
the influence of viral vector delivery of superoxide dismutase
172
1 THE INFLUENCE OF VIRAL VECTOR DELIVERY OF SUPEROXIDE DISMUTASE AND CATALASE TO THE HIPPOCAMPUS ON SPATIAL LEARNING, MEMORY AND NMDA RECEPTOR FUNCTION DURING AGING By WEI-HUA LEE A DISSERTATION PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY UNIVERSITY OF FLORIDA 2012
-
Upload
khangminh22 -
Category
Documents
-
view
3 -
download
0
Transcript of the influence of viral vector delivery of superoxide dismutase

1
THE INFLUENCE OF VIRAL VECTOR DELIVERY OF SUPEROXIDE DISMUTASE AND CATALASE TO THE HIPPOCAMPUS ON SPATIAL LEARNING, MEMORY AND
NMDA RECEPTOR FUNCTION DURING AGING
By
WEI-HUA LEE
A DISSERTATION PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT
OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY
UNIVERSITY OF FLORIDA
2012

2
© 2012 Wei-Hua Lee

3
To my family

4
ACKNOWLEDGMENTS
First, I would like to give special thanks to my mentor Dr. Thomas Foster for his
patience, trust and constant guidance in my Ph.D. training. I appreciate my committee
members, Dr. Jeffrey Harrison, Dr. Christiaan Leeuwenburgh, Dr. Lucia Notterpek, and
Dr. Susan Semple-Rowland for their critiques, comments and advices that polish me to
be a great scientist. I also appreciate all the friends I made in Foster lab, Dr. Ashok
Kumar, Asha Rani, Olga Tchigrinova, Michael Guidi, Xiaoxia (Sylvia) Han, Linda Been,
Dr. Kristiina Aenlle, Dr. Karthik Bodhinathan, Dr. Travis Jackson, Dr. Zane Zeier, Dr. Li
Cui, Jose Herrera, and Katrina Velez. You made my life in the lab wonderful. I would
like to thank my collaborators, Dr. Jinze Xu, Dr. Shinichi Someya, Dr. Eduardo
Candelario-Jalil, Dr. Alfred Lewin, Dr. Nicolas Muzyczka and Marvin Servanez for
helping me with my studies. I thank Ms. Betty J. Streetman at the Neuroscience office
for taking care of me all the time, and Mr. Mark Potter at vector core laboratory for AAV
supply. Last but not least, I thank my parents for being my foundation and for their
prayers. I thank my sister for always be on my side, I thank my husband, Che, and my
daughter, Shin-Ning (Julia) for their love and support along this journey.

5
TABLE OF CONTENTS page
ACKNOWLEDGMENTS.................................................................................................. 4
LIST OF TABLES............................................................................................................ 8
LIST OF FIGURES.......................................................................................................... 9
LIST OF ABBREVIATIONS........................................................................................... 11
ABSTRACT ................................................................................................................... 13
CHAPTER
1 LITERATURE REVIEWS........................................................................................ 15
Aging Effects on Cognitive Function ....................................................................... 15 Animal Models Used in Studying Aging .................................................................. 16 Cognitive Decline during Aging............................................................................... 20 Methods for Detecting Age-Related Changes in Spatial Memory in Rodents ......... 24 Mechanism of Brain Aging ...................................................................................... 30
ROS and Aging ................................................................................................ 30 ROS Markers.................................................................................................... 37
Total ROS .................................................................................................. 37 Superoxide................................................................................................. 37 Hydrogen peroxide..................................................................................... 38 Redox state................................................................................................ 39 Oxidative damages .................................................................................... 39 Antioxidant enzymes.................................................................................. 41
Antioxidant Treatments .................................................................................... 43 Viral Vectors Delivery in Central Nervous System (CNS) ....................................... 45
2 MATERIALS & METHODS ..................................................................................... 51
Animals ................................................................................................................... 51 AAV Viral Vector ..................................................................................................... 52 Construction of Lentiviral Vector and Vector Packaging ......................................... 52 SOD1 Lentiviral Vector Packaging, Concentration and Titration............................. 53 Behavior Testing..................................................................................................... 54 Hippocampal Tissue Dissection.............................................................................. 55 Western Immunoblotting ......................................................................................... 55
Preparation of Lysate from Hippocampal Tissue.............................................. 55 Determination of Protein Concentration ........................................................... 56 Electrophoresis................................................................................................. 56 Transfer of Protein............................................................................................ 56 Blotting ............................................................................................................. 56

6
Stripping ........................................................................................................... 57 Immunofluorescence .............................................................................................. 58 Slice Preparation .................................................................................................... 58 Biochemical Assays................................................................................................ 60
SOD Activity ..................................................................................................... 60 Protein Carbonyls............................................................................................. 60 8-oxodGuo........................................................................................................ 60 Determination of GSH and GSSG .................................................................... 61 Determination of Glutathione Peroxidase Activity............................................. 62 Determination of Glutathione Reductase Activity ............................................. 63
Statistics ................................................................................................................. 63
3 INFLUENCE OF VIRAL VECTOR-MEDIATED DELIVERY OF SUPEROXIDE DISMUTASE AND CATALASE TO THE HIPPOCAMPUS ON SPATIAL LEARNING, MEMORY DURING AGING................................................................ 71
Results.................................................................................................................... 72 Efficiency-Specificity of The Viral Vectors ........................................................ 72 Age-Dependent Influence of Enzyme Overexpression on Spatial Learning ..... 74 Overexpression of SOD1+CAT for Four Months Improves Spatial Learning.... 76
Discussion .............................................................................................................. 79
4 THE INFLUENCE OF AAV DELIVERED SUPEROXIDE DISMUTASE 1 AND CATALASE ON HIPPOCAMPAL SYNAPTIC PLASTICITY AND NMDA RECEPTOR FUNCTION ........................................................................................ 96
Results.................................................................................................................... 99 AAV Delivery of SOD1, CAT and GFP in the Hippocampi of Rats ................... 99 SOD1 Overexpression Impairs Spatial Learning while SOD1+CAT
Overexpression Improves Spatial Learning in Aged Rats. .......................... 100 NMDAR-Mediated Synaptic Potentials are Reduced in Rats with SOD1
Overexpression ........................................................................................... 104 Effect of Overexpression of SOD1 and CAT on Glutathione Redox State ..... 105 Effect of Overexpression of SOD1 and CAT on Activities of GSH
Peroxidase and GSH Reductase ................................................................ 106 Discussion ............................................................................................................ 106
5 GENERAL DISCUSSION ..................................................................................... 122
Summary .............................................................................................................. 122 Discussion ............................................................................................................ 124
H2O2 Produced by Overexpression of SOD1 Affects Synaptic Plasticity and Memory in an Age-Dependent Manner ....................................................... 126
Co-Overexpression of SOD1 and CAT in the Hippocampi is More Beneficial Than Overexpression of SOD1 or CAT Alone............................................. 131
Mitochondrial ROS Has Minimal Effect on Synaptic Plasticity and Memory... 132

7
Reactive Oxygen Species (ROS) and Reactive Nitrogen Species (RNS) as Signal Molecules in Synaptic Plasticity ....................................................... 133
LTP During Aging ........................................................................................... 134 Global Versus Local Redox State................................................................... 135
APPENDIX: ADDITIONAL FIGURES......................................................................... 139
LIST OF REFERENCES ............................................................................................. 143
BIOGRAPHICAL SKETCH.......................................................................................... 172

8
LIST OF TABLES
Table page 1-1 Studies using knock out or overexpression of antioxidant enzymes................... 50
2-1 A list of all primary antibodies used in Chapter 3 and Chapter 4 western blot and immune-fluorescence. ................................................................................. 70

9
LIST OF FIGURES
Figure page 1-1 Mitochondrial respiratory chain and superoxide production................................ 49
2-1 Map of sc-trs-smCBA-Human SOD1-myc (SOD1) ............................................. 65
2-2 Map of pTR-UF11-Human SOD2 (SOD2) .......................................................... 66
2-3 Map of pTR-UF11-Human CAT (CAT) ............................................................... 67
2-4 Cue discrimination .............................................................................................. 68
2-5 Spatial and probe discrimination......................................................................... 69
3-1 Neurons are the primary cell type transduced by the AAV vectors..................... 83
3-2 Overexpression of antioxidant enzymes in the hippocampus reduces markers of oxidative stress............................................................................................... 84
3-3 Overexpression of antioxidant enzymes did not affect cue discrimination in water maze. ........................................................................................................ 86
3-4 Overexpression of SOD1 impaired spatial learning in aged rats. ....................... 87
3-5 Overexpression of SOD1 impaired acquisition of a spatial search strategy in aged rats. ........................................................................................................... 88
3-6 Long-term overexpression of SOD1+CAT improved learning in spatial trials in aged rats......................................................................................................... 90
3-7 Overexpression of SOD1+CAT for four months improves spatial learning in probe test.. ......................................................................................................... 91
3-8 Antioxidant enzymes and oxidative stress markers in hippocampus with 5-month SOD1 and SOD1+CAT overexpression................................................... 93
3-9 DNA oxidative damage is reduced by overexpression of SOD1......................... 95
4-1 Co-overexpression of SOD1+GFP and SOD1+CAT. ....................................... 112
4-2 Antioxidant enzymes and oxidative stress markers in hippocampus with 1-month SOD1+GFP and SOD1+CAT overexpression. ...................................... 113
4-3 Overexpression of SOD1+GFP or SOD1+CAT enhanced cue discrimination in the water maze compared to GFP control.. .................................................. 114

10
4-4 Overexpression of SOD1+CAT improved spatial learning while overexpression of SOD1+GFP impaired spatial learning in aged rats............. 115
4-5 Overexpression of SOD1+CAT improves spatial learning in probe tests.......... 116
4-6 NMDAR-mediated synaptic potentials are reduced in rats with SOD1 overexpression. ................................................................................................ 118
4-7 Decreased level of reduced GSH (GSH), total GSH and oxidized GSH (GSSG) are observed in rats with overexpression of SOD1+GFP. .................. 120
4-8 Decreased glutathione peroxidase (GPx) and glutathione reductase (GR) activities are observed in rats with overexpression of SOD1+GFP .................. 121
5-1 A model for altered redox state in aged rats with overexpression of SOD1...... 138
A-1 Control images for hippocampal immunofluorescence. .................................... 139
A-2 SOD1 and myc expression are co-localized in the hippocampus of a rat with overexpression of SOD1. ................................................................................. 141
A-3 Expression of corresponding antioxidant enzymes are higher in the hippocampus of rats with injection of AAV-SOD1, AAV-SOD2, or AAV-CAT. .. 142

11
LIST OF ABBREVIATIONS
°C degree celsius (unit for expressing temperature)
8-oxodGuo 8-oxo-7,8-dihydro-2-deoxyguanosine
AAV adeno-associated virus
ANOVAs analyses of variance
Ca+ Calcium (ionic form)
CA1 cornu ammonis area 1
cAMP cyclic adenosine monophosphate
CAT catalase
COX OxPhos complex IV subunit I
CREB cAMP response element binding protein
ERK extracellular signal-regulated kinase
Fisher’s PLSD Fisher’s protected least significant difference
GAPDH glyceraldehyde 3-phosphate dehydrogenase
GFAP glial fibrillary acidic protein
GFP green fluorescence protein
GPx glutathione peroxidase
GSH glutathione
HNE 4-hydroxy-2-nonenal
H2O2 hydrogen peroxide
LTP long-term potentiation
MAP2 microtubule-associated protein 2
NeuN neuronal nuclei
NHPs non-human primates
NMDA N-Methyl-D-aspartate

12
NO nitric oxide
μg micro grams (1/1000000 of a gram; unit of mass)
μL micro liters (1/1000000 of a liter; unit of volume)
μm micro meter (1/1000000 of a meter; unit of length)
O2- superoxide anion
OH hydroxyl radical
P-GSH GSH antibody binds to GSH-protein conjugate
PP1 protein phosphatase 1
PKA protein kinase A
PKC protein kinase C
post hoc post hoc ergo propter hoc (Latin for “after this”)
ROS reactive oxygen species
S.E.M. standard error of the mean
SOD superoxide dismutase
tg-SOD SOD transgenic mice

13
Abstract of Dissertation Presented to the Graduate School of the University of Florida in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy
THE INFLUENCE OF VIRAL VECTOR DELIVERY OF SUPEROXIDE DISMUTASE
AND CATALASE TO THE HIPPOCAMPUS ON SPATIAL LEARNING, MEMORY AND NMDA RECEPTOR FUNCTION DURING AGING
By
Wei-Hua Lee
August 2012
Chair: Thomas C Foster Major: Medical Science - Neuroscience
Studies employing transgenic mice indicate overexpression of superoxide
dismutase 1 (SOD1) improves memory during aging. It is unclear whether the
improvement is due to a lifetime of overexpression, decreasing the accumulation of
oxidized molecules, or if increasing antioxidant enzymes in older animals could reduce
oxidative damage and improve cognitive function. The first study tested the hypothesis
that overexpression of antioxidant enzymes in hippocampi will delay age-related
memory decline by reducing oxidative damage. This study used adeno-associated virus
(AAV) to deliver antioxidant enzymes (SOD1, SOD2, CAT and SOD1+CAT) to the
hippocampi of young (4-month) and aged (19-month) F344/BN F1 male rats and
examined memory-related behavioral performance one month and four months post
injection. The second study examined the hypothesis that overexpression of antioxidant
enzymes affects N-Methyl-D-aspartate (NMDA) receptor function via regulating redox
state. AAV was used to deliver SOD1+GFP and SOD1+CAT to the hippocampi of aged
(17-month) F344 rats and examined memory-related behavioral performance one-
month post injection. Following behavioral characterization, hippocampi were removed

14
and used to examine NMDA receptor function, determine antioxidant enzyme
expression, redox state-related antioxidants and enzyme activities.
The first study indicated that overexpression of antioxidant enzymes reduced
oxidative damage; however, memory function was not related to the level of oxidative
damage. Increased expression of SOD1, initiated in advanced age, impaired learning.
Increased expression of SOD1+CAT provided protection from impairments associated
with overexpression of SOD1 alone and appears to guard against cognitive impairments
in advanced age. The second study indicated that SOD1 overexpression was
associated with a decrease in the NMDAR-mediated fEPSP. The decrease in NMDAR
function in SOD1 rats could explain the impaired learning. Biochemical assays indicated
that free glutathione, oxidized glutathione and total glutathione decreased in SOD1 rats.
The reduced level of glutathione could contribute to the decreased NMDAR function.
Glutathione peroxidase and glutathione reductase activities decreased in SOD1 rats,
which might be due to altered redox state of the enzymes. In conclusion, my studies
provide support for the idea that altered redox sensitive signaling rather than the
accumulation of damage may be of greater significance in the emergence of impaired
learning and memory.

15
CHAPTER 1 LITERATURE REVIEWS
Aging Effects on Cognitive Function
The lifespan of humans has increased dramatically over the last century due to the
improvement of sanitation, hygiene, health care, medicine, nutrition, and technology
(Wykle et al., 2005). A longer life has uncovered age-associated deficits in cognitive
function, challenging both personal and public health internationally. Aging is the
greatest risk factor for cognitive decline and neurodegenerative diseases (Bishop et al.,
2010). Understanding the mechanisms of aging provides one perspective or avenue of
research that can potentially prevent or delay the onset of late-life dementias.
Historically, it was once thought that the major contributor to age-related cognitive
decline was massive neuronal loss and deterioration of dendritic branching (Ball, 1977;
Brody, 1955; Coleman and Flood, 1987; Scheibel, 1979; Scheibel et al., 1976).
However, mounting research indicates that changes occurring during normal aging are
more subtle and selective than was once believed (Foster, 2012; Morrison and Hof,
1997).
Due to the limitations of approaches to examine the brains of humans, the work of
solving the mysteries of brain aging still heavily relies on studies using various animal
models. The first part of this thesis will review the use of animal models to study aging
and age-related cognitive decline. This section will focus on rodents as well as the
behavioral tasks employed to test cognitive function in rats. In addition, I will provide the
background for the oxidative stress theory of aging, and how this hypothesized
biological mechanism of aging could explain age-related memory loss. Finally, I will
layout the gaps in our knowledge that my work attempts to address.

16
Animal Models Used in Studying Aging
There are inherent advantages and disadvantages with each animal model used
to study aging. The commonly used animal models of aging include nematode worms
(Caenorhadbitis elegans), fruit flies (Drosophila melanogaster), mice, rats, monkeys and
chimpanzees. C. elegans and Drosophila have the advantage of a short lifespan and
excellent genetic tools for studying how a single gene may affect aging (Olsen et al.,
2006); however, these models are evolutionarily far from humans. Thus, some
important aging genes in the invertebrates species have no mammalian homologs
(Rikke et al., 2000), and many physiologically important systems in mammals are not
found in invertebrates, including the immune system (Johnson, 2003). By contrast, 90%
of the DNA from Rhesus monkeys and almost 99% of the DNA from chimpanzees are
identical to humans; therefore, it may not be surprising that many aspects of aging,
including life course, are very similar across primates. Behaviorally, the testing protocols
used in non-human primates (NHPs) can be adapted for use in humans. With this
similarity to humans in terms of molecular and cognitive measures, the NHP studies
have a further advantage in that experiments can be conducted under tightly controlled
laboratory conditions, such as long-term regulation of diet or environmental constraints
(Colman and Anderson, 2011; Ingram et al., 1990). Unfortunately, the high cost and low
availability of aged NHPs limits the utility of these animals in aging research (Lane,
2000).
Laboratory rodents are commonly used in aging studies because of their smaller
size, ease of maintenance, and prolific reproduction. Moreover, most rodents are
relatively short lived, with the longest recorded lifespan being 4 and 5 years for mice
and rats, respectively. Mice and rats share remarkable homology with humans in terms

17
of the underlying neural basis for a number of complex behaviors (Foster, 2012; LeDoux,
2000). However, before delving deeper into a discussion of the advantages of mice and
rats in aging studies, it should be pointed out that there are also important limitations for
laboratory mice and rats. A few rodent species, such as naked mole rats, beavers,
porcupines and several species of squirrels are long-lived, and can maintain a lifespan
of up to 20 years. Thus, studies limited to laboratory mice and rats with the fastest aging
process, may not provide all the desired clues for aging, particularly when attempting to
model humans, one of the slowest aging mammals (Austad, 2005; Buffenstein and
Jarvis, 2002; Jones, 1992). Indeed, some researchers have suggested that including
slow-aging rodents in studies on mechanisms of aging may provide important insights
(Gorbunova et al., 2008). A similar argument has been made for examining the
mechanisms of successful aging in humans (Murabito et al., 2012; Newman et al., 2011;
Rozing et al., 2010).
Mice are now widely used in aging studies due in part to a well described genome
and the relative ease in generating knock out or transgenic animal models (Capecchi,
2005). For example, mice with genetic changes in a single antioxidant gene have
revolutionized the study of free radicals and oxidative damage in aging and diseases
(Bartke and Brown-Borg, 2004; Hasty et al., 2003; Hasty and Vijg, 2004; Li et al., 1995;
Reaume et al., 1996). Rats, on the other hand, have a long history of being used by
experimental psychologists and behavioral neuroscientists, yielding a great body of
knowledge in cognitive and behavioral neuroscience, laying a solid foundation for the
study of cognitive decline in aging.

18
Rat Strains. Genetic background is an important consideration when choosing a
rat model for aging research. Outbred strains (with breeding schemes that avoid
crosses between closely related individuals in order to maintain the maximal level of
heterozygosity in all offspring) such as Wistar, Sprague Dawley and Long-Evans rats
are genetically diverse and more accurately reflect the genetic diversity of the humans.
However, there are several disadvantages to the use of outbred rats (Lipman, 1997).
The characteristics of outbred rats can vary from one another, especially if they derived
from a small breeding population (Nadon, 2006). As a result, the sample size must be
larger in order to adjust to the genetic diversity. Inbred rats that require the rat strain
being mated brother x sister for 20 or more consecutive generation are considered
genetically identical. The genetic identity allows the experiments to be more easily
replicated in different laboratories (Phelan, 1992). However, the high homozygosity of
inbred rats also increases the chances of the population being affected by recessive or
deleterious traits, resulting in strain-specific lesions. As a result, the F1 hybrids of two
inbred strains can be the solution for both outbred and inbred rats. F1 hybrids are
genetically uniform and heterozygous for all of the genes for which the two parental
strains differ, facilitating comparison between individuals. The increased heterozygosity
in F1 hybrids protects them from the high incidence of inbred strain-specific disorders.
The National Institute on Aging (NIA) offers three strains of rats for biological aging
studies, allowing researchers to easily obtain the rats at advanced ages. The inbred
Fischer 344 (F344) strain which has been available through the NIA since the mid-
1970s is the most commonly used rat strain in aging research due to its moderate body
weight throughout life and the absence of genetic variability (Markowska and

19
Savonenko, 2002; Sprott, 1991; Sprott and Ramirez, 1997). Median survival of F344
rats is approximately 24 months for males and 26 months for females (Turturro et al.,
1999). Due to the nature of this inbred strain, F344 rats have a high incidence of certain
diseases, including glomerulonephropathy and leukemia at advanced ages (Lipman et
al., 1999). The inbred Brown Norway (BN) rat lives longer than the F344 rat, with a
median life expectancy of 32 months for both males and females, and has far fewer
strain specific lesions with age (Turturro et al., 1999). The BN strain is a docile rat and
its behavioral performance has been noted to decline more slowly with age than F344
rats (Spangler et al., 1994). However, one report indicated that adult BN rats have poor
performance in many learning tasks, although their performance on reference memory
tasks was similar to other strains (Van Luijtelaar and Coenen, 1988). The F344 × BN
hybrid (F344BNF1) rat strain expresses the expected hybrid vigor and has lower levels
of the strain-specific pathologies seen in the parent populations. Thus, F344BNF1 rats
are more susceptible to environmental factors such as diet restriction (Lipman, 1997;
Lipman et al., 1999; Markowska and Savonenko, 2002; Sprott and Ramirez, 1997).
Further, adult F344BNF1 rats have been shown to exhibit good performance in various
cognitive tasks including water maze, inhibitory and passive avoidance tasks, delayed
nonmatching to position, and active avoidance (Lipman et al., 1996; Sprott and Ramirez,
1997; van der Staay and Blokland, 1996). They also live longer than F344 with the
median life expectancy for males of 34 months and 29 months for females (Turturro et
al., 1999). The hybrid vigor, ability to perform in most of the cognitive tasks, and longer
lifespan make F344BNF1 a good model to examine age-related cognitive decline in the
presence of maintained physical function (Nadon, 2006).

