p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases
10
REVIEWS NATURE REVIEWS | DRUG DISCOVERY VOLUME 2 | SEPTEMBER 2003 | 717 Cell growth, differentiation and apoptosis are regulated by a plethora of extracellular signals. The type, duration and magnitude of these signals are efficiently transmitted to the inside of the cells, where signalling complexes are assembled for appropriate integration and processing, eventually resulting in a stimulus-specific response. The mitogen-activated protein kinases (MAPKs), which integrate and process various extracellular signals, are primary components of this intracellular signalling circuitry. A series of three protein kinases — a MAPK and two upstream components, MAPK kinase (MAPKK or MKK) and MAPKK kinase (MAPKKK) — consti- tutes the MAPK cascade (FIG. 1). So far, three distinct MAPK pathways have been described in mammalian cells, which seem to be con- served from yeast to man 1 . The extracellular signal- regulated kinases ERK1 and 2 were the first of these MAPKs to be discovered. Subsequently, c-Jun N-terminal kinase 1 (JNK1) and p38α MAPK were identified. These kinases are ~60–70% identical to each other and differ in the sequence and size of their activation loop, as well as in their activation in response to different stimuli. Each MAPK subfamily consists of several iso- forms and members, which often have distinct functions. These kinases are activated by the dual phosphorylation of Thr and Tyr residues in a ‘TXY’ (where X is Glu, Pro and Gly in ERKs, JNKs and p38 MAPKs, respectively) activation motif by a dual-specificity MAPKK 1 . The MAPKK, in turn, is phosphorylated by a serine/threo- nine kinase termed MAPKKK. In general, the ERKs are activated by mitogenic and proliferative stimuli, whereas the JNKs and p38 MAPKs respond to environ- mental stress, including ultraviolet light, heat, osmotic shock and inflammatory cytokines 1 . The human p38α MAPK was originally identified as the molecular target of the pyridinyl imidazole class of compounds that were known to inhibit biosynthesis of inflammatory cytokines such as interleukin-1 (IL-1) and tumour-necrosis factor (TNF) in lipopolysaccha- ride (LPS)-stimulated human monocytes 2 . The murine p38α MAPK was identified as a major phosphoprotein activated by LPS and inflammatory cytokines 3 . So far, four splice variants of p38α MAPK have been identi- fied. Of these, p38α is the best characterized and perhaps the most physiologically relevant kinase involved in inflammatory responses. Searches of gene databases led to the identification of the additional isoforms p38β and p38β2, which have >70% identity to p38α 4,5 . Both p38α and p38β2 are expressed in several tissues, but are thought to have distinct functions. For example, although the activity of p38α and p38β2 increased in a ventricular hypertrophy model in mouse, increased P38 MAP KINASES: KEY SIGNALLING MOLECULES AS THERAPEUTIC TAR- GETS FOR INFLAMMATORY DISEASES Sanjay Kumar, Jeffrey Boehm and John C. Lee The p38 MAP kinases are a family of serine/threonine protein kinases that play important roles in cellular responses to external stress signals. Since their identification about 10 years ago, much has been learned of the activation and regulation of the p38 MAP kinase pathways. Inhibitors of two members of the p38 family have been shown to have anti-inflammatory effects in preclinical disease models, primarily through the inhibition of the expression of inflammatory mediators. Several promising compounds have also progressed to clinical trials. In this review, we provide an overview of the role of p38 MAP kinases in stress-activated pathways and the progress towards clinical development of p38 MAP kinase inhibitors in the treatment of inflammatory diseases. GlaxoSmithKline Pharmaceuticals Research & Development, 709 Swedeland Road, King of Prussia, Pennsylvania 19406, USA. Correspondence to J. C. L. e-mail: [email protected] doi:10.1038/nrd1177
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases

R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 2 | SEPTEMBER 2003 | 717
Cell growth, differentiation and apoptosis are regulatedby a plethora of extracellular signals. The type, durationand magnitude of these signals are efficiently transmittedto the inside of the cells, where signalling complexes areassembled for appropriate integration and processing,eventually resulting in a stimulus-specific response.The mitogen-activated protein kinases (MAPKs), whichintegrate and process various extracellular signals, areprimary components of this intracellular signallingcircuitry. A series of three protein kinases — a MAPKand two upstream components, MAPK kinase (MAPKKor MKK) and MAPKK kinase (MAPKKK) — consti-tutes the MAPK cascade (FIG. 1).
So far, three distinct MAPK pathways have beendescribed in mammalian cells, which seem to be con-served from yeast to man1. The extracellular signal-regulated kinases ERK1 and 2 were the first of theseMAPKs to be discovered. Subsequently, c-Jun N-terminalkinase 1 (JNK1) and p38α MAPK were identified.These kinases are ~60–70% identical to each other anddiffer in the sequence and size of their activation loop,as well as in their activation in response to differentstimuli. Each MAPK subfamily consists of several iso-forms and members, which often have distinct functions.These kinases are activated by the dual phosphorylationof Thr and Tyr residues in a ‘TXY’ (where X is Glu, Pro
and Gly in ERKs, JNKs and p38 MAPKs, respectively)activation motif by a dual-specificity MAPKK1. TheMAPKK, in turn, is phosphorylated by a serine/threo-nine kinase termed MAPKKK. In general, the ERKs areactivated by mitogenic and proliferative stimuli,whereas the JNKs and p38 MAPKs respond to environ-mental stress, including ultraviolet light, heat, osmoticshock and inflammatory cytokines1.
The human p38α MAPK was originally identified asthe molecular target of the pyridinyl imidazole class ofcompounds that were known to inhibit biosynthesis ofinflammatory cytokines such as interleukin-1 (IL-1)and tumour-necrosis factor (TNF) in lipopolysaccha-ride (LPS)-stimulated human monocytes2. The murinep38α MAPK was identified as a major phosphoproteinactivated by LPS and inflammatory cytokines3. So far,four splice variants of p38α MAPK have been identi-fied. Of these, p38α is the best characterized and perhapsthe most physiologically relevant kinase involved ininflammatory responses. Searches of gene databases ledto the identification of the additional isoforms p38βand p38β2, which have >70% identity to p38α4,5. Bothp38α and p38β2 are expressed in several tissues, but arethought to have distinct functions. For example,although the activity of p38α and p38β2 increased in aventricular hypertrophy model in mouse, increased
P38 MAP KINASES: KEY SIGNALLINGMOLECULES AS THERAPEUTIC TAR-GETS FOR INFLAMMATORY DISEASESSanjay Kumar, Jeffrey Boehm and John C. Lee
The p38 MAP kinases are a family of serine/threonine protein kinases that play important roles incellular responses to external stress signals. Since their identification about 10 years ago, muchhas been learned of the activation and regulation of the p38 MAP kinase pathways. Inhibitors oftwo members of the p38 family have been shown to have anti-inflammatory effects in preclinicaldisease models, primarily through the inhibition of the expression of inflammatory mediators.Several promising compounds have also progressed to clinical trials. In this review, we provide anoverview of the role of p38 MAP kinases in stress-activated pathways and the progress towardsclinical development of p38 MAP kinase inhibitors in the treatment of inflammatory diseases.
GlaxoSmithKlinePharmaceuticals Research& Development,709 Swedeland Road,King of Prussia,Pennsylvania 19406, USA.Correspondence to J. C. L.e-mail: [email protected]:10.1038/nrd1177

3′ UNTRANSLATED REGION
Part of the sequence in amessenger RNA molecule at the3′ end of the coding sequencethat is not translated. Thisstructural feature is involved inmRNA stability or turnover.
