroprotection After Closed Head Injury E. Shohami, R. Bass, *D. Wallach, tAo Yamin, and tR. GaUily Departments of Pharmacology and tImmunology School of Pharmacy, Hebrew University, Jerusalem; and *Department of Molecular Genetics and Virology, Weizmann Institute, Rehovot, Israel Summary: We recently demonstrated that closed head in- jury (CHI) in the rat triggers the production of tumor necrosis factor alpha (TNFa) in the contused hemisphere, Other investigations have shown that this cytokine plays a role in the inflammatory response following trauma. The present study was designed to determine whether inhibition of TNFa production or activity affects the de- velopment of cerebral edema as well as neurological dys- function and hippocampal cell loss aſter CHI. To this end, we used two pharmacological agents, each acting via a different mechanism: pentoxifylline (PTX), which atten- uates the production of TNFa, and tumor necrosis factor binding protein (TBP), a physiological inhibitor of TNFa Tumor necrosis factor alpha (TNFa), a potent cy- tokine implicated in inflammation and immunity (Dinarello, 1988), is produced by activated macro- phages (Gallily et aI., 1989; Semenzato, 1990), as well as by astrocytes (Brenner et aI., 1993). The role of cytokines in the cascade of injury has not been fully elucidated, although a recent review (Ott et aI., 1994) describes changes in cytokine production and metabolic dysfunction aſter severe head injury. Cytokine levels appear to be augmented aſter head injury (McClain et aI. , 1991); the elevation is related Received December 7. 1995; final revision revised January 8, 1996; accepted January 8, 1996. Address correspondence and reprint requests to Dr. Esther Shohami, Department of Pharmacology, Hebrew University, School of Pharmacy, Jerusalem, 91120, Israel. Abbreviations used: BBB, blood-brain barrier; CHI, closed head injury; IL, interleukin; LPS, lipopolysaccharides; NSS, neurological severity score; PTX. pentoxifylline; TBP. tumor necrosis factor binding protein; TNFa, tumor necrosis factor alpha; TNFa-R, tumor necrosis factor alpha receptor; rh-TNFa, recombinant human tumor necrosis factor alpha. 378 activity. Both agents significantly lessened peak edema formation at 24 h and facilitated the recovery of motor function for 14 days postinjury. In addition, TBP atten- uated disruption of the blood-brain barrier and protected hippocampal cells. PTX significantly lowered the brain TNFa level (by �80%), and TBP completely abolished the activity of recombinant human TNF when they were added at the same time in the in vitro bioassay. We sug- gest, therefore. that a decrease in TNFa level or the in- hibition of its activity is accompanied by reduced brain damage. Key Words: Tumor necrosi s factor alpha (TNFa)-Brain injury-Pentoxifylline-Tumor necrosis factor binding protein. to morbidity and mortality in a variety of diseases. Tracy and Cerami, in their review of the biology of TNFa, emphasize its potential as a therapeutic tar- get due to its broad scope of injurious and beneficial effects (1994). A rat model of closed head injury (CHI) has been developed in our laboratory (Sha- pira et aI., 1988), and we have described the acti- vation of an inflammatory pathway, namely, phos- pholipase A2, and increased eicosanoid production (Shohami et aI., 1987, 1989). We have demonstrated that following CHI in the rat, there is an increase in TNFa and interleukin-6 (lL-6) production in the cortical tissue adjacent to the site of injury (Sho- hami et aI., 1994). Since cytokine activity was identified as early as 1 h post-CHI, before infiltration of inflammatory cells is detectable (Shapira et aI., 1988), the rise in cytokines has been attributed to increased cytokine production by resident brain cells (astrocytes and microglia) rather than to its release from invading monocytes. Assuming that elevated TNFa levels
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Inhibition of Tumor Necrosis Factor Alpha (TNFα) Activity in Rat Brain Is Associated with...

Journal of Cerebral Blood Flow and Metaboli.,m 16:378-384 © 1996 The International Society of Cerebral Blood Flow and Metabolism Published by Lippincott-Raven Publishers, Philadelphia
Rapid Communication
Inhibition of Tumor Necrosis Factor Alpha (TNFa) Activity in Rat Brain Is Associated with Cerebroprotection
After Closed Head Injury
E. Shohami, R. Bass, *D. Wallach, tAo Yamin, and tR. GaUily
Departments of Pharmacology and tImmunology School of Pharmacy, Hebrew University, Jerusalem; and
*Department of Molecular Genetics and Virology, Weizmann Institute, Rehovot, Israel
Summary: We recently demonstrated that closed head injury (CHI) in the rat triggers the production of tumor necrosis factor alpha (TNFa) in the contused hemisphere, Other investigations have shown that this cytokine plays a role in the inflammatory response following trauma. The present study was designed to determine whether inhibition of TNFa production or activity affects the development of cerebral edema as well as neurological dysfunction and hippocampal cell loss after CHI. To this end, we used two pharmacological agents, each acting via a different mechanism: pentoxifylline (PTX), which attenuates the production of TN Fa, and tumor necrosis factor binding protein (TBP), a physiological inhibitor of TNFa
Tumor necrosis factor alpha (TNFa), a potent cytokine implicated in inflammation and immunity (Dinarello, 1988), is produced by activated macrophages (Gallily et aI., 1989; Semenzato, 1990), as well as by astrocytes (Brenner et aI. , 1993). The role of cytokines in the cascade of injury has not been fully elucidated, although a recent review (Ott et aI. , 1994) describes changes in cytokine production and metabolic dysfunction after severe head injury. Cytokine levels appear to be augmented after head injury (McClain et aI. , 1991); the elevation is related
Received December 7. 1995; final revision revised January 8, 1996; accepted January 8, 1996.
