
Alcohol septal ablation in hypertrophic obstructive cardiomyopathy
Upload
independentCategory
view
0download
0
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
CLINICAL RESEARCHHeart failure/cardiomyopathy
Titin gene mutations are common in familieswith both peripartum cardiomyopathy anddilated cardiomyopathyKarin Y. van Spaendonck-Zwarts1,2*, Anna Posafalvi1, Maarten P. van den Berg3,Denise Hilfiker-Kleiner4, Ilse A.E. Bollen5, Karen Sliwa6, Marielle Alders2,Rowida Almomani1, Irene M. van Langen1, Peter van der Meer3, Richard J. Sinke1,Jolanda van der Velden5, Dirk J. Van Veldhuisen3, J. Peter van Tintelen1,7†,and Jan D.H. Jongbloed1†
1DepartmentofGenetics,Universityof Groningen,UniversityMedical CenterGroningen,Groningen, the Netherlands; 2Departmentof Genetics, Academic Medical Center,UniversityofAmsterdam, PO Box 22660, 1100 DD, Amsterdam, the Netherlands; 3Department of Cardiology, University of Groningen, University Medical Center Groningen, Groningen, theNetherlands; 4Department of Cardiology and Angiology, Medical School Hannover, Hannover, Germany; 5Department of Physiology, VU University Medical Center, Amsterdam, theNetherlands; 6Hatter Institute for Cardiovascular Research in Africa, Department of Medicine and IIDMM, University of Cape Town, Cape Town, South Africa; and 7Durrer Center forCardiogenetic Research, Utrecht, the Netherlands
Received 23 June 2013; revised 13 January 2014; accepted 27 January 2014; online publish-ahead-of-print 20 February 2014
Aim Peripartum cardiomyopathy (PPCM) can be an initial manifestation of familial dilated cardiomyopathy (DCM). We aimedto identify mutations in families that could underlie their PPCM and DCM.
Methodsand results
We collected 18 families with PPCM and DCM cases from various countries. We studied the clinical characteristics of thePPCM patients andaffected relatives, andapplied a targeted next-generation sequencing (NGS) approach todetectmuta-tions in 48 genes known to be involved in inherited cardiomyopathies. We identified 4 pathogenic mutations in 4 of 18families (22%): 3 in TTN and 1 in BAG3. In addition, we identified 6 variants of unknown clinical significance that may bepathogenic in 6 other families (33%): 4 in TTN, 1 in TNNC1, and 1 in MYH7. Measurements of passive force in single car-diomyocytes and titin isoform composition potentially support an upgrade of one of the variants of unknown clinical sig-nificance in TTN to a pathogenic mutation. Only 2 of 20 PPCM cases in these families showed the recovery of leftventricular function.
Conclusion Targeted NGS shows that potentially causal mutations in cardiomyopathy-related genes are common in families withboth PPCM and DCM. This supports the earlier finding that PPCM can be part of familial DCM. Our cohort is particularlycharacterized by a high proportion of TTN mutations and a low recovery rate in PPCM cases.
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -Keywords Cardiomyopathy † Peripartum cardiomyopathy † Genetics † Pregnancy † Titin
IntroductionPeripartum cardiomyopathy (PPCM) is an idiopathic cardiomyop-athy presenting with heart failure secondary to left ventricular systol-ic dysfunction towards the end of pregnancy or in the first monthsfollowing delivery, where no other cause of heart failure is found.The left ventricle may not be dilated but the ejection fraction isnearly always reduced ,45%.1 According to this recent definition,
the time frame is not strictly defined, in contrast to previous defini-tions.2 –4 The severity of PPCM is highly variable, ranging from com-plete recovery to rapid progression to end-stage heart failure.Peripartum cardiomyopathy affects 1:300 to �1:3000 pregnancies,with geographic hot spots of high incidence such as in Haiti andNigeria.4,5
The precise mechanisms that lead to PPCM are not fully known.Several risk factors and possible underlying pathological processes
* Corresponding author. Tel: +31 2056665281, Fax: +31 205669498, Email: [email protected]† The last two authors contributed equally.
Published on behalf of the European Society of Cardiology. All rights reserved. & The Author 2014. For permissions please email: [email protected]
European Heart Journal (2014) 35, 2165–2173doi:10.1093/eurheartj/ehu050
at University of G
roningen on August 21, 2014
http://eurheartj.oxfordjournals.org/D
ownloaded from

have received attention, such as abnormal autoimmune responses,apoptosis, and impaired cardiovascular microvasculature.5,6 Recentwork into the pathogenesis of PPCM has shown involvement of acascade with oxidative stress, the prolactin-cleaving protease cathe-psin D, and the nursing hormone prolactin, which may lead to a targetfor a disease-specific therapy, namely pharmacological blockade ofprolactin by bromocriptine.7– 9 In addition, involvement of cardiacangiogenic imbalance may explain why PPCM is a disease seen inlate pregnancy and why pre-eclampsia and multiple gestation are im-portant risk factors.10 Peripartum cardiomyopathy is probablycaused by a complex interaction of more than one pathogenic mech-anism. The large variation in incidence and clinical characteristics mayreflect the involvement of specific mechanisms, or combinationsthereof, in certain subgroups of PPCM.
We and others recently reported that PPCM can be an initialmanifestation of familial dilated cardiomyopathy (DCM),11,12 indicat-ing that, at least in a subset of cases, genetic predisposition plays arole in the pathophysiology of pregnancy-associated heart failure.Accordingly, Haghikia et al. reported a positive family history forcardiomyopathy in 16.5% (19 of 115) of PPCM cases from aGerman PPCM cohort.13 So far, eight cases with underlying muta-tions in DCM-related genes have been published11,12,14,15 andseveral other cases with familial occurrences of PPCM and DCM,as well as familial clustering of PPCM, have been reported.16–24
Here, we describe ourextensive genetic analysis using next-generationsequencing (NGS) technology to identify potentially causal mutationsin families with both PPCM and DCM from various parts of the world.
Methods
Subjects and clinical evaluationWe collected a cohort of families with cases of both PPCM and DCMfrom various parts of the world (the Netherlands, Germany, and SouthAfrica) and studied their clinical characteristics by reviewing medicalreports. The local institutional review committees approved the study,and all the participants gave their informed consent.
