
THE ROLE OF THE DSB SYSTEM IN ANTIMICROBIAL ...
280
THE ROLE OF THE DSB SYSTEM IN ANTIMICROBIAL RESISTANCE PhD Thesis Submitted to the Department of Life Sciences, Imperial College London in partial fulfilment of the requirements for the degree of Doctor of Philosophy Supervisors: Dr Despoina Mavridou and Professor Alain Filloux NIKOL KADEŘÁBKOVÁ MRC CMBI, Imperial College London September 2020
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of THE ROLE OF THE DSB SYSTEM IN ANTIMICROBIAL ...

THE ROLE OF THE DSB SYSTEM IN
ANTIMICROBIAL RESISTANCE
PhD Thesis
Submitted to the Department of Life Sciences, Imperial College London
in partial fulfilment of the requirements for the degree of
Doctor of Philosophy
Supervisors: Dr Despoina Mavridou and Professor Alain Filloux
NIKOL KADEŘÁBKOVÁ MRC CMBI, Imperial College London
September 2020

2
COPYRIGHT DECLARATION AND DECLARATION OF
ORIGINALITY
The copyright of this thesis rests with the author. Unless otherwise indicated, its contents are licensed
under a Creative Commons Attribution-Non Commercial-No Derivatives 4.0 International Licence.
Researchers may copy and redistribute the thesis in any medium or format on the condition that they
credit the author, that they do not use it for commercial purposes and that they do not distribute modified
versions of the work. When reusing or sharing this work, researchers must ensure that the licence terms
are clear to others by naming the licence and linking to the licence text. Researchers must seek
permission from the copyright holder for uses of this work that are not included in this licence or
permitted under UK Copyright Law.
I hereby declare that all work presented in this thesis, unless detailed below or referenced appropriately
in the text, is my own.
Figure 3.3 A, Figure 3.4 A, Figure 3.5, Figure 3.7, Figure 3.9, Figure 5.2 A, and Figure 5.3 A –
Experiments performed by Dr R. Christopher D. Furniss.
Figure 3.11 – Experiments performed by Evgenia Maslova and Dr Ronan R. McCarthy.
The MG1655, MG1655 acrA, MG1655 tolC strains and pSLTS plasmid were from Dr Jessica M. A.
Blair. The pCB112 plasmid was a kind gift from Professor Thomas G. Bernhardt (Harvard Medical
School). The Pseudomonas and Stenotrophomonas clinical isolates were a kind gift of Dr Laurent
Dortet (Bicêtre Hospital, Le Kremlin-Bicêtre). Antibodies against DsbA, AcrA, and TolC were the
from Professor Jonathan R. Beckwith (Harvard Medical School), Dr Felicity Alcock (Newcastle
University) and Professor Vassilis Koronakis (University of Cambridge), respectively.

3
LIST OF PUBLICATIONS ARISING FROM THIS WORK
Furniss, R.C.D.*, Kadeřábková, N.*, Barker, D., Bernal, P., Maslova, E., Antwi, A.A.A., McNeil,
H.E., Pugh, H.L., Dortet, L., Blair, J.M.A., Larrouy-Maumus, G., McCarthy, R.R., Gonzales, D.,
Mavridou, D.A.I., Breaking antimicrobial resistance by disrupting extracytoplasmic protein folding.
eLife (in revision) - included in this thesis as Chapter 10 - APPENDIX II.
*Equally-contributing first authors
Kadeřábková, N., Furniss, R.C.D., Maslova, E., Bernal, P., Filloux, A., Gonzales, D., McCarthy, R.R.,
Mavridou, D.A.I., Breaking species-specific antimicrobial resistance in Gram-negative pathogens by
targeting disulfide bond formation. (manuscript written)

4
ACKNOWLEDGEMENTS
First and foremost, I would like to thank my main supervisor, Dr Despoina Mavridou. I came to you as
green a microbiologist as one could be, thank you for guiding me, letting me learn and develop at my
own pace. This thesis would not have been written without your trust in me, your endless patience, and
your support. I have learnt so much from you both inside and outside the lab over the past three years
and have grown as a scientist and as a person thanks to all your effort and dedication. Thank you.
I would also like to acknowledge my second supervisor Professor Alain Filloux, for his guidance,
expertise, and advice throughout my PhD. Thank you for believing in me at the beginning.
My deepest thank you goes to Dr Chris Furniss for being there all the way, for teaching me, supporting
me, and letting me make my own mistakes. You answered every single little question and then some,
offered advice and listened to my thoughts and ideas. I cannot express enough how lucky I was to have
such a kind and helpful guide, lab partner and friend. Thank you for everything.
The end of my three years would not have been what they were without the help, support, and hard
work of Dr Alex McCarthy. You gave up your lab time, your office and even your amazing scones to
make my final four months as stress free as you could. Thank you.
My PhD would not be possible without the following people:
Dr Diego Gonzales, for expertise, in silico studies and advice.
Dr Patricia Bernal, for experimental advice, expertise, and guidance in the field of genetic
manipulation of Pseudomonas species.
Helen McNeil, Hannah Pugh, and Dr Jessica Blair, for strains and expertise on efflux.
Evgenia Maslova and Dr Ronan McCarthy, for running my Galleria mellonella in vivo models.
Dr Sabrina Slater, for her expertise and magician’s touch with computer software.

5
A special thank you goes to Amanda Antwi and Declan Barker for their friendship and
companionship at various times in the lab and beyond. Last, but not least thank you to all the
members of the CMBI Level 5 and the Filloux group on CMBI Level 1 for friendship, support, and
feedback over the years.
In the life beyond the lab, there are many who deserve a thank you from me, and I would like to give
special mention to the following:
Suja Moore, Anna Cooke, Magdalena Lemanczyk, Sophie Bennet, Fabia Borrmann, Hannah Moore,
Emma Lambe and Stacey Smiley-Carr, our time together has been short but sweet and I feel I have
known you my entire life. Thank you for getting me out of the lab and into the ‘real’ world on a
regular basis.
Dang Quoc Anh, Charlie, I still cannot pronounce your name despite the 13 years of friendship. This
is a heartfelt thank you for your never-ending snarky humour and support – you have been making
me laugh and despair at the same time for over a decade and your spot-on humour was never more
needed than now. Thank you my friend.
Tony Emmerson and Dr Nicola Howarth, you were there to spark and light the fire for my love of
research and this thesis would never had been written had it not been for you believing in me, pushing
me and challenging me right at the start. Thank you.
Alasdair Keith, thank you for your support and understanding over the many years. I would not have
embarked on so many adventures without you. Thank you.
My brother Filip, who let it be known that there is only one ‘right doctor’, your high regard of me is
humbling and I am grateful for your belief in me. Thank you.
My parents, and my grandparents, thank you for being there throughout the years and opening so
many doors at the start. No price was high enough for you in supporting me and words cannot express
how grateful I am for the opportunities you have given me. Thank you.
This thesis is dedicated to Mirka and Vaclav Kaderabkovi for their unwavering faith in me.

6
ABSTRACT
Extensive use of antibiotics in medicine and agriculture has led to increasing emergence of
antimicrobial resistance in bacterial populations. Dwindling resources in the discovery of novel active
compound leads and the increasing demands for safety and efficacy of new drugs mean that we are now
faced with treatment failures due to multi-drug resistant pathogens. In the quest for new targets that will
enable us to counter antibiotic resistance, it is often ignored that many resistance mechanisms precede
the clinical use of antibiotics. Instead, the ability to adapt, survive and bypass the toxicity of many
chemical compounds is wired within the bacterial genome. Continuous inter-strain and inter-species
competition have given microorganisms tools to thrive under conditions of chemical warfare.
Recognising this is important when characterising mechanisms underpinning bacterial antimicrobial
resistance, as it can lead to novel strategies that can help us bypass it.
The work described here explores the connection between the disulfide bond formation system, a key
oxidative protein folding pathway in the cell envelope of Gram-negative bacteria, and two widespread
antimicrobial resistance mechanisms, -lactamase catalysed hydrolysis of -lactam antibiotics and
efflux-mediated drug expulsion. It is demonstrated that oxidative-protein-folding-mediated proteostasis
is crucial for both resistance mechanisms, and its inhibition can sensitise multidrug-resistant pathogens
to existing antibiotics. Preliminary results from an experimental evolution approach, set the scene for
future exploration of the importance of disulfide linkages for the capacity of -lactamase enzymes to
evolve under selective pressure. Together, these findings aim to address the mechanistic basis of a new
avenue for antibiotic adjuvant therapy, whereby targeting a non-essential process would allow us to
potentiate existing antibiotics towards previously resistant bacterial strains. With novel essential targets
against bacteria being scarce, adjuvant approaches like this one could prolong the use and efficacy of
existing drugs against some of the most resistant Gram-negative pathogens.

7
TABLE OF CONTENTS
Copyright Declaration and Declaration of Originality 2
List of Publications Arising From This Work 3
Acknowledgements 4
Abstract 6
Table of Contents 7
List of Figures and Tables 11
List of Abbreviations 15
1 Introduction 17
1.1 Bacteria, the Causative Agents of Disease 18
1.2 Brief Overview of Antibiotic Development 20
1.3 Antimicrobial Resistance and Its Spread in Bacterial Communities 21
1.3.1 Intrinsic resistance 21
1.3.2 Acquired resistance 28
1.3.3 Foreign DNA acquisition 28
1.3.4 Mutational resistance 34
1.4 Overcoming Antibiotic Resistance By Using Antibiotic Adjuvants 36
1.4.1 Inhibition of antibiotic modification 36
1.4.2 Membrane permeabilising compounds 38
1.4.3 Inhibition of drug efflux 40
1.4.4 Inhibiting the spread of antimicrobial genes 41
1.5 The Bacterial Cell Envelope 44
1.5.1 Protein folding and transport into the cell envelope 45
1.5.2 Protein transport across the inner membrane of Gram-negative bacteria 46
1.6 Oxidative Protein Folding Pathways 49
1.6.1 Disulfide bond formation in eukaryotes 49
1.6.2 Disulfide bond formation in prokaryotes 53
1.6.3 Polymorphisms of the Gram-negative DSB system 74
1.6.4 Gram-positive bacteria 75
1.6.5 Targeting bacterial pathogenicity through inhibition of the DSB system and oxidative
folding 76
1.7 Aims of This Work 81
2 Materials and methods 82

8
2.1 Reagents and Bacterial Growth Conditions 82
2.2 Genetic Manipulation Techniques 82
2.2.1 Genomic DNA extraction, purification of plasmid DNA and PCR products of genes 82
2.2.2 PCR amplification 83
2.2.3 Agarose gel electrophoresis 84
2.2.6 Restriction digestion 84
2.2.7 Ligation 85
2.2.4 Site-directed mutagenesis 85
2.2.5 DNA sequencing 86
2.2.8 Preparation and transformation of chemically competent cells 86
2.2.9 Preparation and transformation of electrocompetent cells 87
2.3 Bacterial Strains and Plasmids 87
2.3.1 Cloning of -lactamase genes 93
2.3.2 Generation of E. coli dsbA, degP and marR mutants 93
2.3.3 Generation of P. aeruginosa dsbA1 mutants 94
2.3.4 Generation of S. maltophilia dsbA1 dsbL1 mutant 94
2.3.5 Triparental conjugation of P. aeruginosa and S. maltophilia 95
2.3.6 Complementation of E. coli MG1655 dsbA 95
2.4 Minimum Inhibitory Concentration (MIC) Assays 96
2.5 SDS-PAGE Analysis and Immunoblotting 97
2.6 -lactam Hydrolysis Assay 98
2.7 NPN Uptake Assay 99
2.8 PI Uptake Assay 99
2.9 CPRG Cell Envelope Integrity Assay 99
2.10 Motility Assay 100
2.11 AMS labelling 100
2.12 Bacterial Growth Assay – dsbA Mutant 100
2.13 Bacterial Growth Assay – DSB System Chemical Inhibitor 101
2.14 In Vivo Clearance Assay 101
2.15 Statistical Analysis of Experimental Data 102
3 The Importance of Disulfide Bond Formation for the Function of Mobile Class D -
lactamases Enzymes of Pseudomonas aeruginosa 103
3.1 Introduction 103
3.2 Results 106

9
3.2.1 Deletion of dsbA substantially decreases -lactamase mediated antibiotic resistance in E.
coli MC1000 106
3.2.2 Deletion of dsbA does not affect the integrity of the cell envelope in E. coli MC1000 109
3.2.3 Deletion of dsbA deletion does not affect the viability of E. coli MC1000 111
3.2.4 Class D -lactamases misfold in absence of DsbA 112
3.2.5 DsbA is a tractable target for class D -lactamases 114
3.3 Discussion 122
4 The Importance of Disulfide Bond Formation for the Function of Chromosomally-resident
-lactamase enzymes 124
4.1 Introduction 124
4.2 Results 127
4.2.1 The activity of cysteine-containing chromosomally encoded β-lactamase enzymes is
dependent on DsbA 127
4.2.2 Chromosomally encoded β-lactamase enzymes degrade or misfold in the absence of DsbA
130
4.2.3 Deletion of dsbA1 compromises the function of the intrinsic β-lactamase OXA-50 in P.
aeruginosa laboratory strains and clinical isolates 132
4.2.4 Deletion of dsbA1 results in sensitization of P. aeruginosa clinical isolates to existing β-
lactam antibiotics 134
4.2.5 Deletion of dsbA1 and dsbL1 results in increased sensitivity of a S. maltophilia clinical
isolate to ceftazidime 136
4.3 Discussion 138
5 The Importance of Disulfide Bond Formation for the Function of Resistance-Nodulation-
Division Efflux Pumps 140
5.1 Introduction 140
5.2 Results 144
5.2.1 Deletion of dsbA in E. coli MG1655 does not affect the outer or the inner membrane
permeability 144
5.2.2 Deletion of dsbA in E. coli MG1655 causes only minor decreases in bacterial viability 146
5.2.3 RND efflux pump function is compromised in the absence of DsbA 147
5.2.4 Compromised function of RND efflux pumps is due to altered periplasmic proteostasis
148
5.2.5 DsbA as a tractable target for RND efflux pumps 150
5.3 Discussion 152

10
6 The Importance Of Disulfide Bond Formation For The Expansion Of The Hydrolytic
Spectrum Of -Lactamase Enzymes 153
6.1 Introduction 153
6.2 Experimental Design 157
6.2.1 Deletion of dsbA does not severely impact the resistance to b-lactams conferred by the
narrow-spectrum b-lactamases SHV-1 and TEM-1 157
6.2.2 Setup of the experimental evolution experiment 158
6.3 SHV-1 Pilot Study Results 163
6.3.1 Absence of DsbA decreases the potential for evolution of antibiotic resistance to
ceftazidime upon exposure to increasing antibiotic concentrations 163
6.3.2 Characterisation of evolved SHV-1 expressing strains 164
6.3.3 Increase of the hydrolytic spectrum of SHV-1 does not affect bacterial fitness 167
6.4 Discussion 169
7 Discussion and Future Work 171
8 References 176
9 APPENDIX I 208
10 APPENDIX II 217

11
LIST OF FIGURES AND TABLES
Table 1 Comparison of the features of the bacterial cell-envelope of the Gram-positive and Gram-
negative bacterial species. 19
Figure 1.1 Examples of the five main efflux pump superfamilies. 24
Figure 1.2 Schematic of cell wall synthesis and the role of -lactam antibiotics in the inhibition of PBP-
catalysed peptidoglycan cross-linking. 25
Figure 1.3 Clinically available -lactamase inhibitors target class A, C and D b-lactamases. 37
Figure 1.4 The first identified oxidative folding catalyst PDI1 carries a classical Trx fold with a
conserved disulfide bond in a CXXC motif, a common feature in all thiol-redox enzymes.
51
Figure 1.5 The oxidative pathway of the disulfide bond formation system. 54
Figure 1.6 Crystal structure of the primary oxidase of the E. coli DSB system, EcDsbA. 55
Figure 1.7 Differential binding of DsbA to substrate peptide or DsbB is directed by the hydrophobic
surfaces surrounding the active site of DsbA and the histidine residue, His32. 57
Figure 1.8 Crystal structure of EcDsbB, the membrane partner protein of DsbA. 61
Figure 1.9 The proposed mechanism of DsbA oxidation by DsbB. 62
Figure 1.10 The isomerase pathway of the DSB system. 65
Figure 1.11 Crystal structure of the primary isomerase of the E. coli DSB system, EcDsbC. 66
Figure 1.12 Crystal structure of E. coli DsbG, EcDsbG, rendered in cartoon representation. 70
Figure 1.13 Crystal structure of the reduced state of the N-terminal domain of E. coli, nDsbD, rendered
in cartoon representation. 71
Figure 1.14 Crystal structures of EcDsbC - nDsbD and cDsbD - nDsbD complexes elucidate the
mechanism behind the transfer of reductive potential through the periplasmic subunits of
DsbD. 72
Figure 1.15 Fragment based screening identified the first active inhibitors of the DSB system. 77
Figure 1.16 Peptidomimetic library screening identified EcDsbA inhibitors. 79
Figure 1.17 Inhibitors of the DsbA partner proteins, DsbB and VKOR. 80
Table 2 Bacterial strains used in this thesis. 87
Table 3 Plasmids used in this thesis. 89
Table 4 Oligonucleotide primers used in this thesis. 91
Figure 3.1 Antimicrobial resistance mediated by OXA-type -lactamases depends on disulfide bond
formation. 107

12
Figure 3.2 Complementation of dsbA restores the β-lactam MIC values for E. coli MC1000 expressing
class D β-lactamases. 108
Figure 3.3 Deletion of dsbA has no effect on outer membrane permeability in E. coli MC1000. 110
Figure 3.4 Deletion of dsbA does not result in damage to the bacterial inner membrane cell envelope.
111
Figure 3.5 Deletion of dsbA does not have drastic effects on the growth of E. coli MC1000. 112
Figure 3.6 Class D -lactamase enzyme levels remain unaffected by the absence of DsbA. 113
Table 5 The hydrolytic activities of tested β-lactamase enzymes are significantly decreased in the
absence of DsbA. 114
Figure 3.7 Chemical inhibition of the DSB system impedes DsbA re-oxidation and flagellar motility
in E. coli MC1000. 116
Figure 3.8 Chemical inhibition of the DSB system phenocopies the β-lactam MIC changes observed
using E. coli MC1000 dsbA mutant. 117
Figure 3.9 Chemical inhibition of the DSB system has no effect on the growth of E. coli MC1000.
118
Table 6 Chemical inhibition of the DSB system via DsbB shows no effects on multidrug-resistant
P. aeruginosa clinical isolates. 119
Figure 3.10 Absence of DsbA1, the principal pseudomonal DsbA analogue, sensitizes multidrug-
resistant clinical P. aeruginosa isolates to first-line and last-resort -lactam antibiotics. 120
Figure 3.11 Absence of DsbA1 from a P. aeruginosa clinical isolate expressing OXA-198 allows it to
be cleared from infected G. mellonella larvae by piperacillin. 121
Figure 4.1 Antimicrobial resistance mediated by chromosomally resident -lactamases depends on
disulfide bond formation. 128
Figure 4.2 Complementation of dsbA restores the β-lactam MIC values for E. coli MC1000 expressing
β-lactamases. 130
Figure 4.3 The majority of tested β-lactamase enzymes become unstable in the absence of DsbA. 131
Table 7 The hydrolytic activities of tested β-lactamase enzymes are significantly decreased in the
absence of DsbA. 132
Figure 4.4 Absence of DsbA1, the principal pseudomonal DsbA analogue, from P. aeruginosa
laboratory strains and clinical isolates expressing OXA-50 results in a two-fold decrease in
their β-lactam MIC values. 133
Figure 4.5 Absence of DsbA1, the principal pseudomonal DsbA analogue, sensitises P. aeruginosa
clinical isolates expressing AIM-1 to penicillins and cephalosporins. 135

13
Figure 4.6 Absence of DsbA1 and of its analogue DsbL1 significantly decreases the MIC of the S.
maltophilia GUE clinical isolate, expressing L2-1 and L1-1, to ceftazidime. 137
Figure 5.1 Structure of the E. coli AcrAB-TolC efflux pump. 141
Figure 5.2 Deletion of dsbA has no effect on the membrane permeability or on the outer membrane
integrity of E. coli MG1655. 144
Figure 5.3 Deletion of dsbA does not result in damage to the bacterial cell envelope. 145
Figure 5.4 Deletion of dsbA causes a small defect in the growth of E. coli MG1655. 146
Figure 5.5 Antimicrobial resistance mediated by a tripartite efflux pump, AcrAB-TolC, of E. coli
MG1655 is affected in absence of DsbA. 147
Figure 5.6 Complementation of dsbA restores efflux-pump substrate MIC values for E. coli MG1655.
148
Figure 5.7 RND efflux pump function is impaired in the absence of DsbA due to accumulation of
unfolded AcrA resulting from insufficient DegP activity. 149
Figure 5.8 Deletion of dsbA sensitizes the efflux-active E. coli MG1655 strain to chloramphenicol.
150
Figure 5.9 Deletion of marR results in increased expression of the AcrAB pump. 151
Table 8 An overview of commonly occurring amino acid substitutions in TEM-1 and SHV-1 -
lactamases that mediate expansion of their hydrolytic activity. 155
Figure 6.1 Antimicrobial activity of narrow-spectrum enzymes, SHV-1 and TEM-1, does not
dependent on DsbA. 158
Table 9 E. coli MC1000 strains constitutively expressing SHV-1 and TEM-1 -lactamases as well
as the single-cysteine variants of these enzymes were plated on ceftazidime containing
plates (0.5-64x MIC). 160
Table 10 Bacterial suspensions resulting from overnight growth of E. coli MC1000 strains
constitutively expressing wild-type SHV-1 and TEM-1 -lactamases. 161
Figure 6.2 Schematic of the experimental evolution method to be used for strains expressing TEM-1
and SHV-1 -lactamases. 162
Table 11 E. coli MC1000 strains expressing SHV-1 and its single-cysteine variant, develop resistance
upon exposure to increasing ceftazidime concentrations. 164
Figure 6.3 Determination of -lactam MIC values (µg/mL) of three isolates of E. coli MC1000 pDM2-
blaSHV-1 obtained during passage II from plates containing 32 g/mL of ceftazidime. 165

14
Figure 6.4 Determination of -lactam MIC values (µg/mL) of three isolates of E. coli MC1000 dsbA
pDM2-blaSHV-1 obtained during passage II from plates containing 32 g/mL of ceftazidime.
166
Figure 6.5 Determination of -lactam MIC values (µg/mL) of three isolates of E. coli MC1000 pDM2-
blaSHV-1 C54A obtained during passage II from plates containing 32 g/mL of ceftazidime.
167
Figure 6.6 The experimental evolution process does not affect the fitness of any of the evolved strains.
168
Supplementary Table 1 Overview of the β-lactamase enzymes investigated in this thesis. 208
Supplementary Table 2 Deletion of dsbA lowers the β-lactam MIC values for E. coli MC1000
expressing diverse β-lactamases. 209
Supplementary Table 3 Chemical inhibition of the DSB system reduces the MIC values of
representative -lactam antibiotics for E. coli MC1000 expressing disulfide-
bond-containing class D β-lactamases in a similar manner to the deletion of
dsbA. 211
Supplementary Table 4 Antibiotic resistance profiles (MIC values in µg/mL) of the clinical isolates
and laboratory strains tested in this study for -lactam compounds. 212
Supplementary Table 5 Antibiotic resistance profiles (MIC values in µg/mL) of the clinical isolates
and laboratory strains tested in this study for a range of commonly used non-
-lactam antibiotics. 213
Supplementary Table 6 Deletion of dsbA does not decrease the β-lactam MIC values for E. coli
MC1000 expressing the narrow-spectrum β-lactamases TEM-1 and SHV-1 at
either 37°C or 42°C. 214
Supplementary Table 7 β-lactam MIC values and MIC fold changes (FC) recorded in evolved and
original backgrounds, after experimental evolution of E. coli MC1000 strains
expressing the narrow-spectrum β-lactamase SHV-1. 215

15
LIST OF ABBREVIATIONS
AAC – aminoglycoside N-acetyltransferase
ABC – ATP-binding cassette efflux pump
AC – amoxicillin
AM – ampicillin
AMR – antimicrobial resistance
AMS – 4-acetamido-4'-maleimidylstilbene-
2,2'-disulfonic acid
ANT – aminoglycoside O-
nucleotidyltransferase
AP – alkaline phosphatase
APH – aminoglycoside O-
phosphotransferase
AT – aztreonam
ATP – adenosine triphosphate
ASST – aryl-sulfate sulfotransferase
BMD – broth microdilution
bp – base pair
cDsbD – C-terminal domain of DsbD
CCCP – carbonyl cyanide m-chlorophenyl
hydrazone
CFU – colony forming unit
CI – ciprofloxacin
CO – colistin
CPRG – chlorophenyl red-β-D-
galactopyranoside
DSB – disulfide bond formation system
DMSO – dimethyl sulfoxide
DNA – deoxyribonucleic acid
DTT – dithiothreitol
EcDsbA/DsbB/DsbC/DsbG – Escherichia
coli DsbA/DsbB/DsbC/DsbG
EDTA – Ethylenediaminetetraacetic acid
EPI – efflux pump inhibitor
ERO1p – ER oxidoreductin 1
Erv1 and Erv2 – protein essential for
respiration and vegetative growth
ER – endoplasmic reticulum
ESBL – extended-spectrum -lactamase
FAD – flavin adenine dinucleotide
FC – fold change
GFP – green fluorescent protein
GM – gentamicin
GTP – guanosine-5'-triphosphate
HEPES – N-2-hydroxyethylpiperazine-N-
ethanesulfonic acid
HGT – horizontal gene transfer
HRP – horseradish peroxidase
Ig – immunoglobulin
IMS – intermembrane space
IPTG – isopropyl β-D-1-
thiogalactopyranoside
IP – imipenem
IR – inverted repeat
IS – insertion sequence
ITS/MISS – intermembrane space targeting
signals
L-Ara4N – 4-amino-4-deoxy-L-arabinose
LB – Lysogeny broth
LPS – lipopolysaccharide
MATE – multidrug and toxic compound
extrusion efflux pump
MCS – multiple cloning site

16
MES – 2-(N-morpholino)ethanesulfonic
acid
MFS – major facilitator superfamily of
efflux pump
MH – Mueller-Hinton
MIC – minimum inhibitory concentration
MOPS – 3-(N-morpholino) propanesulfonic
acid
MQ – menaquinone
mRNA – messenger ribonucleic acid
MtDsbA – Mycobacterium tuberculosis
DsbA
nDsbD – N-terminal domain of DsbD
NmDsbA1/DsbA2/DsbA3 – Neisseria
meningitidis DsbA1/DsbA2/DsbA3
NPN – 1-N-phenylnaphthylamine
NS – narrow-spectrum -lactamase
OD – optical density
ORI – origin of replication
P – periplasmic
PBP – penicillin binding proteins
PBS – phosphate buffered saline
PCR – polymerase chain reaction
PDI – protein disulfide isomerase
PI – propidium iodide
PP – piperacillin
PmDsbA – Proteus mirabilis DsbA
PT – piperacillin/tazobactam combination
QSOX – quiescin sulfhydryl oxidase
RNA – ribonucleic acid
RND – resistance-nodulation-division efflux
pump
RPM – revolutions per minute
SAR – structure-activity relationship
SDS PAGE – sodium dodecyl sulphate-
polyacrylamide gel electrophoresis
Sec – general secretory pathway
SeDsbA – Salmonella enterica serovar
typhimurium DsbA
SMR – small multidrug resistance efflux
pump
SOB – super optimal broth
Tat – twin arginine pathway
TBS – tris-buffered saline
TBS-T – tris-buffered saline – Tween20
TINS – a target-immobilized NMR
screening
TM – transmembrane
tmDsbD – transmembrane domain of DsbD
TS – trimethoprim/sulfamethoxazole
combination
TR – trimethoprim
Trx – thioredoxin
TZ – ceftazidime
UPEC – uropathogenic Escherichia coli
UQ – ubiquinone
VKOR – vitamin K epoxide reductase-like
protein
X-Gal – 5-bromo-4-chloro-3-indolyl--d-
galactopyranoside
XM – cefuroxime

17
1 INTRODUCTION
Part A
Antimicrobial Resistance in Bacteria

18
1.1 BACTERIA, THE CAUSATIVE AGENTS OF DISEASE
Since the discovery of bacteria by Anton van Leeuwenhoek in the late 1600s, human understanding of
infections and disease has evolved significantly. The role of pathogenic microorganisms in disease, as
described by the ‘Germ Theory’, is now seen as common knowledge and is widely accepted by modern
society.1,2 It is thus remarkable to consider that, despite the scientific observations by Leeuwenhoek, it
was not until the mid to late 1800s that the ideological shift from the ‘Miasma theory’ to ‘Germ theory’
begun in Europe, thanks to the work of John Snow, Louis Pasteur and Robert Koch.3 Interestingly,
literature sources show that the first notions of Germ Theory appeared in cultures outside the European
sphere of influence much earlier, dating as far back as ancient Greece, Rome or India, or recently
appearing in accounts of Islamic medicine from the late Middle Ages.2,4,5
Bacteria represent a large fraction of prokaryotic microorganisms that are found in most habitats on
Earth, from temperate soil and water to acidic hot springs.6,7 Their survival, even in the most unlikely
of places, is supported by their fast replication, high gene mutational rates, flexible genomes capable of
quickly incorporating or removing genetic elements, and their ability to form both symbiotic and
parasitic relationships with other species in their vicinity. This remarkable diversity and complexity
mean that, despite decades of intensive research, only a small fraction of bacteria has been characterised
to date.
Bacterial cells show a general lack of intracellular organisation, in comparison to eukaryots, and thus,
structurally, their cytoplasmic composition is fairly uniform. Most distinct differences can be observed
at the membrane and extracellular level, the so called “bacterial cell envelope”. This protective structure
has been intensively studied, using biochemical and biophysical methods, to further understand its
protective effects that allow bacteria to grow and survive in hostile environments. Further, differences
in the envelope can be used to classify bacteria using the Gram stain and morphological appearance
(Table 1). Discovered in late 19th century by Hans Christian Gram, the Gram stain is composed of a
crystal violet stain and a safranin counterstain and it exploits the inherent differences in the bacterial
cell wall compositions to separate the bacterial domain into two distinct groups, the Gram-positive
(stain purple) and Gram-negative (stain red) species (Table 1). Additional classification is then afforded
by cell morphology, with the two most common being the separation between spherical cocci and rod-
shaped bacilli; this gives rise to the commonly used four-group classification system.8 Smaller groups

19
can also be recognised, such as the comma-shaped Vibrio, the spiral-shaped Spirilla, or the tightly-
coiled Spirochetes although their use in bacterial classification is not as prevalent.8
Gram-negative bacteria, such as the Escherichia, Pseudomonas or Neisseria spp, are surrounded by a
well organised three-layered bacterial cell envelope composed of the outer membrane, the cell wall, and
the inner membrane.9 The glycolipid outer membrane, built from lipopolysaccharide (LPS) chains,
protects Gram-negative bacteria against a wide range of toxic compounds; it is this layer that is
recognised by the human immune system.10 Attached to the inner leaflet of the outer membrane is a
thin, cross-linked peptidoglycan layer which forms the rigid cell wall that determines the bacterial
shape, prevents Gram staining and provides physical protection of the cell interior.11 In between this
outer layer and the inner membrane lies the aqueous and highly oxidative periplasmic space. For
bacteria that cause disease, the periplasm is home not only to housekeeping pathways, but also virulence
factors and antimicrobial resistance (AMR) determinants.
Table 1 Comparison of the features of the bacterial cell-envelope of the Gram-positive and Gram-negative bacterial
species.12
Gram-negative bacteria Gram-positive bacteria
Peptidoglycan layer thin (single-layered) thick (multi-layered)
Teichoic acids absent present in many
Periplasmic space present absent
Outer membrane present absent
Lipopolysaccharide (LPS) content high none
Lipid /lipoprotein content
high
(presence of outer
membrane)
Low
(acid-fast bacteria have lipids linked
to peptidoglycan)
Flagellar structure four rings in basal body two rings in basal body
Toxins production primarily endotoxins primarily exotoxins
Resistance to physical disruption low high
Susceptibility to anionic detergents low high
Resistance to sodium azide low high
Resistance to drying low high
Cell wall composition
70-120 Å thick; two layers.
Lipid content is 20-30%
(high), Murein content is 10-
20% (low)
100-120 Å thick; single layer.
Lipid content is low,
Murein content is 70-80% (high).
Intrinsic antibiotic resistance more resistant more susceptible

20
In contrast to Gram-negative species, Gram-positive bacteria, such as Staphylococci or Bacilli, lack an
outer membrane. Instead a thick cell wall composed of many layers of peptidyglycan-techoic/mycolic
acids is anchored onto their cytoplasmic membrane. This permits the entrance of more chemicals,
including the Gram stain, and makes these bacteria more susceptible to several antimicrobial
compounds that Gram-negative species are resistant to.
1.2 BRIEF OVERVIEW OF ANTIBIOTIC DEVELOPMENT
The discovery and successful isolation of the antibiotic compound penicillin in the late 1920s and early
1940s, respectively, has long been considered the major milestone of antibiotic discovery. Historically,
however, the appearance of antibiotic compounds dates much further back, with traces of tetracycline,
a broad-spectrum antibiotic produced by Streptomyces spp., found in human remains as far back as
A.D. 350-550 in line, perhaps, with the first seeds of germ theory described by the ancient Greek,
Roman, and Middle Eastern cultures.2,13,14
Interestingly, the first “modern” hospital use of antibiotics also predates the discovery of penicillin by
over three decades. The extracts of Pyocyanase from Pseudomonas aeruginosa, prepared by Emmerlich
and Löw, were used in 1899 to treat a range of infections.13,15 Even though the practice and the outcomes
of these applications were questionable at best, they sparked interest in the discovery and development
of other antimicrobial compounds.13,15 By the end of the 19th century, Ehrlich and his group had started
to work on large-scale synthesis and screening of hundreds of compounds in an attempt to find a cure
for syphilis.13,16 This was a resounding success; not only did their experiments lead to the identification
of Salvarsan and Neosalvarsan, they also provided the methodological basis for future identification of
other active compounds.13,16 Using a similar systematic approach, the sulfonamide Protonsil was
identified a few decades later.13,17 These antibiotics, in combination with penicillin have formed the
cornerstone of pharmaceutical drug discovery. In the following decades, several additional antibiotic
classes were discovered and introduced to the hospital setting, until progress tapered off in 1960s.18
Despite the major advances in technology in the early 20th century, drug discovery timescales have
remained long and little progress has been made in developing clinically approved novel-target, broad-
spectrum antimicrobials.
Broadly, antibiotic compounds can be separated into bactericidal (killing bacteria) and bacteriostatic
(inhibiting the growth of bacteria). They target a wide range of cellular processes, including, but not

21
limited to, cell-wall synthesis (-lactams, glycopeptides), transcription (quinolones, rifampicin) and
translation (aminoglycosides, macrolides). Although hundreds of active compounds are now available
in clinics, they all belong to a relatively small group of approximately 15 antibiotic classes. Taking into
consideration the ever-growing number of resistance mechanisms, this collection is quickly becoming
insufficient and the treatment of multidrug-resistant infections, for example those caused by P.
aeruginosa, Acinetobacter baumannii or Klebsiella pneumoniae, is becoming increasingly difficult.
Indeed, the World Health Organisation has issued a warning on the rise of multi-drug resistant
pathogens and composed a list of priority ‘ESKAPE’ pathogens for which novel treatments are urgently
required.19,20
1.3 ANTIMICROBIAL RESISTANCE AND ITS SPREAD IN BACTERIAL
COMMUNITIES
Antimicrobial resistance is a multifaceted concept, examples of which can be found for any cellular
process that can be affected by a chemical compound exerting selective pressure. It can arise through
drug-target modifications, decreased cell-wall permeability, increased efflux rate, pathway variation, or
antibiotic inactivation; this list is representative but certainly not comprehensive. Generally, these
resistance mechanisms can be divided into two types, intrinsic and acquired. Intrinsic resistance is
linked to the natural ability of bacteria to survive and thrive in the presence of other organisms that
produce antimicrobial compounds in shared environments, such as soil or water habitats. As expected,
antibiotic-producing species are themselves naturally resistant to the antibiotics they produce,
broadening the pool of resistant organisms.21 By contrast, acquired resistance emerges in previously
susceptible strains under the selective pressure applied by an antimicrobial agent.
1.3.1 Intrinsic resistance
Complex microbial communities, containing bacteria as well as eukaryotic organisms like fungi, are
major sources of antimicrobial compounds. These communities are essentially a cocktail of microbial
species, metabolic by-products, signalling molecules and toxic compounds that form highly complex
environments in which bacteria have co-evolved to survive. Natural fitness advantages required for
bacterial survival in the presence of a specific antibiotic and environmental stressors are termed intrinsic
resistance mechanisms, have developed independently of previous antibiotic exposure without

22
horizontal gene transfer, and are usually present across the entire species.22 Even though these intrinsic
resistance mechanisms have not been evolved in clinical settings, they can contribute to antibiotic
treatment failure, not only because they confer resistance to several opportunistic pathogens but also
because bacteria that harbour such mechanisms can act as reservoirs from which resistance genes can
be mobilised and spread to other species (discussed further in section 1.3.2).21,23 Although this is not a
comprehensive review, the most important of these mechanisms are discussed below.
1.3.1.1 Membrane permeability
The uptake of nutrients such as amino acids, ions, or sugars is essential for bacterial survival. In Gram-
negative species, transfer of these molecules across the outer membrane is mediated by protein
channels, called porins.24 In addition, to allowing important nutrients through, these structures can be
used by some antibiotics, such as -lactams, aminoglycosides or fluoroquinolones, to cross the outer
membrane barrier and access the interior of the cell.24–27 The simplest way for bacteria to protect
themselves from these antimicrobials is to limit their membrane permeability. There are many examples
of this in nature with Pseudomonas spp. offering some of the most impressive ones. P. aeruginosa , an
increasingly critical Gram-negative pathogen, has an intrinsically less permeable outer membrane (up
to 100-fold less permeable than Escherichia coli), which makes it naturally less susceptible to harmful
substances.25 In addition, several Pseudomonas species exhibit deficiency in OprD, a small but specific
porin, which has been shown to be the major route for imipenem uptake; imipenem is a -lactam of
last-resort.28,29 The formation of biofilms, complex structures where the cells are encapsulated by a
robust extracellular matrix, also decreases bacterial permeability as it prevents drugs from reaching the
cell membrane; P. aeruginosa biofilms, especially in cystic fibrosis patients, are known to cause severe
problems during antibiotic treatment.25,30,31
1.3.1.2 Removal of toxic substances
In cases where antibiotics enter the bacterial cell, other mechanisms are in place to ensure survival. One
of the most widespread resistance mechanisms is the expulsion of chemicals from the periplasm and
the cytoplasm through the action of efflux pumps.32 These complex, and often multi-protein, molecular
machines are present in both prokaryotic and eukaryotic organisms and ensure the removal of metabolic
side products and toxic molecules from the cellular interior using either ATP hydrolysis or chemical
gradients for energy.33–35 Their broad substrate scope means that efflux pumps can carry out removal of

23
a wide range of antibiotic classes, which leads to multidrug-resistant phenotypes and can result in
treatment failure.32 Further, overexpression of efflux pumps is commonly observed in pathogenic
bacteria, including P. aeruginosa and Mycobacterium tuberculosis, which further exacerbates antibiotic
resistance.36,37
To date, efflux pumps have been categorised into five main super-families: the ATP-binding cassette
(ABC) family, the major facilitator superfamily (MFS), the multidrug and toxic compound extrusion
(MATE) familyi, the small multidrug resistance (SMR) family and the resistance-nodulation-division
(RND) family (Figure 1.1).24,38 Most efflux pumps are chromosomally encoded; however specialised
machineries have been found on large AMR plasmids, often in conjunction with other AMR
determinants.37,39
The most studied efflux system in bacteria is the tripartite E. coli RND pump AcrAB-TolC, composed
of the periplasmic component AcrA, the cytoplasmic membrane protein AcrB, and the outer membrane
β-barrel TolC; all three components are essential for pump function.49 Its overexpression is commonly
observed as a direct response to antibiotic stress and leads to increased levels of intrinsic resistance.50,51
This prevents the use of entire antibiotic classes, as observed by the major contribution of AcrAB-TolC
to macrolide resistance in E. coli.21,52 Similar phenotypes are observed in other species, such as P.
aeruginosa, which expresses the MexAB-OprM RND pump or K. pneumoniae, which expresses the
AcrAB and KexEF systems.32,53,54 Notably, many organisms encode more than one efflux pump; their
function can be specific, whilst at the same time, redundancy between efflux systems can also be in
play.
i The MATE family of efflux pumps can be subdivided further into DinF, NorM and eukaryotic pump subfamilies that attain the same overall
fold but exhibit great variety of substrate binding sites.157

24
Figure 1.1 Examples of the five main efflux pump superfamilies. (A) The cytoplasmic ABC pump Sav1866 from
Staphylococcus aureus, whose activity is stimulated by doxorubicin and vinblastine (anti-cancer therapeutics).40 PDB code:
2HYD. (B) The tripartite ABC pump MacAB-TolC from E. coli, responsible for resistance to macrolide antibiotics38; it is
composed of a TolC trimer, MacA hexamer and MacB dimer. Removal of MacAB from Salmonella typhimurium leads to
increased susceptibility to oxidative stress and loss of virulence in mice.38,41 PDB code: 5NIK.42 (C) The MFS efflux pump
MdfA from E. coli, shown to remove chloramphenicol.38,43 Another the E. coli MFS pump, EntS, has been linked to secretion
of the siderophore enterobactin.38,43,44 The MFS group is the largest and most diverse family of efflux pumps.38 PDB code:
4ZOW. (D) The MATE pump NorM-VC from Vibrio cholerae, conferring resistance to tigecycline, a last resort antibiotic for
methicillin- and vancomycin-resistant S. aureus.45 PDB code:3MKT. (E) The SMR pump EmrE from E. coli, responsible for
resistance against lipophilic cations.46,47 PDB code: 3B5D. (F) The tripartite RND efflux pump MexAB-OprM from P.
aeruginosa; it is composed of TolC trimer, MexA hexamer and MexB trimer subunits. Its structure is homologous to the model
RND pump AcrAB-TolC of E. coli.24,48 PDB Code: 6TA6. Figure adapted from Du et al.38
1.3.1.3 Inherent pathway variation
Intrinsic resistance mechanisms are commonly due to natural target or pathway variations in some
bacterial species. This abrogates the bacteriostatic or bactericidal effects of drugs despite active
antibiotic uptake. Pathway differentiation resulting in polymyxin B and colistin resistance has been
studied in detail for several species, such as Proteus spp., Serratia spp., and Burkholderia cepacia.7,55–
57 This type of resistance originates from variations in the LPS of the bacterial cell wall of these species.
For example, modification of the lipid A moiety of the LPS through the addition of 4-amino-4-deoxy-

25
L-arabinose (L-Ara4N) results in an overall positively charged cell envelope which leads to decreased
polymyxin binding and antibiotic resistance.57 This modification is a consequence of variation in
specific two-component systems, like PhoP/PhoQ or PmrA/PmrB, that lead to constitutive
overexpression of LPS modifying genes.57–61
In addition to outer membrane modifications, antibiotic resistance at the outer membrane can occur
through expression of diverse penicillin binding proteins (PBPs), membrane associated acyltransferases
that act as catalysts for the final crosslinking reaction in cell wall synthesis (Figure 1.2).62,63 Many
bacterial species express enzymes that introduce functional modifications to PBP substrates. These
variations are reflected in the PBP active site and can lead to abrogation -lactam antibiotic recognition
and binding. For example, the Gram-positive Streptococci express murM and murN, which catalyse the
addition of short dipeptides onto the muropeptide backbone and result in branched peptidoglycan and
penicillin resistance.63–66
Figure 1.2 Schematic of cell wall synthesis and the role of -lactam antibiotics in the inhibition of PBP-catalysed
peptidoglycan cross-linking. The bacterial cell wall is composed of alternating N-acetylmuramic/N-acetyglucosamine sugar
subunits which are cross-linked at the terminal D-alanine residue. The cross-linking reaction is catalysed by penicillin binding
proteins (PBPs) that bind the D-Ala-D-Ala moiety of the nascent NAM subunit and the diamino residue of a neighbouring
polypeptide.63 This residue (represented by ‘X’) can exhibit extensive species-specific variation; with glycine present in S.
aureus, lysine in Streptococcus pneumoniae and a diaminopimelate group in E. coli.63 -lactam antibiotics emulate the D-Ala-

26
D-Ala motif (shown in red) and covalently inhibit PBPs at their active site serine residue. Figure adapted from Zeng & Lin
and Macheboeuf et al.63,72
1.3.1.4 Chemical modification of the key pharmacophore
A large sub-set of antibiotic resistance mechanisms is driven by the binding, break down or modification
of active antibiotic compounds to effectively lower their cellular concentrations. Some of the most
extensively studied examples include the -lactamase or aminoglycoside-modifying enzymes.5 The
former is elaborated upon as an example below.
-lactamase enzymes catalyse the hydrolysis of a strained -lactam peptide bond that is key for the
activity of -lactam antibiotics. These drugs include some of the most commonly used therapeutics due
to their relatively low toxicity levels and activity against both Gram-positive and Gram-negative
species.67 Continuous research has resulted in the development and deployment of numerous -lactam
derivatives that can broadly be separated into four categories, the simplest 1st generation penicillins
(amoxicillin, ampicillin), followed by the increasingly specific cephalosporins (cefuroxime,
ceftazidime), the broad and higher-activity carbapenems (imipenem, meropenem), as well as the
narrow-spectrum monobactam compounds (aztreonam).
Despite their diversity, all -lactam antibiotics disrupt the last step of bacterial cell wall formation by
emulating the highly specific D-Ala-D-Ala moiety of the nascent peptidoglycan chain terminus. This
structural similarity results in competitive binding and interaction with the native PBPs.62 Interaction of
the strained -lactam ring with the catalytic serine of these DD-transpeptidases results in the formation
of an irreversible covalent bond which blocks the entry of native substrates, the nascent N-
acetylmuramic Acid / N-acetylglucosamine peptide subunits.5,68–70 In addition to stopping the synthesis
of new cell wall and thus preventing cellular division, the activity of -lactam antibiotics leads to
accumulation of un-crosslinked precursors prompting increased expression of autolytic hydrolases.5,68–
70 These enzymes degrade both the precursor molecules and the existing peptidoglycan layer, which
enhances the antibiotic effects and lead to further cell swelling and eventual lysis.5,68–70 Notably, the
inclusion of monobactam compounds in the list of classical -lactam antibiotics has recently been called
into question, due to their divergent monocyclic core and evidence suggesting inhibition of a different
enzyme in the cell wall biosynthesis process which, among other things, explains their extensive activity
against P. aeruginosa infections.71

27
Inactivation of -lactam compounds by bacteria poses a threat to many currently available therapies.
Although treatment failure can also arise from species-specific differences in the amounts or affinities
of PBPs, usually resistance to these compounds arises from -lactamase-mediated hydrolysis of the -
lactam moiety. Encoded on the bacterial chromosome, as well as on plasmids and transposable
elements, thousands of -lactamase enzymes have spread rapidly through bacterial populations with
their high mutational rates complicating the development of agents mitigating their activity (more detail
in section 1.3.2).67
-lactamases, first described in the 1940s in Balantidium coli by Fleming as well as Abraham & Chain,
predate the first clinical use of -lactam drugs.13,73 Since then, they have been shown to be produced by
both Gram-positive (secreted into extracellular space) and Gram-negative (excreted into the periplasmic
space) bacteria and their high diversity has given rise to the amino-acid-sequence-based Ambler
classification system (class A-D).67,74 Classes A, C, and D comprise serine-based lactamases which have
a conserved catalytic serine (Ser70) residue in their active site and are characterised by structural
similarity to the naturally-encoded PBPs.67 By contrast, class B lactamases have evolved along a
distinctly different evolutionary pathway from metallo-hydrolase enzymes and depend on the presence
of a catalytically active Zn2+ in their active site.62,67,75,76
Within the Ambler classification system, -lactamases can be further separated by their activity into
narrow-spectrum, extended-spectrum, or carbapenem-hydrolysing enzymes. Narrow-spectrum -
lactamases only hydrolyse penicillins and early generation cephalosporins, while extended-spectrum
enzymes act on most -lactam antibiotics, except for carbapenem drugs. Although most ‘modern’ -
lactamase enzymes are located on mobile genetic elements and can break down even the last-generation
β-lactams, they originate from narrow-spectrum chromosomally-resident species which were acquired
by pathogens through horizontal gene transfer and have mutated into broader-spectrum hydrolases.
Examples of such archetypical chromosomally-resident enzymes include SHV family of enzymes from
K. pneumoniae, capable of hydrolysing penicillin and ampicillin or AmpC from P. aeruginosa which
hydrolyses early-generation cephalosporins.24,77,78
Notably, intrinsic resistance mechanisms are not strictly limited to inherently-resistant species, and
many of them can be attained through mutations (for example porin gene mutations) or acquired via
horizontal gene transfer (discussed further in section 1.3.3). As a matter of fact, the ensemble of intrinsic
resistance mechanisms creates a basis for the multi-factorial way in which antimicrobial resistance can

28
spread and evolve, as can be seen by the multitude of resistance determinants encoded by clinical
isolates of highly-resistant organisms like P. aeruginosa or Stenotrophomonas maltophilia.28,79–81
1.3.2 Acquired resistance
Acquired antimicrobial resistance develops in susceptible strains over time and in response to exposure
to an antimicrobial agent. Although resistance to most antibiotic classes is becoming increasingly
prevalent, the rate of development depends strongly on the target of the antibiotic compound. For
example, resistance to a single-target antibiotic, such as rifampicin, occurs more easily than mechanisms
of resistance to antimicrobial compounds with greater scope. In all cases, however, resistance can arise
through two main routes: a) incorporation of external genetic material from an already-resistant strain,
and b) mutation(s) within the bacterial genome.
1.3.3 Foreign DNA acquisition
Apart from cell division, horizontal gene transfer (HGT) is the most important mechanism for
acquisition of DNA both within and in between bacterial species.82 The majority of DNA acquired
through this route has either no or negative effect on the receiving organism. However, a small
percentage of the acquired material results in gain of beneficial traits, which subsequently become
vertically propagated within a bacterial population.22 Most retained genes drive a simple function, like
the production of a single antimicrobial resistance determinant (for example a -lactamase enzyme),
which may come with its own set of regulatory components to ensure expression.22,83,84 Successful DNA
incorporation events rarely involve central cellular pathways, like transcription or translation, as these
vary substantially between species and any “swaps” would be detrimental to bacterial fitness.83 There
are three main mechanisms driving HGT, which commonly occur in environments where toxic-
compound-producing organisms co-exist with non-producing species: a) transformation, b)
transduction and c) conjugation.
1.3.3.1 Transformation
Natural transformation is a mechanism for external DNA uptake in many bacterial species, although its
contribution to antimicrobial resistance in clinically-relevant bacteria is limited.22 Overall,

29
transformation occurs when a competent receiving strain uptakes DNA from its environment under
normal growth conditions, and subsequently integrates the acquired genetic material into its own
chromosome.83 Unlike Neisseria gonorrhoeae, whose competence appears to be constitutively active,
the majority of bacteria develop competence transiently to achieve this process.22,85 This usually occurs
in response to starvation to specific nutrients, increased cell density or environmental pressures like
temperature.86 This mechanism of HGT is remarkably widespread in the bacterial world, including
Gram-positive (Staphylococcus spp., Streptococcus spp.) as well as Gram-negative (Helicobacter spp.,
Pseudomonas spp.) human pathogens, and it is clear that it contributes greatly to bacterial evolution.22,86
The primary requirement for transformation is the presence of extracellular DNA in the surrounding
environment. Active excretionii, from species such as Acinetobacter calcoaceticus, Bacillus subtilis87,88
or P. aeruginosa87,89 and passive release from decomposing or disrupted cells and virus particles, can
lead to a highly varied mixture of genetic information, ranging from short linear fragments to fully
circularised plasmids. Although the majority of the DNA is likely to be damaged or decomposed by
environmental enzymes, it has been shown that large plasmids and chromosomal DNA can remain
intact for several hours and small plasmids and linear fragments for even longer time periods.90 Non-
covalent binding of DNA to cell surface receptors promotes translocation across the membrane in a
sequence-dependent (Haemophilus influenzae or N. gonorrhoeae) or independent (B. subtilis or S.
pneumoniae) manner.83,86,91 During the DNA import process the double-helix structure of DNA needs
to unwind; only a single strand of DNA is imported.92 This means that plasmid DNA needs to be either
integrated into the chromosome or reconstituted to generate a form that would successfully replicate.
Due to this, more than one copy of plasmid DNA usually needs to be taken up to achieve successful
transformation.83 While plasmid DNA is often self-sufficient, linear fragments of chromosomal DNA
must be incorporated into the chromosome of the recipient cell to confer any function. Mechanisms that
allow this integration will be discussed in more detail in section 1.3.3.4.
ii While active secretion of DNA into the environment may eventually lead to transformation, its role has been implicated in other processes,
such as the formation in biofilms in P. aeruginosa.30

30
1.3.3.2 Transduction
A less common route for DNA acquisition is mediated by phages; these are bacteria-specific viruses
which can be divided into two types, virulent and temperate.82,93,94 The maturation and release of virulent
phages causes host-cell lysis leading to DNA fragmentation.95 Some of this DNA can then be integrated
into the phage head and passed on to a new host during the next infection cycle.93,95,96 This process is
called generalised transduction, reported for example in the case of the P1 phage in E. coli, and results
in a non-specific dissemination of genetic material.95 By contrast, temperate phages insert into the
bacterial chromosome without killing their host. In this case, host DNA incorporation into the virus
occurs due to incorrect excision of the prophage from the bacterial chromosome.93 This results in the
inclusion of surrounding bacterial genes, as observed in phage λ in E. coli which has been shown to
package only genes from the neighbouring galactose or biotin metabolism pathways.97,98
Useful genetic determinants increasing virulence or conferring antimicrobial resistance can be acquired
through transduction. For example, pathogenicity islands in S. aureus have been shown to be excised,
replicated, and integrated into phage particles upon phage infection.93,99 This leads to dissemination to
a range of new host cells. In V. cholerae a key virulence determinant, the cholera toxin, is encoded in
the genome of a CTXΦ phage.100 Transduction has been documented in many environmental settings,
including but not limited to the soil, marine, and fresh water environments, suggesting that it plays an
important role in gene dissemination and evolution.93,98
1.3.3.3 Conjugation
Conjugation is a highly efficient route for DNA transfer occurring at high rates in the human
gastrointestinal tract under antibiotic treatment.22 It is the most prevalent route of HGT responsible for
the spread of antimicrobial resistance and is believed to be the primary mechanism through which
development of hospital-acquired resistance occurs.22,101 In contrast to the previous two modes of HGT,
this mechanism requires two metabolically-active cells to establish cell-to-cell contact and form a
junction that allows them to successfully transfer genetic material from a donor to a receiver cell.83,86
Direct transfer between chromosomes has rarely been reported for bacterial conjugation.102 Instead
mobile genetic elements such as insertion sequences (IS), transposons, genomic islands, plasmids,
integrative and conjugative elements and miniature inverted repeat (IR) transposable elements are
efficiently transferred.22,103–105 The wide range of mobile genetic elements that are transferred through

31
conjugation, allows for gene expression without the need for plasmid reconstruction or homology-based
chromosomal integration in the recipient cells. Some of these mobile genetic elements will be discussed
further below.
Insertion sequences are the simplest and most common examples of mobile genetic elements. Their
inter-species recurrence promotes homology-based matching and efficient insertion into the
chromosome of the receiving cell.83 Classically, they encode their own transposition proteins allowing
them to move easily both within and in between bacterial chromosomes.106 With only minor exceptions,
the open reading frame is flanked by a short terminal IRs that double as transposase binding domains
and strand cleavage/transfer sites.106 The transposase enzyme catalyses the generation of a free plasmid-
like copy containing the IR and the DNA to be transposed by joining together the IS ends and forming
a transient tnpA promoter to increase insertion frequency.83 If two identical IS are present on either side
of a DNA region, mobility of the entire genetic segment is enabled leading to transposon
formation.106,107 This can lead to beneficial or detrimental effects, depending on the final insertion
location and the gene carried. For example, a mobile antibiotic resistance gene cassette can introduce a
survival advantage upon transposition and thus have beneficial effects. On the other hand, insertion of
an IS element into a gene sequence or its promoter region is likely to cause disruptions in protein
expression that may negatively affect bacterial fitness.106 Overall, these mobile genetic elements offer
a convenient route of DNA mobilization. Their effects can be seen in the dissemination of AIM-1, a
newly emerging -lactamase isolated from P. aeruginosa clinical isolates.108 Class B3 -lactamase
enzymes, such as AIM-1, have long been believed to be immobile environmental enzymes. The
characterisation of AIM-1 has demonstrated the presence of ISCR stability elements that have enabled
this enzyme to be mobilised from the genome and transferred between P. aeruginosa strains.108,109 This
case demonstrates how powerful HGT is for the evolution of bacterial populations and the spread of
antimicrobial resistance.
Plasmids are circular mobile genetic elements that are commonly gained through natural transformation
and conjugative transfer and often encode virulence, detoxification or antibiotic resistance genes which
can function autonomouslyiii.110,111 Plasmids transferred via conjugation are acquired fully functional,
bypassing the need for their reconstitution.83 All plasmids contain a replicon region which encodes the
iii Plasmids can also be incorporated into the genome; however, they tend not to be maintained as such for long.110,111

32
origin of replication (ORI) as well as the relevant control elements. The ORI is an AT-rich DNA
sequence from which replication ensues; it determines the copy number, the stability and the
compatibility of the plasmid with its host.110 Despite their ‘independence’, plasmids rely on host
machinery for replication. This means that the resources for replication are limited and any genes
encoded will generally have to be beneficial for both the plasmid and the host in order to be maintained
over time.110,112 Bacterial cells can take up and maintain more than one plasmid at a time if the genes
encoded confer a strong selective advantage to make up for any decrease in fitness caused by their
carriage. However, sharing of the available resources also means that plasmids utilising the same
replication system cannot co-exist in the same cell. Over time, the propagation of one plasmid leads to
the dilution of the other until one is lost from the population.110 Notably, plasmids responsible for
spreading antimicrobial resistance genes often encode their own conjugative machinery, something that
makes their replication and maintenance more robust.101,113
1.3.3.4 DNA integration into the chromosome
The spread of antimicrobial resistance via HGT requires DNA stabilisation (plasmid reconstitution) or
its incorporation into the chromosome of the host (linear fragments of DNA cannot replicate on their
own). This can be achieved in a homology-dependent or independent manner. Homologous
recombination can occur in cases where the donor and the recipient share high sequence identity, as is
the case between members of the same genus or species.83 Studies show that the average maximum
divergence for successful recombination through this mechanism is approximately 25%.83 This route,
unlike many others, ensures that the size of the recombined region remains unchanged and, if integrated
successfully, fully functional.83 In stark contrast, homology-independent recombination, observed for
example in E. coli, occurs via double-strand DNA breaks enabling risky fragment insertions into the
genome.114 As this procedure requires blunt-end joining of DNA fragments it can lead to detrimental
effects due to incorrect recombination. These two mechanisms can also come together in the case of
additive integration. In this instance, two circular DNA molecules, or one circular molecule or linear
fragment and the chromosome integrate together.83,115,116 For this to happen short, high-similarity
regions overlap enabling integration. However, total similarity remains low and leads to new DNA
addition, exchange or even to host DNA deletions. This allows the acquisition and integration of DNA
from phylogenetically distant species.

33
Finally, DNA integration can be achieved by integrons, site-specific recombination systems encoding
an integrase enzyme responsible for incorporation of external DNA into its own binding site.103 An
upstream promoter ensures gene expression, which allows the bacterium to ‘test’ the new DNA for
function.103 In addition, many of these systems carry a LexAiv-binding site nearby, thus allowing the
SOS pathway of the host to influence genome mobility and DNA integration frequency.117 As such,
under stress conditions gene mobility and integration may be upregulated in an attempt to increase the
chance of beneficial gene acquisition. Integrons have classically been divided into two types,
chromosomal ones responsible for a build-up of long-term genome complexity, and mobile ones
associated with integration of plasmids and transposons.118 A gene cassette can be recruited into an
integron of any of the two types; self-mobilisation then makes it dependent on other mobile genetic
elements and associated transposase genesv. Nonetheless, it is the mobile integrons that are mostly
linked to inter-species genome penetration and development of modern antibiotic resistance in
clinically-relevant pathogenic strainsvi.22,118
1.3.3.5 Gene maintenance
The acquisition of a potential resistance gene is only the beginning of an on-going fight for bacterial
survival in the presence of antimicrobial agents. Numerous adaptive changes need to happen in the
recipient cell, and where relevant, in the donor cell to ensure proper gene expression, successful AMR
determinant maturation and future gene propagation. Overall, only a minor proportion of externally
acquired DNA remains actively maintained in the genome across multiple generations, as vertical gene
transfer is strongly dependent on gene stability as well as the net effect associated with the costs and
benefits of its upkeep. Considering this, in the absence of selective pressure large plasmids or integrons
carrying many gene cassettes are less likely to be maintained in the long term.
Though highly adaptable, the ability of bacteria to offset the fitness costs originating from the
acquisition of AMR determinants varies extensively between species. It depends, among other things,
iv LexA is a transcriptional repressor responsible for the repression of SOS genes linked to DNA repair. Its autolytic cleavage leads to the SOS
response, activated among others by the presence of -lactam antibiotics, and to the expression of the integron integrase IntI.117,118
v It can be easily imagined how the integrons’ ability to incorporate DNA can become the springboard for the development of new mobile
elements such as plasmids carrying multiple resistance genes.
vi It has been noted that the ability of integrons to cross horizontally between genomes is somewhat limited to the environmental origin of the
species as specific integron sequences vary between soil and marine environments.118

34
on habitat, nutrient availability at any given timepoint during evolution and even gene expression level
for the AMR gene of interest. While intrinsic resistance mechanisms are likely to be constitutively
active in order to afford protection in the natural environment of the resistant bacterium, acquired genes
may be more prone to stress-mediated induction and might require adaptations to cellular pathways in
order to be expressed.39 Successful development of antimicrobial resistance extends beyond the
genome’s capacity to integrate, stabilise and express the relevant genes. Key additional considerations
include the ability of cells to correctly fold and translocate the synthesised products to appropriate
cellular locations. While exact details of the processes involved may vary between species and gene
products, they will inevitably depend on the same general principles guiding protein synthesis, folding
and transport; these are discussed further in section 1.5.1.
1.3.4 Mutational resistance
The emergence and accumulation of mutational changes in genomes is at the heart of evolution and
natural selection. The high replication rates of most bacterial species allow the generation of a
considerable number of mutations that result in phenotypic changes over a short period of time. In
general, these mutations lead to similar resistance mechanisms as described above for naturally-resistant
bacteria i.e. drug target modifications, decrease in drug uptake through porin mutations, efflux
upregulation or changes to metabolic pathways and their associated regulatory networks.22 Such
adaptations can be caused by single nucleotide and amino acid changes or through more extensive
sequence deletions and insertions.119 This process often generates substitutions which decrease bacterial
fitness under normal conditions, but offer a significant survival advantage under antibiotic pressure. In
this case, clearing of susceptible cells by antibiotics will ensure the propagation of the resistant species,
and maintenance of the given mutation through vertical gene transfer.22 This is particularly relevant in
clinical environments where repetitive treatment with multiple antibiotics can exacerbate the
development of multidrug-resistant strains and lead to poor patient outcomes. Indeed, M. tuberculosis
or P. aeruginosa have been shown to be particularly efficient in developing antibiotic resistance in this
way.119 In such cases, combinatorial therapies are often employed to exploit the different modes of
action of antibiotic compounds in order to decrease the chances of mutations.
In M. tuberculosis, RNA polymerase mutations have been shown to quickly develop during rifampicin
exposure. Discovered in mid-1960s in the soil bacterium Amycolatopsis rifamycinica, rifampicin was
reported as a wide-spectrum antibiotic with strong selectivity for prokaryotic enzymes. The drug targets

35
the prokaryotic DNA-dependent RNA polymerase, RpoB, by binding to its -subunit and sterically
preventing the access of nascent RNA into its binding site, blocking RNA chain extension.120,121 In E.
coli up to 95% of the commonly observed mutations have been shown to be point mutations in the N-
terminus of RpoB, between amino acids 505 and 537, leading to decrease in binding affinity.121–123
Similarly, in M. tuberculosis all mutations occur in nucleotides encoding amino acids 419-451.121,124
Point mutants emerge at high frequencies of up to 10-8 per bacterium per cell division, and their
occurrence leads to the development of varying levels of resistance that is vertically transmitted and
can be enhanced upon further exposure to the antibiotic through accumulation.121,123 Often, the only
option to bypass this development is to use rifampicin, and its derivative compounds, only in
combination therapies, as is done in its clinical use against M. tuberculosis.
Other examples of mutational resistance are common in bacteria. For example, mutations in -
lactamase genes increasing their hydrolytic activity or the aforementioned OprD porin deficiency of P.
aeruginosa are well-reported.24,27 Of particular interest to our work is the OXA family of mobile -
lactamases consisting of over 750 different enzymes. All members reported so far exhibit distinct
hydrolytic profiles originating from a variety of amino acid mutations. Examples include the Ser73Asn
substitution, conferring extended-spectrum activity to enzymes in the OXA-7 family and the Gly157Asp
substitution which leads to high-level of resistance to ceftazidime.125
In summary, bacterial exposure to different stressors and environmental factors results in the activation
of variety of stress-response cascades which drive the differential induction of a wide range of survival
mechanisms.39 These can lead to the development of varying levels of drug resistance, as seen in the
cases of RpoB-mediated rifampicin resistance in M. tuberculosis or the non-enzymatic aminoglycoside
resistance in P. aeruginosa.126 The exact level of resistance to antibiotics and the location of the bacterial
infection will ultimately determine the choice and efficiency of antibiotic therapy, as even resistant
strains may be cleared by a drug that they are resistant to if the bacterial load and the bioavailability
that can be achieved in the infection location of interest are favourable. Susceptibility breakpoints tables
have been developed to correlate these in vivo considerations with the results of in vitro studies to guide
clinicians and scientists in determining the best strategies for antibiotic therapy.22

36
1.4 OVERCOMING ANTIBIOTIC RESISTANCE BY USING ANTIBIOTIC
ADJUVANTS
The development and spread of antimicrobial resistance have been exacerbated by extensive, and
sometimes inappropriate, use of antibiotic compounds in clinical and agricultural settings. Common
antibiotic strategies that target essential bacterial pathways generate strong selective pressure and lead
to rapid emergence of antibiotic-resistant strains.18 Combined with the limited success in the
development of novel antibiotics during the past 60 years, this increase in antimicrobial resistance
creates an urgent need for the discovery of next-generation antibacterial strategies, as well as approaches
that sensitise resistant pathogens to existing antibiotic compounds. With the hope of exerting minimal
selective pressure, an increasingly adopted approach is to target pathways which are not essential for
bacterial survival in the absence of the antibiotic compound of interest. Several examples of such
adjuvant approaches are currently in consideration, whilst some are already in use in clinical therapy.
These can be broadly divided into four types, based on the use of a) resistance enzyme inhibitors, b)
membrane-permeabilising compounds, c) efflux pump inhibitors, and d) horizontal gene transfer
blockers.37,101,127
1.4.1 Inhibition of antibiotic modification
Due to the importance of -lactam antibiotics, routes to disable -lactamase activity have been
extensively investigated, and -lactamase inhibitors form the biggest class of AMR breakers to
date.67,75,76 The development of such compounds is challenging because of the vast chemical and
structural diversity of known -lactamase enzymes. Nonetheless, several inhibitor compounds have
been deployed and can achieve bacterial clearance when co-administered with -lactam antibiotics,
such as clavulanic acid which is routinely co-administered with amoxicillin.62
Clavulanic acid, the first clinically used -lactam inhibitor, is a -lactam derivative that, on its own,
exhibits poor bactericidal properties (Figure 1.3).128,129 It is commonly used in combination with
amoxicillin or ticarcillin to irreversibly inhibit serine -lactamases; its -lactam moiety forms a stable
acyl-enzyme complex with the catalytic Ser70 and blocks the hydrolytic action of the enzyme.37
Although the use of clavulanic acid is a definite success, the high mutational capacity of -lactamase
genes resulted in rapid emergence of adjuvant-resistant strains that prompted the search for additional

37
inhibitors.130 Modification of the oxazolidine in the heterocyclic core of clavulanic acid gave birth to
the sulfone-based sulbactam and tazobactam inhibitors, and expanded our capacity to inhibit -
lactamase activity in highly resistant Gram-negative pathogens, such as Pseudomonas, Acinetobacter,
or Klebsiella spp.37,131 It is interesting to note that both compounds show very distinct activity profiles
despite their similar structure. Sulbactam, for example, exhibits species-specific inhibition of
Acinetobacter spp., while piperacillin-tazobactam combination shows more extensive inhibitory
activity of more Gram-negative pathogens than the cefoperazone-sulbactam combination.37,131,132
Despite the apparent mechanistic similarity of class A and C serine--lactamases, class C enzymes are
particularly resilient to inhibition by the aforementioned classical -lactamase inhibitors. This is due to
an active site variation that enables them to hydrolyse the ‘inhibitory’ serine peptide bond with these
compounds.133,134 A novel non--lactam inhibitor, avibactam, has been shown to substantially decrease
the resistance levels of class C -lactamase-carrying bacteria, and ceftazidime/aztreonam are approved
for use in complicated intra-abdominal and urinary tract infections in combination with this adjuvant
(Figure 1.3).62,133,135,136 Unlike classical inhibitors, avibactam, acts as a semi-reversible inhibitor by
stabilising the carbamoyl complex at the active site and promoting re-cyclisation rather than hydrolytic
release from the active site.134,137 This means, that the inhibitor concentration is not depleted over time
allowing on-going competitive inhibition of the -lactamase enzymes.
Figure 1.3 Clinically available -lactamase inhibitors target class A, C and D -lactamases. Clavulanic acid, sulbactam
and tazobactam emulate the key -lactam ring of -lactam antibiotics and are effective, primarily, against class A and D -
lactamase enzymes. Novel inhibitors avibactam and vaborbactam have been developed to target class C and KPC-type -
lactamases, respectively; these enzymes therapeutically challenging targets.37

38
Another novel inhibitor, vaborbactam, a boronic acid transition state compound, was also recently
approved for use in complicated urinary tract infections as a co-treatment with meropenem.37,138,139 Its
novel mode of action makes it particularly suited for use against KPC-producing Enterobacteriaceae,
but unsuitable for use with A. baumannii or P. aeruginosa.138
Despite the non-lethality of these inhibitors, clinical use in combination with antibiotics has already
been shown to cause resistance development, seen for example with the emergence of mutated KPC-3
(Asp179Tyr/Thr243Met) -lactamase in K. pneumoniae clinical isolates.140 Further, class B metallo--
lactamase enzymes remain unaffected by inhibitor compounds, due to their unique and divergent
hydrolytic mechanism that depends on the use of Zn2+ ions; they are thus of increasing clinical
concern.62
As -lactamase resistance is widespread across pathogenic species, some of the most worrying
representatives of bacterial pathogens, such as K. pneumoniae, Enterobacter cloacae or M. tuberculosis,
are treated with aminoglycoside compounds such as streptomycin, gentamicin or tobramycin.141
Aminoglycoside antibiotics inhibit protein synthesis by targeting the ribosome and strong synergistic
behaviour with other drug classes has been reported.141,142 Structural modification to these antibiotics is
carried out by the often mobile aminoglycoside N-acetyltransferase (AACs), aminoglycoside O-
nucleotidyltransferase (ANTs) or aminoglycoside O-phosphotransferase (APHs) enzymes.143 These
minimise the aminoglycoside binding by respectively acetylating, adenylating, or phosphorylating the
hydroxyl or amine groups.141,143,144 Several strategies for targeting these modifying enzymes have been
suggested and include the repurposing of current protein kinase inhibitors or the synthesis of substrate
and peptide-based analogues. However, no compounds have reached the clinic to date.37
1.4.2 Membrane permeabilising compounds
Decreased membrane permeability affects the treatment of many Gram-negative infections. In addition
to other antibiotic resistance mechanisms, pathogens, such as P. aeruginosa, commonly mutate or
downregulate the expression of transmembrane -barrel porin structures to further non-selectively
exclude small molecules.28,145

39
Polymyxin compounds, like colistin, are key molecules, used to disrupt the Gram-negative outer
membrane. These compounds interact with LPS-associated cations and permeabilise the outer
membrane to facilitate the entrance of small molecules, including antibiotics, into the cell.146 This
strategy has enabled the sensitisation of several ESKAPE pathogens to previously ineffective drugs,
like azithromycin.37,147 The polymyxin colistin is a special case in AMR inhibitors, as it exhibits
antibiotic function and is also used as an antibiotic drug on its own. Despite significant nephrotoxicity,
colistin use is on the rise in an attempt to counteract the loss of other antibiotic classes due to
resistance.148,149
Interestingly, truncated versions of polymyxin antibiotics retain their permeabilising activities and these
compounds could make suitable antibiotic adjuvants.37,150The polymyxin B nonapeptide synthesised by
Vaara et al. was shown to sensitize resistant K. pneumoniae, P. aeruginosa and S. typhimurium strains
to different classes of antibiotic compounds, including the -lactam ampicillin, and the Gram-positive
drug vancomycin.37,151 Research into less cytotoxic derivatives of these cationic peptides has yielded
several adjuvants that are currently in clinical trials and might open new avenues for antimicrobial
peptide strategies.37
Antimicrobial peptides, natural or synthetic, encompass a large family of multipurpose scaffolds that
exhibit promising in vitro activities, while synergising with many clinically used compounds.152,153 As
such, strategies for the use of organic acids, cholic acids, and polyethyleneimines have been proposed;
their clinical use, however, is complicated by a number of factors including lack of in vivo activity, low
specificity and high cytotoxicity.154,155
Overall permeabilising compounds are a promising strategy that could help increase the intracellular
concentrations of existing antibiotics while also potentially exhibiting antibiotic function when used in
isolation. Most importantly, targeting the outer membrane may open the use of compounds previously
unsuitable for the treatment of Gram-negative species due to their exclusion by the cell wall, as is the
case with the antibiotic vancomycin.

40
1.4.3 Inhibition of drug efflux
Inhibition of prokaryotic efflux has long been attempted to prevent antibiotic removal, following the
passage of drugs through the outer membrane. However, development of both active and specific
compounds against efflux is challenging due to the innate role of efflux pumps in the expulsion of
metabolic side-products, which is conserved in both prokaryotic and eukaryotic species.156 Strategies
proposed to date involve energy decoupling, steric inhibition or manipulation of pump expression.37,156
Small-molecule and peptide-based inhibitors designed to enable steric blocking of efflux pumps are
collectively known as EPIs, efflux pump inhibitors.156 Numerous alkaloid, flavonoid, polyphenol,
quinolone, aryl and heterocyclic derivatives have been developed and tested for activity, and while
many promising hits have been identified, cytotoxicity or specificity issues have, in most cases,
prevented their clinical use.37,156 A notable exception is verapamil, a small molecule channel blocker
used in the treatment of hypertension which has been approved for the use in M. tuberculosis infections,
where it competitively inhibits the activity of the multidrug and toxin extrusion pumps.156–159
Similar competitive inhibitors to verapamil have been extensively investigated and yielded several
promising avenues of research. Two representative compounds are the carbonyl cyanide-m-
chlorophenylhydrazone (CCCP) that disrupts the proton motive force and PAN, which competitively
inhibits the efflux activity of RND pumps.160,161 Through different modes of action, both compounds
result in sensitisation of critical Gram-negative pathogens to previously ineffective antibiotics; CCCP
sensitises Klebsiella spp. to tetracycline, whilst PAN allows the use of erythromycin and
chloramphenicol on resistant P. aeruginosa.161,162 Additionally, CCCP exhibits synergistic effects with
a range of other antibiotics, including carbapenems, resulting in metabolic arrest.156,160,163
A slightly different approach for targeting efflux resistance uses peptide mimics of nucleic acids and
synthetic DNA molecules to downregulate or block the expression of essential pump components. This
has led to the sensitisation of Campylobacter jejuni strains to ciprofloxacin and erythromycin.37,164
While successful, this method requires a thorough understanding of the transcriptional and translational
regulation behind efflux pump expression, which is likely to be species and even strain specific. As
such, although in principle effective, it is unlikely to be suitable for the development of broad-acting
inhibitor compounds.

