
Theoretical investigation of stilbene as photochromic spin coupler
11
Theoretical Investigation of Stilbene as Photochromic Spin Coupler Arun K. Pal, Shekhar Hansda, and Sambhu N. Datta* Department of Chemistry, Indian Institute of Technology, Bombay, Powai, Mumbai 400076, India Francesc Illas Department de Químcia Física & Institut de Química Teò rica I Computacional (IQTCUB), Universitat de Barcelona, C/Martí i Franque ́ s 1, 08028 Barcelona, Spain * S Supporting Information ABSTRACT: Density functional theory (DFT) based calculations are used here to investigate the magnetic behavior, spectroscopic transitions, and possible photomagnetic properties of stilbene derivatives using photo- chromicity of cis- and trans-forms of the parent molecule. Nitronyl nitroxide (NN), iminonitroxide (IN), tetrathiafulvalene cation (TTF), and verdazyl (VER) are used as monoradical centers at the p, p′ positions. The B3LYP functional with the usual broken symmetry approach and a sufficiently large basis set is chosen to obtain reliable estimates of the intramolecular exchange coupling constants (J). It is found that, with stilbene as a spacer, the coupling of TTF with NN, IN, and VER is always antiferromagnetic with J being generally large and negative. Although J values obtained for cis- and trans- forms are both negative, the difference in J values is quite large. Spectroscopic transition energies and corresponding oscillator strengths of cis- and trans- stilbene diradicals are estimated by time-dependent (TD)-DFT calculations using the same functional. Interestingly, the spectral features of the diradicals are similar to those of cis- and trans-stilbene, which suggests that stilbene diradicals would have good photoswitching properties. Finally, we show that, when these diradicals are placed in a matrix, photochromicity would be accompanied by a significant change in paramagnetic susceptibility. I. INTRODUCTION The reversible transformation of a chemical species to its isomer and vice versa, under the absorption of light of two different frequencies, is known as photochromism. 1 By irradiating a photochromic material, molecular and crystal geometries as well as physical properties of the material can be changed. This is important for designing photoswitchable chemical species. If a photoswitchable molecule is used as a spin coupler between two magnetic units, then the magnetism of the resulting species can change upon irradiation. 2 Stilbene is a photochromic molecule: Cis-stilbene converts to trans-stilbene when exposed to light. 3 This is schematically illustrated in Figure 1 where the experimental wavelength for cis-stilbene correspond to a solution in n-heptane at room temperature 4 and that for trans-stilbene has been observed by Suzuki. 5 A major part of research on organic molecular magnetism has been on the synthesis and characterization of nitronyl nitroxide (NN), 6 imino nitroxide (IN), 7 and verdazyl (VER). 8 This is because these radical centers are stable at a relatively high temperature, are easily synthesized and have the ability to generate co-operative magnetic properties. 9−17 Relatively less work has been done on the magnetic properties of a potent candidate, namely, the tetrathiafulvalene cation (TTF) that can be used as a building block for mixed diradical systems. It is a strong electron donor, and its electronic properties have drawn attention from various fields of chemistry. 18 In TTF, two routes of conjugation exist from the point of contact with the coupler to the lone π electron in the central carbon−carbon bond. A structural change such as the modification of the dihedral angle between the coupler and TTF planes can favor a specific route over the other, thereby changing the nature of coupling. In fact, Latif et al. 19 have theoretically predicted photoswtiching magnetic properties of NN coupled to TTF via polyene spacers. The monoradical centers are illustrated in Figure 2. The aim of this work is to investigate the photomagnetic nature of diradicals resulting from coupling of two radical centers such as NN-TTF, IN-TTF and VER-TTF via cis- and trans-stilbene fragments as spacer. First, from our theoretical calculations we show that with stilbene spacer the diradicals formed from the interaction of TTF with NN, IN, and VER are all antiferromagnetically coupled. This observation can be accounted for by spin alternation rule 20 that is illustrated in Figure 3. Second, we estimate spectroscopic transition energies and oscillator strengths for both cis- and trans-isomers of these Received: July 6, 2012 Revised: January 31, 2013 Published: February 19, 2013 Article pubs.acs.org/JPCA © 2013 American Chemical Society 1773 dx.doi.org/10.1021/jp306715y | J. Phys. Chem. A 2013, 117, 1773−1783
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Theoretical investigation of stilbene as photochromic spin coupler
Theoretical Investigation of Stilbene as Photochromic Spin CouplerArun K. Pal, Shekhar Hansda, and Sambhu N. Datta*
Department of Chemistry, Indian Institute of Technology, Bombay, Powai, Mumbai 400076, India
Francesc Illas
Department de Químcia Física & Institut de Química Teorica I Computacional (IQTCUB), Universitat de Barcelona,C/Martí i Franques 1, 08028 Barcelona, Spain
*S Supporting Information
ABSTRACT: Density functional theory (DFT) based calculations are usedhere to investigate the magnetic behavior, spectroscopic transitions, andpossible photomagnetic properties of stilbene derivatives using photo-chromicity of cis- and trans-forms of the parent molecule. Nitronyl nitroxide(NN), iminonitroxide (IN), tetrathiafulvalene cation (TTF), and verdazyl(VER) are used as monoradical centers at the p, p′ positions. The B3LYPfunctional with the usual broken symmetry approach and a sufficiently largebasis set is chosen to obtain reliable estimates of the intramolecular exchangecoupling constants (J). It is found that, with stilbene as a spacer, the couplingof TTF with NN, IN, and VER is always antiferromagnetic with J beinggenerally large and negative. Although J values obtained for cis- and trans-forms are both negative, the difference in J values is quite large. Spectroscopictransition energies and corresponding oscillator strengths of cis- and trans-stilbene diradicals are estimated by time-dependent (TD)-DFT calculations using the same functional. Interestingly, the spectralfeatures of the diradicals are similar to those of cis- and trans-stilbene, which suggests that stilbene diradicals would have goodphotoswitching properties. Finally, we show that, when these diradicals are placed in a matrix, photochromicity would beaccompanied by a significant change in paramagnetic susceptibility.
I. INTRODUCTIONThe reversible transformation of a chemical species to its isomerand vice versa, under the absorption of light of two differentfrequencies, is known as photochromism.1 By irradiating aphotochromic material, molecular and crystal geometries as wellas physical properties of the material can be changed. This isimportant for designing photoswitchable chemical species. If aphotoswitchable molecule is used as a spin coupler between twomagnetic units, then the magnetism of the resulting species canchange upon irradiation.2
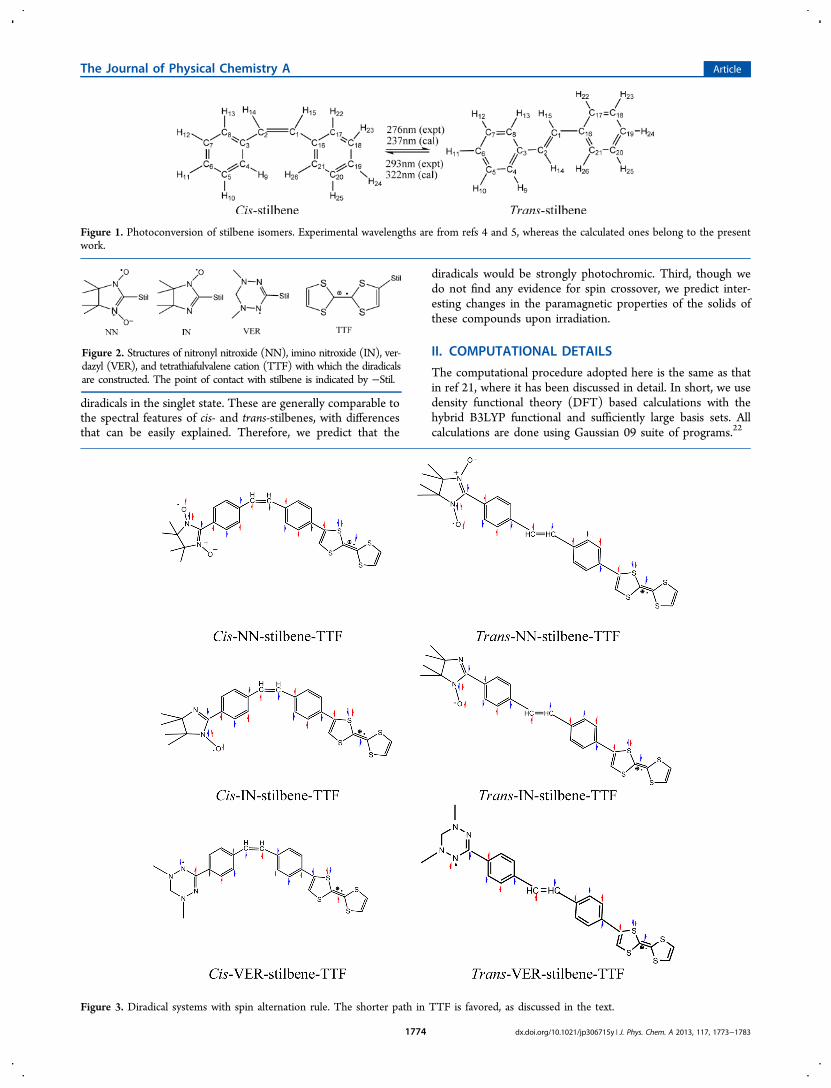
Stilbene is a photochromic molecule: Cis-stilbene converts totrans-stilbene when exposed to light.3 This is schematicallyillustrated in Figure 1 where the experimental wavelength forcis-stilbene correspond to a solution in n-heptane at roomtemperature4 and that for trans-stilbene has been observed bySuzuki.5
A major part of research on organic molecular magnetism hasbeen on the synthesis and characterization of nitronyl nitroxide(NN),6 imino nitroxide (IN),7 and verdazyl (VER).8 This isbecause these radical centers are stable at a relatively hightemperature, are easily synthesized and have the ability togenerate co-operative magnetic properties.9−17 Relatively lesswork has been done on the magnetic properties of a potentcandidate, namely, the tetrathiafulvalene cation (TTF) that canbe used as a building block for mixed diradical systems. It is a
strong electron donor, and its electronic properties have drawnattention from various fields of chemistry.18
In TTF, two routes of conjugation exist from the point ofcontact with the coupler to the lone π electron in the centralcarbon−carbon bond. A structural change such as themodification of the dihedral angle between the coupler andTTF planes can favor a specific route over the other, therebychanging the nature of coupling. In fact, Latif et al.19 havetheoretically predicted photoswtiching magnetic properties ofNN coupled to TTF via polyene spacers. The monoradicalcenters are illustrated in Figure 2.The aim of this work is to investigate the photomagnetic
nature of diradicals resulting from coupling of two radicalcenters such as NN-TTF, IN-TTF and VER-TTF via cis- andtrans-stilbene fragments as spacer. First, from our theoreticalcalculations we show that with stilbene spacer the diradicalsformed from the interaction of TTF with NN, IN, and VER areall antiferromagnetically coupled. This observation can beaccounted for by spin alternation rule20 that is illustrated inFigure 3. Second, we estimate spectroscopic transition energiesand oscillator strengths for both cis- and trans-isomers of these
Received: July 6, 2012Revised: January 31, 2013Published: February 19, 2013
Article
pubs.acs.org/JPCA
© 2013 American Chemical Society 1773 dx.doi.org/10.1021/jp306715y | J. Phys. Chem. A 2013, 117, 1773−1783
diradicals in the singlet state. These are generally comparable tothe spectral features of cis- and trans-stilbenes, with differencesthat can be easily explained. Therefore, we predict that the
diradicals would be strongly photochromic. Third, though wedo not find any evidence for spin crossover, we predict inter-esting changes in the paramagnetic properties of the solids ofthese compounds upon irradiation.