Peripartumcardiomyopathy wasdiagnosed when a patient had an idio-pathic cardiomyopathy presenting with heart failure secondary to leftventricular systolic dysfunction towards the end of pregnancy or in thefirst months following delivery, where no other cause of heart failurewas found.1 Dilated cardiomyopathy was diagnosed when a patient hadboth a reduced systolic function of the left ventricle (left ventricular sys-tolic ejection fraction ,0.45) and dilation of the left ventricle (left ven-tricular end-diastolic dimension .117% of the predicted valuecorrected for body surface area and age) and only after other identifiablecauses such as severe hypertension, coronary artery disease, and system-ic disease had been excluded.25 If only one of the two criteria was fulfilled,the patient was labelled with ‘mild DCM’. If the family history suggestedDCM in a relative but there were no medical reports to confirm this,the relative was labelled as having ‘possible DCM’. Familial PPCM/DCMwas diagnosed when there were ≥2 affected family members, at leastone with PPCM and one with DCM or sudden cardiac death ≤35 years.
Targeted next-generation sequencingof 48 cardiomyopathy-related genesGenomic deoxyribonucleic acid (DNA) was extracted from bloodsamples obtained from all the available PPCM patients and their affectedrelatives.TargetedNGSwasperformed inoneor twoaffected relatives in
the selected families (these individuals are marked with an arrow inFigures 1 and 2).
We developed a kit based on Agilent Sure Select Target Enrichmentfor mutation detection in 48 genes (all exonic and +20 bp of exon-flanking intronic sequences) known to be involved in inherited cardiomy-opathies (ABCC9, ACTC1, ACTN2, ANKRD1, BAG3, CALR3, CRYAB, CSRP3/MLP, DES, DMD, DSC2, DSG2, DSP, EMD, GLA, JPH2, JUP, LAMA4, LAMP2,LMNA, MYBPC3, MYH6, MYH7, MYL2, MYL3, MYPN, MYOZ1, MYOZ2,PKP2, PLN, PRKAG2, PSEN1, PSEN2, RBM20, RYR2, SCN5A, SGCD, TAZ,TBX20, TCAP, TMEM43, TNNC1, TNNI3, TNNT2, TPM1, TTN, VCL, ZASP/LDB3).26 Samples were prepared according to the manufacturer’s proto-cols and multiplexed to an amount still permitting a theoretical coverageof 100 reads per targeted sequence/per patient. All samples weresequenced using 151 bp paired-end reads on an Illumina MiSeq sequen-cer and analysed using the MiSeq Reporter pipeline and Nextgene soft-ware.27 Eleven amplicons with low coverage were also analysed bySanger sequencing. Identified mutations were confirmed by Sanger se-quencing. To study co-segregation, affected relatives were screened forcarriership of the identified mutations by Sanger sequencing.
Sanger sequencing STAT3 geneThe STAT3 gene (all coding exons and flanking intronic sequences) wasanalysed by Sanger sequencing in PPCM patients of the collected families.
Classification of identified mutationsThe criteria used to classify mutations were published recently.28 Briefly,we used a list of mutation-specific features based on in silico analysis usingthe mutation interpretation software Alamut (version 2.2.1). A scorewasgiven depending on the outcome of a prediction test for each feature(i.e. the PolyPhen-2 prediction tool). Then, depending on the totalscore and the presence/absence of the mutation in at least 300 ethnicallymatched control alleles (data obtained from the literature and/or avail-able databases, e.g. http://evs.gs.washington.edu/EVS and http://www.nlgenome.nl, or from our own control alleles), we classified mutationsas: pathogenic, not pathogenic, or as a variant of unknown clinical signifi-cance (VUS; VUS1, unlikely to be pathogenic; VUS2, uncertain; VUS3,likely to be pathogenic). Co-segregation data and/or functional analysiswere needed to classify a mutation as pathogenic.
Functional analysis of TTN mutationPassive force was measured in single membrane-permeabilized cardio-myocytes mechanically isolated from the heart tissue.29,30 Titin isoformcomposition was analysed, as described previously.30
Results
Clinical characteristics: low rate of fullrecovery in peripartum cardiomyopathycases of familial peripartumcardiomyopathy/dilated cardiomyopathyWe collected 18 families with familial PPCM/DCM. These families ori-ginated from the Netherlands (n ¼ 11), Germany (n ¼ 6), and SouthAfrica (n ¼ 1; black). Clinical data of the PPCM cases in these familiesare summarized in Table 1 and of all (likely) affected relatives in Supple-mentary material online, Table S1. The pedigrees of all the families areshown in Figure 1 (NL1-11) and 2 (SA1 and GER1-6). In two families,there were two cases of PPCM (NL1 and SA1). Eight families (NL1-7and SA1) have been described previously.11,31
K.Y. van Spaendonck-Zwarts et al.2166
at University of G
roningen on August 21, 2014
http://eurheartj.oxfordjournals.org/D
ownloaded from

The median age at diagnosis in PPCM patients was 29 years(n ¼ 15; range 20–36 years), with mean parity 2 (n ¼ 13; range1–4). Peripartum cardiomyopathy diagnosis was post-partum in 12of 14 patients. Only 2 of 20 PPCM patients showed a full recoveryof left ventricular function, one of them even had an uneventfulnext pregnancy (NL9 III:1, LVEF still normal 3 years after diagnosis;
and GER1 II:1, full recovery with uneventful second pregnancy 2years later). Another PPCM patient showed the recovery of left ven-tricular function, but only under treatment with ab-blocker and ACEinhibitor (NL10 III:6).
In addition to 20 confirmed PPCM patients in these families, fiverelatives show clinical characteristics suggestive for PPCM (NL4
Figure 1 Pedigrees of the Dutch families (NL1-11). Square symbols indicate men; circles, women; diamonds, unknown sex; and triangles, miscar-riage. Blue symbols indicate a clinical diagnosis of peripartum cardiomyopathy; black symbols, (mild) dilated cardiomyopathy; grey symbols, possibledilated cardiomyopathy; orange symbols, sudden cardiac death. Diagonal lines through symbols indicate deceased; arrows indicate patients selectedfor targeted next-generation sequencing; and the number in a symbol indicates the number of individuals with this symbol (question mark ifunknown).
Titin gene mutations in PPCM/DCM families 2167
at University of G
roningen on August 21, 2014
http://eurheartj.oxfordjournals.org/D
ownloaded from

II:2, GER1 I:1, GER3 I:1, GER4 I:1, GER5 I:1; Supplementary materialonline, Table S1). Peripartum cardiomyopathy could not be con-firmed because clinical data of these relatives was lacking. In addition,two relatives with DCM showed a decline of left ventricular functionafter delivery (NL2 IV:8 and SA1 II:3; Supplementary material online,Table S1).
