
The protein kinase Akt/PKB regulates both prelamin A degradation and Lmna gene expression
11
The FASEB Journal • Research Communication The protein kinase Akt/PKB regulates both prelamin A degradation and Lmna gene expression Jessika Bertacchini,* ,†,1 Francesca Beretti,* ,1 Vittoria Cenni, ‡ Marianna Guida,* ,† Federica Gibellini,* Laura Mediani,* Oriano Marin, § Nadir M. Maraldi, †,‡ Anto de Pol,* Giovanna Lattanzi, ‡ Lucio Cocco, † and Sandra Marmiroli* ,‡,2 *Cellular Signaling Laboratory, Department of Surgery, Medicine, Dentistry, and Morphology, University of Modena and Reggio Emilia, Modena, Italy; † Cellular Signaling Laboratory, Department of Biomedical Sciences, University of Bologna, Bologna, Italy; ‡ National Research Council of Italy (CNR) Institute of Molecular Genetics, Istituto Ortopedico Rizzoli (IOR), Bologna, Italy; and § Department of Biomedical Sciences, University of Padua, Padua, Italy ABSTRACT The serine/threonine kinase Akt/PKB is a major signaling hub integrating metabolic, survival, growth, and cell cycle regulatory signals. The definition of the phospho-motif cipher driving phosphorylation by Akt led to the identification of hundreds of putative substrates, and it is therefore pivotal to identify those whose phosphorylation by Akt is of consequence to biological processes. The Lmna gene products lamin A/C and the lamin A precursor prelamin A are type V intermediate filament proteins forming a filamentous meshwork, the lamina, underneath the inner nuclear membrane, for nuclear envelope structures organization and interphase chromatin anchoring. In our previous work, we reported that A-type lamins are phosphorylated by Akt at S301 and S404 in physiological conditions and are therefore bona fide substrates of Akt. We report here that Akt phosphorylation at S404 targets the precursor prelamin A for degradation. We further demonstrate that Akt also regulates Lmna transcription. Our study unveils a previously unknown function of Akt in the control of prelamin A stability and expression. Moreover, given the large number of diseases related to prelamin A, our findings represent a further important step bridging basic A-type lamin physiology to therapeutic approaches for lamin A-linked disorders.—Bertacchini, J., Beretti, F., Cenni, V., Guida, M., Gibellini, F., Mediani, L., Marin, O., Maraldi, N. M., de Pol, A., Lattanzi, G., Cocco, L., Marmiroli, S. The protein kinase Akt/PKB regulates both prelamin A degradation and Lmna gene expression. FASEB J. 27, 2145–2155 (2013). www.fasebj.org Key Words: 14.3.3 lamina phosphorylation nuclear sig- naling nucleus cell cycle The 63-kDa, AGC, serine/threonine Akt/PKB pro- tein kinase is a major signaling hub integrating signals involved in the regulation of cellular metab- olism, survival, growth, and cell cycle progression (1). These aspects are differentially regulated by Akt iso- forms: Akt1/PKB, Akt2/PKB, and Akt3/PKB. Akt is activated downstream on the phosphoinositide-3 (PI3) kinase signaling pathway. In resting cells, Akt is a predominantly cytosolic enzyme; however, generation of PI3-kinase lipid products recruits Akt to the plasma membrane, resulting in a conformational change that confers full enzymatic activity through the phosphory- lation of the membrane-bound protein at 2 residues, Thr308 and Ser473. Thr308 lies within the T loop of the catalytic domain and is phosphorylated by 3-phos- phoinositide-dependent protein kinase 1 (PDK-1; ref. 2). mTORC2, a rapamycin-insensitive complex of the mTOR protein kinase with Rictor:GL, mediates S473 phosphorylation in vivo (3), though this residue can also be targeted by other kinases, depending on cell type or stimuli (4 – 6). In addition, the PH domain leucine-rich repeat protein phosphatase (PHLPP) de- phosphorylates S473 directly (7), whereas PP2A possi- bly regulates dephosphorylation of Thr308. Although we have a deep understanding of the mechanisms by which Akt is activated by growth factors and other extracellular stimuli, as well as by oncogenic mutations in growth factor receptors, tissue specificity, subcellular location, and activity temporal changes (8, 9), what ultimately qualifies a kinase are its substrates. The definition of the phospho-motif cipher driving phos- phorylation by Akt has led to the identification of 180 putative substrates in a few years (10), and it is now of great importance to verify which of those take place in vivo and to establish their relevance to biological pro- cesses. We have recently identified 2 bona fide substrates of Akt in muscle cells: lamin A/C (11) and Ankrd2 1 These authors contributed equally to this work. 2 Correspondence: Cellular Signaling Laboratory, Depart- ment of Surgery, Medicine, Dentistry, and Morphology, Uni- versity of Modena and Reggio Emilia, Via Del Pozzo 71, I-41124 Modena, Italy. E-mail: [email protected] doi: 10.1096/fj.12-218214 Abbreviations: 2d-IEF, bidimensional isoelectric focusing; EV, empty vector; HA, hemagglutinin-tagged Akt; HEK, hu- man embryonic kidney; PI3, phosphoinositide-3; RPL32, ri- bosomal protein L32; WT, wild type 2145 0892-6638/13/0027-2145 © FASEB
Transcript of The protein kinase Akt/PKB regulates both prelamin A degradation and Lmna gene expression

The FASEB Journal • Research Communication
The protein kinase Akt/PKB regulates both prelamin Adegradation and Lmna gene expression
Jessika Bertacchini,*,†,1 Francesca Beretti,*,1 Vittoria Cenni,‡ Marianna Guida,*,†
Federica Gibellini,* Laura Mediani,* Oriano Marin,§ Nadir M. Maraldi,†,‡
Anto de Pol,* Giovanna Lattanzi,‡ Lucio Cocco,† and Sandra Marmiroli*,‡,2
*Cellular Signaling Laboratory, Department of Surgery, Medicine, Dentistry, and Morphology,University of Modena and Reggio Emilia, Modena, Italy; †Cellular Signaling Laboratory, Departmentof Biomedical Sciences, University of Bologna, Bologna, Italy; ‡National Research Council of Italy(CNR) Institute of Molecular Genetics, Istituto Ortopedico Rizzoli (IOR), Bologna, Italy; and§Department of Biomedical Sciences, University of Padua, Padua, Italy
ABSTRACT The serine/threonine kinase Akt/PKB is amajor signaling hub integrating metabolic, survival,growth, and cell cycle regulatory signals. The definitionof the phospho-motif cipher driving phosphorylationby Akt led to the identification of hundreds of putativesubstrates, and it is therefore pivotal to identify thosewhose phosphorylation by Akt is of consequence tobiological processes. The Lmna gene products laminA/C and the lamin A precursor prelamin A are type Vintermediate filament proteins forming a filamentousmeshwork, the lamina, underneath the inner nuclearmembrane, for nuclear envelope structures organizationand interphase chromatin anchoring. In our previouswork, we reported that A-type lamins are phosphorylatedby Akt at S301 and S404 in physiological conditions andare therefore bona fide substrates of Akt. We report herethat Akt phosphorylation at S404 targets the precursorprelamin A for degradation. We further demonstrate thatAkt also regulates Lmna transcription. Our study unveils apreviously unknown function of Akt in the control ofprelamin A stability and expression. Moreover, given thelarge number of diseases related to prelamin A, ourfindings represent a further important step bridging basicA-type lamin physiology to therapeutic approaches forlamin A-linked disorders.—Bertacchini, J., Beretti, F.,Cenni, V., Guida, M., Gibellini, F., Mediani, L., Marin, O.,Maraldi, N. M., de Pol, A., Lattanzi, G., Cocco, L.,Marmiroli, S. The protein kinase Akt/PKB regulates bothprelamin A degradation and Lmna gene expression.FASEB J. 27, 2145–2155 (2013). www.fasebj.org
Key Words: 14.3.3 � lamina phosphorylation � nuclear sig-naling � nucleus � cell cycle
The 63-kDa, AGC, serine/threonine Akt/PKB pro-tein kinase is a major signaling hub integrating
signals involved in the regulation of cellular metab-olism, survival, growth, and cell cycle progression (1).These aspects are differentially regulated by Akt iso-forms: Akt1/PKB�, Akt2/PKB�, and Akt3/PKB�. Akt isactivated downstream on the phosphoinositide-3 (PI3)kinase signaling pathway. In resting cells, Akt is apredominantly cytosolic enzyme; however, generationof PI3-kinase lipid products recruits Akt to the plasmamembrane, resulting in a conformational change thatconfers full enzymatic activity through the phosphory-lation of the membrane-bound protein at 2 residues,Thr308 and Ser473. Thr308 lies within the T loop ofthe catalytic domain and is phosphorylated by 3-phos-phoinositide-dependent protein kinase 1 (PDK-1; ref.2). mTORC2, a rapamycin-insensitive complex of themTOR protein kinase with Rictor:G�L, mediates S473phosphorylation in vivo (3), though this residue canalso be targeted by other kinases, depending on celltype or stimuli (4–6). In addition, the PH domainleucine-rich repeat protein phosphatase (PHLPP) de-phosphorylates S473 directly (7), whereas PP2A possi-bly regulates dephosphorylation of Thr308. Althoughwe have a deep understanding of the mechanisms bywhich Akt is activated by growth factors and otherextracellular stimuli, as well as by oncogenic mutationsin growth factor receptors, tissue specificity, subcellularlocation, and activity temporal changes (8, 9), whatultimately qualifies a kinase are its substrates. Thedefinition of the phospho-motif cipher driving phos-phorylation by Akt has led to the identification of �180putative substrates in a few years (10), and it is now ofgreat importance to verify which of those take place invivo and to establish their relevance to biological pro-cesses. We have recently identified 2 bona fide substratesof Akt in muscle cells: lamin A/C (11) and Ankrd2
1 These authors contributed equally to this work.2 Correspondence: Cellular Signaling Laboratory, Depart-
ment of Surgery, Medicine, Dentistry, and Morphology, Uni-versity of Modena and Reggio Emilia, Via Del Pozzo 71,I-41124 Modena, Italy. E-mail: [email protected]
doi: 10.1096/fj.12-218214
Abbreviations: 2d-IEF, bidimensional isoelectric focusing;EV, empty vector; HA, hemagglutinin-tagged Akt; HEK, hu-man embryonic kidney; PI3, phosphoinositide-3; RPL32, ri-bosomal protein L32; WT, wild type
21450892-6638/13/0027-2145 © FASEB

(12), whose phosphorylation has been validated byother laboratories (10).
