
Diversity of Glutathione S-Transferases (GSTs) in Cyanobacteria ...
Upload
global-studiesCategory
view
0download
0
J. Mol. Biol. (1996) 255, 289-309
. IMB Three-dimensional Structure of Glutathione
o
S-transferase from Arabidopsis thaliana at 2.2 A Resolution: Structural Characterization of Herbicide-conjugating Plant Glutathione S-transferases and a Novel Active Site Architecture
Peter Reinemer ~,2., Lars Prade 2, Peter Hof 2, Torsten Neuefeind ~,2 Robert Huber 2., Rolf ZettP, Klaus Palme 3, Jeff SchelP, Ingo Koelln 4 Hans D. Bartunik 4 and Barbara Bieseler ~*
IBayer AG, GB Pflanzenschutz (PF-F Biotechnologie) Pflanzenschutzzentrum Monheim, Alfred-Nobel-Str. 50, D-51368 Leverkusen Germany
2Max-Planck-Institut ffir Biochemie, Abt. Strukturforschung, Am Klopferspitz 18a, D-82152 Martinsried, Germany
3Max-Planck-Institut ffir Z fichtungsforsch ung Carl-von-Linnd-Weg 10 D-50829 KiJln, Germany
4Max-Planck-Arbeitsgruppen ffir Strukurelle Molekularbiologie, c/o DESY Notkestr. 85, D-22603 Hamburg, Germany
*Corresponding authors
Glutathione S-transferases (GST) are a family of multifunctional enzymes involved in the metabolization of a broad variety of xenobiotics and reactive endogenous compounds. The interest in plant glutathione S-transferases may be attributed to their agronomic value, since it has been demonstrated that glutathione conjugation for a variety of herbicides is the major resistance and selectivity factor in plants.
The three-dimensional structure of glutathione S-transferase from the plant Arabidopsis thaliana has been solved by multiple isomorphous replacement and multiwavelength anomalous dispersion techniques at 3/~ resolution and refined to a final crystallographic R-factor of 17.5% using data from 8 to 2.2 ~ resolution. The enzyme forms a dimer of two identical subunits each consisting of 211 residues. Each subunit is characterized by the GST-typical modular structure with two spatially distinct domains. Domain I consists of a central four-stranded [3-sheet flanked on one side by two a-helices and on the other side by an irregular segment containing three short 310-helices, while domain II is entirely helical. The dimeric molecule is globular with a prominent large cavity formed between the two subunits. The active site is located in a cleft situated between domains I and II and each subunit binds two molecules of a competitive inhibitor S-hexylglutathione. Both hexyl moieties are oriented parallel and fill the H-subsite of the enzyme's active site. The glutathione peptide of one inhibitor, termed productive binding, occupies the G-subsite with multiple interactions similar to those observed for other glutathione S-transferases, while the glutathione backbone of the second inhibitor, termed unproductive binding, exhibits only weak interactions mediated by two polar contacts. A most striking difference from the mammalian glutathione S-transferases, which share a conserved catalytic tyrosine residue, is the lack of this tyrosine in the active site of the plant glutathione S-transferase.
tD 1996 Academic Press Limited
Keywords: glutathione S-transferase; crystallography; herbicide detoxification; protein structure; glutathione-binding domain
Present address: P. Reinemer, Bayer AG, GB Pharma (PH-R CR NASP), Postfach 10 17 09, D-42096 Wuppertal, Germany
Abbreviations used: GST, glutathione S-transferase; MIR, multiple isomorphous replacement; MAD, multiwavelength anomalous dispersion; r.m.s., root-mean-square; CDNB, 1-chloro-2,4-dinitrobenzene; EPNP, 1,2-epoxy-3-(p-nitrophenoxy) propane.
Introduction The glutathione S-transferases (GST, EC 2.5.1.18)
are a family of multifunctional enzymes found in all vertebrates, plants, insects, nematodes, yeasts and aerobic bacteria. They constitute a complex super- gene family that collectively metabolizes a broad variety of potentially toxic exogenous chemicals
0022-2836/96/020289-21 $12.00/0 iO 1996 Academic Press Limited

290 Structure of A. thaliana Glutathione S-transferase
(xenobiotics) and reactive endogenous compounds resulting from oxidative metabolism. Glutathione S-transferases primarily function as phase-II detox- ification enzymes (Jakoby & Ziegler, 1990) and catalyze the nucleophilic addition of glutathione to numerous chemically diverse compounds includ- ing alkyl- and arylhalides, lactones, epoxides, quin- ones, esters and activated alkenes (Mannervik & Danielson, 1988). As a consequence of the reaction a less reactive and more polar glutathionyl S-conju- gate is formed which, in animals, is further catabolized and subsequently excreted (Boyland & Chasseaud, 1969), while in plants the conjugation reaction is coupled to internal compartmentation or storage due to the lack of effective excretion pathways (Sandermann, 1992). Certain transferases can also catalyze a selenium-independent peroxi- dase activity (Prohaska, 1980) with organic per- oxides as substrates which thereby are reduced to alcohols (Mannervik & Danielson, 1988). In addition to their catalytic activity they can also function as ligand-binding proteins ("ligandins") and thereby facilitate the intracellular storage and transport of a variety of hydrophobic non-substrate compounds, including hormones, metabolites and drugs (Lis- towski, 1993). Due to their key role in xenobiotic metabolism, glutathione S-transferases have also been implicated in the development by organisms of natural and acquired resistance towards xenobiotic compounds such as carcinogens, therapeutic agents and pesticides (Hayes & Wolf, 1988; Tsuchida & Sato, 1992).
The cytosolic mammalian glutathione S-trans- ferases exist in multiple enzyme forms and their isozyme multiplicity, structure and function, and physiological significance has been studied in great detail (for recent reviews see Beckett & Hayes, 1993; Rushmore & Pickett, 1993; Armstrong, 1993; Dirr et al., 1994a). According to sequence simi- larities, they can be catalogued in five species-inde- pendent gene classes, referred to as Alpha, Mu, Pi (Mannervik et al., 1985), Sigma (Buetler & Eaton, 1992; Ji et al., 1995) and Theta (Hiratsuka et al., 1990; Meyer et al., 1991a). All known cytosolic isoenzymes have dimeric quarternary structures (Mr~50,000) assembled from identical or non-identical subunits of the same gene class. A variety of crystal struc- tures for mammalian isoenzymes from class Pi (porcine P1-1: Reinemer et al., 1991; Dirr et al., 1994b; human P1-1: Reinemer et al., 1992; mouse P1-1: Garcia-S~iez et al., 1994), class Mu (rat M1-1: Ji et al., 1992; human M2-2: Raghunathan et al., 1994) and class Alpha (human A1-1: Sinning et al., 1993) have been solved and thus elucidated not only the subunit architecture and quarternary structure but also provided a detailed picture of the active site geometry Each subunit in the dimeric structure has an active site that functions with kinetic indepen- dence (Danielson & Mannervik, 1985) and consists of a highly specific glutathione binding region and a less-specific hydrophobic binding region to accommodate the second substrate. In addition, evidence was provided that a conserved tyrosine
residue, located in the active site, was probably involved in catalysis. Subsequent134 the key role in catalysis of this residue was demonstrated in a variety of site-directed mutagenesis experiments, thereby providing a clearer understanding of the catalytic mechanism (Armstrong, 1993; Dirr et al., 1994a). Recentl~ crystal structures from glutathione S-transferases from Schistosoma japontcum (Lim et al., 1994; McTigue et al., 1995), squid (Ji et al., 1995) and Lucilia cuprina (Wilce et al., 1995) have been determined and provided first insights into non-mammalian glutathione S-transferase structure and function.
In contrast to the well established properties of the mammalian glutathione S-transferases, current knowledge on plant isoenzymes is rather limited and awaits further study The interest in plant glutathione S-transferases may be attributed to their agronomic value, since it has been demonstrated that glutathione conjugation for a variety of herbicides is the major resistance and selectivity factor in plants (Hatzios & Penner, 1982; Lamoureux & Rusness, 1989; Timmerman, 1989). The first glutathione S-transferase reported to participate in herbicide metabolism was isolated from maize (Frear & Swanson, 1970) and was responsible for the conjugation of atrazine (2-chloro-4-ethylamino- 6-isopropylamino-s-triazine) to a water-soluble metabolite. This ability to detoxify atrazine has been shown to be the primary factor in conferring resistance to this herbicide (Shimabukuro et al., 1970). Further evidence for the eminent role of these enzymes in atrazine metabolism was gained from the study of two Mississippi-inbred lines of maize, namely GT 112 RfRf (resistant line) and GT 112 (susceptible line). Shimabukuro et al. (1971) demon- strated that the ability of the sensitive line to metabolize atrazine was reduced significantly due to the lack of glutathione S-transferase activity Moreover, selective phytotoxicity by herbicides may be gained by the action of herbicide antidotes or safeners, that protect plants against toxic effects of herbicides by the selective induction of glutathione S-transferases (Hoffman, 1978). In addition, the phytotoxicity of herbicides can be increased by the action of herbicidal synergists, which inhibit glutathione S-transferase activity (Lamoureux & Rusness, 1987). The best analysed plant glutathione S-transferase system occurs in maize, where four isoforms, termed GST I to IV, have been character- ized in some detail (Mozer et al., 1983; Moore et al., 1986; Shah et al., 1986; Wiegand et al., 1986; Grove et al., 1988; O'Connell et al., 1988; Irzyk & Fuerst, 1993; Jepson et al., 1994; Irzyk et al., 1995). GST I and III are constitutively expressed, while GST II and IV are induced by treatment with herbicide safeners. The isoforms I, III and IV are homodimers com- posed of 29, 26 and 27 kDa subunits, while GST II is a heterodimer composed of a GSTI-29 kDa and a GSTIV-27 kDa subunit. However, at present the relationship at a molecular level between these enzymes and the isoenzyme(s) conjugating atrazine is unclear. Moreover, the relationship between