718 | SEPTEMBER 2003 | VOLUME 2 www.nature.com/reviews/drugdisc
R E V I E W S
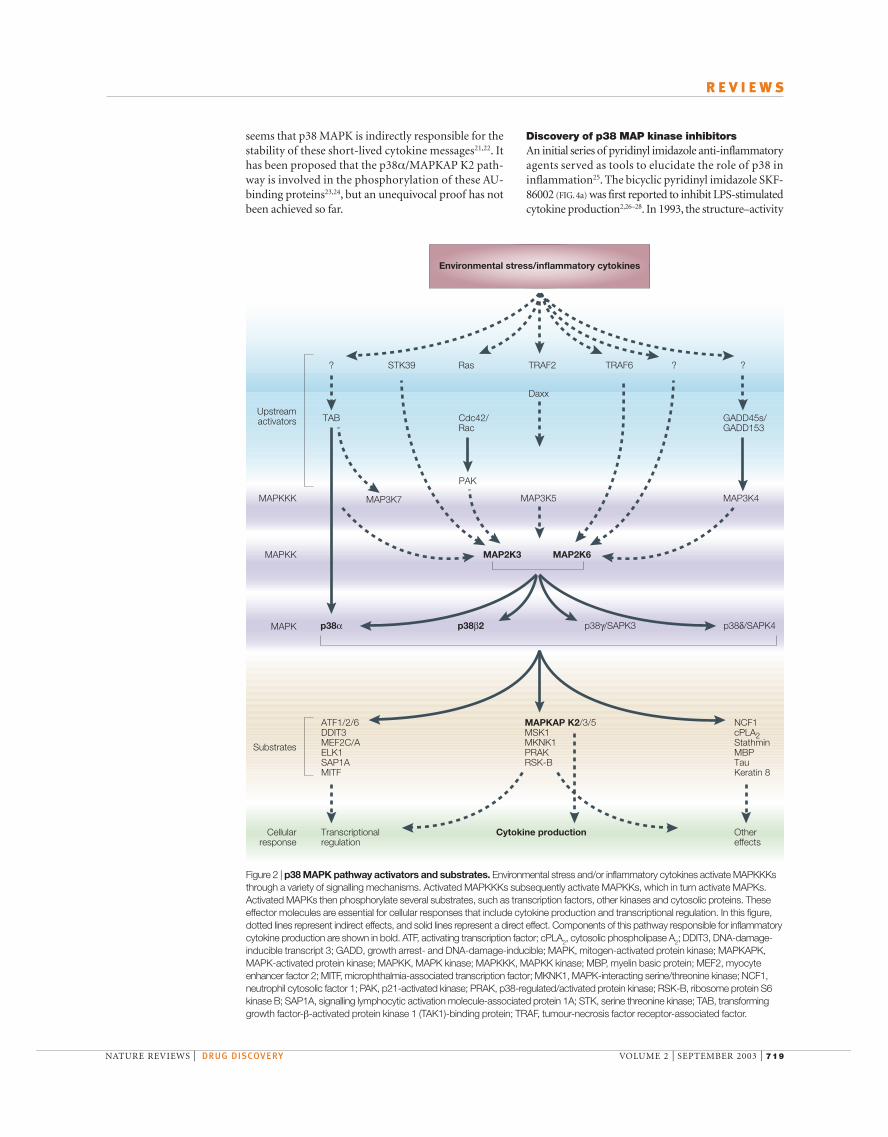
activated by several MAPKKKs, which are activated by awide variety of stimuli11,12. This underscores the complexnature of these signal transduction pathways, whichrequire the integration of multiple signals from differentpathways (FIG. 2). Recently, a MAPKK-independentmechanism of p38 MAPK activation involving TAB1(transforming growth factor-β-activated proteinkinase 1 (TAK1)-binding protein 1) has been des-cribed13. In a yeast two-hybrid screen, TAB1 was iden-tified as a protein that formed a complex with p38α.Association of p38α with TAB1 results in intramolecu-lar autophosphorylation and activation of p38α that isindependent of MAPKKs. This mode of activationseems to be as effective as MAP2K6-mediated activa-tion of p38α13. The substrates of p38 MAPKs includeother kinases, cytosolic proteins and transcription factors(FIG. 2)11,12. The activity of p38 MAPK can be downreg-ulated by dephosphorylation by various protein phos-phatases, such as protein phosphatase 2A. In vitrostudies have shown that p38 MAPK can also bedephosphorylated by dual-specificity phosphatase 16(DUSP16, also known as MAPK phosphatase-7 orMKP7)14,15 and, to a lesser extent, by DUSP10 (alsoknown as MKP5)16.
Role of p38 in regulation of cytokine expressionAlthough p38α MAPK-null mice are not viable,embryonic stem cells taken from these mice show areduced capacity for IL-1-induced IL-6 productionand activation of MAPK-activated protein kinase 2(MAPKAP K2), a downstream substrate of p38αMAPK, in response to chemical stress17. More impor-tantly, MAPKAP K2-deficient mice also show dimin-ished production of IL-6 and TNF18. These effects arethought to be mediated via a mechanism involvingmessenger RNA turnover and protein translation. So,the p38α/MAPKAP K2 pathway is crucial to inflamma-tory cytokine production and signalling. Furthermore,p38α MAPK phosphorylates a variety of transcriptionfactors, some of which are responsible for transcriptionalexpression of genes encoding inflammatory cytokines.
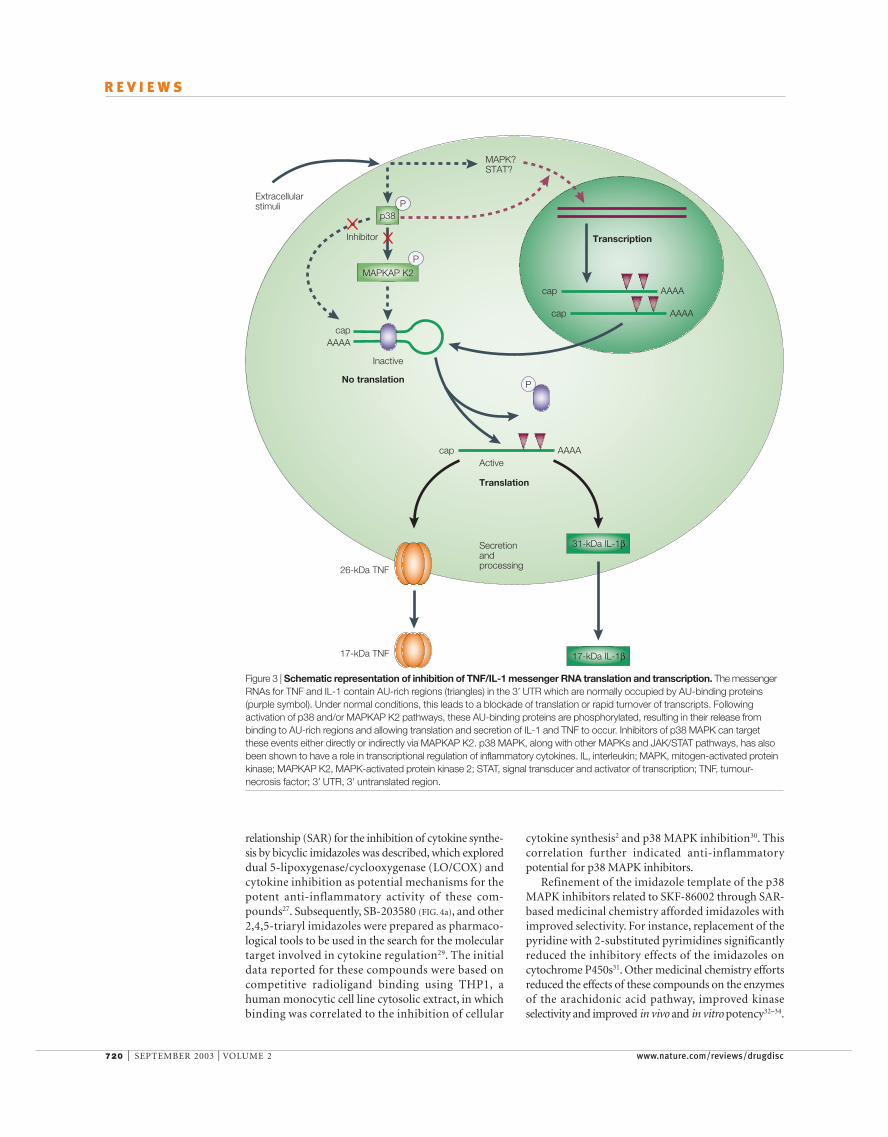
Several p38 MAPK inhibitors have been shown toblock the production of IL-1, TNF and othercytokines11. The inhibition of cytokine productionseems to result from the combined effect at the level oftranscription and translation. Messenger RNAs forinducible cytokines such as IL-1, IL-8 and TNF areshort lived and contain an AU-rich element in their 3′ UNTRANSLATED REGION that is responsible for their highturnover rate19. The transfer of these AU-rich elementsto otherwise stable messages renders them short-lived20. It is believed that under normal conditions, theAU-rich elements of these transcripts are occupied byAU-binding proteins and, as a result, are not translat-able or are rapidly turned over. In response to anappropriate stimulus such as LPS, it is postulated thatthese AU-binding proteins are phosphorylated in ap38 MAPK-dependent manner, which results in therelease, stabilization and translation of these mRNAs(FIG. 3). Because MAPKAP K2 deficiency is also able toblock the production of these short-lived mRNAs, it
p38β2 activity was shown to induce cardiomyocytehypertrophy, whereas elevated p38α activity resulted inan induction of apoptosis of cardiomyocytes in thismodel system6. Further searches of expressed sequencetag (EST) databases also identified p38γ (also known asSAPK3 and MAPK12) and p38δ (also known as SAPK4and MAPK13)4,5,7–9. These kinases have ~60% identityto p38α, but much less is known about their function.p38γ is largely expressed in skeletal muscle, whereasp38δ is expressed more widely in several adult tissuesand during development. Recently, p38γ has beenshown to be expressed in both normal and diseasedhuman heart myocytes, and in normal and hyper-trophic cultured rat cardiac myocytes, indicating thatp38γ might have a role in heart pathophysiology10.
