
Dexamethasone induces apoptosis of multiple myeloma cells in a JNK/SAP kinase independent mechanism
7
Dexamethasone induces apoptosis of multiple myeloma cells in a JNK/SAP kinase independent mechanism Dharminder Chauhan 1 , Pramod Pandey 2 , Atsushi Ogata 1 , Gerrard Teoh 1 , Steven Treon 1 , Mitsuyoshi Urashima 1 , Surender Kharbanda 2 and Kenneth C Anderson 1 1 Divisions of Hematologic Malignancies and 2 Cancer Pharmacology, Dana-Farber Cancer Institute, and the Department of Medicine, Harvard Medical School, Boston, Massachusetts 02215, USA The stress-activated protein kinases (SAPKs), also known as c-Jun amino-terminal kinases (JNKs), are activated in response to diverse stimuli including DNA damage, heat shock, interleukin-1, tumor necrosis factor- a and Fas. Although all these inducers cause apoptosis, whether SAPK/JNK activation is required for apoptosis is controversial. In this study, we demonstrate that ionizing radiation (IR) and dexamethasone (Dex) induce apoptosis in multiple myeloma (MM) derived cell lines, as well as in patient cells. IR-induced apoptosis is associated with activation of SAPK/JNK and p38 kinase, in contrast to Dex-induced apoptosis, which is not associated with activation of stress kinases. More- over, Dex-induced apoptosis is associated with a significant decrease in the activities of mitogen activated protein kinase (MAPK) and p70 S6K , whereas IR- treatment does not alter the activity of these kinases. Both IR and Dex induce poly (ADP ribose) polymerase (PARP) cleavage, a signature event of apoptosis. Finally, interleukin-6 (IL-6) inhibits Dex-induced apoptosis, downregulation of MAP and p70 S6K growth kinases and PARP cleavage; in contrast, IL-6 does not inhibit IR- induced apoptosis, activation of SAPK/JNK, and PARP cleavage. Taken together, our findings suggest that SAPK/JNK activation is not required for apoptosis in MM cells, and that there are at least two distinct apoptotic signaling pathways: (i) SAPK/JNK-associated, which is induced by IR and unaected by IL-6; and (ii) SAPK/JNK-independent, which is induced by Dex, associated with downregulation of MAPK and p70 S6K and inhibited by IL-6. Keywords: multiple myeloma; irradiation; dexametha- sone; apoptosis Introduction Treatment of eukaryotic cells with ionizing radiation (IR) induces cell cycle arrest, activation of DNA repair and apoptosis. IR aects cells either by a direct interaction with DNA or through the formation of hydroxyl radicals and superoxides (Limoli et al., 1993; Hall, 1988). The finding that treatment with IR induces transcription of early response genes, such as c-jun and Egr-1 (Sherman et al., 1990; Hallahan et al., 1991; Datta et al., 1992), suggests the involvement of nuclear signals. To date, multiple protein kinases have been identified which transduce IR-triggered signals to the nucleus. For example, we and others have previously demonstrated that IR induces activation of SAPK/ JNK in diverse cell types (Kharbanda et al., 1995a,b; Chen et al., 1996). The stress activated protein kinase (SAPK/JNK) and p38 kinase belong to a family of serine/threonine protein kinases and share a significant homology with mitogen activated protein (MAP) kinases (Anderson et al., 1990; Boulton et al., 1991; Derijard et al., 1994; Kyriakas et al., 1994; Lee et al., 1994; Sluss et al., 1994; Han et al., 1994; Gupta et al., 1995). However, the biological responses elicited by the activation of each of these kinases are distinct. For example, activation of SAPK/JNK and p38 kinases is asso- ciated with environmental stress, inflammatory cyto- kine and withdrawal of growth factor-induced apoptosis (Derijard et al., 1994; Kyriakis et al., 1994; Raingeaud et al., 1995; Verheij et al., 1996; Xia et al., 1995), whereas activation of MAPK is associated with proliferation and dierentiation triggered by growth factors and phorbol esters (Kharbanda et al., 1994; Kolch et al., 1991). Moreover, recent studies have demonstrated that these kinases have dierent up- stream activators and substrate specificities. For example, transcription factor c-Jun is a substrate of SAPK/JNK (Kyriakis et al., 1994; Lee et al., 1994), AFT2 is phosphorylated by p38 and SAPK/JNK (Raingeaud et al., 1995, 1996; Derijard et al., 1995), and Elk-1 is a substrate of all three members of this family (Marais et al., 1993; Whitmarsh et al., 1995). These kinases may also be coordinately regulated. For example, a recent report demonstrates that apoptosis in pheochromocytoma cells was associated not only with sustained activation of SAPK/JNK and p38 kinase, but also with downregulation of MAPK, suggesting that activation of SAPK/JNK/p38 kinases and concomitant inhibition of MAPK is a determining factor for cells to undergo apoptosis (Xia et al., 1995). Furthermore, we have shown previously that Fas- induced apoptosis in multiple myeloma (MM) cells is associated with activation of both SAPK/JNK and p38 kinase (Chauhan et al., 1997). To date, however, it remains controversial as to whether SAPK/JNK activation is required during apoptosis. Specifically, the role of SAPK/JNK activation during apoptosis in MM cells induced by IR or other agents is not yet defined. Recent emphasis has focused on the family of aspartate-specific cysteine proteases (ASAPs) that mediates all apoptotic cell death (Henkart, 1996). Once activated, ASAPs eciently cleave poly (ADP- ribose) polymerase (PARP), thereby preventing DNA Correspondence: KC Anderson Received 22 January 1997; revised 2 May 1997; accepted 6 May 1997 Oncogene (1997) 15, 837 – 843 1997 Stockton Press All rights reserved 0950 – 9232/97 $12.00
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Dexamethasone induces apoptosis of multiple myeloma cells in a JNK/SAP kinase independent mechanism

Dexamethasone induces apoptosis of multiple myeloma cells in a JNK/SAPkinase independent mechanism
Dharminder Chauhan1, Pramod Pandey2, Atsushi Ogata1, Gerrard Teoh1, Steven Treon1,Mitsuyoshi Urashima1, Surender Kharbanda2 and Kenneth C Anderson1
1Divisions of Hematologic Malignancies and 2Cancer Pharmacology, Dana-Farber Cancer Institute, and the Department ofMedicine, Harvard Medical School, Boston, Massachusetts 02215, USA
