
Theoretical modeling of optical properties of Ag8 and Ag14 silver clusters embedded in an LTA...
12
15404 Phys. Chem. Chem. Phys., 2013, 15, 15404--15415 This journal is c the Owner Societies 2013 Cite this: Phys. Chem. Chem. Phys., 2013, 15, 15404 Theoretical modeling of optical properties of Ag 8 and Ag 14 silver clusters embedded in an LTA sodalite zeolite cavity† Ngo Tuan Cuong,* a Hue Minh Thi Nguyen a and Minh Tho Nguyen* bc Optical properties of silver Ag n nanoclusters are demonstrated to be dependent on their size, structure and charge state. It is found that when being contained in the sodalite cavity of LTA zeolite the tetradecanuclear hexacation silver cluster Ag 14 6+ is stable. Its lower-lying states and optical spectrum are theoretically determined using the quantum chemical TD-DFT method. Its ground state possesses an outer-shell electron configuration of A 1g 2 T 2g 6 mimicking the s 2 p 6 valence of noble gas atoms. These frontier orbitals are constructed from 5s,5p(Ag)-AOs with contributions from framework oxygen atoms. Light absorption of Ag 14 6+ embedded in the sodalite cage which is characterized by strong peaks centered at 331 and 476 nm (transitions 5s,p(Ag) - 5s,p(Ag)) leads to much longer wavelength emission. The sodalite cage, as a container, stabilizes the central Ag 14 6+ cluster by electrostatic attraction. The absorption spectrum of the isovalent neutral Ag 8 cluster embedded inside the same sodalite cavity is also simulated using TD-DFT and CASPT2 methods. This absorption spectrum which is similar to that of the Ag 14 6+ cluster has two absorption bands in the near UV and visible regions. 1. Introduction The oligoatomic silver clusters, consisting of silver atoms and ions, continue to attract considerable attention. In contrast to bulk materials, silver clusters, especially their positively charged derivatives, present a molecule-like behavior featuring discrete energy levels. When interacting with photons, clusters undergo electronic transitions and as a result, absorption and emission occur as in any molecular system. Optical properties of silver Ag n nanoclusters were intensively investigated both experimentally and theoretically. They appear to depend on their sizes, structures and charge states. Due to the strong luminescence in the UV-VIS region, these compounds have promising potential for applications in, among others, bio- medical treatments, production of biological sensors, optical storage, and so on. 1–29 In this context, it is of significant interest to obtain a deeper understanding of the photochemical properties of silver clusters in different environments. To reduce surface energy, silver nanoclusters can irreversibly be fused together to form larger clusters or even bulk materials. Therefore, in order to prevent them from aggregating, they are usually charged or dispersed in media such as noble gases, organic scaffolds or zeolite cavities. Zeolites, aluminosilicate- based porous materials, usually serve as containers of nanoclusters since they can incorporate various cluster types. Zeolites containing transition metal ions have been known to be promising heterogeneous catalysts. Knowledge about the location and structure of the metal centers is of paramount importance for the understanding of the catalytic mechanism of these materials. 30–37 The inherently stable and highly crystal- line structures of zeolites make them unique among high surface area solids in that the active site can well be defined both structurally and electronically. 13,14,19,38 Encapsulation of a molecular system in a restricted environ- ment of a zeolite is expected to change the guest properties. Through host–guest interactions, even an unstable cluster could be stabilized to exist inside a nanometer-size cavity of a host. 39 In zeolites, substitution of Si 4+ in SiO 2 frameworks by Al 3+ cations induces negative charges on the framework oxygen atoms neighboring the Al sites. 40 By requirements of charge balance, various cations are bound in the vicinity of the sub- stitution sites. The negatively charged oxygen atoms can in turn induce an influence on the guest structure. Sometimes the inner cations aggregate to form unusually small clusters, due to the stabilizing interactions with the negatively charged oxygens a Center for Computational Science and Faculty of Chemistry, Hanoi National University of Education, Hanoi, Vietnam. E-mail: [email protected] b Department of Chemistry, University of Leuven, B-3001 Leuven, Belgium. E-mail: [email protected] c Institute for Computational Science and Technology at Ho Chi Minh City (ICST), Quang Trung Software City, Ho Chi Minh City, Vietnam † Electronic supplementary information (ESI) available. See DOI: 10.1039/ c3cp51017d Received 7th March 2013, Accepted 10th July 2013 DOI: 10.1039/c3cp51017d www.rsc.org/pccp PCCP PAPER
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Theoretical modeling of optical properties of Ag8 and Ag14 silver clusters embedded in an LTA...

15404 Phys. Chem. Chem. Phys., 2013, 15, 15404--15415 This journal is c the Owner Societies 2013
Cite this: Phys. Chem.Chem.Phys.,2013,15, 15404
Theoretical modeling of optical properties of Ag8 andAg14 silver clusters embedded in an LTA sodalitezeolite cavity†
Ngo Tuan Cuong,*a Hue Minh Thi Nguyena and Minh Tho Nguyen*bc
Optical properties of silver Agn nanoclusters are demonstrated to be dependent on their size, structure
and charge state. It is found that when being contained in the sodalite cavity of LTA zeolite the
tetradecanuclear hexacation silver cluster Ag146+ is stable. Its lower-lying states and optical spectrum are
theoretically determined using the quantum chemical TD-DFT method. Its ground state possesses an
outer-shell electron configuration of A1g2T2g
6 mimicking the s2p6 valence of noble gas atoms. These
frontier orbitals are constructed from 5s,5p(Ag)-AOs with contributions from framework oxygen atoms.
Light absorption of Ag146+ embedded in the sodalite cage which is characterized by strong peaks
centered at 331 and 476 nm (transitions 5s,p(Ag) - 5s,p(Ag)) leads to much longer wavelength
emission. The sodalite cage, as a container, stabilizes the central Ag146+ cluster by electrostatic
attraction. The absorption spectrum of the isovalent neutral Ag8 cluster embedded inside the same
sodalite cavity is also simulated using TD-DFT and CASPT2 methods. This absorption spectrum which is
similar to that of the Ag146+ cluster has two absorption bands in the near UV and visible regions.
1. Introduction
The oligoatomic silver clusters, consisting of silver atoms andions, continue to attract considerable attention. In contrast tobulk materials, silver clusters, especially their positivelycharged derivatives, present a molecule-like behavior featuringdiscrete energy levels. When interacting with photons, clustersundergo electronic transitions and as a result, absorption andemission occur as in any molecular system. Optical propertiesof silver Agn nanoclusters were intensively investigated bothexperimentally and theoretically. They appear to depend ontheir sizes, structures and charge states. Due to the strongluminescence in the UV-VIS region, these compounds havepromising potential for applications in, among others, bio-medical treatments, production of biological sensors, opticalstorage, and so on.1–29 In this context, it is of significantinterest to obtain a deeper understanding of the photochemicalproperties of silver clusters in different environments.
To reduce surface energy, silver nanoclusters can irreversiblybe fused together to form larger clusters or even bulk materials.Therefore, in order to prevent them from aggregating, they areusually charged or dispersed in media such as noble gases,organic scaffolds or zeolite cavities. Zeolites, aluminosilicate-based porous materials, usually serve as containers ofnanoclusters since they can incorporate various cluster types.Zeolites containing transition metal ions have been known tobe promising heterogeneous catalysts. Knowledge about thelocation and structure of the metal centers is of paramountimportance for the understanding of the catalytic mechanismof these materials.30–37 The inherently stable and highly crystal-line structures of zeolites make them unique among highsurface area solids in that the active site can well be definedboth structurally and electronically.13,14,19,38
Encapsulation of a molecular system in a restricted environ-ment of a zeolite is expected to change the guest properties.Through host–guest interactions, even an unstable clustercould be stabilized to exist inside a nanometer-size cavity of ahost.39 In zeolites, substitution of Si4+ in SiO2 frameworks byAl3+ cations induces negative charges on the framework oxygenatoms neighboring the Al sites.40 By requirements of chargebalance, various cations are bound in the vicinity of the sub-stitution sites. The negatively charged oxygen atoms can in turninduce an influence on the guest structure. Sometimes theinner cations aggregate to form unusually small clusters, due tothe stabilizing interactions with the negatively charged oxygens
a Center for Computational Science and Faculty of Chemistry, Hanoi National
University of Education, Hanoi, Vietnam. E-mail: [email protected] Department of Chemistry, University of Leuven, B-3001 Leuven, Belgium.
