
HijAkt: The PI3K/Akt pathway in virus replication and pathogenesis
Upload
independentCategory
view
2download
0
Cyclooxygenase-2 Promotes Hepatocellular CarcinomaCell Growth Through AKT Activation: Evidence for
AKT Inhibition in Celecoxib-Induced Apoptosis
Jing Leng,1,2 Chang Han,1 A. Jake Demetris,1 George K. Michalopoulos,1 and Tong Wu1
Cyclooxygenase-2 (COX-2)-controlled prostaglandin (PG) metabolism recently has beenimplicated in the pathogenesis of hepatocellular carcinoma (HCC). However, the biologicrole and molecular mechanism of COX-2–mediated PGs in the control of liver cancer growthhave not been established. This study was designed to examine the direct effect of COX-2 andits inhibitor celecoxib on the growth control of liver cancer cells. Human HCC cell linesHep3B and HepG2 transfected with COX-2 expression vector showed increased cell growthand enhanced phosphorylation of serine/threonine protein kinase B (Akt). The level ofCOX-2 expression and Akt phosphorylation is correlated positively in cultured HCC cellsand human liver cancer tissues. Inhibition of Akt activation by phosphatidylinositol 3-ki-nase (PI3-kinase) inhibitor LY294002 significantly decreased the viability of Hep3B andHepG2 cells (P < .01). These results reveal a novel role of Akt activation in COX-2–inducedHCC cell survival. Furthermore, HCC cells treated with the COX-2 inhibitor celecoxibshowed significant reduction of Akt phosphorylation and marked morphologic and bio-chemical characteristics of apoptosis. Overexpression of COX-2 or addition of exogenousPGE2 partially prevented celecoxib-induced apoptosis (P < .01). In conclusion, our resultssuggest the involvement of COX-2–dependent and –independent mechanisms in celecoxib-mediated HCC cell apoptosis. (HEPATOLOGY 2003;38:756-768.)
Hepatocellular carcinoma (HCC) often arisesfrom a background of long-standing chronic in-flammatory liver diseases. Several lines of evi-
dence suggest that mediators of inflammation, such asprostaglandins (PGs), may be involved causally in itspathogenesis.1 Increased cyclooxygenase-2 (COX-2) ex-
pression has been documented recently in humanHCC.2-6 Nonsteroidal anti-inflammatory drugs (NSAIDs)inhibit COX and block the growth of cultured HCCcells.4,7,8 These drugs also exert a chemopreventive effectin hepatocarcinogenesis models.9-14 For example,NSAIDs reduce the number and size of preneoplastic fociin the liver of rats fed a choline-deficient diet9-11; theseinhibitors also antagonize HCC growth promotion in thepostinitiation phase.12-14
The synthesis of PGs is controlled by coordinatedactivation of eicosanoid-forming enzymes includingCOXs.15 Two isoforms of COX have been identified,COX-1 and COX-2, both catalyzing the same enzymaticreaction. Despite the structural similarity, COX-1 andCOX-2 are regulated and function differently.15-21
COX-1 is expressed constitutively in most tissues, and isresponsible for maintaining basic physiologic functions,such as cytoprotection in the stomach and duodenum,vasodilation in kidney, and production of proaggregatoryprostanoid thromboxane in platelets. COX-2 is not con-stitutively expressed, but is induced by a variety of stimulisuch as cytokines, hormones, mitogens, and growth fac-tors,15-21 which explains its up-regulation in various in-flammatory diseases and human cancers. Evidence has
Abbreviations: HCC, hepatocellular carcinoma; PG, prostaglandins; COX, cy-clooxygenase; NSAID, nonsteroidal anti-inflammatory drug; Akt, serine/threonineprotein kinase B; zVAD-fmk, benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone;p-Akt, phosphorylated Akt; PBS, phosphate-buffered saline; PI3-kinase, phosphati-dylinositol 3-kinase; GSK-3�, glycogen synthase kinase-3� PDK1, 3-phosphoino-sitide-dependent protein kinase-1; PTEN, phosphatase and tensin homologue.
From the 1Department of Pathology, University of Pittsburgh School of Medicine,Pittsburgh, PA; and the 2Department of Pathology, Nanjing Medical University,Nanjing, China.
Received January 15, 2003; accepted June 17, 2003.Supported by grants from the American Liver Foundation (T.W.), the Cancer
Research Foundation of America (T.W.), the National Institutes of Health(DK49615 to A.J.D.), and the Department of Pathology, University of PittsburghMedical Center.
Address reprint requests to: Tong Wu, M.D., Ph.D., Department of Pathology,University of Pittsburgh School of Medicine, Presbyterian University HospitalC902, 200 Lothrop St., Pittsburgh, PA 15213. E-mail: [email protected]; fax:412-647-5237.
Copyright © 2003 by the American Association for the Study of Liver Diseases.0270-9139/03/3803-0026$30.00/0doi:10.1053/jhep.2003.50380
756

shown that COX-2–generated PGs promote tumor cellproliferation, survival, and angiogenesis.15-21 For exam-ple, COX-2 overexpression in cultured cells leads to in-creased cell survival.22-25 Knockout of the COX-2 genecan suppress tumorigenesis in mice that have a geneticpredisposition to form polyps.26 In animal studies, en-hanced COX-2 expression in mammary gland27 andskin28 induces tumorigenesis. These findings stronglysuggest the importance of COX-2–controlled PG metab-olism in the development and progression of human can-cers.
NSAIDs exert their antitumor effect by inhibition ofcell proliferation, potentiation of immune response, in-hibition of angiogenesis, and induction of apopto-sis.16,18,29-31 However, the exact mechanisms for NSAID-mediated chemoprevention are not well defined. Severalputative targets have been proposed to account forNSAID-induced growth inhibition of cancer cells, whichinclude inhibition of Bcl-2 expression,32,33 stimulation ofceramide production,34 inhibition of peroxisome prolif-erator activated receptor �,35 and interference with thenuclear factor � B signaling pathway.36 Although accu-mulating evidence points to inhibition of arachidonicacid metabolism via COX enzymes, which in turnmodulates the synthesis of PGs that affect cell pro-liferation, tumor growth, and immune responsiveness,there is evidence suggesting the involvement of COX-2–independent mechanisms in the antitumor effects ofNSAIDs. Celecoxib (SC-58635) belongs to the new gen-eration of NSAIDs that selectively inhibit COX-2 activitywithout inhibition of COX-1, and thus lack the side ef-fects associated with traditional NSAIDs. Recent studieshave shown that celecoxib inhibits the progression of co-lon tumor in human and animal models and inhibits thein vitro growth of several other tumor cell types.37-44 Theobservation that celecoxib induces tumor cell apoptosis incells that lack the COX-2 enzyme also suggests the in-volvement of COX-2–independent mechanism in cele-coxib-induced tumor cell apoptosis.39,40,43,44
Despite increased expression of COX-2 in human livercancer, inhibition of liver cancer growth by NSAIDs, andthe documented role of COX-2 in human cancers in gen-eral, the direct biologic role and molecular mechanism ofCOX-2–mediated prostanoids in the control of liver can-cer growth have not been established with molecularapproach. Furthermore, the potential mechanisms bywhich COX-2 inhibitors prevent liver cancer remainpoorly understood. This study reveals an important roleof COX-2 in liver cancer growth by altering the expres-sion of COX-2 in human liver cancer cells. Our resultsprovide novel evidence for the involvement of serine/thre-onine protein kinase B (Akt) in COX-2–mediated HCC
cell growth. Furthermore, we show that inhibition of Aktis an important mechanism for celecoxib-mediated apo-ptosis, which likely involves both COX-2–dependentand –independent mechanisms.
Materials and Methods
Human HCC Tissues. A total of 20 archival, forma-lin-fixed, paraffin-embedded specimens of human HCCand surrounding nontumor liver tissues were obtainedfrom the University of Pittsburgh Medical Center. Thetissue specimens were used for immunohistochemicalanalysis for COX-2 and p-Akt (Thr308) according to theprotocol approved by the University of Pittsburgh (Insti-tutional Review Board #020134). None of the cases usedin this study had patient identifiers and strict confidenti-ality was maintained in accordance with the approvalgranted by the Institutional Review Board.
Cell Culture. Human HCC cell lines Hep3B andHepG2 were obtained from American Type Culture Col-lection (Manassas, VA). The Hep3B cells were cultured inminimum essential medium (with Eagle’s salt and glu-tamine) with 10% (vol/vol) fetal bovine serum. TheHepG2 cells were cultured in modified minimum essen-tial medium with 2 mmol/L L-glutamine, 1.0 mmol/Lsodium pyruvate, 0.1 mmol/L nonessential amino acids,1.5 g/L sodium bicarbonate supplemented with 10% fetalbovine serum, and antibiotics. The cells were cultured at37°C in a humidified CO2 incubator.