Structure of A. thaliana Glutathione S-transferase 291
maize glutathione S-transferases and those in other higher plants that have been impli- cated in the detoxification of various herbicides, including diphenyle ther (Frear & Swanson, 1973; Shimabukuro et al., 1973; Diesperger & Sandermann, 1979), acetanilide and thiocarbamate herbicides (Leavitt & Penner, 1979), is also in question and awaits fur ther s tudy
Plant glutathione S-transferases have also been shown to be induced by phytohormone treatment (Meyer et al., 1991b; Takahashi & Nagata, 1992; Droog et al., 1993; Zhou & Goldsbrough, 1993) or pathogenetic attack (Hahn & Strittmatter, 1994). Furthermore, photoaffinity labelling demonstrated the binding of indole 3-acetic acid to glutathione S-transferases (Bilang et al., 1993; Zettl et al., 1994), suggesting that they are either regulated by this phytohormone or are involved in its metabolism.
We present here the three-dimensional s tructure of glutathione S-transferase from Arabidopsis thaliana, which was recently identified and cloned (Zhou & Goldsbrough, 1993; Zettl et al., 1994). The Arabidopsis thaliana isoenzyme, the first plant glutathione S-transferase whose structure has been solved, shares significant sequence homology ( ~ 60%) with the maize isoenzymes, suggesting that its s tructure may serve as a model system to s tudy herbicide resistance and selectivity in plants.
Results
Structure determination and quality of the model
The A. thaliana GST amino acid sequence has only little similarity to the Alpha-, Mu- and Pi-class GSTs from mammals; they share a pairwise sequence identi ty of about 20%. Consequently; i somorphous replacement techniques were used to determine the structure, since Patterson search methods using
Alpha-, Mu- and Pi-class GST crystal structures (reviewed by Dirr et al., 1994a) as seai'ch models failed.
Two heavy atom derivatives were prepared by soaking crystals with diazotized 3,5-diiodosul- phanilic acid (Helmkamp & Sears, 1970) and by incorporation of selenomethionine in the recombi- nant enzyme, respectively X-ray reflection data were collected for these two derivatives (DISA and SEME; see Table I for data collection statistics) using CuK~ X-radiation (k = 1.5418 A) and analysed (for details see experimental section) with four iodine and four selenium positions. Each derivative was interpreted with two pairs of local symmetry-re- lated binding sites using a prel iminary orientation for the non-crystallographic 2-fold symmet ry axis (~ = 78 °, qb = 0 °, ~: = 180 ° with a height of 62.5% of the peak originating from the crystallographic 6-fold) derived from a Patterson self-rotation function.
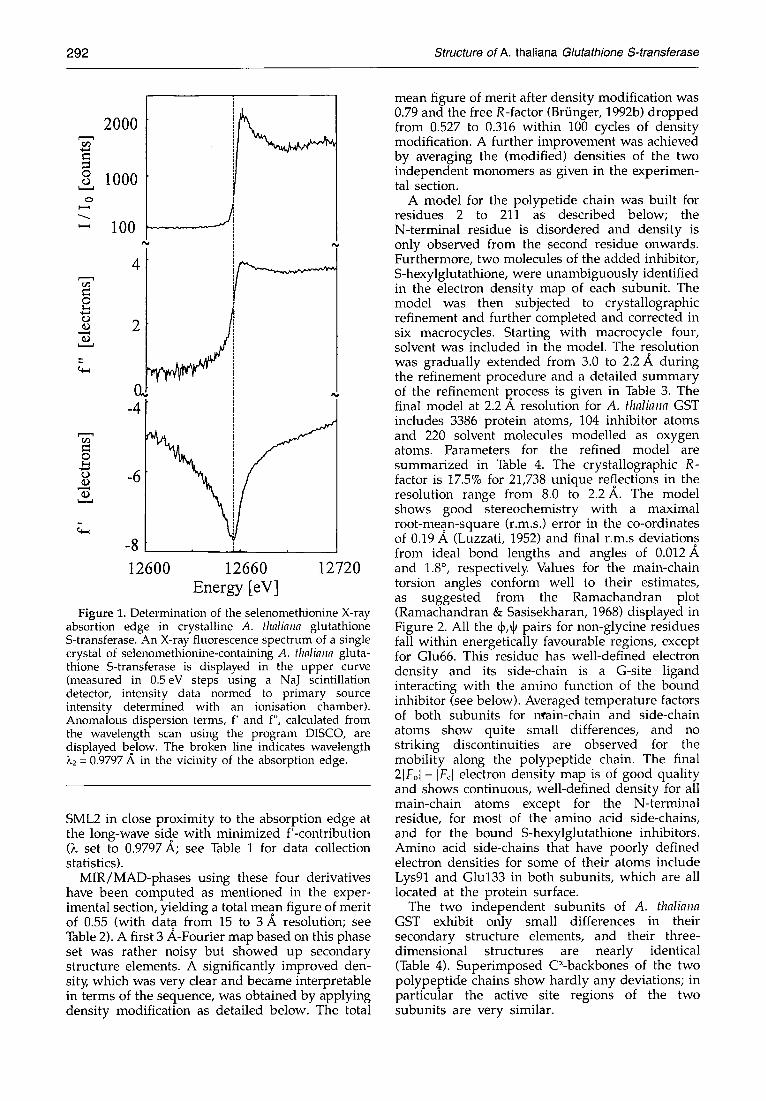
In order to improve the phasing, multiwavelength anomalous data of selenomethionine-containing crystals of A. thaliana GST were collected with synchrotron radiation using beamline BW-6 at the MPG-outstation at DESY (Deutsches Elektronen- synchrotron, Hamburg , Germany). X-ray fluor- escence spectroscopy as described in the experimental section was used to determine the exact location of the selenium K-absorption edge. The X-ray fluorescence spectrum of a selenome- thionine-containing A. thaliana GST crystal, measured through the K-absorption edge of selenium and anomalous scattering factors derived from this spec t rum are shown in Figure 1. The position of the absorption edge was determined to be at an energy of 12,660 eV. Subsequentl34 X-ray reflection data were collected at either side of the absorption edge; SML1 somewhat distinct at the short-wave side of the absorption edge° with maximized f"-contribution (X set to 0.9295 A) and
Table 1. Data collection statistics for native and derivative crystals of glutathione S-transferase from A. thaliana
Derivative ~ NATI DISA SEME SML1 SML2 InstrumenP FAST FAST FAST BW-6 BW-6 Wavelength (.&) 1.5418 1.5418 1.5418 0.9295 0.9797 Unique reflections 23438 15813 15092 34174 32524 Anomalous measurements 13525 12495 32545 30546 Resolution limit (A) 2.2 2.5 2.5 2.0 2.0 Multiplicity c 4.6 4.6 5.0 7.1 4.8 Completeness d 88.9 (46.2) 99.0 (95.6) 97.9 (95.6) 98.9 (96.8) 95.9 (94.7) Rm~,g~ ~ 0.094 0.085 0.108 0.055 0.054 Rico f 0.138 0.082 0.102 0.104
NATI, native A. thaliana GST, DISA, 10 M diazotized 3,5-diiodosulphanilic acid for 14 hours; SEME, SML1, SML2, multiwavelength data for selenomethionine-substituted A. thaliana GST.
b FAST, FAST area television detector, conventional CuK~ rotating anode source; BW-6, Hendrix-Lentfer imaging plate scanner, 300 mm diameter, beamline BW-6 at DORIS (MPG outstation at DESY, Hamburg).
c Redundancy of the data, defined as the ratio of the number of measured and the number of unique reflections. d Ratio of the number of possible unique reflections and the number of present unique reflections in percent;
resolution ranges are o~J to 2.2 .~ (2.23 to 2.20 A) for the native and co to 3.0 .& (3.03 to 3.00 ,/0 for the derivatives, respectively.
e RmCrg~ = ~ . h ~ i l l ( h , i ) - <l(h))l/Y~hl(h, i), where l(h, i) is the intensity value of the ith measurement of h and <I(h)) is the corresponding mean value of h for all i measurements of h; the summation is over all measurements.
r Ri~, = I~l,klllFJ, I- IFPHII/Y-J,~dFP[, where IFPI and IFpHI are the structure factor amplitudes for the native and the heavy-atom derivatized crystal, respectively.
292 Structure of A. thaliana Glutathione S-transferase
o ¢3
2000
1000
100 . . . . . . . . ! ! " .
4
~ 2
-4
o
o -6
-8 i,
12600 12660 12720 Energy [eV]
Figure 1. Determination of the selenomethionine X-ray absortion edge in crystalline A. flmliana glutathione S-transferase. An X-ray fluorescence spectrum of a single crystal of selenomethionine-containing A. thaliana gluta- thione S-transferase is displayed in the upper curve (measured in 0.5eV steps using a NaJ scintillation detector, intensity data normed to primary source intensity determined with an ionisation chamber). Anomalous dispersion terms, f' and f", calculated from the wavelength scan using the program DISCO, are displayed below. The broken line indicates wavelength )~2 = 0.9797 A in the vicinity of the absorption edge.
SML2 in close proximity to the absorption edge at the long-wave side with minimized f'-contribution (k set to 0.9797 A; see Table 1 for data collection statistics).
MIR/MAD-phases using these four derivatives have been computed as mentioned in the exper- imental section, yielding a total mean figure of merit of 0.55 (with data from 15 to 3 A resolution; see Table 2). A first 3 A-Fourier map based on this phase set was rather noisy but showed up secondary structure elements. A significantly improved den- sit)~ which was very clear and became interpretable in terms of the sequence, was obtained by applying density modification as detailed below. The total
mean figure of merit after density modification was 0.79 and the free R-factor (Brfinger, 1992b) dropped from 0.527 to 0.316 within 100 cycles of density modification. A further improvement was achieved by averaging the (modified) densities of the two independent monomers as given in the experimen- tal section.
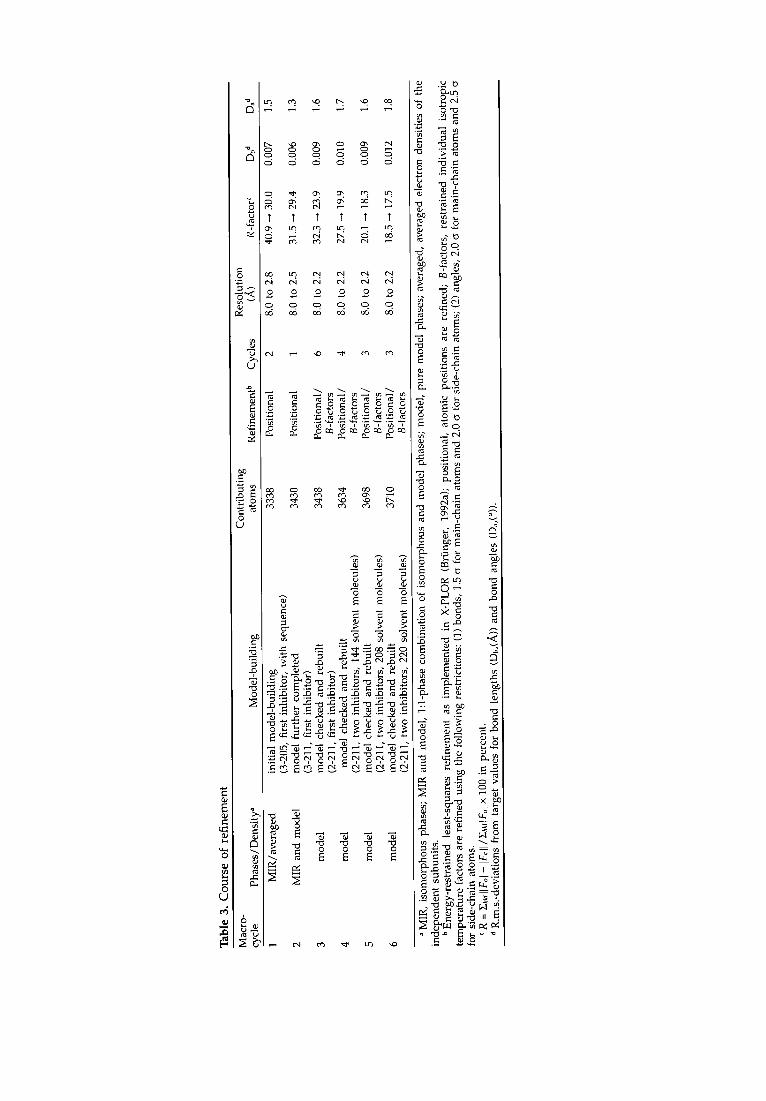
A model for the polypetide chain was built for residues 2 to 211 as described below; the N-terminal residue is disordered and density is only observed from the second residue onwards. Furthermore, two molecules of the added inhibitor, S-hexylglutathione, were unambiguously identified in the electron density map of each subunit. The model was then subjected to crystallographic refinement and further completed and corrected in six macrocycles. Starting with macrocycle four, solvent was included in the model. The resolution was gradually extended from 3.0 to 2.2 A during the refinement procedure and a detailed summary of the refinement process is given in Table 3. The final model at 2.2 A resolution for A. thaliana GST includes 3386 protein atoms, 104 inhibitor atoms and 220 solvent molecules modelled as oxygen atoms. Parameters for the refined model are summarized in Table 4. The crystallographic R- factor is 17.5% for 21,738 unique reflections in the resolution range from 8.0 to 2.2 A. The model shows good stereochemistry with a maximal root-mean-square (r.m.s.) error in the co-ordinates of 0.19 A (Luzzati, 1952) and final r.m.s deviations from ideal bond lengths and angles of 0.012 and 1.8 ° , respectively. Values for the main-chain torsion angles conform well to their estimates, as suggested from the Ramachandran plot (Ramachandran & Sasisekharan, 1968) displayed in Figure 2. All the qb,~ pairs for non-glycine residues fall within energetically favourable regions, except for Glu66. This residue has well-defined electron density and its side-chain is a G-site ligand interacting with the amino function of the bound inhibitor (see below). Averaged temperature factors of both subunits for n, rain-chain and side-chain atoms show quite small differences, and no striking discontinuities are observed for the mobility along the polypeptide chain. The final 21Fol- IFcl electron density map is of good quality and shows continuous, well-defined density for all main-chain atoms except for the N-terminal residue, for most of the amino acid side-chains, and for the bound S-hexylglutathione inhibitors. Amino acid side-chains that have poorly defined electron densities for some of their atoms include Lys91 and Glu133 in both subunits, which are all located at the protein surface.
The two independent subunits of A. thaliana GST exhibit only small differences in their secondary structure elements, and their three- dimensional structures are nearly identical (Table 4). Superimposed C~-backbones of the two polypeptide chains show hardly any deviations; in particular the active site regions of the two subunits are very similar.