Targeted next-generation sequencing:potential causal mutations incardiomyopathy-related genes, inparticular TTN, are common in familialperipartum cardiomyopathy/dilatedcardiomyopathyUsing our validated NGS approach,27 a mean coverage of 220× perindividual patient was reached and, on average, 98.5% of all targetednucleotides were covered at least 20×.
In 4 of 18 families (22%), pathogenic mutations in cardiomyopathy-related genes were identified (3 in TTN and 1 in BAG3). In addition, insix other families (33%) VUS3s were identified (four in TTN, one inTNNC1, and one in MYH7). An overview of these mutations and
VUS3s, and the respective co-segregation analyses are shown inTable 2. All seven TTN mutations/VUS3s were located in the titinA-band, for which over-representation of mutations in DCM patientswas reported previously.32 No potential mutations were identified ineight families (NL2, NL5, NL7, NL8, SA1, GER2, GER3, and GER6).
An overview of the 26 mutations that were not classified as poten-tially disease-causing (VUS1s and VUS2s) identified in the 18 familiesis shown in Supplementary material online, Table S2.
No STAT3 mutations in peripartumcardiomyopathy casesNo STAT3 mutations were identified in 15 PPCM cases (DNA wasavailable from 15 to 20 cases).
Functional and protein analyses supportthe pathogenicity of a likely pathogenicTTN mutationHeart tissue from PPCM patient GER4 II:1 with a VUS3 in TTN wasavailable for functional and protein analyses. Passive force wasmeasured in single cardiomyocytes (n ¼ 4) at sarcomere lengths of
Figure 2 Pedigrees of the South African (SA1) and German families (GER1-6). Square symbols indicate men; circles, women; diamonds, unknownsex. Blue symbols indicate clinical diagnosis of peripartum cardiomyopathy; black symbols, (mild) dilated cardiomyopathy; orange symbol, suddencardiac death. Diagonal lines through symbols indicate deceased; arrows indicate patients selected for targeted next-generation sequencing; thenumber in a symbol indicates the number of individuals with this symbol (question mark if unknown); and SB indicates still birth.
K.Y. van Spaendonck-Zwarts et al.2168
at University of G
roningen on August 21, 2014
http://eurheartj.oxfordjournals.org/D
ownloaded from
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
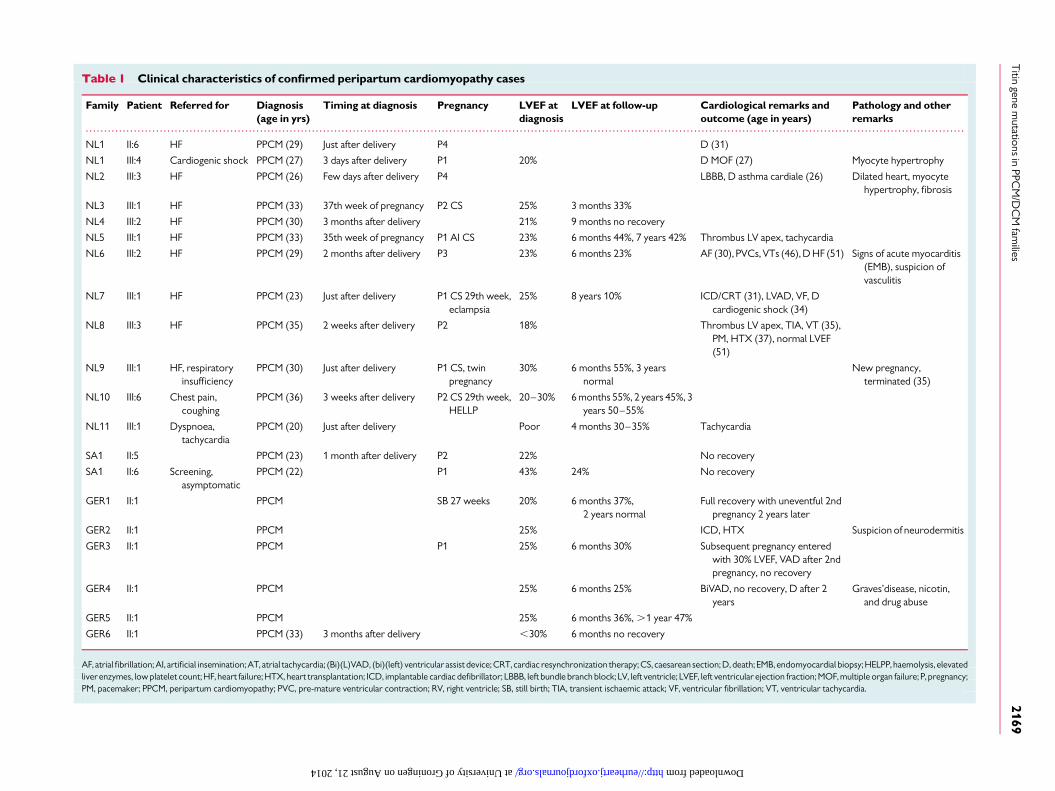
Table 1 Clinical characteristics of confirmed peripartum cardiomyopathy cases
Family Patient Referred for Diagnosis(age in yrs)
Timing at diagnosis Pregnancy LVEF atdiagnosis
LVEF at follow-up Cardiological remarks andoutcome (age in years)
Pathology and otherremarks
NL1 II:6 HF PPCM (29) Just after delivery P4 D (31)
NL1 III:4 Cardiogenic shock PPCM (27) 3 days after delivery P1 20% D MOF (27) Myocyte hypertrophy
NL2 III:3 HF PPCM (26) Few days after delivery P4 LBBB, D asthma cardiale (26) Dilated heart, myocytehypertrophy, fibrosis
NL3 III:1 HF PPCM (33) 37th week of pregnancy P2 CS 25% 3 months 33%
NL4 III:2 HF PPCM (30) 3 months after delivery 21% 9 months no recovery
NL5 III:1 HF PPCM (33) 35th week of pregnancy P1 AI CS 23% 6 months 44%, 7 years 42% Thrombus LV apex, tachycardia
NL6 III:2 HF PPCM (29) 2 months after delivery P3 23% 6 months 23% AF (30), PVCs, VTs (46), D HF (51) Signs of acute myocarditis(EMB), suspicion ofvasculitis
NL7 III:1 HF PPCM (23) Just after delivery P1 CS 29th week,eclampsia
25% 8 years 10% ICD/CRT (31), LVAD, VF, Dcardiogenic shock (34)
NL8 III:3 HF PPCM (35) 2 weeks after delivery P2 18% Thrombus LV apex, TIA, VT (35),PM, HTX (37), normal LVEF(51)
NL9 III:1 HF, respiratoryinsufficiency
PPCM (30) Just after delivery P1 CS, twinpregnancy
30% 6 months 55%, 3 yearsnormal
New pregnancy,terminated (35)
NL10 III:6 Chest pain,coughing
PPCM (36) 3 weeks after delivery P2 CS 29th week,HELLP
20–30% 6 months 55%, 2 years 45%, 3years 50–55%
NL11 III:1 Dyspnoea,tachycardia
PPCM (20) Just after delivery Poor 4 months 30–35% Tachycardia
SA1 II:5 PPCM (23) 1 month after delivery P2 22% No recovery
SA1 II:6 Screening,asymptomatic
PPCM (22) P1 43% 24% No recovery
GER1 II:1 PPCM SB 27 weeks 20% 6 months 37%,2 years normal
Full recovery with uneventful 2ndpregnancy 2 years later
GER2 II:1 PPCM 25% ICD, HTX Suspicion of neurodermitis
GER3 II:1 PPCM P1 25% 6 months 30% Subsequent pregnancy enteredwith 30% LVEF, VAD after 2ndpregnancy, no recovery
GER4 II:1 PPCM 25% 6 months 25% BiVAD, no recovery, D after 2years
Graves’disease, nicotin,and drug abuse
GER5 II:1 PPCM 25% 6 months 36%, .1 year 47%
GER6 II:1 PPCM (33) 3 months after delivery ,30% 6 months no recovery