Address correspondence and reprint requests to Dr. Esther Shohami, Department of Pharmacology, Hebrew University, School of Pharmacy, Jerusalem, 91120, Israel.
Abbreviations used: BBB, blood-brain barrier; CHI, closed head injury; IL, interleukin; LPS, lipopolysaccharides; NSS, neurological severity score; PTX. pentoxifylline; TBP. tumor necrosis factor binding protein; TNFa, tumor necrosis factor alpha; TNFa-R, tumor necrosis factor alpha receptor; rh-TNFa, recombinant human tumor necrosis factor alpha.
378
activity. Both agents significantly lessened peak edema formation at 24 h and facilitated the recovery of motor function for 0%14 days postinjury. In addition, TBP attenuated disruption of the blood-brain barrier and protected hippocampal cells. PTX significantly lowered the brain TNFa level (by �80%), and TBP completely abolished the activity of recombinant human TNF when they were added at the same time in the in vitro bioassay. We suggest, therefore. that a decrease in TN Fa level or the inhibition of its activity is accompanied by reduced brain damage. Key Words: Tumor necrosis factor alpha (TNFa)-Brain injury-Pentoxifylline-Tumor necrosis factor binding protein.
to morbidity and mortality in a variety of diseases. Tracy and Cerami, in their review of the biology of TNFa, emphasize its potential as a therapeutic target due to its broad scope of injurious and beneficial effects (1994). A rat model of closed head injury (CHI) has been developed in our laboratory (Shapira et aI., 1988), and we have described the activation of an inflammatory pathway, namely, phospholipase A2, and increased eicosanoid production (Shohami et aI., 1987, 1989). We have demonstrated that following CHI in the rat, there is an increase in TNFa and interleukin-6 (lL-6) production in the cortical tissue adjacent to the site of injury (Shohami et aI., 1994).
Since cytokine activity was identified as early as 1 h post-CHI, before infiltration of inflammatory cells is detectable (Shapira et aI., 1988), the rise in cytokines has been attributed to increased cytokine production by resident brain cells (astrocytes and microglia) rather than to its release from invading monocytes. Assuming that elevated TNFa levels

TNFa INHIBITION AND CEREBROPROTECTION 379
play a detrimental role in the pathophysiology of CHI, the present study was designed to determine the effects of TNFa inhibition on the development of cerebral edema as well as on neurological dysfunction and hippocampal cell loss after CHI. To this end, two pharmacological agents acting via different mechanisms were used.
Pentoxifylline (PTX), a methylxanthine, has been known for many years for its hemorrheological properties. This drug has been shown to prevent or attenuate the production of TNFa induced by bacterial lipopolysaccharides (LPS) both in vitro (Strieter et aI. , 1988) and in vivo (Zabel et aI. , 1989) at the transcriptional level (Doherty et aI., 1991). TNFa binding protein (TBP) is a cysteine-rich glycoprotein identified and purified from human urine (Peetre et aI. , 1988; Seckinger et aI. , 1988; Engelmann et aI. , 1989). TBP corresponds to a soluble fragment of the cell-surface TNFa receptor (TNFa-R) generated by proteolytic cleavage of the extracellular binding domain (Nophar et aI. , 1990). It displays a sequence homology to the extracellular portions of the TNF-R p80 chain and to nerve growth factor. TBP is a physiological inhibitor of TNFa, which acts in vitro by competing with the cell-surface TN Fa receptor. We hypothesized that if TNFa plays a part in the pathophysiology of CHI, TBP might counteract its effect and find potential use as a therapeutic agent in our CHI. Indeed, our results support the notion that TN Fa is a harmful mediator in CHI, and its suppression by both agents correlates with improved recovery following brain injury.
MATERIALS AND METHODS
Closed head injury model Male Sabra rats (Hebrew University strain) weighing
200-220 g were used. CHI was induced under ether anesthesia by a calibrated weight-drop device that falls over the exposed skull, covering the left hemisphere 1-2 mm lateral to the midline in the mid coronal plane, as described previously (Shapira et a!., 1988). Rats were kept according to the institutional regulations of the Animal Care Committee.