A-type lamins are type V intermediate filament pro-teins, products of the Lmna gene, and include lamin A,its precursor prelamin A, and alternatively spliced prod-ucts, including lamin C. Lamin A and lamin C share acommon architecture, with an amino-terminal globulardomain, a central helical rod domain, and a carboxy-terminal globular domain. A-type lamins dimerize to formparallel coiled-coil homodimers, then associate head-to-tail to form strings, and ultimately form a higher-orderfilamentous meshwork, the nuclear lamina, lining thenucleoplasmic surface of the inner nuclear membraneof all higher-order eukaryotes and thought to provide aframework for organizing nuclear envelope structuresand anchoring the interphase chromatin (13–17). ThusA-type lamins have essential roles in maintaining lam-ina stability, and in regulating transcription factors, andhave been shown to interact with a plethora of bindingpartners associated with both inner and outer nuclearmembrane (18–20).
Unlike lamin C, lamin A is synthetized as a precursor,prelamin A, a 664-aa protein containing also a C-termi-nal CaaX motif, which undergoes multistep post-trans-lational processing involving several enzymes and even-tually produces mature lamin A. Initially, a farnesylmoiety is added to the CaaX-box cysteine by farnesyl-transferase, followed by cleavage of the last 3 aa (-aaX)by either Rce1 or ZmpSte24. The C-terminal cysteine iscarboxymethylated by isoprenylcysteine carboxymethyltransferase to generate farnesylated/carboxymethy-lated-prelamin A (see Fig. 6). Shortly after the synthe-sis, Zmpste24 cleaves prelamin A 14 aa upstream of theC terminus to generate mature lamin A (646 aa; refs.21–24). Although Lmna is not needed for embryonicdevelopment, Lmna�/� mice grow slowly after birthand die by the 6th week of age. Mutations in the Lmnagene encoding A-type lamins cause inherited laminopa-thies, including Emery-Dreifuss muscular dystrophyand lipodystrophies with metabolic alterations andearly cardiovascular disease (25–27). Moreover, pertur-bation in prelamin A processing, which leads to accu-mulation of prelamin A, is associated with prematuresenescence (23, 26, 27).
In our recent work, we established A-type lamins asphysiological substrates of Akt and identified S301and S404 as phosphorylation sites, both contained incanonical RxRxxS/T Akt phosphorylation motifs(11). The purpose of the present work was to inves-tigate the biological function of prelamin A phos-phorylation by Akt under physiological conditions, incultured C2C12 myoblasts. We report here that phos-phorylation by Akt is a key event that targetsprelamin A for degradation. This issue is more thanjust academic, as featuring the physiological fate ofthe precursor prelamin A may help to elucidate thepathogenetic mechanisms of laminopathies and pavethe way to therapeutic intervention.
MATERIALS AND METHODS
Cell culture
C2C12 mouse myoblasts and human embryonic kidney(HEK) epithelial 293T cells were grown in Dulbecco’s mod-ified Eagle’s medium high glucose (Sigma-Aldrich, St. Louis,MO, USA) supplemented with 10% heat-inactivated FCS(EuroClone Ltd., Devon, UK). C2C12 cells were synchronizedby double methionine block and then stimulated to reenterthe cell cycle by serum and methionine readdition. Fordetection and quantification of cell cycle distribution, sam-ples containing 2–5 � 105 cells were harvested by centrifuga-tion, fixed in cold ethanol, and subjected to propidiumiodide staining and Western blotting of cyclin A.
Where indicated, cells were treated as follows: 50 �Mchloroquine, 0.1 �M bafilomycin, 5 �M MG-132, 20 �MzVAD, 100 �M cycloheximide, 1.6 �M insulin, and 25�M mevinolin.
Transfections
Wild-type (WT) or mutant flag-lamin A (10 �g/100-mmplate) and/or WT hemagglutinin-tagged Akt (HA-Akt; 5�g/plate) were transiently expressed in 293T cells by thecalcium phosphate method, or in C2C12 cells by Cell LineNucleofector kit V and Amaxa Nucleoporator (Instrumenta-tion Laboratory, Milan, Italy), following the manufacturer’sinstructions. cDNA of pEGFP (1 �g/plate) was routinelyadded to transfection mixtures for normalization. In cotrans-fection experiments, empty vector (EV) was added to balancethe amount of DNA. On average, C2C12 nucleoporationachieved between 30 and 35% efficiency.
Plasmids
Both lamin A and Akt plasmids were as described previously(11). Flag-tagged lamin A in pCI expression vector was usedas template to generate S301D/S404D mutants by means ofthe QuikChange strategy (Stratagene, La Jolla, CA, USA).Thefollowing primers were used: forward, GCAGTCTCGAATC-CGCATTGACGACCTCTCAGCCCAGCTCAGCCAGCTCCA,and reverse , TGGAGCTGGCTGAGCTGGGCTGA-GAGGTCGTCAATGCGGATTCGAGACTGC. All constructswere verified by a DNA sequencing service (BMR Genomics,Padua, Italy).
Preparation of cell extracts
Cell extracts were obtained as described previously (28).Briefly, subconfluent cells were extracted by addition of RIPAbuffer (20 mM Tris-Cl, pH 7.0; 1% Nonidet P-40; 150 mMNaCl; 10% glycerol; 10 mM EDTA; 20 mM NaF; 5 mM sodiumpyrophosphate; and 1 mM Na3VO4) and freshly added Sigma-Aldrich Protease Inhibitor Cocktail at 4°C for 30 min. Lysateswere cleared by centrifugation and used for immunoprecipi-tation experiments, as described below.
Immunoprecipitation and electrophoresis
Equal amounts of precleared lysates were incubated for 3 hwith anti-flag (3 �g) or mouse IgG to provide a negativecontrol, plus 30 �l of 50% (v/v) of protein A/G agarose slurry(Santa Cruz Biotechnology, Heidelberg, Germany) at 4°Cwith gentle rocking. To separate prelamin A and lamin A,protein extracts were loaded on 4–12% Bis-Tris SDS-poly-acrylamide gels (Bio-Rad, Hercules, CA, USA), transferred to
2146 Vol. 27 June 2013 BERTACCHINI ET AL.The FASEB Journal � www.fasebj.org

Immobilon-P membranes (Millipore, Waltham, MA, USA),probed with the indicated antibody, and detected by chemi-luminescence with a Supersignal kit (Pierce, Rockford, IL,USA) on a Kodak Image Station 440CF equipped with Kodak1D Image software (Kodak, Rochester, NY, USA). Densito-metric analysis was performed on a Bio-Rad GS-800 densitom-eter by Quantity One 4.6 software. For 2D-electrophoresis, atotal volume of 120 �l of TSU (7 M urea; 2 M thiourea; 4%CHAPS; 1% DTT; 20 mM Tris-Cl, pH 7.4; and 0.2% ampho-lines; plus fresh protease inhibitor cocktail, DNase, andRNase) containing nuclear lysates (120 �g) or anti-M2 immu-noprecipitates, as described previously (11).
The following antibodies were used: anti-flag, clone M2(Sigma-Aldrich); anti-prelamin A nonfarnesylated and anti-prelamin A farnesylated (Diatheva, Fano, Italy); anti-laminA/C, anti-14.3.3�, anti-LC3b, anti-actin, anti-tubulin, anti-parp, anti-Akt, anti-cyclin A, and anti-cyclin E (Santa CruzBiotechnology); anti-pS473Akt, anti-pS253FoxO3a, anti-pS21GSK3a, and anti-Akt phosphosubstrate (Cell SignalingTechnology, Danvers, MA, USA); and anti-GFP and anti-HA(Roche, Indianapolis, IN, USA). Anti-phospholamin A S404(pS404) was produced and purified as described previously(11). Anti-prelamin A antibodies were from Loren Fong(University of California, Los Angeles, CA, USA) and havebeen described previously (15).
[35S] metabolic labeling
At 12 h after cotransfection of flag-Lmna with HA-Akt orempty expression vector, cells were incubated for 1 h inmedium without methionine, then fed with 200 �Ci/ml[35S]methionine (Perkin-Elmer, Monza, Italy) for 16 h incomplete medium. At the indicated chase times, cells werelysed, and prelamin A was immunoprecipitated. Pellets weresubject to SDS-PAGE followed by autoradiography. Proteinhalf-life was estimated using densitometry results from the 4chase time points.