Structure of A. thaliana Glutathione S-transferase 293
Table 2. Summary of phasing statistics Derivative SEME SML1 SML2 DISA No. of sites ~ 4 4 4 4 Phasing power s 0.86 1.10 0.78 0.90 Rc',,lli~' 0.87 0.82 0.90 0.86
Resolution (A) 15-I0.0 7.5 6.0 5.0 4.3 3.8 3.3 3.0 No. of phased reflections 279 396 638 947 1309 1737 2205 2752 Mean figure of merit d 0.59 0.56 0.65 0.66 0.59 0.54 0.52 0.49
Total 10263 0.55
Fractional coordinates in multiples of 10 -3 are Se-1, 271, 241, 311; Se-2, 282, 269, 72; Se-3, 323, 282, 122; Se-4, 307, 241,265 for the selenomethionine derivatives SEME, SML1, SM12; and I-1,774, 139, 0; I-2, 396, 323, 390; I-3, 312, 205, 29; I-4, 349, 322, 355 for the iodine derivative D1SA.
b Phasing power, mean heavy-atom contribution over lack of closure, defined as YqFH~I/Y. IIFPHo[--IIFi~qexp(iqb~) + F.~ II, where FH~ is the calculated heavy-atom structure factor, IF.q the corresponding amplitude, [Frol and [F,Hol a r e the observed amplitudes for the protein and heavy-atom derivatives and qb, is the calcutate phase; data were used in the range of 15 to 3 A resolution.
Rcum~, defined as lack of closure over isomorphous difference; data were used in the range of 15 to 3 A re~olution. d Figure of merit, defined as ~P(~))exp(iq~) d~ / J P(qb) d{b, where P is the probability distribution of q~, the phase angle.
Overall structure of A. thaliana glutathione S-transferase
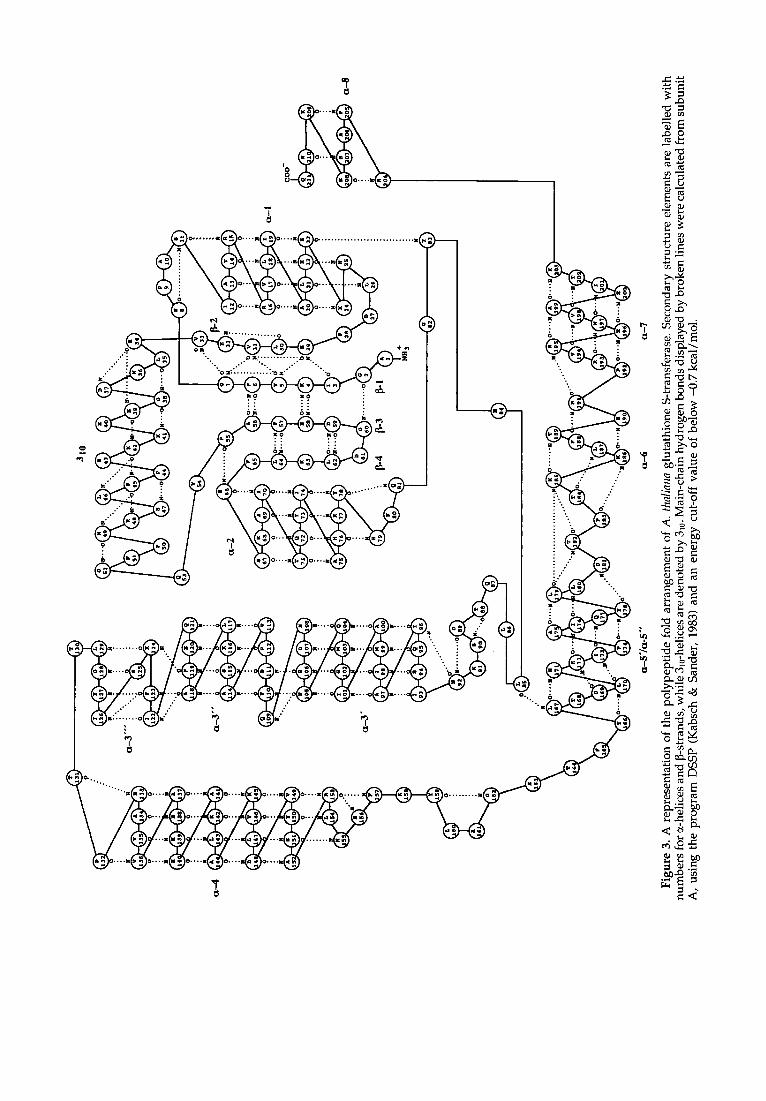
Glutathione S-transferase from A. thaliana forms a dimer of identical subunits each consisting of 211 residues. Although the A. thaliana and the mammalian isoenzymes exhibit only limited se- quence identity (see below) they are topologically similar. The A. thaliana GST subunit is characterized by the typical modular structure with two spatially distinct domains; an N-terminal, smaller one with 77 residues (domain I) and a C-terminal, larger one with 119 residues (domain II) which are covalently connected by a 15-residue linker segment. Sec- ondary structure elements for A. thaliana GST are listed in Table 5; a schematic representation of the main-chain hydrogen-bonding pattern is depicted in Figure 3.
Domain I (residues 1 to 77) has an ~/~-structure comprised of a central four-stranded ~-pleated sheet flanked on one side by two s-helices and on the other side by an irregular segment containing three short 3m-helices (Figure 4). The four ~-strands are arranged in the order ~2 (residues 29 to 32), [31 (residues 3 to 7), D3 (residues 56 to 59) and [34 (residues 62 to 65) with strand ~3 aligned antiparallel to the others. A 1,4-tight turn (residues 59 to 62) connects strands D3 and ~4, while strand ~1 and ~2 are joined by a right-handed crossover connection comprising a D-turn (residues 8 to 11) and s-helix ~.1 (residues 12 to 23). Helix ~1 is associated with that side of the D-sheet that is shielded from the solvent. A second helix, ~2 (residues 67 to 77) is also situated on this side of the p-sheet and has its axis oriented almost parallel to helix ~1 (Figure 4). The ~-strands D2 and D3 are joined by a mainly irregular element consisting of three short 3m-helices each comprising a single 3m-helical turn (3~0, residues 35 to 37; 3~'0, residues 39 to 41; 3~'t~, residues 45 to 47) and two ~-turns (residues 42 to 45 and 49 to 52). The 3m-element is situated at the solvent-exposed side of the ~-sheet with the axis of the quasi-helical element oriented almost perpendicular to the direction of the D-strands. The ~-sheet exhibits a left-handed twist
of about 45 ° when viewed from the side and its topology can be described as - lx , +2, +1 (Richardson, 1977), where x is the ~l-helix crossover connection between strands ~1 and ~2. Residues in a cis-configuration are Pro44 and Pro55, respect- ively. The cis-Pro bend formed at position 55 is situated in the conserved Q(N)V(L,I)P element and is essential for a correct backbone conformation at the glutathione binding site.
The second domain (residues 93 to 211) has an all-~ structure composed of six amphipathic a-helices, which are arranged in a right-handed spiral (Figure 4). Helices a3 to ~7 and their connecting elements pack together to form the main body of domain II, the interior forming a compact hydrophobic core. The first two helical elements, namely ~3 (residues 93 to 126) and ~4 (residues 133 to 154), form an up-down arrangement and comprise 34 and 22 residues, respectively. They are connected by a ~-turn associated with the C terminus of helix ~3 (residues 125 to 128) and a short extended segment (residues 129 to 132). Helix ~3 exhibits a sharp kink at its centre of about 30 ° and should more accurately be described as two helices, termed ~3' (residues 93 to 108) and ~3"/~3"' (residues 111 to 121 and 123 to 126) in order to keep the nomenclature similar to that of the mammalian GSTs (see below). Helix ~3' is straight and oriented nearly parallel to helix ~2, while helix ~3"/~3'" has a bent appearance and projects over the active site located on domain I (Figure 4). The central kink and the bent shape of ~3"/~3"' are due to disruptions of the helical main-chain hydrogen-bonding pattern and backbone conformation at positions 109 and 122, respectively (Figure 3). Helix ~4 is also bent into a banana-like shape. A ~-turn associated with the C terminus of a4 (residues 153-156) followed by an S-shaped bend links helix ~4 to ~5, which is oriented upwards again. It also exhibits a sharp kink at its centre and may be described as two helices, termed ~5' and ~5". Helix ~5' (residues 167 to 170) is oriented almost parallel to helix ~3' and comprises only a single turn. The polypeptide chain then turns away by approximately 45 ° and adopts a 1,4-tight turn (residues 170 to 173) connecting helix
2
u
r-, ~ o o o ° ~ o o. o. . . . . 0 0 0 0 0 0
o. -41. o-, ~ rq.
1" t t t t 1"
c5 " ' t< o6
N N r,,i c,i N N 0 0 0 0 0 0
¢-4 ~ ~ ~ r ¢5 i '~
0 " ~
o -~ r-i
m
o o
"-=" ~ ".5 ~ = ~ ' = ~ ~
o
c ~ o
.#-,~
o ~
~E
~ ~ "~ o X o
"~ ..= o ~ o . . ~ o ~ o . ~ o - ~ o ~ ~ 0 ."'~.c~ 0 ~ 3 ~ . . Q ._o ~ . .c ; ~ . . 0 ~-. ~
-~.~ .- ~.~ ~.~_~-- ~.~ ..
~ ~'~ '-.'- " " ' . . ~ ~ ~ ~ 0 ~ ' - ~
I ' ~ ¢ x l ,Y.,C~I Y~C'-I ~ l "X l , ~ 1 0 t',,I "~ '~=: r t ,-',.-~ r,-', e4 ~ 'c4 c4 c4 "
_ a" ~ ' N
~ 1 ~ ~ ~ ~ ~ ~ I - ~ ~ ~ . ~ "0 "0 "~ "~ I ~n ~ A ~ '.~
, I , " ~ ~ . ~ 0 ~ ~ " ~ , - .~ / :~ ~ ,-

Structure of A. thaliana Glutathione S-transferase 295
Table 4. Final r e f inemen t stat ist ics
Resolution Range (,,~) 8.0 to 2.2 No. of unique reflections in resolution range 21,738 Total number of atoms (excl. H) 3710 Solvent atoms (excl. H) 220 R-factor ~
8.0 to 2.2 A 0.175 2.22 to 2.20 A 0.289
Mean error in coordinates ~ (,d,) 0.19 Average B-factors (~2)
All atoms (3710) 19.5 Main-chain atoms (1722) 16.9 Side-chain atoms (1768) 20.4 Solvent atoms (220) 32.5
R.m.s.-deviations from target values Bonds (A) 0.012 Angles (°) 1.767 Proper dihedrals (o) 23.028 Improper dihedrals (o) 1.029
R.m.s.-deviation in B-factors for bonded atoms < (fi2) 3.7 R.m.s.-deviations between subunits a
for 21(1 (132) pairs of C~-atoms 0.22 (0.13) for 1693 (1527) pairs of all atoms 0.63 (0.26)
R = E~,~ !1 F<.I- IF< II/~:,.. IF<.I. b Luzzati, 1952.
Calculated using MOLEMAN and a probe radius of 2.0 ft. (G. J. Kleywegt & T. A. Jones, unpublished work).
Derived from a least-squares fit of the two subunits using MOLFIT (M. Schneider, unpublished work); values in brackets are given for pairs showing deviations below 1 c~.
~.5' to ~5", which is projecting away from domain I. Another short helix, c~6 (residues 185 to 189), which is linked to ~.5 by two consecutive ]3-turns (residues 179-182 and 182-185) is situated next to helix c~4 at the per iphery of the molecule and is oriented
downwards. A ]]-turn associated with the C terminus of c~6 forms the link to the next helical element, helix cz7 (residues 192 to 202). It runs across and is oriented almost perpendicular to helix ~4, packing against helix ¢z4 and ¢t5'. A last short helix, ct8 (residues 205 to 208) and a [3-turn associated with its C terminus (residues 207 to 210) are slightly separated from the main body of domain II and are situated in an almost upright orientation near helix ¢tl. A short linker segment (residues 78 to 92) composed of a ]]-turn succeeding helix c~2 (residues 78 to 81), of an S-shaped bend and of another 13-turn preceding helix Gt3 (residues 89 to 92) connects both domains. It is stabilized by a hydrogen bond between the side-chains of Glu79 and Ser90 and by the side-chains of Leu85 and Leu86 wedging between helices c~2 and c~,5' and between helices ¢t2 and c~3, respectivel)t Non-covalent contacts between domain I and II are mainly formed by helices 0tl and ~.2 of domain I and by helices c~3'/~3", ~5, 0¢8 and the C-terminal element of domain II. Main h y d r o p h o b i c i n t e r a c t i o n s i n v o l v e I le12, L e u l 8 , I l e l 9 , His22, and His76 from domain I and Tyr96, Leu170, Ile173, Pro174, Ala206, Lys209 and Va1210 from domain II, while polar main-chain to side-chain and side-chain to side-chain interactions are formed between Alal0(O), Arg15(N ~, NH2), Arg16(NH1), Glu23(O d, O°-), Arg68(N c, NH2) and Thr71(O "~) f r o m d o m a i n I a n d G l u 1 0 6 ( O ¢ 1 ) , A s p l 0 7 ( O a 2 ) , His171(O, N~2), Tyr178(OH), Arg204(NH1) and Va1210(O) of domain II.
T h e h o m o d i m e r i c a s s e m b l y o f n a t i v e A. thaliana GST is displayed in Figure 5. The dimer has a globular shape with molecular dimensions of approximately 54 A x 51 A x 48 z~. The symmetry
o ( D
o O _
o
- - i + 1 8 0
I x xX~x x x X l
I x ix,< I I
I x I T -J ' i ]
I
IXCID I
I I
i\ Xx>ll ~I~.,.~ <
. . . . . . . ¢.
I
I I I I
---I- I I I I I
x× I I
-120
I I I I
- - - 4 I I I I I
-60 PH I
I I I I I
. . . . . . . I . I I
S I I I
lxt I
i I I I I I L _ _
B 6 6 i
A 6 6 i I I I I
I I I
I I I I
I I I I I I i
- - - p
I I I I I I
. i _o _ _ I o I
o o l I I
A 6O
i I I I I I I t - - I I I I J I
I I
t I I I I
120
o 0
1 8 0
Figure 2. R a m a c h a n d r a n plot of A. thaliana g lu t a th ione S- t ransferase . Non-g lyc ine res idues are deno t ed by crosses.
.o
o
0
~ : ~ ~ ~ ~ ~ , ~ ~ ~
~ ~--~ ¢'4 t"-I ~ -4" C¢3
v ~
W
~ ~ ~ , .
v
I
' ~ h ~ ~ ~ ~-. h l
~ # ¢ ¢ ¢
N
o.,.-~
x'"'" " ~ : " ' " " ' " ' " " ' " " " " " "'" o z . . . . . . . . . . . . . " "o
? & z o z
z'" ~ ..z o z o . . . . ~ o - . . . z o . , . . ~ o . . . " " .'~
~ . . . . . . . . o a
o .
• 0
j"
°°
~ m" "-°.. °
.~ .
:
~ . r w .,.
,.Q 0
E; U
~J ~ j
U E; = =
~ .~ ~
~ . ~ <

