
Role of Akt/protein kinase B in metabolism
8
TRENDS in Endocrinology & Metabolism Vol.13 No.10 December 2002 http://tem.trends.com 1043-2760/02/$ – see front matter © 2002 Elsevier Science Ltd. All rights reserved. PII: S1043-2760(02)00662-8 444 Review 61 Tian, X. et al. (2001) The development of a potential single photon emission computed tomography (SPECT) imaging agent for the corticotropin-releasing hormone receptor type 1. Bioorg. Med. Chem. Lett. 11, 331–333 62 Gold, P.W. and Chrousos, G.P. (2002) Organization of the stress system and its dysregulation in melancholic and atypical depression: high versus low CRH/NE States. Mol. Psychiatry 7, 254–275 63 Webster, E.L. et al. (1996) In vivo and in vitro characterization of antalarmin, a nonpeptide corticotropin-releasing hormone (CRH) receptor antagonist: suppression of pituitary ACTH release and peripheral inflammation. Endocrinology 137, 5747–5750 64 Deak, T. et al. (1999) The impact of the nonpeptide corticotropin-releasing hormone antagonist antalarmin on behavioral and endocrine responses to stress. Endocrinology 140, 79–86 65 Habib, K.E. et al. (2000) Oral administration of a CRH receptor antagonist significantly attenuates behavioral, neuroendocrine, and autonomic responses to stress in primates. Proc. Natl. Acad. Sci. U. S. A. 10, 6079–6084 66 Wong, M.L. et al. (1999) Chronic administration of the non-peptide CRH type 1 receptor antagonist antalarmin does not blunt hypothalamic–pituitary–adrenal axis responses to acute immobilization stress. Life Sci. 65, 53–58 67 Bornstein, S.R. et al. (1998) Chronic effects of a nonpeptide corticotropin- releasing hormone type 1-receptor antagonist on pituitary-adrenal function, and body weight and metabolic regulation. Endocrinology 139, 1546–1555 68 Vgontzas, A.N. et al. (2002) Endocrine disorders and sleep. In Sleep Medicine (Lee-Chiong, T.L. et al., eds), pp. 477–487, Hanley & Belfus, Philadelphia, PA, USA 69 Vgontzas, A.N. et al. (2001) Middle-aged men show higher sensitivity of sleep to the arousing effects of corticotropin-releasing hormone than young men: clinical implications. J. Clin. Endocrinol. Metab. 86, 1489–1495 70 Vgontzas, A.N. et al. (2001) Chronic insomnia is associated with nyctohemeral activation of the hypothalamic–pituitary–adrenal axis: clinical implications. J. Clin. Endocrinol. Metab. 86, 3787–3794 71 Weiss, S.R.B. et al. (1986) CRF-induced seizures and behavior: interaction with amygdala kindling. Brain Res. 372, 345–351 72 Theoharides, T.C. et al. (1995) Stress-induced intracranial mast cell degranulation: a corticotropin-releasing hormone-mediated effect. Endocrinology 136, 5745–5750 73 Theoharides, T.C. et al. (1998) Corticotropin- releasing hormone induces skin mast cell degranulation and increased vascular permeability; a possible explanation for its proinflammatory effects. Endocrinology 139, 403–413 74 Roe, S.Y. et al. (1998) Evidence for the involvement of corticotropin-releasing hormone in the pathogenesis of traumatic brain injury. Eur. J. Neurosci. 10, 553–559 75 Webster, E.L. et al. (2002) Corticotropin-releasing hormone (CRH) antagonist attenuattes adjuvant-induced arthritis: role of CRH in peripheral inflammation. J. Rheumatol. 29, 1252–1261 76 Habib, K.E. et al. Marked suppression of gastric ulcerogenesis and intestinal respones to stress by a novel class of drugs. Mol. Psychol. (in press) 77 Chrousos, G.P. et al. (1998) Interactions between the hypothalamic–pituitary–adrenal axis and the female reproductive system: clinical implications. Ann. Intern. Med. 129, 229–240 78 McLean, M. and Smith, R. (1999) Corticotropin- releasing hormone in human pregnancy and parturition. Trends Endocrinol. Metab. 10, 174–178 79 Jaffe, R.B. (2001) Role of the human fetal adrenal gland in the initiation of parturition. Front. Horm. Res. 27, 75–85 80 Erickson, K. et al. (2001) Preterm birth: associated neuroendocrine, medical, and behavioral risk factors. J. Clin. Endocrinol. Metab. 86, 2544–2552 81 Chan, E.C. et al. (1998) Corticotropin-releasing hormone type 1 receptor antagonist delays parturition in sheep. Endocrinology 139, 3357–3360 82 Merke, D.P. et al. (2002) Future directions in the study and management of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Ann. Intern. Med. 136, 320–334 83 Vetter, D.E. et al. (2002) Urocortin-deficient mice show hearing impairment and increased anxiety-like behavior. Nat. Genet. 31, 363–369 84 Parkes, G.D. and May, C.N. (2000) Urocortin: a novel player in cardiac control. News Physiol. Sci. 15, 264–268 Review One of the most remarkable aspects of vertebrate physiology is the ability to maintain circulating glucose levels within a very precise range in spite of wide fluctuations in food intake. A tightly regulated system for recognizing the immediate nutritional state of the organism and the concomitant ability to respond appropriately are crucial to this adaptation. Following a meal, simple molecules are redistributed into long-term energy stores, typified by triglyceride in the adipose cell, and glycogen and protein in liver and muscle. Insulin, the secretion of which from the islets of Langerhans depends on an abundance of ingested and circulating nutrients, regulates energy metabolism following food intake by promoting the uptake and storage of glucose, amino acids and fat, whilst simultaneously antagonizing the catabolism of fuel reserves. This basic function of insulin as a signal that informs the organism of nutritional abundance appears to be well conserved among species as disparate as nematodes, fruit flies and mammals. The primary components of the insulin signal transduction pathway are also remarkably similar in invertebrates and mammals, although in mammals there has been an increase in complexity and possible redundancy (Fig. 1). This has led to complex patterns of metabolic regulation within individual cells, Role of Akt/protein kinase B in metabolism Eileen L. Whiteman, Han Cho and Morris J. Birnbaum Since its discovery more than a decade ago, the Ser/Thr kinase Akt/PKB (protein kinase B) has been recognized as being remarkably well conserved across a broad range of species and involved in a diverse array of cellular processes. Among its many roles, Akt appears to be common to signaling pathways that mediate the metabolic effects of insulin in several physiologically important target tissues. Refining our understanding of those pivotal molecular components that normally coordinate insulin action throughout the body is essential for a full understanding of insulin resistance in diabetes mellitus and ultimately the successful treatment of this disease.