The stress-activated protein kinases (SAPKs), alsoknown as c-Jun amino-terminal kinases (JNKs), areactivated in response to diverse stimuli including DNAdamage, heat shock, interleukin-1, tumor necrosis factor-a and Fas. Although all these inducers cause apoptosis,whether SAPK/JNK activation is required for apoptosisis controversial. In this study, we demonstrate thationizing radiation (IR) and dexamethasone (Dex) induceapoptosis in multiple myeloma (MM) derived cell lines,as well as in patient cells. IR-induced apoptosis isassociated with activation of SAPK/JNK and p38kinase, in contrast to Dex-induced apoptosis, which isnot associated with activation of stress kinases. More-over, Dex-induced apoptosis is associated with asigni®cant decrease in the activities of mitogen activatedprotein kinase (MAPK) and p70S6K, whereas IR-treatment does not alter the activity of these kinases.Both IR and Dex induce poly (ADP ribose) polymerase(PARP) cleavage, a signature event of apoptosis. Finally,interleukin-6 (IL-6) inhibits Dex-induced apoptosis,downregulation of MAP and p70S6K growth kinases andPARP cleavage; in contrast, IL-6 does not inhibit IR-induced apoptosis, activation of SAPK/JNK, and PARPcleavage. Taken together, our ®ndings suggest thatSAPK/JNK activation is not required for apoptosis inMM cells, and that there are at least two distinctapoptotic signaling pathways: (i) SAPK/JNK-associated,which is induced by IR and una�ected by IL-6; and (ii)SAPK/JNK-independent, which is induced by Dex,associated with downregulation of MAPK and p70S6K
and inhibited by IL-6.
Keywords: multiple myeloma; irradiation; dexametha-sone; apoptosis
Introduction
Treatment of eukaryotic cells with ionizing radiation(IR) induces cell cycle arrest, activation of DNA repairand apoptosis. IR a�ects cells either by a directinteraction with DNA or through the formation ofhydroxyl radicals and superoxides (Limoli et al., 1993;Hall, 1988). The ®nding that treatment with IR inducestranscription of early response genes, such as c-jun andEgr-1 (Sherman et al., 1990; Hallahan et al., 1991;Datta et al., 1992), suggests the involvement of nuclearsignals. To date, multiple protein kinases have been
identi®ed which transduce IR-triggered signals to thenucleus. For example, we and others have previouslydemonstrated that IR induces activation of SAPK/JNK in diverse cell types (Kharbanda et al., 1995a,b;Chen et al., 1996).The stress activated protein kinase (SAPK/JNK)
and p38 kinase belong to a family of serine/threonineprotein kinases and share a signi®cant homology withmitogen activated protein (MAP) kinases (Anderson etal., 1990; Boulton et al., 1991; Derijard et al., 1994;Kyriakas et al., 1994; Lee et al., 1994; Sluss et al.,1994; Han et al., 1994; Gupta et al., 1995). However,the biological responses elicited by the activation ofeach of these kinases are distinct. For example,activation of SAPK/JNK and p38 kinases is asso-ciated with environmental stress, in¯ammatory cyto-kine and withdrawal of growth factor-inducedapoptosis (Derijard et al., 1994; Kyriakis et al., 1994;Raingeaud et al., 1995; Verheij et al., 1996; Xia et al.,1995), whereas activation of MAPK is associated withproliferation and di�erentiation triggered by growthfactors and phorbol esters (Kharbanda et al., 1994;Kolch et al., 1991). Moreover, recent studies havedemonstrated that these kinases have di�erent up-stream activators and substrate speci®cities. Forexample, transcription factor c-Jun is a substrate ofSAPK/JNK (Kyriakis et al., 1994; Lee et al., 1994),AFT2 is phosphorylated by p38 and SAPK/JNK(Raingeaud et al., 1995, 1996; Derijard et al., 1995),and Elk-1 is a substrate of all three members of thisfamily (Marais et al., 1993; Whitmarsh et al., 1995).These kinases may also be coordinately regulated. Forexample, a recent report demonstrates that apoptosisin pheochromocytoma cells was associated not onlywith sustained activation of SAPK/JNK and p38kinase, but also with downregulation of MAPK,suggesting that activation of SAPK/JNK/p38 kinasesand concomitant inhibition of MAPK is a determiningfactor for cells to undergo apoptosis (Xia et al., 1995).Furthermore, we have shown previously that Fas-induced apoptosis in multiple myeloma (MM) cells isassociated with activation of both SAPK/JNK and p38kinase (Chauhan et al., 1997). To date, however, itremains controversial as to whether SAPK/JNKactivation is required during apoptosis. Speci®cally,the role of SAPK/JNK activation during apoptosis inMM cells induced by IR or other agents is not yetde®ned.Recent emphasis has focused on the family of
aspartate-speci®c cysteine proteases (ASAPs) thatmediates all apoptotic cell death (Henkart, 1996).Once activated, ASAPs e�ciently cleave poly (ADP-ribose) polymerase (PARP), thereby preventing DNA
Correspondence: KC AndersonReceived 22 January 1997; revised 2 May 1997; accepted 6 May 1997
Oncogene (1997) 15, 837 ± 843 1997 Stockton Press All rights reserved 0950 ± 9232/97 $12.00

repair mechanisms during apoptosis. Activation ofASAPs and related PARP cleavage in MM cellsundergoing apoptosis induced by IR or other agentshas also not yet been characterized.In the present study, we characterized the signaling
mechanisms involved during apoptosis induced by IRand by Dexamethasone (Dex) in MM derived cell linesand patient cells. Although both IR and Dex treatmentof MM cells induce apoptosis and PARP cleavage,only IR-induced apoptosis involves activation ofSAPK/JNK and p38 kinase. In contrast, Dex-inducedapoptosis is associated with decreased MAPK andp70S6K activity, whereas IR treatment does not alter theactivity of these kinases. Finally, interleukin-6 (IL-6)blocks Dex-induced apoptosis, downregulation ofMAPK and p70S6K activities, and PARP cleavage inMM cells but does not inhibit IR-induced apoptosis,SAPK/JNK activation, and PARP cleavage. Our datatherefore demonstrate that SAPK/JNK activation isnot required for apoptosis in MM cells and, inparticular, that Dex induces apoptosis in a SAPK/JNK independent mechanism.
