
Emerging roles of ATF2 and the dynamic AP1 network in cancer
Upload
rockefellerCategory
view
1download
0
296
RADIATION RESEARCH 163, 296–306 (2005)0033-7587/05 $15.00q 2005 by Radiation Research Society.All rights of reproduction in any form reserved.
Regulation of Ultraviolet B Radiation-Mediated Activation of AP1Signaling by Retinoids in Primary Keratinocytes
Nadine Darwiche,1 Hisham Bazzi, Lara El-Touni, Ghada Abou-Lteif, Rami Doueiri, Asma Hatoum, Samar Maalouf andHala Gali-Muhtasib1
Department of Biology, American University of Beirut, Beirut, Lebanon
Darwiche, N., Bazzi, H., El-Touni, L., Abou-Lteif, G.,Doueiri, R., Hatoum, A., Maalouf, S. and Gali-Muhtasib, H.Regulation of Ultraviolet B Radiation-Mediated Activation ofAP1 Signaling by Retinoids in Primary Keratinocytes. Radiat.Res. 163, 296–306 (2005).
The main cause of skin cancer and photo-aging is chronicexposure to ultraviolet B (UVB) radiation. Such damage canbe ameliorated by retinoid treatment. UVB-radiation-inducedskin carcinogenesis is associated with the induction of acti-vator protein 1 (AP1) signaling and factors, namely FOS andJUN family members. We investigated the effects of severalretinoids, all-trans-retinoic acid (tRA), 9-cis-retinoic acid(cRA), and N-(4-hydroxyphenyl)-retinamide (HPR), on UVB-induced damage in primary mouse keratinocytes. In addition,the interplay between UVB radiation, retinoid receptors, andAP1 signaling was assessed using Western blot analysis andribonuclease protection and gene reporter assays. Exposure ofkeratinocytes to UVB radiation caused a down-regulation ofthe retinoid receptor protein levels in a proteasome-mediatedmanner. In contrast, FOS and JUN proteins were transientlyinduced shortly after exposure to UVB radiation. Retinoidtreatment caused a dose-dependent reduction in the levels ofretinoid receptor proteins. When irradiated cells were treatedwith retinoids, no significant effects on AP1 protein expressionwere noted. Interestingly, pretreatments with tRA and cRA,but not HPR, suppressed UVB-radiation-induced AP1 activityby more than 50%, whereas post-treatment failed to producesimilar effects. Our findings indicate that the inhibition of AP1activity by retinoids explains, at least in part, the chemopre-ventive potential of retinoids in UV-radiation-associated epi-dermal damage. q 2005 by Radiation Research Society
INTRODUCTION
Skin cancer is one of the most prevalent types of cancerswhose primary cause is chronic exposure to the UVB range(290–320 nm) of the spectrum (1). Ultraviolet B radiationis considered to be a complete carcinogen, acting as aninitiator and a promoter (2) as well as an agent of tumor
1 Address for correspondence: Biology Department, American Univer-sity of Beirut, PO Box 11-0236, Beirut, Lebanon; e-mail: [email protected] or [email protected].
progression (3). The tumorigenic potential of UVB radia-tion is also attributed to its ability to generate highly re-active short-lived oxygen free radicals (4). These high-en-ergy species attack virtually all cellular components includ-ing proteins, lipids and nucleotides. In addition, UVB ra-diation directly activates many cell surface cytokine andgrowth factor receptors and thus adversely affects the in-tracellular milieu (5). At the molecular level, UVB radiationhas been shown to activate the expression of genes involvedin stress responses, inflammation and apoptosis (6) and tu-mor suppressor genes such as TP53 that cause growth arrest(7), and it may induce the expression and activation ofproto-oncogene products such as nuclear factor kB (NFkB,NFKB) (8) and activator protein 1 (AP1) (9). In particular,AP1 transcriptional factors are activated as early as 2 h afterUVB irradiation of human keratinocytes (10).
AP1 is a sequence-specific transcriptional activator com-posed of members of the JUN and FOS families that bindsto DNA-binding motifs required for gene regulation byphorbol ester tumor promoters and UV radiation (9, 11). Inhuman skin in vivo, exposure to low doses of UV radiationresults in the elevated expression of JUN, which heterodi-merizes with the constitutively expressed FOS, forming anactive AP1 complex. AP1 factors are often associated withcell proliferation and tumor progression. In transgenic mice,a skin-targeted Jun mutant blocks skin tumor formation bythe two-stage chemical carcinogenesis protocol (12) or byUVB radiation (13). Moreover, AP1 DNA-binding activityis increased in nuclear extracts from chemically inducedpapillomas that have a high risk for malignant progression(14). Transduction of v-rasHa keratinocytes with v-Fos re-sults in malignant conversion (15), and malignant skin tu-mors do not develop in response to Ras gene activation andtumor promoter treatment in c-Fos null mice (16). There-fore, there is considerable interest in identifying compoundsthat are capable of down-regulating AP1 activity.
Retinoids regulate epidermal proliferation and differen-tiation and have commonly been used in the treatment ofvarious skin disorders including acne, psoriasis, cancer (17)and photo-aging (5). Recently, much emphasis has beenplaced on chemoprevention of cancer by agents such asretinoids (18). Inhibition of tumor formation or progression

297RETINOIDS MODULATE UVB-RADIATION-INDUCED AP1 ACTIVITY
by retinoids occurs at several levels including initiation(19), promotion (20), progression (21) and malignant con-version (22). Vitamin A deficiency in experimental animalscorrelates with an increased susceptibility to chemical car-cinogens (17). Furthermore, epidemiological studies haveindicated that individuals with lower dietary intakes of vi-tamin A are at a higher risk of developing cancer.
Retinoid receptors are members of the steroid/thyroidhormone nuclear receptor superfamily and fall into two ma-jor categories—the retinoic acid receptors (RARs) and theretinoid X receptors (RXRs) (23, 24). Three genes, desig-nated a, b and g, code for RARs and RXRs. Upon tRA orcRA binding, RARs heterodimerize with RXRs and exerttranscriptional regulation of target genes (25). The physi-ological ligand of RXR is cRA, and RXRs act as transcrip-tional factors through homodimerization as well as heter-odimerization with other members of the same superfamilyincluding thyroid hormone and vitamin D3 receptors. Re-cently, some synthetic retinoids, such as HPR, have beendeveloped that function through retinoid receptor-depen-dent and -independent pathways (26).
RARs exert two types of action on gene activity, as tran-sactivators or repressors. Transactivation is due to agonistbinding to the receptor leading to structural transitions thatallow recruitment of nuclear coactivators to the RXR-RARdimer and therefore to the promoter (25). In addition totheir positive regulation of gene transcription, retinoid re-ceptors also function as negative transcriptional factors.One of the well-known transcriptional repressive effects istheir inhibition of AP1 activity (27). In theory, the repres-sion mechanism can take place at several levels and re-mains largely unknown, although a direct protein-proteininteraction between the RARs and AP1 components and acompetition for a common activator has been proposed(28). Transient transfection assays in HeLa cells revealedthat all three RAR subtypes (a, b and g) could effectivelyinhibit TPA-induced AP1 activity on reporter genes in thepresence of RA (27). Furthermore, JUN and FOS are alsopotent inhibitors of the transactivation mechanism of RARs(29).