Transcript of Role of Akt/protein kinase B in metabolism

TRENDS in Endocrinology & Metabolism Vol.13 No.10 December 2002
http://tem.trends.com 1043-2760/02/$ – see front matter © 2002 Elsevier Science Ltd. All rights reserved. PII: S1043-2760(02)00662-8
444 Review
61 Tian, X. et al. (2001) The development of apotential single photon emission computedtomography (SPECT) imaging agent for thecorticotropin-releasing hormone receptor type 1.Bioorg. Med. Chem. Lett. 11, 331–333
62 Gold, P.W. and Chrousos, G.P. (2002)Organization of the stress system and itsdysregulation in melancholic and atypicaldepression: high versus low CRH/NE States.Mol. Psychiatry 7, 254–275
63 Webster, E.L. et al. (1996) In vivo and in vitrocharacterization of antalarmin, a nonpeptidecorticotropin-releasing hormone (CRH) receptorantagonist: suppression of pituitary ACTHrelease and peripheral inflammation.Endocrinology 137, 5747–5750
64 Deak, T. et al. (1999) The impact of the nonpeptidecorticotropin-releasing hormone antagonistantalarmin on behavioral and endocrineresponses to stress. Endocrinology 140, 79–86
65 Habib, K.E. et al. (2000) Oral administration of aCRH receptor antagonist significantly attenuatesbehavioral, neuroendocrine, and autonomicresponses to stress in primates. Proc. Natl. Acad.Sci. U. S. A. 10, 6079–6084
66 Wong, M.L. et al. (1999) Chronic administrationof the non-peptide CRH type 1 receptorantagonist antalarmin does not blunthypothalamic–pituitary–adrenal axis responsesto acute immobilization stress. Life Sci. 65,53–58
67 Bornstein, S.R. et al. (1998) Chronic effects of anonpeptide corticotropin- releasing hormonetype 1-receptor antagonist on pituitary-adrenal
function, and body weight and metabolicregulation. Endocrinology 139, 1546–1555
68 Vgontzas, A.N. et al. (2002) Endocrine disordersand sleep. In Sleep Medicine (Lee-Chiong, T.L.et al., eds), pp. 477–487, Hanley & Belfus,Philadelphia, PA, USA
69 Vgontzas, A.N. et al. (2001) Middle-aged menshow higher sensitivity of sleep to the arousingeffects of corticotropin-releasing hormone thanyoung men: clinical implications.J. Clin. Endocrinol. Metab. 86, 1489–1495
70 Vgontzas, A.N. et al. (2001) Chronic insomnia isassociated with nyctohemeral activation of thehypothalamic–pituitary–adrenal axis: clinicalimplications. J. Clin. Endocrinol. Metab. 86,3787–3794
71 Weiss, S.R.B. et al. (1986) CRF-induced seizuresand behavior: interaction with amygdalakindling. Brain Res. 372, 345–351
72 Theoharides, T.C. et al. (1995) Stress-inducedintracranial mast cell degranulation: acorticotropin-releasing hormone-mediated effect.Endocrinology 136, 5745–5750
73 Theoharides, T.C. et al. (1998) Corticotropin-releasing hormone induces skin mast celldegranulation and increased vascular permeability;a possible explanation for its proinflammatoryeffects. Endocrinology 139, 403–413
74 Roe, S.Y. et al. (1998) Evidence for theinvolvement of corticotropin-releasing hormone inthe pathogenesis of traumatic brain injury. Eur. J. Neurosci. 10, 553–559
75 Webster, E.L. et al. (2002) Corticotropin-releasinghormone (CRH) antagonist attenuattes
adjuvant-induced arthritis: role of CRH inperipheral inflammation. J. Rheumatol. 29,1252–1261
76 Habib, K.E. et al. Marked suppression of gastriculcerogenesis and intestinal respones to stress bya novel class of drugs. Mol. Psychol. (in press)
77 Chrousos, G.P. et al. (1998) Interactions betweenthe hypothalamic–pituitary–adrenal axis and thefemale reproductive system: clinical implications.Ann. Intern. Med. 129, 229–240
78 McLean, M. and Smith, R. (1999) Corticotropin-releasing hormone in human pregnancy andparturition. Trends Endocrinol. Metab. 10,174–178
79 Jaffe, R.B. (2001) Role of the human fetal adrenalgland in the initiation of parturition. Front. Horm.Res. 27, 75–85
80 Erickson, K. et al. (2001) Preterm birth:associated neuroendocrine, medical, andbehavioral risk factors. J. Clin. Endocrinol.Metab. 86, 2544–2552
81 Chan, E.C. et al. (1998) Corticotropin-releasinghormone type 1 receptor antagonist delaysparturition in sheep. Endocrinology 139, 3357–3360
82 Merke, D.P. et al. (2002) Future directions in thestudy and management of congenital adrenalhyperplasia due to 21-hydroxylase deficiency.Ann. Intern. Med. 136, 320–334
83 Vetter, D.E. et al. (2002) Urocortin-deficient miceshow hearing impairment and increasedanxiety-like behavior. Nat. Genet. 31, 363–369
84 Parkes, G.D. and May, C.N. (2000) Urocortin: anovel player in cardiac control. News Physiol. Sci.15, 264–268
Review
One of the most remarkable aspects of vertebratephysiology is the ability to maintain circulatingglucose levels within a very precise range in spite ofwide fluctuations in food intake. A tightly regulatedsystem for recognizing the immediate nutritionalstate of the organism and the concomitant ability torespond appropriately are crucial to this adaptation.Following a meal, simple molecules are redistributed
into long-term energy stores, typified by triglyceridein the adipose cell, and glycogen and protein in liverand muscle. Insulin, the secretion of which from theislets of Langerhans depends on an abundance ofingested and circulating nutrients, regulates energymetabolism following food intake by promoting theuptake and storage of glucose, amino acids and fat,whilst simultaneously antagonizing the catabolismof fuel reserves. This basic function of insulin as asignal that informs the organism of nutritionalabundance appears to be well conserved amongspecies as disparate as nematodes, fruit flies and mammals.
The primary components of the insulin signaltransduction pathway are also remarkably similar ininvertebrates and mammals, although in mammalsthere has been an increase in complexity and possibleredundancy (Fig. 1). This has led to complex patternsof metabolic regulation within individual cells,
Role of Akt/protein kinase B
in metabolism
Eileen L. Whiteman, Han Cho and Morris J. Birnbaum
Since its discovery more than a decade ago, the Ser/Thr kinase Akt/PKB
(protein kinase B) has been recognized as being remarkably well conserved
across a broad range of species and involved in a diverse array of cellular
processes. Among its many roles, Akt appears to be common to signaling
pathways that mediate the metabolic effects of insulin in several
physiologically important target tissues. Refining our understanding of those
pivotal molecular components that normally coordinate insulin action
throughout the body is essential for a full understanding of insulin
resistance in diabetes mellitus and ultimately the successful treatment
of this disease.

in addition to communication among insulin-responsive tissues throughout the organism.Currently, the most commonly recognized defect ininsulin action is type 2 diabetes mellitus (T2DM),which afflicts 5% of humans. Genetic andenvironmental factors contribute to theheterogeneous etiology behind the development ofthis complex metabolic disease, in which elevatedblood glucose is a common denominator. Insulin actsto suppress glucose production in liver and to promoteglucose uptake in muscle and fat, resulting in theclearance of blood glucose to appropriate levels.
In spite of substantial effort over the past 50 years,the signaling pathways that mediate the metabolicactions of insulin remain largely undefined.Accumulating evidence implicates the insulin-activated, phosphatidylinositol 3′-kinase(PI 3K)-dependent Ser/Thr kinase Akt as a regulator ofglucose transport, glycolysis, protein synthesis,lipogenesis, glycogen synthesis, suppression ofgluconeogenesis, cell survival, determination of cell sizeand cell-cycle progression. A description of the currentmodel describing the activation of Akt is summarized inFig. 2. Here, we focus on the effects of insulin thatmight be mediated by Akt in four important targettissues: muscle, fat, liver and pancreas.