Results and discussion
Previous studies have demonstrated that treatment ofdiverse cell types with IR induces apoptosis. We andothers have shown that Fas or glucocorticoids triggersapoptosis in MM cells (Chauhan et al., 1997;Westerndorf et al., 1995; Shima et al., 1995; Hata etal., 1995; Lichtenstein et al., 1995). In the presentstudy, OCI-MY5 MM cells were exposed to IR for48 h and DNA fragmentation assays were performedat various intervals (Figure 1a). Increased cleavage ofDNA by 24 h was noted in IR-treated, but not inuntreated control MM cells. To quantify cell death,¯ow cytometric analyses using propidium iodide(Ormerod et al., 1992) were next performed. Afterexposure of OCI-MY5 MM cells to IR for 48 h, themajority (67+3%, n=3) of cells underwent apoptosis.Acridine orange staining (Duke et al., 1992) con®rmedchanges characteristic of apoptosis.Since IR and U.V. have been shown to triggers
activation of SAPK/JNK and p38 kinase in othersystems (Kharbanda et al., 1995; Derijard et al., 1994),we next assayed for IR-induced activation of SAPK/JNK and p38 kinase in MM cells. OCI-MY5 MM cellswere treated with IR, and in vitro immune complexkinase assays were performed (Figure 1b, upper panel).Analyses of anti-SAPK/JNK immunoprecipitates de-monstrated a transient 8 ± 10-fold activation of SAPK/JNK at 1 h in MM cells. Similar results were obtainedwhen anti-p38 immunoprecipitates from control andIR-treated cells were analysed for phosphorylation ofGST-ATF2 (Figure 1c, upper panel). To determinewhether IR induces SAPK/JNK and p38 kinaseactivity in a dose dependent fashion, we treated OCI-MY5 MM cells for 1 h with IR doses ranging from 5 ±33 Gy. Although IR induces SAPK/JNK and p38kinase activity at 5 Gy, maximal activity of thesekinases was observed when cells were treated with20 Gy (data not shown). In order to con®rm whetherIR a�ects JNK/SAPK and p38 kinase protein levels,total cell lysates were also subjected to immunoblottingwith respective antibodies. The results demonstrated
that IR treatment of MM cells does not alter theprotein level of both JNK/SAPK and p38 kinase(Figure 1b, lower panel and Figure 1c, lower panel).Our ®ndings that IR-induced apoptosis involves
activation of SAPK/JNK suggest a role of SAPK/JNK in at least IR-induced apoptosis in MM cells.These ®ndings are in concert with those of Verheij etal. (1996) which demonstrated a role of SAPK/JNK inceramide- or TNF-induced apoptosis. Furthermore, ithas been demonstrated that coordinate regulation ofSAPK/JNKs, p38 and ERK kinases facilitates apop-tosis induced by withdrawal of growth factor,suggesting that activation of SAPK/JNK may beessential for apoptosis (Xia et al., 1995). In contrast,a recent report demonstrated that TNF-inducedactivation of SAPK/JNK is not linked to TNF-induced apoptosis (Liu et al., 1996). It thereforeremains controversial as to whether activation ofSAPK/JNK is an obligatory event during apoptosisinduced by diverse apoptotic stimuli in all cell types.To determine whether distinct inducers of apoptosis
utilize the SAPK/JNK pathway, we next studied Dex-induced apoptosis in our MM cell line and patientmodel system. OCI-MY5 MM cells were treated with
600 bp -
100 bp -
MY
5
24h
48h
MY
5
1h
3h
6h
kD46 -
31 -
Fold: 1.0 10 1.1 1.1
anti-SAPKIR
GST-Jun
SAPK
MY
5
1h
3h
6h
kD
anti-p38IR
46 -
31 -
Fold: 1.0 8.6 4.1 4.1
IR
GST-ATF2
p38
a b
c
Figure 1 IR induces DNA fragmentation and activation ofSAPK and p38 kinase in OCI-MY5 MM cells. Cells were culturedin media alone for the indicated times with an IR dose of 20 Gy.Genomic DNA was isolated, end-labeled with 0.5 mCi of[g32P]dCTP, and analysed by 1.8% agarose gel electrophoresis(a). OCI-MY5 MM cells were treated with IR (20 Gy) for 1 h,3 h and 6 h. Lysates from control and IR-treated OCI-MY5 MMcells were immunoprecipitated with anti-SAPK antibody (b) andanti-p38 kinase antibody (c). Immune complex kinase assays wereperformed by addition of GST-Jun (b) or GST-ATF2 (c) and[g32P]ATP and incubation for 15 min at 308C. The phosphory-lated proteins were resolved by 10% SDS±PAGE and analysedby Coomassie blue staining and autoradiography (Exposure timefor autoradiography was 30 mins for panel b). Total cell lysatesfrom treated and untreated cells were also subjected toimmunoblotting with either SAPK/JNK (b, lower panel) or p38kinase antibody (c, lower panel)
