
The Role of JNK and p38 MAPK Activities in UVA-Induced Signaling Pathways Leading to AP1 Activation...
11
The Role of JNK and p38 MAPK Activities in UVA-Induced Signaling Pathways Leading to AP-1 Activation and c-Fos Expression 1 Amy L. Silvers, Michael A. Bachelor and G. Timothy Bowden Department of Radiation Oncology, Arizona Cancer Center, The University of Arizona, Tucson, AZ, USA Abstract To further delineate ultraviolet A (UVA) signaling pathways in the human keratinocyte cell line HaCaT, we examined the potential role of mitogen-activated protein kinases (MAPKs) in UVA-induced activator protein-1 (AP-1) transactivation and c-Fos expres- sion. UVA-induced phosphorylation of p38 and c-Jun N-terminal kinase (JNK) proteins was detected im- mediately after irradiation and disappeared after approximately 2 hours. Conversely, phosphorylation of extracellular signal-regulated kinase was signifi- cantly inhibited for up to 1 hour post-UVA irradi- ation. To examine the role of p38 and JNK MAPKs in UVA-induced AP-1 and c-fos transactivations, the selective pharmacologic MAPK inhibitors, SB202190 (p38 inhibitor) and SP600125 (JNK inhibitor), were used to independently treat stably transfected HaCaT cells in luciferase reporter assays. Both SB202190 and SP600125 dose-dependently inhibited UVA-induced AP-1 and c-fos transactivations. SB202190 (0.25– 0.5 MM) and SP600125 (62–125 nM) treatments also primarily inhibited UVA-induced c-Fos expression. These results demonstrated that activation of both JNK and p38 play critical role in UVA-mediated AP-1 transactivation and c-Fos expression in these human keratinocyte cells. Targeted inhibition of these MAPKs with their selective pharmacologic inhibitors may be effective chemopreventive strategies for UVA-induced nonmelanoma skin cancer. Neoplasia (2003) 5, 319 – 329 Keywords: UVA; AP-1; MAPK; c-Fos; HaCaT. Introduction Exposure to sunlight is the major risk factor for the develop- ment of nonmelanoma skin cancer [1]. Because strato- spheric ozone completely absorbs short-wavelength ultraviolet C (<280 nm), the relevant carcinogenic compo- nents of sunlight that reach the Earth’s surface are ultraviolet B (UVB) (280–320 nm) and ultraviolet A (UVA) (320–400 nm) [2,3]. Although UVA is magnitudes less carcinogenic than UVB [4,5], chronic UVA exposure has been shown to induce photoaging [6] and skin tumors (papillomas and squ- amous cell carcinomas) [5,7,8] in experimental animals. The signaling pathways involved in UVA-induced photo- aging and skin tumorigenesis are subjects of intense interest and have not been fully elucidated. UVA has been shown to alter the expression of numerous mammalian genes, such as heme oxygenase-1 [9], intercellular adhesion molecule-1 (ICAM-1) [10], and matrix metalloproteinase-1 [11], through the generation of reactive oxygen intermediates, specifically singlet O 2 [12]. UVA irradiation has also been reported to activate several transcription factors, including activator pro- tein-1 (AP-1) [13 – 15], activator protein-2 (AP-2) [10], nuclear factor kappa-B (NFnB) [16,17], and signal transducer and activator of transcription (STAT) 1 and 3 [18,19]. Both UVA- induced AP-1 and AP-2 activations were mediated through singlet oxygen [10,14]. AP-1 is an important regulatory protein involved in cell growth, differentiation, transformation, and apoptosis, and may also contribute to inflammatory and immune responses [20,21]. It can be induced by growth factors, cytokines, 12-O- tetradecanoylphorbol-13-acetate (TPA), UV radiation, and transforming oncoproteins [20]. The AP-1 complex consists of heterodimers of Fos (c-Fos, Fra-1, Fra-2, and FosB) and Jun (c-Jun, JunB, and JunD) family members, or homodimers and heterodimers of Jun family members that bind to TPA response elements (TREs) in AP-1 – inducible gene promoters, contribu- ting to transcriptional activity or repression of these genes [20]. Abbreviations: AP-1, activator protein-1; AP-2, activator protein-2; ATF-2, activating transcription factor-2; DMEM, Dulbecco’s modified Eagle’s medium; DTT, dithiothreitol; EDTA, ethylenediamine-tetraacetic acid; EGTA, ethylene glycol-bis(h-aminoethly ether)- N,N,N V,N V-tetraacetic acid; ERK, extracellular signal-regulated kinase; HRP, horseradish peroxidase; ICAM-1, intercellular adhesion molecule-1; JNK, c-Jun NH 2 -terminal kinase; MAPK, mitogen-activated protein kinase; MAPKAPK2, MAPK-activated protein kinase-2; MEF, MADS box transcription enhancer factor; MEK, MAPK/ERK kinase; NFnB, nuclear factor kappa-B; PBS, phosphate-buffered saline; SAP, serum response factor accessory protein; SDS, sodium dodecyl sulfate; SRE, serum response element; STAT, signal transducer and activator of transcription; TBST, Tris-buffered saline with 0.05% Tween 20; TPA, 12-O-tetradecanoylphorbol-13-acetate; TRE, TPA response element; UVA, ultraviolet A Address all correspondence to: G. Timothy Bowden, Arizona Cancer Center, Room 4999, 1515 North Campbell Avenue, Tucson, AZ 85724, USA. E-mail: [email protected] 1 This work was supported, in part, by the National Cancer Institute–funded grant R25CA78447 through the Cancer Prevention and Control Program at the Arizona Cancer Center, Tucson, AZ. This work was also supported by the National Institutes of Health grants CA27502 and CA23074. Received 9 February 2003; Revised 18 June 2003; Accepted 18 June 2003. Copyright D 2003 Neoplasia Press, Inc. All rights reserved 1522-8002/03/$25.00 Neoplasia . Vol. 5, No. 4, July/August 2003, pp. 319 – 329 319 www.neoplasia.com RESEARCH ARTICLE
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of The Role of JNK and p38 MAPK Activities in UVA-Induced Signaling Pathways Leading to AP1 Activation...

The Role of JNK and p38 MAPK Activities in UVA-InducedSignaling Pathways Leading to AP-1 Activation andc-Fos Expression1
Amy L. Silvers, Michael A. Bachelor and G. Timothy Bowden
Department of Radiation Oncology, Arizona Cancer Center, The University of Arizona, Tucson, AZ, USA
Abstract
To further delineate ultraviolet A (UVA) signaling
pathways in the human keratinocyte cell line HaCaT,
we examined the potential role of mitogen-activated
protein kinases (MAPKs) in UVA-induced activator
protein-1 (AP-1) transactivation and c-Fos expres-
sion. UVA-induced phosphorylation of p38 and c-Jun
N-terminal kinase (JNK) proteins was detected im-
mediately after irradiation and disappeared after
approximately 2 hours. Conversely, phosphorylation
of extracellular signal-regulated kinase was signifi-
cantly inhibited for up to 1 hour post-UVA irradi-
ation. To examine the role of p38 and JNK MAPKs in
UVA-induced AP-1 and c-fos transactivations, the
selective pharmacologic MAPK inhibitors, SB202190
(p38 inhibitor) and SP600125 (JNK inhibitor), were
used to independently treat stably transfected HaCaT
cells in luciferase reporter assays. Both SB202190 and
SP600125 dose-dependently inhibited UVA-induced
AP-1 and c-fos transactivations. SB202190 (0.25–
0.5 MM) and SP600125 (62–125 nM) treatments also
primarily inhibited UVA-induced c-Fos expression.
These results demonstrated that activation of both
JNK and p38 play critical role in UVA-mediated AP-1
transactivation and c-Fos expression in these human
keratinocyte cells. Targeted inhibition of these MAPKs
with their selective pharmacologic inhibitors may be
effective chemopreventive strategies for UVA-induced
nonmelanoma skin cancer.
Neoplasia (2003) 5, 319 – 329
Keywords: UVA; AP-1; MAPK; c-Fos; HaCaT.
Introduction
Exposure to sunlight is the major risk factor for the develop-
ment of nonmelanoma skin cancer [1]. Because strato-
spheric ozone completely absorbs short-wavelength
ultraviolet C (<280 nm), the relevant carcinogenic compo-
nents of sunlight that reach the Earth’s surface are ultraviolet
B (UVB) (280–320 nm) and ultraviolet A (UVA) (320–400
nm) [2,3]. Although UVA is magnitudes less carcinogenic
than UVB [4,5], chronic UVA exposure has been shown to
induce photoaging [6] and skin tumors (papillomas and squ-
amous cell carcinomas) [5,7,8] in experimental animals.