Cell Viability Measurement. Cell viability was deter-mined using the cell proliferation reagent WST-1, atetrazolium salt that is cleaved by mitochondrial dehydro-genases in viable cells. Briefly, 100 �L of cell suspension(containing 0.5-2 � 104 cells) were plated in each well of96-well plates. After 24-hour culture to allow reattach-ment, the cells then were treated with specific reagentssuch as PGE2, LY294002, celecoxib, or benzyloxycar-bonyl-Val-Ala-Asp-fluoromethylketone (z-VAD-fmk)and vehicle for indicated time points. At the end of eachexperiment, the cell proliferation reagent WST-1 (10 �L)was added to each well and the cells were incubated at37°C for 0.5 to 5 hours. A450nm was measured using anautomatic enzyme-linked immunosorbent assay platereader.
Detection of Cytosolic Cytochrome C. The cellsgrown on 100-mm culture dishes with or without cele-coxib treatment were harvested by 0.05% trypsin and thecell pellets were suspended in 150 �L buffer solution (20mmol/L HEPES-KOH, pH 7.5, 10 mmol/L MgCl2, 250mmol/L sucrose, 1 mmol/L ethylenediaminetetraaceticacid, 1 mmol/L EGTA, 1 mmol/L phenylmethylsulfonylfluoride, 10 �g/mL leupeptin, 10 �g/mL aprotinin). The
HEPATOLOGY, Vol. 38, No. 3, 2003 LENG ET AL. 757
cells were then lysed by passing through a 26-gauge needlefor 10 times and the homogenate was centrifuged at750 � g for 10 minutes at 4°C to remove nuclei andunbroken cells. The samples were then centrifuged at10,000 � g for 15 minutes at 4°C and the supernatantswere collected for sodium dodecyl sulfate–polyacrylamidegel electrophoresis and Western blot analysis using mono-clonal anti-human cytochrome c antibody.
Immunohistochemical Analysis. For immunohisto-chemical staining of COX-2 and phospho-Akt (p-Akt)(Thr308) using human HCC tissue, 5-�m thick tissuesections of formalin-fixed and paraffin-embedded sec-tions were deparaffinized and rehydrated by routine pro-cedure. Endogenous peroxidase activity was quenchedwith 1% hydrogen peroxide and microwave retrieval ofantigen was performed. Nonspecific binding was blockedusing buffer containing normal serum from the speciescorresponding to the specific antibodies. The slides wereincubated at 4°C overnight with 1:100 diluted monoclo-nal anti-human COX-2 (obtained from Cayman Chem-ical, Ann Arbor, MI) and p-Akt (Thr308) (obtained fromUpstate Biotech, Lake Placid, NY) in labeling buffer.After repeated washings, the slides were incubated withbiotin-conjugated secondary antibody (1:200) for 30minutes at room temperature. After probing, the ABCcomplex was added and, finally, 3,3�-diaminobenzidineor 3-amino-9-ethylcarbazole substrate was used for colordevelopment. The slides were then counterstained withhematoxylin. The intensity of staining for COX-2 andp-Akt was scored in each specimen on a scale of 0 to 3, inwhich 0 � negative staining, 1 � weakly positive stain-ing, 2 � moderately positive staining, and 3 � stronglypositive staining. For each sample, 10 random high-power fields were scored. The immunoreactivity forCOX-2 and p-Akt in each sequential section was docu-mented and compared.
Results
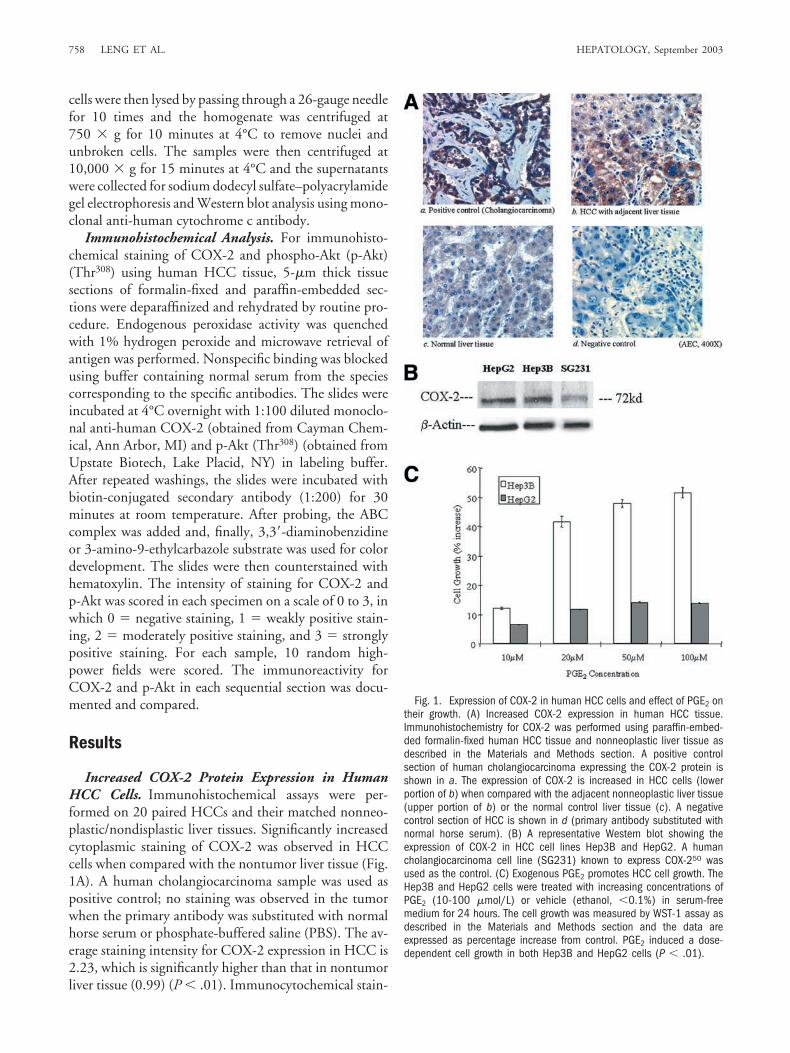
Increased COX-2 Protein Expression in HumanHCC Cells. Immunohistochemical assays were per-formed on 20 paired HCCs and their matched nonneo-plastic/nondisplastic liver tissues. Significantly increasedcytoplasmic staining of COX-2 was observed in HCCcells when compared with the nontumor liver tissue (Fig.1A). A human cholangiocarcinoma sample was used aspositive control; no staining was observed in the tumorwhen the primary antibody was substituted with normalhorse serum or phosphate-buffered saline (PBS). The av-erage staining intensity for COX-2 expression in HCC is2.23, which is significantly higher than that in nontumorliver tissue (0.99) (P � .01). Immunocytochemical stain-
Fig. 1. Expression of COX-2 in human HCC cells and effect of PGE2 ontheir growth. (A) Increased COX-2 expression in human HCC tissue.Immunohistochemistry for COX-2 was performed using paraffin-embed-ded formalin-fixed human HCC tissue and nonneoplastic liver tissue asdescribed in the Materials and Methods section. A positive controlsection of human cholangiocarcinoma expressing the COX-2 protein isshown in a. The expression of COX-2 is increased in HCC cells (lowerportion of b) when compared with the adjacent nonneoplastic liver tissue(upper portion of b) or the normal control liver tissue (c). A negativecontrol section of HCC is shown in d (primary antibody substituted withnormal horse serum). (B) A representative Western blot showing theexpression of COX-2 in HCC cell lines Hep3B and HepG2. A humancholangiocarcinoma cell line (SG231) known to express COX-250 wasused as the control. (C) Exogenous PGE2 promotes HCC cell growth. TheHep3B and HepG2 cells were treated with increasing concentrations ofPGE2 (10-100 �mol/L) or vehicle (ethanol, �0.1%) in serum-freemedium for 24 hours. The cell growth was measured by WST-1 assay asdescribed in the Materials and Methods section and the data areexpressed as percentage increase from control. PGE2 induced a dose-dependent cell growth in both Hep3B and HepG2 cells (P � .01).
758 LENG ET AL. HEPATOLOGY, September 2003
ing in the human HCC cell lines Hep3B and HepG2 alsoshowed expression of COX-2 in the cytoplasm (see below).The expression of COX-2 in Hep3B and HepG2 cellsalso was confirmed by Western blot analysis (Fig. 1B).