Real-time PCR
Total RNA was extracted using the Aurum Total RNA Fattyand Fibrous Tissue Kit (Bio-Rad) according to the manufac-turer’s instructions. Genomic DNA was eliminated by DNasetreatment. Lmna primers were specifically designed to amplifyonly the cDNA of the full-length product of the LmnA genein mice (accession no. NM_170707) and were the following:forward, GCACCGCTCTCATCAACTC, and reverse, CCATC-CTCGTCGTCATCC. Real-time amplifications, using EvaGreen detection chemistry, were run in triplicate on 96-wellreaction plates with the CFX96 machine (Bio-Rad). Baselineand threshold values were automatically determined for allplates using the Bio-Rad CFX Manager 1.5 software. The dataare represented as fold expression � sem, normalized withribosomal protein L32 (RPL32) gene expression.
Immunofluorescence microscopy
Immunofluorescence was as described previously (29).Briefly, myoblasts grown on glass coverslips were fixed in 4%paraformaldehyde at 4°C for 10 min or methanol at �20°Cfor 7 min. Cells were blocked in 4% BSA-PBS and incubatedwith primary antibodies overnight at 4°C. Coverslips werenext washed and incubated with fluorescence-labeled second-ary antibodies (Sigma-Aldrich) for 1 h at room temperature.Slides were washed and mounted with antifade reagent inglycerol and observed with a Nikon E600 fluorescence micro-scope equipped with a digital camera.
All the images shown in this paper are representative of �3
independent experiments carried out under the same condi-tions. Images from immunochemical and immunofluores-cence studies were processed using Adobe Photoshop CS 8.0software (Adobe Systems, San Jose, CA, USA).
RESULTS
Akt phosphorylates prelamin A during G2/M phase ofthe cell cycle
Our previous work established that A-type lamins arephysiological substrates of Akt and identified 2 phos-phorylation sites, S301 and S404, both contained incanonical RxRxxS/T Akt phosphorylation motifs. Akt-dependent phosphorylation of the A-type lamin precur-sor prelamin A was here examined during cell cycleprogression of C2C12 myoblasts by means of a specificanti-phosphoS404 (pS404) lamin A antibody (11).C2C12 cells were synchronized by double methionineblock, followed by refeeding with 10% FBS and methi-onine. Cell cycle distribution was checked by flowcytometry (Fig. 1A, bottom panel) and anti-cyclin AWestern blotting at the indicated time points. Notably,prelamin A phosphorylation increased as cells ap-proached mitosis, peaking 18 h after refeeding, when69% of cells were in G2/M (Fig. 1A). Though Aktactivity was weak in serum-starved cells, it increasedsharply after refeeding and remained sustainedthroughout G1/S and G2/M, in good agreement withprevious reports (30, 31).
Phosphorylation by Akt prompts prelamin A fordegradation
Numerous Akt substrates are clients for 14.3.3 proteinsin diverse signaling pathways (32). According to theScansite algorithm (http://Scansite3.mit.edu), laminphosphorylation by Akt at S404 and/or S301 createsputative binding sites for 14.3.3. Thus, we investigatedwhether the binding takes place in vivo, by coimmuno-precipitation with an anti-prelamin A antibody. To ruleout interference by IgG light chain, the pellets were runon a 4–15% gradient SDS-PAGE. As shown in Fig. 1B,14.3.3 indeed associates with endogenous prelamin A.Moreover, association is promoted by prelamin A phos-phorylation, as it is significantly reduced by expressionof a phosphorylation-resistant mutant S404A (Fig. 1B)as well as by dephosphorylation in vitro of prelamin Apellets with lambda phosphatase prior to 14.3.3 pull-down (Fig. 1C).
To determine whether prelamin A association with14.3.3 adjusts during cell cycle progression, cells weresynchronized as described above to induce accumula-tion at either G0/G1 or G2/M. Endogenous prelamin Awas then immunoprecipitated and probed with anti-14.3.3. Prelamin A binding was dramatically increasedin the sample enriched with G2/M cells compared tothe sample enriched with G0/G1 cells (Fig. 1D).
Based on previous reports on several Akt substrates(1, 32–36), we reasoned that the destiny of prelamin A
2147AKT REGULATES PRELAMIN A DEGRADATION
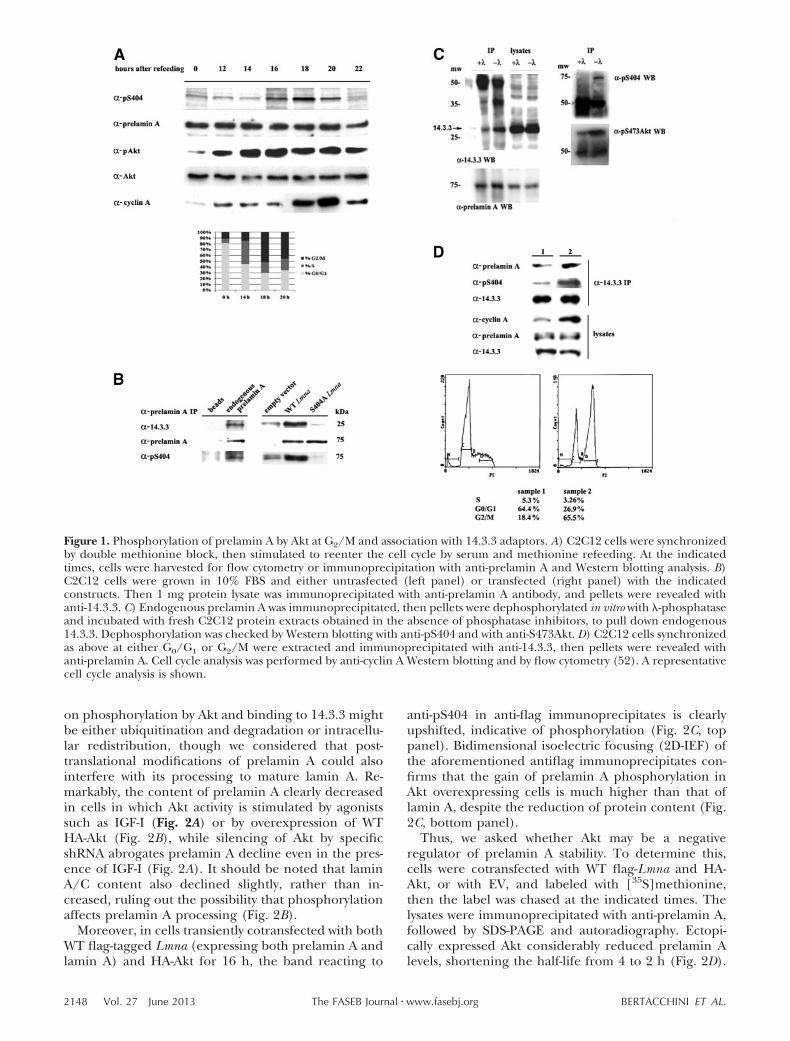
on phosphorylation by Akt and binding to 14.3.3 mightbe either ubiquitination and degradation or intracellu-lar redistribution, though we considered that post-translational modifications of prelamin A could alsointerfere with its processing to mature lamin A. Re-markably, the content of prelamin A clearly decreasedin cells in which Akt activity is stimulated by agonistssuch as IGF-I (Fig. 2A) or by overexpression of WTHA-Akt (Fig. 2B), while silencing of Akt by specificshRNA abrogates prelamin A decline even in the pres-ence of IGF-I (Fig. 2A). It should be noted that laminA/C content also declined slightly, rather than in-creased, ruling out the possibility that phosphorylationaffects prelamin A processing (Fig. 2B).
Moreover, in cells transiently cotransfected with bothWT flag-tagged Lmna (expressing both prelamin A andlamin A) and HA-Akt for 16 h, the band reacting to
anti-pS404 in anti-flag immunoprecipitates is clearlyupshifted, indicative of phosphorylation (Fig. 2C, toppanel). Bidimensional isoelectric focusing (2D-IEF) ofthe aforementioned antiflag immunoprecipitates con-firms that the gain of prelamin A phosphorylation inAkt overexpressing cells is much higher than that oflamin A, despite the reduction of protein content (Fig.2C, bottom panel).
Thus, we asked whether Akt may be a negativeregulator of prelamin A stability. To determine this,cells were cotransfected with WT flag-Lmna and HA-Akt, or with EV, and labeled with [35S]methionine,then the label was chased at the indicated times. Thelysates were immunoprecipitated with anti-prelamin A,followed by SDS-PAGE and autoradiography. Ectopi-cally expressed Akt considerably reduced prelamin Alevels, shortening the half-life from 4 to 2 h (Fig. 2D).
Figure 1. Phosphorylation of prelamin A by Akt at G2/M and association with 14.3.3 adaptors. A) C2C12 cells were synchronizedby double methionine block, then stimulated to reenter the cell cycle by serum and methionine refeeding. At the indicatedtimes, cells were harvested for flow cytometry or immunoprecipitation with anti-prelamin A and Western blotting analysis. B)C2C12 cells were grown in 10% FBS and either untrasfected (left panel) or transfected (right panel) with the indicatedconstructs. Then 1 mg protein lysate was immunoprecipitated with anti-prelamin A antibody, and pellets were revealed withanti-14.3.3. C) Endogenous prelamin A was immunoprecipitated, then pellets were dephosphorylated in vitro with -phosphataseand incubated with fresh C2C12 protein extracts obtained in the absence of phosphatase inhibitors, to pull down endogenous14.3.3. Dephosphorylation was checked by Western blotting with anti-pS404 and with anti-S473Akt. D) C2C12 cells synchronizedas above at either G0/G1 or G2/M were extracted and immunoprecipitated with anti-14.3.3, then pellets were revealed withanti-prelamin A. Cell cycle analysis was performed by anti-cyclin A Western blotting and by flow cytometry (52). A representativecell cycle analysis is shown.