II. COMPUTATIONAL DETAILS
The computational procedure adopted here is the same as thatin ref 21, where it has been discussed in detail. In short, we usedensity functional theory (DFT) based calculations with thehybrid B3LYP functional and sufficiently large basis sets. Allcalculations are done using Gaussian 09 suite of programs.22
Figure 1. Photoconversion of stilbene isomers. Experimental wavelengths are from refs 4 and 5, whereas the calculated ones belong to the presentwork.
Figure 2. Structures of nitronyl nitroxide (NN), imino nitroxide (IN), ver-dazyl (VER), and tetrathiafulvalene cation (TTF) with which the diradicalsare constructed. The point of contact with stilbene is indicated by −Stil.
Figure 3. Diradical systems with spin alternation rule. The shorter path in TTF is favored, as discussed in the text.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp306715y | J. Phys. Chem. A 2013, 117, 1773−17831774

Both cis- and trans-stilbene have closed-shell singlet groundstates that can be directly computed. The triplet−singlet gap isobserved to be very large (≫ kT), and the species would becompletely diamagnetic in their ground states.In the case of the open-shell singlet for the diradicals, the
broken symmetry (BS) approach has been used.23−26 This isbecause the unrestricted Kohn−Sham formalism requires asingle Slater determinant to describe the electron density ofthe “non-interacting” reference system, whereas the minimumacceptable wave function describing the open shell singletinvolves a closed shell determinant (CS) and its doubly excitedconfiguration (D) which are mixed either through configurationinteraction (CI) or, preferably, through a complete active spaceself-consistent-field (CASSCF) calculation with two electronsand two orbitals defining the complete active space, usuallydenoted as CAS(2,2).25 The BS approach offers an avenue forcalculating the magnetic coupling constant J from DFT based(and UHF) calculations as an energy difference and a suitablemapping between N-electron and spin states of the appropriatespin Hamiltonian.25
For a diradical, the spin Hamiltonian is usually written as
= − ·H JS S2ex 1 2 (1)
when no magnetic field is present. The coupling constant thusdefined can be calculated using the Yamaguchi expression26
=−
⟨ ⟩ − ⟨ ⟩J
E ES S
TBS2
T2
BS (2)
where EBS is the total energy of the BS state and ET is the totalenergy of the triplet state. Expression 2 is equivalent to themapping procedure described in detail by Moreira and Illas25
since, in most cases, the expectation values of the square of thetotal spin operator, ⟨S2⟩, are close to 2 and 1 for the triplet andbroken symmetry states, respectively. However, in case oforganic diradicals the expectation value for the BS state oftendeviates from 1 that corresponds to an exact 50% singlet and50% triplet mixing, mainly because of the delocalization of thesingly occupied orbitals. Then, the denominator in eq 2 allowsone to take this effect into account.
Figure 4. Structures of optimized geometries calculated at UB3LYP/6-311G(d,p) level. Legend: S stands for singlet and T is for triplet.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp306715y | J. Phys. Chem. A 2013, 117, 1773−17831775
All relevant molecular geometries are optimized using 6-311G(d,p)basis set. The singlet geometry optimization procedure led to abroken symmetry solution for every diradical. Because the ex-change coupling constant ideally involves only spin degrees offreedom, the J value calculated from the optimized BS geometrywould be compatible with the equilibrium geometry of thesinglet and hence with Curie studies. Henceforth we will referto this J as adiabatic or Curie-compatible coupling constant. Inall present cases, the BS energy is more negative than the tripletenergy, indicating intramolecular antiferromagnetic coupling.It is usual to evaluate the J value that is compatible with the
one obtained from electron paramagnetic resonance (EPR)studies. To achieve this end, one normally considers a verticaltransition and performs a single-point triplet calculation and asingle-point BS calculation, both by employing the optimizedgeometry of triplet. This was also done here using the largerbasis set 6-311++G(3df,3pd). This J will be called as vertical orEPR-compatible coupling constant. However, note that in thepresent case the adiabatic ground state is a singlet. Therefore,for the primary part of our discourse, we take the energydifference corresponding to the optimized geometries for eachstate at the B3LYP/6-311G(d,p) level as stated earlier. Relevantexcitation energies and oscillator strengths are obtained from
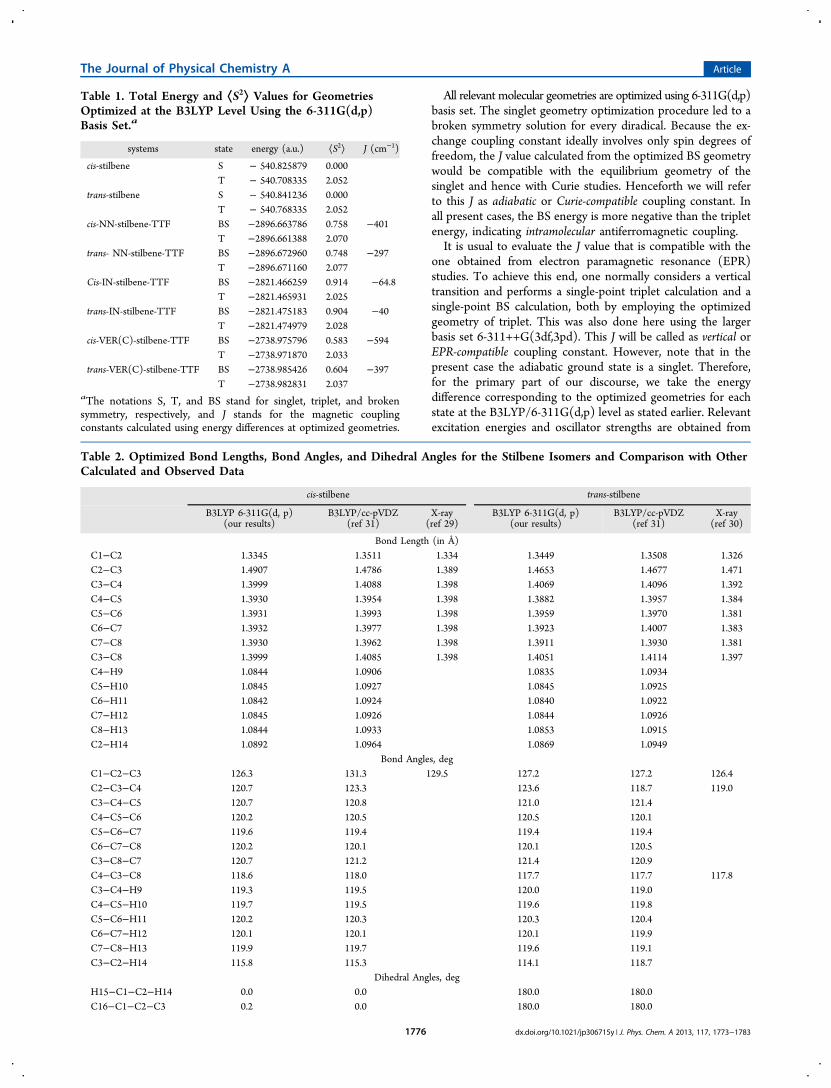
Table 1. Total Energy and ⟨S2⟩ Values for GeometriesOptimized at the B3LYP Level Using the 6-311G(d,p)Basis Set.a
systems state energy (a.u.) ⟨S2⟩ J (cm−1)
cis-stilbene S − 540.825879 0.000T − 540.708335 2.052
trans-stilbene S − 540.841236 0.000T − 540.768335 2.052
cis-NN-stilbene-TTF BS −2896.663786 0.758 −401T −2896.661388 2.070
trans- NN-stilbene-TTF BS −2896.672960 0.748 −297T −2896.671160 2.077
Cis-IN-stilbene-TTF BS −2821.466259 0.914 −64.8T −2821.465931 2.025
trans-IN-stilbene-TTF BS −2821.475183 0.904 −40T −2821.474979 2.028
cis-VER(C)-stilbene-TTF BS −2738.975796 0.583 −594T −2738.971870 2.033
trans-VER(C)-stilbene-TTF BS −2738.985426 0.604 −397T −2738.982831 2.037
aThe notations S, T, and BS stand for singlet, triplet, and brokensymmetry, respectively, and J stands for the magnetic couplingconstants calculated using energy differences at optimized geometries.