In light of the important role of UVB-radiation-inducedAP1 activation in skin carcinogenesis, we examined theinterplay between AP1 and retinoid signaling pathwaystriggered by tRA, cRA and HPR in keratinocytes as thetarget cell type for skin cancer.
MATERIALS AND METHODS
Reagents and Drugs
Retinoids were purchased from Sigma Chemical Co. (St. Louis, MO).Keratinocytes were cultured in Eagle’s minimum essential medium(EMEM) (Bio Whittaker, Cambrex Company). PBS, FBS, L-glutamine,dispase, sodium bicarbonate, penicillin-streptomycin and LipofectAMINE2000y were obtained from Gibco BRL Life Technologies (Bethesda,MD). The Dual-Luciferase Reporter Gene Assay System was obtainedfrom Promega Corp. (Madison, WI). All antibodies, except K14, werepurchased from Santa Cruz Biotechnology (Santa Cruz, CA). K14 anti-
body was kindly provided by Dr. S. H. Yuspa (NIH, Bethesda, MD). b-Galactosidase plasmid (PSV-b-Gal) was from Promega Corp.
Cell Culture
Primary mouse keratinocytes were isolated from newborn BALB/cmice as described previously (30) and was approved by the InstitutionalAnimal Care and Use Committee of the American University of Beirut.One- to 2-day-old mice were killed humanely on ice and their skinsfloated in 0.25% dispase in EMEM overnight at 48C. The epidermis waspeeled from the dermis and incubated in 0.25% trypsin for 15 min at378C. The cell suspension was passed through a 40-mm cell strainer. Cellswere seeded at a density of two mouse equivalents per 100-mm dish andused after 3 days in culture. Keratinocytes were cultured in EMEM con-taining 9% chelexed fetal bovine serum containing 0.05 mM Ca21 tomaintain a basal cell phenotype.
Irradiation Procedure
UV radiation was generated using a parallel bank of two FS40T12fluorescent sunlamps (Light Sources) localized in a sterile hood emittinga continuous spectrum between 280 and 320 nm with a peak emission at312 nm. The UVB output in mW/cm2 was measured with a UVX digitalradiometer equipped with a UVX-31 midrange sensor (San Gabriel, CA)and fitted with a UVB filter sensitive to radiation between 280–340 nm.The total energy output was calibrated by adjusting the time of exposureto the light source. UVB radiant flux to the keratinocytes was restrictedto 30 mJ/cm2. Prior to irradiation, the cell culture dishes containing ke-ratinocytes were washed with PBS, placed without covers in a thin layerof PBS, and irradiated at a distance of 60 cm from the center of thelamps.
Retinoid or Proteasomal Inhibitor Treatment
Cells were treated with the retinoids tRA, cRA and HPR dissolved inDMSO; the final solvent concentration was #0.1%. Retinoids were re-constituted at a stock concentration of 3.3 3 1022 M (tRA), 3.3 3 1023
M (cRA), or 1 3 1022 M (HPR), divided into aliquots, and stored at2708C. Cells were treated with retinoids under yellow safe light (ThomasDuplex super safelights, Calumet Photographic Inc.) to avoid oxidationof these light-sensitive compounds. The proteasomal inhibitor MG132was dissolved in DMSO, and the lysosomal inhibitor E64 was dissolvedin water.
Western Blot Analysis
Cellular protein extracts were prepared from keratinocytes, washedtwice with ice-cold PBS, and scraped into SDS-lysis buffer (0.25 M Tris-HCl, pH 6.8, 20% glycerol, 4% SDS, 0.002% bromophenol blue, 10%b-mercaptoethanol). Protein concentration was determined using the DCProtein Assay from Bio-Rad (Hercules, CA). Cellular protein extracts (30mg) were run on denaturing 10% polyacrylamide gels and transferredonto nitrocellulose membranes (Bio-Rad) by standard blotting proce-dures. Membranes were then blocked in a wash buffer (100 mM Tris-HCl buffer, pH 7.5, 150 mM NaCl, 0.05% Tween 20) with 5% nonfatdried milk. The membranes were probed with RARa (sc-551), RARg(sc-550), RXRa (sc-553), FOS (sc-52), or JUN (sc-44X) polyclonal an-tibodies. Equal protein loading and quality were verified through K14reprobing of membranes and parallel protein gel Coomassie staining. Pro-teins were detected by enhanced chemiluminescence using the ECL sys-tem (Amersham, Buckinghamshire, England) with horseradish peroxi-dase-conjugated secondary antibody. Bands were quantified usingImageQuant software and the Molecular Dynamics Storm 860 System(Molecular Dynamics, Sunnyvale, CA).
RiboQuant Ribonuclease Protection Assay (RPA)
Keratinocytes were plated in 100-mm tissue culture dishes. Total RNAwas extracted using an RNeasy kit from Qiagen (Valencia, CA) according

298 DARWICHE ET AL.
to the manufacturer’s instructions. RPA template (Pharmingen, San Die-go, CA) was radiolabeled using [a-32P]UTP and T7 RNA polymerase at378C for 60 min. The reaction was terminated by adding DNase at 378Cfor 30 min. The radiolabeled RNA templates were extracted using phenoland chloroform isoamyl alcohol (50:1). The upper aqueous layer con-taining RNA was then precipitated in ammonium acetate and ice-coldabsolute ethanol and washed once with 90% ethanol. The RNA pelletwas then air-dried, suspended in 50 ml of hybridization buffer, and quan-tified (cpm/ml). RNA extracts were incubated with the probe (;3 3 105
counts/ml) at 568C for 12–16 h. Unprotected RNA was then digested withRNase A and T1 mix and incubated with proteinase K cocktail for 15min. RNA (protected) was extracted in phenol and chloroform isoamylalcohol at a ratio of 1:0.5:0.5, precipitated in ammonium acetate andabsolute ethanol, and washed once with 90% ethanol. RNA pellets weresuspended in 5 ml of 13 loading buffer, warmed at 908C for 3 min, andthen run on 5% acrylamide-urea gels in TBE buffer at 50 W until theleading edge of bromophenol blue reached 30 cm. Bands were quantifiedusing ImageQuant software and the Molecular Dynamics Storm 860 Sys-tem.
Transfections and Luciferase Reporter Gene Assays
Primary mouse keratinocytes were transiently transfected with pAP1(PathDetectt AP1 cis-Reporting System kit, Stratagene Cloning Systems)or b-galactosidase plasmids using LipofectAMINE 2000y. Keratinocyteswere washed once with serum- and antibiotic-free SMEM containing 1%L-glutamine and then transfected. Eighteen hours after transfection, cellswere exposed to UVB radiation to activate AP1 expression. Transfectedcells were washed twice with cold PBS and scraped on ice with 60 mlof passive lysis buffer provided in the luciferase kit. Luciferase activitywas measured in 20 ml of each extract using Luciferase Assay Reagent.Luminescence was read on a Wallac 1420 Victor plate reader-Luminom-eter (Perkin Elmer Life Sciences, Boston, MA). Another 20-ml fractionof the corresponding extracts was assayed for the normalizing controlplasmid b-galactosidase product activity. Then 20 ml of 23 substrate(Promega Corp.) was added to the cell lysates in a 96-well plate andincubated at 378C for 30 min. The reaction was stopped using 1 M sodiumcarbonate and then absorbance was measured using a microplate reader(iEMS, Labsystems) with a 414-nm filter. Normalization of AP1 lucif-erase activity per microgram proteins yielded the same values as nor-malization using b-Gal values. Statistical analysis was performed usingone-way analysis of variance (ANOVA) followed by post hoc multiplecomparisons Tukey’s test to validate the significance level of the observedresults (P , 0.05).