The signaling pathways involved in UVA-induced photo-
aging and skin tumorigenesis are subjects of intense interest
and have not been fully elucidated. UVA has been shown to
alter the expression of numerous mammalian genes, such as
heme oxygenase-1 [9], intercellular adhesion molecule-1
(ICAM-1) [10], and matrix metalloproteinase-1 [11], through
the generation of reactive oxygen intermediates, specifically
singlet O2 [12]. UVA irradiation has also been reported to
activate several transcription factors, including activator pro-
tein-1 (AP-1) [13–15], activator protein-2 (AP-2) [10], nuclear
factor kappa-B (NFnB) [16,17], and signal transducer and
activator of transcription (STAT) 1 and 3 [18,19]. Both UVA-
induced AP-1 and AP-2 activations were mediated through
singlet oxygen [10,14].
AP-1 is an important regulatory protein involved in cell
growth, differentiation, transformation, and apoptosis, and
may also contribute to inflammatory and immune responses
[20,21]. It can be induced by growth factors, cytokines, 12-O-
tetradecanoylphorbol-13-acetate (TPA), UV radiation, and
transforming oncoproteins [20]. The AP-1 complex consists
of heterodimers of Fos (c-Fos, Fra-1, Fra-2, and FosB) and Jun
(c-Jun, JunB, and JunD) family members, or homodimers and
heterodimers of Jun family members that bind to TPA response
elements (TREs) in AP-1–inducible gene promoters, contribu-
ting to transcriptional activity or repression of these genes [20].
Abbreviations: AP-1, activator protein-1; AP-2, activator protein-2; ATF-2, activating
transcription factor-2; DMEM, Dulbecco’s modified Eagle’s medium; DTT, dithiothreitol;
EDTA, ethylenediamine-tetraacetic acid; EGTA, ethylene glycol-bis(h-aminoethly ether)-
N,N,N V,N V-tetraacetic acid; ERK, extracellular signal-regulated kinase; HRP, horseradish
peroxidase; ICAM-1, intercellular adhesion molecule-1; JNK, c-Jun NH2-terminal kinase;
MAPK, mitogen-activated protein kinase; MAPKAPK2, MAPK-activated protein kinase-2;
MEF, MADS box transcription enhancer factor; MEK, MAPK/ERK kinase; NFnB, nuclear
factor kappa-B; PBS, phosphate-buffered saline; SAP, serum response factor accessory
protein; SDS, sodium dodecyl sulfate; SRE, serum response element; STAT, signal
transducer and activator of transcription; TBST, Tris-buffered saline with 0.05% Tween
20; TPA, 12-O-tetradecanoylphorbol-13-acetate; TRE, TPA response element; UVA,
ultraviolet A
Address all correspondence to: G. Timothy Bowden, Arizona Cancer Center, Room 4999,
1515 North Campbell Avenue, Tucson, AZ 85724, USA. E-mail: [email protected] work was supported, in part, by the National Cancer Institute – funded grant
R25CA78447 through the Cancer Prevention and Control Program at the Arizona Cancer
Center, Tucson, AZ. This work was also supported by the National Institutes of Health grants
CA27502 and CA23074.
Received 9 February 2003; Revised 18 June 2003; Accepted 18 June 2003.
Copyright D 2003 Neoplasia Press, Inc. All rights reserved 1522-8002/03/$25.00
Neoplasia . Vol. 5, No. 4, July/August 2003, pp. 319 – 329 319
www.neoplasia.com
RESEARCH ARTICLE

Deregulated expression of the AP-1 complex has been
shown to play a prominent role in skin tumor promotion. Loss
of AP-1 DNA-binding activity resulted in loss of proliferative
potential and induction of differentiation in human keratino-
cytes [22]. Dong et al. [23] demonstrated that anchorage-
independent growth of JB6 cells required the transactivation
of AP-1.
Deregulated expression of individual AP-1 complex com-
ponents has also been shown to induce malignant trans-
formation in vivo. Transfection of v-fos into murine papilloma
cell lines expressing an activated Ha-ras oncogene resulted
in the malignant conversion of these cells [24]. Similarly, the
development of malignant skin tumors in v-Ha-ras transgenic
mice following TPA treatment was inhibited in c-fos �/� mice
[25]. Young et al. [26] also reported that TPA-mediated
promotion in a two-stage skin carcinogenesis mouse model
was suppressed by the stable expression of an epidermis-
targeted dominant negative c-jun transgene. Similarly,
Thompson et al. [27] demonstrated that the expression of
the same epidermis-targeted dominant negative c-jun trans-
gene inhibited okadaic acid–mediated skin tumor promotion.
The mitogen-activated protein kinase (MAPK) family of
proteins include p38, c-Jun N-terminal kinase (JNK), and
extracellular signal-regulated kinase (ERK). MAPKs are pro-
line-directed serine/threonine kinases that are activated by
dual phosphorylation on threonine and tyrosine residues in
response to a wide variety of extracellular stimuli [28]. They
mediate signal transduction from the cell surface to
the nucleus. Activation of ERK is primarily involved in
growth factor– and phorbol ester–stimulated responses.
Responses to proinflammatory cytokines, UV radiation,
and other stresses are mostly dependent on JNK and p38
activation [21,29]. MAPK signaling pathways have been
shown to affect AP-1 activity by direct phosphorylation
of AP-1 proteins and by influence on the abundance of
individual AP-1 components in a cell [29,30].
c-Jun is directly phosphorylated by JNK at N-terminal
serines 63 and 73, resulting in increased stability and trans-
activation potential [31,32]. c-Jun is also phosphorylated by
ERK1/2 on C-terminal inhibitory sites [33,34]. Although the in
vivo relevance is still unclear, ERK1/2 can also phosphor-
ylate c-Fos and ATF-2 [28,35].
MAPKs also increase the abundance of AP-1 complex
components by transcriptionally activating their promoters.
The ternary complex factors Elk-1 (a substrate of ERK,
p38, and JNK) and serum response factor accessory
protein (SAP) 1a and 2 (substrates of ERK and p38) form
complexes with dimeric serum response factors at the
serum response element (SRE) in the c-fos promoter
[28,30]. Additionally, activation of STAT1 and STAT3 by
JNK [18] and possibly ERK [30,36] at the cis-inducible
element in the c-fos promoter may act in cooperation
with the SRE to influence c-fos expression [30]. Induction
of c-jun expression is predominantly mediated by two
TREs that preferentially bind c-Jun and ATF-2 hetero-
dimers. These proteins are activated by phosphorylation
in their transactivation domains [29]. JNK and p38 phos-
phorylate and activate ATF-2, whereas JNK phosphorylates
the c-Jun activation domain [37,38]. Additionally, ERK and
p38 activation may contribute to c-jun expression through
phosphorylation of MEF2 proteins—transcription factors
that also bind to the c-jun promoter [39,40].
Although a few studies have addressed the potential role
of MAPK activation in UVA signaling, results are not con-
sistent. Klotz et al. [12] reported a rapid and transient
induction of p38 and JNK activity, but not ERK activity, in
human skin fibroblasts. In contrast, UVA irradiation stimu-
lated the activation of all three MAPKs in the NCTC 2544
human keratinocyte cell line [41] and in the mouse epidermal
JB6 promotion-sensitive Cl 41 cell line [42,43]. Additionally,
Djavaheri-Mergny and Dubertret [14] provided evidence that
UVA-induced AP-1 activation required the Raf/ERK pathway
in NCTC 2544 keratinocytes.
Our previous results demonstrated that UVA irradiation of
the human immortalized keratinocyte cell line HaCaT
induced the expression of several AP-1 family members,
including c-Fos and c-Jun, and potentiated the transactiva-
tion of the c-fos promoter and the AP-1–binding site in the
collagenase-1 gene promoter [15]. To further delineate the
UVA signaling pathway(s) in these keratinocytes, we exam-
ined the potential role of MAPKs in UVA-induced AP-1
transactivation and c-fos expression through the use of
specific pharmacologic MAPK inhibitors. We report, for the
first time, that p38 and JNK MAPKs contributed to UVA-
induced AP-1 activation and UVA-induced c-fos transactiva-
tion as well as UVA-induced c-Fos protein expression. The
use of SB202190 and SP600125 to selectively inhibit their
respective UVA-induced stress-activated protein kinases
may be a useful chemopreventive strategy for UVA-induced
nonmelanoma skin cancer.
Materials and Methods
Cell Culture
The human keratinocyte cell line, HaCaT, was stably
transfected with a sequence from the human collagenase-1
gene promoter (�73 to +63) containing one endogenous
AP-1 – binding site driving a luciferase reporter gene
(HCL14 cells), as reported previously [44]. HaCaT cells
were also independently stably transfected with a sequence
from the human c-fos promoter (�404 to +41) driving a
luciferase reporter gene (FL30 cells), as reported previously
[44]. These cells were cultured in Dulbecco’s modified
Eagle’s medium (DMEM) supplemented with 10% fetal
bovine serum and 100 U/ml penicillin/streptomycin at
37jC and 5% CO2. Cells were grown to near confluence
and then serum-starved for 24 to 28 hours prior to treat-
ment. Treatment of cells with selective pharmacologic
MAPK inhibitors involved a 1-hour preirradiation incubation
and variable postirradiation incubations. Each compound
was diluted in serum-free medium prior to use.