Differential apoptotic signals in multiple myelomaD Chauhan et al
838

Dex (10 mM) for 72 h and DNA cleavage was analysedby agarose gel electrophoresis of the 32P-labeledgenomic DNA at various intervals (Figure 2a). Incontrast to control untreated cells, signi®cant DNAfragmentation was detected in OCI-MY5 MM cellstreated with Dex for 72 h. Both FACS analysis andacridine orange staining con®rmed apoptosis in Dextreated MM cells (data not shown). We next examinedwhether Dex-induced apoptosis also involves activationof SAPK/JNK or p38 kinase. OCI-MY5 MM cellswere treated with Dex for the indicated periods andassayed for the activity of these kinases. In contrast toIR, Dex did not induce activation of either SAPK/JNK(Figure 2b, upper panel) or p38 kinase (Figure 2c,upper panel) in OCI-MY5 MM cells. Furthermore,Dex treatment did not alter the protein levels of eitherSAPK/JNK or p38 kinase (Figure 2b, lower panel, andFigure 2c, lower panel, respectively). However, down-regulation of SAPK/JNK activity was observed at12 h, 24 h and 48 h after Dex treatment (Figure 2b,upper panel). The lack of activation of SAPK/JNK orp38 kinase in MM cells treated with Dex suggests thatDex-induced apoptosis in MM cells is independent ofSAPK/JNK and p38 kinase pathways.
We next performed similar experiments using MMpatient (PCL) cells. As noted in OCI-MY5 MM cells,treatment of PCL cells with IR or Dex is associatedwith increased DNA fragmentation (Figure 3a).Treatment of PCL cells with IR activates bothSAPK/JNK and p38 kinase (Figure 3b and c),whereas Dex treatment does not activate either ofthese kinases (Figure 3d and e). In addition, proteinlevels of SAPK/JNK and p38 kinases were not alteredby treatment of PCL cells with either IR or Dex (datanot shown). Taken together, our results thereforesuggest that there are at least two di�erent apoptoticpathways in MM cell lines as well as patient cells: onewhich involves activation of SAPK/JNK and p38kinase, as induced by IR; and another which isindependent of SAPK/JNK and p38 kinase, astriggered by Dex.Since Dex-induced apoptosis in MM cells does not
involve activation of SAP/JNK or p38 kinase, we next
600 bp -
100 bp -anti-p38
Dex
MY
5
1h
3h
6h
12h
24h
48h
MY
5
6h
12h
24h
GST-Jun
SAPK
anti-SAPKDex
kD46 -
31 -
MW
Mar
ker
MY
5
72h
Dex
kD46 -
31 -
GST-ATF2
p38
a b
c
Figure 2 Dex treatment induces DNA fragmentation withouta�ecting SAPK and p38 kinase activity in OCI-MY5 MM cells.Cells were cultured for the indicated times in media alone andwith Dex (10 mM). Genomic DNA was isolated, end-labeled with0.5 mCi of [g32P]dCTP, and analysed by 1.8% agarose gelelectrophoresis (a). MW, molecular weight markers (100 bpladders and HindIII digest of 1-DNA). OCI-MY5 MM cellswere treated with DEX (10 mM) for 1 h, 3 h, 6 h, 12 h, 24 h and48 h. Lysates from control and Dex-treated OCI-MY5 MM cellswere immunoprecipitated with anti-SAPK antibody (b) and anti-p38 kinase antibody (c). Immune complex kinase assays wereperformed by addition of GST-Jun (b) or GST-ATF2 (c) and[g32P]ATP and incubation for 15 min at 308C. The phosphory-lated proteins were resolved by 10% SDS±PAGE and analysedby Coomassie blue staining and autoradiography (Exposure timefor autoradiography was 24 h for panel b). The results arerepresentative of three independent experiments. Total cell lysatesfrom treated and untreated cells were also subjected toimmunoblotting with either SAPK/JNK (b, lower panel) or p38kinase antibody (c, lower panel)