Table 2. Optimized Bond Lengths, Bond Angles, and Dihedral Angles for the Stilbene Isomers and Comparison with OtherCalculated and Observed Data
cis-stilbene trans-stilbene
B3LYP 6-311G(d, p)(our results)
B3LYP/cc-pVDZ(ref 31)
X-ray(ref 29)
B3LYP 6-311G(d, p)(our results)
B3LYP/cc-pVDZ(ref 31)
X-ray(ref 30)
Bond Length (in Å)C1−C2 1.3345 1.3511 1.334 1.3449 1.3508 1.326C2−C3 1.4907 1.4786 1.389 1.4653 1.4677 1.471C3−C4 1.3999 1.4088 1.398 1.4069 1.4096 1.392C4−C5 1.3930 1.3954 1.398 1.3882 1.3957 1.384C5−C6 1.3931 1.3993 1.398 1.3959 1.3970 1.381C6−C7 1.3932 1.3977 1.398 1.3923 1.4007 1.383C7−C8 1.3930 1.3962 1.398 1.3911 1.3930 1.381C3−C8 1.3999 1.4085 1.398 1.4051 1.4114 1.397C4−H9 1.0844 1.0906 1.0835 1.0934C5−H10 1.0845 1.0927 1.0845 1.0925C6−H11 1.0842 1.0924 1.0840 1.0922C7−H12 1.0845 1.0926 1.0844 1.0926C8−H13 1.0844 1.0933 1.0853 1.0915C2−H14 1.0892 1.0964 1.0869 1.0949
Bond Angles, degC1−C2−C3 126.3 131.3 129.5 127.2 127.2 126.4C2−C3−C4 120.7 123.3 123.6 118.7 119.0C3−C4−C5 120.7 120.8 121.0 121.4C4−C5−C6 120.2 120.5 120.5 120.1C5−C6−C7 119.6 119.4 119.4 119.4C6−C7−C8 120.2 120.1 120.1 120.5C3−C8−C7 120.7 121.2 121.4 120.9C4−C3−C8 118.6 118.0 117.7 117.7 117.8C3−C4−H9 119.3 119.5 120.0 119.0C4−C5−H10 119.7 119.5 119.6 119.8C5−C6−H11 120.2 120.3 120.3 120.4C6−C7−H12 120.1 120.1 120.1 119.9C7−C8−H13 119.9 119.7 119.6 119.1C3−C2−H14 115.8 115.3 114.1 118.7
Dihedral Angles, degH15−C1−C2−H14 0.0 0.0 180.0 180.0C16−C1−C2−C3 0.2 0.0 180.0 180.0
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp306715y | J. Phys. Chem. A 2013, 117, 1773−17831776
the time-dependent (TD)-DFT calculation on the singletspecies by using RB3LYP/6-311++G(d,p) computational setup.Before closing this section it is worth mentioning that the
magnitude of the exchange coupling constant is known todepend significantly on the choice of the exchange-correlationfunctional.27,28 A comparison of the calculated values predictedby different functionals has also been carried out, and theresults are included in Supporting Information as Tables S1, S2,and S3. We stress that the hybrid B3LYP functional with a largebasis set has been known to yield qualitatively good magnitudesfor the exchange coupling constant as compared to theexperiment. Besides, the conclusions reached in this work relyon the sign of J and the differences in the values of J betweenisomers, and these are less likely to be affected by the exchange-correlation potential.
III. RESULTS AND DISCUSSIONEquilibrium Optimized Geometries. Singlet and triplet
geometries are optimized for the unsubstituted stilbeneisomers. For the diradicals, triplet geometries are optimized,while the singlet geometry optimization leads to optimized BSgeometries. The optimized structures are shown in Figure 4. Allgeometry optimization are carried out using 6-311G(d,p) basisset. The optimized structure of cis-stilbene is approximatelyplanar with C2 point group whereas trans-stilbene turns out tobe completely planar with C2h symmetry.Computed total energies and ⟨S2⟩ values are given in Table 1.
The closed shell determinant singlet state defines theelectronic ground state for both stilbene isomers. From theoptimized geometries of cis- and trans-isomers, we find all bondlengths, bond angles, and dihedral angles (reported in Table 2)to be more or less comparable to the X-ray crystallographicdata29,30 and also to previously reported B3LYP/cc-PVDZcalculations.31
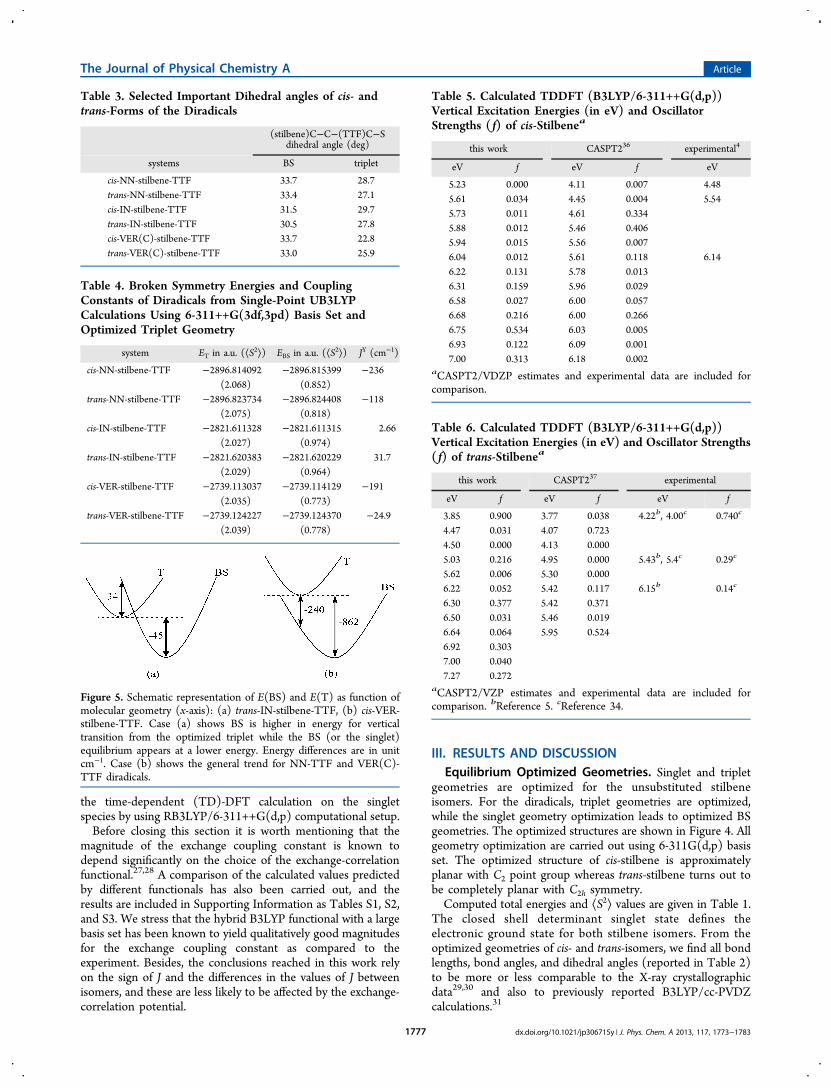
Table 3. Selected Important Dihedral angles of cis- andtrans-Forms of the Diradicals
(stilbene)C−C−(TTF)C−Sdihedral angle (deg)
systems BS triplet
cis-NN-stilbene-TTF 33.7 28.7trans-NN-stilbene-TTF 33.4 27.1cis-IN-stilbene-TTF 31.5 29.7trans-IN-stilbene-TTF 30.5 27.8cis-VER(C)-stilbene-TTF 33.7 22.8trans-VER(C)-stilbene-TTF 33.0 25.9
Table 4. Broken Symmetry Energies and CouplingConstants of Diradicals from Single-Point UB3LYPCalculations Using 6-311++G(3df,3pd) Basis Set andOptimized Triplet Geometry
system ET in a.u. (⟨S2⟩) EBS in a.u. (⟨S2⟩) JY (cm−1)
cis-NN-stilbene-TTF −2896.814092 −2896.815399 −236(2.068) (0.852)
trans-NN-stilbene-TTF −2896.823734 −2896.824408 −118(2.075) (0.818)
cis-IN-stilbene-TTF −2821.611328 −2821.611315 2.66(2.027) (0.974)
trans-IN-stilbene-TTF −2821.620383 −2821.620229 31.7(2.029) (0.964)
cis-VER-stilbene-TTF −2739.113037 −2739.114129 −191(2.035) (0.773)
trans-VER-stilbene-TTF −2739.124227 −2739.124370 −24.9(2.039) (0.778)
Figure 5. Schematic representation of E(BS) and E(T) as function ofmolecular geometry (x-axis): (a) trans-IN-stilbene-TTF, (b) cis-VER-stilbene-TTF. Case (a) shows BS is higher in energy for verticaltransition from the optimized triplet while the BS (or the singlet)equilibrium appears at a lower energy. Energy differences are in unitcm−1. Case (b) shows the general trend for NN-TTF and VER(C)-TTF diradicals.
Table 5. Calculated TDDFT (B3LYP/6-311++G(d,p))Vertical Excitation Energies (in eV) and OscillatorStrengths ( f) of cis-Stilbenea
this work CASPT236 experimental4
eV f eV f eV
5.23 0.000 4.11 0.007 4.485.61 0.034 4.45 0.004 5.545.73 0.011 4.61 0.3345.88 0.012 5.46 0.4065.94 0.015 5.56 0.0076.04 0.012 5.61 0.118 6.146.22 0.131 5.78 0.0136.31 0.159 5.96 0.0296.58 0.027 6.00 0.0576.68 0.216 6.00 0.2666.75 0.534 6.03 0.0056.93 0.122 6.09 0.0017.00 0.313 6.18 0.002
aCASPT2/VDZP estimates and experimental data are included forcomparison.