Neurological evaluation Rats were evaluated on the basis of clinical criteria,
which include reflexes and motor functions, and were graded according to the neurological severity score (NSS) 1 h after CHI (Table 1). Rats with a score of 15-20 (>90% of the injured animals) were considered severely injured and were included in the study. At various times postCHI (24 h to 2 weeks) the animals were reevaluated. The difference between the new score and that at 1 h was defined as the extent of recovery (�NSS) and was used to assess the neuroprotective properties of the drugs. The greater the difference, the better the recovery.
TABLE 1. Neurological severity score
Functions
Inability to exit from circle (50 cm diameter)
For 30 min after injury For 60 min after injury For >60 min after injury
Loss of righting reflex For 20 min after injury For 40 min after injury For >60 min after injury
Hemiplegia-inability of rat to resist forced change in position
Flexion of hind limb when raised by tail Inability to walk straight when placed on
the floor Reflexes
Pinna reflex CQrneal reflex Startle reflex
Clinical grade Loss of seeking behavior Prostration
Limb reflexes Loss of placing reflex
Forelimbs Left Right
Hind limbs Left Right
Functional tests Failure in beam-balancing task (1.5 cm
wide beam) For 20 s For 40 s For >60 s
Failure in beam walking task 8.5 cm wide 5 cm wide 2.5 cm wide
Total (maximum points)
Evaluation of edema
Evaluation time
postinjury
1 h >l h
1 1 1
24
1 1 1
20
At 24 h post-CHI, the rats were decapitated, and their brains were removed. Since we had shown that the region adjacent to the core of injury develops the highest degree of edema, samples of this region were taken to assess drug effectiveness. The water content in a -20-mg segment of cortical tissue adjacent to the site of injury was measured, using the dry/wet weight ratio, as described by Shapira et a!. (1988).
Evaluation of BBB integrity Blood-brain barrier (BBB) integrity was tested using
the Evans Blue dye extravasation method. Evans Blue was injected i.v. 3 or 23 h after CHI, and 1 h later the animals were perfused with saline, their brains were removed, the dye was extracted, and its fluorescence was determined (Uyama et a!., 1988).
TNFa determination TNFa titer was determined by a cytotoxicity assay.
Brain extracts were bioassayed as described previously
J Cereb Blood Flow Metab, Vol. 16, No.3, 1996
380 E. SHOHAMI ET AL.
(Sher et aI., 1990, Shohami et aI., 1994), using the BALB/c CL. 7 line as target cells. Briefly, cells were plated in Dulbecco's Modified Eagle's Medium containing 5% fetal calf serum. After 24 h, threefold dilutions of the brain extracts were added to the target cells, followed by the addition of actinomycin D (Sigma, St. Louis, MO, U.S.A.). The cultures were incubated for 20 h, stained with 2% crystal violet, rinsed, and dried. Destruction of CL. 7 cells was assessed by measuring the absorbance of the stained cells (550 nm) with an MR 700 microplate reader (Dynatech, Farmingdale, NY, U.S.A.).
The TNFex titer (Sso), defined as the reciprocal of the dilution of test extract required to destroy 50% of the target cell monolayer, was determined. Various dilutions of a known concentration of recombinant TNFex were added to the target cells to quantify the Sso, and the data were converted into picograms per milligram of tissue protein. The specificity of TNFex cytotoxicity was confirmed using anti-TNFex antibodies in the same assay, as has been described (Shohami et aI., 1994). The capacity of TBP to neutralize TNFex activity was demonstrated in vitro by coadministration of recombinant human TNFex (rh-TNFex) and TBP in the bioassay.
Neuropathological analysis Fourteen days post-CHI rats were reanesthetized with
ether, and their brains were perfused transcardially with 4% formaldehyde. The brains were removed for histopathological evaluation, and 6-fLm-thick coronal sections were cut using a microtome and stained with hematoxylin and eosin. Hippocampal damage was quantified under a light microscope (Nikon) by an uninformed observer by counting the number of viable and dead cells in the CAI_3 fields of the hippocampus.
Agents PTX (Sigma) was used as a stock solution of 4 mg/ml in
saline. Traumatized rats were injected i. v. with PTX (20 mg/kg) immediately after induction of CHI. TBP was the generous gift of Interpharm Laboratories Ltd. (Rehovot, Israel). A stock solution of 1 mg/ml saline was used, and a dose of 1 mg/kg was administered i.v. within 5 min of CHI. Recombinant human TNFex was obtained from Genetech Inc. (San Francisco, CA, U.S.A.).
Experimental protocol Rats were subjected to CHI and treated with the tested
agent or its vehicle immediately after induction of trauma. NSS was assessed 1 h later, and the animals were returned to their cages. NSS was reevaluated at 24, 48, and 72 h and at 7 and 14 days, and �NSS was calculated at each time point. Other groups of rats were decapitated 24 h post-CHI for determination of water content or at 4 or 24 h for evaluation of· BBB integrity. For measuring TNFex levels, groups of rats were traumatized in parallel, treated with the different agents, and decapitated 4 h post-CHI, when TNFex accumulation peaks (Shohami et aI., 1994).