20
For the first study of the dissertation, F344BNF1 rats were employed. We
employed F344BNF1 male rats as our animal model because of all the benefits for
aging studies as mentioned above. In addition, we chose 18-month old (in the middle of
their life span) as the aged group, because cognitive decline measured in tasks for
spatial reference memory and spatial working memory begin to emerge at this age
(Markowska and Savonenko, 2002). Our second study focused on electrophysiological
markers of aging, therefore, we chose male F344 rats in order to better compare the
results to our previous electrophysiological findings using the same rat strain. In addition,
we started treatment at the age of 17 months, because a significant drop in the level of
performance in spatial reference memory tasks was observed around this age in F344
male rats (Markowska, 1999).
Cognitive Decline during Aging
Memory and the Hippocampus. Senescence is associated with a decline in
memory that depends on the hippocampus, sometimes referred to as explicit memory,
episodic memory, or declarative memory (Singer et al., 2003). Before detailing the type
or form of memory that is processed by the hippocampus, it is important to take a
historical perspective on research focused on the neural systems involved in memory.
From a historical perspective, the most famous case is the patient, H. M., who had
bilateral medial temporal lobe resection because of his severe symptoms of epilepsy.
The surgery involved the removal of large portions of the temporal lobe from both
hemispheres of the brain, including amygdala, the entorhinal and perirhinal cortices,
and about two-thirds of the hippocampus. After the surgery, H. M. had much milder
epilepsy symptoms, but he lost the ability to form long lasting memories of the events in
his life that happened since the surgery. In other words, he could not acquire new

21
factual knowledge about the world around him. He also had difficulty remembering
information acquired several years prior to his surgery. Yet, some aspects of H. M.’s
memory did remain intact, such as language, short-term memory, and learning simple
sensorimotor skills. Psychologist Professor Brenda Milner has studied H. M. more
extensively than any other investigator. Milner’s early work in studying H. M. changed
how people thought about memory. Prior to H. M., one of the dominant theories on the
biological basis of memory was based on the work of Karl Lashley, who trained rats in a
maze and then made incisions in their cerebral cortices to determine whether the site of
the incision affected the animals performance when they were reintroduced to the maze.
Lashley observed that memory for the maze degraded as a function of the amount of
damage to the cortex. However, the site of the damage did not affect memory. These
findings led to Lashley's notions of equipotentiality, the idea that the locus is not
important, and mass action, that the amount or size of damage is the critical factor. It is
important to point out that Lashley only looked at the cerebral cortex, whereas the
hippocampus and the amygdale are deep structures. The case of H. M. conflicted with
Lashley’s idea, emphasizing the importance of hippocampal formation for specific types
of memory. In addition, H. M.’s case appeared to support the dual memory model, as
proposed in 1968 by Richard Atkinson and Richard Shiffrin. According to the dual
memory model, new information needs to be transferred from short-term memory to
long-term memory in order to be remembered at a later time. H. M.’s ability to follow
conversations and retain small amounts of verbal information for ~15 seconds
suggested that his short-term memory was still intact, but he could not transfer the
short-term memory into long-term store. In addition, he maintained memories from

22
before the surgery, indicating that much of long-term memory was intact. In conclusion,
H. M. and other patients with hippocampal lesions were impaired in acquiring or storing
new, consciously accessible memories, whereas procedural learning and some remote
memories that were acquired well before the lesion remained intact (Milner, 1972;
Milner et al., 1998; Rempel-Clower et al., 1996; Scoville and Milner, 1957; Zola-Morgan
et al., 1986).
With respect to the current set of studies, an important aspect of the memory
deficits associated with hippocampal damage is impaired spatial memory. The interest
in spatial processing by the hippocampus was driven to a large extent by the discovery
of ‘place cells’ in the rat hippocampus (O'Keefe and Dostrovsky, 1971). Recordings of
cell discharge activity in freely moving animals demonstrated cells that fire when an
animal was in specific locations in an environment. The ensemble of cells provides a
stable representation of the animal’s location, independent of its orientation. Place cells
are believed to be pyramidal cells in the CA1 and CA3 hippocampal subfields, or
granule cells in the dentate gyrus (Moser et al., 2008). Based on the discovery of place
cells, the hippocampus was proposed to have a primary function in forming cognitive
maps of the environment (O’Keefe & Nadel, 1978). In conclusion, the studies of humans
and animals with hippocampal damage, as well as the discovery of place cells indicate
that the hippocampus is essential in acquiring new information, consolidating short-term
memory into long-term memory, and plays an important role in spatial learning and
memory.
The deficits observed during aging are consistent with impaired hippocampal
functions. Elderly humans exhibit intact remote memories and are able to learn and

23
remember new skills (i.e. procedural learning) to a similar level as that observed in
young adults. Older individuals appear to acquire new information; however, their
learning may be slower and they exhibit increased forgetting as retention delays
increase (Davis et al., 2003; Hogge et al., 2008; Huppert and Kopelman, 1989; Kral,
1962; Macdonald et al., 2006; Mitchell et al., 1990; Park et al., 1988). Interestingly,
spatial memory decline is one of the most common complaints expressed by older
people; for example, they are unable to remember the location of household objects and
frequently suffer from spatial disorientation (Jonker et al., 1996). In addition, it is quite
common for the elderly to have difficulty memorizing maps or recalling the temporo-
spatial context of a memory (Wilkniss et al., 1997).
Despite the similarity in memory deficits, impaired spatial learning and impaired
retention with increasing delays, it is important to point out that the memory deficits
observed during aging in humans are not as severe as that observed following
hippocampal damage. Similar distinctions can be made for animal models of age-
related cognitive decline. Animals with hippocampal lesions are impaired in acquiring
spatial information (Martin and Clark, 2007; Morris et al., 1990). In contrast to
hippocampal lesions, most aged animals can acquire new information with only a mild
impairment in acquisition of spatial discrimination (e.g. slower learning rate). In many
cases, with extensive training, aged animals can acquire a spatial reference memory
(memory for a location that is the same from trial to trial, i.e. trial independent) to the
same extent as young animals. In contrast, age-related deficits become more obvious
for tests that focus on acquisition of trial-dependent (locations that change from trial to
trial) spatial information and memory impairments increase with an increase in the

24
retention delay (Bizon et al., 2009; Driscoll et al., 2006; Foster, 1999; Foster et al., 2003;
Frick et al., 1995). Combined with the fact that age-related memory deficits are not
associated with a loss of neurons (Gazzaley et al., 1997; Keuker et al., 2003; Merrill et
al., 2001; Merrill et al., 2000; Pakkenberg and Gundersen, 1997; Peters et al., 1994;
Rapp and Gallagher, 1996; Rasmussen et al., 1996; West et al., 1994), the differences
suggest that memory deficits during aging are not due to lesions and probably result
from more subtle changes in mechanisms for storing and maintaining information
(Foster, 1999). In addition, the results suggest that the training protocols influence the
sensitivity of each task for detecting cognitive changes during aging. The next section
will review techniques/protocols for using the water maze to access spatial memory
impairment in aged rats.
Methods for Detecting Age-Related Changes in Spatial Memory in Rodents
The Water Maze. The insight that aging affects hippocampus-dependent spatial
memory has received broad support across a range of species using a variety of spatial
tasks. Spatial mazes have been employed since the beginning of 20th century (Vincent,
1915; Watson, 1907) and are commonly used today to evaluate spatial cognition in
animals (Fouquet et al., 2011; Ingram, 1988; Kametani et al., 1989). Conventional maze
learning tasks such as the T-Maze, Barnes circular platform maze and Corridor mazes
(Barnes, 1979) have been frequently used to investigate spatial learning in rodents
(McLay et al., 1999). Generally, animals are food or water deprived and the access to
food or water is used as behavioral reinforcement or the motivation for learning the
maze. These tasks require animals to remember a route (e.g. a series of left and right
turns) or learn the spatial layout (e.g. cognitive map) in order to obtain a water or food
reward. Thus, one important factor to take into account is that animals can use different

25
strategies to try and solve spatial problems. In most cases the behavior is defined in
terms of egocentric and allocentric strategies (Burgess, 2006). An egocentric strategy is
based on the information provided by bodily cues, and therefore it is independent of
spatial cues. For this strategy, the animal functions as its own central point of reference,
and so, all other object positions are defined in relation to the animal’s position in space
(O’Keefe and Nadel, 1978; Klatzky, 1998). An allocentric strategy depends on spatial
cues and their relation to each other. When using this strategy the animal memorizes
the target location in relation to the spatial position of the environmental reference
landmarks (Benhamou and Poucet, 1995).
The water maze is usually considered superior to other conventional mazes for
several reasons. First, learning on the water maze is relatively rapid such that animals
can acquire a spatial search strategy in a single training session. Second, the water
maze does not allow the animals to use aromatic cues to orient themselves in the
escape search, which may happen in the dry mazes. Third, the water maze does not
require water or food deprivation. For food and water deprivation, it may be hard to
determine if the motivation is similar across different age groups. In the case of the
water maze, the animal is motivated to learn the spatial layout of the maze in order to
escape from the water and young and old animals appear to be equally motivated. In
addition, the water maze is more suitable for examining the effects of calorie-restricted
animals. Finally, the water maze can be designed to detect impaired sensory-motor or
motivational differences, and the use of egocentric and allocentric strategies.
The water maze task was first introduced in 1982 by R. Morris’s paper in which he
developed this task for studying the role of the hippocampus in spatial learning in rats

26
(Morris, 1984; Morris et al., 1982). In the task, the rats must use distal spatial cues to
locate a hidden escape platform just under surface of the water. The original protocol
consisted of 8 days of training, one trial on the first day and 4 trials a day during the next
seven days. The rat is released from a different start location along the wall of the tank
or pool during each training trial, so the rat must use environmental cues, rather than a
series of motor responses, to navigate to the hidden escape platform. The spatial
learning is reflected by faster escape latency and shorter distance to reach the platform
over the course of training trials. Importantly, Morris tested three groups of rats in the
water maze, including a group with hippocampal lesions, a group with cortical lesions,
and a control group with intact brain structures. The rats with hippocampal lesions
exhibited a significantly longer path length to reach the platform indicating that acquiring
spatial information in water maze with a hidden platform is hippocampus-dependent.
The rats were also given a probe trial with the platform removed. During the probe trial,
the rat is released from the quadrant opposite from the quadrant that had the platform.
The animal is allowed to swim for 60 seconds in the pool and the time spending
searching each quadrant, total distance from the platform location, and number of times
the animal crosses the location that held the escape platform are recorded. The probe
trials provide a more sensitive measure of the use of a spatial search strategy that
latency or distance to reach the platform during training trials. A decrease in latency and
distance could be observed for animals using a cue-response strategy, e.g., swim in a
certain distance from the wall until they bump into the platform. In the Morris study, the
group with hippocampal lesions performed significantly worse than other two groups in
terms of spending significant less time searching in the location which had previously

27
held the platform, supporting the idea that hippocampus is involved in spatial learning.
In contrast, the rats with hippocampal lesions were not impaired in the cue
discrimination version of water maze task. For the cue discrimination task, a visible
escape platform is used as a local visual cue. This visible escape platform can be
located in different spatial locations across training trials. In addition, the area
surrounding the pool is usually separated from other part of the room by a curtain to
minimize distal spatial cues. Animals use the same sensory-motor and motivational
components as in spatial/hidden platform training; however, the most efficient way to
escape the pool is to employ a cue response strategy by simply locating the visible
platform and swimming to it. One study that examined the effect of brain lesions on the
acquisition of the cue discrimination task found that damage to the basal ganglia, but
not the hippocampus, disrupted acquisition on this task (Packard and McGaugh, 1992).
In summary, the evidence indicates that the hippocampus and basal ganglia are parts of
systems that differ in the type of memory they mediate. Furthermore, performance on
the spatial discrimination version of the task is more sensitive to impairment associated
with aging.
For the current studies, we employed the cue task prior to spatial training. This
protocol consists of cue/visual platform training in which a visibly prominent, moveable
platform with a flag on the top is used. Cue/visual training consisted of 5 blocks of three
trials, with all training delivered in one day. For cue training, the visible platform was
placed in a novel position for each trial. The cue task was used to identify sensory-
motor or motivated deficits, which would affect acquisition of a spatial search strategy.
In addition, animals learn the procedural aspect of the task, including how to swim and

28
learning that the pool wall is not a route of escape (Vorhees and Williams, 2006). When
animals are trained in the cue task first, they may perform better in the spatial task due
to the acquisition of the procedural aspects of the task (Foster, 2012). Similarly, animals
trained initially in the spatial version of water maze may perform relatively poorly, but
exhibit superior performance in subsequent cue training (Gerlai, 2001). As a result, the
initial poorer performance in spatial training may be due to deficits related to procedural
learning or to deficits in spatial learning, or a combination.
For studies presented in this dissertation, spatial training was initiated three days
following completion of the cue discrimination task. Recently it has been emphasized
that training schedules are important in determining the sensitivity of the Morris escape
task for the early detection of age-related deficits in the acquisition and retention of
spatial information in rodents (Foster, 2012). A common spatial training schedule
involves distributed training over days, in which the animals are trained 3~4 trials a day
for several days. An alternative method is to provide all the training in a single episode,
usually by massing all the training trials into a single session. Historically, much of the
research on aging has employed distributed training to examine cognitive decline during
aging. However, distributed training is not as sensitive to the earliest signs of an age-
related decline in hippocampal function. In many cases, the older animals can acquire a
spatial reference memory if provided training over several days. Thus, age differences
in acquisition, which are evident in the first couple days of training, may disappear after
several days of repetitive training. Similarly, studies in mice suggest that distributed
training procedures can mask memory deficits in mice with mutations of memory related
genes, e.g. CREB, PKA, PP1 (Genoux et al., 2002; Gulinello et al., 2009; Jones et al.,

29
2001; Kogan et al., 1997; Malleret et al., 2010). On contrast, due to the fact that the
learning or/and memory deficits are more pronounced in the initial training trials, many
laboratories have massed all the trainings into a single section in order to increase the
sensitivity of the task (Commins et al., 2003; Dash et al., 2002; Lal et al., 1973; Spreng
et al., 2002; Vasquez et al., 1983). Studies using F344BNF1 rats, the same rat strain
employed in the first study (see Chapter 3), also show that distributed testing across
several days is insensitive to cognitive decline between adult (6-month-old) and aged
(21-month-old) F344BNF1 rats (Wu et al., 2004a). In contrast, using a massed training
procedure, age-related cognitive deficits were observed between middle-age (12-15
months), aged (25-27 months), and very old (35-38 months) F344BNF1 rats (Carter et
al., 2009). Due to the sensitivity of the massed training version of the task, particularly
for animals within the age range employed in the studies presented in this dissertation,
we used a spatial training protocol consisting of five blocks of three trials per block with
training massed into a single day, for examining the effect of overexpression of
antioxidant enzymes in the hippocampus on spatial learning and memory in young and
aged F344BNF1 rats.
In the current set of studies, we employed three probe trials with the platform
removed to examine acquisition and retention of spatial information. The first probe trial
(probe 1) was given after the 5th block of spatial training, in which the rat was released
from the opposite quadrant and allowed to swim for up to 60s. This probe trial is used to
examine the acquisition of a spatial search strategy. A refresher-training block is given
immediately following probe 1. The refresher-training block is used to refresh rat’s
memory of platform location and insure that the probe trial does not result in the

30
extinction of spatial memory (Lattal et al., 2003). An hour later, a retention probe trial
(probe 2) was delivered followed by another refresher-training block. This second probe
trial is used to examine the one-hour retention of newly learned spatial search strategy.
Twenty-four hours later, a block of spatial training was given followed by a probed trial
(probe 3). The rats are tested in this third probe trial for their ability to relearn the
platform location. Note that the rats are sent back to their home cages with water and
food during the one-hour and twenty-four time periods between testing.
A camera on the roof tracks the rats’ performance on every trial, and different
measurements can be made using EthoVision software. To assess the performance
during training, latency to find the platform and path length are both used in cue and
spatial training. Using latency to find the platform, as the only measure in cue and
spatial training, may be confounded by an age-related decline in swim speed. Thus, a
longer latency may reflect slower swim speed rather than impaired learning. Similarly,
measuring only path length may ignore the fact that some animals initially tend to float
in the water giving artificially short path lengths but longer latency to find the platform
during the first block of trials. With the same consideration, we also apply multiple
measures in the probe trials, including latency to first reach the platform location,
percentage time in the goal quadrant, discrimination index: (time in goal quadrant-time
in opposite quadrant)/(time in goal quadrant +time in opposite quadrant), and number of
platform crossings.
Mechanism of Brain Aging
ROS and Aging
From the preceding review it is clear that aging is associated with selective
changes in specific cognitive functions. Furthermore, the deficits appear to be selective

31
for a particular neural system involving the hippocampus. This raises the question as to
what might cause the age-related cognitive decline. One possibility is that the age-
related memory loss is due to cellular aging. A prominent theory of cellular aging, the
free-radical theory of aging, indicates that the overproduction of endogenous reactive
oxygen species (ROS), including superoxide, hydrogen peroxide, and hydroxyl radicals
result in a pattern of cumulative damage due to modification of proteins, nucleic acids,
and polyunsaturated fatty acids of the lipid membrane (Oliver et al. 1987; Smith et al.
1991; Leutner et al. 2001; Liu et al. 2002). Normally, the production of ROS can act as a
signaling mechanism in the brain (Bokoch and Knaus, 2003; Infanger et al., 2006).
However, many studies have provided evidence for increased oxidative damage during
normal aging (Halliwell, 1992; Harman, 1992), and in relation to neurodegenerative
diseases of aging, including Alzheimer’s and Parkinson’s disease (Olanow and
Arendash, 1992). Thus, aging can be viewed as the process of accumulation of
oxidative damage.
ROS are produced by a number enzymatic reactions including cytochromes p450
(CYTP450), xanthine oxidase (XOD), phospholipase A2 (PLA2), lipoxygenase (LOX)
and ribonucleotide reductase, and nicotinamide adenine dinucleotide phosphate
(NADPH) oxidase (Noxs) (Muller et al., 2007). For example, NADPH oxidase represents
a family of proteins with oxidase activity, that contain six transmembrane domains. Nox
is found primarily in neutrophils and microglia, playing a protective role in fighting
infection by releasing superoxide (Block et al., 2004; Lambeth, 2004). Superoxide, the
primary product of the Nox protein, forms when NADPH binds to Nox on the protein’s
cytosolic side, where NADPH transfers electrons to flavin adenine dinucleotide (FAD)

32
and the heme centers and finally the oxygen on the outer membrane surface (Brown
and Griendling, 2009).
In addition, a common source of superoxide is through the mitochondria as by-
products of respiration. Superoxide is generated through oxidative phosphorylation; and
forms when an electron leaks from electron transport chain and reacts with oxygen.
During normal respiration, as much as 0.4 - 4% of total oxygen consumed by the
mitochondria can be converted to the free radical superoxide (Boveris, 1984; Boveris
and Chance, 1973; Turrens and Boveris, 1980). The majority of mitochondrial
superoxide is generated from two respiratory chain complexes, complex I (NADH
dehydrogenase) and complex III (ubiquinone-cytochrome c reductase) (Raha et al.,
2000). Complex I produces superoxide in the mitochondrial matrix and complex III
produces superoxide in both the matrix and intermembrane space (Han et al., 2001;
Miwa et al., 2003; Muller et al., 2004) (See Fig. 1-1).
Superoxide can be dismutated to hydrogen peroxide spontaneously or
enzymatically via superoxide dismutase (SOD) (Fridovich, 1983b). The properties of
superoxide and hydrogen peroxide, suggest that they act differently as damaging or
signaling molecules. Because of the short half-life and fast dismutation rate, superoxide
must be produced very close to its target to be effective as a signaling or damaging
molecule. In addition, superoxide is not able to diffuse across biological membranes due
to its negative charge. In this regard, hydrogen peroxide is more stable than superoxide
and is also capable of crossing biological membranes.
Superoxide is also capable of reacting with nitric oxide (NO), forming highly
reactive and potentially damaging peroxynitrite (OONO−). The formation of OONO− from

33
superoxide can then lead to reversible glutathionylation of proteins via reactive
cysteines, as has been described for Na+-K+ pump regulation (Figtree et al., 2009).
Superoxide is also known to react with protein thiols such as cysteine residues.
Nevertheless, the reaction rate of SOD in converting O2- to H2O2 is much faster than
that of O2- with biothiols, which helps to diminish the potential for damage (Forman et
al., 2004). H2O2 also reacts with protein thiols, and although the reaction rate of O2- with
protein thiols is chemically faster than that of H2O2, the greater stability and diffusibility
of H2O2 increase its probability of reacting with the protein thiol groups involved in ROS
signaling. This suggests that physiological protein thiol oxidation is most likely H2O2
dependent. Indeed, although the production of O2- is the main biological function of Nox
proteins and is important in the bactericidal activity of Nox2, much of the signaling that
occurs is directly mediated by its dismutation product, H2O2.
Because of the presence of SOD in the cell, H2O2 is formed rapidly from Nox-
generated O2-, or in the case of Nox4, perhaps prior to the release of O2
- from the
enzyme (Dikalov et al., 2008). H2O2 is also tightly regulated biologically by catalase,
glutathione peroxidase, and peroxiredoxins, which convert H2O2 to water and other
metabolites. H2O2 can reversibly react with low pKa cysteine residues (Winterbourn and
Metodiewa, 1999) on proteins to initially form a disulfide bond (–SSR) and sulfenic acid
(–SOH). Sulfinic acid (–SO2H) and sulfonic acid (–SO3H) can be formed by additional
oxidation; however, these latter reactions are essentially irreversible, and not useful for
signaling (Barford, 2004; Forman et al., 2002).
The balance between the production and removal of ROS is a critical factor in
determining the extent of oxidative stress. ROS mediated oxidative stress can be