41
Overall, broad-acting breakers of antimicrobial resistance, such as -lactamase or efflux inhibitors
represent attractive treatment. However, given the low percentage of successful clinically used
inhibitors, in comparison to the number of compounds in development, new strategies may need to be
considered for the future. The use of repurposed drugs or modified antibiotic substrates and covalent
inhibitors may allow the exploitation of this approach to generate next-generation broad-acting
resistance breakers.
1.4.4 Inhibiting the spread of antimicrobial genes
Conjugation of AMR-gene-carrying plasmids is the most prevalent mechanism for the spread of
resistance determinants in bacterial populations.101,165 Out of the 28 conjugative plasmid types
characterised to date, four families have been strongly linked to the transmission of AMR genes.101,165
Their presence is particularly notable in Enterobacteriaceae, where these large, low-copy and self-
transmitting mobile elements encode multiple resistance determinants including carbapenemases and
extended-spectrum -lactamases (ESBLs).101,127 Further, unlike small-sized mobile elements that are
easily lost in the absence of selective pressure, these ‘AMR plasmids’ appear to be maintained for
extended time periods in E. coli.127,166,167 Thus, inhibition of the spread or maintenance of these gene
pools is a potentially promising approach to mitigate the spread of antimicrobial resistance.82 Inhibition
of plasmid conjugation can, theoretically, be achieved at different levels: a) through inhibition of
pathways in the recipient cell or donor cell, and b) through inhibition of the conjugation machinery.
The generation of inhibitors targeting either the recipient or donor cells is challenging, due to the variety
of recipient-donor combinations possible. However, inhibitors targeting plasmid maintenance
mechanisms open the possibility of clearing donor cells of pre-existing plasmids or, alternatively,
preventing the stabilisation and maintenance of newly acquired plasmids in the recipient cells.127,167,168
These approaches are reminiscent of the current practice of perioperative antibiotics use. Targeted
design of conjugative ‘interference plasmids’ that exploit the ori incompatibility of many closely related
plasmids resulting in plasmid loss in the population, has been successfully applied in vitro and in mouse
models by Kamruzzaman et al.167 While clinical use of these plasmids is unlikely, it provides an
interesting tool for laboratory use and acts as a proof of principle. More relevant is the identification of
two already existing anti-HIV drugs, abacavir and azidothymine, in a medium throughput screen; these
prevented the transmission of two AMR plasmids isolated from E. coli and K. pneumoniae.127 While

42
these two compounds offer species- and plasmid- specific protection, further development may improve
their scope.127
Finally, conjugation can be interrupted through the inhibition of the conjugative machinery, such as the
relaxosome and the type IV secretion system. Several strategies have been employed in the literature,
like the use of intracellular antibodies to target the relaxase or ATP inhibitors against the secretion
system or its chaperones.101,169–172 These anti-HGT approaches are similar to the concepts driving the
development of novel anti-virulence strategies that target non-essential bacterial pathways responsible
for pathogenicity in an attempt to minimise selective pressure while potentially allowing bacterial
clearance. In addition to stringent and rational therapy design, these approaches can help maximise
treatment success and minimise chances of resistance development for challenging resistant pathogens.

43
Part B
Protein Folding in the Cell Envelope

44
1.5 THE BACTERIAL CELL ENVELOPE
The bacterial cell envelope is a complex cellular compartment that protects the cytoplasm from external
stresses. Its composition depends on the bacterial species and its natural environment, nonetheless two
main structural arrangements are largely recognised: the Gram-positive cell envelope, mostly composed
of a thick peptidoglycan layer, and the Gram-negative cell envelope, where an aqueous compartment
along with a thin peptidoglycan cell wall are sandwiched between the inner and outer membranes.
Most of the Gram-negative cell envelope is taken up by the highly oxidative periplasm. This soluble
fraction houses a variety of pathways that safeguard the overall stability of the envelope and ensure the
optimal function of its components.173 Periplasmic chaperones and proteases play an essential role in
managing proteostasis in this part of the cell, either by directing proteins to their final destinations or
by managing misfolding and aggregation events.174–177 Although the activity of the three major
representatives of these folding catalysts, SurA, Skp and DegP, shows a level of functional redundancy,
combined losses (SurA/Skp or SurA/DegP) are not tolerated and result in deleterious effects and
decreased bacterial survival. The widely conserved DegP protease/chaperone, for example, is
responsible for the degradation of a range of membrane-associated and oxidatively-damaged proteins.
It exhibits a temperature-sensitive protease/chaperone activity, with its chaperone activity prevalent at
low temperatures (28°C) and its protease activity appearing at higher temperatures and becoming
essential for survival at temperatures above 42°C.178,179
At the interface between the periplasm and the outer membrane resides a protective layer of rigid
peptidoglycan that is linked to the outer membrane by murein proteins, and that determines the shape
of the cell.5,11 Finally, the outer membrane itself is composed of a lipid bilayer, where outward facing
glycolipids act as a charge exclusion barrier and size-restricting -barrel porin structures, such as OmpF
or OmpC, control the transport of most biomolecules.12 An additional single layer of protein, the S-
layer, is present in some species and encapsulates the cell offering further protection. Overall, the cell
envelope evolved to allow the selective passage of nutrients and waste to and from the cytoplasm,
respectively, to safeguard the cytoplasmic content while ensuring structural integrity of the cell.

45
1.5.1 Protein folding and transport into the cell envelope
Protein folding refers to the ensemble of cellular processes by which proteins attain their biologically
stable and functional three-dimensional structures. As mRNA molecules are translated into nascent
polypeptides by ribosomes in the cytoplasm, co-translational folding driven by non-covalent
intramolecular forces, such as hydrophilic, hydrophobic and ionic interactions or hydrogen bonds,
begins at the N-terminus.180 Protein folding is generally an energetically favourable process, which
often occurs spontaneously; the majority of initial folding interactions are both enthalpically and
entropically favourable and follow the thermodynamic gradient towards the lowest energy
conformation. The three-dimensional topology of approximately 70% of all protein amino acids is
governed by hydrogen bonding and consists of highly regular α-helices, β-sheets, and, less commonly,
- or -loops. Interplay between these discrete structural elements leads to the collapse of hydrophobic
regions to the interior of the protein molecule and the rearrangement of hydrophilic regions to coat the
external surface of the forming structure. Interactions between non-polar protein regions and their
surrounding aqueous environment are, thus, minimised to decrease the total energy of the structure.
Achievement of the correct folding intermediate is critical for the protein’s continued existence at this
stage; misfolding in the secondary and super-secondary structure often leads to costly protease-
mediated degradation to prevent toxicity caused by accumulation and aggregation of misfolded
proteins.180–182
Although most protein amino acids can spontaneously self-arrange into highly ordered states, a lack of
strong secondary structure is generally observed in catalytically active sites. As enzymatic activity is
often controlled by amino acids that are polar or charged in nature, their close spatial proximity causes
steric strain resulting in loss of structural regularity.181,182 Thus, unusual kinks and angles in amino acid
backbones result in above-average representation of random coils in the active-site regions.181,183,184 In
addition, key residues are commonly located within, or in proximity to, hydrophobic pockets to
maximise productive van der Waals surface interactions between an enzyme and its substrate. Given
the exposure of active sites to the surrounding aqueous environment, interactions at these non-polar
surfaces increase the total energy of the system.181 Therefore, high thermodynamic favourability in the
rest of the protein fold is essential to offset these inherently unfavourable interactions and achieve
overall protein stability and functionality. This delicate balance can be disrupted even by a single change
in the primary amino acid sequence giving rise to stability-function trade-offs. Trade-offs of this nature
underpin the ability of enzymes to evolve improved functions as seen, for example, with the effects of

46
single amino acid substitutions on the stability or hydrolytic activity of several class D OXA-family -
lactamase enzymes.181,183,185
While all proteins are synthesised in the cytoplasm, acquisition of their native folds may not be achieved
until their target destination has been reached. Often, protein folding depends on the action of specific
post-translational adaptation machinery, incorporation into a membrane, formation of a higher-order
protein complex, or simply translocation across the cytoplasmic membrane. A key role in all these cases
is performed by cytoplasmic chaperones that slow down or entirely prevent spontaneous protein folding
during and immediately after mRNA translation. These helper molecules trap partially folded protein
intermediates, prevent their misfolding and aggregation or help mediate their post-translational
transport. In Gram-negative bacteria, approximately 20-30% of all expressed gene products is destined
to perform key functions in the bacterial cell envelope, such as swimming and twitching motility,
nutrient transport, or protection of the cell against mechanical stresses and antimicrobial agents. It is
thus essential that all of these protein components efficiently traverse the inner membrane through co-
or post-translational protein translocation pathways.186,187
1.5.2 Protein transport across the inner membrane of Gram-negative bacteria
The bacterial inner membrane serves as a selective barrier that separates the cytoplasm from the
periplasm. In the absence of defined organelle-like structures in bacteria, this divider provides a
platform for all membrane-associated reactions and functions, including energy production, lipid
biosynthesis, and protein secretion and transport. Given the harsh conditions of the extra-cytoplasmic
environment, and the segregation of potentially harmful degradative enzymes such as RNAses or
phosphatases in the intermembrane space of the cell envelope, membrane transport is a tightly
controlled process.188 Polypeptides that are required to cross the inner membrane carry a characteristic
24 to 30 amino-acid-long N-terminal sequence; this signal peptide is composed of a positively-charged
N-terminus, a hydrophobic -helical core (h-region), and a C-terminal domain with a signal peptidase
cleavage site (exported proteins) or a hydrophilic region (transmembrane proteins).188,189 The nature of
the signal peptide along with the post-translational folding state of the protein, determine the
translocation pathway that mediates the peptide’s transport outside the cytoplasm.
The general secretory pathway (Sec) selectively transports unfolded proteins across the inner membrane
during translation, or integrates them into the inner membrane, immediately after they are translated.188

47
Most transmembrane proteins are integrated into the membrane in a co-translational fashion. This helps
prevent their release from the ribosome and minimises unfavourable interactions with the aqueous
cytoplasmic environment that could result either in aggregation or in errors in signal peptide
recognition. Ribosome-bound signal peptide and its signal recognition particle associate with the signal
receptor at the membrane. In turn, the signal receptor hydrolyses GTP to transfer the signal peptide into
the SecYEG translocase channel.187,190–193 This process depends heavily on the hydrophobic h-region in
the signal peptide which drives the efficient interaction of the signal peptide with its signal recognition
particle.187,190–193 Continuing polypeptide synthesis from the ribosome, pushes the unfolded polypeptide
further into the SecYEG channel. The subsequent integration of the protein into the inner membrane
has been shown to be mediated by the membrane chaperone YidC and the ensemble of
hydrophobic/hydrophilic interactions between the h-region of the signal peptide, the transmembrane
helices of the folding protein, the hydrophobic domains of the Sec translocase, and the membrane
itself.187,189,194–197
In contrast, soluble periplasmic proteins are usually transported across the inner membrane post-
translationally. In E. coli, the cytoplasmic chaperone SecB has been shown to direct substrates to the
translocation channel through its interaction with the Zn2+ coordination site of SecA.187,188,198 SecA, a
membrane associated ATPase, drives polypeptide translocation across the membrane. Notably, SecA
interacts with both the C- terminal and the N-terminal parts of the SecYEG channel and this binding is
thought to be responsible for the opening of the channel to the polypeptide chain.187,199 The signal
peptide is looped through the channel such that the N-terminus of the polypeptide remains at the
cytoplasmic side of the membrane while the C-terminus enters into the periplasm.187,188,200 Consecutive
ATP-powered insertion and de-insertion cycles of SecA generate a push-and-slide movement of the
polypeptide across the membrane.187,188,200 Throughout this process, the permeability of the membrane
remains unperturbed due to the polypeptide plugging the SecYFG channel.187,201 To this end, the
association of SecYEG with the SecDF complex has been shown to prevent polypeptide backsliding
and increase transport efficiency in vivo.187 Interestingly, the prokaryotic Sec translocation pathway
shares many similarities with the Sec61/62/63 membrane complex and the associated BiP ATPase of
the endoplasmic reticulum (ER) in eukaryotes, and has only rarely been retained in mitochondria.188,202
The second most common inner membrane protein translocation system in prokaryotic cells is the twin-
arginine translocation (Tat) pathway.203 In E. coli this pathway has specialised to transport folded and
co-factor binding proteins that are too large to pass through the Sec channel and require a larger complex

48
assembly to transverse the inner membrane.186,189,204 Although these proteins represent a smaller fraction
of exported polypeptides, they are often essential in energy metabolism as they form parts of the
respiratory and photosynthetic electron transport chains.186,189,204
In E. coli proteins are targeted to the TatABC translocase-docking complex by a 30 amino acid signal
peptide adapted to avoid the Sec machinery through the inclusion of an essential twin arginine (RR)
motif, a less hydrophobic h-region and a positively-charged Sec-avoidance signal prior to the C-
terminal cleavage site.189,203,204 Recognition of the signal peptide by TatC and deep insertion of the
signal peptide into the protein, leads to interaction of the h-domain of the signal with TatB and exposure
of the cleavage site to the peptidase on the other side of the inner membrane.186 Notably, evidence
suggests that assembly of several TatBC complexes at the membrane allows simultaneous binding of
multiple substrate proteins.186
TatB docking and its interaction with TatC and the signal peptide drives the recruitment of TatA to the
membrane.186,203,205 TatA oligomerisation leads to the formation of large (100 to 500 kDa) multimeric
structures that are thought to translocate proteins across the membrane via two possible mechanisms,
the formation of a translocation pore or through the weakening of the membrane.186,206,207 In both
potential mechanisms, TatA assembly and protein transport are driven by the proton motive force; the
existence of an antiporter mechanism coupled to this process has also been suggested.186 Interestingly,
the Tat system is able to recognise misfolded proteins, prevent their export, and direct them to be
degraded in the cytoplasm.186,203,208–210 While the mechanism behind this activity is not fully understood,
it has been linked to co-factor insertion and thus potentially to conformational stability of the
translocated protein.186,189,204 In the case of B. subtilis, the WprA protease was shown to be essential for
protein translocation by directly interacting with the TatAyCy system; this suggests that Tat
translocases can be linked to specific localised degradation systems.186,203,211
An essential step in achieving full protein export through either the Sec or the Tat pathway is the
exposure of and cleavage at the C-terminal cleavage site when the polypeptide has reached the
periplasm. This liberates the mature protein which is then ready for further post-translational
modifications, folding and transport in the cell envelope.186,188,189,203
Sec and Tat translocation machineries exhibit distinct substrate profiles. However, protein substrates
belonging to the same protein superfamily are not necessarily transported exclusively by one of the two

49
systems. For example, at least 89 signal peptides have been identified in -lactamase enzymes to date,
showcasing the existing diversity in bacterial protein export.189 With PBPs invariably undergoing Sec
transport, the evolutionary-related -lactamases were also initially thought to be only transported via
the Sec pathway.189,212 However, examples from M. tuberculosis (BlaC, BlaS), S. maltophilia (L2-1),
and even Pseudomonas luteola (LUT-1, (SignalP 5.0 likelihood scores: Sec/SPI = 0.0572, Tat/SPI =
0.9312, Sec/SPII (lipoprotein) = 0.0087, other = 0.0029)) have proven that the export of some of these
hydrolases can be Tat dependent.189,212,213 These discrepancies are likely linked to post-translational
processing requirements of some -lactamases. For example, some enzymes undergo Sec-mediated
transport to gain access to periplasmic post-translational modification machinery, like the disulfide
bond formation system, which helps them achieve their biologically active structure. Others, like the
representative enzymes from M. tuberculosis, S. maltophilia or P. luteola do not require such
modifications and are transported into the periplasm in their fully functional state.189,213
1.6 OXIDATIVE PROTEIN FOLDING PATHWAYS
The stability of proteins located in the cell envelope is key for bacterial survival. A plethora of
chaperones and proteases work against the harsh conditions of the extra-cytoplasmic environment (for
example low pH or high salt content) to safeguard the integrity of the cell envelope proteome.214 In
addition, the formation of disulfide bonds in many extra-cytoplasmic proteins improves their stability
by reinforcing their non-covalent intra- and inter-molecular interactions, like van der Waals or
hydrophobic interactions and ionic or hydrogen bonds. The biochemical processes resulting in the
formation and isomerisation of covalent bonds between two spatially proximal cysteine amino acids,
are collectively known as oxidative protein folding and occur post-translationally. The process of
disulfide bond formation, despite the simplicity of the oxidation reaction that entails the removal of two
protons and two electrons from the thiol side groups of the cysteines, is catalysed by dedicated protein
systems in both prokaryotic and eukaryotic species.215,216 A brief overview of oxidative protein folding,
with a focus on Gram-negative bacteria, is given below.
1.6.1 Disulfide bond formation in eukaryotes
The highly compartmentalised nature of eukaryotic cells offers many surfaces where oxidative protein
folding can take place, and as such more than one organelle structure plays a role in disulfide bond

50
formation. Two major and distinct oxidative pathways have been identified in eukaryotes, the protein
disulfide isomerase pathway in the endoplasmic reticulum (ER) and the MIA pathway in the
mitochondrial intermembrane space (IMS).217 Oxidative folding also takes place in thylakoid
membranes of plant chloroplasts, although this field is highly understudied and will not be discussed
further here.218
The existence of a catalyst responsible for oxidative protein folding was first noted in 1963 in the
process of re-oxidation of reduced RNase obtained from rat liver,; this process can occur spontaneously,
but only at a very slow rate and under highly specific conditions.219,220 The rate of RNase oxidation was
shown to accelerate by addition of crude rat liver homogenate. Characterisation of this homogenate by
fractionation let to the discovery of the protein disulfide isomerase (PDI) enzyme.219,221 Since then, the
function and mechanism behind PDI catalysis has been extensively studied in Saccharomyces
cerevisiae, where its isomerase activity was shown to be essential for cell viability, and three key players
were identified, PDI1 (PDI family member), ER oxidoreductin 1 (ERO1p) and a protein essential for
respiration and vegetative growth (Erv2p).220,222–224 Interestingly, although ERO1p and Erv2 do not
share sequence similarity, they both contain two active cysteine pairs, bind a flavin adenine dinucleotide
(FAD) co-factor within a 4-helix core, and carry out similar functions.218,223,225
PDI1 is a soluble, V-shaped enzyme with a classical thioredoxin (Trx) fold that has a highly conserved
CGHC functional motif (Figure 1.4).218,226 In its oxidised state, the protein acts as a disulfide bond donor
and forms a mixed-disulfide bond with nascent polypeptides upon their translocation into the ER
lumen.217 Nucleophilic attack by a secondary thiol leads to the release of the oxidised substrate and
generation of reduced PDI1. In turn, the latter can either act as an isomerase or be re-oxidised by
membrane associated ERO1p/Erv2p.217 These two proteins shuttle electrons through a FAD co-factor
to the respiratory chain.217,223,225 The formation of FAD2H and its subsequent reaction with molecular
oxygen yields hydrogen peroxide and regenerates ERO1p/Erv2p.217,223,225

51
Figure 1.4 The first identified oxidative folding catalyst PDI1 carries a classical Trx fold with a conserved disulfide
bond in a CXXC motif, a common feature in all thiol-redox enzymes. (A) A schematic of the classical thioredoxin fold;
the N-terminal domain carries the catalytically active CXXC motif. An helix connects the N-terminal domain to the C-
terminal domain, where a conserved cis-proline is located; this is a key residue for substrate binding and release. Adapted from
Shouldice et al.218 (B) A crystal structure of the PDI1 enzyme from S. cerevisiae, showing a V-shaped fold composed of four
Trx domains. The N- and C-terminal domains contain a conserved pair of cysteine residues; one of the conserved thioredoxin
domains containing a disulfide bond (Trx domain 1) is highlighted. In the second cysteine-containing Trx domain the cysteines
are in their reduced form. PDB code: 2B5E.66,224
Eukaryotic PDI proteins are remarkably diverse, with five different yeast and over 20 distinct human
homologues.218 It is, thus, not surprising that in more complex mammalian species, additional routes
for PDI re-oxidation have been identified and include numerous peroxidase enzymes or vitamin K
epoxide reductase-like proteins (VKOR is discussed further in section 1.6.3).217,227,228 For example,
peroxiredoxin IV mediates PDI oxidation alongside the ERO-1 pathway in mammals, using hydrogen
peroxide as the electron sink.217,228 Its absence leads to impaired disulfide formation and growth.217,228
Other peroxidase enzymes, such as the glutathione peroxidases 7 and 8, have also been shown to oxidise
members of the PDI family and likely act in concert with the main PDI pathway in order to utilise the
hydrogen-peroxide-to-water conversion and maximising the use of reduced oxygen.217,228
In addition to the oxidative protein folding pathways in the ER, disulfide bond formation also occurs in
the mitochondria where it mitigates the effects of restrictive protein import across the mitochondrial
membrane. As the mitochondrial structure closely copies that of its prokaryotic predecessors, with an
outer membrane, an intermembrane space, an inner membrane, and a matrix, the passage across its outer
membrane is restricted to reduced and unfolded polypeptides.202,218,229 The introduction of disulfide
bonds into these protein precursors ensures their entrapment within the organelle and their functional
activation.218,229 The mitochondrial MIA pathway is composed of two folding catalysts, the

52
oxidoreductase Mia40 and a FAD dependent sulfyhydryl oxidase Erv1, both of which have similar
redox interactions as the prokaryotic DsbA-DsbB system (discussed further in section 1.6.2.1.1).229,230
Mia40 is a highly conserved protein with an -hairpin core that, despite its redox activity, does not
contain the classical Trx redox fold.231,232 Instead, catalytic activity is driven by a CPC functional motif,
which is supported by a hydrophilic binding cleft and stabilised by two oxidised N-terminal CX9C
moieties.229,231 Its substrates include polypeptides from the respiration and protein biogenesis pathways,
such as the Tim family of chaperones, and commonly include twin CX9C or CX3C motifs.229,231,233
Substrate oxidation is thought to occur through the formation of a mixed disulfide between the second
Mia40 cysteine and its substrate.229 Reduced Mia40 is re-oxidised by Erv1 and electrons are passed
through its FAD domain to cytochrome c, and ultimately to the respiratory chain where they generate
water molecules.229,230
Mia40 appears to be able to ‘selectively’ introduce correct disulfide linkages in the presence of more
than two cysteine residues, likely through the recognition and folding of a nine-amino-acid-long IMS-
targeting signal (ITS/MISSvii).229,231,233,234 The presence of proofreading mechanisms has been proposed,
including the utilisation of reduced glutathione or direct Mia40-facilitated disulfide isomerisation.233,234
Additionally, in a fashion that is not too dissimilar from the dual role of the prokaryotic isomerase
DsbC, Mia40 has also been shown to act as a chaperone for numerous cysteine-rich proteins, including
the mitochondrial protease Atp23.229,234
The similar functions of the thiol-oxidase and thiol-oxidoreductase enzymes of the PDI and MIA
pathways result in the formation of the majority of eukaryotic disulfide bonds. However, despite the
mechanistic similarities, their respective substrate profiles are entirely distinct; polypeptides folded in
the ER are targeted to all cellular and extra-cellular locations, while substrates of the MIA pathway are
oxidised for exclusive use in the mitochondria.218
Last but not least, it should be noted that in addition to the PDI and MIA pathways, mammalian cells
are also able to oxidise nascent polypeptides through the action of the quiescin sulfhydryl oxidase
vii Research by Sideris et al. suggests that the IMS-targeting sequence overlaps with the mitochondria IMS-sorting signals previously observed
by Milenkovic et al. in some Mia40 substrates. For ease of reference and limited understanding these targeting sequences are now commonly
and jointly referred to as the ITS/MISS sequences.229,233,459

53
(QSOX).217,235 Identified by Thorpe and Coppock, this enzyme is present in the ER membranes, the
Golgi apparatus and the extra-cellular matrix and given that it contains both an FAD co-factor and a
Trx domain, it has been suggested that its role might be to form disulfide bonds in the extra-cellular
environment.217
1.6.2 Disulfide bond formation in prokaryotes
1.6.2.1 Gram-negative bacteria – the archetypical E. coli system
Bacterial cells lack the clearly organised organelle structures that provide the surface for oxidative
protein folding reactions in eukaryotes. Instead, in Gram-negative bacteria, these processes take place
in the oxidative environment of the outer leaflet of the inner membrane, in the periplasm. The first
bacterial oxidative folding mechanism was discovered in E. coli, and this archetypical system has since
been characterised in depth.236–240 It is composed of two complementary pathways, the oxidative and
the isomerase pathways, which are respectively responsible for the introduction and rearrangement of
disulfide bonds in newly translocated polypeptide chains.
1.6.2.1.1 The oxidative pathway
The oxidative pathway introduces consecutive disulfide bonds into polypeptides during, or immediately
after, their periplasmic translocation through the Sec translocase. As cysteine residues enter the
periplasmic space, they are trapped in a mixed-disulfide bond by the soluble thiol oxidase, DsbA. The
resolution of this intermediate complex occurs via disulfide transfer to the polypeptide substrate.
Reduced DsbA is then recycled by its membrane-bound partner DsbB, a unique quinone reductase
located in the inner membrane (Figure 1.5).

54
Figure 1.5 The oxidative pathway of the disulfide bond formation system. The oxidative pathway introduces disulfide
bonds into polypeptide chains containing two or more cysteine residues, through the activity of the primary oxidase DsbA,
and immediately after protein translocation through the Sec pathway. The removal of two electrons from two consecutive
cysteine residues leads to substrate oxidation and DsbA reduction. DsbA is re-oxidised by its partner, the membrane-embedded
protein DsbB. Two electrons are shuttled from DsbA to DsbB through an intermolecular disulfide. A thiol-disulfide exchange
transfers the acquired charge onto ubiquinone or menaquinone carriers and from there to the electron transport chain. Bond
lengths not to scale. Figure adapted from Messens & Collett.241
1.6.2.1.1.1 DsbA
DsbA is a 21 kDa soluble monomeric protein with a conserved active-site disulfide bond that is
available for transfer to substrates with two or more cysteine residues in their primary sequence.242 The
protein core comprises an extended Trx-like fold that is disrupted by an -helical domain (Figure 1.6
A).242 The classical Trx fold is composed of two sub-domains, the N-terminal and the
C-terminal domains, connected by a single -helix (Figure 1.6 A).218,243 In DsbA, the loop linking the
-helix to the -sheet is broken up by an insertion of a compact helical unit of three clustered -
helices (−) flanked by single -helices ( at the N-terminal end of , Figure 1.6 A).218,242
cytoplasm
periplasm

55
Figure 1.6 Crystal structure of the primary oxidase of the E. coli DSB system, EcDsbA. (A) Structure of EcDsbA rendered
in a cartoon representation. The protein is composed of a Trx core, 1/1/2-2- 3/4/3, disrupted by a compact -helical
domain insertion which is composed of three clustered -helices, 2-4, followed by single -helix, 5.218,242 The active site
disulfide bond lies on the N-terminal side of the helix and is depicted by yellow spheres.242,245,246 (B) A close-up view of
the active site of EcDsbA. The catalytically active motif, Cys30-Pro-His-Cys33, and the substrate binding loop, Gln145-Leu-
Arg-Gly-Val150, are shown in cyan stick representation. Highlighted are the Pro31-His32 dipeptide residues responsible for the
protein’s strong oxidative potential and the conserved cPro151 residue critical to successful substrate release. (C) The
electrostatic surface of EcDsbA generated using PyMol with a normalised hydrophobicity scale developed by Eisenberg et al.;
a sliding colour scale shows the hydrophilic regions in light grey/pink and hydrophobic residues in darker red.248 The active
site residues are shown in cyan (Pro31-His32 dipeptide) and yellow (Cys30 and Cys33). On the left, the hydrophobic patch
responsible for the recognition and binding of substrate polypeptides is prominent in dark red colour to the left of and above
the active site. The DsbB binding groove (Groove 1) runs below the active site.242,246,247 On the right, a side-on view shows
the presence of a hydrophilic groove (Groove 2) of unknown function.242 Information for this figure compiled from Guddat et
al., Shouldice et al. and Martin et al.; PDB code: 1FVK.66,218,242,248–250

56
Interestingly, a similar but much less close-packed insertion is observed in the eukaryotic glutathione
peroxidase enzyme responsible for the formation of disulfide bonds in glutathione.242 The inserted
cluster creates a flexible -turn allowing hinge-like motion to the two DsbA sub-domains.244 At the
interface of the N-terminal end of the Trx domain and the -helical domain lies the active site.242,245,246
The C30XXC33 functional motif (CPHC in E. coli) is surrounded by highly hydrophobic amino acids
responsible for recognition and binding of substrate polypeptides and the partner protein DsbB (Figure
1.6 C).242,246,247 Specifically, a loop connecting the and forms a hydrophobic patch just above the
active site (Figure 1.6 A and C).242,243,246 The C-terminal end of this loop carries a conserved cis-proline
(cPro151, Figure 1.6 B) residue which extends into a deep hydrophobic groove running just below the
active site.242,243,246 An additional prominent groove, lined with polar acidic residues, is located on the
opposite side of the protein, but its function remains unclear.242
DsbA binds and oxidizes a plethora of polypeptides, including virulence factors such as toxins, pili or
secretion system components.216,218,237,251–253 With the exception of an observed bias for an even number
of cysteines, these substrates show no clear conservation of amino acid sequences or structural motifs
proximal to their cysteine residues.246 Instead, substrate recognition seems to be occurring at the DsbA
level; a crystal structure of DsbA in complex with a peptide originating from a native substrate shows
that binding interactions occur in the hydrophobic patch directly above the active site (Figure 1.7
A).246,251 Access to this region in E. coli DsbA (EcDsbA) is dictated by the protein’s oxidation state and
controlled by a histidine residue (His32, Figure 1.6 B, Figure 1.7 C) located in the C30PHC33 dipeptide
motif.246 In particular, in oxidised DsbA, the histidine is positioned across the hydrophobic groove
located under the active site allowing access of the substrate to the hydrophobic patch (Figure 1.7).242,246

57
Figure 1.7 Differential binding of DsbA to substrate peptide or DsbB is directed by the hydrophobic surfaces
surrounding the active site of DsbA and the histidine residue, His32. DsbA is shown either shown in cartoon representation
coloured pink or in an electrostatic surface representation using the normalised hydrophobicity scale developed by Eisenberg
et al.; a sliding colour scale shows the hydrophilic regions in light grey/pink and hydrophobic residues in darker red.248 The
DsbA active site cysteines are coloured in yellow or shown as yellow spheres. The conserved loop of DsbA (QLRGV) known
to affect substrate binding is shown in cyan sticks.246,251 (A) Top panel: DsbA in complex with a substrate polypeptide (dark
blue). Substrate binding is promoted by surface-surface interactions with the hydrophobic patch above the active site. The
active site dipeptide moiety is shown in cyan. Bottom panel: Stick representation of the DsbA-substrate binding site showing
the key interactions with the Gln145- Val150 loop.243,246,251 PDB code: 3DKS. (B) Top panel: DsbA in complex with the P2
periplasmic loop of DsbB (dark blue); DsbB active site cysteines are coloured in orange. The DsbA active site dipeptide moiety
is shown in cyan. Binding is achieved by interaction with both the hydrophobic patch above and the hydrophobic groove below
the active site. Bottom panel: Stick representation of the DsbA-DsbB showing the P2 (Pro100-Phe106) loop in the hydrophobic
groove.247,250,252,262,265 (C) Binding of the substrate or DsbB is directed by the position of the His32 residue which changes its
conformation depending on the oxidative state of DsbA. When the substrate is bound in the active site, the aromatic ring of
His32 (dark blue) blocks the DsbB binding groove. Upon DsbA reduction, a shift of the His32 thiolate to the cyan conformation
results in the opening of the hydrophobic groove to allow DsbB binding.242,246 PDB code: 2HI7. Figure adapted from Paxman
et al. and Inaba et al.66,246,247
DsbA-polypeptide binding occurs primarily through backbone hydrogen bonding with only few direct
surface-surface interactions observed.246 This weak binding is common to many Trx species and is in
)

58
line with the enzyme’s broad substrate scope.246,254 A Gln145-Leu-Arg-Gly-Val150 motif of the Trx loop
was identified as key for substrate recognition (Figure 1.6 B, Figure 1.7).243,246,251 While mutations that
alter these residues do not completely abrogate the enzyme’s disulfide-forming ability, they result in
alteration of the substrate specificity and the redox potential.243,246,251 For example, the single amino
acid substitution Val150Gly leads to complex formation defects for a range of substrate proteins.246,251
Substitution of the entire native E. coli motif (QLRGV, Figure 1.6 B) with the equivalent sequences
from the Neisseria meningitidisviii (QIDGT of DsbA1 or QISGT of DsbA2) affects the ability of DsbA
to confer resistance to the reducing agent dithiothreitol (DTT).246,251 This suggests that additional
interactions and conformational changes may be required to promote correct substrate binding.249
Immediately following the substrate recognition motif is a conserved cis-proline residue (cPro151, Figure
1.6 B) which plays a key role in DsbA stability and its ability to release substrates.251,252 Substitutions
in this residue result in significant loss of activity, likely due to its key position at the end of the Trx
loop and its spatial positioning within the hydrophobic groove.251,252 For example, substitution for
alanine or threonine increases the stability of the reaction intermediates resulting in the accumulation
of the DsbA-polypeptide complex.246,249,252 By contrast, substitution for serine propagates extensive
conformational changes through the Trx loop and results in the accumulation of the DsbA-DsbB
complex.251,252
The process of disulfide bond formation is catalysed by the interaction of the oxidised Cys30XXCys33
motif of DsbA with the cysteine residues of the substrate proteins.242,249 The cysteine pair of DsbA is
strictly conserved across all DsbA homologues, but minor variations are observed in the enclosed
dipeptide moiety.218 While EcDsbA harbours the very common Pro31-His32 combination, DsbA3 of N.
meningitidis, for example, contains a Val-His sequence.218,242,255 In general, His32 is often conserved in
thiol-oxidases, including in the human PDI, as it increases the oxidative power of the enzymes through
stabilisation of their reduced states.256,249 This is due to the residue’s inherently destabilising nature
caused by unfavourable interactions with the nearby -helix dipole in oxidised DsbA.256,249 -
galactosidase functional assays in E. coli by Grauschopf et al. confirm that the dipeptide motif is
viii Multiple homologues of DSB system components have been observed in pathogenic species, such as N. meningitidis. This can result in
more differentiated catalytic profiles (see section 1.6.3 for further discussion of the DSB system polymorphism).

59
responsible for the enzyme’s oxidative properties.257 Substitutions in the CPHC motif in EcDsbA to
CPPC, CPLC or CTRC markedly reduces the protein’s oxidative powerix.250,257
The transfer of the disulfide bond from DsbA to its substrate begins with a nucleophilic attack from the
first, deprotonated cysteine residue of the substrate at the N-terminally located Cys30 of
DsbA.242,246,251,258,259 A stabilising mixed-disulfide bond forms between DsbA and the substrate
polypeptide chain.254 The resultant complex is then resolved by a second nucleophilic attack from the
substrate cysteine to Cys30 of DsbA that leads to the release of the oxidised substrate protein and reduced
DsbA.241,245,251,260,261 DsbA reduction is a favourable process due to the formation of the thiolate anion
of the highly acidic Cys33.218,242,249 Structural comparison of the two redox states of DsbA by Kortemme
et al. and Guddat et al. shows that this thiolate anion forms a favourable electrostatic interaction with
the positive dipole moment of the proximal -helix and a hydrogen bond with the side chain of
His32.249,250,256,257 Jointly, these interactions stabilise reduced DsbA.249,250,256,257 In addition, the thiolate-
His32 interaction causes a conformational change in His32 that results in the opening of the hydrophobic
groove of DsbA in preparation for DsbB binding (Figure 1.7 C).242,246 Unlike substrate binding,
specificity during DsbA-DsbB complex formation is achieved by a great number of interactions
between the two proteins localised at both hydrophobic surfaces that are proximal to the DsbA active
site, the patch above it and the groove underneath it (Figure 1.7 B).246,247,251,254 DsbA is re-oxidised
through a disulfide exchange cascade with DsbB. Mechanistic understanding of this process has been
achieved with the help of four structures of reaction intermediates (three crystal structures and one NMR
structure), which are discussed in more detail in the next section.247,262–264
1.6.2.1.1.2 DsbB
Re-oxidation of DsbA following its catalytic cycle is carried out by a dedicated transmembrane partner
protein, DsbB, which generates the DsbA disulfide de novo. A series of redox reactions allows DsbB
to remove two electrons from DsbA and to re-instate it in its active oxidised state. Electrons removed
from DsbA are then passed from DsbB to terminal oxidases via ubiquinone (UQ, aerobic conditions)
or menaquinone (MQ, anaerobic conditions).241,266 Notably, in comparison to DsbA, DsbB has a low
ix More recent quantum-mechanical simulations of the active site by Carvalho et al. confirmed that highly localised changes in the hydrogen
bonding network affect the protein’s redox activity.260 Reductase species, for example, were observed in the presence of less stabilising
residues, such as glycine or tyrosine.260

60
redox potential, and as a result prevention of electron backflow through the DsbA-DsbB complex is
achieved by substantial conformational changes in the DsbB backbone.247,267
DsbB is a 21 kDA integral membrane protein with an -helical transmembrane domain, two flexible
periplasmic domains and two conserved disulfide-bonded cysteine pairs (Figure 1.8).265,268,269 Four
transmembrane -helices (TM1-4) traverse the inner membrane such that the N- and C-termini of the
protein are located at the cytoplasmic side, whilst the two flexible loops that connect the transmembrane
segments are on the periplasmic side.247 The shorter of the two loops (P1, Gln33-Arg48) links TM1 and
TM2, and the longer loop (P2, Tyr97-Gln144) connects TM3 and TM4 (Figure 1.8).247 Each of the loops
carries a conserved disulfide bond. Cys41-Cys44 is present in the P1 loop and interacts with the quinone
molecule, which acts as an electron sink.240,263–265 Cys104-Cys130 is located in the P2 loop and interacts
with the DsbA thiols as an electron acceptor.240,263–265 The P2 loop is associated with the periplasmic
leaflet of the inner membrane via a short and conserved amphipathic -helix (Leu116-Val120, assignment
is structure-dependent, Figure 1.8).247,263–265 This places the two cysteine residues of the Cys104-Cys130
bond to different protein regions with distinct mobilities. Mutational studies show that this feature
underpins the structural flexibility of DsbB which is central to its ability to oxidise DsbA, despite the
aforementioned unfavourable redox potential.247,263,264 The flexibility of the P2 region makes
crystallisation of isolated DsbB challenging, therefore the protein’s mode of action has mostly been
studied using trapped complexes between DsbA and DsbB cysteine mutants.247,262

61
Figure 1.8 Crystal structure of EcDsbB, the membrane partner protein of DsbA. The left structure shows a side view of
DsbB Cys130Ser variant in complex with UQ, while the structure on the right shows a top view of DsbB. Four helical
transmembrane domains (TM1-4) dock in the inner membrane with the N- and C-termini emerging in the cytoplasm. Flexible
periplasmic loops carry two catalytically active disulfide bonds, Cys41-Cys44 (P1 loop) and Cys104-Cys130 (P2 loop, Cys130Ser
mutant is used in this crystal structure). The second half of the P2 loop, connecting the amphipathic -helix to the TM4 domain,
is very flexible and its position was not determined in the structures above. Thus the Ser130 residue is not shown.240,263–265
Cysteines are shown in blue stick representation, the sulfur atoms are represented by yellow spheres. The ubiquinone (UQ)
co-factor is shown in black sticks. PDB code: 2HI7. Figure adapted from Inaba et al.66,247
DsbA re-oxidation depends on the formation of a DsbA-DsbB complex, which is held together through
interactions of DsbB with the hydrophobic surfaces surrounding the DsbA active site.247 A crystal
structure of DsbA Cys33Ala/DsbB Cys130Ser (Figure 1.7 B, Figure 1.9 A) shows that DsbB interacts
with the hydrophobic patch above the DsbA active site in manner similar to DsbA substrates. In
addition, the specificity of DsbB for DsbA is dictated by an additional interaction with the hydrophobic
groove below the DsbA active site.246,247 Groove binding occurs through the N-terminal end of the P2
periplasmic loop (DsbB, Pro100-Phe106)x and depends primarily on hydrogen bonds, salt bridges and
surface-surface interactions with conserved DsbA residues, such as the cis-proline (cPro151, section
1.6.2.1.1.1) or the Pro63-Phe174 sequence (Figure 1.7 B).247,250,252,262,265 Additional close contacts between
the Ala126-Ala131 sequence of DsbB and DsbA were identified by NMR.263–265 Van der Waals surface
interactions between the active site His32(DsbA) and Ala102-Thr103(DsbB), as well as the formation of a short,
antiparallel -sheet between Arg148-Val150(DsbA) and Cys104-Phe106(DsbB) initiate DsbA re-oxidation
placing Cys104(DsbB) in spatial proximity of Cys30(DsbA) (Figure 1.7).246,247
x Crystal structure by Inaba et al. has shown notable interactions with the Pro163, Gln164, Thr168,Met171 and Phe174.247

62
Figure 1.9 The proposed mechanism of DsbA oxidation by DsbB. DsbA is shown in pink, DsbB is shown in orange. (A)
Crystal structure of DsbA Cys33Ala - DsbB Cys130Ser complex prior to disulfide rearrangement. DsbB binds into the
hydrophobic groove of DsbA via the P2 loop and forms an intermolecular disulfide bond (Cys30(DsbA)-Cys104(DsbB), cyan and
blue, respectively).246,247 The Cys41-Cys44 disulfide bond in the P1 loop of DsbB remains intact (blue stick representation).262–
265 PDB code: 2ZUP.247 (B) Crystal structure of DsbA Cys33Ala - DsbB Cys130Ser after disulfide rearrangement.

63
Conformational shift in the amphipathic -helix leads to the formation of an intramolecular disulfide Cys130-Cys41 (not shown
as Cys130Ser mutant is used). This results in formation of a Cys44 thiolate which interacts with the bound UQ. PDB code:
3E9J.262 (C) NMR structure of the DsbB Cys44Ser/Cys104Ser mutant following DsbA dissociation. A transient intramolecular
Cys130-Cys41 disulfide is shown, trapped by the use of the double cysteine mutant. PDB code: 2K74.263 (D) Crystal structure
of DsbB Cys41Ser mutant showing a Cys104-Cys130 disulfide where full reoxidation has been achieved; the protein can now
undergo another round of DsbA oxidation. PDB code: 2ZUQ.264 Figure adapted from Bushweller et al.66,265
Oxidation of DsbA begins with a nucleophilic attack from the Cys30(DsbA) on Cys104(DsbB); the latter is
located in the P2 loop where also UQ is bound.247,265 This leads to formation of a mixed Cys30(DsbA)-
Cys104(DsbB) disulfide bond leaving Cys130(DsbB) in a thiolate anion form (Figure 1.9 A).247,265,267 At this
point, the C-terminal amphipathic -helix (Figure 1.8) facilitates a conformational shift in the N-
terminus of the P2 loop and increases the distance between the Cys130(DsbB) thiolate and the mixed
disulfide bond to ~8Å.262–265 This prevents the backflow of electrons to DsbA, and places the thiolate in
the vicinity of the Cys41-Cys44 disulfide bond in the P1 loop of DsbB (Figure 1.9 B).262–265
The exact sequence of steps leading to the resolution of this DsbA-DsbB-UQ complex has yet to be
determined, but it invariantly includes a nucleophilic attack on the DsbA-DsbB intermolecular disulfide
bond, rearrangement of DsbB, and an ensuing thiol-disulfide exchange cascade.265 Two non-exclusive
pathways have been described in literature.267 In the rapid pathway, the Cys30(DsbA)-Cys104(DsbB) is
resolved by an immediate attack of Cys33(DsbA) (Figure 1.9 B, C), resulting in release of oxidised DsbA
and a hemi-oxidised state of DsbBxi (Figure 1.9 C, D).262,264,267 By contrast, in the slow pathway, the
DsbA-DsbB covalent bond is retained until the removed electrons have been passed on to the electron
transport chain.267 In both proposed mechanisms, the formation of an interdomain disulfide (Cys130-
Cys41) in DsbB is favoured by the strongly electron-withdrawing Cys41, and this in turns promotes the
interaction of a Cys44 thiolate with UQ (Figure 1.9 B).262–265
While the DsbB-UQ binding site is yet to be fully characterised, available structures suggest that face-
on interactions occur between the quinone ring, the TM1 C-terminus and the TM2 N-terminus, with the
UQ isoprenyl chains extending into a TM1-TM4 groove (Figure 1.8, Figure 1.9).262,263,265,270–272 These
contacts are stabilised by the guanidinium group of an essential Arg48 (P1 loop) and the TM2 helix
xi Cys104-SH, Cys130-SH, Cys41-Cys44 / Cys104-SH, Cys130-Cys41, Cys44-SH / Cys104-Cys130, Cys41-SH, Cys44-SH

64
dipole moment.262 Generally, the proximity of the DsbB Cys41-Cys44 disulfide to the quinone ring
promotes a nucleophilic attack between the Cys44 thiolate and UQ and results in the formation of the
DsbB-UQ charge transfer complex that provides a direct electron gradient between DsbA and the
electron transport chain (Figure 1.9 B).247,262,264,265,270,273
The mechanism behind de novo disulfide bond formation depends on the binding state of DsbB. The
slow pathway proceeds via a cascade of nucleophilic substitution reactions that start with the attack of
Cys33(DsbA) and terminate with the attack of Cys41(DsbB) on the Cys44-UQ complex.267 Conversely, the
rapid pathway (where DsbA is released prior to P2 loop rearrangement) depends on the re-instatement
of the Cys41-Cys44 disulfide mediated by the Cys44-UQ adduct (Figure 1.9 D).267 Experimental evidence
by Inaba et al. suggests that the rapid pathway is likely preferred, in part, due to the transient tethering
of Cys130(DsbB) in an intermediate location between both Cys104 and Cys41, which would facilitate the
electron exchange.247,267 Irrespective of the release pathway mechanism, the DsbB disulfide cascade
results in the release of oxidised DsbA which enables further oxidative protein folding steps to take
place in the cell envelope maintaining its protein homeostasis.
1.6.2.1.2 The isomerase pathway
The oxidative pathway of the DSB system rapidly introduces disulfide bonds between consecutive
cysteine residues of cell envelope proteins as the unfolded polypeptides transverse the inner membrane.
Due to the non-discriminatory nature of DsbA-catalysed cysteine coupling, in cases where a protein has
more than two cysteine amino acids the formation of non-native disulfide bonds can occur. The
detection and correction of oxidatively misfolded proteins is carried out by the isomerase pathway of
the DSB system, which is composed of the freely diffusible isomerase, DsbC, its analogue DsbG, and
their membrane-bound partner protein, DsbD (Figure 1.10).274 The rearrangement of incorrect disulfide
linkages is crucial in preventing protein aggregation which could eventually intoxicate the bacterial
cell.241,275

65
Figure 1.10 The isomerase pathway of the DSB system. The isomerase pathway corrects proteins misfolded by DsbA
through the activity of the isomerase DsbC. The misfolded substrate binds in the wide cleft formed by the N-terminal
dimerisation domains of DsbC. Thiol-disulfide exchange reactions between the reduced cystine pairs of the C-terminal
domains of DsbC and the misfolded substrate results in the release of a reduced substrate and oxidised DsbC. The
transmembrane partner protein DsbD ensures that DsbC is present in its active, reduced form. DsbD itself is kept reduced by
cytoplasmic thioredoxins. The isomerase pathway also encompasses the DsbC homologue, DsbG. The function of this protein
is to protect single thiols from irreversible oxidation. Figure adapted from Messens & Collett and Katzen & Beckwith.241,276
1.6.2.1.2.1 DsbC
DsbC is a 46 kDa V-shaped homo-dimeric protein composed of an N-terminal dimerization domain and
a C-terminal catalytic domain containing two conserved disulfide bonds formed by two conserved pairs
of cysteine residues (Figure 1.11).274 Two monomeric DsbC subunits are held together by hydrogen
bond interactions between their N-terminal domains, each comprising a six-strand antiparallel -sheet
(−) and a single N-terminal -helix () (Figure 1.11 A).274 Interaction and binding of the two
domains is mediated by the exchange of the 4 strand of monomer 1 and the 5’ strand of monomer 2,
cytoplasm
periplasm

66
such that strands 1-4 of monomer 1 overlap with strands 5’-6’ of monomer 2 and vice versa (Figure
1.11 B).274 A cis-proline (Pro50) residue disrupts the -sheet planarity at the C-terminus and, along with
the proximal flexible -helix linker, ensures that the catalytic domain extends away from the
dimerization domain.274 This creates the opposing arms of the protein and minimises undesirable inter-
C-domain interactions between the two monomers (Figure 1.11 A and C).274 A localised 13Å twist
around one of the -helices of the catalytic domain exposes one of the C-terminal active sites to the
periplasmic environment.277
Figure 1.11 Crystal structure of the primary isomerase of the E. coli DSB system, EcDsbC. (A) (top) Cartoon
representation of the V-shaped DsbC homodimer held together by the N-terminal dimerization domains, -−. The catalytic
site is located in the extended Trx core (- −-) at the distal end of the C-terminus which is extended away from the
dimerization domain via the helix.274 (bottom) The structure of a DsbC monomer in cartoon representation. The active
cysteine residues of the C98-GY-C101 motif are shown as sticks in cyan with the sulfur atoms represented by yellow spheres.
(B) Close up view of the N-terminal dimerization domain of EcDsbC. Hydrogen bond interactions occur through the exchange
of strand of monomer 1 (pink) and the ' strand of monomer 2 (dark red); 1-4 of monomer 1 overlap with strands ’−’ of
monomer 2 and vice versa.274 (C) The electrostatic surface of EcDsbC generated using PyMol (top) and using PyMol with a
normalised hydrophobicity scale developed by Eisenberg et al. (bottom; a sliding colour scale representation shows the
hydrophilic regions in light grey/pink and hydrophobic residues in darker red.66,248 Hydrophobic residues line the wide binding
cleft formed by the N-terminal dimerization domains.274,281 PDB code: 1EEJ. Figure adapted from McCarthy et al.274

67
The cysteine-containing C-terminal domains are, in their monomeric form, structurally similar to DsbA
(Figure 1.6 A, Figure 1.11 A).274 Interestingly, early studies by Rietsch et al. and Missiakas, Schwager
& Raina showed that DsbC can partially complement DsbA activity under suppression of DsbD and in
the absence of DsbA.239,245,278 This behaviour is not observed under standard conditions, due to kinetic
and structural restrictions introduced by the dimerization domain and the -helix linker ()xii.279–281
The C-terminal domains are formed from an extended Trx core ( − and Figure 1.11 A), which
is disrupted by an -helical insertion () in the − connecting loop.242,274 This -helical insertion
is smaller in comparison to that of DsbA, something that likely prevents the formation of the
hydrophobic groove observed under the active site of DsbA.274 Instead, the wide hydrophobic cleft
formed by the -sheets of the dimerization domain enables binding of misfolded substrates, while the
two extended C-terminal domains jointly interact with their partner protein DsbD.274,281 The great
conformational flexibility in DsbC promotes interactions with a wide range of partially folded and
misfolded polypeptides and additionally supports the secondary role of DsbC as a periplasmic
chaperone.245,253,274,278,281 In further contrast to DsbA, the catalytically active disulfide bond C98XXC101
(CGYC in E. coli) of DsbC is reduced to enable interactions with disulfide bonds rather than with free
thiols.279 Stabilisation of the reduced form of DsbC has been shown to be partially supported by
interactions between the Cys98 and Thr182 sidechains.282 An additional disulfide bond, located at Cys141-
Cys163, is catalytically inactive and its loss causes increased DsbC sensitivity to denaturation, thus
showing a role in protein stability.277,283
The isomerisation process begins by substrate binding into the uncharged hydrophobic cleft of DsbC,
likely mediated by the loop residues of the Trx fold, Thr182-cPro183, which are located in an equivalent
position as the loop residues shown to affect DsbA-substrate/DsbB and DsbC/DsbD complex formation
(see section 1.6.2.1.1.1).243,246,249,252,274,281 This results in the exposure of the misfolded disulfide to the
C-terminal active site and subsequent nucleophilic attack by the essential and reactive N-terminal Cys98
of DsbC forming an intermolecular disulfide bond .245,274,278,279,283 While the mechanism of the resultant
DsbC-substrate complex resolution has yet to be conclusively established, two mutually non-exclusive
xii Research by Rozhkova et al. showed that native DsbA-DsbB or DsbC-DsbD interactions are kinetically 3-7 orders of magnitude more
favourable than cross-pathway interactions of DsbA-DsbD or DsbC-DsbB.277,292 Steric hindrance and lack of a hydrophobic groove under the
DsbC active site prevents DsbC association with DsbB, while the -helical domain of DsbA is believed to prevent interactions with
DsbD.245,247,280,295 Segatori et al. show that the absence of the -helix linker () in DsbC also results in the loss of resistance to DsbB.245,274,280

68
pathways have been proposed in literature. In similar manner to PDI isomerisation in eukaryotic cells,
resolution could occur via a nucleophilic attack from a second free substrate cysteine to the
intermolecular disulfide, thus resulting in the release of the isomerised substrate and reduced DsbC.226
Alternatively, in the second proposed mechanism, the mixed disulfide undergoes a nucleophilic attack
by Cys101(DsbC) to release a fully reduced substrate and oxidised DsbC. A second round of DsbA mediated
oxidation reaction would then be required to introduce new disulfide bonds to the now semi-folded
substrate, while oxidised DsbC would be reduced by DsbD.245,284
Experimental evidence suggests that the second, stepwise process is more likely to occur as DsbC
accumulates in its oxidised form in the absence of DsbD.279 Further, DsbC cysteine substitution studies
by Liu and Wang and Rietsch et al. suggest that both active site cysteines, Cys98 and Cys101, are involved
in the isomerisation process; the loss of Cys101 causes extensive defects in the folding of a model
urokinase substrate, a eukaryotic protease with several non-consecutive disulfide bonds.245,279,283,284 As
expected, while variations in the Cys98-XX-Cys101 motif greatly affect the catalytic activity of DsbC,
they have no effect on its stability or its role as a periplasmic chaperone.277,283
Further supporting evidence comes from evolution experiments on antibiotic resistance, where an
artificial variant of an isomerisation-dependent TEM -lactamase was studied in dsbC mutants of
Legionella pneumophila.245,285 Presence of multiple non-consecutive disulfide bonds in the TEM -
lactamase enzyme resulted in overexpression of DsbA under antibiotic selection pressure.245,285 Limited
availability of DsbB, in comparison to the amount of DsbA produced, led to accumulation of the
reduced form of DsbA in the periplasm and enabled this non-oxidising form to interact with and reduce
misfolded disulfide bonds in the -lactamase.245,285,286 In this case, isomerisation of the TEM variant
was achieved in a stepwise fashion and, given the structural similarity between monomeric DsbC and
DsbA, it is likely that the classical E. coli isomerisation route follows a similar mechanism.
Irrespective of its mechanism of action, the ability of DsbC to break incorrectly formed disulfide bonds
plays an essential role in processes even beyond new protein synthesis and folding, like copper
detoxification; the presence of copper in the periplasm results in fast and uncontrolled catalysis of
disulfide bond formation.278,287 Although the ability of DsbC to identify misfolded substrates is not yet
fully understood, its activity is key to maintaining proper oxidative folding of numerous components of
the cell envelope.

69
1.6.2.1.2.2 DsbG
In addition to correcting wrongly-formed disulfide linkages catalysed by indiscriminate DsbA folding,
the isomerase pathway plays a key role in bacterial survival under oxidative stress conditions.287,288 The
presence of reactive oxygen species catalyses the formation of unwanted disulfide bonds as well as the
oxidation of thiols into sulfenic, sulfinic and sulfonic acids.288 While DsbC reverses the former, its
ability to reduce fully oxidised cysteine residues is limited. As the conversion into sulfinic and sulfonic
acids results in terminal protein misfolding and necessitates protease-mediated degradation, it is
important that the intermediate sulfenic acids are promptly reduced to thiols.245 Reduction of these side
chains is driven by DsbG, whose action safeguards key periplasmic mechanisms such as the cell wall
synthesis; DsbG prevents the loss of catalytic single cysteine residues in several transpeptidase
enzymes, such as YbiS and ErfK.241,287,288
DsbG, a V-shaped homo-dimeric protein with monomers composed of an N-terminal dimerization
domain and C-terminal catalytic domain, exhibits many structural similarities to DsbC (Figure 1.12).282
Primary differences arise from variation in size, as well as the polarity of the wide cleft which is formed
by the interacting N-terminal dimerization domains.282 In DsbG the substrate binding cleft is lined with
several acidic residues and an additional highly conserved, charged groove is formed by the −
sheets; together these polar traits promote the binding of the oxidised single cysteine residuesxiii.282 The
Trx-like C-terminal domains of DsbG are extended further away from the dimerization domain, in
comparison to DsbC.282 They carry the catalytically active Cys109-XX-Cys112 motif (Cys109-Pro-Tyr-
Cys112 in E. coli) and it has been shown that substitutions of the two amino acid dipeptide between the
cysteines result in stabilisation of the DsbG-substrate complex which prevents its resolution and he
release of reduced substrate (Figure 1.12).241,288
xiii This contrasts with DsbC, where the binding cleft is purely hydrophobic in nature.

70
Figure 1.12 Crystal structure of E. coli DsbG, EcDsbG, rendered in cartoon representation. Two homodimers form a V-
shaped homo-dimeric protein through interactions at the -sheets of the N-terminal dimerization domains (−) in a EcDsbC-
like fashion. Yellow spheres represent the sulfur atoms of the active cysteine residues. PDB code: 1V58. Figure adapted from
Heras et al.66,282
Together the isomerase proteins of the DSB system exhibit enough structural and functional flexibility
to allow a comprehensive protection of periplasmic cysteine residues under oxidative stress conditions.
Their contribution to a several periplasmic protein modifications/processes is in stark contrast to the
more restricted DsbA-DsbB oxidative pathway, which only catalyses introduction of disulfide bonds,
albeit to more substrates than the isomerase pathway.
1.6.2.1.2.3 DsbD
In the highly oxidative environment of the periplasm, disulfide bond isomerisation depends on the
activity of the reduced forms of DsbC or DsbG. This is ensured by a transmembrane protein, DsbD,
which was initially identified for its role in the reduction of CcmGxiv and which transfers reductive
equivalents across the inner membrane.265,289 DsbD is a 59 kDa protein composed of three domains.
Two of these domains are in the periplasm and are formed by the N- and C-terminal ends of the protein
(nDsbD and cDsbD, Figure 1.13 and Figure 1.14 D). These two soluble domains flank a third
transmembrane domain (tmDsbD).290,291 Each domain carries an essential pair of cysteine residues that,
xiv CcmG is an enzyme responsible for cytochrome c maturation.

71
Figure 1.13 Crystal structure of the reduced state of N-terminal domain of E. coli, nDsbD, rendered in cartoon
representation. Cysteine residues are shown as blue sticks or yellow spheres. (A) The Ig fold of nDsbD is formed by two
antiparallel -sheets, 1/2/8/5 and 4/9/12, with the active-site cysteines located in the randomly coiled region perpendicular to the
Ig fold. (B) A close-up view of the active site of nDsbD. The catalytically active cysteines are protected by a Glu69-Phe-Tyr-
Gly-Lys73, and particularly by the Phe70 and Tyr71 aromatic sidechains (blue). On the right a partially rendered electrostatic
surface of the cap residues shows the steric hindrance effects caused by the protective aromatic ring moieties which limit
access to the active site in absence of DsbC/DsbG. PDB code: 3PFU.66,297

72
Figure 1.14 Crystal structures of EcDsbC - nDsbD and cDsbD - nDsbD complexes elucidate the mechanism behind the
transfer of reductive potential through the periplasmic subunits of DsbD. (A) Crystal structure of the DsbC Cys101Ser –
nDsbD Cys103Ala complex; DsbC is shown in pink with reactive sulfur shown as yellow spheres and nDsbD is shown in orange
with its reactive sulfur moiety shown in orange spheres. Extensive conformational changes in the cap-loop region of nDsbD,
enable Cys98(DsbC) and Cys109(nDsbD) to come into contact with the cysteine residues of substrate proteins like DsbC/G, in order
to transfer reductive potential; this eventually leads to the formation of an intermolecular disulfide in nDsbD. (B) A close-up
view of the active site of nDsbD Cys103Ser - cDsbD Cys464Ser complex showing the proximity of Cys109(nDsbD) and Cys98(DsbC)
prior to formation of the disulfide bond. PDB code: 1JZD. (C) Crystal structure of nDsbD Cys103Ser - cDsbD Cys464Ser shows
the electron transfer step between the two periplasmic domains occurring through an interdomain disulfide between Cys109
and Cys461 (orange spheres). PDB code: 1SE1. (D) Crystal structure of reduced cDsbD in cartoon representation. PDB code:
2FWH. Figure adapted from Rozhkova et al., Haebel et al., and Stirnimann et al.66,281,292,299

73
through progressively increasing redox potentials, mediate the transfer of reductant from the cytoplasm
to the periplasm via a series of thiol-disulfide exchange reactionsxv.265,292 Co-operation between the three
domains of DsbD enables the movement of electrons from cytoplasmic thioredoxins to periplasmic
substrates.293 The exact mechanism of electron transfer is not fully understood due to the lack of
structural data on tmDsbD.276 Nonetheless, crystal structures of the two periplasmic domains along with
other biochemical evidence suggest that significant conformational changes occur both in DsbD and its
partners, DsbC and DsbG, in order for this electron cascade to take place.265,291,294
nDsbD has an immunoglobulin-like (Ig) fold composed of two antiparallel -sheets ( and 4/9/12),
something quite uncommon in redox-active enzymes (Figure 1.13 A).281,295–297 The catalytic C103-X5-
C109 motif is located in a -sheet perpendicular to the Ig fold ( ) and is protected by a cap
structure formed by the flexible Glu69-Phe-Tyr-Gly-Lys73 loop, and especially by the sidechains of Phe70
and Tyr71 (Figure 1.13 B).281,292 The interaction of oxidised DsbC/G with reduced nDsbD leads to
extensive conformational changes in the cap-loop region of nDsbD, enabling electrostatic and hydrogen
bonding interactions with the cPro183(DsbC) that lead to the opening of the cap loop and exposure of the
active site of nDsbD (Figure 1.13 A vs Figure 1.14 A).281
Binding and recognition of oxidised DsbC/G in turn leads to the formation of an intermolecular disulfide
bridge between Cys98(DsbC) and Cys109(nDsbD) (Figure 1.14 A and B).281,295,296 The DsbC-DsbD complex is
rapidly resolved by a second nucleophilic attack from Cys103(nDsbD) and reduced DsbC is promptly
released.293,296 Electrons from the nDsbD Cys103-Cys109 disulfide bond are then transferred to the Trx-
like cDsbD domain (Figure 1.14 C and D). Rapid formation and breakdown of an inter-domain Cys109-
Cys461 complex shows the favourability of this disulfide exchange reaction and results in a Cys461-Cys464
disulfide in cDsbD (Figure 1.14 C).292,298 Interestingly, direct reduction of DsbC by cDsbD can also
occur, but happens almost five times slower than the nDsbD catalysed process.292
The last steps of the cascade require the interaction of cDsbD with tmDsbD followed by the interaction
of tmDsbD with cytoplasmic thioredoxins. tmDsbD, predicted to be composed of 8 membrane-inserted
helices, carries two conserved cysteine residues, Cys163 (TM1) and Cys285 (TM4).276,292,294,300,301
Formation of the Cys163-Cys285 disulfide is counteracted by productive interaction with cytoplasmic
xv nDsbD is also known as DsbD, cDsbD is also known as DsbD, tmDsbD is also known as DsbD

74
thioredoxin (TrxA) which leaves Cys163 and Cys285 in their reduced thiol form.290,293,296 These steps
depend extensively on conformational changes that are yet to be fully elucidated.291 Characterisation of
a functional homolog of DsbD, CcdA, which comprises six transmembrane helices, homologous to
TM1-6 of DsbD, provides an insight into this process. Recently, an NMR structure of Thermus
thermophilus CcdA has suggested that this protein uses an elevator-type transport mechanism.265,302,303
Rotational movement of the transmembrane helices results in conformational shift between an inward
and an outward state protecting the inner membrane integrity and enabling the vertical movement of
the active cysteines across the inner membrane.265,302 Interaction with the cytoplasmic thioredoxins is
mediated by the Cys20(CcdA) of TM1 (equivalent to Cys163(DsbD)) while Cys127(CcdA) (equivalent to
Cys285(DdsbD)) interacts with periplasmic substrates.265,290,302,304,305
Along with glutathione, DsbD is the only other source of reductant for the E. coli cell envelope. Its
ability to transfer reductant obtained from the cytoplasmic compartment to periplasmic substrates, is
key to several periplasmic processes.275,290 For example, the loss of DsbD results in a pleiotropic
phenotype, which includes increased susceptibility to benzylpenicillin and temperature sensitivity.239
Overall, the role of the DSB isomerase pathway extends beyond the isomerisation of disulfide bonds
performed by DsbC, to the synthesis and maturation of c-type cytochromes via DsbD/CcmG, or the
protection of single-cysteine residues from oxidation via DsbG.275,290
1.6.3 Polymorphisms of the Gram-negative DSB system
The prototypical E. coli DSB system described here, was long thought to be conserved across Gram-
negative bacterial species. However, bioinformatic studies by several groups unveiled great diversity in
the DSB protein players.215,216,306 Interestingly, DSB polymorphisms are often found in bacterial species
that are human pathogens, such as N. meningitidis, P. aeruginosa or Salmonella enterica.307–309 Many
of these species encode an extended set of DSB proteins, some of which are thought to fold specialised
substrates.215,216
The archetypical E. coli DsbA (EcDsbA) is a promiscuous catalyst that introduces disulfide bonds into
any polypeptide with more than one cysteine residue. However, significant substrate specificity has
been observed in the additional DsbA components of species with extended DSB systems, like the
oxidative pathway of the causative agent of cerebrospinal meningitis, N. meningitidis. N. meningitidis
encodes three DsbA proteins, two of which are membrane bound lipoproteins (NmDsbA1 and 2) and a

75
third one that is a soluble periplasmic protein (NmDsbA3).216,255,310 NmDsbA1 and 2 are responsible for
the formation of pili through the folding of PilQ, which in turn ensures successful binding and uptake
of extracellular DNA.255 While both of these proteins can complement the activity of EcDsbA to a
certain degree, NmDsbA2 has been shown to be unable to restore strain motility suggesting that the two
proteins have distinct catalytic profiles.218,246,255
Another example of extended DSB systems has been described for E. coli strains associated with extra-
intestinal infections, such as uropathogenic E. coli (UPEC), which often encode a second pair of
functional redox enzymes, EcDsbL (EcDsbA-like) and EcDsbI (EcDsbB-like), alongside the classical
oxidative pathway.218,311 The DsbL/I pair is responsible for the folding of a single periplasmic substrate,
arylsulfate sulfotransferase (ASST), an enzyme linked to urinary tract colonisation.216,311 Notably, DSB
system variations are not limited to chromosomally resident enzymes. S. enterica serovars, for example,
carry a EcDsbA-like enzyme, SeSrgA, encoded on their virulence plasmid in addition to the classical
DSB proteins and the EcDsbL-EcDsbI pair.216,312 This DsbA-like protein is responsible for the folding
of structural subunits of PefA fimbrae.216,312
1.6.4 Gram-positive bacteria
While disulfide bonds are formed in the periplasmic space of Gram-negative bacteria, Gram-positive
species lack this cellular compartment. In its absence, the peptidoglycan-teichoic/mycolic-acid cell wall
of Gram-positive bacteria creates a periplasm-like environment suitable for disulfide bond
formation.11,236,313,314 Unlike the archetypical DSB system of E. coli, the formation of these bonds in
Gram-positive organisms, is not carried out by a conserved protein pathway but is rather species-
dependent.
A well-studied disulfide bond formation pathway in Gram-positive bacteria is that of the MtDsbA-
VKOR oxidation system in M. tuberculosis, where a membrane-anchored MtDsbA catalyses disulfide
formation.236,315 Re-oxidation of this oxidase is mediated by an analogue of human vitamin K epoxide
reductase, VKOR, often encoded directly next or fused to its MtDsbA protein partner.236,315 This is
interesting in light of the fact that unlike VKOR, DsbB is specific to prokaryotic cells.236 In addition, a
secondary pair of oxidoreductase enzymes, DsbE and DsbF, has been identified in M.
tuberculosis.236,316–318 However, unlike the primary pathway, these proteins are not essential for M.
tuberculosis survival and their function remains unclear.236,316–318

76
Other examples in Gram-positive bacteria include the MdbA-VKOR functional pairs identified in
Actinomyces and Corynebacterium diphtheriae or the BsBdb proteins of B. subtilis.236 Notably, the
four-protein Bdb system in B. subtilis closely resembles the oxidative pathway of E. coli, with BsBdbA
and BsBdbD being EcDsbA-like and BsBdbB and BsBdbC being similar to EcDsbB-like proteins.236,319
In other species, such as S. aureus or Listeria monocytogenes, disulfide formation occurs through
EcDsbA orthologues that appear to be functionally independent of other enzymes.216,320 Despite
numerous studies of disulfide bond formation in Gram-positive species, no isomerisation pathway has
been identified to date, pointing to a more limited disulfide bond formation system or to a lack of
proteins with more than two cysteines in these organisms.
1.6.5 Targeting bacterial pathogenicity through inhibition of the DSB system and oxidative
folding
While polymorphisms in the DSB proteins across the bacterial phylogeny are not yet fully characterised,
the conserved function of archetypical DSB proteins, which fold hundreds of proteins in the cell
envelope, has been well established. In the case of pathogenic species, DsbA is essential for the stability
and folding of numerous virulence factors, including secretion systems, toxins and pili that are required
for bacterial pathogenicity.215,216,236,246,251 Most notably, some human pathogens, such as Shigella
flexnerii or P. aeruginosa, depend on oxidative protein folding for intracellular survival, pointing to a
more generalized role of the DSB system in the survival of bacteria in their environmental niches.309,321
These observations open the avenue of abrogating the dangers posed by bacterial pathogens through the
use of the DSB system as a target for broad-acting anti-virulence strategies that do not impair bacterial
viability.216,236,306,322–324
Several strategies for targeting bacterial virulence through inhibiting the DSB system have been
proposed in the literature. These include both DsbA and DsbB as targets, and can be broadly divided
into three approaches: a) fragment-based discovery for targeting DsbA or DsbB, b) development of
peptide and peptidomimetic inhibitors of DsbA and c) high-throughput screening methods for DsbB
inhibitors.322,325–327 Whilst several chemical inhibitors have been identified for both DsbA and DsbB,
no clinically suitable compounds have been developed to date.

77
1.6.5.1 Fragment-based discovery of small molecule inhibitors against DsbA and DsbB
Fragment-based drug discovery is a commonly used technique that allows the screening of a library of
compounds against a pharmaceutical target of interest. Historically, membrane-embedded proteins such
as EcDsbB were unsuitable for this technique as it required large quantities of purified and solubilised
target protein.322,326,328 Früh et al. have bypassed these limitations by employing a target-immobilized
NMR screening (TINS) strategy. With this technique, EcDsbB was solubilised in detergent micelles
prior to its immobilisation onto a resin support. Normalisation of the results enabled the identification
of several candidate compounds with two distinct modes of action, compounds that were perturbing the
DsbB-UQ binding or molecules that were disrupting the binding of EcDsbB to both UQ and EcDsbA
(Figure 1.15).326,328 The work of Früh et al., and in particular Compound 2 (Figure 1.15 A), was used
for structure-activity-relationship (SAR)-based optimisation studies by Halili et al. and resulted in the
development of Compound 19.326,328,329 These molecules represent the first active inhibitors against the
DSB system. Though, the exact mode of action of Compound 19 is unconfirmed, Halili et al. show that
this UQ derivative binds a reduced cysteine residue and covalently inhibits either EcDsbA and/or
EcDsbB.329 Further, and more importantly, this work confirmed the possibility of developing
prokaryote-specific anti-DSB compounds and gave rise to further DSB anti-virulence efforts.329
Figure 1.15 Fragment based screening identified the first active inhibitors of the DSB system.326,328,330 Compound 2 and
19 by identified by Halili et al. and Adams et al. target the transmembrane EcDsbB while compound 40 acts as a competitive
inhibitor of EcDsbA.326,328,330 These small molecules confirmed the possibility of inhibiting the DSB system without affecting
cell viability and provide the a platform for further drug design.