p38 MAPK is activated by dual phosphorylation onThr180 and Tyr182 by an upstream MAPKK termedMAP2K6 (also known as MKK6). Other MAPKKs,such as MAP2K3 (also known as MKK3), have alsobeen suggested to activate p38 MAPK. MAP2K6 is
Growth factors/environmental stressStimuli
Outcome
MAPKKK
MAPKK(MEK1–7)
MAPK(ERKs, JNKs, p38s)
Substrates(cytosolic proteins/transcription factors)
Cellular responses(survival, cell cycle, proliferation, apoptosis,
cytoskeletal changes, gene expression)
MAPKcascade
Figure 1 | Generic MAP kinase pathways. Mitogen-activatedprotein kinases (MAPKs), which integrate and process variousextracellular signals. The MAPK cascade consists of a seriesof three protein kinases — a MAPK and two upstream components, MAPK kinase (MAPKK or MEK) and MAPKKkinase (MAPKKK). ERK, extracellular signal-regulatedkinase; JNK, c-Jun N-terminal kinase.
NATURE REVIEWS | DRUG DISCOVERY VOLUME 2 | SEPTEMBER 2003 | 719
R E V I E W S
Discovery of p38 MAP kinase inhibitorsAn initial series of pyridinyl imidazole anti-inflammatoryagents served as tools to elucidate the role of p38 ininflammation25. The bicyclic pyridinyl imidazole SKF-86002 (FIG. 4a) was first reported to inhibit LPS-stimulatedcytokine production2,26–28. In 1993, the structure–activity
seems that p38 MAPK is indirectly responsible for thestability of these short-lived cytokine messages21,22. Ithas been proposed that the p38α/MAPKAP K2 path-way is involved in the phosphorylation of these AU-binding proteins23,24, but an unequivocal proof has notbeen achieved so far.
Environmental stress/inflammatory cytokines
Cellularresponse
Transcriptional regulation
ATF1/2/6DDIT3MEF2C/AELK1SAP1AMITF
NCF1cPLA2StathminMBPTauKeratin 8
MAPKAP K2/3/5MSK1MKNK1PRAKRSK-B
Cytokine production Othereffects
MAPK p38α p38β2 p38γ/SAPK3 p38δ/SAPK4
MAPKK MAP2K3 MAP2K6
MAPKKK
STK39 Ras TRAF2
Daxx
TRAF6? ? ?
GADD45s/GADD153
MAP3K4
Substrates
Upstreamactivators
MAP3K5
PAK
Cdc42/Rac
MAP3K7
TAB
Figure 2 | p38 MAPK pathway activators and substrates. Environmental stress and/or inflammatory cytokines activate MAPKKKsthrough a variety of signalling mechanisms. Activated MAPKKKs subsequently activate MAPKKs, which in turn activate MAPKs.Activated MAPKs then phosphorylate several substrates, such as transcription factors, other kinases and cytosolic proteins. Theseeffector molecules are essential for cellular responses that include cytokine production and transcriptional regulation. In this figure,dotted lines represent indirect effects, and solid lines represent a direct effect. Components of this pathway responsible for inflammatorycytokine production are shown in bold. ATF, activating transcription factor; cPLA2, cytosolic phospholipase A2; DDIT3, DNA-damage-inducible transcript 3; GADD, growth arrest- and DNA-damage-inducible; MAPK, mitogen-activated protein kinase; MAPKAPK,MAPK-activated protein kinase; MAPKK, MAPK kinase; MAPKKK, MAPKK kinase; MBP, myelin basic protein; MEF2, myocyteenhancer factor 2; MITF, microphthalmia-associated transcription factor; MKNK1, MAPK-interacting serine/threonine kinase; NCF1,neutrophil cytosolic factor 1; PAK, p21-activated kinase; PRAK, p38-regulated/activated protein kinase; RSK-B, ribosome protein S6kinase B; SAP1A, signalling lymphocytic activation molecule-associated protein 1A; STK, serine threonine kinase; TAB, transforminggrowth factor-β-activated protein kinase 1 (TAK1)-binding protein; TRAF, tumour-necrosis factor receptor-associated factor.
720 | SEPTEMBER 2003 | VOLUME 2 www.nature.com/reviews/drugdisc
R E V I E W S
cytokine synthesis2 and p38 MAPK inhibition30. Thiscorrelation further indicated anti-inflammatorypotential for p38 MAPK inhibitors.
Refinement of the imidazole template of the p38MAPK inhibitors related to SKF-86002 through SAR-based medicinal chemistry afforded imidazoles withimproved selectivity. For instance, replacement of thepyridine with 2-substituted pyrimidines significantlyreduced the inhibitory effects of the imidazoles oncytochrome P450s31. Other medicinal chemistry effortsreduced the effects of these compounds on the enzymesof the arachidonic acid pathway, improved kinaseselectivity and improved in vivo and in vitro potency32–34.
relationship (SAR) for the inhibition of cytokine synthe-sis by bicyclic imidazoles was described, which exploreddual 5-lipoxygenase/cyclooxygenase (LO/COX) andcytokine inhibition as potential mechanisms for thepotent anti-inflammatory activity of these com-pounds27. Subsequently, SB-203580 (FIG. 4a), and other2,4,5-triaryl imidazoles were prepared as pharmaco-logical tools to be used in the search for the moleculartarget involved in cytokine regulation29. The initialdata reported for these compounds were based oncompetitive radioligand binding using THP1, ahuman monocytic cell line cytosolic extract, in whichbinding was correlated to the inhibition of cellular
p38
MAPKAP K2
Inactive
Inhibitor
MAPK?STAT?
P
P
capAAAA
Transcription
cap AAAA
cap AAAA
31-kDa IL-1β
17-kDa IL-1β
No translation
Translation
Secretionandprocessing
Active
P
cap AAAA
26-kDa TNF
17-kDa TNF
Extracellularstimuli
Figure 3 | Schematic representation of inhibition of TNF/IL-1 messenger RNA translation and transcription. The messengerRNAs for TNF and IL-1 contain AU-rich regions (triangles) in the 3′ UTR which are normally occupied by AU-binding proteins(purple symbol). Under normal conditions, this leads to a blockade of translation or rapid turnover of transcripts. Followingactivation of p38 and/or MAPKAP K2 pathways, these AU-binding proteins are phosphorylated, resulting in their release frombinding to AU-rich regions and allowing translation and secretion of IL-1 and TNF to occur. Inhibitors of p38 MAPK can targetthese events either directly or indirectly via MAPKAP K2. p38 MAPK, along with other MAPKs and JAK/STAT pathways, has alsobeen shown to have a role in transcriptional regulation of inflammatory cytokines. IL, interleukin; MAPK, mitogen-activated proteinkinase; MAPKAP K2, MAPK-activated protein kinase 2; STAT, signal transducer and activator of transcription; TNF, tumour-necrosis factor; 3′ UTR, 3′ untranslated region.