AF, atrial fibrillation; AI, artificial insemination; AT, atrial tachycardia; (Bi)(L)VAD, (bi)(left) ventricular assist device; CRT, cardiac resynchronization therapy; CS, caesarean section; D, death; EMB, endomyocardial biopsy; HELPP, haemolysis, elevatedliverenzymes, low platelet count; HF, heart failure; HTX, heart transplantation; ICD, implantable cardiac defibrillator; LBBB, left bundle branch block; LV, left ventricle; LVEF, left ventricular ejection fraction; MOF, multiple organ failure; P, pregnancy;PM, pacemaker; PPCM, peripartum cardiomyopathy; PVC, pre-mature ventricular contraction; RV, right ventricle; SB, still birth; TIA, transient ischaemic attack; VF, ventricular fibrillation; VT, ventricular tachycardia.
Titin
genem
utationsin
PPCM
/DC
Mfam
ilies2169
at University of Groningen on August 21, 2014 http://eurheartj.oxfordjournals.org/ Downloaded from

1.8 to 2.2 mm (see Figure 3). Our functional measurements of passivestiffness, which is largely based on titin composition in the heart,revealed a very low passive force development (1.0+ 0.3 kN/m2)at a sarcomere length of 2.2 mm in the PPCM sample comparedwith previously reported values in control hearts (�2.5 kN/m2).29,30
Analysis of titin isoform composition showed a shift towards themore compliant N2BA isoform evident from a higher N2BA/N2Bratio (0.72+0.02; mean of triplo) in the PPCM heart comparedwith the previously reported ratio (0.39+0.05) in control hearts.30
DiscussionThis is the first report of a comprehensive genetic analysis in a largeseries of cases with familial occurrences of PPCM and DCM. Weidentified pathogenic mutations in cardiomyopathy-related genes in4 of 18 families (22%) and VUSs that may be pathogenic in 6 otherfamilies (33%). These data support the earlier finding that PPCMcan be part of familial DCM.11,12
Cascade genetic screening can identify relatives at risk in thosefamilies in which an underlying mutation has been identified. Ourdata also specifically show a low recovery rate in our cohort (only10%) compared with reports in other groups not selected for familialcases (recovery rates of around 25 to 50%),33 –36 indicating that thepresence of an underlying mutation or positive family history for car-diomyopathy in a patient with PPCM may be a prognostic factor for alow recovery rate.
The targeted NGS approach that we have developed provideshigh-throughput, rapid and affordable molecular analysis for cardio-myopathies.27 As accurate annotation of mutations in cardiomyop-athies is of the utmost importance,37 we were extremely careful inclassifying these.28 Our study has several advantages: one is the inclu-sion of some large families, where co-segregation analysis addedvalue to the classification of mutations. Another was the largenumber of genes we tested, including the large TTN gene, for whichmutation analyses on a large scale were impossible before NGSbecame available, because exclusion of pathogenic mutations in 47
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Table 2 Pathogenic mutations and variants of unknown clinical significance that may be pathogenic in 10 of 18 families
Family Testedpatient
Gene Amino acidchange
Nucleotide change Classification Co-segregation Affected relativescarrierYes/unknown
NL1 II:3 TTN p.Arg27373* c.82117C.T Pathogenic Yes II:1, II:3, II:4, III:4, III:5, III:6
NL3 II:2 BAG3 p.Gln340* c.1018C.T Pathogenic Yes II:2, III:1
NL4 III:2 TNNC1 p.Gln50Arg c.149T.C VUS3 Yes II:5, III:2, III:5, IV:1
NL6 II:3 TTN p.Asn28726Lysfs*3 c.86171_86174dupAAAG VUS3 Yes II:1, II:3
NL9 III:1 TTN p.Arg17599* c.52795C.T Pathogenic Yes III:1, III:5
NL10 II:6 TTN p.Arg23956Thrfs*9a c.71867_71876delGAGTTCTGGAa Pathogenic Yes II:1, II:2, II:6, III:2, III:5, III:6
NL11 III:1 TTN p.Ser27317Lysfs*10 c.81949dupA VUS3 Yes II:1, III:1
GER1 II:1 TTN p.Trp18357* c.55070G.A VUS3 Unknown No samples available
GER4 II:1 TTN p.Lys15664Valfs*13 c.46990_46993delAAGG VUS3 Unknown No samples available
GER5 II:1 MYH7 p.Arg1303Gly c.3907C.G VUS3 Yes I:1, II:1
Nomenclature according to HGVS (Human Genome Variation Society) using the reference sequences: TTN (NM_001256850.1; Q8WZ42-1), BAG3 (NM_004281.3), TNNC1(NM_003280.2), MYH7 (NM_000257.2).VUS indicates variant of unknown clinical significance (VUS3, likely to be pathogenic; VUS2, uncertain).aVUS2 p.Arg279Trp (c.835C.T) on same allele.
Figure3 Forcemeasurements in heart tissue of GER4 II:1. Single cardiomyocyteof theperipartum cardiomyopathy heart sample (A). Passive forcedevelopment was measured at sarcomere lengths of 1.8, 2.0, and 2.2 mm (B).