2148 Vol. 27 June 2013 BERTACCHINI ET AL.The FASEB Journal � www.fasebj.org

To confirm this result, the decay of prelamin A wasdetermined also in cells transfected with phospho-mimetic S404D and nonphosphorylatable S404A mu-tants. In cells treated with the protein translationinhibitor cycloheximide, the phosphorylation-resistantmutant was more stable, while the phospho-mimeticmutant was dramatically reduced, compared with theWT counterpart (Fig. 2E).
To determine whether phosphorylation selectively
targets prelamin A for degradation, cells were treatedwith cycloheximide alone or in combination with eithercaspase or protease inhibitors. Unexpectedly, however,pretreatment with the broad proteasome inhibitor MG-132 or with the caspase inhibitor zVAD led to anincomplete recover of prelamin A (Fig. 2F). Conversely,well-known inhibitors of lysosomal hydrolases, such aschloroquine or bafilomycin, rescued the amount ofprelamin A entirely, indicating that phosphorylation
Figure 2. Phosphorylation by Akt drives prelamin A degradation. A) Akt was silenced with shRNA targeting Akt1/2 for 72 h, thencells were stimulated with IGF-I for 2 h. plko, EV. The filter was probed with anti-Akt and anti-pS473Akt to confirm silencingand activation by IGF-I, respectively. B) C2C12 cells were transfected with EV (lane 1) or WT HA-Akt for 16 h (lane 2) or 24 h(lane 3), then prelamin A content was monitored by Western blotting. C) Cells were transfected for 16 h with EV (lane 1) orWT flag-lamin A, expressing prelamin A and lamin A, alone (lane 2) or combined with HA-Akt (lane 3), then protein extractswere immunoprecipitated with anti-flag. The pellets were halved, and one half was loaded on SDS-PAGE, the other was resolvedby 2D-IEF onto 5–8 IP strips for the first dimension. Both samples were run on a 4–12% bis-tris SDS-PAGE for the seconddimension, then transferred to Immobilon P and probed with the indicated antibodies. D) Cells were incubated for 1 h inmedium without methionine, then fed with 200 �Ci/ml [35S]methionine for 16 h in complete medium. At the indicated chasetimes, cells were lysed, and prelamin A was immunoprecipitated. Pellets were subjected to SDS-PAGE, followed by autoradiog-raphy. Total lysates were probed with anti-flag and anti-HA to check transfection efficiency. E) C2C12 cells were transientlytransfected with WT or nonphosphorylatable or phospho-mimetic flag-lamin A, then treated with cycloheximide, as indicated,to establish whether phosphorylation affects protein stability. F) Cells were treated with cycloheximide for 2 h, followed byMG-132 or zVAD for 1 h, to assess their efficacy on endogenous prelamin A degradation.
2149AKT REGULATES PRELAMIN A DEGRADATION
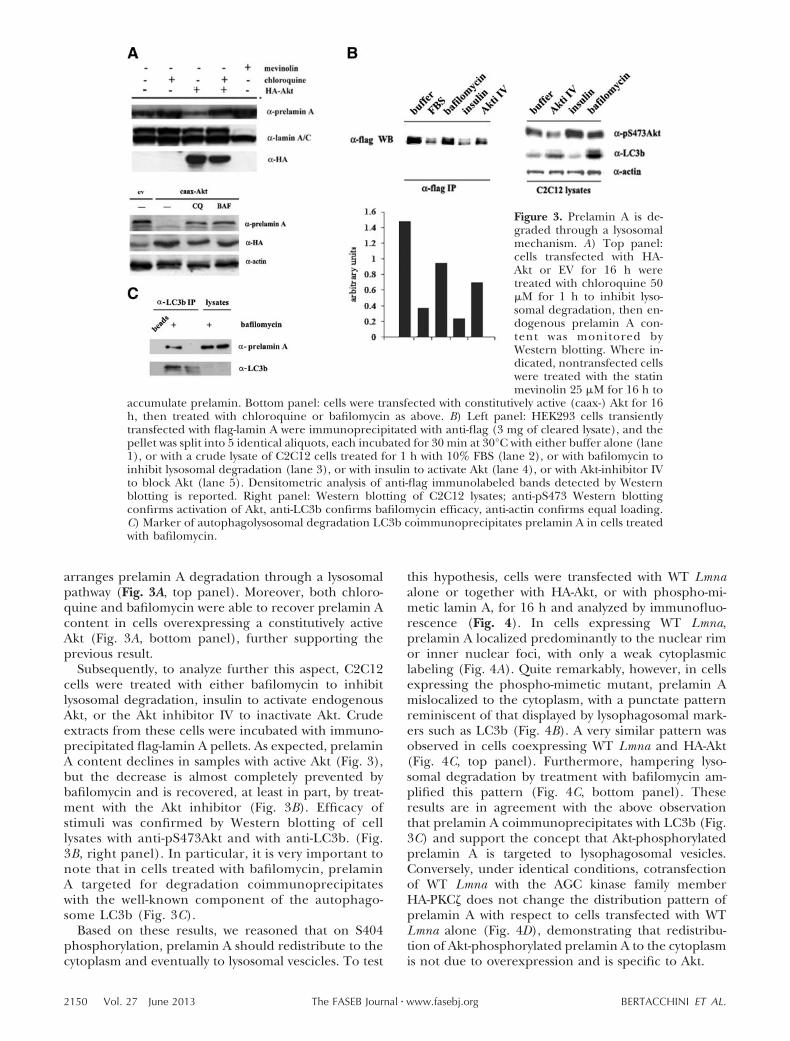
arranges prelamin A degradation through a lysosomalpathway (Fig. 3A, top panel). Moreover, both chloro-quine and bafilomycin were able to recover prelamin Acontent in cells overexpressing a constitutively activeAkt (Fig. 3A, bottom panel), further supporting theprevious result.
Subsequently, to analyze further this aspect, C2C12cells were treated with either bafilomycin to inhibitlysosomal degradation, insulin to activate endogenousAkt, or the Akt inhibitor IV to inactivate Akt. Crudeextracts from these cells were incubated with immuno-precipitated flag-lamin A pellets. As expected, prelaminA content declines in samples with active Akt (Fig. 3),but the decrease is almost completely prevented bybafilomycin and is recovered, at least in part, by treat-ment with the Akt inhibitor (Fig. 3B). Efficacy ofstimuli was confirmed by Western blotting of celllysates with anti-pS473Akt and with anti-LC3b. (Fig.3B, right panel). In particular, it is very important tonote that in cells treated with bafilomycin, prelaminA targeted for degradation coimmunoprecipitateswith the well-known component of the autophago-some LC3b (Fig. 3C).
Based on these results, we reasoned that on S404phosphorylation, prelamin A should redistribute to thecytoplasm and eventually to lysosomal vescicles. To test
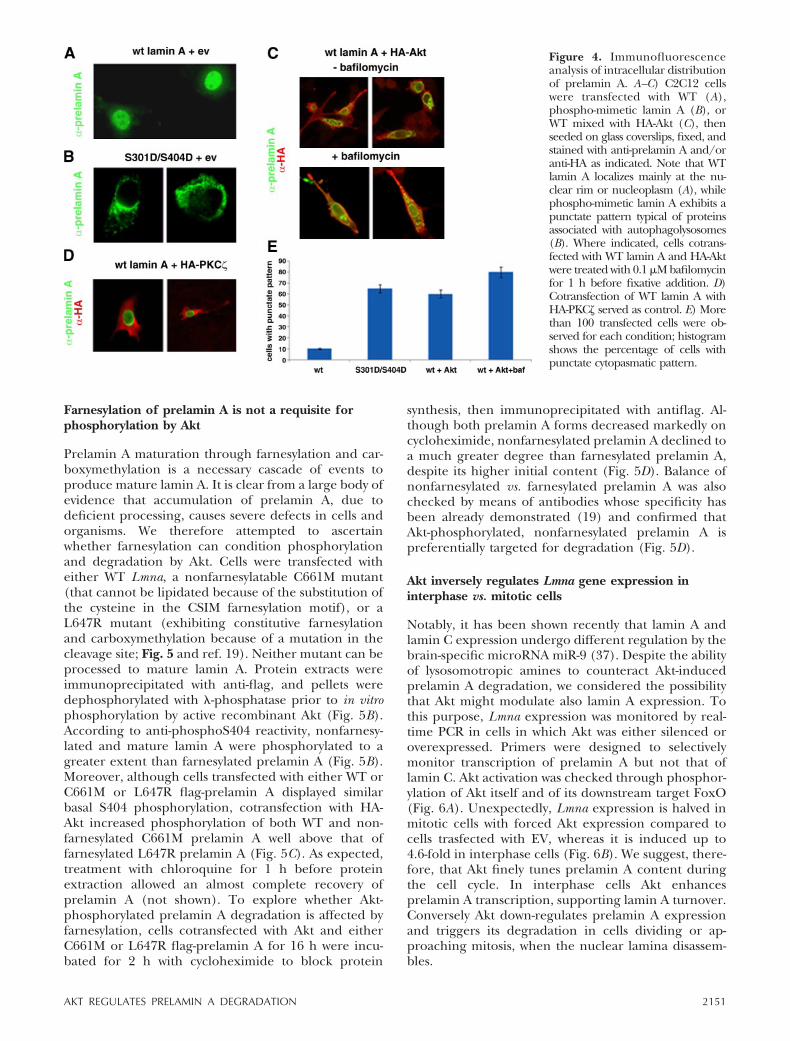
this hypothesis, cells were transfected with WT Lmnaalone or together with HA-Akt, or with phospho-mi-metic lamin A, for 16 h and analyzed by immunofluo-rescence (Fig. 4). In cells expressing WT Lmna,prelamin A localized predominantly to the nuclear rimor inner nuclear foci, with only a weak cytoplasmiclabeling (Fig. 4A). Quite remarkably, however, in cellsexpressing the phospho-mimetic mutant, prelamin Amislocalized to the cytoplasm, with a punctate patternreminiscent of that displayed by lysophagosomal mark-ers such as LC3b (Fig. 4B). A very similar pattern wasobserved in cells coexpressing WT Lmna and HA-Akt(Fig. 4C, top panel). Furthermore, hampering lyso-somal degradation by treatment with bafilomycin am-plified this pattern (Fig. 4C, bottom panel). Theseresults are in agreement with the above observationthat prelamin A coimmunoprecipitates with LC3b (Fig.3C) and support the concept that Akt-phosphorylatedprelamin A is targeted to lysophagosomal vesicles.Conversely, under identical conditions, cotransfectionof WT Lmna with the AGC kinase family memberHA-PKC does not change the distribution pattern ofprelamin A with respect to cells transfected with WTLmna alone (Fig. 4D), demonstrating that redistribu-tion of Akt-phosphorylated prelamin A to the cytoplasmis not due to overexpression and is specific to Akt.