PC
L
24h
48h
72h
48h
72h
600 bp —
100 bp —
IR Dexa
31 —P
CL
1h
3h
6h
24h
PC
L 1h
3h
6h
24
h
GST- Jun
GST- ATF2
Dex Dex
PC
L
1h
3h
6h
10h
46 —
31 —
kD
GST- Jun
46 —kD
GST- ATF2
PC
L 1h
3h
6h
IRIR
anti-SAPK anti-p38
anti-SAPK anti-p38
b c
d e
Figure 3 E�ects of IR or DEX on DNA fragmentation as wellas on SAPK and p38 kinase activity in PCL patient cells. Cellswere cultured for the indicated times in media alone and witheither IR (20 Gy) or Dex (10 mM). Genomic DNA was isolated,end-labeled with 0.5 mCi of [g32P]dCTP and analysed by 1.8%agarose gel electrophoresis (a). PCL patient cells were treatedwith IR (20 Gy) or Dex (10 mM) for the indicated time period.Lysates from control and IR or Dex-treated PCL patient cellswere immunoprecipitated with anti-SAPK antibody (b and d), oranti-p38 kinase antibody (c and e). Immune complex kinaseassays were performed by addition of GST-Jun (b and d) or GST-ATF2 (c and e) and [g32P]ATP and incubated for 15 min at 308C.The phosphorylated proteins were resolved by 10% SDS±PAGEand analysed by Coomassie blue staining and autoradiography.The results are representative of three independent experiments
Differential apoptotic signals in multiple myelomaD Chauhan et al
839

determined whether Dex a�ects growth-related kinases,such as MAPK and p70S6K. Treatment of OCI-MY5cells with Dex downregulates MAPK activity, assayedby MBP phosphorylation (Figure 4a, upper panel).Reprobing of these blots with anti-MAPK antibodydemonstrated equal amounts of protein in each lane(Figure 4a, lower panel). We also studied p70S6K sinceprevious studies have demonstrated that activation ofp70S6K is associated with G1 to S phase transition(Chung et al., 1992). Moreover, the signaling cascaderegulating p70S6K is distinct from Ras44Raf44MEK44MAPK and RSK (Blenis, 1993) and ismediated directly by phosphatidylinositol 3-kinase(PI3K) (Cheatham et al., 1994; Chung et al., 1994).In the present study, Dex treatment of OCI-MY5 MMcells is associated with signi®cant inhibition of p70S6K
activity at 24 h, as measured directly by S6 peptidephosphorylation (Figure 4b). Furthermore, whetherinhibition of p70S6K activity by Dex treatment is PI3Kdependent or independent requires further studies. Arecent study which demonstrates that Dex blocks theability of NF-kB to activate transcription of genes thatregulate cell proliferation (Scheiman et al., 1995) isconsistent with these ®ndings. IR treatment of OCI-MY5 MM cells did not downregulate either MAPK orp70S6K (data not shown and Figure 4b).In the present study we next examined whether IR
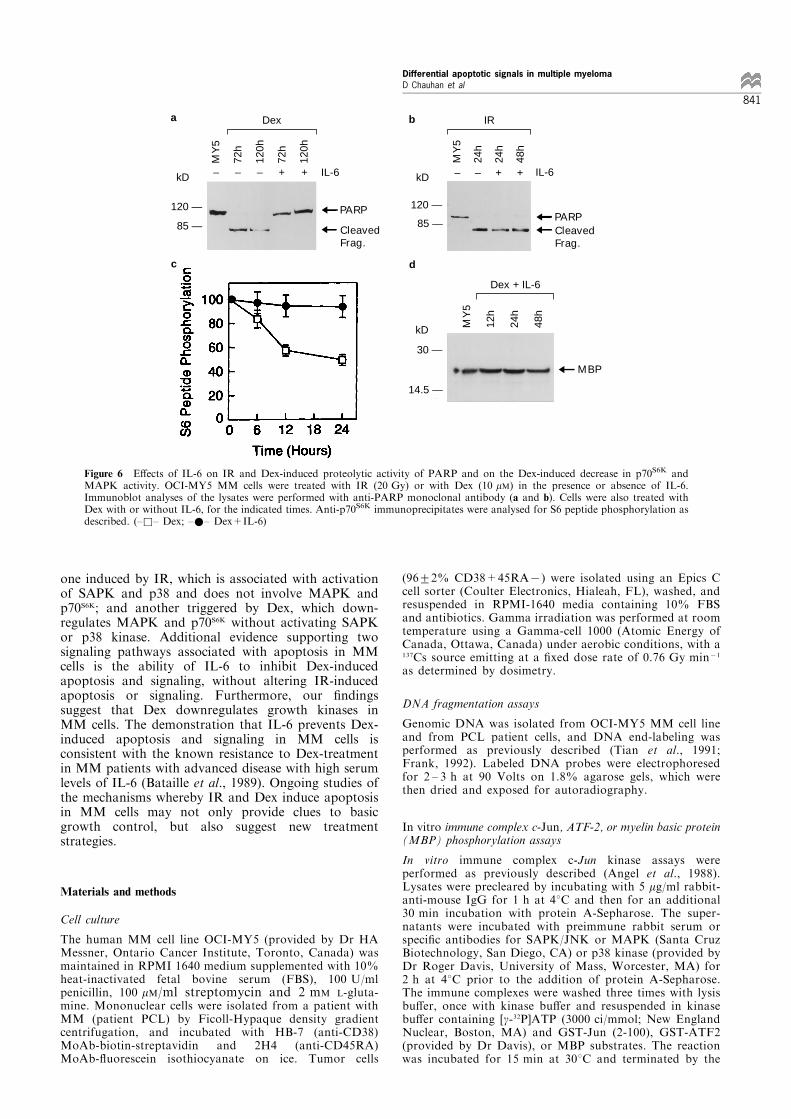
and Dex induce poly (ADP-ribose) polymerase (PARP)cleavage, the hallmark of apoptosis which indicatesactivation of the protease cascade (Henkart, 1996;Desnoyers et al., 1994). Both IR and Dex inducedPARP cleavage, consistent with apoptosis and activa-tion of proteases (Figure 5a and b). Taken together,these ®ndings suggest that Dex-induced apoptosis isindependent of SAPK/JNK activation, and involvesinhibition of growth-related kinases as well asactivation of proteases. In contrast, IR-inducedapoptosis is associated with activation of SAP/JNKand proteases, without a�ecting growth kinases.Finally we attempted to con®rm the existence of
distinct pathways using inhibitors of apoptosis. SinceIL-6 is an anti-apoptotic factor for MM cells(Lichtenstein et al., 1995), we studied the e�ect of IL-6 on the IR and Dex induced apoptotic signalingevents. Treatment of OCI-MY5 MM cells with IL-6(100 ng/ml) prior to Dex resulted in complete blockingof apoptosis and PARP cleavage (Figure 6a), but didnot prevent IR-induced apoptosis and PARP cleavage(Figure 6b). Moreover, IL-6 prevented Dex-induceddownregulation of p70S6K and MAPK activities in theabsence of any changes in the protein levels of eitherkinase (Figure 6c, d, and data not shown). In contrast,IR-induced apoptosis and SAP/JNK activation is notinhibited by IL-6 (data not shown).The present study therefore provides evidence for at