RESULTS
Ultraviolet B Radiation Reduces Retinoid Receptors andStimulates AP1 Factor Gene Expression
To investigate the effects of UVB radiation on retinoidand AP1 signaling pathways in keratinocytes, we first as-sessed the effect of 30 mJ/cm2 of UVB radiation, a dosethat lies within one minimum erythemal dose, on the pro-tein kinetics of retinoid receptors and AP1 factors. UVBirradiation leads to a significant reduction in the levels ofRXRa protein, the most abundant retinoid receptor in theepidermis (Fig. 1A). A 20% decrease was evident as earlyas 4 h, through the appearance of a set of RXRa bandswith higher molecular weights, and was complete at 24 h(Fig. 1B). The same UVB-radiation treatment caused a 40%decrease in RARa protein levels at 16 h (Fig. 1B). On theother hand, RARg protein, the most abundant RAR in theepidermis, was consistently unaffected by 30 mJ/cm2 of
UVB radiation (Fig. 1B). Conversely, AP1 factors exhibiteddifferent kinetics from retinoid receptors upon exposure tothe same dose of UVB radiation. In normal nonirradiatedproliferating keratinocytes, we were able to detect JUN butno FOS proteins (Fig. 1A). Several JUN bands were ob-served since the JUN antibody used detects three JUNmembers, JUN, JUNB and JUND. Upon exposure to UVBradiation, FOS and JUN protein levels were induced as ear-ly as 2 h postirradiation, remained elevated up to 16 h, anddeclined by 24 h (Fig. 1B).
We also tested by RNase protection assay whether UVBirradiation of keratinocytes resulted in an altered expressionof FOS and JUN transcripts. We found that JUNB and FOSwere the only transcripts induced at 12 h postirradiation(Fig. 1C). However at 24 h, a twofold induction of all JUNand FOS mRNA was noted. These findings indicate thatexposure of keratinocytes to UVB radiation decreasesRXRa and RARa proteins while it increases the transcriptand protein levels of AP1.
Effects of Ultraviolet B Radiation on Retinoid ReceptorDegradation are Dependent on Dose and Mediated byProteasomes
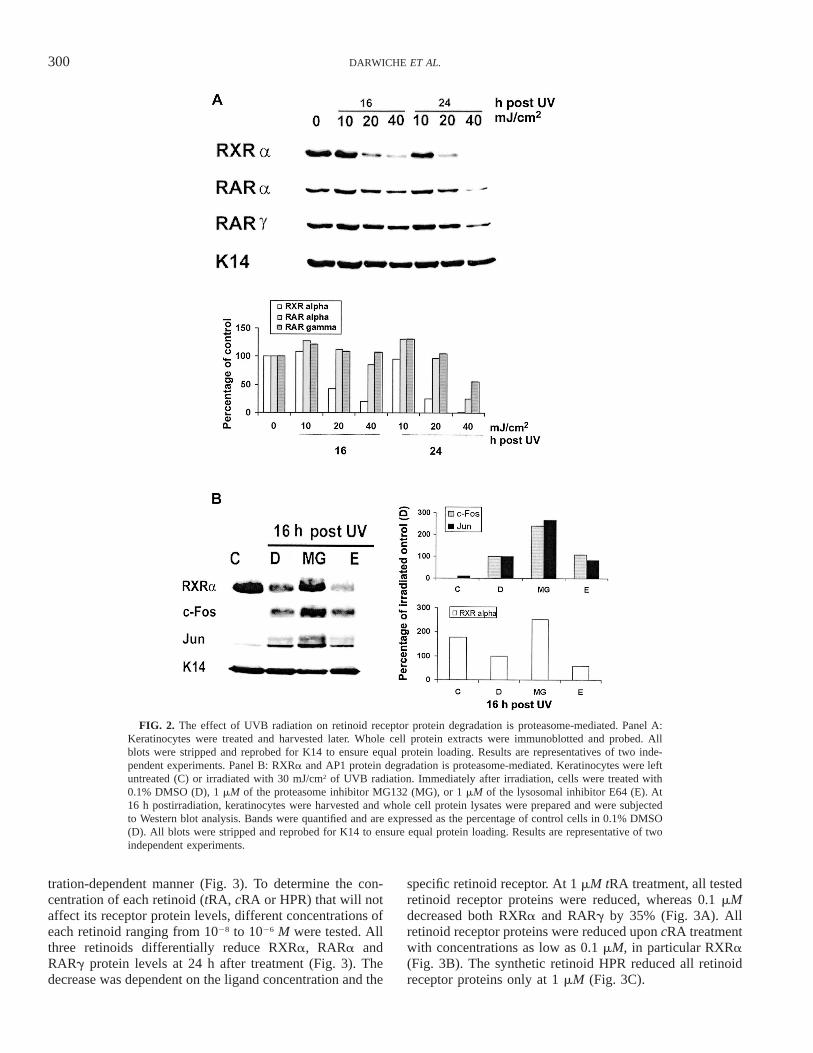
To test whether the decrease in retinoid receptor proteinlevels is dependent on the dose of radiation, we treatedkeratinocytes with several doses of UVB radiation and de-termined protein levels at 16 and 24 h after treatment. Alow dose of 10 mJ/cm2 caused no significant detectablechanges in RARa, RARg and RXRa proteins at either time(Fig. 2A). However, at 24 h after 40 mJ/cm2, RXRa pro-teins were completely degraded, whereas RARa and RARgproteins were reduced by 75% and 45%, respectively (Fig.2A).
In eukaryotic cells, the proteasome is the major organelleresponsible for the targeted selective degradation of short-lived regulatory proteins and transcription factors (31). Todetermine whether the degradation of RXRa and AP1 pro-teins was proteasome-mediated, we irradiated keratinocyteswith 30 mJ/cm2 of UVB radiation and then treated themwith the specific proteasomal inhibitor MG132 (32). Inter-estingly, treatment with MG132 prevented the UVB-radi-ation-induced degradation of RXRa, FOS and JUN proteins16 h postirradiation (Fig. 2B). Such protein degradationwas specifically mediated by proteasomes since treating theirradiated keratinocytes with the lysosomal inhibitor E64(33) failed to reverse the UVB-radiation-induced RXR andAP1 protein degradation (Fig. 2B).
Retinoids Down-regulate their own Receptors’ Proteins
Recent studies have shown that retinoid pretreatment ofhuman skin alleviates the UVB-radiation-induced down-regulation of retinoid receptors (34). In an attempt to re-verse the in vitro effects of UVB radiation on the differentretinoid receptor protein levels, we unexpectedly found thatretinoids down-regulate their receptor protein in a concen-

299RETINOIDS MODULATE UVB-RADIATION-INDUCED AP1 ACTIVITY
FIG 1. Effects of UVB radiation on retinoid receptors and AP1 factor gene expression after UVB irradiation.Panel A: keratinocytes were irradiated with 30 mJ/cm2 of UVB radiation and whole cell protein lysates were preparedand subjected to immunoblot analysis. The antibody for JUN is broadly reactive with other members of the JUNfamily (JUNB and JUND). All blots were stripped and reprobed for K14 to ensure equal protein loading. Panel B:Quantification of bands. Similar trends in protein levels were obtained in at least three independent trials. Panel C:UVB radiation increases the transcript levels of AP1 family members. Keratinocytes were irradiated with 30 mJ/cm2
UVB radiation and harvested, and an RNase protection assay was performed. Results are expressed as a percentageof control cells for each group. These values are normalized to GAPD. Similar results were obtained if normalizedto L32. Results are representative of three independent experiments.