34
defined or measured as irreversible oxidative damage of critical biological molecules,
causing DNA strand breaks, destruction of ion transporters or other specific proteins,
and membranous lipid peroxidation (Halliwell, 1992). In this case, the term irreversible
indicates that repair of damaged molecules will require cellular cleaning
process/degradation. There are many oxidative lesions in endogenous mammalian DNA,
of which 8-hydroxyguanine (8-oxodG) is one of the most abundant (Ames, 1989).
Oxidative DNA damage can be repaired through base excision repair (BER) and
nucleotide excision repair (NER) mechanisms. BER can recognize specific lesions in
the DNA, and thus correct only damaged bases that can be removed by a specific
glycosylase (Friedberg, 1995; Seeberg et al., 1995). For example, 8-oxo-dG is
recognized by the DNA glycosylases, 8-oxoguanine glycosylase/AP lyase (Ogg1) (Xie
et al., 2004). An additional repair mechanism in the BER system involves removing dA
mispaired to 8-oxo-dG to prevent 8-oxo-dG from forming transverse G to T mutations.
NER employs a complex set of proteins that recognize large distortions in the shape of
the DNA double helix, and remove a short single-stranded DNA segment that includes
the lesion (Sancar, 1996; Wood, 1996).
Irreversible oxidation products of amino acids are most frequently observed as
hydroxylated and carbonylated amino acid derivatives and detection of protein-
associated carbonyls represents a common way of assessing protein oxidation after
carbonyl derivatization by dinitrophenyl hydrazine (Levine et al., 1994). Oxidized
proteins are generally less active, less thermo-stable and are likely to expose
hydrophobic amino acids at their surface. In addition, protein damage can result from
protein adduct formation with lipid peroxidation products such 4-hydroxy-2-nonenal

35
(HNE) and from oxidation of glycation products leading to the formation of glycoxidation
adducts such as pentosidine or carboxymethyllysine (Berlett and Stadtman, 1997).
These latter modifications often bring carbonyl groups and/or cross-links within the
protein. In the cytosol, oxidized proteins have been shown to be preferentially degraded
by the 20S proteasome in an ATP- and ubiquitin-independent manner (Davies, 2001;
Shringarpure et al., 2003). Moreover, upon oxidative stress chaperone-mediated
autophagy can participate, to accelerate degradation of oxidized proteins carrying a
KFERQ motif (Kiffin et al., 2004).
Lipid peroxidation refers to the oxidative degradation of lipids, a process in which
free radicals steal electrons from the lipids in cell membranes, resulting in cell damage.
Polyunsaturated fatty acids are the most common lipids affected by free radicals
because they contain multiple double bonds formed by methylene-CH2-groups. The
brain is particularly susceptible to free radical attack because the brain is enriched with
polyunsaturated fatty acids. In addition, the brain is low in antioxidant capacity and has
high utilization of molecular oxygen relative to other organs (Retter, 1995). Damaged
lipids cannot be repaired; rather, they must be removed and replaced. Thus, damage of
cell membranes may accumulate. In blood, damage to lipids such as cholesterol, will
accumulate along cardiovascular vessels, which is associated with atherosclerosis
(Pratico, 2001). The end product of lipid peroxidation, such as 4-hydroxy-2-nonenal
(HNE) and malondialdehyde (MDA) can also cause damage to DNA (Marnett, 1999)
and proteins (Kaemmerer et al., 2007). As a result, lipid peroxidation has been
suggested to lead to carcinogenesis (Hu et al., 2002), Alzheimer’s disease (Dei et al.,

36
2002; Montine et al., 2002) and Parkinson’s disease (Dexter et al., 1986; Gupta et al.,
2010; Kilinc et al., 1988).
Reducing-oxidizing (redox) state is a term that has historically been used to
describe the ratio of the interconvertible oxidized and reduced form of a specific redox
couple. For example, Sir Hans Krebs defined the redox state of NAD+/ NADH couple as
[free NAD+]/[ NADH] (Krebs, 1967; Krebs and Gascoyne, 1968; Krebs and Veech,
1969). In recent years, the term redox state has been used not only to describe the
state of a particular redox pair, but also to more generally describe the redox
environment of a cell. This more general use of the term redox state is not very well
defined and differs considerably from historical uses. Schafer and Buettner suggest that
the term redox environment should be used when a general description of a linked set
of redox couples is intended. Schafer and Buettner define this kind of redox state/redox
environment as:
The redox environment of a linked set of redox couples as found in a biological fluid, organelle, cell, or tissue is the summation of the products of the reduction potential and reducing capacity of the linked redox couples present.
There are many redox couples in a cell that work together to maintain the redox
environment. The redox state of the glutathione disulfide-glutathione couple, oxidized
form of GSH (GSSG) and reduced form of GSH (GSH) can serve as an important
indicator of redox environment because the GSSG/2GSH couple is the most abundant
redox couple in a cell (Schafer and Buettner, 2001).
From the above discussion it is apparent that measures of ROS levels, oxidative
damage, and redox state are essential for examining the free radical theory of brain
aging and the possible link between these processes and cognitive decline. The

37
following section will discuss different methods for measuring ROS, redox state,
oxidative damage, and antioxidant enzymes.
ROS Markers
Total ROS
Dihydro-compounds such as 2′,7′-dichlorodihydrofluorescein (DCFH) and its
derivatives, have traditionally been used for detecting oxidative stress in vivo (LeBel et
al., 1992). Two big drawbacks for using these probes including lack of specificity for
different ROS and photosensitivity which tends to autoxidize DCFH and causes high
background fluorescence (Afzal et al., 2003; Marchesi et al., 1999; Rota et al., 1999;
Soh, 2006).
Superoxide
With one unpaired electron, superoxide is a free radical, and is toxic biologically. A
number of methods have been developed to detect superoxide. Cytochrome c, lucigenin
and luminol are the most commonly used scavenger indicators, which react with
superoxide and produce detectable products. All three chemicals have drawbacks.
While cytochrome c has low sensitivity with superoxide, both lucigenin and luminol
generate superoxide as they react to produce light, which raise an objection for using
them to quantify superoxide. More recently, several florescent probes have been
developed to detect the overall level of superoxide or superoxide specifically localized in
mitochondria. Dihydroethidium (DHE) is one such superoxide-sensitive dye used to
localize superoxide in tissues (Al-Mehdi et al., 1997; Benov et al., 1998; Murakami et al.,
1998). MitoSOX-Red is a mitochondrial specific superoxide indicator and can be paired
with a mitochondria-specific dye such as MitoTracker-Green FM to detect mitochondria
produced superoxide (Hu et al., 2007a). The fluorescent probe bis (2,4-

38
dinitrobenzenesulfonyl) fluorescein, is the only probe that is highly selective to
superoxide over other ROS, and can be used to detect real-time production of
superoxide by living cells (Maeda et al., 2005). It should be noted that, although many
methods and probes have been developed to detect superoxide, many people still
doubt that the steady state concentration of superoxide can be measured directly
because of its very short half-life at 37C (1 x 10-6 seconds). As a result, the levels of
superoxide should be determined indirectly by measuring the rates of production and
removal and the relationship between these two processes; or by measuring the
damage caused by superoxide.
Hydrogen peroxide
The classical method for measuring H2O2 concentration is through directly
measuring the absorbance at 330nm of the H2O2 molecule or through reaction with
ferrous iron (Fe2+), monitored via a subsequent reaction with dye Xylenol Orange and
detected as an increase in absorbance in solution at 550nm. Several more sensitive
assays have been developed based on the horseradish peroxidase (HRP) mediated
reaction between H2O2 and some indicating reactant. For example, in the presence of
H2O2, HRP will oxidize luminol and produce light. The light output can be proportionally
compared to the H2O2 present. Regardless of the assay used, two possible sources of
error must be controlled. The first is that other redox active compounds can also create
a signal in the assay. These signals can still be detected after treating the samples with
catalase, which specifically remove H2O2. Thus, any signal lost after treating with
catalase can be assigned to H2O2. The second possible error interfering with the assay
often comes from sample with high peroxide scavenging compounds, which can

39
continue to be active after the sample preparation. It is therefore important to work
under the conditions for minimizing peroxide degradation, e.g. shorter sample storage
time, prepare and keep the samples and all reagents used in the assay at a lower
temperature.
Redox state
Glutathione is considered to be the major thiol-disulfide redox buffer of the cell
(Gilbert, 1990). On average, the GSH concentration in the cytosol is 1–11 mM (Smith et
al., 1996). This is far higher than most other redox active compounds. Measurements of
total GSH and/or GSSG levels have been used to estimate the redox environment of
cells. Many researchers estimate the redox state of the system by taking the ratio of
[GSH]/[GSSG]. This is convenient as the units divide out, so it is not necessary to
determine an absolute concentration. A measurement in mg/mg protein, arbitrary
fluorescence units, or the area under an HPLC peak can be entered into the ratio and a
useful estimate calculated (Schafer and Buettner, 2001). Based on the results from the
first study (Chapter 3), we hypothesized that overexpression of SOD1 created a more
oxidized redox environment. Therefore, in the second study (Chapter 4), GSH and
GSSG levels were measured in hippocampal tissue following overexpression of
SOD1+GFP, SOD1+CAT, or GFP to determine the redox state. The methods for
measuring GSH and GSSG are described in more detail in Chapter 2.
Oxidative damages
For studies examining ROS induced damage to macromolecules, the most
common measures related to damage of nucleotides, proteins, and lipids. The
measurement of each is discussed below.

40
Among all the lesions discovered thus far, one of the most abundant in DNA and
RNA is the 8-hydroxyguanine due to the high oxidation potential of this base relative
to cytosine, thymine, and adenine (Ames and Gold, 1991; Gajewski et al., 1990).
Besides its abundance, 8-hydroxydeoxyguanosine (8-oxodG) and 8-hydroxyguanosine
(8-oxoG) are identified as the most detrimental oxidation lesions due to their mutagenic
effect (Ames and Gold, 1991) in which this damaged molecule can pair with both
adenine and cytosine with the same efficiency (Shibutani et al., 1991). This mis-pairing
brings about the alteration of genetic information through the synthesis of DNA and
RNA. In RNA, oxidation levels are mainly estimated through 8-oxoG-based assays. So
far, approaches developed to directly measure 8-oxoG level include HPLC-based
analysis and assays employing monoclonal anti-8-oxoG antibody. The HPLC-based
method measures 8-oxoG with an electrochemical detector (ECD) and total G with
a UV detector (Weimann et al., 2002). The ratio that results from comparing the two
numbers provides the extent that the total G is oxidized. Monoclonal anti-8-oxoG mouse
antibody is broadly applied to directly detect this residue on either tissue sections or
membrane, offering a more visual way to study its distribution in tissues and in discrete
subsets of DNA or RNA.
Protein carbonyl content is the most general indicator for protein oxidation. Several
approaches have been taken to detect and quantify the carbonyl content in protein
preparation. The most convenient procedure is the reaction between 2,4-
dinitrophenylhydrazine (DNPH) and protein carbonyls. DNPH reacts with protein
carbonyls, forming a Schiff base to produce the corresponding hydrazine, which can be
analyzed spectrophotometrically (Reznick and Packer, 1994).

41
Two lipid peroxidation products are commonly used as markers for lipid damage,
4-hydroxy-2-nonenal (HNE) and malondialdehyde (MDA). HNE is formed by
peroxidation of linoleic acid. HNE is more stable than free radicals and it is able to
migrate to sites that are distant from its formation, resulting in greater damage. The
most damaging effect of HNE is its ability to form covalent adducts with histidine, lysine,
and cysteine residues in proteins, enabling a modification in the activity of proteins
(Butterfield et al., 1997). It has been shown that the HNE-modified proteins, along with
neurofibrillary tangles, are present in the senile plaques in aged dogs (Papaioannou et
al., 2001). Increased levels of HNE have also been found in Alzheimer’s and
Parkinson’s disease (Yoritaka et al., 1996; Zarkovic, 2003). These findings support the
idea that oxidative stress/damage is elevated in neurodegenerative diseases and lipid
peroxidation may contribute to the deterioration of central nervous system (CNS)
function. MDA is formed by peroxidation of arachidomic acid. MDA induces DNA
damage by reacting with amino acids in protein to form adducts that disrupt DNA base-
pairing. Increased levels of MDA have been found in the aged canine brain (Head et al.,
2002). In the aged human brain, increased levels of MDA have been found in inferior
temporal cortex and in the cytoplasm of neurons and astrocytes (Dei et al., 2002), as
well as in the hippocampus and cerebellum of aged rodents (Cini and Moretti, 1995;
Gemma et al., 2002). Lipid peroxidation from biological samples can be detected by
antibodies (Opalach et al., 2010) or through thiobarbituric acid reaction (Cui et al., 2009;
Dawn-Linsley et al., 2005).
Antioxidant enzymes
Antioxidative enzymes, including superoxide dismutase (SOD), catalase (CAT)
and glutathione peroxidase (GPx) (Halliwell, 1991; Brigelius-Flohe, 1999) are the

42
primary factors to protect neural tissue from toxic free radicals. Thus, a change in
antioxidant enzyme activity can be used as biomarkers for detecting a possible change
of ROS. Certainly, SOD activity is a major defense mechanism against oxygen toxicity
(McCord and Fridovich, 1969; Fridovich, 1983) by converting superoxide to the less
reactive hydrogen peroxide. Catalase (CAT) and glutathione peroxidase (GPx)
cooperate with SOD to decompose H2O2 to water and O2. Although age-related
changes in the antioxidant enzymatic system have been described, results on the SOD,
GPx, and CAT activities are contradictory. Total SOD activity was reported to be
unchanged during aging in whole brain from older Wistar rats (Sahoo and Chainy, 1997)
and in older Fisher 344 rats (Tian et al., 1998). Other studies reported that the activity of
SOD was decreased in cerebrum, hypothalamus, hippocampus, cerebellum, brain stem
and spinal cord with increasing age from Charles Foster rats (Gupta et al., 1991) and in
whole brain from Fischer 344 rats (Semsei et al., 1991). Catalase activity in the whole
brain was significantly decreased in aged Fisher 344 rats (Tian et al., 1998) and Wistar
rats (Sahoo and Chainy., 1997). As described above, catalase has very low activity in
the brain, and it seems to play a secondary role in converting H2O2 compared with other
organs. The activity of GPx was reported to be unchanged in the cortex (Cand and
Verdetti, 1989; Dogru-Abbasoglu et al., 1997), and in whole brain extracts (Sahoo and
Chainy, 1997) from Wistar rats; whole brain (Tian et al., 1998) and several brain regions
including hippocampus, striatum, substantia nigra, and three different cortical regions of
the cerebrum (frontal, parietotemporal and occipital regions) (Carrillo et al., 1992) from
Fischer 344 rats; however, a decrease activity during aging has also been reported
(Benzi et al., 1989). In addition, changes in the activity of antioxidant enzymes have

43
been reported to be markedly dependent on the sex of animals (Guo et al., 1993; Tam
et al., 2003; Banos et al., 2005; Ehrenbrink et al., 2006). Furthermore, one paper
suggest that it is the cooperation of antioxidant enzyme network, rather than their
individual levels that is crucial for the overall oxidant/antioxidant status during aging
process (Sobocanec et al., 2008). In conclusion, although the conflicting results may
arise from different factors like animal strains, conditions of animal maintenance, or
experimental procedures, the results still indicate that the activities of antioxidant
enzymes are age- and tissue-specific.
Antioxidant Treatments
Previous studies have shown that in both young and aged rats, dietary
supplements of fruit and vegetable extracts, with high antioxidant activity, retarded age-
related declines in neuronal and cognitive function (Joseph et al., 1999). Several studies
have also addressed the effects of antioxidant dietary supplementation on humans. The
most common antioxidants, for example, tocopherol (Vitamin E) and ascorbate (vitamin
C) are usually taken through supplementation or vitamin-enriched food. However,
vitamin E only operates under strong oxidative condition, i.e. Alzheimer’s disease,
whereas it can evolve into pro-oxidant under mild oxidative conditions (Bowry et al.
1992). Vitamin C, on the other hand, has little effect on improving cognition
(Mendelsohn et al., 1998; Grodstein et al., 2003; Maxwell et al., 2005). Vitamin C is
excreted rapidly after ingestion, and the hydrophilic character of vitamin C makes it
difficult for it to penetrate through blood brain barrier (BBB). Though some studies
indicated that vitamin C can reduce the amount of metal ions, which may down-regulate
the free radicals production from the fenton reaction (Stohs and Bagchi , 1995; Carr and
Frei, 1999). Thus, although adding antioxidant supplements or an antioxidant enriched

44
diet may seem like a relatively simple therapy, it may not act at the source of ROS in the
nervous system; as such it may have little effect on normal cognitive aging.
Different invertebrate animal models that overexpress antioxidant enzymes have
been created as tools for studying the relationship between oxidative stress and
longevity. The literature concerning whether increasing antioxidant enzymes can delay
aging is controversial. Overexpression of Cu/Zn SOD, MnSOD or catalase extend life
span in the short-lived strain Drosophila melanogaster but, the same phenomena was
not observed in a long-lived strain (Mockett et al., 2003; Orr et al., 2003; Orr and Sohal,
1993, 1994; Sun et al., 2002). Overexpression of catalase, targeted to mitochondria,
extends life span in mice (Schriner et al., 2005). Administration of synthetic superoxide
dismutase/ catalase mimetics have been shown to extend the life span of
Caenorhabditis elegans (Melov et al., 2000; Sampayo et al., 2003a; Sampayo et al.,
2003b), but the effect may depend on the experimental conditions (Keaney and Gems,
2003; Keaney et al., 2004).
The hypothesis that age-related cognitive decline could be reversed by increasing
antioxidant defenses has been tested in transgenic mouse models expressing different
forms of SOD. Relative to wild-type controls, the enzymatic activity in the brain of
transgenic mice represents a 6-fold increase for SOD1 (Kamsler and Segal, 2003b), a
2.3-fold increase for SOD2 (Maragos et al., 2000), and about a 10-fold increase for
SOD3 (Thiels et al., 2000). Depending on the type of SOD examined, genetic
manipulations of SOD expression can have a profound effect on age-related cognitive
impairment, and an age-dependent influence on synaptic plasticity. Young adult mice
overexpressing SOD1 or extracellular SOD exhibit impairment in long-term potentiation

45
(LTP), a form of synaptic plasticity that is thought to be involved in memory. These mice
also exhibit impaired hippocampus-dependent memory (Gahtan et al., 1998; Kamsler
and Segal, 2003b; Levin et al., 1998; Thiels et al., 2000). In contrast, aged transgenic
mice over expressing SOD1 or SOD3 exhibit improved LTP and SOD3 transgenic mice
exhibit better performance in the Morris water maze compared with wild-type littermates
(Kamsler and Segal, 2003b; Levin et al., 2002). Up regulation of SOD2, on the other
hand, has no obvious effect on hippocampal LTP (Hu et al., 2007a). Taken together,
these results indicate that overexpression of SOD1 and SOD3 affect synaptic plasticity
and memory in an age-dependent fashion (See Table 1-1 for a summary of antioxidant
enzymes studies in mice).
Viral Vectors Delivery in Central Nervous System (CNS)
Adeno-associated virus (AAV) vectors have been utilized in pre-clinical and clinical
gene delivery studies, primarily because the vectors can transduce non-dividing cells
and confer long-term stable gene expression, without associated inflammation or
toxicity (Bessis et al., 2004; Goncalves, 2005; Haberman et al., 1998). AAV is a small,
icosahedral, nonenveloped virus that belongs to Parvoviridae family. The name
“associated” came from the fact that Adenovirus is usually served as helper virus for a
productive infection of AAV to occur. The wild AAV has a linear single-stranded DNA
genome of about 4.7 kb of either plus or minus polarity. There are two inverted terminal
repeats (ITRs) flanking the two viral genes, rep (replication) and cap (capsid). The rep
gene encodes four regulatory proteins (Rep78, Rep68, Rep52 and Rep40) required for
the AAV life cycle. The cap gene encodes three structural proteins (VP1, VP2 and VP3)
to form a capsid that is approximately 22 nm. In our studies, we used recombinant AAV
(rAAV) to deliver antioxidant enzymes to the hippocampus. rAAV does not contain any

46
AAV rep and cap genes. Rather, rep and cap genes are replaced with a gene or
construct of interest, which is flanked by the ITRs containing all the cis-acting elements
necessary for replication and packaging. Use of rAAV does have some disadvantage.
The cloning capacity of the vector is relatively limited. Efficient packaging of rAAV can
be performed with constructs ranging from 4.1 kb to 4.9 kb in size. Large genes are,
therefore, not suitable for use in a standard rAAV vector.
There have been at least 11 AAV capsid serotypes described. The surface of the
AAV capsid is involved in cell binding, internalization, and trafficking within the targeted
cell; therefore, each capsid exhibits a unique tissue tropism and transduction efficiency
(Van Vliet et al., 2008). Serotype 2 (AAV2) has been the most extensively examined
(Bartlett et al., 1998; Burger et al., 2004; Fischer et al., 2003; Nicklin et al., 2001). AAV2
is used to deliver genes to cells in CNS (Flannery et al., 1997; Kaplitt et al., 1994;
McCown, 2005, 2011; McCown et al., 1996) and primarily transduces neurons (Bartlett
et al., 1998; Burger et al., 2004; Klein et al., 1998) through the interaction of AAV2
capsid proteins with heparin sulphate proteoglycan (HSPG) moieties on the neuronal
cell surface. In our current studies, we used pseudotyped rAAV2/5 (putting the genome
of rAAV2 into the capsid of rAAV5) because rAAV2/5 has been shown to transduce
hippocampus with high efficiency in CA1-CA3 and the dentate gyrus (Burger et al.,
2004). We also take the advantage of AAV that can affect various genes simultaneously
by using multiple vectors (Mandel et al., 1998; Muramatsu et al., 2002; Rendahl et al.,
1998; Shen et al., 2000). We transduced hippocampal neurons with two separate AAV
vectors to overexpress SOD1 and CAT or GFP simultaneously in the same cell.