78
Simultaneously to Halili et al., Adams et al. used fragment-based discovery to identify EcDsbA specific
inhibitors.330 Positive hits were narrowed down to a single halogen-substituted phenyl thiazole
compound which was investigated further using SAR studies and resulted in the isolation of a
competitive inhibitor binding within the hydrophobic groove below the EcDsbA active site (Compound
40, Figure 1.15).330 Chemical treatment using this molecule resulted in the inhibition of cell motility but
did not affect bacterial viability, thus confirming the second key consideration of anti-virulence
therapies.324,326,330
More recently Totsika et al. expanded on this work to consider DSB system inhibition in pathogens
with multiple copies of DsbA.322 Use of phenylthiophene- and phenoxyphenol-based inhibitors of
EcDsbA resulted in more variable and less pronounced motility effects in UPEC (EcDsbA and EcDsbL)
and S. typhimurium (SeDsbA and SeSrgA) strains.322 This highlighted the potential challenges ahead
for the development of broad-acting inhibitors against the DSB pathway.
1.6.5.2 Designing peptide and peptidomimetic inhibitors of DsbA
A different approach for achieving DsbA inhibition was taken by Duprez et al. who took advantage of
our in-depth structural understanding of the mechanism leading to DsbA-DsbB complex formation,
supported by high-resolution structures, in an attempt to develop peptide-based DsbA inhibitors.327,331
A synthetic peptide, ‘Pro-Phe-Ala-Thr-Cys-Asp-Ser’, that mimics the P2 loop of DsbB was used to
further the understanding behind DsbA-DsbB complex formation and was confirmed to bind in the
hydrophobic groove of DsbA.327 Targeted design led to the development of the DsbA-specific peptide
‘Pro-Trp-Ala-Thr-Cys-Asp-Ser’.327 SAR studies showed that the cysteine residue was critical for
binding suggesting that covalent peptides may be suitable and potent candidates for DSB inhibition.327
Interestingly, this peptide was also active against the Proteus mirabilis DsbA (PmDsbA).332
Virtual screening of a peptidomimetic library, using the ‘Pro-Trp-Ala-Thr-Cys-Asp-Ser’ as a template,
yielded additional scaffolds suitable for DsbA inhibition. In comparison to the rational drug design, like
the strategy described above, this method identified several small molecule fragments that acted as weak
non-covalent inhibitors (Figure 1.16).331 Though not investigated further, these peptide-like scaffolds
could benefit from mimicking the now known protein-protein interactions, while evading the issues
associated with many peptide-based therapeutics, including poor oral bioavailability, low stability or
high toxicity.326,331

79
Figure 1.16 Peptidomimetic library screening identified EcDsbA inhibitors. Scaffold design took advantage of our in-
depth knowledge of the DsbA-DsbB interaction, gleaned from multiple high-resolution structures, and is based on a ‘Pro-Trp-
Ala-Thr-Cys-Asp-Ser’ template. These preliminary inhibitors open up the possibility of SAR modifications that could results
in improved specificity, bioavailability, and stability - issues commonly observed with peptide-based therapeutic
candidates.326,331
1.6.5.3 High throughput screening for small molecule inhibitors of DsbB
Cell-based high-throughput screening methods have allowed the identification of in vivo DSB system
inhibitors from large libraries of compounds. 250, 000 potential inhibitor compounds were screened
using a chromogenic -galactosidase assay.325,333 Here, the usually cytoplasmic -galactosidase enzyme
was targeted to the periplasm where it became oxidised by DsbA.325 Introduction of a non-native
disulfide bond inhibits enzymatic function and prevents X-Gal hydrolysis.325 Absence, or inhibition of
the DSB system, rescues -galactosidase activity and results in blue pigment formation.325
A first study resulted in the identification of several EcDsbB inhibitors, and their further development
led to the identification of Compound 12 (Figure 1.17).325 The activity of this compound was specific
to Gram-negative bacteria, and inhibition of the VKOR of M. tuberculosis or the human PDI were not
observed.325 Notably, Compound 12 displayed different inhibition levels for DsbB enzymes from
several Gram-negative pathogens such as A. baumannii, S. enterica, or P. aeruginosa.325 Further
investigation as part a second study showed that species-specific inhibition of P. aeruginosa and M.
tuberculosis is possible through targeted development, and resulted in Compounds PA1 and MT17,
respectively (Figure 1.17).333

80
Figure 1.17 Inhibitors of the DsbA partner proteins, DsbB and VKOR. Described in two studies by Landeta et al., inhibitor
Compounds 12 and PA1 are specific to the Gram-negative EcDsbB and PaDsbB1, while MT17 showed selective activity
against the Gram-positive MtVKOR.325,333 In all cases, no cross-activity was observed with the human PDI enzyme showing
the potential for the development of prokaryotic and species-specific inhibitors of the DSB system.325,333
It should be noted that despite the development of functional DsbB inhibitors by Landeta et al.,
successful abrogation of oxidative folding in vivo will most likely require inhibitors targeting
DsbA.325,327,333 This arises from the fact that DsbA re-oxidation due to molecular oxygen or the dipeptide
cystine, which is abundant in most biological fluids, can occur spontaneously, albeit slowly, in the
absence of DsbB, and thus partially mitigate the inhibitory effects of DsbB inhibition.334 This would be
evaded through development of efficient DsbA inhibitors, as these exogenous oxidants cannot
efficiently oxidise the substrates of DsbA.
Therapeutic approaches targeting non-essential bacterial pathways are believed to be useful in
decreasing the chance of resistance development as well as minimise deleterious effects on the host’s
natural microbiota.82,216,236,306,322–324 To date, several strategies targeting bacterial virulence have been
proposed but were limited by narrow activities and high target specificities and their use would likely
depend on the development of complex drug cocktails.82A desirable alternative would be to inhibit a
conserved broad-acting pathway, such as the DSB system in Gram-negative bacteria. The combined
work of Totsika et al., Kurth et al. and Landeta et al. identified several inhibitor compounds with activity
against both EcDsbA and EcDsbB-like proteins.322,325,327,329–333 Their work confirms the potential behind
the use of the DSB system as a target for both narrow and broad-spectrum anti-virulence agents that do
not inhibit bacterial viability, and at the same time highlights several key challenges behind this

81
approach. It is clear that to target the DSB system with the purpose of abrogating bacterial virulence, a
good understanding of its inherent polymorphisms, which are particularly prominent in pathogenic
organisms, is required.216 In addition, if species-specific DSB inhibitors are more promising, their
deployment into clinical practice will hinge on the ability to rapidly detect and identify the bacterial
species responsible for the infections of interest.
1.7 AIMS OF THIS WORK
The Gram-negative DSB system has long been perceived as a non-essential house-keeping-only system.
More recently, this system has been implicated in bacterial pathogenesis due to its role in folding and
safeguarding the stability and function of many key virulence factors across different bacterial species.
For example, oxidative folding, catalysed by the primary oxidase DsbA, has been shown to be essential
for the function of V. cholerae toxins, the type III secretion in S. enterica and P. aeruginosa as well as
the formation of fimbriae in uropathogenic E. coli.309,335–337
While the role of the DSB system proteins in bacterial virulence is now broadly accepted, and chemical
inhibitors of the DSB proteins have even been generated with the hope to abrogate virulence, very little
is known about the contribution of the DSB proteins to mechanisms of antimicrobial resistance, despite
the fact that several of these mechanisms are protein-based, contain more than one cysteine residue, and
are located in the cell envelope. Thus, the aims of this work are to:
1. Investigate the effects of loss of DsbA on antibiotic resistance mediated by cysteine-containing
mobile class D OXA-type -lactamase enzymes.
2. Investigate the role of DsbA in intrinsic antibiotic resistance conferred by cysteine-containing
chromosomally-resident -lactamase enzymes.
3. Characterise the relationship between efflux-pump mediated resistance and cell envelope
protein homeostasis safeguarded by the DSB proteins.
4. Understand the role of disulfide bonds in the mutational evolution capacity of extended-
spectrum -lactamase enzymes.

82
2 MATERIALS AND METHODS
2.1 REAGENTS AND BACTERIAL GROWTH CONDITIONS
Unless otherwise stated, chemicals and reagents were acquired from Sigma Aldrich, growth media were
purchased from Oxoid and antibiotics were obtained from Melford Laboratories. Lysogeny broth (LB)
(10 g/L peptone, 5 g/L yeast extract, 10 g/L NaCl) and agar (1.5% w/v) was used for routine growth of
all organisms at 37°C with shaking at 220 RPM, except for S. maltophilia strains which were routinely
grown in low-salt LB (Luria protocol, 10 g/L peptone, 5 g/L yeast extract, 0.5 g/L NaCl).
Mueller-Hinton (MH, 300 g/L beef dehydrate infusion, 17.5 g/L casein hydrolysate, 1.5 g/mL starch)
broth and agar (1.5% w/v) were used for Minimum Inhibitory Concentration (MIC) assays. Growth
media were supplemented with the following, as required: 0.25 mM Isopropyl β-D-1-
thiogalactopyranoside (IPTG) (for strains harbouring β-lactamase-encoding pDM1 plasmids), 12.5
g/mL tetracycline, 33 g/mL chloramphenicol, 100 g/mL ampicillin, 50 g/mL kanamycin, 10
g/mL gentamicin, 50 g/mL streptomycin (for cloning purposes), 2000-6000 g/mL streptomycin (for
the construction of P. aeruginosa mutants), and 6000 g/mL streptomycin (for the construction of a S.
maltophilia mutant).
2.2 GENETIC MANIPULATION TECHNIQUES
2.2.1 Genomic DNA extraction, purification of plasmid DNA and of PCR products
Extraction of genomic DNA was carried out from a single bacterial colony harbouring the β-lactamase
gene of interest. The colony was resuspended in 100 L of molecular-biology-grade H2O and boiled at
100°C for 10 minutes. The lysed cells were centrifuged on a tabletop centrifuge (13 500 rpm, 3 min)
and the DNA-containing supernatant was used as template for PCR amplification.
Purification of plasmid DNA was carried out using the QIAprep Spin Miniprep Kit (Qiagen), according
to the manufacturer’s instructions. Briefly, overnight cultures were centrifuged to remove the culture
media and resultant cell pellets were resuspended in the P1 Resuspension Buffer containing a RNase I.

83
Subsequently, the alkaline P2 Lysis Buffer was added to lyse the cells. Addition of the N3 Neutralisation
Buffer followed by a 10-minute centrifugation at 13, 000 rpm was used to separate the cellular debris,
including membranes, proteins and genomic DNA, from the plasmid DNA. Supernatants were applied
to QIAquick silica membrane spin columns allowing binding of plasmid DNA. Bound plasmid DNA
was washed with Buffer PB and ethanol-based Buffer PE and then eluted into molecular-biology-grade
H2O.
Purification of PCR products was carried out using the QIAquick PCR Purification Kit PCR according
to the manufacturer’s instructions. Briefly, amplified DNA was mixed with Buffer PB, in a 1:5 ratio,
and applied to a QIAquick silica membrane spin column. The column was then washed with Buffer PE
and eluted in molecular-biology-grade H2O.
Purification of restriction digestion products was carried out using the QIAquick Gel Extraction Kit
(Qiagen) according to the manufacturer’s instructions. Briefly, DNA molecules were separated using
agarose gel electrophoresis and the fragment of interest was excised using a clean scalpel. The gel piece
was dissolved in Buffer QG in 1:3 w/v ratio at 50°C and mixed with 1 gel volume of isopropanol. The
solution was applied to a QIAquick silicon membrane spin column which was washed with Buffers PB
and PE and the DNA was eluted using molecular-biology-grade H2O.
2.2.2 PCR amplification
KOD DNA polymerase (Merck) was used for all the cloning steps described in this thesis. Generally,
the PCR reactions were carried out using the following reaction mixture composition: 5 L 10X buffer,
5 L dNTP mix (2mM of each base) mix, 3 L 2mM MgCl2, 1 L KOD DNA polymerase, 1.5 L of
each primer (in an appropriate dilution), 2 L genomic DNA or 1 L plasmid DNA. The reaction
mixture was made up to 50 L with molecular-biology-grade H2O. For cloning of genes with high %GC
content 5 L DMSO was included in the reaction mixture. For cloning using genomic DNA from
Pseudomonas or Stenotrophomonas isolates, 5 L betaine was added.

84
Standard thermo-cycling conditions used were as follows:
1. Hot start denaturation - 95°C, 2 minutes, 1 cycle
2. Denaturation - 95°C, 30 seconds, 30 cycles
3. Annealing - 50-65°C, 20 seconds, 30 cycles
4. Extension - 70°C, 20 seconds per amplified kb, 30 cycles
5. Final extension - 95°C, 5-10 minutes, 1 cycle
6. Storage - 4°C, up to 14 hours
OneTaq DNA polymerase (New England BioLabs) was standardly used for PCR amplification during
colony screening for the construction of dsbA1 and dsbA1 dsbL1 mutants of P. aeruginosa and S.
maltophilia clinical isolates, respectively. These reactions were carried out using the following reaction
mixture composition: 7.5 L OneTaq DNA Polymerase, 2.5 L betaine, 0.75 L of each primer (in
appropriate dilution), 1 L genomic DNA or 1 colony. The reaction mixture was made up to 25 L with
molecular-biology-grade H2O. Standard thermo-cycling conditions were used as described above,
except for with increased extension time of 1 minute per amplified kb.
For both PCR reaction setups, the annealing temperature and time, as well as the extension time were
adapted as required for specific genes and primer combinations.
2.2.3 Agarose gel electrophoresis
Agarose gel slabs were made in TAE buffer (40 mM Tris-acetate (pH 8.0), 1 mM EDTA) by adding 1
% agarose and 50 nL/ml SybrSafe while DNA samples were added into 6 x gel loading dye (10 mM
EDTA, 50 % v/v glycerol, 0.5 % bromophenol blue). Electrophoresis was performed in TAE buffer at
a constant current of 70 mA. DNA bands were revealed by SybrSafe fluorescence under UV light.
2.2.4 Restriction digestion
DNA restriction digestions were standardly performed with high-fidelity restriction endonuclease
enzymes (New England Biolabs) and the following reaction mixture composition: 5 L CutSmart®
buffer, 1000 ng DNA template, 1 L of each restriction digestion enzyme. The reaction mixture was
made up to 50 L with molecular-biology-grade H2O. Reactions were incubated at 37°C for 2 hours,

85
analysed using agarose gel electrophoresis and purified before further use. Where high fidelity enzymes
were not available, stepwise digestion with standard restriction digestion enzymes followed by gel
extraction purification after every step was performed.
2.2.5 Ligation
DNA ligations were standardly performed at 16°C overnight using T4 DNA ligase (New England
Biolabs) and the following reaction mixture composition: 5 L 1x T4 DNA Ligase Reaction Buffer, 0.2
L T4 DNA ligase, 50 ng vector, DNA insert at a 5:1 ratio to the vector. The reaction mixture was made
up to 20 L with molecular-biology-grade H2O. Ligation reactions were stored at 4°C and used directly
for transformation into competent cells.
2.2.6 Site-directed mutagenesis
Site-directed mutagenesis was performed using the QuickChange site-directed mutagenesis method
(Stratagene) as per the manufacturer’s instruction manual. Briefly, KOD DNA polymerase was used to
amplify plasmid DNA using QuickChange primers and the following reaction mixture composition: 5
L 10X buffer, 5 L dNTP mix (2mM of each base), 3 L 2mM MgCl2, 1 L KOD DNA polymerase,
125 ng per primer, 1/5/10 ng plasmid DNA. The reaction mixture was made up to 50 L with molecular-
biology-grade H2O.
Standard thermo-cycling conditions used were as follows:
1. Hot start - 95°C, 2 minutes, 1 cycle
2. Denaturation - 95°C, 30 seconds, 18 cycles
3. Annealing - 55°C, 20 seconds, 18 cycles
4. Extension - 70°C, 3-5 minutes, 18 cycles
5. Final extension - 95°C, 10 minutes, 1 cycle
6. Storage - 4°C, up to 14 hours
Following thermal cycling, the methylated template DNA was digested by addition of 1 L Dpn I (New
England Biolabs) and one-hour incubation at 37°C. Resultant amplified plasmids were directly

86
transformed into chemically competent E. coli DH5, selected on antibiotic-supplemented LB, and
confirmed by DNA sequencing.
2.2.7 DNA sequencing
All the plasmids used in this work were sequenced to confirm that only the desired genes had been
incorporated. Plasmid DNA Sanger sequencing was carried out externally by Eurofins Genomics.
2.2.8 Preparation and transformation of chemically competent cells
E. coli strains to be made chemically competent were grown in Super Optimal Broth (SOB, 20 g/L
peptone, 5 g/L yeast extract, 584 mg/L NaCl, 184 mg/L KCl, 2.03 g/L MgCl2, 2.44 g/L MgSO4)
overnight, sub-cultured 1:100 in SOB and grown to optical density at 600 nm (OD600) 0.45. The cultures
were incubated on ice for 30 minutes before centrifugation at 4°C and 2 000 x g for 15 minutes. Cell
pellet was resuspended in 66 mL of Buffer RF1 (12 g/L RbCl, 9.9 g/L MnCl2•H2O, 2.95 g/L K(OAc),
1.5 g/L CaCl2•H2O, 150 g/L glycerol; pH 5.8 with AcOH; filter sterilised) and incubated on ice for 1
hour. Cell pellets obtained from a second centrifugation step were resuspended in 16 mL of Buffer RF2
(2.09 g/L, MOPS, 1.2 g/L RbCl, 11 g/L CaCl2•H2O, 150 g/L glycerol; pH 6.8 with NaOH; filter
sterilised) and incubated on ice for 15 minutes. The cell suspension was aliquoted in 220 L aliquots
and frozen on liquid N2.
E. coli DH5 chemically competent cells were routinely used for transformations for cloning and site-
directed mutagenesis purposes. E. coli MC1000, MC1000 dsbA, and MC1000 dsbA atTn7 dsbA
competent cells were used for transformation with plasmids carrying -lactamase genes for experiments
described in Chapters 3, 4, and 6. In both cases the following steps were performed: 10 L of ligation
reaction mixture / 1 L of plasmid DNA / 50 ng linear DNA were added to 50 L of cells and incubated
on ice for 15 minutes. Cells were then subjected to a 30-45 second heat shock at 42 °C and cooled on
ice for 2 minutes. After addition of 900 L SOB, a one-hour recovery step was carried out at 37°C
before transformants were selected on antibiotic-supplemented LB agar overnight.

87
2.2.9 Preparation and transformation of electrocompetent cells
Overnight cultures of cells to become electrocompetent were centrifuged at 4°C and 4000 x g for 10
minutes in order to remove culture media. Cell pellets were then washed three times with ice-cold sterile
MQ H2O, resuspended in 100 L ice-cold sterile MQ H2O and used immediately. 2 L of plasmid or
linear DNA were routinely electroporated into E. coli MG1655, Pseudomonas or Stenotrophomonas
strains in 1mm electroporation cuvettes at 25 FD, 200 , and 1.5 V. Cells were recovered in 900 L
SOB at 30°C for 3 hours or at 37°C for 1 hour as required. Transformants were selected for on antibiotic-
supplemented LB agar overnight.
2.3 BACTERIAL STRAINS AND PLASMIDS
Bacterial strains, plasmids and oligonucleotides described in this thesis are listed in Table 2, Table 3
and Table 4, respectively.
Table 2. Bacterial strains used in this thesis. All listed strains in the “Clinical isolates / laboratory strains” section are clinical
strains except for P. aeruginosa PAO1LA and P. aeruginosa PAO1LD, which are laboratory strains. FNRCAR refers to the
French National Reference Centre for Antibiotic Resistance in Le Kremlin-Bicêtre, France. CRBIP stands for Centre de
Ressources Biologiques de l’Institut Pasteur, France.
Name Description Source
Escherichia coli
DH5α F– endA1 glnV44 thi-1 recA1 relA1
gyrA96 deoR nupG purB20
φ80dlacZ∆M15 ∆(lacZYA-argF)U169
hsdR17(rK–mK
+) λ–
344
CC118λpir araD Δ(ara, leu) ΔlacZ74 phoA20 galK
thi-1 rspE rpoB argE recA1 λpir 345
HB101 supE44 hsdS20 recA13 ara-14 proA2
lacY1 galK2 rpsL20 xyl-5 mtl-1 346
MC1000 araD139 ∆(ara, leu)7697 ∆lacX74
galU galK strA 347
MC1000 dsbA dsbA::aphA, KanR 251
MC1000 dsbA attTn7::Ptac-dsbA dsbA::aphA attTn7::dsbA, KanR 338
MG1655 K-12 F– λ– ilvG– rfb-50 rph-1 348
MG1655 dsbA dsbA::aphA, KanR This study
MG1655 dsbA attTn7::Ptac-dsbA dsbA::aphA attTn7::dsbA, KanR This study
MG1655 acrA acrA 338

88
MG1655 tolC tolC 338
MG1655 degP degP::strAB, StrR This study
MG1655 marR marR::accC, GentR This study
MG1655 dsbA marR dsbA::aphA marR::accC, KanR, GentR This study
Clinical isolates / laboratory strains
Pseudomonas aeruginosa SOF-1 blaOXA-4 349
Pseudomonas aeruginosa PU21 blaOXA-10 350
Pseudomonas aeruginosa PAe191 blaOXA-19 351
Pseudomonas aeruginosa PA43417 blaOXA-198 352
Pseudomonas aeruginosa PA43417 dsbA1 dsbA1 blaOXA-198 This study
Pseudomonas aeruginosa 51170 blaBEL-1 353
Pseudomonas luteola CIP 102067 blaLUT-1 CRBIP
Pseudomonas aeruginosa G4R7 blaAIM-1 FNRCAR
Pseudomonas aeruginosa G4R7 dsbA1 dsbA1 blaAIM-1 This study
Pseudomonas aeruginosa G6R7 blaAIM-1 FNRCAR
Pseudomonas aeruginosa G6R7 dsbA1 dsbA1 blaAIM-1 This study
Stenotrophomonas maltophilia GUE blaL2-1 blaL1-1 354
Stenotrophomonas maltophilia GUE dsbA dsbL dsbA dsbL blaL2-1 blaL1-1 This study
Pseudomonas otitidis CIP 109236T blaPOM-1 CRBIP
Pseudomonas aeruginosa PA14 blaOXA-50 355
Pseudomonas aeruginosa PA14 dsbA1 dsbA1 blaOXA-50 This study
Pseudomonas aeruginosa PAO1 LA blaOXA-50 356
Pseudomonas aeruginosa PAO1 LA dsbA1 dsbA1 blaOXA-50 This study
Pseudomonas aeruginosa PAO1 LD blaOXA-50 350
Pseudomonas aeruginosa PAO1 LD dsbA1 dsbA1 blaOXA-50 This study

89
Table 3. Plasmids used in this thesis.
Name Description Source
pDM1 pDM1 vector (GenBank MN128719), p15A ori, Ptac
promoter, lacI, MCS, TetR
Mavridou
lab
pDM2 pDM1 derivative, p15A ori, BioFab promoter, MCS, TetR Mavridou
lab
pDM1-blaL2-1 blaL2-1 cloned into pDM1, TetR 338
pDM1-blaOXA-4 blaOXA-4 cloned into pDM1, TetR This study
pDM1-blaOXA-10 blaOXA-10 cloned into pDM1, TetR This study
pDM1-blaOXA-198 blaOXA-198 cloned into pDM1, TetR This study
pDM1-blaBEL-1 blaBEL-1 cloned into pDM1, TetR This study
pDM1-blaBPS-1m blaBPS-1m cloned into pDM1, TetR This study
pDM1-blaCARB-2 blaCARB-2 cloned into pDM1, TetR This study
pDM1-blaFTU-1 blaFTU-1 cloned into pDM1, TetR This study
pDM1-blaLUT-1 blaLUT-1 cloned into pDM1, TetR This study
pDM1-blaAIM-1 blaAIM-1 cloned into pDM1, TetR This study
pDM1-blaPOM-1 blaPOM-1 cloned into pDM1, TetR This study
pDM1-blaSMB-1 blaSMB-1 cloned into pDM1, TetR This study
pDM1-blaL1-1 blaL1-1 cloned into pDM1, TetR 338
pDM1-blaOXA-50 blaOXA-50 cloned into pDM1, TetR This study
pDM1-StrepII-blaOXA-4 blaOXA-4 encoding OXA-4 with an N-terminal StrepII tag
cloned into pDM1, TetR This study
pDM1-blaOXA-10-StrepII blaOXA-10 encoding OXA-10 with a C-terminal StrepII tag
cloned into pDM1, TetR This study
pDM1-blaOXA-198-StrepII blaOXA-198 encoding OXA-198 with a C-terminal StrepII
tag cloned into pDM1, TetR This study
pDM1-blaBEL-1-StrepII blaBEL-1 encoding BEL-1 with a C-terminal StrepII tag
cloned into pDM1, TetR This study
pDM1-blaBPS-1m-StrepII blaBPS-1m encoding BPS-1m with a C-terminal StrepII tag
cloned into pDM1, TetR This study
pDM1-blaCARB-2-StrepII blaCARB-2 encoding CARB-2 with a C-terminal StrepII tag
cloned into pDM1, TetR This study
pDM1-blaFTU-1-StrepII blaFTU-1 encoding FTU-1 with a C-terminal StrepII tag
cloned into pDM1, TetR This study
pDM1-blaLUT-1-StrepII blaLUT-1 encoding LUT-1 with a C-terminal StrepII tag
cloned into pDM1, TetR This study
pDM1-blaAIM-1-StrepII blaAIM-1 encoding AIM-1 with a C-terminal StrepII tag
cloned into pDM1, TetR This study
pDM1-blaPOM-1-StrepII blaPOM-1 encoding POM-1 with a C-terminal StrepII tag
cloned into pDM1, TetR This study
pDM1-blaSMB-1-StrepII blaSMB-1 encoding SMB-1 with a C-terminal StrepII tag
cloned into pDM1, TetR This study
pDM1-blaL2-1-StrepII blaL2-1 encoding L2-1 with a C-terminal StrepII tag cloned
into pDM1, TetR 338
pDM1-blaL1-1-StrepII blaL1-1 encoding L1-1 with a C-terminal StrepII tag cloned
into pDM1, TetR 338

90
pDM1-blaOXA-50-StrepII blaOXA-50 encoding OXA-50 with a C-terminal StrepII tag
cloned into pDM1, TetR This study
pDM2-blaSHV-1 blaSHV-1 cloned into pDM2, TetR This study
pDM2-blaSHV-1 C54A blaSHV-1 C54A cloned into pDM2, TetR This study
pDM2-blaTEM-1 blaTEM-1 cloned into pDM2, TetR This study
pDM2-blaTEM-1 C86A blaTEM-1 C86A cloned into pDM2, TetR This study
pGRG25-Ptac::dsbA
Ptac::dsbA fragment cloned within the Tn7 of pGRG25;
when inserted into the chromosome and the plasmid
cured, the strain expresses DsbA upon IPTG induction,
AmpR
338
pSLTS Thermosensitive pSC101ori, ParaB for λ-Red, PtetR for
I-SceI, AmpR 339
pUltraGFP-GM
Constitutive sfGFP expression from a strong Biofab
promoter, p15A ori, (template for the accC cassette),
GentR
357
pKNG101 Gene replacement suicide vector, oriR6K, oriTRK2,
sacB, (template for the strAB cassette), StrR 342
pKNG101-dsbA-PA
PCR fragment containing the regions upstream and
downstream P. aeruginosa dsbA1 cloned in pKNG101;
when inserted into the chromosome the strain is a
merodiploid for dsbA1 mutant, StrR
338
pKNG101-dsbA dsbL-SM GUE
PCR fragment containing the regions upstream and
downstream S. maltophilia GUE dsbA and dsbL genes
cloned in pKNG101; when inserted into the chromosome
the strain is a merodiploid for dsbA dsbL mutant, StrR
This study
pRK600 Helper plasmid, ColE1 ori, mobRK2, traRK2, ClR 358
pCB112 CPRG membrane integrity assay vector, lacIq Plac ::lacZ ,
MCS, ClR 359
pMK-RQ bps-1m GeneArt® cloning vector containing bps-1m, ColE1 ori,
(template for bps-1m), KanR This study
pMK-RQ carb-2 GeneArt® cloning vector containing carb-2, ColE1 ori,
(template for carb-2), KanR This study
pMK-T ftu-1 GeneArt® cloning vector containing ftu-1, ColE1 ori,
(template for ftu-1), KanR This study
pMK-RQ smb-1 GeneArt® cloning vector containing smb-1, ColE1 ori,
(template for smb-1), KanR This study
pMQ-RQ shv-1 GeneArt® cloning vector containing shv-1, ColE1 ori,
(template for shv-1), KanR This study
pMK-T tem-1 GeneArt® cloning vector containing tem-1, ColE1 ori,
(template for tem-1), KanR This study

91
Table 4. Oligonucleotide primers used in this thesis. The “Brief description” column provides basic information on the
primer design (restriction enzyme used for cloning, encoded protein or gene replaced by antibiotic resistance cassette, forward
or reverse orientation of the primer (F or R); QC stands for QuickChange primers and SQ stands for sequencing primers).
Number Brief description Sequence (5ˊ-3ˊ)
P1 SacI.OXA-4.F ctggagctcaaaaacacaatacatatcaacttcgc
P2 KpnI.OXA-4.R cagggtaccttataaatttagtgtgtttagaatggtg
P3 SacI.OXA-10.F ctggagctcaaaacatttgccgcatatgtaattatcgc
P4 KpnI.OXA-10.R cagggtaccttagccaccaatgatgccctc
P5 SacI.OXA-18.F ctggagctccaacggagcctgtccatga
P6 KpnI.OXA-18.R cagggtacctcagaagttttccgacagggc
P7 NdeI.OXA-198.F actgcatatgcataaacacatgagtaagctcttc
P8 KpnI.OXA-198.R ctgggtaccttattcgatcccctttgctt
P9 SacI.BEL-1.F ctggagctcaaactgctctacccgttattgc
P10 PstI.BEL-1.R cagctgcagtcagtgaacatattgacgtgc
P11 SacI.BPS-1m.F ctggagctcaatcattctccgttgcgccgctc
P12 XmaI.BPS-1m.R caacccgggtcaggcgaacgcccgcgcg
P13 SacI.CARB-2.F ctggagctcaagtttttattggcattttcgc
P14 KpnI.CARB-2.R cagggtacctcagcgcgactgtgatgta
P15 SacI.FTU-1.F ctggagctccgtctattagttacaactttatc
P16 XmaI.FTU-1.R ctgcccgggttatttataagtgttagtcagatc
P17 SacI.LUT-1.F ctggagctcaatgtcatcctgaaccgtcga
P18 PstI.LUT-1.R cagctgcagtcagcctgtcacccattcag
P19 SacI.AIM-1.F ctggagctcaaacgtcgcttcaccctgg
P20 KpnI.AIM-1.R ctgggtacctcaaggccgcgcgccgctg
P21 SacI.POM-1.F ctggagctccgtaccctgaccctcg
P22 KpnI.POM-1.R cagggtaccttatgcgtcatcagagacctc
P23 NdeI.SMB-1.F cagctccatatgaaaatcatcgcttccctgatcc
P24 XmaI.SMB-1.R ctgcccgggtcagcgtttctcgctggcca
P25 SacI.OXA-50.F ctggagctccgccctctcttcagtg
P26 KpnI.OXA-50.R cagggtacctcagggcagtatcccgagag
P27 QC.BPS-1m.F gaattcgcccttctgcccgggtcagcgtttc
P28 QC.BPS-1m.R gaaacgctgacccgggcagaagggcgaattc
P29 QC.FTU-1.F acccgggctatttttcaaattgcggatggctcc
P30 QC.FTU-1.R ggagccatccgcaatttgaaaaatagcccgggt
P31 QC.LUT-1.F ttgggtgacatgctggctgcggataa
P32 QC.LUT-1.R ttatccgcagccagcatgtcacccaa
P33 KpnI.StrepII.OXA-4.R cagggtaccttatttttcaaattgcggatggctccaagcgct
ccctaaatttagtgtgtttagaatggtgatc

92
P34 signalseq.StrepII.OXA-4.body.R tgcaacagtagagatatctgttgatttttcaaattgcggatggctccaagcgctcc
ctgcactggcgctgctgta
P35 OXA-4.body.F tcaacagatatctctactgttgca
P36 KpnI.StrepII.OXA-10.R cagggtaccttatttttcaaattgcggatggctccaagcgctcccgccaccaatg
atgccctcacttg
P37 KpnI.StrepII.OXA-18.R cagggtaccttatttttcaaattgcggatggctccaagcgctcccgaagttttccg
acagggcc
P38 KpnI.StrepII.OXA-198.R ctgggtaccttatttttcaaattgcggatggctccaagcgctcccttcgatcccctt
tgcttg
P39 PstI.StrepII.BEL-1.R cagctgcagttatttttcaaattgcggatggctccaagcgctcccgtgaacatatt
gacgtgctaac
P40 XmaI.StrepII.BPS-1m.R ctgcccgggctatttttcaaattgcggatggctccaagcgctcccggcgaacgc
ccgcgcggcg
P41 KpnI.StrepII.CARB-2.R cagggtaccttatttttcaaattgcggatggctccaagcgctcccgcgcgactgt
gatgtataa
P42 XmaI.StrepII.FTU-1.R ctgcccgggctatttttcaaattgcggatggctccaagcgctccctttataagtgtt
agtcagatcattag
P43 KpnI.StrepII.AIM-1.R cagggtaccttatttttcaaattgcggatggctccaagcgctcccaggccgcgc
gccgctggag
P44 KpnI.StrepII.POM-1.R cagggtaccttatttttcaaattgcggatggctccaagcgctcccgccgcgctgc
ttc
P45 XmaI.StrepII.SMB-1.R ctgcccgggctatttttcaaattgcggatggctccaagcgctcccgcgtttctcgc
tggccag
P46 KpnI.StrepII.OXA-50.R cagggtaccttatttttcaaattgcggatggctccaagcgctcccgggcagtatc
ccgagagcc
P47 QC.SHV-1.C54A.F cacgtgccagaactgcaccagccagaacaactttaaaggtg
P48 QC.SHV-1.C54A.R cacctttaaagttgttctggctggtgcagttctggcacgtg
P49 QC.TEM-1.C86A.F cggctcagaactgcaccagccagcagaactttaaagg
P50 QC.TEM-1.C86.R cctttaaagttctgctggctggtgcagttctgagccg
P51 NotI.Ptac.EcDsbA.F
ctggcggccgctgacaattaatcatcggctcgtataatgtgtggaattgtgacta
gtcgaggtccaggacctcggatcgctaagataggatgattgtatgaaaaagattt
ggctggc
P52 XhoI.EcDsbA.R ctgctcgagttattttttctcggacagatatttc
P53 EcdsbA::aphA.F atgaaaaagatttggctggcgctggctggtttagttttagcgtttagcgcgtgtag
gctggagctgcttc
P54 EcdsbA::aphA.R ttattttttctcggacagatatttcactgtatcagcatactgctgaacaagggaatta
gccatggtccat
P55 EcdegP::strAB.F atgaaaaaaaccacattagcactgagtgcactggctctgagtttaggtttggaac
tgcacattcgggatatttctc
P56 EcdegP::strAB.R ttactgcattaacaggtagatggtgctgtcgccgcgctgaatgttgagtgccagg
ccggatctagatatctagtatga
P57 EcmarR::accC.F atggttaatcagaagaaagatcgcctgcttaacgagtatctgtctccgctggtga
agttcctatactttctagagaataggaacttcaagatcccctg
P58 EcmarR::accC.R ttacggcaggactttcttaagcaaatactcaagtgttgccacttcgtccgcgaagt
tcctattctctagaaagtataggaacttcacttactcaatggaattctagatcg
P59 SQ.dsbA1.Paeruginosa.F tacctgctcaagcagatgcatg
P60 SQ.dsbA1.Paeruginosa.R ggtgttcatgtcgcccatca
P61 XbaI.dsbA1.Paeruginosa F ggttcctctagagcctacttcgccagccagaa
P62 dsbA1.Paeruginosa.body.R ctacttcttgttacgcatcgttcactc
P63 dsbA1.Paeruginosa.body.F atgcgtaacaagaagtaggcaaggtga

93
P64 BamHI.dsbA1.Paeruginosa.R aattaaggatcctcatcactaccaccagcgcg
P65 SQ.dsbAdsbL.Smaltophilia.F atggtgccgttcgtgcaga
P66 SQ.dsbAdsbL.Smaltophilia.R acagcacctgcatttccgg
P67 XbaI.dsbAdsbL.Smaltophilia.F ggttcctctagatcttctggtacagcacctgcatttccg
P68 dsbAdsbL.Smaltophilia.body.R tgcgtgtcgatgaggttggctcactga
P69 dsbAdsbL.Smaltophilia.body.F tctcttggatcagtgagccaacctcat
P70 BamHI.dsbAdsbL.Smaltophilia.R aattaaggatcctcgctggaggtggatttcagcaagacc
P71 SQ.pKNG101.vector.upstream ccctggatttcactgatgag
P72 SQ.pKNG1010.vector.downstream catatcacaacgtgcgtgga
2.3.1 Cloning of -lactamase genes
Genes for β-lactamase enzymes were amplified from genomic DNA extracted from clinical isolates,
with the exception of bps-1, carb-2, ftu-1, smb-1, shv-1, and tem-1, which were synthesized by GeneArt
Gene Synthesis (ThermoFisher Scientific). All β-lactamase genes, except shv-1 and tem-1, were cloned
into the IPTG-inducible plasmid pDM1 using primers P1-26 (Table 4).338 All StrepII-tag fusions of
these β-lactamases (constructed using primers P37-46, Table 4) bear a C-terminal StrepII tag
(GSAWSHPQFEK), except OXA-4. The latter has an N-terminal StrepII tag inserted between the Sec
signal peptide and the body of the protein using the primers P33-36 (Table 4). Synthesized shv-1 and
tem-1 genes were designed to include a StrepII tag sequence which was placed after the STOP codon.
The STOP codon was preceded by an AvrII restriction site and followed by a LLAGAVLCGAVLLSX
linker sequence. In this way, if needed, single restriction digestion and re-ligation steps on each
construct would generate plasmids expressing StrepII tagged versions of the enzymes. The synthesised
sequences were digested out of their respective cloning vectors and ligated into pDM2 (using SacI/XmaI
sites), whilst point mutants of each gene were obtained by QuickChange mutagenesis (using primers
P47-50, Table 4).
2.3.2 Generation of E. coli dsbA, degP and marR mutants
E. coli MG1655 dsbA, degP, and marR gene mutants were constructed using a pSLTS vector and a
modified lambda-Red recombination method, as previously described.339 Briefly, the pSLTS plasmid
was transformed into chemically competent E. coli and 10 mL of LB were innoculated with 100 L of
an E. coli pSLTS overnight and grown at 30°C for 1 hour. Lambda-Red recombinase expression was
induced using L-arabinose at final concentration of 2 mM and the culture was grown to OD600 0.7-0.9.

94
Cells were harvested by centrifugation (4500 x g), washed twice with ice-cold MQ H2O, and
resuspended in 100 L of ice cold MQ H2O. 50-bp DNA fragments upstream and downstream of the
dsbA, degP, marR genes were amplified from E. coli gDNA (primers P53-58, Table 4). 100 ng of the
linear mutation cassettes were electroporated into the prepared electro-competent E. coli pSLTS cells
and after 1 hour of incubation at 30°C the outgrowth was plated on kanamycin- and ampicillin-
supplemented LB agar for overnight incubation at 30°C. Western blot analysis was used to screen single
colonies and identify/confirm protein loss and gene deletion.
2.3.3 Generation of P. aeruginosa dsbA1 mutants
The dsbA1 mutants of P. aeruginosa laboratory and clinical strains (Table 2) were constructed by allelic
exchange, as previously described.341 Briefly, the dsbA1 gene area of all P. aeruginosa strains (including
the dsbA1 gene and 600 bp on either side of this gene) was amplified (primers P59-60, Table 4) and the
obtained DNA was sequenced to allow for accurate primer design for the ensuing cloning step.
Subsequently, 500-bp DNA fragments upstream and downstream of the dsbA1 gene were amplified
using P. aeruginosa PA43417 genomic DNA (primers P61-62 (upstream) and P63-64 (downstream),
Table 4). A fragment containing both of these regions was obtained by overlapping PCR (primers P61
and P64, Table 4) and inserted into the XbaI/BamHI sites of the pKNG101 vector. The suicide vector
pKNG101342 is not replicative in P. aeruginosa; it was maintained in E. coli CC118λpir and mobilized
into the P. aeruginosa strains by triparental conjugation (see section 2.3.5).
2.3.4 Generation of the S. maltophilia dsbA1 dsbL1 mutant
The dsbA1 dsbL1 mutant of the S. maltophilia GUE clinical isolate was constructed by allelic exchange,
as previously described.341,343 Briefly, the dsbA dsbL gene area of S. maltophilia strains (including the
dsbA dsbL genes and 600 bp on either side of these genes) was amplified (primers P65-66, Table 4) and
the obtained DNA was sequenced to allow for accurate primer design for the ensuing cloning step.
Subsequently, 600-800-bp DNA fragments upstream and downstream of the dsbA dsbL genes were
amplified using S. maltophilia genomic DNA (primers P67-68 (upstream) and P69-70 (downstream),
Table 4). A fragment containing both of these regions was obtained by overlapping PCR (primers P67
and P70) and inserted into the XbaI/BamHI sites of pKNG101. The suicide vector pKNG101342 is not
replicative in S. maltophilia; it was maintained in E. coli CC118λpir and mobilized into S. maltophilia
strains by triparental conjugation (see section 2.35) on low salt LB (Luria protocol).

95
2.3.5 Triparental conjugation of P. aeruginosa and S. maltophilia
Triparental conjugations of Pseudomonas or Stenotrophomonas clinical isolates was carried out using
an E. coli HB101 helper strain and an E. coli CC118pir donor strain transformed with the helper
pRK600 plasmid and the mutation-cassette-harbouring pKNG101 plasmid, respectively. 20 L of the
helper and donor strains were spotted on LB agar, both separately and on top of one another, and
incubated at 37°C for 2 hours. Concurrently, the recipient isolate to be conjugated was cultured at 43°C.
20 L of the recipient strain was then spotted on top of the combined helper + donor spot, as well as on
its own. The plate was incubated facing up at 37°C overnight. The donor + helper + recipient spot and
the individual spots were scraped using a sterile loop and resuspended into 1 mL of sterile PBS. Then
successful conjugants were selected on streptomycin-supplemented VBM (2% technical agar
supplemented with 200 mg/L MgSO4.7H2O; 2 g/L anhydrous citric acid; 10 g/L K2HPO4; 3.5 g/L
H5NNaO4P.4H2O; pH 7.0; filter-sterilised) media at 37°C for 24 -72 hours. Single colonies were
patched on LB agar supplemented with 20% w/v sucrose and LB agar supplemented with streptomycin
and incubated at room temperature for 24-48 hours. Colonies susceptible to sucrose, but resistant to
streptomycin, were screened by PCR (primers P61, 64, 67, 70, 71, 72; Table 4). PCR reactions could
yield two distinct band sizes, each representative of the direction in which the pKNG101 mutator
plasmid had incorporated into the bacterial chromosome. A colony where the pKNG101 mutator
plasmid was incorporated was selected and the less common orientation was selected and incubated at
30°C until turbidity was observed. 100 L was then plated on LB agar supplemented with 20% sucrose
and incubated at room temperature for 24-72 hours. Single colonies were patched on LB agar
supplemented with streptomycin, LB and Pseudomonas Isolation Agar (1.4 g/L MgCl2, 20 g/L peptic
digest of animal tissue, 10 g/L K2SO4, 0.025 g/L triclosan, 10 mL/L glycerol, 13.6 g/L agar), and
incubated at 37°C overnight. Colonies with good growth on LB and PIA with susceptibility to
streptomycin were screened for the absence of dsbA1 and dsbA1 dsbL1 (primers P61, 64, 67, 70; Table
4) and mutants were confirmed by DNA sequencing.
2.3.6 Complementation of E. coli MG1655 dsbA
To complement the E. coli dsbA mutant, dsbA was reintroduced into the E. coli chromosome at the
attTn7 site, as previously described using the pGRG25 plasmid carrying dsbA under a Ptac promoter
(Table 3).340 Briefly, pGRG25 plasmid was transformed into E. coli dsbA and a single transformant was
grown in LB supplemented with 0.3% w/v L-arabinose and ampicillin at 30°C overnight. The overnight

96
culture was diluted to 2 x 10-6, 20 L of this dilution was plated on LB agar and incubated at 42°C
overnight. Single colonies were sub-cultured on ampicillin-supplemented LB to check for plasmid
absence. Western blot analysis was then used to confirm dsbA complementation.
2.4 MINIMUM INHIBITORY CONCENTRATION (MIC) ASSAYS
Unless otherwise stated, β-lactam MIC assays were carried out in accordance with the EUCAST
recommendations using E-test strips (BioMérieux). Briefly, overnight cultures of each strain to be tested
were standardized to OD600 0.063 in 0.85% NaCl (equivalent to McFarland standard 0.5) and distributed
evenly across the surface of MH agar plates. E-test strips were placed on the surface of the plates, evenly
spaced, and the plates were incubated for 18-24 hours at 37°C. MICs were read according to the
manufacturer’s instructions. β-lactam and vancomycin MICs were also determined using the Broth
dilution (BD) method, as required. Briefly, a series of antibiotic concentrations was prepared by two-
fold serial dilution in MH broth in a clear-bottomed 96-well microtiter plate (Corning) or 7 mL bijou
containers. The strain to be tested was added to the wells/containers at approximately 5 x 105 colony
forming units (CFU) per well and incubated for 18-24 hours at 37°C. When 7mL bijou containers were
used, shaking of the cultures was performed at 220 RPM. The MIC was defined as the lowest antibiotic
concentration with no visible bacterial growth. All colistin sulphate (Acros Organics) MIC assays were
also performed using the BMD method as described above using MH broth supplemented with CaCl2
at a final concentration of 0.223mM. When used, tazobactam was included in broth or agar at a fixed
concentration of 4 g/mL, in accordance with the EUCAST guidelines.
The covalent DsbB inhibitor 4,5-dichloro-2-(2-chlorobenzyl)pyridazin-3-one325 was used to chemically
impair the function of the DSB system. Inactivation of DsbB results in abrogation of DsbA function360
only in media free of small-molecule oxidants, such as the dipeptide cystine.238 Therefore, MIC assays
involving chemical inhibition of the DSB system were performed using M63 broth (15.1 mM
(NH4)2SO4, 100 mM KH2PO4, 1.8 mM FeSO4.7H2O, adjusted to pH 7.4 with KOH) and agar (1.5%
w/v) supplemented with 1 mM MgSO4, 0.02% w/v glucose, 0.005% w/v thiamine, 31 µM FeCl3.6H2O,
6.2 μM ZnCl2, 0.76 µM CuCl2.2H2O, 1.62 µM H3BO3, 0.081 µM MnCl2.4H2O, 84.5 mg/L alanine, 19.5
mg/L arginine, 91 mg/L aspartic acid, 65 mg/L glutamic acid, 78 mg/L glycine, 6.5 mg/L histidine, 26
mg/L isoleucine, 52 mg/L leucine, 56.34 mg/L lysine, 19.5 mg/L methionine, 26 mg/L phenylalanine,
26 mg/L proline, 26 mg/L serine, 6.5 mg/L threonine, 19.5 mg/L tyrosine, 56.34 mg/L valine, 26 mg/L
tryptophan, 26 mg/L asparagine and 26 mg/L glutamine for E. coli strains. For P. aeruginosa strains

97
MOPS medium (40 mM 3-(N-morpholino)propanesulfonic acid, 4 mM Tricine, 9.53 mM NH4Cl, 0.5
μM CaCl2, 0.52 mM MgCl2.7H2O, 50 mM NaCl, 0.01 mM FeSO4.7H2O and 1.32 mM K2HPO4,
adjusted to pH 7.2 with KOH) and agar (1.5%) supplemented with 0.02% w/v glucose, 500 mg/L L-
glutamine, 0.03 μM (NH4)6Mo7O24.4H2O, 4 μM H3BO3, 0.3 μM CoCl2, 0.01 μM CuSO4, 0.081 μM
MnCl2.4H2O, 0.01 μM ZnSO4, and 4.2 μM NiCL2.6H2O were used. Either DMSO (vehicle control) or
the covalent DsbB inhibitor 4,5-dichloro-2-(2-chlorobenzyl)pyridazin-3-one (final concentration of 50
μM) (Enamine)325 were added to the M63 or MOPS medium, as required. The strain to be tested was
added at an inoculum that recapitulated the MH medium MIC values obtained for each strain.
2.5 SDS-PAGE ANALYSIS AND IMMUNOBLOTTING
All samples for immunoblotting were prepared as follows with the exception of samples for strains
expressing FTU-1. Strains to be tested were grown on LB agar plates as lawns in the same manner as
for MIC assays described above. Bacteria were collected using an inoculation loop and resuspended in
LB to OD600 2.0 (except for strains expressing OXA-4, where OD600 6.0 was used). The cell suspensions
were spun at 10,000 x g for 10 minutes and bacterial pellets were lysed by addition of BugBuster Master
Mix (Merck Millipore) for 25 minutes at room temperature with gentle agitation. Subsequently, lysates
were spun at 10,000 x g for 10 minutes at 4 °C and the supernatant was added to 4 x Laemmli buffer.
Samples were boiled for 5 minutes before separation by SDS-PAGE. Strains expressing FTU-1 were
grown in LB broth supplemented with 12.5 μg/mL tetracycline until OD600 0.4. FTU-1 -lactamase
expression was induced with 0.5 mM IPTG for 4 hours. Bacteria were then diluted in LB to OD600 2
and spun at 10,000 x g for 10 minutes. Bacterial pellets were lysed by addition of BugBuster Master
Mix (Merck Millipore) for 5 minutes at room temperature with gentle agitation. Subsequently, lysates
were added to 4 x Laemmli buffer and separated by SDS-PAGE.
Unless otherwise stated, SDS-PAGE analysis was carried out using 10% BisTris NuPAGE gels
(ThermoFisher Scientific) using MES/SDS running buffer prepared according to the manufacturer’s
instructions and including pre-stained protein markers (SeeBlue Plus 2, ThermoFisher Scientific).
Proteins were transferred to Amersham Protran nitrocellulose membranes (0.45 µm pore size, GE Life
Sciences) using a Trans-Blot Turbo transfer system (Bio-Rad) before blocking in 3% w/v Bovine Serum
Albumin (BSA)/TBS-T (0.1 % v/v Tween 20) or 5% w/v skimmed milk/TBS-T and addition of primary
and secondary antibodies. The following primary antibodies were used in this study: Strep-Tactin-HRP
conjugate (Iba Lifesciences) (dilution 1:3,000 in 3 w/v % BSA/TBS-T), Strep-Tactin-AP conjugate (Iba

98
Lifesciences) (dilution 1:3,000 in 3 w/v % BSA/TBS-T), rabbit anti-DsbA antibody (dilution 1:1,000
in 5 w/v % skimmed milk/TBS-T), rabbit anti-AcrA antibody (dilution 1:10,000 in 5 w/v % skimmed
milk/TBS-T), rabbit anti-TolC antibody (dilution 1:5,000 in 5 w/v % skimmed milk/TBS-T), rabbit
anti-HtrA1 (DegP) antibody (Abcam) (dilution 1:1,000 in 5 w/v % skimmed milk/TBS-T) and mouse
anti-DnaK 8E2/2 antibody (Enzo Life Sciences) (dilution 1:10,000 in 5% w/v skimmed milk/TBS-T).
The following secondary antibodies were used in this study: goat anti-rabbit IgG-AP conjugate (Sigma
Aldrich) (dilution 1:6,000 in 5% w/v skimmed milk/TBS-T), goat anti-rabbit IgG-HRP conjugate
(Sigma Aldrich) (dilution 1:6,000 in 5% w/v skimmed milk/TBS-T), goat anti-mouse IgG-AP conjugate
(Sigma Aldrich) (dilution 1:6,000 in 5% w/v skimmed milk/TBS-T) and goat anti-mouse IgG-HRP
conjugate (Sigma Aldrich) (dilution 1:6,000 in 5% w/v skimmed milk/TBS-T). Membranes were
washed three times for 5 minutes with TBS-T prior to development. Development for AP conjugates
was carried out using a SigmaFast BCIP/NBT tablet, while HRP conjugates were visualized with the
Novex ECL HRP chemiluminescent substrate reagent kit (ThermoFisher Scientific) or the Luminata
Crescendo chemiluminescent reagent (Merck) using a Gel Doc XR+ Imager (Bio-Rad).
2.6 -LACTAM HYDROLYSIS ASSAY
β-lactam hydrolysis measurements were carried out using the chromogenic β-lactam nitrocefin
(Abcam). Briefly, overnight cultures of strains to be tested were centrifugated, pellets were weighed
and resuspended in 150 L of 100 mM sodium phosphate buffer (pH 7) per 1 mg of wet-cell pellet, and
cells were lysed by sonication. Lysates were transferred into clear-bottomed 96-well microtiter plates
(Corning) and used at the following loadings: strains harbouring pDM1, pDM1-blaL2-1, pDM1-blaL1-1,
pDM1-blaOXA-10 and pDM1-blaOXA-50 (lysates equivalent to 0.34 mg of cell pellet); pDM1-blaOXA-4
(lysates equivalent to 0.2 mg of cell pellet); pDM1-blaBEL-1, pDM1-blaAIM-1 and pDM1-blaSMB-1 (lysates
equivalent to 0.17 mg of cell pellet); pDM1-blaPOM-1 (lysates equivalent to 0.1 mg of cell pellet); pDM1-
blaOXA-198 (lysates equivalent to 0.015 mg of cell pellet); pDM1-blaBPS-1m (lysates equivalent to 0.07 mg
of cell pellet); and pDM1-blaCARB-2 (lysates equivalent to 0.03 mg of cell pellet). In all cases, nitrocefin
was added at a final concentration of 400 M, and the final reaction volume was made up to 100 L
using 100 mM sodium phosphate buffer (pH 7). Nitrocefin hydrolysis was monitored at 25°C by
recording absorbance at 490 nm at 60-second intervals for 15 minutes using an Infinite M200 Pro
microplate reader (Tecan). The amount of nitrocefin hydrolysed by each lysate in 15 minutes was
calculated using a standard curve generated by acid hydrolysis of nitrocefin standards.

99
2.7 NPN UPTAKE ASSAY338
1-N-phenylnaphthylamine (NPN) (Arcos Organics) uptake assays were performed as described by
Helander & Mattila-Sandholm.361 Briefly, mid-log phase cultures of strains to be tested were diluted to
OD600 0.5 in 5 mM HEPES (pH 7.2) before transfer to clear-bottomed 96-well microtiter plates
(Corning) and addition of NPN at a final concentration of 10 M. Colistin sulphate (Arcos Organics)
was included at a final concentration of 0.5 g/mL, as required. Immediately after the addition of NPN,
fluorescence was measured at 60-second intervals for 10 minutes using an Infinite M200 Pro microplate
reader (Tecan); the excitation wavelength was set to 355 nm and emission was recorded at 405 nm.
2.8 PI UPTAKE ASSAY338
Exponentially growing (OD600 0.4) E. coli strains harbouring pUltraGFP-GM357 were diluted to OD600
0.1 in phosphate buffered saline (PBS) (pH 7.4) and cecropin A was added to a final concentration of
20 M, as required. Cell suspensions were incubated at room temperature for 30 minutes before
centrifugation and resuspension of the pellets in PBS. Propidium iodide (PI) was then added at a final
concentration of 3 M. Suspensions were incubated for 10 minutes at room temperature and analysed
on a two-laser, four colour BD FACSCalibur flow cytometer (BD Biosciences). 50,000 events were
collected for each sample and data were analysed using FlowJo v.10.0.6 (Treestar).
2.9 CPRG CELL ENVELOPE INTEGRITY ASSAY
Exponentially growing (OD600 0.4) E. coli MG1655 and MC1000 pCB112 strains were diluted to 1:105
in MH broth and plated on MH agar containing CPRG and IPTG at final concentrations of 20 g/mL
and 50 M, respectively. Plates were incubated at 37°C for 18 hours. Plates images were analysed using
Adobe Photoshop CS4 (Adobe).362 Briefly, plate images were converted to CMYK colour space format,
colonies were manually selected using consistent tolerance (26, anti-alias, contiguous) and edge
refinement (32 px, 100% contrast). Magenta colour was quantified for each image and used to assess
changes in the cell envelope integrity of the tested strains.

100
2.10 MOTILITY ASSAY338
500 μL of overnight culture of each strain to be tested were centrifuged and the pellets were washed in
M63 broth before resuspension in the same medium to achieve a final volume of 25 L. Bacterial
motility was assessed by growth in M63 medium containing 0.25% w/v agar supplemented as described
above. DMSO (vehicle control) or the covalent DsbB inhibitor 4,5-dichloro-2-(2-
chlorobenzyl)pyridazin-3-one (final concentration of 50 M) (Enamine) were added to the medium, as
required. 1 L of the washed cell suspension was inoculated into the centre of a 90 mm diameter agar
plate, just below the surface of the semi-solid medium. Plates were incubated at 37 °C in a humidified
environment for 16-18 hours and growth halo diameters were measured.
2.11 AMS LABELLING338
Bacterial strains to be tested were grown for 18 hours in M63 broth supplemented as described above.
DMSO (vehicle control) or the covalent DsbB inhibitor 4,5-dichloro-2-(2-chlorobenzyl)pyridazin-3-
one (final concentration of 50 μM) (Enamine) were added to the medium, as required. Cultures were
standardized to OD600 2.0 in M63 broth, were spun at 10,000 x g for 10 minutes and bacterial pellets
were lysed by addition of BugBuster Master Mix (Merck Millipore) for 25 minutes at room temperature
with gentle agitation. Subsequently, lysates were spun at 10,000 x g for 10 minutes at 4 °C prior to
reaction with 4-acetamido-4ˊ-maleimidyl-stilbene-2,2ˊ-disulfonic acid (AMS) (ThermoFisher
Scientific). AMS alkylation was performed by vortexing the lysates in 15 mM AMS, 50 mM Tris-HCl,
3% w/v SDS and 3 mM EDTA (pH 8.0) for 30 minutes at 25°C, followed by incubation at 37°C for 10
minutes. SDS-PAGE analysis and immunoblotting was carried out as described above, except that 12%
BisTris NuPAGE gels (ThermoFisher Scientific) and MOPS/SDS running buffer were used. DsbA was
detected using a rabbit anti-DsbA primary antibody and an AP-conjugated secondary antibody, as
described above.
2.12 BACTERIAL GROWTH ASSAY – DSBA MUTANT
Overnight cultures of the strains to be tested were centrifuged and the pellets were washed in LB broth
before transfer to clear-bottomed 96-well microtiter plates (Corning) at approximately 5 x 107 CFU/well
(starting OD600 ~ 0.03). LB broth, supplemented with antibiotics as required, was used as a growth

101
medium. Plates were incubated at 37 °C with orbital shaking (amplitude 3 mm, equivalent to ~ 220
RPM) and OD600 was measured at 900-second intervals for 18 hours using an Infinite M200 Pro
microplate reader (Tecan).
2.13 BACTERIAL GROWTH ASSAY – DSB SYSTEM CHEMICAL INHIBITOR338
Overnight cultures of the strains to be tested were centrifuged and the pellets were washed in M63 broth
before transfer to clear-bottomed 96-well microtiter plates (Corning) at approximately 5 x 107 CFU/well
(starting OD600 ~ 0.03). M63 broth supplemented as described above was used as a growth medium.
DMSO (vehicle control) or the covalent DsbB inhibitor 4,5-dichloro-2-(2-chlorobenzyl)pyridazin-3-
one (final concentration of 50 M) (Enamine) were added to the medium, as required. Plates were
incubated at 37°C with orbital shaking (amplitude 3 mm, equivalent to ~ 220 RPM) and OD600 was
measured at 900-second intervals for 18 hours using an Infinite M200 Pro microplate reader (Tecan).
2.14 IN VIVO CLEARANCE ASSAY338
The wax moth model Galleria mellonella was used for in vivo clearance assays.363 Briefly, overnight
cultures of the strains to be tested were standardized to OD600 1.0. Suspensions were centrifuged and
the pellets were washed three times in PBS and serially diluted. 10 l of a 10–5 dilution of each bacterial
suspension was injected into the last right abdominal proleg of 5 G. mellonella larvae per condition; a
second, equal-size group of larvae were injected with PBS as negative control. 3 hours after infection,
larvae were injected with 13 l of piperacillin to a final concentration of 12 g/mL in the last left
abdominal proleg. 24 hours after infection larvae were euthanized and macerated individually in 1 ml
of PBS by vortexing for 15 minutes. The larval suspension was then serially diluted and 20 l of each
dilution plated on Pseudomonas Isolation Agar. Plates were incubated at 37°C for 16 hours before CFU
counting.

102
2.15 STATISTICAL ANALYSIS OF EXPERIMENTAL DATA
For MIC assays, all recorded values were plotted; bar charts were predominantly used to display MIC
results. For experiments where 3 independent experiments were performed, the bar indicates the median
of the recorded values, whilst for experiments where 2 independent experiments were performed the
bar indicates the most conservative of the two recorded values.
For all other assays, statistical analysis was performed in GraphPad PRISM v8.3.1 using an unpaired
T-test with Welch’s correction, a one-way ANOVA with correction for multiple comparisons, or a
Kruskal-Wallis test with correction for multiple comparisons, as appropriate. Statistical significance
was defined as p < 0.05. Outliers were defined as any technical repeat >2 SD away from the average of
the other technical repeats within the same biological experiment. Such data were excluded, and all
remaining data were included in the analysis. Detailed information for each analysis is provided in the
relevant figures.

103
3 THE IMPORTANCE OF DISULFIDE BOND FORMATION FOR
THE FUNCTION OF MOBILE CLASS D -LACTAMASE
ENZYMES OF PSEUDOMONAS AERUGINOSA
3.1 INTRODUCTION
Resistance determinants encoded on mobile elements are a common trait of multi-drug resistant
organisms. The dissemination potential of such genes contributes to the increase in resistance of
pathogenic bacteria and creates a great challenge for current antibiotic therapies. Mobilizable -
lactamases are easily transferred, both among clinically significant species and across environmental
strains. Several families of these enzymes have become prevalent in some of the most concerning
bacterial pathogens, such as K. pneumoniae, P. aeruginosa or A. baumannii. These include the SHV,
CTX-M and KPC (class A -lactamases), the NDM and VIM (class B -lactamases) or the OXA (class
D -lactamases) families of enzymes.62
P. aeruginosa is a highly resistant opportunistic pathogen, primarily affecting immunocompromised
individuals. Infections result in a variety of nosocomial diseases that often have poor clinical outcomes,
including sepsis, pneumonia, urinary tract and soft-tissue infections.28,75 In addition to its virulence
traits, P. aeruginosa strains have multiple resistance mechanisms, including, for example the reduction
of membrane permeability via loss of OprD proteins, structural modifications of topoisomerase
enzymes, overexpression of efflux pumps or production of many endogenous and acquired -lactamase
enzymes belonging to different Ambler classes.28 Due to a highly flexible and diverse genome, P.
aeruginosa is uniquely adapted to retain its essential genes while it incorporates new ones that are
typically acquired in hostile environments, such as hospital surfaces.20,81 For these reasons P.
aeruginosa is considered a high priority organism for novel intervention therapies.20,81
Although a comprehensive overview of the overall prevalence of the OXA -lactamase family in P.
aeruginosa has not been carried out, several country-specific studies have noted the appearance of these
oxacillinase enzymes in hospital infections. For example, samples from 12 clinical laboratories across
South Korea in 2005 identified 252 P. aeruginosa strains with enzymes from the OXA-7 (OXA-10,

104
17), OXA-36 (OXA-2) and OXA-224 (OXA-1, 4) families.364 A more recent study in Warsaw, Poland
noted that 110 out of 900 tested strains of the pathogen were phenotypically-positive for extended-
spectrum -lactamases, with many expressing members from the OXA-7 and OXA-36 families.365 It is
notable that the enzymes identified in these two epidemiological studies originate from several
phylogenetic families, confirming their wide dissemination and importance in antibiotic resistance.
Predominantly encoded near mobilizable elements or on plasmids, class D OXA enzymes are active
serine -lactamases with a conserved catalytic serine (Ser70) and a critical carboxylated lysine residue
in their active site.366,367 With some exceptions, for example OXA-198, they can be inhibited by
clinically available compounds, such as clavulanic acid or tazobactam.62 The OXA-family enzymes
form the largest group of class D -lactamases with over 750 members, sub-divided into over 50
phylogenetic sub-families that display a diverse range of hydrolytic profiles. These often originate from
single of amino acid variations that result in high levels of resistance to all classes of -lactam
antibiotics. Commonly observed mutations include the Ser73Asn replacement, responsible for extended-
spectrum activity in the OXA-7 sub-family, or the Gly157Asp substitution leading to ceftazidime
resistance.28,125
A characteristic trait of these serine hydrolases is the Tyr144-Gly-Asn146 or Phe144-Gly-Asn146 functional
motif.352,368 This structural element, denoted as the loop, is composed of a pair of NH hydrogen-bond
donors located next to an acyl group carbonyl oxygen. The arrangement of the hydrogen-bonding
partners is known as the “oxyanion hole” and has been shown to play a critical role in stabilising the
formation of enzymatic intermediates, while it is also believed to affect the hydrolytic efficiency of the
catalytic site.352,366,369 Computed molecular simulations by Szarecka et al. and Simakov et al. suggest
that further stabilisation in this region could stem from the presence of a disulfide bridge which shapes
the highly defined loop conformational arrangement in OXA-198 or OXA-10-like enzymes.366,368
Further, they suggest that the stabilising effect of this disulfide bond could drive the evolution of new
hydrolytic functions in these enzymes.181,366
Previous work in the Mavridou lab has shown a link between DsbA activity and the ability of disulfide-
bond containing class A -lactamases, and one class B enzyme, to confer resistance to a range of -
lactam antibiotics. The importance of oxidative protein folding for class D OXA enzymes, many of
which have highly conserved disulfide bridges, was not investigated. The wide dissemination of these
-lactamases in the accessory genomes of clinically isolated P. aeruginosa strains, along with the fact

105
that resistance to classical inhibitors is often observed, offered an incentive to examine their dependence
on DsbA-catalysed folding.364,365 Four enzymes, OXA-4, 10, 18, and 198, commonly encoded by P.
aeruginosa strains causing hospital-acquired infections, were selected as representative members of
phylogenetically distinct sub-families of the OXA group of enzymes. All of these proteins have a
cysteine pair at different positions in their primary sequence, and exhibit varied hydrolytic profiles
(Supplementary Table 1).364,365 Testing the importance of DsbA for their activity, thus offers a
comprehensive overview of the role of disulfide bond formation for cysteine-containing members of
the class D OXA family.

106
3.2 RESULTS
3.2.1 Deletion of dsbA substantially decreases -lactamase mediated antibiotic resistance
in E. coli MC1000
The selected -lactamase enzymes, OXA-4, 10, 18, and 198, were expressed in the E. coli MC1000 K-
12 strain and its isogenic dsbA mutant. A panel of -lactam antibiotics was selected for each enzyme
according to its known hydrolytic capabilities, and MIC values were recorded. An aminoglycoside
antibiotic, gentamicin, which cannot be neutralised by -lactamase enzymes, as well as strains
harbouring the pDM1 empty vector or expressing the class A -lactamase L2-1, were included as
negative controls. The class A β-lactamase L2-1 from S. maltophilia contains three cysteine residues,
but lacks a disulfide bond (PDB ID: 5NE1).137 The reason for this is that it is transported to the periplasm
pre-folded via the Tat pathway, rather than by the Sec system, and thus is not a DsbA substrate.189
The absence of DsbA resulted in a substantial decrease in MIC values for all tested enzymes across
multiple -lactam compounds (Figure 3.1). In addition, the observed effects were specific to the tested
resistance proteins and their interaction with DsbA and not a result of a general inability of the dsbA
mutant to resist antibiotic stress; no decreases in MIC values were recorded for the aminoglycoside
antibiotic gentamicin nor strains carrying the disulfide-free -lactamase L2-1 or the pDM1 empty vector
(Figure 3.1).

107
Figure 3.1 Antimicrobial resistance mediated by OXA-type -lactamases depends on disulfide bond formation. β-lactam
minimum inhibitory concentration (MIC) values for E. coli MC1000 expressing class D disulfide-bond-containing β-
lactamases from P. aeruginosa are substantially reduced in the absence of DsbAxvi. No changes in MIC values are observed
for the aminoglycoside antibiotic gentamicin (white bars, MIC fold changes: < 2) confirming that absence of DsbA does not
compromise the general ability of this strain to resist antibiotic stress. Further, no changes in MIC values are observed for
strains harbouring the empty vector control (pDM1) or those expressing a class A β-lactamase L2-1, which contains three
cysteines but no disulfide bond (PDB ID: 5NE1; top row). Graphs show MIC fold changes (fold change is defined as MC1000
MIC (µg/mL) / MC1000 dsbA (µg/mL),) for β-lactamase-expressing E. coli MC1000 and its dsbA mutant and represent three
independent experiments; black dotted lines indicate an MIC fold change of 2.
It should be noted that although the overall fold changes in MIC values recorded for the extended-
spectrum -lactamase OXA-18 were not higher than 2, the cut off below which we considered observed
effects insignificant, MIC values for this enzyme in the absence of DsbA decreased by 500 or even
1000 MIC points for the compounds tested (Supplementary Table 2). This suggests that its hydrolytic
activity is affected in the absence of disulfide bond formation. Nonetheless, this -lactamase is
extremely efficient in hydrolysing all of the tested -lactam antibiotics (MIC values of 1000-2000
xvi Where broth dilution experiments were required (pDM1-blaOXA-4, pDM1-blaOXA-10, pDM1-blaOXA-18, pDM1-blaOXA-198,) Ampicillin was used
instead of amoxicillin. Both antibiotics are 2nd generation amino-benzyl penicillins but have varying salt-solubility properties. Amoxicillin hydrate is insoluble at the concentrations required for broth dilution experiments. The more soluble amoxicillin sodium salt is markedly more
expensive, and thus unsuitable for use in large-scale experiments. Ampicillin trihydrate is soluble at the required concentrations and its
structural similarity to amoxicillin makes it a good substitute for our purposes.
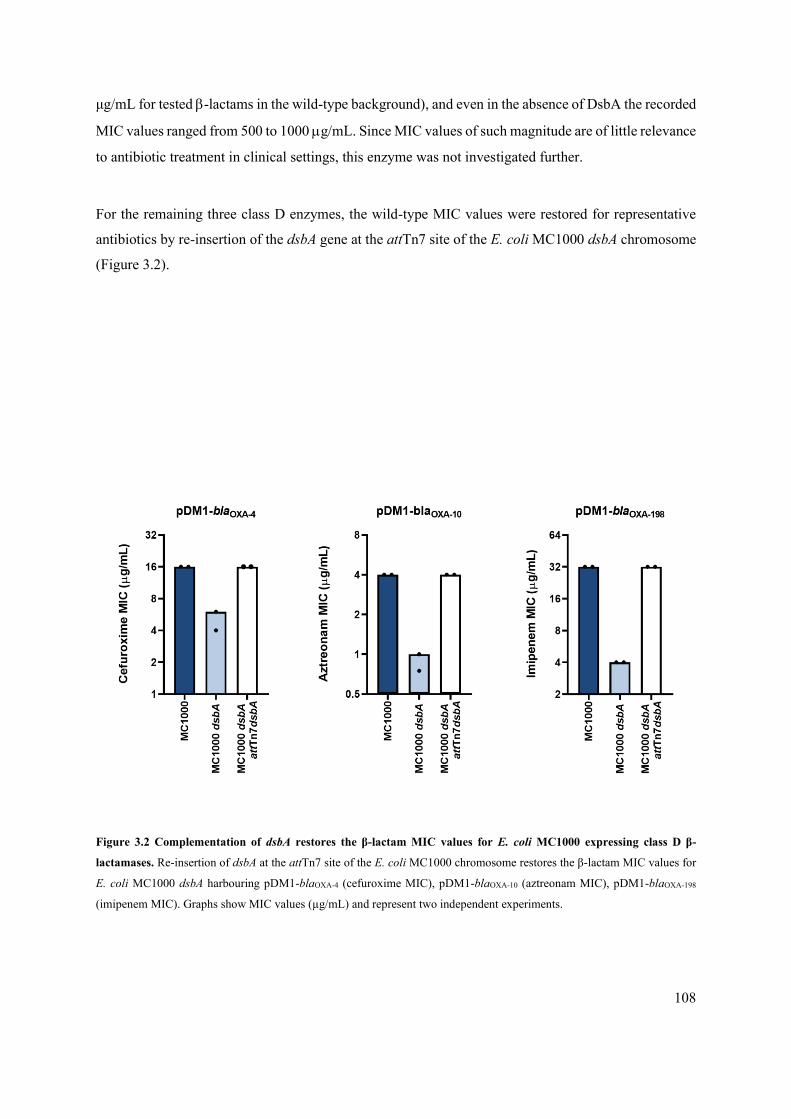
108
μg/mL for tested -lactams in the wild-type background), and even in the absence of DsbA the recorded
MIC values ranged from 500 to 1000 g/mL. Since MIC values of such magnitude are of little relevance
to antibiotic treatment in clinical settings, this enzyme was not investigated further.
For the remaining three class D enzymes, the wild-type MIC values were restored for representative
antibiotics by re-insertion of the dsbA gene at the attTn7 site of the E. coli MC1000 dsbA chromosome
(Figure 3.2).
Figure 3.2 Complementation of dsbA restores the β-lactam MIC values for E. coli MC1000 expressing class D β-
lactamases. Re-insertion of dsbA at the attTn7 site of the E. coli MC1000 chromosome restores the β-lactam MIC values for
E. coli MC1000 dsbA harbouring pDM1-blaOXA-4 (cefuroxime MIC), pDM1-blaOXA-10 (aztreonam MIC), pDM1-blaOXA-198
(imipenem MIC). Graphs show MIC values (µg/mL) and represent two independent experiments.

109
3.2.2 Deletion of dsbA does not affect the integrity of the cell envelope in E. coli MC1000
The gentamicin, pDM1 and pDM1-blaL2-1 controls described above (Figure 3.1), showed that the effects
on the recorded MIC values were specific to the interaction of the -lactamases with DsbA. In addition
to these tests, and since DsbA assists the folding of hundreds of cell envelope proteins, a series of
experiments were performed to assess whether altered outer-membrane permeability or cell-envelope
integrity of the dsbA mutant strain could confound our results.215,251
The permeability of the outer membrane of the dsbA mutant was measured using the hydrophobic
fluorescent dye 1-N-phenylnaphthylamine (NPN), and was found to be no different to that of the
parental strain (Figure 3.3 A).338,361 Additionally, a vancomycin sensitivity assay was used to confirm
this result. Vancomycin is a bactericidal antibiotic targeting cell wall synthesis. Outer membrane porins
of Gram-negative bacteria are too small to allow the passage of large glycopeptides, such as
vancomycin, across the membrane and, thus sensitivity to this antibiotic is only observed in Gram-
positive bacteria.370 Any perturbation to the integrity of the outer membrane in a dsbA mutant, would
thus enable the passage of vancomycin to the periplasmic space and result in major decreases in MIC
values. With the MIC values of MC1000 dsbA being comparable to those of the parental strain, it was
concluded that DsbA absence does not result in any outer membrane defects (Figure 3.3 B), consistent
with the data reported by Denoncin et al.370

110
Figure 3.3 Deletion of dsbA has no effect on outer membrane permeability in E. coli MC1000. (A) The bacterial outer
membrane acts as a selective permeability barrier to hydrophobic molecules. Deletion of dsbA has no effect on the outer
membrane integrity of E. coli MC1000, as the hydrophobic fluorescent dye NPN crosses the outer membrane of E. coli
MC1000 and its dsbA mutant to the same extent. Conversely, exposure to the outer-membrane-permeabilizing antibiotic
colistin results in a significant increase in NPN uptake. Graph shows means ± SD, significance is indicated by *** = p < 0.001,
ns = non-significant. Significance was determined using one-way ANOVA with Bonferroni’s multiple comparison test; n=3;
6 degrees of freedom; F value=39.22; p=0.0007 (significance), p=0.99 (non-significance). (B) No major differences are
observed in the MIC values for the aminoglycoside antibiotic vancomycin confirming that the outer membrane of E. coli
MC1000 dsbA is not compromised, and hence blocks the entry of the antibiotic. For both experiments n=3; NPN assay data is
courtesy of Dr R. Christopher D. Furniss (APPENDIX II).338
The integrity of the entire cell envelope of the dsbA mutant was also assessed using the fluorescent dye
propidium iodide (PI). PI is a cationic hydrophilic dye that fluoresces upon intercalation with nucleic
acids. Under normal conditions PI freely crosses the outer membrane but is unable to cross the inner
membrane.357 The cell envelope integrity of the dsbA mutant was found to be no different to that of the
parental strain (Figure 3.4 A). Further, a chromogenic -galactosidase assay based on the substrate
CPRG was used to confirm these results.359 CPRG is excluded from the cytoplasm by the cell envelope,
and therefore its hydrolysis by the cytosolic -galactosidase is prevented. If both the inner and outer
membranes are compromised, release of -galactosidase results in CPRG breakdown and the
appearance of red colour. The red colouration of the dsbA mutant colonies was comparable to those of
the parent strain showing that the cell envelope is not compromised in the mutant strain (Figure 3.4 B).

111
Figure 3.4 Deletion of dsbA does not result in damage to the bacterial inner membrane cell envelope. (A) No difference
in basal PI uptake is seen between E. coli MC1000 and its dsbA mutant when both strains express superfolder GFP (sfGFP).
Fluorescence was used to distinguish live from dead cells. Addition of the inner-membrane-permeabilizing antimicrobial
peptide cecropin A371 to E. coli MC1000 induces robust inner-membrane permeabilization in the sfGFP-positive population
indicating compromised inner membrane. Graph shows means ± SD, significance is indicated by *** = p < 0.001, ns = non-
significant. Significance determined using one-way ANOVA with Bonferroni’s multiple comparison test; n=3; 6 degrees of
freedom; F value=61.84; p=0.0002 (significance), p=0.99 (non-significance). (B) No difference in the red colouration of E.
coli MC1000 and its dsbA mutant colonies is seen, suggesting that CPRG is excluded from the colonies to the same extent.
This confirms that the integrity of the cell envelope is not compromised in the mutant strain. Images of plates were converted
to the CMYK colour space in Adobe Photoshop CS4. Colonies were selected using the magic wand tool with consistent
tolerance and edge refinement, and their magenta levels were compared. Graph shows means ± SD, significance is indicated
by ns = non-significant. Significance determined using unpaired T-test with Welch’s correction; n=3; 4 degrees of freedom; t-
value=0.1136 p=0.9150 (non-significance). PI uptake data is courtesy of Dr R. Christopher D. Furniss (APPENDIX II).338
3.2.3 Deletion of dsbA does not affect the viability of E. coli MC1000
Bacterial growth of the E. coli MC1000 strains was assessed under standard conditions to determine
whether DsbA absence causes any significant growth defects that could potentially confound our
results. No differences in growth were observed for the dsbA mutant strain, when compared to its wild-
type counterpart (Figure 3.5).