Figure 3. Prelamin A is de-graded through a lysosomalmechanism. A) Top panel:cells transfected with HA-Akt or EV for 16 h weretreated with chloroquine 50�M for 1 h to inhibit lyso-somal degradation, then en-dogenous prelamin A con-tent was monitored byWestern blotting. Where in-dicated, nontransfected cellswere treated with the statinmevinolin 25 �M for 16 h to
accumulate prelamin. Bottom panel: cells were transfected with constitutively active (caax-) Akt for 16h, then treated with chloroquine or bafilomycin as above. B) Left panel: HEK293 cells transientlytransfected with flag-lamin A were immunoprecipitated with anti-flag (3 mg of cleared lysate), and thepellet was split into 5 identical aliquots, each incubated for 30 min at 30°C with either buffer alone (lane1), or with a crude lysate of C2C12 cells treated for 1 h with 10% FBS (lane 2), or with bafilomycin toinhibit lysosomal degradation (lane 3), or with insulin to activate Akt (lane 4), or with Akt-inhibitor IVto block Akt (lane 5). Densitometric analysis of anti-flag immunolabeled bands detected by Westernblotting is reported. Right panel: Western blotting of C2C12 lysates; anti-pS473 Western blottingconfirms activation of Akt, anti-LC3b confirms bafilomycin efficacy, anti-actin confirms equal loading.C) Marker of autophagolysosomal degradation LC3b coimmunoprecipitates prelamin A in cells treatedwith bafilomycin.
2150 Vol. 27 June 2013 BERTACCHINI ET AL.The FASEB Journal � www.fasebj.org
Farnesylation of prelamin A is not a requisite forphosphorylation by Akt
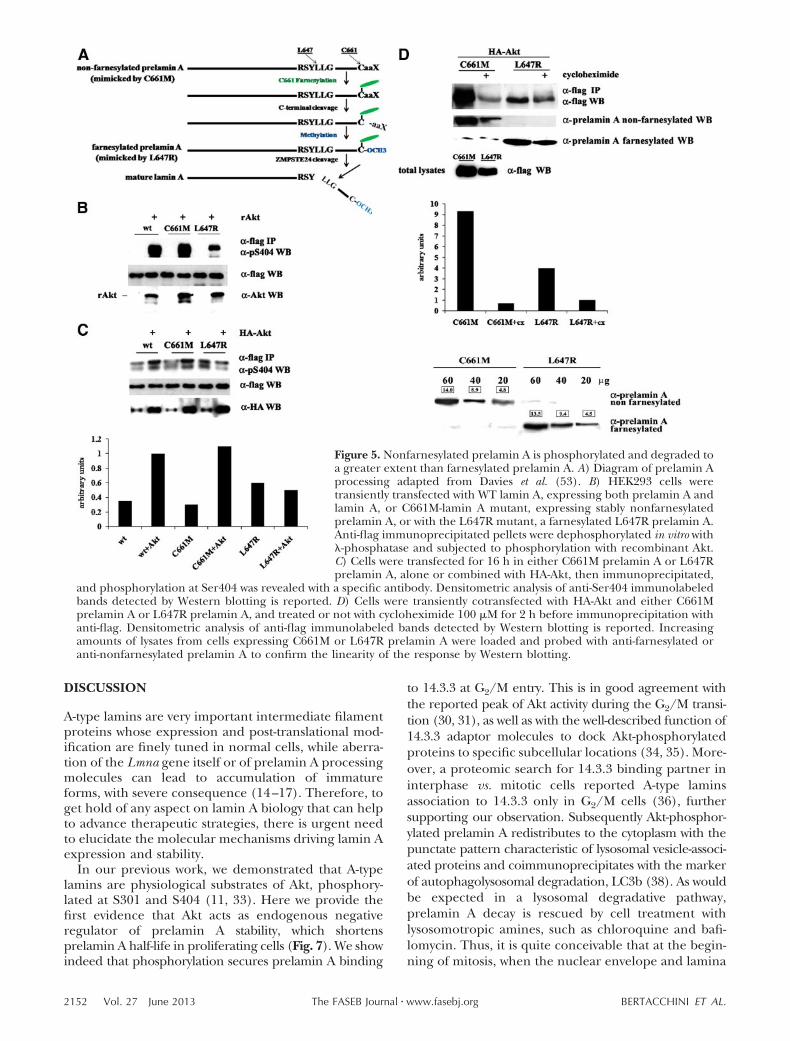
Prelamin A maturation through farnesylation and car-boxymethylation is a necessary cascade of events toproduce mature lamin A. It is clear from a large body ofevidence that accumulation of prelamin A, due todeficient processing, causes severe defects in cells andorganisms. We therefore attempted to ascertainwhether farnesylation can condition phosphorylationand degradation by Akt. Cells were transfected witheither WT Lmna, a nonfarnesylatable C661M mutant(that cannot be lipidated because of the substitution ofthe cysteine in the CSIM farnesylation motif), or aL647R mutant (exhibiting constitutive farnesylationand carboxymethylation because of a mutation in thecleavage site; Fig. 5 and ref. 19). Neither mutant can beprocessed to mature lamin A. Protein extracts wereimmunoprecipitated with anti-flag, and pellets weredephosphorylated with -phosphatase prior to in vitrophosphorylation by active recombinant Akt (Fig. 5B).According to anti-phosphoS404 reactivity, nonfarnesy-lated and mature lamin A were phosphorylated to agreater extent than farnesylated prelamin A (Fig. 5B).Moreover, although cells transfected with either WT orC661M or L647R flag-prelamin A displayed similarbasal S404 phosphorylation, cotransfection with HA-Akt increased phosphorylation of both WT and non-farnesylated C661M prelamin A well above that offarnesylated L647R prelamin A (Fig. 5C). As expected,treatment with chloroquine for 1 h before proteinextraction allowed an almost complete recovery ofprelamin A (not shown). To explore whether Akt-phosphorylated prelamin A degradation is affected byfarnesylation, cells cotransfected with Akt and eitherC661M or L647R flag-prelamin A for 16 h were incu-bated for 2 h with cycloheximide to block protein
synthesis, then immunoprecipitated with antiflag. Al-though both prelamin A forms decreased markedly oncycloheximide, nonfarnesylated prelamin A declined toa much greater degree than farnesylated prelamin A,despite its higher initial content (Fig. 5D). Balance ofnonfarnesylated vs. farnesylated prelamin A was alsochecked by means of antibodies whose specificity hasbeen already demonstrated (19) and confirmed thatAkt-phosphorylated, nonfarnesylated prelamin A ispreferentially targeted for degradation (Fig. 5D).
Akt inversely regulates Lmna gene expression ininterphase vs. mitotic cells
Notably, it has been shown recently that lamin A andlamin C expression undergo different regulation by thebrain-specific microRNA miR-9 (37). Despite the abilityof lysosomotropic amines to counteract Akt-inducedprelamin A degradation, we considered the possibilitythat Akt might modulate also lamin A expression. Tothis purpose, Lmna expression was monitored by real-time PCR in cells in which Akt was either silenced oroverexpressed. Primers were designed to selectivelymonitor transcription of prelamin A but not that oflamin C. Akt activation was checked through phosphor-ylation of Akt itself and of its downstream target FoxO(Fig. 6A). Unexpectedly, Lmna expression is halved inmitotic cells with forced Akt expression compared tocells trasfected with EV, whereas it is induced up to4.6-fold in interphase cells (Fig. 6B). We suggest, there-fore, that Akt finely tunes prelamin A content duringthe cell cycle. In interphase cells Akt enhancesprelamin A transcription, supporting lamin A turnover.Conversely Akt down-regulates prelamin A expressionand triggers its degradation in cells dividing or ap-proaching mitosis, when the nuclear lamina disassem-bles.