Role of Akt in muscle and adipose tissue
Glucose transportMuscle and fat, the two primary sites of insulin-dependent glucose disposal, synthesize the
insulin-sensitive glucose transporter isoform, GLUT4,which redistributes from intracellular storage vesiclesto the plasma membrane following insulin stimulation.The physiological significance of this process is a net ten- to 40-fold increase in glucose flux into the cells.There is substantial evidence that Akt plays a role indirecting GLUT4 vesicles to the plasma membrane andthus promotes glucose transport. Insulin rapidly andpersistently activates Akt in traditional target tissues.The initial observation that strongly implicated Akt asa mediator of the metabolic actions of insulin was thatAkt rendered constitutively active by membrane-targeting mimics insulin in eliciting high levels ofglucose transport and GLUT4 translocation inadipocytes in the absence of hormone [1]. Insulinstimulates the association of Akt2 with GLUT4 vesicles in rat adipocytes, and this results in thephosphorylation of several associated proteins [2],whereas the inhibition of Akt activity by interferingantibodies, substrate peptides or synthesis ofdominant-negative Akt partially blocks insulin-stimulated GLUT4 translocation in fat and musclecells [3,4]. In addition, there is much circumstantialevidence indicating that changes in Akt activitycorrelate with glucose transport. As one example, thesphingomyelin derivative ceramide dramaticallyantagonizes insulin-activated glucose transport andGLUT4 translocation in adipocytes and myotubes, andthis correlates with a 60% reduction in insulin-stimulated Akt activity [5,6]. By contrast, severalstudies have found a lack of correlation between Aktand glucose transport in insulin-responsive cells. In themost striking example, synthesis in cultured murineadipocytes of a dominant-negative Akt that suppresses80% of the endogenous kinase activity does not impairinsulin-stimulated glucose transport [7]. However, it ispossible that residual Akt activity might be sufficient tosupport insulin-stimulated glucose uptake, aninterpretation supported by parallel experiments incultured myoblasts [4]. Other instances in which Aktand glucose transport do not correlate well include theintegrin-dependent activation of PI 3K signalingpathways in adipocytes and in muscle from diabeticindividuals [8,9]. One plausible model suggests thatthe increase in Akt activity is necessary but notsufficient for stimulation of glucose transport, but thatunder the artificial conditions engendered byoverproduction of a membrane-targeted Akt the kinaseis capable of driving transport. Other signalingprocesses that have been proposed to function in parallel with Akt include the atypical protein kinase C(PKCλ/ζ) [10], p38 mitogen-activated protein kinase [11]and a novel cascade dependent on phosphorylation ofthe Cbl proto-oncogene product and recruitment of asignaling complex to caveolae [12].
Indirect regulation of glucose transport: endothelialnitric oxide synthaseNew studies suggest that the vasodilatation of bloodvessels by insulin may be integral in promoting
TRENDS in Endocrinology & Metabolism Vol.13 No.10 December 2002
http://tem.trends.com
445Review
Eileen L. Whiteman
Han Cho
Morris J. Birnbaum*
Dept Medicine andHoward Hughes MedicalInstitute, University ofPennsylvania School ofMedicine, 415 Curie Blvd,322 Clinical ResearchBuilding, Philadelphia,PA 19104, USA.*e-mail: [email protected]
TRENDS in Endocrinology & Metabolism
p85 p110
PI 3KAPS
CblCAP
CrkIIC3GTC10
GTP
IRS
PtdIns(3,4,5)P3
PDKs
GLUT4vesicle
Glucose
Insulin receptor
Insulin
TC10 effectors
Actin rearrangements
?
?
?
Caveolin
Flotillin
Caveola
P
P
P
P PP
Akt PKCλ/ζ
Fig. 1. A schematic model for insulin-stimulated glucose transport in adipocytes, depicting theinvolvement of the classic, PI 3K-dependent pathway and the proposed PI 3K-independent, alternativepathway. The activated insulin receptor catalyzes IRS tyrosine phosphorylation, leading torecruitment of downstream effectors, the most crucial for glucose transport being PI 3K. Akt and theatypical PKC isoforms have both been implicated as PtdIns(3,4,5)P3 effectors involved in GLUT4translocation, although it has been proposed that these two Ser/Thr kinases target separate GLUT4compartments. In a parallel pathway, the CAP–Cbl dimeric complex is recruited to the insulin receptor,and Cbl is also Tyr phosphorylated by the insulin receptor kinase. The CAP–Cbl complex is recruited tolipid raft domains in the plasma membrane, and phosphorylated Cbl assembles a signaling complexconsisting of the scaffolding protein CrkII, the guanine nucleotide exchange factor C3G, and the smallGTP-binding protein TC10. Activation of TC10 is proposed to mediate insulin-stimulated actinrearrangement. Abbreviations: APS, adapter protein with PH and SH2 domains; CAP, Cbl-associatedprotein; Cbl, Cas B-lineage lymphoma; GLUT4, glucose transporter type 4; IRS, insulin receptorsubstrate; PDK, phosphoinositide-dependent kinase; PI 3K, phosphatidylinositol 3′-kinase; PKC,protein kinase C; PtdIns(3,4,5)P3, phosphatidylinositol-3,4,5-trisphosphate.
glucose disposal in peripheral tissues [13]. Aktincreases nitric oxide (NO) production in thevascular endothelium by phosphorylating Ser1179and activating endothelial NO synthase; NOfacilitates vasodilatation [14]. The precise,quantitative contribution of NO generation toaugmented glucose uptake in muscle remains largely undefined.
Protein synthesisInsulin promotes protein synthesis in muscle and fatcells by stimulating the initiation and elongationsteps in protein translation. There is evidence thatAkt modulates the activity of translationalcomponents (reviewed in [15,16]). Constitutivelyactive Akt increases protein synthesis in L6 musclecells and 3T3-L1 adipocytes [17,18], and a putativedominant-inhibitory Akt mutant protein blocksinsulin-stimulated protein synthesis in 3T3-L1adipocytes [7]. In addition, Akt has been shown toincrease system A amino acid transport inL6 myotubes [18].
A limiting initiation factor, the 7-methylguanosinecap-binding protein eIF-4E, recruits mRNAto the 40S ribosomal subunit and, in quiescent
cells, is sequestered by the 4E-binding protein(4E-BP1)/PHAS-I. Insulin stimulatesphosphorylation of 4E-BP1 on multiple residues: onemodel posits that Akt phosphorylates and activatesthe 4E-BP1 kinase, mammalian target of rapamycin(mTOR), on Ser2448 [19]. This phosphorylationreleases eIF-4E and increases translation initiation.Dominant-inhibitory Akt partially blocks 4E-BP1phosphorylation by insulin in L6 muscle cells, andconstitutively active, membrane targeted Akt resultsin robust phosphorylation of this protein [20,21]. Inaddition, the guanine nucleotide exchange factor foreIF2, eIF-2B, is phosphorylated on its ε subunit atSer540 and thus inactivated by glycogen synthasekinase-3 (GSK-3) [22]. Akt-mediated phosphorylationand inactivation of GSK-3 on Ser23 (GSK-3α) andSer9 (GSK-3β) increases the exchange activity ofeIF-2B and promotes the recruitment of initiatortRNA to the 40S ribosome.