112
Figure 3.5 Deletion of dsbA does not have drastic effects on the growth of E. coli MC1000. Growth curves of E. coli
MC1000 and its dsbA mutant show that bacterial growth remains largely unaffected by the absence of DsbA. SD is marked by
the light blue shaded area in each graph. Significance determined using unpaired T-test with Welch’s correction; n=3; 4 degrees
of freedom; t-value=3.049; p=0.0381 (significance). Courtesy of Dr R Christopher D Furniss (APPENDIX II).338
3.2.4 Class D -lactamases misfold in absence of DsbA
To understand the underlying mechanism resulting in the decreased MIC values observed for the dsbA
mutant strains, -lactamase protein levels were assessed by immunoblotting. When expressed in the
dsbA mutant all of the tested class D enzyme levels remained unchanged (Figure 3.6 A), in a similar
fashion to the amount of the control enzyme L2-1, which also remained unaffected when DsbA was
absent (Figure 3.6 B).

113
Figure 3.6 Class D -lactamase enzyme levels remain unaffected by the absence of DsbA. (A) Protein levels for the
cysteine-containing β-lactamases OXA-4, OXA-10, and OXA-198 are not affected when expressed in E. coli MC1000 dsbA.
OXA-4 is detected as two bands at ~ 28 kDa. (B) The amount of the control enzyme L2-1, which contains three cysteines but
no disulfide bonds, remains unaffected in the absence of DsbA.137,189 Protein levels of StrepII-tagged β-lactamases were
assessed using a Strep-Tactin-AP conjugate (OXA-10 and OXA-198) or a Strep-Tactin-HRP conjugate (OXA-4).
Representative blots from three independent experiments are shown. Molecular weight markers (M) are on the left, DnaK was
used as a loading control and solid black lines indicate where the membrane was cut.
To assess the activity of the tested -lactamase enzymes when they lacked their disulfide bonds,
nitrocefin hydrolysis assays were performed. Nitrocefin is a chromogenic cephalosporin with a -
lactam ring that can be easily hydrolysed. The opening of this key pharmacophore leads to a detectable
bathochromic shift from =390 nM to =486 nM.
Direct comparison of -lactamase lysates of E. coli MC1000 and its dsbA mutant showed that the
hydrolytic activity of these β-lactamases was significantly decreased in absence of DsbA (Table 5),
suggesting a folding defect that leads to loss of function. E. coli MC1000 carrying the pDM1 empty
vector showed minimal nitrocefin hydrolysis, consistent with the lack of an enzyme that can perform
-lactam ring breakdown. Both E. coli MC1000 and its dsbA mutant expressing the disulfide-free L2-
1 enzyme showed high levels of nitrocefin hydrolysis, but the concentration of hydrolysed nitrocefin
did not vary (Table 5). These results are consistent with the MIC assays (Figure 3.1). Notably, the
hydrolytic activity of -lactamases expressed in the dsbA deletion strain remained higher than that of
the empty vector. Thus, in the absence of DsbA, the -lactamases appear to be partially misfolded, and
as a result not as effective as when they have undergone oxidative folding in the parental strain, but not
entirely inactive; the fact that their protein levels do not decrease in the absence of DsbA is likely crucial
to basal activity maintenance (Figure 3.6). Together the MIC and functional data show that for class D

114
Table 5 The hydrolytic activities of tested β-lactamase enzymes are significantly decreased in the absence of DsbA. The
hydrolysis of the chromogenic β-lactam nitrocefin by cysteine-containing class D β-lactamases is impaired when these
enzymes are expressed in E. coli MC1000 dsbA. The hydrolytic activities of strains harbouring the empty vector or expressing
the control enzyme L2-1, containing three cysteines but no disulfide bond, show no dependence on DsbA. The “Enzyme
stability” column informs on the stability of each enzyme when lacking its disulfide bond; this was inferred from the
immunoblotting experiments presented in Figure 3.6. The “Nitrocefin hydrolysis” column shows the amount of nitrocefin
hydrolysed per 1 mg of bacterial cell pellet in 15 minutes. n=3, table shows means ±SD, significance is indicated by * = p <
0.05, ns = non-significant. Significance was determined using unpaired T-test with Welch’s correction; n=3; 3.417 degrees of
freedom, t-value=0.3927, p=0.7178 (non-significance) (for pDM1 strains); 2.933 degrees of freedom, t-value=0.3296,
p=0.7639 (non-significance) (for pDM1-blaL2-1 strains); 2.011 degrees of freedom, t-value=6.825, p=0.0205 (significance) (for
pDM1-blaOXA-4 strains); 2.005 degrees of freedom, t-value=6.811, p=0.0208 (significance) (for pDM1-blaOXA-10 strains); 2.025
degrees of freedom, t-value=5.629, p=0.0293 (significance) (for pDM1-blaOXA-198 strains).
Strain (MC1000) Enzyme stability Nitrocefin hydrolysis†
pDM1
dsbA pDM1 -
3.57±4.40
4.51±3.15ns
pDM1-blaL2-1
dsbA pDM1-blaL2-1 not degrading
56.17±4.90
57.83±3.73ns
pDM1-blaOXA-4
dsbA pDM1-blaOXA-4 not degrading
96.93±20.22
17.16±1.05*
pDM1-blaOXA-10
dsbA pDM1-blaOXA-10 not degrading
1420±3.11
1059.78±91.67*
pDM1-blaOXA-198
dsbA pDM1-blaOXA-198 not degrading
790.75±137.07
343.90±10.78* †nM.mg-1 pellet.15 min-1
cysteine-containing AMR determinants, absence of DsbA leads to reduced levels of enzymatic activity
resulting in the inability of these enzymes to confer resistance.
3.2.5 DsbA is a tractable target for class D -lactamases
Given the role of DsbA in folding of many virulence factors, the inhibition of the DSB system has been
proposed as a promising anti-virulence approach and some efforts have been made to develop inhibitors
for DsbA, its redox partner DsbB or both (see also section 1.6.5).216,236,306,322,325,327,329,331 These studies
have made the first steps towards the production of chemical compounds that inhibit the function of the
DSB proteins and provided a laboratory tool to test our DsbA inhibition strategy against AMR.
4,5-dichloro-2-(2-chlorobenzyl)pyridazin-3-one, termed “Compound 12” in Landeta et al., is a potent
laboratory inhibitor of E. coli DsbB and its analogues from closely related organisms.325 It was
developed through further optimization of lead compounds discovered in an adapted, disulfide-sensitive

115
-galactosidase assay in an effort to target DsbB. In particular, the reaction of the chromogenic substrate
X-Gal (5-bromo-4-chloro-3-indolyl--d-galactopyranoside) with the native form of -galactosidase
results in the formation of blue pigment called 5,5’-dibromo-4,4’-dichloro-indigo. In this case, -
galactosidase-MalF fusions were used to ensure enzyme translocation into the periplasm. Since -
galactosidase contains cysteines, DsbA acts on it in the cell envelope and the introduction of non-native
disulfide bonds leads to enzyme deactivation and loss of the blue pigment. Compounds capable of
inhibiting the DSB system by blocking the function of DsbB, were identified through the re-appearance
of blue pigmentation to the otherwise white colonies.325
Although the reoxidation of DsbA is primarily carried out by DsbB, in the absence of DsbB function
this process can also occur via small-molecule oxidants, like oxygen and cystine. The presence of
cystine in most laboratory media, including Mueller-Hinton (the gold standard for MIC measurments),
interferes with chemical inhibition of the DSB system, since DsbB-independent DsbA re-oxidation can
occur. For this reason, defined cysteine-free M63 media was used for any assays involving the chemical
inhibitor of DsbB. Furthermore, the bacterial load was adjusted for E. coli MC1000 so that the same
MIC values as the ones recorded in MH media were achieved in M63 media. Using these adjustments,
the effect of chemical inhibition of the DSB system on the activity of class D OXA enzymes was probed.
Before conducting MIC experiments, the ability of Compound 12 to chemically inhibit the function of
the DSB system was established. First, the motility of E. coli MC1000 in the presence of this compound
was tested, as impairment of DSB function is known to prevent the formation of the flagellar P-ring
component FlgI rendering cells immotile. Chemical inhibition of DsbB function, indeed impaired
bacterial motility, similar to a dsbA deletion (Figure 3.7 A, B).238,372 Further, the redox state of DsbA in
the presence of the compound was also assessed to probe whether it was being re-oxidized by DsbB.
This is a necessary step that occurs after each round of oxidative protein folding and allows DsbA to
remain active. Under normal growth conditions, DsbA was in its active oxidized form in the bacterial
periplasm (i.e. Cys30 and Cys33 formed a disulfide bond), showing that it was efficiently regenerated by
DsbB (Figure 3.7 C).360 By contrast, addition of the inhibitor to growing E. coli MC1000 cells resulted
in accumulation of inactive reduced DsbA and confirmed that DsbB function was impeded.

116
Figure 3.7 Chemical inhibition of the DSB system impedes DsbA re-oxidation and flagellar motility in E. coli MC1000.
(A) A functional DSB system is necessary for flagellar motility in E. coli. In the absence of DsbA, or upon addition of a
chemical inhibitor of the DSB system, the motility of E. coli MC1000 is significantly impeded. Representative images of
motility plates are shown. (B) Quantification of the growth halo diameters in the motility assays shown in panel. n=3, graph
shows means ±SD, significance is indicated by **** = p < 0.0001. Significance was determined using one-way ANOVA with
Bonferroni’s multiple comparison test; n=3; 6 degrees of freedom; F value=1878; p=0.000000002 (significance). (C) Addition
of the reducing agent DTT to E. coli MC1000 bacterial lysates allows the detection of DsbA in its reduced form (DsbAred)
during immunoblotting; this redox state of the protein, when labelled with the cysteine-reactive compound AMS, shows a 1
kDa size difference (lane 2) compared to oxidized DsbA as found in AMS-labelled but not reduced lysates of E. coli MC1000
(lane 3). Addition of a small-molecule inhibitor of DsbB to growing E. coli MC1000 cells also results in accumulation of
reduced DsbA (lane 4). E. coli MC1000 dsbA was used as a negative control for DsbA detection (lane 1). A representative blot
from two independent experiments is shown; DsbA was visualized using an anti-DsbA primary antibody and an AP-conjugated
secondary antibody. Molecular weight markers (M) are shown on the left. Courtesy of Dr R Christopher D Furniss
(APPENDIX II).338
Having confirmed the efficacy of the DsbB inhibitor, chemical inhibition of the DSB system was tested
to determine whether it could be a suitable strategy to impair the function of class D OXA-type -
lactamases. The addition of the inhibitor compound during MIC testing phenocopied the effects of a
dsbA deletion (Figure 3.8, Supplementary Table 3). The fold changes observed for OXA-4 and -198
were comparable to those recorded for the E. coli MC1000 dsbA mutant, while the OXA-10 fold
changes were of greater magnitude (Figure 3.1). The MIC values of for the aminoglycoside antibiotic
gentamicin, the pDM1 empty vector and the disulfide-free L2-1 -lactamase controls remained

117
unchanged upon the addition of the DsbB inhibitor to the growth medium. These results were not due
to altered cell growth, as addition of the compound did not affect the bacterial growth (Figure 3.9).
Moreover, the DSB inhibitor has previously been tested in the Mavridou lab to ensure that it specifically
inhibits the DSB system, and no off-target effects were observed.338
Figure 3.8 Chemical inhibition of the DSB system phenocopies the β-lactam MIC changes observed using E. coli
MC1000 dsbA mutant. Chemical inhibition of the DSB system reduces the MIC values of representative -lactam antibiotics
for E. coli MC1000 expressing disulfide-bond-containing β-lactamases in a similar manner to the deletion of dsbA (Figure
3.1). Graphs show MIC fold changes (i.e. MC1000 MIC (µg/mL) / MC1000 + DSB system inhibitor MIC (µg/mL)) for β-
lactamase-expressing E. coli MC1000 with and without addition of a DSB system inhibitor to the culture medium and show
three independent experiments. Black dotted lines indicate an MIC fold change of 2. No changes in MIC values are observed
for the aminoglycoside antibiotic control, gentamicin (white bars, MIC fold changes: < 2). In addition, no changes in MIC
values are observed for strains harbouring the empty vector control (pDM1).

118
Figure 3.9 Chemical inhibition of the DSB system has no effect on the growth of E. coli MC1000. Growth curves for E.
coli MC1000 with and without chemical inhibition of the DSB show that bacterial growth remains unaffected by the inhibitor
compound on the supplemented M63 media. DMSO was used a carrier. n=3, curves show SD as light blue shaded area.
3.2.5.1 Sensitisation of multidrug-resistant P. aeruginosa clinical isolates to existing
antibiotics can be achieved by compromising the DSB system
Following the successful chemical inhibition of the DSB system in E. coli MC1000 strains (Figure 3.8),
the same inhibitor was used on clinical P. aeruginosa isolates. Resistance to β-lactam antibiotics in P.
aeruginosa is partially caused by the interplay between resident β-lactamase(s) and the MexAB-OprM
efflux pump.373 All P. aeruginosa possess two resident β-lactamases, the inducible class C enzyme
AmpC and the constitutively expressed class D enzyme OXA-50 (Figure 3.11). However, compared to
-lactamases like the OXA family enzymes, these enzymes do not represent a major resistance
determinant in clinical P. aeruginosa and, in combination with efflux pumps, play a relatively minor
role in β-lactam resistance.374
Clinical isolates expressing a single OXA-family -lactamase were selected. Their resistance profiles
were determined for both -lactam and non--lactam antibiotics using MH agar and E-test strips, with
the exception of colistin MICs for which broth microdilution was used (Supplementary Table 4). All
isolates were multi-drug resistant. For these strains defined cysteine-free MOPS media was used and
bacterial loads were adjusted to achieve the same MIC values for representative -lactam antibiotics,

119
as recorded on MH media. Only two of the P. aeruginosa isolates (expressing OXA-198 and OXA-28,
a member of the OXA-10 phylogenetic family) could be taken forward for MIC testing using the DSB
system inhibitor (Table 6), as the defined nature of the MOPS media strongly impacted the bacterial
viability of the remaining clinical isolates. For both isolates tested, the addition of Compound 12 to the
growth media did not result in reduction of the recorded MIC values for any of the -lactam antibiotics
tested.
These results reflect the challenges presented when attempting to target the DSB system in some
pathogenic bacterial species. The fact that Compound 12, which was developed to inhibit the E. coli
DsbB protein, was not effective against P. aeruginosa either due to a lower P. aeruginosa membrane
permeability in comparison to E. coli, the low sequence similarity between PaDsbA and EcDsbA, as
well as the polymorphisms that are quite common in this system, (see section 1.6.3).215 In particular,
the P. aeruginosa genome carries at least two functionally redundant DsbB analogues.375 As Compound
12, has been shown to be effective against DsbB1, but less so against DsbB2 of P. aeruginosa PA14,
the DsbB protein redundancy along with the initial high levels of resistance of the clinical isolates tested
mean that any effects of DsbB inhibition were likely masked.325
In the absence of a suitable chemical inhibitor, to investigate the consequences of compromising the
DSB system for the resistance of P. aeruginosa clinical strains to -lactam drugs, dsbA1, the principal
pseudomonal dsbA gene, was deleted from the P. aeruginosa PA43417 and PAe191 clinical isolates,
expressing OXA-198 and OXA-19 (a member of the OXA-10 phylogenetic family and the most
disseminated OXA enzyme in clinical strains), respectively (Figure 3.10).351
Table 6 Chemical inhibition of the DSB system via DsbB shows no effects on multidrug-resistant P. aeruginosa clinical
isolates. Addition of a small-molecule inhibitor of DsbB does not result in MIC drop for the two P. aeruginosa strains tested.
MIC values determined using MH agar in accordance with the EUCAST guidelines are comparable to the values obtained
using defined cysteine-free media (MOPS agar, recorded below). The use of growth media lacking small-molecule oxidants
is required for the DSB system inhibitor to be effective. MIC values (g/ml) are representative of two independent experiments.
DMSO was used as a vehicle control; DsbB inhibitor 4,5-dichloro-2-(2-chlorobenzyl)pyridazin-3-one was used at final
concentration of 50 M. The following abbreviations are used: XM, cefuroxime; IP, imipenem; AT, aztreonam.
Strain Additive XM IP AT
P. aeruginosa EDI
(blaOXA-28)
DMSO
DMSO + inhibitor - -
16
16
P. aeruginosa PA43417
(blaOXA-198)
DMSO
DMSO + inhibitor
>256
>256
>32
>32
6
6

120
Figure 3.10 Absence of DsbA1, the principal pseudomonal DsbA analogue, sensitizes multidrug-resistant clinical P.
aeruginosa isolates to first-line and last-resort -lactam antibiotics. (A) Removal of DsbA1 from P. aeruginosa PA43417
expressing OXA-198 sensitises the strain to the first-line antibiotic piperacillin (median MIC of 12 g/mL). (B) Removal of
DsbA1 from P. aeruginosa PAe191 expressing OXA-19 sensitises the strain to the aztreonam (median MIC of 12 g/mL).
The graphs show MIC values (g/ml) from 3 independent experiments, the red dotted lines indicate the EUCAST clinical
breakpoints (16 g/mL for piperacillin and aztreonam, 8 g/mL for ceftazidime and 4 g/mL for imipenem).
Deletion of dsbA1 in P. aeruginosa 41437 led to sensitization of the isolate to the first-line β-lactam
piperacillin (Figure 3.10 A), while its absence in P. aeruginosa PAe191 resulted in significant drops in
the MIC values for ceftazidime (16-fold drop) and imipenem (5.33-fold drop) and sensitisation for
aztreonam (Figure 3.10 B).These results suggest that targeting disulfide bond formation could be useful
for the sensitization of many more clinically important Gram-negative species to existing -lactam
compounds.
3.2.5.2 In vivo clearance experiments in the Galleria mellonella infection model
To test this strategy in an infection context we performed in vivo clearance assays using the wax moth
model G. mellonella (Figure 3.11). Larvae were infected with the P. aeruginosa PA43417 clinical
isolate producing OXA-198 and its dsbA1 mutant, and infections were treated once with piperacillin at
final concentrations below the EUCAST breakpoint. Neither deletion of dsbA1 nor treatment with
piperacillin were sufficient to reliably clear the infection when applied alone. In some cases, deletion
of dsbA1 led to a significant decrease in the recovered bacterial load due to the fact that absence of the

121
principal DsbA protein affects the virulence of the pathogen.333 Nonetheless, in other cases, the infection
with the dsbA1 mutant was only ~40%-70% cleared. On the other hand, treatment of the dsbA1 mutant
with piperacillin resulted in a drastic (> 99% on average) reduction in bacterial load in the infected
larvae in agreement with the fact that in the absence of DsbA the ability of OXA-198 to hydrolyse β-
lactams is impaired (Figure 3.1). As OXA-198 is a broad-spectrum β-lactamase that cannot be
neutralized by classical β-lactamase inhibitors (Supplementary Table 1) and piperacillin is a first-line
antibiotic, these results further highlight the promise of our approach for future clinical applications.
Figure 3.11 Absence of DsbA1 from a P. aeruginosa clinical isolate expressing OXA-198 allows it to be cleared from
infected G. mellonella larvae by piperacillin. Neither deletion of dsbA1, nor treatment with Piperacillin (at a concentration
of 12 g/mL) is sufficient to reliably clear P. aeruginosa PA4317 from infected G. mellonella larvae. However, the
combination of both results in an average reduction in bacterial load that is greater than 99%. The graph shows the average
number of colony-forming units (CFU) recovered from infected larvae for each condition relative to the CFU recovered for
the untreated P. aeruginosa PA4317 strain. n = 8 groups infected on eight different days; each group contains five G. mellonella
larvae per condition except for one group which contains three G. mellonella larvae per condition. Graph shows means ±SD,
significance is indicated by ns = p > 0.05 * = p < 0.05, ** = p < 0.01, *** = p <0.001. Significance was determined using
Kruskal-Wallis test with Dunn’s multiple comparisons test; n=8; Kruskal-Wallis H=25.24, 3 degrees of freedom; p=<0.0001.
For multiple comparisons, p=0.0029 (P. aeruginosa versus P. aeruginosa dsbA1), p=<0.0001 (P. aeruginosa versus P.
aeruginosa dsbA1 treated with piperacillin), p=0.0369 (P. aeruginosa treated with piperacillin versus P. aeruginosa dsbA1
treated with piperacillin). Data courtesy of Dr Ronan R. McCarthy and Evgenia Maslova (Brunel University, APPENDIX
II).338

122
3.3 DISCUSSION
Mobile resistance genes create a serious problem for the use of clinically available therapeutics, due to
their potential for spreading amongst bacterial pathogens. OXA-type -lactamases, which were initially
isolated from Acinetobacter species represent an example of the threat that mobile resistance
determinants pose to clinical therapy. Their prevalence in P. aeruginosa isolates resulted in the
emergence of clinical strains with very high levels of resistance, such as the OXA-19-expressing P.
aeruginosa PAe191 isolate that was tested in this study.67 To explore the potential of targeting the DSB
system as a strategy for mitigating antibiotic resistance, the activity of representative class D -
lactamases from P. aeruginosa was tested in an E. coli MC1000 strain and its isogenic dsbA mutant.
Clinically meaningful decreases in their ability to confer resistance in the absence of DsbA was
observed in three out of four enzymes, OXA-4, OXA-10 and OXA-198 (Figure 3.1)xvii. While the
absence of the disulfide bond did not notably decrease the protein stability of the tested -lactamases at
physiological conditions (Figure 3.6), their hydrolytic efficiency was found to be impaired (Table 5).
This is in agreement with the computer simulations carried out by Simakov et al. who proposed that the
disulfide bond is responsible for the correct conformation of one of the enzyme’s key catalytic
components, the omega loop.368
A potent chemical inhibitor of DSB activity, Compound 12, which covalently attaches to one of the
essential cysteines of DsbB blocking its function, was tested on model E. coli MC1000 strains
expressing OXA-4, -10, and -198.325 In all cases the trends observed for the recorded MIC values
(Figure 3.8) were in agreement with the results obtained from the dsbA mutant strain (Figure 3.1)
suggesting that the DSB system could be targeted chemically, thus offering a potential route for
abrogating antimicrobial resistance. While direct chemical inhibition of the DSB system was also
attempted on several P. aeruginosa clinical isolates expressing disulfide-bond-containing OXA-family
enzymes, no changes in their MIC values were observed. This was likely due to the presence of two
functionally redundant DsbB analogues in P. aeruginosa; Compound 12 is an efficient inhibitor of only
one of these two proteins.39 In combination with the high levels of resistance of these strains, any
xvii Absence of the native disulfide bond of the OXA-18 -lactamase also led to decreased MIC values, nonetheless the basal resistance of this
enzyme, even in the absence of its disulfide, was at levels that were not clinically meaningful (MIC values of 500-1000 g/mL for all tested
-lactams), and thus was not pursued further.

123
Compound-12-mediated effects were likely masked. Notably, however, deletion of dsbA1 in P.
aeruginosa isolates expressing OXA-198 and OXA-19 led to sensitization to first- and last-line β-
lactam antibiotics (Figure 3.11). As such, targeting of DsbA1 in P. aeruginosa is a promising strategy
for breaking class D -lactamase-mediated resistance.
Overall, these observations provide evidence that inhibition of DsbA, a non-essential cell envelope
protein which is unique to bacteria, could be useful for the sensitization of clinically important P.
aeruginosa strains to existing antibiotics. In addition, previous work by Dr R. Christopher D. Furniss
focusing on class A and B plasmid-encoded -lactamases from pathogenic species such as E. coli,
Citrobacter freundii, K. pneumoniae or E. cloacae, also showed that targeting the DSB system impairs
-lactamase function in other highly-resistant Gram-negative organisms. To our knowledge, this is the
first report of a strategy that can target -lactamase enzymes from three different Ambler classes.
Moreover, it is noteworthy that with 25% of β-lactamases found in bacterial pathogens and organisms
capable of causing opportunistic infections containing two or more cysteine residues,338 many more
clinically relevant β-lactamases are likely to depend on DsbA. This opens a new avenue to reverse -
lactamase-mediated resistance in Gram-negative organisms, through the development of DsbA
inhibitors acting as broad-acting resistance breakers.338

124
4 THE IMPORTANCE OF DISULFIDE BOND FORMATION FOR
THE FUNCTION OF CHROMOSOMALLY-RESIDENT -
LACTAMASE ENZYMES
4.1 INTRODUCTION
Chromosomally-encoded resistance genes evolve naturally in communities of environmental bacteria,
as their presence offers fitness advantages against competing microorganisms.22 High levels of genetic
variation can lead to intrinsic multi-drug resistance through a range of mechanisms, including disparities
in metabolic pathways, divergent membrane permeabilities, and expression of efflux pumps or -
lactamase enzymes. In addition to directly causing infections, intrinsically resistant environmental
bacteria can act as reservoirs for the dissemination of novel antimicrobial resistance genes. This appears
to be the case with broadly disseminated -lactamase or mcr colistin resistance genes, which are
currently spreading with an alarming speed in clinical strains.376
S. maltophilia, one of the most intrinsically resistant bacterial species, is an opportunistic pathogen
primarily associated with serious nosocomial respiratory infections. Although the clinical prevalence
of this organism is smaller compared to its closely related P. aeruginosa, S. maltophilia infections are
associated with extremely poor clinical prognosis stemming from the activity of multiple efflux pumps
and the presence of an extensive array of other resistance mechanisms.79,137 As a result, S. maltophilia
infections are resistant to most currently available antibiotics, including -lactams, macrolides,
fluoroquinolones, aminoglycosides, chloramphenicol, tetracyclines and colistin.79 S. maltophilia
resistance to -lactam antibiotics is predominantly driven by the expression of two chromosomally-
encoded -lactamases, the disulfide-free class A L2-1 enzyme and the disulfide-bond-bearing class B3
metallo--lactamase L1-1.79,137 Interestingly, the hydrolytic spectra of these two enzymes appear to
have co-evolved to confer comprehensive protection against all available -lactam classes. The ESBL
L2-1 can efficiently degrade penicillins and monobactams, whilst the carbapenemase L1-1 breaks down
penicillins and cephalosporins.79,137,338 The current clinically used therapy relies on sensitivity of the
bacterium to the third generation cephalosporin Ceftazidime, arising from low L1-1 expression levels,
or on the combination of Trimethoprim and Sulfamethoxazole.137,377–379 However, both therapy options

125
suffer from mutational and horizontally acquired resistance.137,377,378 Although extreme, the traits of S.
maltophilia infections are often recapitulated in infections caused by other environmental bacteria that
act as opportunistic human pathogens, for example saprophytic species like Burkholderia and
Pseudomonas, which become near impossible to treat.79,373,380,381
Previous work in the Mavridou lab has shown that mobile class A and D β-lactamases rely on DsbA-
mediated disulfide bond formation for their stability and function (Chapter 3 and APPENDIX II).338
Additionally, two important chromosomally resident enzymes were also observed to be DsbA
dependent, the class A enzyme SME-1 from Serratia marcescens and the aforementioned class B3
metallo-β-lactamase L1-1 from S. maltophilia.79,338,382 The DsbA dependence of these two
representative chromosomal enzymes formed the basis for the work carried out in this chapter aiming
to investigate the potential of targeting the DSB system of inherently resistant organisms as a strategy
to sensitize them against existing β-lactam compounds.
Eight phylogenetically distinct and chromosomally encoded β-lactamase enzymes from P. aeruginosa
(BEL-1, CARB-2, AIM-1, OXA-50), Pseudomonas otitidis (POM-1), Burkholderia spp (BPS-1m),
Franciscella tularensis (FTU-1), and Serratia spp (SMB-1) were selected for this work. These proteins
cover the full range of hydrolytic spectra of -lactamase enzymes, while their expression, which causes
considerable treatment complications, originates from diverse chromosomal settings (Supplementary
Table 1).109,338,353,383–386 For example, Burkholderia pseudomallei is the causative agent of
melioidosisxviii, a disease with limited treatment options due to intrinsic multi-drug resistance of the
bacterium to penicillin, first- and second- generation cephalosporins, aminoglycosides, macrolides, and
colistin.380 The mutation of its resident -lactamase BPS-1 gene into the clinically observed BPS-1m
results in resistance to ceftazidime which means the loss of the choice treatment option for this
organism.213,380,385
In addition to testing enzymes from bacterial species with high levels of intrinsic resistance, this study
also expands on class B metallo--lactamases which were underrepresented in previous investigations
(Chapter 3 and APPENDIX II); the requirement for oxidative protein folding for three representative
class B enzymes, AIM-1, POM-1, and SMB-1, was investigated here.338 These -lactamases are of
xviii Melioidosis is a severe disease endemic to Southeast Asia and Northern Australia.

126
increasing clinical concern due to the fact that they cannot be neutralised by currently available classical
inhibitor compounds, since their hydrolytic mechanism is discrete compared to serine -lactamases
(Section 1.3.1.4).67,75,76 To date, most class B enzymes have been found on the chromosomes of
environmental bacteria that are usually responsible for opportunistic infections, such as the
aforementioned S. maltophilia, P. otitidis, or S. marcescens. However, increasing evidence on the
mobility of these genes is recently coming to light.76,79,109,387,388 For example the carbapenemase SMB-
1 has been isolated upstream of an ISCR1 insertion sequence in the genome of S. marcescens, a rare
opportunistic pathogen which causes, among others, urinary and respiratory infections, septicaemia and
meningitis. Sequence comparison showed 75% sequence identity of SMB-1 with a -lactamase from
an uncultured soil bacterium, AMO-1, while its flanking region was 84% identical to the gene upstream
of AMO-1.388 Given that this ISCR1 mobility element in S. marcescens has previously been shown to
be associated with a class 1 integron carrying kanamycin and chloramphenicol resistance genes, it is
likely that SMB-1 has originated and was mobilised from a similar environmental reservoir to the one
AMO-1 was found in.386 More generally, ISCR elements have been implicated in the mobilisation of
other class B3 -lactamases outside S. marcescens, including SPM-1, NDM-1, or AIM-1.109,389,390
AIM-1, an emerging class B metallo-carbapenemase, is of particular importance in this study, given
both its unusually high number of disulfide bonds (all six of its cysteine residues are covalently linked
by disulfide bridges) and its location on the accessory part of the P. aeruginosa chromosome something
that increases its mobility potential.81 Similar chromosomal positions have been reported for the genes
of pseudomonal enzymes BEL-1 and CARB-2, which are associated with mobile genetic elements.353,391
Altogether the capacity of these -lactamases to spread to other strains and species, becomes
increasingly important in opportunistic pathogens and further exacerbates their already marked
resistance to antimicrobials in clinical settings.109,386,392 Further corroborating this, the genomes of
multidrug resistant S. maltophilia strains have also been observed to support great environmental
adaptability and have exhibited the ability to include and retain novel antimicrobial genes even after the
loss of selective pressure, a trait shared with Pseudomonas spp.79,80,393

127
4.2 RESULTS
4.2.1 The activity of cysteine-containing chromosomally encoded β-lactamase enzymes is
dependent on DsbA
In a similar manner to the work carried out with class D mobile -lactamase enzymes (Chapter 3), the
eight selected chromosomal β-lactamases were cloned and expressed in the E. coli MC1000 K-12 strain
and its isogenic dsbA mutant. MIC values against a panel of -lactam antibiotics, in accordance with
the known hydrolytic profiles of each enzyme, were recorded for the resulting strains. The
aminoglycoside antibiotic gentamicin, which cannot be inactivated by -lactamase enzymes, as well as
strains carrying the pDM1 empty vector or expressing the -lactamases L2-1 (from S. maltophilia) or
LUT-1 (from P. luteola) were included as negative controls. Both control enzymes contain two or more
cysteine residues but lack disulfide bonds. This is because they are transported pre-folded to the
periplasm via the Tat pathway, rather than by the Sec system. In the case of L2-1, Tat-dependent
transport has been experimentally confirmed189, whilst LUT-1 contains a predicted Tat signal peptide
(SignalP 5.0213 likelihood scores: Sec/SPI = 0.0572, Tat/SPI = 0.9312, Sec/SPII (lipoprotein) = 0.0087,
other = 0.0029).
It the absence of DsbA markedly reduced MIC values (>2-fold) were observed for at least one antibiotic
for all tested enzymes, except FTU-1, when compared to the parental strain (Figure 4.1, Supplementary
Table 2). Importantly, the sub-set of tested enzymes associated with mobile genetic elements were
dependent on DsbA, as were all three representatives of class B3 metallo-β-lactamases, POM-1, AIM-
1 and SMB-1. The latter is consistent with previous results where the class B3 L1-1 enzyme encoded
on the chromosome of S. maltophilia showed dependence on DsbA (APPENDIX II).338 Most of the
affected enzymes cannot be inhibited by classical -lactamase inhibitors, further expanding the group
of -lactamases that can be targeted by compromising the process of oxidative protein folding in the
cell envelope.338,394

128
Figure 4.1 Antimicrobial resistance mediated by chromosomally resident -lactamases depends on disulfide bond
formation. β-lactam minimum inhibitory concentration (MIC) values for E. coli MC1000 expressing a range of disulfide-
bond-containing β-lactamases (Ambler classes A, B and D) decrease in the absence of DsbA. For E. coli MC1000 expressing
FTU-1 and that of its dsbA mutant, the graph shows only a two-fold decrease between the ampicillin MIC value. This effect
corresponds to a reduction in MIC of 250 g/mL (Supplementary Table 2). No changes in MIC values are recorded for the
aminoglycoside antibiotic gentamicin (white bars, MIC fold changes: <2) confirming that absence of DsbA does not
compromise the general ability of MC1000 dsbA strain to resist antibiotic stress. No changes in MIC values are observed for
strains harbouring the empty vector control (pDM1) or those expressing the disulfide free β-lactamase controls L2-1 and LUT-
1. Graphs show MIC fold changes (MIC fold changes: > 2, fold change defined as MC1000 MIC (µg/mL) / MC1000 dsbA

129
(µg/mL),) for β-lactamase-expressing E. coli MC1000 and its dsbA mutant and show three independent experiments. Black
dotted lines indicate an MIC fold change of 2.
MIC values recorded for strains expressing the class A -lactamase FTU-1 from the human pathogen
F. tularensis only decreased by 2-fold in the absence of DsbA (Figure 4.1).384 Nonetheless, this effect
corresponded to a reduction in the Amoxicillin MIC value by 250 g/mL (Supplementary Table 2),
indicating that the function of this enzyme is affected by the disulfide bond loss even though substantial
hydrolytic activity is retained in the mutant strain. This -lactamase was not investigated further.
Finally, the MIC values for the aminoglycoside antibiotic gentamicin, E. coli strains harbouring the
pDM1 empty vector or strains expressing the disulfide-free -lactamases L2-1 or LUT-1 remained
unaffected (Figure 4.1, Supplementary Table 2).These results, along with the cell envelope integrity
checks presented in section 3.2.2 for this background, confirm that the observed MIC effects were
specific to the tested resistance proteins and their interaction with DsbA and not a result of a general
inability of the dsbA mutant to resist antibiotic stress. This was further supported by the fact that
complementation of dsbA restores the recorded MIC values to wild-type levels for all of the affected
enzymes (Figure 4.2).

130
Figure 4.2 Complementation of dsbA restores the β-lactam MIC values for E. coli MC1000 expressing β-lactamases.
Re-insertion of dsbA at the attTn7 site of the E. coli MC1000 chromosome restores the β-lactam MIC values for E. coli
MC1000 dsbA harbouring pDM1-blaBEL-1 (ceftazidime MIC), pDM1-blaBPS-1m (ceftazidime MIC), pDM1-blaCARB-2
(cefuroxime MIC), pDM1-blaFTU-1 (ampicillin MIC), pDM1-blaAIM-1 (ceftazidime MIC), pDM1-blaPOM-1 (imipenem MIC),
pDM1-blaSMB-1 (ceftazidime MIC) and pDM1-blaOXA-50 (amoxicillin MIC). Graphs show MIC values (µg/mL) from two
independent experiments.
4.2.2 Chromosomally encoded β-lactamase enzymes degrade or misfold in the absence of
DsbA
To gain insight into how impairment of disulfide bond formation impacts the production or activity of
the seven enzymes further tested in this chapter, we assessed their protein levels by immunoblotting in
the presence and absence of DsbA. For five of the tested β-lactamases (AIM-1, BEL-1, OXA-50,
CARB-2 and SMB-1) deletion of dsbA resulted in drastically reduced protein levels demonstrating that
without their disulfide bonds these proteins are unstable and ultimately degrade (Figure 4.3 A). In the

131
case of BPS-1m and POM-1, enzyme levels remained unchanged in the absence of dsbA (Figure 4.3
B).
In all tested cases, β-lactamase enzymes were also significantly less able to hydrolyse the chromogenic
β-lactam substrate nitrocefin (Table 7), consistent with the reduced MIC values recorded in the absence
of DsbA (Figure 4.1). For BPS-1m and POM-1, in particular, this shows that the absence of their
disulfide bond leads to a folding defect resulting in loss of function, despite the fact that enzyme
degradation was not observed (Figure 1.16). The protein levels of the control enzyme L2-1 and its
hydrolytic activity remained unaffected by the dsbA deletion (Figure 4.3, Table 7). Further, the strain
carrying the empty vector control, pDM1, was also unaffected by the absence of DsbA (Table 7).
Figure 4.3 The majority of tested β-lactamase enzymes become unstable in the absence of DsbA. (A) Protein levels were
drastically reduced for the cysteine-containing β-lactamases AIM-1, BEL-1, OXA-50, CARB-2, and SMB-1 when expressed
in E. coli MC1000 dsbA. The amount of the control enzyme L2-1, which contains three cysteines but no disulfide bonds
remains unaffected. (B) Protein levels of BPS-1m, and POM-1 β-lactamases are largely unaffected by the absence of DsbA.
Protein levels of StrepII-tagged β-lactamases were assessed using a Strep-Tactin-AP conjugate; representative blots from three
independent experiments are shown. Molecular weight markers (M) are on the left, DnaK was used as a loading control and
solid black lines indicate where the membrane was cut.

132
Table 7 The hydrolytic activities of tested β-lactamase enzymes are significantly decreased in the absence of DsbA. The
hydrolysis of the chromogenic β-lactam nitrocefin by cysteine-containing β-lactamases is impaired when these enzymes are
expressed in E. coli MC1000 dsbA. The hydrolytic activities of strains harbouring the empty vector or expressing the control
enzyme L2-1, containing three cysteines but no disulfide bonds, show no dependence on DsbA. The “Enzyme stability”
column informs on the stability of each enzyme when it is lacking its disulfide bond(s); this was inferred from the
immunoblotting experiments presented in Figure 4.3. The “Nitrocefin hydrolysis” column shows the amount of nitrocefin
hydrolysed per mg of bacterial cell pellet in 15 minutes. n=3, table shows means ±SD, significance is indicated by * = p <
0.05, ns = non-significant. Significance was determined using unpaired T-test with Welch’s correction; n=3; 3.417 degrees of
freedom, t-value=0.3927, p=0.7178 (non-significance) (for pDM1 strains); 2.933 degrees of freedom, t-value=0.3296,
p=0.7639 (non-significance) (for pDM1-blaL2-1 strains); 2.021 degrees of freedom, t-value=7.549, p=0.0166 (significance) (for
pDM1-blaBEL-1 strains); 2.146 degrees of freedom, t-value=9.153, p=0.0093 (significance) (for pDM1-blaCARB-1 strains); 2.320
degrees of freedom, t-value=5.668, p=0.0210 (significance) (for pDM1-blaAIM-1 strains); 2.345 degrees of freedom, t-
value=15.02, p=0.0022 (significance) (for pDM1-blaSMB-1 strains); 3.316 degrees of freedom, t-value=4.353, p=0.0182
(significance) (for pDM1-blaOXA-50 strains); 3.416 degrees of freedom, t-value=13.68, p=0.0004 (significance) (for pDM1-
blaBPS-1m strains); 3.998 degrees of freedom, t-value=4.100, p=0.0149 (significance) (for pDM1-blaPOM-1 strains).
Strain (MC1000) Enzyme stability Nitrocefin hydrolysis†
pDM1
dsbA pDM1 -
1.82 ± 3.20
0.96 ± 2.06ns
pDM1-blaL2-1
dsbA pDM1-blaL2-1 not degrading
82.66 ± 1.26
82.99 ± 0.77ns
pDM1-blaBEL-1
dsbA pDM1-blaBEL-1 degrading
92.66 ± 17.99
14.05 ± 1.31*
pDM1-blaCARB-2
dsbA pDM1-blaCARB-2 degrading
484.67 ± 72.09
96.79 ± 13.80*
pDM1-blaAIM-1
dsbA pDM1-blaAIM-1 degrading
109.45 ± 5.14
47.77 ± 18.13*
pDM1-blaSMB-1
dsbA pDM1-blaSMB-1 degrading
144.96 ± 14.69
12.11 ± 4.33*
pDM1-blaOXA-50
dsbA pDM1-blaOXA-50 degrading
37.30 ± 7.36
15.59 ± 4.51*
pDM1-blaBPS-1m
dsbA pDM1-blaBPS-1m not degrading
30.55 ± 1.14
13.99 ± 1.76*
pDM1-blaPOM-1
dsbA pDM1-blaPOM-1 not degrading
27.87 ± 1.70
22.23 ± 1.67* †nM.mg-1 pellet.15 min-1
4.2.3 Deletion of dsbA1 compromises the function of the intrinsic β-lactamase OXA-50 in
P. aeruginosa laboratory strains and clinical isolates
The resistance to some β-lactam antibiotics in P. aeruginosa isolates is a consequence of the interplay
between the MexAB-OprM efflux pump and the resident β-lactamase(s) AmpC and OXA-50.373 When
expressed in E. coli, OXA-50 conferred increased amoxicillin and cefuroxime MICs, which were
reduced in the absence of DsbA (Figure 4.1, Supplementary Table 2) due to enzyme degradation (Figure
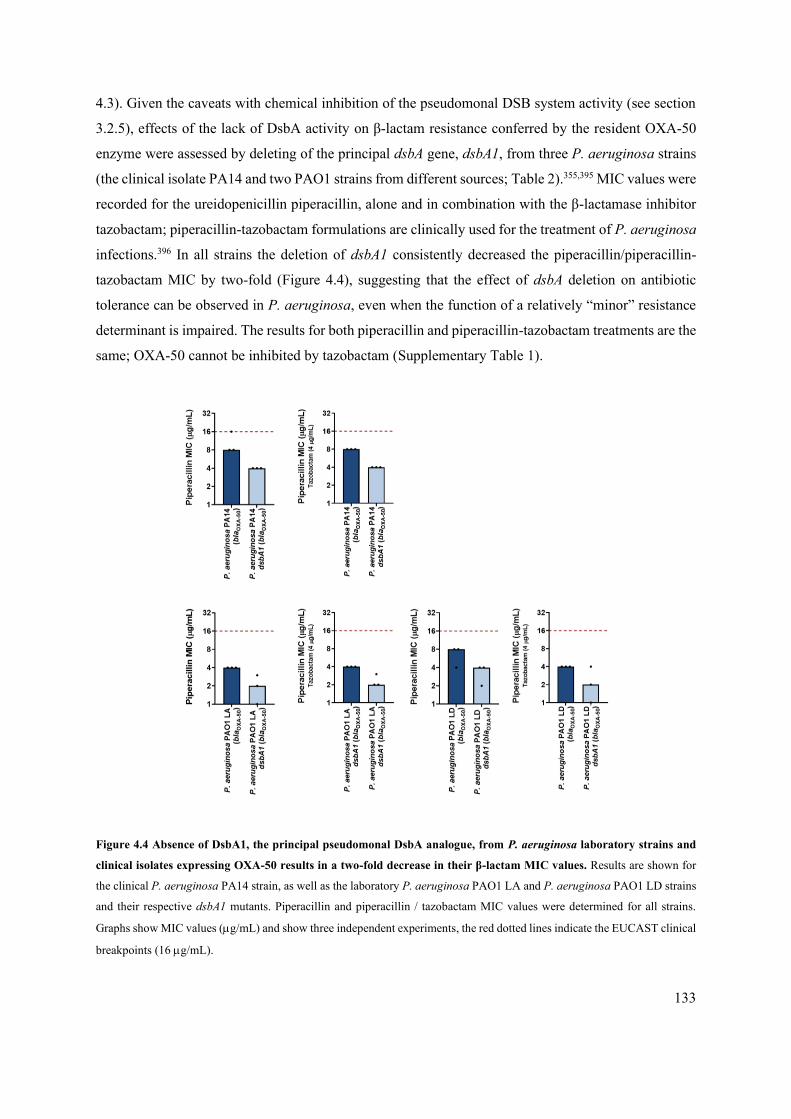
133
4.3). Given the caveats with chemical inhibition of the pseudomonal DSB system activity (see section
3.2.5), effects of the lack of DsbA activity on β-lactam resistance conferred by the resident OXA-50
enzyme were assessed by deleting of the principal dsbA gene, dsbA1, from three P. aeruginosa strains
(the clinical isolate PA14 and two PAO1 strains from different sources; Table 2).355,395 MIC values were
recorded for the ureidopenicillin piperacillin, alone and in combination with the β-lactamase inhibitor
tazobactam; piperacillin-tazobactam formulations are clinically used for the treatment of P. aeruginosa
infections.396 In all strains the deletion of dsbA1 consistently decreased the piperacillin/piperacillin-
tazobactam MIC by two-fold (Figure 4.4), suggesting that the effect of dsbA deletion on antibiotic
tolerance can be observed in P. aeruginosa, even when the function of a relatively “minor” resistance
determinant is impaired. The results for both piperacillin and piperacillin-tazobactam treatments are the
same; OXA-50 cannot be inhibited by tazobactam (Supplementary Table 1).
Figure 4.4 Absence of DsbA1, the principal pseudomonal DsbA analogue, from P. aeruginosa laboratory strains and
clinical isolates expressing OXA-50 results in a two-fold decrease in their β-lactam MIC values. Results are shown for
the clinical P. aeruginosa PA14 strain, as well as the laboratory P. aeruginosa PAO1 LA and P. aeruginosa PAO1 LD strains
and their respective dsbA1 mutants. Piperacillin and piperacillin / tazobactam MIC values were determined for all strains.
Graphs show MIC values (g/mL) and show three independent experiments, the red dotted lines indicate the EUCAST clinical
breakpoints (16 g/mL).

134
4.2.4 Deletion of dsbA1 results in sensitization of P. aeruginosa clinical isolates to existing
β-lactam antibiotics
Given the promising results above, deletion of dsbA1 in P. aeruginosa was carried out for two
multidrug-resistant clinical isolates expressing the class B3 metallo-β-lactamase AIM-1. In contrast to
OXA-50, this β-lactamase confers high-level resistance to piperacillin-tazobactam as well as to the third
generation cephalosporin ceftazidime, an anti-pseudomonal β-lactam used for the treatment of critically
ill patients (Supplementary Table 4).109 MICs for piperacillin, piperacillin-tazobactam and ceftazidime
were determined for the two P. aeruginosa isolates (strains G4R7 and G6R7; Supplementary Table 4)
and their dsbA1 mutants (Figure 4.5).
For both strains the deletion of dsbA1 resulted in decrease MIC values for all tested antibiotics (Figure
4.5). The G4R7 strain was less affected by the loss of DsbA1, which resulted in a substantial 4-fold
decrease in piperacillin MIC values but did not sensitise the isolate to the first-line antibiotic.
Nonetheless, sensitisation to the third-generation cephalosporin ceftazidime was observed. The deletion
of DsbA1 was less tolerated by the G6R7 strain, where sensitisation to all three tested treatments was
observed (Figure 4.5). Consistent with the fact that currently available β-lactamase inhibitors do not
counteract metallo-β-lactamases, the MICs of both strains remained identical in the presence of
tazobactam in the media.62

135
Figure 4.5 Absence of DsbA1, the principal pseudomonal DsbA analogue, sensitises P. aeruginosa clinical isolates
expressing AIM-1 to penicillins and cephalosporins. Deletion of dsbA1 in the AIM-1 expressing P. aeruginosa G4R7
clinical isolate sensitizes this strain to ceftazidime (ceftazidime MIC of 8 g/mL) and results in reduction of the piperacillin
MIC value by over 192 g/mL. Deletion of dsbA1 in the AIM-1 expressing P. aeruginosa G6R7 clinical isolate sensitizes this
strain to piperacillin (piperacillin MIC of 8 g/mL) and ceftazidime (ceftazidime MIC of 6 g/mL). For both AIM-1 expressing
strains the piperacillin MIC values remain unchanged by the addition of tazobactam to the growth media, since AIM-1 is not
inhibited by tazobactam (Supplementary Table 1). Graphs show MIC values (g/mL) and show three independent experiments,
the red dotted lines indicate the EUCAST clinical breakpoints (16 g/mL for piperacillin and 8 g/mL for ceftazidime).

136
4.2.5 Deletion of dsbA1 and dsbL1 results in increased sensitivity of a S. maltophilia clinical
isolate to ceftazidime
To investigate whether the strategy of abrogating oxidative protein folding, which showed promise in
decreasing the resistance of P. aeruginosa clinical strains, could be used for other resistant bacterial
pathogens, S. maltophilia was tested. This organism, often found to colonise the lungs of cystic fibrosis
patients along with P. aeruginosa, was selected because of its high levels of resistance and because of
the fact that its resident L1-1 carbapenemase is dependent on DsbA; for the disulfide-harbouring L1-1
enzyme significant drops in MIC values were observed when expressed in E. coli K-12 lacking dsbA
(Supplementary Table 2).79,338
Two adjacent DSB oxidase genes, dsbA1 and dsbL1, were deleted from the multidrug-resistant S.
maltophilia GUE clinical isolate expressing both L2-1 and L1-1 -lactamases (Supplementary Table
4). Ceftazidime MIC values were recorded as previous work has attributed the high aztreonam MIC
values of this organism exclusively to the activity of the disulfide-free L2-1 -lactamase.137,338,378,379
The deletion of both dsbA1 and dsbL1 resulted in robust drops (8-fold) in the ceftazidime MIC value
for the clinical strain (Figure 4.6). No ceftazidime breakpoint is available for S. maltophilia, due to the
inherently high levels of resistance of this bacterium to cephalosporins afforded by L1-1. Nonetheless,
the recorded MICs for the dsbA dsbL mutant strain were 1-2 g/mL, values that are 4-8-fold lower than
the ceftazidime EUCAST clinical breakpoint for P. aeruginosa (8 g/mL), a closely related organism.
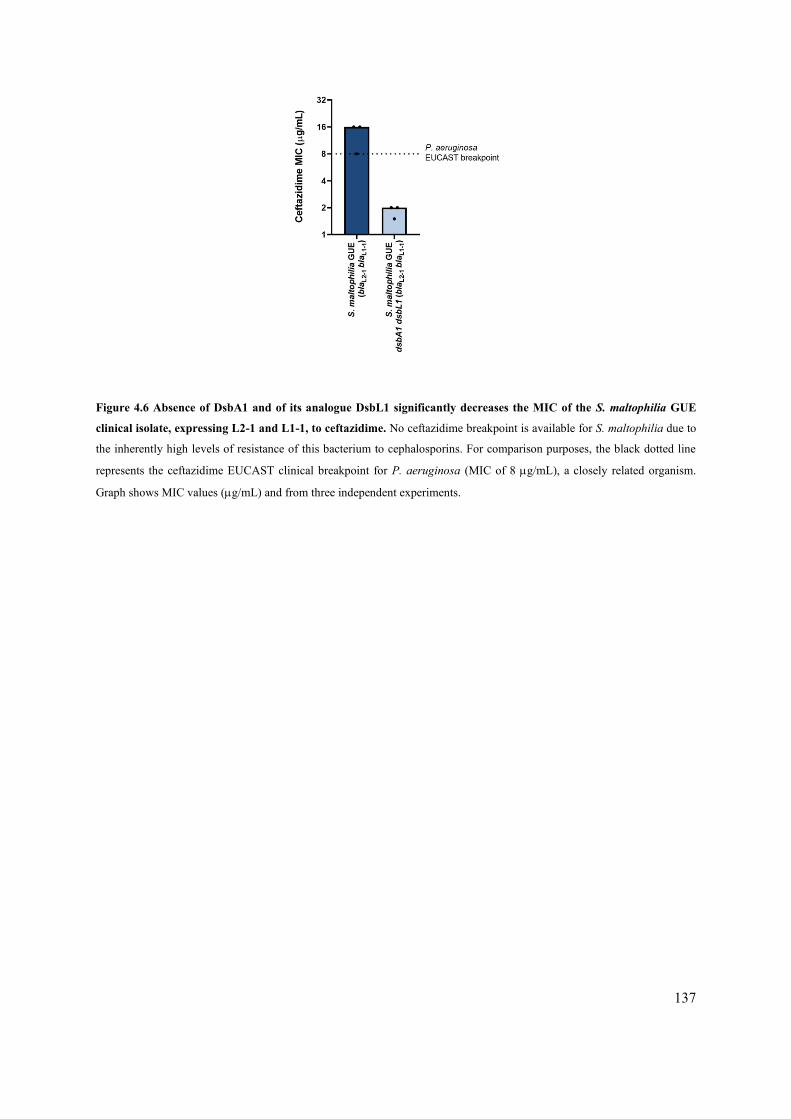
137
Figure 4.6 Absence of DsbA1 and of its analogue DsbL1 significantly decreases the MIC of the S. maltophilia GUE
clinical isolate, expressing L2-1 and L1-1, to ceftazidime. No ceftazidime breakpoint is available for S. maltophilia due to
the inherently high levels of resistance of this bacterium to cephalosporins. For comparison purposes, the black dotted line
represents the ceftazidime EUCAST clinical breakpoint for P. aeruginosa (MIC of 8 g/mL), a closely related organism.
Graph shows MIC values (g/mL) and from three independent experiments.

138
4.3 DISCUSSION
A panel of chromosomally-resident -lactamases exhibiting a range of hydrolytic spectra and driving
intrinsic resistance in their respective host organisms were investigated for their dependence on the
DSB system. Understanding the importance of the native disulfide bonds of these enzymes for their
stability and function allows us to assess the potential of targeting the DSB system as a strategy to
overcome species-specific resistance and treat infections with high-case fatality rates.
Eight -lactamase genes, encoding the BEL-1, BPS-1m, CARB-2, FTU-1, AIM-1, POM-1, SMB-1 and
OXA-50 enzymes, were cloned into an IPTG-inducible vector and expressed in the E. coli MC1000
K12 strain and its isogenic dsbA mutant. Their ability to confer resistance to -lactam antibiotics in the
absence of DsbA was drastically decreased for the majority of the enzymes tested (Figure 4.1). These
included -lactamases associated with mobile genetic elements (BEL-1, CARB-2, AIM-1, and SMB-
1), as well as metallo-β-lactamases found in environmental bacteria (POM-1, AIM-1, SMB-1, and L1-
1338) that cannot be neutralized via classical -lactamase inhibitors (Figure 4.1).109,394 Notably, the
deletion of dsbA, led to substantial ceftazidime MIC drops for the clinically observed extended-
spectrum enzyme BPS-1m which is responsible for the failure of one of the few viable treatments for
melioidosis (Figure 4.1).397,398 Further, the absence of their native disulfide bonds decreased the protein
stability of five out of the seven affected enzymes (Figure 4.3), and impaired the hydrolytic efficiency
of all tested -lactamases (Figure 4.3, Table 7). This is in line with the effects observed previously in
the Mavridou lab338 as well as our understanding of the role of disulfide bonds in the cell envelope.
In addition to BPS-1m, substantial MIC drops were also observed for the class B3 metallo-β-lactamase
AIM-1 (Figure 4.1), which is of clinical concern.76 Moreover, deletion of the primary dsbA gene from
the chromosomes of two multidrug-resistant P. aeruginosa clinical strains expressing this enzyme,
resulted in sensitisation to piperacillin and ceftazidime (Figure 4.5), which are drugs that are routinely
used to treat pseudomonal infections. Further promise of this approach can be seen in the results using
an almost pan-resistant clinical isolate of S. maltophilia expressing the resident L2-1 and L1-1 enzymes.
In this case, robust 8-fold drops in the ceftazidime MIC values were recorded when the genes encoding
the DSB oxidases DsbA and DsbL were deleted (Figure 4.6). This brought down the ceftazidime MIC
of the mutant strain at values that are 4-8-fold lower than the EUCAST clinical breakpoint of P.
aeruginosa, a closely related bacterium to S. maltophilia. Since the DSB system of S. maltophilia is
largely uncharacterized, and the dsbA and dsbL oxidase genes are located next to each other on its

139
chromosome, it was more practical to delete both oxidase genes simultaneously for this first test of the
role of DSB proteins in β-lactamase-mediated resistance; performing gene deletions in clinical isolates
of S. maltophilia is particularly challenging. Further experiments using single-gene deletions would be
required to investigate whether the activity of only one or both oxidases are necessary for the folding
of L1-1.
Together the results presented in this chapter show that the use of future DsbA inhibitors as antibiotic
adjuvants has the potential to help overcome intrinsic resistance of several bacterial species to existing
-lactam antibiotics.

140
5 THE IMPORTANCE OF DISULFIDE BOND FORMATION FOR
THE FUNCTION OF RESISTANCE-NODULATION-DIVISION
EFFLUX PUMPS
5.1 INTRODUCTION
Efflux pumps are crucial for the upkeep of cellular homeostasis in Gram-negative bacteria. They protect
the cytoplasm of the cell by removing a range of toxic substances, including solvents, metabolic by-
products or antimicrobial agents, from the periplasm.32,49,399 In the case of some pathogens, effluxed
substrates also play an active role in the development of biofilms, extracellular structures that exacerbate
antibiotic resistance by limiting antibiotic access and decreasing the achievable intracellular
concentrations of useful drugs.37,400 The majority of characterised antibiotic efflux pump protein
families are localised in the cytoplasmic membrane, where they counteract the spontaneous diffusion
of deleterious compounds into the cytoplasm (Figure 1.1).399 There are a few notable exceptions, with
the most important one being Resistance-Nodulation-Division (RND) efflux pumps, complex molecular
machineries that span the entire cell envelope with their tripartite structure.399,401 In addition to the
cytoplasmic pump, these macromolecular assemblies also have an outer membrane channel and a
periplasmic adaptor protein.402,403 Bacteria often encode more than one RND pump, and partial
redundancy between different pumps or their components, allows for extensive protection of the cell
against cytoplasmically-active antibiotics, which are efficiently expelled from the periplasm to the
extracellular millieu.404
The best characterised RND efflux pump is the E. coli AcrAB-TolC, the assembled structure of which
has just recently been observed in situ.402,403 AcrAB-TolC is composed of the periplasmic component
AcrA, the cytoplasmic membrane protein AcrB, and an outer-membrane channel, TolC (Figure
5.1).402,403,405 All three proteins are essential for the assembly of a functional pump with activity against
substrates like tetracycline, erythromycin, nalidixic acid or chloramphenicol.38,52,402,403,405 Pump
assembly is mediated by AcrA interactions with AcrB, TolC and a small conserved protein AcrZ.47,406,407
Substrate efflux begins by its binding on a loose protomer of the AcrB trimer, which is embedded in
the inner membrane.47,406 Subsequent binding on the tight protomer results in a conformational change
that allows substrate binding on the last of the three AcrB protomers, the open protomer. This in turn

141
results in expulsion of the substrate into the AcrA pump tunnel.406,408 Ultimately, the substrate exits the
cell through the TolC outer-membrane channel, a large trimeric -barrel structure composed of 12
transmembrane -sheets and 12 periplasmic -helices as well as an periplasmic domain.408
Figure 5.1 Structure of the E. coli AcrAB-TolC efflux pump. The RND pump spans across the whole length of the periplasm
and expels a wide range of substrates, including macrolides, chloramphenicol or nalidixic acid, to the extracellular
environment. It is composed of the outer-membrane-embedded TolC trimer, the inner-membrane-embedded AcrB trimer and
the periplasmic hexamer AcrA. PDB code: 5V5S.66,403

142
Expression of the AcrAB-TolC pump in E. coli is dependent on many factors and is, more importantly,
highly responsive to environmental parameters such as the presence of bile, fatty acids, antibiotics or
cationic peptides.409 Genetically, at the local level, acrAB falls under the control of two local repressors,
AcrS/EnvR and AcrR, the genes of which are both located upstream of the acrAB operon.50,51,409 Li et
al. have shown that a variety of antimicrobial compounds are responsible for repression of AcrR, leading
to pump expression.32,51 At the global level, pump expression control is also driven from the multiple
antibiotic resistance operon, Mar, and its transcriptional activator MarA.50,51,409 In addition, a high
degree of sequence homology between MarA, SoxS and Rob (activators implicated in antibiotic and
superoxide resistance) also allows SoxS/Rob-associated pump expression.410 Under normal growth
conditions, MarA, and as a result efflux pump expression, is repressed by MarR.410 Overexpression of
SoxS or Rob, as well as repression or mutation of MarR have been commonly observed in resistant E.
coli isolates with increased AcrAB-TolC expression.409,411,412 Interestingly, these genome mutations
often appear together, demonstrating how complex regulation of efflux pump expression is. Despite
their complex nature, the regulatory control networks characterised in E. coli are broadly conserved in
other species expressing analogues of AcrAB-TolC.409
Although RND pumps are large macromolecular assemblies, they contain very few or sometimes no
cysteine residues in Enterobacteriaceae. As such, they do not contain any disulfide bonds that would
clearly link them to the function of the DSB system.406 However, given the role of DsbA in the folding
of more than 300 extra-cytoplasmic proteins and in the maintenance of cell envelope homeostasis, it is
possible that changes in periplasmic proteostasis occurring in its absence could indirectly influence
efflux pump function.215,242 An examination of the literature on this topic revealed very little. An
unbiased genetic screen by Weatherspoon-Griffin et al., using the Keio collection and an effluxed
antimicrobial peptide called protamine, showed a significant drop in colony forming units (CFU) and
in the survival of E. coli in a disulfide bond formation impaired background (dsbA deletion).179 The
effects recorded in this study were of comparatively lesser impact than those caused by the deletion of
the essential pump components, AcrA and TolC. Further, results from this work also indicated that the
potential link between DsbA and efflux activity relates to the periplasmic chaperone/protease DegP,
although a clear justification for this connection was not given.179 In addition, a study by Hayashi et al.
reported that antibiotic resistance in B. cepacia, an opportunistic Gram-negative pathogen commonly
found in chronic lung infections, is dependent on its DSB system.413,414 A dsbA deletion in B. cepacia
lowered its resistance levels to several antibiotics that are likely efflux substrates for efflux pumps
homologous to those found in Alcaligenes eutrophus or B. pseudomallei.413,415–417 Together, these

143
observations suggest that, despite the absence of disulfide bonds in efflux pumps, there might be a link
between RND pump-mediated resistance and the DSB system.
To investigate the importance of DSB activity for the function of RND efflux pumps, AcrAB-TolC was
selected as a model system. The reason for this choice is that this particular efflux pump has been
extensively characterised and is known to be homologous to RND pumps of other highly resistant
organisms, such as P. aeruginosa (MexAB-OprM) and A. baumannii (AdeABC).48,418,419 MIC values
were measured for a panel of AcrAB-TolC antibiotic substrates, erythromycin, chloramphenicol, and
nalidixic acid, in the wild-type E. coli MG1655 strain and its isogenic dsbA mutant.21,52 Immunoblotting
analysis performed on key pump components and the periplasmic protease/chaperone DegP, which is
known to be important for AcrA proteostasis, to further tease apart the mechanism of the effects
observed.420,421

144
5.2 RESULTS
5.2.1 Deletion of dsbA in E. coli MG1655 does not affect the outer or the inner membrane
permeability
Assessment of the effects of the disruption of periplasmic proteostasis caused by the absence of DsbA
on efflux pump function was carried out in the efflux-active E. coli MG1655 K-12 strain and its isogenic
dsbA mutant. As with the E. coli MC1000 strains (Chapters 3 and 4), the permeability of the outer
membrane of the dsbA mutant was examined using the fluorescent dye 1-N-phenylnaphthylamine
(NPN) and the obtained results were confirmed using a vancomycin MIC assay.361,370 Both experiments
showed that the outer-membrane permeability of the dsbA mutant is no different from that of the
parental strain (Figure 5.2).
Figure 5.2 Deletion of dsbA has no effect on the membrane permeability or on the outer membrane integrity of E. coli
MG1655. (A) The hydrophobic fluorescent dye NPN crosses the outer membrane of E. coli MG1655 and its dsbA mutant to
the same extent. Conversely, exposure to the outer-membrane-permeabilizing antibiotic colistin results in a significant increase
in NPN uptake Graph shows means ± SD, significance is indicated by *** = p < 0.001, ns = non-significant. Significance was
determined using a one-way ANOVA with Bonferroni’s multiple comparison test; n=3; 6 degrees of freedom; F value=261.4;
p=0.00000055 (significance), p=0.0639 (non-significance). (B) No major differences are observed in the MIC values for the
aminoglycoside antibiotic vancomycin confirming that the outer membrane of E. coli MG1655 dsbA is not compromised, and
hence blocks the entry of the antibiotic. Graph shows n=3. NPN assay data is courtesy of Dr R. Christopher D. Furniss
(APPENDIX II).338

145
The integrity of the entire cell envelope of the dsbA mutant was also tested using the fluorescent dye
propidium iodide (PI) as well as a chromogenic CPRG assay.357,359 In both cases, there were no
significant differences between the cell envelope integrity of the wild-type strain and its dsbA mutant
(Figure 5.3).
Figure 5.3 Deletion of dsbA does not result in damage to the bacterial cell envelope. (A) No difference in basal PI uptake
is seen between E. coli MG1655 and its dsbA mutant. Both strains express sfGFP, and fluorescence was used to distinguish
live from dead cells. Addition of the inner-membrane-permeabilizing antimicrobial peptide cecropin A371 to E. coli MG1655
induces robust inner membrane permeabilization in the sfGFP-positive population indicating that the inner membrane becomes
compromised. Graph shows means ±SD, significance is indicated by **** = p < 0.0001, ns = non-significant. Significance
was determined using a one-way ANOVA with Bonferroni’s multiple comparison test; n=3; 6 degrees of freedom; F
value=77.49; p=0.0001 (significance), p=0.9999 (non-significance). (B) No difference in the red colouration of E. coli
MG1655 and its dsbA mutant colonies is seen, suggesting that CPRG is excluded from the colonies to the same extent. This
confirms that the integrity of the cell envelope is not compromised in the dsbA mutant strain. Images of plates were converted
to the CMYK colour space in Adobe Photoshop CS4. Colonies were selected using the magic wand tool with consistent
tolerance and edge refinement, and their magenta levels were compared. Graph shows means ± SD, significance is indicated
by ns = non-significant. Significance determined using an unpaired T-test with Welch’s correction; n=3; 4 degrees of freedom;
t-value=0.02647 p=0.9801 (non-significance). PI uptake data is courtesy of Dr R. Christopher D. Furniss (APPENDIX II).338
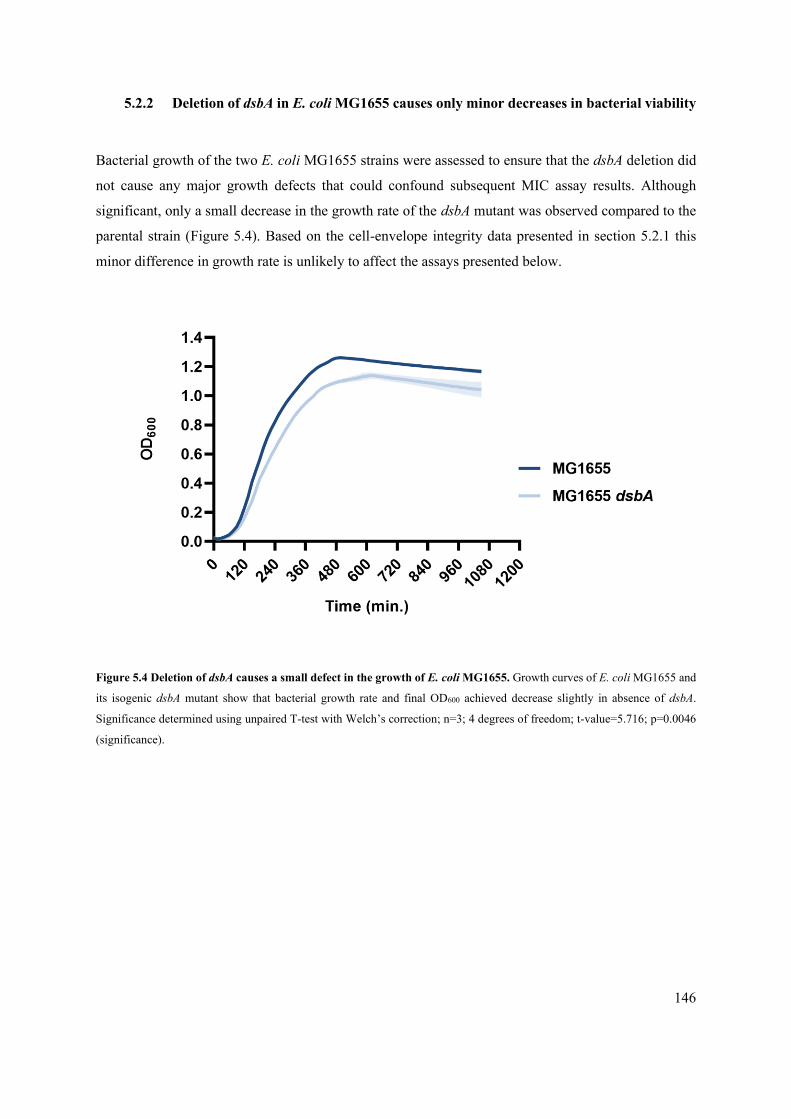
146
5.2.2 Deletion of dsbA in E. coli MG1655 causes only minor decreases in bacterial viability
Bacterial growth of the two E. coli MG1655 strains were assessed to ensure that the dsbA deletion did
not cause any major growth defects that could confound subsequent MIC assay results. Although
significant, only a small decrease in the growth rate of the dsbA mutant was observed compared to the
parental strain (Figure 5.4). Based on the cell-envelope integrity data presented in section 5.2.1 this
minor difference in growth rate is unlikely to affect the assays presented below.
Figure 5.4 Deletion of dsbA causes a small defect in the growth of E. coli MG1655. Growth curves of E. coli MG1655 and
its isogenic dsbA mutant show that bacterial growth rate and final OD600 achieved decrease slightly in absence of dsbA.
Significance determined using unpaired T-test with Welch’s correction; n=3; 4 degrees of freedom; t-value=5.716; p=0.0046
(significance).

147
5.2.3 RND efflux pump function is compromised in the absence of DsbA
Erythromycin, chloramphenicol and nalidixic acid are known AcrAB-TolC substrates.21,38,52 MIC
values for all three antibiotics were determined for the E. coli MG1655, its dsbA mutant, as well as
mutant in acrA which is an essential AcrAB-TolC pump component. The aminoglycoside antibiotic
gentamicin is not an AcrAB-TolC substrate and was used as control to assess the general ability of the
mutant strains to tolerate antibiotic stress. Decreases in MIC values were observed for both mutant
strains, with the acrA mutant being much more affected than the dsbA one; the MIC values measured
for the dsbA mutant were visibly lower than those of the parental strain, but higher than the ones
recorded for the acrA mutant. (Figure 5.5). Although less substantial than the MIC drops for E. coli
MG1655 acrA, the decreases in MIC value for the dsbA mutant were robust and reproducible, and
importantly were not observed for the non-substrate gentamycin. These effects are also in agreement
with previous studies reporting that deletion of dsbA increases the sensitivity of E. coli to dyes like
acridine orange and pyronin Y which are known substrates of AcrAB-TolC.237
The observed phenotype could be fully reversed by complementation of dsbA into the Tn7 site of the
E. coli MG1655 chromosome (Figure 5.6), further indicating a link between DsbA-mediated protein
homeostasis and efflux pump function.
Figure 5.5 Antimicrobial resistance mediated by a tripartite efflux pump, AcrAB-TolC, of E. coli MG1655 is affected
in absence of DsbA. Deletion of dsbA decreases the erythromycin, chloramphenicol and nalidixic acid MIC values for E. coli
MG1655, but no effects are detected for the non-substrate antibiotic gentamicin. The essential pump component AcrA serves
as a positive control. Graphs show MIC values (µg/mL) and shows three independent experiments.

148
Figure 5.6 Complementation of dsbA restores efflux-pump substrate MIC values for E. coli MG1655. Re-insertion of
dsbA at the attTn7 site of the E. coli MG1655 chromosome restores erythromycin, chloramphenicol and nalidixic acid MIC
values for MG1655 dsbA. Graphs show MIC values (µg/mL) and shows two independent experiments.
5.2.4 Compromised function of RND efflux pumps is due to altered periplasmic
proteostasis
To further elucidate the role of DsbA activity in efflux pump function we performed immunoblotting.
Since RND efflux pump proteins do not contain any disulfide bonds, the decreases in MIC values for
pump substrates in the absence of dsbA (Figure 5.5) are likely mediated by additional cell-envelope
components. As indicated by Weatherspoon-Griffin et al. the protease DegP, a previously identified
DsbA substrate, was a promising candidate. DegP degrades a range of misfolded extra-cytoplasmic
proteins including, but not limited to, subunits of higher order protein complexes and proteins lacking
their native disulfide bonds.179,422,423 In a dsbA mutant the substrate burden on DegP would likely be
dramatically increased. Additionally, DegP itself would not function optimally due to absence of its
disulfide bond.424 Consequently, protein turn over in the cell envelope would be less efficient. AcrA, an
essential component of AcrAB-Tolc RND efflux pump, is cleared by DegP when it becomes
misfolded.420,421 Thus, reduction in DegP efficiency in a dsbA mutant could result in accumulation of
non-functional AcrA in the periplasm and interfere with pump function.424

149
Immunoblotting of E. coli MG1655 and its mutants showed DegP degradation leading to the reduction
in the pool of active enzyme at any given time (Figure 5.7 A). In both, dsbA and degP backgrounds,
accumulation of AcrA was observed (Figure 5.7 B) showing that in both strains AcrA was not cleared
efficiently. No accumulation was detected for the control outer-membrane protein TolC (Figure 5.7 C),
which is not a DegP substrate.425 Thus, in the absence of DsbA, inefficient DegP-mediated periplasmic
proteostasis impacts RND efflux pump function through accumulation of non-functional AcrA. These
results are consistent with the study by Weatherspoon-Griffin et al., who did not observe any additive
effects in a dsbA/degP mutant and suggested that these two proteins are part of the same pathway.179
Figure 5.7 RND efflux pump function is impaired in the absence of DsbA due to accumulation of unfolded AcrA
resulting from insufficient DegP activity. (A) In the absence of DsbA the pool of active DegP is reduced. In E. coli MG1655
(lane 1), DegP is detected as a single band, corresponding to the intact active enzyme. In E. coli MG1655 dsbA (lane 2), an
additional lower molecular weight band of equal intensity is present, indicating that DegP is degraded in the absence of its
disulfide bond.423,424 DegP protein levels were assessed using an anti-DegP primary antibody and an HRP-conjugated
secondary antibody. E. coli MG1655 degP was used as a negative control for DegP detection (lane 3); the red arrow indicates
the position of intact DegP. (B) The RND pump component AcrA accumulates to the same extent in the E. coli MG1655 dsbA
and degP strains, indicating that in both strains protein clearance is affected. AcrA protein levels were assessed using an anti-
AcrA primary antibody and an HRP-conjugated secondary antibody. E. coli MG1655 acrA was used as a negative control for
AcrA detection; the red arrow indicates the position of the AcrA band. (C) TolC, the outer-membrane channel of the AcrAB-
TolC pump, does not accumulate in a dsbA or a degP mutant. TolC is not a DegP substrate, hence similar TolC protein levels
are detected in E. coli MG1655 (lane 1) and its dsbA (lane 2) and degP (lane 3) mutants.425 TolC protein levels were assessed
using an anti-TolC primary antibody and an HRP-conjugated secondary antibody. E. coli MG1655 tolC was used as a negative
control for TolC detection (lane 4); the red arrow indicates the position of the bands originating from TolC. For all panels, a
representative blot from three independent experiments is shown. Molecular weight markers (M) are on the left, DnaK was
used as a loading control and solid black lines indicate where the membrane was cut.