300 DARWICHE ET AL.
FIG. 2. The effect of UVB radiation on retinoid receptor protein degradation is proteasome-mediated. Panel A:Keratinocytes were treated and harvested later. Whole cell protein extracts were immunoblotted and probed. Allblots were stripped and reprobed for K14 to ensure equal protein loading. Results are representatives of two inde-pendent experiments. Panel B: RXRa and AP1 protein degradation is proteasome-mediated. Keratinocytes were leftuntreated (C) or irradiated with 30 mJ/cm2 of UVB radiation. Immediately after irradiation, cells were treated with0.1% DMSO (D), 1 mM of the proteasome inhibitor MG132 (MG), or 1 mM of the lysosomal inhibitor E64 (E). At16 h postirradiation, keratinocytes were harvested and whole cell protein lysates were prepared and were subjectedto Western blot analysis. Bands were quantified and are expressed as the percentage of control cells in 0.1% DMSO(D). All blots were stripped and reprobed for K14 to ensure equal protein loading. Results are representative of twoindependent experiments.
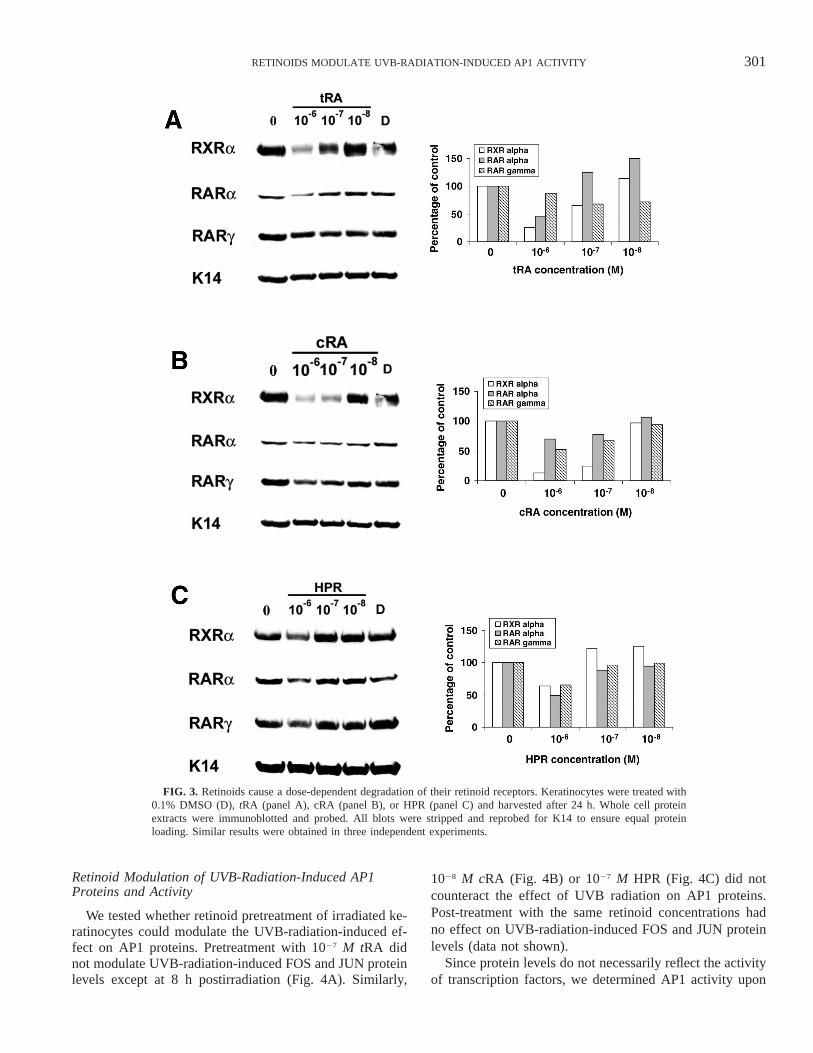
tration-dependent manner (Fig. 3). To determine the con-centration of each retinoid (tRA, cRA or HPR) that will notaffect its receptor protein levels, different concentrations ofeach retinoid ranging from 1028 to 1026 M were tested. Allthree retinoids differentially reduce RXRa, RARa andRARg protein levels at 24 h after treatment (Fig. 3). Thedecrease was dependent on the ligand concentration and the
specific retinoid receptor. At 1 mM tRA treatment, all testedretinoid receptor proteins were reduced, whereas 0.1 mMdecreased both RXRa and RARg by 35% (Fig. 3A). Allretinoid receptor proteins were reduced upon cRA treatmentwith concentrations as low as 0.1 mM, in particular RXRa(Fig. 3B). The synthetic retinoid HPR reduced all retinoidreceptor proteins only at 1 mM (Fig. 3C).
301RETINOIDS MODULATE UVB-RADIATION-INDUCED AP1 ACTIVITY
FIG. 3. Retinoids cause a dose-dependent degradation of their retinoid receptors. Keratinocytes were treated with0.1% DMSO (D), tRA (panel A), cRA (panel B), or HPR (panel C) and harvested after 24 h. Whole cell proteinextracts were immunoblotted and probed. All blots were stripped and reprobed for K14 to ensure equal proteinloading. Similar results were obtained in three independent experiments.
Retinoid Modulation of UVB-Radiation-Induced AP1Proteins and Activity
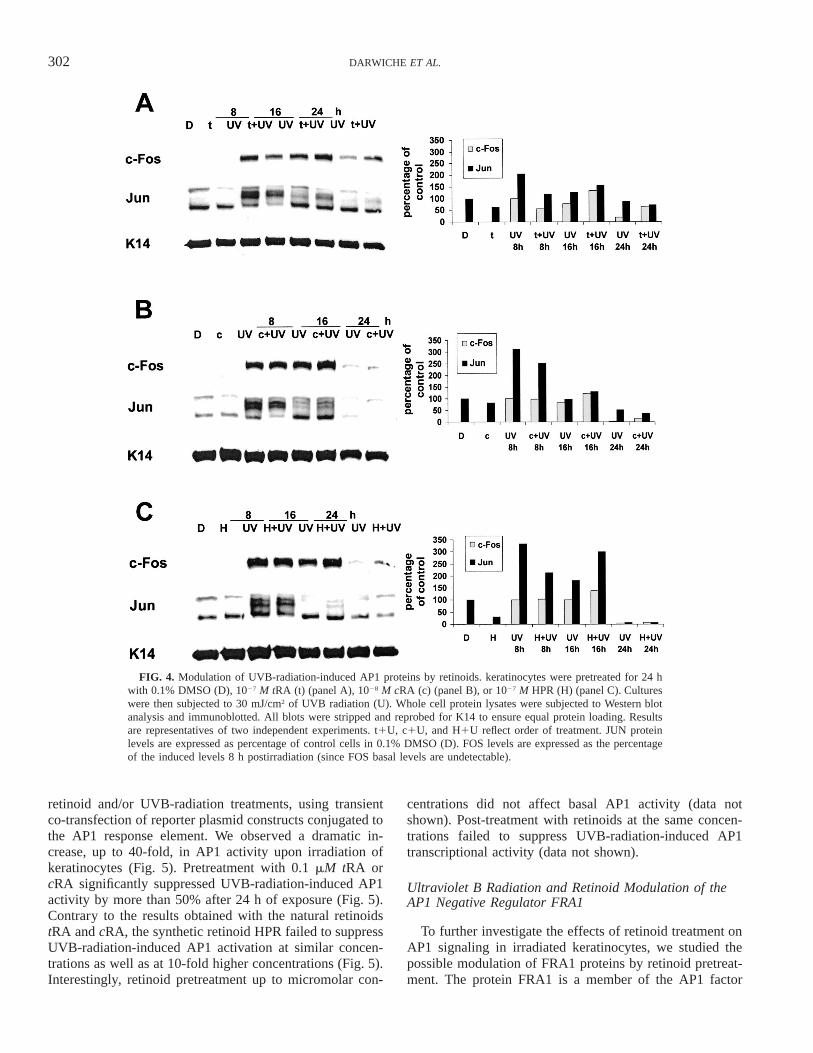
We tested whether retinoid pretreatment of irradiated ke-ratinocytes could modulate the UVB-radiation-induced ef-fect on AP1 proteins. Pretreatment with 1027 M tRA didnot modulate UVB-radiation-induced FOS and JUN proteinlevels except at 8 h postirradiation (Fig. 4A). Similarly,
1028 M cRA (Fig. 4B) or 1027 M HPR (Fig. 4C) did notcounteract the effect of UVB radiation on AP1 proteins.Post-treatment with the same retinoid concentrations hadno effect on UVB-radiation-induced FOS and JUN proteinlevels (data not shown).