To further assess the role of endogenous TNFex in the pathophysiology of CHI, a group of rats were injected intracerebrally, into the right lateral ventricle, with 24 fLg of rh-TNFex (in 30 fLl of saline) 5 min before CHI. Within 5 min of CHI the rats were injected i.v. with PTX, and their NSS and water content were evaluated 24 h later.
Statistical analysis Percentage water content, TNFex levels, and percent
age cell loss are presented as the mean ± SD and com-
J Cereb Blood Flow Metab, Vol. 16. No.3, 1996
pared using the two-tailed Student's t test. Clinical recovery, expressed as �NSS, is shown as the median (range) and compared by the two tailed nonparametric Mann Whitney test. The level of significance was p < 0.05.
RESULTS
Effect of PTX
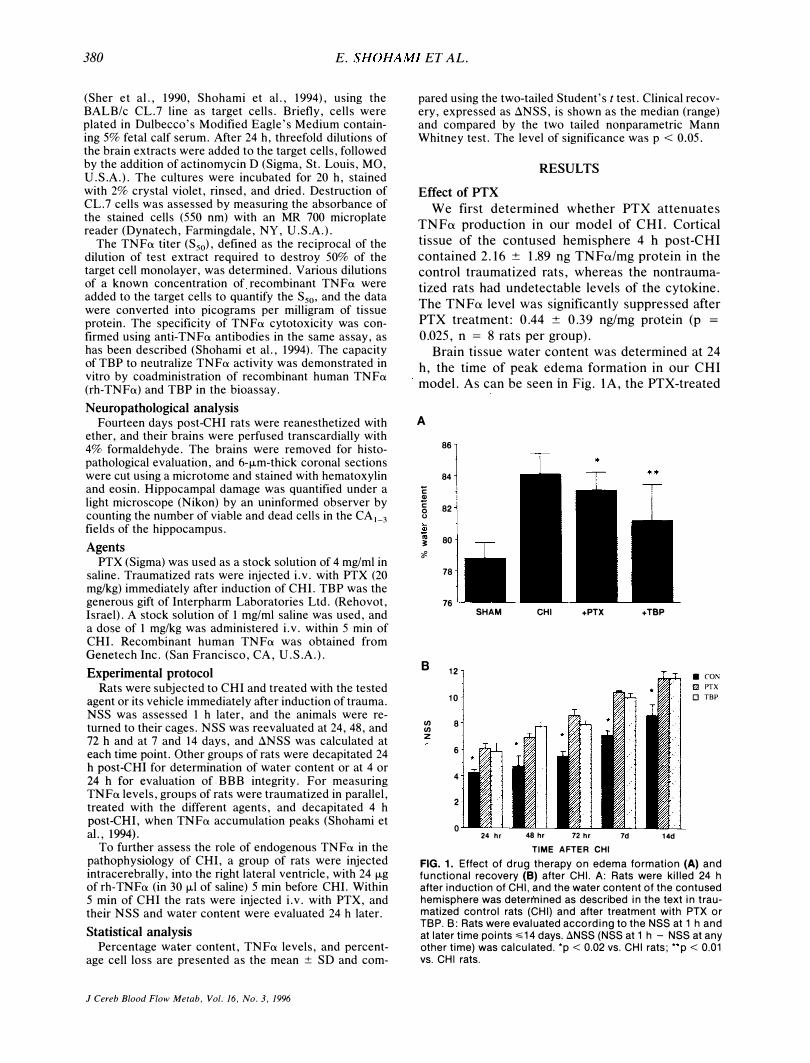
We first determined whether PTX attenuates TNFa production in our model of CHI. Cortical tissue of the contused hemisphere 4 h post-CHI contained 2.16 ± 1.89 ng TNFa/mg protein in the control traumatized rats, whereas the nontraumatized rats had undetectable levels of the cytokine. The TNFa level was significantly suppressed after PTX treatment: 0.44 ± 0.39 ng/mg protein (p =
0.025, n = 8 rats per group). Brain tissue water content was determined at 24
h, the time of peak edema formation in our CHI model. As can be seen in Fig. lA, the PTX-treated
A
86
84 E CI) E 82 0 u
! '" 80 �
#.
78
76 SHAM CHI +PTX +TBP
B 12 • CON rn PTX
10 IJ TBP
CIl 8 CIl z
6
4
2
0 24 hr 48 hr 72 hr 7d 14d
TIME AFTER CHI
FIG. 1. Effect of drug therapy on edema formation (A) and functional recovery (8) after CHI. A: Rats were killed 24 h after induction of CHI, and the water content of the contused hemisphere was determined as described in the text in traumatized control rats (CHI) and after treatment with PTX or TBP. B: Rats were evaluated according to the NSS at 1 h and at later time points �14 days. 8NSS (NSS at 1 h - NSS at any other time) was calculated. 'p < 0.02 vs. CHI rats; "p < 0.01 vs. CHI rats.