E-mail: [email protected] Institute for Computational Science and Technology at Ho Chi Minh City (ICST),
Quang Trung Software City, Ho Chi Minh City, Vietnam
† Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp51017d
Received 7th March 2013,Accepted 10th July 2013
DOI: 10.1039/c3cp51017d
www.rsc.org/pccp
PCCP
PAPER

This journal is c the Owner Societies 2013 Phys. Chem. Chem. Phys., 2013, 15, 15404--15415 15405
of zeolites. For example, small silver clusters consisting of 2–8atoms have been reported to exist inside zeolite cavities.41–58
Depending on the size and the charge of enclosed silverspecies, the embedded zeolites exhibit quite unique opto-electronic properties.53 For instance, Ag3
n+ and Ag6m+ clusters
can be formed within FAU- and LTA-type zeolites and becomefluorescent after calcinations.54
Recently, the Ag14 cluster has been found to be formedinside a sodalite cavity of LTA zeolite.59 However, the actualcharge of the Ag14 cluster formed in this zeolite environment,the nature of such association and metal–metal interactions, aswell as the role of sodalite cavity incarcerating silver atomsand/or ions in its center are not fully understood yet. Theoryplays here a crucial role in the interpretation of optical spectra ofzeolites containing transition metals, and can yield quantitativeinformation regarding the electronic structure of intra-zeolite‘‘active centers’’.60 In this context, we set out to investigatetheoretically the electronic structures and optical properties ofAg14 silver clusters embedded inside a sodalite cage of LTAzeolite aiming at revealing the preferable charge of the encapsu-lated Ag14 and its interaction with the environment. Further, weshall investigate the optical properties of the neutral isovalentsilver octamer Ag8 as a typical case of a complete electronic shellcluster embedded in the same sodalite cavity.
2. Methods of calculation
We first construct a non-substituted zeolite model including asix-membered ring (6-MR) cavity, a composite building unit, ora sodalite (SOD 24T) cavity, as shown in Fig. 1. This simplestmodel has a Si24O36 framework whose silicon atoms are allsaturated by hydrogen atoms. As a result, the chemical formulaof the host is Si24H24O36. To construct the embedded Ag14-SODmodel, we put fourteen Ag atoms (Ag14) inside the cavity of theSi24H24O36 framework, and then optimize the geometries of thepossible structures using density functional theory (DFT) com-putations. Following extensive geometry optimizations with afixed zeolite framework, the most plausible configuration of theresulting cluster structure can be determined. In the latter,eight silver atoms are located on the eight six-membered ringswhereas the six remaining silver atoms are pointed toward thefour-membered rings of the sodalite cavity. The formula of thecluster thus becomes [Ag14(Si24H24O36)]. The Ag14 clusterembedded in a half–half Al-substituted SOD cage, namely the[Ag14(Al12Si12H24O36)] cluster is also investigated.
In constructing the neutral silver octamer cluster embeddedAg8 model, we put the eight silver atoms inside the cavity ofthe Si24H24O36 framework. The most plausible structure of theembedded Ag8 is in the Td point group in which each of theeight silver atoms points toward one of the eight six-memberedrings of the sodalite cavity. This embedded Ag8 structure iscomparable to the most stable structure of the pure neutralsilver octamer. The chemical formula of the resulting cluster isthus [Ag8(Si24H24O36)].
Using DFT methods,61 we optimize the geometries of[Ag14(Si24H24O36)], [Ag14(Al12Si12H24O36)] and [Ag8(Si24H24O36)]
structures in such a way that the geometry of the surroundingsodalite cage is fixed and the embedded silver clusters arerelaxed converging to an energy minimum in the groundelectronic state. Harmonic vibrational frequency calculationsare followed to confirm the optimized stationary points asminima (with all real frequencies). In the following stage, thetime-dependent density functional theory (TD-DFT)62 calcula-tions are performed in order to identify the electron transitionsfrom the ground to excited states that are responsible for thecorresponding absorption spectra.
The popular hybrid B3LYP functional63 is employed sincethis functional is well known to reproduce relatively well theexperimental absorption spectra of silver clusters.64 Due to therelatively large size of the systems considered, the LANL2DZbasis set65 is used and all calculations are performed usingGaussian 09 package.66 In the last stage of calculations, we usethe methods of a complete active space self-consistent fieldfollowed by a second-order perturbation theory treatment(CASSCF/CASPT2 method), and the restricted active space state
Fig. 1 (a) A composite building unit SOD 24T cage, and (b) the structure of theSi24H24O36 host. The red circles represent O atoms; the small grey circles H atomsand dark-green circles the Si atoms.
Paper PCCP

15406 Phys. Chem. Chem. Phys., 2013, 15, 15404--15415 This journal is c the Owner Societies 2013
interaction (RASSI method) to simulate the emission processesof some of the [Ag8(Si24H24O36)] investigated clusters usingMOLCAS package.67 The selection of methods for treatmentof excited states is due to the fact that when using the CASPT2method, we could identify and select the specific excited states.Therefore CASPT2 calculations have been carried out only forspecific lower-lying excited states from which the emissionprocesses occur that we could identify. In contrast, the absorp-tion involves transitions from the ground state to the energyhigher-lying excited states; therefore they can also be identifiedby TD-DFT calculations that are much more convenient andeconomical to be carried out than CASPT2 computations.
3. Results and discussion3.1. Ag14 cluster embedded inside a half–half substituted LTAsodalite cavity: the [Ag14(Al12Si12H24O36)] cluster
The substitution of Si4+ by Al3+ in the SiO2 framework inducesgeneration of negative charges on framework oxygen atomsneighboring the Al sites. Therefore, for the sake of chargecompensation, cations such as Na+ are incorporated. The Na+
cations are then replaced by Ag+ ions.59,68 The Ag+ ions can bepartially or fully reduced to form the silver clusters encapsu-lated in the sodalite cavity of the zeolite. We thus consider theresulting Ag14 moiety, namely the [Ag14(Al12Si12H24O36)]p� clusterswith different negative charges p = 0, 2, 4, 6, 8.
Geometries of [Ag14(Al12Si12H24O36)]p� structures are opti-mized, followed by harmonic vibrational frequency calculationsfor the singlet spin state. In each [Ag14(Al12Si12H24O36)]p�
system, we consider the most likely possibilities of Al positionswhere 12 Al atoms and 12 Si atoms are alternating to eachother. TD-DFT calculations are subsequently performed usingthese optimized geometries to evaluate the transition energiesin going from the ground to excited states, and thereby tosimulate the absorption spectra. The detailed results on theenergy gaps and absorption spectra are illustrated in Fig. 2.
The behavior of an Ag14 cluster inside a sodalite cavity of thehalf–half exchange zeolite LTA(Al12) can now be examined witha focus on formal charges of clusters and zeolite frameworks.With the aim of finding the charges, or the population ofelectrons, of different compositions of the whole clusters wehave performed an NBO analysis. The changes of the net chargeof Ag14 cluster as well as that of the [Al12Si12H24O36] frameworkwith respect to the negative charge p of the whole[Ag14(Al12Si12H24O36)]p� system are probed by the calculatedNBO charges. The results are listed in Table 1. It should bestressed that similar to other types of charge, the NBO chargesalso have inherent advantages and failures. However, the NBOcharges perform much better for clusters than other types suchas the classical Mulliken charges. The relative values of differentcharges are more meaningful than their absolute values.
The [Al12Si12H24O36] framework with twelve substituted Alatoms has a �12 formal charge. Conversely, the formal chargeof an Ag14 cluster inside the [Al12Si12H24O36] sodalite cageshould be +12. Because of some charge transfer from thenegatively charged framework to the silver block, the
[Al12Si12H24O36] framework has less than �12 negative charge,as can be seen in Table 1. NBO charge results suggest that the[Ag14(Al12Si12H24O36)]p� ion is basically decomposed into twointeracting fragments, namely the [Al12Si12H24O36]12� frameworkand the Ag14
(12�p)+ cluster, that is [Ag14(Si12H36O24Al12)] p� =[(Si12H36O24Al12)]12� + Ag14
(12�p)+.As an example, let us consider the case of
[Ag14(Si12H36O24Al12)]6� = [(Si12H36O24Al12)]12� + Ag146+
The characteristics of frontier orbitals of an Ag14 clusterinside a [Al12Si12H24O36] framework basically depend on thecharge p-value. The much negatively charged [Al12Si12H24O36]
Fig. 2 (a) The energy gaps between the ground and excited states of the[Ag14(Al12Si12H24O36)]p� clusters calculated using TD-DFT at the B3LYP/LANL2DZlevel. (b) The absorption spectra of the [Ag14(Al12Si12H24O36)]p� clusters calcu-lated using TD-DFT at the B3LYP/LANL2DZ level.