Since protein levels do not necessarily reflect the activityof transcription factors, we determined AP1 activity upon
302 DARWICHE ET AL.
FIG. 4. Modulation of UVB-radiation-induced AP1 proteins by retinoids. keratinocytes were pretreated for 24 hwith 0.1% DMSO (D), 1027 M tRA (t) (panel A), 1028 M cRA (c) (panel B), or 1027 M HPR (H) (panel C). Cultureswere then subjected to 30 mJ/cm2 of UVB radiation (U). Whole cell protein lysates were subjected to Western blotanalysis and immunoblotted. All blots were stripped and reprobed for K14 to ensure equal protein loading. Resultsare representatives of two independent experiments. t1U, c1U, and H1U reflect order of treatment. JUN proteinlevels are expressed as percentage of control cells in 0.1% DMSO (D). FOS levels are expressed as the percentageof the induced levels 8 h postirradiation (since FOS basal levels are undetectable).
retinoid and/or UVB-radiation treatments, using transientco-transfection of reporter plasmid constructs conjugated tothe AP1 response element. We observed a dramatic in-crease, up to 40-fold, in AP1 activity upon irradiation ofkeratinocytes (Fig. 5). Pretreatment with 0.1 mM tRA orcRA significantly suppressed UVB-radiation-induced AP1activity by more than 50% after 24 h of exposure (Fig. 5).Contrary to the results obtained with the natural retinoidstRA and cRA, the synthetic retinoid HPR failed to suppressUVB-radiation-induced AP1 activation at similar concen-trations as well as at 10-fold higher concentrations (Fig. 5).Interestingly, retinoid pretreatment up to micromolar con-
centrations did not affect basal AP1 activity (data notshown). Post-treatment with retinoids at the same concen-trations failed to suppress UVB-radiation-induced AP1transcriptional activity (data not shown).
Ultraviolet B Radiation and Retinoid Modulation of theAP1 Negative Regulator FRA1
To further investigate the effects of retinoid treatment onAP1 signaling in irradiated keratinocytes, we studied thepossible modulation of FRA1 proteins by retinoid pretreat-ment. The protein FRA1 is a member of the AP1 factor

303RETINOIDS MODULATE UVB-RADIATION-INDUCED AP1 ACTIVITY
FIG. 5. Differential modulation of UVB-radiation-induced AP1 activ-ity by retinoids. keratinocytes were co-transfected with AP1-LUC and b-gal plasmids using lipofectAMINE reagent. Then cells were treated witheither 0.1% DMSO or retinoids for 24 h: 1027 M tRA (t7), 1028 M tRA(t8), 1027 M cRA (c7), 1028 M cRA (c8), 1027 M HPR (H7), or 1026 MHPR (H6). Cultures were then irradiated with 30 mJ/cm2 of UVB radi-ation and left for an additional 24 h. Whole cell protein lysates wereassayed for LUC or b-gal activity. b-Galactosidase activity was used tonormalize for transfection efficiency. The units on the y axis are arbitrary.The values represent averages of duplicate measurements from one oftwo independent experiments. Bars, SE. *Statistically significant (P ,0.01) difference from control UV-irradiated cells (t test).
family and is a negative regulator of AP1 signaling. Ex-posure of keratinocytes to UVB radiation caused a transientincrease in FRA1 protein expression (Fig. 6A). A threefoldincrease in FRA1 protein was observed 16 h after UVBirradiation (Fig. 6B). In addition, a two- to threefold in-crease in FRA1 transcript levels was evident at 12–24 hpostirradiation (Fig. 6C).
The effect of tRA and HPR treatments on UVB-radia-tion-induced increases in FRA1 expression was then inves-tigated. Treatment with 0.1 mM tRA prior to UVB irradi-ation caused a sharp transient increase in FRA1 proteinlevels as early as 2 h after UVB irradiation (Fig. 6A andB). Cells pretreated with 1 mM HPR showed a similar in-crease in FRA1 protein at 2 h postirradiation, and this in-crease was sustained for up to 16 h postirradiation (Fig. 6Aand B). A similar increase in FRA1 transcripts was ob-served upon treatment with both retinoids with no signifi-cant changes detected in FRA2 mRNA levels (Fig. 6C).Both tRA and HPR pretreatment of keratinocytes furtherelevated the UVB-radiation-induced increases in FRA1gene expression.
DISCUSSION
Intense research efforts are directed toward developingagents capable of combating the onset or progression ofUV-radiation-induced skin malignancies. One of the mostextensively studied classes of chemopreventive agents isthe retinoid family. In the present study, we report thatUVB irradiation of keratinocytes reduces retinoid receptorproteins and stimulates AP1 signaling pathways. Further-
more, we show that tRA, cRA and HPR down-regulate theirown receptors levels, yet only tRA and cRA reduce UVB-radiation-induced AP1 activity. Primary mouse keratino-cytes recapitulate some of the recently reported effects ofUVB radiation in human skin and keratinocytes such asinduced loss of retinoid receptors (34, 35) and up-regulationof AP1 factors (9) as well as tRA repression of AP1 activity(28, 36).
Subjecting keratinocytes to UVB radiation mostly down-regulates the abundant epidermal retinoid receptor, RXRa,in accordance with earlier findings in human keratinocytesin vitro (35). Similar UVB-radiation-induced down-regu-lation of RXRa has been observed in human skin; however,this was transient (34). Contrary to what has been observedin human skin and primary keratinocytes (34, 35), we foundthat RARg proteins in keratinocytes are resistant to down-regulation by UVB radiation. An accumulating body of ev-idence shows that retinoid receptors are down-regulatedduring skin tumor progression (30, 37, 38), upon exposureto chemical (e.g. TPA or mezerein) or physical (e.g. UVBradiation) tumor promoters (30, 34, 35, 39), or upon trans-duction of a v-rasHa oncogene in primary keratinocytes(30).