150
5.2.5 DsbA as a tractable target for RND efflux pumps
The intrinsic resistance mediated by RND efflux pumps is regulated by the MarA transcriptional
activator, which, in turn, is under the control of a the MarR repressor. Mutations in marR that derepress
MarA and that cause constitutive expression of AcrAB are commonly observed in clinical isolates with
increased efflux.405,410,426,427 An E. coli MG1655 marR mutant with de-regulated efflux pump expression
was used to investigate whether the negative effects observed in the efflux pump activity of E. coli dsbA
strain could also be recapitulated in cases of increased efflux pump expression.
While MIC drops were recorded for all efflux substrates tested on E. coli MG1655 dsbA (Figure 5.5),
chloramphenicol was selected to further test the ability of the de-regulated E. coli MG1655 strain to
resist antibiotic stress. The reason for this choice was that a EUCAST clinical breakpoint is available
for this antibiotic when used against Gram-negative bacteria (E. coli strains with MIC of 8 μg/mL or
below are classified as sensitive).428 Notably sensitisation to chloramphenicol was already observed for
E. coli MG1655 upon deletion of dsbA (Figure 5.5, MIC value = 6, sensitive compared to MIC value =
12, resistant),428 indicating that even the indirect effects resulting from compromising disulfide bond
formation are potentially clinically important. This sensitisation trend also held true when
chloramphenicol MIC values were compared between the de-repressed E. coli MG1655 marR strain
and its dsbA mutant (Figure 5.8).
Figure 5.8 Deletion of dsbA sensitizes the efflux-active E. coli MG1655 strain to chloramphenicol. Sensitization is also
observed for the dsbA mutant of the deregulated E. coli MG1655 marR strain (chloramphenicol MIC of 6 g/mL). The data
presented in the shaded blue and light blue bars were also used to generate Figure 5.7. The graph shows MIC values (g/ml)
from two independent experiments.

151
The already high level of efflux activity in the parental E. coli MG1655 meant that deletion of marR
did not result in a change in the recorded chloramphenicol MIC value compared to the parental strain
(Figure 5.8). Nonetheless, immunoblotting analysis showed that the expression of the AcrA component
of the pump was increased in the absence of MarR in comparison to the parental strain (Figure 5.9),
further validating that abrogating DsbA activity robustly compromises efflux pump function.
Figure 5.9 Deletion of marR results in increased expression of the AcrAB pump. MarR deletion increases the amount of
AcrA (lane 2) compared to the parental strain (lane 1). Expression of the AcrAB pump was assessed using an anti-AcrA
primary antibody and an HRP-conjugated secondary antibody. E. coli MG1655 acrA was used as a negative control for AcrA
detection (lane 3); the red arrow indicates the position of the AcrA band. A representative blot from two independent
experiments is shown; molecular weight markers (M) are shown on the left and DnaK was used as a loading control.

152
5.3 DISCUSSION
RND efflux pumps, which effectively decrease cell envelope concentrations of useful antibiotic
compounds, are an important contributor to the antimicrobial resistance profiles of many Gram-negative
bacterial pathogens. The dependence of the function of E. coli AcrAB-TolC-like efflux pumps on DsbA
was tested using E. coli MG1655 and its dsbA, degP, and acrA mutants. In agreement with our initial
hypothesis, modest but robust decreases in MIC values were observed for all three efflux pump
substrates tested (Figure 5.5). In addition, loss of dsbA sensitised E. coli MG1655 to chloramphenicol,
both in the wild-type background and in the absence of the marR, a mutation that was used to simulate
an efflux-pump-overexpression phenotype commonly observed in clinical strains (Figure 5.8). The
defects observed in the absence of DsbA were driven by the accumulation of non-functional AcrA in a
DegP-dependent manner.
By extension, other efflux pumps containing AcrA-like components are also likely to depend on DegP
for their homeostasis in the periplasm, and their function could be compromised by DsbA or DegP
inhibition. More importantly though, the results presented in this chapter demonstrate that cell envelope
protein homeostasis is crucial for the optimal function of RND efflux pumps and indicate that
periplasmic proteostasis pathways could have significant yet untapped potential for the development of
novel antibacterial strategies against these resistant determinants. The development of broad-acting
clinically applicable efflux pump inhibitors has been challenging to date.32,37 As an alternative,
inhibition of DsbA, DegP or of other cell envelope proteostasis pathways (chaperones and proteases)
could offer a new avenue for the development of efficient strategies to compromise the function of these
elusive macromolecular apparatuses.

153
6 THE IMPORTANCE OF DISULFIDE BOND FORMATION FOR
THE EXPANSION OF THE HYDROLYTIC SPECTRUM OF -
LACTAMASE ENZYMES
6.1 INTRODUCTION
Mutation-driven evolution of narrow-spectrum -lactamases into extended-spectrum and carbapenem-
hydrolysing enzymes is a major contributor to the increasing emergence of resistance to -lactam
compounds, the most prescribed antibiotics worldwide. Numerous studies have shown that vertical
evolution, driven by incremental changes in the primary sequence of narrow-spectrum -lactamases,
can lead to the rapid emergence of enzymes with higher hydrolytic capabilities. Increases in protein
expression levels aside, resistance to -lactam antibiotics has been shown to expand mostly through
two types of amino-acid changes, ‘new-function’ and ‘non-functional’ point substitutions and sequence
modifications.181 This, in addition to the extensive mobility of these promiscuous resistance
determinants, poses critical challenges to modern infection treatment strategies.
Stability-function trade-offs determine the evolutionary success of mutational evolution.181 Key
catalytic residues are evidently retained to prevent detrimental loss of enzymatic activity.181–183,185 New-
function substitutions drive structural changes in the generally unstructured parts of the sequence
surrounding the active site of the enzyme, often causing protein instability, due to the already inherent
strain in the active-site area.181–183,185,429 By contrast, ‘non-functional’ substitutions often act as
compensatory changes that allow protein stabilisation.181,183,430 Both types of primary sequence changes
frequently occur, albeit one at a time, and their functional effects are, overall, additive.181,431
The narrow-spectrum class A -lactamase TEM-1 only confers resistance to first- and second-
generation penicillins. However, the Arg162Ser substitution, which transforms TEM-1 to TEM-12, leads
to resistance to third-generation cephalosporins, like ceftazidime, while the addition of the stability-
inducing substitution Glu104Lys (TEM-26) further expands the spectrum of this enzyme to
monobactams, such as aztreonam.432 Interestingly, mutations resulting in the Glu104Lys on its own, as
observed in TEM-17, do not result in efficient hydrolysis of either ceftazidime or aztreonam.432 It should

154
be noted that expansion of the hydrolytic activity of -lactamases through new-function mutations often
concurrently results in increased affinity for -lactamase inhibitors.133,140,433–436 Increased sensitivity to
sulbactam, clavulanic acid, or tazobactam has been observed in the case of both TEM-12 and TEM-
26.432 Unfortunately, occurrence of gene mutations that allow enzymes to escape mutation-driven
sensitivity to classical inhibitors have also been reported for several -lactamases, including AmpC, a
resident enzyme of P. aeruginosa, or KPC-3 and OXA-48 from K. pneumoniae.133,140,437 In particular,
Fröhlich et al. showed that gradual laboratory exposure of the class D carbapenemase OXA-48 to
ceftazidime-avibactam combinations resulted in the development of ceftazidime (Pro68Ala) as well as
ceftazidime-avibactam (Pro68Ala/Tyr211Ser) resistance through increased flexibility of the active site.437
A 1987 study by Schultz et al. showed that the activity of TEM-1 did not depend on its native disulfide
bond at low temperatures, although substrate hydrolysis was impacted at 42°C.438 A similar evaluation
by Dr R. Christopher D. Furniss (postdoctoral research associate, Mavridou lab) on SHV-27 showed
that its hydrolytic activity against a second-generation cephalosporin, cefuroxime, is unaffected at 37°C,
but decreases under temperature stress (42°C) (APPENDIX II).338 SHV-27 has a single amino acid
substitution (Asp156Gly) with respect to the narrow-spectrum enzyme SHV-1.78,439 By contrast the
activity of a range of both mobile and chromosomal -lactamases with broader hydrolytic spectra
clearly depends on the disulfide-forming protein DsbA even at physiological conditions (Chapters 3, 4,
and APPENDIX II).338 More importantly, strains expressing enzymes capable of hydrolysing more
complex -lactams, such as ceftazidime, imipenem or aztreonam, exhibit larger decreases in their MIC
values when disulfide bond formation is abrogated through the deletion of dsbA, compared to strains
expressing narrower spectrum enzymes (Chapters 3, 4, and APPENDIX II). These observations suggest
that disulfide bond formation is important for the enzymatic function of β-lactamases with broader
hydrolytic activities, i.e. enzymes that present the greatest clinical challenge. This was further
confirmed by the work of Dr R. Christopher D. Furniss on class A -lactamases from the GES
phylogenetic family, for which he showed that expansion of the hydrolytic spectrum of these enzymes
increased their dependence on their native disulfide bonds (APPENDIX II).338
We hypothesized that in narrow-spectrum enzymes non-covalent interactions are sufficient to maintain
the integrity of their native structures, and thus to preserve their hydrolytic activities. Further, we posited
that disulfide bonds become crucial for function when the structures of -lactamases are challenged by
de-stabilising new-function substitutions occurring during expansion of their hydrolytic spectra through
mutation-driven evolution. Given that ‘new-function’ variations generally have de-stabilising effects

155
on evolving enzymes, the presence of a covalent bond (for example a disulfide bridge) could act as a
key stabilising element holding the active site together.440 This is supported by computational studies
by Szarecka et al. and Tokuriki et al., which showed that the disulfide bond stabilizes the structure of
OXA -lactamases during their evolution into enzymes with new hydrolytic functions.181,366 In this
chapter, narrow-spectrum -lactamases SHV-1 and TEM-1 were used to test these hypotheses on the
role of oxidative protein folding in the evolution of enzymes of broad hydrolytic spectra.
Laboratory evolution of TEM-1 and SHV-1 was to be performed by the gradual exposure of E. coli
strains expressing each enzyme to a -lactam compound that could not be efficiently hydrolysed by the
starting -lactamase protein. This process has been documented to lead to the emergence of extended-
spectrum enzymes for several -lactamase families, including TEM and SHV. In the case of SHV-1, a
narrow-spectrum -lactamase first identified in K. pneumoniae only capable of hydrolysing penicillin
and ampicillin,441 point mutants SHV-6 (Asp179Ala) and SHV-2 (Gly238Ser) hydrolyse extended-
spectrum cephalosporins, such as ceftazidime and cefotaxime.77,441,442 An Asp179Asn substitution in the
same site as in SHV-6, expands the hydrolytic activity even more, and allows hydrolysis of
monobactams.443 An overview of key amino acid variations reported to enable the expansion of the
hydrolytic spectra of TEM-1 and SHV-1 is shown in Table 8.
Table 8 An overview of commonly occurring amino acid substitutions in TEM-1 and SHV-1 -lactamases that mediate
expansion of their hydrolytic activity.78,432,436,444–448 The “Spectrum” column refers to the hydrolytic spectrum of each variant
enzyme; narrow-spectrum β-lactamases (NS), extended spectrum β-lactamases (ESBL) or carbapenemases.
Enzyme Spectrum Substitution
TEM-1 NS -
TEM-12 ESBL Arg162Ser
TEM-15 ESBL Gly236Ser/Glu104Lys
TEM-17 ESBL Glu104Lys
TEM-19 ESBL Gly236Ser
TEM-26 ESBL Arg162Ser/Glu104Lys
SHV-1 NS -
SHV-2 ESBL Gly238Ser
SHV-5 ESBL Gly238Ser /Glu256Lys
SHV-38 carbapenemase Ala157Val
SHV-31 ESBL Glu256Lys
SHV-144 ESBL Ala157Val/Leu33Gln

156
To determine whether the emergence of broader spectrum enzymes depends on the presence of their
native disulfide bonds, and by extension on the activity of the DSB oxidase DsbA, generation of some
of these naturally occurring mutants of cysteine-containing -lactamases by way of experimental
evolution was carried out. Application of antibiotic pressure and increase in the hydrolytic activity of
each enzyme through the occurrence of de-stabilising new-function mutations, was expected to render
native disulfide bonds increasingly more important for enzyme stability and function. One can imagine
that as the active site “loosens up” in order to efficiently accommodate a wider range of antibiotic
substrates,185,436,443 the flexibility in this protein region increases and the presence of the disulfide bond
could be important for stabilising the overall enzyme fold and preserving its hydrolytic activity.

157
6.2 EXPERIMENTAL DESIGN
6.2.1 Deletion of dsbA does not severely impact the resistance to -lactams conferred by
the narrow-spectrum -lactamases SHV-1 and TEM-1
The genes for the wild-type TEM-1 and SHV-1 enzymes, as well as their single-cysteine variants
(Cys86Ala TEM-1 and Cys54Ala SHV-1) were codon optimised and cloned into the pDM2 vector, where
constitutive protein expression is driven by a strong synthetic Biofab promoter. The -lactamases were
expressed in the E. coli MC1000 K-12 strain and its isogenic dsbA mutant, and their MIC values were
recorded for a panel of -lactam antibiotics. The aminoglycoside antibiotic gentamicin, which is not
hydrolysed by -lactamase enzymes, and strains carrying the pDM2 empty vector were also included,
as negative controls.
At 37°C the absence of DsbA did not result in a substantial decrease in the -lactam MIC values for
either of the tested enzymes (Figure 6.1 A, MIC fold changes: < 2). At 42°C, MIC values for SHV-1
remained unaffected by the deletion of dsbA, but the ceftazidime MIC for E. coli MC1000 dsbA
harbouring pDM2-blaTEM-1 was 3-fold lower than that of the parental strain (Figure 6.1 B). As expected,
no decreases in MIC values were recorded for the aminoglycoside antibiotic gentamicin, nor strains
carrying the pDM2 empty vector at either tested temperature (Figure 6.1). These observations are
consistent with the results of Schultz et al. (TEM-1) and Furniss et al. (SHV-27, APPENDIX II); the
disulfide bonds do not seem to play a major role for protein function at 37°C, but at higher temperatures,
at which protein stability is challenged, their presence becomes more important for substrate
hydrolysis.338,438
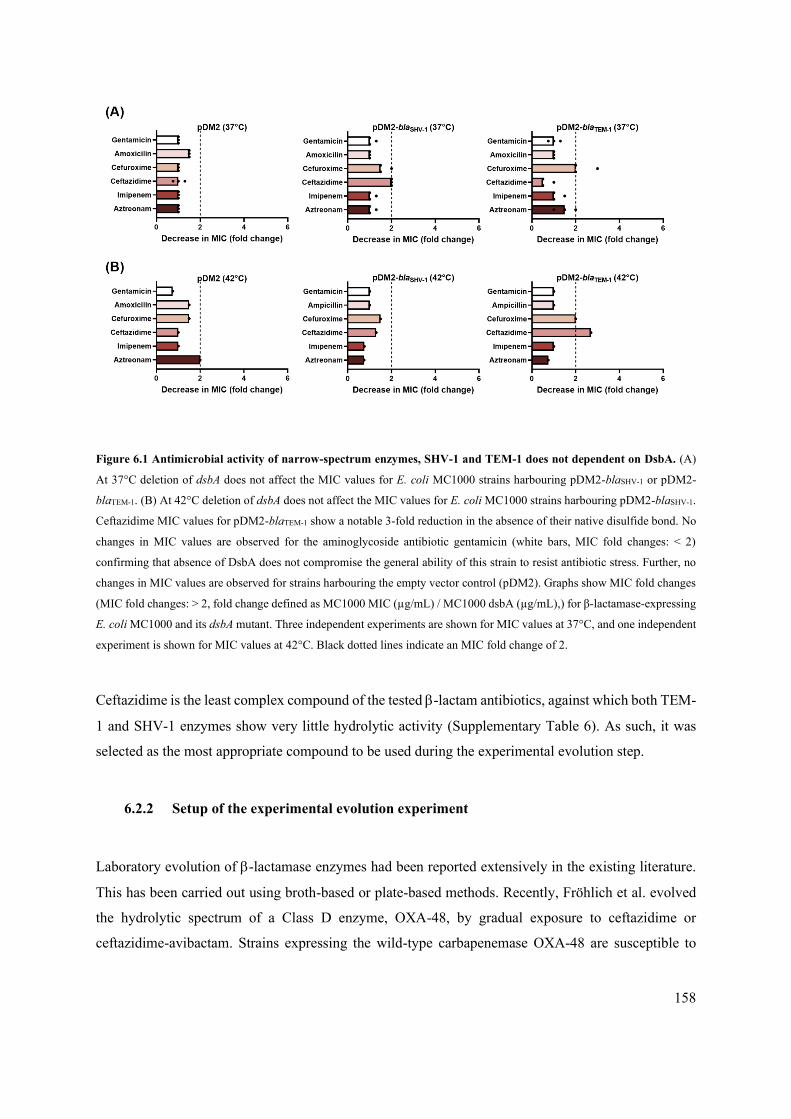
158
Figure 6.1 Antimicrobial activity of narrow-spectrum enzymes, SHV-1 and TEM-1 does not dependent on DsbA. (A)
At 37°C deletion of dsbA does not affect the MIC values for E. coli MC1000 strains harbouring pDM2-blaSHV-1 or pDM2-
blaTEM-1. (B) At 42°C deletion of dsbA does not affect the MIC values for E. coli MC1000 strains harbouring pDM2-blaSHV-1.
Ceftazidime MIC values for pDM2-blaTEM-1 show a notable 3-fold reduction in the absence of their native disulfide bond. No
changes in MIC values are observed for the aminoglycoside antibiotic gentamicin (white bars, MIC fold changes: < 2)
confirming that absence of DsbA does not compromise the general ability of this strain to resist antibiotic stress. Further, no
changes in MIC values are observed for strains harbouring the empty vector control (pDM2). Graphs show MIC fold changes
(MIC fold changes: > 2, fold change defined as MC1000 MIC (µg/mL) / MC1000 dsbA (µg/mL),) for β-lactamase-expressing
E. coli MC1000 and its dsbA mutant. Three independent experiments are shown for MIC values at 37°C, and one independent
experiment is shown for MIC values at 42°C. Black dotted lines indicate an MIC fold change of 2.
Ceftazidime is the least complex compound of the tested -lactam antibiotics, against which both TEM-
1 and SHV-1 enzymes show very little hydrolytic activity (Supplementary Table 6). As such, it was
selected as the most appropriate compound to be used during the experimental evolution step.
6.2.2 Setup of the experimental evolution experiment
Laboratory evolution of -lactamase enzymes had been reported extensively in the existing literature.
This has been carried out using broth-based or plate-based methods. Recently, Fröhlich et al. evolved
the hydrolytic spectrum of a Class D enzyme, OXA-48, by gradual exposure to ceftazidime or
ceftazidime-avibactam. Strains expressing the wild-type carbapenemase OXA-48 are susceptible to

159
both -lactam treatments, nonetheless, resistance development has been observed in clinical strains of
Enterobacteriaceae.437,449 In the work of Fröhlich et al., overnight cultures of strains expressing OXA-
48 were grown in MH broth, spun down and resuspended in a set volume of fresh MH media. 100 L
of that bacterial suspension was plated on MH agar containing increasing concentrations of ceftazidime
or ceftazidime-avibactam (up to 64x MIC).437 After overnight incubation, colonies from the plate with
the highest concentration of ceftazidime were recovered, grown in fresh MH broth and used as a starter
culture for a second round of passaging.437 The emergence of resistant colonies stopped when the
evolving strains reached a ceftazidime MIC of 32 g/mL; at this stage isolated colonies were tested for
the ceftazidime susceptibility.437
An experimental evolution experiment for TEM-1 and SHV-1 was set up using the above method.
However, plating of the bacterial suspensions of E. coli MC1000 strains expressing TEM-1 and SHV-
1 without performing a normalisation step, as was done in the study by Fröhlich et al., resulted in
extensive bacterial growth at all concentrations of ceftazidime used (Table 9).437 Given that the load
used by Fröhlich et al. was optimised for the experimental evolution of a carbapenemase enzyme, and
resulted in no growth at above 32 g/mL of ceftazidime, further inoculum optimisation for strains
expressing narrow-spectrum enzymes was needed.437 Nonetheless, it should be noted that, despite the
high bacterial loads on our selection plates, trends showing a reduced ability for resistance evolution
had started to emerge for SHV-1, especially at high ceftazidime concentrations. More specifically, the
ability to evolve resistance against ceftazidime is lower in the absence of DsbA (50-fold decrease in the
number of resistant colonies at 256 g/mL of ceftazidime) or for the single-cysteine SHV-1 variant,
which can also not form a disulfide bond (25-fold decrease in the number of resistant colonies at 256
g/mL of ceftazidime) (Table 9).
Parental E. coli MC1000 background expressing either wild-type TEM-1 or wild-type SHV-1 were
used to determine the most suitable bacterial load for each set of bacterial strains. The obtained OD600
of bacterial suspensions after an overnight growth were standardised and dilutions were performed.
Subsequently, test inocula were plated on ceftazidime-containing plates of increasing concentrations
and incubated at 37°C for 18 hours (Table 10). Enumeration of the recovered colonies on each plate
allowed the identification of the optimal inoculum to be used for the subsequent experimental evolution
step.

160
Table 9 E. coli MC1000 strains constitutively expressing SHV-1 and TEM-1 -lactamases as well as the single-cysteine
variants of these enzymes were plated on ceftazidime containing plates (0.5-64x MIC). High bacterial loads were
recovered for all concentration of ceftazidime for all strains. “L” strands for “bacterial lawns and “U” stands for “uncountable”
i.e. too many to count.
In contrast to the approach employed by Fröhlich et al., standardisation of the OD600 of the overnight
cultures and selection of an optimal bacterial load for each set of strains ensures the reproducibility of
our results and allows tailoring of the experimental setup to the hydrolytic profile of each enzyme.437 A
schematic presenting our optimised method for performing experimental evolution on strains
expressing TEM-1 and SHV-1 enzymes using ceftazidime pressure is shown in Figure 6.2.
Strain (MC1000) 1 2 4 8 16 32 64 128 256
pDM2-blaSHV-1 L L L L L L U U Est. 300
dsbA pDM2-blaSHV-1 L L L L L L U Est. 200 6
pDM2-blaSHV-1 C54A L L L L L L U Est. 180 12
- 0.125 0.25 0.5 1 2 4 8 16
pDM2-blaTEM-1 - L L L L L L L L
dsbA pDM2-blaTEM-1 - L L L L L L L L
pDM2-blaTEM-C86A - L L L L L L L L

161
Table 10 Bacterial suspensions resulting from overnight growth of E. coli MC1000 strains constitutively expressing
wild-type SHV-1 and TEM-1 -lactamases. Wild-type MC1000 strains harbouring the constitutively expressed pDM2
plasmid encoding the SHV-1 and TEM-1 -lactamases were standardised to OD600 0.5, 1.0 or 2.0 and 10-100L of the neat
suspensions or of further dilutions were plated on ceftazidime-containing plates at 0.5-64x MIC. Enumeration of colonies on
each plate allowed the selection of the optimal bacterial load for the experimental evolution step; bacterial loads resulting in
isolated colonies on plates containing ceftazidime at lower concentrations per plate were counted and loads for the evolution
were selected such that the highest concentration plates were less than 4x MIC and single colonies could be isolated to allow
enumeration and characterisation. “L” strands for “bacterial lawns and “U” stands for “uncountable” i.e. too many to count.
MC1000 pDM2-blaSHV-1 Ceftazidime (g/mL)
OD600 Dilution Load
(L) 1 2 4 8 16 32 64 128 256
2.0
NA
100
L
L L U U 19 1 - - -
NA
10 L L U 107 - - - - -
1:100
100 L L 125 12 - - - - -
1:200
100 L L 57 4 - - - - -
1:400 100 L L 45 1 - - - - -
MC1000 pDM2-blaTEM-1 Ceftazidime (g/mL)
OD600 Dilution Load
(L) 0.125 0.25 0.5 1 2 4 8 16
2.0
NA 100 L L L L L L L L
NA 10 L L L L L L L L
1:100 100 L L L L L L L L
1:200 100 L L L L L L L L
1:400 100 L L L L L L L L
1.0
NA 100 L L L L L L L -
NA 10 L L L L L L L -
1:100 100 L L L L L L L -
1:200 100 L L L L L L L -
1:400 100 L L L L L L L -
0.5
NA 100 U U U 4 1 1 - -
NA 10 U U U 1 - - - -
1:100 100 Est.
350 U
Est.
500 - - - - -
1:200 100 U U 250 - - - - -
1:400 100 U Est.
200 1 - - - - -
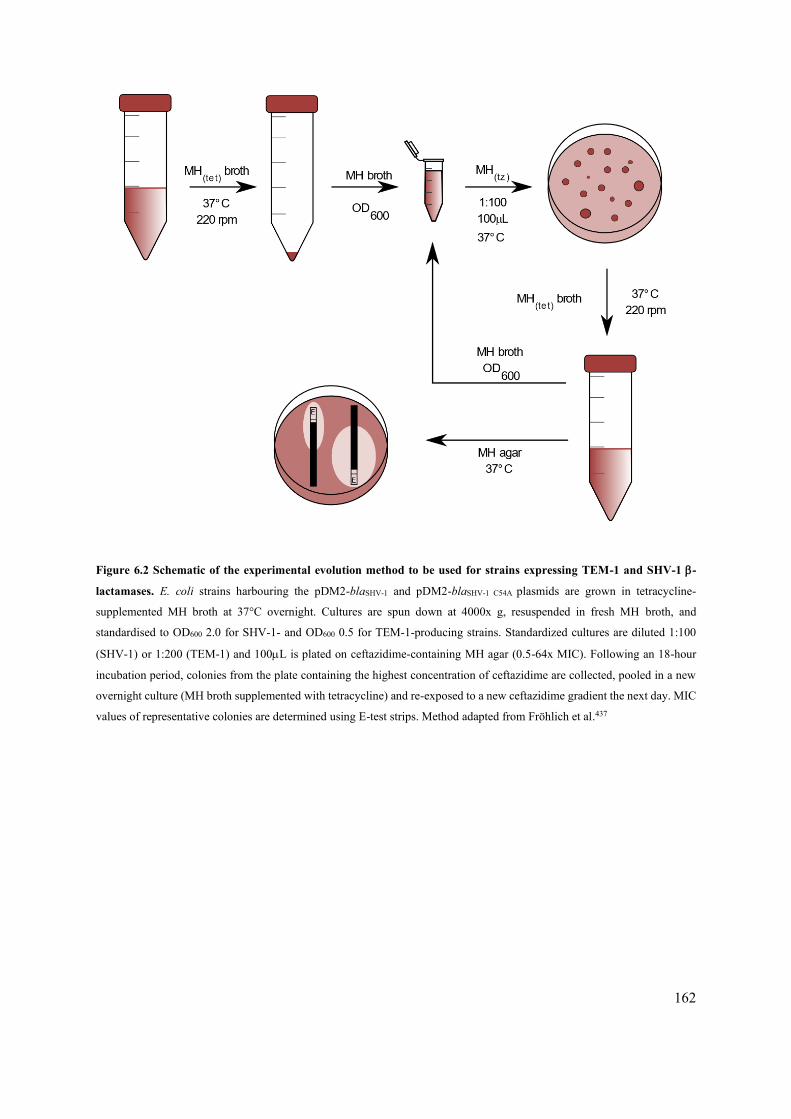
162
Figure 6.2 Schematic of the experimental evolution method to be used for strains expressing TEM-1 and SHV-1 -
lactamases. E. coli strains harbouring the pDM2-blaSHV-1 and pDM2-blaSHV-1 C54A plasmids are grown in tetracycline-
supplemented MH broth at 37°C overnight. Cultures are spun down at 4000x g, resuspended in fresh MH broth, and
standardised to OD600 2.0 for SHV-1- and OD600 0.5 for TEM-1-producing strains. Standardized cultures are diluted 1:100
(SHV-1) or 1:200 (TEM-1) and 100L is plated on ceftazidime-containing MH agar (0.5-64x MIC). Following an 18-hour
incubation period, colonies from the plate containing the highest concentration of ceftazidime are collected, pooled in a new
overnight culture (MH broth supplemented with tetracycline) and re-exposed to a new ceftazidime gradient the next day. MIC
values of representative colonies are determined using E-test strips. Method adapted from Fröhlich et al.437

163
6.3 SHV-1 PILOT STUDY RESULTS
6.3.1 Absence of DsbA decreases the potential for evolution of antibiotic resistance to
ceftazidime upon exposure to increasing antibiotic concentrations
E. coli MC1000 strains constitutively expressing the narrow-spectrum enzyme SHV-1 were subjected
to increasing concentrations of ceftazidime, ranging from 0.5x to 64x of the initial MIC values (4 g/mL
for E. coli MC1000 pDM2-blaSHV-1, and 2 g/mL for E. coli MC1000 dsbA pDM2-blaSHV-1 and MC1000
pDM2-blaSHV-1 C54A). The number of recovered colonies per plate was enumerated, and colonies from
the plate with the highest ceftazidime concentration were pooled for the next round of passaging. In
total three passaging steps were carried out (I-III, Table 11), before colonies with increased ability to
withstand ceftazidime stress stopped emerging. Evolution of resistance to ceftazidime was observed in
all three tested backgrounds, nonetheless varying efficiency in resistance evolution was recorded for
each strain (Table 11).
E. coli MC1000 expressing wild-type SHV-1 produced the greatest number of colonies on increasing
concentrations of ceftazidime throughout rounds I and II of passaging (Table 11). By contrast, resistance
evolution in the absence of DsbA was less efficient, and although resistant colonies emerged at the same
ceftazidime concentration as for the E. coli MC1000 strain, their numbers were greatly decreased.
Surprisingly, the strain expressing the single-cysteine SHV-1 mutant showed a similar behaviour to the
E. coli MC1000 dsbA background for round I of passaging but seemed to behave like the parental strain
during round II. Despite inter-strain differences, these results indicate that oxidative protein folding is
important for the ability of strains expressing narrow-spectrum -lactam to evolve resistance to more
complex -lactam antibiotics.

164
Table 11 E. coli MC1000 strains expressing SHV-1 and its single-cysteine variant, develop resistance upon exposure to
increasing ceftazidime concentrations. Three rounds of passaging were performed (I-III) in each of which 0.5-64x MIC
concentrations of ceftazidime were used. Background MIC values for the strains were as follows: E. coli MC1000 pDM2-
blaSHV-1, MIC of 4 g/mL; E. coli MC1000 dsbA pDM2-blaSHV-1 and E. coli MC1000 pDM2-blaSHV-1 C54A, MIC of 2 g/mL.
The following abbreviations are used: “L” strands for “bacterial lawns and “U” stands for “uncountable” i.e. too many to
count. The star ‘*’ denotes poor bacterial growth.
Passage Strain (MC1000) 1 2 4 8 16 32 64 128 256
I
pDM2-blaSHV-1 NA U U 172 - - - - -
dsbA pDM2-blaSHV-1 L U 13 21 - - - - -
pDM2-blaSHV-1 C54A L U 8 15 1 - - - -
8 16 32 64 128 256 512 1024 2048
II
pDM2-blaSHV-1 L U Est. 750 - - - - - -
dsbA pDM2-blaSHV-1 L U 19 - - - - - -
pDM2-blaSHV-1 C54A NA U Est.950 3 - - - - -
32 64 128 256 512 1024 2048 4096 8192
III
pDM2-blaSHV-1 U 18* - - - - - - -
dsbA pDM2-blaSHV-1 U 10* - - - - - - -
pDM2-blaSHV-1 C54A U 4* - - - - - - -
6.3.2 Characterisation of evolved SHV-1 expressing strains
Three evolved colonies per strain background generated during passage II, were isolated from the plate
containing 32g/mL ceftazidime, and their MICs values were determined for a panel of -lactam
antibiotics (henceforth referred to as “Evolved” strains). In addition, their -lactamase-expressing
plasmids were isolated and transformed into the original E. coli MC1000 or E. coli MC1000 dsbA
backgrounds; MIC values for these strains were also determined for the same antibiotics (henceforth
referred to as “Original” strains). For all nine characterised evolved isolates, increased MIC values for
complex -lactam antibiotics were recorded.
Evolution of E. coli MC1000 pDM2-blaSHV-1 resulted in two to four-fold increase in ceftazidime MIC
values, while only modest changes in the MIC values for other antibiotics, such as cefuroxime or
aztreonam (Figure 6.3, Supplementary Table 7) were recorded. Importantly, MIC values were
comparable for both the evolved and the original strains, suggesting that increased resistance emerged

165
from mutational changes occurring on the resistance plasmid, and likely either on the -lactamase
sequence or its promoter region.
Figure 6.3 Determination of -lactam MIC values (µg/mL) of three isolates of E. coli MC1000 pDM2-blaSHV-1 obtained
during passage II from plates containing 32 g/mL of ceftazidime. Graphs labelled “Evolved” show the MIC values of the
three isolates (#1, #2, #3) directly isolated from the experimental evolution experiment. Graphs labelled “Original” graphs
show the MIC values of the un-evolved E. coli MC1000 harbouring the plasmids obtained from the three evolved isolates.
“Background MICs” are the MIC values of un-evolved E. coli MC1000 pDM2-blaSHV-1. No changes to the MIC values were
observed for the aminoglycoside, gentamicin (white bars), which served as a negative control. Three independent experiments
are shown for the background MIC values, and one independent experiment is shown for Evolved and Original strains.
In the absence of DsbA, emerging resistant colonies had increased MIC values for cefuroxime (6-8-
fold), ceftazidime (8-16-fold), and aztreonam (4-8-fold) (Figure 6.4, Supplementary Table 7). However,
these high MIC values were not recapitulated when the evolved plasmids were transformed into the
original dsbA mutant background (Figure 6.4). Moreover, transformation of the evolved plasmids into
the parental strain, which can introduce the native disulfide bond into the SHV enzyme, also failed to
reproduce the high MICs recorded for the evolved dsbA mutants. Together, these results suggest that
resistance in this background is not only less likely to evolve (Table 11) but is also mostly arising from
changes to the strain background rather than modifications to the enzyme sequence.
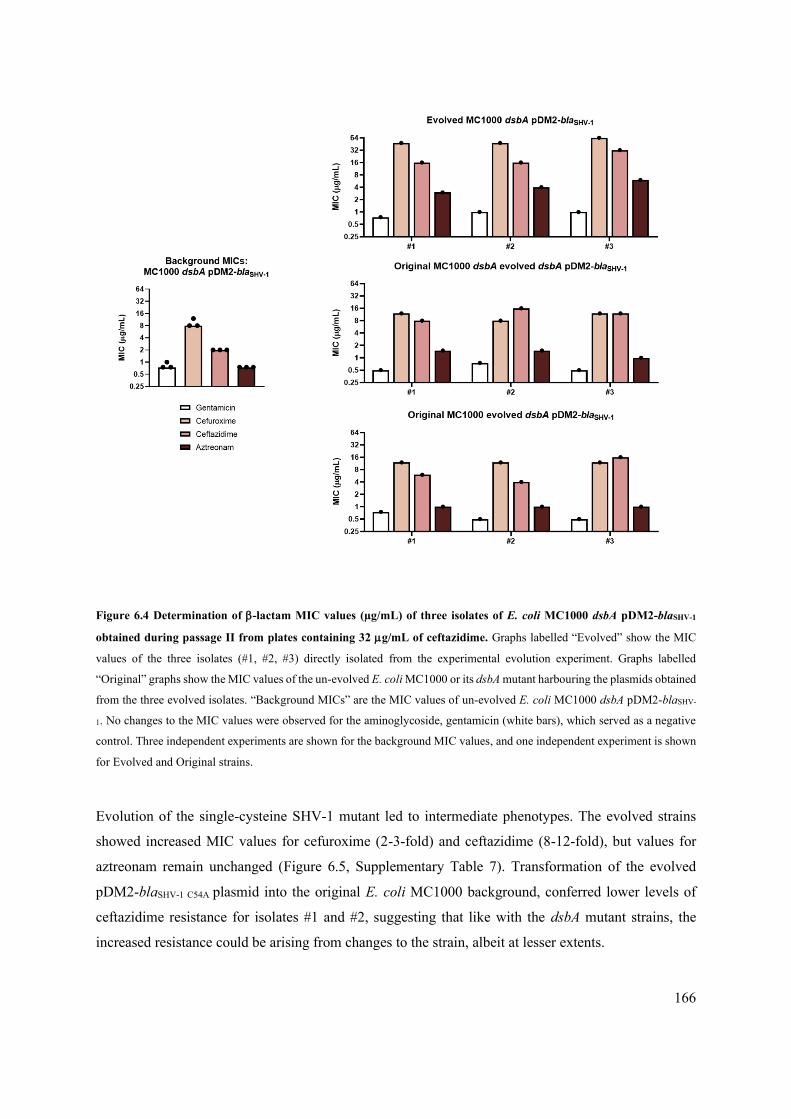
166
Figure 6.4 Determination of -lactam MIC values (µg/mL) of three isolates of E. coli MC1000 dsbA pDM2-blaSHV-1
obtained during passage II from plates containing 32 g/mL of ceftazidime. Graphs labelled “Evolved” show the MIC
values of the three isolates (#1, #2, #3) directly isolated from the experimental evolution experiment. Graphs labelled
“Original” graphs show the MIC values of the un-evolved E. coli MC1000 or its dsbA mutant harbouring the plasmids obtained
from the three evolved isolates. “Background MICs” are the MIC values of un-evolved E. coli MC1000 dsbA pDM2-blaSHV-
1. No changes to the MIC values were observed for the aminoglycoside, gentamicin (white bars), which served as a negative
control. Three independent experiments are shown for the background MIC values, and one independent experiment is shown
for Evolved and Original strains.
Evolution of the single-cysteine SHV-1 mutant led to intermediate phenotypes. The evolved strains
showed increased MIC values for cefuroxime (2-3-fold) and ceftazidime (8-12-fold), but values for
aztreonam remain unchanged (Figure 6.5, Supplementary Table 7). Transformation of the evolved
pDM2-blaSHV-1 C54A plasmid into the original E. coli MC1000 background, conferred lower levels of
ceftazidime resistance for isolates #1 and #2, suggesting that like with the dsbA mutant strains, the
increased resistance could be arising from changes to the strain, albeit at lesser extents.

167
Figure 6.5 Determination of -lactam MIC values (µg/mL) of three isolates of E. coli MC1000 pDM2-blaSHV-1 C54A
obtained during passage II from plates containing 32 g/mL of ceftazidime. Graphs labelled “Evolved” show the MIC
values of the three isolates (#1, #2, #3) directly isolated from the experimental evolution experiment. Graphs labelled
“Original” graphs show the MIC values of the un-evolved E. coli MC1000 harbouring the plasmids obtained from the three
evolved isolates. “Background MICs” are the MIC values of un-evolved E. coli MC1000 pDM2-blaSHV-1 C54A. No changes to
the MIC values were observed for the aminoglycoside, gentamicin (white bars), which served as a negative control. Three
independent experiments are shown for the background MIC values, and one independent experiment is shown for Evolved
and Original strains.
In all three strain backgrounds, no changes to carbapenem susceptibility were recorded (Supplementary
Table 7). Further, no changes in the MIC values of the gentamicin control were recorded (Figure 6.3,
Figure 6.4, Figure 6.5), indicating that any changes to the strains during evolution are not affecting the
general ability of bacteria to withstand antibiotic stress. It should be noted that due to time limitations,
the MIC values reported here represent a single MIC experiment, and biological replicates would be
needed to fully confirm the reliability of the presented data.
6.3.3 Increase of the hydrolytic spectrum of SHV-1 does not affect bacterial fitness
Evolution of antibiotic resistance is often linked to fitness costs.450 For this reason bacterial growth of
the evolved, as well as the original E. coli MC1000 strains harbouring the evolved plasmids, was

168
assessed under standard growth conditions to determine whether the experimental evolution process
resulted in the generation of strains with significant growth defects. No differences in growth were
observed for any of the evolved strains, when compared to their original background counterparts
carrying evolved plasmids that were generated during evolution (Figure 6.6).
Figure 6.6 The experimental evolution process does not affect the fitness of any of the evolved strains. Growth rates of
all three evolved strain backgrounds under standard growth conditions were compared to the growth of the original strain
backgrounds harbouring the plasmids obtained from the evolved strains. SD is marked by the light blue and light grey shaded
area in each graph. Significance determined using unpaired T-test with Welch’s correction; n=3; 4 degrees of freedom. For
multiple comparisons: t-value=2.118, p=0.1016 (non-significance, Original MC1000 evolved pDM2-blaSHV-1 #3 versus
Evolved MC1000 pDM2-blaSHV-1 #3 ); t-value=1.783, p=0.149 (non-significance, Original MC1000 evolved dsbA pDM2-
blaSHV-1 #3 versus Evolved MC1000 dsbA pDM2-blaSHV-1 #3); t-value=0.03789, p=0.9716 (non-significance, Original
MC1000 dsbA evolved dsbA pDM2-blaSHV-1 #3 versus Evolved MC1000 dsbA pDM2-blaSHV-1 #3); t-value=1.360, p=0.2455
(non-significance, Original MC1000 evolved pDM2-blaSHV-1 C54A #3 versus Evolved MC1000 pDM2-blaSHV-1 C54A #3).

169
6.4 DISCUSSION
Native disulfide bonds play a key role in the stability, folding and function of many extra-cytoplasmic
proteins in Gram-negative bacteria.236 Further, the importance of the DSB system for the activity and
stability of class A, B, and D -lactamases has been investigated in the previous chapters (chapter 3, 4
and APPENDIX II). These investigations showed an increased dependence of broad-spectrum -
lactamase enzymes on the DSB oxidase DsbA, compared to narrow-spectrum ones. We hypothesized
that this was due to the fact that the looser active site of broader-spectrum enzymes, necessary to allow
the binding of more complex -lactams, might lead to increased protein flexibility and thus greater
dependence on the stability afforded by the presence of a covalent linkage, like a disulfide bond.185,436,443
Amino acid substitutions in the primary sequences of -lactamase enzymes are one of the key factors
behind the emergence of resistance evolution in this family of proteins. Point changes in numerous
narrow-spectrum hydrolases have been observed to give rise to extended-spectrum and carbapenemase
enzymes that are of great concern to current antibiotic therapy. New-function gene mutations driving
the expansion of the hydrolytic spectrum of the enzymes, are commonly supported by secondary non-
functional mutations, which result in increased protein stability. Extending this to disulfide bonds,
which are known to generally increase protein stability, this chapter focused on testing whether the
process of oxidative protein folding is key for the evolution of broad-spectrum cysteine-containing -
lactamases.451
A pilot experimental evolution study of the narrow-spectrum SHV-1 -lactamase from K. pneumoniae
was performed in presence and absence of its native disulfide bond, using an adaptation of the method
developed by Fröhlich et al.437 Strains expressing SHV-1 were exposed to gradually increasing
concentrations of the complex -lactam ceftazidime, which they could not hydrolyse prior to this
exposure. By abrogating the capacity for disulfide bond formation via deletion of dsbA or removal of
one of the two conserved cysteine residues of the enzyme, 8- to 40-fold decrease in the number of
breakthrough colonies was observed at ceftazidime concentrations higher than the initial strain MIC,
compared to the parental strain.
-lactam MIC values were recorded for three evolved colonies per strain background and in all cases
increase in resistance to complex -lactams was confirmed. However, further MIC testing showed that

170
in the absence of the enzyme’s native disulfide bond this increased capacity to survive -lactam
antibiotic pressure may be arising from mutations on the strain background rather than the -lactamase
gene. This is particularly notable in the dsbA mutant strain, where evolved strains seem to have the
highest -lactam MIC values, but transformation of the evolved plasmids into the original strain
backgrounds does not recapitulate these MICs. This suggests that evolutionary pressure on disulfide-
free SHV-1 enzymes, not only leads to the emergence of drastically fewer resistant colonies, but also
that resistance does not originate from the evolution of the hydrolytic enzyme.
Despite these promising results, further work is required to fully elucidate the role of the DSB system
in -lactamase evolution. Briefly, this includes 1) DNA sequencing of the evolved plasmids for at least
10% of the resistant colonies obtained; 2) characterisation of the resistant colonies used for sequencing
in a similar manner as performed in this chapter (-lactam MIC value determination in the evolved and
original strain backgrounds); 3) repeating the experimental evolution step on strains expressing SHV-1
using ceftazidime, but also another -lactam antibiotic, like cefuroxime or aztreonam; 4) testing of the
experimental evolution process on the second narrow-spectrum enzyme, TEM-1.

171
7 DISCUSSION AND FUTURE WORK
The increasing emergence of resistant bacterial strains represents a silent pandemic that results in
hundreds of thousands of deaths every year.452 Despite extensive efforts over the past decades, few
novel therapeutics are in development and even fewer are successfully reaching the clinics. Generation
of new antibiotic compounds is particularly challenging for many Gram-negative bacteria, such as E.
coli, P. aeruginosa, K. pneumoniae and A. baumannii, which are currently classified as microorganisms
of critical importance.452 Therefore, in a race to extend the lifetime of already approved drugs, new
strategies aiming to break antimicrobial resistance are of growing interest. In this work the DSB system,
a conserved oxidative protein folding pathway in the cell envelope of Gram-negative bacterial species,
is characterised for its role in intrinsic and acquired resistance caused by -lactamase enzymes and
multi-drug efflux pumps, in order to establish whether it could be a novel target against antimicrobial
resistance.
-lactamase enzymes are expressed from a variety of genetic locations and are major contributors to
multidrug-resistant phenotypes due to their ability to hydrolyse different classes of -lactam antibiotics,
from penicillins to carbapenems and monobactams. As such, new approaches that would allow us to
inhibit their hydrolytic activity have great potential for targeting antibiotic resistance. In this work a
total of 13 phylogenetically distinct -lactamase enzymes from different Ambler classes and organisms
(B. pseudomallei, F. tularensis, P. aeruginosa, P. otitidis, S. marcescens and S. maltophilia) were
characterised in the presence and absence of DsbA to assess their functional dependence on this key
oxidative folding catalyst. For most of these enzymes, decrease in protein stability and/or hydrolytic
activity resulted in decreased MIC values against multiple -lactam antibiotics, both in model E. coli
strains and in multidrug-resistant P. aeruginosa and S. maltophilia clinical isolates. Notably the greatest
effects were observed for enzymes with broader hydrolytic activities, whilst enzymes of narrower
spectra, such as CARB-2 or FTU-1, were less dependent on their disulfide bonds (Figure 4.1). In vivo
clearance assays using the wax moth model confirmed that in infections of G. mellonella larvae with a
P. aeruginosa isolate expressing the carbapenemase OXA-198, deletion of dsbA in combination with
piperacillin treatment robustly decreased the bacterial load of the infection by more than 99% (Figure
3.11). This result demonstrates the promise of this approach as a strategy to break antimicrobial
resistance. Time constraints prevented further investigation of the effects of abrogating the function of
the DSB system in vivo. Future work should include the use of the same G. mellonella infection model

172
to investigate whether these observations can be recapitulated in other clinical isolates, for example P.
aeruginosa strains expressing AIM-1 or OXA-19, which were sensitised to -lactam antibiotics when
dsbA was deleted, or inherently resistant organisms, like S. maltophilia, where deletion of DSB genes
resulted in decrease of its ability to tolerate cephalosporin antibiotics (Figure 4.5, Figure 4.6).
Ultimately, a true trial of this strategy would require testing it in a relevant murine infection model,
such as the sub-cutaneous abscess model.453 Implementation of this model is straightforward, enables
highly reproducible chronic infections, and has been shown to be compatible with most organisms of
interest, like P. aeruginosa. The model involves an injection of bacteria underneath the thin skeletal
muscle at the right dorsum on the back of shaved mice. This results in bacterial growth at high densities
within a raised lump around the inoculation point, and subsequent formation of an abscess above the
lump. Bacteria can persist in the abscess for days and resultant infections can be treated by injection of
antibiotics directly into the inflamed tissue. Reduction of bacterial load results in decrease of the size
of the abscess allowing visual monitoring of the treatment; additionally, infection progress can also be
assessed by CFU counts per abscess after sacrificing the animals. To design such in vivo experiments,
especially ones involving vertebrate infection models, it is also important to consider that dsbA gene
deletions are not an ideal strategy for abrogating DSB function; loss of oxidative protein folding can
often cause misfolding of virulence factors and affect the ability of pathogens to establish an
infection.216,245 This means that the use of gene deletions could introduce confounding factors that would
prevent the investigation of the role of DsbA in antibiotic resistance. In the absence of an appropriate
DSB system chemical inhibitor for in vivo experiments, a solution to this problem would come from
using inducible dsbA knockdowns, which could be induced at the point of antibiotic treatment. The
dsbA knockdowns would be based on inducible Mobile-CRISPRi gene silencing, which has been shown
to be effective in both P. aeruginosa and K. pneumoniae infections.454 This way the experimental setup
would overall be simulating, as closely as possible, the treatment of human resistant infections.
The importance of the activity of the DSB system for the folding and function of -lactamase enzymes
demonstrated previously in the Mavridou lab, and further proven in this work, raised the question of
whether other AMR determinants, located in the bacterial cell envelope, might depend on this oxidative
folding pathway (Chapters 3, 4, and APPENDIX II).338 A prominent periplasmic mechanism of
resistance in Gram-negative species is the expulsion of toxic compounds from the cell envelope through
the activity of RND efflux pumps. MIC value determination for antibiotics that are pump substrates
showed that the function of the E. coli AcrAB-TolC RND pump is compromised when dsbA is deleted

173
(Figure 5.5). Despite its considerable size, the AcrAB-TolC assembly does not contain any disulfide
bonds. As such, it is not directly acted on by DsbA, and the observed effects had to be the result of
additional protein interactions. The chaperone/protease DegP was identified as the link between efflux
pump efficiency and oxidative folding (Figure 5.7). DegP is a substrate of DsbA and plays a key role
in periplasmic proteostasis under temperature or reductive stress conditions. Absence of its native
disulfide results in its decreased stability and leads to suboptimal clearing of its substrates, including
AcrA, especially in the presence of reductive stress caused by the absence of DsbA that results in overall
increased DegP substrate load.420 Immunoblotting experiments using a labile AcrA counterpart, AcrA
Leu222Gln, in the presence and absence of DsbA/DegP would provide independent confirmation of the
inability of DegP to deal with its substrate load when oxidative protein folding is abrogated, and
strengthen the link between DsbA function and efflux pump activity.420 In addition to the fact that efflux
activity is compromised due to insufficient periplasmic proteostasis when DsbA is absent, the
possibility that DsbA might act as a chaperone for efflux pump components needs to also be considered.
Disulfide-independent DsbA chaperone activity has been shown in the literature, although experimental
validation is scarce.455 To this end, a DsbA variant where the active site cysteines would be replaced by
alanine or serine residues could be used to test whether any efflux pump components are chaperoned
by DsbA. This would be assessed by immunoblotting of each efflux pump component in the absence
of DsbA or in the presence of cysteine-free DsbA. As AcrA accumulates when dsbA is deleted, here we
would be looking for decrease of AcrB or TolC protein levels in the absence of DsbA and reversal of
this phenotype in the presence of cysteine-free DsbA.
Deletion of dsbA was found to sensitise an E. coli strain to chloramphenicol (Figure 5.8). This indicates
that indirectly compromising resistant determinants via affecting periplasmic proteostasis might be a
strategy worth considering, in particular, for resistance targets such as efflux pumps, for which years of
research have generated very few successful inhibitors.156 As such, it would generally be worth to
investigate the role of other cell envelope folding catalysts (for example chaperones or proteases like
SurA, Skp, FkpA, YfgM, PpiA/D, Spy, HdeA/B, DegQ) in the context of antibiotic resistance.214
Surprisingly, despite the proteinaceous nature of many resistance determinants that are localized in the
cell envelope, the role of these folding catalysts in safeguarding their integrity remains largely
unexplored. This is likely due to the fact that most studies on the function of these proteins have been
carried out in susceptible model strains, and more specifically the E. coli K-12. Therefore, it would be
crucial that any future studies on the role of these proteostasis systems in mechanisms of resistance are
carried out on strains that have either been engineered to express specific resistance proteins, as we did
here, or are resistant clinical isolates.

174
In addition to new interventions that would abrogate the function of intrinsic or mobile resistance
mechanisms, it is also important to find ways to delay or prevent antibiotic resistance evolution. -
lactamase enzymes encompass almost 4,000 discrete proteins of diverse hydrolytic capabilities.456 Some
-lactamase families have several hundreds of members and are thus ideal to test such anti-evolution
approaches. In a pilot study using the narrow-spectrum -lactamase SHV-1, it was shown that absence
of the native disulfide bond of the enzyme decreased the efficiency of mutational acquisition of
resistance by 88-98% in response to the applied antibiotic pressure (Table 11). This indicates that the
DSB system is not only a promising target for the abrogation of resistance, but its inhibition would also
block the evolution of narrow-spectrum enzymes to enzymes that can hydrolyse complex -lactams,
which are drugs of clinical importance.
The experiments presented in Chapter 6 of this thesis are only preliminary and numerous additional
tests need to be carried out to confirm the importance of disulfide bond formation for the evolution of
-lactamase-mediated resistance. Namely the following steps will be taken:
1) The experiments on SHV-1 will be repeated using antibiotic pressure applied by ceftazidime;
additionally, other complex -lactams like aztreonam will be tested. In addition to a dsbA mutant and a
strain expressing a single-cysteine variant of SHV-1, a strain producing a double-cysteine -lactamase
variant will also be included. These experiments will confirm the reproducibility of our original
observations on the effects of the absence of disulfide bond formation for the evolution of -lactamase
resistance.
2) For at least 10% of the evolved resistant colonies emerging for each of the tested strains, next
generation whole-plasmid sequencing will be performed using the Illumina MiSeq platform in order to
identify mutations, both in the -lactamase DNA sequence and in the rest of the plasmid, that are
responsible for evolution of resistance. This will be complemented by determination of -lactam MIC
values of evolved strains and of original strains harbouring the evolved plasmids. In cases of
discrepancy between the two sets of MIC values, something expected to happen mostly in strains
expressing enzymes without their native disulfide which are struggling to expand their hydrolytic
spectra, whole-genome sequencing of the evolved strains will be carried out. This will provide
information on the origins of the resistance in strains expressing disulfide-free -lactamases.

175
3) Specific amino acid substitutions that expand the hydrolytic spectrum of SHV-1 have been both
generated in the lab and identified in clinical samples.439,444,446,447 Representative broad-spectrum SHV-
1 point mutants (Gly255Ser, Ala157Val, Glu256Lys, Glu255Ser/Gln256Lys, Ala157Val/Leu33Gln) will be
constructed in the same expression system and their hydrolytic activity will be determined using MIC
assays for a panel of different classes of β-lactam drugs, in the presence and absence of DsbA. This
approach will identify the point during the reported evolution of this enzyme at which disulfide bonds
might become essential for function and thus for evolution of resistance.
4) All of the above experiments will also be performed for strains expressing the narrow-spectrum
enzyme TEM-1. In this case, the following point mutants will be used to increase its hydrolytic activity:
Glu104Lys, Gly236Ser, Arg162Ser, Gly236Ser/Glu104Lys and Arg162Ser/Glu104Ser. Like with SHV-1, these
rationally mutated enzymes with extended hydrolytic spectra are expected to have increased
dependence on their the native disulfide bond, as seen with other enzymes tested in this thesis or
previously (APPENDIX II).338
In summary, this work characterises further the role of the DSB system, and especially of the principal
oxidase DsbA, in antimicrobial resistance with the aim to assess its potential uses as a novel target for
the simultaneous inhibition of several periplasmic resistance determinants. Further, it hints at the
importance of disulfide bonds for the evolution of narrow-spectrum -lactamases to enzymes that can
hydrolyse complex -lactams, including antibiotics of last resort. Given the already known contribution
of the DSB system in bacterial virulence, targeting this non-essential bacterial pathway could have
multiple beneficial outcomes in decreasing bacterial pathogenicity, breaking antibiotic resistance and
blocking resistance evolution.216,236

176
8 REFERENCES
1. Susser, M. & Susser, E. Choosing a future for epidemiology: I. Eras and paradigms. Am. J.
Public Health 86, 668–673 (1996).
2. Nutton, V. The seeds of disease: An explanation of contagion and infection from the Greeks to
the Renaissance. Med. Hist. 27, 1–34 (1983).
3. Committee National Research Council (US). Science, Medicine and Animals. CWL Publishing
Enterprises, Inc., Madison (National Academies Press, 2004).
4. Byrne, J. P. Encyclopedia of the Black Death. (2012).
5. Kong, K.-F., Schneper, L. & Mathee, K. Beta-lactam Antibiotics: From Antibiosis to Resistance
and Bacteriology. Apmis 118, 1–36 (2010).
6. Young, K. D. The Selective Value of Bacterial Shape. Microbiol. Mol. Biol. Rev. 70, 660–703
(2006).
7. Loutet, S. A. & Valvano, M. A. Extreme antimicrobial peptide and polymyxin B resistance in
the genus Burkholderia. Front. Microbiol. 1, 1–8 (2011).
8. Yang, D. C., Blair, K. M. & Salama, N. R. Staying in Shape: the Impact of Cell Shape on
Bacterial Survival in Diverse Environments. Microbiology Mol. Biol. Rev. 80, 187–203 (2016).
9. Glauert, A. M. & Thornley, M. J. The topography of the bacterial cell wall. Annu. Rev.
Microbiol. 23, 159–198 (1969).
10. Raetz, C. R. H. & Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71, 635–
700 (2002).
11. Vollmer, W., Blanot, D. & De Pedro, M. A. Peptidoglycan structure and architecture. FEMS
Microbiol. Rev. 32, 149–167 (2008).
12. Silhavy, T. J., Kahne, D. & Walker, S. The Bacterial Cell Envelope. Cold Spring Harb Perspect
Biol 2, 1–16 (2010).
13. Aminov, R. I., Otto, M. & Sommer, A. A brief history of the antibiotic era: lessons learned and
challenges for the future. Front. Microbiol. 1, 1–7 (2010).
14. Bassett, E. J., Keith, M. S., Armelagos, G. J., et al. Tetracycline-Labeled Human Bone from
Ancient Sudanese Nubia (A.D. 35). Science. 209, 94–95 (1980).
15. Emmerich, R. & Low, O. Bakteriotytische Enzyme als Ursaehe der erworbenen Immunitat und
die Heilung yon Infeetionskrankheiten durch dieselben. Zeitschrift für Hyg. und Infekt. 31, 1–65
(1899).
16. Ehrlich, P. & Hata, S. Experimentelle Chemotherapie der Spirilosen. (Julius Springer, 1910).

177
17. Domagk, G. Ein Beitrag zur Chemotherapie der bakteriellen Infektionen. Dtsch. Med.
Wochenschr 61, (1935).
18. Davies, J. & Davies, D. Origins and Evolution of Antibiotic Resistance. Microbiology Mol. Biol.
Rev. 74, 417–433 (2010).
19. Harbarth, S., Kahlmeter, G., Kluytmans, J., et al. Global priority list of antibiotic-resistant
bacteria to guide research, discovery, and development of new antibiotics. (2017).
20. Tacconelli, E., Carrara, E., Savoldi, A., et al. Discovery, research, and development of new
antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect.
Dis. 18, 318–327 (2018).
21. Olivares, J., Bernardini, A., Garcia-Leon, G., et al. The intrinsic resistome of bacterial
pathogens. Front. Microbiol. 4, 1–15 (2013).
22. Munita, J. M. & Arias, C. A. Mechanisms of Antibiotics Resistance. Microbiol Spectr 4, 1–37
(2016).
23. Bengtsson-Palme, J., Kristiansson, E. & Larsson, D. G. J. Environmental factors influencing the
development and spread of antibiotic resistance. FEMS Microbiol. Rev. 42, 68–80 (2018).
24. Lister, P. D., Wolter, D. J. & Hanson, N. D. Antibacterial-resistant Pseudomonas aeruginosa:
Clinical impact and complex regulation of chromosomally encoded resistance mechanisms.
Clin. Microbiol. Rev. 22, 582–610 (2009).
25. Gellatly, S. L. & Hancock, R. E. W. Pseudomonas aeruginosa: New insights into pathogenesis
and host defenses. Pathog. Dis. 67, 159–173 (2013).
26. Nikaido, H. Outer Membrane Barrier as a Mechanism of Antimicrobial Resistance. Antimicrob.
Agents Chemother. 33, 1831–1836 (1989).
27. Yoshimura, F. & Nikaido, H. Diffusion of -Lactam Antibiotics Through the Porin Channels of
Escherichia coli K-12. Antimicrob. Agents Chemother. 27, 84–92 (1985).
28. Strateva, T. & Yordanov, D. Pseudomonas aeruginosa - A phenomenon of bacterial resistance.
J. Med. Microbiol. 58, 1133–1148 (2009).
29. Pechdre, J.-C. & Kohler, T. Patterns and modes of -lactam resistance in Pseudomonas
aeruginosa. Clin. Microbiol. Infect. 5, 515–518 (1999).
30. Whitchurch, C. B., Tolker-Nielsen, T., Ragas, P. C., et al. Extracellular DNA Required for
Bacterial Biofilm Formation. Science (80-. ). 295, 1487 (2002).
31. Costerton, J. W., Stewart, P. S. & Greenberg, E. P. Bacterial Biofilms : A Common Cause of
Persistent Infections. Science (80-. ). 284, 1318–1323 (1999).
32. Sun, J., Deng, Z. & Yan, A. Bacterial multidrug efflux pumps: Mechanisms, physiology and
pharmacological exploitations. Biochem. Biophys. Res. Commun. 453, 254–267 (2014).

178
33. Aeschlimann, J. R. The role of multidrug efflux pumps in the antibiotic resistance of
Pseudomonas aeruginosa and other Gram-negative bacteria. Pharmacotherapy 23, 916–924
(2003).
34. Bazzini, S., Udine, C., Sass, A., et al. Deciphering the role of RND efflux transporters in
Burkholderia cenocepacia. PLoS One 6, 1–15 (2011).
35. Yonehara, R., Yamashita, E. & Nakagawa, A. Crystal structures of OprN and OprJ, outer
membrane factors of multidrug tripartite efflux pumps of Pseudomonas aeruginosa. Proteins
Struct. Funct. Bioinforma. 84, 759–769 (2016).
36. Aínsa, J. A., Blokpoel, M. C. J., Otal, I., et al. Molecular cloning and characterization of Tap, a
putative multidrug efflux pump present in Mycobacterium fortuitum and Mycobacterium
tuberculosis. J. Bacteriol. 180, 5836–5843 (1998).
37. Laws, M., Shaaban, A. & Rahman, K. M. Antibiotic resistance breakers: Current approaches
and future directions. FEMS Microbiol. Rev. 43, 490–516 (2019).
38. Du, D., Wang-Kan, X., Neuberger, A., et al. Multidrug efflux pumps: structure, function and
regulation. Nat. Rev. Microbiol. 16, 523–539 (2018).
39. Poole, K. Bacterial stress responses as determinants of antimicrobial resistance. J. Antimicrob.
Chemother. 67, 2069–2089 (2012).
40. Dawson, R. J. P. & Locher, K. P. Structure of a bacterial multidrug ABC transporter. Nature
443, 180–185 (2006).
41. Bogomolnaya, L. M., Andrews, K. D., Talamantes, M., et al. The ABC-type efflux pump
MacAB protects Salmonella enterica serovar typhimurium from oxidative stress. MBio 4, 1–11
(2013).
42. Fitzpatrick, A. W. P., Llabrés, S., Neuberger, A., et al. Structure of the MacAB-TolC ABC-type
tripartite multidrug efflux pump. Nat. Microbiol. 2, 1–20 (2017).
43. ZhHeng, J., ZHao, Y., Liu, M., et al. Substrate-bound structure of the E. coli multidrug resistance
transporter MdfA. Cell Res. 25, 1060–1073 (2015).
44. Horiyama, T. & Nishino, K. AcrB, AcrD, and MdtABC multidrug efflux systems are involved
in enterobactin export in Escherichia coli. PLoS One 9, 1–7 (2014).
45. He, X., Szewczyk, P., Karyakin, A., et al. Structure of a cation-bound multidrug and toxic
compound extrusion transporter. Nature 467, 991–994 (2010).
46. Chen, Y. J., Pornillos, O., Lieu, S., et al. X-ray structure of EmrE supports dual topology model.
Proc. Natl. Acad. Sci. U. S. A. 104, 18999–19004 (2007).
47. Du, D., van Veen, H. W., Murakami, S., et al. Structure, mechanism and cooperation of bacterial
multidrug transporters. Curr. Opin. Struct. Biol. 33, 76–91 (2015).

179
48. Daury, L., Orange, F., Taveau, J. C., et al. Tripartite assembly of RND multidrug efflux pumps.
Nat. Commun. 7, 1–8 (2016).
49. Elkins, C. a & Nikaido, H. Substrate Specificity of the RND-Type Multidrug Ef ux Pumps AcrB
and AcrD of Escherichia coli is Determined Predominantly by Two Large Periplasmic Loops.
J. Bacteriol. 184, 6490–6498 (2002).
50. Ma, D., ALberti, M., Lynch, C., et al. The local repressor AcrR plays a modulating role in the
regulation of acrAB genes of Escherichia coli by global stress signals. Mol. Microbiol. 101–112
(1996).
51. Li, M., Gu, R., Su, C., et al. Crystal Structure of the Transcriptional Regulator AcrR from
Escherichia coli. J. Mol. Biol. 374, 591–603 (2007).
52. Chollet, R., Chevalier, J., Bryskier, A., et al. The AcrAB-TolC pump is involved in macrolide
resistance but not in telithromycin efflux in Enterobacter aerogenes and Escherichia coli.
Antimicrob. Agents Chemother. 48, 3621–3624 (2004).
53. Wilke, M. S., Heller, M., Creagh, A. L., et al. The crystal structure of MexR from Pseudomonas
aeruginosa in complex with its antirepressor ArmR. Proc. Natl. Acad. Sci. U.S.A. 105, 14832–
15837 (2008).
54. Ni, R. T., Onishi, M., Mizusawa, M., et al. The role of RND-type efflux pumps in multidrug-
resistant mutants of Klebsiella pneumoniae. Sci. Rep. 10, (2020).
55. Biswas, S., Brunel, J.-M., Dubus, J.-C., et al. Colistin : an update on the antibiotic of the 21st
century. Expert Rev. Anti. Infect. Ther. 10, 917–934 (2012).
56. Rozalski, A., Sidorczyk, Z. & Kotełko, K. Potential Virulence Factors of Proteus Bacilli.
Microbilogy Mol. Biol. Rev. 61, 65–89 (1997).
57. Olaitan, A. O., Morand, S. & Rolain, J.-M. Mechanisms of polymyxin resistance: acquired and
intrinsic resistance in bacteria. Front. Microbiol. 5, 1–18 (2014).
58. Gunn, J. S., Ryan, S. S., Velkinburgh, J. C. V. A. N., et al. Genetic and Functional Analysis of
a PmrA-PmrB-Regulated Locus Necessary for Lipopolysaccharide Modification, Antimicrobial
Peptide Resistance, and Oral Virulence of Salmonella enterica Serovar Typhimurium. Infect.
Immun. 68, 6139–6146 (2000).
59. Gunn, J. S. & Miller, S. I. PhoP-PhoQ Activates Transcription of pmrAB, Encoding a Two-
Component Regulatory System Involved in Salmonella typhimurium Antimicrobial Peptide
Resistance. J. Bacteriol. 178, 6857–6864 (1996).
60. Abraham, N. & Kwon, D. H. A single amino acid substitution in PmrB is associated with
polymyxin B resistance in clinical isolate of Pseudomonas aeruginosa. FEMS Microbiol. Lett.
298, 249–254 (2009).

180
61. Barrow, K. & Kwon, D. H. Alterations in Two-Component Regulatory Systems of phoPQ and
pmrAB Are Associated with Polymyxin B Resistance in Clinical Isolates of Pseudomonas
aeruginosa. Antimicrob. Agents Chemother. 53, 5150–5154 (2009).
62. Tooke, C. L., Hinchliffe, P., Bragginton, E. C., et al. β-Lactamases and β-Lactamase Inhibitors
in the 21st Century. J. Mol. Biol. 431, 3472–3500 (2019).
63. Macheboeuf, P., Contreras-Martel, C., Job, V., et al. Penicillin binding proteins: Key players in
bacterial cell cycle and drug resistance processes. FEMS Microbiol. Rev. 30, 673–691 (2006).
64. Van Heijenoort, J. Recent advances in the formation of the bacterial peptidoglycan monomer
unit. Nat. Prod. Rep. 18, 503–519 (2001).
65. Filipe, S. R. & Tomasz, A. Inhibition of the expression of penicillin resistance in Streptococcus
pneumoniae by inactivation of cell wall muropeptide branching genes. Proc. Natl. Acad. Sci.
U.S.A. 97, 4891–4896 (2000).
66. Schrödinger, L. The PyMOL Molecular Graphics System v.2.1.0.
67. Bozcal, E. & Dagdeviren, M. Toxicity of β-Lactam Antibiotics: Pathophysiology, Molecular
Biology and Possible Recovery Strategies. Poisoning - From Specif. Toxic Agents to Nov. Rapid
Simpl. Tech. Anal. 87–105 (2017) doi:10.5772/intechopen.70199.
68. Tipper, D. J. & Strominger, J. L. Mechanism of action of penicillins: A proposal based on their
structural similarity to acyl-D-alanyl-D-alanine. Proc. Natl. Acad. Sci. U.S.A. 54, 1133–1140
(1965).
69. Tipper, D. J. & Strominger, J. L. Biosynthesis of the Peptidoglycan of Bacterial Cell Walls. J.
Biol. Chem. 243, 3169–3179 (1968).
70. Wise, E. M. & Park, J. T. Penicillin: its basic site of action as an inhibitor of a peptide cross-
linking reaction in cell wall mucopeptide synthesis. Proc. Natl. Acad. Sci. U.S.A. 54, 75–81
(1965).
71. Patrick, G. L. Antibiotic Agents. in Introduction to Medicinal Chemistry 413–467 (2013).
72. Zeng, X. & Lin, J. Beta-lactamase induction and cell wall metabolism in Gram-negative
bacteria. Front. Microbiol. 4, 1–9 (2013).
73. Abraham, E. P. & Chain, E. An Enzyme from Bacteria able to Destroy Penicillin. Nature 146,
837–837 (1940).
74. Ambler, R. P. The Structure of -lactamases. Phil TransR Soc L. 289, 321–331 (1980).
75. Hong, D. J., Bae, I. K., Jang, I. H., et al. Epidemiology and characteristics of metallo--
lactamase-producing Pseudomonas aeruginosa. Infect. Chemother. 47, 81–97 (2015).
76. Selleck, C., Larrabee, J. A., Harmer, J., et al. AIM-1: An Antibiotic-Degrading Metallohydrolase
That Displays Mechanistic Flexibility. Chem. A Eur. J. 22, 17704–17714 (2016).

181
77. Barthelemy, M., Peduzzi, J., Yaghlane, H. Ben, et al. Single amino acid substitution between
SHV-1 -lactamase and cefotaxime-hydrolyzing SHV-2 enzyme. FEBS Lett. 231, 217–220
(1988).
78. Chang, F.-Y., Siu, L. K., Fung, C.-P., et al. Diversity of SHV and TEM -Lactamases in
Klebsiella pneumoniae : Gene Evolution in Northern Taiwan and Two Novel -Lactamases,
SHV-25 and SHV-26. Antimicrob. Agents Chemother. 45, 2407–2413 (2001).
79. Brooke, J. S. Stenotrophomonas maltophilia: An emerging global opportunistic pathogen. Clin.
Microbiol. Rev. 25, 2–41 (2012).
80. Berg, G., Roskot, N. & Smalla, K. Genotypic and phenotypic relationships between clinical and
environmental isolates of Stenotrophomonas maltophilia. J. Clin. Microbiol. 37, 3594–3600
(1999).
81. Mathee, K., Narasimhan, G., Valdes, C., et al. Dynamics of Pseudomonas aeruginosa genome
evolution. Proc. Natl. Acad. Sci. U.S.A. 105, 3100–3105 (2008).
82. Ogawara, H. Possible drugs for the treatment of bacterial infections in the future: anti-virulence
drugs. J. Antibiot. (Tokyo). (2020) doi:10.1038/s41429-020-0344-z.
83. Thomas, C. M., Nielsen, K. M. & N, S. P. Mechanisms of, and barriers to, horizontal gene
transfer between bacteria. Nat. Rev. Microbiol. 3, 711–721 (2005).
84. Ravi, J., Rivera, M. C. & Lake, J. A. Horizontal gene transfer among genomes: The complexity
hypothesis. Proc. Natl. Acad. Sci. U.S.A. 96, 3801–3806 (1999).
85. Sparling, P. F. Genetic Transformation of Neisseria gonorrhoeae Streptomycin Resistance. J.
Bacteriol. 92, 1364–1371 (1966).
86. Lorenz, M. G. & Wackernagel, W. Bacterial Gene Transfer by Natural Genetic Transformation
in the Environment. Microbiol. Rev. 58, 563–602 (1994).
87. Paget, E. & Simonet, P. On the track of natural transformation in soil. FEMS Microbiol. Ecol.
15, 109–118 (1994).
88. Lorenz, M. G., Gerjets, D. & Wackernagel, W. Release of transforming plasmid and
chromosomal DNS from two cultured soil bacteria. Arch Micrbiol 4, 319–326 (1991).
89. Ueda, S. & Hara, T. Studies on nucleic acid production and application. I. Production of
extracellular DNA by Pseudomonas sp. KYU-1 [industrial fermentations]. J. Appl. Biochem. 3,
1–10 (181AD).
90. Rozenberg-Arska, M., Salters, E. C., Van Strijp, J. A., et al. Degradation of Escherichia coli
Chromosomal and Plasmid DNA in Serum. J. Gen. Microbiol. 130, 217–222 (1983).
91. Cehovin, A., Simpson, P. J., Mcdowell, M. A., et al. Specific DNA recognition mediated by a
type IV pilin. Proc. Natl. Acad. Sci. U.S.A. 110, 3065–3070 (2013).