NATURE REVIEWS | DRUG DISCOVERY VOLUME 2 | SEPTEMBER 2003 | 721
R E V I E W S
in p38 MAPK and slow association kinetics of binding(see next section). The binding modes of some of theother p38 MAPK inhibitor classes (Scios indoles, forinstance) have not been disclosed. The structures ofa series of tetra-substituted imidazoles (Merck,WO9712876) were published in conjunction withpreclinical data, which identified these molecules aspotent, bioavailable and selective p38α/β inhibitors34.Recently, GlaxoSmithKline (WO02059083) andMerck (WO02058695) (FIG. 4b) have published patentsthat claim very closely related heteroaryl fused pyridi-none structures.
Crystal and co-crystal structures of p38 inhibitorsTwo groups initially described the X-ray crystallo-graphic structure of native unactivated p38α (REFS 38,39)
(FIG. 5a). More recently, several crystal structures ofpyridinyl-imidazole–p38α MAPK complexes of SB-203580 and related analogues have become available40–42
(FIG. 5). The pyridinyl imidazoles bind to both the activeand inactive enzyme in an ATP-competitive manner.On the basis of the structural evidence, as well as thatfrom mutagenesis studies, it has been proposed that theselectivity of this class of p38 MAPK inhibitors resultsfrom the binding of the imidazole 4-phenyl moiety to ahydrophobic pocket of p38 MAPK in the region con-taining Thr106. The binding interactions defined bythese structures are in good agreement with the pub-lished SAR data for this class of compounds30.Important features observed in all these structures
In addition, the imidazole core was replaced by otherheteroaryl scaffolding. Provided that the requisite rela-tionships between the pyridinyl, aryl and one of thecore heterocycle heteroatoms is maintained, the newaryl-pyridinyl-heterocycles generally exhibit in vitropotency similar to the pyridinyl imidazoles35. The aryl-pyridinyl-heterocycle p38 MAPK inhibitors haverecently been reviewed36.
New p38 MAP kinase inhibitorsSeveral structurally diverse and notable new p38MAPK inhibitors are shown in FIG. 4b. Cirillo et al. com-piled a detailed review of a subset of novel non-aryl-pyridinyl compounds, including triazanapthalenones,N,N′-diaryl ureas, N,N-diaryl ureas, benzophenones,pyrazole ketones, indole amides, diamides, quinazoli-nones, pyrimido[4,5-d]pyrimidinones and pyridyl-amino-quinazolines37. It is of interest to note that achemically diverse set of compounds inhibits p38α/βpotently and in an ATP-competitive manner. In anumber of cases, the common PHARMACOPHORE is thepresence of a carbonyl group, which acts as a hydrogen-bond acceptor to Met109, and a substituted group thatfits into the aryl selectivity pocket (see next section).Some of the first examples of compounds interacting inthis way included the Vertex triazanapthalenonesexemplified by the clinical candidate VX-745.
The Boehringer N,N′-diaryl ureas related to BIRB-796uniquely bind to the ATP-binding site and kinase-specificity pocket, and result in a conformational change
PHARMACOPHORE
The ensemble of steric andelectronic features that isnecessary to ensure optimalinteractions with a specificbiological target structure andto trigger (or to block) itsbiological response.
F
N
N
Cl N
N
O
O
OO
HN
H3C
N
N
N NHCH3
FF
F
Cl
Cl
N
N
NO NH N
Cl Cl
NN
NO
Cl
1 SCIOSWO0071535
2 MerckWO9712876
3 GlaxoSmithKlineWO02059083
4 MerckWO02058695
N
N
S
N
F
N
N
HN
SO
F
a
b
SKF-86002 SB-203580
Figure 4 | Structures of representative classes of p38 MAP kinase inhibitors. a | Prototypical pyridinyl imidazoles. b | Additionalcompounds of interest.

722 | SEPTEMBER 2003 | VOLUME 2 www.nature.com/reviews/drugdisc
R E V I E W S
residues larger than Thr. Whereas the native kinase wassensitive to SB-203580 (IC
50= 0.3 µM), substituting
Thr106 with a smaller residue, Ala, further enhancedsensitivity (IC
50= 0.01 µM). Taken together, these data
identify Thr106 as a key residue responsible for formingthe aryl-specificity pocket and are fully consistent withthe crystallographic data.
Recently, researchers from Boehringer described aseries of urea-containing p38α inhibitors46. Crystallo-graphic studies of one of the lead compounds revealedthat it binds at a site remote from the ATP pocket, andinduces a major conformational change in the enzyme(FIG. 5c). The molecule caused a significant movement ofp38 MAPK Phe169, such that this residue filled the ATPpocket, preventing ATP binding. Lead optimizationefforts resulted in the identification of BIRB-796. Thishighly potent molecule binds much like the lead com-pound, as revealed by the co-crystal structures, with thep38α MAPK conformational change preserved.However, several structural modifications in BIRB-796improved potency compared with the original structure.Among the additional interactions is an H-bond toMet109. Also, a structural modification provides addi-tional hydrophobic interactions with the imidazole p38MAPK inhibitor aryl-specificity pocket, which con-tributes to improvement in potency, and possibly selec-tivity. Unlike the imidazole-based p38 MAPK inhibitors,BIRB-796 binds in a time-dependent manner46,47.
Preclinical studies with p38 MAP kinase inhibitorsA large body of evidence from preclinical studies indicatesa crucial role of p38α MAPK in inflammation. Severalgroups have reported that specific and selective p38α/βMAPK inhibitors block the production of IL-1, TNF andIL-6 in vitro and in vivo. In addition, the p38 MAPK path-way is involved in the induction of several other inflam-matory molecules, such as COX2 and inducible nitricoxide synthase (iNOS)48,49. Recently, p38α-dependent his-tone H3 phosphorylation has been shown to mark andrecruit nuclear factor-κB to otherwise CRYPTIC PROMOTERS,resulting in increased expression of several inflammatorycytokines and chemokines50.
include: the presence of an aryl-specificity pocketbehind and orthogonal to the site normally occupied bythe adenine ring of ATP; a well-formed hydrogen bondbetween the 4-pyridinyl nitrogen and the amide N-H ofMet109; and an H-bond-like interaction between Lys53and the unalkylated imidazole N-H. The interactionbetween the 4-pyridinyl group and the N-H of Met109is analogous to the N-1 adenine of ATP. Moreover, thishydrogen bond acceptor–donor pair occurs in all of theimidazole inhibitor kinase crystal structures.