Table 1 NBO charges of Ag14 cluster encapsulated in the [Al12Si12H24O36]framework
p� 0� 2� 4� 6� 8�
Charge of Ag14, electron 10.0+ 8.8+ 7.1+ 5.5+ 3.7+Charge of framework, electron 10.0� 10.8� 11.1� 11.5� 11.7�HOMO–LUMO gap (eV) 1.9 1.2 1.0 2.9 0.2
PCCP Paper

This journal is c the Owner Societies 2013 Phys. Chem. Chem. Phys., 2013, 15, 15404--15415 15407
framework that emerged from 12 Al-substitution is likely toinduce a strong electrostatic interaction with the Ag14 cluster.
In order to probe the question of the p-value with which anAg14
(12�p)+ cluster can be embedded in a kinetically stable[Al12Si12H24O36]12� framework, we consider the HOMO–LUMOgaps of the investigated clusters, as well as the transitionenergies between the ground electronic and excited states.The frontier energy gap is usually an indicator for kineticstability. In addition, because in the model systems consideredhere, the structures include fragments of the zeolites withadded hydrogen atoms for the saturation of chemical bondsof Si and Al, any thermodynamical parameter for these systemsshall not reflect the real stability of the zeolite. The calculatedresults are also shown in Table 1 and Fig. 2.
The [Ag14(Si12H36O24Al12)]p� cluster with p = 6 turns out tohave the largest HOMO–LUMO gap among the investigatedclusters, suggesting the highest stability. It also has the largestexcitation energy. Therefore, we pursue further the investiga-tion of the [Ag14(Si12H36O24Al12)]6� systems in which a Ag14
6+
polycation is encapsulated in the sodalite cavity. Let usstress that the polycation Ag14
6+ is found by NBO chargesand frontier orbital analysis to prefer to be embedded insidethe sodalite cage. This does not depend on the level ofAl-substitution or the number of Al atoms in the frameworkinstead of Si.
3.2. Ag146+ cluster encapsulated in the LTA sodalite cavity
The free standing Ag146+ cation is not stable in the gas phase
but it becomes stabilized when being encapsulated in thesodalite cage, in part due to the inherent host–guest inter-actions. To confirm this view, we again consider the Ag14
6+
cation to be embedded in some kinds of LTA cavitiesincluding both non-Al-substituted and Al-substituted cages,such as the [Ag14(Si12H36O24)]6+, [Ag14(Al6Si18H36O24)]0 and[Ag14(Al12Si12H36O24)]6�.
The geometries of the latter clusters are thus optimized withfixed frameworks, followed by vibrational frequency calcula-tions. TD-DFT calculations are subsequently performed attheir ground state optimized geometries in order to modeltheir absorption spectra. Results of the average Ag–Ag andAg(ring)–O(framework) bond lengths along with the NBOcharges of Ag14 are listed in Table 2. The calculated transitionenergies between the ground and excited states for the threeclusters considered are summarized in Fig. 3, and the theore-tical absorption spectra are simulated in Fig. 4.
From the results listed in Table 2 and absorption spectraillustrated in Fig. 4, the [Ag14(Si12H36O24)]6+, [Ag14(Al6Si18H36O24)]0
and [Ag14(Al12Si12H36O24)]6� clusters can be considered as the
Ag146+ cluster encapsulated in the sodalite cages. Accordingly, we
now investigate in more detail the absorption spectrum of[Ag14(Si12H36O24)]6+ as a representative case.
The singlet ground state of [Ag14(Si24H24O36)]6+ in its highsymmetry Oh point group has the following orbital configu-ration for the valence shell:
1A1g: . . . [(A1g)2(T2g)2(T2g)2(T2g)2(T1u)0(T1u)0(T1u)0(E)0(E)0(A1g)0. . .].
This electron configuration could be considered as amimic of the s2p6 electron configuration of noble gasatoms. The geometrical shape of the global minimum of[Ag14(Si24H24O36)]6+ (Oh) is displayed in Fig. 5, and the energies
Table 2 NBO charges (electron) of embedded Ag14 cluster and the average Ag–Ag and Ag(ring)–O(framework) bond lengths in three different clusters
[Ag14(Si12H36O24)]6+ [Ag14(Al6Si18H36O24)]0 [Ag14(Al12Si12H36O24)]6�
NBO Charge of Ag14 +5.4 +5.4 +5.5NBO Charge of the framework +0.6 �5.4 �11.5d(Ag–Ag) (Å) 2.8 2.8 2.8d(Ag(ring)–O(framework)) (Å) 2.3 2.3 2.3
Fig. 3 (a) Energy (eV) gap between the ground and excited states for the[Ag14(Si12H36O24)]6+, [Ag14(Al6Si18H36O24)]0 and [Ag14(Al12Si12H36O24)]6� clusters.(b) Energy diagram for the [Ag14(Si12H36O24)]6+ cluster. The green line denotesground states, black lines denote singlet lowest excited states and red linesdenote triplet lowest excited states. The up arrows show vertical transitionscorresponding to the absorption peaks A and B in Fig. 4. The down arrows showpossible emissions. Results are calculated using TD-DFT (B3LYP/LANL2DZ).
Paper PCCP

15408 Phys. Chem. Chem. Phys., 2013, 15, 15404--15415 This journal is c the Owner Societies 2013
of the valence Kohn–Sham orbitals and their correspondingshapes are shown in Fig. 6.
The average Ag–Ag and Ag(ring)–O bond lengths of theoptimized structure amount to 2.83 and 2.30 Å, respectively.The net NBO charge of the Ag14 component in[Ag14(Si24H24O36)]6+ is 5.4 electrons (B3LYP/LANL2DZ). Of thefourteen Ag atoms present, eight are capped onto the six-membered ring of the Si24H36O24 cavity and have a positivecharge of +6.4 electrons. Six other silver atoms are located
inside the cavity and form an Ag6 moiety with a NBO chargeof �1.0 electron.
TD-DFT calculations using the B3LYP/LANL2DZ method atthe optimized geometry allow several singlet excited states of[Ag14(Si24H24O36)]6+ to be identified. The energy levels ofground 1A1g and singlet excited states are found to be locatedin a range of B4.0 eV. The corresponding optical absorptionspectra are simulated and plotted in Fig. 4. These excited statesare basically generated upon electronic transitions fromHOMOs to LUMOs of the [Ag14(Si24H24O36)]6+ (1A1g) cluster.The HOMOs and LUMOs are three-fold degenerate T orbitalswithin the Oh point group.
A pertinent question which now arises is about the nature ofthe interaction between the polycation Ag14
6+ and the sodaliteframework: is it due to covalent bonding or rather an ionicinteraction?
Calculated densities of states (DOS) for the [Ag14(Si12H36O24)]6+
cluster, which are illustrated in Fig. 7, suggest that there is asmall contribution of AOs of oxygen atoms of about 20% to theformation of frontier orbitals of the encapsulated ion Ag14
6+.The analysis reveals no significant sharing of electrons betweenthe Ag14 entity and the framework. This finding suggests thatinteraction between the Ag14
6+ cluster and the framework is ofan ionic nature. The cavity could be considered as a ligandexerting a strong stabilizing effect on the inherently unstablepolycation Ag14
6+. The latter can in turn be considered as acentral ion, and such a specific effect can be represented solelyby a simple point charge with an electrostatic potential.Covalent bonding between the metallic entity and the sodalitecage can thus be disregarded.