2 9 8 Structure of A. thal iana Glutathione S-transferase
( - - x -. . W ~ ~ - .
k--> '" ' ~ \ ~j f ~
t k L '1' f'l
Figure 4. Stereo-ribbon diagram of a subunit of A. thalhTna glutathione S-transferase with only the model of the productively bound inhibitor included.
relation between both subunits is given in Table 6. A prominent feature in the dimeric structure is the GST-typical very large cleft formed in the centre of the dimer, which is open to the active sites and to the bulk solvent (Figures 5 and 6). Inter-subunit contacts are predominantly mediated by hydro- phobic contacts involving residues in the element connecting 3'~'~; and 134 (Phe51 and Gln53), in strand 134 (Leu62, Leu64 and Phe65), in helix ~2 (Gln66, Arg68, Ala69, Tyr73 and His76) and in helix ~3 (Gin104 and Asp107) of one subunit and in helix ~_3 (Iie93, Ser94, Ala97, Ilel01, Glnl04, Vall05, Asp107, His108 and Glnl09) and in helix u4 (Val149) of the other subunit. A single hydrogen-bonded inter- action linking both subunits is observed between Gln66(O") and Hisl08(N"-).
Active site and inhibitor binding
Dimeric A. thaliana glutathione S-transferase binds four molecules of S-hexylglutathione, a substrate-product analogue and competitive inhibi- tor, as illustrated in Figures 5 and 7. Interpretation of well-defined electron density located in a cleft
formed between domains I and II, which could not be accounted for by protein during refinement, was clear and two molecules of the inhibitor per subunit could be fitted to this density (Figure 7).
The electrophilic substrate binding site (H-site) of A. thaliana glutathione S-transferase's active site is occupied by the hexyl moieties of the two inhibitor molecules, while its glutathione binding site (G-site) is occupied by the peptide backbone of one inhibitor (left inhibitor molecule in Figures 7 and 8), hereafter referred to as the productively bound inhibitor. The peptide backbone of the second inhibitor (right inhibitor rnolecule in Figures 7 and 8), termed the unproductively bound inhibitor, is located next to the G-site and exhibits only few interactions with the protein.
The inhibitors are located in a cleft formed between the 3~,-helical element of domain I and the core of domain II. One side of this cleft is lined by the 3,rhelical element and the segments connecting it to strands ]32 and 133, while the other side is mainly formed by helices ~3" and at5". The bottom of the cleft is created by the segment connecting strand [31 to helix al and by the N-terminal element
l
~: ~1 "' ' " , ,d - " I ~ . . . . j ,
" / ' 7 1 .,, ,/ t l I,~
! 1 . ~ . : I~ ,
Figure 5. Stereo-ribbon diagram of a dimer of A. thaliaua glutathione S-transferase viewed perpendicular to the non-crystallographic 2-fold symmetry axis with only the model of the productively bound inhibitor included.

Structure of A. thaliana Glutathione S-transferase 299
Table 6. Transfornation of monomer B to A" Matrix: 0.92622 0.37690 -0.00802 Vector: -24.32921
0.37692 -0.92625 0.00080 126.08283 -0.00713 -0.00377 -0.99997 97,89299
Calculated from a least squares fit of 1527 pairs of equivalent atoms from both subunits exhibiting deviations below 1 o" using the program MOLFIT (M. Schneider, unpublished work). Rotation in polar coordinates; ~ = 78.93 °, ~ = 0.23 °, ~: = -179.87 °, corresponding to an almost perfect 2-fold rotation through the point X=-12.144, Y= 63.046, Z =48.946 (orthogonal coordinates) with a screw component of t =-0.042 ,~..
of helix ~1 (Figures 4 and 8). The active site cleft opens to the bulk solvent over the segment connecting strand 131 and helix ~1, and through a small cleft lined by the 3,~-helical element of domain I and by helix ~.4 of domain II from the neighbouring subunit. In addition, it opens out over the N-terminal element of helix ~2 into the internal cavity formed between the subunits (Figure 6). The glutathione peptide of the productively bound inhibitor is oriented around the side-chain of Gln53 with its y-Glu moiety pointing downwards to the internal cavity and its glycine residue oriented upwards, projecting into the bulk solvent. The productively bound inhibitor's hexyl group is in an extended conformation pointing towards the bulk solvent. It is situated above the segment connecting strand 131 and helix ~.1 (Figure 8, left inhibitor molecule). Orientation and conformation of the productively bound inhibitor at the active site of A. thaliana GST resembles that of S-hexylglutathione bound to the active site of human GST PI-1 (Reinemer et al., 1992). The peptide backbone of the unproductively bound inhibitor is situated next to the productively bound inhibitor's glutathione peptide. Its ~,-Glu moiety is oriented upwards, pointing towards the bulk solvent, while its glycine residue projects downward to the N-terminal element of helix ~1. Its hexyl group exhibits a slightly kinked conformation and is located next to the productively bound inhibitor's hexyl moiet)4 wedging between the side-chains of helices
~.3"/~3"' and ~.5", respectively (Figure 8, right inhibitor molecule).
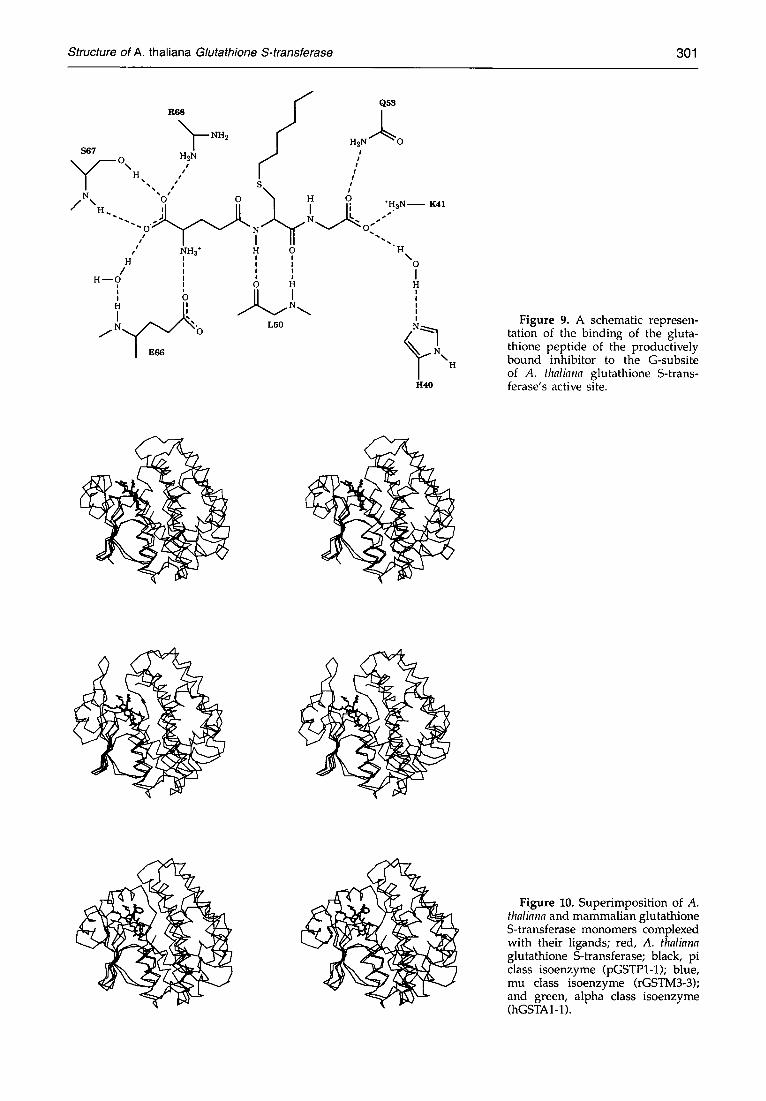
The G-subsite of A. thaliana GST's active site is created by the 3,~-helical element, the segment connecting it to strand 133, and by the element linking strand 134 to helix :z2 (Figure 8). Recognition and binding of the glutathione peptide of the productively bound inhibitor, as displayed in Figures 8 and 9, involves several direct and solvent-mediated side- and main-chain polar inter- actions. G-site ligands include residues His40 (G8; for G-subsite nomenclature see Dirr et al., 1994a), Lys41 (G8), Glu53 (G8), Va154 (G4 and G6), Glu66 (G1 and G2), Ser67 (G2), and Arg68 (G2). The electrophilic binding site is situated next to the G-site and is formed by the segment connecting strand 131 to helix ~.1, by the N-terminal part of the 3~0-helical element, and by the helices ~.3"/~.3"' and ~5", respectively Residues coating the H-subsite of A. thaliana GST's active site include His8, Alal0, Serll , Leu35, Phell9, Phe123, Tyr127 and Tyr178 (Figure 8).
Discussion
Comparison of amino acid sequences and tertiary structures
The primary and tertiary structures of the A. thaliana glutathione S-transferase are compared to those determined for mammalian isoenzymes from
Figure 6. Connolly-like molecular surface of the top region of dimeric A. thaliana glutathione S-transferase as displayed with GRASP (A. Nicholls & B. Honig, Columbia Universit~ unpublished work). The view is along the local dyad showing the large cavity formed between the subunits and both active sites, each of which is occupied by two molecules of S-hexylglutathione.

300 Structure of A. thaliana Glutathione S-transferase
Figure 7. Model of the produc- tively binding (left molecule) and the unproductively binding (right molecule) inhibitor S-hexyl- glutathione with an overlayed IFol-IF<l-omit map contoured at 1.5 c~.
class pi (pGSTPI-1; Reinemer et al., 1991; Dirr et al., 1994b), class mu (rGSTM3-3; Ji et al., 1992) and class alpha (hGSTAI-1; Sinning et al., 1993) and to the non-mammalian enzymes from squid (sigma class GST from the squid digestive gland, Ji et al., 1995), S. japonicum (Lim et al., 1994; McTigue et al., 1995) and from L. cuprina (theta class GST from the Australian sheep blowfl~ Wilce et al., 1995). In addition, on the basis of its primary structure, the A. thaliana enzyme is related to other plant glutathione S-transferases, namely to those of maize GST-I (Shah et al., 1986; Grove et al., 1988), maize GST-II (Jepson et al., 1994), and maize GST-III (Grove et al., 1988).
The A. thaliana glutathione S-transferase and the listed mammalian isoenzymes exhibit only limited sequence similarities and only very few residues are strictly conserved. A sequence alignment resulting from structural comparison of all four enzymes is shown in Figure 11. A pairwise comparison reveals sequence identities of 25.3% and 19.9% (domain I and entire molecule, respectively) for the ara-alpha- pair, 19.0% and 13.3% for the ara-mu-pair, 17.7%
and 15.5% for the ara-pi-pair, and only 11 residues are conserved throughout the four structures. Despite their low sequence identit34 A. thaliana glutathione S-transferase and the mammalian isoforms share a clear topological relationship and a significant structural homology (Table 5) with a decreasing degree of structural similarity in the order domain I > monomer > dimer. Superimposi- tions of the monomers of all four enzymes are displayed in Figure 10. Taken pairwise, the A. thaliana and the mammalian monomers can be aligned with r.m.s, deviations of ~ 1.75 A for 150 to 160 pairs of related C~-atoms. A more detailed inspection reveals that the central four-stranded p-sheet and the ~.-helices (~1 and ~2) are very similar in all four structures, while striking discrepancies are observed in the region of the (quasi-) helical element joining strands ~2 and [33. The conformations adopted here differ in all four structures and no mu-loop is present in the plant-type subunit. Unlike the three mammalian structures the element connecting strand 131 and helix ~1, which thereby forms the bottom of the
180 80
/
Figure 8. Models of the inhibitors S-hexylglutathione (black; left inhibitor molecule is the productively binding, right inhibitor molecule is the unproductively binding) and its next neighbours (blue) in the active site of A. thaliana glutathione S-transferase.
Structure of A. thaliana Glutathione S-transferase 301
R 6 8
S67 ~ N'H2 H2N /'--- ON M /
%x I / N ~0' 0
/ "~H.
o
/ t N t t 3 *
H /
H ~ O i
: o H i , J J, / N~"'v/"~b
I Eee
/ Q53
I H 2 N / ~ O i #
i I
s / H O .
N ~ s o:
I II "'.. H O H : : % * ' I ! !
o H H
I
I
~ N\ H
It4.0
F i g u r e 9. A schematic represen- tation of the binding of the gluta- thione peptide of the productively bound inhibitor to the G-subsite of A. thaliana glutathione S-trans- ferase's active site.
<
!
F i g u r e 10. Superimposition of A. thaliana and mammalian glutathione S-transferase monomers complexed with their ligands; red, A. thaliana glutathione S-transferase; black, pi class isoenzyme (pGSTPI-1); blue, mu class isoenzyme (rGSTM3-3); and green, alpha class isoenzyme (hGSTAI-1).