TNFa INHIBITION AND CEREBROPROTECTION 381
rats had a significantly lower water content than did the controls (83.1 ± 1.13 versus 84.7 ± 1.33%, p =
0.0125). We then addressed the functional state of the traumatized rats and compared the ilNSS of control and PTX-treated rats for 14 days postinjury. NSS was assessed at 24, 48, and 72 h as well as 1 and 2 weeks post-CHI. At each of these time points, the ilNSS of the PTX-treated rats was significantly higher than that of the control, untreated CHI rats, indicating better recovery (Fig. IB).
The effect of PTX on BBB integrity was evaluated at 4 h and 24 h, according to the amount of Evans Blue accumulated in the brain. At 4 h postCHI, in the contused hemisphere of the PTXtreated rats, there was 75 ± 17.9 ng dye/mg tissue (n = 7), compared with 117 ± 22.6 (n = 7) in the controls (this difference did not reach significance). At 24 h post-CHI the level of dye was identical in the two groups (229 ± 131 and 233 ± 90 ng dye/mg tissue in the PTX-treated and untreated rats, respectively).
We next tested whether rh-TNFa injected before CHI would reverse the protective effect of PTX given 5 min after injury. Indeed, the ilNSS of the PTX-treated rats at 24 h was 7 (range 5-9, n = 8), while that of the traumatized rats treated with rhTNFa and PTX was 6 (range 5-7, n = 9), p = 0.02, Mann Whitney test. This value was not significantly different from that obtained for the control (traumatized, untreated) rats, indicating that rhTNFa reversed the beneficial effect of PTX on the clinical outcome. In contrast, the water content of these two groups was similar and significantly lower than that of the control, indicating that rh-TNF a did not abolish the effect of PTX on edema.
The effect exerted by PTX on neurons in the hippocampal CA2 and CA3 regions was assessed by counting the number of viable cells following treatment with this agent. In CA2: 23 ± 16.7% dead cells were found in the controls vs 10.8 ± 18.3% in the PTX-treated rats. In CA3: cell death was 32.7 ±
18.9% in the control non-treated rats (n = 5), vs 20.5 ± 14.5% in the PTX-treated rats (n = 8). The differences were not significant.
Effect of TBP
The effect of TBP on edema formation and functional recovery (ilNSS) after CHI is evident in Fig. I. Less water accumulated at the site of injury in the TBP-treated rats compared with that in untreated rats after a similar degree of injury (81.16 ± 2.32, n = 11, versus 84.17 ± 1.33%, n = 9; p = 0.007,
two-tailed t test; Fig. 1 A). In addition, recovery of motor functions was significantly facilitated in the TBP-treated rats during the 2-week follow-up (Fig.
IB). At all time points, the ilNSS of the TBPtreated rats was significantly higher (p < 0.05) than that of the controls, indicating better recovery of motor functions. The effect of TBP on the disruption of the BBB was examined at 4 and at 24 h post-CHI. Whereas at 4 h the levels of Evans Blue in the TBP-treated rats were similar to those in the control, at 24 h, they were significantly lower: 76.6 ± 42.8 (n = 6) versus 233.4 ± 117.9 (n = 9, p <
0.01). Examining cell viability in the hippocampus
showed that there was significantly less cell damage in the CA2 and CA3 regions of the TBP-treated rats (n = 7) compared with that in the untreated rats (n