Table 6. Calculated TDDFT (B3LYP/6-311++G(d,p))Vertical Excitation Energies (in eV) and Oscillator Strengths( f) of trans-Stilbenea
this work CASPT237 experimental
eV f eV f eV f
3.85 0.900 3.77 0.038 4.22b, 4.00c 0.740c
4.47 0.031 4.07 0.7234.50 0.000 4.13 0.0005.03 0.216 4.95 0.000 5.43b, 5.4c 0.29c
5.62 0.006 5.30 0.0006.22 0.052 5.42 0.117 6.15b 0.14c
6.30 0.377 5.42 0.3716.50 0.031 5.46 0.0196.64 0.064 5.95 0.5246.92 0.3037.00 0.0407.27 0.272
aCASPT2/VZP estimates and experimental data are included forcomparison. bReference 5. cReference 34.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp306715y | J. Phys. Chem. A 2013, 117, 1773−17831777
Two main observations from Table 1 are as follows. First, thetrans-form is lower in energy than the cis- form for all thespecies in each spin statesinglet, triplet, and BS. Second, thesinglet is always more stable than the triplet in bare stilbenes,and the BS states are more stable for the diradicals. It transpiresthat the actual ground states of the diradicals are in fact openshell singlets. These can be described by linear combinations ofthe CS and D determinants as discussed in the previous section.The methyl groups in NN have almost free rotation. The localsymmetries of the cis- and trans- diradicals are C2 and C2hrespectively.The important dihedral angles of cis- and trans-forms of NN-
stilbene-TTF, IN-stilbene-TTF, and VER-stilbene-TTF aregiven in Table 3. The calculated magnetic coupling constants(J) are reported in Table 1. All coupling constants are negative, inagreement with spin alternation rule as illustrated in Figure 3.The Yamaguchi eq 2 is applied to evaluate the magnetic
coupling. It takes care of the error in energy difference due tospin contamination. However, the spin contamination is nottaken into account while the geometry is optimized. This castssome doubt on the validity of the optimized BS geometry,particularly for the NN-TTF and VER(C)-TTF diradicalswhere the BS solutions show significant spin contamination.Kitagawa et al. have addressed this issue and presented ageometry optimization method based on Yamaguchi’s approx-imate spin projection procedure.32
In this context, we show in Table 4 the energy values ob-tained from single point calculations using the much larger basisset 6-311++G(3df,3pd) and the optimized triplet geometry.The calculated J values in Tables 1 and 4 are generally of thesame order. They also retain the same sign except for the cis-and trans-isomers of IN-stilbene-TTF. The sign difference ofthe calculated J for the IN-stilbene-TTF isomers in these twotables is schematically illustrated in Figure 5 where the BS−Tenergy difference is shown to be small and of different signs atthe two (T and BS) optimized geometries. It is also of interestto note that Malrieu and Trinquier have recently suggested amethod to obtain the singlet geometry from calculated BSgeometry33 and have explained the situation by using a similarillustration for the energy curves.Although both cis- and trans-forms of all diradicals have negative
values of adiabatic J and hence intramolecular antiferromagneticcoupling, the difference between the J values for the cis andtrans isomers are 104 cm−1 for NN-TTF, 25 cm−1 for IN-TTF,and 197 cm−1 for VER(C)-TTF diradicals, respectively. This
would leave mark on the change in paramagnetic property of anensemble of these diradicals on photoconversion at roomtemperature. Of course, the fractional change would be lessprominent between the IN-TTF diradicals.
Spin Alternation. The intramolecular antiferromagneticcoupling predicted by the B3LYP/6-311G(d,p) calculations canbe justified by spin alternation rule20 (Figure 3) where for spinpropagation to the unpaired electron in TTF we select theshorter path (through S) and not the longer path (through−C−S−). The reason for this has been explained in ref 19: adihedral angle equal to about 35° makes one of the nonbondingorbitals of each sulfur atom in TTF almost parallel to the πframework of stilbene, but the carbon pz orbital deviates fromthe parallel position. This makes the shorter route morefavorable for spin wave propagation. The longer path is favoredby a zero dihedral angle, but it causes a mismatch in con-jugation through the next sulfur. Since all of the dihedral anglesare considerably large and in fact close to 35° in the BS state(Table 3), the shorter path is active in spin propagation. Thisis the reason for the antiferromagnetic coupling within eachdiradical.In fact, as noticed in ref 19, the sign and magnitude of J is
sensitive to the dihedral angle subtended by TTF to thecoupler. This indicates a very strong competition between thespin flows along the two possible paths in TTF. Table 3 showsthat the dihedral angles in the BS state of the IN-TTF diradicalare relatively small. This leads to a reduced coupling constantfor these species as indicated in Table 1.The vertical J values reported in Table 4 are significantly
reduced to the extent that for cis-IN-stilbene-TTF and trans-IN-stilbene-TTF the coupling constant becomes almost zero andsmall positive, respectively. Table 3 shows that the dihedralangle considerably deviates from 35° in the triplet state. Thisexplains the reduced intramolecular antiferromagnetic nature inTable 4 for all diradicals, as the triplet geometry is used in bothtriplet and BS calculations.
Spectroscopic Transitions. The absorption spectrum forcis-stilbene in n-heptane at room temperature was studied byBeale and Roe4 and that for trans-stilbene was examined bySuzuki.5 In 3-methylpentane, one-electron absorption spectrumof trans-stilbene was investigated by Hohlneicher and Dick34
and Gudipati et al.35 Molina et al.36,37 made an extensive com-putational study on cis- and trans-stilbene and compared theirresults with the experimental measurements of Beale et al.4 forcis-stilbene and those of Hohlneicher et al.34 for trans-stilbene.
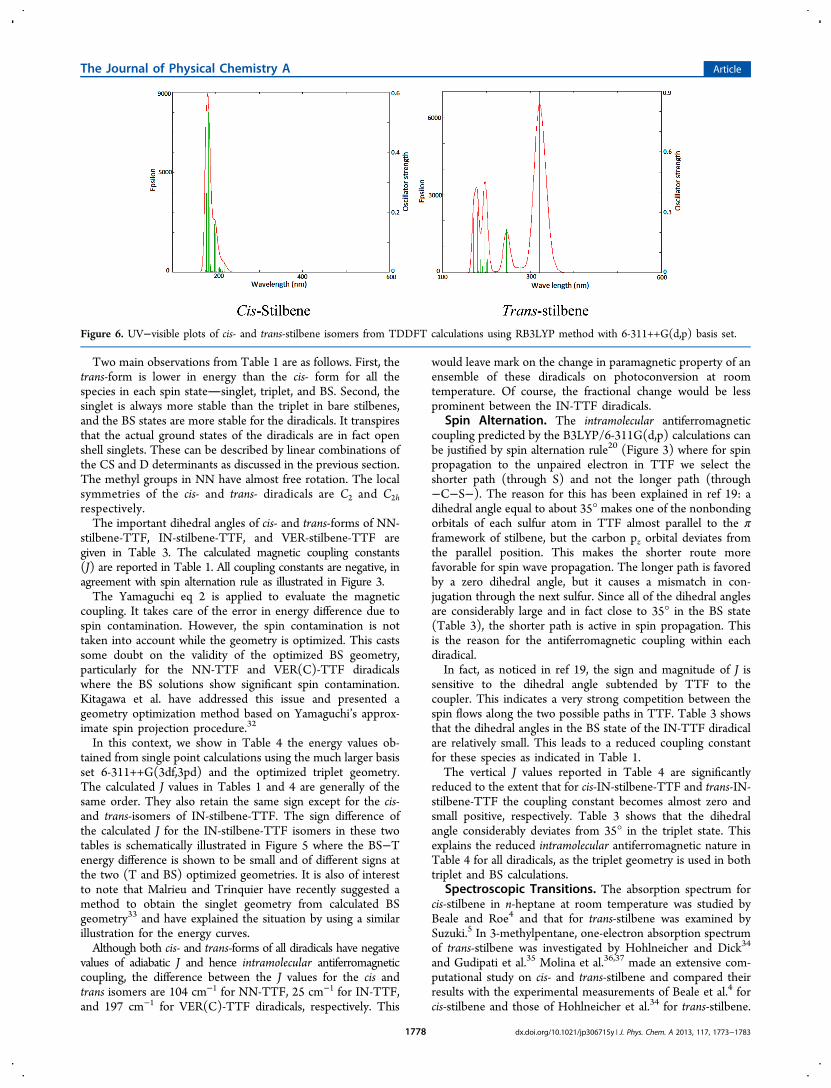