302 Structure of A. thaliana Glutathione S-transferase
At: --AGIKVFGHPASIATRRVLIALHEKNLDFELVHVE ikdg ...... EHKK epflsr Mal: --APMKLYGAVMSWNLTRCATALEEAGSDYEIVPIN fata ...... EHKS pehlvr
Ma2: ATPAVKVYGWAISPFVSRALLALEEAGVDYELVPMS rqdg ...... DHRR pehlar Ma3: APLKLYGMPLSPNVVRVATVLNEKGLDFEIVPVD ittg ...... AHKQ pdflal
Lc: .... MDFYYLPGSA/~CRSVLMT;tKALGIELTKKLLN lqag ...... EHLK peflki
A: -AEKPKLHYFNARGRMESTRWLLAAAGVEFEEKFIK sae ....... DLDK irmdgy
M: ---PMILGYWNVRGLTHPIRLLLEYTDSSYEEKRYA mgdapdydrs QWLN ekfklg
Sj: ---SPILGYWKIKGLVQPTRLLLEYLEEKYEEHLYE redgd ..... KWRN kkfelg
p: --ppYTITYFPVRGRCEAMRMLLADQDQSWKEEVVT me ........ TWPP ikps--
Sq: --PKYTLHYFPLMGRAELCRFVLAHHGEEFTDRVVE ma ........ DWPN ikatm-
NPFGQVPAFEDGDLKLFESRAITQYIA (75)
NPFGQVPALQDGDLYLFESRAICKYAA (75)
NPFGKVPVLEDGDLTLFESRAIAR/{VL (77)
NPFGQIPALVDGDEVLFESRAINRYIA (75)
NPQHTIPTLVDGDFALWESRAIMVYLV (73)
LMFQQVPMVEIDGMKLVQTRAILNYIA (75)
LDFPNLPYLIDGSRKITQSNAIMRYLA (80)
LEFPNLPYYIDGDVKLTQSMAIIRYIA (75)
CLFRQLPKFQDGDLTLYQSNAILRHLG (71)
YS-NAMPVLDIDGTKMSQSMCIARHLA (71)
At: HRYENQGTNLLQTDSKNISQYAIMAIGMQVEDHQFDPVASKLAFE
Mal: RKNKPE---LLREG--NLEEAAMVDVWIEVEANQYTAALNPILFQ
Ma2: RKHKPE---LLGGG--RLEQTAMVDVWLEVEAHQLSPPAIAIVVE
Ma3: SKYASEGTDLLPAT .... ASAAKLEVWLEVESHHFHPNASPLVFQ
Lc: EKYGKIqDS-LFP---KCPKFdZAVINQRLYFDMGTLYKSFADYYYP
A: SKYN ..... LYG---KDIKERALIDMYIEGIAD-LGEMILLLPVC
M: RLHH ..... LCG---ETEEETIRADIVENQVMD-NRMQLIMLCYN
Sj: DK}[N ..... MLG---GCPKERAEISMLEGAVLD-IRYGVSRIAYS
P: RSFG ..... LYG---KDQKEAALVDMVNDGVED-LRCKYATLIYT
Sq: REFG ..... LDG---KTSLEKYLVDEITETLQD-IFNDVVKIKFA
qifksiygltt
vlispmlggtt
cvfapflgrer llvrpllggap
qifa .......
ppee .......
pd .........
kd .........
n ..........
peaa .......
DEAVVAEEEAK- LAK - VLDVYEARLK ( 155 )
DQKVVDENLEK- LKK- VLEVYEARLT ( 150 )
NQAVVDENVEK - LKK - VLEVYEARLA ( 152 )
DA~AVVE KHAE Q - LAK - VLDVYEAHLA (151)
KAPADPEL-YKKMEA- AFDFLNTFL- ( 141 )
KDAKLALI KEK - I KNRYFPAFEKVLK (140)
FEKQKPEFLKT- IPE- KMKLYSEFLG ( 142 )
FETLKVDFLS K- LPE - MLKMFEDRLC (137)
YEAGKEKYVKE- LPE- HLKPFETLLS ( 132 )
KEAVQQNYEKS - CK - RLAP FLEGLLV ( 135 )
At : E .... FKYLAGETFTLTDLH~IPPIQYLLGT-- PTKKLFTERPRVIqEWVAEITKRPASEKVQ* (211)
Mal : K .... CKYLAGDFLSLADLNI/VSVTLCLFAT- - PYASVLDAYPHVKAWWSGLMERPSVQKVAALMKPSA* ( 213 )
Ma2 : T .... CTYLAGDFLSLADLSPFTIMHCLMAT--EYA/LLVHALPHVSAWWQGLAARPAANKVAQFMPVGAGAPKEQE* (222)
Ma3 : R .... NKYLAGDEFTLADAIqHA-LLPALTSARPPRPGCVAARPHVKAWWEAIA/LRPAFQKTVAAIPLPPPPSSSA* (221)
Lc: - EG- - HQYVAGDS LTVADLALLASVSTFEVA- - G- FDF- SKYANVAKWYANAKTVAPGFDENWEGCLE FKKFFN* (208)
A : SHG- - QDYLVGIqKLS RAD I HLVELLYYVE EL - -D- S S L I S S FPLLKALKTRI SNLPTVKKFLQPGS PRKPPMI)EKS LEEARKI FRF* ( 221 )
M: --K- -RPWFAGDKVTYVDFLAYDILDQYHIF- -E- PKCLDAFPNLKDFLARFEGLKKISAYMKSSRYLSTPIFSKLAQWSNK* (217)
Sj : --H--KTLYNGDHVTHPDFMLYDALDVAFLYM--D- PMCLDAFPKLVCFKKRIEAIPQIDKYLKSSKYIAWPLQGWQATFGGGDHPPK* (217)
p: QNQGGQAFVVGSQ I S FADYNLLDLLR IHQVL- - N- PS CLDAFPLLSAYVARLSARPKI KAFLAS PEHVNRP INGNGKQ * (207)
Sq : SNGGGDGFFVGNSMTLADLHCYVALEVPLKH - - T- PELLKDCPKIVALRKRVAECPKIAAYLKKRPVRDF* ( 202 )
Figure 11. Sequence alignment of A. thaliana (At, Zhou & Goldsbrough, 1993; Zettl et al., 1994), maize (Mal, GST-I, Shah et al., 1986; Grove et al., 1988; Ma2, GST-II, Jepson et al., 1994; Ma3, GST-III, Grove et al., 1988), mammalian (A, hGSTAI-1, Tu & Qian, 1986; M, rGSTM3-3, Ding et al., 1985; P, pGSTPI-1, Dirr et al., 1991) and non-mammalian (Sq, sigma class GST from squid digestive gland, Tomarev et al., 1993; Sj, Schistosoma japonicum GST, Smith et al., 1986, 1987; Lc, theta class GST from Lucilia cuprina, Board et al., 1994) glutathione S-transferases. The alignment is based on the structural superposition of the crystal structures from A. thaliana (this work) and the mammalian (A, Sinning et al., 1993; M, Ji et al., 1992; P, Reinemer et al., 1991; Dirr et al., 1994b) and non-mammalian (Sj, Lira et al., 1994; McTigue et al., 1995; S, Ji et al., 1995; Lc, Wilce et al., 1995) glutathione S-transferases, respectively. The sequence alignment of the maize glutathione S-transferases is based solely on sequence similarities with the A. thaliana enzyme since their structures are not known. Residues involved in glutathione binding are shown in boldface. Lower-case characters are used to identify sequences in regions with low structural similarity.
H-subsite, adopts a different conformation in the A. thaliana structure. In contrast to domain I, substan- tial differences are observed for the linking segment and domain II, demonstrating a more distant relationship. The linker element comprises 15 residues in the A. thaliana enzyme compared to a length of six residues in the mammalian isoforms. Despite their remarkably different lengths, a central segment comprising hydrophobic residues involved in the stabilization of the loop structure adopts a very similar conformation in all four subtypes (Figures 10 and 11). Major variations in domain II are observed for the ¢~-helices ~3/~4 and for the C-terminal element. The structural superimposition (Figure 10) reveals that the helical element a3 in the A. thaliana structure is quite long when compared to its corresponding elements in the mammalian structures. In addition, this helix is kinked and protrudes over the active site on domain I, while the corresponding elements in the mammalian isoforms are straight. As a result of these differences the element connecting helices ~3 and ~.4 is also distinct
in the A. thaliana isoform. Here it comprises six residues and adopts an extended conformation, while it is a single residue in a turn-like con- formation in the mammalian subtypes (Figure 10). Helix ~4 and the connecting element to helix ~5 have a similar conformation in all four structures. However, their positions relative to each other differ in each gene class. The highest degree of conformity in conformation and relative position for this element is observed for the ara-mu-pair (Figure 10, middle), while the degree of similarity is worse for the ara-alpha-pair, presumably due to an additional helical turn in helix ~5 in the alpha isoenzyme (Figure 10 bottom, Figure 11). No striking differences are observed for helices ~5 to ~8, which in principle resemble their mammalian counter- parts in conformation. In contrast to these elements, dramatic structural deviations are recognized for the C-terminal region in all four structures. In the mammalian glutathione S-transferases the last helix of domain II is followed by a segment in extended conformation which ranges to the upper part of

Structure of A. thaliana Glutathione S-transferase 303
domain II. There, in the pi- and the mu-class enzymes, the chain turns back to the core of domain II, while in the alpha-class enzyme it turns to domain I to create an additional C-terminal a-helix (Figure 10). Unlike the mammalian structures, no "C-terminal element" is present in the A. thaliana structure. Here, the chain terminates immediately after helix ~8 and the C terminus is situated at the solvent exposed surface of domain II, packing with its preceding helix (~8) onto helix ~2 of domain I (Figure 10). With respect to the dimeric assemblies, the structural differences between plant and mammalian enzymes are even greater; the best dimer fit is observed for the ara-pi-pair (r.m.s.-devi- ation is 1.74 A for 327 pairs of related C~-atoms), while the relationship is more distant for the ara-alpha-pair (1.84 A for 305 pairs) and worse for the ara-mu-pair (1.82 A for 265 pairs). The contact areas in the dimer interface are similar, but the relative orientation of interacting monomers is altered.
The S. japonicum GST structure shares a noticeable resemblance with that of the mammalian mu class isoenzyme as indicated by sequence identities of 57.0% and 41.9% for domain I and for the entire molecule, respectively. The squid GST structure reveals a similarity to the mammalian class pi isoenzyme and exhibits sequence identities of 38.7% for domain I and of 31.2% for the entire molecule. Consequently, they also share only limited sequence identity with the A. thaliana enzyme, namely 17.9% for the ara-Sjap-pair and 13.9% for the ara-squid-pair compared to 13.3% and 15.5% for the ara-mu and ara-pi-pairs, respectively. Their structural superimposition reveals significant variations for ~2 (3~0-helices in A. thaliana; Table 5), the linker region, the ~4-turn-~5-motif (~3/~4 in A. thaliana; Table 5) and for the C-terminal element, while other regions are more similar. The positions of the tip of the ~4-turn-~5-motif in S. japonicum and squid glutathione S-transferases and of the corre- sponding ~3/a4-element in the A. thaliana enzyme are approximately 10 A apart and their C-terminal elements differ dramatically in length. Together, these variations are mainly responsible for the structural differences observed for their H-subsites.
A comparison of the primary structure of A. thaliana glutathione S-transferase with that of the maize isoenzymes resulting from a computerized alignment followed by manual optimization is given in Figure 11. The four enzymes share a significant similarity with the highest pairwise identity for the ara-maize-III-pair (93 residues out of 211 are identical, which corresponds to 44.1% identity) followed by the pairs ara-maize-II and ara-maize-I (83 and 81 identical residues corresponding to 39.3 and 38.4% identity). Out of the 211 amino acid residues, 56 residues are totally conserved in all four enzymes. A closer inspection reveals a greater degree of identity for domain I, where 26 out of 77 residues are conserved (33.8% identity), while the identity comprises only 28 out of 119 residues (23.5%) for domain II. In addition, the seven
residues of domain I which are involved in the recognition of glutathione in the enzyme's active site are strictly conserved or conservatively re- placed. In contrast, remarkable variations are observed for the C-terminal regions following helix ~8, which differ significantly in length and sequence (Figure 11). In conclusion, a substantial structural relationship of other plant glutathione S-trans- ferases with the A. thaliana enzyme is expected. In particular, domain I and the G-site architecture are anticipated to be quite similar, while structural differences corresponding to sequence variations are expected for domain II and their H-site architectures.
Plant glutathione S-transferases have also been implicated to belong to the theta class, a very heterogenous class of GSTs that comprises enzymes from organisms as divergent as bacteria, yeasts, insects, plants and mammals (Buetler & Eaton, 1992; Pemble & Taylor, 1992). Recently, the structure of an insect theta class GST (from the Australian sheep blowfly L. cuprina, Wilce et al., 1995) has been determined and it is now possible to compare the structure of A. thaliana GST with that of L. cuprina. Sequence identities observed for both enzymes are 33.8% for domain I and 20.2% for the entire molecule. As indicated in Table 5, their three-dimen- sional structures also exhibit a number of obvious differences. Major variations include the element connecting ~2 and ~3, the linker, the ~4/~5-motif (~3/~4 in A. thaliana), the element connecting ~6 and a7 (~5 and a7 in A. thaliana) and helix ~8 and the C terminus (see Figure 4 and Figure 3 in Wilce et al., 1995). It is important to note that overall sequence identities and structural differences observed between A. thaliana and L. cuprina GSTs are roughly in the same range as those found for the comparison of A. thaliana and mammalian GSTs or for comparison of GSTs from different mammalian gene classes (see above and Sinning et al., 1993). Although certain regions (N-terminal elements, G-site, see below) exhibit a substantial resemblance, the overall structural relationship shared by theta-class members is not as close as those observed for enzymes within the alpha-, mu- and pi-class. In consequence, the theta class shows more divergence than any of the other gene classes and may have to be further subdivided.
Comparison of active sites and implications for the enzymatic mechanism
The relationship of active site arrangements from glutathione S-transferases from different species is discussed on the basis of the three-dimensional structures of enzymes from A. thaliana, from mammals (Reinemer et al., 1991, 1992; Ji et al., 1992; Sinning et al., 1993; Dirr et al., 1994b; Garcia-S~ez et al., 1994; Raghunathan et al., 1994), from S. japonicum (Lim et al., 1994), from squid (Ji et al., 1995) and from L. cuprina glutathione S-transferase (Wilce et al., 1995).
A close structural relationship is observed for all