182
92. Chen, I. & Dubnau, D. DNA Uptake During Bacterial Transformation. Nat. Rev. Microbiol. 2,
1–9 (2004).
93. Griffiths, A. J., Miller, J. H., Suzuki, D. T., et al. Transduction. in An Introduction to Genetic
Analysis (2000).
94. Torres-Barceló, C. The disparate effects of bacteriophages on antibiotic-resistant bacteria.
Emerg. Microbes Infect. 7, 1–12 (2018).
95. Ikeda, H. & Tomizawa, J. Transducing Fragments in Generalized Transduction by Phage PI. J.
Mol. Biol. 14, 85–109 (1965).
96. Zinder, N. D. & Lederberg, J. Genetic Exchange in Salmonella. J. Bacteriol. 64, 679–699
(1952).
97. Morse, M. L., Lederberg, E. M. & Lederberg, J. Transduction in Escherichia coli K-12. Genetics
41, 142–156 (1955).
98. Lang, A. S., Zhaxybayeva, O. & Beatty, J. T. Gene transfer agents: phage-like elements of
genetic exchange. Nat. Rev. Microbiol. 10, 472–482 (2013).
99. Haaber, J., Leisner, J. J., Cohn, M. T., et al. Bacterial viruses enable their host to acquire
antibiotic resistance genes from neighbouring cells. Nat. Commun. 7, 1–8 (2016).
100. Waldor, M. K. & Mekalanos, J. J. Lysogenic Conversion by a Filamentous Phage Encoding
Cholera Toxin. Science (80-. ). 272, 1910–1914 (1996).
101. Graf, F. E., Palm, M., Warringer, J., et al. Inhibiting conjugation as a tool in the fight against
antibiotic resistance. Drug Dev. Res. 80, 19–23 (2019).
102. Manson, J. M., Hancock, L. E. & Gilmore, M. S. Mechanism of chromosomal transfer of
Enterococcus faecalis pathogenicity island, capsule, antimicrobial resistance, and other traits.
Proc. Natl. Acad. Sci. U.S.A. 107, 12269–12274 (2010).
103. Domingues, S., Silva, G. J. & Nielsen, K. M. Integrons - Vehicles and pathways for horizontal
dissemination in bacteria. Mob. Genet. Elem. 2 5, 211–223 (2012).
104. Domingues, S., Nielsen, K. M. & da Silva, G. J. Various pathways leading to the acquisition of
antibiotic resistance by natural transformation. Mob. Genet. Elem. 2 6, 257–260 (2012).
105. Stokes, H. W. & Gillings, M. R. Gene flow, mobile genetic elements and the recruitment of
antibiotic resistance genes into Gram-negative pathogens. FEMS Microbiol. Rev. 35, 790–819
(2011).
106. Mahillon, J. & Chandler, M. Insertion Sequences. Microbiology Mol. Biol. Rev. 62, 725–774
(1998).
107. Siguier, P., Gourbeyre, E. & Chandler, M. Bacterial insertion sequences: their genomic impact
and diversity. FEMS Microbiol. Rev. 38, 865–891 (2014).

183
108. Leiros, H. K. S., Borr, P. S., Brandsdal, B. O., et al. Crystal structure of the mobile metallo-β-
lactamase AIM-1 from Pseudomonas aeruginosa: Insights into antibiotic binding and the role
of Gln157. Antimicrob. Agents Chemother. 56, 4341–4353 (2012).
109. Yong, D., Toleman, M. A., Bell, J., et al. Genetic and biochemical characterization of an
acquired subgroup B3 metallo-β-lactamase gene, blaAIM-1, and its unique genetic context in
Pseudomonas aeruginosa from Australia. Antimicrob. Agents Chemother. 56, 6154–6159
(2012).
110. Sherratt, D. J. Plasmids Review. Cell 3, 189–195 (1974).
111. Smillie, C., Garcilla, M. P., Francia, M. V., et al. Mobility of Plasmids. Microbilogy Mol. Biol.
Rev. 74, 434–452 (2010).
112. Couturier, M., Bex, F., Bergquist, P. L., et al. Identification and Classification of Bacterial
Plasmids. Microbiol. Rev. 52, 375–395 (1988).
113. Johnson, T. J. & Nolan, L. K. Pathogenomics of the Virulence Plasmids of Escherichia coli.
Microbiol. Mol. Biol. Rev. 73, 750–774 (2009).
114. Heinemann, J. A. & Traavik, T. Problems in monitoring horizontal gene transfer in field trials
of transgenic plants. Nat. Biotechnol. 22, 1105–1110 (2004).
115. Dempsey, L. A. & Dubnau, D. A. Identification of Plasmid and Bacillus subtilis Chromosomal
Recombination Sites Used for pE194 Integration. J. Bacteriol. 171, 2856–2865 (1989).
116. Majewski, J. & Cohan, F. M. DNA Sequence Similarity Requirements for Interspecific
Recombination in Bacillus. Genetics 153, 1525–1533 (1999).
117. Guerin, É., Cambray, G., Sanchez-Alberola, N., et al. The SOS Response Controls Integron
Recombination. Science (80-. ). 324, 1034 (2009).
118. Gillings, M. R. Integrons : Past , Present , and Future Structure of Integrons. Microbiology Mol.
Biol. Rev. 78, 257–277 (2014).
119. Woodford, N. & Ellington, M. J. The emergence of antibiotic resistance by mutation. Clin.
Microbiol. Infect. 13, 5–18 (2006).
120. Sensi, P. SESSION I History of the Development of Rifampin. Rev. Infect. Dis. 5, 5402–5406
(1983).
121. Floss, H. G. & Yu, T. Rifamycin - Mode of Action, Resistance, and Biosynthesis. Chem. Rev.
106, 621–632 (2005).
122. Heep, M., Beck, D., Bayerdo, E., et al. Rifampin and Rifabutin Resistance Mechanism in
Helicobacter pylori. Antimicrob. Agents Chemother. 43, 1497–1499 (1999).
123. Wehrli, W. Rifampin: Mechanisms of Action and Resistance. Rev. Infect. Dis. 5, 407–411
(1983).

184
124. Ramaswamy, S. & Musser, J. M. Molecular genetic basis of antimicrobial agent resistance in
Mycobacterium tuberculosis: 1998 update. Tuber. Lung Dis. 79, 3–29 (1998).
125. Bradford, P. A. Extended-Spectrum -Lactamases in the 21st Century: Characterization,
Epidemiology, and Detection of This Important Resistance Threat. Clin. Microbiol. Rev. 14,
933–951 (2001).
126. El’Garch, F., Jeannot, K., Hocquet, D., et al. Cumulative effects of several nonenzymatic
mechanisms on the resistance of Pseudomonas aeruginosa to aminoglycosides. Antimicrob.
Agents Chemother. 51, 1016–1021 (2007).
127. Buckner, M. M., Cisa, M. L., Meek, R. W., et al. HIV Drugs Inhibit Transfer of Plasmids
Carrying Extended-Spectrum -Lactamase and Carbapenemase Genes. MBio 11, 1–18 (2020).
128. Reading, C. & Cole, M. Clavulanic acid: a beta lactamase inhibiting beta lactam from
Streptomyces clavuligerus. Antimicrob. Agents Chemother. 11, 852–857 (1977).
129. Reading, C., Farmer, T. & Cole, M. The β-lactamase stability of amoxycillin with the β-
lactamase inhibitor, clavulanic acid. J. Antimicrob. Chemother. 11, 27–32 (1983).
130. Leflon-Guibout, V., Speldooren, V., Heym, B., et al. Epidemiological survey of amoxicillin-
clavulanate resistance and corresponding molecular mechanisms in Escherichia coli isolates in
France: New genetic features of bla(TEM) genes. Antimicrob. Agents Chemother. 44, 2709–
2714 (2000).
131. Mohanty, S., Singhal, R., Sood, S., et al. Comparative in vitro activity of beta-lactam/beta-
lactamase inhibitor combinations against gram negative bacteria. Indian J. Med. Res. 122, 425–
428 (2005).
132. Penwell, W. F., Shapiro, A. B., Giacobbe, R. A., et al. Molecular mechanisms of sulbactam
antibacterial activity and resistance determinants in Acinetobacter baumannii. Antimicrob.
Agents Chemother. 59, 1680–1689 (2015).
133. Russ, D., Glaser, F., Tamar, E. S., et al. Escape mutations circumvent a tradeoff between
resistance to a beta-lactam and a beta-lactamase inhibitor. Nat. Commun. 11, 1–9 (2020).
134. Ehmann, D. E., Jahić, H., Ross, P. L., et al. Avibactam is a covalent, reversible, non-β-lactam
β-lactamase inhibitor. Proc. Natl. Acad. Sci. U.S.A. 109, 11663–11668 (2012).
135. Zhanel, G. G., Lawson, C. D., Adam, H., et al. Ceftazidime-avibactam: A novel
cephalosporin/β-lactamase inhibitor combination. Drugs 73, 159–177 (2013).
136. Tuon, F. F., Rocha, J. L. & Formigoni-Pinto, M. R. Pharmacological aspects and spectrum of
action of ceftazidime–avibactam: a systematic review. Infection 46, 165–181 (2018).
137. Calvopiña, K., Hinchliffe, P., Brem, J., et al. Structural/mechanistic insights into the efficacy of
nonclassical β-lactamase inhibitors against extensively drug resistant Stenotrophomonas

185
maltophilia clinical isolates. Mol. Microbiol. 106, 492–504 (2017).
138. Petty, L. A., Henig, O., Patel, T. S., et al. Overview of meropenem-vaborbactam and newer
antimicrobial agents for the treatment of carbapenem-resistant Enterobacteriaceae. Infect. Drug
Resist. 11, 1461–1472 (2018).
139. US Food and Drug Administration. VABOMERE (meropenem and vaborbactam) for injection.
US FDA 1–24 (2017).
140. Shields, R. K., Chen, L., Cheng, S., et al. Emergence of Ceftazidime-Avibactam Mutations
during Treatment of Carbapenem-Resistant Klebsiella pneumoniae Infections. Antimicrob
Agents Chemother. 61, 1–11 (2017).
141. Krause, K. M., Serio, A. W., Kane, T. R., et al. Aminoglycosides: An overview. Cold Spring
Harb. Perspect. Med. 6, 1–18 (2016).
142. Kotra, L. P., Haddad, J. & Mobashery, S. Aminoglycosides: Perspectives on mechanisms of
action and resistance and strategies to counter resistance. Antimicrob. Agents Chemother. 44,
3249–3256 (2000).
143. Ramirez, M. S. & Tolmasky, M. E. Aminoglycoside Modifying Enzymes. Drug Resist Updat
13, 151–171 (2010).
144. Llano-Sotelo, B., Azucena, E. F., Kotra, L. P., et al. Aminoglycosides modified by resistance
enzymes display diminished binding to the bacterial ribosomal aminoacyl-tRNA site. Chem.
Biol. 9, 455–463 (2002).
145. Livermore, D. M. Of Pseudomonas, porins, pumps and carbapenems. J. Antimicrob. Chemother.
47, 247–250 (2001).
146. Landman, D., Georgescu, C., Martin, D. A., et al. Polymyxins revisited. Clin. Microbiol. Rev.
21, 449–465 (2008).
147. Lin, L., Nonejuie, P., Munguia, J., et al. Azithromycin Synergizes with Cationic Antimicrobial
Peptides to Exert Bactericidal and Therapeutic Activity Against Highly Multidrug-Resistant
Gram-Negative Bacterial Pathogens. EBioMedicine 2, 690–698 (2015).
148. Vaara, M. Covalent structure of two novel neutrophile leucocyte-derived proteins of porcine and
human origin. Microbiol. Rev. 56, 395–411 (1992).
149. Falagas, M. E. & Kasiakou, S. K. Toxicity of polymyxins: A systematic review of the evidence
from old and recent studies. Crit. Care 10, (2006).
150. Viljanen, P. & Vaara, M. Susceptibility of gram-negative bacteria to polymyxin B nonapeptide.
Antimicrob. Agents Chemother. 25, 701–705 (1984).
151. Ofek, I., Cohen, S., Rahmani, R., et al. Antibacterial synergism of polymyxin B nonapeptide
and hydrophobic antibiotics in experimental Gram-negative infections in mice. Antimicrob.

186
Agents Chemother. 38, 374–377 (1994).
152. Giacometti, A., Cirioni, O., Kamysz, W., et al. In vitro activity and killing effect of temporin A
on nosocomial isolates of Enterococcus faecalis and interactions with clinically used antibiotics.
J. Antimicrob. Chemother. 55, 272–274 (2005).
153. Simmaco, M., Mignogna, G., Canofeni, S., et al. Temporins, antimicrobial peptides from the
European red frog Rana temporaria. Eur. J. Biochem. 242, 788–792 (1996).
154. Li, C., Lewis, M. R., Gilbert, A. B., et al. Antimicrobial activities of amine- and guanidine-
functionalized cholic acid derivatives. Antimicrob. Agents Chemother. 43, 1347–1349 (1999).
155. Alakomi, H. L., Paananen, A., Suihko, M. L., et al. Weakening effect of cell permeabilizers on
Gram-negative bacteria causing biodeterioration. Appl. Environ. Microbiol. 72, 4695–4703
(2006).
156. Sharma, A., Gupta, V. K. & Pathania, R. Efflux pump inhibitors for bacterial pathogens: From
bench to bedside. Indian J. Med. Res. 149, 129–145 (2019).
157. Radchenko, M., Symersky, J., Nie, R., et al. Structural basis for the blockade of MATE
multidrug efflux pumps. Nat. Commun. 6, 1–11 (2015).
158. Gupta, S., Cohen, K. A., Winglee, K., et al. Efflux inhibition with verapamil potentiates
bedaquiline in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 58, 574–576
(2014).
159. Singh, M., Ramdas, J., Sristava, K., et al. Effect of efflux pump inhibitors on the susceptibility
of Mycobacterium tuberculosis to isoniazid. Indian J. Med. Res. 133, 535–540 (2011).
160. Sekyere, J. O. & Amoako, D. G. Carbonyl cyanide m-chlorophenylhydrazine (CCCP) reverses
resistance to colistin, but not to carbapenems and tigecycline in multidrug-resistant
Enterobacteriaceae. Front. Microbiol. 8, 1–9 (2017).
161. Lomovskaya, O., Warren, M. S., Lee, A., et al. Evaluation of antibacterial effect of Vernonia
anthelmintica seed extract and its synergistic effect with antibiotics on resistant. Antimicrob
Agents Chemother. 45, 105–116 (2001).
162. Bhattacharyya, T., Sharma, A., Akhter, J., et al. The small molecule IITR08027 restores the
antibacterial activity of fluoroquinolones against multidrug-resistant Acinetobacter baumannii
by efflux inhibition. Int. J. Antimicrob. Agents 50, 219–226 (2017).
163. Fenosa, A., Fusté, E., Ruiz, L., et al. Role of TolC in Klebsiella oxytoca resistance to antibiotics.
J. Antimicrob. Chemother. 63, 668–674 (2009).
164. Jeon, B. & Zhang, Q. Sensitization of Campylobacter jejuni to fluoroquinolone and macrolide
antibiotics by antisense inhibition of the CmeABC multidrug efflux transporter. J. Antimicrob.
Chemother. 63, 946–948 (2009).

187
165. Rozwandowicz, M., Brouwer, M. S. M., Fischer, J., et al. Plasmids carrying antimicrobial
resistance genes in Enterobacteriaceae. J. Antimicrob. Chemother. 73, 1121–1137 (2018).
166. Rogers, B. A., Sidjabat, H. E., Paterson, D. L., et al. Prolonged carriage of resistant E. coli by
returned travellers: Clonality, risk factors and bacterial characteristics. Eur. J. Clin. Microbiol.
Infect. Dis. 31, 2413–2420 (2012).
167. Kamruzzaman, M., Shoma, S., Thomas, C. M., et al. Plasmid interference for curing antibiotic
resistance plasmids in vivo. PLoS One 12, 1–20 (2017).
168. Buckner, M. M. C., Ciusa, M. L. & Piddock, L. J. V. Strategies to combat antimicrobial
resistance: Anti-plasmid and plasmid curing. FEMS Microbiol. Rev. 42, 781–804 (2018).
169. Garcillán-Barcia, M. P., Jurado, P., González-Pérez, B., et al. Conjugative transfer can be
inhibited by blocking relaxase activity within recipient cells with intrabodies. Mol. Microbiol.
63, 404–416 (2007).
170. Ripoll-Rozada, J., Garcia-Cazoria, Y., Getino, M., et al. Type IV traffic ATPase TrwD as
molecular target to inhibit bacterial conjugation. Mol. Microbiol. 100, 912–921 (2016).
171. Sayer, J. R., Walldén, K., Pesnot, T., et al. 2- and 3-substituted imidazo[1,2-a]pyrazines as
inhibitors of bacterial type IV secretion. Bioorganic Med. Chem. 22, 6459–6470 (2014).
172. Shaffer, C. L., Good, J. A. D., Kumar, S., et al. Peptidomimetic small molecules disrupt type IV
secretion system activity in diverse bacterial pathogens. MBio 7, 1–10 (2016).
173. Anfinsen, C. B. Principles that Govern the Folding of Protein Chains. Science (80-. ). 181, 223–
230 (1973).
174. Krojer, T., Sawa, J., Schäfer, E., et al. Structural basis for the regulated protease and chaperone
function of DegP. Nature 453, 885–890 (2008).
175. Behrens, S., Maier, R., De Cock, H., et al. The SurA periplasmic PPIase lacking its parvulin
domains functions in vivo and has chaperone activity. EMBO J. 20, 285–294 (2001).
176. Shen, Q. T., Bai, X. C., Chang, L. F., et al. Bowl-shaped oligomeric structures on membranes
as DegP’s new functional forms in protein quality control. Proc. Natl. Acad. Sci. U. S. A. 106,
4858–4863 (2009).
177. Walton, T. A., Sandoval, C. M., Fowler, C. A., et al. The cavity-chaperone Skp protects its
substrate from aggregation but allows independent folding of substrate domains. Proc. Natl.
Acad. Sci. U. S. A. 106, 1772–1777 (2009).
178. Weski, J. & Ehrmann, M. Genetic Analysis of 15 Protein Folding Factors and Proteases of the
Escherichia coli Cell Envelope. J. Bacteriol. 194, 3225–3233 (2012).
179. Weatherspoon-griffin, N., Yang, D., Kong, W., et al. The CpxR / CpxA Two-component
Regulatory System Up-regulates the Multidrug Resistance Cascade to Facilitate Escherichia coli

188
Resistance to a Model Antimicrobial Peptide. J. Biol. Chem. bio 289, 32571–32582 (2014).
180. Sabate, R., De Groot, N. S. & Ventura, S. Protein folding and aggregation in bacteria. Cell. Mol.
Life Sci. 67, 2695–2715 (2010).
181. Tokuriki, N., Stricher, F., Serrano, L., et al. How protein stability and new functions trade off.
PLoS Comput. Biol. 4, 35–37 (2008).
182. Bartlett, G. J., Porter, C. T., Borkakoti, N., et al. Analysis of catalytic residues in enzyme active
sites. J. Mol. Biol. 324, 105–121 (2002).
183. Beadle, B. M. & Shoichet, B. K. Structural bases of stability-function tradeoffs in enzymes. J.
Mol. Biol. 321, 285–296 (2002).
184. Herzberg, O. & Moult, J. Analysis of the steric strain in the polypeptide backbone of protein
molecules. Proteins Struct. Funct. Bioinforma. 11, 223–229 (1991).
185. Thomas, V. L., McReynolds, A. C. & Shoichet, B. K. Structural Bases for Stability-Function
Tradeoffs in Antibiotic Resistance. J. Mol. Biol. 396, 47–59 (2010).
186. Frain, K. M., Robinson, C. & van Dijl, J. M. Transport of Folded Proteins by the Tat System.
Protein J. 38, 377–388 (2019).
187. Rapoport, T. A., Li, L. & Park, E. Structural and Mechanistic Insights into Protein Translocation.
Annu. Rev. Cell Dev. Biol. 33, 369–390 (2017).
188. Hiroyuki, M. & Koreaki, I. The Sec protein-translocation pathway. TRENDS Microbiol. 9, 494–
499 (2001).
189. Pradel, N., Delmas, J., Wu, L. F., et al. Sec- and Tat-dependent translocation of β-lactamases
across the Escherichia coli inner membrane. Antimicrob. Agents Chemother. 53, 242–248
(2009).
190. Ast, T., Cohen, G. & Schuldiner, M. A network of cytosolic factors targets SRP-independent
proteins to the endoplasmic reticulum. Cell 152, 1134–1145 (2013).
191. Josefsson, L. G. & Randall, L. L. Different exported proteins in E. coli show differences in the
temporal mode of processing in vivo. Cell 25, 151–157 (1981).
192. Schibich, D., Gloge, F., Pöhner, I., et al. Global profiling of SRP interaction with nascent
polypeptides. Nature 536, 219–223 (2016).
193. Egea, P. F., Shan, S. O., Napetschnig, J., et al. Substrate twinning activates the signal recognition
particle and its receptor. Nature 427, 215–221 (2004).
194. Dilks, K., Rose, R. W., Hartmann, E., et al. Prokaryotic utilization of the twin-arginine
translocation pathway: A genomic survey. J. Bacteriol. 185, 1478–1483 (2003).
195. Urbanus, M. L., Scotti, P. A., Fröderberg, L., et al. Sec-dependent membrane protein insertion:
Sequential interaction of nascent FtsQ with SecY and YidC. EMBO Rep. 2, 524–529 (2001).

189
196. Nielsen, H. & Engelbrecht, J. Identification of prokaryotic and eukaryotic signal peptides and
prediction of their cleavage sites Artificial neural networks have been used for many biological.
Protein Eng. 10, 1–6 (1997).
197. Hessa, T., Kim, H., Bihlmaier, K., et al. Recognition of transmembrane helices by the
endoplasmic reticulum translocon. Nature 433, 377–381 (2005).
198. Driessen, A. J. M. SecB, a molecular chaperone with two faces. Trends Microbiol. 9, 193–196
(2001).
199. Allen, W. J., Corey, R. A., Oatley, P., et al. Two-way communication between SecY and SecA
suggests a brownian ratchet mechanism for protein translocation. Elife 5, 1–23 (2016).
200. Bauer, B. W., Shemesh, T., Chen, Y., et al. A ‘push and slide’ mechanism allows sequence-
insensitive translocation of secretory proteins by the SecA ATPase. Cell 157, 1416–1429 (2014).
201. Park, E. & Rapoport, T. A. Preserving the membrane barrier for small molecules during bacterial
protein translocation. Nature 473, 239–242 (2011).
202. Petru, M., Wideman, J., Moore, K., et al. Evolution of mitochondrial TAT translocases
illustrates the loss of bacterial protein transport machines in mitochondria. BMC Biol. 16, 1–14
(2018).
203. Palmer, T. & Berks, B. C. The twin-arginine translocation (Tat) protein export pathway. Nat.
Rev. Microbiol. 10, 483–496 (2012).
204. Berks, B. C., Sargent, F. & Palmer, T. The Tat protein export pathway. Mol. Microbiol. 35, 260–
274 (2000).
205. Cline, K. Mechanistic aspects of folded protein transport by the twin arginine translocase (Tat).
J. Biol. Chem. 290, 16530–16538 (2015).
206. Patel, R., Smith, S. M. & Robinson, C. Protein transport by the bacterial Tat pathway. Biochim.
Biophys. Acta 1843, 1620–1628 (2014).
207. Gohlke, U., Pullan, L., McDevitt, C. A., et al. The TatA component of the twin-arginine protein
transport system forms channel complexes of variable diameter. Proc. Natl. Acad. Sci. U.S.A.
102, 10482–10486 (2005).
208. DeLisa, M. P., Tullman, D. & Georgiou, G. Folding quality control in the export of proteins by
the bacterial twin-arginine translocation pathway. Proc. Natl. Acad. Sci. U. S. A. 100, 6115–
6120 (2003).
209. Maurer, C., Panahandeh, S., Moser, M., et al. Impairment of twin-arginine-dependent export by
seemingly small alterations of substrate conformation. FEBS Lett. 583, 2849–2853 (2009).
210. Ize, B., Stanley, N. R., Buchanan, G., et al. Role of the Escherichia coli Tat pathway in outer
membrane integrity. Mol. Microbiol. 48, 1183–1193 (2003).

190
211. Krishnappa, L., Monteferrante, C. G. & van Dijl, J. M. Degradation of the twin-arginine
translocation substrate YwbN by extracytoplasmic proteases of Bacillus subtilis. Appl. Environ.
Microbiol. 78, 7801–7804 (2012).
212. McDonough, J. A., Hacker, K. E., Flores, A. R., et al. The twin-arginine translocation pathway
of Mycobacterium smegmatis is functional and required for the export of mycobacterial β-
lactamases. J. Bacteriol. 187, 7667–7679 (2005).
213. Almagro Armenteros, J. J., Tsirigos, K. D., Sønderby, C. K., et al. SignalP 5.0 improves signal
peptide predictions using deep neural networks. Nat. Biotechnol. 37, 420–423 (2019).
214. Goemans, C., Denoncin, K. & Collet, J. F. Folding mechanisms of periplasmic proteins.
Biochim. Biophys. Acta - Mol. Cell Res. 1843, 1517–1528 (2014).
215. Dutton, R. J., Boyd, D., Berkmen, M., et al. Bacterial species exhibit diversity in their
mechanisms and capacity for protein disulfide bond formation. Proc. Natl. Acad. Sci. U.S.A.
105, 11933–11938 (2008).
216. Heras, B., Shouldice, S. R., Totsika, M., et al. DSB proteins and bacterial pathogenicity. Nat.
Rev. Microbiol. 7, 215–225 (2009).
217. Bulleid, N. J. Disulfide Bond Formation in the Mammalian Endoplasmic Reticulum. Cold
Spring Harb. Perspect. Biol. 1–12 (2012).
218. Shouldice, S. R., Heras, B., Walden, P. M., et al. Structure and function of DsbA, a key bacterial
oxidative folding catalyst. Antioxidants Redox Signal. 14, 1729–1760 (2011).
219. Goldberger, R. F., Epstein, C. J. & Anfinsen, C. B. Acceleration of reactivation of reduced
bovine pancreatic ribonuclease by a microsomal system from rat liver. J. Biol. Chem. 238, 628–
635 (1963).
220. Frand, A. R., Cuozzo, J. W. & Kaiser, C. A. Pathways for protein disulphide bond formation.
Cell Biol. 10, 203–210 (2000).
221. Fuchs, S., De Lorenzo, F. & Anfinsen, C. B. Studies on the mechanism of the enzymic catalysis
of disulfide interchange in proteins. J. Biol. Chem. 242, 398–402 (1967).
222. Pollard, M. G., Travers, K. J. & Weissman, J. S. Ero1p : A Novel and Ubiquitous Protein with
an Essential Role in Oxidative Protein Folding in the Endoplasmic Reticulum. Mol. Cell 1, 171–
182 (1998).
223. Gross, E., Sevier, C. S., Vala, A., et al. A new FAD-binding fold and intersubunit disulfide
shuttle in the thiol oxidase Erv2p. Nat. Struct. Biol. 9, 61–67 (2002).
224. Tian, G., Xiang, S., Noiva, R., et al. The crystal structure of yeast protein disulfide isomerase
suggests cooperativity between its active sites. Cell 124, 61–73 (2006).
225. Frand, A. R. & Kaiser, C. A. Two pairs of conserved cysteines are required for the oxidative

191
activity of Ero1p in protein disulfide bond formation in the endoplasmic reticulum. Mol. Biol.
Cell 11, 2833–2843 (2000).
226. Schwaller, M., Wilkinson, B. & Gilbert, H. F. Reduction-reoxidation cycles contribute to
catalysis of disulfide isomerization by protein-disulfide isomerase. J. Biol. Chem. 278, 7154–
7159 (2003).
227. Wang, X., Wang, L., Wang, X., et al. Structural insights into the peroxidase activity and
inactivation of human peroxiredoxin 4. Biochem. J. 441, 113–118 (2012).
228. Zito, E., Melo, E. P., Yang, Y., et al. Oxidative Protein Folding by an Endoplasmic Reticulum-
Localized Peroxiredoxin. Mol. Cell 40, 787–797 (2010).
229. Chatzi, A., Manganas, P. & Tokatlidis, K. Oxidative folding in the mitochondrial intermembrane
space: A regulated process important for cell physiology and disease. Biochim. Biophys. Acta
1863, 1298–1306 (2016).
230. Allen, S., Balabanidou, V., Sideris, D. P., et al. Erv1 mediates the Mia40-dependent protein
import pathway and provides a functional link to the respiratory chain by shuttling electrons to
cytochrome c. J. Mol. Biol. 353, 937–944 (2005).
231. Banci, L., Bertini, I., Cefaro, C., et al. MIA40 is an oxidoreductase that catalyzes oxidative
protein folding in mitochondria. Nat. Struct. Mol. Biol. 16, 198–206 (2009).
232. Kawano, S., Yamano, K., Naoé, M., et al. Structural basis of yeast Tim40/Mia40 as an oxidative
translocator in the mitochondrial intermembrane space. Proc. Natl. Acad. Sci. U.S.A. 106,
14403–14407 (2009).
233. Sideris, D. P., Petrakis, N., Katrakili, N., et al. A novel intermembrane space-targeting signal
docks cysteines onto Mia40 during mitochondrial oxidative folding. J. Cell Biol. 187, 1007–
1022 (2009).
234. Banci, L., Bertini, I., Cefaro, C., et al. Molecular chaperone function of Mia40 triggers
consecutive induced folding steps of the substrate in mitochondrial protein import. Proc. Natl.
Acad. Sci. U.S.A. 107, 20190–20195 (2010).
235. Thorpe, C. & Coppock, D. L. Generating Disulfides in Multicellular Organisms : Emerging
Roles for a New Flavoprotein Family. J. Biol. Chem. 282, 13929–13933 (2007).
236. Landeta, C., Boyd, D. & Beckwith, J. Disulfide bond formation in prokaryotes. Nat. Microbiol.
3, 270–280 (2018).
237. Bardwell, J. C., McGovern, K. & Beckwith, J. Identification of a protein required for disulfide
bond formation in vivo. Cell 67, 581–589 (1991).
238. Dailey, F. E. & Berg, H. C. Mutants in disulfide bond formation that disrupt flagellar assembly
in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 90, 1043–1047 (1993).

192
239. Missiakas, D., Schwager, F. & Raina, S. Identification and characterization of a new disulfide
isomerase-like protein (DsbD) in Escherichia coli. EMBO J. 14, 3415–3424 (1995).
240. Bardwell, J. C., Lee, J. O., Jander, G., et al. A pathway for disulfide bond formation in vivo.
Proc. Natl. Acad. Sci. U.S.A. 90, 1038–42 (1993).
241. Messens, J. & Collet, J.-F. Pathways of disulfide bond formation in Escherichia coli. Int. J.
Biochem. Cell Biol. 38, 1050–1062 (2006).
242. Martin, J. L., Bardwell, J. C. A. & Kuriyan, J. Crystal structure of the DsbA protein required for
disulphide bond formation in vivo. Nature 365, 464–468 (1993).
243. Ren, G., Stephan, D., Xu, Z., et al. Properties of the thioredoxin fold superfamily are modulated
by a single amino acid residue. J. Biol. Chem. 284, 10150–10159 (2009).
244. Guddat, L. W., Bardwell, J. C. A., Zander, T., et al. The uncharged surface features surrounding
the active site of Escherichia coli DsbA are conserved and are implicated in peptide binding.
Protein Sci. 6, 1148–1156 (1997).
245. Hatahet, F., Boyd, D. & Beckwith, J. Disulfide bond formation in prokaryotes: History, diversity
and design. Biochim. Biophys. Acta 1844, 1402–1414 (2014).
246. Paxman, J. J., Borg, N. A., Horne, J., et al. The structure of the bacterial oxidoreductase enzyme
DsbA in complex with a peptide reveals a basis for substrate specificity in the catalytic cycle of
DsbA enzymes. J. Biol. Chem. 284, 17835–17845 (2009).
247. Inaba, K., Murakami, S., Suzuki, M., et al. Crystal Structure of the DsbB-DsbA Complex
Reveals a Mechanism of Disulfide Bond Generation. Cell 127, 789–801 (2006).
248. Eisenberg, D., Schwarz, E., Komaromy, M., et al. Analysis of membrane and surface protein
sequences with the hydrophobic moment plot. J. Mol. Biol. 179, 125–142 (1984).
249. Guddat, L. W., Bardwell, J. C. A. & Martin, J. L. Crystal structures of reduced and oxidized
DsbA: Investigation of domain motion and thiolate stabilization. Structure 6, 757–767 (1998).
250. Guddat, L. W., Bardwell, J. C. A., Glockshuber, R., et al. Structural analysis of three His32
mutants of DsbA: Support for an electrostatic role of His32 in DsbA stability. Protein Sci. 6,
1893–1900 (1997).
251. Kadokura, H., Tian, H., Zander, T., et al. Snapshots of DsbA in Action: Detection of Proteins in
the Process of Oxidative Folding. Science (80-. ). 303, 534–537 (2004).
252. Charbonnier, J.-B., Belin, P., Moutiez, M., et al. On the role of the cis-proline residue in the
active site of DsbA. Protein Sci. 8, 96–105 (2008).
253. Chen, J., Song, J. L., Zhang, S., et al. Chaperone activity of DsbC. J. Biol. Chem. 274, 19601–
19605 (1999).
254. Couprie, J., Vinci, F., Dugave, C., et al. Investigation of the DsbA mechanism through the

193
synthesis and analysis of an irreversible enzyme-ligand complex. Biochemistry 39, 6732–6742
(2000).
255. Tinsley, C. R., Voulhoux, R., Beretti, J. L., et al. Three homologues, including two membrane-
bound proteins, of the disulfide oxidoreductase DsbA in Neisseria meningitidis: Effects on
bacterial growth and biogenesis of functional type IV pili. J. Biol. Chem. 279, 27078–27087
(2004).
256. Kortemme, T. & Creighton, T. E. Ionisation of cysteine residues at the termini of model α-helical
peptides. Relevance to unusual thiol pKa values in proteins of the thioredoxin family. J. Mol.
Biol. 253, 799–812 (1995).
257. Grauschopf, U., Winther, J. R., Korber, P., et al. Why is DsbA such an oxidizing disulfide
catalyst? Cell 83, 947–955 (1995).
258. Holmgren, A. Thioredoxin structure and mechanism: conformational changes on oxidation of
the active-site sulfhydryls to a disulfide. Structure 3, 239–243 (1995).
259. Jeng, M. F., Reymond, M. T., Tennant, L. L., et al. NMR characterization of a single-cysteine
mutant of Escherichia coli thioredoxin and a covalent thioredoxin-peptide complex. Eur. J.
Biochem. 257, 299–308 (1998).
260. Carvalho, A. T. P., Fernandes, P. A., Swart, M., et al. Role of the Variable Active Site Residues
in the Function of Thioredoxin Family Oxidoreductases. J. Comput. Chem. 30, 710–724 (2008).
261. Kadokura, H. & Beckwith, J. Detecting Folding Intermediates of a Protein as It Passes Through
the Bacterial Translocation Channel. Cell 138, 1164–1173 (2009).
262. Malojčić, G., Owen, R. L., Grimshaw, J. P. A., et al. Preparation and structure of the charge-
transfer intermediate of the transmembrane redox catalyst DsbB. FEBS Lett. 582, 3301–3307
(2008).
263. Zhou, Y., Cierpicki, T., Flores Jimenez, R. H., et al. NMR Solution Structure of the Integral
Membrane Enzyme DsbB: Functional Insights into DsbB-Catalyzed Disulfide Bond Formation.
Mol. Cell 31, 896–908 (2008).
264. Inaba, K., Murakami, S., Nakagawa, A., et al. Dynamic nature of disulphide bond formation
catalysts revealed by crystal structures of DsbB. EMBO J. 28, 779–791 (2009).
265. Bushweller, J. H. Protein Disulfide Exchange by the Intramembrane Enzymes DsbB, DsbD, and
CcdA. J. Mol. Biol. (in Press. (2020) doi:10.1016/j.jmb.2020.04.008.
266. Łasica, A. M. & Jagusztyn-Krynicka, E. K. The role of Dsb proteins of Gram-negative bacteria
in the process of pathogenesis. FEMS Microbiol. Rev. 31, 626–636 (2007).
267. Inaba, K., Takahashi, Y. H., Ito, K., et al. Critical role of a thiolate-quinone charge transfer
complex and its adduct form in de novo disulfide bond generation by DsbB. Proc. Natl. Acad.

194
Sci. U.S.A. 103, 287–292 (2006).
268. Jander, G., Martin, N. L. & Beckwith, J. Two cysteines in each periplasmic domain of the
membrane protein DsbB are required for its function in protein disulfide bond formation. EMBO
J. 13, 5121–5127 (1994).
269. Kobayashi, T. & Ito, K. Respiratory chain strongly oxidizes the CXXC motif of DsbB in the
Escherichia coli disulfide bond formation pathway. EMBO J. 18, 1192–1198 (1999).
270. Bader, M. W., Xie, T., Yu, C. A., et al. Disulfide bonds are generated by quinone reduction. J.
Biol. Chem. 275, 26082–26088 (2000).
271. Inaba, K. & Ito, K. Paradoxical redox properties of DsbB and DsbA in the protein disulfide-
introducing reaction cascade. EMBO J. 21, 2646–2654 (2002).
272. Inaba, K., Takahashi, Y. H. & Ito, K. Reactivities of quinone-free DsbB from Escherichia coli.
J. Biol. Chem. 280, 33035–33044 (2005).
273. Kadokura, H. & Beckwith, J. Four cysteines of the membrane protein DsbB act in concert to
oxidize its substrate DsbA. EMBO J. 21, 2354–2363 (2002).
274. McCarthy, A. A., Haebel, P. W., Törrönen, A., et al. Crystal structure of the protein disulfide
bond isomerase, DsbC, from Escherichia coli. Nat. Struct. Biol. 7, 196–199 (2000).
275. Ito, K. & Inaba, K. The disulfide bond formation (Dsb) system. Curr. Opin. Struct. Biol. 18,
450–458 (2008).
276. Katzen, F. & Beckwith, J. Role and location of the unusual redox-active cysteines in the
hydrophobic domain of the transmembrane electron transporter DsbD. Proc. Natl. Acad. Sci. U.
S. A. 100, 10471–10476 (2003).
277. Gleiter, S. & Bardwell, J. C. A. Disulfide bond isomerization in prokaryotes. Biochim. Biophys.
Acta 1783, 530–534 (2008).
278. Rietsch, A., Belin, D., Martin, N., et al. An in vivo pathway for disulfide bond isomerization in
Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 93, 13048–13053 (1996).
279. Rietsch, A., Bessette, P., Georgiou, G., et al. Reduction of the periplasmic disulfide bond
isomerase, DsbC, occurs by passage of electrons from cytoplasmic thioredoxin. J. Bacteriol.
179, 6602–6608 (1997).
280. Segatori, L., Murphy, L., Arredondo, S., et al. Conserved role of the linker α-helix of the
bacterial disulfide isomerase DsbC in the avoidance of misoxidation by DsbB. J. Biol. Chem.
281, 4911–4919 (2006).
281. Haebel, P. W., Goldstone, D., Katzen, F., et al. The disulfide bond isomerase DsbC is activated
by an immunoglobulin-fold thiol oxidoreductase: Crystal structure of the DsbC-DsbDα
complex. EMBO J. 21, 4774–4784 (2002).

195
282. Heras, B., Edeling, M. A., Schirra, H. J., et al. Crystal structures of the DsbG disulfide isomerase
reveal an unstable disulfide. Proc. Natl. Acad. Sci. U.S.A. 101, 8876–8881 (2004).
283. Liu, X. Q. & Wang, C. C. Disulfide-dependent folding and export of Escherichia coli DsbC. J.
Biol. Chem. 276, 1146–1151 (2001).
284. Shouldice, S. R., Cho, S. H., Boyd, D., et al. In vivo oxidative protein folding can be facilitated
by oxidation-reduction cycling. Mol. Microbiol. 75, 13–28 (2010).
285. Kpadeh, Z. Z., Jameson-Lee, M., Yeh, A. J., et al. Disulfide bond oxidoreductase DsbA2 of
Legionella pneumophila exhibits protein disulfide isomerase activity. J. Bacteriol. 195, 1825–
1833 (2013).
286. Bader, M. W., Hiniker, A., Regeimbal, J., et al. Turning a disulfide isomerase into an oxidase :
DsbC mutants that imitate DsbA. EMBO Kournal 20, 1555–1562 (2001).
287. Hiniker, A., Collet, J. F. & Bardwell, J. C. A. Copper stress causes an in vivo requirement for
the Escherichia coli disulfide isomerase DsbC. J. Biol. Chem. 280, 33785–33791 (2005).
288. Depuydt, M., Leonard, S. E., Vertommen, D., et al. A Periplasmic Reducing System Protects
Single Cysteine Residues from Oxidation. Science (80-. ). 326, 1109–1111 (2009).
289. Crooke, H. & Cole, J. The biogenesis of c-type cytochromes in Escherichia coli requires a
membrane-bound protein, DipZ, with a protein disulphide isomerase-like domain. Mol.
Microbiol. 15, 1139–1150 (1995).
290. Katzen, F. & Beckwith, J. Transmembrane electron transfer by the membrane protein DsbD
occurs via a disulfide bond cascade. Cell 103, 769–779 (2000).
291. Hiniker, A., Vertommen, D., Bardwell, J. C. A., et al. Evidence for conformational changes
within DsbD: Possible role for membrane-embedded proline residues. J. Bacteriol. 188, 7317–
7320 (2006).
292. Rozhkova, A., Stirnimann, C. U., Frei, P., et al. Structural basis and kinetics of inter- and
intramolecular disulfide exchange in the redox catalyst DsbD. EMBO J. 23, 1709–1719 (2004).
293. Cho, S. H., Parsonage, D., Thurston, C., et al. A new family of membrane electron transporters
and its substrates, including a new cell envelope peroxiredoxin, reveal a broadened reductive
capacity of the oxidative bacterial cell envelope. MBio 3, 1–11 (2012).
294. Porat, A., Cho, S. H. & Beckwith, J. The unusual transmembrane electron transporter DsbD and
its homologues: A bacterial family of disulfide reductases. Res. Microbiol. 155, 617–622 (2004).
295. Goulding, C. W., Sawaya, M. R., Parseghian, A., et al. Thiol-disulfide exchange in an
immunoglobulin-like fold: Structure of the N-terminal domain of DsbD. Biochemistry 41, 6920–
6927 (2002).
296. Goldstone, D., Haebel, P. W., Katzen, F., et al. DsbC activation by the N-terminal domain of

196
DsbD. Proc. Natl. Acad. Sci. U.S.A. 98, 9551–9556 (2001).
297. Mavridou, D. A. I., Saridakis, E., Kritsiligkou, P., et al. Oxidation state-dependent protein-
protein interactions in disulfide cascades. J. Biol. Chem. 286, 24943–24956 (2011).
298. Kim, J. H., Kim, S. J., Jeong, D. G., et al. Crystal structure of DsbDγ reveals the mechanism of
redox potential shift and substrate specificity. FEBS Lett. 543, 164–169 (2003).
299. Stirnimann, C. U., Rozhkova, A., Grauschopf, U., et al. High-resolution Structures of
Escherichia coli cDsbD in Different Redox States: A Combined Crystallographic, Biochemical
and Computational Study. J. Mol. Biol. 358, 829–845 (2006).
300. Stewart, E. J., Katzen, F. & Beckwith, J. Six conserved cysteines of the membrane protein DsbD
are required for the transfer of electrons from the cytoplasm to the periplasm of Escherichia coli.
EMBO J. 18, 5963–5971 (1999).
301. Chung, J., Chen, T. & Missiakas, D. Transfer of electrons across the cytoplasmic membrane by
DsbD, a membrane protein involved in thiol-disulphide exchange and protein folding in the
bacterial periplasm. Mol. Microbiol. 35, 1099–1109 (2000).
302. Zhou, Y. & Bushweller, J. H. Solution Structure and Elevator Mechanism of the Membrane
Electron Transporter CcdA. Physiol. Behav. 25, 163–169 (2018).
303. Drew, D. & Boudker, O. Shared Molecular Mechanisms of Membrane Transporters. Annu. Rev.
Biochem. 85, 543–572 (2016).
304. Cho, S. H. & Beckwith, J. Two snapshots of electron transport across the membrane: Insights
into the structure and function of DsbD. J. Biol. Chem. 284, 11416–11424 (2009).
305. Cho, S. H., Porat, A., Ye, J., et al. Redox-active cysteines of a membrane electron transporter
DsbD show dual compartment accessibility. EMBO J. 26, 3509–3520 (2007).
306. Heras, B., Scanlon, M. J. & Martin, J. L. Targeting virulence not viability in the search for future
antibacterials. Br. J. Clin. Pharmacol. 79, 208–215 (2015).
307. Miki, T., Okada, N. & Danbara, H. Two periplasmic bisulfide oxidoreductases, DsbA and SrgA,
target outer membrane protein SpiA, a component of the Salmonella pathogenicity island 2 type
III secretion system. J. Biol. Chem. 279, 34631–34642 (2004).
308. Dacheux, D., Epaulard, O., De Groot, A., et al. Activation of the Pseudomonas aeruginosa type
III secretion system requires an intact pyruvate dehydrogenase aceAB operon. Infect. Immun.
70, 3973–3977 (2002).
309. Ha, U., Wang, Y. & Jin, S. DsbA of Pseudomonas aeruginosa Is Essential for Multiple
Virulence Factors. Infect. Immun. 71, 1590–1595 (2003).
310. Sinha, S., Langford, P. R. & Kroll, J. S. Functional diversity of three different DsbA proteins
from Neisseria meningitidis. Microbiology 150, 2993–3000 (2004).

197
311. Grimshaw, J. P. A., Stirnimann, C. U., Brozzo, M. S., et al. DsbL and DsbI Form a Specific
Dithiol Oxidase System for Periplasmic Arylsulfate Sulfotransferase in Uropathogenic
Escherichia coli. J. Mol. Biol. 380, 667–680 (2008).
312. Bouwman, C. W., Kohli, M., Killoran, A., et al. Characterization of SrgA, a Salmonella enterica
Serovar typhimurium Virulence Plasmid-Encoded Paralogue of the Disulfide Oxidoreductase
DsbA, Essential for Biogenesis of Plasmid-Encoded Fimbriae. Microbiology 185, 991–1000
(2003).
313. Matias, V. R. F. & Beveridge, T. J. Cryo-electron microscopy reveals native polymeric cell wall
structure in Bacillus subtilis 168 and the existence of a periplasmic space. Mol. Microbiol. 56,
240–251 (2005).
314. Zuber, B., Haenni, M., Ribeiro, T., et al. Granular layer in the periplasmic space of gram-positive
bacteria and fine structures of Enterococcus gallinarum and Streptococcus gordonii septa
revealed by cryo-electron microscopy of vitreous sections. J. Bacteriol. 188, 6652–6660 (2006).
315. Walden, P., Halili, M., Kurth, F., et al. Rv2969c, essential for optimal growth in Mycobacterium
tuberculosis, is a DsbA-like enzyme that interacts with VKOR-derived peptides and has atypical
features of DsbA-like disulfide oxidases research papers. Acta Crystallogr. Sect. D D6, 1981–
1994 (2013).
316. Goulding, C. W., Apostol, M. I., Gleiter, S., et al. Gram-positive DsbE Proteins Function
Differently from Gram-negative DsbE homologs. J. Biol. Chem. 279, 3516–3524 (2004).
317. Chim, N., Riley, R., The, J., et al. An extracellular disulfide bond forming protein (DsbF) from
Mycobacterium tuberculosis: Structurel, biochemical, and gene expression analysis. J. Mol.
Biol. 396, 1211–1226 (2011).
318. Sassetti, C. M. & Rubin, E. J. Genetic requirements for mycobacterial survival during infection.
Proc. Natl. Acad. Sci. U.S.A. 100, 12989–12994 (2003).
319. Bolhuis, A., Venema, G., Quax, W. J., et al. Functional Analysis of Paralogous Thiol-disulfide
Oxidoreductases in Bacillus subtilis. J. Biol. Chem. 274, 24531–24538 (1999).
320. Heras, B., Kurz, M., Jarrott, R., et al. Staphylococcus aureus DsbA does not have a destabilizing
disulfide: A new paradigm for bacterial oxidative folding. J. Biol. Chem. 283, 4261–4271
(2008).
321. Yu, J. Inactivation of DsbA, but not DsbC and DsbD, affects the intracellular survival and
virulence of Shigella flexneri. Infect. Immun. 66, 3909–3917 (1998).
322. Totsika, M., Vagenas, D., Paxman, J. J., et al. Inhibition of Diverse DsbA Enzymes in Multi-
DsbA Encoding Pathogens. Antioxidants Redox Signal. 29, 653–666 (2018).
323. Denoncin, K. & Collet, J.-F. Disulfide Bond Formation in the Bacterial Periplasm: Major

198
Achievements and Challenges Ahead. Antioxid. Redox Signal. 19, 63–71 (2013).
324. Allen, R. C., Popat, R., Diggle, S. P., et al. Targeting virulence: Can we make evolution-proof
drugs? Nat. Rev. Microbiol. 12, 300–308 (2014).
325. Landeta, C., Blazyk, J. L., Hatahet, F., et al. Compounds targeting disulfide bond forming
enzyme DsbB of Gram-negative bacteria. Nat. Chem. Biol. 11, 292–298 (2015).
326. Smith, R. P., Paxman, J. J., Scanlon, M. J., et al. Targeting bacterial Dsb proteins for the
development of anti-virulence agents. Molecules 21, (2016).
327. Duprez, W., Premkumar, L., Halili, M. A., et al. Peptide inhibitors of the Escherichia coli DsbA
oxidative machinery essential for bacterial virulence. J. Med. Chem. 58, 577–587 (2015).
328. Früh, V., Zhou, Y., Chen, D., et al. Application of fragment-based drug discovery to membrane
proteins: Identification of ligands of the integral membrane enzyme DsbB. Chem. Biol. 17, 881–
891 (2010).
329. Halili, M. A., Bachu, P., Lindahl, F., et al. Small molecule inhibitors of disulfide bond formation
by the bacterial DsbA-DsbB dual enzyme system. ACS Chem. Biol. 10, 957–964 (2015).
330. Adams, L. A., Sharma, P., Mohanty, B., et al. Application of fragment-based screening to the
design of inhibitors of Escherichia coli DsbA. Angew. Chemie - Int. Ed. 54, 2179–2184 (2015).
331. Duprez, W., Bachu, P., Stoermer, M. J., et al. Virtual screening of peptide and peptidomimetic
fragments targeted to inhibit bacterial dithiol oxidase DsbA. PLoS One 10, 1–16 (2015).
332. Kurth, F., Duprez, W., Premkumar, L., et al. Crystal structure of the dithiol oxidase DsbA
enzyme from Proteus mirabilis bound non-covalently to an active site peptide ligand. J. Biol.
Chem. 289, 19810–19822 (2014).
333. Landeta, C., McPartland, L., Tran, N. Q., et al. Inhibition of Pseudomonas aeruginosa and
Mycobacterium tuberculosis disulfide bond forming enzymes. Mol. Microbiol. 111, 918–937
(2019).
334. Meehan, B. M., Landeta, C., Boyd, D., et al. The disulfide bond formation pathway is essential
for anaerobic growth of Escherichia coli. J. Bacteriol. 199, 1–9 (2017).
335. Altmeyer, R. M., McNern, J. K., Bossio, J. C., et al. Cloning and molecular characterization of
a gene involved in Salmonella adherence and invasion of cultured epithelial cells. Mol.
Microbiol. 7, 89–98 (1993).
336. Yu, J., Webb, H. & Hirst, T. R. A homologue of the Escherichia coli DsbA protein involved in
disulphide bond formation is required for enterotoxin biogenesis in Vibrio cholerae. Mol.
Microbiol. 6, 1949–1958 (1992).
337. Totsika, M., Heras, B., Wurpel, D. J., et al. Characterization of two homologous disulfide bond
systems involved in virulence factor biogenesis in uropathogenic Escherichia coli CFT073. J.

199
Bacteriol. 191, 3901–3908 (2009).
338. Furniss, R. C. D., Kadeřábková, N., Barker, D., et al. Breaking antimicrobial resistance by
disrupting extracytoplasmic protein folding. ELife (in Revis. 1–33.
339. Kim, J., Webb, A. M., Kershner, J. P., et al. A versatile and highly efficient method for scarless
genome editing in Escherichia coli and Salmonella enterica. BMC Biotechnol. 14, 1–13 (2014).
340. Mckenzie, G. J. & Craig, N. L. Fast, easy and efficient: site-specific insertion of transgenes into
Enterobacterial chromosomes using Tn 7 without need for selection of the insertion event. BMC
Microbiol. 6, 1–7 (2006).
341. Vasseur, P., Vallet-Gely, I., Soscia, C., et al. The pel genes of the Pseudomonas aeruginosa
PAK strain are involved at early and late stages of biofilm formation. Microbiology 151, 985–
997 (2005).
342. Kaniga, K., Delor, I. & Cornelis, G. R. A wide-host-range suicide vector for improving reverse
genetics in Gram-negative bacteria: inactivation of the blaA gene of Yersinia enterocolitica.
Gene 137–141 (1991).
343. Welker, E., Domfeh, Y., Tyagi, D., et al. Genetic manipulation of Stenotrophomonas
maltophilia. Curr. Protoc. Microbiol. 37, 6F.2.1-6F.2.14 (2015).
344. Hanahan, D. DNA Cloning: A Practical Approach 1: IRL. (McLean Press, Virginia, 1985).
345. Herrero, M., de Lorenzo, V. & Timmis, K. Transposon Vectors Containing Non-Antibiotic
Resistance Selection Markers for Cloning and Stable Chromosomal Insertion of Foreign Genes
in Gram-Negative Bacteria. J. Bacteriol. 172, 6557–6567 (1990).
346. Boyer, H. W. & Roulland-Dussoix, D. Complementation analysis of the restriction and
modification of DNA in Escherichia coli. J. Mol. Biol. 41, 459–472 (1969).
347. Casadaban, M. J. & Cohen, S. N. Analysis of gene control signals by DNA fusion and cloning
in Escherichia coli. J. Mol. Biol. 138, 179–207 (1980).
348. Blattner, F. R., Plunkett, G., Bloch, C. A., et al. The complete genome sequence of Escherichia
coli K-12. Science (80-. ). 277, 1453–1462 (1997).
349. Aubert, D., Poirel, L., Chevalier, J., et al. Oxacillinase-mediated resistance to cefepime and
susceptibility to ceftazidime in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 45,
1615–1620 (2001).
350. Dortet, L., Poirel, L. & Nordmann, P. Rapid detection of carbapenemase-producing
Pseudomonas spp. J. Clin. Microbiol. 50, 3773–3776 (2012).
351. Mugnier, P., Casin, I., Bouthors, A. T., et al. Novel OXA-10-derived extended-spectrum β-
lactamases selected in vivo or in vitro. Antimicrob. Agents Chemother. 42, 3113–3116 (1998).
352. El Garch, F., Bogaerts, P., Bebrone, C., et al. OXA-198, an acquired carbapenem-hydrolyzing

200
class D β-lactamase from Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 55, 4828–
4833 (2011).
353. Poirel, L., Brinas, L., Verlinde, A., et al. BEL-1, a novel clavulanic acid-inhibited extended-
spectrum β-lactamase, and the class 1 integron In120 in Pseudomonas aeruginosa. Antimicrob.
Agents Chemother. 49, 3743–3748 (2005).
354. Emeraud, C., Escaut, L., Boucly, A., et al. Aztreonam plus clavulanate, tazobactam, or
avibactam for treatment of infections caused by metallo-lactamase-producing gram-negative
bacteria. Antimicrob. Agents Chemother. 63, 1–7 (2019).
355. Rahme, L. G., Stevens, E. J., Wolfort, S. F., et al. Common virulence factors for bacterial
pathogenicity in plants and animals. Science (80-. ). 268, 1899–1902 (1995).
356. Holloway, B. W. Genetics of Pseudomonas. Bacteriol. Rev. 33, 419–443 (1969).
357. Mavridou, D. A. I., Gonzalez, D., Clements, A., et al. The pUltra plasmid series: A robust and
flexible tool for fluorescent labeling of Enterobacteria. Plasmid 87–88, 65–71 (2016).
358. Kessler, B., de Lorenzo, V. & Timmis, K. N. A general system to integrate lacZ fusions into the
chromosomes of gram-negative eubacteria: regulation of the Pm promoter of the TOL plasmid
studied with all controlling elements in monocopy. Mol. Gen. Genet. 233, 293–301 (1992).
359. Paradis-Bleau, C., Kritikos, G., Orlova, K., et al. A Genome-Wide Screen for Bacterial Envelope
Biogenesis Mutants Identifies a Novel Factor Involved in Cell Wall Precursor Metabolism. PLoS
Genet. 10, (2014).
360. Kishigami, S., Akiyama, Y. & Ito, K. Redox states of DsbA in the periplasm of Escherichia coli.
FEBS Lett. 364, 55–58 (1995).
361. Helander, I. M. & Mattila-Sandholm, T. Fluorometric assessment of Gram-negative bacterial
permeabilization. J. Appl. Microbiol. 88, 213–219 (2000).
362. Adobe. Photoshop CS4 Extended v 11.0.
363. McCarthy, R. R., Mazon-Moya, M. J., Moscoso, J. A., et al. Cyclic-di-GMP regulates
lipopolysaccharide modification and contributes to Pseudomonas aeruginosa immune evasion.
Nat. Microbiol. 2, 1–10 (2017).
364. Lee, S., Park, Y. J., Kim, M., et al. Prevalence of Ambler class A and D β-lactamases among
clinical isolates of Pseudomonas aeruginosa in Korea. J. Antimicrob. Chemother. 56, 122–127
(2005).
365. Laudy, A. E., Róg, P., Smolinska-Król, K., et al. Prevalence of ESBL-producing Pseudomonas
aeruginosa isolates in Warsaw, Poland, detected by various phenotypic and genotypic methods.
PLoS One 12, 1–15 (2017).
366. Szarecka, A., Lesnock, K. R., Ramirez-Mondragon, C. A., et al. The Class D β-lactamase

201
family: Residues governing the maintenance and diversity of function. Protein Eng. Des. Sel.
24, 801–809 (2011).
367. Poirel, L., Naas, T. & Nordmann, P. Diversity, epidemiology, and genetics of class D β-
lactamases. Antimicrob. Agents Chemother. 54, 24–38 (2010).
368. Simakov, N., Leonard, D. A., Smith, J. C., et al. A Distal Disulfide Bridge in OXA-1 β-
Lactamase Stabilizes the Catalytic Center and Alters the Dynamics of the Specificity
Determining Ω Loop. J. Phys. Chem. B 121, 3285–3296 (2017).
369. Curley, K. & Pratt, R. F. The oxyanion hole in serine β-lactamase catalysis: Interactions of
thiono substrates with the active site. Bioorg. Chem. 28, 338–356 (2000).
370. Denoncin, K., Vertommen, D., Paek, E., et al. The protein-disulfide isomerase DsbC cooperates
with SurA and DsbA in the assembly of the essential β-barrel protein LptD. J. Biol. Chem. 285,
29425–29433 (2010).
371. Silvestro, L., Weiser, J. N., Axelsen, P. H., et al. Antibacterial and Antimembrane Activities of
Cecropin A in Escherichia coli. Antimicrob Agents Chemother. 44, 602–607 (2000).
372. Hizukuri, Y., Yakushi, T., Kawagishi, I., et al. Role of the intramolecular disulfide bond in FlgI,
the flagellar P-ring component of Escherichia coli. J. Bacteriol. 188, 4190–4197 (2006).
373. Nakae, T., Nakajima, A., Ono, T., et al. Resistance to β-lactam antibiotics in Pseudomonas
aeruginosa due to interplay between the MexAB-OprM efflux pump and β-lactamase.
Antimicrob. Agents Chemother. 43, 1301–1303 (1999).
374. Girlich, D., Naas, T. & Nordmann, P. Biochemical Characterization of the Naturally Occurring
Oxacillinase OXA-50 of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 48, 2043–
2048 (2004).
375. Arts, I. S., Ball, G., Leverrier, P., et al. Dissecting the machinery that introduces disulfide bonds
in Pseudomonas aeruginosa. MBio 4, (2013).
376. Wang, R., Van Dorp, L., Shaw, L. P., et al. The global distribution and spread of the mobilized
colistin resistance gene mcr-1. Nat. Commun. 9, 1–9 (2018).
377. Toleman, M. A., Bennett, P. M., Bennett, D. M. C., et al. Global emergence of
trimethoprim/sulfamethoxazole resistance in Stenotrophomonas maltophilia mediated by
acquisition of sul genes. Emerg. Infect. Dis. 13, 559–565 (2007).
378. Okazaki, A. & Avison, M. B. Induction of L1 and L2 β-lactamase production in
Stenotrophomonas maltophilia is dependent on an AmpR-type regulator. Antimicrob. Agents
Chemother. 52, 1525–1528 (2008).
379. Lemmen, S. W., Häfner, H., Reinert, R. R., et al. Comparison of serum bactericidal activity of
ceftazidime, ciprofloxacin and meropenem against Stenotrophomonas maltophilia. J.

202
Antimicrob. Chemother. 47, 113–124 (2001).
380. Wiersinga, W. J., van der Poll, T., White, N. J., et al. Melioidosis: Insights into the pathogenicity
of Burkholderia pseudomallei. Nat. Rev. Microbiol. 4, 272–282 (2006).
381. Cox, G. & Wright, G. D. Intrinsic antibiotic resistance: Mechanisms, origins, challenges and
solutions. Int. J. Med. Microbiol. 303, 287–292 (2013).
382. Queenan, A. M., Torres-Viera, C., Gold, H. S., et al. SME-type carbapenem-hydrolyzing class
A β-lactamases from geographically diverse Serratia marcescens strains. Antimicrob. Agents
Chemother. 44, 3035–3039 (2000).
383. Borgianni, L., De Luca, F., Thaller, M. C., et al. Biochemical characterization of the POM-1
metallo-β-lactamase from Pseudomonas otitidis. Antimicrob. Agents Chemother. 59, 1755–1758
(2015).
384. Antunes, N. T., Frase, H., Toth, M., et al. The class A β-lactamase FTU-1 is native to Francisella
tularensis. Antimicrob. Agents Chemother. 56, 666–671 (2012).
385. Ho, P. L., Cheung, T. K. M., Yam, W. C., et al. Characterization of a laboratory-generated
variant of BPS β-lactamase from Burkholderia pseudomallei that hydrolyses ceftazidime. J.
Antimicrob. Chemother. 50, 723–726 (2002).
386. Wachino, J. I., Yoshida, H., Yamane, K., et al. SMB-1, a novel subclass B3 metallo-β-lactamase,
associated with ISCR1 and a class 1 integron, from a carbapenem-resistant Serratia marcescens
clinical isolate. Antimicrob. Agents Chemother. 55, 5143–5149 (2011).
387. Thaller, M. C., Borgianni, L., Di Lallo, G., et al. Metallo-β-lactamase production by
Pseudomonas otitidis: A species-related trait. Antimicrob. Agents Chemother. 55, 118–123
(2011).
388. Donato, J. J., Moe, L. A., Converse, B. J., et al. Metagenomic analysis of apple orchard soil
reveals antibiotic resistance genes encoding predicted bifunctional proteins. Appl. Environ.
Microbiol. 76, 4396–4401 (2010).
389. Poirel, L., Magalhaes, M., Lopes, M., et al. Molecular Analysis of Metallo--Lactamase Gene.
Antimicrob. Agents Chemother. 48, 1406–1409 (2004).
390. Yong, D., Toleman, M. A., Giske, C. G., et al. Characterization of a new metallo-β-lactamase
gene, blaNDM-1, and a novel erythromycin esterase gene carried on a unique genetic structure
in Klebsiella pneumoniae sequence type 14 from India. Antimicrob. Agents Chemother. 53,
5046–5054 (2009).
391. Shakibaie, M. R. & Moradie, M. Detection of Carbenicillin Hydrolysing (CARB) Type of ESBL
Enzyme in Acinetobacter baumannii Strains Isolated from Bacterimic and UTI Patients. Int. J.
Biol. Life Sci. 1, 1–5 (2011).

203
392. Toleman, M. A., Bennett, P. M. & Walsh, T. R. ISCR Elements: Novel Gene-Capturing Systems
of the 21st Century? Microbiol. Mol. Biol. Rev. 70, 296–316 (2006).
393. Berg, G., Eberl, L. & Hartmann, A. The rhizosphere as a reservoir for opportunistic human
pathogenic bacteria. Environ. Microbiol. 7, 1673–1685 (2005).
394. Ju, L.-C., Cheng, Z., Fast, W., et al. The Continuing Challenge of Metallo-β-Lactamase
Inhibition: Mechanism Matters. Trends Pharmacol Sci 39, 635–647 (2018).
395. He, J., Baldini, R. L., Déziel, E., et al. The broad host range pathogen Pseudomonas aeruginosa
strain PA14 carries two pathogenicity islands harboring plant and animal virulence genes. Proc.
Natl. Acad. Sci. U.S.A. 101, 2530–2535 (2004).
396. Lodise, T. P., Lomaestro, B. & Drusano, G. L. Piperacillin-tazobactam for Pseudomonas
aeruginosa infection: Clinical implications of an extended-infusion dosing strategy. Clin. Infect.
Dis. 44, 357–363 (2007).
397. Godfrey, A. J., Wong, S., Dance, D. A. B., et al. Pseudomonas pseudomallei resistance to β-
lactam antibiotics due to alterations in the chromosomally encoded β-lactamase. Antimicrob.
Agents Chemother. 35, 1635–1640 (1991).
398. Tribuddharat, C., Moore, R. A., Baker, P., et al. Burkholderia pseudomallei class a β-lactamase
mutations that confer selective resistance against ceftazidime or clavulanic acid inhibition.
Antimicrob. Agents Chemother. 47, 2082–2087 (2003).
399. Nikaido, H. Structure and mechanism of RND-type multidrug efflux pumps. Adv. Enzymol.
Relat. Areas Mol. Biol. 77 1, 1–60 (2010).
400. Alav, I., Sutton, J. M. & Rahman, K. M. Role of bacterial efflux pumps in biofilm formation. J.
Antimicrob. Chemother. 73, 2003–2020 (2018).
401. Lomovskaya, O. & Lewis, K. Emr, an Escherichia coli locus for multidrug resistance. Proc.
Natl. Acad. Sci. U. S. A. 89, 8938–8942 (1992).
402. Wang, Z., Chen, M., Shi, X., et al. In situ structure of the AcrAB-TolC efflux pump at
subnanometer resolution. bioRxiv (preprint) (2020) doi:10.1101/2020.06.10.144618 .
403. Shi, X., Chen, M., Yu, Z., et al. In situ structure and assembly of the multidrug efflux pump
AcrAB-TolC. Nat. Commun. 10, 4–9 (2019).
404. Lee, A., Mao, W., Warren, M. S., et al. Interplay between efflux pumps may provide either
additive or multiplicative effects on drug resistance. J. Bacteriol. 182, 3142–3150 (2000).
405. Okusu, H., Ma, D. & Nikaido, H. AcrAB efflux pump plays a major role in the antibiotic
resistance phenotype of Escherichia coli multiple-antibiotic-resistance (Mar) mutants. J.
Bacteriol. 178, 306–308 (1996).
406. Wang, Z., Fan, G., Hryc, C. F., et al. An allosteric transport mechanism for the AcrAB-TolC

204
multidrug efflux pump. Elife 6, 1–19 (2017).
407. Hobbs, E. C., Yin, X., Paul, B. J., et al. Conserved small protein associates with the multidrug
efflux pump AcrB and differentially affects antibiotic resistance. Proc. Natl. Acad. Sci. U. S. A.
109, 16696–16701 (2012).
408. Luisi, B., Koronakis, V., Hughes, C., et al. Crystal structure of the bacterial membrane protein
TolC central to multidrug efflux and protein export. Nature 405, 914–919 (2002).
409. Weston, N., Sharma, P., Ricci, V., et al. Regulation of the AcrAB-TolC efflux pump in
Enterobacteriaceae. Res. Microbiol. 169, 425–431 (2018).
410. Martin, R. G., Jair, K., Wolf, R. E., et al. Autoactivation of the marRAB Multiple Antibiotic
Resistance Operon by the MarA Transcriptional Activator in Escherichia coli. J. Bacteriol. 178,
2216–2223 (1996).
411. Webber, M. A., Talukder, A. & Piddock, L. J. V. Contribution of mutation at amino acid 45 of
AcrR to acrB expression and ciprofloxacin resistance in clinical and veterinary Escherichia coli
isolates. Antimicrob. Agents Chemother. 49, 4390–4392 (2005).
412. Everett, M. J., Jin, Y. F., Ricci, V., et al. Contributions of individual mechanisms to
fluoroquinolone resistance in 36 Escherichia coli strains isolated from humans and animals.
Antimicrob. Agents Chemother. 40, 2380–2386 (1996).
413. Hayashi, S., Nakazawa, T., Kimoto, M., et al. The DsbA-DsbB Disulfide Bond Formation
System of Burkholderia cepacia Is Involved in the Production of Protease and Alkaline Metal
Resistance, and Multi-Drug Resistance. Microbiology 44, 41–50 (2000).
414. Manno, G., Dalmastri, C., Tabacchioni, S., et al. Epidemiology and Clinical Course of
Burkholderia cepacia Complex Infections, Particularly Those Caused by Different Burkholderia
cenocepacia Strains, among Patients Attending an Italian Cystic Fibrosis Center. J. Clin.
Microbiol. 42, 1491–1497 (2004).
415. Jones, A. L., DeShazer, D. & Woods, D. E. Identification and characterization of a two-
component regulatory system involved in invasion of eukaryotic cells and heavy-metal
resistance in Burkholderia pseudomallei. Infect. Immun. 65, 4972–4977 (1997).
416. Nies, D. H., Nies, A., Chu, L., et al. Expression and nucleotide sequence of a plasmid-
determined divalent cation efflux system from Alcaligenes eutrophus (heavy metal plasmid
resistance/cation transport system). Proc. Natl. Acad. Sci. U.S.A. 86, 7351–7355 (1989).
417. Rensing, C., Pribyl, T. & Nies, D. H. New functions for the three subunits of the CzcCBA cation-
proton antiporter. J. Bacteriol. 179, 6871–6879 (1997).
418. Nicoloff, H., Perreten, V., McMurry, L. M., et al. Role for tandem duplication and lon protease
in AcrAB-TolC-dependent multiple antibiotic resistance (Mar) in an Escherichia coli mutant

205
without mutations in marRAB or acrRAB. J. Bacteriol. 188, 4413–4423 (2006).
419. Abdi, S. N., Ghotaslou, R., Ganbarov, K., et al. Acinetobacter baumannii efflux pumps and
antibiotic resistance. Infect. Drug Resist. 13, 423–434 (2020).
420. Leiser, O. P., Charlson, E. S., Gerken, H., et al. Reversal of the degP Phenotypes by a Novel
rpoE Allele of Escherichia coli. PLoS One 7, (2012).
421. Gerken, H. & Misra, R. Genetic evidence for functional interactions between TolC and AcrA
proteins of a major antibiotic efflux pump of Escherichia coli. Mol. Microbiol. 54, 620–631
(2004).
422. Clausen, T., Southan, C. & Ehrmann, M. The HtrA family of proteases: Implications for protein
composition and cell fate. Mol. Cell 10, 443–455 (2002).
423. Hiniker, A. & Bardwell, J. C. A. In Vivo Substrate Specificity of Periplasmic Disulfide
Oxidoreductases. J. Biol. Chem. 279, 12967–12973 (2004).
424. Skorko-Glonek, J., Zurawa, D., Tanfani, F., et al. The N-terminal region of HtrA heat shock
protease from Escherichia coli is essential for stabilization of HtrA primary structure and
maintaining of its oligomeric structure. Biochim. Biophys. Acta 1649, 171–182 (2003).
425. Werner, J., Augustus, A. M. & Misra, R. Assembly of TolC, a structurally unique and
multifunctional outer membrane protein of Escherichia coli K12. J. Bacteriol. 185, 6540–6547
(2003).
426. Ariza, R. R., Cohen, S. P., Bachhawat, N., et al. Repressor mutations in the marRAB operon that
activate oxidative stress genes and multiple antibiotic resistance in Escherichia coli. J. Bacteriol.
176, 143–148 (1994).
427. Keeney, D., Ruzin, A., Mcaleese, F., et al. MarA-mediated overexpression of the AcrAB efflux
pump results in decreased susceptibility to tigecycline in Escherichia coli. J. Antimicrob.
Chemother. 61, 46–53 (2008).
428. European Committee on Antimicrobial Susceptibility Testing. Breakpoint tables for
interpretation of MICs and zone diameters. (2020).
429. Bloom, J. D., Labthavikul, S. T., Otey, C. R., et al. Protein stability promotes evolvability. Proc.
Natl. Acad. Sci. U. S. A. 103, 5869–5874 (2006).
430. Schreiber, G., Buckle, A. M. & Fersht, A. R. Stability and function: two constraints in the
evolution of barstar and other proteins. Structure 2, 945–951 (1994).
431. Zhang, X. jun, Baase, W. A., Shoichet, B. K., et al. Enhancement of protein stability by the
combination of point mutations in T4 lysozyme is additive. Protein Eng. Des. Sel. 8, 1017–1022
(1995).
432. Giakkoupi, P., Hujer, A. M., Miriagou, V., et al. Substitution of Thr for Ala-237 in TEM-17,

206
TEM-12 and TEM-26: Alterations in β-lactam resistance conferred on Escherichia coli. FEMS
Microbiol. Lett. 201, 37–40 (2001).
433. Cabot, G., Bruchmann, S., Mulet, X., et al. Pseudomonas aeruginosa ceftolozane-tazobactam
resistance development requires multiple mutations leading to overexpression and structural
modification of Ampc. Antimicrob. Agents Chemother. 58, 3091–3099 (2014).
434. Rosenkilde, C. E. H., Munck, C., Porse, A., et al. Collateral sensitivity constrains resistance
evolution of the CTX-M-15 β-lactamase. Nat. Commun. 10, 1–10 (2019).
435. Helfand, M. S., Bethel, C. R., Hujer, A. M., et al. Understanding resistance to β-lactams and β-
lactamase inhibitors in the SHV β-lactamase: Lessons from the mutagenesis of Ser-130. J. Biol.
Chem. 278, 52724–52729 (2003).
436. Wang, X., Minasov, G. & Shoichet, B. K. Evolution of an antibiotic resistance enzyme
constrained by stability and activity trade-offs. J. Mol. Biol. 320, 85–95 (2002).
437. Fröhlich, C., Sørum, V., Thomassen, A. M., et al. OXA-48-Mediated Ceftazidime-Avibactam
Resistance Is Associated with Evolutionary Trade-Offs. mSphere 4, 1–15 (2019).
438. Schultz, S. C., Farland, G. D. & Richards, J. H. Stability of Wild-Type and Mutant RTEM-1 ,
8-Lactamases: Effect of the Disulfide Bond. PROTEINS Struct. Funct. Genet. 2, 290–297
(1987).
439. Salverda, M. L. M., de Visser, J. A. G. M. & Barlow, M. Natural evolution of TEM-1 -
lactamase: experimental reconstruction and clinical relevance. FEMS Microbiol. Rev. 34, 1015–
1036 (2010).
440. Zhu, F., He, B., Gu, F., et al. Improvement in organic solvent resistance and activity of
metalloprotease by directed evolution. J. Biotechnol. 309, 68–74 (2020).
441. Matthew, M., Hedges, R. W. & Smith, J. T. Types of -Lactamase Determined by Plasmids in
Gram-Negative Bacteria. J. Bacteriol. 138, 657–662 (1979).
442. Arlet, G., Rouveau, M. & Philippon, A. Substitution of alanine for aspartate at position 179 in
the SHV-6 extended-spectrum -lactamase. FEMS Microbiol. Lett. 152, 163–167 (1997).
443. Rasheed, J. K., Jay, C., Metchock, B., et al. Evolution of Extended-Spectrum -Lactam
Resistance ( SHV-8 ) in a Strain of Escherichia coli during Multiple Episodes of Bacteremia.
Antimicrob. Agents Chemother. 41, 647–653 (1997).
444. Barlow, M. & Hall, B. G. Predicting Evolutionary Potential: In Vitro Evolution Accurately
Reproduces Natural Evolution of the TEM -Lactamase. Genetics 160, 823–832 (2002).
445. Billot-Klein, D., Gutmann, L. & Collatz, E. Nucleotide sequence of the SHV-5 β-lactamase gene
of a Klebsiella pneumoniae plasmid. Antimicrob. Agents Chemother. 34, 2439–2441 (1990).
446. Gniadkowski, M. Evolution of extended-spectrum β-lactamases by mutation. Clin. Microbiol.