The exposure of keratinocytes to UVB radiation tran-siently up-regulates FOS and JUN family proteins, whereastheir transcripts increase at later times. AP1 factors are up-regulated and/or activated during skin carcinogenesis (14),in response to treatment with chemical promoters such asTPA (40) or physical tumor-promoting agents such as UVBradiation (5, 41), or upon transduction of oncogenes suchas v-rasHa in primary keratinocytes (15, 42). AP1 factoractivation is necessary for the transformation in epidermalcells (42). Interestingly, AP1 DNA-binding activity is high-er in nuclear extracts from papillomas at high risk of ma-lignant progression (14), and conversely, retinoid receptorprotein levels are more reduced in skin tumors at higherrisk for malignant progression (30). Apparently, retinoid re-ceptor and AP1 signaling pathways are interrelated. In fact,retinoid receptors and AP1 factors have been shown to re-press the function of each other (43, 44).
UVB-radiation-induced degradation of RXRa, FOS andJUN proteins seems to be proteasome-mediated. Similarfindings have been reported in human keratinocytes (35,45). RXRa proteins show an early gradual electrophoreticmobility shift that has been observed by others and reflectsthe phosphorylation of the receptor prior to its degradation(46). This may be due to ubiquitination prior to the protea-some-mediated protein degradation in human keratinocytesor to phosphorylation. Retinoid receptor proteins are con-stitutively phosphorylated (47) and are hyperphosphorylat-ed by JNK during UVB-radiation exposure (48).
Each of the retinoids, tRA, cRA and HPR, down-regu-lates its own receptor protein in a concentration-dependentmanner. These efforts were not observed in human skin(34), but they have been reported in vitro to be proteasome-mediated (49, 50). Even though the steroid/thyroid nuclear

304 DARWICHE ET AL.
FIG. 6. Induction of FRA1 expression by treatment with UVB radiation and retinoids. Panel A: Keratinocytes were pretreated with 0.1% DMSO,1027 M tRA, or 1026 M HPR for 24 h and then were irradiated with 30 mJ/cm2 of UVB radiation. Whole SDS cell protein lysates were subjected toWestern blot analysis and immunoblotted. All blots were stripped and reprobed for K14 to ensure equal protein loading. Results are representative oftwo independent experiments. Panel B: Quantification of bands. Panel C: Keratinocytes were pretreated with 0.1% DMSO, 1027 M tRA or 1026 MHPR for 24 h. Cells were then subjected to 30 mJ/cm2 of UVB radiation and harvested and the RNase protection assay was performed. Results areexpressed as a percentage of control cells for each group. These values are normalized to GAPD. Similar results were obtained when normalized toL32. Results are representative of two independent experiments.
receptor superfamily members share several common fea-tures with the retinoid receptors, they differ in that respect.For example, vitamin D stabilizes its own receptor by in-hibiting its ubiquitination and subsequent degradation (51).
The transcription factor AP1 plays an important role inUV-radiation-mediated skin tumor promotion and is there-fore a logical target for chemopreventive strategies (52).Results from several clinical trials have shown that reti-noids have chemopreventive effects. Pre- or post-treatmentof irradiated keratinocytes with concentrations of retinoidsthat do not significantly reduce retinoid receptors did notprotect against UVB-radiation-induced retinoid receptordegradation and increased AP1 protein levels. This is incontrast to the stabilization effect of tRA pretreatment onretinoid receptors in UV-irradiated human skin (34). Inter-estingly, UV-radiation-induced AP1 activity was sup-pressed upon pretreatment with the two natural retinoids,tRA and cRA, but not with the synthetic retinoid, HPR.
Several models have been proposed to explain the sup-pression of AP1 activity by retinoids such as direct protein/protein interaction between the retinoid receptors and AP1factors (29); competitive binding or squelching of commoncoactivators like CBP (28), or inhibition of AP1 protein
phosphorylation (5). The inhibition of AP1-dependent tran-scription is mediated by RA-activated RARs, whereasRXRa failed to repress AP1 activity in retinoid receptor-transfected HeLa cells (43). Therefore, RXRa is not in-volved in the effect observed on AP1 activity in irradiatedkeratinocytes and AP1 components can interact directlywith RAR monomers (29). Recent reports have demonstrat-ed the involvement of FRA1, a member of the FOS family,in antagonizing its own family members (42). Ascorbate,or vitamin C, can inhibit UVB-radiation-induced AP1 ac-tivity by elevating FRA1 protein levels prior to irradiationand maintain them elevated postirradiation (53). We ob-served that exposure of keratinocytes to UVB radiationcauses an increase in FRA1 gene expression and that bothtRA and HPR pretreatments further elevate FRA1 levels.Therefore, the differential effect of these two former reti-noids on UVB-radiation-induced AP1 activity is not ex-plained solely by their effects on FRA1 gene expression.
The two functions of retinoid receptors in transcriptionalactivation and AP1 antagonism can be separated. Severalnew synthetic retinoids have been developed that specifi-cally activate one of the two retinoid functions without af-fecting the other (36, 54). Such compounds are crucial to

305RETINOIDS MODULATE UVB-RADIATION-INDUCED AP1 ACTIVITY
dissect the role of retinoid receptor antagonism of AP1 ac-tivity independently of transcriptional activation and viceversa.
In summary, we have shown that tRA and cRA pretreat-ment but not post-treatment of epidermal keratinocytes pri-or to UVB-radiation exposure reduces AP1 activity andthus has photoprotective effects. Chemopreventive com-pounds such as theaflavins from black tea (55), (-)-epigal-locatechin gallate from green tea (56), the iron chelatordeferoxamine (57), and the polyphenolic lignan nordihy-droguaiaretic acid (58) were also shown to inhibit UVB-radiation-induced AP1 activation in keratinocytes. Furthertesting of naturally occurring or synthetic retinoids mayidentify other promising chemopreventive agents for UVB-radiation-induced skin carcinogenesis.
ACKNOWLEDGMENTS
We are grateful to Dr. Fadia Homaidan for her critical review of themanuscript. This work was supported by grants from the American Uni-versity of Beirut University Research Board and the Third World Acad-emy of Science Award.
Received: January 15, 2004; accepted: October 26, 2004
REFERENCES
1. R. Marks, An overview of skin cancers. Incidence and causation.Cancer 75, 607–612 (1995).
2. D. E. Brash, A. Ziegler, A. S. Jonason, J. A. Simon, S. Kunala andD. J. Leffell, Sunlight and sunburn in human skin cancer: p53, ap-optosis, and tumor promotion. J. Investig. Dermatol. Symp. Proc. 1,136–142 (1996).
3. J. P. Ortonne, Photobiology and genetics of malignant melanoma. Br.J. Dermatol. 146, 11–16 (2002).
4. K. Scharffetter-Kochanek, M. Wlaschek, P. Brenneisen, M. Schauen,R. Blaudschun and J. Wenk, UV-induced reactive oxygen species inphotocarcinogenesis and photoaging. Biol. Chem. 378, 1247–1257(1997).
5. G. Fisher and J. J. Vorhees, Molecular mechanisms of photoagingand its prevention by retinoic acid: Ultraviolet irradiation inducesMAP kinase signal transduction cascades that induce AP-1-regulatedmatrix metalloproteinases that degrade human skins in vivo. J. In-vestig. Dermatol. Symp. Proc. 3, 61–68 (1998).