K.Y. van Spaendonck-Zwarts et al.2170
at University of G
roningen on August 21, 2014
http://eurheartj.oxfordjournals.org/D
ownloaded from

other candidate genes makes it more likely that the identified VUS3shave a pathogenic nature. Accordingly, the previously reportedTNNC1 mutation is still the only potential genetic cause in familyNL4.11 And although the pathogenicity of truncating TTN mutationsis still under debate due to these types of mutations being found inapparently healthy controls (up to 3%) and the general popula-tion,32,38 the pathogenicity of TTN VUS3s identified in our familiesalso becomes more likely after excluding pathogenic mutations in47 other cardiomyopathy-related genes. Possible exclusion of muta-tions in other genes in patients carrying truncating TTNmutations wasnotexplicitly addressedby Hermanet al.32 Asexpected, we identifiedseveral mutations in the majorityof patients, however, we focused onthe pathogenic mutations and VUS3s. Other identified mutations(VUS1s and VUS2s; see Supplementary material online, Table S2)might be benign genetic variations, but some may also contributeto the development of disease in these families. Some of theseVUSs might even be independently pathogenic, but additionaltesting is needed to confirm this (this might be the case for twoVUS2s in TTN [p.Arg1408Cys (c.4222C.T) in GER2, andp.Glu2076Gly (c.6227A.G) in GER6]). Other possibilities are thatthese VUSs may act as modifiers, or that they are risk factors with alow penetrance.
The great majority of pathogenic mutations and VUS3s (7 of 10)were in the TTN gene, which encodes the giant sarcomeric proteintitin. It was recently reported that truncating mutations in TTNaccount for a significant portion (�25%) of the genetic aetiology infamilial DCM.32 The high yield of pathogenic mutations and VUS3sin TTN in our cohort of familial PPCM/DCM cases (39%; 7 of 18) sug-gests that TTN mutations are specifically related to PPCM. Changes inisoform expression and phosphorylation status of titin have beenreported in acquired forms of heart failure (reviewed by Hildalgoand Granzier).39 We were able to measure functional propertiesand titin isoform composition in heart tissue from one of thePPCM patients with a VUS3 in TTN. The passive force was twice aslow as the value previously reported in control groups, and was asso-ciated with a shift towards the more compliant N2BA titin isoform.The shift towards more compliant N2BA has been reported inhuman heart failure.30,40,41 Overall, our data from functional andprotein analyses support the pathogenicity of this particular TTN mu-tation. We still classify this mutation as VUS3; however, extended ex-perience with these functional analyses might drive us to re-classifythis VUS3 towards a pathogenic mutation. Recent studies indicatedthat titin phosphorylation is indirectly altered by increased oxidativestress42 and, as such, may represent a likely pathomechanism inPPCM. Future studies will need to reveal the functional deficitsinduced by mutations in the TTN gene in relation to high oxidativestress, as present in PPCM.
There may be genetic factors specific for PPCM development, forexample a factor tentatively underlying the geographical hotspot ofincidence in Haiti, and a locus near the PTHLH gene reportedby Horne et al.43 We only focused on the STAT3 gene as a possiblespecific genetic factor for PPCM. Because mice with cardiomyo-cyte-specific deletion of STAT3 develop PPCM,7 STAT3 might alsobe involved in human PPCM, but there are no human genetic datasupporting this yet. STAT3 mutations are so far only known tocause hyper-IgE syndrome.44 In contrast to the PPCM cases,some women in our PPCM/DCM families went through several
pregnancies without developing PPCM. We therefore hypothesizedthat STAT3 mutations in the PPCM cases of these families contributedto the development of PPCM, in addition to an underlyingcardiomyopathy-related mutation. However, we found no STAT3pathogenic mutations or VUSs in these PPCM cases, which was con-sistent with previous findings.7 Exome sequencing of rare familialPPCM cases could lead to identifying novel genetic factors specificfor PPCM. However, this approach is limited by the fact that familialPPCM cases with more than two affected relatives or with affecteddistant relatives are lacking. An alternative strategy could be tocompare the data from exome sequencing on different PPCMcases in order to identify a shared genetic cause, but this might notlead to a result because the causal genetic factor may well beunique to each family.
LimitationsOne limitation of our study is that it does not provide data on the fre-quency of familial disease in PPCM. Currently, we only have data froma German cohort reporting a positive family history for cardiomyop-athy in 16.5% of PPCM cases,13 but we hope to gain more informationvia the Peripartum Cardiomyopathy Registry of EURObservationalResearch Programme (www.eorp.org) (unpublished data, 2013,manuscript submitted to European Journal of Heart Failure). Anotherlimitation is that retrieving information on larger deletions/duplica-tions from NGS data is not possible yet, although software toenable such analysis is being developed. We may therefore havemissed that type of mutation in our analyses. A further limitation isthe difficulty of judging which TTN mutations are pathogenic, giventhe presence of truncating TTN mutations in the general populationand reported truncating mutations that do not segregate withdisease in DCM families.32,38,45 In contrast to the latter observation,we were able to show co-segregation of truncating TTN mutations/VUS3s in five of our families (NL1, NL6, NL9, NL10, and NL11;Table 2), and we have data from functional and protein analyses sup-porting the pathogenicity of one likely pathogenic TTN mutation(GER4 II:1). Additional functional studies on TTN mutations and col-lection of large families carrying these mutations are needed. More-over, although our findings suggest a specific role for TTN mutationsin families with PPCM and DCM, we do realize that the number offamilies studied is currently too small to definitely conclude this.Finally, we were lacking some clinical data, especially of cases thatshowed clinical characteristics suggestive of PPCM.
Conclusions and practical implicationsPotentially causal mutations in cardiomyopathy-related genes arecommon in families with both PPCM and DCM, in particular TTNmutations. The targeted NGS approach we applied has beenshown to be suitable for identifying such mutations. Functionalstudies as performed in the present study may provide a futuretool to confirm pathogenicity of TTN mutations. Our resultsprovidemoresupport for the earlierfinding thatPPCMcanbeamani-festation of familial DCM. Cascade genetic screening can identify rela-tives at risk in those families in which an underlying mutation has beenidentified. Moreover, the presence of an underlying mutation or apositive family history for cardiomyopathy in a PPCM patient maybe a prognostic factor for low recovery rate.
Titin gene mutations in PPCM/DCM families 2171
at University of G
roningen on August 21, 2014
http://eurheartj.oxfordjournals.org/D
ownloaded from

Supplementary materialSupplementary material is available at European Heart Journal online.