207
Infect. 14, 11–32 (2008).
447. Matagne, A., Lamotte-Brasseur, J. & Frere, J.-M. Catalytic properties of class A β-lactamases:
efficiency and diversity. Biochem. J. 330, 581–598 (1998).
448. Liakopoulos, A., Mevius, D. & Ceccarelli, D. A review of SHV extended-spectrum β-
lactamases: Neglected yet ubiquitous. Front. Microbiol. 7, 1–27 (2016).
449. Gaibani, P., Campoli, C., Lewis, R. E., et al. In vivo evolution of resistant subpopulations of
KPC-producing Klebsiella pneumoniae during ceftazidime/avibactam treatment. J. Antimicrob.
Chemother. 73, 1525–1529 (2018).
450. Andersson, D. I. & Hughes, D. Antibiotic resistance and its cost: Is it possible to reverse
resistance? Nat. Rev. Microbiol. 8, 260–271 (2010).
451. Heras, B., Kurz, M., Shouldice, S. R., et al. The name’s bond......disulfide bond. Curr. Opin.
Struct. Biol. 17, 691–698 (2007).
452. Cassini, A., Högberg, L. D., Plachouras, D., et al. Attributable deaths and disability-adjusted
life-years caused by infections with antibiotic-resistant bacteria in the EU and the European
Economic Area in 2015: a population-level modelling analysis. Lancet Infect. Dis. 19, 56–66
(2019).
453. Pletzer, D., Mansour, S. C., Wuerth, K., et al. New mouse model for chronic infections by gram-
negative bacteria enabling the study of anti-infective efficacy and host-microbe interactions.
MBio 8, 1–16 (2017).
454. Peters, J. M., Koo, B. M., Patino, R., et al. Enabling genetic analysis of diverse bacteria with
Mobile-CRISPRi. Nat. Microbiol. 4, 244–250 (2019).
455. Zheng, W. D., Quan, H., Song, J. L., et al. Does DsbA have chaperone-like activity? Arch.
Biochem. Biophys. 337, 326–331 (1997).
456. Naas, T., Ouslati, S., Bonnin, R., et al. -lactamase database (BLDB) -structure and function. J
Enzym. Inhib Med Chem 32, 917–919 (2017).
457. Doublet, B., Robin, F., Casin, I., et al. Molecular and biochemical characterization of the natural
chromosome-encoded class A β-lactamase from Pseudomonas luteola. Antimicrob. Agents
Chemother. 54, 45–51 (2010).
458. Aubert, D., Poirel, L., Ali Ben, A., et al. OXA-35 is an OXA-10-related -lactamase from
Pseudomonas aeruginosa. J. Antimicrob. Chemother. 48, 717–721 (2001).
459. Milenkovic, D., Ramming, T., Muller, J. M., et al. Identification of the Signal Directing Tim9
and Tim10 into the Intermembrane Space of Mitochondria. Mol. Biol. Cell 20, 2530–2539
(2009).

208
9 APPENDIX I
Supplementary Table 1 Overview of the β-lactamase enzymes investigated in this thesis. All tested enzymes belong to
distinct phylogenetic clusters.338 The “Cysteine positions” column states the positions of cysteine residues after position 30
and hence, does not include amino acids that would be part of the periplasmic signal peptide which is cleaved after protein
translocation. All β-lactamase enzymes except L2-1 and LUT-1, which are used as negative controls in this study (top two
rows), have one or more disulfide bonds. The “Mob.” (mobilizable) column refers to the possibility for the β-lactamase gene
to be mobilized from the chromosome; “yes” indicates that the gene of interest is located on a mobile element, while “no”
refers to immobile chromosomally-encoded enzymes. The “Spectrum” column refers to the hydrolytic spectrum of each tested
enzyme; tested enzymes are narrow-spectrum β-lactamases (NS), extended spectrum β-lactamases (ESBL) or carbapenemases.
The “Inh.” (inhibition) column refers to classical inhibitor susceptibility i.e. susceptibility to inhibition by clavulanic acid,
tazobactam or sulbactam. The “Organism” column refers to the bacterial species that most commonly express the tested β-
lactamase enzymes.
Enzyme Cysteine
positions
Ambler
class Mob. Spectrum Inh. Organism
L2-1 C82, C136 C233 A no ESBL yes S. maltophilia
LUT-1 C54 C129 A no457 NS yes P. luteola
OXA-4 C43, C63 D yes ESBL yes P. aeruginosa
OXA-10 C44, C51 D yes ESBL no458 P. aeruginosa
OXA-18 C25, C39, C57 D yes ESBL yes P. aeruginosa
OXA-19 C12, C44, C51 D yes ESBL no351 P. aeruginosa
OXA-198 C116, C119 D yes carbapenemase no352 P. aeruginosa
BEL-1 C61 C231 A yes353 ESBL yes P. aeruginosa
BPS-1m C75 C83 C129 A no385 ESBL yes B. pseudomallei
CARB-2 C72 C118 A yes NS yes Pseudomonas. spp.
FTU-1 C60 C230 A no384 NS yes F. tularensis
AIM-1 C31 C56 C194
C199 C234 C274 B3 yes109 carbapenemase no62 P. aeruginosa
L1-1 C239 C265 B3 no62 carbapenemase no62 S. maltophilia
POM-1 C237 C265 B3 no387 carbapenemase no62 P. otitidis
SMB-1 C180 C 185 C226
C260 B3 yes386 carbapenemase no62 Serratia spp.
OXA-50 C208 C211 D no374 NS no374 Pseudomonas spp.

209
Supplementary Table 2 Deletion of dsbA lowers the β-lactam MIC values for E. coli MC1000 expressing diverse β-lactamases. In the absence of DsbA the β-lactam MICs for E. coli
MC1000 expressing disulfide-bond-containing β-lactamases are reduced. This table shows the MIC data used to generate Figure 3.1, Figure 3.2, Figure 4.1, and Figure 4.2. The aminoglycoside
antibiotic gentamicin and E. coli MC1000 strains harbouring pDM1 (vector alone), pDM1-blaL2-1 or pDM1-blaLUT-1 (cysteine-containing β-lactamases which lack disulfide bonds) serve as negative
controls. Combinations for which MICs were not recorded are marked with a dash (-). MIC values (µg/mL) show three independent experiments. The following abbreviations are used: GM,
gentamicin; AC, amoxicillin; AM, ampicillin; XM, cefuroxime; TZ, ceftazidime; IP, imipenem; AT, aztreonam.
Strain (MC1000) GM AC AM XM TZ IP AT
pdM1
dsbA pdM1
1.00, 1.00, 0.50
1.00, 1.00, 0.50
4.00, 6.00, 6.00
6.00, 6.00, 6.00 -
6.00, 6.00, 6.00
4.00, 6.00, 6.00
0.38, 0.38, 0.19
0.19, 0.19, 0.19
0.19, 0.19, 0.19
0.19, 0.25, 0.19
0.125, 0.125, 0.19
0.125, 0.125, 0.19
pDM1-blaL2-1 CF
dsbA pDM1-blaL2-1 CF
0.38, 0.25, 0.75
0.50, 038, 1.00 -
2000, 2000, 2000
2000, 2000, 2000 -
12.0, 8.00, 16.0
8.00, 8.00, 12.0 -
500, 500, 500
500, 1000, 500
pDM1-blaLUT-1
dsbA pDM1-blaLUT-1
0.50, 0.75, 0.38
0.75, 0.75, 0.38 -
2000, 2000, 2000
1000, 2000, 4000 -
1.50, 1.50, 0.75
1.00, 1.00, 0.75 -
32.0, 16.0, 24.0
32.0, 16.0, 12.0
pDM1-blaOXA-4
dsbA pDM1-blaOXA-4
0.25, 0.50, 1.00
0.50, 0.75, 1.00 -
1000, 1000, 1000
250, 250, 250
24.0, 16.0, 16.0
4.00, 6.00, 4.00 - - -
pDM1-blaOXA-10
dsbA pDM1-bla OXA-10
0.50, 1.00, 0.75
0.75, 1.00, 1.00 -
8000, 8000, 8000
2000, 2000, 2000
256, 256, 256
16.0, 24.0, 32.0 - -
4.00, 6.00, 4.00
0.75, 1.00, 0.75
pDM1-bla OXA-18
dsbA pDM1-bla OXA-18
0.75, 0.75, 1.00
0.75, 0.75, 1.00 -
1000, 1000, 1000
500, 500, 500 -
1000, 1000, 1000
500, 500, 500 -
2000, 2000, 2000
1000, 1000, 1000
pDM1-bla OXA-198
dsbA pDM1-bla OXA-198
0.19, 0.75, 1.00
0.38, 1.00, 1.00 -
4000, 4000, 4000
1000, 2000, 1000
16.0, 12.0, 12.0
6.00, 4.00, 6.00 -
32.0, 32.0, 32.0
3.00, 2.00, 4.00 -
pDM1-blaBEL-1
dsbA pDM1-blaBEL-1
0.75, 0.75, 1.00
1.00, 1.00, 1.50 -
2000, 4000, 2000
500, 2000, 1000 -
12.0, 6.00, 8.00
4.00, 2.00, 1.50 -
24.0, 12.0, 16.0
6.00, 4.00, 4.00
pDM1-blaBPS-1m
dsbA pDM1-blaBPS-1m
1.00, 1.00, 0.75
0.75, 0.75, 0.75 -
500, 500, 500
250, 250, 250 -
256, 256, 256
24.0, 24.0, 12.0 -
3.00, 2.00, 1.50
0.75, 0.25, 0.38

210
pDM1-blaCARB-2
dsbA pDM1-blaCARB-2
0.50, 0.75, 1.00
0.75, 0.75, 0.75 -
16000, 16000,
16000
8000, 8000, 8000
12.0, 12.0, 12.0
4.00, 4.00, 4.00 - - -
pDM1-blaFTU-1
dsbA pDM1-blaFTU-1
1.00, 1.00, 075
1.00, 1.00, 1.00 -
500, 500, 500
250, 250, 250 - - - -
pDM1-blaAIM-1
dsbA pDM1-blaAIM-1
0.50, 0.75, 1.00
0.50, 0.75, 1.00 -
4000, 4000, 4000
250, 250, 500 -
6.00, 6.00, 6.00
0.50, 0.75, 0.5
1.00, 2.00, 1.50
0.25, 0.38, 0.75 -
pDM1-blaL1-1CF
dsbA pDM1-blaL1-1CF
0.125, 0.380, 0.75
0.19, 0.380, 1.00
256, 256, 256
16.0, 16.0, 96.0 - -
128, 48, 48
0.38, 0.75, 2
1.00, 1.50, 0.75
0.25, 0.19, 0.19
0.19, 0.125, 0.125
0.094, 0.50, 0.125
pDM1-blaPOM-1
dsbA pDM1-blaPOM-1
1.00, 0.75, 1.00
1.00, 0.75 1.50 -
4000, 4000, 4000
2000, 2000, 2000
256, 256, 256
64.0, 48.0, 32.0 -
4.00, 6.00, 4.00
1.50, 3.00, 2.00 -
pDM1-blaSMB-1
dsbA pDM1-blaSMB-1
1.00, 0.75, 1.00
1.50, 0.75, 1.50 -
2000, 4000, 4000
250, 1000, 1000
6.00, 4.00, 4.00
2.00, 1.50, 1.50 -
1.00, 1.00, 1.00
0.25, 0.38, 0.38 -
pDM1-blaOXA-50
dsbA pDM1-blaOXA-50
1.00, 0.75, 0.75
0.75, 0.75, 1.00
12.0, 8.00, 8.00
6.00, 6.00, 4.00 -
16.0, 24.0, 16.0
3.00, 8.00, 3.00 - - -
CF – Courtesy of Dr R. Christopher D. Furniss.

211
Supplementary Table 3 Chemical inhibition of the DSB system reduces the MIC values of representative -lactam
antibiotics for E. coli MC1000 expressing disulfide-bond-containing class D β-lactamases in a similar manner to the
deletion of dsbA. This table shows the MIC data used to generate Figure 3.8. The aminoglycoside antibiotic gentamicin and
the E. coli MC1000 strain, harbouring the pDM1 empty vector, serve as negative controls. Combinations for which MICs were
not recorded are marked with a dash (-). MIC values (µg/mL) show three independent experiments. The following
abbreviations are used: GM, gentamicin; XM, cefuroxime; IP, imipenem.
Strain (MC1000) Media GM XM IP
pdM1
MHA
M63 + DMSO
M63 +inhibitor
1.00
1.00, 2.00, 1.50
1.00, 1.00, 1.00
6.00
2.00, 2.00, 3.00
2.00, 2.00, 2.00
0.19
0.19, 0.19, 0.19
0.19, 0.19, 0.19
pDM1-blaOXA-4
MHA
M63 + DMSO
M63 +inhibitor
1.00
1.50, 2.00, 2.00
1.50, 1.50, 1.50
16.0
16.0, 12.0, 12.0
3.00, 2.00, 4.00
-
pDM1-blaOXA-10
MHA
M63 + DMSO
M63 +inhibitor
0.50
1.50, 2.00, 1.50
1.50, 1.50, 1.50
256
12.0, 16.0, 24.0
1.00, 2.00, 2.00
-
pDM1-bla OXA-198
MHA
M63 + DMSO
M63 +inhibitor
1.00
1.50, 1.50, 1.50
1.50, 1.50, 1.50
12.0
12.0, 16.0, 8.00
6.00, 6.00, 3.00
32.0
32.0, 16.0, 32.00
4.00, 3.00, 6.00

212
Supplementary Table 4 Antibiotic resistance profiles (MIC values in µg/mL) of the clinical isolates and laboratory
strains tested in this study for -lactam compounds. Values highlighted in pink indicate resistance, as defined by the
EUCAST clinical breakpoint guidelines, whilst values highlighted in light blue indicate antibiotics for which there is no
EUCAST clinical breakpoint. The remaining values (white cells) indicate sensitivity to the tested antibiotic compound.
Combinations for which MIC values were not recorded (yellow cells) are marked with a dash (-). The following abbreviations
are used: AC, amoxicillin; PP, piperacillin; PT, piperacillin/tazobactam; XM, cefuroxime; TZ, ceftazidime; IP, imipenem; AT,
aztreonam.
Strain AC PP PT XM TZ IP AT
Pseudomonas aeruginosa SOF-1
(blaOXA-4) >256 - - >256 2 3 8
Pseudomonas aeruginosa PU21
(blaOXA-10) >256 - - >256 3 6 16
Pseudomonas aeruginosa
(blaOXA-19) >256 >256 - >256 >256 8 >256
Pseudomonas aeruginosa PA41437
(blaOXA-198) >256 24 32CF >256 2 >32 6
Pseudomonas aeruginosa PA14
(blaOXA-50) >256 8 8 >256 1.5 0.38 6
Pseudomonas aeruginosa PAO1 LA
(blaOXA-50) - 4 4 >256 1 2 3
Pseudomonas aeruginosa PAO1 LD
(blaOXA-50) - 4 4 >256 1.5 2 4
Pseudomonas aeruginosa G4R7
(blaOXA-50 blaAIM-1) >256 >256 >256 >256 24 >32 6
Pseudomonas aeruginosa G6R7
(blaOXA-50 blaAIM-1) >256 32 32 >256 24 >32 6
Stenotrophomonas maltophilia GUE
(blaL2-1 blaL1-1) >256 32 8 >256 12 >32 >256
CF – Courtesy of Dr R. Christopher D. Furniss.

213
Supplementary Table 5 Antibiotic resistance profiles (MIC values in µg/mL) of the clinical isolates and laboratory
strains tested in this study for a range of commonly used non--lactam antibiotics. Values highlighted in pink indicate
resistance, as defined by the EUCAST clinical breakpoint guidelines, whilst values highlighted in light blue indicate antibiotics
for which there is no EUCAST clinical breakpoint. The remaining values (white cells) indicate sensitivity to the tested
antibiotic compound. Combinations for which MIC values were not recorded (yellow cells) are marked with a dash (-). The
following abbreviations are used: GM, gentamicin; CO, colistin; CI, ciprofloxacin; TR, trimethoprim; TS,
trimethoprim/sulfamethoxazole.
Strain GM CO CI TR TS
Pseudomonas aeruginosa SOF-1
(blaOXA-4) >256 - >32 >32 -
Pseudomonas aeruginosa PU21
(blaOXA-10) >256 - 0.38 >32 -
Pseudomonas aeruginosa
(blaOXA-19) >256 1 >32 >32 -
Pseudomonas aeruginosa PA41437
(blaOXA-198) 16 1CF >32 >32 -
Pseudomonas aeruginosa PA14
(blaOXA-50) 1.5 1 1 - -
Pseudomonas aeruginosa PAO1 LA
(blaOXA-50) 1.5 2 0.125 - -
Pseudomonas aeruginosa PAO1 LD
(blaOXA-50) 1.5 1 0.125 - -
Pseudomonas aeruginosa G4R7
(blaOXA-50 blaAIM-1) >256 0.5 >32 - -
Pseudomonas aeruginosa G6R7
(blaOXA-50 blaAIM-1) >256 2 >32 - -
Stenotrophomonas maltophilia GUE
(blaL2-1 blaL1-1) 1 2 - >32 0.06
CF – Courtesy of Dr R. Christopher D. Furniss

214
Supplementary Table 6 Deletion of dsbA does not decrease the β-lactam MIC values for E. coli MC1000 expressing the narrow-spectrum β-lactamases TEM-1 and SHV-1 at either
37°C or 42°C. This table shows the MIC data used to generate Figure 6.3, Figure 6.4, and Figure 6.5. The aminoglycoside antibiotic gentamicin and the E. coli MC1000 strains harbouring pDM2
(vector alone) serve as controls. Strains in this table are referred to as Background strains throughout Chapter 6, MIC values highlighted in bold were used to calculate fold change values presented
in Three independent experiments are shown for MIC values recorded at 37°C and one independent experiment is shown for MIC values recorded 42°C. MIC values (µg/mL), abbreviations used:
GM, gentamicin; AC, amoxicillin; XM, cefuroxime; TZ, ceftazidime; IP, imipenem; AT, aztreonam.
Strain (MC1000) Temp (°C) GM AC XM TZ IP AT
pDM2
dsbA pDM2 37
1.00, 0.75, 1.00
1.00, 0.75, 0.75
6.00, 6.00, 6.00
4.00, 4.00, 4.00
6.00, 6.00, 6.00
6.00, 6.00, 6.00
0.25, 0.19, 0.25
0.19, 0.25, 0.25
0.38, 0.38, 0.38
0.38, 0.38, 0.38
0.19, 0.19, 0.19
0.19, 0.19, 0.19
pDM2
dsbA pDM2 42
0.75
1.00
6.00
4.00
6.00
4.00
0.25
0.25
0.38
0.38
0.25
0.19
pDM2-blaSHV-1
dsbA pDM2- blaSHV-1
pDM2- blaSHV-1 C54
37
1.00, 0.75, 1.00
1.00, 0.75, 0.75
0.50, 1.00, 1.00
>256, >256, >256
>256, >256, >256
>256, >256, >256
24.0, 12.0, 12.0
12.0, 8.00, 8.00
12.0, 8.00, 8.00
4.0, 4.0, 4.0
2.0, 2.0, 2.0
2.0, 2.0, 2.0
0.50, 0.38, 0.50
0.50, 0.38, 0.38
0.38, 0.38, 0.38
1.0, 0.75, 0.75
0.75, 0.75, 0.75
1.0, 0.75, 0.75
pDM2-blaSHV-1
dsbA pDM2- blaSHV-1
pDM2- blaSHV-1 C54
42
1.00
1.00
0.50
>256
>256
>256
24.0
16.0
12.0
4.0
3.0
2.0
0.38
0.50
0.50
0.75
1.0
1.0
pDM2-blaTEM-1
dsbA pDM2-blaTEM-1
pDM2-blaTEM-1 C86A
37
0.75, 1.00, 1.00
1.00, 0.75, 1.00
0.75, 1.00, 1.00
>256, >256, >256
>256, >256, >256
>256, >256, >256
12.0, 12.0, 12.0
6.00, 4.00, 6.00
8.00, 6.00, 6.00
0.25, 0.25, 0.25
0.25, 0.50, 0.25
0.50, 0.25, 0.50
0.38, 0.25, 0.25
0.25, 0.25, 0.25
0.25, 0.25, 0.25
0.19, 0.25, 0.19
0.19, 0.125, 0.125
0.25, 0.094, 0.19
pDM2-blaTEM-1
dsbA pDM2-blaTEM-1
pDM2-blaTEM-1 C86A
42
0.75
0.75
0.75
>256
>256
>256
12.0
6.00
8.00
0.25
0.094
0.50
0.38
0.38
0.38
0.19
0.25
0.25

215
Supplementary Table 7 β-lactam MIC values and MIC fold changes (FC) recorded in evolved and original backgrounds, after experimental evolution of E. coli MC1000 strains
expressing the narrow-spectrum β-lactamase SHV-1. This table shows the MIC data used to generate Figure 6.3, Figure 6.4, and Figure 6.5. The fold changes were calculate using Background
strain MIC values (bold font, Supplementary Table 6). The aminoglycoside antibiotic gentamicin serves as a control and shows no changes in MIC values for any of the tested strains. MIC fold
changes (MIC fold changes: > 2, fold change defined as Evolved or Original MC1000 MIC (µg/mL) / median background MC1000 (µg/mL); MIC values (µg/mL); abbreviations used: GM,
gentamicin; AC, amoxicillin; XM, cefuroxime; TZ, ceftazidime; IP, imipenem; AT, aztreonam.
Construct Colony GM AC XM TZ IP AT
MIC FC MIC FC MIC FC MIC FC MIC FC MIC FC
Evolved MC1000 pDM2-blaSHV-1
#1 0.75 0.75 >256 1.0 24.0 2.0 16.0 4.0 0.25 0.50 2.00 2.6
#2 1.00 1.0 >256 1.0 16.0 1.3 8.00 2.0 0.25 0.50 1.00 1.3
#3 1.00 1.0 >256 1.0 16.0 1.3 16.0 4.0 0.25 0.50 1.50 2.0
Original MC1000 evolved pDM2-blaSHV-1
#1 0.75 0.75 >256 1.0 16.0 1.3 160 4.0 0.25 0.50 1.00 1.3
#2 0.75 0.75 >256 1.0 16.0 1.3 8.00 2.0 0.25 0.50 1.00 1.3
#3 0.50 0.50 >256 1.0 12.0 1.0 120. 3.0 0.25 0.50 1.00 1.3
Evolved MC1000 dsbA pDM2-blaSHV-1
#1 0.75 1.0 >256 1.0 48.0 6.0 16.0. 8.0 0.25 0.66 3.00 4.0
#2 1.00 1.0 >256 1.0 48.0 6.0 16.0 8.0 0.38 1.0 4.00 5.3
#3 1.00 1.3 >256 1.0 64.0 8.0 32.0 16 0.50 1.3 6.00 8.0
Original MC1000 dsbA pDM2-blaSHV-1
#1 0.50 0.66 >256 1.0 12.0 1.5 8.00 4.0 0.25 0.66 1.50 2.0
#2 0.75 1.0 >256 1.0 8.00 1.0 16.0 8.0 0.25 0.66 1.50 2.0
#3 0.50 0.66 >256 1.0 12.0 1.5 12.0 6.0 0.25 0.66 1.00 1.3
Original MC1000 pDM2-blaSHV-1
#1 0.75 1.0 >256 1.0 12.0 1.5 6.0 3.0 0.25 0.66 1.00 1.3
#2 0.50 0.66 >256 1.0 12.0 1.5 4.0 2.0 0.25 0.66 1.00 1.3
#3 0.50 0.66 >256 1.0 12.0 1.5 16.0 8.0 0.25 0.66 1.00 1.3
Evolved MC1000 pDM2-blaSHV-1 C54A
#1 1.00 1.0 >256 1.0 24.0 3.0 24.0 12 0.19 0.50 0.75 1.0
#2 0.75 0.75 >256 1.0 16.0 2.0 16.0 8.0 0.19 0.50 0.50 0.66
#3 0.50 0.50 >256 1.0 16.0 2.0 16.0 8.0 0.19 0.50 0.50 0.66

216
Original MC1000 evolved pDM2-blaSHV-1 C54A
#1 0.75 0.75 >256 1.00 8.00 1.0 12.0 6.0 0.19 0.50 0.38 0.50
#2 0.50 0.50 >256 1.00 16.0 2.0 8.00 4.0 0.25 0.66 0.38 0.50
#3 0.50 0.50 >256 1.00 16.0 2.0 12.0 6.0 0.19 0.50 0.38 0.50

217
10 APPENDIX II
Breaking antimicrobial resistance by disrupting extracytoplasmic protein folding
R. Christopher D. Furniss2,†, Nikol Kadeřábková2,†, Declan Barker2, Patricia Bernal3, Evgenia
Maslova4, Amanda A.A. Antwi2, Helen E. McNeil5, Hannah L. Pugh5, Laurent Dortet2,6,7,8, Jessica M.A.
Blair5, Gerald Larrouy-Maumus2, Ronan R. McCarthy4, Diego Gonzalez9, Despoina A.I. Mavridou1,2,*
1Department of Molecular Biosciences, University of Texas at Austin, Austin, 78712, Texas, USA 2MRC Centre for Molecular Bacteriology and Infection, Department of Life Sciences, Imperial College
London, London, SW7 2AZ, UK 3Department of Biology, Faculty of Sciences, Universidad Autónoma de Madrid, Madrid, 28049, Spain 4Division of Biosciences, Department of Life Sciences, College of Health and Life Sciences, Brunel
University London, Uxbridge, UB8 3PH, UK 5Institute of Microbiology and Infection, College of Medical and Dental Sciences, University of
Birmingham, Birmingham, B15 2TT, UK 6Department of Bacteriology-Hygiene, Bicêtre Hospital, Assistance Publique - Hôpitaux de Paris, Le
Kremlin-Bicêtre, 94270, France 7EA7361 “Structure, Dynamics, Function and Expression of Broad-spectrum β-lactamases", Paris-Sud
University, LabEx Lermit, Faculty of Medicine, Le Kremlin-Bicêtre, 94270, France 8French National Reference Centre for Antibiotic Resistance, Le Kremlin-Bicêtre, 94270, France 9Laboratoire de Microbiologie, Institut de Biologie, Université de Neuchâtel, Neuchâtel, 2000,
Switzerland
*Correspondence: [email protected] †These authors have contributed equally to this work
ABSTRACT
Antimicrobial resistance in Gram-negative bacteria is one of the greatest threats to global health. New
antibacterial strategies are urgently needed, and the development of antibiotic adjuvants that either
neutralize resistance proteins or compromise the integrity of the cell envelope is of ever-growing
interest. Most available adjuvants are only effective against specific resistance proteins from the same
class. Here we demonstrate that disruption of cell envelope protein homeostasis simultaneously
incapacitates three major classes of resistance determinants. In particular, we find that impairing DsbA-
mediated disulfide bond formation incapacitates β-lactamases and mobile colistin resistance enzymes,
whilst also compromising Resistance-Nodulation-Division efflux pumps. Furthermore, we show that
chemical inhibition of DsbA sensitizes multidrug-resistant clinical isolates to existing antibiotics and
that absence of DsbA allows clearance of a multidrug-resistant Pseudomonas aeruginosa strain from
the Galleria mellonella infection model. This work lays the foundation for the development of novel
antibiotic adjuvants that function as broad-acting resistance breakers.
IMPACT STATEMENT: Disruption of disulfide bond formation sensitizes resistant Gram-negative
bacteria expressing β-lactamases, mobile colistin resistance enzymes and efflux pumps to currently
available antibiotics.

218
INTRODUCTION
Antimicrobial resistance (AMR) is one of the most important public health concerns of our time (1).
With few new antibiotics in the pharmaceutical pipeline and multidrug-resistant bacterial strains
continuously emerging, it is more important than ever to develop novel antibacterial strategies and find
alternative ways to break resistance. The development of new treatments for Gram-negative bacteria is
considered critical by the WHO (2), however identifying novel approaches to target these organisms is
particularly challenging due to their unique double-membrane permeability barrier and the vast range
of AMR determinants they produce. For this reason, rather than targeting cytoplasmic processes,
antimicrobial strategies that inhibit cell-envelope components or impair the activity of resistance
determinants are being increasingly pursued (3-7).
The Gram-negative cell envelope is home to many different AMR determinants, with β-lactamase
enzymes currently posing a seemingly insurmountable problem. Almost 4,000 unique enzymes capable
of degrading β-lactam compounds have been identified to date (File S1). Despite the development of
more advanced β-lactams, for example carbapenems and monobactams, resistance has continued to
emerge through the evolution of many broad-acting β-lactamases (8). This constant emergence of
resistance not only threatens β-lactams, the most commonly prescribed antibiotics worldwide (9, 10),
but increases the use of last-resort agents like the polymyxin antibiotic colistin for the treatment of
multidrug-resistant infections (11). As a result, resistance to colistin is also on the rise, due in part to
the spread of alarming novel cell-envelope colistin resistance determinants. These proteins, called
mobile colistin resistance (MCR) enzymes, represent the only mobilizable mechanism of polymyxin
resistance reported to date (12). Since their discovery in 2015, nine families of MCR proteins have been
identified and these enzymes are quickly becoming a major threat to the longevity of colistin (13).
Alongside β-lactamases and MCR enzymes, Resistance-Nodulation-Division (RND) efflux pumps
further enrich the repertoire of AMR determinants in the cell envelope. These multi-protein assemblies
span the periplasm and remove many antibiotics (14, 15) rendering Gram-negative bacteria inherently
resistant to important existing antimicrobials.
Inhibition of AMR determinants has traditionally been achieved through the development of antibiotic
adjuvants. These molecules impair the function of resistance proteins and are used in combination with
existing antibiotics to eliminate challenging infections (4). Whilst this approach has proven successful
and has led to the deployment of several β-lactamase inhibitors that are used clinically (4), it cannot be
used to incapacitate multiple different AMR determinants. This is because antibiotic adjuvants bind to
the active site of a resistance enzyme and thus are only effective for specific protein families. To disrupt
AMR more broadly, new strategies have to be developed that target the biogenesis or stability, rather
than the activity, of resistance determinants. In this way, the formation of multiple resistance proteins
can be inhibited at once, instead of developing specific compounds that inactivate individual AMR
enzymes after they are already in place.
In extracytoplasmic environments protein stability largely relies on the formation of disulfide bonds
between cysteine residues (16, 17). Notably, in the cell envelope of Gram-negative bacteria this process
is performed by a single pathway, the DSB system, and more specifically by a single protein, the thiol
oxidase DsbA (18-22). DsbA has been shown to assist the folding of hundreds of proteins in the
periplasm (21, 23, 24) (Figure 1A), including a vast range of virulence factors (25, 26). As such,
inhibition of DSB proteins has been proposed as a promising broad-acting strategy to target bacterial
pathogenesis without impairing bacterial viability (19, 25-27). Nonetheless, the role of oxidative protein
folding in AMR has never been examined. Since several cell envelope AMR determinants contain
multiple cysteines (18, 28), we hypothesized that interfering with the function of DsbA, would not only
compromise bacterial virulence (27), but might also offer a broad approach to break resistance across
different mechanisms by affecting the stability of resistance proteins. Here we test this hypothesis by

219
investigating the contribution of disulfide bond formation to three of the most important resistance
mechanisms in the cell envelope of Enterobacteria: the breakdown of β-lactam antibiotics by β-
lactamases, polymyxin resistance arising from the production of MCR enzymes and intrinsic resistance
to multiple antibiotic classes due to RND efflux pumps. We find that all these resistance mechanisms
depend on DsbA and we demonstrate that when DsbA activity is chemically inhibited, resistance is
abrogated. Our findings prove that it is possible to simultaneously incapacitate multiple classes of AMR
determinants and therefore hold great promise for the development of next-generation therapeutic
approaches that would abolish resistance.

220
RESULTS
The activity of cysteine-containing resistance determinants is dependent on DsbA
DsbA has been shown to assist the folding of numerous periplasmic and surface-exposed proteins in
Gram-negative bacteria (Figure 1A) (25-27). As many AMR determinants also transit through the
periplasm, we postulated that inactivation of the DSB system may affect their folding, and therefore
impair their function. To test this, we first focused on resistance proteins that are present in the cell
envelope and contain two or more cysteine residues, since they may depend on the formation of
disulfide bonds for their stability and folding (18, 28). We selected a panel of twelve clinically important
β-lactamases from different Ambler classes (classes A, B and D), most of which are encoded on
plasmids (Table S1). The chosen enzymes represent different protein structures, belong to discrete
phylogenetic families (File S1) and have distinct hydrolytic activities ranging from the degradation of
penicillins and first, second and third generation cephalosporins (extended spectrum β-lactamases,
ESBLs) to the inactivation of last-resort β-lactams (carbapenemases). In addition to β-lactamases, we
selected five representative phosphoethanolamine transferases from throughout the MCR phylogeny
(Figure S1) to gain a comprehensive overview of the contribution of DsbA to the activity of these
colistin-resistance determinants.
We expressed our panel of 17 discrete resistance enzymes in an Escherichia coli K-12 strain and its
isogenic dsbA mutant and recorded minimum inhibitory concentration (MIC) values for β-lactam or
polymyxin antibiotics, as appropriate. We found that the absence of DsbA resulted in a substantial
decrease in MIC values (>2-fold) for all but one of the tested β-lactamases (Figure 1B, Figure S2). In
addition, deletion of dsbA led to clinically meaningful decreases in colistin MIC values for all MCR
enzymes (Figure 1C), especially given the narrow therapeutic window of this antibiotic (29, 30). More
specifically, expression of all MCR enzymes in our wild-type E.coli K-12 strain resulted in colistin
resistance, however in most cases absence of DsbA caused re-sensitization of the strain as defined by
the EUCAST breakpoint (E. coli strains with an MIC of 2 μg/mL or below are classified as susceptible)
(Figure 1C). For the only enzyme included in this screen that seemed unaffected by the absence of
DsbA, the SHV-27 β-lactamase, we performed the same experiment under temperature stress conditions
(at 42 °C rather than 37 °C). Under these conditions the lack of DsbA also resulted in a noticeable drop
in the cefuroxime MIC value (Figure S3).
Wild-type MIC values could be restored for all tested enzymes by complementation of dsbA (Figures
S4, S5). Moreover, since DsbA acts on its substrates post-translationally, we performed a series of
control experiments designed to assess whether the recorded effects were specific to the interaction of
the resistance proteins with DsbA, and not a result of a general inability of the dsbA mutant strain to
resist antibiotic stress. We found that no decreases in MIC values were observed for the aminoglycoside
antibiotic gentamicin, which is not affected by the tested enzymes (Figure 1B, Figure S6). Furthermore,
the β-lactam MIC values of strains harboring the empty-vector alone, or a plasmid encoding L2-1
(Figure 1B), a β-lactamase containing three cysteine residues, but no disulfide bond (PDB ID: 5NE1)
remained unchanged. Finally, to rule out the possibility that deletion of dsbA caused changes in
membrane permeability that might confound our results, we measured the permeability of the outer and
inner membrane of the dsbA mutant using the fluorescent dyes 1-N-phenylnaphthylamine (NPN) and
propidium iodide (PI), respectively and found it to be no different from that of the parental strain (Figure
S7).
Together, these results indicate that many cell envelope AMR determinants that contain more than one
cysteine residue are substrates of DsbA and that the process of disulfide bond formation is essential for
their activity.

221
The function of E. coli RND efflux pumps is compromised in the absence of DsbA
Unlike β-lactamases and MCR enzymes, none of the components of the six E. coli RND efflux pumps
contain periplasmic cysteine residues (31), thus they are not substrates of the DSB system. Nonetheless,
as DsbA assists the folding of approximately 300 extracytoplasmic proteins and, hence plays a central
role in maintaining the homeostasis of the cell envelope proteome (21, 23, 24), we wanted to assess
whether changes in periplasmic proteostasis that occur in its absence could indirectly influence efflux
pump function. To do this we determined the MIC values of three antibiotics that are RND efflux pump
substrates using E. coli MG1655, a model strain for efflux studies, its dsbA mutant and a mutant lacking
acrA, an essential component of the major E. coli RND pump AcrAB-TolC. MIC values for the dsbA
mutant were lower than for the parental strain for all tested substrate antibiotics, but not for the non-
substrate gentamicin (Figure 1D). As before, the observed phenotype could be reversed by
complementation of dsbA (Figure S8) and the recorded effects were not due to changes in membrane
permeability (Figure S9). The indirect effect of DsbA absence on efflux efficiency, although less
substantial than that measured for a mutant lacking acrA (Figure 1D), is robust and in agreement with
previous studies reporting that deletion of dsbA increases the sensitivity of E. coli to dyes like acridine
orange and pyronin Y (18), which are known substrates of AcrAB-TolC.
β-lactamases and MCR enzymes degrade or misfold in the absence of DsbA
To understand the underlying mechanisms that result in the decreased MIC values observed for the
dsbA mutant strains, we assessed the protein levels of a representative subset of β-lactamases (GES-1,
L1-1, KPC-3, FRI-1, OXA-4, OXA-10, OXA-198) and all tested MCR enzymes by immunoblotting.
When expressed in the dsbA mutant, all Ambler class A and B β-lactamases (Table S1), except GES-1
which we were not able to visualize by immunoblotting, exhibited drastically reduced protein levels
whilst the amount of the control enzyme L2-1 remained unaffected (Figure 2A). This suggests that
when these enzymes lack their disulfide bond, they are unstable and ultimately are degraded. We did
not detect any decrease in protein amounts for Ambler class D enzymes (Table S1, Figure 2B).
However, the hydrolytic activity of these β-lactamases was significantly lower in the dsbA mutant
(Figure 2C), suggesting a folding defect that leads to loss of function.
Like with class A and B β-lactamases, MCR enzymes were undetectable when expressed in a dsbA
mutant (Figure 3A) suggesting that their stability is severely compromised when they lack their
disulfide bonds. We further confirmed this by directly monitoring the lipid A profile of all MCR-
expressing strains with substantial MIC drops (i.e. strains expressing MCR-3, -4, -5 and -8, Figure 1C)
using MALDI-TOF mass spectrometry (Figure 3BC). MCR activity leads to the addition of
phosphoethanolamine to the lipid A portion of bacterial lipopolysaccharide (LPS), resulting in reduced
binding of colistin to LPS and thus resistance. In E. coli the major lipid A peak detected by mass
spectrometry is present at m/z 1796.2 (Figure 3B, first spectrum). This peak corresponds to hexa-acyl
diphosphoryl lipid A (native lipid A). The lipid A profile of E. coli MC1000 dsbA was identical to that
of the parental strain (Figure 3B, second spectrum). In the presence of MCR enzymes two additional
peaks were observed, at m/z 1821.2 and 1919.2 (Figure 3B, third spectrum). The peak at m/z 1919.2
corresponds to the addition of a phosphoethanolamine moiety to the phosphate group at position 1 of
native lipid A, and the peak at m/z 1821.2 corresponds to the addition of a phosphoethanolamine moiety
to the 4ˊ phosphate of native lipid A and the concomitant loss of the phosphate group at position 1 (32).
For dsbA mutants expressing MCR-3, -5 and -8 (Figure 3C), the peaks at m/z 1821.2 and m/z 1919.2
could no longer be detected, whilst the native lipid A peak at m/z 1796.2 remained unchanged (Figure

222
3B, fourth spectrum); dsbA mutants expressing MCR-4 retain some basal lipid A-modifying activity,
nonetheless this is not sufficient for this strain to efficiently evade colistin treatment (Figure 1C).
Together these data suggest that in the absence of DsbA, MCR enzymes are unstable (Figure 3A) and
therefore no longer able to efficiently catalyze the addition of phosphoethanolamine to native lipid A
(Figure 3BC); as a result, they cannot confer resistance to colistin (Figure 1C).
The data presented above validate our initial hypothesis. Absence of DsbA affects the stability and
folding of cysteine-containing resistance proteins and in most cases leads to drastically reduced levels
of the tested enzyme. As a result, and in agreement with the recorded decreases in MIC values (Figure
1BC), these folding defects impede the ability of AMR determinants to confer resistance. Therefore, by
compromising cell envelope protein folding we can impair a broad range of AMR proteins and abrogate
resistance across multiple mechanisms.
RND efflux pump function is compromised in the absence of DsbA due to altered periplasmic
proteostasis
As RND efflux pump proteins do not contain any disulfide bonds, the decreases in MIC values for
pump substrates in the absence of dsbA (Figure 1D) are likely mediated by additional cell-envelope
components. The protease DegP, previously found to be a DsbA substrate (20), seemed a promising
candidate for linking DsbA to efflux pump function. DegP degrades a range of misfolded
extracytoplasmic proteins including, but not limited to, subunits of higher order protein complexes and
proteins lacking their native disulfide bonds (33). We hypothesized that in a dsbA mutant the substrate
burden on DegP would be dramatically increased, whilst DegP itself would not function optimally due
to absence of its disulfide bond (34). Consequently, protein turn over in the cell envelope would not
occur efficiently. Since the essential RND efflux pump component AcrA needs to be cleared by DegP
when it becomes misfolded or nonfunctional (35), we expected that the reduced DegP efficiency in a
dsbA mutant would result in accumulation of nonfunctional AcrA in the periplasm, which would then
interfere with pump function. In agreement with our hypothesis we found that in the absence of DsbA
degradation of DegP occurred, reducing the pool of active enzyme (Figure 4A) (34). In addition, AcrA
accumulated to the same extent in a dsbA and in a degP mutant (Figure 4B), suggesting that in both
these strains AcrA was not efficiently cleared. Finally, no accumulation was detected for the outer-
membrane protein TolC, which is not a DegP substrate (Figure 4C) (36). Thus, in the absence of DsbA,
inefficient DegP-mediated periplasmic proteostasis impacts RND efflux pump function (Figure 1D)
through accumulation of AcrA that should have been degraded and removed from the cell envelope.
These results demonstrate that changes in cell envelope protein homeostasis have a profound effect on
protein function in this compartment, which could be exploited in the design of antibacterial strategies.
Here, disrupting periplasmic proteostasis by preventing disulfide bond formation indirectly impairs
efflux pump activity in addition to incapacitating direct substrates of DsbA, like β-lactamases and MCR
proteins. This allows us to simultaneously abrogate three distinct resistance mechanisms (Figure 4D).
DsbA is a tractable AMR target
DsbA is essential for the folding of many virulence factors. As such, inhibition of the DSB system has
been proposed as a promising anti-virulence strategy (25-27) and efforts have been made to develop
inhibitors for DsbA (37, 38), its redox partner DsbB (Figure 1A) (39) or both (40). These studies have
made the first steps towards the production of chemical compounds that inhibit the function of the DSB
proteins, providing us with a laboratory tool to test our approach against AMR.

223
4,5-dichloro-2-(2-chlorobenzyl)pyridazin-3-one, termed “compound 12” in Landeta et al. (39) is a
potent laboratory inhibitor of E. coli DsbB and its analogues from closely related organisms. Using this
molecule, we could chemically inhibit the function of the DSB system. We first tested the motility of
E. coli MC1000 in the presence of the inhibitor and found that cell were significantly less motile (Figure
S10), consistent with the fact that impairing DSB function prevents the formation of the flagellar P-ring
component FlgI (41, 42). Furthermore, we directly assessed the redox state of DsbA in the presence of
the compound to probe whether it was being re-oxidized by DsbB, a necessary step that occurs after
each round of oxidative protein folding and allows DsbA to remain active (Figure 1A). Under normal
growth conditions, DsbA was in its active oxidized form in the bacterial periplasm (i.e. C30 and C33
form a disulfide bond), showing that it was efficiently regenerated by DsbB (43) (Figure 5A). By
contrast, addition of the inhibitor to growing E. coli MC1000 cells resulted in accumulation of inactive
reduced DsbA, thus confirming that DsbB function was impeded (Figure 5A).
After testing the efficacy of the DsbB inhibitor, we proceeded to examine whether chemical inhibition
of the DSB system could be used to broadly impair the function of AMR determinants. We found that
addition of the compound during MIC testing phenocopied the effects of a dsbA deletion on β-lactamase
and MCR activity (Figure 5BCD) by determining MIC values for the latest generation β-lactam that
each β-lactamase can hydrolyze, and colistin, as appropriate The observed effects are not a result of
altered cell growth, as addition of the molecule does not affect the growth profile of the bacteria (Figure
S11A), in agreement with the fact that deletion of dsbA does not affect cell viability (Figure S11B).
Furthermore, the changes in the recorded MIC values are due solely to inhibition of the DSB system as
no additive effects on MIC values were observed when the dsbA mutant harboring a β-lactamase or mcr
gene was exposed to the compound (Figure S12).
Sensitization of clinical isolates to existing antibiotics can be achieved by chemical inhibition of DsbA
activity
Having shown that the DSB system is a tractable target in the context of AMR, we examined the effect
of chemical inhibition on several species of β-lactamase- and MCR-expressing Enterobacteria (Table
S2). Tested clinical isolates from four different species, including multidrug-resistant E. coli and
Citrobacter freundii strains, showed clinically relevant decreases in their MIC values to last-resort
antibiotics when their DSB system was chemically inhibited. In all but one case this led to sensitization
as defined by EUCAST breakpoints (Figure 6AB, Figure S13). In the one case where sensitization was
not achieved, chemical inhibition of the DSB system of an Enterobacter cloacae isolate expressing
FRI-1 caused a drastic reduction in the aztreonam MIC value by over 180 µg/mL, resulting in
intermediate resistance as defined by EUCAST. These results obtained using clinical strains provide
further validation of the significance of our data from heterologously expressed β-lactamase and MCR
enzymes in E. coli K-12 strains (Figure 1BC), and showcase the potential of this approach for clinical
applications.
Regarding intrinsic resistance mediated by RND efflux pumps, chloramphenicol is the only antibiotic
from the efflux pump substrates that were tested in this study that has a EUCAST breakpoint for Gram-
negative bacteria (E. coli strains with an MIC of 8 μg/mL or below are classified as sensitive). It is
notable that the MIC drop for this pump substrate (Figure 1D) caused by deletion of dsbA sensitized E.
coli to chloramphenicol (Figure 6C), showing that even the indirect effects of compromising disulfide
bond formation are potentially clinically important. Since mutations in marR that derepress MarA and
cause constitutive expression of AcrAB (44, 45) are observed in clinical isolates with increased efflux
(46), we recorded the chloramphenicol MIC for the dsbA mutant of an E. coli MG1655 marR strain and
found that sensitization to chloramphenicol occurred (Figure 6C) even when efflux pump components
were overexpressed (Figure S14).

224
To determine if our approach for Enterobacteria would be appropriate for other multidrug-resistant
pathogens we tested it on Pseudomonas aeruginosa. This bacterium has two DsbB analogues which are
functionally redundant (47). The chemical inhibitor used in this study has been shown to be effective
against DsbB1, but less effective against DsbB2 of P. aeruginosa PA14 (39), making it unsuitable for
MIC assays on P. aeruginosa clinical isolates. Nonetheless, deletion of dsbA1 in the multidrug-resistant
P. aeruginosa PA43417 clinical isolate expressing OXA-198, led to sensitization of this strain to the
antipseudomonal β-lactam piperacillin (Figure 6D), suggesting that targeting disulfide bond formation
could be useful for the sensitization of many more clinically important Gram-negative species.
Finally, to test our approach in an infection context we performed in vivo clearance assays using the
wax moth model Galleria mellonella (Figure 6E). Larvae were infected with the P. aeruginosa
PA43417 clinical isolate producing OXA-198 and its dsbA1 mutant and infections were treated once
with piperacillin at a final concentration below the EUCAST breakpoint. Neither deletion of dsbA1 nor
treatment with piperacillin was sufficient to clear the infection reliably when applied alone, although
the former led to a significant decrease in the recovered bacterial load due to the fact that absence of
the principal DsbA likely affects the virulence of the pathogen (48). However, treatment of the dsbA1
mutant with piperacillin resulted in a drastic (> 99% on average) reduction in bacterial load in the
infected larvae, in agreement with the fact that in the absence of DsbA the ability of OXA-198 to
hydrolyze β-lactams is impaired (Figure 1B, 2C). As OXA-198, in this case produced by a multi-drug
resistant clinical strain (Table S2 and Figure 6DE), is a broad-spectrum β-lactamase that cannot be
neutralized by classical β-lactamase inhibitors (Table S1) and piperacillin is a first-line antibiotic, these
results further highlight the promise of our approach for future clinical applications.

225
DISCUSSION
This work is the first report of a strategy capable of simultaneously impairing multiple types of AMR
determinants by compromising the function of a single target. By inhibiting DsbA, a non-essential cell
envelope protein which is unique to bacteria, we can overcome three entirely distinct resistance
mechanisms and sensitize critically important pathogens to multiple classes of existing antibiotics. This
proof of principle opens a new avenue to reverse AMR in Gram-negative organisms, through the
development of DsbA inhibitors that would function as broad-acting resistance breakers.
We have shown that targeting DsbA incapacitates broad-spectrum β-lactamases from three of the four
Ambler classes (class A, B and D, Figure 1B). This includes enzymes that are not susceptible to classical
β-lactamase inhibitors (Table S1), such as members of the KPC and OXA families, as well as metallo-
β-lactamases like L1-1 from the often pan-resistant organism Stenotrophomonas maltophilia. The
function of these proteins is impaired without a small molecule binding to their active site, unlike
currently-used β-lactamase inhibitors which often generate resistance (4). As DsbA dependence is
conserved within phylogenetic groups (see Figure S2, GES-1, -2, -11 and KPC-2, -3), based on the
number of enzymes belonging to the same phylogenetic family as the β-lactamases tested in this study
(File S1), we anticipate that a total of 149 discrete enzymes rely on DsbA for their stability and function.
DsbA is widely conserved (25), thus targeting the DSB system should not only compromise β-
lactamases in Enterobacteria, but as demonstrated by our experiments using a P. aeruginosa clinical
isolate (Figure 6DE), could also be a promising avenue for impairing the function of AMR determinants
expressed by other highly-resistant Gram-negative organisms. As such, together with the fact that
approximately 25% of β-lactamases found in pathogens and organisms capable of causing opportunistic
infections contain two or more cysteines (File S1), we expect many more clinically relevant β-
lactamases, beyond those already tested in this study, to depend on DsbA.
MCR enzymes are rapidly becoming a grave threat to the use of colistin (13), a drug of last resort often
needed for the treatment of multidrug-resistant infections (11). Currently, experimental inhibitors of
these proteins are sparse and poorly characterized (49). As all MCR members contain multiple disulfide
bonds, inhibition of the DSB system provides a broadly applicable solution for combating MCR-
mediated colistin resistance (Figure 1C and Figure 6B) that would likely extend to novel MCR proteins
emerging in the future. As for MCR enzymes, no clinically applicable efflux pump inhibitors have been
identified to date (50) despite many efforts to use these macromolecular assemblies as targets against
intrinsic resistance. Deletion of dsbA sensitizes the tested E. coli strain to chloramphenicol (Figure 6C)
offering a novel way of targeting RND efflux pumps. Efflux pumps containing AcrA-like components
are dependent on DegP for their homeostasis in the periplasm, and hence their function would be
compromised if DsbA was inhibited. This means that the generation of clinically useful DsbA inhibitors
to combat β-lactamase- and MCR-mediated resistance offers a new platform for exploring the potential
of efflux pump inhibition.
More generally, our findings demonstrate that cell envelope proteostasis pathways have significant yet
untapped potential for the development of novel antibacterial strategies. The example of the DSB
system presented here is particularly telling. This pathway, initially considered merely a housekeeping
system (51), plays a major role in clinically relevant bacterial niche adaptations. In addition to assisting
the folding of 40% of the cell-envelope proteome (23, 24), the DSB system is essential for virulence
(25, 26), has a key role in the formation and awakening of bacterial persister cells (52) and, as seen in
this work, is required for bacterial survival in the presence of a broad range of antibiotic compounds.
As shown in our in vivo experiments (Figure 6E), targeting such a system in Gram-negative pathogens
could lead to adjuvant approaches that inactivate multiple AMR determinants whilst simultaneously
incapacitating an arsenal of virulence factors. Therefore, this study not only lays the groundwork for
future clinical applications, such as the development of broad-acting antibiotic adjuvants, but also

226
serves as a paradigm for exploiting other accessible cell envelope proteostasis processes for the design
of next-generation therapeutic strategies.

227
MATERIALS AND METHODS
Reagents and bacterial growth conditions. Unless otherwise stated, chemicals and reagents were
acquired from Sigma Aldrich, growth media were purchased from Oxoid and antibiotics were obtained
from Melford Laboratories. Lysogeny broth (LB) (10 g/L NaCl) and agar (1.5% w/v) were used for
routine growth of all organisms at 37 °C with shaking at 220 RPM, as appropriate. Unless otherwise
stated, Mueller-Hinton (MH) broth and agar (1.5% w/v) were used for Minimum Inhibitory
Concentration (MIC) assays. Growth media were supplemented with the following, as required: 0.25
mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) (for strains harboring β-lactamase-encoding pDM1
plasmids), 0.5 mM IPTG (for strains harboring MCR-encoding pDM1 plasmids), 12.5 μg/mL
tetracycline, 100 μg/mL ampicillin, 50 μg/mL kanamycin, 10 μg/mL gentamicin, 33 μg/mL
chloramphenicol and 50 μg/mL streptomycin.
Construction of plasmids and bacterial strains. Bacterial strains, plasmids and oligonucleotides used
in this study are listed in Tables S3, S4 and S5, respectively. DNA manipulations were conducted using
standard methods. KOD Hot Start DNA polymerase (Merck) was used for all PCR reactions according
to the manufacturer’s instructions, oligonucleotides were synthesized by Sigma Aldrich and restriction
enzymes were purchased from New England Biolabs. All constructs were DNA sequenced and
confirmed to be correct before use.
Genes for β-lactamase and MCR enzymes were amplified from genomic DNA extracted from clinical
isolates (Table S6) with the exception of mcr-3 and mcr-8, which were synthesized by GeneArt Gene
Synthesis (ThermoFisher Scientific). β-lactamase and MCR genes were cloned into the IPTG-inducible
plasmid pDM1 using primers P1-P36. pDM1 (GenBank accession number MN128719) was constructed
from the p15A-ori plasmid pACYC184 (53) to contain the Lac repressor, the Ptac promoter, an
optimized ribosome binding site and a multiple cloning site (NdeI, SacI, PstI, KpnI, XhoI and XmaI)
inserted into the NcoI restriction site of pACYC184. All StrepII-tag fusions of β-lactamase and MCR
enzymes (constructed using primers P1, P3, P9, P11, P13, P15, P17, P21, P23, P25, P27, P29, P37, P38
and P41-P50) have a C-terminal StrepII tag (GSAWSHPQFEK) except for OXA-4, where an N-
terminal StrepII tag was inserted between the periplasmic signal sequence and the body of the protein
using the primer pairs P7/P40, P39/P9 and P7/P8. Plasmids encoding ges-1, kpc-3 and mcr-3.2 were
obtained by performing QuickChange mutagenesis on pDM1 constructs encoding ges-5, kpc-2 and mcr-
3, respectively (primers P31-P36).
E. coli gene mutants were constructed using a modified lambda-Red recombination method, as
previously described (54) (primers P53-P62). To complement the dsbA mutant, a DNA fragment
consisting of dsbA preceded by the Ptac promoter was inserted into the NotI/XhoI sites of pGRG25
(primers P51/P52) and was reintroduced into the E. coli chromosome at the attTn7 site, as previously
described (55). The dsbA1 mutant of the Pseudomonas aeruginosa PA43417 clinical isolate was
constructed by allelic exchange, as previously described (56). Briefly, the dsbA1 gene area of P.
aeruginosa PA43417 (including the dsbA1 gene and 600 bp on either side of this gene) was amplified
(primers P63/P64) and the obtained DNA was sequenced to allow for accurate primer design for the
ensuing cloning step. Subsequently, 500-bp DNA fragments upstream and downstream of the dsbA1
gene were amplified using P. aeruginosa PA43417 genomic DNA (primers P65/P66 (upstream) and
P67/P68 (downstream)). A fragment containing both of these regions was obtained by overlapping PCR
(primers P65/P68) and inserted into the XbaI/BamHI sites of pKNG101. The suicide vector pKNG101
(57) is not replicative in P. aeruginosa; it was maintained in E. coli CC118λpir and mobilized into P.
aeruginosa PA43417 by triparental conjugation.
Minimum inhibitory concentration (MIC) assays. Unless otherwise stated, antibiotic MIC assays were
carried out in accordance with the EUCAST recommendations using E-test strips (BioMérieux).

228
Briefly, overnight cultures of each strain to be tested were standardized to OD600 0.063 in 0.85% NaCl
(equivalent to McFarland standard 0.5) and distributed evenly across the surface of MH agar plates. E-
test strips were placed on the surface of the plates, evenly spaced, and the plates were incubated for 18-
24 hours at 37 °C. MICs were read according to the manufacturer’s instructions. β-lactam MICs were
also determined using the Broth Microdilution (BMD) method, as required. Briefly, a series of antibiotic
concentrations was prepared by two-fold serial dilution in MH broth in a clear-bottomed 96-well
microtiter plate (Corning). When used, tazobactam was included at a fixed concentration of 4 μg/mL in
every well, in accordance with the EUCAST guidelines. The strain to be tested was added to the wells
at approximately 5 x 104 colony forming units (CFU) per well and plates were incubated for 18-24 hours
at 37 °C. The MIC was defined as the lowest antibiotic concentration with no visible bacterial growth
in the wells. All colistin sulphate MIC assays were performed using the BMD method as described
above except that instead of two-fold serial dilutions, the following concentrations of colistin (Acros
Organics) were prepared individually in MH broth: 4 μg/mL, 3.5 μg/mL, 3 μg/mL, 2.5 μg/mL, 2 μg/mL,
1.5 μg/mL, 1 μg/mL, 0.5 μg/mL.
The covalent DsbB inhibitor 4,5-dichloro-2-(2-chlorobenzyl)pyridazin-3-one (39) was used to
chemically impair the function of the DSB system. Inactivation of DsbB results in abrogation of DsbA
function (43) only in media free of small-molecule oxidants (41). Therefore, MIC assays involving
chemical inhibition of the DSB system were performed using M63 broth (15.1 mM (NH4)2SO4, 100
mM KH2PO4, 1.8 mM FeSO4.7H2O, adjusted to pH 7.2 with KOH) and agar (1.5% w/v) supplemented
with 1 mM MgSO4, 0.02% w/v glucose, 0.005% w/v thiamine, 31 µM FeCl3.6H2O, 6.2 μM ZnCl2, 0.76
µM CuCl2.2H2O, 1.62 µM H3BO3, 0.081 µM MnCl2.4H2O, 84.5 mg/L alanine, 19.5 mg/L arginine, 91
mg/L aspartic acid, 65 mg/L glutamic acid, 78 mg/L glycine, 6.5 mg/L histidine, 26 mg/L isoleucine,
52 mg/L leucine, 56.34 mg/L lysine, 19.5 mg/L methionine, 26 mg/L phenylalanine, 26 mg/L proline,
26 mg/L serine, 6.5 mg/L threonine, 19.5 mg/L tyrosine, 56.34 mg/L valine, 26 mg/L tryptophan, 26
mg/L asparagine and 26 mg/L glutamine. CaCl2 was also added at a final concentration of 0.223 mM
for colistin sulfate MIC assays. Either DMSO (vehicle control) or the covalent DsbB inhibitor 4,5-
dichloro-2-(2-chlorobenzyl)pyridazin-3-one (final concentration of 50 μM) (Enamine) (39) were added
to the M63 medium, as required. The strain to be tested was added at an inoculum that recapitulated the
MH medium MIC values obtained for that strain.
SDS-PAGE analysis and immunoblotting. Samples for immunoblotting were prepared as follows.
Strains to be tested were grown on LB or MH agar plates as lawns in the same manner as for MIC
assays described above. Bacteria were collected using an inoculating loop and resuspended in 0.85%
NaCl or LB to OD600 2.0 (except for strains expressing OXA-4, where OD600 6.0 was used). For strains
expressing β-lactamase enzymes, the cell suspensions were spun at 10,000 x g for 10 minutes and
bacterial pellets were lysed by addition of BugBuster Master Mix (Merck Millipore) for 25 minutes at
room temperature with gentle agitation. Subsequently, lysates were spun at 10,000 x g for 10 minutes
at 4 °C and the supernatant was added to 4 x Laemmli buffer. For strains expressing MCR enzymes cell
suspensions were directly added to 4 x Laemmli buffer, while for E. coli MG1655 and its mutants, cells
were lysed as above and lysates were added to 4 x Laemmli buffer. All samples were boiled for 5
minutes before separation by SDS-PAGE.
Unless otherwise stated, SDS-PAGE analysis was carried out using 10% BisTris NuPAGE gels
(ThermoFisher Scientific) using MES/SDS running buffer prepared according to the manufacturer’s
instructions and including pre-stained protein markers (SeeBlue Plus 2, ThermoFisher Scientific).
Proteins were transferred to Amersham Protran nitrocellulose membranes (0.45 µm pore size, GE Life
Sciences) using a Trans-Blot Turbo transfer system (Bio-Rad) before blocking in 3% w/v Bovine Serum
Albumin (BSA)/TBS-T (0.1 % v/v Tween 20) or 5% w/v skimmed milk/TBS-T and addition of primary
and secondary antibodies. The following primary antibodies were used in this study: Strep-Tactin-HRP
conjugate (Iba Lifesciences) (dilution 1:3,000 in 3 w/v % BSA/TBS-T), Strep-Tactin-AP conjugate (Iba

229
Lifesciences) (dilution 1:3,000 in 3 w/v % BSA/TBS-T), rabbit anti-DsbA antibody (dilution 1:1,000
in 5 w/v % skimmed milk/TBS-T), rabbit anti-AcrA antibody (dilution 1:10,000 in 5 w/v % skimmed
milk/TBS-T), rabbit anti-TolC antibody (dilution 1:5,000 in 5 w/v % skimmed milk/TBS-T), rabbit
anti-HtrA1 (DegP) antibody (Abcam) (dilution 1:1,000 in 5 w/v % skimmed milk/TBS-T) and mouse
anti-DnaK 8E2/2 antibody (Enzo Life Sciences) (dilution 1:10,000 in 5% w/v skimmed milk/TBS-T).
The following secondary antibodies were used in this study: goat anti-rabbit IgG-AP conjugate (Sigma
Aldrich) (dilution 1:6,000 in 5% w/v skimmed milk/TBS-T), goat anti-rabbit IgG-HRP conjugate
(Sigma Aldrich) (dilution 1:6,000 in 5% w/v skimmed milk/TBS-T), goat anti-mouse IgG-AP conjugate
(Sigma Aldrich) (dilution 1:6,000 in 5% w/v skimmed milk/TBS-T) and goat anti-mouse IgG-HRP
conjugate (Sigma Aldrich) (dilution 1:6,000 in 5% w/v skimmed milk/TBS-T). Membranes were
washed three times for 5 minutes with TBS-T prior to development. Development for AP conjugates
was carried out using a SigmaFast BCIP/NBT tablet, while HRP conjugates were visualized with the
Novex ECL HRP chemiluminescent substrate reagent kit (ThermoFisher Scientific) or the Luminata
Crescendo chemiluminescent reagent (Merck) using a Gel Doc XR+ Imager (Bio-Rad).
β-lactam hydrolysis assay. β-lactam hydrolysis measurements were carried out using the chromogenic
β-lactam nitrocefin (Abcam). Briefly, overnight cultures of strains to be tested were centrifugated,
pellets were weighed and resuspended in 150 μL of 100 mM sodium phosphate buffer (pH 7.0) per 1
mg of wet-cell pellet, and cells were lysed by sonication. For strains harboring pDM1, pDM1-blaL2-1,
pDM1-blaOXA-10 and pDM1-blaGES-1, lysates corresponding to 0.34 mg of bacterial pellet were
transferred into clear-bottomed 96-well microtiter plates (Corning). For strains harboring pDM1-
blaOXA-4 and pDM1-blaOXA-198, lysates corresponding to 0.2 mg and 0.014 mg of bacterial pellet were
used, respectively. In all cases, nitrocefin was added at a final concentration of 400 μM and the final
reaction volume was made up to 100 μL using 100 mM sodium phosphate buffer (pH 7.0). Nitrocefin
hydrolysis was monitored at 25 °C by recording absorbance at 490 nm at 60-second intervals for 15
minutes using an Infinite M200 Pro microplate reader (Tecan). The amount of nitrocefin hydrolyzed by
each lysate in 15 minutes was calculated using a standard curve generated by acid hydrolysis of
nitrocefin standards.
NPN uptake assay. 1-N-phenylnaphthylamine (NPN) (Acros Organics) uptake assays were performed
as described by Helander & Mattila-Sandholm (58). Briefly, mid-log phase cultures of strains to be
tested were diluted to OD600 0.5 in 5 mM HEPES (pH 7.2) before transfer to clear-bottomed 96-well
microtiter plates (Corning) and addition of NPN at a final concentration of 10 μM. Colistin sulphate
(Acros Organics) was included at a final concentration of 0.5 μg/mL, as required. Immediately after the
addition of NPN, fluorescence was measured at 60-second intervals for 10 minutes using an Infinite
M200 Pro microplate reader (Tecan); the excitation wavelength was set to 355 nm and emission was
recorded at 405 nm.
PI uptake assay. Exponentially-growing (OD600 0.4) E. coli strains harboring pUltraGFP-GM (59) were
diluted to OD600 0.1 in phosphate buffered saline (PBS) (pH 7.4) and cecropin A was added to a final
concentration of 20 μM, as required. Cell suspensions were incubated at room temperature for 30
minutes before centrifugation and resuspension of the pellets in PBS. Propidium iodide (PI) was then
added at a final concentration of 3 μM. Suspensions were incubated for 10 minutes at room temperature
and analyzed on a two-laser, four color BD FACSCalibur flow cytometer (BD Biosciences). 50,000
events were collected for each sample and data were analyzed using FlowJo v.10.0.6 (Treestar).
MALDI-TOF Mass spectrometry. Lipid A profiles of strains to be tested were determined using intact
bacteria, as previously described (60). The peak for E. coli native lipid A is detected at m/z 1796.2,
whereas the lipid A profiles of strains expressing functional MCR enzymes have two additional peaks,
at m/z 1821.2 and 1919.2. These peaks result from MCR-mediated modification of native lipid A
through addition of phosphoethanolamine moieties (32). The ratio of modified to unmodified lipid A

230
was calculated by summing the intensities of the peaks at m/z 1821.2 and 1919.2 and dividing this value
by the intensity of the native lipid A peak at m/z 1796.2.
Motility assay. 500 μL of overnight culture of each strain to be tested were centrifuged and the pellets
were washed in M63 broth before resuspension in the same medium to achieve a final volume of 25
μL. Bacterial motility was assessed by growth in M63 medium containing 0.25% w/v agar
supplemented as described above. DMSO (vehicle control) or the covalent DsbB inhibitor 4,5-dichloro-
2-(2-chlorobenzyl)pyridazin-3-one (final concentration of 50 μM) (Enamine) were added to the
medium, as required. 1 μL of the washed cell suspension was inoculated into the center of a 90 mm
diameter agar plate, just below the surface of the semi-solid medium. Plates were incubated at 37 °C in
a humidified environment for 16-18 hours and growth halo diameters were measured.
AMS labelling. Bacterial strains to be tested were grown for 18 hours in M63 broth supplemented as
described above. DMSO (vehicle control) or the covalent DsbB inhibitor 4,5-dichloro-2-(2-
chlorobenzyl)pyridazin-3-one (final concentration of 50 μM) (Enamine) were added to the medium, as
required. Cultures were standardized to OD600 2.0 in M63 broth, were spun at 10,000 x g for 10 minutes
and bacterial pellets were lysed by addition of BugBuster Master Mix (Merck Millipore) for 25 minutes
at room temperature with gentle agitation. Subsequently, lysates were spun at 10,000 x g for 10 minutes
at 4 °C prior to reaction with 4-acetamido-4ˊ-maleimidyl-stilbene-2,2ˊ-disulfonic acid (AMS)
(ThermoFisher Scientific). AMS alkylation was performed by vortexing the lysates in 15 mM AMS,
50 mM Tris-HCl, 3% w/v SDS and 3 mM EDTA (pH 8.0) for 30 minutes at 25 °C, followed by
incubation at 37 °C for 10 minutes. SDS-PAGE analysis and immunoblotting was carried out as
described above, except that 12% BisTris NuPAGE gels (ThermoFisher Scientific) and MOPS/SDS
running buffer were used. DsbA was detected using a rabbit anti-DsbA primary antibody and an AP-
conjugated secondary antibody, as described above.
Bacterial growth assays. To assess the effect of DSB system inhibition of the growth of E. coli,
overnight cultures of the strains to be tested were centrifuged and the pellets were washed in M63 broth
before transfer to clear-bottomed 96-well microtiter plates (Corning) at approximately 5 x 107 CFU/well
(starting OD600 ~ 0.03). M63 broth supplemented as described above was used as a growth medium.
DMSO (vehicle control) or the covalent DsbB inhibitor 4,5-dichloro-2-(2-chlorobenzyl)pyridazin-3-
one (final concentration of 50 μM) (Enamine) were added to the medium, as required. Plates were
incubated at 37 °C with orbital shaking (amplitude 3 mm, equivalent to ~ 220 RPM) and OD600 was
measured at 900-second intervals for 18 hours using an Infinite M200 Pro microplate reader (Tecan).
The same experimental setup was also used for recording growth curves of E. coli strains and their
isogenic mutants, except that overnight cultures of the strains to be tested were diluted 1:100 into clear-
bottomed 96-well microtiter plates (Corning) (starting OD600 ~ 0.01) and that LB was used as the growth
medium.
In vivo clearance assay. The wax moth model Galleria mellonella was used for in vivo clearance assays
(61). Individual G. mellonella larvae were randomly allocated to experimental groups; no masking was
used. Overnight cultures of the strains to be tested were standardized to OD600 1.0. Suspensions were
centrifuged and the pellets were washed three times in PBS and serially diluted. 10 μl of the 10–5 dilution
of each bacterial suspension were injected into the last right abdominal proleg of 3 to 5 G. mellonella
larvae per condition; a second, equal-size group of larvae were injected with PBS as negative control.
3 hours after infection, larvae were injected with 13 μl of piperacillin to a final concentration of 12
μg/mL in the last left abdominal proleg. 24 hours after infection larvae were euthanized and macerated
individually in 1 ml of PBS by vortexing for 15 minutes. The larval suspension was then serially diluted
and 20 μl of each dilution plated on Pseudomonas Isolation Agar. Plates were incubated at 37 °C for 16
hours before CFU counting.

231
Statistical analysis of experimental data. The total numbers of performed biological experiments and
technical repeats are mentioned in the figure legend of each display item. Biological replication refers
to completely independent repetition of an experiment using different biological and chemical
materials. Technical replication refers to independent data recordings using the same biological sample.
For MIC assays, according to common practice, we show representative MIC values from one of the
performed biological experiments. For all other assays, statistical analysis was performed in GraphPad
PRISM v8.0.2 using an unpaired T-test with Welch’s correction, a one-way ANOVA with correction
for multiple comparisons, or a Kruskal-Wallis test with correction for multiple comparisons, as
appropriate. Statistical significance was defined as p < 0.05. Outliers were defined as any technical
repeat >2 SD away from the average of the other technical repeats within the same biological
experiment. Such data were excluded and all remaining data were included in the analysis. Detailed
information for each figure is provided below:
Figure 2C: unpaired T-test with Welch’s correction; n=3; 3.621 degrees of freedom, t-value=0.302,
p=0.7792 (non-significance) (for pDM1 strains); 3.735 degrees of freedom, t-value=0.4677, p=0.666
(non-significance) (for pDM1-blaL2-1 strains); 2.273 degrees of freedom, t-value=5.069, p=0.0281
(significance) (for pDM1-blaGES-1 strains); 2.011 degrees of freedom, t-value=6.825, p=0.0205
(significance) (for pDM1-blaOXA-4 strains); 2.005 degrees of freedom, t-value=6.811, p=0.0208
(significance) (for pDM1-blaOXA-10 strains); 2.025 degrees of freedom, t-value=5.629, p=0.0293
(significance) (for pDM1-blaOXA-198 strains)
Figure 3C: one-way ANOVA with Tukey’s multiple comparison test; n=4; 24 degrees of freedom; F
value=21.00; p=0.000000000066 (for pDM1-mcr-3 strains), p=0.0004 (for pDM1-mcr-4 strains),
p=0.000000000066 (for pDM1-mcr-5 strains), p=0.00066 (for pDM1-mcr-8 strains)
Figure 6E: Kruskal-Wallis test with Dunn’s multiple comparisons test; n=8; Kruskal-Wallis H=25.24,
3 degrees of freedom; p=<0.0001. For multiple comparisons, p=0.0029 (P. aeruginosa versus P.
aeruginosa dsbA1), p=<0.0001 (P. aeruginosa versus P. aeruginosa dsbA1 treated with piperacillin),
p=0.0369 (P. aeruginosa treated with piperacillin versus P. aeruginosa dsbA1 treated with piperacillin)
Figure S7A: one-way ANOVA with Bonferroni’s multiple comparison test; n=3; 6 degrees of freedom;
F value=39.22; p=0.0007 (significance), p=0.99 (non-significance)
Figure S7B: one-way ANOVA with Bonferroni’s multiple comparison test; n=3; 6 degrees of freedom;
F value=61.84; p=0.0002 (significance), p=0.99 (non-significance)
Figure S9A: one-way ANOVA with Bonferroni’s multiple comparison test; n=3; 6 degrees of freedom;
F value=261.4; p=0.00000055 (significance), p=0.0639 (non-significance)
Figure S9B: one-way ANOVA with Bonferroni’s multiple comparison test; n=3; 6 degrees of freedom;
F value=77.49; p=0.0001 (significance), p=0.9999 (non-significance)
Figure S10B: one-way ANOVA with Bonferroni’s multiple comparison test; n=3; 6 degrees of freedom;
F value=1878; p=0.000000002 (significance)
Bioinformatics. The following bioinformatics analyses were performed in this study. Short scripts and
pipelines were written in perl (version 5.18.2) and executed on macOS Sierra 10.12.5.
β-lactamase enzymes. All available protein sequences of β-lactamases were downloaded from
http://www.bldb.eu (62). Sequences were clustered using the ucluster software with a 90% identity
threshold and the cluster_fast option (usearch v.7.0 (63)); the centroid of each cluster was used as a
cluster identifier for every sequence. All sequences were searched for the presence of cysteine residues
using a perl script. Proteins with two or more cysteines after the first 30 amino acids of their primary
sequence were considered potential substrates of the DSB system for organisms where oxidative protein
folding is carried out by DsbA and provided that translocation of the β-lactamase outside the cytoplasm
is performed by the Sec system. The first 30 amino acids of each sequence were excluded to avoid
considering cysteines that are part of the signal sequence mediating the translocation of these enzymes
outside the cytoplasm. The results of the analysis can be found in File S1.

232
MCR enzymes. E. coli MCR-1 (AKF16168.1) was used as a query in a blastp (64) search limited to
Proteobacteria on the NCBI Reference Sequence (RefSeq) proteome database (21-04-2019) (evalue <
10e-5). 17,503 hit sequences were retrieved and clustered using the ucluster software with a 70%
identity threshold and the cluster_fast option (usearch v.7.0 (63)). All centroid sequences were retrieved
and clustered again with a 20% identity threshold and the cluster_fast option. Centroid sequences of all
clusters comprising more than five sequences (809 sequences retrieved) along with the sequences of
the five MCR enzymes tested in this study were aligned using muscle (65). Sequences which were
obviously divergent or truncated were manually eliminated and a phylogenetic tree was built from a
final alignment comprising 781 sequences using fasttree with the wag substitution matrix and default
parameters (63). The assignment of each protein sequence to a specific group was done using
hmmsearch (HMMER v.3.1b2)(66) with Hidden Markov Models built from confirmed sequences of
MCR-like and EptA-like proteins.
Data availability. All data generated during this study that support the findings are included in the
manuscript or in the Supplementary Information. All materials are available from the corresponding
author upon request.