The phosphorylation of ribosomal S6 protein byp70S6K on five serine residues enhances translation ofa select population of mRNAs containing 5′-TOP(terminal oligopyrimidine) regions by increasing theinteraction of the 40S ribosomal subunit with mRNA.In addition, p70S6K has been implicated in blockingthe activity of the inhibitory eEF2 kinase, and thisresults in the activation of eEF2, the enzyme thatcatalyzes the translocation of both the mRNA andpeptidyl-tRNA along the 80S ribosome [23].Production of a constitutively active Akt leads toactivation of p70S6K and an increase in S6 proteinphosphorylation, although the physiologicalrelevance of Akt in this process has been questioned [21,24]. In addition, genetic experimentsin Drosophila have shown that the activity of S6kinase does not depend on the presence of a functionalAkt gene [25]. One possibility is that, under someconditions, mammalian p70S6K can be activatedindependently of Akt, but that in other cell typesselected agonists require Akt for stimulation of theenzyme. This stimulation could be mediated by thephosphorylation and activation of mTOR by Akt.
TRENDS in Endocrinology & Metabolism Vol.13 No.10 December 2002
http://tem.trends.com
446 Review
PH
Kinase
TRENDS in Endocrinology & Metabolism
+ Insulin
PP
PP
PDK1
PDK2
Akt/PKB
P PP
PP
PP
PP
PP
Kinase
PH
PH
Kinase
PH
Kinase
PP
PP
P PP
PH
Kinase
PH
Kinase
Extracellular
Cytoplasm
P
PIRS
PI 3K
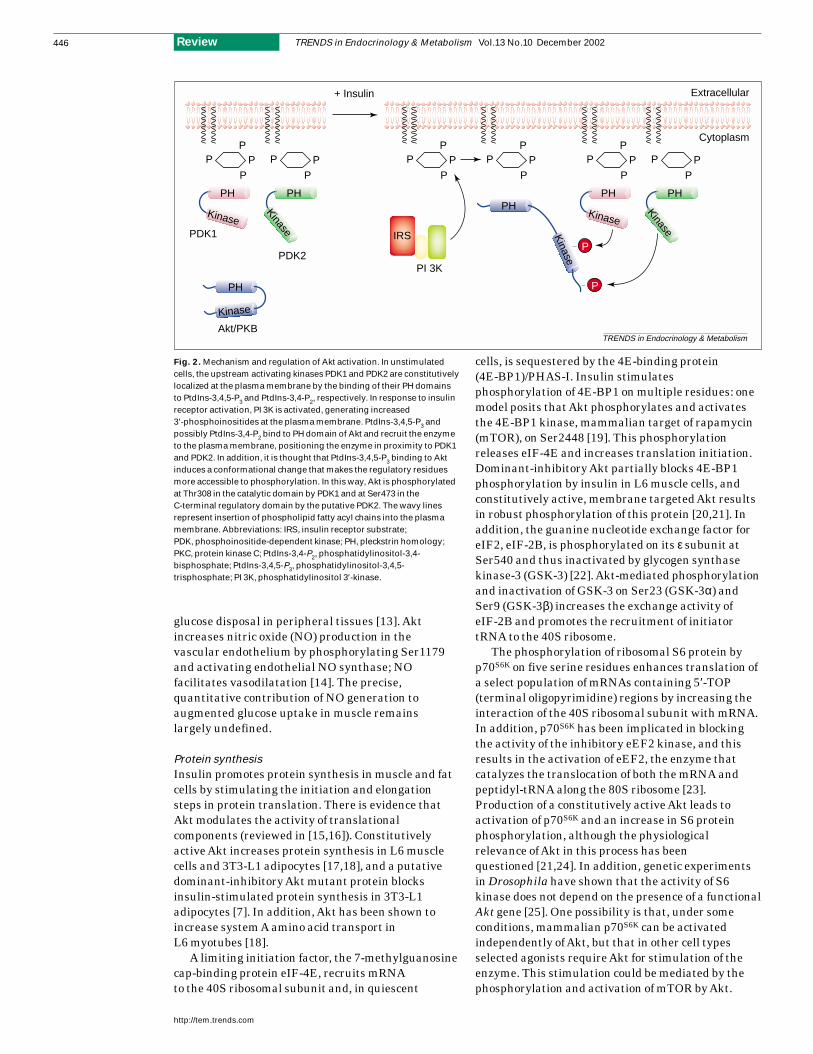
Fig. 2. Mechanism and regulation of Akt activation. In unstimulatedcells, the upstream activating kinases PDK1 and PDK2 are constitutivelylocalized at the plasma membrane by the binding of their PH domainsto PtdIns-3,4,5-P3 and PtdIns-3,4-P2, respectively. In response to insulinreceptor activation, PI 3K is activated, generating increased3′-phosphoinositides at the plasma membrane. PtdIns-3,4,5-P3 andpossibly PtdIns-3,4-P2 bind to PH domain of Akt and recruit the enzymeto the plasma membrane, positioning the enzyme in proximity to PDK1and PDK2. In addition, it is thought that PtdIns-3,4,5-P3 binding to Aktinduces a conformational change that makes the regulatory residuesmore accessible to phosphorylation. In this way, Akt is phosphorylatedat Thr308 in the catalytic domain by PDK1 and at Ser473 in theC-terminal regulatory domain by the putative PDK2. The wavy linesrepresent insertion of phospholipid fatty acyl chains into the plasmamembrane. Abbreviations: IRS, insulin receptor substrate;PDK, phosphoinositide-dependent kinase; PH, pleckstrin homology;PKC, protein kinase C; PtdIns-3,4-P2, phosphatidylinositol-3,4-bisphosphate; PtdIns-3,4,5-P3, phosphatidylinositol-3,4,5-trisphosphate; PI 3K, phosphatidylinositol 3′-kinase.

The understanding of this pathway has beencomplicated by the recent appreciation that aminoacids are potent regulators of p70S6K, leading to a re-evaluation of the physiological role ofinsulin-regulated, mTOR-dependent events.
Lipogenesis and antilipolysisInsulin promotes the rate at which incoming glucoseis converted to fatty acids, and Akt-mediatedinhibition of GSK-3 might contribute to thisphenomenon. GSK-3 phosphorylates ATP citratelyase, an enzyme that catalyzes the conversion ofcitrate to acetyl-coA in the cytosol, on Thr446 andSer450, and decreases its activity [26]. In addition,Akt has been shown to increase the transcription offatty acid synthase, an enzyme that catalyzesseveral steps in the conversion of malonyl-CoAand acetyl-CoA to long-chain fatty acids [27]. In fact,the presence of constitutively active Akt results inhigh levels of lipogenesis in quiescent 3T3-L1adipocytes [28].
Insulin also blocks lipolysis in adipocytes byinhibiting hormone-sensitive lipase (HSL). HSL isactivated by protein kinase A (PKA)-mediatedphosphorylation, and PKA activity is increased bycAMP. Akt stimulates the cyclic nucleotidephosphodiesterase PDE-3B by phosphorylation onSer273, and activated PDE-3B reduces cAMPconcentrations [29,30]. By contrast, recentexperiments using a putative inhibitor of Akt havechallenged the idea that the kinase is involved ineither the activation of PDE-3B or the inhibition oflipolysis [31].