6. A. Sesto, M. Navarro, F. Burslem and J. L. Jorcano, Analysis of theultraviolet B response in primary human keratinocytes using oligo-nucleotide microarrays. Proc. Natl. Acad. Sci. USA 99, 2965–2970(2002).
7. J. Cotton and D. F. Spandau, Ultraviolet B-radiation dose influencesthe induction of apoptosis and p53 in human keratinocytes. Radiat.Res. 147, 148–155 (1997)
8. M. Adachi, A. Gazel, G. Pintucci, A. Shuck, S. Shifteh, D. Ginsburg,L. S. Rao, T. Kaneko, I. M. Freedberg and M. Blumenberg, Speci-ficity in stress response: Epidermal keratinocytes exhibit specializedUV-responsive signal transduction pathways. DNA Cell Biol. 22,665–677 (2003).
9. K. Isoherranen, J. Westermarck, V. M. Kahari, C. Jansen and K.Punnonen, Differential regulation of the AP-1 family members byUV irradiation in vitro and in vivo. Cell Signal 10, 191–195 (1998).
10. D. Li, T. G. Turi, A. Schuck, I. M. Freedberg, G. Khitrov and M.Blumenberg, Rays and arrays: The transcriptional program in theresponse of human epidermal keratinocytes to UVB illumination. FA-SEB J. 15, 2533–2535 (2001).
11. M. Karin, The regulation of AP-1 activity by mitogen-activated pro-tein kinases. J. Biol. Chem. 270, 16483–16486 (1995).
12. M. R. Young, J. J. Li, M. Rincon, R. A. Flavell, B. K. Sathyanar-ayana, R. Hunziker and N. Colburn, Transgenic mice demonstrateAP-1 (activator protein-1) transactivation is required for tumor pro-motion. Proc. Natl. Acad. Sci. USA 96, 9827–9832 (1999).
13. S. J. Cooper, J. MacGowan, J. Ranger-Moore, M. R. Young, N. H.Colburn and G. T. Bowden, Expression of dominant negative c-juninhibits ultraviolet B-induced squamous cell carcinoma number andsize in an SKH-1 hairless mouse model. Mol. Cancer Res. 1, 848–854 (2003).
14. S. E. Rutberg, E. J. Lee, L. H. Hansen, A. B. Glick and S. H. Yuspa,Identification of differentially expressed genes in chemically inducedskin tumors. Mol. Carcinog. 20, 88–98 (1997).
15. D. A. Greenhalgh, D. J. Welty, A. Player and S. H. Yuspa, Twooncogenes, v-fos and v-ras, cooperate to convert normal keratino-cytes to squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 87,643–647 (1990).
16. E. Saez, S. E. Rutberg, E. Mueller, H. Oppenheim, J. Smoluk, S. H.Yuspa and B. M. Spiegelman, c-fos is required for malignant pro-gression of skin tumors. Cell 82, 721–732 (1995).
17. R. Lotan, Retinoids in cancer chemoprevention. FASEB J. 10, 1031–1039 (1996).
18. S. J. Freemantle, M. J. Spinella and E. Dmitrovsky, Retinoids incancer therapy and chemoprevention: Promise meets resistance. On-cogene 22, 7305–7315 (2003).
19. M. I. Dawson, J. H. Park, G. Chen, W. Chao, L. Dousman, N. Waleh,P. D. Hobbs, L. Jong, L. Toll and D. L. Hill, Retinoic acid (RA)receptor transcriptional activation correlates with inhibition of 12-O-tetradecanoylphorbol-13-acetate-induced ornithine decarboxylase(ODC) activity by retinoids: a potential role for trans-RA-inducedZBP-89 in ODC inhibition. Int. J. Cancer 91, 8–21 (2001).
20. A. K. Verma, Inhibition of tumor promoter 12-O-tetradecanoylphor-bol-13-acetate-induced synthesis of epidermal ornithine decarboxyl-ase messenger RNA and diacylglycerol promoted mouse skin tumorformation by retinoic acid. Cancer. Res. 48, 2168–2173 (1988).
21. M. Athar, R. Agarwal, Z. Y. Wang, J. R. Lloyd, D. R. Bickers andH. Mukhtar, All-trans retinoic acid protects against conversion ofchemically induced and ultraviolet B radiation-induced skin papil-lomas to carcinomas. Carcinogenesis 12, 2325–2329 (1991).
22. R. Agarwal, R. R. Mohan, N. Ahmad and H. Mukhtar, Protectionagainst malignant conversion in SENCAR mouse skin by all transretinoic acid: Inhibition of the ras p21-processing enzyme farnesyl-transferase and Ha-ras p21 membrane localization. Mol. Carcinog.17, 13–22 (1996).
23. D. J. Mangelsdorf, E. S. Ong, J. A. Dyck and R. M. Evans, Nuclearreceptor that identifies a novel retinoic acid response pathway. Nature345, 224–229 (1990).
24. P. Chambon, A decade of molecular biology of retinoic acid recep-tors. FASEB J. 10, 940–954 (1996).
25. H. G. Stunnenberg, Mechanisms of transactivation by retinoic acidreceptors. Bioessays 15, 309–315 (1993).
26. F. C. Zusi, M. V. Lorenzi and V. Vivat-Hannah, Selective retinoidsand rexinoids in cancer therapy and chemoprevention. Drug Discov.Today 7, 1165–1174 (2002).
27. F. Lin, D. Xiao, S. K. Kolluri and X. Zhang, Unique anti-activatorprotein-1 activity of retinoic acid receptor b. Cancer Res. 60, 3271–3280 (2000).
28. Y. Kamei, L. Xu, T. Heinzel, J. Torchia, R. Kurokawa, B. Gloss,S. C. Lin, R. A. Heyman, D. W. Rose and M. G. Rosenfeld, A CBPintegrator complex mediates transcriptional activation and AP-1 in-hibition by nuclear receptors. Cell 85, 403–414 (1996).
29. H. F. Yang-Yen, X. K. Zhang, G. Graupner, M. Tzukerman, B. Sak-amoto, M. Karin and M. Pfahl, Anatgonism between retinoic acidreceptors and AP-1: Implications for tumor promotion and inflam-mation. New Biol. 3, 1206–1219 (1991).
30. N. Darwiche, G. Scita, C. Jones, S. Rutberg, E. Greenwald, T. Ten-nenbaum, S. J. Collins, L. M. De Luca and S. H. Yuspa, Loss of

306 DARWICHE ET AL.
retinoic acid receptors in mouse skin and skin tumors is associatedwith activation of the rasHa oncogene and high risk for premalignantprogression. Cancer Res. 56, 4942–4959 (1996).
31. C. M. Pickart, Targeting of substrates to the 26S proteasome. FASEBJ. 11, 1055–1066 (1997).
32. K. L. Rock, C. Gramm, L. Rothstein, K. Clark, R. Stein, L. Dick, D.Hwang and A. L. Goldberg, Inhibitors of the proteasome block thedegradation of most cell proteins and the generation of peptides pre-sented on MHC class I molecules. Cell 78, 761–71 (1994)
33. A. J. Barrett, A. A. Kembhavi, M. A. Brown, H. Kirschke, C. G.Knight, M. Tamai and K. Hanada, L-trans-Epoxysuccinyl-leucylam-ido(4-guanidino)butane (E-64) and its analogues as inhibitors of cys-teine proteinases including cathepsins B, H and L. Biochem J. 201,189–198 (1982).