In order to find out the factors governing the clusterformation inside a cavity, we turn again to their frontier orbitalsdisplayed in Fig. 6. Frontier orbitals of [Ag14(Si24H24O36)]6+ aremade up mostly of their 5s,p(Ag) orbitals. The HOMOs have onenode, whereas the LUMOs have two nodes. Both HOMOs and
Fig. 4 Theoretical absorption spectra of [Ag14(Si12H36O24)]6+, [Ag14(Al6Si18H36O24)]0
and [Ag14(Al12Si12H36O24)]6� clusters. Black curve is the absorption spectrum of[Ag14(Si24H24O36)]6+, Green curve of [Ag14(Al6Si18H24O36)]0 and Blue curve of[Ag14(Al12Si12H24O36)]6�. Results are obtained using TD-DFT (B3LYP/LANL2DZ).
Fig. 5 Geometry of the [Ag14(Si12H36O24)]6+ cluster optimized at B3LYP/LANL2DZ. The cluster belongs to Oh point group. The Ag14
6+ block is located atthe center of the (Si12H36O24) cavity. Of the 14 Ag atoms, six ones form the Ag6
cluster locating at the center of the (Si12H36O24) cavity with the average Ag–Agbond length equal to 2.78 Å; eight Ag atoms are located at the vicinities of six-member rings of the cavity with average distant to the nearest oxygen atomsd(Ag–O) = 2.30 Å.
Fig. 6 Diagram for Kohn–Sham orbital energy levels of the [Ag14(Si24H24O36)]6+
cluster. The HOMOs and LUMOs are identified in the insert.
PCCP Paper

This journal is c the Owner Societies 2013 Phys. Chem. Chem. Phys., 2013, 15, 15404--15415 15409
LUMOs are in the T irreducible representation of the pointgroup. Those orbitals have in-phase interactions betweenadjacent silver atoms, and constitute a major source for forminga consistent Ag14
6+ block.The HOMO� 2, HOMO� 1 and HOMO, which belong to T1u
irreducible representation of the Oh point group, have a similarshape mimicking the 2p type atomic orbitals. The LUMO,LUMO + 1 and LUMO + 2, which belong to T2g irreduciblerepresentation as well, have a similar shape mimicking the 3dtype atomic orbitals. The HOMO–LUMO gap amounts to 2.8 eVand as a result, the low-lying excited states are composedmostly of HOMO to LUMO transitions, and the resultingabsorptions occur in the visible region.
The lower-lying excited states which are formed by HOMO -
LUMO electron transitions, or in other words from T1u to T2g
orbitals, belong to the A2u, Eu, T1u and T2u irreducible repre-sentations of the same Oh point group.
The absorption spectrum of [Ag14(Si24H24O36)]6+ spanned inthe visible and near ultra-violet regions is characterized by twomain peaks. The longest wavelength absorption peak centeredat 476 nm (peak (B) in Fig. 4), is due to a 1A1g -
1T1u transitionwith an oscillator strength of f = 0.12. The most intense peaklocated at 331 nm (being peak (A) in Fig. 4), having the largestoscillator strength f = 0.61, is again due to another 1A1g -
1T1u
electron transition. In the visible region, one other 1T1u state,and two 1T2u, one 1A2u and one 1Eu singlet excited states doexist, but the electronic transitions to these states from theground state are formally forbidden with the oscillator strengthbeing f = 0 (see Fig. 4).
The three lowest-lying singlet excited states identified byTD-DFT calculations arise mostly from HOMO - LUMO tran-sitions, and are located at 2.08 (1Eu), 2.16 (1T2u), 2.22 (1A2u) and2.61 eV (1T1u) with respect to the ground state. These electronictransitions could be considered as movements from 5s,p(Ag) to
5s,p(Ag) orbitals, since the contributions of 5s,p(Ag) to theHOMO and LUMO are predominant, being 61.3 and 61.6%for the HOMO and LUMO, respectively (cf. Fig. 7). They repre-sent the characteristic intra-atomic electronic transitionswithin the Ag14
6+ moiety inside the cavity.As seen in the energy diagram for ground and singlet excited
states for the enclosed Ag146+ shown in Fig. 3, electronic
transitions will not occur from ground state to the lowestexcited state, namely the 1E state, but rather to the higherexcited states – states 1T1u. The two corresponding 1T1u stateslie at 2.61 and 3.74 eV above the ground state, respectively.These states will rapidly decay to the lowest excited state 1E bynon-radiative processes, such as vibrational relaxation, or lossof excited state energy as heat to the surrounding due to theintermediate states, and the small energy gaps between them.A luminescence is expected to occur from the lowest-lyingexcited state 1E, which is located at B2.0 eV above the groundstate, to the ground state on a long time scale due to the factthat it is an orbit forbidden transition.
Overall, it can be concluded that the tetradeca-silver hexa-cation Ag14
6+ is stabilized by electrostatic interactions when it isembedded in a sodalite cavity of LTA zeolite. Its ground statepossesses an outer shell electron configuration of A1g
2T2g6
mimicking the s2p6 eight valence electrons of noble gas atoms.
3.3. Ag8 neutral cluster encapsulated in the LTA sodalitecavity
To probe further the optical properties of Ag clusters, we nowconsider the neutral Ag8 cluster whose valence electrons arecomparable to the polycation Ag14
6+. In the [Kr](4d10)(5s)1
electron configuration of a neutral silver atom, the rather poor4d–5s hybridization is well known as compared to the isovalentelements copper and gold;69 the silver octamer has thus eightelectrons in its valence shell. In this section we describe theoptical properties of the free neutral silver octamer Ag8 and itsencapsulation inside the sodalite cavity of LTA-type zeolite.Although the electronic structure and absorption spectrum ofthe pure silver octamer Ag8 have been analyzed in the litera-ture,64,70 for the sake of comprehension and calibration, wefirst present again some calculated results for the pure silveroctamer, followed by its incorporation in the zeolites. For thepure silver neutral octamer Ag8, two stable isomers have beenfound by our calculations and both have 3D structures. Themost stable isomer is in tetrahedral Td and the other in the D2d
point group, which locates at only B0.1 eV higher energy thanthe most stable one. This finding is in line with previouslycalculated results.64,70 Of the two structures, only the Td onematches the sodalite cavity of LTA zeolite. Therefore we shallinvestigate in more detail the optical response of the neutralAg8 cluster in the Td structure.
3.3.1. Pure silver octamer. The singlet ground state of Ag8
in its Td global minimum has the following orbital configu-ration: 1A1: [. . .(A1)2(T2)2(T2)2(T2)2(T2)0(T2)0(T2)0(A1)0(E)0(E)0. . .]for the valence shell. This electron configuration could againbe considered as a mimic of the s2p6 electron configuration ofnoble gas atoms and is thus comparable to that of Ag14
6+
Fig. 7 Densities of states of Ag146+ encapsulated in the sodalite cavity of zeolite
LTA: the [Ag14(Si12H36O24)]6+ cluster. The black curve is the total density of state,the green curve the contribution of oxygen atoms, the blue curve the contri-bution of d(Ag), the bright blue curve the contribution of p(Ag) and the pinkcurve the contribution of s(Ag) to the total density of state. The contributions inpercentage to the HOMOs and LUMOs of O atoms, d(Ag), p(Ag) and s(Ag) aregiven in the inset.
Paper PCCP

15410 Phys. Chem. Chem. Phys., 2013, 15, 15404--15415 This journal is c the Owner Societies 2013
described above. The geometrical shape of the global minimumof Ag8 (Td) is displayed in Fig. 8a; the Kohn–Sham energies ofthe valence orbitals and their corresponding shapes are shownin Fig. 8b. The Ag–Ag bond length of the optimized octamerstructure amounts to 2.845 Å.
TD-DFT calculations are performed at the Ag8 optimizedgeometry using the B3LYP/LANL2DZ method and several lower-lying singlet excited states can thereby be identified. The energylevels of the ground 1A1 and singlet excited states located at upto B4.0 eV above the ground state and the correspondingoptical absorption spectrum are shown in Fig. 9, and theelectron transitions summarized in Table 3. These low-lyingexcited states are basically generated by electronic transitionsfrom the HOMOs to the LUMOs of the Ag8 (1A) cluster. TheHOMOs and LUMOs are each three-fold degenerate T orbitalswithin the Td point group (see Fig. 8b).