Pharmacologic Inhibitors of MAPKs
SB202190 (Calbiochem, San Diego, CA) is a potent, cell-
permeable, selective, and reversible inhibitor of p38a and
320 UVA-Induced Signaling Pathways Silvers et al.
Neoplasia . Vol. 5, No. 4, 2003

h1 isoforms. Inhibition is competitive with ATP and requires
substrates to accommodate the fluorophenyl ring structure of
the pyridinyl imidazole in their ATP-binding pocket. Struc-
tures of other p38 isoforms, JNKs, and ERK1/2 do not allow
inhibitor binding at equivalent positions [45].
SP600125 (Calbiochem) is a potent, cell-permeable,
selective, and reversible inhibitor of JNK1, JNK2, and
JNK3 isoforms. Inhibition is competitive with ATP and may
involve the interaction of the nitrogen-containing ring system
of the anthrapyrazolone with key residues in the kinase
active site of substrates [46].
PD98059 (Alexis Biochemicals, Carlsbad, CA) is a selec-
tive, cell-permeable inhibitor of MEK1, MEK2, and MEK5 that
does not directly inhibit kinase activity but rather prevents the
activation of the kinase [45,47,48]. It has also been shown to
directly inhibit cyclooxygenase-1 and cyclooxygenase-2
enzyme activities [49].
UVA Irradiation
Cells were irradiated with a bank of four F20T12/BL/HO
UVA bulbs (National Biological, Twinsburg, OH) that were
powered by an Advance electronic ballast REL-4P32-RH-TP
(120 V, 60 Hz, 90 A) (Advance Transformer Co., Chicago, IL).
Spectral emission was reported previously [15]. A UVX
radiometer equipped with a UVX-36 sensor (UVP, Upland,
CA) was used to measure radiation doses. Plate glass
(4 mm) was used to filter wavelengths below 320 nm.
Irradiation was performed in a sterile, well-ventilated laminar
flow hood to eliminate thermal stimulation. Cells were irradi-
ated in phosphate-buffered saline (PBS) supplemented with
0.01% MgCl2 and 0.01% CaCl2 at room temperature. Control
cells were mock-irradiated under similar conditions. Cultures
were continued in serum-free DMEM until harvest.
Luciferase Assay for AP-1 and c-fos Transactivations
Total cellular protein from stably transfected HaCaT
cells was extracted in lysis buffer (15 mM MgSO4, 25 mM
glycylglycine, 4 mM EGTA, 1% vol/vol Triton X-100, and 1
mM DTT) and was quantitated using the Bio-Rad Dc
Protein Assay kit (Bio-Rad Laboratories, Hercules, CA).
Luciferase activity of 20 to 30 Ag of total cellular protein
was measured using the TD-20/20 Luminometer (Turner
Designs, Sunnyvale, CA).
Western Analysis
Total cellular protein was extracted in lysis buffer (150 mM
NaCl, 20 mM Tris, pH 7.5, 1 mM EDTA, 1 mM EGTA, 2.5 mM
Na4P2O7, 1 mM h-glycerol phosphate, 1 mM Na3VO4, 1 Ag/
ml leupeptin, and 1% Triton X-100) supplemented with addi-
tional phosphatase inhibitors (1 mM Na3VO4, 2 Ag/ml leu-
peptin, and 10 Ag/ml aprotinin) and was quantitated using the
Bio-Rad Dc Protein Assay kit (Bio-Rad Laboratories).
Lysates (40 Ag) were resolved on 12.5% sodium dodecyl
sulfate (SDS) polyacrylamide gels and were then transferred
to Immobilon-P nylon membranes (Millipore, Bedford, MA).
Membranes were blocked with 5% evaporated milk in 1�TBS/0.05% Tween 20 (TBST) for 2 hours at room temper-
ature. Primary antibodies against AP-1 family members
Figure 1. Western time course for UVA-induced MAPK activation. HCL14 cells were mock-irradiated or irradiated with 250 kJ/m2 UVA and harvested at the
appropriate time points postirradiation. Forty micrograms of total cell lysate was electrophoresed on 12.5% SDS polyacrylamide gels, transferred to Immobilon-P
membranes, and immunodetected using optimal primary and secondary antibody concentrations for each MAPK. Western blot data are representative of at least
two independent experiments. (A) p38; (B) JNK; and (C) ERK.
UVA-Induced Signaling Pathways Silvers et al. 321
Neoplasia . Vol. 5, No. 4, 2003
(Santa Cruz Biotechnology, Santa Cruz, CA) were diluted
at 1:1000 (c-Fos) or 1:500 (c-Jun), primary antibody against
a-tubulin (Oncogene Research Products, Boston, MA) was
diluted at 1:2500, and primary antibodies against p38, JNK,
and ERK MAPKs (Cell Signaling, Beverly, MA) were diluted at
1:2000 in 5% evaporated milk/TBST and were incubated
with membranes for 2 hours at room temperature. Primary
antibodies against phospho-MAPKs (Cell Signaling)
and phospho-c-Jun (Cell Signaling) were diluted at 1:800
(phospho-p38) or 1:1000 in 5% evaporated milk/TBST
and were incubated with membranes overnight at 4jC.
Anti– rabbit HRP–conjugated secondary antibodies (Cell
Signaling) were diluted at 1:2000, whereas anti–mouse
HRP–conjugated secondary antibodies (Santa Cruz
Biotechnology) were diluted at 1:5000 (c-Fos) and 1:8000
(a-tubulin) in 5% evaporated milk/TBST and were incubated
with membranes for 1 hour at room temperature. Membranes
were washed three times for 10 minutes in TBST after each
antibody incubation. Protein bands were visualized using the
ECL kit (Amersham Pharmacia Biotech, Piscataway, NJ).
In Vivo p38 Activity Assay
The in vivo p38 activity assay was reported previously
[50]. Briefly, total cellular protein was extracted in lysis buffer
(150 mM NaCl, 20 mM Tris, pH 7.5, 1 mM EDTA, 1 mM
EGTA, 2.5 mM Na4P2O7, 1 mM h-glycerol phosphate, 1 mM
Na3VO4, 1 Ag/ml leupeptin, and 1% Triton X-100) supple-
mented with additional phosphatase inhibitors (1 mM
Na3VO4, 2 Ag/ml leupeptin, and 10 Ag/ml aprotinin) and
was quantitated using the Bio-Rad Dc Protein Assay kit
(Bio-Rad Laboratories). Lysates (10 Ag) were resolved on
12.5% SDS polyacrylamide gels overnight at 4jC and
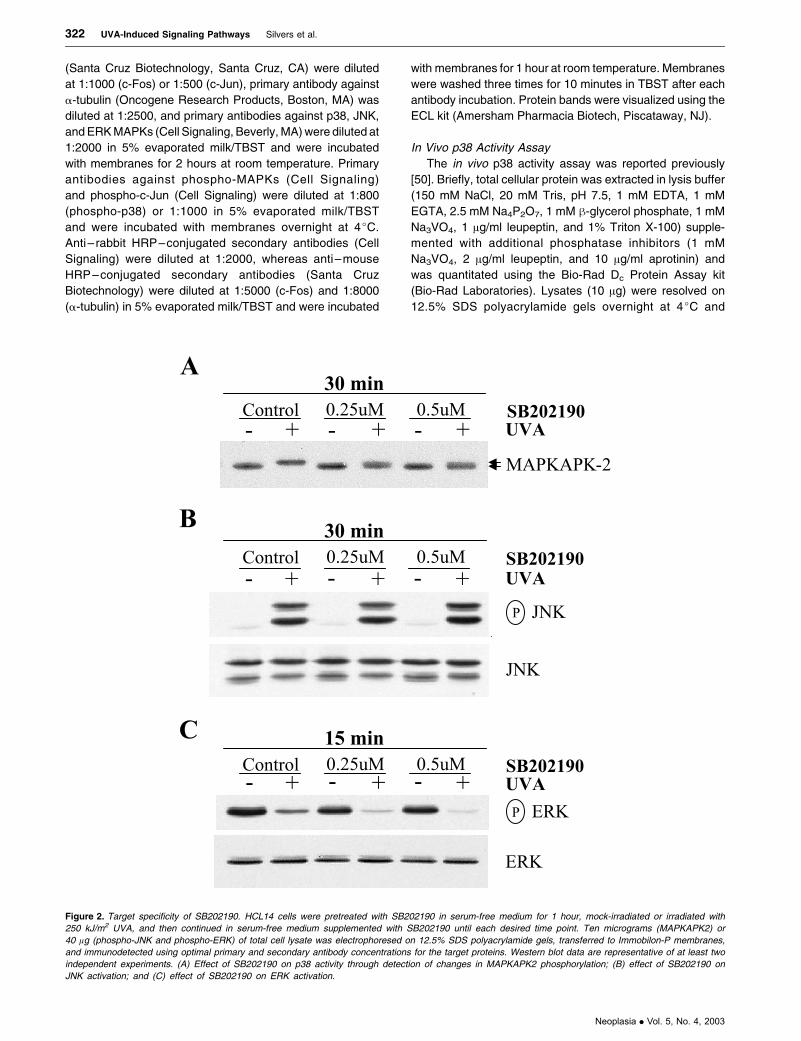
Figure 2. Target specificity of SB202190. HCL14 cells were pretreated with SB202190 in serum-free medium for 1 hour, mock-irradiated or irradiated with
250 kJ/m2 UVA, and then continued in serum-free medium supplemented with SB202190 until each desired time point. Ten micrograms (MAPKAPK2) or
40 lg (phospho-JNK and phospho-ERK) of total cell lysate was electrophoresed on 12.5% SDS polyacrylamide gels, transferred to Immobilon-P membranes,
and immunodetected using optimal primary and secondary antibody concentrations for the target proteins. Western blot data are representative of at least two
independent experiments. (A) Effect of SB202190 on p38 activity through detection of changes in MAPKAPK2 phosphorylation; (B) effect of SB202190 on
JNK activation; and (C) effect of SB202190 on ERK activation.