304 Structure of A. thaliana Glutathione S-transferase
Table 7. G-site ligands and interactions with glutathione for glutathione S-transferases from different species Glutathione S-transferase from class
Glutathione Arabidopsis Schistosoma Lucilia moieties thaliana PP Mu b Alpha' japonica Squid ~ cupriua f
7-Glu NH~ Glu66 Gln62 Gin71 Gln66 Gln66 Asp96* Asp105* Asp100* Asp100*
CO0- Glu66" Arg13 Ser72 Thr67 Gln66 Set67 Ser63 Ser67 Arg68
C~----O - - Gln49 - - - - - - Cys NH Va154 Leu50 Leu59 Val55 Leu54
C~------O Va154 Leu50 Trp7 Va155 Trp7 Gly NH - - - - Asn58 - - Asn53
COO- His40 * Trp38 Arg42 Arg44 Lys44 Lys41 Lys42 Trp45 Argl30* Gln53 Gin49 Lys49
Gin62 Glu64 Asp96* Gin62 Ser65 Ser63 Arg66
m m
Met50 Ile52 Met50 [1e52
Trp38 His38 Lys42 His50 Asn48
° Reinemer et al., 1991, 1992; Dirr et al., 1994b; Garcfa-S~iez et al., 1994. bJi et al., 1992; Raghunathan et al., 1994.
Sinning et al., 1993. d Lim et al., 1994; McTigue et al., 1995. ~Ji et al., 1995. fWilce et al., 1995.
Water mediated interaction. * Interaction contributed from residue from neighbouring subunit.
enzymes with respect to their general active site topology Each enzyme exhibits a conserved overall active site architecture, resulting from the utilization of corresponding secondary structure elements as building blocks. A more thorough analysis, compar- ing G-site ligands and complexes with glutathione moieties, reveals that glutathione backbone confor- mations adopted in each enzyme are quite similar. In addition, although displaying some divergence, the nature of residues acting as G-site ligands and interactions facilitated with glutathione may be considered as analogous in all enzymes (Table 7). A different situation is encountered when H-sites are compared. Here, a unique structure is exhibited for each enzyme. In particular, the A. thaliana enzyme has a broader and quite deep H-subsite when compared to the mammalian isoforms, mainly due to the kinked appearance of helix c~3 and the lack of a C-terminal element. Elements from domain I lining the A. thaliana H-site resemble that of the pi class enzyme, since no mu-loop and no C-terminal helix are present. In contrast, the cavity created in domain II, which is spacious enough to accommodate large aromatic substrates, is unique (Figure 10). In accordance with this observation, specific activities of A. thaliana GST assayed with 1-chloro-2,4-dinitrobenzene (CDNB) and 1,2-epoxy-3-(p-nitrophenoxy)propane (EPNP) are rather low (0.16 lamol min -1 mg -1 for CDNB and 0.04 lamol min -~ mg -~ for EPNP; Zettl et al., 1994; B. Bieseler, unpublished results), while preliminary data (studies are currently in progress) suggest a preference for larger substrates. In addition, two hexyl moieties of the inhibitor S-hexylglutathione are found in the H-site of the A. thaliana enzyme, demonstrating the unique character of its architec- ture and its preference for large substrates.
Of particular importance are active site residues which are involved in the activation of glutathione
during catalysis. The variety of available crystal structures of different glutathione S-transferases (Reinemer et al., 1991, 1992; Ji et al., 1992; Sinning et al., 1993; Dirr et al., 1994b; Garcfa-S~ez et al., 1994; Raghunathan et al., 1994; Lim et al., 1994; McTigue et al., 1995; Ji et al., 1995) has identified an active site tyrosine residue situated within hydrogen-bonding distance of the thiol group of glutathione. This tyrosine residue is located at or near the C terminus of strand 131 and is invariant in almost all published sequences of glutathione S-transferases. Evidence for the pivotal role of this residue for the activation of glutathione has been provided by numerous mutagenesis studies of mammalian isoenzymes (reviewed by Dirr et al., 1994a). Surprisingly; the structurally corresponding residue in the A. thaliana enzyme is a glycine residue (Figures 8 and 11), namely Gly7, which is conserved in the plant glutathione S-transferases (Figure 11) and unam- biguously is not part of the enzyme's active site. An aromatic residue, located at position 6, which is a phenylalanine in residue A. thaliana but tyrosine in other plant enzymes (Figure 11) is oriented away from the active site and is also not part of it (Figure 8). It is generally accepted that the glutathione reacts predominantly as the thiolate anion and that its formation is facilitated by the active site tyrosine residue either through a general base catalytic mechanism or by donation of a hydrogen bond (Armstrong, 1993; Dirr et al., 1994a). It is important to point out that there is only one alternative residue, namely Ser11, that may take over the catalytic tyrosine residue's role in the plant glutathione S-transferases. In support of this hypothesis, Ser11 is situated at the bottom of the active site and is in close proximity to the S~-atom of the productively bound inhibitor. Although the 0 "/ to S '~ distance is about 4.9/k in the complex with S-hexylglutathione, other conformations are

Structure of A. thaliana Glutathione S-transferase 305
possible in a complex with glutathione and will al low a closer approach. In addition, no other residue with a suitable functionali ty is par t of the active site, nor is there any other suitable residue in the vicinity of the inhibitor 's SY-atom (Figure 8). On super impos i - tion of the glutathione S-transferase crystal struc- tures, the hydroxyl g roup of Ser11 supe r imposes close to the posit ion of the catalytically impor tan t tyrosine residue of the m a m m a l i a n enzymes. Finall)~ it is impor tan t to note that Ser11 is conserved in mos t of the plant glutathione S-transferases (Figure 11 and Droog et al., 1993). In addition, a corresponding serine residue is conserved in other theta class GSTs (Hussey & Hayes, 1992; Wilce et al., 1995) and was also repor ted to be par t of the active site of L. cuprina GST (Wilce et al., 1995). It is worth noting that N- te rmina l amino acid sequences of m a m m a l i a n theta class GSTs (Hussey & Hayes, 1992) are similar to those of A. thaliana and L. cuprina GST. In consequence, Se r l l is conserved and the aromat ic residue at posi t ion 5 (phenylalanine in h u m a n GSTT2, tyrosine in the others; Hussey & Hayes, 1992) is likely to equate to Phe6 in A. thaliana GST and is not par t of the active site of these enzymes.
Experimental Methods Purification
Native A. thaliana GST was obtained by heterologous expression in Escherichia coli as previously described (Zettl et al., 1994). Briefly the coding region of the A. thaliana GST was inserted into pET3a resulting in pET-GSTex. The E. coli host BL21 (DE3; Studier et al., 1990) was transformed with pET-GSTex, and after isopropyl-~- D-thiogalactopyranoside induction, A. thaliana GST was expressed as a soluble protein (Zettl et al., 1994).
Bacterial cells were harvested by centrifugation (600 g for 20 minutes), and may be stored at -70°C for later use. A 2g quantity of the frozen bacterial pellet was resuspended in 20 ml 0.1 M potassium phosphate buffer (pH 7.3) and subjected to sonification. The homogenate was then centrifuged at 6370g for 20 minutes. Subse- quently, the supernatant was dialysed against 50 mM potassium phosphate buffer (pH 7.3) and then subjected to S-sulphobromophthaleinglutathione-agarose (Sigma, Deisenhofen, Germany) affinity chromatography as described by Mozer et al. (1983): using 5 mM reduced glutathione in 50mM potassium phosphate buffer (pH 7.3) to elute the adsorbed activity. Fractions con- taining glutathione S-transferase activity were pooled, concentrated by ultrafiltration (PM-10, Amicon, Witten, Germany) and the buffer changed to 10 mM Hepes/ NaOH (pH 7.0) by filtration through Sephadex G-25 (Pharmacia, Freiburg, Germany).
Throughout the purification, glutathione S-transferase activity was monitored at 30°C spectrophotochemically at 340 nm using 1 mM 1-chloro-2,4-dinitro-benzene and 5 mM reduced glutathione in 0.1 M potassium phosphate buffer (pH 6.5), containing 3% (v/v) ethanol (Habig & Jakob)4 1981; Mozer et al., 1983). The enzyme was purified to apparent homogeneity as judged by SDS-PAGE.
Incorporation of selenomethionine
A. thaliana GST containing selenomethionine was obtained by heterologous expression in a methionine-defi-
cient E. coli strain using a method designed by Budisa et al. (1995). Briefl)4 the E. coli host B834 (DE3; Studier & Moffart, 1986), which is auxotrophic for methionine, was transformed with pET-GSTex and cultured in a newly designed minimal media containing 0.8 mM selenome- thionine as described by Budisa et al. (1995). After induction with isopropyl [~-d-thiogalactopyranoside se- lenomethionine A. thaliana GST was expressed as a soluble protein and purified as described above.
Selenomethionine incorporation was monitored using electrospray mass spectroscopy and indicated complete substitution; the observed mass difference of 93(+ 2)Da for native and selenomethionine containing A. thaliana GST subunits is in very good agreement with the expected mass difference of 93.8 Da for two substituted methionine residues.
Crystallization and crystallographic data
The native enzyme was crystallized by vapour diffusion using the sitting drop method with multi-well crystal growth chambers (C. Supper Inc., Natick, MA 01760, USA). Sitting droplets were made by mixing 3 ~tl protein solution (20mg/ml protein in 10 mM Hepes- NaOH buffer, 0.02% (w/v) NaN3 (pH 7.0)), 1 ~1 inhibitor solution (30mM S-hexylglutathione in 10mM Hepes- NaOH buffer, 0.02% NaN~ (pH 7.0)) and 2 ~tl precipitating buffer (2.8 M (NH4)2SO4, 0.2% (w/v) polyethyleneglycol 4000 in 0.2 M Hepes-NaOH buffer, 0.02% NaN3 (pH 7.0)). Droplets were equilibrated against 400 ~1 of precipitating buffer at room temperature. Crystals of maximal dimen- sions 0.3 mm x 0.3 mm x 1.0 mm were obtained within (approximately) five days and were harvested into the reservoir solution.
The crystals are hexagonal and belong to the space group P63 with lattice constants a = b= 113.30A, c = 69.96 A, ~ = ~ = 90 °, y = 120 ° and diffract to at least 2.2 A resolution. The asymmetric unit contains a dimer, the Matthews coefficient is 2.7,~3/Da (Matthews, 1968). Isomorphous crystals of the selenomethionine-containing enzyme were obtained under identical conditions.
Preparation of heavy atom derivatives and determination of the selenomethionine X-ray absorption edge
Selenomethionine-containing heavy atom derivatives were prepared as described previously. Other heavy-atom derivatives were prepared by soaking the crystals under the conditions given in Table 1.
An X-ray fluorescence spectrum of a single crystal of selenomethionine containing A. thaliana GST was measured through the K-absorption edge of selenium using synchrotron X-radiation obtained from beamline BW-6 (MPG outstation at DESY, Hamburg, Germany; for details see below) and a NaJ(Ta) scintillation detector with ORTEC electronics. Anomalous dispersion terms, f' and f", derived from the wavelength scan of the X-ray fluorescence spectrum (Figure 1), were calculated using the program DISCO (K. Eichhorn, unpublished work). The dependence in these experimentally derived dis- persion terms on the X-ray photon energy is displayed in Figure 1.
Data collection and evaluation
The X-ray reflection data for the native enzyme and two heavy atom derivatives (NATI, SEME, DISA; see Table 1) were collected on a computer-controlled CAD-4