PGE2 and COX-2 Promote Human HCC CellGrowth. We next evaluated the potential effect ofCOX-2 and PG on HCC cell growth in vitro. Because
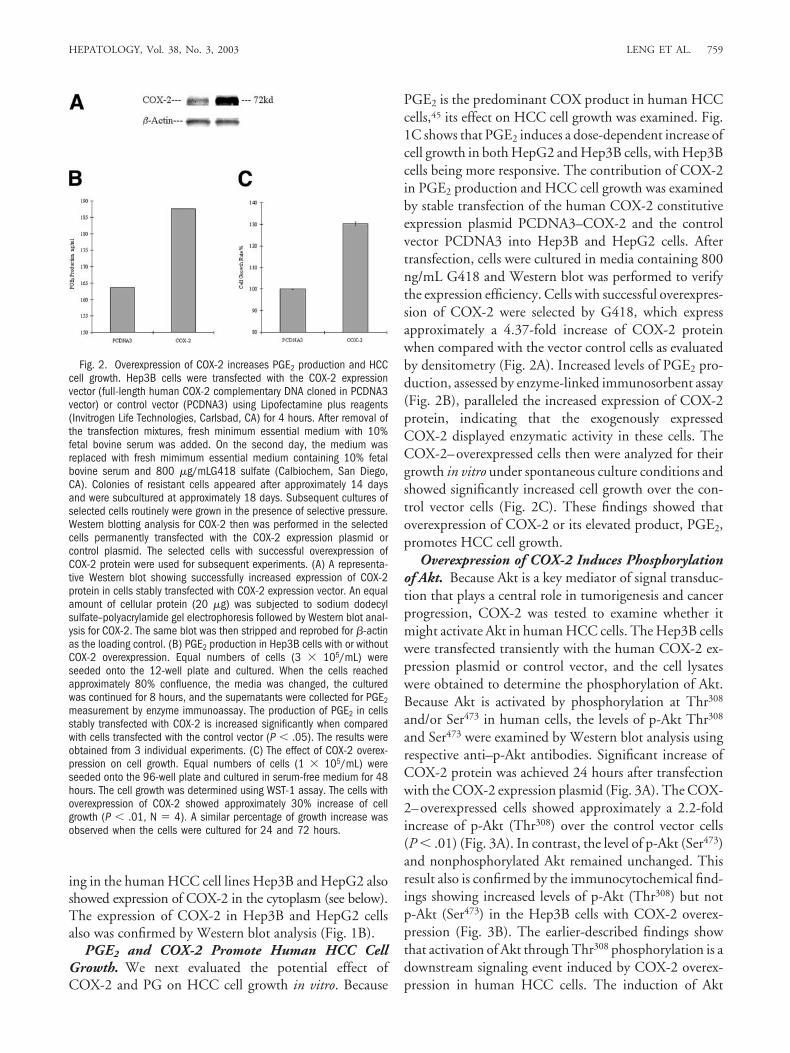
PGE2 is the predominant COX product in human HCCcells,45 its effect on HCC cell growth was examined. Fig.1C shows that PGE2 induces a dose-dependent increase ofcell growth in both HepG2 and Hep3B cells, with Hep3Bcells being more responsive. The contribution of COX-2in PGE2 production and HCC cell growth was examinedby stable transfection of the human COX-2 constitutiveexpression plasmid PCDNA3–COX-2 and the controlvector PCDNA3 into Hep3B and HepG2 cells. Aftertransfection, cells were cultured in media containing 800ng/mL G418 and Western blot was performed to verifythe expression efficiency. Cells with successful overexpres-sion of COX-2 were selected by G418, which expressapproximately a 4.37-fold increase of COX-2 proteinwhen compared with the vector control cells as evaluatedby densitometry (Fig. 2A). Increased levels of PGE2 pro-duction, assessed by enzyme-linked immunosorbent assay(Fig. 2B), paralleled the increased expression of COX-2protein, indicating that the exogenously expressedCOX-2 displayed enzymatic activity in these cells. TheCOX-2–overexpressed cells then were analyzed for theirgrowth in vitro under spontaneous culture conditions andshowed significantly increased cell growth over the con-trol vector cells (Fig. 2C). These findings showed thatoverexpression of COX-2 or its elevated product, PGE2,promotes HCC cell growth.
Overexpression of COX-2 Induces Phosphorylationof Akt. Because Akt is a key mediator of signal transduc-tion that plays a central role in tumorigenesis and cancerprogression, COX-2 was tested to examine whether itmight activate Akt in human HCC cells. The Hep3B cellswere transfected transiently with the human COX-2 ex-pression plasmid or control vector, and the cell lysateswere obtained to determine the phosphorylation of Akt.Because Akt is activated by phosphorylation at Thr308
and/or Ser473 in human cells, the levels of p-Akt Thr308
and Ser473 were examined by Western blot analysis usingrespective anti–p-Akt antibodies. Significant increase ofCOX-2 protein was achieved 24 hours after transfectionwith the COX-2 expression plasmid (Fig. 3A). The COX-2–overexpressed cells showed approximately a 2.2-foldincrease of p-Akt (Thr308) over the control vector cells(P � .01) (Fig. 3A). In contrast, the level of p-Akt (Ser473)and nonphosphorylated Akt remained unchanged. Thisresult also is confirmed by the immunocytochemical find-ings showing increased levels of p-Akt (Thr308) but notp-Akt (Ser473) in the Hep3B cells with COX-2 overex-pression (Fig. 3B). The earlier-described findings showthat activation of Akt through Thr308 phosphorylation is adownstream signaling event induced by COX-2 overex-pression in human HCC cells. The induction of Akt
Fig. 2. Overexpression of COX-2 increases PGE2 production and HCCcell growth. Hep3B cells were transfected with the COX-2 expressionvector (full-length human COX-2 complementary DNA cloned in PCDNA3vector) or control vector (PCDNA3) using Lipofectamine plus reagents(Invitrogen Life Technologies, Carlsbad, CA) for 4 hours. After removal ofthe transfection mixtures, fresh minimum essential medium with 10%fetal bovine serum was added. On the second day, the medium wasreplaced with fresh mimimum essential medium containing 10% fetalbovine serum and 800 �g/mLG418 sulfate (Calbiochem, San Diego,CA). Colonies of resistant cells appeared after approximately 14 daysand were subcultured at approximately 18 days. Subsequent cultures ofselected cells routinely were grown in the presence of selective pressure.Western blotting analysis for COX-2 then was performed in the selectedcells permanently transfected with the COX-2 expression plasmid orcontrol plasmid. The selected cells with successful overexpression ofCOX-2 protein were used for subsequent experiments. (A) A representa-tive Western blot showing successfully increased expression of COX-2protein in cells stably transfected with COX-2 expression vector. An equalamount of cellular protein (20 �g) was subjected to sodium dodecylsulfate–polyacrylamide gel electrophoresis followed by Western blot anal-ysis for COX-2. The same blot was then stripped and reprobed for �-actinas the loading control. (B) PGE2 production in Hep3B cells with or withoutCOX-2 overexpression. Equal numbers of cells (3 � 105/mL) wereseeded onto the 12-well plate and cultured. When the cells reachedapproximately 80% confluence, the media was changed, the culturedwas continued for 8 hours, and the supernatants were collected for PGE2measurement by enzyme immunoassay. The production of PGE2 in cellsstably transfected with COX-2 is increased significantly when comparedwith cells transfected with the control vector (P � .05). The results wereobtained from 3 individual experiments. (C) The effect of COX-2 overex-pression on cell growth. Equal numbers of cells (1 � 105/mL) wereseeded onto the 96-well plate and cultured in serum-free medium for 48hours. The cell growth was determined using WST-1 assay. The cells withoverexpression of COX-2 showed approximately 30% increase of cellgrowth (P � .01, N � 4). A similar percentage of growth increase wasobserved when the cells were cultured for 24 and 72 hours.
HEPATOLOGY, Vol. 38, No. 3, 2003 LENG ET AL. 759

phosphorylation at Thr308, but not Ser473, by overexpres-sion of COX-2 suggests that phosphorylation of Akt atThr308 is sufficient for COX-2–induced Akt activation inHCC cells.
In light of the recent evidence indicating involvementof Bcl-2 in COX-2 and PGE2-mediated cell survival incolon cancer and intestinal epithelial cells,22,32 the level ofBcl-2 also was examined in cells with and without COX-2overexpression (Fig. 3A). A similar level of Bcl-2 proteinwas noted in the COX-2–overexpressed cells and controlvector cells. Thus, COX-2 overexpression induces Aktphosphorylation at Thr308, but not Bcl-2 expression inHep3B cells.