least two cascades involved in apoptosis in MM cells:
30 —
14.5 —
30 —
14.5 —
MY
5
3h
6h
12h
24h
48h
72h
120h
MBP
MAPK
Dex
anti-MAPK
kD
a
b
Figure 4 Dex downregulates MAPK and p70S6K activity in OCI-MY5 cells. Cells were cultured for the indicated times in mediaalone or with Dex (10 mM). Lysates from control and Dex-treatedOCI-MY5 cells were immunoprecipitated with anti-MAPKantibody. Immune complex kinase assays were performed byaddition of MBP as a substrate (a, upper panel) and incubated for15 min at 308C. The phosphorylated proteins were resolved by10% SDS±PAGE and analysed by Coomassie blue staining andautoradiography. Anti-MAPK immunoprecipitates were alsoanalysed by immunoblotting with anti-MAPK antibody (a,lower panel). Lysates from control and Dex-treated OCI-MY5cells were immunoprecipitated with anti-p70S6K antibody and invitro immune complex kinase assays were performed utilizing S6peptide as an exogenous substrate. The results are expressed aspercent inhibition in p70S6K activity (mean+s.d. of threeindependent experiments; indicated as ±&± Dex; ±*± IR)
120 —
85 —
kD MY
5
24h
48h
72h
120h
120 —
85 —
MY
5
24h
48h
72h
120h
Cleaved Frag.
Cleaved Frag.
PARP
PARP
Dex
IR
a
b
kD
Figure 5 IR and Dex induce proteolytic cleavage of PARP. OCI-MY5 MM cells were treated with IR (20 Gy) and Dex (10 mM)for the indicated time periods. Immunoblot analyses of the lysateswas performed with anti-PARP antibody
Differential apoptotic signals in multiple myelomaD Chauhan et al
840
one induced by IR, which is associated with activationof SAPK and p38 and does not involve MAPK andp70S6K; and another triggered by Dex, which down-regulates MAPK and p70S6K without activating SAPKor p38 kinase. Additional evidence supporting twosignaling pathways associated with apoptosis in MMcells is the ability of IL-6 to inhibit Dex-inducedapoptosis and signaling, without altering IR-inducedapoptosis or signaling. Furthermore, our ®ndingssuggest that Dex downregulates growth kinases inMM cells. The demonstration that IL-6 prevents Dex-induced apoptosis and signaling in MM cells isconsistent with the known resistance to Dex-treatmentin MM patients with advanced disease with high serumlevels of IL-6 (Bataille et al., 1989). Ongoing studies ofthe mechanisms whereby IR and Dex induce apoptosisin MM cells may not only provide clues to basicgrowth control, but also suggest new treatmentstrategies.
Materials and methods
Cell culture
The human MM cell line OCI-MY5 (provided by Dr HAMessner, Ontario Cancer Institute, Toronto, Canada) wasmaintained in RPMI 1640 medium supplemented with 10%heat-inactivated fetal bovine serum (FBS), 100 U/mlpenicillin, 100 mM/ml streptomycin and 2 mM L-gluta-mine. Mononuclear cells were isolated from a patient withMM (patient PCL) by Ficoll-Hypaque density gradientcentrifugation, and incubated with HB-7 (anti-CD38)MoAb-biotin-streptavidin and 2H4 (anti-CD45RA)MoAb-¯uorescein isothiocyanate on ice. Tumor cells
(96+2% CD38+45RA7) were isolated using an Epics Ccell sorter (Coulter Electronics, Hialeah, FL), washed, andresuspended in RPMI-1640 media containing 10% FBSand antibiotics. Gamma irradiation was performed at roomtemperature using a Gamma-cell 1000 (Atomic Energy ofCanada, Ottawa, Canada) under aerobic conditions, with a137Cs source emitting at a ®xed dose rate of 0.76 Gy min71
as determined by dosimetry.
DNA fragmentation assays
Genomic DNA was isolated from OCI-MY5 MM cell lineand from PCL patient cells, and DNA end-labeling wasperformed as previously described (Tian et al., 1991;Frank, 1992). Labeled DNA probes were electrophoresedfor 2 ± 3 h at 90 Volts on 1.8% agarose gels, which werethen dried and exposed for autoradiography.