The most intense transition which occurs at B306 nm(4.04 eV) with a large oscillator strength of f = 3.3 is due to a1A1 -
1T2 transition (Fig. 9). The longer wavelength absorptionpeak centered at 403 nm (3.08 eV) with a weaker intensity off = 0.93 is due to another 1A1 -
1T2 transition. These values arein good agreement with the previously calculated results. Harbet al. using the TDDFT/BP86/LANL2DZ method obtained thevalues of 3.96 and 3.04 eV; Bonacic-Koutecky et al. used theEOM-CCSD method and derived the values of 4.16 and 3.27 eV,respectively, for these transitions.11,12 The following 1A1 - 1T2
transition found at 487 nm (2.54 eV) is nearly forbidden with asmall oscillator strength f = 0.0108. There are two other 1T1
excited states located at 2.46 and 3.50 eV, whose transitionsfrom the ground state 1A1 are forbidden with the vanishedoscillator strength f = 0.
The lowest-lying singlet excited state of Ag8 is identified tobe a 1E state with a transition energy of 2.42 eV, but thecorresponding transition 1A1 - 1E is again forbidden (withoscillator strength being f = 0). The singlet excited 2 1A1 state,having the same irreducible representation with the groundstate, lies at 2.62 eV but again this X 1A1 - 2 1A1 is alsoforbidden with oscillator strength being f = 0. From the
Fig. 8 (a) Optimized geometry of the most stable isomer of Ag80 cluster which
is found in Td point group; (b) diagram for Kohn–Sham orbital energy levels ofthe pure Ag8
0 cluster.
Fig. 9 (a) Energy levels of the ground electronic state 1A1 and several singletexcited states of Ag8
0 pure cluster which are calculated by TD-DFT and CASPT2methods. The dark lines represent singlet states; the red lines represent the tripletstates; (b) the absorption spectrum of the Ag8
0 pure cluster calculated by TD-DFTmethod using the B3LYP/LANL2DZ functional/basis set. The absorption spectrumis broadened with the full width at half maximum equal to 0.03 eV.
Table 3 Peak positions in nm (eV) and oscillator strengths of the calculatedoptical transitions of the investigated Ag8
0 and [Ag8(Si24H24O36)] clusters
Peak Peak position Oscillator strength ( f )
Ag8 (Fig. 5) I 306 (4.04) 3.27II 403 (3.08) 0.92III 487 (2.54) 0.01
[Ag8-(Si24H24O36)] I 347 (3.57) 2.10(Fig. 5) II 410 (3.02) 0.29
III 449 (2.76) 0.03
PCCP Paper

This journal is c the Owner Societies 2013 Phys. Chem. Chem. Phys., 2013, 15, 15404--15415 15411
calculated results, the 1T2 excited states located at B4.0 andB3.0 eV can be observed with high intensities.
In order to evaluate more quantitatively the emission of thepure silver octamer, we perform the relevant CASPT2/CASSCFcalculations67 using its DFT optimized geometry and anANO-RCC basis set for silver.71 For initial CASSCF calculations,the active space chosen includes eight electrons in the valenceshell distributed among twelve MOs including the HOMOs andLUMOs of Ag8 (CASSCF(8,12)). The shapes, irreducible repre-sentations and the occupation numbers of MOs in the activespace are shown in Fig. 10.
The CASPT2/CASSCF(8,12) energies for nine lowest-lyingsinglet and eight lowest-lying triplet states are calculated. Therestricted active space state interaction (RASSI)67 is performed toevaluate matrix elements of electronic transition dipole momentsand then oscillator strengths for electronic transitions. The spin–orbit coupling is taken into account in the RASSI calculation.
Transition energies from the ground to excited states andthe associated oscillator strengths for electronic transitions areshown in Fig. 9a. The energies of excited states calculated byTD-DFT are about 0.3 eV smaller than those by CASPT2. Afterabsorption, the excited states which lie at B3.1 and 4.0 eVbecome populated. These will rapidly decay to the lower-lyingstates due to the intermediate states and the small energy gapsbetween them. According to both TD-DFT and CASPT2 results,the luminescence of the Ag8 octamer is predicted to occur in awide range of wavelengths from its lowest excited states, whichare located at from B2.0 to 2.5 eV, to the ground state.
3.3.2. Ag8 cluster embedded inside the sodalite cavity ofLTA-type zeolite: the [Ag8(Si24H24O36)]0 cluster. We extensivelyexplore the most plausible configurations of the [Ag8(Si24H24O36)]0
cluster and find that in the converged optimal structure, eachof the eight silver atoms is pointing toward one six-memberedring thus forming a distorted cube inside the sodalite cavity ofLTA-type zeolite. That the geometry of the [Ag8(Si24H24O36)]
cluster fully optimized to a T point group is confirmed bysubsequent harmonic vibrational frequency calculations. Wehave also performed more geometry optimization followed byvibrational frequency calculations in order to confirm the low-est energy lying isomers of the embedded Ag8 cluster. Thestarting geometries for optimization calculations are taken asfollows: silver atoms located at the vicinities of the six- andfour-membered rings in all possible ways. According to ourcalculations, the most stable isomer is the Td structure that isdescribed above. Other isomers found have quite high relativeenergies as compared to the most stable one.
Let us also mention that although the Ag8 cluster also has alow-lying D2d symmetry structure, it does not match the sym-metry of the sodalite cavity. We have constructed the startinggeometry for optimization calculations of the Ag8 D2d clusterembedded inside the sodalite. Optimization calculations forthis starting structure invariably converged to the Td structure –the most stable isomer. This may be due to a flexibility of thebonding amongst silver atoms in the silver cluster in such a waythat the Ag8 cluster is in its most stable structure that matchesthe point group of the sodalite cage rather than retaining a D2d
point group to withstand the cluster–cage interaction.Similar to the pure Ag8 cluster, the ground state of the
[Ag8(Si24H24O36)] cluster in the T point group has the orbitalconfiguration as:
1A: [. . . (A)2(T)2(T)2(T)2(T)0(T)0(T)0(A)0(E)0(E)0. . .]
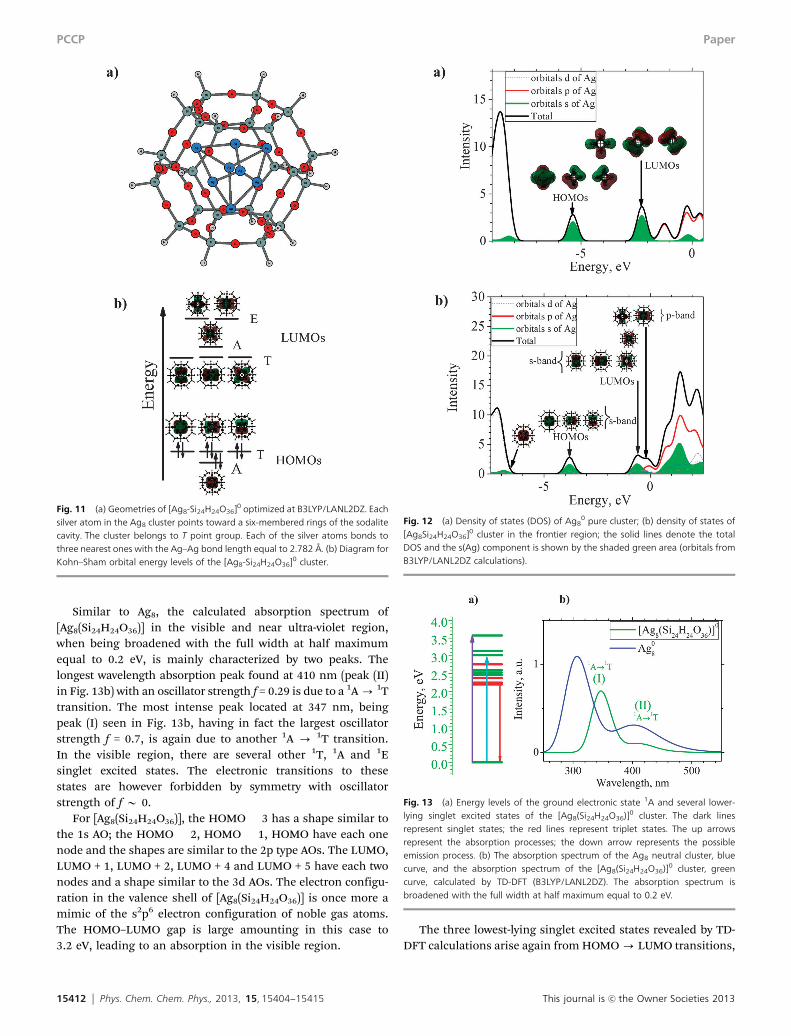
for the valence shell. Optimized geometry of [Ag8(Si24H24O36)] isshown in Fig. 11a whereas the Kohn–Sham energies of frontierorbitals and their corresponding shapes are displayed in Fig. 11b.They are in general similar to those of the pure Ag8 cluster.