4™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™Fig. 3. Overexpression of COX-2 induces phosphorylation of Akt in
HCC cells. (A) Western blot showing increased p-AktThr308 but notp-AktSer473 or Bcl-2 in cells with overexpression of COX-2. The Hep3Bcells grown in 6-well culture dishes were transfected with 1.5 �g each ofCOX-2 or PCDNA3 vectors using Lipofectamine plus reagents (InvitrogenLife Technologies). After exposure to the mixture of transfection reagentsand plasmids for 4 hours, the cultures were continued for 16 to 24 hours.Equal amount of cellular proteins isolated from cells transfected with theCOX-2 expression plasmid or control plasmid were subjected to sodiumdodecyl sufate–polyacrylamide gel electrophoresis followed by Westernblot using antibodies against COX-2, p-Akt (Thr308), p-Akt (Ser473),nonphosphorylated Akt, and Bcl-2. Western blot for �-actin was shownas the loading control. (B) Immunocytochemical staining for COX-2 andp-Akt (Thr308) in cells transfected with COX-2 complementary DNA orcontrol vector (PCDNA3). The cells were trypsinized and washed twicewith PBS and then dripped onto aminopropylethoxysilane-coated slides.Air-dried sample slides were fixed with 4% paraformaldehyde for 30minutes. The slides were treated with 0.1% Triton X-100 (in 0.1 mol/Lcitrate buffer, pH 6) on ice for 4 minutes for cell permeability. Afterwashing with PBS, the slides were subjected to immunocytochemicalstains for COX-2 and p-Akt. To compare the COX-2 and p-Akt expressionin the same cells, the immunostains were performed in the same slideafter stripping the first labeled signal. Briefly, at the end of the firstimmunostain, the 3-amino-9-ethylcarbazole color was stripped by wash-ing the slide in 100% ethanol for 5 minutes, followed by incubation instripping buffer (100 mmol/L 2-mercaptoethanol, 2% sodium dodecylsulfate, 62.5 mmol/L Tris-HCl pH 6.7) for 10 minutes in room temper-ature. After 2 washes with PBS, the slide was ready for the secondimmunostain. (a) Hep3B cells transfected with PCDNA3, probed withanti–COX-2 antibody; (b) Hep3B cells transfected with COX-2, probedwith anti–COX-2 antibody; (c) Hep3B cells transfected with PCDNA3,probed with anti-p-Akt (Thr308) antibody; (d) Hep3B cells transfected withCOX-2, probed with anti–p-Akt (Thr308) antibody. Panels C and D showthe expression of Akt in cells transfected with the control PCDNA3 andCOX-2 expression plasmid, respectively. Increased p-Akt (Thr308) is ob-served in cells transfected with COX-2 expression plasmid. Successfulexpression of COX-2 in these cells was confirmed by immunocytochem-ical analysis for COX-2, which was performed in the same cells afterstripping the initial signals. Individual cells with a relatively high level ofCOX-2 protein exhibit a relatively high level of p-Akt (Thr308) (indicated byarrows). The level of p-Akt (Ser473) was not altered in cells with COX-2overexpression (data not shown). (C) Increased p-Akt in human HCCtissue. Sequential sections of human liver cancer tissues were obtainedand stained by ABC immunohistochemistry for COX-2 and p-Akt (Thr308).A representative photograph is shown to highlight increased levels ofboth COX-2 and p-Akt in the same tumor nodule (shown in left portionof each section) when compared with the adjacent nonneoplastic livertissue (shown in right portion of each section). (a) COX-2 expression(3-amino-9-ethylcarbazole 200X); (b) p-Akt expression (3,3�-diamino-benzidine �200); (c) COX-2 expression (3-amino-9-ethylcarbazole�400); (d) p-Akt expression (3,3�-diaminobenzidine �400).
760 LENG ET AL. HEPATOLOGY, September 2003
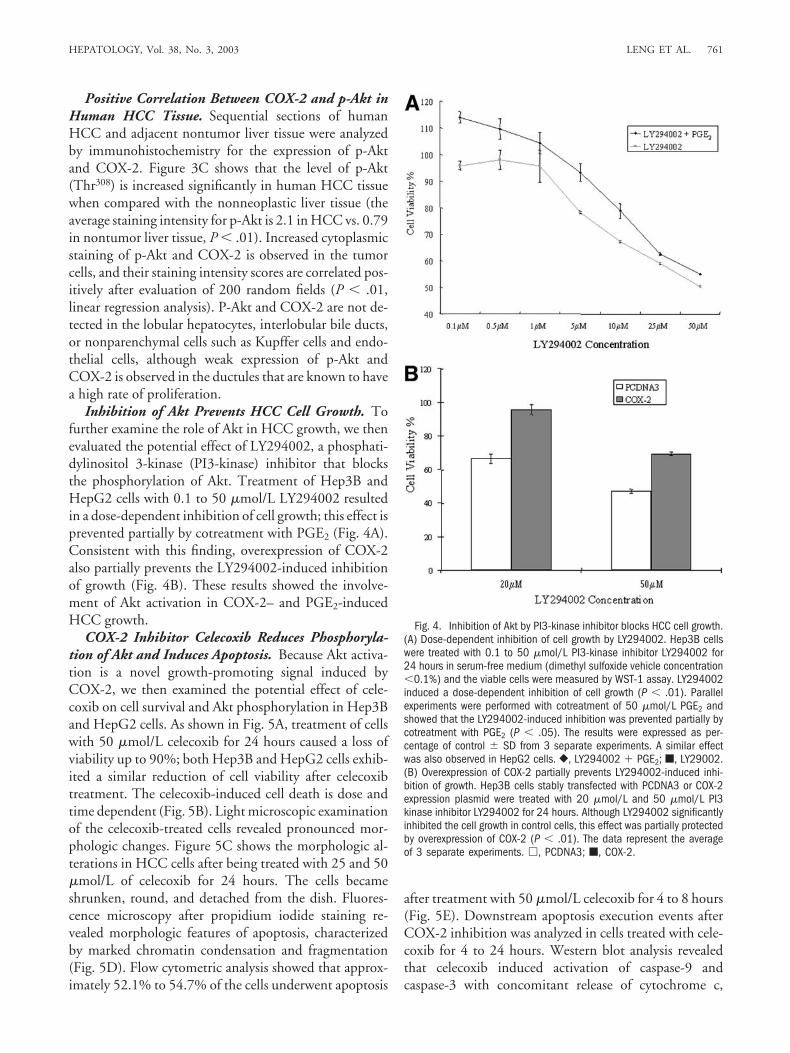
Positive Correlation Between COX-2 and p-Akt inHuman HCC Tissue. Sequential sections of humanHCC and adjacent nontumor liver tissue were analyzedby immunohistochemistry for the expression of p-Aktand COX-2. Figure 3C shows that the level of p-Akt(Thr308) is increased significantly in human HCC tissuewhen compared with the nonneoplastic liver tissue (theaverage staining intensity for p-Akt is 2.1 in HCC vs. 0.79in nontumor liver tissue, P � .01). Increased cytoplasmicstaining of p-Akt and COX-2 is observed in the tumorcells, and their staining intensity scores are correlated pos-itively after evaluation of 200 random fields (P � .01,linear regression analysis). P-Akt and COX-2 are not de-tected in the lobular hepatocytes, interlobular bile ducts,or nonparenchymal cells such as Kupffer cells and endo-thelial cells, although weak expression of p-Akt andCOX-2 is observed in the ductules that are known to havea high rate of proliferation.
Inhibition of Akt Prevents HCC Cell Growth. Tofurther examine the role of Akt in HCC growth, we thenevaluated the potential effect of LY294002, a phosphati-dylinositol 3-kinase (PI3-kinase) inhibitor that blocksthe phosphorylation of Akt. Treatment of Hep3B andHepG2 cells with 0.1 to 50 �mol/L LY294002 resultedin a dose-dependent inhibition of cell growth; this effect isprevented partially by cotreatment with PGE2 (Fig. 4A).Consistent with this finding, overexpression of COX-2also partially prevents the LY294002-induced inhibitionof growth (Fig. 4B). These results showed the involve-ment of Akt activation in COX-2– and PGE2-inducedHCC growth.