Glycogen synthesisIn humans, >70% of insulin-stimulated whole-bodyglucose uptake is ultimately stored as glycogen inskeletal muscle [32]. Insulin promotes glycogensynthesis both by increasing rates of glucosetransport and by activating glycogen synthase (GS),which adds activated glucosyl groups to growingpolysaccharide chains and thus catalyzes the finalstep in glycogen synthesis. The regulation of GS iscomplex and involves allosteric activators,translocation of GS to the plasma membrane in thepresence of glucose metabolites and insulin,inhibitory phosphorylation on serine residues bydifferent kinases, and activating dephosphorylationby the Ser/Thr phosphatase PP1. The relativeimportance of these mechanisms varies among celltypes. An Akt-dependent mechanism contributesdirectly to the activity of GS: GSK-3 phosphorylatesfour serine residues on the C-terminal portion ofGS (sites 3a, 3b, 3c, and 4), and Akt-mediatedinactivation of GSK-3 contributes to a reduction inthe net phosphorylation of GS. Synthesis of a GSK-3βmutant S9A, which is insensitive to Akt, in adipocytesresults in suppression of insulin-stimulated GSactivity [33]. Similarly, the presence of constitutivelyactive Akt in L6 muscle cells significantly blocks
GSK-3 activity and leads to GS activation in theabsence of insulin, whereas dominant-inhibitory Aktresults in 50% inhibition of insulin-stimulatedGS activation in these cells [17,20]. Nonetheless, theabsolute contribution of Akt to the regulation ofglycogen synthase remains uncertain because severalsignaling molecules that are not influenced by Aktmodulate this process.
Role of Akt in liver
Glucose disposal into muscle and adipose tissueaccounts for only part of the insulin-dependentreduction in circulating glucose levels becausesuppression of hepatic glucose output alsocontributes significantly. The kidney and liverproduce most of the glucose for extrahepaticdemands, but only the liver responds acutely toinsulin by reducing glucose output.
Glucose, glucose metabolites and insulincollectively reduce glycogenolysis by inhibition ofphosphorylase a activity and subsequentincorporation of UDP-glucose into glycogen by GS.Thus, the balance is shifted synergistically fromglycogen breakdown to synthesis. One mechanism bywhich Akt might contribute to the insulin-mediatedsuppression of glycogenolysis is by driving glycogensynthesis, and this effect is probably accomplished inthe liver by mechanisms similar to those seen inmyocytes and adipocytes. Nevertheless, it is unlikelyto account fully for the complex regulation ofglycogenolysis and glycogen synthesis.
Insulin can acutely suppress glucocorticoid- and cAMP-stimulated glucose production bydownregulating the enzymes that control therate-limiting steps of gluconeogenesis, whichaccounts for much of the glucose production after amore prolonged fast. Phosphoenolpyruvatecarboxykinase (PEPCK) catalyzes the earlycommitted step for gluconeogenesis, whereas G6Pase(glucose-6-phosphatase) regulates the terminal stepcommon to gluconeogenesis and glycogenolysis beforeglucose release from the liver. No allostericmodification has been ascribed to PEPCK andG6Pase. Instead, PEPCK and G6Pase appear to bepositively regulated principally at the level oftranscription by cAMP or glucocorticoid signaling.The insulin-mediated downregulation of PEPCKtranscription has been mapped within the promoterto a cis-acting element named the insulin-responsiveelement (IRE). IRE is conserved in many insulin-responsive genes, including those encoding G6Paseand the insulin-like growth factor-binding protein 1(IGFBP-1), and Akt has been proposed to link thesignal from the insulin receptor to the IRE. Theability of insulin to suppress PEPCK transcription issensitive to PI 3K inhibition [34,35]. Nonetheless,experiments in which genes encoding constitutivelyactive and inactive Akt mutants were overexpressedin liver-derived cell lines have led to conflictingconclusions. [10,36,37].
TRENDS in Endocrinology & Metabolism Vol.13 No.10 December 2002
http://tem.trends.com
447Review

In spite of these inconsistencies, the pursuit ofAkt as a transcriptional regulator of PEPCK hasbeen sustained by the elucidation of anevolutionarily conserved pathway by which Aktantagonizes positive regulation of transcription.Genetic screens for modifiers of longevity and dauerformation in Caenorhabditis elegans have led to thedelineation of an insulin-like signaling cascadeinvolving homologs of the insulin receptor, PI 3K, Akt and the forkhead transcription factor,DAF-16 [38]. Closely related mammalian homologs of DAF-16, such as FKHR (Foxo1),FKHRL1 (Foxo3) and AFX (Foxo4), can benegatively regulated directly by Aktphosphorylation at three crucial residues [39].Mutagenesis of these sites results in the loss ofAkt-dependent phosphorylation, and an inability of Akt to antagonize FKHR-dependent transcriptionof an IGFBP-1 IRE reporter construct [40].Phosphorylation of FKHR by Akt leads to theredistribution of forkhead transcription factors fromthe nucleus to the cytoplasm, although there mightbe other mechanisms for DAF-16 function. Recentdata have more directly implicated Akt regulatedFKHR in mammalian cells as a possibletranscriptional regulator of the genes encodingG6Pase, and possibly PEPCK, although otherinvestigators have questioned this conclusion,specifically with regard to PEPCK [41–44].
Most recently, a novel transcriptional coactivatorPGC-1 (peroxisome proliferative activated receptor γcoactivator) has been implicated as a globalregulator of gluconeogenesis during fasting [45].Whether Akt might antagonize the PGC-1-mediatedgluconeogenic program in liver remains speculative,but in heart, constitutive activation of Akt leads to a downregulation of PGC-1 levels, providing thefirst suggestion that Akt might modulate PGC-1function [46].
Role of Akt in pancreas
Recent work utilizing genetically modified murinemodels is consistent with the notion that insulinresistance alone is insufficient to lead to diabetesmellitus, which in humans is tightly associated withrelative or absolute failure of insulin secretion.Experiments from several laboratories havesuggested that the insulin-signaling pathwaypositively regulates pancreatic β-cell growth and/orfunction, and thus Akt has become an attractivecandidate as a crucial regulator of insulin secretorycapacity. For example, insulin receptor substrate 2(IRS-2)-deficient mice develop diabetes as a result ofreduction in β-cell mass, and Akt is activateddownstream of the IRS family of proteins [47].Consistent with this idea, pancreatic regenerationis associated with an increase in IRS-2 andactivated Akt in proliferating duct cells [48]. Akt isalso synthesized in β cells, and is activated byinsulin-like growth factor I (IGF-I) in β-cell lines [49]. Lastly, overexpression of active Akt1 inβ cells of transgenic mice leads to a significantexpansion of β-cell mass, owing to an increase inboth cell size and number [50].
In vivo models
An invariant feature of T2DM is insulin resistance;that is, an inability of maximally effectiveconcentrations of insulin to stimulate appropriateglucose uptake in muscle and to suppress hepaticglucose output. Identification of the precise molecular
TRENDS in Endocrinology & Metabolism Vol.13 No.10 December 2002
http://tem.trends.com
448 Review
TRENDS in Endocrinology & Metabolism
Fig. 3. A family of highly related mammalian Akt paralogs. A total ofthree Akt isoforms (Akt 1, 2, 3 or PKB α, β, γ), each transcribed from adistinct gene, have been identified in humans and rodents. The threemouse isoforms aligned here share 74% sequence identity andadditional 12% amino acid similarity. Much of the small sequencedivergence between the isoforms lies between the PH domain and theprotein kinase domain, conserved among all isoforms, and acrossspecies as distinct as Dictyostelium. The L residue (asterisk) stabilizesATP for catalysis. Akt is constitutively phosphorylated at residuesSer124 and Thr450 (in Akt1). However, phosphorylation of Thr308 (red)within the catalytic loop and Ser473 (purple) in the C-terminalregulatory region are highly regulated, and are required for fullactivation of Akt. The PH, kinase and extension domains were identifiedby Conserved Domain Database Search (NCBI). Abbreviation:PH, pleckstrin homology.