Figure 4. Immunofluorescenceanalysis of intracellular distributionof prelamin A. A–C) C2C12 cellswere transfected with WT (A),phospho-mimetic lamin A (B), orWT mixed with HA-Akt (C), thenseeded on glass coverslips, fixed, andstained with anti-prelamin A and/oranti-HA as indicated. Note that WTlamin A localizes mainly at the nu-clear rim or nucleoplasm (A), whilephospho-mimetic lamin A exhibits apunctate pattern typical of proteinsassociated with autophagolysosomes(B). Where indicated, cells cotrans-fected with WT lamin A and HA-Aktwere treated with 0.1 �M bafilomycinfor 1 h before fixative addition. D)Cotransfection of WT lamin A withHA-PKC served as control. E) Morethan 100 transfected cells were ob-served for each condition; histogramshows the percentage of cells withpunctate cytopasmatic pattern.
2151AKT REGULATES PRELAMIN A DEGRADATION
DISCUSSION
A-type lamins are very important intermediate filamentproteins whose expression and post-translational mod-ification are finely tuned in normal cells, while aberra-tion of the Lmna gene itself or of prelamin A processingmolecules can lead to accumulation of immatureforms, with severe consequence (14–17). Therefore, toget hold of any aspect on lamin A biology that can helpto advance therapeutic strategies, there is urgent needto elucidate the molecular mechanisms driving lamin Aexpression and stability.
In our previous work, we demonstrated that A-typelamins are physiological substrates of Akt, phosphory-lated at S301 and S404 (11, 33). Here we provide thefirst evidence that Akt acts as endogenous negativeregulator of prelamin A stability, which shortensprelamin A half-life in proliferating cells (Fig. 7). We showindeed that phosphorylation secures prelamin A binding
to 14.3.3 at G2/M entry. This is in good agreement withthe reported peak of Akt activity during the G2/M transi-tion (30, 31), as well as with the well-described function of14.3.3 adaptor molecules to dock Akt-phosphorylatedproteins to specific subcellular locations (34, 35). More-over, a proteomic search for 14.3.3 binding partner ininterphase vs. mitotic cells reported A-type laminsassociation to 14.3.3 only in G2/M cells (36), furthersupporting our observation. Subsequently Akt-phosphor-ylated prelamin A redistributes to the cytoplasm with thepunctate pattern characteristic of lysosomal vesicle-associ-ated proteins and coimmunoprecipitates with the markerof autophagolysosomal degradation, LC3b (38). As wouldbe expected in a lysosomal degradative pathway,prelamin A decay is rescued by cell treatment withlysosomotropic amines, such as chloroquine and bafi-lomycin. Thus, it is quite conceivable that at the begin-ning of mitosis, when the nuclear envelope and lamina
Figure 5. Nonfarnesylated prelamin A is phosphorylated and degraded toa greater extent than farnesylated prelamin A. A) Diagram of prelamin Aprocessing adapted from Davies et al. (53). B) HEK293 cells weretransiently transfected with WT lamin A, expressing both prelamin A andlamin A, or C661M-lamin A mutant, expressing stably nonfarnesylatedprelamin A, or with the L647R mutant, a farnesylated L647R prelamin A.Anti-flag immunoprecipitated pellets were dephosphorylated in vitro with-phosphatase and subjected to phosphorylation with recombinant Akt.C) Cells were transfected for 16 h in either C661M prelamin A or L647Rprelamin A, alone or combined with HA-Akt, then immunoprecipitated,
and phosphorylation at Ser404 was revealed with a specific antibody. Densitometric analysis of anti-Ser404 immunolabeledbands detected by Western blotting is reported. D) Cells were transiently cotransfected with HA-Akt and either C661Mprelamin A or L647R prelamin A, and treated or not with cycloheximide 100 �M for 2 h before immunoprecipitation withanti-flag. Densitometric analysis of anti-flag immunolabeled bands detected by Western blotting is reported. Increasingamounts of lysates from cells expressing C661M or L647R prelamin A were loaded and probed with anti-farnesylated oranti-nonfarnesylated prelamin A to confirm the linearity of the response by Western blotting.
2152 Vol. 27 June 2013 BERTACCHINI ET AL.The FASEB Journal � www.fasebj.org
disassemble, cells dispose of superfluous, newly synthe-sized prelamin A, rather than undertake a complex andunnecessary post-translational modification process.
Moreover, though the mechanism described hererefers to physiological prelamin A turnover in musclecells, it is in good agreement with a very recent reportthat the macrolide antibiotic rapamycin, a potent in-hibitor of the mTORC1 kinase, stimulates clearance ofthe mutant prelamin A protein progerin in Hutchin-son-Gilford progeria syndrome fibroblasts through in-creased autophagy and might therefore be a proposedtreatment in progeria (39–41). Since the effect ofrapamycin was observed after 2 wk administration, it istempting to speculate that it may be mediated by thewell-known negative feedback loop that, in prolongedrapamycin treatment, results in the up-regulation ofAkt activity (42), which could eventually lead to phos-phorylation of Ser301 and Ser404.
Prelamin A maturation through farnesylation andcarboxymethylation is a necessary cascade of events toproduce mature lamin A, highly conserved duringvertebrate evolution (43, 44). The reason for this com-plex processing is not clear, though, considering that inlaboratory mice, direct production of mature lamin A,thus bypassing prelamin A synthesis and maturation, doesnot cause discernable pathology (45, 46). However, it isnow clear from a large body of evidence that failure toprocess prelamin A causes severe defects in cells andorganisms (26, 47, 48). For instance, the in-framesplice-variant protein progerin, responsible for mostcases of Hutchinson-Gilford progeria syndrome, lacksthe 50 aa containing the clipping site at the C terminusand is thus regarded as a permanently farnesylatedvariant of lamin A (49). Very importantly, we demon-strate here that nonfarnesylated prelamin A is Akt-phosphorylated, and then degraded, to a much greaterextent than farnesylated prelamin A or mature lamin A.We speculate that the farnesyl residue could be addedto the CaaX motif of prelamin A to provide a pool ofstable precursors promptly available at the end ofmitosis. This result has very important implication fortreatment of diseases characterized by accumulation ofprelamin A. In this sense, our data suggest that thetoxicity of farnesylated prelamin A may be related notonly to gain of deleterious intermolecular interactions,but also to a higher protein stability. In this context,farnesylation inhibitors such as statins, currently usedin clinical trials of laminopathies, could efficientlyreduce toxic effects by triggering accumulation of thenonfarnesylated isoform, more prompt to lysosomaldegradation.
Remarkably, Akt not only triggers prelamin A degra-dation, but also rules Lmna expression. By real-timePCR in synchronized cells, we show that Akt indeedfinely tunes prelamin A content during the cell cycle. Ininterphase cells, Akt enhances prelamin A transcrip-tion, supporting lamin A turnover. Conversely Aktdown-regulates prelamin A expression and triggers itsdegradation in cells dividing or approaching mitosis,when the nuclear lamina disassembles. Possibly, Aktexerts this control through regulation of effectors such
Figure 6. Real-time PCR analysis of Akt-regulated prelamin A expression in interphase vs. mitotic cells. A) Real-timeamplifications of Lmna, using Eva Green detection chemistry, were run in triplicate. Data are represented as fold expression �sem, normalized with RPL32 gene expression. B) C2C12 myoblasts were transfected with EV or HA-Akt. Akt activity wasmonitored by anti-pS473Akt and by anti-pFoxO3 phosphorylation. C) Percentage of cell cycle distribution in each sample.
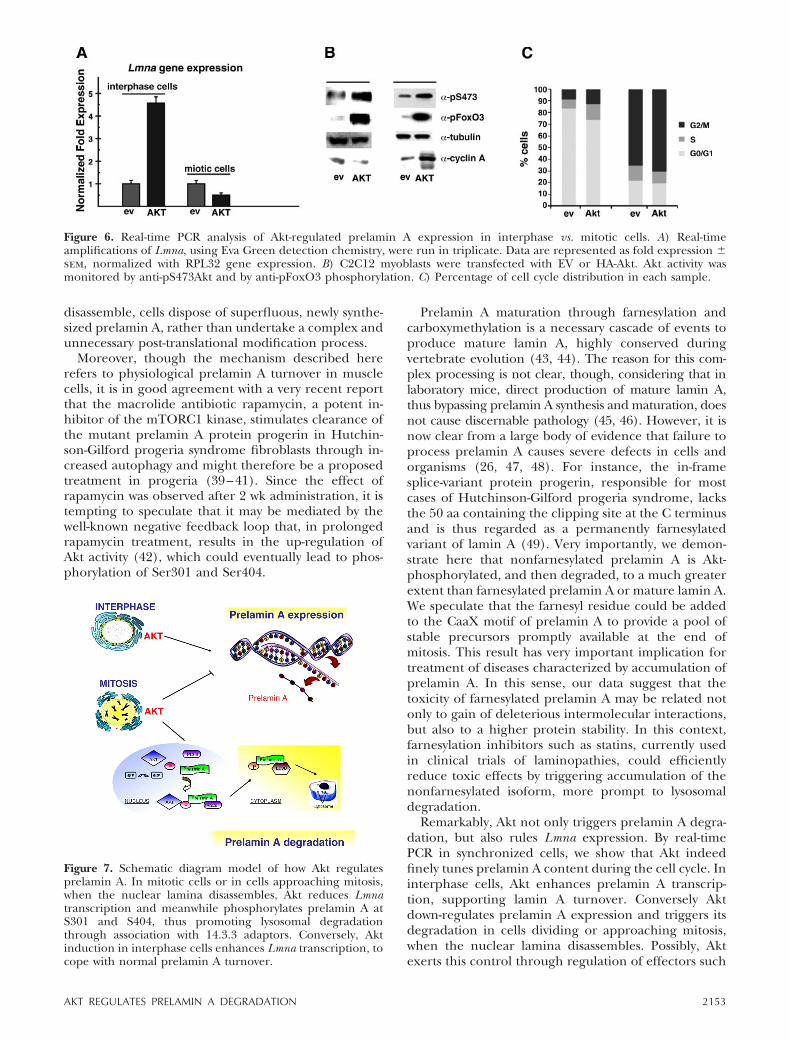
Figure 7. Schematic diagram model of how Akt regulatesprelamin A. In mitotic cells or in cells approaching mitosis,when the nuclear lamina disassembles, Akt reduces Lmnatranscription and meanwhile phosphorylates prelamin A atS301 and S404, thus promoting lysosomal degradationthrough association with 14.3.3 adaptors. Conversely, Aktinduction in interphase cells enhances Lmna transcription, tocope with normal prelamin A turnover.