AcknowledgementsWe thank all the patients who participated in this study; the StudyGroup on PPCM of the Heart Failure Association of the EuropeanSociety of Cardiology, Birgit Sikkema-Raddatz for her help in validat-ing and implementing the targeted enrichment kit; Ludolf Boven,Eddy de Boer, and Lennart Johansson for technical assistance; WiesLommen for assistance with functional analyses; Nicolaas de Jonge,cardiologist, for cardiac evaluation of family NL10; Wilma van derRoest, genetic counsellor, for counselling some of the Dutch families;and Jackie Senior for editing this manuscript.
FundingRowida Almomani was supported by the Netherlands Heart Foundation(grant 2010B164).
Conflict of interest: none declared.
References1. Sliwa K, Hilfiker-Kleiner D, Petrie MC, Mebazaa A, Pieske B, Buchmann E,
Regitz-Zagrosek V, Schaufelberger M, Tavazzi L, van Veldhuisen DJ, Watkins H,Shah AJ, Seferovic PM, Elkayam U, Pankuweit S, Papp Z, Mouquet F, McMurray JJ.Current state of knowledge on aetiology, diagnosis, management, and therapy ofperipartum cardiomyopathy: a position statement from the Heart Failure Associ-ation of the European Society of Cardiology Working Group on peripartum cardio-myopathy. Eur J Heart Fail 2010;12:767–778.
2. Demakis JG, Rahimtoola SH. Peripartum cardiomyopathy. Circulation 1971;44:964–968.
3. Hibbard JU, Lindheimer M, Lang RM. A modified definition for peripartum cardiomy-opathy and prognosis based on echocardiography. J Obstet Gynecol 1999;94:311–316.
4. Pearson GD, Veille JC, Rahimtoola S, Hsia J, Oakley CM, Hosenpud JD, Ansari A,Baughman KL. Peripartum cardiomyopathy. National Heart, Lung, and Blood Insti-tute and Office of Rare Diseases (National Institutes of Health) workshop recom-mendations and review. JAMA 2000;283:1183–1188.
5. Sliwa K, Fett J, Elkayam U. Peripartum cardiomyopathy. Lancet 2006;368:687–693.6. Hilfiker-Kleiner D, Sliwa K, Drexler H. Peripartum cardiomyopathy: recent insights
in its pathophysiology. Trends Cardiovasc Med 2008;18:173–179.7. Hilfiker-Kleiner D, Kaminski K, Podewski E, Bonda T, Schaefer A, Sliwa K, Forster O,
Quint A, Landmesser U, Doerries C, Luchtefeld M, Poli V, Schneider MD,Balligand J-L, Desjardins F, Ansari A, Struman I, Nguyen NQN, Zschemisch NH,Klein G, Heusch G, Schulz R, Hilfiker A, Drexler H. A cathepsin D-cleaved 16 kDaform of prolactin mediates postpartum cardiomyopathy. Cell 2007;128:589–600.
8. Hilfiker-Kleiner D, Meyer GP, Schieffer E, Goldmann B, Podewski E, Struman I,Fischer P, Drexler H. Recovery from postpartum cardiomyopathy in 2 patients byblocking prolactin releasewith bromocriptine. J Am Coll Cardiol 2007;50:2354–2345.
9. Sliwa K, Blauwet L, Tibazarwa K, Libhaber E, Smedema JP, Becker A, McMurray J,Yamac H, Labidi S, Struman I, Hilfiker-Kleiner D. Evaluation of bromocriptine inthe treatment of acute severe peripartum cardiomyopathy: a proof-of-conceptpilot study. Circulation 2010;121:1465–1473.
10. Patten IS, Rana S, Shahul S, Rowe GC, Jang C, Liu L, Hacker MR, Rhee JS, Mitchell J,Mahmood F, Hess P, Farrell C, Koulisis N, Khankin EV, Burke SD, Tudorache I,Bauersachs J, del Monte F, Hilfiker-Kleiner D, Karumanchi SA, Arany Z. Cardiacangiogenic imbalance leads to peripartum cardiomyopathy. Nature 2012;485:333–338.
11. van Spaendonck-Zwarts KY, van Tintelen JP, van Veldhuisen DJ, van der Werf R,Jongbloed JD, Paulus WJ, Dooijes D, van den Berg MP. Peripartum cardiomyopathyas a part of familial dilated cardiomyopathy. Circulation 2010;121:2169–2175.
12. Morales A, Painter T, Li R, Siegfried JD, Li D, Norton N, Hershberger RE. Rare variantmutations in pregnancy-associated or peripartum cardiomyopathy. Circulation 2010;121:2176–2182.
13. Haghikia A, Podewski E, Libhaber E, Labidi S, Fischer D, Roentgen P, Tsikas D,Jordan J, Lichtinghagen R, von Kaisenberg CS, Struman I, Bovy N, Sliwa K,Bauersachs J, Hilfiker-Kleiner D. Phenotyping and outcome on contemporary man-agement in a German cohort of patients with peripartum cardiomyopathy. Basic ResCardiol 2013;108:366.
14. Toib A, Grange DK, Kozel BA, Ewald GA, White FV, Canter CE. Distinct clinical andhistopathological presentations of Danon cardiomyopathy in young women. J AmColl Cardiol 2010;55:408–410.
15. Møller DV, Andersen PS, Hedley P, Ersbøll MK, Bundgaard H, Moolman-Smook J,Christiansen M, Køber L. The role of sarcomere gene mutations in patients with idio-pathic dilated cardiomyopathy. Eur J Hum Genet 2009;17:1241–1249.
16. Ntusi NB, Wonkam A, Shaboodien G, Badri M, Mayosi BM. Frequency and clinicalgenetics of familial dilated cardiomyopathy in Cape Town: implications for the evalu-ation of patients with unexplained cardiomyopathy. S Afr Med J 2011;101:394–398.
17. Baruteau AE, Leurent G, Schleich JM, Gervais R, Daubert JC, Mabo P. Can peripar-tum cardiomyopathy be familial? Int J Cardiol 2009;137:183–185.
18. Pierce JA, Price BO, Joyce JW. Familial occurrence of postpartal heart failure. ArchIntern Med 1963;111:151–155.
19. Voss EG, Reddy CVR, Detrano R, Virmani R, Zabriskie JB, Fotino M. Familial dilatedcardiomyopathy. Am J Cardiol 1984;54:456–457.
20. Massad LS,ReissCK,Mutch DG, HaskelEJ. Familial peripartum cardiomyopathyaftermolar pregnancy. Obstet Gynecol 1993;81:886–888.
21. Pearl W. Familial occurrence of peripartum cardiomyopathy. Am Heart J 1995;129:421–422.
22. Fett JD, Sundstrom BJ, King ME, Ansari AA. Mother-daughter peripartum cardiomy-opathy. Int J Cardiol 2002;86:331–332.