On the basis of the X-ray crystallographic dataimplicating Thr106 as a key residue conferring selectivity,several groups have used a mutagenesis approach toestablish the importance of this residue. Following theinitial study by Wilson et al., a more extensive study byGum et al. focused on Thr106 and two adjacentresidues at the back of the ATP pocket41,43. These threeresidues (Thr106, His107 and Leu108) are identical inp38α and p38β, but are different in the more distanthomologues, p38γ and p38δ (Met106, Pro107 andPhe108). These latter homologues differ from p38αand p38β in being insensitive to SB-203580. Whenthese three residues in p38α are changed to the Met-Pro-Phe seen in p38γ and p38δ, the mutant kinase is nolonger inhibited by SB-203580. A single change ofThr106 to Met resulted in a ten-fold reduction in thesensitivity to SB-203580 in mammalian expressedp38α. By contrast, the introduction of the p38αsequence (Thr-His-Leu) into p38γ and p38δ, or eventhe more distantly related JNK1 (REF. 43) and ERK44, issufficient to render these mutant kinases as sensitive toSB-203580 as p38α. The single change to Thr106 led toa partial increase in sensitivity. Interestingly, the singlechange to or from Thr106 seems to be all that is neededto confer sensitivity to SB-203580 when these enzymesare expressed in bacteria or yeast, which indicates thatthe other two residues might interact with other pro-teins in mammalian cells43,45. In a scanning mutagenesisstudy, position 106 has been examined for the effect often different amino acids at this position in p38α.Kinases with Thr106 mutated to Met were insensitive(IC
50>100 µM)to SB-203580, as were all mutants having
CRYPTIC PROMOTERS
Promoter regions that do notcontain recognized sequenceelements corresponding to aclassical promoter sequence.
a b c
Met109Met109 Met109
Glu71
Glu71
Glu71
Arg67Arg67 Arg67
Thr106
Thr106
Thr106
Phe169 Phe169
Phe169
Figure 5 | Crystallographic data for p38 MAPK. a | A high-resolution crystal structure of the unphosphorylated apo-p38 . b | Co-crystal structure of binding of an ATP-competitive inhibitor (SB-220025) to p38 MAPK. c | Co-crystal structure of binding of anon-competitive p38 MAPK inhibitor (BIRB-796) to p38 MAPK. For the sake of clarity, only the key interacting residues are shown.

NATURE REVIEWS | DRUG DISCOVERY VOLUME 2 | SEPTEMBER 2003 | 723
R E V I E W S
and in vivo models. p38 MAPK inhibitors have alsobeen shown to be efficacious in animal models ofrheumatoid arthritis, such as collagen-induced arthritisin the mouse and adjuvant-induced arthritis in the rat(TABLE 1)34,47,54–60.
Given that p38 MAPK inhibitors block the produc-tion and action of inflammatory cytokines, it is postu-lated that the use of such compounds in diseases wherethese cytokines are elevated might be beneficial. Muchof the evidence for this has come from animal models inwhich p38 MAPK inhibitors have been evaluated(TABLE 1). p38 MAPK inhibitors have been shown to beefficacious in a rat model of cardiac hypertrophy anddysfunction61. Some evidence also exists for the impor-tance of p38 MAPK in Alzheimer’s disease62. Severalreports suggest an important role of p38 MAPK invascular injury in rabbit and in ischaemia-reperfusioninjury in rat liver and lung63–65. Increased activity of p38MAPK has also been observed in patients sufferingfrom inflammatory bowel disease66.
Clinical studies with p38 MAP kinase inhibitorsTwo imidazole analogues based on the structures of theoriginal p38 MAPK inhibitors have been reported toreach clinical studies in humans (FIG. 6). Healthy malevolunteers were orally dosed with SB-242235 rangingfrom 1 mg to 500 mg. All doses of the drug were gener-ally well tolerated and pharmacokinetics were linear inthe dose range 25–500 mg. Peak plasma levels werereached in a mean of 1.5–6 hours and a long half-lifewas seen (14.7–18.3 hours). Ex vivo, SB-242235 dose-dependently inhibited endotoxin-induced TNF-α, IL-1β,IL-6 and IL-8 production, with a mean suppression ofmore than 75% at three hours following doses of at least150 mg. Plasma concentrations of SB-242235 and inhi-bition of cytokine production seemed to be related67.
The effects of RWJ-67657 on the clinical and cytokineresponse to endotoxaemia were assessed in a study thatincluded 62 healthy volunteers who were randomized
Rheumatoid arthritis is the most common inflam-matory disease of synovial joints, and is characterized bythe production of pro-inflammatory mediators byimmune cells that infiltrate synovium. This causes theproliferation of synovial fibroblasts, further release ofinflammatory molecules and formation of pannus tissuethat eventually degrades cartilage and subchondralbone, leading to joint destruction, pain and disability.IL-1 and TNF are the two most important inflamma-tory cytokines in stimulating the destructive cascade ofinflammation via the production of secondary media-tors, such as prostaglandin E
2(PGE
2), matrix metallo-
proteinases and vascular cell adhesion molecule, amongothers. Agents that restrict the availability of either IL-1or TNF have been shown to be efficacious both in ani-mal models and in the clinic for rheumatoid arthritisand Crohn’s disease51,52. The anti-TNF antibody inflix-imab (Remicade; Centocor) and the TNF receptor–Fcfusion protein etanercept (Enbrel; Immunex) bind toTNF and prevent it binding to cell-surface receptors,thereby inhibiting its biological activity. Anakinra(Kineret; Amgen), a soluble IL-1 receptor antagonist,has recently been approved for the treatment ofrheumatoid arthritis by the US FDA. However, thesebiological agents are relatively expensive, difficult toproduce and need to be parenterally administered.Furthermore, a combination treatment with both IL-1and TNF inhibitors is likely to be more effective thaneither cytokine alone51. Therefore, an orally active phar-macological agent that can inhibit either the productionor activity of these inflammatory cytokines would be anideal therapeutic agent for rheumatoid arthritis. This,along with other therapeutic approaches for rheuma-toid arthritis, has recently been reviewed in detail53. Alarge body of genetic and pharmacological evidencepoints to p38α MAPK as one of the most validated ther-apeutic targets for rheumatoid arthritis. The availabilityof p38 MAPK inhibitors such as SB-203580 has led totheir widespread use in many different in vitro systems
Table 1 | Summary of preclinical studies with various p38 MAP kinase inhibitors
Compound Company Study/Model References
SB-203580 GlaxoSmithKline AA rat and CIA mice, ischaemia/reperfusion 54,55in rat heart
SB-242235 GlaxoSmithKline AA rats 57
Selective imidazole Merck AA rats 34(L-167307)
RWJ-67657 RW Johnson AA rats 56Pharmaceutical
Pyridinyloxazole inhibitor Novartis CIA in rats 58
RPR-200765A Aventis Streptococcal cell wall-induced arthritis in rats 59
RPR-238677 Aventis Streptococcal cell wall-induced arthritis in rats 60
BIRB-796 Boehringer Ingelheim LPS challenge in mice 47Pharmaceuticals
FR167653 Fujisawa Ischaemia/reperfusion of lung and liver in rats 63,64Pharmaceuticals
SB-239063 GlaxoSmithKline Cardiac hypertrophy and dysfunction in rats, cerebral 61,65,75focal ischaemia in rats, balloon injury in rabbit
AA, adjuvant-induced arthritis; CIA, collagen-induced arthritis; LPS, lipopolysaccharide; MAP, mitogen-activated protein.

724 | SEPTEMBER 2003 | VOLUME 2 www.nature.com/reviews/drugdisc
R E V I E W S
smaller, transient increases in AST or ALT and one onthe lowest dose had a minimal increase in ALT. Noinhibition of LPS-induced TNF-α production wasobserved, but the compound inhibited ex vivo neu-trophil activation at 4 hours after doses of 50 mg ormore, with mixed neutrophil inhibition/activation at 24hours70. Another Phase I study involved the administra-tion of single escalating doses of BIRB-796BS in a poly-ethylene glycol 400 solution to 64 healthy volunteers.The pharmacodynamic effects of the drug — that is,ex vivo inhibition of LPS-induced TNF-α productionand neutrophil activation — were related to plasmalevels and it was much more effective in inhibitingneutrophil activation than TNF-α production71.Elevated transaminases were detected in 9 of 48 subjects,2 in placebo, 3 in 15 mg and 4 in 30 mg BIRB-796BS,although all remained asymptomatic72.
The Hoffman-La Roche compound Ro-320-1195has also been reported to be in Phase I clinical trials73.SCIO-469 is the lead compound under development byScios as an oral treatment for rheumatoid arthritis.Results from Phase I clinical studies have been reportedin several press releases from Scios, which indicate thatSCIO-469 is well tolerated. To date, SCIO-469 seems tobe the most advanced of the p38 MAPK inhibitorspresently in clinical trials.