Figure 6. UV−visible plots of cis- and trans-stilbene isomers from TDDFT calculations using RB3LYP method with 6-311++G(d,p) basis set.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp306715y | J. Phys. Chem. A 2013, 117, 1773−17831778
In this work, we first use TD-DFT to calculate the transitionenergies for the cis- and trans-stilbene isomers which haveclosed shell ground states, and then explore the possibility ofusing this methodology to estimate the transition energies ofthe geometrical isomers of the diradicals with stilbene ascoupler. However, most of the diradicals exhibit an open-shellsinglet ground state which implies some caution as discussedlater.The experimentally observed n−π*, π−π*, and σ−σ*
transition energies are 4.48, 5.54, and 6.14 eV for cis-stilbene,respectively,4 and 4.22, 5.43, and 6.15 eV for trans-stilbene.5
Somewhat smaller values were found by the CASPT2calculations of Molina et al.36,37 Tables 5 and 6 show thetransition energies and oscillator strengths calculated for thestilbene isomers by the RB3LYP methodology. It is easy tonotice that the average error of TDDFT excitation energy isalways less than the error calculated from the CASPT2calculations, except in the case of the n−π* transitions of cis-stilbene. In addition, the transitions of cis-isomer are of lowintensity and that of trans-isomer are highly intense, which is inagreement with experiment. The TDDFT calculations showthe n−π* transition of cis-stilbene at somewhat higher energy(5.2 eV) and that for trans-stilbene at lower energy (3.85 eV)compared to the experimental data.4,5,34 The calculated π−π*transitions are found in the range 5.6−5.9 eV (for cis-) and5.0−5.6 eV (for trans-) and are moderately intense in bothcases. The σ−σ* transitions are found at ≥6.0 eV for the cis-form and ≥6.2 eV for the trans-isomer. The simulated spectraof cis- and trans-stilbene are given in Figure 6.Stilbene is a stable system as there is delocalization of
π-electrons between two phenyl rings. By addition of twomonoradicals at the positions (p,p′), the π-electron delocaliza-tion becomes more extensive which leads to an increasednumber of π and π* states with a concomitant broader band fortransitions involving these orbitals. This leads to a red shif taccompanied by a large increase in intensity. To check thesetrends, we have used TDDFT to estimate the excited states ofthe corresponding diradicals.However, TDDFT is based on a closed shell reference state
(which is the ground state for a closed shell species) whileTable 1 shows that the electronic ground state of each diradicalis an open shell singlet. Therefore, TDDFT calculations on thediradicals must correspond to transitions from a closed shellexcited state. Hence these transitions are expected to show notonly increased band widths and oscillator strengths but also anextra large red shift. The additional largeness of TDDFT red shiftowes its origin to the energy difference between the TDDFT-modified CS state and the ground state.Table 7 gives the TDDFT excitation energies for all diradical
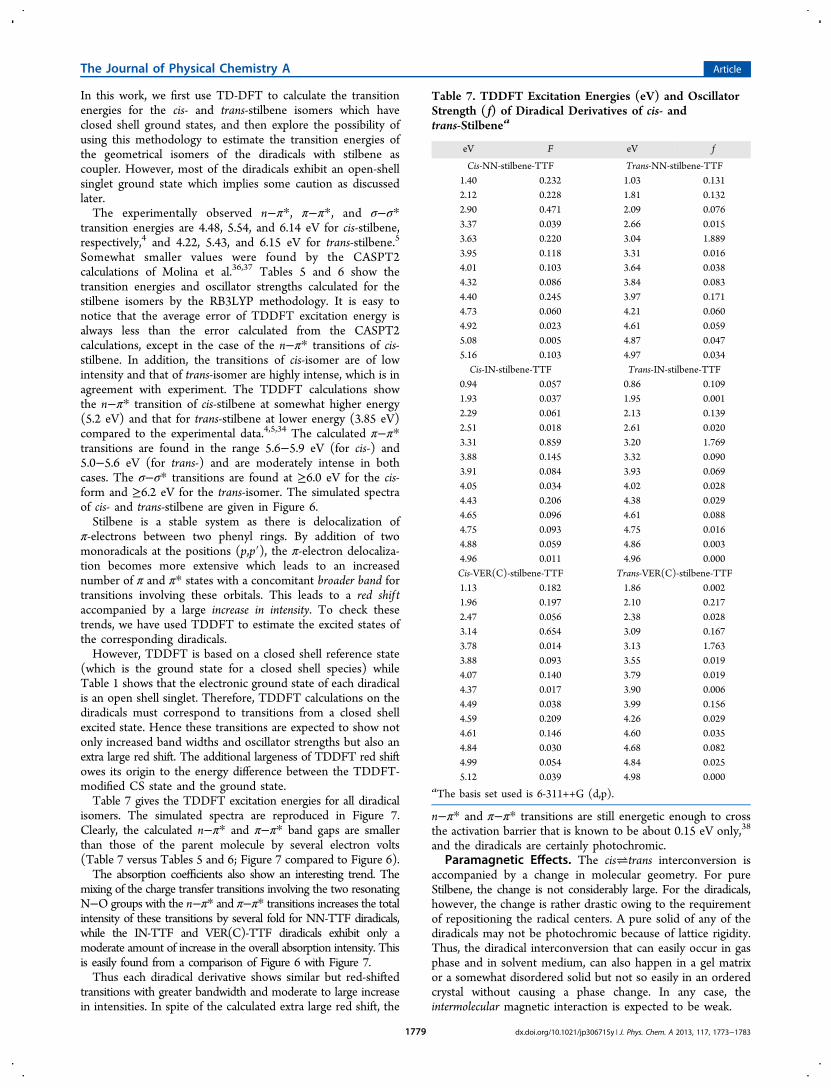
isomers. The simulated spectra are reproduced in Figure 7.Clearly, the calculated n−π* and π−π* band gaps are smallerthan those of the parent molecule by several electron volts(Table 7 versus Tables 5 and 6; Figure 7 compared to Figure 6).The absorption coefficients also show an interesting trend. The
mixing of the charge transfer transitions involving the two resonatingN−O groups with the n−π* and π−π* transitions increases the totalintensity of these transitions by several fold for NN-TTF diradicals,while the IN-TTF and VER(C)-TTF diradicals exhibit only amoderate amount of increase in the overall absorption intensity. Thisis easily found from a comparison of Figure 6 with Figure 7.Thus each diradical derivative shows similar but red-shifted
transitions with greater bandwidth and moderate to large increasein intensities. In spite of the calculated extra large red shift, the
n−π* and π−π* transitions are still energetic enough to crossthe activation barrier that is known to be about 0.15 eV only,38
and the diradicals are certainly photochromic.Paramagnetic Effects. The cis⇌trans interconversion is
accompanied by a change in molecular geometry. For pureStilbene, the change is not considerably large. For the diradicals,however, the change is rather drastic owing to the requirementof repositioning the radical centers. A pure solid of any of thediradicals may not be photochromic because of lattice rigidity.Thus, the diradical interconversion that can easily occur in gasphase and in solvent medium, can also happen in a gel matrixor a somewhat disordered solid but not so easily in an orderedcrystal without causing a phase change. In any case, theintermolecular magnetic interaction is expected to be weak.
Table 7. TDDFT Excitation Energies (eV) and OscillatorStrength ( f) of Diradical Derivatives of cis- andtrans-Stilbenea
eV F eV f
Cis-NN-stilbene-TTF Trans-NN-stilbene-TTF1.40 0.232 1.03 0.1312.12 0.228 1.81 0.1322.90 0.471 2.09 0.0763.37 0.039 2.66 0.0153.63 0.220 3.04 1.8893.95 0.118 3.31 0.0164.01 0.103 3.64 0.0384.32 0.086 3.84 0.0834.40 0.245 3.97 0.1714.73 0.060 4.21 0.0604.92 0.023 4.61 0.0595.08 0.005 4.87 0.0475.16 0.103 4.97 0.034Cis-IN-stilbene-TTF Trans-IN-stilbene-TTF
0.94 0.057 0.86 0.1091.93 0.037 1.95 0.0012.29 0.061 2.13 0.1392.51 0.018 2.61 0.0203.31 0.859 3.20 1.7693.88 0.145 3.32 0.0903.91 0.084 3.93 0.0694.05 0.034 4.02 0.0284.43 0.206 4.38 0.0294.65 0.096 4.61 0.0884.75 0.093 4.75 0.0164.88 0.059 4.86 0.0034.96 0.011 4.96 0.000Cis-VER(C)-stilbene-TTF Trans-VER(C)-stilbene-TTF1.13 0.182 1.86 0.0021.96 0.197 2.10 0.2172.47 0.056 2.38 0.0283.14 0.654 3.09 0.1673.78 0.014 3.13 1.7633.88 0.093 3.55 0.0194.07 0.140 3.79 0.0194.37 0.017 3.90 0.0064.49 0.038 3.99 0.1564.59 0.209 4.26 0.0294.61 0.146 4.60 0.0354.84 0.030 4.68 0.0824.99 0.054 4.84 0.0255.12 0.039 4.98 0.000
aThe basis set used is 6-311++G (d,p).