322 UVA-Induced Signaling Pathways Silvers et al.
Neoplasia . Vol. 5, No. 4, 2003
were then transferred to Immobilon-P nylon membranes
(Millipore). The membrane was blocked with 5% evaporated
milk/1� TBS/0.1% Tween 20 for 1 hour at room temperature
and was then washed three times for 5 minutes in 1� TBS/
0.1% Tween 20. MAPK-activated protein kinase-2 (MAP-
KAPK2) primary antibody (Cell Signaling) was diluted at
1:1000 in 5% BSA/1� TBS/0.1% Tween 20 and was incu-
bated with the membrane overnight at 4jC. HRP-conjugated
secondary antibody (Cell Signaling) was diluted at 1:2000 in
5% evaporated milk/1� TBS/0.1% Tween 20 and was incu-
bated with the membrane for 1 hour at room temperature.
The membrane was washed three times for 5 minutes in 1�TBS/0.1% Tween 20 after each antibody incubation. Protein
bands were visualized using the ECL kit (Amersham Phar-
macia Biotech). Both phosphorylated and unphosphorylated
bands were detected.
Results
Effects of UVA Irradiation on the Activation of MAPKs
As reported previously, maximal AP-1 transactivation in
the stably transfected HaCaT cells was achieved with a dose
of 250 kJ/m2 UVA. This dose of UVA increased AP-1 trans-
activation levels between 2 and 8 hours post-UVA [15].
Therefore, time courses for Western analyses to detect the
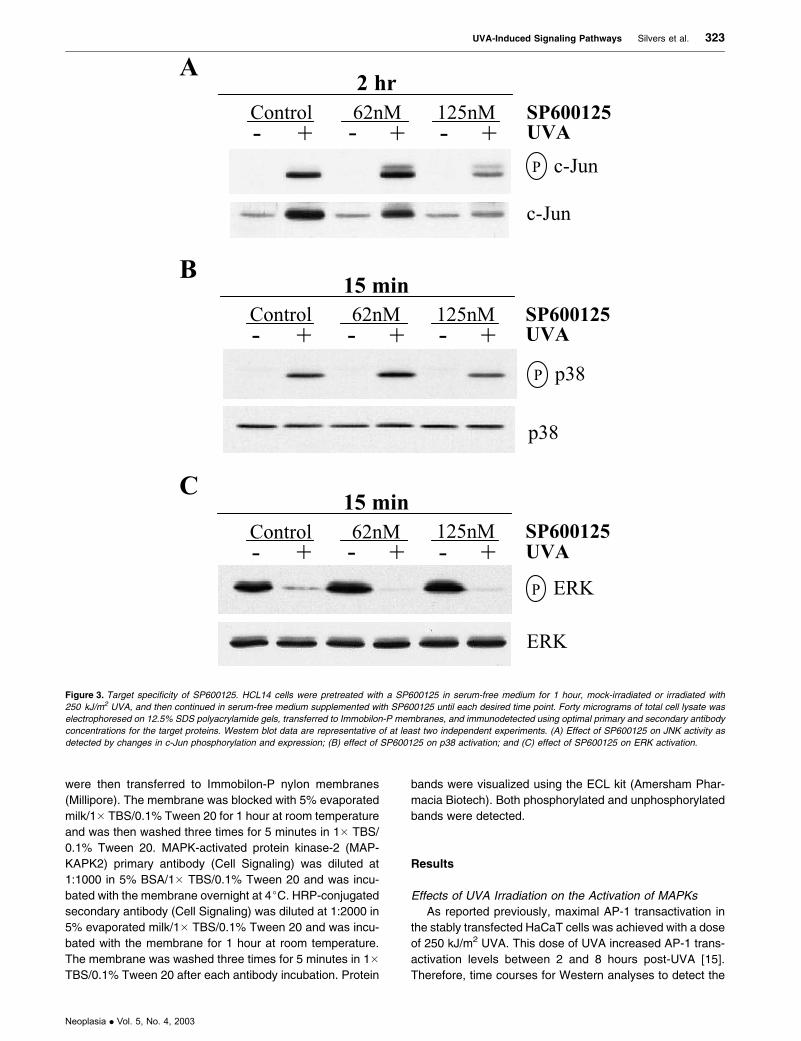
Figure 3. Target specificity of SP600125. HCL14 cells were pretreated with a SP600125 in serum-free medium for 1 hour, mock-irradiated or irradiated with
250 kJ/m2 UVA, and then continued in serum-free medium supplemented with SP600125 until each desired time point. Forty micrograms of total cell lysate was
electrophoresed on 12.5% SDS polyacrylamide gels, transferred to Immobilon-P membranes, and immunodetected using optimal primary and secondary antibody
concentrations for the target proteins. Western blot data are representative of at least two independent experiments. (A) Effect of SP600125 on JNK activity as
detected by changes in c-Jun phosphorylation and expression; (B) effect of SP600125 on p38 activation; and (C) effect of SP600125 on ERK activation.
UVA-Induced Signaling Pathways Silvers et al. 323
Neoplasia . Vol. 5, No. 4, 2003
UVA-induced activation of MAPKs were performed on these
cells using 250 kJ/m2 UVA, beginning immediately after
irradiation and spanning 8 hours post-UVA irradiation
(Figure 1).
Phosphorylation of p38 was detected immediately after
UVA irradiation and persisted for approximately 1 hour, with
only very weak activation detected at 2, 4, and 8 hours
postirradiation (Figure 1A). Total p38 levels were unaffected
by UVA irradiation. Increased phosphorylation of JNK was
detected immediately after irradiation and disappeared after
approximately 2 hours, with maximal expression between 15
and 30 minutes postirradiation (Figure 1B). UVA irradiation
did not affect basal levels of JNK. Interestingly, UVA irradi-
ation transiently but consistently inhibited the activation of
ERK. This inhibition was detected immediately after irradi-
ation and persisted for up to 1 hour post-UVA (Figure 1C).
Conversely, ERK was transiently activated in mock-treated
cells. This activation may have been due to the environ-
mental change experienced by the cells after irradiation,
specifically the switch from supplemented PBS to serum-
free medium. Despite this activation, ERK phosphorylation
was consistently lower in irradiated lysates compared to
controls. Basal levels of ERK expression were unaffected
by UVA irradiation.