23. Ferguson JE, Harney KS, Bachicha JA. Peripartum maternal cardiomyopathy withidiopathic cardiomyopathy in the offspring. A case report. J Reprod Med 1986;31:1109–1112.
24. Strung P. Familial cardiomyopathy. Peripartum and primary congestive cardiomyop-athy in a sister and brother. Ugeskr Laeger 1976;138:2567–2569.
25. Mestroni L, Maisch B, McKenna WJ, Schwartz K, Charron P, Rocco C, Tesson F,Richter A, Wilke A, Komajda M. on behalf of the Collaborative Research Group ofthe European Human and Capital Mobility Project on Familial Dilated Cardiomyop-athy. Guidelines for the study of familial dilated cardiomyopathy. Eur Heart J 1999;20:93–102.
26. Posafalvi A, Herkert JC, Sinke RJ, van den Berg MP, Mogensen J, Jongbloed JD, vanTintelen JP. Clinical utility gene card for: dilated cardiomyopathy (CMD). Eur JHum Genet 2013;21:doi:10.1038/ejhg.2012.276.
27. Sikkema-Raddatz B, Johansson LF, de Boer EN, Almomani R, Boven LG, van denBerg MP, van Spaendonck-Zwarts KY, van Tintelen JP, Sijmons R, Jongbloed JDH,Sinke RJ. Targeted next generation sequencing can replace sanger sequencing in clin-ical diagnostics. Human Mutation 2013;34:1035–1042.
28. van Spaendonck-Zwarts KY, van Rijsingen IA, van den Berg MP, Lekanne Deprez RH,Post JG, van Mil AM, Asselbergs FW, Christiaans I, van Langen IM, Wilde AA, deBoer RA, Jongbloed JD, Pinto YM, van Tintelen JP. Genetic analysis in 418 indexpatients with idiopathic dilated cardiomyopathy: overview of 10 years’ experience.Eur J Heart Fail 2013;15:628–636.
29. van Dijk SJ, Paalberends ER, Najafi A, Michels M, Sadayappan S, Carrier L,Boontje NM, Kuster D, van Slegtenhorst M, Dooijes D, dos Remedios C, tenCate FJ, Stienen GJM, van der Velden J. Contractile dysfunction irrespective of themutant protein in human hypertrophic cardiomyopathy with normal systolic func-tion. Circ Heart Fail 2012;5:36–46.
30. Borbely A, Falcao-Pires I, van Heerebeek L, Hamdani N, Edes I, Gavina C,Leite-Moreira AF, Bronzwaer JGF, Papp Z, van der Velden J, Stienen GJM,Paulus WJ. Hypophosphorylation of the stiff N2B titin isoform raises cardiomyocyteresting tension in failing human myocardium. Circ Res 2009;104:780–786.
31. Tibazarwa K, Sliwa K, Wonkam A, Mayosi BM. Peripartum cardiomyopathy and fa-milial dilated cardiomyopathy: a tale of two cases. Cardiovasc J Afr 2013;24:e4–e7.
32. Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L,DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR,Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ,Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJ, Cook SA, Mestroni L,Seidman JG, Seidman CE. Truncations of titin causing dilated cardiomyopathy. NEngl J Med 2012;366:619–628.
33. Blauwet LA, Libhaber E, Forster O, Tibazarwa K, Mebazaa A, Hilfiker-Kleiner D,Sliwa K. Predictors of outcome in 176 South African patients with peripartum car-diomyopathy. Heart 2013;99:308–313.
34. Goland S, Bitar F, Modi K, Safirstein J, Ro A, Mirocha J, Khatri N, Elakayam U. Evalu-ationof the clinical relevance of baseline left ventricularejection fractionas a predict-or of recovery or persistence of severe dysfunction in women in the United Stateswith peripartum cardiomyopathy. J Card Fail 2011;17:426–430.
35. Fett JD, Christie LG, Carraway RD, Murphy JG. Five-year prospective study of theincidence and prognosis of peripartum cardiomyopathy at a single institution.Mayo Clin Proc 2005;80:1602–1606.
36. Duran N, Gunes H, Duran I, Biteker M, Ozkan M. Predictors of prognosis in patientswith peripartum cardiomyopathy. Int J Gynaecol Obstet 2008;101:137–140.
37. Norton N, Robertson PD, Rieder MJ, Zuchner S, Rampersaud E, Martin E, Li D,Nickerson DA, Hershberger RE. National Heart, Lung and Blood Institute GO
K.Y. van Spaendonck-Zwarts et al.2172
at University of G
roningen on August 21, 2014
http://eurheartj.oxfordjournals.org/D
ownloaded from
Exome Sequencing Project. Evaluating pathogenicity of rare variants from dilatedcardiomyopathy in the exome era. Circ Cardiovasc Genet 2012;5:167–174.
38. Golbus JR, Puckelwartz MJ, Fahrenbach JP, Dellefave-Castillo LM, Wolfgeher D,McNally EM. Population-based variation in cardiomyopathy genes. Circ CardiovascGenet 2012;5:391–399.
39. Hidalgo C, Granzier H. Tuning the molecular giant titin through phosphorylation:Role in health and disease. Trends Cardiovasc Med 2013;23:165–171.
40. Makarenko I, Opitz CA, Leake MC, Kulke M, Gwathmey JK, del Monte F, Hajjar RJ,Linke WA. Passive stiffness changes caused by upregulation of compliant titin iso-forms in human dilated cardiomyopathy hearts. Circ Res 2004;95:708–716.
41. Nagueh SF, Shah G, Wu Y, Torre-Amione G, King NM, Lahmers S, Witt CC, Becker K,Labeit S, Granzier HL. Altered titin expression, myocardial stiffness, and left ventricularfunction in patients with dilated cardiomyopathy. Circulation 2004;110:155–162.
42. van Heerebeek L, Hamdani N, Falcao-Pires I, Leite-Moreira AF, Begieneman MP,Bronzwaer JG, van der Velden J, Stienen GJ, Laarman GJ, Somsen A, Verheugt FW,
Niessen HW, Paulus WJ. Low myocardial protein kinase G activity in heart failurewith preserved ejection fraction. Circulation 2012;126:830–839.
43. Horne BD, Rasmusson KD, Alharethi R, Budge D, Brunisholz KDMetz T, Carlquist JF, Connolly JJ, Porter TF, Lappe DL, Muhlestein JB, Silver R,Stehlik J, Park JJ, May HT, Bair TL, Anderson JL, Renlund DG, Kfoury AG. Genome-wide significance and replication of the chromosome 12p11.22 locus near thePTHLH gene for peripartum cardiomyopathy. Circ Cardiovasc Genet 2011;4:359–366.