47
Lentiviruses are a subclass of Retroviruses, developed based on the human
immunodeficiency virus (HIV). Unlike other retroviruses that can only infect dividing cells,
lentivirus has the capacity to integrate into the genome of non-dividing cells, which
makes it an attractive CNS gene transfer tool (Blomer et al., 1997; Naldini et al., 1996a).
Lentivirus has relatively large cloning capacity compared to AAV. The capacity is
generally considered to be 8-10 kb (Naldini et al., 1996b; Sinn et al., 2005).
A process known as pseudotyping can modify the host range of lentiviral vectors.
Psudotyped lentiviral vectors consist of vector particles carrying glycoproteins derived
form other enveloped viruses. Such particle has the tropism of the virus from which the
glycoproteins were obtained. For our initial studies, we chose the most widely used
glycoproteins for pseudotyping lentiviral vector, the vesicular stomatitis virus
glycoproteins (VSV-G) (Barsoum et al., 1997; Naldini et al., 1996b; Reiser et al., 1996;
Russell and Cosset, 1999). VSV-G pseudotyped lentiviral vector has been shown to
have broader transduction range (Coil and Miller, 2004), increased transduction
efficiency (Barsoum et al., 1997), and stronger viral particles to make concentration by
ultracentrifugation possible (Burns et al., 1993). However, the virion transport of VSV-G
pesudotyped lentivirus is limited to relatively shorter distance (Akli et al., 1993; Alisky et
al., 2000). Lentivirus has been demonstrated to transduce most cell types in CNS in
vivo, including neurons, astrocytes, adult neuronal stem cells, oligodendrocytes, and
glioma cells with desired cell type promoters or modified envelop proteins that bind to
the receptors only found in the desired cell type (Blomer et al., 1997; Consiglio et al.,
2004; Geraerts et al., 2006; Jakobsson et al., 2003; Miletic et al., 2004). For safety
reasons, lentivirus never carries the genes for replication (Dull et al., 1998; Zufferey et

48
al., 1998; Zufferey et al., 1997). As a result, to produce a lentivirus, several plasmids
need to be transfected into a packaging cell; including the packaging plasmids that
encode virion proteins, such as the capsid and the reverse transcriptase and the
plasmid contains genetic material (See Chapter 2 for SOD1 lentiviral vector packaging,
concentration and titration).
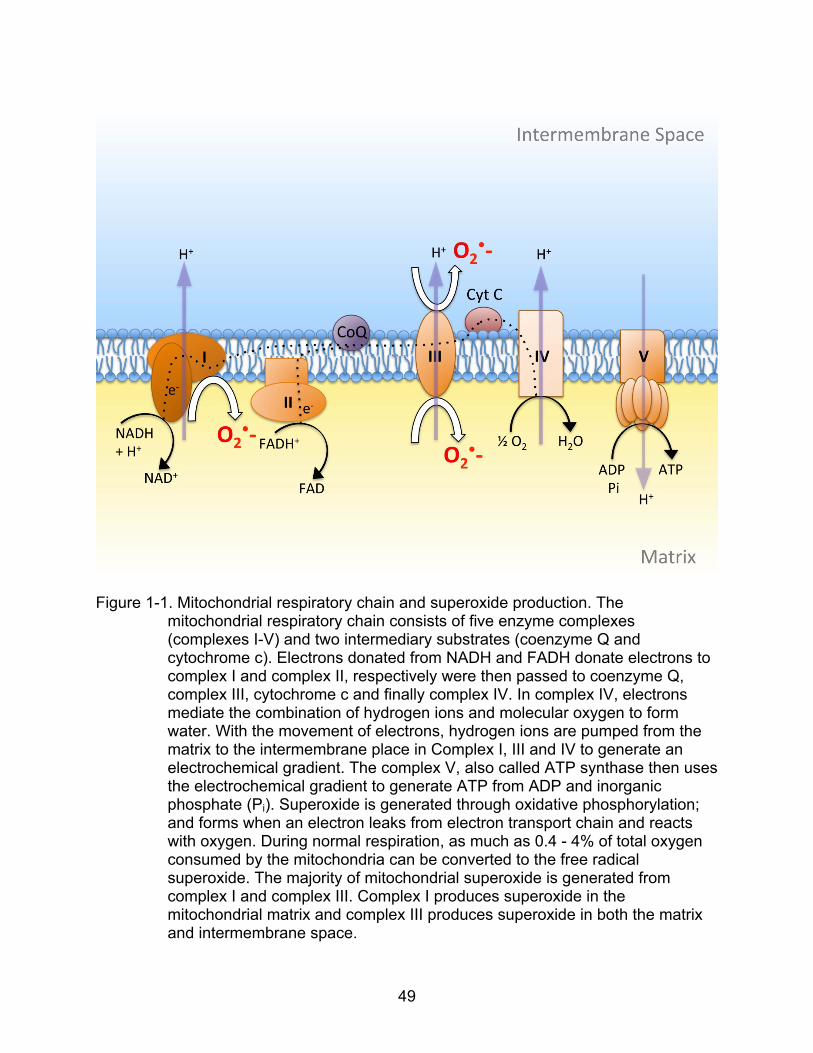
49
Figure 1-1. Mitochondrial respiratory chain and superoxide production. The mitochondrial respiratory chain consists of five enzyme complexes (complexes I-V) and two intermediary substrates (coenzyme Q and cytochrome c). Electrons donated from NADH and FADH donate electrons to complex I and complex II, respectively were then passed to coenzyme Q, complex III, cytochrome c and finally complex IV. In complex IV, electrons mediate the combination of hydrogen ions and molecular oxygen to form water. With the movement of electrons, hydrogen ions are pumped from the matrix to the intermembrane place in Complex I, III and IV to generate an electrochemical gradient. The complex V, also called ATP synthase then uses the electrochemical gradient to generate ATP from ADP and inorganic phosphate (Pi). Superoxide is generated through oxidative phosphorylation; and forms when an electron leaks from electron transport chain and reacts with oxygen. During normal respiration, as much as 0.4 - 4% of total oxygen consumed by the mitochondria can be converted to the free radical superoxide. The majority of mitochondrial superoxide is generated from complex I and complex III. Complex I produces superoxide in the mitochondrial matrix and complex III produces superoxide in both the matrix and intermembrane space.

50
Table 1-1. Studies using knock out or overexpression of antioxidant enzymes Gene symbol
Gene name (localization)
Cognition Life span
Main antioxidant function
Reference
Sod1-/- CuZn superoxide dismutase (C, M)
Not reported
Mutiple pathologies, ~30% shortened life span
Scavenger of O2
-
Elchuri et al., 2005
Sod2-/- Mn superoxide dismutase (M)
Not reported
Neonatal lethal Scavenger of O2
- Li et al., 1995
Sod3-/- Extracellular superoxide dismutase (E)
Not reported
“Normal,” no reduction in life span
Scavenger of O2
- Sentman et al., 2006
Cat-/- Catalase (C) Not reported
“Normal,” life span not reported
Scavenges H2O2
Ho et al. 2004
Gpx1-/- Glutathione peroxidase 1 (C)
Not reported
“Normal,” life span not reported
Scavenges H2O2
Ho et al. 1997
Gpx2-/- Glutathione peroxidase 2 (E)
Not reported
“Normal,” life span not reported
Scavenges H2O2
Esworthy et al. 2001
Gpx4-/- Phospholipid glutathione peroxidase (M, C)
Not reported
Embryonic lethal Scavenges H2O2
Yant et al., 2003
Tg-SOD1 CuZn superoxide dismutase (C, M)
Impaired in young Enhanced in old
“Normal,” does not extend life span
Scavenger of O2
- Gahtan et al., 1998; Kamsler and Segal, 2003; Kamsler et al., 2007; Huang et al., 2000
Tg-SOD2 Mn superoxide dismutase (M)
No effect Growth retardation and decreased fertility, life span not reported
Scavenger of O2
- Hu et al., 2007; Raineri et al., 2001
Tg-SOD3 Extracellular superoxide dismutase (E)
Impaired in young Enhanced in old
Not reported Scavenger of O2
- Thiels et al., 2000; Levin et al., 1998; Hu et al., 2007; Levin et al., 2002;
MCAT; Tg-CAT
Catalase (M); Catalase (Peroxision)
Not reported
Extend medium and maximal life span (MCAT) Does not extend maximal life span (PCAT)
Scavenges H2O2
Schrier et al., 2005; Chen et al., 2004

51
CHAPTER 2
MATERIALS & METHODS
Animals
All treatments were approved by the Institutional Animal Care and Use Committee
and were in accordance with guidelines established by the United States Public Health
Service Policy on Human Care and Use of Laboratory Animals. Young tg-SOD1 mice
exhibit impaired spatial learning (Gahtan et al., 1998); therefore, young rats were
included to examine age effects and specificity of gene overexpression in the first study.
In the first study, AAV was injected into the hippocampus of young (4-month) and aged
(19-month) male Fischer 344/Brown Norway F1 rats (Harlan Laboratories) in order to
express SOD1 (young: n = 14; aged: n = 13), SOD2 (young: n = 9; aged: n = 9), CAT
(young: n = 10; aged: n = 9), mix of SOD1+CAT (young: n = 11; aged: n = 12) or GFP
(young: n = 8; aged: n=8). We also included young (n = 4) and aged (n = 4) no surgery
controls. No behavioral difference was observed between GFP and no surgery controls;
therefore, these animals were combined for behavioral analyses. In the second study,
AAV was stereotactically injected bilaterally into dorsal hippocampi of 17-month old
male Fischer 344 rats (Harlan Laboratories). Rats were injected with GFP as control (n
= 10); SOD1+GFP (n = 6); or SOD1+CAT (n = 7). In the second study, AAV containing
antioxidant enzymes genes were stereotactically injected bilaterally into dorsal
hippocampi of 17-month old male Fischer 344 rats. Rats were injected with GFP as
control (n = 14); SOD1+GFP (n = 11); or SOD1+CAT (n = 12). Behavioral testing in the
water maze was initiated 1 month following virus injections.

52
AAV Viral Vector
Virus particles were produced and quantified by dot blot analysis, through the
Powell Gene Therapy Center at the University of Florida (Gainesville, FL). Human
SOD1 gene with a c-terminal myc tag (myc) was cloned into, self-complementary AAV
(scAAV), containing a cytomegalovirus/chicken β-actin promoter with a
cytomegalovirus-immediate early (CMV-IE) enhancer (Figure 2-1). Plasmids pTR-UF11-
hSOD2 and pTR-UF11-hCAT were gifts from Dr. Nicolas Muzyczka (Department of
Molecular Genetics and Microbiology, University of Florida). These plasmids express
the human SOD2 gene, which codes for Mn-superoxide dismutase, or the human CAT,
which encodes catalase, respectively (Figure 2-2 and 2-3). Plasmid UF11-GFP was
used as control vector. Expression of each gene was driven by the chicken β-actin
proximal promoter and the CMV immediate early enhancer (CBA promoter). All vectors
contain AAV2 inverted terminal repeats for packaging in pseudotyped AAV5 capsid.
Rats were anesthetized with ketamine/xylazine (90/10 mg/kg) and virus was
stereotactically injected at 2 sites bilaterally in the hippocampus using glass pipettes.
Each injection consisted of 2 μL of GFP (dot blot titer: 1.02 x 1013 vg/mL), SOD1 (dot
blot titer: 1.14 x 1013 vg/mL), SOD2 (dot blot titer: 6.99 x 1012 vg/mL), CAT (dot blot
titer: 2.53 x 1013 vg/mL), or 3 uL 2:1 mix of SOD1 and CAT.
Construction of Lentiviral Vector and Vector Packaging
pTYF-EF1-CuZnSOD-IRES-GFP. The open reading frame encoding the human
Cu/Zn SOD (SOD1) was amplified from cDNA clone using primers that introduced NheI
and ClaI sites on the 5`- and 3`-ends of the coding region ( 5’t a a g c t a g c t t t g c g t c
g t a g t c t c c t g c 3’ and 5’t a t a t a t c g a t a a g g g a a t g t t t a t t g g g c g 3’). The
resulting 567-base pair SOD1 PCR products were subcloned into TOPO TA cloning

53
vector. The human SOD1 cDNA was removed from TOPO vector and ligated into pTYF-
EF1a-IRES-eGFP linker vector by NheI and ClaI to create pTYF-EF1-CuZnSOD-IRES-
GFP (LV-SOD1/eGFP).
SOD1 Lentiviral Vector Packaging, Concentration and Titration
SOD1 transducing vectors were packaged into vesicular stomatitis virus envelop G
(VSV G)-pseudotyped vector to make lentivirus using a three plasmid packaging system.
The 293 FT cells (Invitrogen, no. R70007) were plated at a density of 3 x 107 cells in
T225 flasks and were transfected the next day (80-90 % cell confluence) with the three
plasmid vectors, pNHP, pHEF-VSVG and the SOD-1 transducing vector using
Superfect (Qiagen). Media containing virus were collected at 30 hours and 45 hours
post-transfection. The medium collected 30 hours after transfection were centrifuged at
2000 rpm for 5min at 4 °C to remove cell debris and stored at 4 °C until the next day.
The medium collected 45 hours after transfection were similarly centrifuged, mixed with
the 30 hour sample and filtered through a 0.45 um low-protein-binding durapore filter
device (Millipore). All but 26 mL of the filtrate were centrifuged at 2500 rcf for 1 h at 4 °C.
The retentate recovered from the column were mixed with the 26 mL of unconcentrated
medium, loaded into a 30 mL conical polyallomer tube (Beckman), and centrifuged at
24,000 rpm in a SW28 rotor (Beckman) for 1.5 hr at 4°C. The resulting viral pellet was
overlayed with DMEM and stored at 4 °C overnight. The next day, the viral solution was
aliquoted and stored at -80 °C until use. Particle titers were determined using a HIV p24
ELISA kit (Cell biolabs, no. VPK-108-HIV p24), according to the protocols provided.
Biological titration was determined by transfecting HeLa cells with serial diluted
lentivirus, and counting the cells expressing GFP by flow cytometry.

54
Behavior Testing
Methods for water maze testing have been published previously (Foster et al.,
2001). Rats were first trained to find a visible platform using 15 trials separated to 5
blocks. Rats that did not reach the platform within 60 seconds on all trials during the 5th
block were removed from the experiment. Standard water maze testing across several
days is insensitive to cognitive decline across this age span in Fischer 344/Brown
Norway F1 rats (Wu et al., 2004b). Therefore, the task difficulty was increased by
employing a single day massed training protocol, which is sensitive to age (Carter et al.,
2009). Spatial discrimination testing occurred three days later and consisted of 18 trials
separated into 6 blocks. A free swim probe trial was inserted between blocks 5 and 6.
For the probe trial, the number of platform crossings was counted and the time spent in
both goal and opposite quadrants was recorded.
The water maze protocol used in the second study was similar to the one used in
the first study but with some modifications. Rats were first trained to find a visible
platform using 15 trials separated to five blocks (Table 2-5). Rats that did not reach the
platform within 60 s on more than one trial during the fifth block were removed from the
experiment. Four days later, a single-day massed training protocol, which is sensitive to
age was used to test spatial discrimination (Table 2-6). The spatial training protocol
consists 15 trials separated into five blocks. A free swim probe trial with the platform
removed (probe 1) was inserted between blocks 5 and 6. For the probe trial, the number
of platform crossings was counted and the time spent in both goal and opposite
quadrants was recorded. A refresher training block was given following probe 1. An
hour later, a retention probe trial (probe 2) was delivered followed by a refresher training
block. Twenty-four hours later, the rats were tested for their ability to relearn the

55
platform location. A single block of spatial training was given followed by a probed trial
(probe 3). Note that the rats were sent back to their home cages with water and food
during the one-hour and twenty-four time periods.
Hippocampal Tissue Dissection
Starting one week after finishing behavioral testing, animals were euthanized
using isoflurane (Webster, Sterling, MA) and decapitated with a guillotine (Myneurolab,
St Louis, MO). The brains were rapidly removed and transferred to a beaker containing
ice-cold artificial cerebrospinal fluid (ACSF). For biochemical studies, the hippocampi
were removed and flash frozen in liquid nitrogen. The samples were then stored at –
80C until further processing. For the animals used in electrophysiology, one
hippocampus from each animal was flash frozen in liquid nitrogen and stored at –80C
for biochemical analysis and the other hippocampus from the same animal was
dissected out and prepared for electrophysiological recording (see slice preparation).
Western Immunoblotting
Preparation of Lysate from Hippocampal Tissue
The hippocampi were frozen in liquid nitrogen, and stored at -80°C. Hippocampal
tissues were grinded into powder by mortar and pestle with liquid nitrogen. Half of the
tissue powder was homogenized with lysis buffer [Radioimmunoprecipitation assay
buffer (RIPA buffer) supplemented with protease inhibitor, phosphatase inhibitor and
EDTA (Thermo scientific, Rockford, IL)] and incubated for 1 hour on ice with intermittent
vortexing. The remaining tissue powder was gathered in 1.5 mL tube and stored in -
80°C. The lysates were centrifuged at 20,000 g for 30 minutes at 4 °C.

56
Determination of Protein Concentration
The lysates were first diluted 1/20 in a total of 50 L of ddH2O, and were used to
measure protein content using a BCA kit (Pierce, Rockford, IL). The protein
concentration was measured on microplate reader (Bio-Rad Laboratories, Hercules, CA)
at 560 nm. The samples were diluted with lysis buffer to the same protein concentration.
The samples were then mixed with 4X LaemmLi’s SDS-sample buffer [Tris-HCL (250
mM, pH 6.8), SDS (8%), glycerol (40%), -mercaptoethonol (8%) and bromophenol
(0.02%), Boston BioProducts, Ashland, MA] and heated to 100 °C for 5 minutes .
Electrophoresis
Samples (20 g/lane) were loaded on 4-15 % gradient polyacrylamide gels (Bio-
Rad Laboratories, Hercules, CA) and ran at 90 Volts in electrophoresis buffer (25 mM
Tris, 192 mM glycine, 0.1% SDS, pH 8.3, Bio-Rad Laboratories, Hercules, CA) until the
blue indicator line reached the bottom of the gel.
Transfer of Protein
Proteins were transferred to PVDF membranes (GE Healthcare) overnight at 50
Volts, 4 °C in transfer buffer (25 mM Tris-HCl, 192 mM glycine, 20% methanol). Blots
were then stained with Ponceau S and examined for transfer quality (e.g. roughly how
much protein was transferred to the membrane, verifying no air bubbles, or other signs
of transfer failure were present).
Blotting
Blots were washed 5 min in TBST and subsequently blocked in 5% milk in TBST
for 1 hour. Primary antibodies were then applied to blots overnight at 4 °C (Table 2-1
for a list of all antibodies used), washed 3 times (10 min each time) with TBST, and
secondary antibodies applied for 1~2 hours at room temperature. Blots were again

57
washed 4 times (10 min each time) with TBST and developed using Amersham ECL
Plus Western Blot Detection Kit (GE Health Care) on biomax film (Kodak). Blots were
scanned using 6500 scanner (Bio-Rad), and densitometry determined using Image J
Stripping
Stripping of the membranes allowed us to re-probe with different antibodies, thus
avoiding inconsistent sample loading between each electrophoresis blotting, and
conserved the samples for other assays. Typically, the blot was first probed with
antibody with lowest level of signal. This blot was then stripped, blocked with 5% milk in
TBST and re-probed with another primary antibody made from different species. For
example, if the first blot was probed with mouse antibody, then the second blot would be
re-probed with antibody made from another species rather than mouse. This prevented
using of the same secondary antibody in the first and second blotting, which may result
to additional signals from the residual primary antibody after stripping. The blot was then
stripped again and re-probed with either a third antibody or with anti-GAPDH antibody
as loading control. Each membrane was stripped for up to 3 times.
A good stripping should remove the antibodies, but leave transferred proteins. The
recipe for stripping buffer varied from simple as 0.2~0.5 M NaOH or 0.2 M glycine (HCl)
in ddH2O to a more complex buffer adding SDS, Tris, Tween 20, and -mercaptoethanol.
The tripping process can be done at room temperature or at 37C for better results. The
stripping buffer used in current studies was from Thermo Scientific. The membrane was
incubated with the stripping buffer for 15 min at 37C, and 3X washed with TBST before
blocked with 5% milk in TBST.