In vitro immune complex c-Jun, ATF-2, or myelin basic protein(MBP) phosphorylation assays
In vitro immune complex c-Jun kinase assays wereperformed as previously described (Angel et al., 1988).Lysates were precleared by incubating with 5 mg/ml rabbit-anti-mouse IgG for 1 h at 48C and then for an additional30 min incubation with protein A-Sepharose. The super-natants were incubated with preimmune rabbit serum orspeci®c antibodies for SAPK/JNK or MAPK (Santa CruzBiotechnology, San Diego, CA) or p38 kinase (provided byDr Roger Davis, University of Mass, Worcester, MA) for2 h at 48C prior to the addition of protein A-Sepharose.The immune complexes were washed three times with lysisbu�er, once with kinase bu�er and resuspended in kinasebu�er containing [g-32P]ATP (3000 ci/mmol; New EnglandNuclear, Boston, MA) and GST-Jun (2-100), GST-ATF2(provided by Dr Davis), or MBP substrates. The reactionwas incubated for 15 min at 308C and terminated by the
120 —
85 —
– – – + +
PARP
Cleaved Frag.
kD
MY
5
72h
120h
72h
120h
Dex
IL-6 – – + +
PARPCleaved Frag.
120 —
85 —
kD IL-6
MY
5
24h
24h
48h
IR
30 —
14.5 —
MY
5
12h
24h
48h
MBP
Dex + IL-6
c
a b
d
kD
Figure 6 E�ects of IL-6 on IR and Dex-induced proteolytic activity of PARP and on the Dex-induced decrease in p70S6K andMAPK activity. OCI-MY5 MM cells were treated with IR (20 Gy) or with Dex (10 mM) in the presence or absence of IL-6.Immunoblot analyses of the lysates were performed with anti-PARP monoclonal antibody (a and b). Cells were also treated withDex with or without IL-6, for the indicated times. Anti-p70S6K immunoprecipitates were analysed for S6 peptide phosphorylation asdescribed. (±&± Dex; ±*± Dex+IL-6)
Differential apoptotic signals in multiple myelomaD Chauhan et al
841

addition of SDS sample bu�er. Proteins were analysed bySDS ± PAGE, Coomassie blue staining and autoradio-graphy.
Immunoprecipitation and immunoblotting
Immunoprecipitations were performed as previouslydescribed (Chauhan et al., 1995). Brie¯y, cell lysates wereprepared by resuspending OCI-MY5 MM cells for 30 minon ice in lysis bu�er containing 50 mM Tris (pH 7.6),150 mM NaCl, 1 mM PMSF, 1 mM sodium vanadate, 1 mM
DTT, 10 mM sodium ¯uoride, 1% NP-40 and 10 mg/mleach of leupeptin and aprotinin. Equal amounts of proteins(250 ± 300 mg) were subjected to immunoprecipitation withthe indicated antibodies and immune complexes wereprecipitated with protein A-Sepharose. The resultingprotein precipitates were washed three times with lysisbu�er and resolved by SDS ± PAGE. Proteins were thentransferred to nitrocellulose ®lters, blocked by incubationin 5% dry milk in PBST (0.05% Tween-20 in PBS), andprobed with the indicated antibodies. p70S6K antibody wasprovided by Dr John Blenis (Harvard Medical School,Boston, MA). Blots were then developed by enhanced
chemiluminescence (ECL; Amersham, Arlington Heights,IL). Preparation of cell lysates for PARP immunoblotanalyses was performed as described using C-2-10 anti-PARP monoclonal antibody (Desnoyers et al., 1994).
Immune complex protein kinase assays for p70S6K
Total cell lysates were prepared with anti-p70S6K antiserum;immune complexes were separated and subjected to in vitrokinase assays using synthetic S6 (RRRLSSLRA) peptide assubstrate (Chung et al., 1992). In brief, the immunecomplexes were washed twice with hypotonic lysis bu�er,twice with LiCl bu�er (0.5M LiCl, 100 mM Tris, pH 7.6)and twice with assay bu�er (20 mM Tris, pH 7.2, 10 mM
MgCl2). After washing, they were suspended in kinasebu�er (20 mM Tris, pH 7.2, 10 mM MgCl2) and thereaction (50 ml) was started by the addition of 20 mM
ATP (1 mCi of [g32-P]ATP) and 100 mM S6 peptide. Afterincubation for 15 min at 308C, 25 ml was spotted ontophosphocellulose paper (P81, Whatman), followed bywashing with 1% phosphoric acid and then distilledwater. Incorporation of [32P]phosphate was assayed byscintillation counting.
References
Anderson NG, Maller JL, Tonks NK and Sturgill TW.(1990). Nature, 343, 651 ± 653.
Angel P, Allegretto EA, Okino ST, Hattori K, Boyle WJ,Hunter T and Karin M. (1988). Nature, 332, 166 ± 171.
Bataille R, Jourdan M, Zang X-G and Klein B. (1989). J.Clin. Invest., 84, 2008 ± 2014.
Blenis J. (1993). Proc. Natl. Acad. Sci. USA, 90, 5889 ± 5892.Boulton TG, Nye SH, Robbins DJ, Ip Ny, Radziejewska E,Morgenbesser SD, DePinho R, Panayotatos N, Cobb Mand Yancopoulos GD. (1991). Cell, 65, 663 ± 665.
Chauhan D, Kharbanda S, Ogata A, Urashima M, Malik N,Frank D, Kufe DW and Anderson KC. (1995). J. Exp.Med., 182, 1801 ± 1806.
Chauhan D, Kharbanda S, Ogata A, Urashima M, Teoh G,Robertson MJ, Kufe DW and Anderson KC. (1997).Blood, 89, 227 ± 234.
Cheatham B, Vlahos C-J, Cheatham L, Wang L, Blenis J andKahn C-R. (1994). Mol. Cell. Biol., 14, 4902 ± 4911.
Chen Y-R, Meyer CF and Tan T-H. (1996). J. Biol. Chem.,271, 631 ± 634.
Chung J, Kuo CJ, Crabtree GR and Blenis J. (1992). Cell, 69,1227 ± 1236.