34. Z. Wang, M. Boudjelal, S. Kang, J. J. Voorhees and G. J. Fisher,Ultraviolet irradiation of human skin causes functional vitamin Adeficiency, preventable by all-trans retinoic acid pre-treatment. Nat.Med. 5, 418–422 (1999)
35. M. Boudjelal, Z. Wang, I. J. Voorhees and G. J. Fisher, Ubiquitin/proteasome pathway regulates levels of retinoic acid receptor gammaand retinoid X receptor alpha in human keratinocytes. Cancer Res.60, 2247–2252 (2000).
36. J. J. Li, Z. Dong, M. I. Dawson and N. H. Colburn, Inhibition oftumor promoter-induced transformation by retinoids that transrepressAP-1 without transactivating retinoic acid response element. CancerRes. 56, 483–489 (1996).
37. N. Darwiche, G. Celli, T. Tennenbaum, A. B. Glick, S. H. Yuspa andL. M. De Luca, Mouse skin tumor progression results in differentialexpression of retinoic acid and retinoid X receptors. Cancer Res. 55,2774–2782 (1995).
38. X. C. Xu, W. Y. Wong, L. Goldberg, S. C. Baer, J. E. Wolf, W. M.Ramsdell, D. S. Alberts, S. M. Lippman and R. Lotan, Progressivedecreases in nuclear retinoid receptors during skin squamous carci-nogenesis. Cancer Res. 61, 4306–4310 (2001).
39. R. Kumar, A. R. Shoemaker and A. K. Verma, Retinoic acid nuclearreceptors and tumor promotion: Decreased expression of retinoic acidnuclear receptors by the tumor promoter 12-O-tetradecanoylphorbol-13-acetate. Carcinogenesis 15, 701–705 (1995).
40. Z. Dong, M. Birrer, R. Watts, L. Matrisian and N. H. Colburn, Block-ing of tumor promoter-induced AP-1 activity inhibits induced trans-formation in JB6 mouse epidermal cells. Proc. Natl. Acad. Sci. USA91, 609–613 (1994).
41. W. Chen, A. H. Borchers, Z. Dong, M. B. Powell and G. T. Bowden,UVB irradiation-induced activator protein-1 activation correlateswith increased c-Fos gene expression in a human keratinocyte cellline. J. Biol. Chem. 273, 32176–32181 (1998).
42. S. E. Rutberg, T. L. Adams, A. Glick, M. T. Bonovich, C. Vinsonand S. H. Yuspa, Activator protein-1 transcription factors are fun-damental to v-rasHa-induced changes in gene expression in neoplastickeratinocytes. Cancer Res. 60, 6332–6338 (2000).
43. R. Schule, P. Rangarajan, N. Yang, S. Kliewer, L. J. Ransone, J.Bolado, I. M. Verma and R. M. Evans, Retinoic acid is a negativeregulator of AP-1 responsive genes. Proc. Natl. Acad. Sci. 88, 6092–6096 (1991).
44. L. Yang, H. T. Kim, D. Munoz Medellin, P. Reddy and P. H. Brown,
Induction of retinoid resistance in breast cancer cells by overexpres-sion of c-Jun. Cancer Res. 57, 4652–4661 (1997).
45. M. Boudjelal, J. J. Voorhees and G. J. Fisher, Retinoid signaling isattenuated by proteasome-mediated degradation of retinoid receptorsin human keratinocyte HaCaT cells. Exp. Cell Res. 274, 130–137(2002).
46. M. Gianni, A. Tarrade, E. A. Nigro, E. Garattini and C. Rochette-Egly, The AF-1 and AF-2 domains of RAR gamma 2 and RXR alphacooperate for triggering the transactivation and the degradation ofRAR gamma 2/RXR alpha heterodimers. J. Biol. Chem. 278, 34458–34466 (2003).
47. M. P. Gaub, C. Rochette-Egly, Y. Lutz, S. Ali, H. Matthes, I. Scheuerand P. Chambon, Immunodetection of multiple species of retinoicacid receptor alpha: Evidence for phosphorylation. Exp. Cell. Res.201, 335–346 (1992).
48. S. Adam-Stitah, L. Penna, P. Chambon and C. Rochette-Egly, Hy-perphosphorylation of the retinoid X receptor alpha by activated c-Jun NH2-terminal kinases. J. Biol. Chem. 274, 18932–18941 (1999).
49. D. L. Osburn, G. Shao, H. M. Seidel and I. G. Schulman, Ligand-dependent degradation of retinoid X receptors does not require tran-scriptional activity or coactivator interactions. Mol. Cell. Biol. 21,4909–4918 (2001).
50. T. Tanaka, M. L. Rodriguez de la Concepcion and L. M. De Luca,Involvement of all-trans-retinoic acid in the breakdown of retinoicacid receptors alpha and gamma through proteasomes in MCF-7 hu-man breast cancer cells. Biochem. Pharmacol. 61, 1347–1355 (2001).
51. X. Y. Li, M. Boudjelal, J. H. Xiao, Z. H. Peng, A. Asuru, S. Kang,G. J. Fisher and J. J. Voorhees, 1,25-Dihydroxyvitamin D3 increasesnuclear vitamin D3 receptors by blocking ubiquitin/proteasome-me-diated degradation in human skin. Mol. Endocrinol. 13, 1686–1694(1999).
52. J. G. Einspahr, G. T. Bowden and D. S. Alberts, Skin cancer che-moprevention: Strategies to save our skin. Recent Results CancerRes. 163, 151–164 (2003).
53. M. V. Catani, A. Rossi, A. Costanzo, S. Sabatini, M. Levrero, G.Melino and L. Avigliano, Induction of gene expression via activatorprotein-1 in the ascorbate protection against UV-induced damage.Biochem. J. 356, 77–85 (2001).
54. A. Fanjul, M. I. Dawson, P. D. Hobbs, L. Jong, J. F. Cameron, E.Harlev, G. Graupner, X. P. Lu and M. Pfahl, A new class of retinoidswith selective inhibition of AP-1 inhibits proliferation. Nature 372,107–110 (1994).
55. M. Nomura, W. Y. Ma, C. Huang, C. S. Yang, G. T. Bowden, K.Miyamoto and Z. Dong, Inhibition of ultraviolet B-induced AP-1activation by theaflavins from black tea. Mol. Carcinog. 28, 148–155(2000).
56. M. Barthelman, W. B. Bair 3rd, K. K. Stickland, W. Chen, B. N.Timmermann, S. Valcic, Z. Dong and G. T. Bowden, (-)-Epigallo-catechin-3-gallate inhibition of ultraviolet B-induced AP-1 activity.Carcinogenesis 19, 2201–2204 (1998).
57. K. Kramer-Stickland, A. Edmonds, W. B. Bair 3rd and G. T. Bowden,Inhibitory effects of deferoxamine on UVB-induced AP-1 transacti-vation. Carcinogenesis 20, 2137–2142 (1999).
58. M. Gonzales and G. T. Bowden, Nordihydroguaiaretic acid-mediatedinhibition of ultraviolet B-induced activator protein-1 activation inhuman keratinocytes. Mol. Carcinog. 34, 102–111 (2002).
Copyright © 2022 FDOKUMEN




