58
Immunofluorescence
Brains were postfixed in 4% paraformaldehyde followed by 30% sucrose in PBS
(4°C). Sections (10-20 μm) were incubated with primary antibodies overnight at 4°C.
(See Table 2-1 for a list of primary antibodies used in western blot and
immunofluorescence). The brain sections were then washed and incubated in
corresponding Alexa 488 or 594 secondary antibodies (Molecular Probes, Eugene, OR)
for 1 hour at room temperature. Sections were washed and counterstained with 4'-6-
Diamidino-2-phenylindole (DAPI) solution (0.1 μg/mL in PBS) before mounting.
Expression was confirmed using fluorescent microscopy (Zeiss Axioplan 2 upright
fluorescent microscope, equipped with a QImaging Retiga 4000R Camera with RGB-
HM-5 Color Filter and QImaging QCapture Pro 6.0 software; QImaging Surrey, BC
Canada). No primary antibody control and isotype control were made to be compared
with SOD1 and c-myc staining. In no primary antibody control, the tissue was incubated
with antibody diluent (3% goat serum in PBS), without the primary antibody included.
The tissue was then incubated with secondary antibody and DAPI before mounting. In
isotype control, the tissue was incubated with antibody diluent, supplemented with a
non-immune immunoglobulin of the same isotype (for example, IgG2a for c-myc control)
and concentration as the primary antibody. The tissue was then incubated with the
secondary antibody and DAPI before mounting. The isotype control helped ensure that
what appeared to be specific staining was not caused by non-specific interactions of
immunoglobulin molecules with the sample.
Slice Preparation
For physiology experiments, one animal per day was killed, staring were one week
after completing the water maze testing. Recordings were obtained from hippocampi of

59
aged, SOD1+GFP, SOD1+CAT or GFP transduced animals. Rats were anesthetized
with isoflurane (Halocarbon Laboratories, River Edge, NJ) and the hippocampi were
collected from rats after decapitation. Transverse hippocampal slices (400 m) were cut
parallel to the alvear fibers using a tissue chopper. Slices were maintained in an
interface chamber (Harvard Apparatus, Boston, MA) at 30 °C and perfused with an
oxygenated artificial cerebrospinal fluid (ACSF) (in mM: 124.0 NaCl, 2.0 KCl, 1.25
KH2PO4, 1.5 MgSO4, 2.4 CaCl2, 26.0 NaHCO3, and 10 glucose. Slices were permitted
to recover for 120 min before recording. Extracellular field excitatory post synaptic
potentials (fEPSPs) were recorded from Schaffer collateral-CA1 synapses using glass
micropipettes (4–6 MΩ) filled with recording medium (ACSF). Two concentric bipolar
stimulating electrodes (outer pole: stainless steel, 200 m diameter; inner pole:
platinum/iridium, 25 m diameter, FHC, Bowdoinham, ME) were positioned
approximately 1 mm from either side of the recording electrode localized in the middle
of the stratum radiatum.
Baseline fEPSP responses to stimuli of a given intensity (input-output curves)
were measured to examine basal synaptic transmission at the Schaffer collateral CA3–
CA1 synapse in hippocampal slices. Stimulus intensity was from 0 V to 28 V when
examining the total fEPSP, and from 0 V to 32 V when examining the NMDAR-mediated
fEPSP in order to achieve the maximum response. To obtain the NMDAR-mediated
fEPSP, slices were incubated in ACSF containing low extracellular Mg2+, (0.5 mM),
AMPA receptor antagonist, 6,7-dinitroquinoxaline-2,3-dione (DNQX, 30 M), and GABA
receptor antagonist, picrotoxin (PTX, 10 M).

60
For induction of LTP, the stimulus current was set to produce a response 50–60%
of the maximal fEPSP slope (from input-output curve). After stable baseline recording at
0.033 Hz for at least 20 min, high-frequency stimulation (HFS) was delivered to the
pathway with 4 episodes of 100 Hz (each episode was 1 sec with an inter episode time
of 1 sec) at the baseline stimulus current, and recorded for 60 min after HFS. A
simultaneously recorded control (non-HFS) pathway received the test stimulus but not
the HFS.
The signals were amplified, filtered between 1 Hz and 1 kHz, and stored for off-line
analysis. For analysis, two cursors were placed around the initial descending phase of
the EPSP waveform, and the maximum slope (mV/ms) of the EPSP was determined by
a computer algorithm that found the maximum change across all sets of 20
consecutively recorded points (20 kHz sampling rate) between the two cursors.
Biochemical Assays
SOD Activity
SOD activity was measured using the HT superoxide dismutase assay kit
(Trevigen Inc, Gaithersburg, MD) according to the manufacturer’s instructions.
Protein Carbonyls
Protein carbonyls were measured using a commercial ELISA (Zentech PC Test,
Protein Carbonyl Enzyme Immuno-Assay Kit, Biocell Corp, Papatoetoe, NZ) according
to the manufacturer’s instructions.
8-oxodGuo
8-oxo-7,8-dihydro-2-deoxyguanosine (8-oxodGuo) levels were determined
according to the neutral guanidine isothiocyanate (GTC) method previously described
(Cui et al., 2009; Hofer et al., 2006). Briefly, after homogenization in GTC in the

61
presence of the metal chelator deferoxamine mesylate (DFOM; Sigma), proteins and
lipids were removed using organic solvents and centrifugation. After salt/isopropanol
precipitation of nucleic acid at 80 °C and washing in 70% ethanol, nucleic acids were
dissolved in 30 μM DFOM and hydrolyzed using 4 U Nuclease P1 (MP Biomedicals,
Irvine, CA) and 5 U alkaline phosphatase (Sigma) in 30 mM sodium acetate, 20 μM
ZnCl2, pH 5.3, at 50 °C for 60 min. After filtration to remove enzymes, nucleosides were
separated using HPLC-EC-UV and analyzed for dGuo and 8-oxodGuo
electrochemically using a Coulochem III detector from ESA Inc. (Chelmsford, MA).
HPLC peaks were quantified against daily made calibration curves of standards from
Sigma and Calbiochem (San Diego, CA).
Determination of GSH and GSSG
A glutathione fluorescent detection kit (Arbor Assays LLC, Ann Arbor, MI) was
used to measure reduced/oxidized glutathione (GSH/GSSG) ratio in rat hippocampus
samples. Rat hippocampi were frozen by liquid nitrogen and stored in -80 C until use.
The tissues were homogenized in ice cold RIPA buffer supplemented with protease
inhibitor, phosphatase inhibitor and EDTA (Thermo scientific, Rockford, IL), and
centrifuged at 14,000 rpm for 10 minutes at 4°C. An aliquot of the supernatant was
removed for protein determination. A second aliquot of the supernatant containing 50 g
of protein was precipitated with an equal volume of ice cold 5% (w/v) 5-sulfosalicylic
acid (SSA) solution. After 10 minutes incubation with SSA at 4°C, the samples were
centrifuged at 14,000 rpm for 10 minutes at 4°C to remove precipitated protein. The
supernatant was collected and diluted with assay buffer to reduce the SSA
concentration to 1%. After mixing the sample or standard with a proprietary

62
nonfluorescent molecule, ThioStar®, that covalently binds to the free thiol group on
GSH to yield a highly fluorescent product, the samples were incubated at room
temperature for 15 minutes, and the fluorescent product was read at 510 nm on a
fluorescent plate reader with excitation at 370-410 nm. Free glutathione, GSH, was read
first, followed by addition of a reaction mixture that converts all the oxidized glutathione,
GSSG, into free GSH, which then reacts with the excess ThioStar® to yield the signal
related to total GSH content. The concentration of GSSG was estimated by subtracting
the measured free GSH from the measured total GSH.
Determination of Glutathione Peroxidase Activity
Glutathione peroxidase (GPx) activity was measured by using a glutathione
peroxidase assay kit (Cayman, Ann Arbor, MI). Briefly, frozen rat hippocampal tissues
were homogenized in cold RIPA buffer supplemented with protease inhibitor,
phosphatase inhibitor and EDTA (Thermo scientific, Rockford, IL), and centrifuged at
10,000 g for 15 minutes at 4°C. The supernatants were removed for analyses. The 20
uL samples containing 40 μg total proteins were added to a solution containing GSH,
glutathione reductase, and nicotinamide adenine dinucleotide phosphate (NADPH). The
reaction was initiated by adding substrate cumene hydroperoxide (final concentration:
0.22 mM), and the reduction was recorded at 340 nm using a kinetic program (6
readings at 1-minute intervals). The GPx activity was determined by the rate of
decrease in absorbance at 340 nm. The GPx activity is calculated using the following
formula: GPx activity = (A340/min)/(0.00373 M-1) x 0.19 mL/0.02 mL =
nmol/min/mL/20 μg protein.

63
Determination of Glutathione Reductase Activity
A glutathione reductase (GR) fluorescent activity kit (Arbor Assays LLC, Ann Arbor,
MI) was used to measure GR activity in rat hippocampus samples. Rat hippocampi
were frozen by liquid nitrogen and stored in -80 C until use. The tissues were
homogenized in ice cold RIPA buffer supplemented with protease inhibitor,
phosphatase inhibitor and EDTA (Thermo scientific, Rockford, IL), and centrifuged at
14,000 rpm for 10 minutes at 4°C. An aliquot of the supernatant was removed for
protein determination. 25 L of GR standards or 25 L of samples containing 50 g of
protein were added to the 96-well plate followed by addition of 15 L nonfluorescent
molecule, ThioStar®, that covalently bind to the free thiol group on GSH to yield a highly
fluorescent product. The plate was incubated at room temperature for 5 minutes, and
the fluorescent product was read at 510 nm in a fluorescent plate reader with excitation
at 370-410 nm. This data was used to subtract any background thiol signal in samples.
By adding the oxidized glutathione, GSSG and NADPH to the samples, the GR activity
of the sample was determined by measuring the amount of GSH generated from the
reduction of GSSG by reacting the GSH with ThioStar® to covalently bind the free thiol
group on GSH and yield a highly fluorescent product, which was used to compared to
the readings from standards.
Statistics
All statistical analyses were performed using StatView 5.0 (SAS Institute).
Analyses of variance (ANOVAs) were used to establish main effects and interactions.
Follow-up ANOVAs and/or Fisher’s protected least significant difference (PLSD) post
hoc comparisons with p < 0.05 were employed to determine specific differences.

64
Student t tests were used to determine whether quadrant search behavior was different
than that expected by chance. Western blots were analyzed using Image J (National
Institutes of Health), a Java-based image processing program for densitometry; data
significant at *p<0.05. Data from western blots were analyzed using an ANOVA. Post-
hoc analysis was determined using Fisher’s PLSD test. All graphs were produced in
GraphPad Prism software (GraphPad Prism Inc).
65
Figure 2-1. Map of sc-trs-smCBA-Human SOD1-myc (SOD1)
66
Figure 2-2. Map of pTR-UF11-Human SOD2 (SOD2)
67
Figure 2-3. Map of pTR-UF11-Human CAT (CAT)

68
Figure 2-4. Cue discrimination

69
Figure 2-5. Spatial and probe discrimination
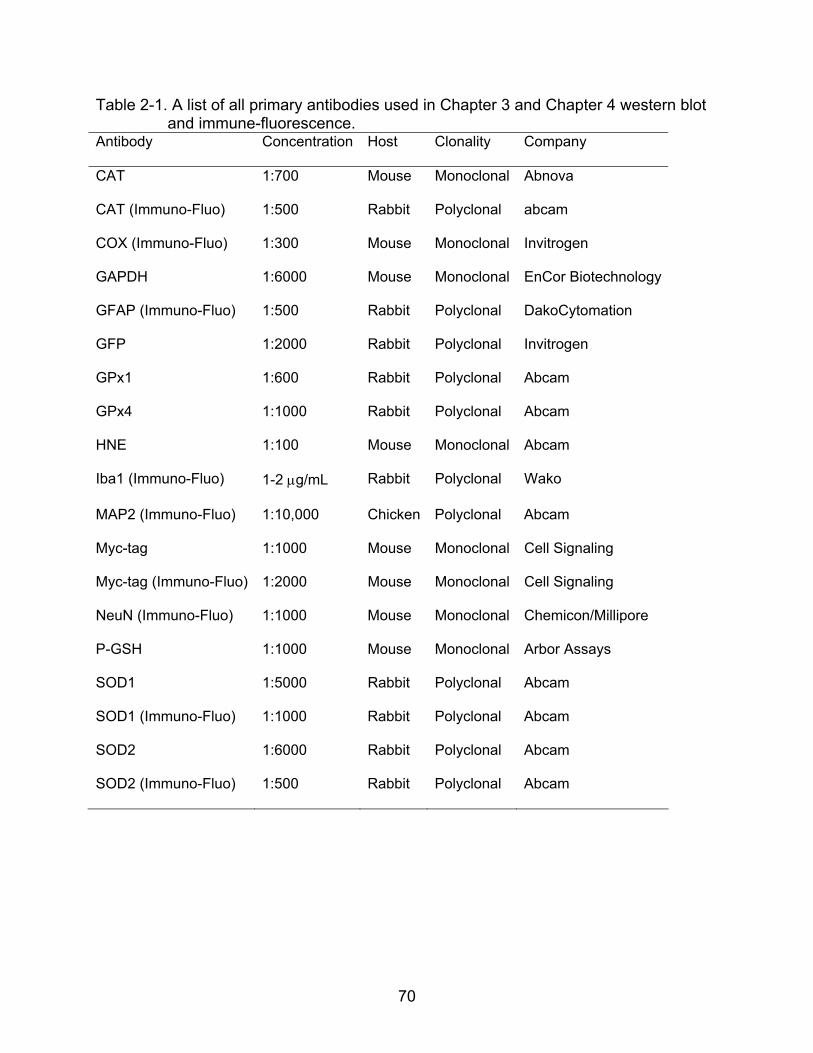
70
Table 2-1. A list of all primary antibodies used in Chapter 3 and Chapter 4 western blot and immune-fluorescence.
Antibody Concentration Host Clonality Company
CAT 1:700 Mouse Monoclonal Abnova
CAT (Immuno-Fluo) 1:500 Rabbit Polyclonal abcam
COX (Immuno-Fluo) 1:300 Mouse Monoclonal Invitrogen
GAPDH 1:6000 Mouse Monoclonal EnCor Biotechnology
GFAP (Immuno-Fluo) 1:500 Rabbit Polyclonal DakoCytomation
GFP 1:2000 Rabbit Polyclonal Invitrogen
GPx1 1:600 Rabbit Polyclonal Abcam
GPx4 1:1000 Rabbit Polyclonal Abcam
HNE 1:100 Mouse Monoclonal Abcam
Iba1 (Immuno-Fluo) 1-2 g/mL Rabbit Polyclonal Wako
MAP2 (Immuno-Fluo) 1:10,000 Chicken Polyclonal Abcam
Myc-tag 1:1000 Mouse Monoclonal Cell Signaling
Myc-tag (Immuno-Fluo) 1:2000 Mouse Monoclonal Cell Signaling
NeuN (Immuno-Fluo) 1:1000 Mouse Monoclonal Chemicon/Millipore
P-GSH 1:1000 Mouse Monoclonal Arbor Assays
SOD1 1:5000 Rabbit Polyclonal Abcam
SOD1 (Immuno-Fluo) 1:1000 Rabbit Polyclonal Abcam
SOD2 1:6000 Rabbit Polyclonal Abcam
SOD2 (Immuno-Fluo) 1:500 Rabbit Polyclonal Abcam

71
CHAPTER 3 INFLUENCE OF VIRAL VECTOR-MEDIATED DELIVERY OF SUPEROXIDE
DISMUTASE AND CATALASE TO THE HIPPOCAMPUS ON SPATIAL LEARNING, MEMORY DURING AGING
The brain is highly sensitive to oxidative stress (Halliwell, 1992), and the
accumulation of damaged molecules may contribute to age-related memory
impairments (Butterfield and Sultana, 2007; Forster et al., 1996; Knoferle et al., 2010;
Navarro et al., 2002; Nicolle et al., 2001). Antioxidant molecules and enzymes balance
the biological activity of reactive oxygen species (ROS), superoxide (O2·-) and hydrogen
peroxide (H2O2). Superoxide dismutase (SOD) catalyzes O2·- into H2O2 and catalase
(CAT) or glutathione peroxidase (GPx) converts H2O2 to water and oxygen. SODs are
classified according to their metal cofactors and cellular localization. The Cu/Zn-SOD1
(SOD1) is distributed throughout the cytoplasm, nucleus, and inner membrane space of
mitochondria, Cu/Zn-SOD3 (SOD3) is located in the extracellular space, and Mn-SOD
(SOD2) is restricted to the mitochondrial matrix (Chang et al., 1988; Fridovich, 1983a;
Keller et al., 1991).
SOD1 overexpression differentially influences brain function over the course of
aging (Hu et al., 2007b). Enhanced long-term potentiation (LTP) and spatial memory are
observed in aged transgenic SOD1 (tg-SOD1) mice (Kamsler et al., 2007; Kamsler and
Segal, 2003b). In contrast, tg-SOD2 mice do not exhibit altered synaptic plasticity or
memory (Hu et al., 2007b). It is unclear if increased SOD1 activity is required over the
lifespan in order to prevent the accumulation of oxidative damage; or whether enhanced
SOD1 activity, initiated in aged animals, might be therapeutic. To test this hypothesis,
we used adeno-associated virus (AAV) to deliver antioxidant enzymes (SOD1, SOD2,
CAT, or SOD1+CAT) to the hippocampus of young and aged rats. The results

72
demonstrate a dissociation of learning from measures of oxidative stress and suggest
that changes in redox sensitive signaling may mediate cognitive impairment.
Results
Efficiency-Specificity of The Viral Vectors
Following behavioral testing (see below) a subset of animals was killed for
examination of vector expression. Figure 3-1 illustrates the pattern of AAV-mediated
transduction. Strong expression was observed throughout the dorsal hippocampus,
extending > 3,000 μm along the anterior-posterior axis and included all major cell layers
(Fig. 3-1A). Transduction was limited to the hippocampus in confirmation of our previous
work (Foster et al., 2008) and mainly observed in neurons. Immunostaining for the myc
tag of SOD1-myc revealed transduction in neurons identified by NeuN (Fig. 3-1B) or
MAP2 (Fig. 3-1C) staining. Myc immunofluorescence was not localized to astrocytes or
microglia, assessed by immunostaining for GFAP (Fig. 3-1D) and Iba1 (Fig. 3-1E).
Injections of SOD1+CAT resulted in co-localization within the soma and dendrites of
neurons (Fig. 3-1F). SOD2 was observed in the soma, but not the nucleus and co-
localized with COX consistent with mitochondrial localization (Fig. 3-1G).
Figure 3-2 shows the increase in enzyme expression and decrease in oxidative
damage associated with viral vector treatment. For quantification, band intensities were
normalized to GAPDH, and then normalized to the mean value for young animals. Two
SOD1 bands were observed for young and aged SOD1-myc injected rats (Fig. 3-2A).
The band representing endogenous SOD1 was observed at 19 kDa and a larger band
representing the myc tagged human SOD1 was located at 23 kDa. Densitometry
indicated that rats injected with SOD1-myc exhibited a fourfold increase in total SOD1
[F(1,16)=45.59, p<0.0001) (Fig. 3-2E). Measurement of the 19 kDa band indicated that

73
SOD1-myc expression did not modify the level of endogenous SOD1 (data not shown).
An increase in the relevant vector was observed for SOD2 [F(1,11)=7.9, p<0.05] (Figs.
3-2B, 3-2F) and CAT [F(1,8)=7.14, p<0.05] (Figs. 3-2C, 3-2G). Finally, injection of
SOD1+CAT increased the expression of SOD1 [F(1,10)=14.54, p<0.01] and CAT
[F(1,12)=7.83, p<0.05] (Figs. 3-2D, 3-2H). It should be noted that the level of expression
for SOD1 under this condition was reduced relative to injection of SOD1-myc (i.e. 2
versus 4 fold).
As shown in Figure 3-2, SOD1, SOD2, CAT, or SOD1+CAT overexpression was
associated with reduced HNE-staining. Quantification confirmed a decrease in HNE-
reactive protein for SOD1 [F(1,8)=9.26, p<0.05], SOD2 [F(1,12)=11.45, p<0.01, CAT
[F(1,8)=18.53, p>0.01], and SOD1+CAT [F(1,8)=16.53, p<0.01]. A decrease in protein
S-glutathionylation was observed for rats overexpressing SOD1 [F(1,12)=15.17,
p<0.005], SOD2 [F(1,12)=7.83, p< 0.05], CAT [F(1,8)=22.54, p<0.01], and SOD1+CAT
[F(1,8)=7.3, p<0.05]. Examination of SOD activity indicated that SOD1-myc rats
exhibited a two-fold increase in SOD activity compared to GFP-injected rats (p<0.05; n
= 4/group, data not shown).
SOD overexpression can increase the level of H2O2 and the expression of
downstream antioxidant enzymes (de Haan et al., 1996; Fullerton et al., 1998; Kelner et
al., 1995; Liochev and Fridovich, 2007). Western blots indicate that GPx4 and CAT
levels did not differ among groups; however, there was a tendency for a treatment effect
for GPx1 [F(1,15)=3.46, p=0.08] and post hoc comparisons indicated GPx1 expression
significantly increased in aged SOD1 rats (Fig. 3-2E).