306 Structure of A. thaliana Glutathione S-transferase
diffractometer equipped with a FAST area television detector (Enraf-Nonius, Delft, Netherlands). Nickel- filtered CuK~ X-radiation (~. = 1.5418 A), obtained from a Rigaku-Denki (Tokyo, Japan) rotating anode generator operated at 5.4kW, was used. X-ray intensities were evaluated with the MADNES system (Messersclm'ddt & Pflugrath, 1987) and further processed using ABSCOR (Messerschmidt et al., 1990) for loading to the PROTEIN program suite (Steigemann, 1991) and using ABSURD (P. Evans) and ROTAVATA-AGROVATA (J. M. Smith & A. J. Wonacott) from the CCP4 program package (Collaborative Computational Project No. 4, 1994) for further use with CCP4 programs, respectively
Resonant X-ray diffraction data were measured on the beamline BW-6 (H. D. Bartunik, G. Buth, W. Fechte, M. Kobarg, I. Koelln & T. Wroblewski, unpublished results), located on a 56-pole wiggler at DORIS (MPG-outstation at DESY, Hamburg, Germany). The X-ray optics include a water-cooled Si(111) double monochromator which is positioned between a flat mirror and a double-focussing toroidal mirror. Both mirrors consist of SiC coated with Au and are water-cooled. X-ray reflection data were measured for selenomethionine-containing A. thaliana GST crystals at two different wavelengths, namely at )~ = 0.9295 A and K2 = 0.9797 A (SML1, SML2; Table 2) in the vicinity of the experimentally derived selenium K-absorption edge. The wavelength bandwidth was determined to be 3.7eV, corresponding to a relative wavelength resol- ution of approximately 3 x 10 -~ A. The diffraction data were collected by the rotation method using a Hendrix- Lentfer-imaging-plate detector (MAR Research, Ham- burg, Germany) with a diameter of 300 mm. The X-ray intensity incident on the sample was monitored with ionization chambers, and the speed of the rotation was adjusted as a function of the ionization current (Bartunik et al., 1981). X-ray intensities were evaluated with the MOSFLM program package (Leslie, 1991) and scaled and averaged using ROTAVATA-AGROVATA. Data collection statistics for all data sets are given in Table 1.
MIR-phasing, density modification and averaging
Heavy-atom derivatives were analysed by difference Patterson (for SEME, SML2 and DISA) and anomalous Patterson maps (for SML1) using data in the range of 25 to 3 A resolution. These maps were interpreted with the aid of a vector verification program (Colman et al., 1975) as implemented in PROTEIN (Steigemann, 1991). Heavy-atom sites obtained were refined and SIR-phases were calculated using ML-PHARE (Z. Otwinowski) from the CCP4 suite (Collaborative Computational Project No. 4, 1994). Subsequently, SIR-cross-phased difference Fourier maps were used to find a common origin. The positions and occupancies of the heavy atom sites of all four derivatives were refined and MIR-phases were computed with ML-PHARE using data in the range of 15 to 3 A resolution. Anomalous dispersion data were included and the correct handedness of the heavy-atom structure was derived from the obtained anomalous occupancies.
The phases were further improved by solvent flattening (Wang, 1985), histogram matching (Zhang & Main, 1990) using the program DM (K. D. Cowtan) from the CCP4 program package and by non-crystallographic symmetry averaging using routines from MAIN (Turk, 1992). The solvent mask was calculated assuming a solvent content of 55%. A preliminary orientation for the
non-crystallographic 2-fold symmetry axis was obtained by analyzing local symmetry elements in the Patterson function using a rotation function operated in direct space (Huber, 1965) as implemented in PROTEIN. The symmetry axis relating the two independent monomers was determined from related heavy atom sites and refined by picking density peaks above 2.0 cy in the range of 15 A around the centre of the dimer and searching for the optimal correlation with (local) symmetry-related density points by alternate three-dimensional rotation and translation grid searches. The phasing statistics are given in Table 2.
Model building and refinement
An almost-complete model was built into an averaged electron density map with data from 15 to 3 A resolution and phases obtained from ML-PHARE and DM using the interactive graphics program FRODO (Jones, 1978), implemented on an ESV-30 graphics workstation (Evans & Sutherland, Salt Lake City, USA). The model was crystallographically refined using X-PLOR (Br/inger, 1992a) in combination with an accurate parameter set derived by Engh & Huber (1991). Subsequently, the model was progressively improved in macrocycles consisting of energy-restrained crystallo- graphic refinement until energy and R-factor had converged and manual intervention using 2tF,,]-]F¢] electron density maps based on a 1:1 phase combination of MIR and model phases and on pure model phases, respectively; resolution was gradually extended to 2.2 A. In later refinement steps, constrained individual isotropic temperature factors were refined. Water mol- ecules were added at stereochemically reasonable positions, if appropriate density was present in a ]G] - ]Fd electron density map contoured at 2.0 c~ and were accepted if they reappeared with appropriate density in a 2IFo]- IF~] electron density map contoured at 1.0 c~ and refined to have a temperature factor of no more then 70 ~2. A detailed summary of the refinement process is given in Table 3; final refinement parameters are given in Table 4. The refined coordinates of glutathione S-transferase from A. thaliana will be deposited with the Protein Data Bank (Brookhaven National Laboratory, Chemistry Department, Upton NY 11973, USA) and are available directly from the authors on request until they have been processed and released.
Miscellaneous
Main-chain hydrogen bonds were analysed with the program DSSP (Kabsch & Sander, 1983) using an energy cut-off value of -0 .7kca l /mol ( lcal =4.184J). The stereochemical quality of the model was assessed using the program PROCHECK (Laskowski et al., 1993). Ribbon plots were prepared using MOLSCRIPT (Kraulis, 1991) and RASTER3D (Merrit & Murphy, 1994); other plots were made using the program GRASP (A. Nicholls & B. Honig, Columbia University, unpublished work) and the graphics facilities of FRODO (Jones, 1978). Algorithms from the MAIN program suite (Turk, 1992) have been used to compare structures. An initial transformation was generated from a pairwise manual fitting of structures at the display Subsequently, a pair list definingoatOms that match to within a defined range (0 to 2.6 A) was generated and a new transformation matrix was deter- mined. This process was repeated until convergence was reached and r.m.s.-deviations between protein units were calculated.

Structure of A. thaliana Glutathione S-transferase 307
Acknowledgements We are grateful to Professor D. Berg (BAYER AG, PF-F
Biotechnologie), Dr P. Reinecke (BAYER AG, PF-F Fungizide) and Professor K. Findeisen (BAYER AG, PF-F Leitung) for their valuable support and their continuous interest in the project. We thank C. Krebs and H. Fr6hlich for excellent technical assistance and S. Steinbacher (MPI f6r Biochemie) and M. Kobarg (MPG Hamburg) for their help with the data collection. We are grateful to Dr R. Ficner (EMBL) for some helpful suggestions on scientific matters and to N. Budisa (MPI fiir Biochemie) for his help with the expression of the selenomethionine-containing enzyme.
References Armstrong, R. N. (1993). Glutathione S-transferases:
structure and mechanism of an archetypical detoxi- cation enzyme. Adv. Enzymol. 69, 1--44.
Bartunik, H. D., Clout, P. N. & Robrahn, B. (1981). Rotation data collection for protein crystallography with time-variable incident intensity from synchro- tron radiation sources. J. Appl. Crystallog. 14, 134-136.
Beckett, G. J. & Hayes, J. D. (1993). Glutathione S-transferase: biomedical applications. Adv. Clin. Chem. 30, 281-380.
Bilang, J., Macdonald, H., King, P. J. & Sturm, A. (1993). A soluble auxin-binding protein from Hyoscyamus muticus is a glutathione S-transferase. Plant Physiol. 102, 29-34.
Board, P. G., Russel, R. J., Marano, R. J. & Oakeshot, J. G. (1994). Purification, molecular cloning and heter- ologous expression of a glutathione S-transferase from the Australian sheep blowfly Lucilia cuprina. Biochem. J. 299, 425-430.
Boyland, E. & Chasseaud, L. F. (1969). The role of glutathione and glutathione S-transferase in mercap- turic acid biosynthesis. Adv. Enzymol. 32, 173-219.
Br~inger, A. T. (1992a). X-PLOR (Version 3.1)--A System for X-ray Crystallography and NMR. Yale University Press, New Haven.
Bri.inger, A. T. (1992b). Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature, 355, 472-475.
Budisa, N., Steipe, B., Demange, P., Eckerskorn, C., Kellermann, S. & Huber, R. (1995). High-level biosynthetic substitution of methionine in proteins by its analogs norleucine, selenomethionine, tel- luromethionine and ethionine in Escherichia coli. Eur. J. Biochem. 230, 788-796.
Buetler, T. M. & Eaton, D. L. (1992). Glutathione S-transferases: amino acid sequence comparison, classification and phylogenetic relationship. Environ. Carcino. Ecotox. Rev. C10, 181-203.
Collaborative Computational Project No. 4 (1994). The CCP4 suite: programs for protein crystallography. Acta Crystallog. D50, 760-763.
Colman, P. M., Fehlhammer, H. & Huber, R. (1975). Patterson search methods in protein structure determination: ~-trypsin and immunoglobolin frag- ments. In Crystallographic Computing Techniques (Ahmed, F. R., Huml, K. & Sedlacek, B., eds), pp. 248-258, Munksgaard, Copenhagen.
Danielson, U. H. & Mannervik, B. (1985). Kinetic independence of the subunits of cytosolic glutathione transferase from the rat. Biochem. J. 231, 263-267.
Diesperger, H. & Sandermann, H., Jr (1979). Soluble and microsomal glutathione S-transferase activities in
pea seedlings (Pisum satiwlm L.)..Planta, 146, 643-648.
Ding, G. J.-F., Lu, A. Y. H. & Pickett, C. B. (1985). Rat liver glutathione transferases--nucleotide sequence analy- sis of a Ybl cDNA clone and prediction of the complete amino acid sequence of the Ybl subunit. J. Biol. Chem. 260, 13268-13271.
Dirr, H. W., Mann, K., Huber, R., Ladenstein, R. & Reinemer, E (1991). Class n glutathione S-transferase from pig lung--purification, biochemical characteriz- ation, primary structure and crystallization. Eur. J. Biochem. 196, 693-698.
Dirt, H. W., Reinemer, P. & Huber, R. (1994a). X-ray crystal structures of cytosolic glutathione S-trans- ferases. Implications for protein architecture, sub- strate recognition and catalytic function. Eur. J. Biochem. 220, 645-661.
Dirr, H. W., Reinemer, P. & Huber, R. (1994b). Refined crystal structure of porcine class pi glutathione S-transferase (pGST P1-1) at 2.1 A resolution. J. Mol. Biol. 243, 72-92.
Droog, F. N. J., Hooykaas, E J. J., Libbenga, K. R. & van der Zaal, E. J. (1993). Proteins encoded by an auxin regulated gene family of tobacco share limited but significant homology with glutathione S-transferases and one member indeed shows in vitro GST acitivity. Plant. Mol. Biol. 21, 965-972.
Engh, R. A. & Huber, R. (1991). Accurate bond and angle parameters for X-ray protein structure refinement. Acta Crystallog. A47, 392-400.
Frear, D. S. & Swanson, H. R. (1970). Biosynthesis of S-(4-ethylamino-6-isopropylamino-2-s-tri- azino)glutathione: partial purification and properties of glutathione S-transferase from corn. Phytochem- istry, 9, 2123-2132.
Frear, D. S. & Swanson, H. R. (1973). Metabolism of substituted diphenylether herbicides in plants. 1. Enzymatic cleavage of fluoridifen in peas (Pisum sativum L.). Pestic. Biochem. Physiol. 3, 473-482.
Garcia-S~ez, I., P~irraga, A., Phillips, M. F., Mantle, T. J. & Coll, M. (1994). Molecular structure at 1.8 A of mouse liver class pi glutathione S-transferase complexed with S-(p-nitrobenzyl)glutathione and other inhibi- tors. J. Mol. Biol. 237, 298-314.
Grove, G., Zarlengo, R. E, Timmerman, K. P, Li, N.-Q., Tam, M. F. & Tu, C.-E D. (1988). Characterization and heterospecific expression of cDNA clones of genes in the maize GSH S-transferase multigene family Nucl. Acids Res. 16, 425-438.
Habig, W. H. & Jakoby, W. B. (1981). Assays for differentiation of glutathione S-transferases. Methods Enzymol. 77, 398-405.
Hahn, K. & Strittmatter, G. (1994). Pathogen-defence gene prpl-1 from potato encodes an auxin-responsive glutathione S-transferase. Eur. J. Biochem. 226, 619-626.
Hatzios, K. K. & Penner, D. (1982). Metabolism of Herbicides in Higher Plants. Burgess Publishing, Minneapolis.
Hayes, J. D. & Wolf, C. R. (1988). Role of glutathione transferase in drug resistance. In Glutathione Conju- gation: Mechanisms and Biological Significance (Sies, H. & Ketterer, B., eds), pp. 316-356, Academic Press, London.
Helmkamp, R. W. & Sears, D. A. (1970). A label for the red cell membrane: diazotized diiodosulfanilic acid. Int. J. Rad. Isotopes, 21, 683-685.
Hiratsuka, A., Sebata, N., Kawashima, K., Okuda, H., Ogura, K., Watabe, T., Satoh, K., Hatayama, I., Tsuchida, S., Ishikawa, T. & Sato, K. (1990). A new