ConclusionThe p38 MAPKs have been the subject of intense scien-tific study across multiple disciplines. The discovery ofselective and potent inhibitors of p38 MAPKs has aidedthe understanding of their role in signal transductionand the regulation of a multitude of cellular responses.p38α MAPK in particular has been implicated in theregulation of inflammatory mediators, including thecytokines, PGE
2, iNOS and so on. Since the discovery of
to receive either placebo or a single oral dose of 350, 700or 1400 mg of RWJ-67657. In placebo-treated patients,administration of 4 ng per kg body weight of endotoxinhalf an hour after the study treatment induced a flu-likesyndrome and increased serum levels of TNF-α, IL-6and IL-8. Treatment with RWJ-67657 dose-dependentlydecreased the symptoms and elevated cytokine levels,indicating that it might be efficacious in the treatment ofsepsis and cytokine-mediated diseases such as Crohn’sdisease or rheumatoid arthritis68.
A dose-escalation trial was designed to study thesafety, pharmacokinetics and clinical activity of VX-745dosed orally in patients with active rheumatoidarthritis69.VX-745 was well tolerated and produced a sig-nificant clinical effect compared with placebo. The pri-mary end point (area under the curve of the ACR20 scoresduring the 12 weeks of treatment) was significantlyhigher (t-test, p value = 0.015) in the group receivingVX-745 versus placebo. The most frequently reportedadverse event was elevation in liver transaminases (maxi-mum aspartate transaminase (AST) 82–258 IU (normallevel, 9–34); maximum alanine transaminase (ALT)30–297 IU (normal level, 6–34)), which was reported insix patients on VX-745 starting at weeks 4–12, comparedwith none on placebo. Elevations declined promptlywith treatment discontinuation.Vertex suspended devel-opment of VX-745, after preclinical safety evaluationsindicated that the agent crosses from the blood into thecentral nervous system and, in animals receiving highdose VX-745, causes adverse neurological effects.
The safety, pharmacokinetics and pharmacodynam-ics of BIRB-796 have been assessed in a series of double-blind, randomized, placebo-controlled studies inhealthy volunteers. Asymptomatic increases in transam-inases (that is, AST, ALT) were observed in all subjects atthe highest dose, two subjects receiving 50 mg had
ACR20
A 20% response rate accordingto American College ofRheumatology clinicalclassification criteria.
NN
O
N
N
HN
F
N
F
N
N
OH
Cl Cl
N NN S
F
FO
NN
HN NH
O
ON
O
O OH
OHO
N N
F
H2N
Aryl-pyridyl heterocycles
Non-aryl-pyridyl heterocycles
SB-242235
VX-745 BIRB-796 RO3201195
RWJ-67657
Figure 6 | Structures of p38 MAP kinase inhibitors that have advanced to clinical trials. MAP, mitogen-activated protein.

NATURE REVIEWS | DRUG DISCOVERY VOLUME 2 | SEPTEMBER 2003 | 725
R E V I E W S
regard, new inhibitors that are non-ATP-competitiveand, in principle, are not likely to target other kinasesmight have an improved selectivity profile. Similarly,the finding that p38 MAPK can be activated by its asso-ciation with TAB1 suggests that inhibitors designed todisrupt this interaction may have distinct advantagesover ATP competitors13. So, an alternative approach isto target other molecules in the p38 MAPK pathway.Finally, the role of other p38 MAPK homologues suchas p38β2, p38δ and p38γ should also be explored, asthey might have a critical role in specific disease states.Indeed, it has been reported that all four p38 MAPKhomologues are activated with similar kinetics inmyeloid cells exposed to inflammatory stimuli74. Theattractive and compelling pharmacological profile ofp38 MAPK inhibitors warrants further studies to per-sistently pursue not only the best compound but alsothe appropriate therapeutic indication.
p38α MAPK in the early 1990s, there has been greatinterest in identifying compounds targeted against it forthe treatment of a variety of inflammatory diseases.Recently, several of the more promising compoundshave entered human clinical trials and have shown goodpharmacokinetic and pharmacodynamic properties.
It is widely anticipated that p38 MAPK inhibitorswill have efficacy in arthritic and inflammatory dis-eases, and some compounds have reached Phase II andIII trials. A marked reduction in the levels of inflamma-tory cytokines has been observed following oral admin-istration of drug and an endotoxic challenge using in vivoor ex vivo protocols. In several cases, the clinical trialshave been stopped due to safety issues. One of theunderlying reasons for these undesirable effects mightbe the cross-reactivities against other kinases or othercellular signalling molecules. Almost all p38α/βinhibitors described herein are ATP competitors. In this
1. Pearson, G. et al. Mitogen-activated protein (MAP) kinasepathways: Regulation and physiological functions. Endocr.Rev. 22, 153–183 (2001).A comprehensive review of all three MAPK pathways.
2. Lee, J. C. et al. A protein kinase involved in the regulation ofinflammatory cytokine biosynthesis. Nature 372, 739–746(1994).Describes in detail the identification of the moleculartarget of a class of pyridinyl imidazole compoundsknown to inhibit cytokine production as p38 MAPK.
3. Han, J., Lee, J. D., Bibbs, L. & Ulevitch, R. J. A MAP kinasetargeted by endotoxin and hyperosmolarity in mammaliancells. Science 265, 808–811 (1994).This paper identifies a prominent phosphoproteinextracted from LPS-stimulated mouse macrophagecell line as murine p38 MAPK.
4. Jiang, Y. et al. Characterization of the structure and function ofa new mitogen-activated protein kinase (p38β). J. Biol. Chem.271, 17920–17926 (1996).
5. Kumar, S. et al. Novel homologues of CSBP/p38 MAPkinase-activation, substrate specificity and sensitivity toinhibition by pyridinyl imidazoles. Biochem. Biophys. Res.Commun. 235, 533–538 (1997).
6. Wang, Y. B. et al. Cardiac muscle cell hypertrophy andapoptosis induced by distinct members of the p38mitogen-activated protein kinase family. J. Biol. Chem.273, 2161–2168 (1998).An interesting paper describing the opposing role ofvarious p38 MAPKs in cardiac cells.
7. Li, Z., Jiang, Y., Ulevitch, R. J. & Han, J. The primarystructure of p38γ: a new member of p38 group of MAPkinases. Biochem. Biophys. Res. Commun. 228, 334–340(1996).
8. Mertens, S., Craxton, M. & Goedert, M. SAP kinase-3, anew member of the family of mammalian stress-activatedprotein kinases. FEBS Lett. 383, 273–276 (1996).
9. Cuenda, A., Cohen, P., Buee-Scherrer, V. & Goedert, M.Activation of stress-activated protein kinase-3 (SAPK3) bycytokines and cellular stresses is mediated via SAPKK3(MKK6); comparison of the specificities of SAPK3 andSAPK2 (RK/p38). EMBO J. 16, 295–305 (1997).
10. Court, N. W., dos Remedios, C. G., Cordell, J. &Bogoyevitch, M. A. Cardiac expression and subcellularlocalization of the p38 mitogen-activated protein kinasemember, stress-activated protein kinase-3 (SAPK3). J. Mol. Cell. Cardiol. 34, 413–426 (2002).
11. Adams, J. L., Badger, A. M., Kumar, S. & Lee, J. C. p38MAP kinase: molecular target for the inhibition of pro-inflammatory cytokines. Prog. Med. Chem. 38, 1–60 (2001).A comprehensive review of the p38 MAPK pathway,its biology and discovery of various inhibitors.
12. Kyriakis, J. M. & Avruch, J. Mammalian mitogen-activatedprotein kinase signal transduction pathways activated bystress and inflammation. Physiol. Rev. 81, 807–869 (2001).A good review of JNK and p38 MAPK pathways.
13. Ge, B. X. et al. MAPKK-independent activation of p38αmediated by TAB1-dependent autophosphorylation ofp38α. Science 295, 1291–1294 (2002).An interesting paper describing TAB1-dependentphosphorylation and activation of p38α MAPK.
14. Masuda, K., Shima, H., Watanabe, M. & Kikuchi, K. MKP-7,a novel mitogen-activated protein kinase phosphatase,functions as a shuttle protein. J. Biol. Chem. 276,39002–39011 (2001).