= 5). In the CA2 it was 3.42 ± 1.44 versus 23.02 ±
16.7% (p < 0.05), and in the CA3 it was 14.5 ±
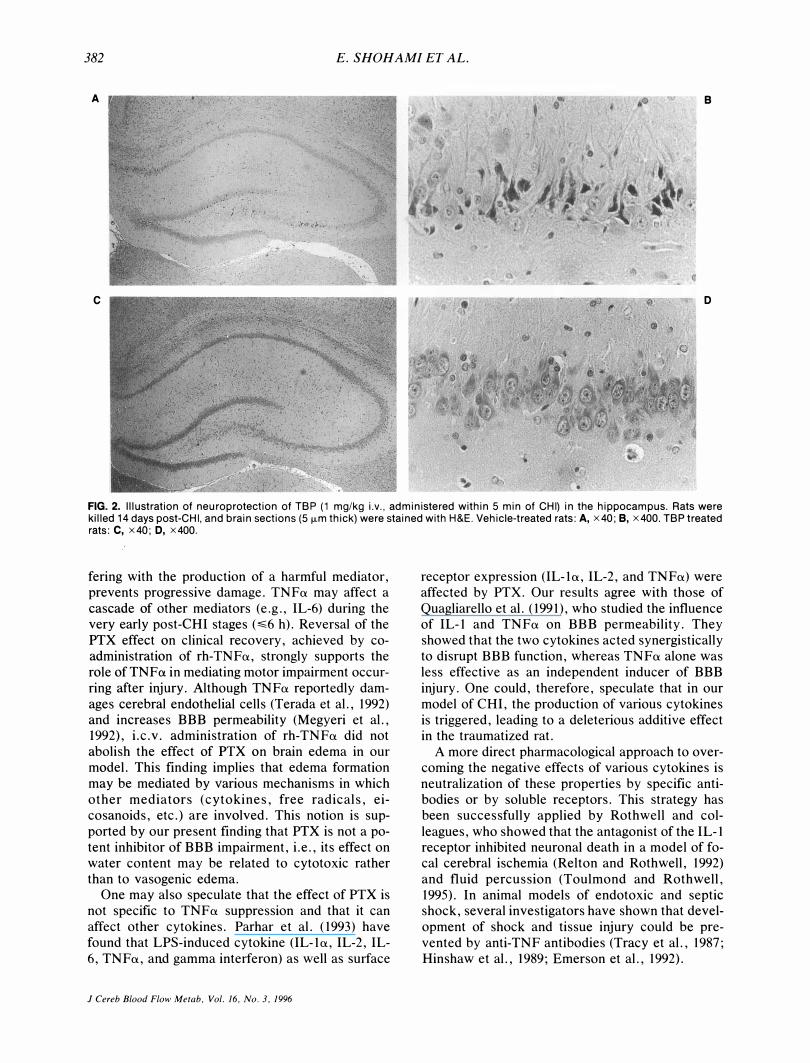
10.4% versus 32.7 ± 18.9% (p = 0.056, marginally si&nificant). The protective effect of TBP on hippocampal cells is shown in Fig. 2.
In an attempt to verify that TBP treatment indeed reduced brain TNFa activity, brain extracts were prepared from the traumatized, TBP-treated rats and tested in the bioassay. The results show that there was no reduction in TNFa activity in the brain homogenates of the treated rats (SSG of 61 ± 35, n =
12, in the control, untreated rats, versus 151 ± 106, n = 7, in the TBP-treated rats, which was not significant). However, the activity was totally abolished when TBP was added in vitro along with rhTNFa (SSG of 1,919 for the rh-TNFa, and 0 for the rh-TNFa added together with TBP).
DISCUSSION
The results of the present study support the notion that TNFa mediates at least some of the harmful events occurring following traumatic brain injury. Indeed, PTX reduced the early and transient surge of TN Fa activity in the brain following CHI. This finding concurs with earlier reports by a number of investigators regarding the drug's mechanism of action (Strieter et aI., 1988; Doherty et aI., 1991). We showed that PTX also ameliorated motor dysfunction, even as late as 14 days postinjury, and reduced brain edema at 24 h, the time of maximal edema (Shapira et aI., 1988). We, therefore, suggest that the low level of TNFa is associated with the cerebroprotection observed under these conditions. We previously showed that there is a transient increase in TN Fa production after CHI, starting at 1 h, peaking at 4 h, and terminating at � 12 h postinjury (Shohami et al. 1994).
In the present study, PTX was administered only once, immediately after CHI, whereas the protective effect was evident even as late as 2 weeks postinjury. It is conceivable that the drug, by inter-
J Cereb Blood Flow Metab, Vol. 16, No.3, 1996
382 E. SHOHAMI ET AL.
A e
c D
FIG. 2. Illustration of neuroprotection of TBP (1 mg/kg i.v., administered within 5 min of CHI) in the hippocampus. Rats were killed 14 days post-CHI, and brain sections (5 fLm thick) were stained with H&E. Vehicle-treated rats: A, x40; e, x400. TBP treated rats: C, x40; D, x400.
fering with the production of a harmful mediator, prevents progressive damage. TNFa may affect a cascade of other mediators (e.g., IL-6) during the very early post-CHI stages (�6 h). Reversal of the PTX effect on clinical recovery, achieved by coadministration of rh-TNFa, strongly supports the role of TN Fa in mediating motor impairment occurring after injury. Although TNFa reportedly damages cerebral endothelial cells (Terada et aI., 1992) and increases BBB permeability (Megyeri et aI. , 1992), i.c.v. administration of rh-TNFa did not abolish the effect of PTX on brain edema in our model. This finding implies that edema formation may be mediated by various mechanisms in which other mediators (cytokines, free radicals, eicosanoids, etc.) are involved. This notion is supported by our present finding that PTX is not a potent inhibitor of BBB impairment, i.e., its effect on water content may be related to cytotoxic rather than to vasogenic edema.