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp306715y | J. Phys. Chem. A 2013, 117, 1773−17831779
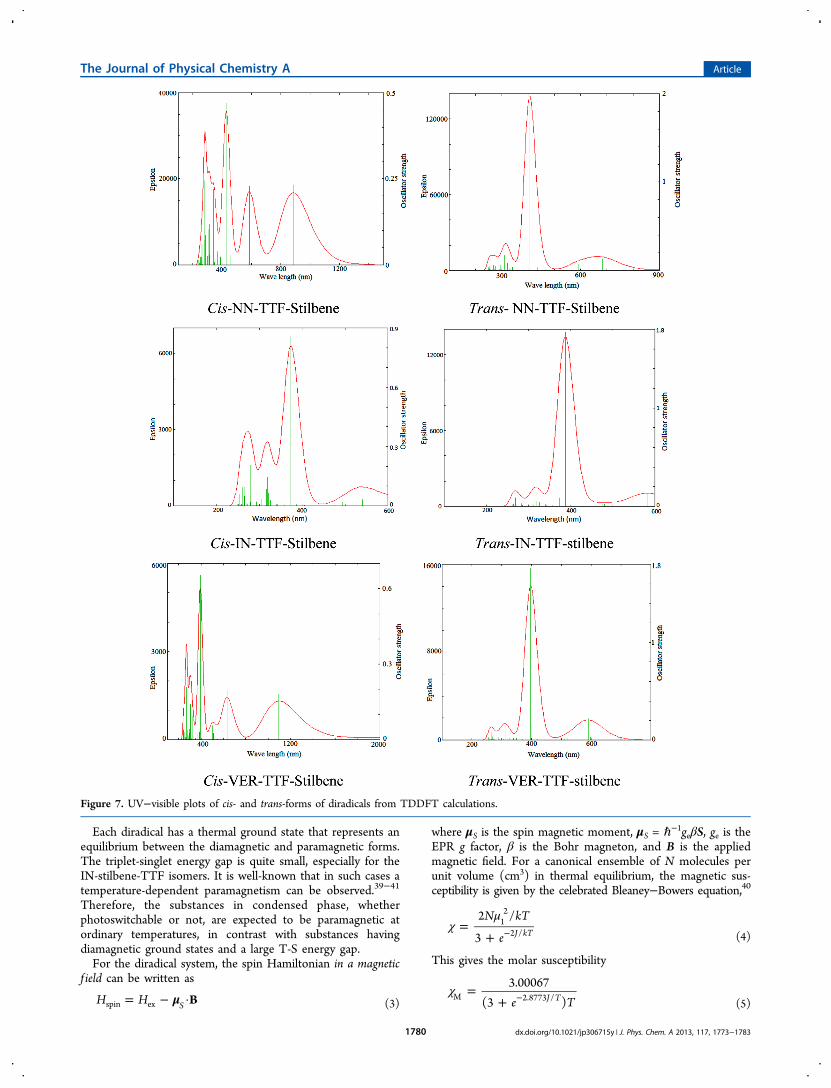
Each diradical has a thermal ground state that represents anequilibrium between the diamagnetic and paramagnetic forms.The triplet-singlet energy gap is quite small, especially for theIN-stilbene-TTF isomers. It is well-known that in such cases atemperature-dependent paramagnetism can be observed.39−41
Therefore, the substances in condensed phase, whetherphotoswitchable or not, are expected to be paramagnetic atordinary temperatures, in contrast with substances havingdiamagnetic ground states and a large T-S energy gap.For the diradical system, the spin Hamiltonian in a magnetic
f ield can be written as
μ= − ·H H BSspin ex (3)
where μS is the spin magnetic moment, μS = ℏ−1geβS, ge is theEPR g factor, β is the Bohr magneton, and B is the appliedmagnetic field. For a canonical ensemble of N molecules perunit volume (cm3) in thermal equilibrium, the magnetic sus-ceptibility is given by the celebrated Bleaney−Bowers equation,40
χμ
=+ −
N kT
e
2 /
3 J kT12
2 / (4)
This gives the molar susceptibility
χ =+ −e T
3.00067(3 )J TM 2.8773 /
(5)
Figure 7. UV−visible plots of cis- and trans-forms of diradicals from TDDFT calculations.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp306715y | J. Phys. Chem. A 2013, 117, 1773−17831780
where J is in cm−1 and T is in Kelvin. Note, however, that evenif J corresponds to magnetic coupling within the diradical as inTables 1 and 4, the magnetic susceptibility arises from thecontribution of the total magnetic moment of the isolateddiradicals in the triplet state neglecting intermolecularcoupling which will be very weak due to the distance betweenthe diradicals in such ensemble. The dimensions of the molarsusceptibility in eq 5 are cm3 mol−1 and is in the 0.75/T ≤χM ≤ 1/T when J ≥ 0 and 0 ≤ χM ≤ 0.75/T when J ≤ 0.The temperature dependence of susceptibility for these
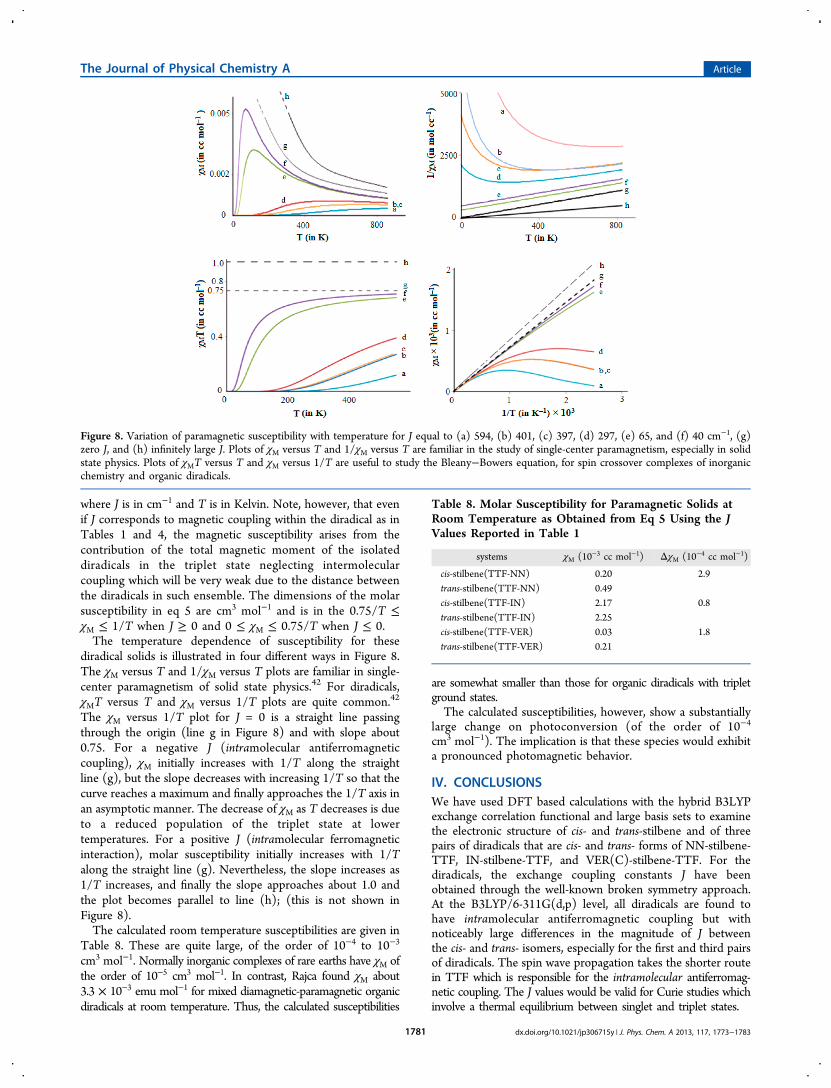
diradical solids is illustrated in four different ways in Figure 8.The χM versus T and 1/χM versus T plots are familiar in single-center paramagnetism of solid state physics.42 For diradicals,χMT versus T and χM versus 1/T plots are quite common.42
The χM versus 1/T plot for J = 0 is a straight line passingthrough the origin (line g in Figure 8) and with slope about0.75. For a negative J (intramolecular antiferromagneticcoupling), χM initially increases with 1/T along the straightline (g), but the slope decreases with increasing 1/T so that thecurve reaches a maximum and finally approaches the 1/T axis inan asymptotic manner. The decrease of χM as T decreases is dueto a reduced population of the triplet state at lowertemperatures. For a positive J (intramolecular ferromagneticinteraction), molar susceptibility initially increases with 1/Talong the straight line (g). Nevertheless, the slope increases as1/T increases, and finally the slope approaches about 1.0 andthe plot becomes parallel to line (h); (this is not shown inFigure 8).The calculated room temperature susceptibilities are given in
Table 8. These are quite large, of the order of 10−4 to 10−3
cm3 mol−1. Normally inorganic complexes of rare earths have χM ofthe order of 10−5 cm3 mol−1. In contrast, Rajca found χM about3.3 × 10−3 emu mol−1 for mixed diamagnetic-paramagnetic organicdiradicals at room temperature. Thus, the calculated susceptibilities
are somewhat smaller than those for organic diradicals with tripletground states.The calculated susceptibilities, however, show a substantially
large change on photoconversion (of the order of 10−4
cm3 mol−1). The implication is that these species would exhibita pronounced photomagnetic behavior.
IV. CONCLUSIONSWe have used DFT based calculations with the hybrid B3LYPexchange correlation functional and large basis sets to examinethe electronic structure of cis- and trans-stilbene and of threepairs of diradicals that are cis- and trans- forms of NN-stilbene-TTF, IN-stilbene-TTF, and VER(C)-stilbene-TTF. For thediradicals, the exchange coupling constants J have beenobtained through the well-known broken symmetry approach.At the B3LYP/6-311G(d,p) level, all diradicals are found tohave intramolecular antiferromagnetic coupling but withnoticeably large differences in the magnitude of J betweenthe cis- and trans- isomers, especially for the first and third pairsof diradicals. The spin wave propagation takes the shorter routein TTF which is responsible for the intramolecular antiferromag-netic coupling. The J values would be valid for Curie studies whichinvolve a thermal equilibrium between singlet and triplet states.
Figure 8. Variation of paramagnetic susceptibility with temperature for J equal to (a) 594, (b) 401, (c) 397, (d) 297, (e) 65, and (f) 40 cm−1, (g)zero J, and (h) infinitely large J. Plots of χM versus T and 1/χM versus T are familiar in the study of single-center paramagnetism, especially in solidstate physics. Plots of χMT versus T and χM versus 1/T are useful to study the Bleany−Bowers equation, for spin crossover complexes of inorganicchemistry and organic diradicals.
Table 8. Molar Susceptibility for Paramagnetic Solids atRoom Temperature as Obtained from Eq 5 Using the JValues Reported in Table 1
systems χM (10−3 cc mol−1) ΔχM (10−4 cc mol−1)
cis-stilbene(TTF-NN) 0.20 2.9trans-stilbene(TTF-NN) 0.49cis-stilbene(TTF-IN) 2.17 0.8trans-stilbene(TTF-IN) 2.25cis-stilbene(TTF-VER) 0.03 1.8trans-stilbene(TTF-VER) 0.21
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp306715y | J. Phys. Chem. A 2013, 117, 1773−17831781

The excitation characteristics of the diradicals have beenstudied using TD-DFT at the B3LYP/6-311++G(d,p) level.These calculations agree with the expected trends of red shift,increased bandwidth, and increased absorption intensity andpredict that the diradical isomers are strongly photochromic.From examination of paramagnetism for a canonical ensemble of
the diradicals, we conclude that photochromicity would accompanya considerable change of magnetization in the condensed phase.Finally, as shown by Table 4, the B3LYP/6-311++G(3df,3pd)
single point calculations using optimized triplet geometries givesmaller values for coupling constants. These values would berelevant to vertical transitions in EPR. This EPR can be obtainedonly for the IN-TTF diradicals for which there would be asignificant population in the triplet state at higher temperatures. Inthermal equilibrium, the cis-isomer has an estimated 33% tripletwith ΔST about −130 cm−1 while the trans-isomer has 40% tripletwith ΔST about −80 cm−1 (Table 1). The cis→trans inter-conversion would involve a transition from a rather EPR silence(Jvertical ∼ 0) to EPR activity with Jvertical ∼ 30 cm−1.