2153AKT REGULATES PRELAMIN A DEGRADATION

as FoxO, Sp1/Sp3, AP1, and CREB. Indeed, binding-domain-specific transcription factors, such as SP1/3 orAP1, have been reported in prelamin A promoter (50,51). However, we cannot exclude the possibility thatAkt modulates prelamin A expression through an indi-rect mechanism, and this aspect deserves deeper inves-tigation.
Collectively, in this study, we provide the first evi-dence that Akt operates as a physiological controller ofboth prelamin A stability and expression.
The authors are grateful to Maria Ruzzene for helpfuldiscussion, to Loren Fong (University of California, LosAngeles, CA, USA) for antibodies against prelamin A, and toGiacomo Spinsanti for skillful technical assistance with real-time PCR. S.M. and J.B. acknowledge the “International ShortTerm Mobility Program” from Italian Consiglio Nazionaledelle Ricerche (CNR) and Fondazione CaRiMo, respectively.M.G. was supported in part by Fondazione Angela Serra forCancer Research (Modena, Italy). This work was supported bygrants from Italian Ministero dell’Istruzione, dell’Universita edella Ricerca (MIUR)–Programma di Ricerca di RilevanteInteresse Nazionale (PRIN), MIUR–Fondo per gli Investi-menti della Ricerca di Base (FIRB; Human Proteome Net),MIUR-FIRB (Accordi di Programma 2010), FondazioneCarisbo, and Associazione Italiana Progeria Sammy Basso(AIProSaB) Italy.
REFERENCES
1. Manning, B. D., and Cantley, L. C. (2007) AKT/PKB signaling:navigating downstream. Cell 129, 1261–1274
2. Alessi, D. R., James, S. R., Downes, C. P., Holmes, A. B., Gaffney,P. R., Reese, C. B., and Cohen, P. (1997) Characterization of a3-phosphoinositide-dependent protein kinase which phosphor-ylates and activates protein kinase B alpha. Curr. Biol. 7, 261–269
3. Sarbassov, D. D., Guertin, D. A., Siraj, M. A., and Sabatini, D. M.(2005) Phosphorylation and regulation of Akt/PKB by therictor-mTOR complex. Science 307, 1098–1101
4. Toker, A., and Newton, A. C. (2000) Akt/protein kinase B isregulated by autophosphorylation at the hypothetical PDK-2site. J. Biol. Chem. 275, 8271–8274
5. Persad, S., Attwell, S., Gray, V., Mawji, N., Deng, J. T., Leung, D.,Yan, J., Sanghera, J., Walsh, M. P., and Dedhar, S. (2001)Regulation of protein kinase B/Akt-serine 473 phosphorylationby integrin-linked kinase: critical roles for kinase activity andamino acids arginine 211 and serine 343. J. Biol. Chem. 276,27462–27469
6. Feng, J., Park, J., Cron, P., Hess, D., and Hemmings, B. A. (2004)Identification of a PKB/Akt hydrophobic motif Ser-473 kinaseas DNA-dependent protein kinase. J. Biol. Chem. 279, 41189–41196
7. Gao, T., Furnari, F., and Newton, A. C. (2005) PHLPP: aphosphatase that directly dephosphorylates Akt, promotes apo-ptosis, and suppresses tumor growth. Mol. Cell 18, 13–24
8. Cenni, V., Sirri, A., Riccio, M., Lattanzi, G., Santi, S., de Pol, A.,Maraldi, N. M., and Marmiroli, S. (2003) Targeting of theAkt/PKB kinase to the actin skeleton. Cell. Mol. Life Sci. 60,2710–2720
9. Gonzalez, E., and McGraw, T. E. (2009) Insulin-modulated Aktsubcellular localization determines Akt isoform-specific signal-ing. Proc. Natl. Acad. Sci. U. S. A. 106, 7004–7009
10. Moritz, A., Li, Y., Guo, A., Villén, J., Wang, Y., MacNeill, J.,Kornhauser, J., Sprott, K., Zhou, J., Possemato, A., Ren, J. M.,Hornbeck, P., Cantley, L. C., Gygi, S. P., Rush, J., and Comb,M. J. (2010) Akt-RSK-S6 kinase signaling networks activated byoncogenic receptor tyrosine kinases. Sci. Signal. 3, ra64
11. Cenni, V., Bertacchini, J., Beretti, F., Lattanzi, G., Bavelloni, A.,Riccio, M., Ruzzene, M., Marin, O., Arrigoni, G., Parnaik, V.,Wehnert, M., Maraldi, N. M., de Pol, A., Cocco, L., and
Marmiroli, S. (2008) Lamin A Ser404 is a nuclear target of Aktphosphorylation in C2C12 cells. J. Proteome Res. 7, 4727–2735
12. Cenni, V., Bavelloni, A., Beretti, F., Tagliavini, F., Manzoli, L.,Lattanzi, G., Maraldi, N. M., Cocco, L., and Marmiroli, S. (2011)Ankrd2/ARPP is a novel Akt2 specific substrate and regulatesmyogenic differentiation upon cellular exposure to H(2)O(2).Mol. Biol. Cell 22, 2946–2956
13. Prokocimer, M., Davidovich, M., Nissim-Rafinia, M., Wiesel-Motiuk, N., Bar, D. Z., Barkan, R., Meshorer, E., and Gruen-baum, Y. (2009) Nuclear lamins: key regulators of nuclearstructure and activities. J. Cell. Mol. Med. 13, 1059–1085
14. Dechat, T., Pfleghaar, K., Sengupta, K., Shimi, T., Shumaker,D. K., Solimando, L., and Goldman, R. D. (2008) Nuclearlamins: major factors in the structural organization and functionof the nucleus and chromatin. Genes Dev. 22, 832–853
15. Fong, L. G., Ng, J. K., Lammerding, J., Vickers, T. A., Meta, M.,Coté N, Gavino, B., QiaoX, Chang, S. Y., Young, S. R., Yang,S. H., Stewart, C. L., Lee, R. T., Bennett, C. F., Bergo, M. O., andYoung, S. G. (2006) Prelamin A and lamin A appear to bedispensable in the nuclear lamina. J. Clin. Invest. 116, 743–752
16. Eriksson, J. E., Dechat, T., Grin, B., Helfand, B., Mendez, M.,Pallari, H. M., and Goldman, R. D. (2009) Introducing interme-diate filaments: from discovery to disease. J. Clin. Invest. 119,1763–1771
17. Maraldi, N. M., Capanni, C., Cenni, V., Fini, M., and Lattanzi, G.(2011) Laminopathies and lamin-associated signaling pathways.J. Cell. Biochem. 112, 979–992
18. Naetar, N., Korbei, B., Kozlov, S., Kerenyi, M. A., Dorner, D.,Kral, R., Gotic, I., Fuchs, P., Cohen, T. V., Bittner, R., Stewart,C. L., and Foisner, R. (2008) Loss of nucleoplasmic LAP2alpha-lamin A complexes causes erythroid and epidermal progenitorhyperproliferation. Nat. Cell Biol. 10, 1341–1348
19. Mattioli, E., Columbaro, M., Capanni, C., Maraldi, N. M., Cenni,V., Scotlandi, K., Marino, M. T., Merlini, L., Squarzoni, S., andLattanzi, G. (2011) Prelamin A-mediated recruitment of SUN1to the nuclear envelope directs nuclear positioning in humanmuscle. Cell Death Differ. 18, 1305–1315
20. Margalit, A., Brachner, A., Gotzmann J., and Foisner, R. Y. G.(2007) Barrier-to-autointegration factor—a BAFfling little pro-tein. Trends Cell Biol. 17, 202–208
21. Butin-Israeli, V., Adam, S. A., Goldman, A. E., and Goldman,R. D. (2012) Nuclear lamin functions and disease. Trends Genet.28, 464–471
22. Rusiñol, A. E., and Sinensky, M. S. (2006) Farnesylated lamins,progeroid syndromes and farnesyl transferase inhibitors. J. CellSci. 119, 3265–3272
23. Varela, I., Pereira, S., Ugalde, A. P., Navarro, C. L., Suárez, M. F.,Cau, P., Cadiñanos, J., Osorio, F. G., Cobo, J., de Carlos, F., Lèvy,N., Freije, J. M. P., and López-Otín, C. (2008) Combinedtreatment with statins and aminobisphosphonates extends lon-gevity in a mouse model of human premature aging. Nat. Med.14, 767–772
24. Worman, H. J. (2012) Nuclear lamins and laminopathies. J.Pathol. 226, 316–325
25. Goldman, R. D., Shumaker, D. K., Erdos, M. R., Eriksson, M.,Goldman, A. E., Gordon, L. B., Gruenbaum, Y., Khuon, S.,Mendez, M., Varga, R., and Collins, F. S. (2004) Accumulationof mutant lamin A causes progressive changes in nucleararchitecture in Hutchinson-Gilford progeria syndrome. Proc.Natl. Acad. Sci. U. S. A. 101, 8963–8968