sites that are responsible for insulin resistance is achallenge of substantial clinical importance, andnumerous candidates have been proposed. Variousstudies assessing a possible link betweendysregulation of Akt and the insulin resistance ofT2DM have resulted in inconsistent findings.Although one study reported normal Akt activity evenin the presence of reduced PI 3K activation in musclesobtained from diabetic patients [9], other experimentshave demonstrated a reduction in Akt activity inadipocytes [51] and muscle [52] obtained frompatients with T2DM. Such confounding observationshave been recapitulated in rodent models of obesityand insulin resistance [53,54]. Thus, evidenceindicating that impairment in Akt activation has amajor role in the etiology of spontaneous diabetesmellitus is currently absent, but this is notinconsistent with the well-appreciated heterogeneousetiology of diabetes in the general population.
Surprisingly, the result of targeted disruption inAkt2 in mice is a phenotype bearing a strikingresemblance to human impaired glucose tolerance [55].Three highly similar paralogs of Akt (Akt1, Akt2 andAkt3) comprise a family of Akt proteins known to besynthesized in mammals (Fig. 3). Akt2 is highlyexpressed in the insulin-responsive tissues, muscleand fat. Akt2-knockout mice display a severeimpairment in insulin-stimulated whole-bodyglucose disposal, which is associated withinsulin-resistant glucose output by the liver andreduced glucose uptake in muscle and fat. It ispossible that the other Akt isoforms or otherPI 3K-dependent enzymes are responsible for theresidual transport. Although β-cell function was notdirectly examined for defects in glucose sensitivity orsecretion in the Akt2 study, the glucose intoleranceimplicates relative β-cell dysfunction. Severalimportant caveats should be considered wheninterpreting the phenotype of Akt2-knockout mice or other genetic models of insulin resistance.
Most importantly, although these data indicate quiteclearly that Akt2 is required for normal glucosehomeostasis, they do not prove that the kinase actsas an obligate insulin signaling intermediate in acell-autonomous manner. An alternative explanationmight be that Akt2 regulates the secretion of acrucial circulating regulatory molecule [56]. Yetanother mechanism for the observed Akt2-nullphenotype might be that the encoded kinase affectsthe synthesis of another protein, which is itself arate-determining signaling intermediate.Nonetheless, the results of experiments usingAkt2-knockout mice focus in on this enzyme as themost downstream signaling molecule shown thus farto be required for the metabolic actions of insulin.
Interestingly, Akt1-knockout mice display noobvious indication of insulin resistance, revealingthat Akt isoforms are assigned distinct physiologicalroles. Akt1-deficient mice display growth reductionbut no metabolic defects [57,58]. This suggests aprincipal role for Akt1 in transmitting thegrowth-promoting signals triggered by IGF.
Conclusions
The original realization of the role of Akt as amediator of the metabolic actions of insulin derivedfrom a unique convergence of several seeminglyindependent lines of investigation. At the time, it wasknown that PI 3K was an obligate intermediate in theinsulin pathway, but Akt was an unrelated ‘orphan’proto-oncogene product and protein kinase. Thedemonstration that Akt was activated by growthfactors in a PI 3K-dependent manner led to theseminal experiments described above examining Aktin insulin-responsive tissues (Fig. 4). In spite of this,biochemical studies failed to establish definitively anecessary role for Akt in insulin action, and this hasonly come as a result of genetic approaches. Atpresent, Akt stands as the furthest knowndownstream component of the insulin-signalingpathway that leads to metabolic regulation. Eventhough there are no compelling data indicating adefect in Akt linked to T2DM, the diabetes-likephenotype of Akt2-null mice will focus considerableattention on the identification of new members of thispathway and their rapid evaluation as T2DMcandidate genes. This is of paramount importance,because, in recent years, the number of pathwaysactivated by insulin has been rapidly expanding,with, until now, little appreciation of which ones linkto physiological targets. Moreover, the ratherdivergent physiological roles of each of the Aktisoforms, at least as defined by knockoutexperiments, raise the hope that future therapeuticapproaches targeting Akt2 or other members of thepathway might achieve sufficient specificity to provepractical. Again, this issue becomes exceedinglyimportant when one considers modifying the activityof a target with the protean signaling potential of theAkt family.
TRENDS in Endocrinology & Metabolism Vol.13 No.10 December 2002
http://tem.trends.com
449Review
TRENDS in Endocrinology & Metabolism
Liver Muscle
AdipocytePancreas
Gluconeogenesis
Glycogenesis
Glycogenolysis
Glucose transport
Glycogenesis
Protein synthesis
β-cell growth
Insulin secretion
Glucose transport
Protein synthesis
Lipogenesis
Lipolysis
Akt
Fig. 4. Summary of Akt action in insulin-responsive metabolictissues.
Acknowledgements
E.L.W. is supported by aHoward Hughespredoctoral fellowship forthe biological sciences.M.J.B. is the recipient ofNIH grants DK56886 andDK49210.