44. MinegishiY, SaitoM,TsuchiyaS,Tsuge I,TakadaH,HaraT,KawamuraN,ArigaT,Pasic S,StojkovicO,MetinA,KarasuyamaH.Dominant-negativemutations in theDNA-bindingdomain of STAT3 cause hyper-IgE syndrome. Nature 2007;448:1058–1062.
45. Norton N, Li D, Rampersaud E, Morales A, Martin ER, Zuchner S, Guo S,Gonzalez M, Hedges DJ, Robertson PD, Krumm N, Nickerson DA,Hershberger RE. Exome sequencing and genome-wide linkage in 17 families illus-trates the complex contribution of TTN truncating variants to dilated cardiomyop-athy. Circ Cardiovasc Genet 2013;6:144–153.
CARDIOVASCULAR FLASHLIGHT. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
doi:10.1093/eurheartj/ehu086Online publish-ahead-of-print 26 February 2014
Multiparametric assessment of myocarditis using simultaneous positronemission tomography/magnetic resonance imagingFelix Nensa1*, Thorsten D. Poeppel2, Peter Krings3, and Thomas Schlosser1
1Department of Diagnostic and Interventional Radiology and Neuroradiology, University Hospital Essen, Essen, Germany; 2Department of Nuclear Medicine, University HospitalEssen, Essen, Germany; and 3Department of Cardiology, University Hospital Essen, Essen, Germany
* Corresponding author. Tel: +49 20172384595, Fax: +49 2017239471528, Email: [email protected]
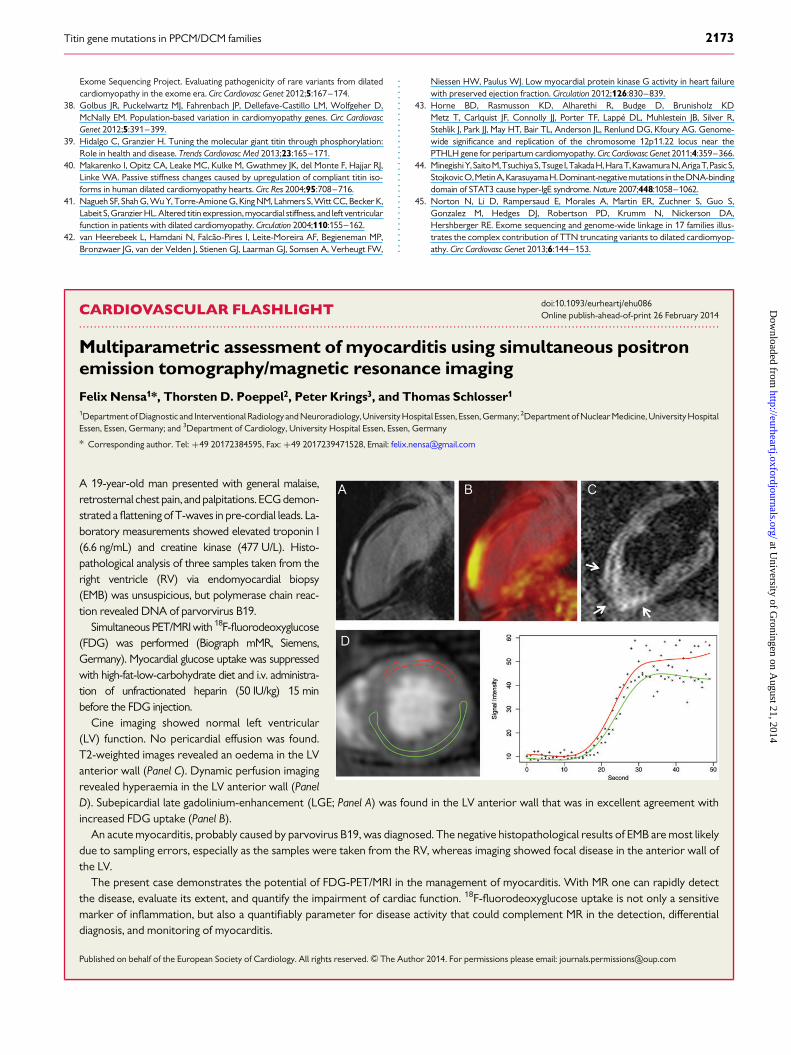
A 19-year-old man presented with general malaise,retrosternal chest pain, and palpitations. ECG demon-strated a flattening of T-waves in pre-cordial leads. La-boratory measurements showed elevated troponin I(6.6 ng/mL) and creatine kinase (477 U/L). Histo-pathological analysis of three samples taken from theright ventricle (RV) via endomyocardial biopsy(EMB) was unsuspicious, but polymerase chain reac-tion revealed DNA of parvorvirus B19.
Simultaneous PET/MRI with 18F-fluorodeoxyglucose(FDG) was performed (Biograph mMR, Siemens,Germany). Myocardial glucose uptake was suppressedwith high-fat-low-carbohydrate diet and i.v. administra-tion of unfractionated heparin (50 IU/kg) 15 minbefore the FDG injection.
Cine imaging showed normal left ventricular(LV) function. No pericardial effusion was found.T2-weighted images revealed an oedema in the LVanterior wall (Panel C). Dynamic perfusion imagingrevealed hyperaemia in the LV anterior wall (PanelD). Subepicardial late gadolinium-enhancement (LGE; Panel A) was found in the LV anterior wall that was in excellent agreement withincreased FDG uptake (Panel B).
An acute myocarditis, probably caused by parvovirus B19, was diagnosed. The negative histopathological results of EMB are most likelydue to sampling errors, especially as the samples were taken from the RV, whereas imaging showed focal disease in the anterior wall ofthe LV.
The present case demonstrates the potential of FDG-PET/MRI in the management of myocarditis. With MR one can rapidly detectthe disease, evaluate its extent, and quantify the impairment of cardiac function. 18F-fluorodeoxyglucose uptake is not only a sensitivemarker of inflammation, but also a quantifiably parameter for disease activity that could complement MR in the detection, differentialdiagnosis, and monitoring of myocarditis.
Published on behalf of the European Society of Cardiology. All rights reserved. & The Author 2014. For permissions please email: [email protected]
Titin gene mutations in PPCM/DCM families 2173
at University of G
roningen on August 21, 2014
http://eurheartj.oxfordjournals.org/D
ownloaded from
Copyright © 2022 FDOKUMEN




