COX-2 Inhibitor Celecoxib Reduces Phosphoryla-tion of Akt and Induces Apoptosis. Because Akt activa-tion is a novel growth-promoting signal induced byCOX-2, we then examined the potential effect of cele-coxib on cell survival and Akt phosphorylation in Hep3Band HepG2 cells. As shown in Fig. 5A, treatment of cellswith 50 �mol/L celecoxib for 24 hours caused a loss ofviability up to 90%; both Hep3B and HepG2 cells exhib-ited a similar reduction of cell viability after celecoxibtreatment. The celecoxib-induced cell death is dose andtime dependent (Fig. 5B). Light microscopic examinationof the celecoxib-treated cells revealed pronounced mor-phologic changes. Figure 5C shows the morphologic al-terations in HCC cells after being treated with 25 and 50�mol/L of celecoxib for 24 hours. The cells becameshrunken, round, and detached from the dish. Fluores-cence microscopy after propidium iodide staining re-vealed morphologic features of apoptosis, characterizedby marked chromatin condensation and fragmentation(Fig. 5D). Flow cytometric analysis showed that approx-imately 52.1% to 54.7% of the cells underwent apoptosis
after treatment with 50 �mol/L celecoxib for 4 to 8 hours(Fig. 5E). Downstream apoptosis execution events afterCOX-2 inhibition was analyzed in cells treated with cele-coxib for 4 to 24 hours. Western blot analysis revealedthat celecoxib induced activation of caspase-9 andcaspase-3 with concomitant release of cytochrome c,
Fig. 4. Inhibition of Akt by PI3-kinase inhibitor blocks HCC cell growth.(A) Dose-dependent inhibition of cell growth by LY294002. Hep3B cellswere treated with 0.1 to 50 �mol/L PI3-kinase inhibitor LY294002 for24 hours in serum-free medium (dimethyl sulfoxide vehicle concentration�0.1%) and the viable cells were measured by WST-1 assay. LY294002induced a dose-dependent inhibition of cell growth (P � .01). Parallelexperiments were performed with cotreatment of 50 �mol/L PGE2 andshowed that the LY294002-induced inhibition was prevented partially bycotreatment with PGE2 (P � .05). The results were expressed as per-centage of control � SD from 3 separate experiments. A similar effectwas also observed in HepG2 cells. }, LY294002 � PGE2; �, LY29002.(B) Overexpression of COX-2 partially prevents LY294002-induced inhi-bition of growth. Hep3B cells stably transfected with PCDNA3 or COX-2expression plasmid were treated with 20 �mol/L and 50 �mol/L PI3kinase inhibitor LY294002 for 24 hours. Although LY294002 significantlyinhibited the cell growth in control cells, this effect was partially protectedby overexpression of COX-2 (P � .01). The data represent the averageof 3 separate experiments. �, PCDNA3; �, COX-2.
HEPATOLOGY, Vol. 38, No. 3, 2003 LENG ET AL. 761

Fig. 5. Celecoxib inhibits HCC cell growth by induction of apoptosis. (A) The dose-dependent inhibition of HCC cell growth by celecoxib. The Hep3Bcells and HepG2 cells were treated with different concentrations of celecoxib in serum-free medium for 24 hours. The viable cells were measuredby WST-1 assay and the results were compared with cells treated with vehicle alone (dimethyl sulfoxide �0.1%). The data are expressed aspercentage of inhibition � SD from 3 individual experiments (P � .01). �, Hep3B; �, HepG2. (B) The time-dependent inhibition of HCC cell growthby celecoxib. The Hep3B cells were treated with 25 to 100 �mol/L celecoxib in serum-free medium for 4, 8, 12, and 24 hours. The viable cells weremeasured by WST-1 assay and the results were compared with cells treated with vehicle alone (dimethyl sulfoxide �0.1%). Progressive inhibitionof cell growth was observed at all concentrations of celecoxib (25-100 �mol/L) over 4 to 24 hours (P � .01). The results were obtained from 3separate experiments. (C) Light microscopy showing the typical morphologic changes of Hep3B cells after celecoxib treatment for 24 hours. 25�mol/L celecoxib resulted in decreased cell number; 50 �mol/L celecoxib resulted in cell floating with nuclear condensation. A similar effect wasobserved in HepG2 cells. (D) Fluorescence microscopy after propidium iodide staining showing nuclear features of apoptosis in Hep3B cells. The cellswith or without celecoxib treatment were trypsinized and the cell suspension was centrifuged at 500 � g. After washing with cold PBS twice, the cellpellets were suspended in ice cold 75% ethanol with gentle vortex. After ethanol fixation at �20°C for at least 4 hours, the ethanol was removedand cells were washed twice with PBS via repeated centrifugation at 500 � g. The cells were then resuspended in 1 mL propidium iodide stainingsolution (0.01% Triton X-100, 0.2 mg/mL RNase, 0.1% sodium citrate, 0.05 mg/mL propidium iodide) for 30 minutes at room temperature in a darkplace. The stained cells were collected by centrifugation and dissolved in 50% glycerol PBS solution for fluorescence microscopy. (a) Control cellswith normal nuclear morphology; (b) cells treated with 50 �mol/L celecoxib for 8 hours showing the typical features of apoptosis characterized bynuclear shrinkage, chromatin condensation, fragmentation, and attachment to nuclear envelope. A similar effect was observed in HepG2 cells.
762 LENG ET AL. HEPATOLOGY, September 2003

Fig. 5 (Cont’d.). (E) FACS analysis. Hep3B cells were treated with 50 �mol/L celecoxib in serum-free medium for 4 and 8 hours and the cellswere collected for flow cytometry analysis after Annexin-V-fluorescein isothiocyanate and propidium iodide double staining using the Annexin-V-FluosStaining kit purchased from Roche Applied Science (Indianapolis, IN). The cells (approximately 2 � 106 cells for each sample) were incubated in100 �L of Annexin-V-fluorescein isothiocyanate (with propidium iodide) Labeling Solution for 15 minutes at room temperature, followed by additionof 0.5 mL incubation buffer. The samples were analyzed on a flow cytometer using 488-nm excitation and 515-nm bandpass filter for fluoresceindetection and a filter greater than 600 nm for propidium iodide detection. Those cells positively stained by Annexin-V but negatively stained bypropidium iodide were counted as apoptotic cells. The results were confirmed by 2 separate experiments. (F) Western blot showing activation ofapoptosis-associated proteins and enzymes after celecoxib treatment. Activation of caspase-3 and caspase-9 was observed 4 to 24 hours after 50�mol/L celecoxib treatment. Activation of caspase-3 was reflected by a precipitous disappearance of the 32-kd precursor accompanied by aconcomitant increase in the 20-kd cleaved band; activation of caspase-9 was reflected by conversion of the 47-kd enzymatically inactive precursorto the active 35- and 37-kd proteolytic cleavage products. Western blot analysis using cytosolic protein showed increased cytochrome c release 4to 24 hours after 50 �mol/L celecoxib treatment. A similar effect also was observed in HepG2 cells.
HEPATOLOGY, Vol. 38, No. 3, 2003 LENG ET AL. 763

which was observed 4 to 24 hours after 50 �mol/L cele-coxib treatment (Fig. 5F). Consistent with these findings,assays for caspase protease activity showed an 8.2- to 12.6-fold increase of caspase-3 activity and a 5.5- to 6.7-foldincrease of caspase-9 activity in Hep3B cells treated with50�mol/L celecoxib for 4 to 8 hours (P � .01, n � 3). Tofurther determine whether the cytotoxic effect of cele-coxib was caused by apoptosis, the Hep3B cells weretreated with celecoxib for 24 hours in the presence of thewide-spectrum caspase inhibitor Z-VAD-fmk. The cele-coxib-induced reduction of cell viability was significantlyprevented by cotreatment with Z-VAD-fmk (67% celldeath after 50 �mol/L celecoxib vs. 38% cell death after50 �mol/L celecoxib plus 100 �mol/L Z-VAD-fmk; P �.01; n � 3). These findings showed that celecoxib reducedHCC viability by induction of apoptosis.