References
1 Kohn, A.D. et al. (1996) Expression of aconstitutively active Akt Ser/Thr kinase in 3T3-L1adipocytes stimulates glucose uptake and GLUT4translocation. J. Biol. Chem. 271, 31372–31378
2 Calera, M.R. et al. (1998) Insulin increases theassociation of Akt-2 with Glut4-containingvesicles. J. Biol. Chem. 273, 7201–7204
3 Hill, M.M. et al. (1999) A role for protein kinaseB/Akt2 in insulin-stimulated GLUT4translocation in adipocytes. Mol. Cell. Biol. 19,7771–7781
4 Wang, Q. et al. (1999) Protein kinase B/Aktparticipates in GLUT4 translocation by insulin inL6 myoblasts. Mol. Cell. Biol. 19, 4008–4018
5 Summers, S.A. et al. (1998) Regulation ofinsulin-stimulated glucose transporter GLUT4translocation and Akt kinase activity byceramide. Mol. Cell. Biol. 18, 5457–5464
6 Hajduch, E. et al. (2001) Ceramide impairs theinsulin-dependent membrane recruitment ofprotein kinase B leading to a loss in downstreamsignalling in L6 skeletal muscle cells.Diabetologia 44, 173–183
7 Kitamura, T. et al. (1998) Requirement foractivation of the serine-threonine kinase Akt(protein kinase Bβ) in insulin stimulation ofprotein synthesis but not of glucose transport.Mol. Cell. Biol. 18, 3708–3717
8 Guilherme, A. and Czech, M.P. (1998) Stimulationof IRS-1-associated phosphatidylinositol 3-kinaseand Akt/protein kinase B but not glucosetransport by β1-integrin signaling in ratadipocytes. J. Biol. Chem. 273, 33119–33122
9 Kim, Y.B. et al. (1999) Normal insulin-dependentactivation of Akt/protein kinase B, withdiminished activation of phosphoinositide3-kinase, in muscle in type 2 diabetes. J. Clin.Invest. 104, 733–741
10 Kotani, K. et al. (1998) Requirement of atypicalprotein kinase Cγfor insulin stimulation ofglucose uptake but not for Akt activation in3T3-L1 adipocytes. Mol. Cell. Biol. 18, 6971–6996
11 Sweeney, G. et al. (1999) An inhibitor of p38mitogen-activated protein kinase preventsinsulin-stimulated glucose transport but notglucose transporter translocation in 3T3-L1adipocytes and L6 myotubes. J. Biol. Chem. 274,10071–10078
12 Baumann, C.A. et al. (2000) CAP defines a secondsignalling pathway required for insulin-stimulatedglucose transport. Nature 407, 202–207
13 Shankar, R.R. et al. (2000) Mice with genedisruption of both endothelial and neuronal nitricoxide synthase exhibit insulin resistance.Diabetes 49, 684–687
14 Fulton, D. et al. (1999) Regulation ofendothelium-derived nitric oxide production bythe protein kinase Akt. Nature 399, 597–601
15 Shah, O.J. et al. (2000) 4E-BP1 and S6K1:translational integration sites for nutritional andhormonal information in muscle. Am. J. Physiol.Endocrinol. Metab. 279, E715–E729
16 Gingras, A.C. et al. (2001) Regulation oftranslation initiation by FRAP/mTOR. Genes Dev.15, 807–826
17 Ueki, K. et al. (1998) Potential role of proteinkinase B in insulin-induced glucose transport,glycogen synthesis, and protein synthesis. J. Biol.Chem. 273, 5315–5322
18 Hajduch, E. et al. (1998) Constitutive activation ofprotein kinase Bα by membrane targeting
promotes glucose and system A amino acidtransport, protein synthesis, and inactivation ofglycogen synthase kinase 3 in L6 muscle cells.Diabetes 47, 1006–1013
19 Nave, B.T. et al. (1999) Mammalian target ofrapamycin is a direct target for protein kinase B:identification of a convergence point for opposingeffects of insulin and amino-acid deficiency onprotein translation. Biochem. J. 344, 427–431
20 Takata, M. et al. (1999) Requirement for Akt(protein kinase B) in insulin-induced activation ofglycogen synthase and phosphorylation of 4E-BP1(PHAS-1). J. Biol. Chem. 274, 20611–20618
21 Kohn, A.D. et al. (1998) Construction andcharacterization of a conditionally active versionof the serine/threonine kinase Akt. J. Biol. Chem.273, 11937–11943
22 Welsh, G.I. and Proud, C.G. (1993) Glycogensynthase kinase-3 is rapidly inactivated inresponse to insulin and phosphorylateseukaryotic initiation factor eIF-2B. Biochem. J.294, 625–629
23 Redpath, N.T. et al. (1996) Regulation oftranslation elongation factor-2 by insulin via arapamycin-sensitive signalling pathway.EMBO J. 15, 2291–2297
24 Dufner, A. et al. (1999) Protein kinase Blocalization and activation differentially affect S6kinase 1 activity and eukaryotic translationinitiation factor 4E-binding protein 1phosphorylation. Mol. Cell. Biol. 19, 4525–4534
25 Radimerski, T. et al. (2002) dS6K-regulated cellgrowth is dPKB/dPI(3)K-independent, butrequires dPDK1. Nat. Cell Biol. 4, 251–255
26 Potapova, I.A. et al. (2000) Phosphorylation ofrecombinant human ATP:citrate lyase bycAMP-dependent protein kinase abolisheshomotropic allosteric regulation of the enzyme bycitrate and increases the enzyme activity. Allostericactivation of ATP:citrate lyase by phosphorylatedsugars. Biochemistry 39, 1169–1179
27 Wang, C.N. et al. (1998) Effects of cell-permeableceramides and tumor necrosis factor-α on insulinsignaling and glucose uptake in 3T3-L1adipocytes. Diabetes 47, 24–31
28 Kohn, A.D. et al. (1996) Akt, a pleckstrin homologydomain containing kinase, is activated primarilyby phosphorylation. J. Biol. Chem. 271,21920–21926
29 Kitamura, T. et al. (1999) Insulin-inducedphosphorylation and activation of cyclic nucleotidephosphodiesterase 3B by the serine-threoninekinase Akt. Mol. Cell. Biol. 19, 6286–6296
30 Wijkander, J. et al. (1998) Insulin-inducedphosphorylation and activation ofphosphodiesterase 3B in rat adipocytes: possiblerole for protein kinase B but not mitogen-activated protein kinase or p70 S6 kinase.Endocrinology 139, 219–227
31 Smith, U. et al. (2000) PKB inhibition preventsthe stimulatory effect of insulin on glucosetransport and protein translocation but not theantilipolytic effect in rat adipocytes.Biochem. Biophys. Res. Commun. 268, 315–320
32 Shulman, G.I. et al. (1990) Quantitation of muscleglycogen synthesis in normal subjects andsubjects with non-insulin-dependent diabetes by13C nuclear magnetic resonance spectroscopy.N. Engl. J. Med. 322, 223–228
33 Summers, S.A. et al. (1999) The role of glycogensynthase kinase 3β in insulin-stimulated glucosemetabolism. J. Biol. Chem. 274, 17934–17940
34 Gabbay, R.A. et al. (1996) Insulin regulation ofphosphoenolpyruvate carboxykinase geneexpression does not require activation of theRas/mitogen-activated protein kinase signalingpathway. J. Biol. Chem. 271, 1890–1897
35 Sutherland, C. et al. (1995) Phosphatidylinositol3-kinase, but not p70/p85 ribosomal S6 proteinkinase, is required for the regulation ofphosphoenolpyruvate carboxykinase (PEPCK)gene expression by insulin. Dissociation ofsignaling pathways for insulin and phorbol esterregulation of PEPCK gene expression. J. Biol.Chem. 270, 15501–15506
36 Liao, J. et al. (1998) Activation of protein kinaseB/Akt is sufficient to repress the glucocorticoidand cAMP induction of phosphoenolpyruvatecarboxykinase gene. J. Biol. Chem. 273,27320–27324
37 Agati, J.M. et al. (1998) Assessment of the roles of mitogen-activated protein kinase,phosphatidylinositol 3-kinase, protein kinase B,and protein kinase C in insulin inhibition ofcAMP-induced phosphoenolpyruvatecarboxykinase gene transcription. J. Biol. Chem.273, 18751–18759
38 Paradis, S. and Ruvkun, G. (1998) CaenorhabditiselegansAkt/PKB transduces insulin receptor-likesignals from AGE-1 PI3 kinase to the DAF-16transcription factor. Genes Dev. 12, 2488–2498