The optimized Ag–Ag bond length is now compressed to2.782 Å, being slightly shorter than the Ag–Ag bond in Ag8. The5s atomic orbitals of Ag atoms have energies higher as com-pared to 4d(Ag) and other AOs of Si and O atoms. Therefore, in[Ag8(Si24H24O36)], the 5s(Ag) atomic orbitals preferred to com-bine with each other rather than to other AOs to form frontiermolecular orbitals, as could be seen in their densities of states(DOS shown in Fig. 12).
Several low-lying singlet excited states of the embeddedsilver octamer [Ag8(Si24H24O36)] are identified from TD-DFT/B3LYP/LANL2DZ calculations. The energy levels are again up toB4.0 eV and the corresponding optical absorption spectrum isplotted in Fig. 13, and the main transitions summarized inTable 3.
The frontier molecular orbitals of [Ag8Si24H24O36] are againof T symmetry, and electronic transitions from the degenerateHOMOs to the degenerate LUMOs lead to discrete electronicexcited states, and thereby define the absorption spectrum. Dueto the rather poor combination between AO(Ag) and MOs of thesurrounding (Si24H24O36) cavity, the frontier orbitals of[Ag8(Si24H24O36)] are again constructed mainly from the 5sand 5p AOs of silver atoms, as can be seen in Fig. 12 andTable 4. Therefore, the electronic transitions which are respon-sible for the absorption peaks in the visible region occur withinthe silver octamer.
Fig. 10 Shapes of MOs used in the active space in the CASPT2/CASSCF calcula-tion, the irreducible representation and their occupation numbers for the Ag8 in1A1 ground state.
Paper PCCP
15412 Phys. Chem. Chem. Phys., 2013, 15, 15404--15415 This journal is c the Owner Societies 2013
Similar to Ag8, the calculated absorption spectrum of[Ag8(Si24H24O36)] in the visible and near ultra-violet region,when being broadened with the full width at half maximumequal to 0.2 eV, is mainly characterized by two peaks. Thelongest wavelength absorption peak found at 410 nm (peak (II)in Fig. 13b) with an oscillator strength f = 0.29 is due to a 1A - 1Ttransition. The most intense peak located at 347 nm, beingpeak (I) seen in Fig. 13b, having in fact the largest oscillatorstrength f = 0.7, is again due to another 1A - 1T transition.In the visible region, there are several other 1T, 1A and 1Esinglet excited states. The electronic transitions to thesestates are however forbidden by symmetry with oscillatorstrength of f B 0.
For [Ag8(Si24H24O36)], the HOMO � 3 has a shape similar tothe 1s AO; the HOMO � 2, HOMO � 1, HOMO have each onenode and the shapes are similar to the 2p type AOs. The LUMO,LUMO + 1, LUMO + 2, LUMO + 4 and LUMO + 5 have each twonodes and a shape similar to the 3d AOs. The electron configu-ration in the valence shell of [Ag8(Si24H24O36)] is once more amimic of the s2p6 electron configuration of noble gas atoms.The HOMO–LUMO gap is large amounting in this case to3.2 eV, leading to an absorption in the visible region.
The three lowest-lying singlet excited states revealed by TD-DFT calculations arise again from HOMO - LUMO transitions,
Fig. 11 (a) Geometries of [Ag8-Si24H24O36]0 optimized at B3LYP/LANL2DZ. Eachsilver atom in the Ag8 cluster points toward a six-membered rings of the sodalitecavity. The cluster belongs to T point group. Each of the silver atoms bonds tothree nearest ones with the Ag–Ag bond length equal to 2.782 Å. (b) Diagram forKohn–Sham orbital energy levels of the [Ag8-Si24H24O36]0 cluster.
Fig. 12 (a) Density of states (DOS) of Ag80 pure cluster; (b) density of states of
[Ag8Si24H24O36]0 cluster in the frontier region; the solid lines denote the totalDOS and the s(Ag) component is shown by the shaded green area (orbitals fromB3LYP/LANL2DZ calculations).
Fig. 13 (a) Energy levels of the ground electronic state 1A and several lower-lying singlet excited states of the [Ag8(Si24H24O36)]0 cluster. The dark linesrepresent singlet states; the red lines represent triplet states. The up arrowsrepresent the absorption processes; the down arrow represents the possibleemission process. (b) The absorption spectrum of the Ag8 neutral cluster, bluecurve, and the absorption spectrum of the [Ag8(Si24H24O36)]0 cluster, greencurve, calculated by TD-DFT (B3LYP/LANL2DZ). The absorption spectrum isbroadened with the full width at half maximum equal to 0.2 eV.
PCCP Paper

This journal is c the Owner Societies 2013 Phys. Chem. Chem. Phys., 2013, 15, 15404--15415 15413
and are found to be close to each other in the region of 2.47,2.53 and 2.60 eV. These electronic transitions are mainly ofintra-silver character, going from 5s(Ag) to 5s(Ag) orbitals, andare forbidden transitions with oscillator strength equal to zero.We can consider the electronic transitions from HOMO � 2,HOMO � 1, HOMO to LUMO + 4 and LUMO + 5 as the 5s(Ag) to5p(Ag) transitions since the 5p(Ag) AOs contribute mostly to theLUMO + 4 and LUMO + 5 (Table 2). These excitations turn out tobe responsible for the absorption peaks (I), (II) and (III)illustrated in Fig. 13b. Some low-lying triplet excited statesare located at 2.2 eV as can be seen in Fig. 13.
From the calculated energy diagram which is displayed inFig. 13, we propose that the Ag8 cluster encapsulated in asodalite cavity is expected to have a luminescence in a widerange of wavelengths in the red part of visible region due to
emissions from both singlet and triplet excited states to thesinglet ground state.
4. Concluding remarks
In the present study, we investigated the electronic structureand optical spectra of two silver clusters in different environ-ments and charge states. The tetradecanuclear hexacationAg14
6+ was proposed for the first time to be a stabilizedpolycation when it is embedded in the sodalite cavity of azeolite LTA, giving the Ag14
6+@ZA system. The sodalite cage,which typically acts as a container, tends to stabilize the ioniccentral moiety by exerting an electrostatic attraction on it.