74
Age-Dependent Influence of Enzyme Overexpression on Spatial Learning
Spatial learning in the water maze was tested one month following virus injections.
Figure 3-3 shows the mean escape latency and escape path length across the five
training blocks during cue discrimination training. All groups exhibited a decrease in
escape latency and path length during training, and young animals exhibited superior
latency and path length relative to aged rats. An ANOVA on latency indicated an effect
of training [F(4,404 =107.21, p<0.0001] and an age effect [F(1,404)=34.71, p<0.0001] in
the absence of a treatment effect (Figs. 3-3A, 3-3B). Similarly, an ANOVA on path
length indicated an effect of training [F(4,404)=71.23, p<0.0001] and age
[F(1,404)=40.084, p<0.0001] in the absence of a treatment effect (Figs. 3-3C, 3-3D).
For spatial discrimination, an ANOVA on escape latency indicated significant main
effects of training [F(5,505)=68.03, p<0.0001] and age [F(1,505)=37.35, p<0.0001], with
a tendency for a treatment effect [F(4,505)=2.10, p=0.08] (Figs. 3-4A, 3-4B). Analysis of
escape path length indicated effects of training [F(5, 505)=81.19, p<0.0001] and age
[F(1,505)=28.83, p<0.0001], with a tendency for a treatment effect [F(4,505)=2.27,
p=0.06] (Figs. 3-4C, 3-4D). To localize treatment effects, the data for the last two
training blocks were averaged and an ANOVA was run within each age group. No
treatment effects were found in young rats. A tendency [F(4,50)=1.92, p=0.12] for a
treatment effect on latency was observed for aged rats, and post hoc analyses indicated
that aged SOD1+CAT rats exhibited significantly less time to reach platform compared
to aged rats expressing SOD2 or SOD1 alone (Fig. 3-4E). The results were confirmed
for distance (Fig. 3-4F). An ANOVA on the mean for the last two blocks revealed
tendency for a treatment effect in aged rats [F(4,50)=2.5, p=0.05] and post hoc analysis

75
indicated that aged SOD1+CAT rats had a shorter path length compared to aged SOD2
or SOD1 rats.
A 60 sec probe trial was delivered between training blocks 5 and 6. Analyses were
performed on two consecutive 30 sec segments. An ANOVA on the percent time in the
goal quadrant during the first 30 seconds indicated young spent more time in the goal
quadrant than aged rats [F(1,100)=4.82, p<0.05] and there was a tendency for a
treatment effect [F(4,100)=2.07, p=0.08] (Figs. 3-5A, 3-5B). ANOVAs within each age
group revealed a treatment effect for aged animals [F(4,50)=3.07, p<0.05] and post hoc
comparisons indicated that aged control rats spent more time in the goal quadrant
compared to aged rats with SOD1 or CAT overexpression. Aged SOD1+CAT rats spent
significantly more time in the goal quadrant relative to aged SOD1 rats. The percent
time in the goal quadrant was compared to that expected by chance (i.e. 25%). For the
first 30 seconds of the probe trial, all groups except for aged SOD1 or CAT performed
above chance. Examination of time spent searching the goal quadrant for the last 30
sec of the probe trial indicated a tendency for an age effect [F(1,100)=5.06, p=0.08] in
the absence of a treatment effect (Figs. 3-5C, 3-5D). An examination of the percent time
in the goal quadrant indicated that all groups except aged controls and aged SOD1 rats
spent significantly greater time in the goal quadrant relative to chance (Figs. 3-5C, 3-
5D). The fact that aged SOD1 rats did not spend a significant portion of their search
behavior in the goal quadrant for either segment of the probe trial indicates that these
animals did not acquire a spatial search strategy.
The aged SOD1 impairment was confirmed by measuring the latency to the first
platform crossing (Figs. 3-5E, 3-5F). An ANOVA indicated an age [F(1,100)=10.95,

76
p<0.01] and treatment [F(4,100)=2.52, p<0.05] effect. ANOVAs within each age group
indicated a tendency for a treatment effect in aged animals [F(4,49)=2.38, p=0.06] and
post hoc comparisons indicated that aged SOD1 rats required a longer time to cross the
platform location compared to age matched controls, CAT, and SOD1+CAT expressing
groups. There was a tendency (p=0.08) for a difference between SOD1 and SOD2
groups. An ANOVA on the total number of platform crossing indicated an age difference
[F(1,100)=13.15, p<0.001] (Figs. 3-5G, 3-5H), in the absence of a treatment effect. In
sum, the results indicate that SOD1 overexpression for one month was associated with
impaired spatial learning in aged rats. The fact that the SOD1+CAT exhibited superior
performance relative to SOD1 group indicates that the impairment may be rescued to
co-overexpressing CAT.
Overexpression of SOD1+CAT for Four Months Improves Spatial Learning
Our results contrast with studies in tg-SOD1 mice, which indicate that young tg-
SOD1 mice exhibit impaired spatial learning while aged tg-SOD1 mice show enhanced
spatial memory (Gahtan et al., 1998; Kamsler et al., 2007). The difference may result
from an interaction of age and the duration of overexpression. To examine this
possibility, a subset of animals tested at one month was retested at four months post-
injection. The experimental groups included young rats (8 mo) injected with SOD1 (n =
6), SOD1+CAT (n = 10), GFP (n = 2), no surgery controls (n = 4), and aged rats (23 mo)
injected with SOD1 (n = 6), SOD1+CAT (n = 10), GFP (n = 2) or no surgery controls (n
= 4).
For examination of spatial learning 4 months after viral vector injections, the
platform location in the maze was shifted to a new quadrant. An ANOVA on escape
latency across training blocks indicated main effects of training [F(5,185)=40.53,

77
p<0.0001] and age [F(1,185) =5.19, p<0.05], with a tendency for a treatment effect
[F(2,185)=5.19, p=0.07] (Figs. 6A, 6B). Analysis of escape path length indicated effects
of training [F(5, 185)=36.12, p<0.0001] and age [F(1,185)=5.20, p<0.05] (Figs. 6C, 6D).
To localize treatment effects, the data for the last two training blocks were averaged and
an ANOVA for treatment effects was run within each age group. An ANOVA for young
animals indicated a tendency for a treatment effect [F(2,18)=7.12, p=0.14], however;
post hoc comparisons did not reach significance; although there was a trend for young
SOD1 rats to exhibit an increase in the escape latency relative to young control rats
(p=0.05). An ANOVA for aged animals indicated a tendency for a treatment effect
[F(2,19)=3.24, p=0.06] and post hoc comparisons indicated that aged SOD1+CAT rats
required less time to reach platform compared to aged SOD1 rats (Fig. 3-6B). Analysis
of the mean distance for the last two training blocks also indicated a tendency for a
treatment effect in aged rats [F(2,19)=2.67, p=0.09) (Figs. 3-6C, 3-6D) and post hoc
comparisons confirmed that aged SOD1+CAT rats had a shorter path length to escape
compared to aged SOD1 rats (Fig. 3-6D).
Examination of the first 30 seconds of the probe trial delivered between blocks 5
and 6, indicated a tendency for young rats to spend more time in the goal quadrant
relative to aged rats [F(1,37)=3.33, p=0.07] in the absence of a treatment effect (Figs. 3-
7A, 3-7B). The percent time searching the goal quadrant was significantly above chance
for all groups, indicating that all groups had acquired a spatial search strategy. A
tendency for a treatment effect [F(2, 37)=2.47, p=0.09] in the absence of an age
difference was observed for the second half-minute of the probe trial (Figs. 3-7C, 3-7D).
Again, all groups spent a significantly longer time in goal quadrant than expected by

78
chance (Figs. 3-7C, 3-7D). An ANOVA on the latency to the first platform crossing
indicated a significant age effect [F(1,37)=12.73, p<0.01], and a tendency for a
treatment effect [F(2,37)=2.24, p=0.1]. An ANOVA within each age group indicated a
tendency for a treatment effect only in the aged group [F(2,19)=3.29, p=0.05]. Post hoc
comparison indicated that aged SOD1+CAT rats required less time for the first platform
crossing compared to aged SOD1 rats and there was a tendency (p=0.08) for
SOD1+CAT rats to cross quicker than aged controls. Finally, an ANOVA on total
platform crossing indicated an age effect [F(1,37)=12.67, p<0.005] and a tendency for a
treatment effect [F(2,37)=2.54, p=0.09]. An ANOVA within each age group indicated a
tendency for a treatment effect in aged rats [F(2,19)=3.45, p=0.05] and post hoc
comparisons indicated that aged SOD1+CAT rats exhibited significantly more platform
crossings than aged controls (Fig. 3-7H). In fact, aged SOD1+CAT rats exhibited
performance similar to young animals.
Figure 3-8 shows western blot data on enzyme expression and lipid peroxidation
in the hippocampus for animals that overexpressed viral vectors for ~5 months. As
shown in Figure 8A, the two SOD1 bands (19 and 23 kDa) were observed for SOD1-
myc injected rats. Densitometry confirmed that rats injected with SOD1-myc exhibited
~3 fold increase in total SOD1 [F(1,12)=133.08, p<0.0001) (Figs. 3-8A, 3-8C). Again,
the 19 kDa band was not influenced by SOD1-myc expression (data not shown).
Injection of SOD1+CAT increased the expression of SOD1 [F(1,12)=31.31, p<0.001]
and CAT [F(1,8)=10.40, p<0.05] (Figs. 3-8B, 3-8D). Similar to 2-month overexpression,
the level of expression for SOD1 under this condition was reduced relative to injection of
SOD1-myc alone (i.e. 2 versus 2.5 fold).

79
A decrease in HNE-reactive protein was observed for rats that overexpress SOD1
[F(1,12)=24.23, p<0.001] or SOD1+CAT [F(1,12)=7.63, p<0.05] (Fig. 3-8). For SOD1
rats, there was an age effect on CAT expression [F(1,12)=6.58, p<0.05] in the absence
of a treatment effect. Post hoc tests within each treatment group indicated an age-
related decrease in expression in animals overexpressing SOD1 (Fig. 3-8C).
Examination of GPx1 indicated no effect of age or treatment for SOD1 animals and
SOD1+CAT animals exhibited an effect of age [F(1,16)=15.00, p<0.005] and treatment
[F(1,16)=4.27, p<0.05]. Post hoc analyses indicated that GPx1 expression was reduced
in aged rats that overexpressed SOD1+CAT [F(1,8)=7.9, p<0.05] compared to aged
controls (Figs. 3-8B, 3-8D). Levels of protein carbonyls were measured; however, the
results did not reveal any information concerning the effect of SOD1 overexpression,
possibly due to the lack of specificity of carbonyl measures (Adams et al., 2001) or the
fact that H2O2 is a poor mediator of stable oxidative damage for protein carbonyls
(Shacter, 2000b). Furthermore, previous research indicates that lipid peroxidation may
be more sensitive since SOD overexpression in mice markedly reduces lipid
peroxidation in the absence of a significant effect on protein carbonyls (Jang et al.,
2009). Consistent with the lipid peroxidation results, overexpression of SOD1 resulted in
a decrease in Oxo8dG [F(1,21)=16.58, p<0.0005] in the absence of an age difference
(Fig 3-9), confirming that SOD1 overexpression decreased oxidative damage.
Discussion
While it is clear that oxidative stress is a likely component of aging, it is unclear
whether increased production of ROS or the accumulation of oxidative damage is the
primary cause of functional decline. Consistent with previous work in tg-mice,
overexpression of SOD1, SOD2, and catalase reduced markers of oxidative stress

80
(Perez et al., 2009a). However, decreased oxidative damage was not associated with
improved cognitive function. Moreover, SOD1 overexpression impaired spatial learning
in aged rats. The impairment was specific to SOD1 and spatial learning in aged animals.
The results indicate that accumulation of oxidative damage is not the only mechanism
for cognitive decline. Other processes influenced by oxidative stress may be more
relevant to age-related cognitive decline, including mitochondrial function, redox
signaling, and inflammation.
SOD1 can be toxic to motor neurons (Bosco et al., 2010). However, in the current
study, the deficit was limited to aged SOD1 animals, indicating that it was not simply
due to SOD1 overexpression. Further, one would expect that toxicity should increase
over time. On the contrary, we observed that the spatial learning deficit decreased
following four months of SOD1 overexpression in our oldest rats. Although, there was a
tendency for the emergence of impairment as young SOD1 rats approached middle-age
(Fig. 3-6). SOD1 catalyzes O2·- into H2O2 and the levels of O2
·- and H2O2 in brains of Tg-
SOD1 mice are decreased and increased, respectively (Malinska et al., 2009) and tg-
SOD1 overexpressing mice exhibit age-dependent effects on cognition and
hippocampal synaptic plasticity which are thought to result from elevation of ambient
H2O2 levels (Kamsler and Segal, 2003a). Thus, the specificity may be due to ambient
H2O2 levels. One indication that H2O2 was increased in the current study is that aged
SOD1 animals exhibited increased GPx1 expression (Fig. 3-2A). In other tissues, H2O2
can provoke an increase in GPx activity (Noack et al., 1998; Rohrdanz et al., 2001;
Wijeratne et al., 2005). Finally, aged SOD1+CAT rats did not exhibit cognitive

81
impairments, suggesting improved cognition resulted from better homeostatic regulation
of redox signaling due to CAT processing of excess H2O2.
How can SOD1 overexpression reduce oxidative damage if H2O2 levels increase?
H2O2 is a relatively mild oxidizing agent. Irreversible oxidative damage results from O2·-;
SOD1 removes O2·-and overexpression of SOD1 reduces the level of O2
·- in the brain
(Malinska et al., 2009). If free iron is available, H2O2 can produce hydroxyl radicals (OH.)
through Fenton-type reactions. However, a large body of evidence indicates that H2O2
per se may not induce irreversible oxidative damage (Catala, 2010a; Leutner et al.,
2001b; Linden et al., 2008a; Shacter, 2000b). In contrast, H2O2 can readily and
reversibly influence redox sensitive signaling cascades. Age-related changes in redox
state disrupt calcium homeostasis, increasing the release of calcium from internal stores,
decreasing CaMKII activity, and impairing LTP (Bodhinathan et al., 2010a, b; Foster,
2007). H2O2 application to hippocampal slices mimics age-related changes, promoting
calcium release from internal stores (Gonzalez et al., 2006), decreasing synaptic
CaMKII activity (Shetty et al., 2008), and impairing LTP (Auerbach and Segal, 1997;
Kamsler and Segal, 2003a; Pellmar et al., 1991). The LTP impairment in tg-SOD1 mice
is attenuated by catalase suggesting the involvement of elevated H2O2 (Gahtan et al.,
1998). Together the results suggest that SOD1 overexpression could impair learning
through increased H2O2 acting on redox signaling pathways involved in memory
formation.
Adaptation to Altered Redox State: The effect of SOD overexpression on
memory changes with age (Hu et al., 2007b). SOD1 overexpression impaired or had no
effect on learning in young mice and enhanced spatial memory in older mice (Kamsler

82
et al., 2007; Kamsler and Segal, 2003b). The authors suggest that aged mice adapt to
elevated H2O2. Transcription of genes regulating redox homeostasis increase during
middle-age (Aenlle et al., 2009; Blalock et al., 2003; Zeier et al., 2011). Thus, the
increase in GPx1 expression in aged SOD1 mice may represent an adaptive response.
Other adaptive changes may underlie the improvement in behavior following four-
months of SOD1 overexpression. Finally, aged SOD1+CAT rats exhibited superior
water maze performance, indicating that treatment to reduce O2·- and H2O2 may be
more beneficial than simply enhancing SOD1. Indeed, administration of a superoxide
dismutase/catalase mimetic improved memory in aging mice (Liu et al., 2003). Future
studies should employ redox proteomics to identify specific changes related to cognitive
decline.
In summary, memory function was not related to the level of oxidative damage.
Increased SOD1 expression impaired learning in older rats. In contrast, increased
expression of SOD1+CAT provided protection from age-related cognitive impairments.
The results extend a growing body of evidence indicating the importance of H2O2 and
the redox signaling for regulating cognitive function during aging.
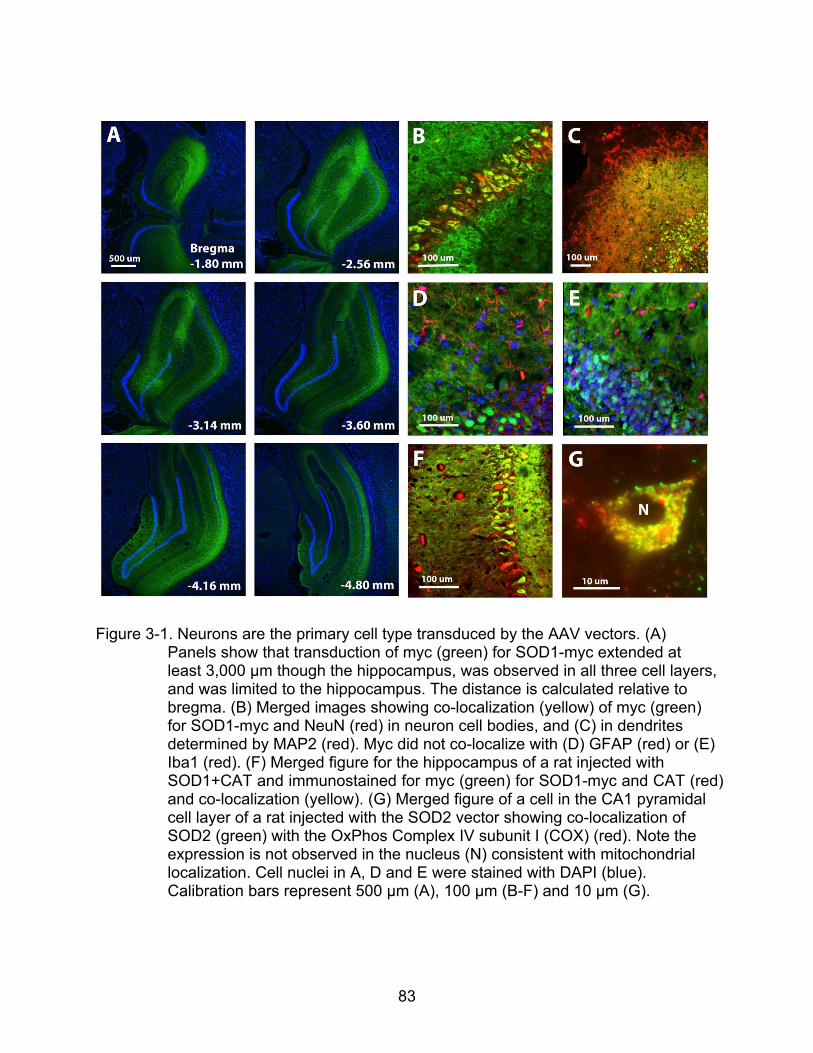
83
Figure 3-1. Neurons are the primary cell type transduced by the AAV vectors. (A) Panels show that transduction of myc (green) for SOD1-myc extended at least 3,000 μm though the hippocampus, was observed in all three cell layers, and was limited to the hippocampus. The distance is calculated relative to bregma. (B) Merged images showing co-localization (yellow) of myc (green) for SOD1-myc and NeuN (red) in neuron cell bodies, and (C) in dendrites determined by MAP2 (red). Myc did not co-localize with (D) GFAP (red) or (E) Iba1 (red). (F) Merged figure for the hippocampus of a rat injected with SOD1+CAT and immunostained for myc (green) for SOD1-myc and CAT (red) and co-localization (yellow). (G) Merged figure of a cell in the CA1 pyramidal cell layer of a rat injected with the SOD2 vector showing co-localization of SOD2 (green) with the OxPhos Complex IV subunit I (COX) (red). Note the expression is not observed in the nucleus (N) consistent with mitochondrial localization. Cell nuclei in A, D and E were stained with DAPI (blue). Calibration bars represent 500 µm (A), 100 µm (B-F) and 10 µm (G).

84
Figure 3-2. Overexpression of antioxidant enzymes in the hippocampus reduces
markers of oxidative stress. Western blot analysis of hippocampal lysates from young and aged rats injected with viral vectors to express (A, E) SOD1-myc, (B, F) SOD2, (C, G) CAT, or (D, H) SOD1+CAT. For quantification, band intensities were normalized to expression of GAPDH and this value was

85
normalized to the mean value of young controls from the same blot. In each case, transduction resulted in an increase in the expression of the antioxidant enzyme. For SOD1-myc (A, D), two bands were observed representing endogenous rat SOD1 (rSOD1) and the human myc tagged SOD1 (hSOD1). Lipid peroxidation (HNE) and S-glutathionylated proteins (GSH) were decreased by enzyme overexpression. To determine whether overexpression of antioxidant enzymes would influence expression of downstream antioxidant enzymes, immunostaining was performed for glutathione peroxidase 1 (GPx1), glutathione peroxidase 4 (GPx4), and CAT. Overexpression of SOD1-myc was associated with an increase in GPx1 (E). Each bar represents the normalized mean + SEM (n = 3-6). Asterisks indicate significant (p < 0.05) differences determined by post hoc Fisher PLSD.

86
Figure 3-3. Overexpression of antioxidant enzymes did not affect cue discrimination in
water maze. All groups learned to reach the visible platform, as indicated by a significant overall decrease in latency (A, B) and distance (C, D) across blocks. Y = 5-month and A = 20-month old rats injected with SOD1, SOD2, CAT, or SOD1+CAT. Age matched controls (Cont) included rats injected with GFP and no surgery controls.

87
Figure 3-4. Overexpression of SOD1 impaired spatial learning in aged rats. Behavioral
measures for young and aged rats during training on the spatial version of the water escape task. Mean latency (A, B) and mean path length (C, D) to escape during spatial discrimination training. No treatment effects were found for young rats. Examination of the mean latency (E) and mean path length (F) averaged across the last two training blocks in aged animals indicated an effect of treatment. Due to better performance by the SOD1+CAT group relative to the SOD1 and SOD2 groups. Asterisk indicates a significant (p < 0.05) difference.