15. Tanoue, T., Yamamoto, T., Maeda, R. & Nishida, E. A novelMAPK phosphatase MKP-7 acts preferentially onJNK/SAPK and p38α and β MAPKs. J. Biol. Chem. 276,26629–26639 (2001).
16. Theodosiou, A., Smith, A., Gillieron, C., Arkinstall, S. &Ashworth, A. MKP5, a new member of the MAP kinasephosphatase family, which selectively dephosphorylatesstress-activated kinases. Oncogene 18, 6981–6988(1999).
17. Allen, M. et al. Deficiency of the stress kinase p38α results inembryonic lethality: Characterization of the kinasedependence of stress response of enzyme-deficientembryonic stem cells. J. Exp. Med. 191, 859–869 (2000).This paper describes the phenotype of embryoniccells derived from p38-null mice.
18. Kotlyarov, A. et al. MAPKAP kinase 2 is essential for LPS-induced TNF-α biosynthesis. Nature Cell Biol. 1, 94–97(1999).
19. Caput, D. et al. Identification of a common nucleotidesequence in the 3’-untranslated region of mRNA moleculesspecifying inflammatory mediators. Proc. Natl Acad. Sci.USA 83, 1670–1674 (1986).
20. Shaw, G. & Kamen, R. A conserved AU sequence from the3’ untranslated region of GM-CSF mRNA mediates selectivemRNA degradation. Cell 46, 659–667 (1986).
21. Winzen, R. et al. The p38 MAP kinase pathway signals forcytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targetedmechanism. EMBO J. 18, 4969–4980 (1999).This paper, together with references 22–24, providesevidence for a role of the p38 MAPK pathway instabilization of short-lived mRNAs.
22. Ming, X. F., Stoecklin, G., Lu, M., Looser, R. & Moroni, C.Parallel and independent regulation of interleukin-3 mRNAturnover by phosphatidylinositol 3-kinase and p38 mitogen-activated protein kinase. Mol. Cell. Biol. 21, 5778–5789(2001).
23. Neininger, A. et al. MK2 targets AU-rich elements andregulates biosynthesis of tumor necrosis factor andinterleukin-6 independently at different post-transcriptionallevels. J. Biol. Chem. 277, 3065–3068 (2002).
24. Frevel, M. A. E. et al. p38 mitogen-activated protein kinase-dependent and -independent signaling of mRNA stability ofAU-rich element-containing transcripts. Mol. Cell. Biol. 23,425–436 (2003).
25. Lantos, I. et al. Antiinflammatory activity of 5,6-diaryl-2,3-dihydroimidazo[2,1-b]thiazoles. Isomeric 4-pyridyl and 4-substituted phenyl derivatives. J. Med. Chem. 27, 72–75(1984).
26. Lee, J. C., Griswold, D. E., Votta, B. & Hanna, N. Inhibition ofmonocyte IL-1 production by the anti-inflammatorycompound, SK&F 86002. Int. J. Immunopharmacol. 10,835–843 (1988).
27. Lee, J. C. et al. Bicyclic imidazoles as a novel class ofcytokine biosynthesis inhibitors. Ann. NY Acad. Sci. 696,149–170 (1993).
28. Lee, J. C., Kassis, S., Kumar, S., Badger, A. & Adams, J. L.p38 mitogen-activated protein kinase inhibitors. Mechanismand therapeutic potentials. Pharmacol. Ther. 82, 389–397(1999).
29. Gallagher, T. F. et al. 2,4,5-Triarylimidazole inhibitors of IL-1biosynthesis. Bioorg. Med. Chem. Lett. 5, 1171–1176(1995).
30. Gallagher, T. et al. Regulation of stress-induced cytokineproduction by pyridinylimidazoles; inhibition of CSBP kinase.Bioorg. Med. Chem. 5, 49–64 (1997).
31. Adams, J. L. et al. Pyrimidinylimidazole inhibitors ofCSBP/p38 kinase demonstrating decreased inhibition ofhepatic cytochrome P450 enzymes. Bioorg. Med. Chem.Lett. 8, 3111–3116 (1998).
32. Boehm, J. C. et al. 1-substituted 4-aryl-5-pyridinyl-imidazoles: a new class of cytokine suppressive drugs withlow 5-lipoxygenase and cyclooxygenase inhibitory potency.J. Med. Chem. 39, 3929–3937 (1996).
33. Adams, J. L. et al. Pyrimidinylimidazole inhibitors of p38:Cyclic N-1 imidazole substituents enhance p38 kinaseinhibition and oral activity. Bioorg. Med. Chem. Lett. 11,2867–2870 (2001).
34. Liverton, N. J. et al. Design and synthesis of potent,selective, and orally bioavailable tetrasubstituted imidazoleinhibitors of p38 mitogen-activated protein kinase. J. Med.Chem. 42, 2180–2190 (1999).
35. Boehm, J. C. & Adams, J. L. New inhibitors of p38 kinase.Expert Opin. Ther. Patents 10, 25–37 (2000).This review of the p38 MAPK inhibitor patentliterature through 1999 includes a summary of the SAR of the pyridinyl imidazole-based analogues. The molecular basis of the SAR is also highlighted.
36. Jackson, P. F. & Bullington, J. L. Pyridinylimidazole basedp38 MAP kinase inhibitors. Curr. Top. Med. Chem. 2,1011–1020 (2002).
37. Cirillo, P. F., Pargellis, C. & Regan, J. The non-diaryl hetero-cycle classes of p38 MAP kinase inhibitors. Curr. Top. Med.Chem. 2, 1021–1035 (2002).This is a thorough review of potent p38 MAPKinhibitors unrelated to pyridinyl imidazole-based p38MAPK inhibitors.
38. Wilson, K. P. et al. Crystal structure of p38 mitogen-activated protein kinase. J. Biol. Chem. 271, 27696–27700(1996).First published report on the crystal structure of thep38 MAPK apoenzyme.
39. Wang, Z. et al. The structure of mitogen-activated proteinkinase p38 at 2.1 Å resolution. Proc. Natl Acad. Sci. USA94, 2327–2332 (1997).
40. Wang, Z. L. et al. Structural basis of inhibitor selectivity inMAP kinases. Structure 6, 1117–1128 (1998).
41. Wilson, K. P. et al. The structural basis for the specificity ofpyridinylimidazole inhibitors of p38 MAP kinase. Chem. Biol.4, 423–431 (1997).
42. Tong, L. et al. A highly specific inhibitor of human p38 MAPkinase binds in the ATP pocket. Nature Struct. Biol. 4,311–316 (1997).First report of a co-crystal structure of an inhibitorbound to the p38 MAPK.

726 | SEPTEMBER 2003 | VOLUME 2 www.nature.com/reviews/drugdisc
R E V I E W S
43. Gum, R. J. et al. Acquisition of sensitivity of stress-activatedprotein kinases to the p38 inhibitor, SB 203580, by alterationof one or more amino acids within the ATP binding pocket.J. Biol. Chem. 273, 15605–15610 (1998).A comprehensive analysis of various amino acidsubstitutions at Thr106 of p38 MAPK and sensitivityto SB-203580.
44. Fox, T. et al. A single amino acid substitution makes ERK2susceptible to pyridinyl imidazole inhibitors of p38 MAPkinase. Protein Sci. 7, 2249–2255 (1998).
45. Eyers, P. A., Craxton, M., Morrice, N., Cohen, P. & Goedert, M. Conversion of SB 203580-insensitive MAPkinase family members to drug-sensitive forms by a singleamino-acid substitution. Chem. Biol. 5, 321–328 (1998).Describes the importance of the Thr106 residue indefining the space requirement for binding by thediaryl imidazole inhibitors of p38 MAPK.
46. Pargellis, C. et al. Inhibition of p38 MAP kinase by utilizinga novel allosteric binding site. Nature Struct. Biol. 9,268–272 (2002).
47. Regan, J. et al. Pyrazole urea-based inhibitors of p38 MAPkinase: From lead compound to clinical candidate. J. Med.Chem. 45, 2994–3008 (2002).