One may also speculate that the effect of PTX is not specific to TN Fa suppression and that it can affect other cytokines. Parhar et al. (1993) have found that LPS-induced cytokine (IL-1a, IL-2, IL-6, TNFa, and gamma interferon) as well as surface
J Cereb Blood Flow Metab, Vol. 16, No.3, 1996
receptor expression (IL-1a, IL-2, and TNFa) were affected by PTX. Our results agree with those of Quagliarello et aL (1991), who studied the influence of IL-1 and TNFa on BBB permeability. They showed that the two cytokines acted synergistically to disrupt BBB function, whereas TNFa alone was less effective as an independent inducer of BBB injury. One could, therefore, speculate that in our model of CHI, the production of various cytokines is triggered, leading to a deleterious additive effect in the traumatized rat.
A more direct pharmacological approach to overcoming the negative effects of various cytokines is neutralization of these properties by specific antibodies or by soluble receptors. This strategy has been successfully applied by Rothwell and colleagues, who showed that the antagonist of the IL-1 receptor inhibited neuronal death in a model of focal cerebral ischemia (ReIton and Rothwell, 1992) and fluid percussion (Toulmond and Rothwell, 1995). In animal models of endotoxic and septic shock, several investigators have shown that development of shock and tissue injury could be prevented by anti-TNF antibodies (Tracy et aI., 1987; Hinshaw et aI. , 1989; Emerson et aI., 1992),

TNFa INHIBITION AND CEREBROPROTECTION 383
In the present study we treated the traumatized rats with TBP within 1 h of CHI and found reduced edema and BBB impairment at 24 h, a significant amelioration of neurological dysfunction � 14 days post-CHI, and protection of hippocampal neurons. TBP, a specific anti-TN Fa compound, acts as a soluble fragment of the cell-surface TN Fa receptor and binds this cytokine, thus neutralizing its activity. TBP, a natural molecule that interferes with TNFa activity, prevents the interaction of the latter with the target cell. The apparent ineffectiveness of TBP in the bioassay of brain extracts of TBPtreated rats may be attributed to the fact that the brain homogenates were prepared by sonicating the brain tissue. This procedure may have dissociated the complex and liberated the free TN Fa that had accumulated in the brain after injury, yielding the high activity observed in the bioassay.
In contrast to the PTX-treated brains, where TNFa production was inhibited, in the TBP-treated rats, TNFa was produced in response to injury, and was probably neutralized thereafter, resulting in the improved in vivo outcome. However, the complex TNF-TBP may be unstable under the extraction conditions, leading to the release of free TNFa and the expression of its activity in the in vitro assay system. Indeed, upon co-administration of rhTNFa and TBP to the target cells, there was a loss of activity. We therefore assume that TBP acts to neutralize the direct or indirect actions of TNFa in the brain.
In their study of the pharmacokinetics and tissue distribution of TBP in mice, Gascon et al. (1992) found that only a minor fraction of the i. v .-injected TBP was present in the brain, probably owing to its inability to cross the BBB. In our model of CHI, there is early impairment of the BBB, which may facilitate the penetration of nonpermeable molecules and could account for the observed effect of TBP on brain function. Our results, showing the ameliorative effects of PTX and TBP on CHI outcome, could be interpreted as showing the involvement of TNFa, and probably of other cytokines activated by TNFa, in the pathophysiology of brain irijury. If so, neutralization of its deleterious effects could be a novel approach to the treatment of CHI patients.
Acknowledgment: This study was partly supported by the Israel Ministry of Absorption and by the David R. Bloom Center for Pharmacy. We gratefully acknowledge the support of the Friends of the Lautenberg Center and the Concern Foundations of Los Angeles. We appreciate the skillful assistance of Semion Breiterman Ph.D., from the Department of Experimental Surgery, Hadassah Medical Center.
REFERENCES
Brenner T. Yamin A, Abramsky 0, Gallily R (1993) Stimulation of tumor necrosis factor-production by and inhibition by dexamethasone in cultured astrocytes. Brain Res 608:273-279
Dinarello CA (1988) Cytokines: interleukin-I and tumor necrosis factor (cachectin). In: Injlammation: Basic Principles and Clinical Correlates (Gallin JI, Goldstein ML, Snyderman R, eds), New York, Raven Press, pp 1195-208
Doherty GM, Jensen JC, Alexander R, Buresh CM, Nortron JA (1991) Pentoxifylline suppression of tumor necrosis factor gene transcription. SurRery 110:192-198