Target Specificity of SB202190
Concentrations of SB202190 as high as 15 AM have
been used in HaCaT cells to assess the role of p38 a/hisoforms in UV-induced signaling pathways [50–53]. How-
ever, much lower doses of this compound (2 AM) have
been shown to inhibit UVA-induced p38 activity [53]. In
vivo p38 activity assays were performed using even lower
doses of SB202190 (0.25 and 0.5 AM) in HCL14 cells to
ensure that these doses were also inhibitory. At 30 minutes
post-UVA irradiation, the p38 MAPK substrate MAPKAPK2
was primarily phosphorylated, whereas MAPKAPK2 in
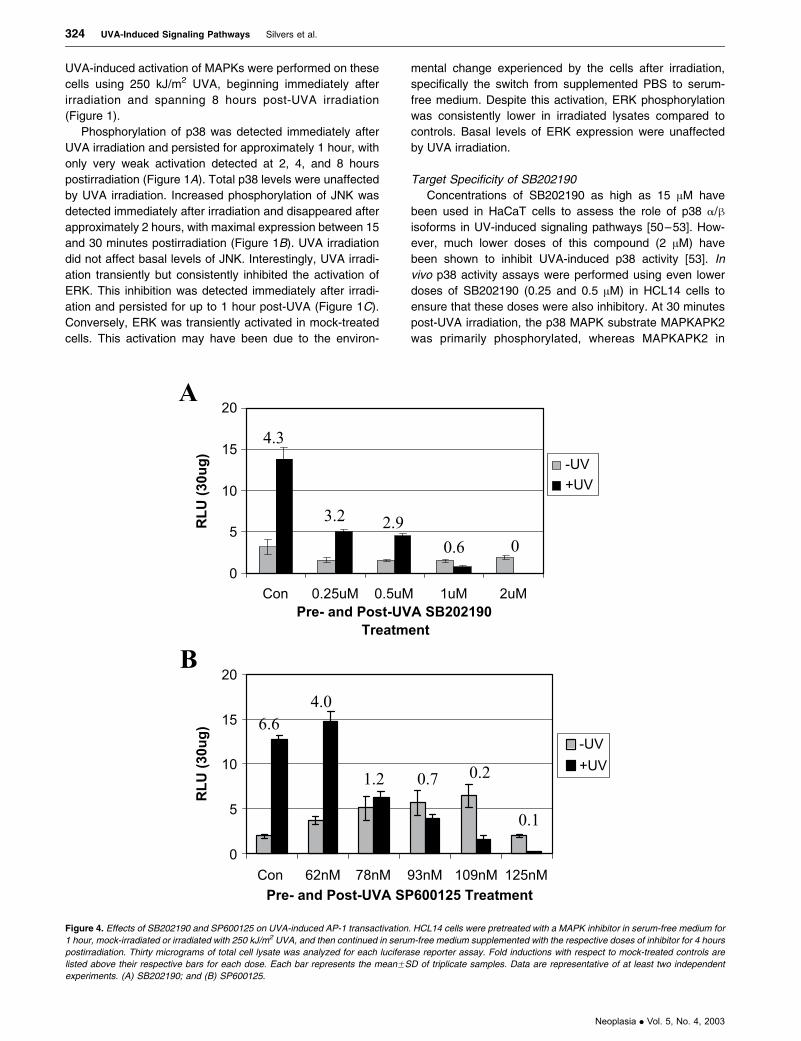
Figure 4. Effects of SB202190 and SP600125 on UVA-induced AP-1 transactivation. HCL14 cells were pretreated with a MAPK inhibitor in serum-free medium for
1 hour, mock-irradiated or irradiated with 250 kJ/m2 UVA, and then continued in serum-free medium supplemented with the respective doses of inhibitor for 4 hours
postirradiation. Thirty micrograms of total cell lysate was analyzed for each luciferase reporter assay. Fold inductions with respect to mock-treated controls are
listed above their respective bars for each dose. Each bar represents the meanFSD of triplicate samples. Data are representative of at least two independent
experiments. (A) SB202190; and (B) SP600125.
324 UVA-Induced Signaling Pathways Silvers et al.
Neoplasia . Vol. 5, No. 4, 2003
mock-treated cells was unphosphorylated. Treatment with
a combination of UVA and SB202190 resulted in detection
of both phosphorylated and unphosphorylated forms of
MAPKAPK2, indicating that SB202190 reduced the
capacity of p38 to phosphorylate and activate MAPKAPK2
(Figure 2A).
To determine the target specificity of SB202190, a time
point was selected for each MAPK that would allow for the
detection of inhibition of UVA-mediated forms of the
kinases by the pharmacologic inhibitor. No effects on
JNK phosphorylation were detected with 0.25 or 0.5 AM
SB202190 at 30 minutes post-UVA irradiation (Figure 2B).
Minimal effects of SB202190 on ERK phosphorylation were
observed, with band intensity decreasing slightly in both
unirradiated and irradiated groups at 15 minutes postirra-
diation (Figure 2C).
Target Specificity of SP600125
The anthrapyrazolone SP600125 has been recently mar-
keted as a potent, selective, and reversible inhibitor of all
three JNK isoforms. The inhibitory activity of SP600125 in
Jurkat T cells and primary human monocytes required IC50
doses between 5 and 10 AM [46]. However, in HaCaT cells,
much lower doses were sufficient to inhibit UVA-induced
JNK activity and c-Jun expression. We previously showed
that c-Jun was biphasically activated by UVA irradiation.
Preexisting c-Jun protein was phosphorylated at early time
points, followed by maximal c-Jun expression between 2 and
4 hours after UVA irradiation [15]. The JNK inhibitor
SP600125 diminished UVA-induced JNK activity as well as
c-Jun expression with a dose of 125 nM at 2 hours postirra-
diation (Figure 3A). The effect on total c-Jun levels might be
a consequence of c-Jun autoregulation through the TRE-
binding sites in the c-Jun promoter [54]. SP600125 treatment
of these human keratinocytes in combination with UVA
resulted in the appearance of a slower-migrating, unidenti-
fied band that may represent a modified form of c-Jun. These
doses of SP600125 did not significantly affect p38 or ERK1/2
phosphorylation at 15 minutes postirradiation (Figure 3, B
and C, respectively).
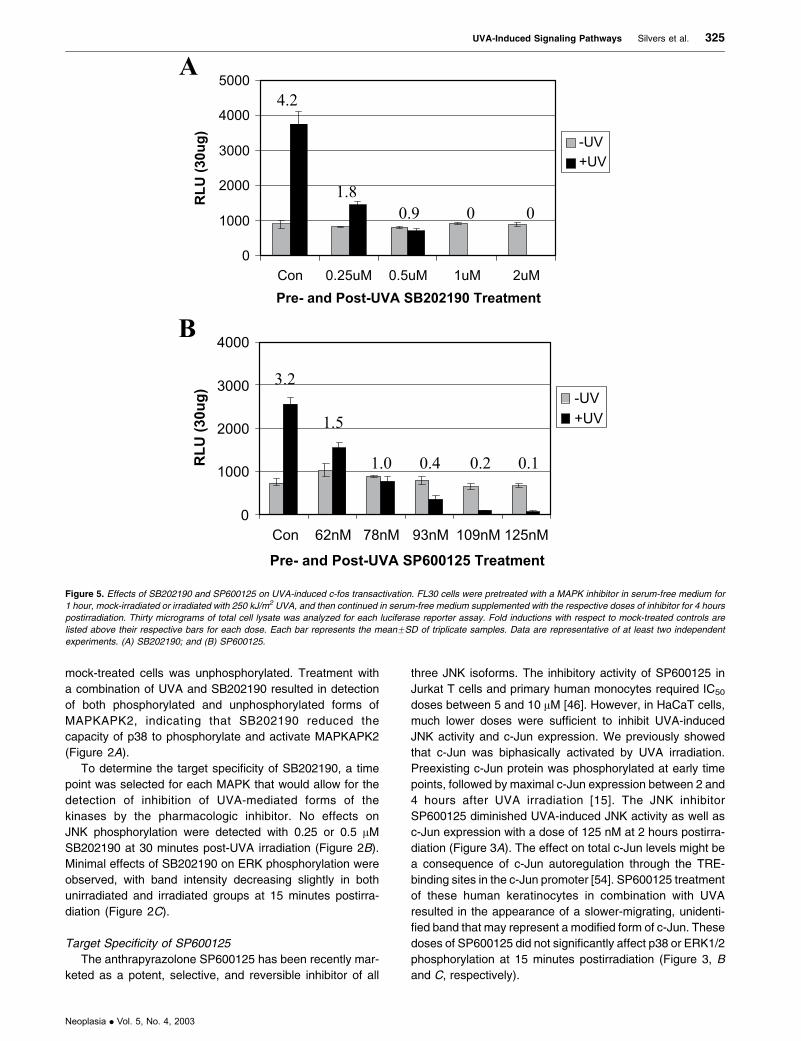
Figure 5. Effects of SB202190 and SP600125 on UVA-induced c-fos transactivation. FL30 cells were pretreated with a MAPK inhibitor in serum-free medium for
1 hour, mock-irradiated or irradiated with 250 kJ/m2 UVA, and then continued in serum-free medium supplemented with the respective doses of inhibitor for 4 hours
postirradiation. Thirty micrograms of total cell lysate was analyzed for each luciferase reporter assay. Fold inductions with respect to mock-treated controls are
listed above their respective bars for each dose. Each bar represents the meanFSD of triplicate samples. Data are representative of at least two independent
experiments. (A) SB202190; and (B) SP600125.
UVA-Induced Signaling Pathways Silvers et al. 325
Neoplasia . Vol. 5, No. 4, 2003
Effects of SB202190 and SP600125 on UVA-Induced AP-1
Transactivation
As previously reported, maximal AP-1 transactivation was
achieved with a dose of 250 kJ/m2 at 4 hours post-UVA
irradiation [15]. Therefore, these experimental conditions
were used to examine the downstream effects of p38 and
JNK MAPK inhibition. Independent dose responses with
SB202190 and SP600125 were performed to determine
the potential involvement of p38 and/or JNK in UVA-induced
AP-1 transactivation using HaCaT cells stably transfected
with the collagenase promoter driving a luciferase reporter
gene (HCL14 cells). As shown in Figure 4A, UVA-induced
AP-1 transactivation was significantly and dose-dependently
blocked with SB202190 treatment at 4 hours post-UVA
irradiation. Doses as low as 0.25 AM inhibited UVA-induced
AP-1 transactivation by as much as 64% compared to
controls. SP600125 also significantly and dose-dependently
blocked UVA-induced AP-1 transactivation at this time point
(Figure 4B). A 78-nM dose of SP600125 resulted in an
approximately 51% reduction in UVA-induced AP-1 trans-
activation compared to controls. The increased AP-1 activity
in unirradiated cells between 62 and 109 nM SP600125 in
this representative experiment was not consistently
observed. Basal levels of AP-1 activity tended to bobble
around control levels in additional experiments.