48. Badger, A. M. et al. Differential effects of SB 242235, aselective p38 mitogen-activated protein kinase inhibitor, onIL-1 treated bovine and human cartilage/chondrocytecultures. Osteoarthr. Cartil. 8, 434–443 (2000).
49. Guan, Z. H., Buckman, S. Y., Pentland, A. P., Templeton, D. J. & Morrison, A. R. Induction of cyclo-oxygenase-2 by the activated MEKK1-/SEK1/MKK4-/p38mitogen-activated protein kinase pathway. J. Biol. Chem.273, 12901–12908 (1998).
50. Saccani, S., Pantano, S. & Natoli, G. p38-dependentmarking of inflammatory genes for increased NF-κBrecruitment. Nature Immunol. 3, 69–75 (2002).An interesting paper describing a p38 MAPK-dependent increase in expression of inflammatorycytokines and chemokines.
51. Bendele, A. M. et al. Combination benefit of treatment withthe cytokine inhibitors interleukin-1 receptor antagonist andPEGylated soluble tumor necrosis factor receptor type I inanimal models of rheumatoid arthritis. Arthritis Rheum. 43,2648–2659 (2000).
52. Kalden, J. R. How do the biologics fit into the currentDMARD armamentarium? J. Rheumatol. 28, 27–35(2001).
53. Smolen, J. S. & Steiner, G. Therapeutic strategies forrheumatoid arthritis. Nature Rev. Drug Discov. 2, 473–488(2003).A recent comprehensive review on rheumatoidarthritis.
54. Badger, A. M. et al. Pharmacological profile of SB 203580,a selective inhibitor of cytokine suppressive bindingprotein/p38 kinase, in animal models of arthritis, boneresorption, endotoxin shock and immune function. J. Pharmacol. Exp. Therap. 279, 1453–1461 (1996).
55. Ma, X. L. et al. Inhibition of p38 mitogen-activated proteinkinase decreases cardiomyocyte apoptosis and improvescardiac function after myocardial ischemia and reperfusion.Circulation 99, 1685–1691 (1999).
56. Wadsworth, S. A. et al. RWJ 67657, a potent, orally activeinhibitor of p38 mitogen-activated protein kinase. J. Pharmacol. Exp. Ther. 291, 680–687 (1999).
57. Badger, A. M. et al. Disease-modifying activity of SB242235, a selective inhibitor of p38 mitogen-activatedprotein kinase, in rat adjuvant-induced arthritis. ArthritisRheum. 43, 175–183 (2000).Describes the efficacy of a p38 MAPK inhibitor in a ratin vivo model of rheumatoid arthritis.
58. Revesz, L. et al. SAR of 4-hydroxypiperidine and hydroxy-alkyl substituted heterocycles as novel p38 MAP kinaseinhibitors. Bioorg. Med. Chem. Lett. 10, 1261–1264 (2000).
59. McLay, I. M. et al. The discovery of RPR 200765A, a p38MAP kinase inhibitor displaying a good oral anti-arthriticefficacy. Bioorg. Med. Chem. 9, 537–554 (2001).
60. McKenna, J. M. et al. An algorithm-directed two-component library synthesized via solid-phase methodologyyielding potent and orally bioavailable p38 MAP kinaseinhibitors. J. Med. Chem. 45, 2173–2184 (2002).
61. Behr, T. M. et al. Hypertensive end-organ damage andpremature mortality are p38 mitogen-activated proteinkinase-dependent in a rat model of cardiac hypertrophy anddysfunction. Circulation 104, 1292–1298 (2001).Describes the role of p38 MAPK in an in vivo model ofcardiac hypertrophy.
62. Hull, M., Lieb, K. & Fiebich, B. L. Pathways of inflammatoryactivation in Alzheimer’s disease: Potential targets for diseasemodifying drugs. Curr. Med. Chem. 9, 83–88 (2002).
63. Kawashima, Y. et al. FR167653 attenuates ischemia andreperfusion injury of the rat lung with suppressing p38mitogen-activated protein kinase. J. Heart Lung Transplant.20, 568–574 (2001).
64. Kobayashi, M., Takeyoshi, I., Yoshinari, D., Matsumoto, K. &Morishita, Y. P38 mitogen-activated protein kinase inhibitionattenuates ischemia-reperfusion injury of the rat liver.Surgery 131, 344–349 (2002).Describes the role of p38 MAPK in ischaemia andreperfusion.
65. Ju, H. S. et al. Sustained activation of p38 mitogen-activatedprotein kinase contributes to the vascular response to injury.J. Pharmacol. Exp. Ther. 301, 15–20 (2002).
66. Waetzig, G. H., Seegert, D., Rosenstiel, P., Nikolaus, S. &Schreiber, S. p38 mitogen-activated protein kinase isactivated and linked to TNF-α signaling in inflammatorybowel disease. J. Immunol. 168, 5342–5351 (2002).
67. Fullerton, T. et al. Suppression of ex vivo cytokine productionby SB-242235, a selective inhibitor of p38 MAP kinase.101st Ann. Meet. Am. Soc. Clin. Pharmacol. 67, 114 (2000).
68. Fijen, J. W. et al. Suppression of the clinical and cytokineresponse to endotoxin by RWJ-67657, a p38 mitogen-activated protein-kinase inhibitor, in healthy humanvolunteers. Clin. Exp. Immunol. 124, 16–20 (2001).
Describes the inhibition of inflammatory cytokines bya p38 MAPK inhibitor in healthy human volunteers.
69. Weisman, M. et al. Double-blind, placebo-controlled trial of VX-745, an oral p38 mitogen activated protein kinaseinhibitor, in patients with rheumatoid arthritis (RA). Ann. European Congress Rheumatol. A10018 (2002).
70. Wood, C. C., Yong, C. L., Madwed, J. B. & Gupta, A.Pharmacokinetics and pharmacodynamics of an oral p38MAP kinase inhibitor (BIRB 796 BS), administered once dailyfor 7 days. 58th Ann. Meet. Am. Acad. Allergy, AsthmaImmunol. 109, S321 A993 (2002).
71. Gupta, A. et al. Safety, pharmacokinetics, andpharmacodynamics of single doses of an oral p38 MAPkinase inhibitor (BIRB 796 BS) in healthy human males, aplacebo controlled, randomized study, double blinded ateach dose level. Ann. Meet. Am. Acad. Allergy, AsthmaImmunol. 109, S67 A158 (2002).
72. Polmar, S. H. et al. Safety and pharmacokinetics of an oral p38 MAP kinase inhibitor (BIRB 796 BS), administeredtwice daily for 14 days to healthy volunteers. 58th Ann.Meet. Am. Acad. Allergy, Asthma Immunol. 109, S66 A167 (2002).
73. Goldstein, D. M. The discovery and development ofselective inhibitors of p38 MAP kinase from distinct chemicalclasses. J. Inflamm. Res. 51, S114 (2002).
74. Fearns, C. et al. Coordinate activation of endogenous p38α, β, γ, and δ by inflammatory stimuli. J. Leukoc. Biol. 67,705–711 (2000).
75. Barone, F. C. et al. SB 239063, a second-generation p38mitogen-activated protein kinase inhibitor, reduces braininjury and neurological deficits in cerebral focal ischemia. J. Pharmacol. Exp. Ther. 296, 312–321 (2001).
AcknowledgementThe authors wish to acknowledge N. Nevins for her contributionto the illustration in FIG. 5 and S. Blake for critical reading of the manuscript.
Online links
DATABASESThe following terms in this article are linked online to:LocusLink: http://www.ncbi.nlm.nih.gov/LocusLink/DUSP10 | DUSP16 | ERK1 | ERK2 | JNK1 | MAP2K3 | MAP2K6 | p38α MAPK | p38δ | p38γ | TAB1Online Mendelian Inheritance in Man:http://www.ncbi.nlm.nih.gov/Omim/Alzheimer’s disease | Crohn’s disease | inflammatory boweldisease | rheumatoid arthritis
FURTHER INFORMATIONEncyclopedia of Life Sciences: http://www.els.netcytokinesAccess to this interactive links box is free online.