The effect of celecoxib on Akt phosphorylation atThr308 and Ser473 was evaluated using specific phospho-antibodies. The cells were treated with 50 �mol/L cele-coxib for 4 to 24 hours and phosphorylation of Akt wasdetermined by Western blot analysis. Figure 6A depictsa dramatic impact of celecoxib treatment on the levelof p-Akt in HCC cells, whereas the control showed nosignificant changes within a 4- to 24-hour period. Incelecoxib-treated cells, p-Akt Thr308 was undetectablethroughout the 4- to 24-hour time periods. The level ofp-Akt Ser473 also was reduced significantly after celecoxibtreatment, with negligible levels of p-Akt Ser473 remain-ing 4 hour posttreatment and no detectable amountthereafter. Consistent with the marked inhibition of Aktphosphorylation, a concomitant increase of nonphospho-rylated Akt was observed at all time periods after celecoxibtreatment, which most likely was caused by reduced con-version of non–p-Akt to p-Akt by celecoxib. The inhibi-tory effect of celecoxib on Akt phosphorylation andactivation was confirmed further by measuring the Akt-mediated phosphorylation of glycogen synthase ki-nase-3� (GSK-3�), a direct in vivo downstream substrateof Akt. As shown in Fig. 6A, the reduction of Akt phos-phorylation correlated with an equivalent decreaseof GSK-3� phosphorylation, although inhibition ofGSK-3� phosphorylation appeared at a delayed time pe-riod. The latter observation is most likely due to thepresence of other signaling mechanisms for GSK-3�phosphorylation that is not altered by celecoxib treat-ment.
The phosphorylation status of Akt is controlled by thePI3 kinase and 3-phosphoinositide–dependent proteinkinase-1 (PDK1) pathway. Phosphorylation of PDK1 byPI3K increases its enzyme activity, which subsequentlyactivates Akt via phosphorylation at Thr308. This processis inhibited by a phosphatase and tensin homolog(PTEN), a phosphatase that converts phosphatidylinosi-tol (3,4,5) triphosphate 3 back to phosphatidylinositol(3,4,5) triphosphate 2, thus inhibiting PI3-kinase/PDK1activation (phosphorylation of PTEN down-regulates itsphosphatase activity leading to increased PI3-kinase/PDK activity). In contrast, phosphorylation of Akt atSer473 is mediated by PDK2.46 To further document themechanism by which celecoxib inhibits Akt phosphoryla-tion in HCC cells, the effect of celecoxib on the phos-phorylation of PDK1 and PTEN also was examined. Asshown in Fig. 6B, the cells treated with 50 �mol/L cele-coxib showed significantly decreased phosphorylation ofPDK1 and PTEN. These results indicate that celecoxibmay inhibit Akt phosphorylation at Thr308 in HCC cellsthrough mechanisms involving inhibition of PDK1 andPTEN phosphorylation. The observation that celecoxib
Fig. 6. Celecoxib inhibits phosphorylation of Akt, PDK1, and PTEN inHCC cells. (A) Celecoxib inhibits Akt phosphorylation in HCC cells. Hep3Bcells were treated with celecoxib (50 �mol/L) for 4 to 24 hours and thecell lysates were obtained for Western blot analysis. A total of 20 �g ofcellular protein was used for each sample and the immunoblot for�-actin was used as loading control. Celecoxib treatment significantlyinhibited the phosphorylation of Akt at Thr308 and Ser473. The level ofnonphosphorylated Akt was increased after celecoxib treatment. Consis-tent with inhibition of Akt, celecoxib also inhibited the p-Akt–dependentphosphorylation of GSK-3�. The results are representative of at least 3separate experiments. A similar effect was observed in HepG2 cells. (B)Celecoxib inhibits phosphorylation of PDK1 and PTEN in HCC cells.Hep3B cells were treated with celecoxib (50 �mol/L) for 8 hours and thecell lysates were obtained for Western blot analysis for p-PDK1 andp-PTEN. A total of 20 �g of cellular protein was used for each sampleand the immunoblot for �-actin was used as loading control. Celecoxibtreatment significantly inhibited the phosphorylation of PDK1 and PTEN.The results are representative of at least 3 individual experiments. Asimilar effect was observed in HepG2 cells.
764 LENG ET AL. HEPATOLOGY, September 2003

decreases Akt phosphorylation at Thr308 and Ser473 sug-gests participation of both PDK1 and PDK2 in celecoxib-mediated inhibition of Akt phosphorylation.
Recent studies in prostate and colon cancers have asso-ciated Bcl-2 regulation with COX-2 inhibition.32,33 Toexamine whether Bcl-2 was involved in the control of
celecoxib-induced apoptosis in HCC cells, the level ofBcl-2 protein was evaluated by Western blot analysis. Al-though Bcl-2 was expressed in Hep3B and HepG2 cells,its level of expression remained unchanged throughoutthe course of apoptotic death in response to 50 �mol/Lcelecoxib treatment. Furthermore, protein concentra-tions of Bax, another proapoptotic Bcl-2 family member,also was unchanged after celecoxib treatment.
Celecoxib-Induced Inhibition of HCC Cell GrowthIs Reversed Partially by COX-2 and PGE2. Furtherexperiments were performed to examine if COX-2 andPGE2 could protect HCC cells from celecoxib-inducedcell death. As shown in Fig. 7A, overexpression of COX-2in Hep3B cells partially prevented the celecoxib-mediatedreduction of cell viability. Consistent with this result,Hep3B cells treated with 10 to 50 �mol/L celecoxib for 8hours showed approximately 20% to 23% reduction ofPGE2 production (P � .05, n � 3), indicating an inhibi-tion of COX-2 by celecoxib in these cells. Furthermore,exogenous PGE2 partially prevented the celecoxib-in-duced caspase-3 activation and cell death (Figs. 7B and7C). These observations suggest that celecoxib may in-hibit cell growth partially through inhibition of COX-2in HCC cells. However, the incomplete prevention ofcelecoxib-induced HCC cell death by COX-2 and PGE2
also suggests the existence of a COX-2–independentmechanism.
DiscussionThe findings in this study establish a direct relationship
between COX-2 expression and cell survival in HCC
4™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™Fig. 7. Effect of COX-2 and PGE2 on celecoxib-induced cell death. (A)
Overexpression of COX-2 partially prevents the celecoxib-induced inhibi-tion of cell growth. Hep3B cells stably transfected with the PCDNA3 orCOX-2 expression plasmid were treated with 1 to 30 �mol/L celecoxibfor 24 hours and the viable cells were measured by WST-1 assay. Thecelecoxib-induced inhibition of cell growth is protected partially byoverexpression of COX-2 and this effect is seen at all 4 concentrations (P� .01). The data are the average of 3 separate experiments. },PCDNA3; �, COX-2. (B) Exogenous PGE2 partially prevents the celecoxib-induced inhibition of cell growth. Hep3B cells were treated with 35�mol/L celecoxib in serum-free medium with or without 10 �mol/L and25 �mol/L PGE2 for 24 hours. The viable cells were measured by WST-1assay. The celecoxib-induced decrease of cell viability was preventedpartially by 10 �mol/L and 25 �mol/L PGE2 (P � .01). The data wereobtained from 3 separate experiments. A similar effect was observed inHepG2 cells. (C) A representative Western blot showing that PGE2partially prevents the celecoxib-induced caspase-3 activation. Hep3Bcells were treated with celecoxib (50 �mol/L) in serum-free medium inthe presence or absence of PGE2 (10 and 25 �mol/L) for 4 hours. Thecell lysates were obtained for Western blot analysis for caspase-3. Thebar graph indicates the relative amounts of activated caspase 3 afterbeing normalized to the respective actin level; the band density wasanalyzed by densitometry software ImageJ (version 1.28) provided by theNational Institutes of Health (Bethesda, MD).
HEPATOLOGY, Vol. 38, No. 3, 2003 LENG ET AL. 765

cells. Hepatocellular carcinoma cells expressing higherlevels of COX-2 have increased PGE2 production andgrowth advantage. Furthermore, our data provide novelevidence for the involvement of Akt activation in COX-2–induced HCC cell growth. The latter is attested by theobservations that COX-2 overexpression increases the p-Akt level and that inhibition of Akt activation by PI3Kinhibitor LY294002 decreases HCC cell growth. Otherinvestigators have observed that forced expression ofCOX-222 in intestinal cells or PGE2 exposure32 to coloncancer cells caused up-regulation of the Bcl-2 protein.The level of Bcl-2 protein, however, was unchanged inour cell system, suggesting that induction of Bcl-2 byCOX-2 may depend on specific cell types.
Recent evidence indicates that Akt plays a key role intumorigenesis and cancer progression by stimulating cellproliferation and inhibiting apoptosis.46-48 The Akt iscomposed of an N-terminal pleckstrin homology domainand a C-terminal kinase catalytic domain. It is activatedby a dual regulatory mechanism that requires both trans-location to the plasma membrane and phosphorylation atThr308 and Ser473. The generation of phosphatidylinositol(3,4,5) triphosphate on the inner layer of the plasmamembrane, after PI3-kinase activation, recruits Akt bydirect interaction with its PH domain. At the membrane,Akt is phosphorylated on Thr308 by PDK1. Thr308 phos-phorylation is necessary and sufficient for Akt activation,although maximal activation is achieved through addi-tional phosphorylation at Ser473 by PDK2, a kinase thathas been characterized biochemically but the molecularidentity of which remains undetermined.46 The phos-phorylated Akt dissociates from the membrane and entersthe cytoplasm and nucleus, where it phosphorylates sev-eral key proteins resulting in promotion of cell cycle andinhibition of apoptosis. In this study, we observed thatoverexpression of COX-2 induces Akt phosphorylation atThr308, but not at Ser473 in HCC cells, indicating that theCOX-2–induced growth promotion is mediated mainlyby PDK1-controlled Akt phosphorylation at Thr308 inHCC cells.