88
Figure 3-5. Overexpression of SOD1 impaired acquisition of a spatial search strategy in
aged rats. A single probe trial was given between block 5 and block 6 of spatial training. Percent time spent searching the goal quadrant during the first 30 seconds of for (A) young and (B) aged rats. The search behavior of aged rats with overexpression of SOD1 or CAT was not different from chance

89
levels (25% dashed line). Aged SOD1 rats spent significant less time compared to aged control and aged rats with overexpression of SOD1+CAT. Aged rats with overexpression of CAT spent significant less time than aged control. Examination of the percent of time spent in goal quadrant during the second 30 seconds of probe trial for (C) young and (D) aged rats indicated that aged rats with overexpression of SOD1 continued to perform at chance levels. Examination of the latency for first platform crossing for (E) young and (F) aged rats indicated a treatment effect for aged animals due to an extended latency for the SOD1 group. Age differences were observed for total number of platform crossings between (G) young and (H) aged rats. Asterisk = p < 0.05 group difference. Pound sign indicates a significance (p < 0.05) difference from chance.
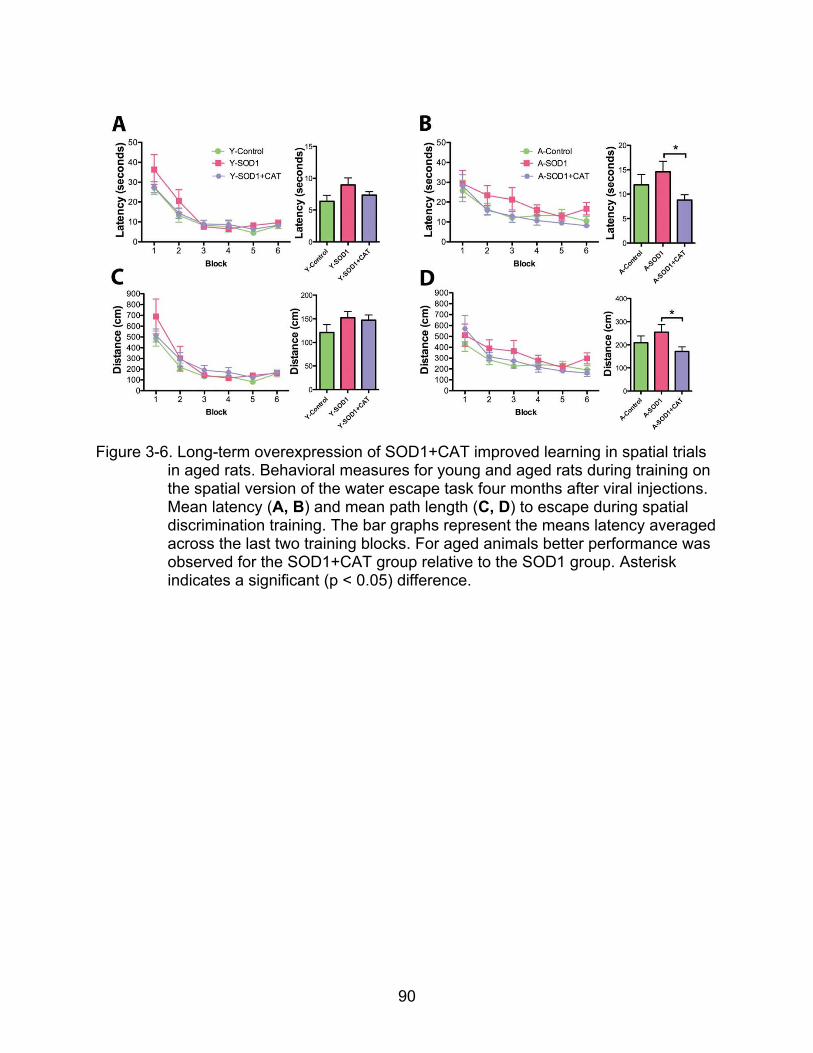
90
Figure 3-6. Long-term overexpression of SOD1+CAT improved learning in spatial trials
in aged rats. Behavioral measures for young and aged rats during training on the spatial version of the water escape task four months after viral injections. Mean latency (A, B) and mean path length (C, D) to escape during spatial discrimination training. The bar graphs represent the means latency averaged across the last two training blocks. For aged animals better performance was observed for the SOD1+CAT group relative to the SOD1 group. Asterisk indicates a significant (p < 0.05) difference.

91
Figure 3-7. Overexpression of SOD1+CAT for four months improves spatial learning in probe test. A single probe trial was given between block 5 and block 6 of spatial training. Mean percent time spent searching the goal quadrant during

92
the first 30 seconds of for (A) young and (B) aged rats. Mean percent of time spent in goal quadrant during the second 30 seconds of probe trial for (C) young and (D) aged rats. Examination of the latency for first platform crossing for (E) young and (F) aged rats indicated a treatment effect for aged animals due to an superior performance by the SOD1+CAT group compared to the SOD1 group. Asterisk = p < 0.05 group difference. Pound sign indicates a significance (p < 0.05) difference from chance.

93
Figure 3-8. Antioxidant enzymes and oxidative stress markers in hippocampus with 5-
month SOD1 and SOD1+CAT overexpression. Western blot analysis of hippocampal lysates from young and aged rats injected with viral vectors to express (A) SOD1, and (B) SOD1+CAT. For SOD1 (A, C), two bands were observed representing endogenous rat SOD1 (rSOD1) and the human myc

94
tagged SOD1 (hSOD1). CAT level did not change in SOD1 rats but increased in SOD1+CAT rats. Lipid peroxidation measured with anti-4-hydroxy-2-nonenal (HNE) antibody was decreased by SOD1 and SOD1+CAT overexpression. Expression of SOD1 did not change the level of GPx1 but expression of SOD1+CAT in aged rats was associated with a decrease in GPx1 (D). GAPDH was used as a loading control. Asterisk indicates a significant (p < 0.05) difference in treatment. ω indicates a significant (p < 0.05) difference in age.
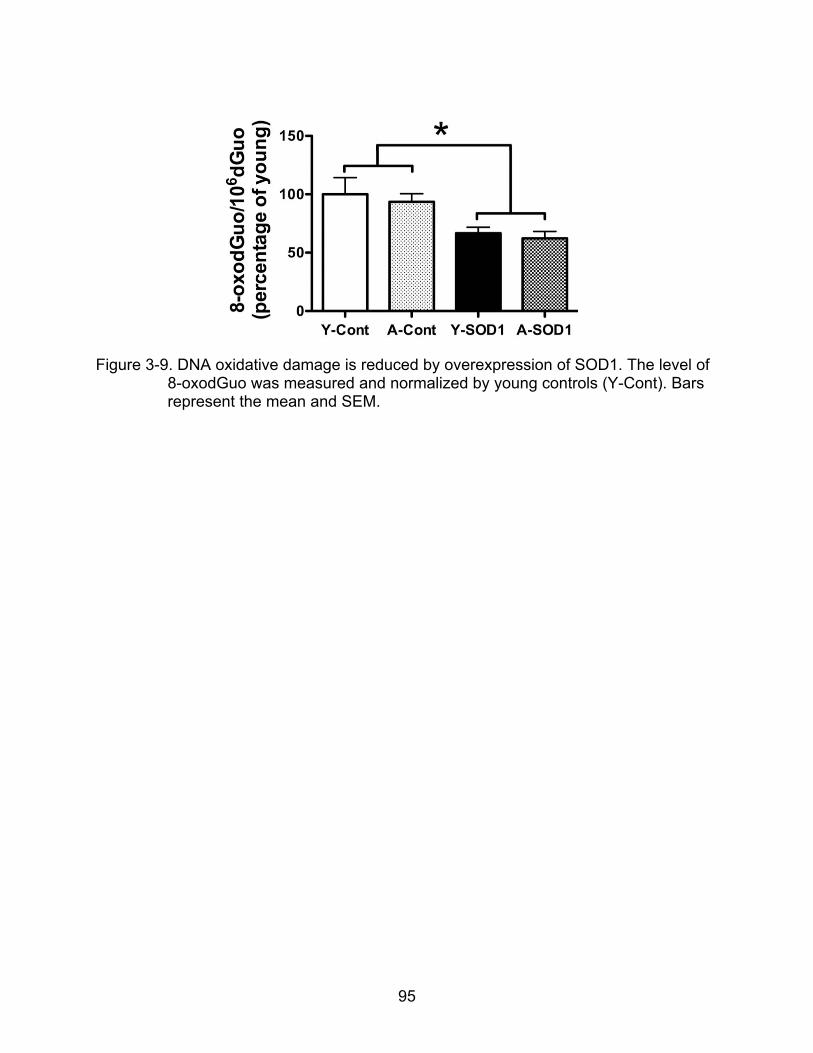
95
Figure 3-9. DNA oxidative damage is reduced by overexpression of SOD1. The level of
8-oxodGuo was measured and normalized by young controls (Y-Cont). Bars represent the mean and SEM.

96
CHAPTER 4
THE INFLUENCE OF AAV DELIVERED SUPEROXIDE DISMUTASE 1 AND CATALASE ON HIPPOCAMPAL SYNAPTIC PLASTICITY AND NMDA RECEPTOR
FUNCTION
Unlike neurodegenerative disease, cognitive decline in normal aging is not
associated with a significant loss of neurons (Gallagher et al., 1996). Rather, cognitive
decline in normal aging is associated with senescent physiology including changes in
synaptic plasticity (Foster, 2002; Foster and Kumar, 2002; Kumar et al., 2009). Long-
term potentiation (LTP) has been widely considered as a cellular mechanism that
underlies learning and memory because of the characteristics of synaptic modification
(Bliss and Collingridge, 1993; Malenka and Nicoll, 1999). In experimental settings, LTP
is usually initiated by high frequency afferent stimulation (>50 Hz), in order to activate N-
methyl-D-aspartate glutamate receptors (NMDARs). NMDARs play a central role in
LTP-induction and the specificity of synaptic modification. The activation of NMDAR
starts from the neurotransmitters binding to NMDAR and the postsynaptic depolarization
removing magnesium that blocks the channel. Ca2+ thus enters the NMDAR channel at
the synaptic site. Ca2+ will then initiate the signaling cascade that results in LTP-
induction.
During aging, the induction of LTP becomes more difficult, there is an increase in
the level of synaptic activation needed to induce LTP (Foster, 1999, 2012; Kumar, 2011;
Kumar et al., 2007; Milner et al., 2004). The synaptic plasticity shift is due to disruption
of Ca2+ homeostasis (Burke and Barnes, 2010; Foster, 2007; Thibault et al., 2001). The
major sources of intracellular Ca2+ include influx into the cell through NMDA receptors or
voltage-dependent Ca2+ channels (VDCC) and release of Ca2+ from intracellular Ca2+

97
stores. Work done in our lab and others indicate that, in the hippocampus, aging is
associated with a decreased involvement of NMDA receptors and an increased role for
VDCC and internal Ca2+ stores in regulating synaptic modifiability (Barnes et al., 1997;
Foster, 2012; Foster and Norris, 1997; Kumar and Foster, 2005; Thibault and Landfield,
1996). The exact mechanism for ROS-mediated regulation of synaptic plasticity is
unknown; however, recent work demonstrates that an age-related shift in the
intracellular oxidation-reduction (redox) state decreases NMDAR function and increases
release of Ca2+ from intracellular Ca2+ stores (Bodhinathan et al., 2010a, b).
The nervous system is highly sensitive to oxidative stress (Halliwell, 1992). The
highly active free radical, superoxide can make irreversible oxidative damage on DNA,
RNA, lipids and proteins. To prevent the irreversible oxidative damage on tissues,
superoxide dismutase catalyzes O2·- into less active H2O2. Hydrogen peroxide per se
does not induce irreversible oxidative damage (Catala, 2010b; Leutner et al., 2001a;
Linden et al., 2008b; Shacter, 2000a). The relatively milder hydrogen peroxide induces
the reversible formation of disulfide bonds between pairs of cysteine residues (thiol
groups: R-S-H) in proteins, shifting protein structure and function, and influencing
multiple signaling cascades including Ca2+ signaling (Foster, 2007). The redox state of
cysteine residues on the extracellular portion of the NMDAR have been implicated in
regulating NMDAR function in cortical neuronal culture and hippocampal slices
(Aizenman et al., 1990; Aizenman et al., 1989; Bernard et al., 1997; Choi and Lipton,
2000; Lipton et al., 2002; Reynolds et al., 1990), suggesting that the age-related decline
in NMDAR function could be regulated by altered redox state acting on extracellular
cysteine residues. Indeed, application of the disulfide reducing agent, dithiothreitol

98
(DTT), selectively increased the NMDAR-mediated synaptic responses to a greater
extent and enhanced LTP in hippocampal slides from older rats (Bodhinathan et al.,
2010a). Surprisingly, applying the membrane-impermeable reducing agent, L-
glutathione directly to the hippocampal slices failed to increase the NMDAR response;
instead, an increase in the NMDAR response was observed if L-glutathione was
delivered through the intracellular recording pipette. The results indicate that
intracellular redox state, rather than disulfide bonds of extracellular cysteine residues,
mediates the suppression of NMDAR responses and impaired LTP. The intracellular
redox state mediated NMDAR function decline during aging has recently been
confirmed by other labs (Robillard et al., 2011; Yang et al., 2010).
We recently provided evidence that overexpression of SOD1 in the hippocampi
impaired spatial learning in aged F344BN F1 rats. In addition, overexpression of CAT
together with SOD1 rescued the spatial learning deficit caused by overexpression of
SOD1 alone. The results suggested that the impaired spatial learning in older SOD1
could be due to over production of H2O2 and impaired NMDAR function. The current
study used AAV to deliver SOD1+GFP and SOD1+CAT to the hippocampus of aged
(17-month) F344 rats and examined memory-related behavioral performance one-
month post injection. Following collection of behavioral data, one hippocampus from
each rat was used to examine NMDA receptor function, and the other hippocampus
from each rat was used to determine antioxidant enzyme expression and activity as well
as the level of a major redox buffer, glutathione.

99
Results
AAV Delivery of SOD1, CAT and GFP in the Hippocampi of Rats
Animals were killed for examination of vector expression after behavioral testing.
Figure 4-1 illustrates the pattern of AAV-mediated transduction. Immunofluorescence on
brain slides from rats injected with AAV-SOD1 and AAV-GFP revealed that neurons
were co-transduced with AAV-SOD1 (showing in red with anti-myc antibody) and AAV-
GFP (showing in green) (Fig. 4-1a). Injections of AAV-SOD1+ AAV-CAT resulted in co-
transduction of SOD1 and CAT in hippocampal neurons (SOD1 was shown with anti-
myc antibody in red, and CAT was shown with anti-CAT antibody in green) (Fig. 4-1b).
Figure 4-2 shows the increase in enzyme expression and decrease in oxidative
damage associated with viral vector treatment. For quantification, band intensities were
normalized to GAPDH. Two SOD1 bands were observed for SOD1-myc injected rats
(Fig. 4-2A). The band representing endogenous SOD1 was observed at 19 kDa and a
larger band representing the myc tagged human SOD1 was located at 23 kDa.
Densitometry indicated that rats injected with SOD1-myc exhibited a ~2.3 fold increase
in total SOD1 in both SOD1+GFP [F(1,5)=14.36, p <0.05] and SOD1+CAT [F(1,6)=43.0,
p<0.001] rats. Measurement of the 23 kDa band indicated that hSOD1 expression is not
different between SOD1+GFP rats and SOD1+CAT rats [F(1,5)=2.0, p=0.22].
Measurement of the 19 kDa band indicated that SOD1-myc expression did not modify
the level of endogenous SOD1. rSOD1 expression is not different in SOD1+GFP rats
[F(1,6)=0.65, p=0.45] or SOD1+CAT rats [F(1,6)=2.25, p=0.18] compared to rSOD1
expression in GFP rats. C-myc was only detected in rats injected with SOD1+GFP and
SOD1+CAT viruses. GFP expression was observed in rats injected with GFP and
SOD1+GFP virus. In addition, SOD1+GFP and SOD1+CAT overexpression were

100
associated with a tendency of reduced HNE-staining. Quantification indicated a
tendency of decrease in HNE-reactive protein for SOD1+GFP [F(1,6)=3.46, p=0.11] and
SOD1+CAT [F(1,6)=2.63, p=0.16]. A tendency for increased CAT expression was
observed in SOD1+CAT rats [F(1,5)=2.78, p=0.16], and no difference was observed for
Cat expression in SOD1+GFP rats compared to GFP rats [F(1,4)=0.025, p=0.88].
SOD overexpression can increase the level of H2O2 and the expression of
downstream antioxidant enzymes (de Haan et al., 1996; Fullerton et al., 1998; Kelner et
al., 1995; Liochev and Fridovich, 2007). A tendency of increase of GPx1 was observed
in SOD1+GFP rats compared to GFP rats [F(1,5)=4.54, p=0.08]; however, no difference
in GPx1 expression was observed in SOD1+CAT rats compared to GFP rats
[F(1,5)=0.001, p=0.98].
SOD1 Overexpression Impairs Spatial Learning while SOD1+CAT Overexpression Improves Spatial Learning in Aged Rats.
Figure 4-3 shows the mean escape latency and escape path length across the five
training blocks during cue discrimination training. A repeated measures analysis of
variance (ANOVA) on latency indicated an effect of training [F(4,136) = 26.25, p <
0.0001] and a treatment effect [F(2,136) = 12.22, p < 0.0005] (Fig 4-3a). Post hoc
analysis indicated that GFP rats required significant longer time to find platform than
SOD1+GFP rats (p<0.0001) and SOD1+CAT rats (p<0.005). ANOVA on mean escape
latency in individual block indicated that the treatment effect was only seen in the early
phase of training including block 2 [F(2, 34)=8.69, p<0.001] and block 3 [F(2, 34)=11.72,
p<0.005]; but not the late phase of training including block 4 [F(2,34)=2.05, p=0.14] and
block 5 [F(2,34)=1.88, p=0.16]. Repeated measures ANOVAS within each group
indicated that all groups exhibited a decrease in escape latency during training (GFP:

101
F(4,52)=11.25, p<0.0001; SOD1+GFP: F(4,44)=7.74, p<0.0001; SOD1+CAT:
F(4,40)=10.13, p<0.0001). Similarly, a repeated measure ANOVA on path length
indicated an effect of training [F(4, 136) = 11.43, p < 0.0001] and a treatment effect
[F(2,136)=5.97, p<0.01] (Fig. 4-3b). Post hoc analysis indicated that GFP rats required
significant longer path length to find platform than SOD1+GFP rats (p<0.05). ANOVA on
mean escape path length in individual block indicated that the treatment effect was only
seen in block 2 [F(2, 34)=3.84, p<0.05] and block 3 [F(2,34)=7.54, p<0.005]; but not the
late phase of training like block 4 [F(2,34)=0.47, p=0.63] and block 5 [F(2,34)=1.11,
p=0.34]. Finally, repeated measures ANOVAs within each group indicated that all
groups exhibited a decrease in path length during training (GFP: F(4,52)=11.25,
p<0.0001; SOD1+GFP: F(4,44)=7.74, p<0.0001; SOD1+CAT: F(4,40)=3.64, p<0.05).
Spatial discrimination was initiated 3 days later. A repeated ANOVA on escape
latency indicated significant main effects of training [F(4,124) = 7.96, p < 0.0001] and
treatment [(F4,124)=6.54, p<0.005] (Fig. 4-4a). Post hoc analysis indicated that the
treatment effect was due to decreased escape latency for SOD1+CAT rats, which
exhibited a significant decrease in escape latency relative to GFP (p<0.05) and
SOD1+GFP rats (p<0.05). Repeated measures ANOVAs within each treatment group
indicated that GFP rats (p<0.05) and SOD1+CAT rats (p<0.005) reduced their latency to
find platform during spatial training. In contrast, no effect of training on latency was
observed for SOD1+GFP rats (p=0.51).
Analysis of escape path length indicated effects of training [F(4,124)= 8.87,
p<0.0001], and a tendency of treatment effect [F(2,124)=3.27, p=0.05] (Fig. 4-4b). Post
hoc analysis indicated that the treatment effect was due to decreased escape path

102
length for SOD1+CAT rats relative to GFP (p<0.05) rats and a tendency for a decrease
in path length relative to SOD1+GFP rats (p=0.10). Repeated measures ANOVAs within
each treatment group indicated that GFP rats (p<0.005) and SOD1+CAT rats (p<0.005)
exhibited a significant decrease of path length to find platform during spatial training. In
contrast, no effect of training on path length was observed for SOD1+GFP rats (p=0.20).
To determine whether animals acquired a spatial search strategy, a 60 s probe
trial (probe1: acquisition) was delivered immediately following the fifth block of spatial
training. Following probe 1, a refresher training block was delivered, followed one hour
later by a retention probed trial (Probe 2) and a second reminder spatial training block
(block 7). Twenty-four hours later, a training block was delivered followed by a
relearning probed trial (Probe 3). Probe trails were measured by latency to first reach
platform, number of platform crossing and discrimination index score (DI): (% time in
goal quadrant - % time in opposite quadrant) / (% time in goal quadrant + % time in
opposite quadrant). A repeated measure ANOVA across three probe trials on latency to
platform indicated a significant effect on treatment [F(2,64)=3.96, p<0.05] (Fig. 4-5a).
Post hoc analysis indicated that the treatment effect was due to better performance by
SOD1+CAT rats, which reached the platform significant faster than SOD1+GFP rats
(p<0.01). SOD1+CAT rats also had a tendency to reach the platform faster than GFP
rats (p=0.08).
The superior performance of SOD1+CAT rats was confirmed by a repeated
measure ANOVA across three probe trials for platform crossing. A significant effect was
observed for treatment [F(2,68)=4.20, p<0.05] (Fig. 4-5b). Post hoc analysis indicated

103
that SOD1+CAT rats made significant more platform crossings than GFP (p<0.05) and
SOD1+GFP (p<0.05) rats.
An ANOVA on DI score from three groups of animals indicated no treatment
effects over the course of testing. To determine if the animals acquired a spatial search
strategy, the DI scores were compared with that expected by chance (i.e., when the
animal spent equal amount of time in goal and opposite quadrants, then DI=0). One
group student t-tests indicated that only GFP and SOD1+CAT rats exhibited DI score
above chance (p<0.05) in probe 1 (acquisition) (Fig. 4-5c). The results indicate that
following the initial training, only GFP and SOD1+CAT rats acquired a spatial search
strategy, focused on the goal quadrant. In probe 2 (one-hour retention), only
SOD1+CAT rats had a DI score above chance (p<0.05) indication that SOD1+CAT rats
retained the spatial search strategy over the one-hour retention interval. In probe 3
(relearning), all groups exhibited DI score above chance (p<0.05), indicating that all
animals, including SOD1+GFP rats can acquire a spatial search strategy with repeated
training. In sum, the results indicate that SOD1 overexpression for 1 month was
associated with impaired spatial learning in aged rats. The fact that the SOD1 +CAT
exhibited superior performance relative to SOD1 group indicates that the impairment
maybe rescued to co-overexpressing CAT. In addition, SOD1+CAT rats exhibited some
benefits relative to GFP animals, exhibiting a decrease in the escape latency during
training, more platform crossings during the probe trial and better memory during the
one hour retention interval.