Effects of SB202190 and SP600125 on UVA-Induced c-fos
Transactivation
To determine whether p38 and JNK were involved in UVA-
induced c-fos transactivation, HaCaT cells that were stably
transfected with the human c-fos promoter driving a lucifer-
ase reporter gene (FL30 cells) were treated with varying
doses of their pharmacologic inhibitors in combination with
250 kJ/m2 UVA. As shown in Figure 5A, UVA-induced c-fos
transactivation was significantly and dose-dependently
blocked with SB202190 at 4 hours post-UVA irradiation.
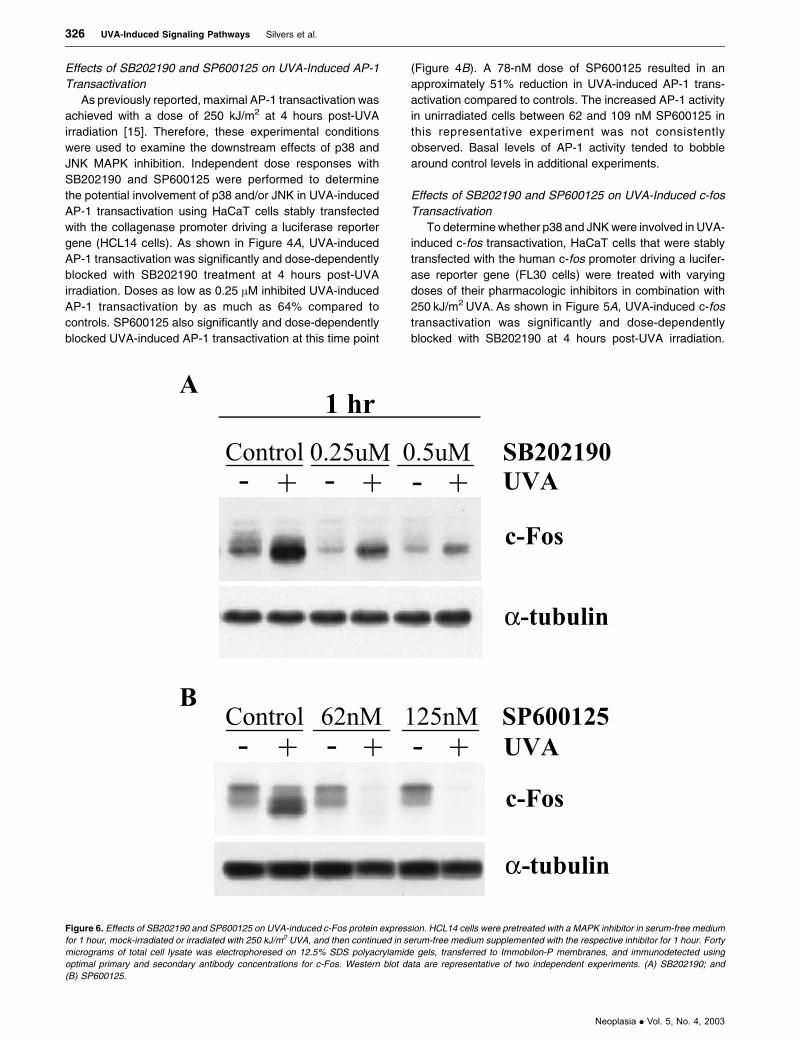
Figure 6. Effects of SB202190 and SP600125 on UVA-induced c-Fos protein expression. HCL14 cells were pretreated with a MAPK inhibitor in serum-free medium
for 1 hour, mock-irradiated or irradiated with 250 kJ/m2 UVA, and then continued in serum-free medium supplemented with the respective inhibitor for 1 hour. Forty
micrograms of total cell lysate was electrophoresed on 12.5% SDS polyacrylamide gels, transferred to Immobilon-P membranes, and immunodetected using
optimal primary and secondary antibody concentrations for c-Fos. Western blot data are representative of two independent experiments. (A) SB202190; and
(B) SP600125.
326 UVA-Induced Signaling Pathways Silvers et al.
Neoplasia . Vol. 5, No. 4, 2003

The effects of drug combined with UVA irradiation paral-
leled the effects observed with this combination treatment
on AP-1 transactivation, with a dose of 0.25 AM SB202190
reducing UVA-induced c-fos transactivation by 61% com-
pared to controls. SP600125 also significantly and dose-
dependently blocked UVA-induced c-fos transactivation
(Figure 5B). Although SP600125 dose-dependently inhibited
both AP-1 and c-fos transactivations, 62 nM SP600125
consistently blocked UVA-induced c-fos transactivation
(approximately 39% of control values) but did not effectively
block UVA-induced AP-1 transactivation. In fact, in some
experiments, this dose of SP600125 appeared to slightly
enhance AP-1 transactivation.
Effects of SB202190 and SP600125 on c-Fos Expression
To further explore the downstream effects of p38 and JNK
inhibition in irradiated and unirradiated keratinocytes,
changes in c-Fos expression were examined. At 1 hour
postirradiation, the dose of SB202190 that blocked both
AP-1 and c-fos transactivations also significantly inhibited
UVA-induced c-Fos expression and only marginally affected
basal c-Fos expression (Figure 6A). Doses of SP600125 that
reduced UVA-induced c-fos transactivation were also able to
block UVA-induced c-Fos expression, whereas basal c-Fos
expression was not affected by drug treatment (Figure 6B).
The observed effects of these MAPK inhibitors on activation
of the c-fos promoter and c-Fos protein expression were
highly correlative in these human keratinocytes.
Discussion
A prominent role for UVA in skin tumor promotion has
recently emerged, involving altered expression of mamma-
lian genes through activation of transcription factors such as
AP-1, AP-2, NFnB, and STATs [10,13–19]. UVA irradiation
was previously shown to induce AP-1 DNA binding and AP-1
transactivation in the human keratinocyte cell line HaCaT.
This induction correlated with UVA-induced c-fos promoter
activation and c-Fos protein expression [15]. To further
delineate the UVA signaling pathway(s) in these immortal-
ized keratinocytes, we examined potential roles for p38,
JNK, and ERK MAPKs in the mediation of UVA-induced
AP-1 and c-fos promoter activations as well as UVA-induced
c-Fos expression.
UVA irradiation (250 kJ/m2) rapidly and transiently
induced p38 and JNK activation but inhibited ERK activa-
tion. Although peak activation or inhibition of UVA-induced
MAPK activities differed slightly, all three kinases returned
to basal levels by 2 hours postirradiation. These combined
effects of UVA on MAPK activities may have temporarily
halted proliferative signaling and allowed the keratinocytes
to respond to the environmental stress through the p38
and JNK pathways before progressing through the cell
cycle. Interestingly, basal levels of ERK activity were
induced up to 15 minutes post-UVA irradiation. This induc-
tion may have been due to the environmental change
experienced by the cells after the long irradiation period.
Despite this induction, ERK activity was consistently lower
in UVA-irradiated samples compared to controls. Basal
levels of activated p38 and JNK were not affected by the
transition. Total MAPK protein levels were not affected by
UVA irradiation.
Studies that have addressed the potential roles of
MAPKs in UVA signaling have been inconsistent. In the
NCTC 2544 human keratinocyte cell line, UVA-induced
ERK, JNK, and p38 activation through the generation of
oxidative stress [14,41]. UVA-mediated AP-1 DNA binding
and transcriptional activity through the Raf/ERK cascade,
with dose-dependent ERK activation considerably declining
up to 240 kJ/m2 UVA [14]. In mouse epidermal JB6
promotion-sensitive Cl 41 cells, UVA also activated ERK,
JNK, and p38 MAPKs at doses between 40 and 160 kJ/m2
[42,43]. Klotz et al. examined MAPK activation patterns
induced by singlet oxygen and UVA in human skin fibro-
blasts. With 300 kJ/m2 UVA, the authors reported a rapid and
transient induction of p38 and JNK but no effect on ERK
activation [12,55]. Although UVA irradiation did not appear to
mediate ERK activity, unirradiated controls for each time
point were not reported. Interestingly, intracellularly gener-
ated singlet oxygen activated p38 and JNK but appeared to
inhibit ERK activity [12]. Similarly, Zhang et al. observed the
UVA-induced activation of p38 and JNK but no activation of
ERK in the normal human lymphoblast cell line JY. At
60 minutes post-UVA irradiation, however, these cells
appeared to have decreased levels of ERK phosphorylation
in comparison to controls [56].
The pyridinyl imidazole SB202190 and the anthrapy-
razolone SP600125 were selected to implicate p38a/hand JNK, respectively, as components in the UVA-induced
signaling pathways leading to transcriptional activation of
AP-1–responsive genes. As potential modulators of AP-1
transactivation and c-Fos expression, these MAPK pharma-
cologic inhibitors could be clinically useful in skin cancer
chemopreventive strategies. As reported previously, UVA
irradiation of HaCaT cells significantly induced both AP-1
transactivation and c-fos transactivation [15]. Suppression
of JNK with SP600125 and of p38a/h with SB202190
resulted in the abrogation of both AP-1 and c-fos activities
in dose-dependent manners. Additionally, SB202190 and
SP600125 dose-dependently decreased UVA-induced lev-
els of c-Fos protein, the primary component in UVA-induced
AP-1 DNA binding in these cells [15]. The doses of
SB202190 and SP600125 that inhibited these downstream
targets were shown to specifically reduce their respective
UVA-induced MAPK activities.