The exact mechanisms by which COX-2–derivedprostaglandins activate Akt in HCC cells remain to bedefined. Two classes of PG receptors transduce signalsupon binding of the ligand, the G protein–coupled cyto-plasmic receptor class (EP receptors for PGE2) and thenuclear receptor class peroxisome proliferator activatedreceptor, which acts as a transcription factor. Because thereceptor-coupled G protein is known to transduce thesignal to the PI3-kinase,49 the cytoplasmic EP recep-tors appear to be the preferred mediator for the COX-2–induced Akt-dependent cell survival. Further studies areneeded to determine whether the EP receptor or peroxi-
some proliferator activated receptor is involved in theCOX-2–mediated liver cancer cell survival.
Celecoxib belongs to the new generation of NSAIDsthat selectively inhibit COX-2 activity. Recent studieshave shown that celecoxib inhibits the growth of severalhuman cancers.37-44 However, it remains unclear howcelecoxib exerts its preventive effect on tumor growth andwhat the relative importance of COX-2 inhibition is inthis process. In the present study, we showed that cele-coxib induces morphologic and biochemical changescharacteristic of apoptosis in HCC cells. In contrast to theprevious reports that correlated the apoptotic effect ofCOX-2 inhibitors with Bcl-2 down-regulation in othercancer cells,32,33 our data indicate that the induction ofapoptosis by celecoxib is independent of Bcl-2 in HCCcells. This assertion is consistent with the recent observa-tions by Kern et al.8 showing that celecoxib treatment didnot alter Bcl-2 level in HCC cells. These investigators alsoobserved a reduction of p-Akt Ser473 in HCC cells treatedwith celecoxib, although they did not show a direct par-ticipation of p-Akt Ser473 in celecoxib-induced apoptosis.The study by Kern et al.,8 however, did not provide suf-ficient evidence for the contribution of Akt in COX-2–mediated liver cancer growth because the effect of COX-2on Akt activation or COX-2 inhibitor on p-Akt Thr308
was not examined. In this study, we showed that celecoxibinhibits Akt phosphorylation at both Thr308 and Ser473,with more prominent inhibition on Thr308 phosphoryla-tion. Furthermore, we showed that celecoxib inhibits Aktactivation through inhibition of PDK1 and PTEN phos-phorylation. This finding is noteworthy because our dataalso revealed that Akt phosphorylation is enhanced byCOX-2 overexpression in HCC cells. The observationsthat celecoxib inhibits the production of PGE2, that over-expression of COX-2 and addition of exogenous PGE2
provide partial protection against celecoxib-induced apo-ptosis, and that overexpression of COX-2 induces thephosphorylation of Akt suggest the involvement ofCOX-2 inhibition in celecoxib-induced inhibition ofHCC cell growth. However, the incomplete protection ofcelecoxib-induced inhibition by PGE2 or COX-2 overex-pression and the relatively high concentrations of cele-coxib (25-50 �mol/L) required for induction of apoptosisalso indicate the possible involvement of COX-2–inde-pendent mechanisms. In addition, the difference in theamino acid residues of phospho-Akt (decrease of Aktphosphorylation at Thr308 and Ser473 by celecoxib; in-crease of Akt phosphorylation at Thr308 but not Ser473 byCOX-2 overexpression) in HCC cells also suggests aCOX-2–independent mechanism in celecoxib-mediatedinhibition of growth. Taken together, it is plausiblethat celecoxib induces apoptosis through both COX-2
766 LENG ET AL. HEPATOLOGY, September 2003

activity-dependent and -independent pathways in HCCcells.
In summary, this study showed the direct role ofCOX-2 in the growth control of human liver cancer cells.Our data provide evidence for the involvement of Aktactivation in COX-2–mediated HCC growth. Further-more, we have shown that inhibition of Akt phosphory-lation is an important mechanism by which the COX-2inhibitor celecoxib inhibits the growth of liver cancercells. Thus, the role of COX-2–controlled prostaglandinmetabolism in hepatocarcinogenesis and the mechanismsby which COX-2 inhibitors prevent liver cancer warrantfurther investigation.
Acknowledgment: The authors thank Dr. TimothyHla at the University of Connecticut Health Centerfor providing the human COX-2 expression plasmidand Pharmacia Corp. (St. Louis, MO) for providingcelecoxib.
References1. Okuda K, Kojiro M, Okuda H. Neoplasms of the liver. In: Schiff L, Schiff
ER, eds. Diseases of the Liver. Philadelphia: Lippincott, 1993;1236-1296.2. Shiota G, Okubo M, Noumi T, Noguchi N, Oyama K, Takano Y,
Yashima K, et al. Cyclooxygenase-2 expression in hepatocellular carci-noma. Hepatogastroenterology 1999;46:407-412.
3. Koga H, Sakisaka S, Ohishi M, Kawaguchi T, Taniguchi E, Sasatomi K,Harada M, et al. Expression of cyclooxygenase-2 in human hepatocellularcarcinoma: relevance to tumor dedifferentiation. HEPATOLOGY 1999;29:688-696.
4. Bae SH, Jung ES, Park YM, Kim BS, Kim BK, Kim DG, Ryu WS. Ex-pression of cyclooxygenase-2 (COX-2) in hepatocellular carcinoma andgrowth inhibition of hepatoma cell lines by a COX-2 inhibitor, NS-398.Clin Cancer Res 2001;7:1410-1418.
5. Rahman MA, Dhar DK, Yamaguchi E, Maruyama S, Sato T, Hayashi H,Ono T, et al. Coexpression of inducible nitric oxide synthase and COX-2in hepatocellular carcinoma and surrounding liver: possible involvement ofCOX-2 in the angiogenesis of hepatitis C virus-positive cases. Clin CancerRes 2001;7:1325-1332.
6. Kondo M, Yamamoto H, Nagano H, Okami J, Ito Y, Shimizu J, Eguchi H,et al. Increased expression of COX-2 in nontumor liver tissue is associatedwith shorter disease-free survival in patients with hepatocellular carcinoma.Clin Cancer Res 1999;5:4005-4012.
7. Bayer BM, Beaven MA. Evidence that indomethacin reversibly inhibits cellgrowth in the G1 phase of the cell cycle. Biochem Pharmacol 1979;28:441-443.
8. Kern MA, Schubert D, Sahi D, Schoneweiss MM, Moll I, Haugg AM,Dienes HP, et al. Proapoptotic and antiproliferative potential of selectivecyclooxygenase-2 inhibitors in human liver tumor cells. HEPATOLOGY
2002;36:885-894.9. Endoh T, Tang Q, Denda A, Noguchi O, Kobayashi E, Tamura K,
Horiguchi K, et al. Inhibition by acetylsalicylic acid, a cyclo-oxygenaseinhibitor, and p-bromophenacylbromide, a phospholipase A2 inhibitor, ofboth cirrhosis and enzyme-altered nodules caused by a choline-deficient,L-amino acid-defined diet in rats. Carcinogenesis 1996;17:467-475.
10. Denda A, Tang Q, Endoh T, Tsujiuchi T, Horiguchi K, Noguchi O,Mizumoto Y, et al. Prevention by acetylsalicylic acid of liver cirrhosis andcarcinogenesis as well as generations of 8-hydroxydeoxyguanosine andthiobarbituric acid-reactive substances caused by a choline-deficient, L-amino acid-defined diet in rats. Carcinogenesis 1994;15:1279-1283.
11. Denda A, Endoh T, Tang Q, Tsujiuchi T, Nakae D, Konishi Y. Preventionby inhibitors of arachidonic acid cascade of liver carcinogenesis, cirrhosis
and oxidative DNA damage caused by a choline-deficient, L-amino acid-defined diet in rats. Mutat Res 1998;402:279-288.
12. Denda A, Ura H, Tsujiuchi T, Tsutsumi M, Eimoto H, Takashima Y,Kitazawa S, et al. Possible involvement of arachidonic acid metabolism inphenobarbital promotion of hepatocarcinogenesis. Carcinogenesis 1989;10:1929-1935.