233
ACKNOWLEDGEMENTS: We thank J. Rowley for assistance with flow cytometry, IHMA Inc.
Schaumburg for the kind gift of the E. coli 1144230 isolate and J. Beckwith, F. Alcock and V. Koronakis
for the kind gifts of the anti-DsbA, the anti-AcrA and the anti-TolC antibodies, respectively. This study
was funded by the MRC Career Development Award MR/M009505/1 (to D.A.I.M.), the institutional
BBSRC-DTP studentships BB/M011178/1 (to N.K.) and BB/M01116X/1 (to H.L.P.), the BBSRC
David Philips Fellowship BB/M02623X/1 (to J.M.A.B.), the ISSF Wellcome Trust grant
105603/Z/14/Z (to G.L.-M.), the Brunel Research Innovation and Enterprise Fund, Innovate UK and
British Society for Antimicrobial Chemotherapy grants 2018-11143, 37800 and BSAC-2018-0095,
respectively (to R.R.MC) and the Swiss National Science Foundation Postdoc Mobility and Ambizione
Fellowships P300PA_167703 and PZ00P3_180142, respectively (to D.G.).
AUTHOR CONTRIBUTIONS: R.C.D.F. and D.A.I.M. designed the research. R.C.D.F. and N.K.
performed most of the experiments. D.B. performed colistin MIC assays and prepared samples for
MALDI-TOF analysis. P.B. provided genetic tools and advice on P. aeruginosa molecular biology.
A.A.A.A. performed β-lactam MIC assays. L.D. provided laboratory materials and strains. H.E.M.,
H.L.P. and J.M.A.B. constructed strains and provided advice on RND efflux pump biology and
experimental design. G.L.-M. performed MALDI-TOF experiments and analyzed the data. E.M and
R.R.MC performed in vivo clearance assays. D.G. performed in silico analyses and advised on several
aspects of the project. R.C.D.F. and D.A.I.M. wrote the manuscript with input from all authors.
D.A.I.M. directed the project.
DECLARATION OF INTERESTS: The authors declare no competing interests.

234
FIGURES
Figure 1. Several antimicrobial resistance mechanisms depend on disulfide bond
formation. (A) DsbA introduces disulfide bonds into extracytoplasmic proteins containing two
or more cysteine residues. After each round of oxidative protein folding, DsbA is regenerated
by the quinone (Q)-containing protein DsbB, which in turn transfers the reducing equivalents
to the respiratory chain (RC) (51). DsbA substrates (in dark blue) are distributed throughout
the extracytoplasmic space of Gram-negative bacteria. Disulfides are introduced to 1) soluble
periplasmic proteins (e.g. alkaline phosphatase, β-lactamases (18)), 2) periplasmic domains of
inner-membrane proteins (e.g LptA-like enzymes (28)), 3) periplasmic domains of outer-

235
membrane proteins (e.g. RcsF (19)), 4) outer-membrane proteins (e.g. OmpA, LptD (19, 25)),
5) secreted proteins (e.g. toxins or enzymes (25)), 6-9) protein components of macromolecular
assemblies like secretion systems, pili or flagella (25) (e.g. 6) GspD, 7) EscC, 8) BfpA, 9)
FlgI); all examples are E. coli proteins with the exception of LptA. (B) β-lactam minimum
inhibitory concentration (MIC) values for E. coli MC1000 expressing diverse disulfide-bond-
containing β-lactamases (Ambler classes A, B and D) are substantially reduced in the absence
of DsbA (MIC fold changes: > 2, fold change of 2 is indicated by the black dotted lines); no
effect is observed for SHV-27, which is further discussed in Figure S3. DsbA dependence is
conserved within phylogenetic groups (see Figure S2, GES-1, -2, -11 and KPC-2, -3). No
changes in MIC values are observed for the aminoglycoside antibiotic gentamicin (white bars)
confirming that absence of DsbA does not compromise the general ability of this strain to resist
antibiotic stress. No changes in MIC values are observed for strains harboring the empty vector
control (pDM1) or those expressing the class A β-lactamase L2-1, which contains three
cysteines but no disulfide bond (top row). Graphs show MIC fold changes for β-lactamase-
expressing E. coli MC1000 and its dsbA mutant and are representative of three biological
experiments each conducted as a single technical repeat; the MIC values used to generate this
panel are presented in Figure S2. (C) Colistin MIC values for E. coli MC1000 expressing
diverse MCR enzymes (Figure S1) are substantially reduced in the absence of DsbA. Graphs
show MIC values (µg/mL) from four biological experiments, each conducted in technical
quadruplicate, to demonstrate the robustness of the observed effects. Gentamicin control data
are presented in Figure S6. (D) Deletion of dsbA reduces the erythromycin, chloramphenicol
and nalidixic acid MIC values for E. coli MG1655, but no effects are detected for the non-
substrate antibiotic gentamicin. The essential pump component AcrA serves as a positive
control. Graphs show MIC values (µg/mL) and are representative of three biological
experiments, each conducted as a single technical repeat.

236
Figure 2. β-lactamase enzymes from most classes become unstable in the absence of DsbA.
(A) Protein levels of disulfide-bond-containing Ambler class A and B β-lactamases are
drastically reduced when these enzymes are expressed in E. coli MC1000 dsbA; the amount of
the control enzyme L2-1 is unaffected. (B) Protein levels of Class D disulfide-bond-containing
β-lactamases are unaffected by the absence of DsbA. OXA-4 is detected as two bands at ~ 28
kDa. For panels (A) and (B) protein levels of StrepII-tagged β-lactamases were assessed using
a Strep-Tactin-AP conjugate or a Strep-Tactin-HRP conjugate. A representative blot from three
biological experiments, each conducted as a single technical repeat, is shown; molecular weight
markers (M) are on the left, DnaK was used as a loading control and solid black lines indicate
where the membrane was cut. (C) The hydrolytic activities of the tested Class D β-lactamases
and of the Class A enzyme GES-1, which could not be detected by immunoblotting, are

237
significantly reduced in the absence of DsbA. The hydrolytic activities of strains harboring the
empty vector or expressing the control enzyme L2-1 show no dependence on DsbA. n=3 (each
conducted in technical triplicate), table shows means ±SD, significance is indicated by * = p <
0.05, ns = non-significant.
238
Figure 3. MCR enzymes become unstable in the absence of DsbA. (A) The amounts of
MCR proteins are drastically reduced when they are expressed in E. coli MC1000 dsbA; the
red arrow indicates the position of the MCR-specific bands. Protein levels of StrepII-tagged
MCR enzymes were assessed using a Strep-Tactin-AP conjugate. A representative blot from
three biological experiments, each conducted as a single technical repeat, is shown; molecular
weight markers (M) are on the left, DnaK was used as a loading control and solid black lines
indicate where the membrane was cut. (B) The ability of MCR enzymes to transfer
phoshoethanolamine to the lipid A portion of LPS is either entirely abrogated or significantly
reduced in the absence of DsbA. This panel shows representative MALDI-TOF mass spectra
of unmodified and MCR-modified lipid A in the presence and absence of DsbA. In E. coli
MC1000 and MC1000 dsbA the major peak for native lipid A peak is detected at m/z 1796.2
(first and second spectrum, respectively). In the presence of MCR enzymes (E. coli MC1000
expressing MCR-3 is shown as a representative example), two additional peaks are observed,
at m/z 1821.2 and 1919.2 (third spectrum). For dsbA mutants expressing MCR enzymes (E.
coli MC1000 dsbA expressing MCR-3 is shown), these additional peaks are not present, whilst
the native lipid A peak at m/z 1796.2 remains unchanged (fourth spectrum). Mass spectra are
representative of the data generated from four biological experiments each conducted as a
technical duplicate. (C) Quantification of the intensities of the lipid A peaks recorded by
MALDI-TOF mass spectrometry for all tested MCR-expressing strains. n=4 (each conducted

239
in technical duplicate), table shows means ±SD, significance is indicated by *** = p < 0.001
or **** = p <0.0001.

240
Figure 4. (A,B, C) RND efflux pump function is impaired in the absence of DsbA due to
accumulation of unfolded AcrA resulting from insufficient DegP activity. (A) In the
absence of DsbA the pool of active DegP is reduced. In E. coli MG1655 (lane 1), DegP is
detected as a single band, corresponding to the intact active enzyme. In E. coli MG1655 dsbA
(lane 2), an additional lower molecular weight band of equal intensity is present, indicating
that DegP is degraded in the absence of its disulfide bond (20, 34). DegP protein levels were
assessed using an anti-DegP primary antibody and an HRP-conjugated secondary antibody. E.
coli MG1655 degP was used as a negative control for DegP detection (lane 3); the red arrow
indicates the position of intact DegP. (B) The RND pump component AcrA accumulates to the
same extent in the E. coli MG1655 dsbA and degP strains, indicating that in both strains protein
clearance is affected. AcrA protein levels were assessed using an anti-AcrA primary antibody
and an HRP-conjugated secondary antibody. E. coli MG1655 acrA was used as a negative
control for AcrA detection; the red arrow indicates the position of the AcrA band. (C) TolC,
the outer-membrane channel of the AcrAB pump, does not accumulate in a dsbA or a degP
mutant. TolC is not a DegP substrate (36), hence similar TolC protein levels are detected in E.
coli MG1655 (lane 1) and its dsbA (lane 2) and degP (lane 3) mutants. TolC protein levels were
assessed using an anti-TolC primary antibody and an HRP-conjugated secondary antibody. E.
coli MG1655 tolC was used as a negative control for TolC detection (lane 4); the red arrow
indicates the position of the bands originating from TolC. For all panels a representative blot

241
from three biological experiments, each conducted as a single technical repeat, is shown;
molecular weight markers (M) are on the left, DnaK was used as a loading control and solid
black lines indicate where the membrane was cut. (D) Impairing disulfide bond formation
in the cell envelope simultaneously incapacitates three distinct classes of AMR
determinants. (Left) When DsbA is present, i.e. when disulfide bond formation occurs,
degradation of β-lactam antibiotics by β-lactamases (marked “bla”), modification of lipid A by
MCR proteins and active efflux of RND pump substrates lead to resistance. The major E. coli
RND efflux pump AcrAB-TolC is depicted in this schematic as a characteristic example.
(Right) In the absence of DsbA, i.e. when the process of disulfide bond formation is impaired,
most cysteine-containing β-lactamases as well as MCR proteins are unstable and degrade,
making bacteria susceptible to β-lactams and colistin. Absence of DsbA also affects
proteostasis in the cell envelope which results in reduced clearance of nonfunctional AcrA-like
proteins (termed “AcrA'” and depicted in dark red color) by periplasmic proteases. Insufficient
clearance of these damaged AcrA components from the pump complex makes efflux
ineffective and offers a way to bypass intrinsic resistance.

242
Figure 5. Chemical inhibition of the DSB system impedes DsbA re-oxidation in E. coli
MC1000 and phenocopies the β-lactam and colistin MIC changes that were observed
using a dsbA mutant. (A) Addition of the reducing agent DTT to E. coli MC1000 bacterial
lysates allows the detection of DsbA in its reduced form (DsbAred) during immunoblotting; this
redox state of the protein, when labelled with the cysteine-reactive compound AMS, shows a
1 kDa size difference (lane 2) compared to oxidized DsbA as found in AMS-labelled but not
reduced lysates of E. coli MC1000 (lane 3). Addition of a small-molecule inhibitor of DsbB to
growing E. coli MC1000 cells also results in accumulation of reduced DsbA (lane 4). E. coli
MC1000 dsbA was used as a negative control for DsbA detection (lane 1). A representative
blot from two biological experiments, each conducted as a single technical repeat, is shown;
DsbA was visualized using an anti-DsbA primary antibody and an AP-conjugated secondary
antibody. Molecular weight markers (M) are shown on the left. (B) MIC experiments using
representative β-lactam antibiotics show that chemical inhibition of the DSB system reduces
the MIC values for E. coli MC1000 expressing disulfide-bond-containing β-lactamases in a

243
similar manner to the deletion of dsbA (compare with Figure 1B). Graphs show MIC fold
changes (i.e. MC1000 MIC (µg/mL) / MC1000 + DSB system inhibitor MIC (µg/mL)) for β-
lactamase-expressing E. coli MC1000 with and without addition of a DSB system inhibitor to
the culture medium and are representative of two biological experiments, each conducted as a
single technical repeat. Black dotted lines indicate an MIC fold change of 2. The
aminoglycoside antibiotic gentamicin serves as a control for all strains; gentamicin MIC values
(white bars) are unaffected by chemical inhibition of the DSB system (MIC fold changes: < 2).
No changes in MIC values (MIC fold changes: < 2) are observed for strains harboring the
empty vector control (pDM1) or expressing the class A β-lactamase L2-1, which contains three
cysteines but no disulfide bond (PDB ID: 5NE1) (top row). (C) Colistin MIC experiments
show that chemical inhibition of the DSB system reduces the MIC values for E. coli MC1000
expressing MCR enzymes in a similar manner to the deletion of dsbA (compare with Figure
1C). Colistin MIC values for strains harboring the empty vector control (pDM1) are unaffected
by chemical inhibition of the DSB system. Graphs show MIC values (µg/mL) from four
biological experiments, each conducted in technical quadruplicate, to demonstrate the
robustness of the observed effects. (D) The aminoglycoside antibiotic gentamicin serves as a
control for all strains tested in panel (C); gentamicin MIC values are unaffected by chemical
inhibition of the DSB system. Graphs show MIC values (µg/mL) from two biological
experiments, each conducted as a single technical repeat.

244
Figure 6. Chemical inhibition of the DSB system sensitizes multidrug-resistant clinical
isolates to currently available antibiotics. (A) Addition of a small-molecule inhibitor of
DsbB results in sensitization of Klebsiella pneumoniae, E. coli and Citrobacter freundii clinical
isolates to imipenem. Chemical inhibition of the DSB system of an Enterobacter cloacae
clinical isolate harboring blaFRI-1 results in reduction of the aztreonam MIC value by over 180

245
µg/mL, resulting in intermediate resistance as defined by EUCAST. MIC values determined
using Mueller-Hinton agar (MHA) in accordance with the EUCAST guidelines (light blue bars)
are comparable to the values obtained using defined media (M63 agar, white bars); use of
growth media lacking small-molecule oxidants is required for the DSB system inhibitor to be
effective. Graphs show MIC values (μg/ml) representative of two biological experiments, each
conducted as a single technical repeat. (B) Application of the same chemical inhibitor to
colistin-resistant E. coli expressing MCR enzymes results in sensitization of all tested clinical
isolates to colistin. Graphs show MIC values (µg/mL) from four biological experiments, each
conducted in technical quadruplicate, to demonstrate the robustness of the observed effects.
(C) Deletion of dsbA sensitizes the efflux-active E. coli MG1655 strain to chloramphenicol;
the data presented in the blue and light blue bars were also used to generate part of Figure 1D.
Sensitization is also observed for the dsbA mutant of the deregulated E. coli MG1655 marR
strain (chloramphenicol MIC of 6 μg/mL). The graph shows MIC values (μg/ml) from 2
biological experiments, each conducted as a single technical repeat. (D) Absence of the
principal pseudomonal DsbA analogue (DsbA1) sensitizes the P. aeruginosa PA43417 clinical
isolate expressing OXA-198 to the first-line antibiotic piperacillin (piperacillin MIC of 12
μg/mL). The graph shows MIC values (μg/ml) from 2 biological experiments, each conducted
as a single technical repeat. For all panels, red dotted lines indicate the EUCAST clinical
breakpoint for each antibiotic. (E) Absence of the principal DsbA analogue (DsbA1) from
a P. aeruginosa clinical isolate expressing OXA-198 allows it to be cleared from infected
G. mellonella larvae by piperacillin. Neither deletion of dsbA1, nor treatment with
piperacillin (at a concentration of 12 μg/mL) is sufficient to clear P. aeruginosa PA4317 from
infected G. mellonella larvae, but the combination of both results in an average reduction in
bacterial load that is greater than 99%. The graph shows the average number of colony forming
units (CFU) recovered from infected larvae for each condition relative to the CFU recovered
for the untreated P. aeruginosa PA4317 strain. n = 8 groups infected on eight different days;
each group contains five G. mellonella larvae per condition except for one group which
contains three G. mellonella larvae per condition. Graph shows means ±SD, significance is
indicated by * = p < 0.05, ** = p < 0.01, *** = p <0.001.

246
REFERENCES
1. Rochford C, Sridhar D, Woods N, Saleh Z, Hartenstein L, Ahlawat H, et al. Global
governance of antimicrobial resistance. Lancet. 2018;391(10134):1976-8.
2. Tacconelli E, Carrara E, Savoldi A, Harbarth S, Mendelson M, Monnet DL, et al.
Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-
resistant bacteria and tuberculosis. Lancet Infect Dis. 2018;18(3):318-27.
3. Hart EM, Mitchell AM, Konovalova A, Grabowicz M, Sheng J, Han X, et al. A small-
molecule inhibitor of BamA impervious to efflux and the outer membrane permeability barrier.
Proc Natl Acad Sci U S A. 2019;116(43):21748-57.
4. Laws M, Shaaban A, Rahman KM. Antibiotic resistance breakers: current approaches
and future directions. FEMS Microbiol Rev. 2019;43(5):490-516.
5. Luther A, Urfer M, Zahn M, Muller M, Wang SY, Mondal M, et al. Chimeric
peptidomimetic antibiotics against Gram-negative bacteria. Nature. 2019;576(7787):452-8.
6. Nicolas I, Bordeau V, Bondon A, Baudy-Floc'h M, Felden B. Novel antibiotics
effective against Gram-positive and -negative multi-resistant bacteria with limited resistance.
PLoS Biol. 2019;17(7):e3000337.
7. Srinivas N, Jetter P, Ueberbacher BJ, Werneburg M, Zerbe K, Steinmann J, et al.
Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa.
Science. 2010;327(5968):1010-3.
8. Bush K. Past and present perspectives on β-lactamases. Antimicrob Agents Chemother.
2018;62(10).
9. Meletis G. Carbapenem resistance: overview of the problem and future perspectives.
Ther Adv Infect Dis. 2016;3(1):15-21.
10. Versporten A, Zarb P, Caniaux I, Gros MF, Drapier N, Miller M, et al. Antimicrobial
consumption and resistance in adult hospital inpatients in 53 countries: results of an internet-
based global point prevalence survey. Lancet Glob Health. 2018;6(6):e619-e29.
11. Li J, Nation RL, Turnidge JD, Milne RW, Coulthard K, Rayner CR, et al. Colistin: the
re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet
Infect Dis. 2006;6(9):589-601.
12. Poirel L, Jayol A, Nordmann P. Polymyxins: antibacterial activity, susceptibility
testing, and resistance mechanisms encoded by plasmids or chromosomes. Clin Microbiol Rev.
2017;30(2):557-96.
13. Sun J, Zhang H, Liu YH, Feng Y. Towards understanding MCR-like colistin resistance.
Trends Microbiol. 2018;26(9):794-808.
14. Blair JMA, Richmond GE, Piddock LJV. Multidrug efflux pumps in Gram-negative
bacteria and their role in antibiotic resistance. Future Microbiol. 2014;9(10):1165-77.

247
15. Cox G, Wright GD. Intrinsic antibiotic resistance: mechanisms, origins, challenges and
solutions. Int J Med Microbiol. 2013;303(6-7):287-92.
16. Goemans C, Denoncin K, Collet JF. Folding mechanisms of periplasmic proteins.
Biochim Biophys Acta. 2014;1843(8):1517-28.
17. Heras B, Kurz M, Shouldice SR, Martin JL. The name's bond......disulfide bond. Curr
Opin Struct Biol. 2007;17(6):691-8.
18. Bardwell JC, McGovern K, Beckwith J. Identification of a protein required for disulfide
bond formation in vivo. Cell. 1991;67(3):581-9.
19. Denoncin K, Collet JF. Disulfide bond formation in the bacterial periplasm: major
achievements and challenges ahead. Antioxid Redox Signal. 2013;19(1):63-71.
20. Hiniker A, Bardwell JC. In vivo substrate specificity of periplasmic disulfide
oxidoreductases. J Biol Chem. 2004;279(13):12967-73.
21. Kadokura H, Tian H, Zander T, Bardwell JC, Beckwith J. Snapshots of DsbA in action:
detection of proteins in the process of oxidative folding. Science. 2004;303(5657):534-7.
22. Martin JL, Bardwell JC, Kuriyan J. Crystal structure of the DsbA protein required for
disulphide bond formation in vivo. Nature. 1993;365(6445):464-8.
23. Dutton RJ, Boyd D, Berkmen M, Beckwith J. Bacterial species exhibit diversity in their
mechanisms and capacity for protein disulfide bond formation. Proc Natl Acad Sci U S A.
2008;105(33):11933-8.
24. Vertommen D, Depuydt M, Pan J, Leverrier P, Knoops L, Szikora JP, et al. The
disulphide isomerase DsbC cooperates with the oxidase DsbA in a DsbD-independent manner.
Mol. Microbiol. 2008;67(2):336-49.
25. Heras B, Shouldice SR, Totsika M, Scanlon MJ, Schembri MA, Martin JL. DSB
proteins and bacterial pathogenicity. Nat Rev Microbiol. 2009;7(3):215-25.
26. Landeta C, Boyd D, Beckwith J. Disulfide bond formation in prokaryotes. Nature
Microbiol. 2018;3(3):270-80.
27. Heras B, Scanlon MJ, Martin JL. Targeting virulence not viability in the search for
future antibacterials. Br J Clin Pharmacol. 2014;79(2):208-15.
28. Piek S, Wang Z, Ganguly J, Lakey AM, Bartley SN, Mowlaboccus S, et al. The role of
oxidoreductases in determining the function of the neisserial lipid A phosphoethanolamine
transferase required for resistance to polymyxin. PloS One. 2014;9(9):e106513.
29. Nation RL, Garonzik SM, Li J, Thamlikitkul V, Giamarellos-Bourboulis EJ, Paterson
DL, et al. Updated US and European dose recommendations for intravenous colistin: how do
they perform? Clin Infect Dis. 2016;62(5):552-8.
30. Plachouras D, Karvanen M, Friberg LE, Papadomichelakis E, Antoniadou A, Tsangaris
I, et al. Population pharmacokinetic analysis of colistin methanesulfonate and colistin after
intravenous administration in critically ill patients with infections caused by Gram-negative
bacteria. Antimicrob Agents Chemother. 2009;53(8):3430-6.

248
31. Wang Z, Fan G, Hryc CF, Blaza JN, Serysheva, II, Schmid MF, et al. An allosteric
transport mechanism for the AcrAB-TolC multidrug efflux pump. eLife. 2017;6.
32. Dortet L, Bonnin RA, Pennisi I, Gauthier L, Jousset AB, Dabos L, et al. Rapid detection
and discrimination of chromosome- and MCR-plasmid-mediated resistance to polymyxins by
MALDI-TOF MS in Escherichia coli: the MALDIxin test. J Antimicrob Chemother.
2018;73(12):3359-67.
33. Clausen T, Southan C, Ehrmann M. The HtrA family of proteases: implications for
protein composition and cell fate. Mol Cell. 2002;10(3):443-55.
34. Skorko-Glonek J, Zurawa D, Tanfani F, Scire A, Wawrzynow A, Narkiewicz J, et al.
The N-terminal region of HtrA heat shock protease from Escherichia coli is essential for
stabilization of HtrA primary structure and maintaining of its oligomeric structure. Biochim
Biophys Acta. 2003;1649(2):171-82.
35. Gerken H, Misra R. Genetic evidence for functional interactions between TolC and
AcrA proteins of a major antibiotic efflux pump of Escherichia coli. Mol Microbiol.
2004;54(3):620-31.
36. Werner J, Augustus AM, Misra R. Assembly of TolC, a structurally unique and
multifunctional outer membrane protein of Escherichia coli K-12. J Bacteriol.
2003;185(22):6540-7.
37. Duprez W, Premkumar L, Halili MA, Lindahl F, Reid RC, Fairlie DP, et al. Peptide
inhibitors of the Escherichia coli DsbA oxidative machinery essential for bacterial virulence.
J Med Chem. 2015;58(2):577-87.
38. Totsika M, Vagenas D, Paxman JJ, Wang G, Dhouib R, Sharma P, et al. Inhibition of
diverse DsbA enzymes in multi-DsbA encoding pathogens. Antioxid Redox Signal.
2018;29(7):653-66.
39. Landeta C, Blazyk JL, Hatahet F, Meehan BM, Eser M, Myrick A, et al. Compounds
targeting disulfide bond forming enzyme DsbB of Gram-negative bacteria. Nat Chem Biol.
2015;11(4):292-8.
40. Halili MA, Bachu P, Lindahl F, Bechara C, Mohanty B, Reid RC, et al. Small molecule
inhibitors of disulfide bond formation by the bacterial DsbA-DsbB dual enzyme system. ACS
Chem Biol. 2015;10(4):957-64.
41. Dailey FE, Berg HC. Mutants in disulfide bond formation that disrupt flagellar
assembly in Escherichia coli. Proc Natl Acad Sci U S A. 1993;90(3):1043-7.
42. Hizukuri Y, Yakushi T, Kawagishi I, Homma M. Role of the intramolecular disulfide
bond in FlgI, the flagellar P-ring component of Escherichia coli. J Bacteriol.
2006;188(12):4190-7.
43. Kishigami S, Akiyama Y, Ito K. Redox states of DsbA in the periplasm of Escherichia
coli. FEBS Lett. 1995;364(1):55-8.

249
44. Ariza RR, Cohen SP, Bachhawat N, Levy SB, Demple B. Repressor mutations in the
marRAB operon that activate oxidative stress genes and multiple antibiotic resistance in
Escherichia coli. J Bacteriol. 1994;176(1):143-8.
45. Okusu H, Ma D, Nikaido H. AcrAB efflux pump plays a major role in the antibiotic
resistance phenotype of Escherichia coli multiple-antibiotic-resistance (Mar) mutants. J
Bacteriol. 1996;178(1):306-8.
46. Keeney D, Ruzin A, McAleese F, Murphy E, Bradford PA. MarA-mediated
overexpression of the AcrAB efflux pump results in decreased susceptibility to tigecycline in
Escherichia coli. J Antimicrob Chemother. 2008;61(1):46-53.
47. Arts IS, Ball G, Leverrier P, Garvis S, Nicolaes V, Vertommen D, et al. Dissecting the
machinery that introduces disulfide bonds in Pseudomonas aeruginosa. mBio. 2013;4(6).
48. Landeta C, McPartland L, Tran NQ, Meehan BM, Zhang Y, Tanweer Z, et al. Inhibition
of Pseudomonas aeruginosa and Mycobacterium tuberculosis disulfide bond forming
enzymes. Mol Microbiol. 2019;111(4):918-37.
49. Zhou YL, Wang JF, Guo Y, Liu XQ, Liu SL, Niu XD, et al. Discovery of a potential
MCR-1 inhibitor that reverses polymyxin activity against clinical mcr-1-positive
Enterobacteriaceae. J Infect. 2019;78(5):364-72.
50. Sharma A, Gupta VK, Pathania R. Efflux pump inhibitors for bacterial pathogens: From
bench to bedside. Indian J Med Res. 2019;149(2):129-45.
51. Kadokura H, Katzen F, Beckwith J. Protein disulfide bond formation in prokaryotes.
Annu Rev Biochem. 2003;72:111-35.
52. Wilmaerts D, Dewachter L, De Loose PJ, Bollen C, Verstraeten N, Michiels J. HokB
monomerization and membrane repolarization control persister awakening. Mol Cell.
2019;75(5):1031-42.
53. Chang AC, Cohen SN. Construction and characterization of amplifiable multicopy
DNA cloning vehicles derived from the P15A cryptic miniplasmid. J Bacteriol.
1978;134(3):1141-56.
54. Kim J, Webb AM, Kershner JP, Blaskowski S, Copley SD. A versatile and highly
efficient method for scarless genome editing in Escherichia coli and Salmonella enterica. BMC
Biotechnol. 2014;14:84.
55. McKenzie GJ, Craig NL. Fast, easy and efficient: site-specific insertion of transgenes
into enterobacterial chromosomes using Tn7 without need for selection of the insertion event.
BMC Microbiol. 2006;6:39.
56. Vasseur P, Vallet-Gely I, Soscia C, Genin S, Filloux A. The pel genes of the
Pseudomonas aeruginosa PAK strain are involved at early and late stages of biofilm formation.
Microbiology. 2005;151:985-97.
57. Kaniga K, Delor I, Cornelis GR. A wide-host-range suicide vector for improving
reverse genetics in Gram-negative bacteria: inactivation of the blaA gene of Yersinia
enterocolitica. Gene. 1991;109(1):137-41.

250
58. Helander IM, Mattila-Sandholm T. Fluorometric assessment of Gram-negative
bacterial permeabilization. J Appl Microbiol. 2000;88(2):213-9.
59. Mavridou DA, Gonzalez D, Clements A, Foster KR. The pUltra plasmid series: a robust
and flexible tool for fluorescent labeling of Enterobacteria. Plasmid. 2016;87-88:65-71.
60. Larrouy-Maumus G, Clements A, Filloux A, McCarthy RR, Mostowy S. Direct
detection of lipid A on intact Gram-negative bacteria by MALDI-TOF mass spectrometry. J
Microbiol Methods 2016;120:68-71.
61. McCarthy RR, Mazon-Moya MJ, Moscoso JA, Hao Y, Lam JS, Bordi C, et al. Cyclic-
di-GMP regulates lipopolysaccharide modification and contributes to Pseudomonas
aeruginosa immune evasion. Nat Microbiol. 2017;2:17027.
62. Naas T, Oueslati S, Bonnin RA, Dabos ML, Zavala A, Dortet L, et al. β-lactamase
database (BLDB) - structure and function. J Enzyme Inhib Med Chem. 2017;32(1):917-9.
63. Edgar RC. Search and clustering orders of magnitude faster than BLAST.
Bioinformatics. 2010;26(19):2460-1.
64. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search
tool. J Mol Biol. 1990;215(3):403-10.
65. Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high
throughput. Nucleic Acids Res. 2004;32(5):1792-7.
66. Finn RD, Clements J, Arndt W, Miller BL, Wheeler TJ, Schreiber F, et al. HMMER
web server: 2015 update. Nucleic Acids Res. 2015;43(W1):W30-8.

251
SUPPLEMENTARY INFORMATION FOR
Breaking antimicrobial resistance by disrupting extracytoplasmic
protein folding
R. Christopher D. Furniss2,†, Nikol Kadeřábková2,†, Declan Barker2, Patricia Bernal3, Evgenia
Maslova4, Amanda A.A. Antwi2, Helen E. McNeil5, Hannah L. Pugh5, Laurent Dortet2,6,7,8,
Jessica M.A. Blair5, Gerald Larrouy-Maumus2, Ronan R. McCarthy4, Diego Gonzalez9,
Despoina A.I. Mavridou1,2,*
1Department of Molecular Biosciences, University of Texas at Austin, Austin, 78712, Texas,
USA 2MRC Centre for Molecular Bacteriology and Infection, Department of Life Sciences, Imperial
College London, London, SW7 2AZ, UK 3Department of Biology, Faculty of Sciences, Universidad Autónoma de Madrid, Madrid,
28049, Spain 4Division of Biosciences, Department of Life Sciences, College of Health and Life Sciences,
Brunel University London, Uxbridge, UB8 3PH, UK 5Institute of Microbiology and Infection, College of Medical and Dental Sciences, University
of Birmingham, Birmingham, B15 2TT, UK 6Department of Bacteriology-Hygiene, Bicêtre Hospital, Assistance Publique - Hôpitaux de
Paris, Le Kremlin-Bicêtre, 94270, France 7EA7361 “Structure, Dynamics, Function and Expression of Broad-spectrum β-lactamases",
Paris-Sud University, LabEx Lermit, Faculty of Medicine, Le Kremlin-Bicêtre, 94270, France 8French National Reference Centre for Antibiotic Resistance, Le Kremlin-Bicêtre, 94270,
France 9Laboratoire de Microbiologie, Institut de Biologie, Université de Neuchâtel, Neuchâtel, 2000,
Switzerland
*Correspondence: [email protected]
†These authors have contributed equally to this work
This PDF file includes:
Figures S1 toS14
Tables S1 to S6
Legend for File S1
Supplementary references

252
SUPPLEMENTARY FIGURES
Figure S1. Phylogenetic analysis of MCR- and EptA-like enzymes found in
Proteobacteria. A phylogenetic tree was built based on the alignment of 781 sequences from
Proteobacteria. The assignment of each sequence to a specific group was done using Hidden
Markov Models built from confirmed sequences of MCR- and EptA-like proteins; EptA-like
enzymes are chromosomal phosphoethanolamine transferases that belong to the same extended
protein superfamily as MCR enzymes, but do not give rise to polymyxin resistance in
Enterobacteria (1). The different MCR groups are broadly indicated in different colors,
however it should be noted that there is significant overlap between groups. Open circles mark
the enzymes tested in this study which are distributed throughout the MCR phylogeny.

253
Figure S2. Deletion of dsbA lowers the β-lactam MIC values for E. coli MC1000
expressing diverse β-lactamases. In the absence of DsbA the β-lactam MICs (for multiple
classes of β-lactam antibiotics) for E. coli MC1000 expressing disulfide-bond-containing β-
lactamases are drastically reduced. This figure shows the MIC data used to generate Figure 1B,
and the MIC values for the additional GES and KPC family members GES-2, GES-11 and
KPC-2. The aminoglycoside antibiotic gentamicin serves as a control for all strains. (A)
MC1000 pDM1 (vector alone) (B) MC1000 pDM1-blaL2-1 (cysteine-containing β-lactamase
which lacks a disulfide bond) (C) MC1000 pDM1-blaGES-1, (D) MC1000 pDM1-blaGES-2, (E)
MC1000 pDM1-blaGES-11, (F) MC1000 pDM1-blaSHV-27, (G) MC1000 pDM1-blaOXA-4, (H)
MC1000 pDM1-blaOXA-10, (I) MC1000 pDM1-blaOXA-198, (J) MC1000 pDM1-blaFRI-1, (K)
MC1000 pDM1-blaL1-1, (L) MC1000 pDM1-blaKPC-2, (M) MC1000 pDM1-blaKPC-3, (N)
MC1000 pDM1 blaSME-1. Graphs show MIC values (µg/mL) and are representative of three
biological experiments, each conducted as a single technical repeat.

254

255

256
Figure S3. SHV-27 function is dependent on DsbA at temperatures higher than 37 °C.
The ESBL SHV-27 differs from the canonical SHV-1 enzyme by a single amino acid
substitution (D156G) (2). At 37 °C deletion of dsbA does not affect the cefuroxime MIC for E.
coli MC1000 harboring pDM1-blaSHV-27. However, at 42 °C the cefuroxime MIC for E. coli
MC1000 dsbA harboring pDM1-blaSHV-27 is notably reduced. Thus, in a similar manner to
TEM-1 (3), the SHV-27 disulfide bond becomes important for enzyme function under stress
conditions (temperature stress). As SHV-27 has the narrowest hydrolytic spectrum out of all
the enzymes tested in this study, this result suggests that there could be a correlation between
the hydrolytic spectrum of the β-lactamase and its dependence on DsbA for conferring
resistance. The graph shows MIC values (µg/mL) and is representative of three biological
experiments, each conducted as a single technical repeat.

257
Figure S4. Complementation of dsbA restores the β-lactam MIC values for E. coli
MC1000 dsbA expressing β-lactamases. Re-insertion of dsbA at the attTn7 site of the
chromosome restores the β-lactam MIC values for E. coli MC1000 dsbA harboring (A) pDM1-
blaGES-1 (ceftazidime MIC), (B) pDM1-blaOXA-4 (cefuroxime MIC), (C) pDM1-blaOXA-10
(aztreonam MIC), (D) pDM1-blaOXA-198 (imipenem MIC), (E) pDM1-blaL1-1 (ceftazidime
MIC), (F) pDM1-blaFRI-1 (aztreonam MIC) and (G) pDM1-blaKPC-3 (ceftazidime MIC). Graphs
show MIC values (µg/mL) and are representative of two biological experiments, each
conducted as a single technical repeat.

258
Figure S5. Complementation of dsbA restores the colistin MIC values for E. coli MC1000
dsbA expressing MCR enzymes. Re-insertion of dsbA at the attTn7 site of the chromosome
restores the colistin MIC values for E. coli MC1000 dsbA harboring (A) pDM1-mcr-1 (B)
pDM1-mcr-3 (C) pDM1-mcr-4 (D) pDM1-mcr-5 (E) pDM1-mcr-8. Graphs show MIC values
(µg/mL) from four biological experiments, each conducted in technical quadruplicate, to
demonstrate the robustness of the observed effects.

259
Figure S6. Gentamicin MIC values for E. coli MC1000 strains expressing MCR enzymes.
Deletion of dsbA does not affect the gentamicin MIC values for E. coli MC1000 strains
expressing MCR enzymes, confirming that absence of DsbA does not compromise the general
ability of this strain to resist antibiotic stress. Graphs show MIC values (µg/mL) and are
representative of two biological experiments, each conducted as a single technical repeat.

260
Figure S7. Deletion of dsbA has no effect on membrane permeability in E. coli MC1000.
(A) The bacterial outer membrane acts as a selective permeability barrier to hydrophobic
molecules. Deletion of dsbA has no effect on the outer membrane integrity of E. coli MC1000,
as the hydrophobic fluorescent dye NPN crosses the outer membrane of E. coli MC1000 and
its dsbA mutant to the same extent. Conversely, exposure to the outer-membrane-
permeabilizing antibiotic colistin results in a significant increase in NPN uptake. (B) PI is a
cationic hydrophilic dye that fluoresces upon intercalation with nucleic acids. Under normal
conditions PI freely crosses the outer membrane but is unable to cross the inner membrane.
Deletion of dsbA does not result in damage to the bacterial inner membrane, as no difference
in basal PI uptake is seen between E. coli MC1000 and its dsbA mutant. Both strains express
superfolder GFP (sfGFP), and fluorescence was used to distinguish live from dead cells.
Addition of the inner-membrane-permeabilizing antimicrobial peptide cecropin A (4) to E. coli
MC1000 induces robust inner-membrane permeabilization in the sfGFP-positive population
indicating that the inner membrane becomes compromised. For both experiments n=3 (each
conducted in technical triplicate), graph shows means ±SD, significance is indicated by *** =
p < 0.001, ns = non-significant.

261
Figure S8. Complementation of dsbA restores efflux-pump substrate MIC values for E.
coli MG1655 dsbA. Re-insertion of dsbA at the attTn7 site of the chromosome restores (A)
erythromycin, (B) chloramphenicol and (C) nalidixic acid MIC values for MG1655 dsbA.
Graphs show MIC values (µg/mL) and are representative of two biological experiments, each
conducted as a single technical repeat.

262
Figure S9. Deletion of dsbA has no effect on membrane permeability in E. coli MG1655.
(A) Deletion of dsbA has no effect on the outer membrane integrity of E. coli MG1655, as the
hydrophobic fluorescent dye NPN crosses the outer membrane of E. coli MG1655 and its dsbA
mutant to the same extent. Conversely, exposure to the outer-membrane-permeabilizing
antibiotic colistin results in a significant increase in NPN uptake. (B) Deletion of dsbA does
not result in damage to the bacterial inner membrane, as no difference in basal PI uptake is
seen between E. coli MG1655 and its dsbA mutant. Both strains express sfGFP, and
fluorescence was used to distinguish live from dead cells. Addition of the inner-membrane-
permeabilizing antimicrobial peptide cecropin A (4) to E. coli MG1655 induces robust inner
membrane permeabilization in the sfGFP-positive population indicating that the inner
membrane becomes compromised. For both experiments n=3 (each conducted in technical
triplicate), graph shows means ±SD, significance is indicated by **** = p < 0.0001, ns = non-
significant.

263
Figure S10. Chemical inhibition of the DSB system impedes flagellar motility in E. coli
MC1000. (A) A functional DSB system is necessary for flagellar motility in E. coli because
folding of the P-ring component FlgI requires DsbA-mediated disulfide bond formation (5). In
the absence of DsbA, or upon addition of a chemical inhibitor of the DSB system, the motility
of E. coli MC1000 is significantly impeded. Representative images of motility plates are
shown. (B) Quantification of the growth halo diameters in the motility assays shown in panel
(A). n=3 (each conducted as a single technical repeat), graph shows means ±SD, significance
is indicated by **** = p < 0.0001.

264
Figure S11. Chemical inhibition of the DSB system or deletion of dsbA does not
compromise the growth of E. coli MC1000. Growth curves of (A) E. coli MC1000 with and
without chemical inhibition of the DSB system and (B) E. coli MC1000 and its dsbA mutant
show that bacterial growth remains unaffected by the DSB system inhibitor compound used in
this study, or by the absence of DsbA. n=3 (each conducted as a technical triplicate), solid lines
indicate mean values, shaded areas indicate SD.

265
Figure S12. Changes in MIC values observed using the DSB system inhibitor are due
solely to inhibition of the DSB system. (A) E. coli MC1000 harboring pDM1-blaKPC-3 has an
imipenem MIC value of 24 μg/mL. Upon chemical inhibition of the DSB system the imipenem
MIC for this strain drops to 4 μg/mL, and accordingly the imipenem MIC for E. coli MC1000
dsbA harboring pDM1-blaKPC-3 is 2 μg/mL. The imipenem MIC for E. coli MC1000 dsbA
harboring pDM1-blaKPC-3 when exposed to the chemical inhibitor of the DSB system is also 2
μg/mL, indicating that the chemical compound used in this study does not have any off-target
effects and only affects the function of the DSB system proteins. (B) Chemical inhibition of
the DSB system does not lead to any cumulative effects when tested on an E. coli MC1000
strain expressing MCR-5. The colistin MIC for E. coli MC1000 harboring pDM1-mcr-5 is 3
μg/mL and it drops to 1 μg/mL when the DSB system is chemically inhibited or dsbA is deleted.
The same drop in colistin MIC is observed when the E. coli MC1000 dsbA strain harboring
pDM1-mcr-5 is exposed to the chemical inhibitor of the DSB system. Data shown in both
panels are representative of two biological experiments, each conducted as a single technical
repeat.

266
Figure S13. Deletion of dsbA results in reduced MIC values for E. coli MC1000 expressing
MCR-3.2. When cloned into pDM1 and expressed in E. coli MC1000, MCR-3.2 confers
colistin resistance as expected (MIC of 3.0-3.5 μg/ml). Deletion of dsbA reduces the colistin
MIC values for E. coli MC1000 expressing MCR-3.2 (MIC ≤ 2 μg/mL). Graphs show MIC
values (µg/mL) from four biological experiments, each conducted in technical quadruplicate,
to demonstrate the robustness of the observed effects.

267
Figure S14. Deletion of marR results in increased expression of the AcrAB pump (lane 2)
compared to the parental strain (lane 1) even though the chloramphenicol MIC for both
strains is the same (Figure 6C). E. coli MG1655 has an already high level of efflux activity
and therefore deletion of marR likely does not result in a drastic change in the observed
chloramphenicol MIC value. Expression of the AcrAB pump was assessed using an anti-AcrA
primary antibody and an HRP-conjugated secondary antibody. E. coli MG1655 acrA was used
as a negative control for AcrA detection (lane 3); the red arrow indicates the position of the
AcrA band. A representative blot from two biological experiments, each conducted as a single
technical repeat, is shown; molecular weight markers (M) are shown on the left and DnaK was
used as a loading control.
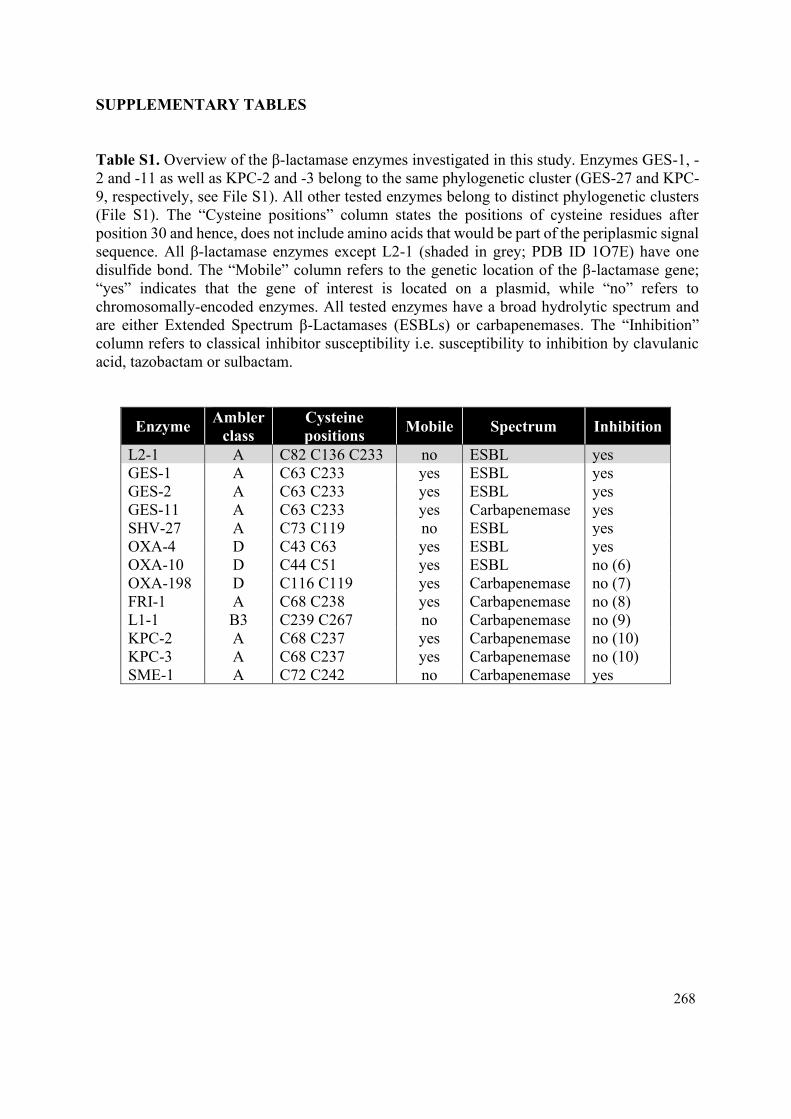
268
SUPPLEMENTARY TABLES
Table S1. Overview of the β-lactamase enzymes investigated in this study. Enzymes GES-1, -
2 and -11 as well as KPC-2 and -3 belong to the same phylogenetic cluster (GES-27 and KPC-
9, respectively, see File S1). All other tested enzymes belong to distinct phylogenetic clusters
(File S1). The “Cysteine positions” column states the positions of cysteine residues after
position 30 and hence, does not include amino acids that would be part of the periplasmic signal
sequence. All β-lactamase enzymes except L2-1 (shaded in grey; PDB ID 1O7E) have one
disulfide bond. The “Mobile” column refers to the genetic location of the β-lactamase gene;
“yes” indicates that the gene of interest is located on a plasmid, while “no” refers to
chromosomally-encoded enzymes. All tested enzymes have a broad hydrolytic spectrum and
are either Extended Spectrum β-Lactamases (ESBLs) or carbapenemases. The “Inhibition”
column refers to classical inhibitor susceptibility i.e. susceptibility to inhibition by clavulanic
acid, tazobactam or sulbactam.
Enzyme Ambler
class
Cysteine
positions Mobile Spectrum Inhibition
L2-1 A C82 C136 C233 no ESBL yes
GES-1 A C63 C233 yes ESBL yes
GES-2 A C63 C233 yes ESBL yes
GES-11 A C63 C233 yes Carbapenemase yes
SHV-27 A C73 C119 no ESBL yes
OXA-4 D C43 C63 yes ESBL yes
OXA-10 D C44 C51 yes ESBL no (6)
OXA-198 D C116 C119 yes Carbapenemase no (7)
FRI-1 A C68 C238 yes Carbapenemase no (8)
L1-1 B3 C239 C267 no Carbapenemase no (9)
KPC-2 A C68 C237 yes Carbapenemase no (10)
KPC-3 A C68 C237 yes Carbapenemase no (10)
SME-1 A C72 C242 no Carbapenemase yes

269
Table S2. Antibiotic resistance profiles of the clinical isolates tested in this study. The table
shows MIC values (µg/mL) for a range of commonly used antibiotics. Values highlighted in
pink indicate resistance, as defined by the EUCAST clinical breakpoint guidelines, whilst
values highlighted in light blue indicate antibiotics for which there is no EUCAST clinical
breakpoint. The remaining values (white cells) indicate sensitivity to the tested antibiotic
compound. Strains shaded in yellow are multidrug resistant. The following abbreviations are
used: AC, amoxicillin; XM, cefuroxime; TZ, ceftazidime; IP, imipenem; AT, aztreonam; PT,
piperacillin/tazobactam; GM, gentamicin; CO, colistin; CI, ciprofloxacin; NF, nitrofurantoin;
TR, trimethoprim.
Strain AC XM TZ IP AT PT GM CO CI NF TR
E. coli BM16
(blaTEM-1b blaKPC-
2)
>256 >256 192 12 >256 >64 8 1 >32 >512 >32
E. coli LIL-1
(blaTEM-1 blaOXA-9
blaKPC-2)
>256 >256 8 3 192 >64 1.5 2 >32 6 >32
E. coli CNR1790 (blaTEM-15 mcr-1)
>256 >256 32 0.5 16 <2 1 4 >32 16 >32
E. coli
CNR20140385
(blaOXA-48 mcr-1)
>256 96 1 0.25 0.38 32 2 4 >32 8 >32
E. coli WI2
(blaOXA-48 blaKPC-
28 mcr-1)
>256 >256 >256 1.5 32 >64 1.5 4 0.016 6 0.25
E. coli 27841 (blaCTX-M-55 mcr-
3.2)
>256 >256 16 0.19 64 <2 32 3 >32 8 >32
E. coli 1144230 (blaCMY-2 mcr-5)
>256 96 12 0.5 6 8 1.5 4 0.025 48 1
K. pneumoniae
ST234
(blaSHV-27 blaKPC-
2)
>256 >256 48 16 128 >64 0.38 2 0.047 96 1
C. freundii
BM19
(blaKPC-2)
>256 >256 128 4 64 >64 24 2 >32 8 >32
E. cloacae DUB
(blaFRI-1) >256 >256 16 12 >256 >64 1.5 >4 0.016 48 0.75
P. aeruginosa
PA43417
(blaOXA-198)
>256 >256 2 >32 6 32 16 1 >32 >256 >32

270
Table S3. Bacterial strains used in this study. All listed isolates are clinical strains except for
Escherichia coli 27841 (ST744), which is an environmental strain. For clinical and
environmental isolates, the multi-locus sequence types (ST) are given in parenthesis, where
available.
Name Description Source
Escherichia coli
DH5α
F– endA1 glnV44 thi-1 recA1 relA1
gyrA96 deoR nupG purB20
φ80dlacZ∆M15 ∆(lacZYA-argF)U169
hsdR17(rK–mK
+) λ–
(11)
CC118λpir araD Δ(ara, leu) ΔlacZ74 phoA20
galK thi-1 rspE rpoB argE recA1 λpir (12)
HB101 supE44 hsdS20 recA13 ara-14 proA2
lacY1 galK2 rpsL20 xyl-5 mtl-1 (13)
MC1000 araD139 ∆(ara, leu)7697 ∆lacX74
galU galK strA (14)
MC1000 dsbA dsbA::aphA, KanR (15)
MC1000 dsbA attTn7::Ptac-dsbA dsbA::aphA attTn7::dsbA, KanR This study
MG1655 K-12 F– λ– ilvG– rfb-50 rph-1 (16)
MG1655 dsbA dsbA::aphA, KanR This study
MG1655 dsbA attTn7::Ptac-dsbA dsbA::aphA attTn7::dsbA, KanR This study
MG1655 acrA acrA This study
MG1655 tolC tolC This study
MG1655 degP degP::strAB, SmR This study
MG1655 marR marR::accC, GentR This study
MG1655 dsbA marR dsbA::aphA marR::accC, KanR, GentR This study
Clinical / environmental isolates
Escherichia coli BM16 blaTEM-1b blaKPC-2 (17)
Escherichia coli LIL-1 blaTEM-1 blaOXA-9 blaKPC-2 (17)
Escherichia coli CNR1790 blaTEM-15 mcr-1 (18)
Escherichia coli CNR20140385 blaOXA-48 mcr-1 (18)
Escherichia coli WI2 (ST1288) blaOXA-48 blaKPC-28 mcr-1 (19)
Escherichia coli 27841 (ST744) blaCTX-M-55 mcr-3.2 (20)
Escherichia coli 1144230 (ST641) blaCMY-2 mcr-5 (21)
Klebsiella pneumoniae (ST234) blaSHV-27 blaKPC-2 (22)
Citrobacter freundii BM19 blaKPC-2 (17)
Enterobacter cloacae DUB blaFRI-1 (8)
Pseudomonas aeruginosa PA43417 blaOXA-198 (7)
Pseudomonas aeruginosa PA43417
dsbA1 dsbA1 blaOXA-198 This study

271
Table S4. Plasmids used in this study.
Name Description Source
pDM1 pDM1 vector (GenBank MN128719), p15A
ori, Ptac promoter, MCS, TetR Lab stock
pDM1-blaL2-1 blaL2-1 cloned into pDM1, TetR This study
pDM1-blaGES-1 blaGES-1 cloned into pDM1, TetR This study
pDM1-blaGES-2 blaGES-2 cloned into pDM1, TetR This study
pDM1-blaGES-11 blaGES-11 cloned into pDM1, TetR This study
pDM1-blaSHV-27 blaSHV-27 cloned into pDM1, TetR This study
pDM1-blaOXA-4 blaOXA-4 cloned into pDM1, TetR This study
pDM1-blaOXA-10 blaOXA-10 cloned into pDM1, TetR This study
pDM1-blaOXA-198 blaOXA-198 cloned into pDM1, TetR This study
pDM1-blaFRI-1 blaFRI-1 cloned into pDM1, TetR This study
pDM1-blaL1-1 blaL1-1 cloned into pDM1, TetR This study
pDM1-blaKPC-2 blaKPC-2 cloned into pDM1, TetR This study
pDM1-blaKPC-3 blaKPC-3 cloned into pDM1, TetR This study
pDM1-blaSME-1 blaSME-1 cloned into pDM1, TetR This study
pDM1-mcr-1 mcr-1 cloned into pDM1, TetR This study
pDM1-mcr-3 mcr-3 cloned into pDM1, TetR This study
pDM1-mcr-3.2 mcr-3.2 cloned into pDM1, TetR This study
pDM1-mcr-4 mcr-4 cloned into pDM1, TetR This study
pDM1-mcr-5 mcr-5 cloned into pDM1, TetR This study
pDM1-mcr-8 mcr-8 cloned into pDM1, TetR This study
pDM1-blaL2-1-StrepII blaL2-1 encoding L2-1 with a C-terminal
StrepII tag cloned into pDM1, TetR This study
pDM1-blaGES-1-StrepII blaGES-1 encoding GES-1 with a C-terminal
StrepII tag cloned into pDM1, TetR This study
pDM1-StrepII-blaOXA-4 blaOXA-4 encoding OXA-4 with an N-terminal
StrepII tag cloned into pDM1, TetR This study
pDM1-blaOXA-10-StrepII blaOXA-10 encoding OXA-10 with a C-terminal
StrepII tag cloned into pDM1, TetR This study
pDM1-blaOXA-198-StrepII blaOXA-198 encoding OXA-198 with a C-
terminal StrepII tag cloned into pDM1, TetR This study
pDM1-blaFRI-1-StrepII blaFRI-1 encoding FRI-1 with a C-terminal
StrepII tag cloned into pDM1, TetR This study
pDM1-blaL1-1-StrepII blaL1-1 encoding L1-1 with a C-terminal
StrepII tag cloned into pDM1, TetR This study
pDM1-blaKPC-3-StrepII blaKPC-3 encoding KPC-3 with a C-terminal
StrepII tag cloned into pDM1, TetR This study
pDM1-mcr-1-StrepII blaMCR-1 encoding MCR-1 with a C-terminal
StrepII tag cloned into pDM1, TetR This study
pDM1-mcr-3-StrepII blaMCR-3 encoding MCR-3 with a C-terminal
StrepII tag cloned into pDM1, TetR This study
pDM1-mcr-4-StrepII blaMCR-4 encoding MCR-4 with a C-terminal
StrepII tag cloned into pDM1, TetR This study

272
pDM1-mcr-5-StrepII blaMCR-5 encoding MCR-5 with a C-terminal
StrepII tag cloned into pDM1, TetR This study
pDM1-mcr-8-StrepII blaMCR-8 encoding MCR-8 with a C-terminal
StrepII tag cloned into pDM1, TetR This study
pGRG25
Encodes a Tn7 transposon and tnsABCD under
the control of ParaB, thermosensitive pSC101
ori, AmpR
(23)
pGRG25-Ptac::dsbA
Ptac::dsbA fragment cloned within the Tn7 of
pGRG25; when inserted into the chromosome
and the plasmid cured, the strain expresses
DsbA upon IPTG induction, AmpR
This study
pSLTS Thermosensitive pSC101ori, ParaB for λ-Red,
PtetR for I-SceI, AmpR (24)
pUltraGFP-GM
Constitutive sfGFP expression from a strong
Biofab promoter, p15A ori, (template for the
accC cassette), GentR
(25)
pKD4 Conditional oriRγ ori, (template for the aphA
cassette), AmpR (26)
pKNG101
Gene replacement suicide vector, oriR6K,
oriTRK2, sacB, (template for the strAB
cassette), StrR
(27)
pKNG101-dsbA1
PCR fragment containing the regions upstream
and downstream P. aeruginosa dsbA1 cloned
in pKNG101; when inserted into the
chromosome the strain is a merodiploid for
dsbA1 mutant, StrR
This study
pRK600 Helper plasmid, ColE1 ori, mobRK2, traRK2,
CamR (28)
pMA-T mcr-3 GeneArt® cloning vector containing mcr-3,
ColE1 ori, (template for mcr-3), AmpR This study
pMK-T mcr-8 GeneArt® cloning vector containing mcr-8,
ColE1 ori, (template for mcr-8), KanR This study

273
Table S5. Oligonucleotide primers used in this study. The “Brief description” column provides
basic information on the primer design (restriction enzyme used for cloning, encoded protein
or gene replaced by antibiotic resistance cassette, forward or reverse orientation of the primer
(F or R); QC stands for QuickChange primers and SQ stands for sequencing primers).
Numbe
r
Brief description Sequence (5ˊ-3ˊ)
P1 SacI.L2.F ctggagctcctcgcccgtcgccgatt
P2 XmaI.L2.R ctgcccgggtcatccgatcaaccggtcggca
P3 SacI.GES.F ctggagctccgcttcattcacgcac
P4 XmaI.GES.R ctgcccgggctatttgtccgtgctcaggatg
P5 SacI.SHV.F ctggagctccgttatattcgcctgtg
P6 XmaI.SHV.R ctgcccgggttagcgttgccagtgctcga
P7 SacI.OXA-4.F ctggagctcaaaaacacaatacatataacttcgc
P8 KpnI.OXA-4.R cagggtaccttataaatttagtgtgtttagaatggtg
P9 SacI.OXA-10.F ctggagctcaaaacatttgccgcatatgtaattatcgc
P10 KpnI.OXA-10.R cagggtaccttagccaccaatgatgccctc
P11 NdeI.OXA-198.F actgcatatgcataaacacatgagtaagctcttc
P12 KpnI.OXA-198.R ctgggtaccttattcgatgatcccctttgctt
P13 SacI.FRI-1.F ctggagctctttttttttaaaaaaggtgcaagtac
P14 XmaI.FRI-1.R ctgcccgggttatttataacttccataaactgcctttatagc
P15 SacI.L1.F ctggagctccgttctaccctgctcgc
P16 XhoI.L1.R actgagctctcagcgggccccggccgt
P17 SacI.KPC.F ctggagctctcactgtatcgccgtc
P18 KpnI.KPC.R ctgccatggttactgcccgttgacgccca
P19 SacI.SME-1.F ctggagctctcaaacaaagtaaattttaaaacgg
P20 XmaI.SME-1.R ctgcccgggttaatcaattgcctgaattgcaatacg
P21 SacI.MCR-1.F ctggagctcatgcagcatacttctgtgtggtac
P22 XmaI.MCR-1.R ctgcccgggtcagcggatgaatgcggtgc
P23 NdeI.MCR-3.F ctgatacatatgccttcccttataaaaataaaaattgttccg
P24 XmaI.MCR-3.R cagcccgggttattgaacattacgacattgactgaaaatatctag
P25 SacI.MCR-4.F ctggagctccgtgctgacgagatttaaaaccc
P26 XmaI.MCR-4.R ctgcccgggttaaccgcggcagcgggcaaaaatatc
P27 SacI.MCR-5.F ctggagctccggttgtctgcatttatcac
P28 XmaI.MCR-5.R ctgcccgggtcattgtggttgtccttttctg
P29 SacI.MCR-8.F ctggagctcttcaagtatcttttatctttcaaact aacc
P30 XmaI.MCR-8.R ctgcccgggctaaccattcccatctgttttctc
P31 QC.GES5-GES1.F aaagagccggagatgggcgacaacacacctg
P32 QC.GES5-GES1.R caggtgtgttgtcgcccatctccggctcttt
P33 QC.KPC2-KPC3.F ctaacaaggatgacaagtacagcgaggccgtcatc
P34 QC.KPC2-KPC3.R gatgacggcctcgctgtacttgtcatccttgttag
P35 QC.MCR3-3.2.F caacgcctttctctttgataaatccaggtgacatcc
P36 QC.MCR3-3.2.R ggatgtcacctggatttatcaaagagaaaggcgttg
P37 XmaI.StrepII.L2.R ctgcccgggttatttttcaaattgcggatggctccaagcgctccctccg
atcaaccggtcggca

274
P38 XmaI.StrepII.GES.R ctgcccgggctatttttcaaattgcggatggctccaagcgctccctttg
tccgtgctcaggatgag
P39 OXA-4.body.F tcaacagatatctctactgttgca
P40 OXA-4.StrepII.R tgcaacagtagagatatctgttgatttttcaaattgcggatggctccaa
gcgctccctgcactggcgctgctgta
P41 KpnI.StrepII.OXA-10.R cagggtaccttatttttcaaattgcggatggctccaagcgctcccgcc
accaatgatgccctcacttg
P42 KpnI.StrepII.OXA-198.R ctgggtaccttatttttcaaattgcggatggctccaagcgctcccttcga
tgatcccctttgcttg
P43 XmaI.StrepII.FRI-1.R ctgcccgggttatttttcaaattgcggatggctccaagcgctccctttat
aacttccataaactgcctttatagc
P44 KpnI.StrepII.L1.R gggggtacctcatttttcaaattgcggatggctccaagcgctcccgcg
ggccccggccgtttccttggccaactgc
P45 KpnI.StrepII.KPC.R ctgccatggttatttttcaaattgcggatggctccaagcgctcccctgc
ccgttgacgcccaatc
P46 XmaI.StrepII.MCR-1.R cagcccgggttatttttcaaattgcggatggctccaagcgctcccgcg
gatgaatgcggtgcggt
P47 XmaI.StrepII.MCR-3.R cagcccgggttatttttcaaattgcggatggctccaagcgctcccttga
acattacgacattgactgaaaatatctag
P48 XmaI.StrepII.MCR-4.R ctgcccgggctatttttcaaattgcggatggctccaagcgctcccacc
gcggcagcgggcaaaaatatc
P49 XmaI.StrepII.MCR-5.R ctgcccgggctatttttcaaattgcggatggctccaagcgctcccttgt
ggttgtccttttctgca
P50 XmaI.StrepII.MCR-8.R ctgcccgggctatttttcaaattgcggatggctccaagcgctcccacc
attcccatctgttttctctcttac
P51 NotI.Ptac.EcDsbA.F
ctggcggccgctgacaattaatcatcggctcgtataatgtgtggaatt
gtgactagtcgaggtccaggacctcggatcgctaagataggatgatt
gtatgaaaaagatttggctggc
P52 XhoI.EcDsbA.R ctgctcgagttattttttctcggacagatatttc
P53 EcdsbA::aphA.F atgaaaaagatttggctggcgctggctggtttagttttagcgtttagcg
cgtgtaggctggagctgcttc
P54 EcdsbA::aphA.R ttattttttctcggacagatatttcactgtatcagcatactgctgaacaag
ggaattagccatggtccat
P55 EcacrA::aphA.F atgaacaaaaacagagggtttacgcctctggcggtcgttctggtgtag
gctggagctgcttc
P56 EcacrA::aphA.R ttaagacttggactgttcaggctgagcaccgcttgcggcttggggaat
tagccatggtccat
P57 EctolC::aphA.F ttttacagtttgatcgcgctaaatactgcttcaccacaaggaatgcaag
tgtaggctggagctgcttc
P58 EctolC::aphA.R tcgtcgtcatcagttacggaaagggttatgatgggaattagccatggt
cc
P59 EcdegP::strAB.F atgaaaaaaaccacattagcactgagtgcactggctctgagtttaggt
ttggaactgcacattcgggatatttctc
P60 EcdegP::strAB.R ttactgcattaacaggtagatggtgctgtcgccgcgctgaatgttgagt
gccaggccggatctagatatctagtatga

275
P61 EcmarR::accC.F
atggttaatcagaagaaagatcgcctgcttaacgagtatctgtctccg
ctggtgaagttcctatactttctagagaataggaacttcaagatcccct
g
P62 EcmarR::accC.R
ttacggcaggactttcttaagcaaatactcaagtgttgccacttcgtcc
gcgaagttcctattctctagaaagtataggaacttcacttactcaatgg
aattctagatcg
P63 SQ.dsbA1.Paeruginosa.F tacctgctcaagcagatgcatg
P64 SQ.dsbA1.Paeruginosa.R ggtgttcatgtcgcccatca
P65 XbaI.dsbA1.F ggttcctctagagcctacttcgccagccagaa
P66 dsbA1.body.R ctacttcttgttacgcatcgttcactc
P67 dsbA1.body.F atgcgtaacaagaagtaggcaaggtga
P68 BamHI.dsbA1.R aattaaggatcctcatcactaccaccagcgcg

276
Table S6. Sources of genomic DNA used for amplification of β-lactamase and MCR genes in
this study.
Strain Gene(s) Source
Stenotrophomonas maltophilia ATCC 13637 blaL2-1 blaL1-1 ATCC
Pseudomonas aeruginosa GW-1 blaGES-2 (29)
Enterobacter cloacae CHE-2 blaGES-5 (30)
Acinetobacter baumannii K45 blaGES-11 (31)
Klebsiella pneumoniae ST234 blaSHV-27 blaKPC-2 (22)
Pseudomonas aeruginosa SOF1 blaOXA-4 (32)
Pseudomonas aeruginosa PU21 blaOXA-10 (33)
Pseudomonas aeruginosa PA41437 blaOXA-198 (7)
Enterobacter cloacae DUB blaFRI-1 (8)
Serratia marcescens blaSME-1 (22)
Escherichia coli CNR1790 mcr-1 (18)
Shewanella bicestrii JAB-1 mcr-4 (34)
Escherichia coli 1144230 mcr-5 (21)

277
LEGENDS FOR SUPPLEMENTARY DATA FILES
File S1. Analysis of the cysteine content and phylogeny of all identified β-lactamases.
3,947 unique β-lactamase protein sequences were clustered with a 90% identity threshold and
the centroid of each cluster was used as a phylogenetic cluster identified for each sequence
(“Phylogenetic cluster” column). All sequences were searched for the presence of cysteine
residues (“Total number of cysteines” and “Positions of all cysteines” columns). Proteins with
two or more cysteines after the first 30 amino acids of their primary sequence (cells shaded in
grey in the “Number of cysteines after position 30” column) are potential substrates of the DSB
system for organisms where oxidative protein folding is carried out by DsbA and provided that
translocation of the β-lactamase outside the cytoplasm is performed by the Sec system. The
first 30 amino acids of each sequence were excluded to avoid considering cysteines that are
part of the signal sequence mediating the translocation of these enzymes outside the cytoplasm.
Cells shaded in grey in the “Reported in pathogens” column mark β-lactamases that are found
in pathogens or organisms capable of causing opportunistic infections. The Ambler class of
each enzyme is indicated in the “Ambler class column” and each class (A, B1, B2, B3, C and
D) is highlighted with a different color.

278
SUPPLEMENTARY REFERENCES
1. Zhang H, Srinivas S, Xu Y, Wei W, Feng Y. Genetic and biochemical mchanisms for
bacterial lipid A modifiers associated with polymyxin resistance. Trends Biochem Sci.
2019;44(11):973-88.
2. Corkill JE, Cuevas LE, Gurgel RQ, Greensill J, Hart CA. SHV-27, a novel cefotaxime-
hydrolysing β-lactamase, identified in Klebsiella pneumoniae isolates from a Brazilian
hospital. J Antimicrob Chemother. 2001;47(4):463-5.
3. Schultz SC, Dalbadie-McFarland G, Neitzel JJ, Richards JH. Stability of wild-type and
mutant RTEM-1 β-lactamases: effect of the disulfide bond. Proteins. 1987;2(4):290-7.
4. Silvestro L, Weiser JN, Axelsen PH. Antibacterial and antimembrane activities of
cecropin A in Escherichia coli. Antimicrob Agents Chemother. 2000;44(3):602-7.
5. Dailey FE, Berg HC. Mutants in disulfide bond formation that disrupt flagellar
assembly in Escherichia coli. Proc Natl Acad Sci U S A. 1993;90(3):1043-7.
6. Aubert D, Poirel L, Ali AB, Goldstein FW, Nordmann P. OXA-35 is an OXA-10-
related β-lactamase from Pseudomonas aeruginosa. J Antimicrob Chemother. 2001;48(5):717-
21.
7. El Garch F, Bogaerts P, Bebrone C, Galleni M, Glupczynski Y. OXA-198, an acquired
carbapenem-hydrolyzing class D β-lactamase from Pseudomonas aeruginosa. Antimicrob
Agents Chemother. 2011;55(10):4828-33.
8. Dortet L, Poirel L, Abbas S, Oueslati S, Nordmann P. Genetic and biochemical
characterization of FRI-1, a carbapenem-hydrolyzing class A β-lactamase from Enterobacter
cloacae. Antimicrob Agents Chemother. 2015;59(12):7420-5.
9. Palzkill T. Metallo-β-lactamase structure and function. Ann N Y Acad Sci.
2013;1277:91-104.
10. Papp-Wallace KM, Bethel CR, Distler AM, Kasuboski C, Taracila M, Bonomo RA.
Inhibitor resistance in the KPC-2 β-lactamase, a preeminent property of this class A β-
lactamase. Antimicrob Agents Chemother. 2010;54(2):890-7.
11. Hanahan D. In: Glover DM, editor. DNA Cloning: A Practical Approach. 1: IRL Press,
McLean, Virginia; 1985. p. 109.
12. Herrero M, de Lorenzo V, Timmis KN. Transposon vectors containing non-antibiotic
resistance selection markers for cloning and stable chromosomal insertion of foreign genes in
Gram-negative bacteria. J Bacteriol. 1990;172(11):6557-67.
13. Boyer HW, Roulland-Dussoix D. A complementation analysis of the restriction and
modification of DNA in Escherichia coli. J Mol Biol. 1969;41(3):459-72.
14. Casadaban MJ, Cohen SN. Analysis of gene control signals by DNA fusion and cloning
in Escherichia coli. J Mol Biol. 1980;138(2):179-207.

279
15. Kadokura H, Tian H, Zander T, Bardwell JC, Beckwith J. Snapshots of DsbA in action:
detection of proteins in the process of oxidative folding. Science. 2004;303(5657):534-7.
16. Blattner FR, Plunkett G, 3rd, Bloch CA, Perna NT, Burland V, Riley M, et al. The
complete genome sequence of Escherichia coli K-12. Science. 1997;277(5331):1453-62.
17. Dortet L, Brechard L, Poirel L, Nordmann P. Rapid detection of carbapenemase-
producing Enterobacteriaceae from blood cultures. Clin Microbiol Infect. 2014;20(4):340-4.
18. Dortet L, Bonnin RA, Pennisi I, Gauthier L, Jousset AB, Dabos L, et al. Rapid detection
and discrimination of chromosome- and MCR-plasmid-mediated resistance to polymyxins by
MALDI-TOF MS in Escherichia coli: the MALDIxin test. J Antimicrob Chemother.
2018;73(12):3359-67.
19. Beyrouthy R, Robin F, Lessene A, Lacombat I, Dortet L, Naas T, et al. MCR-1 and
OXA-48 in vivo acquisition in KPC-producing Escherichia coli after colistin treatment.
Antimicrob Agents Chemother. 2017;61(8).
20. Haenni M, Beyrouthy R, Lupo A, Chatre P, Madec JY, Bonnet R. Epidemic spread of
Escherichia coli ST744 isolates carrying mcr-3 and blaCTX-M-55 in cattle in France. J Antimicrob
Chemother. 2018;73(2):533-6.
21. Wise MG, Estabrook MA, Sahm DF, Stone GG, Kazmierczak KM. Prevalence of mcr-
type genes among colistin-resistant Enterobacteriaceae collected in 2014-2016 as part of the
INFORM global surveillance program. PloS One. 2018;13(4):e0195281.
22. Nordmann P, Poirel L, Dortet L. Rapid detection of carbapenemase-producing
Enterobacteriaceae. Emerg Infect Dis. 2012;18(9):1503-7.
23. McKenzie GJ, Craig NL. Fast, easy and efficient: site-specific insertion of transgenes
into enterobacterial chromosomes using Tn7 without need for selection of the insertion event.
BMC Microbiol. 2006;6:39.
24. Kim J, Webb AM, Kershner JP, Blaskowski S, Copley SD. A versatile and highly
efficient method for scarless genome editing in Escherichia coli and Salmonella enterica. BMC
Biotechnol. 2014;14:84.
25. Mavridou DA, Gonzalez D, Clements A, Foster KR. The pUltra plasmid series: a robust
and flexible tool for fluorescent labeling of Enterobacteria. Plasmid. 2016;87-88:65-71.
26. Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia
coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97(12):6640-5.
27. Kaniga K, Delor I, Cornelis GR. A wide-host-range suicide vector for improving
reverse genetics in Gram-negative bacteria: inactivation of the blaA gene of Yersinia
enterocolitica. Gene. 1991;109(1):137-41.
28. Kessler B, Delorenzo V, Timmis KN. A general system to integrate lacZ fusions into
the chromosomes of Gram-negative eubacteria: regulation of the Pm Promoter of the TOL
plasmid studied with all controlling elements in monocopy. Mol Gen Genet. 1992;233(1-
2):293-301.

280
29. Poirel L, Weldhagen GF, Naas T, De Champs C, Dove MG, Nordmann P. GES-2, a
class A β-lactamase from Pseudomonas aeruginosa with increased hydrolysis of imipenem.
Antimicrob Agents Chemother. 2001;45(9):2598-603.
30. Poirel L, Carrer A, Pitout JD, Nordmann P. Integron mobilization unit as a source of
mobility of antibiotic resistance genes. Antimicrob Agents Chemother. 2009;53(6):2492-8.
31. Bonnin RA, Rotimi VO, Al Hubail M, Gasiorowski E, Al Sweih N, Nordmann P, et al.
Wide dissemination of GES-type carbapenemases in Acinetobacter baumannii isolates in
Kuwait. Antimicrob Agents Chemother. 2013;57(1):183-8.
32. Aubert D, Poirel L, Chevalier J, Leotard S, Pages JM, Nordmann P. Oxacillinase-
mediated resistance to cefepime and susceptibility to ceftazidime in Pseudomonas aeruginosa.
Antimicrob Agents Chemother. 2001;45(6):1615-20.
33. Dortet L, Poirel L, Nordmann P. Rapid detection of carbapenemase-producing
Pseudomonas spp. J Clin Microbiol. 2012;50(11):3773-6.
34. Jousset AB, Dabos L, Bonnin RA, Girlich D, Potron A, Cabanel N, et al. CTX-M-15-
producing Shewanella species clinical isolate expressing OXA-535, a chromosome-encoded
OXA-48 variant, putative progenitor of the plasmid-encoded OXA-436. Antimicrob Agents
Chemother. 2018;62(1).




