The JNK inhibitor SP600125 consistently blocked c-fos
transactivation at a dose of 62 nM, whereas AP-1 trans-
activation consistently required doses of 125 nM. Although
c-Jun was a component of the UVA-induced AP-1 DNA-
binding complex in these cells, other Jun family members
that could compete for dimerization and AP-1 site binding
were also present [15]. We demonstrated that JNK inhibition
by SP600125 reduced not only c-Jun activity and expres-
sion, but also c-Fos expression. Preexisting c-Fos protein,
however, could still be activated by c-Fos–regulating kinase
[30,57]. Therefore, higher concentrations of inhibitors may
UVA-Induced Signaling Pathways Silvers et al. 327
Neoplasia . Vol. 5, No. 4, 2003

have been required to effectively inhibit AP-1 activation. In
contrast, JNK activates ternary complex factor proteins such
as Elk-1 and STAT proteins, which bind directly to transcrip-
tional control elements in the c-fos promoter [18,28,30].
Therefore, lower doses of SP600125 may be sufficient to
see an effect on this promoter construct.
SP600125 treatment of HaCaT cells with UVA irradia-
tion not only inhibited the phosphorylation of c-Jun on
serines 63 and 73 but also decreased total c-Jun protein
levels. The c-Jun promoter contains two AP-1–binding sites
that allow c-Jun protein to regulate its own transcription [54].
Therefore, inhibition of c-Jun phosphorylation may also
affect total c-Jun protein levels.
The doses of SP600125 that inhibited UVA-induced c-Jun
activity and expression were considerably lower than IC50
doses reported in primary human monocytes stimulated with
lipopolysaccharide and Jurkat T cells stimulated with phor-
bol-12-myristate-13-acetate plus anti-CD3 and anti-CD28
(5–10 AM) [46]. Additionally, although doses of SB202190
as high as 15 AM have been used in HaCaT cells to
determine the role of p38a/h in UV signaling, much lower
doses were capable of inhibiting UVA-induced p38 activity. In
addition to potentially diverse signaling pathways between
cell types, different stimuli can also alter the effects of an
inhibitor on cells. The combination of 250 kJ/m2 UVA with low
doses of either SB202190 or SP600125 resulted in dramatic
morphologic changes and increased cell death that were
visibly detectable approximately 4 hours post-UVA irradia-
tion (unpublished observations). We are currently exploring
the biologic consequences and significance of these syner-
gistic effects in HaCaT cells as well as in primary human
keratinocytes.
Although ERK activation did not play a role in the UVA-
induced AP-1 transactivation in these human keratino-
cytes, treatment with the MEK1/2 inhibitor PD98059
dose-dependently blocked both basal and UVA-induced
c-fos activation and c-Fos protein levels (data not shown).
The dose-dependent drop in c-fos activity observed in the
UVA-treated groups may have reflected the modulation of
basal c-fos activity. We suggest that ERK activity may
contribute to basal levels of c-fos expression in HaCaT cells.
Our increased elucidation and understanding of cellular
signaling pathways allow mechanistic approaches to chemo-
prevention and chemotherapy. The availability of potent,
selective inhibitors of specific signaling proteins may
allow researchers to further dissect cellular signaling path-
ways, to individually genetically tailor preventive strategies,
and to reduce adverse toxicologic responses elicited by the
screening of natural products with multiple mechanisms of
action [58,59].
Our results demonstrated for the first time that activa-
tion of the stress-activated kinases p38 and JNK played
important roles in UVA-mediated AP-1 transactivation and
c-fos transactivation, and that these kinases are involved in
UVA-induced c-Fos expression, a primary component of
the UVA-induced AP-1 DNA-binding complex [15].
Because deregulated expression of AP-1 has been shown
to play an important role in the neoplastic transformation
of epidermal cells in both cell culture and animal models
[23–26], targeted inhibition of the upstream kinases p38
and JNK with the pyridinyl imidazole SB202190 and the
anthrapyrazolone SP600125, respectively, may be effec-
tive chemopreventive strategies for UVA-induced nonme-
lanoma skin cancer.
References[1] Hall EJ, Astor M, Bedford J, Borek C, Curtis SB, Fry M, Geard C, Hei T,
Mitchell J, Oleinick N, Rubin J, Tu A, Ullrich R, Waldren C, and Ward J
(1988). Basic radiobiology. Am J Clin Oncol 11, 220 – 52.
[2] Moan J (1994). UV-A radiation, melanoma induction, sunscreens, so-
laria and ozone reduction. J Photochem Photobiol B Biol 24, 201 – 203.
[3] Madronich S (1993). The atmosphere and UV-B radiation at ground
level. In Environmental UV Photobiology. AR Young, LO Bjorn, J Moan,
W Nultsch (Eds.). Plenum, New York, pp. 1– 39.
[4] Kelfkens G, de Gruijl FR, and van der Leun JC (1990). Ozone depletion
and increase in annual carcinogenic ultraviolet dose. Photochem Pho-
tobiol 52, 819 – 23.
[5] de Laat A, van der Leun JC, and de Gruijl FR (1997). Carcinogenesis
induced by UVA (365-nm) radiation: the dose – time dependence of
tumor formation in hairless mice. Carcinogenesis 18, 1013 –1020.
[6] Kligman LH (1991). The hairless mouse and photoaging. Photochem
Photobiol 54, 1109 –118.
[7] Kelfkens G, de Gruijl FR, and van der Leun JC (1991). Tumorigenesis
by short-wave ultraviolet A: papillomas versus squamous cell carcino-
mas. Carcinogenesis 12, 1377 – 382.
[8] Sterenborg HJ, and van der Leun JC (1990). Tumorigenesis by a long
wavelength UV-A source. Photochem Photobiol 51, 325 – 30.
[9] Basu-Modak S, and Tyrrell RM (1993). Singlet oxygen: a primary
effector in the ultraviolet A/near-visible light induction of the human
heme oxygenase gene. Cancer Res 53, 4505 –510.
[10] Grether-Beck S, Olaizola-Horn S, Schmitt H, Grewe M, Jahnke A,
Johnson JP, Briviba K, Sies H, and Krutmann J (1996). Activation of
transcription factor AP-2 mediates UVA radiation – and singlet oxy-
gen – induced expression of the human intercellular adhesion molecule
1 gene. Proc Natl Acad Sci USA 93, 14586 –591.
[11] Wlaschek M, Briviba K, Stricklin GP, Sies H, and Scharffetter-Kochanek
K (1995). Singlet oxygen may mediate the ultraviolet A – induced
synthesis of interstitial collagenase. J Invest Dermatol 104, 194 – 98.
[12] Klotz LO, Pellieux C, Briviba K, Pierlot C, Aubry JM, and Sies H (1999).
Mitogen-activated protein kinase (p38-, JNK-, ERK-) activation pattern
induced by extracellular and intracellular singlet oxygen and UVA. Eur J
Biochem 260, 917 – 22.
[13] Djavaheri-Mergny M, Mergny JL, Bertrand F, Santus R, Maziere C,
Dubertret L, and Maziere JC (1996). Ultraviolet-A induces activation
of AP-1 in cultured human keratinocytes. FEBS Lett 384, 92 –96.
[14] Djavaheri-Mergny M, and Dubertret L (2001). UV-A – induced AP-1
activation requires the Raf/ERK pathway in human NCTC 2544 kerati-
nocytes. Exp Dermatol 10, 204 –10.
[15] Silvers AL, and Bowden GT (2002). UVA irradiation-induced activation
of activator protein-1 is correlated with induced expression of AP-1
family members in the human keratinocyte cell line HaCaT. Photochem
Photobiol 75, 302 –10.
[16] Djavaheri-Mergny M, Gras MP, Mergny JL, and Dubertret L (1999). UV-
A– induced decrease in nuclear factor-kappaB activity in human kera-
tinocytes. Biochem J 338, 607 –13.
[17] Vile GF, Tanew-Ilitschew A, and Tyrrell RM (1995). Activation of NF-
kappa B in human skin fibroblasts by the oxidative stress generated by
UVA radiation. Photochem Photobiol 62, 463 – 68.
[18] Zhang Y, Liu G, and Dong Z (2001). MSK1 and JNKs mediate phos-
phorylation of STAT3 in UVA-irradiated mouse epidermal JB6 cells.
J Biol Chem 276, 42534 –542.
[19] Maziere C, Dantin F, Dubois F, Santus R, and Maziere J (2000). Bi-
phasic effect of UVA radiation on STAT1 activity and tyrosine phos-
phorylation in cultured human keratinocytes. Free Radic Biol Med 28,
1430 – 437.