■ ASSOCIATED CONTENT*S Supporting InformationLog files of all calculations and ref 22. This material is availablefree of charge via the Internet at http://pubs.acs.org.
■ AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected] authors declare no competing financial interest.
■ ACKNOWLEDGMENTSS.N.D. and A.K.P. are grateful to DST Grant SR-S1-PC-19-2010 for financial support of this work. S.N.D. and F.I. thankIndo-Spain Collaborative Program in ScienceNanotechnology(DST Grant INT-Spain-P42-2012 and Spanish Grant PRI-PIBIN-2011-1028) for financial support. S.H. thanks UGC fora research fellowship, and F.I. acknowledges additional financialsupport from Spanish MICINN through research grantsFIS2008-02238 and CTQ2012-30751 and partial supportfrom Generalitat de Catalunya through grants 2009SGR1041,XRQTC, and the 2009 ICREA Academia award for excellencein research. We acknowledge IIT Bombay computer center formaking their facilities available to us.
■ REFERENCES(1) Irie, M. Photochromism: Memories and SwitchesIntroduction.Chem. Rev. 2000, 100, 1683−1684.(2) (a) Matsuda, K.; Irie, M. Effective Photoswitching of Intra-molecular Magnetic Interaction by Diarylethene: Backgrounds andApplications. Polyhedron 2005, 24, 2477−2483. (b) Tanifuji, N.;Matsuda, K.; Irie, M. Synthesis of a New Diarylethene Diradical whichhas Dxtended π-conjugated Dhains from the 2,5-position of DneDhiophene Ring. Polyhedron 2005, 24, 2484−2490. (c) Matsuda, K.Photoswitching of Intramolecular Magnetic Interaction Using Diary-lethene Photochromic Spin Couplers. Bull. Chem. Soc. Jpn. 2005, 78,383−392. (d) Tanifuji, N.; Irie, M.; Matsuda, K. New PhotoswitchingUnit for Magnetic Interaction: Diarylethene with 2,5-Bis(arylethynyl)-3-thienyl Group. J. Am. Chem. Soc. 2005, 127, 13344−13353.(e) Tanifuji, N.; Matsuda, K. M.; Irie, M. Effect of Imino Nitroxyland Nitronyl Nitroxyl Groups on the Photochromic Reactivity ofDiarylethenes. Org. Lett. 2005, 7, 3777−3780. (f) Matsuda, K.; Irie, M.Diarylethene as a Photoswitching Unit. J. Photochem. Photobiol., C:Photochem. Rev. 2004, 5, 169−182. (g) Matsuda, K.; Matsuo, M.; Irie,
M. Photoswitching of Intramolecular Magnetic Interaction UsingDiarylethene with Oligothiophene π-Conjugated Chain. J. Org. Chem.2001, 66, 8799−8803.(3) Dou, Y.; Allen, R. E. Detailed Dynamics of a ComplexPhotochemical Reaction: Cis−trans photoisomerization of stilbene. J.Chem. Phys. 2005, 119, 10658−10666.(4) Beale, R. N.; Roe, E. M. F. Ultra-violet Absorption Spectra ofTrans- and Cis-Stilbenes and their Derivatives. Part I. Trans- and Cis-Stilbenes. J. Chem. Soc. 1953, 116, 2755−2763.(5) Suzuki, H. Relations between Electronic Absorption Spectra andSpatial Configurations of Conjugated Systems. V. Stilbene. Bull. Chem.Soc. Jpn. 1960, 33, 379−388.(6) (a) Romero, F. M.; Ziessel, R.; Bonnet, M.; Pontillon, Y.;Ressouche, E.; Schweizer, J.; Delley, B.; Grand, A.; Paulsen, C.Evidence for Transmission of Ferromagnetic Interactions throughHydrogen Bonds in Alkyne-Substituted Nitroxide Radicals: Magneto-structural Correlations and Polarized Neutron Diffraction Studies. J.Am. Chem. Soc. 2000, 122, 1298−1309. (b) Caneschi, A.; Ferraro, F.;Gatteschi, D.; Lirzin, A. L.; Rentschler, E. Ferromagnetic Intermo-lecular Coupling in the Nitronyl Nitroxide Radical 2-(4-thiomethyl-phenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide, NIT(Sme)Ph.Inorg. Chim. Acta 1995, 235, 159−164.(7) (a) Lescope, C.; Luneau, D.; Rey, P.; Bussiere, G.; Reber, C.Synthesis, Structures, and Magnetic and Optical Properties of a Seriesof Europium(III) and Gadolinium(III) Complexes with ChelatingNitronyl and Imino Nitroxide Free Radicals. Inorg. Chem. 2002, 41,5566−5574. (b) Oshio, H.; Watanabe, T.; Ohto, A.; Ito, T. A One-Dimensional Helical Copper(II) Imino Nitroxide. Inorg. Chem. 1997,36, 1608−1610.(8) (a) Gilroy, J. B.; McKinnon, S. D. J.; Kennepohl, P.; Zsombor, M.S.; Ferguson, M. J.; Thompson, L. K.; Hicks, R. G. Probing ElectronicCommunication in Stable Benzene-Bridged Verdazyl Diradicals. J. Org.Chem. 2007, 72, 8062−8069. (b) Lemaire, M. T.; Barclay, T. M.;Thompson, L. K.; Hicks, R. G. Synthesis, Structure, and Magnetism ofa Binuclear Cobalt(II) Complex of a Potentially Bis-tridentateVerdazyl Radical Ligand. Inorg. Chim. Acta 2006, 359, 2616−2621.(9) (a) Ullman, E. F.; Boocock, D. G. B. “Conjugated” Nitronyl-Nitroxide and Imino-Nitroxide Biradicals. J. Chem. Soc., Chem.Commun. 1969, 20, 1161−1162. (b) Ullman, E. F.; Osiecki, J. H.;Boocock, D. G. B.; Darcy, R. Stable Free Radicals. X. NitronylNitroxide Monoradicals and Biradicals as Possible Small MoleculeSpin Labels. J. Am. Chem. Soc. 1972, 94, 7049−7059.(10) Castell, O.; Caballol, R.; Subra, R.; Grand, A. Ab Initio Study ofUllman’s Nitroxide Biradicals. Exchange Coupling versus StructuralCharacteristics Analysis. J. Phys. Chem. 1995, 99, 154−157.(11) Barone, V.; Bencini, A.; Matteo, A. Intrinsic and EnvironmentalEffects in the Structure and Magnetic Properties of Organic MolecularMagnets: Bis(imino)nitroxide. J. Am. Chem. Soc. 1997, 119, 10831−10837.(12) Vyas, S.; Ali, Md. E.; Hossain, E.; Patwardhan, S.; Datta, S. N.Theoretical Investigation of Intramolecular Magnetic Interactionthrough an Ethylenic Coupler. J. Phys. Chem. A 2005, 109, 4213−4217.(13) Fischer, P. H. H. LCAO-MO Calculations on Verdazyls.Tetrahedron 1967, 23, 1939−1952.(14) Markovsky, L. N.; Polumbrik, O. M.; Nesterenko, A. M.Quantum-Chemical Investigation of Spatial and Electronic Structureof Verdazyl and its Derivatives. Int. J. Quantum Chem. 1979, 16, 891−899.(15) Green, M. T.; McCormick, T. A. Controlling the Singlet−Triplet Splitting in Bisverdazyl Diradicals: Steps toward MagneticPolymers. Inorg. Chem. 1999, 38, 3061−10.(16) Ciofini, I.; Daul, C. A. DFT Calculations of Molecular MagneticProperties of Coordination Compounds. Coord. Chem. Rev. 2003, 238,187−209.(17) (a) Takui, T.; Sato, K.; Shiomi, D.; Ito, K.; Nishizawa, M.; Itoh,K. Magnetic Coupling of Nitronyl Nitroxide-based Biradical Salts.Synth. Met. 1999, 103, 2271−2272. (b) Romero, F. M.; Ziessel, R.;Bonnet, M.; Pontillon, Y.; Ressouche, E.; Schweizer, J.; Delley, B.;Grand, A.; Paulsen, C. Evidence for Transmission of Ferromagnetic
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp306715y | J. Phys. Chem. A 2013, 117, 1773−17831782