The energy levels of the ground state and some lower-lyingsinglet excited states of the resulting enclosed Ag14
6+ silver
Table 4 Calculated electronic transitions of [Ag8(Si24H24O36)] (symmetry, transition energy and oscillator strength)a
Excitation Initial orbitals Final orbitals Coefficient
d-/p-/s of Ag contribution to Total d-/p-/s of Ag contribution to
Initial orbitals Final orbitals Initial orbitals Final orbitals
(I) (T, 3.57 eV, 3.27) HOMO � 2 LUMO �0.23 14/14/56 4/15/55 84.2 74.4HOMO � 1 LUMO + 1 �0.23 14/14/56 4/15/55 84.2 74.4HOMO LUMO + 4 �0.22 14/14/56 1/66/0 84.2 66.4HOMO LUMO + 5 0.54 14/14/56 1/66/0 84.2 66.4HOMO � 2 LUMO + 3 0.22 14/14/56 2/21/34 84.2 57.4HOMO � 2 LUMO + 4 0.49 14/14/56 1/66/0 84.2 66.4HOMO � 2 LUMO + 5 0.22 14/14/56 1/66/0 84.2 66.4HOMO � 1 LUMO + 2 0.23 14/14/56 4/15/55 84.2 74.4HOMO LUMO 0.23 14/14/56 4/15/55 84.2 74.4HOMO � 2 LUMO + 2 �0.23 14/14/56 4/15/55 84.2 74.4HOMO � 1 LUMO + 3 �0.22 14/14/56 2/21/34 84.2 57.4HOMO � 1 LUMO + 4 0.44 14/14/56 1/66/0 84.2 66.4HOMO � 1 LUMO + 5 �0.32 14/14/56 1/66/0 84.2 66.4HOMO LUMO + 1 �0.23 14/14/56 4/15/55 84.2 74.4
(II) (T, 3.02 eV, 0.29) HOMO � 2 LUMO + 3 0.54 14/14/56 2/21/34 84.2 57.4HOMO � 2 LUMO + 4 �0.33 14/14/56 1/66/0 84.2 66.4HOMO � 2 LUMO + 5 �0.15 14/14/56 1/66/0 84.2 66.4HOMO � 1 LUMO + 2 0.16 14/14/56 4/15/55 84.2 74.4HOMO LUMO 0.16 14/14/56 4/15/55 84.2 74.4HOMO � 2 LUMO 0.16 14/14/56 4/15/55 84.2 74.4HOMO � 1 LUMO + 1 0.16 14/14/56 4/15/55 84.2 74.4HOMO LUMO + 3 0.54 14/14/56 2/21/34 84.2 57.4HOMO LUMO + 5 0.36 14/14/56 1/66/0 84.2 66.4HOMO � 2 LUMO + 2 0.16 14/14/56 4/15/55 84.2 74.4HOMO � 1 LUMO + 3 0.55 14/14/56 2/21/34 84.2 57.4HOMO � 1 LUMO + 4 0.30 14/14/56 1/66/0 84.2 66.4HOMO � 1 LUMO + 5 �0.22 14/14/56 1/66/0 84.2 66.4HOMO LUMO + 1 0.16 14/14/56 4/15/55 84.2 74.4
(III) (T, 2.76 eV, 0.03) HOMO � 2 LUMO 0.40 14/14/56 4/15/55 84.2 74.4HOMO � 1 LUMO + 1 0.40 14/14/56 4/15/55 84.2 74.4HOMO LUMO + 3 �0.37 14/14/56 2/21/34 84.2 57.4HOMO LUMO + 5 0.20 14/14/56 1/66/0 84.2 66.4HOMO � 2 LUMO + 3 �0.37 14/14/56 2/21/34 84.2 57.4HOMO � 2 LUMO + 4 �0.19 14/14/56 1/66/0 84.2 66.4HOMO � 1 LUMO + 2 0.40 14/14/56 4/15/55 84.2 74.4HOMO LUMO 0.40 14/14/56 4/15/55 84.2 74.4HOMO � 2 LUMO + 2 0.40 14/14/56 4/15/55 84.2 74.4HOMO � 1 LUMO + 3 �0.37 14/14/56 2/21/34 84.2 57.4HOMO � 1 LUMO + 4 0.17 13/14/56 1/66/0 84.2 66.4HOMO � 1 LUMO + 5 �0.12 14/14/56 1/66/0 84.2 66.4HOMO LUMO + 1 0.40 14/14/56 4/15/55 84.2 74.4
a Obtained at the B3LYP/LANL2DZ level, initial and final MOs participating in these transitions as well as percentage of total s-, p-, d-type characterof the initial and final orbitals. MO excitations contributing mainly to the electronic transitions are listed in bold.
Paper PCCP

15414 Phys. Chem. Chem. Phys., 2013, 15, 15404--15415 This journal is c the Owner Societies 2013
cluster were determined using time-dependent density func-tional theory (TD-DFT) with the hybrid B3LYP functional andthe LANL2DZ basis set. Several lower-lying singlet excited statesof the Ag14
6+ encapsulated cluster located at up to B4.0 eV wereidentified. The ground state possesses an outer-shell electronconfiguration A1g
2T2g6 mimicking the noble gas atoms s2p6
with eight valence electrons, and frontier orbitals are con-structed from 5s,5p(Ag) atomic orbitals with some contribu-tions from oxygen atoms present in the cavity framework.
The predicted absorption spectrum of the Ag146+@ZA in the
visible region is characterized by two strong absorption peakscentered at 331 and 476 nm, respectively, and arises from intra-atomic electron transitions from 5s,p(Ag) domain to 5s,p(Ag)domain of the silver atoms. The light absorption will not occurto the lowest excited state but to the higher located onesresulting in a large difference in wavelength between thepositions of the band maxima of the absorption and emissionspectra. The luminescence is expected to occur at B2.0 eV andin a long time scale.
Along with the tetradeca-silver hexacation Ag146+ cluster, the
optical properties of another silver cluster also having eightelectrons in the valence shell but without charge, namely theAg8 neutral cluster, embedded inside the sodalite cavity ofLTA-type zeolite was also investigated. Several lowest-lyingsinglet and triplet excited states located at up to B4.0 eV werefound. Again, the [Ag8(Si24H24O36)] compound possesses anouter-shell electron configuration A2T6 which also mimics theelectron configuration of s2p6 of noble gas atoms, and thecorresponding frontier orbitals are basically constructed fromintra-atomic 5s(Ag) orbitals.
The predicted absorption spectra of the bare Ag8 andembedded [Ag8(Si24H24O36)] clusters in the visible region aresimilar to each other, and characterized by strong absorptionpeaks at around 306 and 347 nm, respectively, and arise fromelectron transitions from the 5s(Ag) to 5p(Ag) domain of silveratoms. Furthermore, the absorption spectrum of the Ag8 neutralcluster embedded inside the sodalite cavity of LTA-type zeolite issimilar to that of the tetradeca-silver hexacation Ag14
6+ clusterembedded in the same environment, bearing two absorptionbands in the visible region that include one strong intensityband in the violet region and another weak intensity band in thelight blue region. Both absorption processes are expected to leadto luminescence in the subsequent light emission in red color.
Acknowledgements
NTC and HMTN are grateful to the Vietnamese Ministry ofEducation and Training under the project B2012-17-28. MTNthanks the KU Leuven Research Council (GOA, BOF and IDOprograms) and the ICST for supporting his stays in Vietnam.
References
1 J. Ruamps, Compt. Rend., 1954, 238, 1489.2 P. J. Ruamps, Ann. Phys., 1959, 4, 1111.3 R. C. Maheshwari, Indian J. Phys., 1963, 37, 368.
4 H. Handschuh, C. Y. Cha, P. S. Bechthold, G. Gantefor andW. Eberhardt, J. Chem. Phys., 1995, 102, 6406.
5 V. Beutel, H.-G. Kramer, G. L. Bhale, M. Kuhn, K. Weyersand W. Demtroder, J. Chem. Phys., 1993, 98, 2699.
6 C. Jackschath, I. Rabin and W. Schulze, Z. Phys. D: At., Mol.Clusters, 1992, 22, 517.
7 C. Shin-Piaw, W. Loong-Seng and L. Yoke-Seng, Nature,1966, 209, 1300.
8 T. Diederich, J. Tiggesbaumker and K.-H. Meiwes-Broer,J. Chem. Phys., 2002, 116, 3263.
9 E. Janssens, X. J. Hou, M. T. Nguyen and P. Lievens, J. Chem.Phys., 2006, 124, 184319.
10 X. J. Hou, E. Janssens, P. Lievens and M. T. Nguyen, Chem.Phys., 2006, 330, 365.
11 V. Bonacic-Koutecky, J. Burda, R. Mitric, M. Ge, G. Zampellaand P. Fantucci, J. Chem. Phys., 2002, 117, 3120.
12 V. Bonacic-Koutecky, V. Veyret and R. Mitric, J. Chem. Phys.,2001, 115, 10450.
13 N. T. Cuong, V. K. Timikhomirov, L. F. Chibotaru,A. Stesmans, V. D. Rodriguez, M. T. Nguyen andV. V. Moschalkov, J. Chem. Phys., 2012, 136, 174108.
14 J. Velaquez, V. Tikhomirov, L. Chibotaru, N. T. Cuong,A. S. Kuznetsov, V. D. Rodriguez, V. Moschalkov andM. T. Nguyen, Opt. Express, 2012, 20, 13582.
15 M. Harb, F. Rabilloud, D. Simon, A. Rydlo, S. Lecoultre,F. Conus, V. Rodrigues and C. Felix, J. Chem. Phys., 2008,129, 194108.