13. Tanaka T, Kojima T, Okumura A, Sugie S, Mori H. Inhibitory effect ofthe non-steroidal anti-inflammatory drugs, indomethacin and piroxicamon 2-acetylaminofluorene-induced hepatocarcinogenesis in male ACI/Nrats. Cancer Lett 1993;68:111-118.
14. Gupta C, Banks M, Shinozuka H. Elevated levels of prostaglandin E2 inthe liver of rats fed a choline deficient diet: possible involvement in livertumor promotion. Cancer Lett 1989;46:129-135.
15. Fitzpatrick FA, Soberman R. Regulated formation of eicosanoids. J ClinInvest 2001;107:1347-1351.
16. Gupta RA, Dubois RN. Colorectal cancer prevention and treatment byinhibition of cyclooxygenase-2. Nat Rev Cancer 2001;1:11-21.
17. Fosslien E. Molecular pathology of cyclooxygenase-2 in neoplasia. AnnClin Lab Sci 2000;30:3-21.
18. Fournier DB, Gordon GB. COX-2 and colon cancer: potential targets forchemoprevention. J Cell Biochem Suppl 2000;34:97-102.
19. Gately S. The contributions of cyclooxygenase-2 to tumor angiogenesis.Cancer Metastasis Rev 2000;19:19-27.
20. Williams CS, Mann M, DuBois RN. The role of cyclooxygenases in in-flammation, cancer, and development. Oncogene 1999;18:7908-7916.
21. Smith WL, Langenbach R. Why there are two cyclooxygenase isozymes.J Clin Invest 2001;107:1491-1495.
22. Tsujii M, DuBois RN. Alterations in cellular adhesion and apoptosis inepithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell1995;83:493-501.
23. DuBois RN, Shao J, Tsujii M, Sheng H, Beauchamp RD. G1 delay in cellsoverexpressing prostaglandin endoperoxide synthase-2. Cancer Res 1996;56:733-737.
24. McGinty A, Chang YW, Sorokin A, Bokemeyer D, Dunn MJ. Cyclooxy-genase-2 expression inhibits trophic withdrawal apoptosis in nerve growthfactor-differentiated PC12 cells. J Biol Chem 2000;275:12095-12101.
25. Lin MT, Lee RC, Yang PC, Ho FM, Kuo ML. Cyclooxygenase-2 inducingMcl-1-dependent survival mechanism in human lung adenocarcinomaCL1.0 cells. Involvement of phosphatidylinositol 3-kinase/Akt pathway.J Biol Chem 2001;276:48997-49002.
26. Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E,Trzaskos JM, et al. Suppression of intestinal polyposis in Apc delta716knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell 1996;87:803-809.
27. Liu CH, Chang SH, Narko K, Trifan OC, Wu MT, Smith E, Haudens-child C, et al. Overexpression of cyclooxygenase-2 is sufficient to inducetumorigenesis in transgenic mice. J Biol Chem 2001;276:18563-18569.
28. Muller-Decker K, Neufang G, Berger I, Neumann M, Marks F, Fursten-berger G. Transgenic cyclooxygenase-2 overexpression sensitizes mouseskin for carcinogenesis. Proc Natl Acad Sci U S A 2002;99:12483-12488.
29. Verburg KM, Maziasz TJ, Weiner E, Loose L, Geis GS, Isakson PC.Cox-2-specific inhibitors: definition of a new therapeutic concept. Am JTher 2001;8:49-64.
30. Thun MJ, Henley SJ, Patrono C. Nonsteroidal anti-inflammatory drugs asanticancer agents: mechanistic, pharmacologic, and clinical issues. J NatlCancer Inst 2002;94:252-266.
31. Patrono C, Patrignani P, Garcia Rodriguez LA. Cyclooxygenase-selectiveinhibition of prostanoid formation: transducing biochemical selectivityinto clinical read-outs. J Clin Invest 2001;108:7-13.
32. Sheng H, Shao J, Morrow JD, Beauchamp RD, DuBois RN. Modulationof apoptosis and Bcl-2 expression by prostaglandin E2 in human coloncancer cells. Cancer Res 1998;58:362-366.
33. Liu XH, Yao S, Kirschenbaum A, Levine AC. NS398, a selective cycloox-ygenase-2 inhibitor, induces apoptosis and down-regulates bcl-2 expres-sion in LNCaP cells. Cancer Res 1998;58:4245-4249.
HEPATOLOGY, Vol. 38, No. 3, 2003 LENG ET AL. 767

34. Chan TA, Morin PJ, Vogelstein B, Kinzler KW. Mechanisms underlyingnonsteroidal antiinflammatory drug-mediated apoptosis. Proc Natl AcadSci U S A 1998;95:681-686.
35. He TC, Chan TA, Vogelstein B, Kinzler KW. PPARdelta is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell 1999;99:335-345.
36. Yin MJ, Yamamoto Y, Gaynor RB. The anti-inflammatory agents aspirinand salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 1998;396:77-80.
37. Steinbach G, Lynch PM, Phillips RK, Wallace MH, Hawk E, Gordon GB,Wakabayashi N, et al. The effect of celecoxib, a cyclooxygenase-2 inhibi-tor, in familial adenomatous polyposis. N Engl J Med 2000;342:1946-1952.
38. Williams CS, Watson AJ, Sheng H, Helou R, Shao J, DuBois RN. Cele-coxib prevents tumor growth in vivo without toxicity to normal gut: lack ofcorrelation between in vitro and in vivo models. Cancer Res 2000;60:6045-6051.
39. Arico S, Pattingre S, Bauvy C, Gane P, Barbat A, Codogno P, Ogier-DenisE. Celecoxib induces apoptosis by inhibiting 3-phosphoinositide-depen-dent protein kinase-1 activity in the human colon cancer HT-29 cell line.J Biol Chem 2002;277:27613-27621.
40. Grosch S, Tegeder I, Niederberger E, Brautigam L, Geisslinger G. COX-2independent induction of cell cycle arrest and apoptosis in colon cancercells by the selective COX-2 inhibitor celecoxib. FASEB J 2001;15:2742-2744.
41. Harris RE, Alshafie GA, Abou-Issa H, Seibert K. Chemoprevention ofbreast cancer in rats by celecoxib, a cyclooxygenase 2 inhibitor. Cancer Res2000;60:2101-2103.
42. Grubbs CJ, Lubet RA, Koki AT, Leahy KM, Masferrer JL, Steele VE,Kelloff GJ, et al. Celecoxib inhibits N-butyl-N-(4-hydroxybutyl)-nitro-samine-induced urinary bladder cancers in male B6D2F1 mice and femaleFischer-344 rats. Cancer Res 2000;60:5599-5602.
43. Hsu AL, Ching TT, Wang DS, Song X, Rangnekar VM, Chen CS. Thecyclooxygenase-2 inhibitor celecoxib induces apoptosis by blocking Aktactivation in human prostate cancer cells independently of Bcl-2. J BiolChem 2000;275:11397-11403.
44. Waskewich C, Blumenthal RD, Li H, Stein R, Goldenberg DM, BurtonJ. Celecoxib exhibits the greatest potency amongst cyclooxygenase (COX)inhibitors for growth inhibition of COX-2-negative hematopoietic andepithelial cell lines. Cancer Res 2002;62:2029-2033.
45. Han C, Demetris AJ, Michalopoulos G, Shelhamer JH, Wu T. 85-kDacPLA(2) plays a critical role in PPAR-mediated gene transcription in hu-man hepatoma cells. Am J Physiol 2002;282:G586-G597.
46. Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathwayin human cancer. Nat Rev Cancer 2002;2:489-501.
47. Testa JR, Bellacosa A. AKT plays a central role in tumorigenesis. Proc NatlAcad Sci U S A 2001;98:10983-10985.
48. Lawlor MA, Alessi DR. PKB/Akt: a key mediator of cell proliferation,survival and insulin responses? J Cell Sci 2001;114:2903-2910.
49. Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Coffer P,Downward J, Evan G. Suppression of c-Myc-induced apoptosis by Rassignalling through PI(3)K and PKB. Nature 1997;385:544-548.
50. Wu T, Han C, Lunz JG 3rd, Michalopoulos G, Shelhamer JH, DemetrisAJ. Involvement of 85-kd cytosolic phospholipase A(2) and cyclooxygen-ase-2 in the proliferation of human cholangiocarcinoma cells. HEPATOL-OGY 2002;36:363-373.
768 LENG ET AL. HEPATOLOGY, September 2003
Copyright © 2022 FDOKUMEN




