[20] Angel P, and Karin M (1991). The role of Jun, Fos and the AP-1 com-
plex in cell-proliferation and transformation. Biochim Biophys Acta
1072, 129 – 57.
[21] Shaulian E, and Karin M (2001). AP-1 in cell proliferation and survival.
Oncogene 20, 2390 –400.
[22] Briata P, D’Anna F, Franzi AT, and Gherzi R (1993). AP-1 activity
328 UVA-Induced Signaling Pathways Silvers et al.
Neoplasia . Vol. 5, No. 4, 2003

during normal human keratinocyte differentiation: evidence for a
cytosolic modulator of AP-1/DNA binding. Exp Cell Res 204, 136 – 46.
[23] Dong Z, Birrer MJ, Watts RG, Matrisian LM, and Colburn NH (1994).
Blocking of tumor promoter – induced AP-1 activity inhibits induced
transformation in JB6 mouse epidermal cells. Proc Natl Acad Sci
USA 91, 609 –13.
[24] Greenhalgh DA, and Yuspa SH (1988). Malignant conversion of mur-
ine squamous papilloma cell lines by transfection with the fos onco-
gene. Mol Carcinog 1, 134 – 43.
[25] Saez E, Rutberg SE, Mueller E, Oppenheim H, Smoluk J, Yuspa SH,
and Spiegelman BM (1995). c-fos is required for malignant progression
of skin tumors. Cell 82, 721 – 32.
[26] Young MR, Li JJ, Rincon M, Flavell RA, Sathyanarayana BK, Hunziker
R, and Colburn N (1999). Transgenic mice demonstrate AP-1 (activator
protein-1) transactivation is required for tumor promotion. Proc Natl
Acad Sci USA 96, 9827 – 832.
[27] Thompson EJ, MacGowan J, Young MR, Colburn N, and Bowden GT
(2002). A dominant negative c-jun specifically blocks okadaic acid –
induced skin tumor promotion. Cancer Res 62, 3044 –3047.
[28] Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M,
Berman K, and Cobb MH (2001). Mitogen-activated protein (MAP)
kinase pathways: regulation and physiological functions. Endocr Rev
22, 153 – 83.
[29] Karin M (1995). The regulation of AP-1 activity by mitogen-activated
protein kinases. J Biol Chem 270, 16483 – 486.
[30] Whitmarsh AJ, and Davis RJ (1996). Transcription factor AP-1 regula-
tion by mitogen-activated protein kinase signal transduction pathways.
J Mol Med 74, 589 – 607.
[31] Hibi M, Lin A, Smeal T, Minden A, and Karin M (1993). Identification
of an oncoprotein- and UV-responsive protein kinase that binds and
potentiates the c-Jun activation domain. Genes Dev 7, 2135 – 148.
[32] Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, and Davis
RJ (1994). JNK1: a protein kinase stimulated by UV light and Ha-ras
that binds and phosphorylates the c-Jun activation domain. Cell 76,
1025 – 1037.
[33] Minden A, Lin A, Smeal T, Derijard B, Cobb M, Davis R, and Karin M
(1994). c-Jun N-terminal phosphorylation correlates with activation of
the JNK subgroup but not the ERK subgroup of mitogen-activated pro-
tein kinases. Mol Cell Biol 14, 6683 – 688.
[34] Chou SY, Baichwal V, Ferrell Jr, JE (1992). Inhibition of c-Jun DNA
binding by mitogen-activated protein kinase. Mol Biol Cell 3, 1117 –130.
[35] Chen RH, Abate C, and Blenis J (1993). Phosphorylation of the c-Fos
transrepression domain by mitogen-activated protein kinase and
90-kDa ribosomal S6 kinase. Proc Natl Acad Sci USA 90, 10952 –956.
[36] Ihle JN (1996). STATs and MAPKs: obligate or opportunistic partners in
signaling. Bioessays 18, 95 –98.
[37] Gupta S, Campbell D, Derijard B, and Davis RJ (1995). Transcription
factor ATF2 regulation by the JNK signal transduction pathway.
Science 267, 389 – 93.
[38] Wilhelm D, van Dam H, Herr I, Baumann B, Herrlich P, and Angel P
(1995). Both ATF-2 and c-Jun are phosphorylated by stress-activated
protein kinases in response to UV irradiation. Immunobiology 193,
143 – 48.
[39] Han J, Jiang Y, Li Z, Kravchenko VV, and Ulevitch RJ (1997). Activation
of the transcription factor MEF2C by the MAP kinase p38 in inflamma-
tion. Nature 386, 296 –99.
[40] Han TH, and Prywes R (1995). Regulatory role of MEF2D in serum
induction of the c-jun promoter. Mol Cell Biol 15, 2907 –915.
[41] Maziere C, Conte MA, Leborgne L, Levade T, Hornebeck W, Santus R,
and Maziere JC (2001). UVA radiation stimulates ceramide production:
relationship to oxidative stress and potential role in ERK, JNK, and p38
activation. Biochem Biophys Res Commun 281, 289 –94.
[42] Zhang Y, Dong Z, Nomura M, Zhong S, Chen N, and Bode AM (2001).
Signal transduction pathways involved in phosphorylation and activa-
tion of p70S6K following exposure to UVA irradiation. J Biol Chem 276,
20913 – 923.
[43] Zhang Y, Zhong S, Dong Z, Chen N, Bode AM, and Ma W (2001). UVA
induces Ser381 phosphorylation of p90RSK/MAPKAP-K1 via ERK and
JNK pathways. J Biol Chem 276, 14572 – 580.
[44] Chen W, Borchers AH, Dong Z, Powell MB, and Bowden GT (1998).
UVB irradiation– induced activator protein-1 activation correlates with
increased c-fos gene expression in a human keratinocyte cell line.
J Biol Chem 273, 32176 –181.
[45] English JM, and Cobb MH (2002). Pharmacological inhibitors of MAPK
pathways. Trends Pharmacol Sci 23, 40 –45.
[46] Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W,
Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning
AM, and Anderson DW (2001). SP600125, an anthrapyrazolone in-
hibitor of Jun N-terminal kinase. Proc Natl Acad Sci USA 98,
13681 – 686.
[47] Davies SP, Reddy H, Caivano M, and Cohen P (2000). Specificity and
mechanism of action of some commonly used protein kinase inhibitors.
Biochem J 351, 95 –105.
[48] Mody N, Leitch J, Armstrong C, Dixon J, and Cohen P (2001). Effects of
MAP kinase cascade inhibitors on the MKK5/ERK5 pathway. FEBS Lett
502, 21 –24.
[49] Borsch-Haubold AG, Pasquet S, and Watson SP (1998). Direct inhib-
ition of cyclooxygenase-1 and -2 by the kinase inhibitors SB 203580
and PD 98059. SB 203580 also inhibits thromboxane synthase. J Biol
Chem 273, 28766 – 772.
[50] Chen W, and Bowden GT (1999). Activation of p38 MAP kinase and
ERK are required for ultraviolet-B induced c-fos gene expression in
human keratinocytes. Oncogene 18, 7469 – 476.
[51] Chen W, and Bowden GT (2000). Role of p38 mitogen-activated pro-
tein kinases in ultraviolet-B irradiation – induced activator protein 1
activation in human keratinocytes. Mol Carcinog 28, 196 –202.
[52] Chen W, Tang Q, Gonzales MS, and Bowden GT (2001). Role of
p38 MAP kinases and ERK in mediating ultraviolet-B induced cyclo-
oxygenase-2 gene expression in human keratinocytes. Oncogene 20,
3921 – 926.
[53] Bachelor MA, Silvers AL, and Bowden GT (2002). The role of p38 in
UVA-induced cyclooxygenase-2 expression in the human keratinocyte
cell line, HaCaT. Oncogene 21, 7092 – 7099.
[54] Angel P, Hattori K, Smeal T, and Karin M (1988). The jun proto-
oncogene is positively autoregulated by its product, Jun/AP-1. Cell
55, 875 – 85.
[55] Klotz LO, Briviba K, and Sies H (1997). Singlet oxygen mediates the
activation of JNK by UVA radiation in human skin fibroblasts. FEBS Lett
408, 289 – 91.
[56] Zhang Y, Mattjus P, Schmid PC, Dong Z, Zhong S, Ma WY, Brown RE,
Bode AM, and Schmid HH (2001). Involvement of the acid sphingo-
myelinase pathway in UVA-induced apoptosis. J Biol Chem 276,
11775 – 782.
[57] Deng T, and Karin M (1994). c-Fos transcriptional activity stimulated by
H-Ras – activated protein kinase distinct from JNK and ERK. Nature
371, 171 – 75.
[58] Shureiqi I, Reddy P, and Brenner DE (2000). Chemoprevention: gen-
eral perspective. Crit Rev Oncol Hematol 33, 157 – 67.
[59] Stratton SP (2001). Prevention of non-melanoma skin cancer. Curr
Oncol Rep 3, 295 – 300.
UVA-Induced Signaling Pathways Silvers et al. 329
Neoplasia . Vol. 5, No. 4, 2003




