Interactions through Hydrogen Bonds in Alkyne-Substituted NitroxideRadicals: Magnetostructural Correlations and Polarized NeutronDiffraction Studies. J. Am. Chem. Soc. 2000, 122, 1298−1309.(c) Nagashima, H.; Irisawa, M.; Yoshioka, N.; Inoue, H. Mol. Cryst.Liq. Cryst. Sci. Technol. Sect. A 2002, 376, 371. (d) Rajadurai, C.;Ivanova, A.; Enkelmann, V.; Baumgarten, M. Study on the HeteroatomInfluence in Pyridine-Based Nitronyl Nitroxide Biradicals withPhenylethynyl Spacers on the Molecular Ground State. J. Org. Chem.2003, 68, 9907−9915. (e) Wautelet, P.; Le Moigne, J.; Videva, V.;Turek, P. Spin Exchange Interaction through Phenylene-EthynyleneBridge in Diradicals Based on Iminonitroxide and NitronylnitroxideRadical Derivatives. 1. Experimental Investigation of the Through-Bond Spin Exchange Coupling. J. Org. Chem. 2003, 68, 8025−8036.(f) Deumal, M.; Robb, M. A.; Novoa, J. J. The Mechanism of theMagnetic Interaction in the β Phase of the p-(nitro)phenyl NitronylNitroxide (KAXHAS). A Bottom-up Study Using Only Ab Initio Data.Polyhedron 2003, 22 (14−17), 1935−1944. (g) Zoppellaro, G.;Ivanova, A.; Enkelmann, V.; Geies, A.; Baumgarten, M. Synthesis,Magnetic Properties and Theoretical Calculations of Novel NitronylNitroxide and Imino Nitroxide Diradicals Grafted on TerpyridineMoiety. Polyhedron 2003, 22, 2099−2110.(18) (a) Segura, J. L.; Martin, N. New Concepts in TetrathiafulvaleneChemistry. Angew. Chem., Int. Ed. 2001, 40, 1372−1409. (b) Chahma,M.; Wang, X. S.; van der Est, A.; Pilkington, M. Synthesis andCharacterization of a New Family of Spin Bearing TTF Ligands. J. Org.Chem. 2006, 71, 2750−2755. (c) Chahma, M.; Macnamara, K.; van derEst, A.; Alberola, A.; Polo, V.; Pilkington, M. Synthesis andCharacterization of a TTF-π-verdazyl Radicala New BuildingBlock for Conducting and/or Magnetic Systems. New J. Chem. 2007,31, 1973−1978. (d) Polo, V.; Alberola, A.; Andres, J.; Anthony, J.;Pilkington, M. Towards Understanding of Magnetic Interactionswithin a Series of Tetrathiafulvalene−π Conjugated-Verdazyl DiradicalCation System: A Density Functional Theory Study. Phys. Chem.Chem. Phys. 2008, 10, 857−864.(19) (a) Latif, A. I.; Singh, V. P.; Bhattacharjee, U.; Panda, A.; Datta,S. N. Very Strongly Ferromagnetically Coupled Diradicals from MixedRadical Centers. II. Nitronyl Nitroxide Coupled to Tetrathiafulvalenevia Spacers. J. Phys. Chem. A 2010, 114, 6648−6656. (b) Saha, A.;Latif, I. A.; Datta, S. N. Photoswitching Magnetic Crossover in OrganicMolecular Systems. J. Phys. Chem. A 2011, 115, 1371−1379.(20) (a) Trindle, C.; Datta, S. N. Molecular Orbital Studies on theSpin States of Nitroxide Species: Bis- and Tris-nitroxymetaphenylene,1,1-bisnitroxyphenylethylene, and 4,6-dimethoxy-1,3-dialkylnitroxy-benzenes. Int. J. Quantum Chem. 1996, 57, 781−799. (b) Trindle,C.; Datta, S. N.; Mallik, B. Phenylene Coupling of Methylene Sites. TheSpin States of Bis(X-methylene)-p-phenylenes and Bis(chloromethylene)-m-phenylene. J. Am. Chem. Soc. 1997, 119, 12947−12951.(21) Datta, S. N.; Pal, A. K.; Hansda, S.; Latif, I. A. On thePhotomagnetism of Nitronyl Nitroxide, Imino Nitroxide, and Verdazyl-Substituted Azobenzene. J. Phys. Chem. A 2012, 116, 3304−3311.(22) Frisch, M. J.; Trucks, G. W.; et al. Gaussian 09, Revision A.1;Gaussian, Inc.: Wallingford, CT, 2009.(23) (a) Noodleman, L. Valence Bond Description of Antiferro-magnetic Coupling in Transition Metal Dimers. J. Chem. Phys. 1981,74, 5737−5744. (b) Noodleman, L.; Baerends, E. J. ElectronicStructure, Magnetic Properties, ESR, and Optical Spectra for 2-ironFerredoxin Models by LCAO-X.alpha. Valence Bond Theory. J. Am.Chem. Soc. 1984, 106, 2316−2327. (c) Noodleman, L.; Davidson, E. R.Ligand Spin Polarization and Antiferromagnetic Coupling inTransition Metal Dimers. Chem. Phys. 1986, 109, 131−143.(d) Noodleman, L.; Peng, C. Y.; Case, D. A.; Mouesca, J. M. OrbitalInteractions, Electron Delocalization and Spin Coupling In Iron-SulfurClusters. Coord . Chem . Rev. 1995, 144, 199−244.(24) (a) Caballol, R.; Castell, O.; Illas, F.; Malrieu, J. P.; Moreira, I.de P. R. Remarks on the Proper Use of the Broken SymmetryApproach to Magnetic Coupling. J. Phys. Chem. A 1997, 101, 7860−7866. (b) Illas, F.; Moreira, I. de P. R.; De Graaf, C.; Barone, V.Magnetic Coupling in Biradicals, Binuclear Complexes and Wide GapInsulators: A Survey of Ab Initio Wave Function and Density
Functional Theory Approaches. Theor. Chem. Acc. 2000, 104, 265−272.(25) Moreira, I. de P. R.; Illas, F. A Unified View of the TheoreticalDescription of Magnetic Coupling in Molecular Chemistry and SolidState Physics. Phys. Chem. Chem. Phys. 2006, 8, 1645−1659.(26) (a) Yamaguchi, K.; Takahara, Y.; Fueno, T.; Nasu, K. Ab InitioMO Calculations of Effective Exchange Integrals between Transition-Metal Ions via Oxygen Dianions: Nature of the Copper-Oxygen Bondsand Superconductivity. Jpn. J. Appl. Phys. 1987, 26, L1362−L1364.(b) Yamaguchi, K.; Jensen, F.; Dorigo, A.; Houk, K. N. A SpinCorrection Procedure for Unrestricted Hartree-Fock and Møller-Plesset Wavefunctions for Singlet Diradicals and Polyradicals. Chem.Phys. Lett. 1988, 149, 537−542. (c) Yamaguchi, K.; Takahara, Y.;Fueno, T.; Houk, K. N. Extended Hartree-Fock (EHF) Theory ofChemical Reactions. Theor. Chim. Acta 1988, 73, 337−364.(27) Valero, R.; Costa, R.; Moreira, I. de P. R.; Truhlar, D. G.; Illas, F.Performance of the M06 Family of Exchange-Correlation Functionalsfor Predicting Magnetic Coupling in Organic and Inorganic Molecules.J. Chem. Phys. 2008, 128, 114103(1−8).(28) Rivero, P.; Moreira, I. de P. R.; Illas, F.; Scuseria, G. E.Reliability of Range-Separated Hybrid Functionals for DescribingMagnetic Coupling in Molecular Systems. J. Chem. Phys. 2008, 129,184110(1−7).(29) Bouwstra, J. A.; Schouten, A.; Roon, J. K. Structural studies ofthe system trans-azobenzene/trans-stilbene. II. A Reinvestigation ofthe Disorder in the Crystal Structure of trans-Stilbene, C14H12. ActaCrystallogr. 1984, C40, 428−431.(30) Traetteberg, M.; Frantsen, E. B. A Gas Electron DiffractionStudy of the Molecular Structure of cis-Stilbene. J. Mol. Struct. 1975,26, 69−76.(31) Chen, P. C.; Chieh, Y. C. Azobenzene and Stilbene: AComputational Study. J. Mol. Struct.: THEOCHEM 2003, 624, 191−200.(32) Kitagawa, Y.; Saito, T.; Ito, M.; Shoji, M.; Koizumi, K.; Yamanaka,S.; Kawakami, T.; Okumura, M.; Yamaguchi, K. Approximately Spin-Projected Geometry Optimization Method and its Application to di-Chromium Systems. Chem. Phys. Lett. 2007, 442, 445−450.(33) Malrieu, J. P.; Trinquier, G. A Recipe for GeometryOptimization of Diradicalar Singlet States from Broken-SymmetryCalculations. J. Phys. Chem. A. 2012, 116, 8226−8237.(34) Hohlneicher, G.; Dick, B. Experimental Determination of theLow-Lying Excited a States of Trans-Stilbene. J. Photochem. 1984, 27,215−231.(35) Gudipati, M. S.; Maus, M.; Daverkausen, J.; Hohlneicher, G.Higher Excited States of Aromatic Hydrocarbons. III. Assigning the In-Plane Polarized Transitions of Low-Symmetry Molecules: Chryseneand E-Stilbene. Chem. Phys. 1995, 192, 37−47.(36) Molina, V.; Merchan, M.; Roos, B. O. A Theoretical Study of theElectronic Spectrumo Cis-Stilbene. Spectrochim. Acta, Part A 1999, 55,433−446.(37) Molina, V.; Merchan, M.; Roos, B. O. Theoretical Study of theElectronic Spectrum of trans-Stilbene. J. Phys. Chem. A 1997, 101,3478−3487.(38) Syage, J. A.; Felker, P. M.; Zewail, A. H. Picosecond Dynamicsand Photoisomerization of Stilbene in Supersonic Beams. II. ReactionRates and Potential Energy Surface. J. Chem. Phys. 1984, 81, 4706−4723.(39) Rajca, A.; Takahashi, M.; Pink, M.; Spangnol, G.; Rajca, S.Conformationally Constrained, Stable, Triplet Ground State (S=1)Nitroxide Diradicals. Antiferromagnetic Chains of S = 1 Diradicals. J.Am. Chem. Soc. 2007, 129, 10159−10170.(40) Bleany, B.; Bowers, K. D. Anomalous Paramagnetism of CopperAcetate. Proc. R. Soc. London, Ser. A 1952, 214, 451−465.(41) Deumal, M.; Robb, M. A.; Novoa, J. J. Quantitative Analysis ofthe Magnetism of the Meta-(Methoxy)Phenyl Nitronyl NitroxideCrystal: A Bottom−Up Analysis of a Crystal Presenting CompetingFerro and Antiferromagnetic Interactions. Polyhedron 2005, 24, 2368−2376.(42) Kittel, C. Introduction to solid state physics, 6th ed.; Wiley: NewYork, 1991.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp306715y | J. Phys. Chem. A 2013, 117, 1773−17831783




