39 Nakae, J. et al. (1999) Insulin stimulatesphosphorylation of the forkhead transcriptionfactor FKHR on serine 253 through awortmannin-sensitive pathway. J. Biol. Chem.274, 15982–15985
40 Tang, E.D. et al. (1999) Negative regulation of theforkhead transcription factor FKHR by Akt.J. Biol. Chem. 274, 16741–16746
41 Nakae, J. et al. (2001) The forkhead transcriptionfactor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J. Clin.Invest. 108, 1359–1367
42 Barthel, A. et al. (2001) Differential regulation ofendogenous glucose-6-phosphatase andphosphoenolpyruvate carboxykinase geneexpression by the forkhead transcription factorFKHR in H4IIE-hepatoma cells. Biochem.Biophys. Res. Commun. 285, 897–902
43 Yeagley, D. et al. (2001) Gene- and activation-specific mechanisms for insulin inhibition of basaland glucocorticoid-induced insulin-like growthfactor binding protein-1 andphosphoenolpyruvate carboxykinasetranscription. Roles of forkhead and insulinresponse sequences. J. Biol. Chem. 276,33705–33710
44 Hall, R.K. et al. (2000) Regulation ofphosphoenolpyruvate carboxykinase andinsulin-like growth factor-binding protein-1 geneexpression by insulin. The role of wingedhelix/forkhead proteins. J. Biol. Chem. 275,30169–30175
45 Yoon, J.C. et al. (2001) Control of hepaticgluconeogenesis through the transcriptionalcoactivator PGC-1. Nature 413, 131–138
46 Cook, S.A. et al. (2002) Transcriptional effects ofchronic akt activation in the heart. J. Biol. Chem.277, 22528–22533
47 Burks, D.J. and White, M.F. (2001) IRS proteinsand β-cell function. Diabetes 50, S140–S145
48 Jetton, T.L. et al. (2001) Enhanced expression ofinsulin receptor substrate-2 and activation ofprotein kinase B/Akt in regenerating pancreatic
TRENDS in Endocrinology & Metabolism Vol.13 No.10 December 2002
http://tem.trends.com
450 Review

duct epithelium of 60%-partial pancreatectomyrats. Diabetologia 44, 2056–2065
49 Holst, L.S. et al. (1998) Protein kinase B isexpressed in pancreatic β cells and activatedupon stimulation with insulin-like growth factorI. Biochem. Biophys. Res. Commun. 250,181–186
50 Tuttle, R.L. et al. (2001) Regulation of pancreaticβ-cell growth and survival by the serine/threonineprotein kinase Akt1/PKBα. Nat. Med. 7,1133–1137
51 Rondinone, C.M. et al. (1999) Impaired glucosetransport and protein kinase B activation byinsulin, but not okadaic acid, in adipocytes from
subjects with type II diabetes mellitus.Diabetologia 42, 819–825
52 Krook, A. et al. (1998) Insulin-stimulated Akt kinase activity is reduced in skeletal muscle from NIDDM subjects. Diabetes 47,1281–1286
53 Nadler, S.T. et al. (2001) Normal Akt/PKB withreduced PI3K activation in insulin-resistant mice.Am. J. Physiol. Endocrinol. Metab. 281,E1249–E1254
54 Kim, Y.B. et al. (2000) Divergent regulation of Akt1 and Akt2 isoforms in insulin targettissues of obese Zucker rats. Diabetes 49,847–856
55 Cho, H. et al. (2001) Insulin resistance and adiabetes mellitus-like syndrome in mice lackingthe protein kinase Akt2 (PKBβ). Science 292,1728–1731
56 Birnbaum, M.J. (2001) Diabetes. Dialoguebetween muscle and fat. Nature 409, 672–673
57 Chen, W.S. et al. (2001) Growth retardation andincreased apoptosis in mice with homozygousdisruption of the Akt1 gene. Genes Dev. 15,2203–2208
58 Cho, H. et al. (2001) Akt1/PKBα is required fornormal growth but dispensable for maintenanceof glucose homeostasis in mice. J. Biol. Chem. 276,38349–38352
TRENDS in Endocrinology & Metabolism Vol.13 No.10 December 2002
http://tem.trends.com 1043-2760/02/$ – see front matter © 2002 Elsevier Science Ltd. All rights reserved. PII: S1043-2760(02)00667-7
451Review
Since the discovery of somatostatin (SS) in 1973,knowledge of its roles in neurotransmission, as aninhibitor of endocrine and exocrine secretoryprocesses, and its vasoconstrictor andimmunomodulatory properties has increasedsignificantly [1–3]. The diverse biological effects of SSare mediated by a family of G-protein-coupledreceptors (GPCRs), the SS receptors (sst), whichcharacteristically comprise a single polypeptide chain with seven transmembrane-spanning domains.At present, five different human sst subtypes (sst1, sst2, sst3, sst4 and sst5) have been cloned andcharacterized [2]. sst are encoded by five non-allelicgenes, located on separate chromosomes [2]. Althoughthe different sst subtypes are 40–60% structurallyhomologous, each subtype mediates differentbiological actions of SS. Depending on the cell type, thefive sst are coupled to a variety of signal transductionpathways, including adenylate and guanylate cyclase,
phospholipase A2 and C, K+ and Ca2+ channels, Na+–H+ exchanger, Src, Erk1/2, p38 mitogen-activatedprotein kinases, and tyrosine phosphatases [2].
Although the evidence is preliminary, somespecific physiological regulatory roles can beattributed to sst subtypes. For example, in humans,sst2 and sst5 are involved in the control of growthhormone (GH) release, and sst5 appears to beimportant in the control of insulin and possibly alsoglucagon release (for Refs see [4]). The earlyobservation that SS might also control cellproliferation in normal and tumorous tissues has notyet resulted in clinically important applications [5].However, in recent years, important new data havebeen presented on sst-activated signal transductionpathways that are responsible for inhibition of cellgrowth and induction of apoptosis: sst3, and to a lesserextent sst2, can induce apoptosis, and sst1, sst4 andsst5 have an inhibitory effect on the cell cycle. For asummary of these effects see Table 1 and for moredetailed information see Refs [6–10].
Several other preclinical and clinical advanceshave brought the potential use of SS analogs in thediagnosis and treatment of patients with cancer muchcloser to reality. Here, we summarize the currentthoughts, in addition to the developments andexpectations for practical application in the diagnosisand treatment of cancer in the immediate future.
Experimental tumor models
SS analogs inhibit the growth of severaltransplantable tumors in rodents [5]: (transplantable)osteo- and chondrosarcomas, acinar and ductalpancreatic adenocarcinomas, pituitary tumors,insulinomas and various mammary and prostate
Somatostatin analogs in the
diagnosis and treatment of cancer
Steven W.J. Lamberts, Wouter W. de Herder and Leo J. Hofland
Over the past few years, significant progress has been made in our
understanding of the biology and functional significance of somatostatin
receptors (sst) on human tumors. Somatostatin analogs, such as octreotide,
bind predominantly to sst2
and successfully control hormone hypersecretion in
patients with acromegaly, islet cell tumors and carcinoids, and (temporary)
control of tumor growth is often also seen. Furthermore, sst2
on tumors can be
imaged in vivo after the injection of radionuclide-coupled octreotide. Targeted
chemo- and radiotherapy, in which somatostatin analogs coupled to a
chemotherapeutic agent or a radionuclide are selectively internalized by
sst-positive tumors, are now being studied for their effect on tumor growth.
Knowledge about the differential anti-tumor effects of the sst subtypes on
tumor cells might have clinical significance after the development of new
subtype-specific somatostatin analogs.
Published online: 14 October 2002
Steven W.J. Lamberts*
Wouter W. de Herder
Leo J. Hofland
Dept Medicine, ErasmusMedical Centre, 40 Dr. Molewaterplein,3015 GD Rotterdam, The Netherlands.*e-mail: [email protected]




