16 J. C. Idrobo, S. Ogut and J. Jellinek, Phys. Rev. B: Condens.Matter Mater. Phys., 2005, 72, 085445.
17 G. F. Zhao, Y. Lei and Z. Zeng, Chem. Phys., 2006, 327, 261.18 V. A. Spasov, T. H. Lee, J. P. Maberry and K. M. Ervin,
J. Chem. Phys., 1999, 110, 5208.19 M. V. Shestakov, N. T. Cuong, V. K. Tikhomirov,
M. T. Nguyen, L. F. Chibotaru and V. V. Moshchalkov,J. Phys. Chem. C, 2013, 17, 7796.
20 S. Liu, Y. Li, X. Zhao, X. Liu and M. Chen, Spectrochim. Acta,Part A, 2011, 82, 205.
21 S. Zhao, Z.-H. Li, W.-N. Wang, Z.-P. Liu, K.-N. Fan, Y. Xie andH. Schaefer, J. Chem. Phys., 2006, 124, 184102.
22 R. Fournier, J. Chem. Phys., 2001, 115, 2165.23 K. A. Bosnick, T. L. Haslett, S. Fedrigo, M. Moskovits,
W.-T. Chan and R. Fournier, J. Chem. Phys., 1999, 111, 8867.24 E. Janssens, S. Neukermans, H. M. T. Nguyen, M. T. Nguyen
and P. Lievens, Phys. Rev. Lett., 2005, 94, 11304.25 Y. Shi, V. A. Spasov and K. M. Ervin, J. Chem. Phys., 1999,
111, 938.26 M. Yang, K. A. Jackson and J. Jellinek, J. Chem. Phys., 2006,
125, 144308.27 D. W. Silverstein and L. Jensen, J. Chem. Phys., 2010,
132, 194302.28 S. Lecoultre, A. Rydlo, J. Buttet, C. Felix, S. Gilb and
W. Harbich, J. Chem. Phys., 2011, 134, 184504.29 C. Felix, C. Sieber, W. Harbich, J. Buttet, I. Rabin,
W. Schulze and G. Ertl, Chem. Phys. Lett., 1999, 313, 105.30 E. Bayram, J. Lu, C. Aydin, A. Uzun, N. D. Browning,
B. C. Gates and R. G. Finke, ACS Catal., 2012, 2, 1947.
PCCP Paper

This journal is c the Owner Societies 2013 Phys. Chem. Chem. Phys., 2013, 15, 15404--15415 15415
31 S. Wannakao, C. Warakulwit, K. Kongpatpanich, M. Probstand J. Limtrakul, ACS Catal., 2012, 2, 986.
32 S. M. T. Almutairi, B. Mezari, P. C. M. M. Magusin,E. A. Pidko and E. J. M. Hensen, ACS Catal., 2012, 2, 71.
33 W. F. Schneider, K. C. Hass, R. Ramprasad and J. B. Adams,J. Phys. Chem., 1996, 100, 6032.
34 A. M. Ferrari, K. M. Neyman, M. Mayer, M. Staufer,B. C. Gates and N. Rosch, J. Phys. Chem. B, 1999, 103, 5311.
35 K. Pierloot, A. Delabie, M. H. Groothaert and R. A.Schoonheydt, Phys. Chem. Chem. Phys., 2001, 3, 2174.
36 K. Pierloot, A. Delabie and C. Ribbing, J. Phys. Chem. B,1998, 102, 10789.
37 A. Delabie, K. Pierloot, M. H. Grootheart, R. A. Schoonheydtand L. G. Vanquickenborne, Eur. J. Inorg. Chem., 2002, 515.
38 K. Klier, Langmuir, 1988, 4, 13.39 T. Yumura, Phys. Chem. Chem. Phys., 2011, 13, 337.40 T. Yumura, T. Nanba, H. Torigoe, Y. Kuroda and
H. Kobayashi, Inorg. Chem., 2011, 50, 6533.41 P. A. Jacobs, J. B. Uytterhoeven and K. H. J. Beyer, J. Chem.
Soc., Faraday Trans. 1, 1979, 75, 56.42 S. Y. Kim, Y. Kim and K. Seff, J. Phys. Chem. B, 2003,
107, 6938.43 R. A. Schoonheydt and H. Leeman, J. Phys. Chem., 1989,
93, 93.44 M. D. Baker, G. A. Ozin and J. Godber, J. Phys. Chem., 1985,
89, 305.45 B. Xu and L. Kevan, J. Phys. Chem., 1991, 95, 95.46 J. Texter, R. Kellerman and T. Gonsiorowski, J. Phys. Chem.,
1986, 90, 2118.47 E. Gachard, J. Belloni and M. A. Subramanian, J. Mater.
Chem., 1996, 6, 867.48 W. S. Ju, M. Matsuoka, K. Iino, H. Yamashita and M. Anpo,
J. Phys. Chem. B, 2004, 108, 2128.49 S. M. Kanan, M. C. Kanan and H. H. Patterson, J. Phys.
Chem. B, 2001, 105, 7508.50 K. Sawabe, T. Hiro, K. Shimizu and A. Satsuma, Catal.
Today, 2010, 153, 90.51 T. Baba, H. Sawada, T. Takahashi and M. Abe, Appl. Catal.,
A, 2002, 231, 55.52 H. Yoshida, T. Hamajima, Y. Kato, J. Shibata, A. Satsuma
and T. Hattori, Res. Chem. Intermed., 2003, 29, 897.
53 S. Miao, Y. Wang, D. Ma, Q. Zhu, S. Zhou, L. Su, D. Tan andX. Bao, J. Phys. Chem. B, 2004, 108, 17866.
54 G. De Cremer, E. Coutino-Gonzalez, M. B. Roeffaers,D. E. De Vos, J. Hofkens, T. Vosch and B. F. Sels, Chem-PhysChem, 2010, 11, 1627.
55 A. Royon, K. Bourhis, M. Bellec, G. Papon, B. Bousquet,Y. Deshayes, T. Cardinal and L. Canioni, Adv. Mater., 2010,22, 5282.
56 G. De Cremer, E. Coutino-Gonzalez, M. B. J. Roeffaers,B. Moens, J. Ollevier, M. Van der Auweraer, R. Schoonheydt,P. A. Jacobs, F. C. De Schryver, J. Hofkens, D. E. De Vos,B. F. Sels and T. Vosch, J. Am. Chem. Soc., 2009, 131, 3049.
57 O. M. Bakr, V. Amendola, C. M. Aikens, W. Wenseleers,R. Li, L. Dal Negro, G. C. Schatz and F. Stellacci, Angew.Chem., Int. Ed., 2009, 48, 5921.
58 I. Dıez and R. H. A. Ras, Nanoscale, 2011, 3, 1963.59 A. Mayoral, T. Carey, P. A. Anderson, A. Lubk and I. Diaz,
Angew. Chem., Int. Ed., 2011, 50, 1.60 K. Lier, Stud. Surf. Sci. Catal., 2005, 157, 205.61 P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, 864.62 R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996,
256, 454.63 A. D. Becke, J. Chem. Phys., 1993, 98, 5648.64 S. Lecoultre, A. Rydlo, J. Buttet, C. Felix, S. Gilb and
W. Harbich, J. Chem. Phys., 2011, 134, 184504.65 P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270.66 M. J. Frisch, et al., Gaussian 09, Revision A.02, Gaussian, Inc.,
Wallingford, CT, 2009.67 G. Karlstrom, R. Lindh, P.-Å. Malmqvist, B. O. Roos,
U. Ryde, V. Veryazov, P.-O. Widmark, M. Cossi,B. Schimmelpfennig, P. Neogrady and L. Seijo, Comput.Mater. Sci., 2003, 28, 222.
68 P. A. Anderson, Molecular Sieves, Springer, Berlin, 2002,vol. 3.
69 H. Hakkinen, M. Moseler and U. Landman, Phys. Rev. Lett.,2002, 89, 033401.
70 M. Harb, F. Rabilloud, D. Simon, A. Rydlo, S. Lecoultre,F. Conus, V. Rodrigues and C. Felix, J. Chem. Phys., 2008,129, 194108.
71 B. O. Roos, R. Lindh, P.-Å. Malmqvist, V. Veryazov andP.-O. Widmark, J. Phys. Chem. A, 2005, 108, 6575.
Paper PCCP




