Emerson TE, Lindsey DC, Jesmok GJ, et al. (1992) Efficacy of monoclonal antibody against TNF-a in an endotoxemic baboon model. Circ Shock 38:75-84
Engelmann H, Aderka D, Rubinstein M, Rotman D, Wallach D (1989) A tumor necrosis factor-binding protein purified to homogeneity from urine protects cells from tumor necrosis factor toxicity. J Bioi Chem 264:11974-11980
Gallily R, Sher T, Ben-Av P, Loewenstein J (1989) Tumor necrosis factor as a mediator of mycoplasma ovale-induced tumor cell lysis in macrophages. Cell ImmunoI121:141-153
Gascon MP, Porchet HC, Le Cotonnec YJY, Ythier A, Wallach D, Piguet PF, Grau GE (1992') Pharmacokinetics and tissue distribution of human urinary tumor necrosis factor binding protein in mice. Drug Metab Dispos 20:592-595
Hinshaw L, Olson P, Kuo G (1989) Efficacy of post-treatment with anti-TNF monoclonal antibody in preventing the pathophysiology and lethality of sepsis in baboon. Circ Shock 27:362-369
McClain CJ. Cohen D, Phillips R, Ott L, Young B (1991) Increased plasma and ventricular fluid interleukin-6 levels in patients with head injury. J Lab Clin Med 118:225-231
Megyeri p, Abraham CS, Temesvari P, Kovacs J, Vas T, Speer CP (1992) Recombinant human tumor necrosis factor alpha constricts pial arterioles and increases blood brain barrier permeability in newborn piglets. Neurosci Lett 148: 137-140
Nophar Y, Kemper 0, Brakebusch C, et al. (1990) Soluble forms of tumor necrosis factor receptors (TNF-Rs): The cDNA for the type 1 TNF-R, cloned using amino acid sequence data of its soluble form, encodes both the cell surface and a soluble form of the receptor. EMBO J 9:3269-3278
Ott L, McClain CJ, Gillespie M, Toung B (1994) Cytokines and metabolic dysfunction after severe head injury. J Neurotrauma 11 :447-472
Parhar PA, Ernst P, Sheth K, Einspenner M, AI-Sedairy S (1993) Mechanism of pentoxifylline mediated down-regulation of killer lineage cell function. Mediators Injlamm 2:379-384
Peetre C, Thysell H, Grubb A, Olsson I (1988) A tumor necrosis factor binding protein is present in human biological fluids. Eur J Haematol 41 :414-423
Quagliarello VJ, Wispelwey B, Long WJ, Scheid WM (1991) Recombinant human interleukin-l induces meningitis and blood brain barrier injury in the rat: characterization and comparison with tumor necrosis factor. J Clin Invest 87: 1360-1366.
Relton JK, Rothwell NJ (1992) Interleukin-l receptor antagonist inhibits ischaemic and excitotoxic neuronal damage in the rat. Brain Res Bull 29:243-246
Seckinger P, Isaaz S, Dayer JM (1988) A human inhibitor of tumor necrosis factor a. J Bioi Chem 264: 11966--11973
Semenzato G (1990) Tumor necrosis factor: a cytokine with multiple biological activity (a review). Br J Cancer 61 :354-361
Shapira Y, Shohami E, Sidi A, Soffer D, Freeman S, Cotev S. (1988) Experimental closed head injury in rats: mechanical, pathophysiologic, and neurologic properties. Crit Care Med 16:258-265
Sher T, Rottem S, Gallily R (1990) Mycoplasma capricolum membranes induce tumor necrosis factor by a mechanism different from that of lipopolysaccharide. Cancer Immunol Immunother 31:86--92
Shohami E, Novikov M, Bass R, Yamin A, Gallily R (1994)
J Cereb Blood Flow Metab. Vol. 16. No. 3. 1996

384 E. SHOHAMI ET AL.
Closed head injury triggers early production of TNF()[ and IL-6 by brain tissue. J Cereb Blood Flow Metab 14:615-619
Shohami E, Shapira Y, Sidi A, Cotev S. (1987) Head injury induces increased prostaglandin synthesis in rat brain. J Cereb Blood Flow Metab 7:58-63
Shohami E, Shapira Y, Yadid G, Reisfeld N, Yedgar S. (1989) Brain phospholipase Az is activated after experimental closed head injury in rats. J Neurochem 53: 1541-1546
Strieter RM, Remick DG, Ward PA, et al. (1988) Cellular and molecular regulation of tumor necrosis factor-alpha production by pentoxifylline. Biochem Biophys Res Commun 155: 1230--1236
Terada LS, Willingham IR, Guidot DM, Shibao GN, Kindt GW, Repine JE (1992). Tungsten treatment prevents tumor necrosis factor-induced injury of brain endothelial cells. Inflammation 16:13-19
J Cereb Blood Flow Metab, Vol. /6, No.3, 1996
Toulmond S, Rothwell NJ (1995) Interleukin-I receptor antagonist inhibits neuronal damage caused by fluid percussion injury in the rat. Brain Res 671 :261-266
Tracy KJ, Cerami A (1994) Tumor necrosis factor: a pleiotropic cytokine and therapeutic target. Ann Rev Med 45:491-503
Tracy KJ, Fong Y, Hesse DG, et al. (1987) Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature 330:662-664
Uyama 0, Okamura N, Yamase M, Narita M, Kawabata K, Sugita M (1988) Quantitative evaluation of vascular permeability in the gerbil brain after transient ischemia using Evans blue fluorescence. J Cereb Blood Flow Metab 8:282-284
Zabel P, Schonharting MM, Wolter DT, Schade UF (1989) Oxpentifylline in endotoxemia. Lancet 2: 1474-1477




