308 Structure of A. thaliana Glutathione S-transferase
class of rat glutathione S-transferase Yrs-Yrs inacti- vating reactive sulfate esters as metabolites of carcinogenic arylmethanols. J. Biol. Chem. 265, 11973-11981.
Hoffman, O. L. (1978). Herbicide antidots: from concept to practice. In Chemistry and Action of Herbicide Antidotes (Pallos, F. M. & Casida, J. E., eds), pp. 1-13, Academic Press, New York.
Huber, R. (1965). Die automatisierte FaltmolkLilmethode. Acta Crystallog. 19, 293-297.
Hussey, A. J. & Hayes, J. D. (1992). Characterization of human class-theta glutathione S-transferase with activity towards 1-menaphthyl sulphate. Biochem. J. 286, 929-935.
Irzyk, G. P. & Fuerst, E. P. (1993). Purification and characterization of a glutathione S-transferase from benoxacor-treated maize (Zea mays). Plant Physiol. 102, 803-810.
Irzyk, G., Potter, S., Ward, E. and Fuerst, E. P. (1995). A cDNA clone encoding the 27-kilodalton subunits of glutathione S-transferase IV from Zea mays. Plant Physiol. 107, 311-312.
Jakoby, W. B. & Ziegler, D. M. (1990). The enzymes of detoxication. J. Biol. Chem. 265, 20715-20718.
Jepson, I., Lay, V. J., Holt, D. C., Bright, S.-W. J. & Greeland, A. J. (1994). Cloning and characterization of maize herbicide safener-induced cDNAs encoding subunits of glutathione S-transferase isoforms I, II and IV. Plant Mol. Biol. 26, 1855-1866.
Ji, X., Zhang, P., Armstrong, R. N. & Gilliland, G. L. (1992). The three-dimensional structure of a glutathione S-transferase from the mu gene class. Structural analysis of the binary complex of isoenzyme 3-3 and glutathione at 2.2A resolution. Biochemistry, 31, 10169-10184.
Ji, X., von Rosenvinge, E. C., Johnson, W. W., Tomarev, S. I., Piatigorsky, J., Armstrong, R. N. & Gilliland, G. L. (1995). Three-dimensional structure, catalytic properties, and evolution of a sigma class glutathione transferase from squid, a progenitor of the lens S-crystallins of cephalopods. Biochemistry, 34, 5317- 5328.
Jones, T. A. (1978). A graphics model building and refinement system for macromolecules. J. Appl. Crystallog. 15, 23-31.
Kabsch, W. & Sander, C. (1983). Dictionary of protein secondary structure: pattern recognition of hydrogen bonded and geometrical features. Biopohdmers 22, 2577-2637.
Kraulis, P. J. (1991). MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallog. 24, 946-950.
Lamoureux, G. L. & Rusness, D. G. (1987). Synergism of diazinon toxicity and inhibition of diazinon metab- olism in the house fly by tridiphane: inhibition of glutathione S-transferase activity. Pestic. Biochem. Physiol. 27, 318-329.
Lamoureux, G. L. & Rusness, D. G. (1989). The role of glutathione and glutathione S-transferases in pesti- cide metabolism, selectivity and mode of action in plants and insects. In Coenzymes and Cofactors, Vol. 3: Glutathione: Chemical, Biochemical and Medical Aspects (Dolphin, D., Poulson, R. & Avramovic, O., eds), Part B, pp. 153-196, Wiley, New York.
Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). PROCHECK: a program to check the stereochemical quality of protein struc- tures. J. Appl. Crystallog. 26, 283-291.
Leavitt, J. R. C. & Penner, D. (1979). In vitro conjugation
of glutathione and other thiols with acetanilide herbicides and EPTC sulfoxide and the action of the herbicide antidote R-25788. J. Agric. Food Chem. 27, 533-536.
Leslie, A. G. W. (1991). Recent changes to the MOSFLM package for processing film and image plate data. CCP4 and ESF-EACMB Newsletter on Protein Crystal- Iography (available from the Librarian, SERC Labora- tory, Daresbury, Warrington, WA4 4AD, UK).
Lim, K., Ho, J. X., Keeling, K., Gilliland, G. L., Ji, X., R~iker, F. & Carter, D. C. (1994). Three-dimensional structure of Schistosoma japonicum glutathione S- transferase fused with a six-amino acid conserved neutralizing epitope of gp41 from HIV. Protein Science, 3, 2233-2244.
Listowski, I. (1993). Glutathione S-transferases: intracellu- lar binding, detoxification, and adaptive responses. In Hepatic Transport and Bile Secretion: Physiology and Pathophysiology (Tavolini, N. & Beck, P. D., eds), pp. 397-405, Raven Press, New York.
Luzzati, P. V. (1952). Traitement statistique des erreurs dans la determination des structures cristallines. Acta Crystallog. 5, 802-810.
Mannervik, B. & Danielson, U. H. (1988). Glutathione transferases: structure and catalytic activity. CRC Crit. Rev. Biochem. Mol. Biol. 23, 283-337.
Mannervik, B., Alin, P., Guthenberg, C., Jensson, H., Tahir, M. K., Warholm, M. & J6rnvall, H. (1985). Identification of three classes of cytosolic glutathione transferases common to several mammalian species: correlation between structural data and enzymatic properties. Proc. Natl Acad. Sci. USA, 82, 7202-7206.
Matthews, B. W. (1968). Solvent content of protein crystals. ]. Mol. Biol. 33, 491-497.
McTigue, M., Williams, D. R. & Tainer, J. A. (1995). Crystal structures of a schistosomal drug and vaccine target: glutathione S-transferase from Schistosoma japonicum and its complex with the leading antischis- tosomal drug Praziquantel. ]. Mol. Biol. 246, 21-27.
Merrit, E. A. & Murphy, M. E. P. (1994). RASTER3D Version 2.0. A program for photorealistic molecular graphics. Acta Crystallog. D50, 869-873.
Messerschmidt, A. & Pflugrath, J. W. (1987). Crystal orientation and X-ray pattern prediction routines for area detector diffractometer systems in macromol- ecular crystallography. J. Appl. Crystallog. 20, 306-315.
Messerschmidt, A., Schneider, M. & Huber, R. (1990). ABSCOR: a scaling and absorption correction program for the FAST area detector diffractometer. J. Appl. Crystallog. 23, 436-439.
Meyer, D. J., Coles, B., Pemble, S. E., Gilmore, K. S., Frazer, G. M. & Ketterer, B. (1991a). Theta, a new class of glutathione transferases purified from rat and man. Biochem. J. 274, 409-414.
Meyer, R. C., Goldsbrough, P. B. & Woodson, W. R. (1991b). An ethylene responsive flower senescence- related gene from carnation encodes a protein homologous to glutathione S-transferases. Plant. Mol. Biol. 17, 277-281.
Moore, R. E., Davies, M. S., O'Connell, K. M., Harding, E. I., Wiegand, R. C. & Tiemeier, D. C. (1986). Cloning and expression of a cDNA encoding a maize glutathione S-transferase in E. coll. Nucl. Acids Res. 14, 7227-7235.
Mozer, T. J., Tiemeier, D. C. & Jaworski, E. G. (1983). Purification and characterization of corn glutathione S-transferase. Biochemistry, 22, 1068-1072.
O'Connell, K. M., Breaux, E. J. & Fraley, R. T. (1988). Different rates of metabolism of two chloroac-

Structure of A. thaliana Glutathione S-transferase 309
etanilide herbicides in Pioneer 3320 corn. Plant Physiol. 86, 359-363.
Pemble, S. E. & Taylor, J. B. (1992). An evolutionary perspective on glutathione transferases inferred from class-theta glutathione transferase cDNA sequences. Biochem. J. 287, 957-963.
Prohaska, J. R. (1980). The glutathione peroxidase activity of glutathione S-transferases. Biochim. Biophys. Acta, 611, 87-98.
Raghunathan, S., Chandross, R. J., Kretsinger, R. H., Allison, T. J., Penington, C. J. & Rule, G. S. (1994). Crystal structure of human class mu glutathione transferase GSTM2-2. Effects of lattice packing on conformational heterogeneit~ J. Mol. Biol. 238, 815-832.
Ramachandran, G. N. & Sasisekharan, V. (1968). Conformation of polypeptides and proteins. Adv. Protein Chem. 23, 283-437.
Reinemer, P., Dirr, H. W., Ladenstein, R., Sch~iffer, J., Galla~ O. & Huber, R. (1991). The three-dimensional structure of class 7r glutathione S-transferase in complex with glutathione sulfonate at 2.3 A resol- ution. EMBO J. 10, 1997-2005.
Reinemer, P., Dirr, H. W., Ladenstein, R., Huber, R., Lo Bello, M., Federici, G. & Parker, M. W. (1992). Three- dimensional structure of class ~ glutathione S-trans- ferase from human placenta in complex with S-hexylglutathione at 2.8 A resolution. J. Mol. Biol. 227, 214-226.
Richardson, J. S. (1977). ~-sheet topology and the relatedness of proteins. Nature, 268, 495-500.
Rushmore, T. H. & Pickett, C. B. (1993). Glutathione S-transferases, structure, regulation, and therapeutic implications. ]. Biol. Chem. 268, 11475-11478.
Sandermann, H., Jr (1992). Plant metabolism of xeno- biotics. Trends Biochem. Sci. 17, 82-84.
Shah, D. M., Hironaka, C. M., Wiegand, R. C., Harding, E. I., Krivi, G. G. & Tiemeier, D. C. (1986). Structural analysis of a maize gene coding for glutathione S-transferase involved in herbicide detoxification. Plant Mol. Biol. 6, 203-211.
Shimabukuro, R. H., Swanson, H. R. & Walsh, W. C. (1970). Glutathione conjugation: atrazine detoxifica- tion mechanism in corn. Plant Physiol. 46, 103-107.
Shimabukuro, R. H., Frear, D. S., Swanson, H. R. & Walsh, W. C. (1971). Glutathione conjugation. An enzymatic basis for atrazine resistance in corn. Plant. Physiol. 47, 10-14.
Shimabukuro, R. H., Lamoureux, G. L., Swanson, H. R., Walsh, W. C., Stafford, L. E. & Frear, D. S. (1973). Metabolism of substituted diphenylether herbicides in plants. 2. Identification of a new fluorodifen metabolite S-(2-nitro-4-trifluoromethyl- phenyl)glutathione in peanut. Pestic. Biochem. Physiol. 3, 483-494.
Sinning, I., Kleywegt, G. J., Cowan, S. W., Reinemer, P., Dirr, H. W., Huber, R., Gilliland, G. L., Armstrong, R. N., Ji, X., Board, P. G., Olin, B., Mannervik, B. & Jones, T. A. (1993). Structure determination and refinement of human alpha class glutathione trans- ferase A1-1, and a comparison with the mu and pi class enzymes. J. Mol. Biol. 232, 192-212.
Smith, D. B., Davern, K. M., Board, P. G., Tiu, W. U., Garcia, E. G. & Mitchell, G. F. (1986). Mr 26,000 antigen of
Schistosoma japonicum recognized by resistant WEHI 129/J mice is a parasite glutathione S-transferase. Proc. Natl Acad. Sci. USA, 83, 8703-8707.
Smith, D. B., Davern, K. M., Board, P. G., Tiu, W. U., Garcia, E. G. & Mitchell, G. F. (1987). Correction. Proc. Natl Acad. Sci. USA, 84, 6541.
Steigemann, W. (1991). Recent advances in the PROTEIN program system for the X-ray structure analysis of biological macromolecules. In Crystallographic Com- puting 5 (Moras, D., Podjarn34 A. D. & Thiery, J. C., eds), pp. 115-125, Oxford University Press, Oxford.
Studier, F. W. & Moffart, B. (1986). Use of bacteriophage T7 polymerase to direct selective high-level ex- pression of cloned gene. J. Mol. Biol. 189, 113-130.
Studier, F. W., Rosenberg, A. H., Dunn, J. J., Dubendorff, J. W. (1990). Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 185, 60-89.
Takahashi, Y. & Nagata, T. (1992). ParB: an auxin- regulated gene encoding glutathione S-transferase. Proc. Natl Acad. Sci. USA, 89, 56-59.
Timmerman, K. E (1989). Molecular characterization of corn glutathione S-transferase isoenzymes involved in herbicide detoxification. Physiol. Plant. 77, 465-471.
Tomarev, S. I., Zinovieva, R. D., Guo, K. & Piatigorsk)4 J. (1993). Squid glutathione S-transferase. Relationships with other glutathione S-transferases and S-crys- tallins of cephalopods. J. Biol. Chem. 268, 4534-4542.
Tsuchida, S. & Sato, K. (1992). Glutathione S-transferases and cancer. CRC Crit. Rev. Biochem. Mol. Biol. 27, 337-384.
Tu, C.-E D. & Qian, B. (1986). Human liver glutathione S-transferase: complete primary seqeunce of an Ha subunit cDNA. Biochem. Biophys. Res. Commun. 141, 229-237.
Turk, D. (1992). Weiterentwicklung eines Programms f/Jr Molekiilgraphik und Elektronen-dichte-Manipu- lation und seine Anwendung auf verschiedene Protein-strukturaufkl~irungen. Ph.D. thesis, Technical Universit~ Munich.
Wang, B.-C. (1985). Resolution of phase ambiguity in macromolecular crystallograph~ Methods Enzymol. 115, 90-122.
Wiegand, R. C., Shah, D. M., Mozer, T. J., Harding, E. I., Diaz-Collier, J., Sanders, C., Jaworski, E. G. & Tiemeier, D. C. (1986). Messenger RNA encoding a glutathione S-transferase responsible for herbicide tolerance in maize is induced in response to safener treatment. Plant Mol. Biol. 7, 235-243.
Wilce, M. C. J., Board, E G., Feil, S. C. & Parker, M. W. (1995). Crystal structure of a theta-class glutathione transferase. EMBO J. 14, 2133-2143.
Zettl, R., Schell, J. & Palme, K. (1994). Photoaffinity labelling of Arabidopsis thaliana plasma membrane vesicles by 5-azido-[7-3H]indole-3-acetic acid: identification of a glutathione S-transferase. Proc. Natl Acad. Sci. USA, 91, 689-693.
Zhang, K. Y. J. & Main, P. (1990). Histogram matching as a new density modification technique for phase refinement and extension of protein molecules. Acta Crystallog. A46, 41-46.
Zhou, J. & Goldsbrough, P. B. (1993). An Arabidopsis gene with homology to glutathione S-transferases is regulated by ethylene. Plant Mol. Biol. 22, 517-523.
Edited by L A. Wilson
(Received 14 July 1995; accepted in revised form 5 October 1995)
Copyright © 2022 FDOKUMEN




