26. Scaffidi, P., and Misteli, T. (2005) Reversal of the cellularphenotype in the premature aging disease Hutchinson-Gilfordprogeria syndrome. Nat. Genet. 11, 440–445
27. Muchir, A., and Worman, H. J. (2010) Signaling defects and thenuclear envelope in progeria. Dev. Cell. 19, 355–356
28. Maraldi, T., Bertacchini, J., Benincasa, M., Guida, M., De Pol, A.,Liotta, L. A., Petricoin, E., Cocco, L., and Marmiroli, S. (2011)Reverse-phase protein microarrays (RPPA) as a diagnostic andtherapeutic guide in multidrug resistant leukemia. Int. J. Oncol.38, 427–435
29. Maraldi, T., Riccio, M., Sena, P., Marzona, L., Nicoli, A., LaMarca, A., Marmiroli, S., Bertacchini, J., La Sala, G., and De Pol,A. (2009) MATER protein as substrate of PKCepsilon in humancumulus cells. Mol. Hum. Reprod. 15, 499–506
30. Shtivelman, E., Sussman, J., and Stokoe, D. (2002) A role for PI3-kinase and PKB activity in the G2/M phase of the cell cycle.Curr. Biol. 12, 919–924
2154 Vol. 27 June 2013 BERTACCHINI ET AL.The FASEB Journal � www.fasebj.org

31. Kandel, E. S., Skeen, J., Majewski, N., Di Cristofano, A., Pandolfi,P. P., Feliciano, C. S., Gartel, A., and Hay, N. (2002) Activationof Akt/protein kinase B overcomes a G(2)/M cell cycle check-point induced by DNA damage. Mol. Cell. Biol. 22, 7831–7841
32. Christofk, H., Wu, N., Cantley, L. C., and Asara, J. (2011)Proteomic screening method for phosphopeptide motif bindingproteins using peptide libraries. J. Proteome Res. 10, 4158–4164
33. Marmiroli, S., Bertacchini, J., Beretti, F., Cenni, V., Guida, M.,De Pol, A., Maraldi, N. M., and Lattanzi, G. (2009) A-type laminsand signaling: the PI 3-kinase/Akt pathway moves forward. J.Cell. Physiol. 220, 553–561
34. Lin, H. K., Wang, G., Chen, Z., Teruya-Feldstein, J., Liu, Y.,Chan, C. H., Yang, W. L., Erdjument-Bromage, H., Nakayama,K. I., Nimer, S., Tempst, P., and Pandolfi, P. P. (2009) Phospho-rylation-dependent regulation of cytosolic localization and on-cogenic function of Skp2 by Akt/PKB. Nat. Cell Biol. 4, 420–432
35. Brunet, A., Kanai, F., Stehn, J., Xu, J., Sarbassova, D., Frangioni,J. V., Dalal, S. N., DeCaprio, J. A., Greenberg, M. E., and Yaffe,M. B. (2002) 14-3-3 transits to the nucleus and participates indynamic nucleocytoplasmic transport. J. Cell Biol. 156, 817–828
36. Meek, S. E., Lane, W. S., and Piwnica-Worms, H. (2004)Comprehensive proteomic analysis of interphase and mitotic14-3-3-binding proteins. J. Biol. Chem. 279, 32046–32054
37. Jung, H. J., Coffinier, C., Choe, Y., Beigneux, A. P., Davies, B. S.,Yang, S. H., Barnes, R. H., 2nd, Hong, J., Sun, T., Pleasure, S. J.,Young, S. G., and Fong, L. G. (2012) Regulation of prelamin Abut not lamin C by miR-9, a brain-specific microRNA. Proc. Natl.Acad. Sci. U. S. A. 109, E423–E431
38. Barth, S., Glick, D., and Macleod, K. F. (2010) Autophagy: assaysand artifacts. J. Pathol. 221, 117–124
39. Cao, K., Graziotto, J. J., Blair, C. D., Mazzulli, J. R., Erdos, M. R.,Krainc, D., and Collins, F. S. (2011) Rapamycin reverses cellularphenotypes and enhances mutant protein clearance in Hutchin-son-Gilford progeria syndrome cells. Sci. Transl. Med. 29, 89ra58
40. Graziotto, J. J., Cao, K., Collins, F. S., and Krainc, D. (2012)Rapamycin activates autophagy in Hutchinson-Gilford progeriasyndrome: implications for normal aging and age dependentneurodegenerative disorders. Autophagy 8, 147–151
41. Cenni, V., Capanni, C., Columbaro, M., Ortolani, M., D’Apice,M. R., Novelli, G., Fini, M., Marmiroli, S., Scarano, E., Maraldi,N. M., Squarzoni, S., Prencipe, S., and Lattanzi, G. (2011)Autophagic degradation of farnesylated prelamin A as a thera-peutic approach to laminlinked progeria. Eur. J. Histochem. 55,e36
42. Wan, X., Harkavy, B., Shen, N., Grohar, P., and Helman, L. J.(2007) Rapamycin induces feedback activation of Akt signalingthrough an IGF-1R-dependent mechanism. Oncogene 26, 1932–4043.
43. Weber, K., Plessmann, U., and Traub, P. (1989) Maturation ofnuclear lamin A involves a specific carboxy-terminal trimming,
which removes the polyisoprenylation site from the precursor;implications for the structure of the nuclear lamina. FEBS Lett.257, 411–414
44. Corrigan, D. P., Kuszczak, D., Rusinol, A. E., Thewke, D. P.,Hrycyna, C. A., Michaelis, S., and Sinensky, M. S. (2005)Prelamin A endoproteolytic processing in vitro by recombinantZmpste24. Biochem. J. 387, 129–138
45. Davies, B. S., Coffinier, C., Yang, S. H., Barnes, R. H., II, Jung,H. J., Young, S. G., and Fong, L. G. (2011) Investigating thepurpose of prelamin A processing. Nucleus 2, 4–9
46. Coffinier, C., Jung, H. J., Li, Z., Nobumori, C., Yun, U. J., Farber,E. A., Davies, B. S., Weinstein, M. M., Yang, S. H., Lammerding,J., Farahani, J. N., Bentolila, L. A., Fong, L. G., and Young, S. G.(2010) Direct synthesis of lamin A, bypassing prelamin a pro-cessing, causes misshapen nuclei in fibroblasts but no detectablepathology in mice. J. Biol. Chem. 285, 20818–20826
47. Stewart, C. L., Roux, K. J., and Burke, B. (2007) Blurring theboundary: the nuclear envelope extends its reach. Science 318,1408–1412
48. Eriksson, M., Brown, W. T., Gordon, L. B., Glynn, M. W., Singer,J., Scott, L., Erdos, M. R., Robbins, C. M., Moses, T. Y., Berglund,P., Dutra, A., Pak, E., Durkin, S., Csoka, A. B., Boehnke, M.,Glover, T. W., and Collins, F. S. (2003) Recurrent de novo pointmutations in lamin A cause Hutchinson-Gilford progeria syn-drome. Nature 423, 293–298
49. Bergo, M. O., Gavino, B., Ross, J., Schmidt, W. K., Hong, C.,Kendall, L. V., Mohr, A., Meta, M., Genant, H., Jiang, Y., Wisner,E. R., Van Bruggen, N., Carano, R. A., Michaelis, S., Griffey,S. M., and Young, S. G. (2002) Zmpste24 deficiency in micecauses spontaneous bone fractures, muscle weakness, and aprelamin A processing defect. Proc. Natl. Acad. Sci. U. S. A. 99,13049–13054
50. Okumura, K., Hosoe, Y., and Nakajima, N. (2004) c-Jun and Sp1family are critical for retinoic acid induction of the lamin A/Cretinoic acid-responsive element. Biochem. Biophys. Res. Commun.320, 487–492
51. Muralikrishna, B., and Parnaik, V. K. (2001) SP3 and AP-1mediate transcriptional activation of the lamin A proximalpromoter. Eur. J. Biochem. 268, 3736–3743
52. Mirandola, P., Sponzilli, I., Gobbi, G., Marmiroli, S., Rinaldi, L.,Binazzi, R., Piccari, G., Ramazzotti, G., Gaboardi, G. C., Cocco,L., and Vitale, M. (2006) Anticancer agents sensitize osteosar-coma cells to TNF-related apoptosis-inducing ligand down-modulating IAP family proteins. Int. J. Oncol. 28, 127–133
53. Davies, B. S., Fong, L. G., Yang, S. H., Coffinier, C., and Young,S. G. (2009) The posttranslational processing of prelamin A anddisease. Annu. Rev. Genomics Hum. Genet. 10, 153–174
Received for publication September 10, 2012.Accepted for publication February 4, 2013.
2155AKT REGULATES PRELAMIN A DEGRADATION




