Chung J, Grammer T-C, Lemon K-P, Kazlauskas A andBlenis J. (1994). Nature, 370, 71 ± 75.
Datta R, Rubin E, Sukhatme V, Qureshi S, Weichselbaum Rand Kufe D. (1992). Proc. Natl. Acad. Sci. USA, 89,10149 ± 10153.
Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, KarinM and Davis RJ. (1994). Cell, 76, 1025 ± 1037.
Derijard B, Raingeaud J, Barrett T, Wu I-H, Han J, UlevitchRJ and Davis RJ. (1995). Science, 267, 682 ± 685.
Desnoyers S, Shah GM and Poirier GG. (1994). Ann.Biochem., 218, 470 ± 473.
Duke RC and Cohen JJ. (1992). In: Current Protocols inImmunology, Coligan JE, Kruisbeek AM, Margulies DH,Shevach EM and Strober W, (eds). 1, pp. 3.17.1 ± 3.17.16,John Wiley and Sons, Inc., New York.
Frank R. (1992). Nuc. Acid. Res., 20, 5243 ± 5244.Gupta S, Campbell D, De rijard B and Davis RJ. (1995).
Science, 267, 389 ± 399.Hall EJ. (1988). In: Radiobiology for the Radiologist, Hall EJ,(ed.), Lippincott, Philadelphia, pp. 128 ± 135.
Hallahan D, Sukhatme V, Sherman M, Virudachalam S andKufe D. (1991). Proc. Natl. Acad. Sci. USA, 88, 2156 ±2160.
Han J, Lee J-D, Bibbs L and Ulevitch RJ. (1994). Science,265, 808 ± 811.
Hata H, Hiromitsu M, Takeya M, Yoshida M, Sonoki T,Nagasaki A, Kuribayashi N, Kawano F and Takatsuki K.(1995). Blood, 86, 1939 ± 1945.
Henkart PA. (1996). Immunity, 4, 195 ± 201.Kharbanda S, Saleem A, Emoto Y, Stone R, Rapp U andKufe DW. (1994). J. Biol. Chem., 269, 872 ± 878.
Kharbanda S, Ren R, Pandey P, Shafman TD, Feller SM,Weichselbaum R and Kufe DW. (1995a). Nature, 376,785 ± 788.
Kharbanda S, Saleem A, Shafman T, Emoto Y, Weichsel-baum R, Woodgett J, Avruch Y, Kyriakis J and Kufe D.(1995b). J. Biol. Chem., 270, 18871 ± 18874.
Kolch W, Heidecker G, Lyd P and Rapp U. (1991). Nature,349, 426 ± 428.
Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA,Ahmad MF, Avruch J and Woodgett JR. (1994). Nature,369, 156 ± 160.
Lee JC, Laydon JT, McConnell PC, Gallagher TF, Kumar S,Green D, McNulty D, Blumenthal MJ, Heys JR, Land-vatter SW, Strikler JE, McLaughlin MM, Siemens IR,Fisher SM, Livi GP, White JR, Adams JL and Young PR.(1994). Nature, 372, 739 ± 746.
Lichtenstein A, Tu Y, Fady C, Vescio R and Berenson J.(1995). Cell Immunol., 162, 248 ± 255.
Limoli CL and Ward JF. (1993). Radiat. Res., 134, 160 ± 169.Liu ZG, Hsu H, Goddel DV and Karin M. (1996). Cell, 87,565 ± 576.
Marais R, Wynne J and Treisman R. (1993). Cell, 73, 381 ±393.
Ormerod MG, Collins MK, Rodriguez-Tarduchy G andRobertson D. (1992). J. Immunol. Methods., 153, 57 ± 60.
Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J,Ulevitch RJ and Davis RJ. (1995). J. Biol. Chem., 270,7420 ± 7426.
Raingeaud J, Whitmarsh AJ, Barrett T, Derijard B andDavis RJ. (1996). Mol. Cell. Biol., 16, 1247 ± 1255.
Scheiman RI, Gualberto A, Jewell CM, Cidlowski JA andBaldwin Jr, AS. (1995). Mol. Cell. Biol., 15, 943 ± 953.
Sherman M, Datta R, Hallahan D, Weichselbaum R andKufe D. (1990). Proc. Natl. Acad. Sci. USA, 87, 5663 ±5666.
Shima Y, Nishimoto N, Ogata A, Fuji Y, Yoshizaki K andKishimoto T. (1995). Blood, 85, 757 ± 764.
Differential apoptotic signals in multiple myelomaD Chauhan et al
842

Sluss H, Barett T, Derijard B and Davis RJ. (1994). Mol.Cell. Biol., 14, 8376 ± 8384.
Tian Q, Streuli M, Saito H, Schlossman SF and Anderson P.(1991). Cell, 67, 629 ± 639.
Verheij M, Bose R, Lin X-H, Yao B, Jarvis WD, Grant S,Birrer MJ, Szabo E, Zon LI, Kyriakis JM, Haimovitz-Friedman A, Fuks Z and Kolesnick RN. (1996). Nature,380, 75 ± 79.
Westerndorf JJ, Lammeret J and Jelinek DF. (1995). Blood,85, 3566 ± 3576.
Whitmarsh AJ, Shore P, Sharrocks AD and Davis RJ.(1995). Science, 269, 403 ± 407.
Zia Z, Dickens M, Raingeaud J, Davis RJ and GreenbergME. (1995). Science, 270, 1326 ± 1331.
Differential apoptotic signals in multiple myelomaD Chauhan et al
843




