104
NMDAR-Mediated Synaptic Potentials are Reduced in Rats with SOD1 Overexpression
Starting two weeks after finishing water maze study, animals were killed (one per
day) to examine hippocampal synaptic function. SOD1+GFP (n = 8 rats, 12 slices),
SOD1+CAT (n =10 rats, 15 slices), and GFP (n = 12 rats, 20 slices) were used to
examine the input-output function (stimulation intensity-synaptic response) for the total
synaptic response and the N-methyl-D-aspartic acid receptor (NMDAR) mediated
component of the synaptic response, and the level of LTP. Examination of the input-
output curves for the synaptic responses indicated no group difference in baseline
synaptic strength [F(2,444)=0.038, p=0.96] (Fig. 4-6a) suggesting no difference in the -
amino-3-hydroxy-5-methy-4-isoxazolepropionic acid (AMPA) receptor component of
synaptic transmission. Repeated ANOVA on the input-output curves of the NMDAR
synaptic response indicated a significant interaction between treatment and stimulation
intensity [F(24,624)=2.20, p<0.001]. The treatment effect was apparent for the higher
stimulation intensities. In fact, ANOVA on fEPSP at the highest stimulation intensity
indicates a tendency of treatment effect [F(2,52)=2.35, p=0.10] and a post hoc
comparison indicated that SOD1+GFP rats had a significantly reduced NMDAR synaptic
response relative to GFP rats (p<0.05) (Fig. 4-6b). The results suggest that the
influence of redox state regulated by SOD1 and CAT is specific to NMDAR but not
AMPAR.
For the study on LTP, slices were bathed in normal ACSF to record the AMPA and
NMDAR component of the synaptic response. LTP was induced by four episodes of
high-frequency stimulation (HFS; 100 Hz, 1 s with 1 s interval). One group t-tests
indicated that the fEPSP was significantly greater in HFS-induced LTP from GFP

105
(p<0.0001), SOD1+GFP (p<0.005) and SOD1+CAT (p<0.005) compared to the baseline
(=100) (Fig. 4-6c). However, there was no difference in the levels of HFS-induced LTP
between treatment groups [F(2,44)=0.242, p=0.78] (Fig. 4-6c).
Effect of Overexpression of SOD1 and CAT on Glutathione Redox State
The level of GSH, GSSG were measured in hippocampus of rats with
overexpression of SOD1+GFP (n = 11), SOD1+CAT (n = 10), or GFP (n = 13) for the
purpose of determining the effects of overexpression of SOD1 or CAT on glutathione
redox state. ANOVA revealed a tendency for a treatment effect for GSH levels
[F(2,31)=1.87, p=0.17]. Post hoc comparison indicated a tendency (p = 0.08) for
decreased GSH in hippocampi of SOD1+GFP compared to GFP rats (Fig. 4-7a). Total
GSH was measured when all the GSSG in the samples were reduced to GSH by adding
extra glutathione reductase to the samples. An ANOVA revealed a treatment effect in
total GSH [F(2,31)=7.55, p<0.005] and post hoc comparisons indicated that
overexpression of SOD1+GFP resulted in a significant decrease (22% compared to
GFP and 17% compared to SOD1+CAT) in total GSH compared to GFP and
SOD1+CAT rats (p<0.001 and p<0.05, respectively). In addition, an ANOVA revealed a
treatment effect in GSSG [F(2,30)=7.449, p<0.005]. Post hoc comparisons indicated
that overexpression of SOD1+GFP resulted in a significant decrease of GSSG
compared to GFP and SOD1+CAT rats (p<0.005 and p<0.005 respectively) (Fig. 4-7c).
Furthermore, an ANOVA revealed a treatment effect in the GSH: GSSG ratio
[F(2,30)=5.19, p<0.05]. Post hoc comparisons indicated that overexpression of
SOD1+GFP in hippocampi resulted in a significant increase in GSH: GSSG ratio
compared to GFP (71% increase; p<0.05) and SOD1+CAT (99% increase; p<0.05) rats
(Fig. 4-5d).

106
Effect of Overexpression of SOD1 and CAT on Activities of GSH Peroxidase and GSH Reductase
The activity of glutathione peroxidase (GPx) and glutathione reductase (GR), were
measured in hippocampus of rats with overexpression of SOD1+GFP (n = 12),
SOD1+CAT (n = 10), or GFP (n = 14) for the purpose of determining other enzymes
involved in glutathione redox state affected by overexpression of SOD1 or CAT. GPx
catalyzes the reduction of hydrogen peroxide by reducing glutathione. An ANOVA
revealed a significant treatment effect on GPx activity [F(2,33)=5.96, p=0.006]. Post hoc
comparisons indicated that SOD1+GFP rats had a significant decrease in GPx activity
compared to SOD1+CAT (28 % decrease; p < 0.005) and GFP rats (20 % decrease; p <
0.05). The GPx activity was not different between GFP rats and SOD1+CAT rats (p =
0.23) (Fig. 4-8a). GR catalyzes the reduction of oxidized glutathione (GSSG) to reduced
glutathione (GSH), using β-nicotinamide dinucleotide phosphate (NADPH) as the
hydrogen donor. An ANOVA revealed a significant treatment effect on GR activity
[F(2,33)=4.14, p < 0.05]. Post hoc comparisons indicated that SOD1+GFP rats had a
significant decrease in GR activity relative to GFP rats (10 % decrease; p<0.01);
SOD1+CAT rats exhibited a tendency to have decreased GR activity relative to
SOD1+CAT rats (8 % decrease; p = 0.05). The GR activity was not different between
GFP rats and SOD1+CAT rats (p=0.52) (Fig. 4-8b).
Discussion
In this study we have examined the hypothesis that overexpression of antioxidant
enzymes affects NMDA receptor function via regulating redox state. We used AAV to
deliver SOD1+GFP and SOD1+CAT to the hippocampus of aged (17-month) F344 rats
and examined memory-related behavioral performance one-month post injection. The

107
results indicated that SOD1 overexpression was associated with 1) improved cue
discrimination, 2) impaired spatial learning in water maze, 3) a decrease in input-output
curve with NMDAR-mediated fEPSP, 4) reductions in free GSH, total GSH, and GSSG
contents and 5) reductions in GPx and GR activities. On the other hand, overexpression
of SOD1+CAT was associated with improved cue discrimination, spatial learning and 1
hour retention in water maze. In addition, overexpression of SOD1+CAT did not affect
input-output curve of NMDAR-mediated fEPSP. Finally, free GSH and total GSH
contents were decreased in SOD1+CAT rats, but GSSG content did not change. The
activities of GPx and GR were also not altered in SOD1+CAT rats.
Our previous study with viral vector delivery of SOD1, SOD2 and CAT in the
hippocampi of Fischer 344 x Brown Norway F1 (F344BN F1) rats indicated that
overexpression of SOD1 impaired spatial learning and overexpression of CAT together
with SOD1 rescued the learning deficits caused by SOD1 overexpression alone (Lee et
al., 2012). To test the hypothesis that the benefit of SOD1+CAT was not due to
decreased SOD1 expression, as a result of using two viruses, we used a mix of two
viruses containing SOD1 and GFP, respectively, to transduce hippocampal cells. The
results demonstrate spatial learning deficits in SOD1+GFP rats, indicating that the
rescue by CAT was not due to using two viruses. Furthermore, Western blot results
indicate that the expression of hSOD1 was not different between SOD1+GFP rats and
SOD1+CAT rats (Fig. 4-2). In the cue discrimination task, SOD1+GFP and SOD1+CAT
animals exhibited enhanced performance in block 2 and block 3 compared to GFP rats.
The superior performance of the SOD1+GFP rats might due to impaired hippocampal
function, and a reliance or compensation by other neural systems (e.g. basal ganglia).

108
One study that examined the effect of brain lesions on the acquisition of the cue
discrimination task found that damage to the basal ganglia, but not the hippocampus,
disrupted acquisition on this task (Packard and McGaugh, 1992). Importantly, for the
current study, no effect of viral treatment was found for behavioral measures after block
4, indicating that all the animals ultimately employed the cue response strategy, the
animals did not exhibit a sensory motor deficit that would prevent them from learning on
the spatial task, and all animals learned the cue task to about the same extent.
Consistent with our previous study, overexpression of SOD1+GFP and
SOD1+CAT reduced the marker of lipid peroxidation, and the decrease in lipid
peroxidation was not associated with improved cognitive function (Lee et al., 2012). The
results indicate that accumulation of oxidative damage is not the only mechanism of
cognitive decline. In this case, other processes influenced by ROS may be more
relevant to age-related cognitive decline, such as intracellular redox state-regulated
synaptic plasticity. Our laboratory previously demonstrated that an age-related shift in
the intracellular redox state was associated with decreased NMDAR function and
increased release of Ca2+ from intracellular Ca2+ stores (Bodhinathan et al., 2010a, b).
Age-related impairment in NMDAR function, including impaired induction of LTP was
reversed by treating the slices with reducing agents. These results have recently been
replicated by several other groups, suggesting that NMDAR function is sensitive to age-
related changes in redox state (Robillard et al., 2011; Yang et al., 2010). To test the
hypothesis that SOD1 overexpression results in redox changes, similar to that observed
during aging, we measured the input-output curve for total fEPSP, NMDAR-mediated
fEPSP, and the level of LTP. Consistent with the idea that SOD1 overexpression

109
resulted in a more oxidized redox environment; input-output curve for NMDAR-mediated
fEPSP was reduced in SOD1+GFP rats. In contrast, the input-output curve for total
fEPSP was not affected by overexpression of SOD1+GFP. Since total fEPSP is mainly
due to AMPARs, the result is consistent with previous reports that the reducing agent
DTT had no effect on the AMPAR function of aged neurons (Abele et al., 1998;
Bodhinathan et al., 2010a; Gozlan et al., 1995). Together, the data suggests that the
effects of SOD1 overexpression, like altered redox state, are specific for NMDARs.
The overexpression of SOD1 is thought to influence the redox state through an
increase in H2O2 (Lee et al., 2012). As noted above, viral delivery of SOD1 reduces
oxidative damage in all age groups, but cognitive impairments are age-dependent. Our
previous work involving repeated cognitive testing suggested that deficits began to
emerge starting about 8-9 mo. of age, a time when markers of oxidative stress increase
(Gilmer et al., 2010; Martins et al., 2012). In addition, an increased expression of GPx1
is only observed in older SOD1 animals (Fig. 4-2). Previous work in other tissues
indicates that elevated H2O2 induces an increase in GPx activity (Noack et al., 1998;
Rohrdanz et al., 2001; Wijeratne et al., 2005). Finally, the cognitive impairments
associated with SOD1 overexpression in older animals were ameliorated by co-
expression of SOD1+CAT. Since CAT is one mechanism for the elimination of H2O2, the
results suggest improved cognition resulted from better homeostatic regulation of redox
state due to CAT processing of excess H2O2.
The redox state of tissues shifts towards a more oxidized state during aging
(Rebrin et al., 2004). Many human diseases conditions such as Parkinson’s, HIV/AIDs,
and cystic fibrosis, the more oxidized redox environment is associated with low GSH

110
levels (Townsend et al., 2003). Glutathione is considered to be the major thiol-disulfide
redox buffer of the cell (Gilbert, 1990). Measurements of total GSH and/or GSSG levels
have been used to estimate the redox environment of cells. In the current study, we
observed a decrease in glutathione levels in hippocampal tissue that overexpressed
SOD1. The reduction of free GSH, total GSH in SOD1+GFP and SOD1+CAT rats
suggested that the intracellular environments were in a more oxidized state. Normally, a
reduction of GSSG indicates a more reduced environment. However, the combination of
decreased GSSG in addition to loss of free GSH and total GSH in SOD1+GFP rats
suggests a serious loss of GSH, possibly due to deficits in GSH redox cycling.
Glutathione levels are maintained by the GSH redox cycle consisting of glutathione
peroxidase and glutathione reductase. The redox cycle has a high capacity to provide a
constant supply of reduced glutathione through the conversion of GSSG by glutathione
reductase. However, in SOD1+GFP rats, the activities of glutathione peroxidase and
glutathione reductase were both decreased (Fig. 4-8). The results suggest that
glutathione peroxidase and glutathione reductase, which are both redox sensitive
enzymes, might be disrupted/ modified due to the oxidized environment caused by high
level of H2O2 in SOD1+GFP rats. In this regard, overexpression of CAT may provide an
important alternate pathway for elimination of H2O2, preserving glutathione peroxidase
and glutathione reductase activity.
Another possibility for the decrease in both GSH and GSSG in SOD1+GFP rats is
that GSSG may have been actively exported out of the cells. As a result of this transport
process, severe oxidative stress can deplete both cellular GSH and GSSG (Lu, 1999).
Finally, overexpression of SOD1+GFP may have influenced GSH synthesis to decrease

111
GSH and GSSG levels. It has been reported that a decrease in GSH in several tissues
during aging is associated with a corresponding fall in the gene expression of glutamate
cysteine ligase and GSH synthase, two enzymes that are important in GSH synthesis
(Liu et al., 2004; Wang et al., 2003a). Future, studies may want to examine GSH
synthesis under conditions of SOD1 overexpression.

112
Figure 4-1. Co-overexpression of SOD1+GFP and SOD1+CAT. (A) A rat with co-transduction of AAV-SOD1 and AAV-GFP. A merged image of myc staining (red) and GFP (green). (B) A rat with co-transduction of AAV-SOD1 and AAV-CAT. A merged image of myc staining (red) and CAT staining (green). Calibration bars represent 100 µm

113
Figure 4-2. Antioxidant enzymes and oxidative stress markers in hippocampus with 1-month SOD1+GFP and SOD1+CAT overexpression. (A) Western blots of hippocampal lysates from 17-month old rats injected with viral vectors to express SOD1+GFP and SOD1+CAT. For SOD1, two bands were observed representing endogenous rat SOD1 (rSOD1) and the human myc tagged SOD1 (hSOD1 and c-myc). GFP was detected in GFP and SOD1+GFP rats but not in SOD1+CAT rats. CAT level did not change in SOD1+GFP rats but increased in SOD1+CAT rats. Lipid peroxidation measured with anti-4-hydroxy-2-nonenal (HNE) antibody was decreased by SOD1+GFP and SOD1+CAT overexpression. The level of GPx1 was increased in SOD1+GFP rats but was not changed in SOD1+CAT rats. GAPDH was used as a loading control. (B) Quantification of western blot data from SOD1+GFP and GFP rats. (C) Quantification of western blot data from SOD1+CAT and GFP rats. All densitometry was normalized by GAPDH. Asterisk indicates a significant (p < 0.05) difference in treatment. All densitometry was normalized by GAPDH. Error bars represent S.E.M.

114
Figure 4-3. Overexpression of SOD1+GFP or SOD1+CAT enhanced cue discrimination in the water maze compared to GFP control. All groups (GFP rats: black filled circles, n=14; SOD1+GFP rats: red square, n=11; SOD1+CAT rats: blue triangle, n=12) learned to reach the visible platform, as indicated by a significant overall decrease in latency (A) and distance (B) across blocks. Asterisk indicates significant difference (p<0.05) from GFP control in a block of cue training by ANOVA. Error bars represent S.E.M.
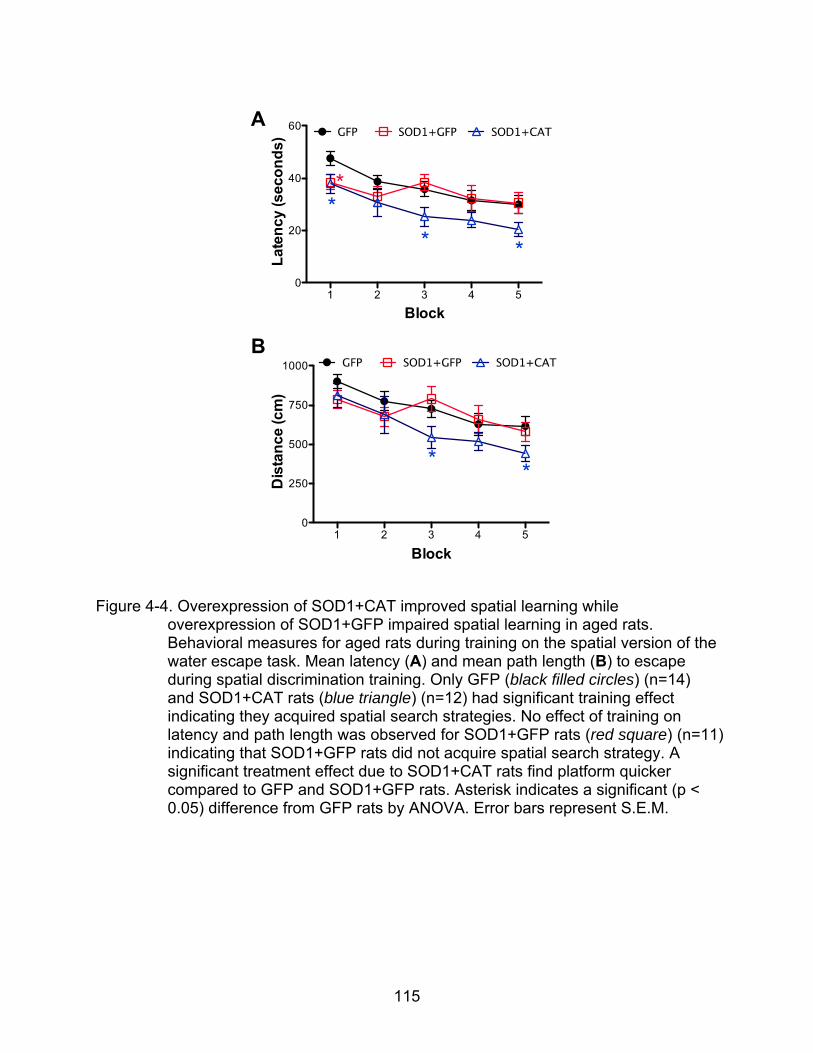
115
Figure 4-4. Overexpression of SOD1+CAT improved spatial learning while overexpression of SOD1+GFP impaired spatial learning in aged rats. Behavioral measures for aged rats during training on the spatial version of the water escape task. Mean latency (A) and mean path length (B) to escape during spatial discrimination training. Only GFP (black filled circles) (n=14) and SOD1+CAT rats (blue triangle) (n=12) had significant training effect indicating they acquired spatial search strategies. No effect of training on latency and path length was observed for SOD1+GFP rats (red square) (n=11) indicating that SOD1+GFP rats did not acquire spatial search strategy. A significant treatment effect due to SOD1+CAT rats find platform quicker compared to GFP and SOD1+GFP rats. Asterisk indicates a significant (p < 0.05) difference from GFP rats by ANOVA. Error bars represent S.E.M.

116
GFP
SOD1+GFP
SOD1+CAT
0
10
20
30
40
50 GFP SOD1+GFP SOD1+CAT
1 2 3 1 2 3 1 2 3Probe
Lat
ency
(sec
on
ds)
GFP SOD1+GFP SOD1+CAT
0
1
2
3
4
5GFP SOD1+GFP SOD1+CAT
Probe1 2 3 1 2 3 1 2 3
Pla
tfo
rm c
ross
ing
s
GFP SOD1+GFP SOD1+CAT
0.0
0.1
0.2
0.3
0.4
0.5
0.6GFP SOD1+GFP SOD1+CAT
1 2 3 1 2 3 1 2 3
Probe
**
* **
*
Dis
crim
inat
ion
ind
ex
A
B
C
Figure 4-5. Overexpression of SOD1+CAT improves spatial learning in probe tests. Three single probe trial was given between block 5 and block 6 of spatial training, 1 hour after block 6 followed by a refresher training block (block 7), and 24 hours later following block 8, respectively. (A) Examination of the latency for first platform crossing across three probe trials, indicated a treatment effect due to an superior performance by the SOD1+CAT group

117
(blue triangle) (n=12) compared to the SOD1+GFP (red square) (n=11) (p<0.01) and GFP (black filled circles) (n=14) (p=0.08) group. (B) Examination of the number of platform crossings across three probe trials indicated a significant treatment effect due to SOD1+CAT rats made more crossings than GFP (p<0.05) and SOD1+GFP (p<0.05) rats. (C) No treatment effect was found when examining discrimination index (DI). To determine if the animals acquired a spatial search strategy, the DI scores were compared with that expected by chance. Asterisk sign indicates a significance (p < 0.05) difference from chance (one-group t-test). Error bars represent S.E.M.

118
Figure 4-6. NMDAR-mediated synaptic potentials are reduced in rats with SOD1 overexpression. (A) Total field potential (total-fEPSP) in GFP rats (black filled circles) (n=11 rats, 15 slices) was not different from total-fEPSP in SOD1+GFP rats (red square) (n=9 rats, 14 slices) or SOD1+CAT rats (blue

119
triangle) (n=8 rats, 11 slices). (B) An interaction of stimulation intensity and treatment for the NMDAR mediated synaptic response (NMDAR-fEPSP) was due a decrease in the response for rats with overexpression of SOD1+GFP (n=10 rats, 21 slices) compared to GFP rats (n=10 rats, 18 slices) at the highest stimulation intensities. (C) Quantification of the mean percent change in the fEPSP slope recorded from the control (white bars) and HFS (grey bars) pathway of the hippocampal slices from GFP (n=12 rats, 20 slices), SOD1+GFP (n=8 rats, 12 slices) or SOD1+CAT rats (blue triangle) (n=10 rats, 15 slices). Asterisk sign indicates that the fEPSP is significantly greater in HFS-induced LTP from GFP (p<0.0001), SOD1+GFP (p<0.005) and SOD1+CAT (p<0.005) compared to the baseline (=100). However, overexpression of SOD1+GFP or SOD1+CAT does not affect LTP in hippocampal area CA1. Error bars represent S.E.M.

120
Figure 4-7.Decreased level of reduced GSH (GSH), total GSH and oxidized GSH (GSSG) are observed in rats with overexpression of SOD1+GFP. (A) Free GSH level was measured in hippocampal samples from GFP, SOD1+GFP and SOD1+CAT rats. SOD1+GFP rats had significantly reduced free GSH level than GFP rats. (B) Total GSH level was significantly reduced in SOD1+GFP compared to GFP and SOD1+CAT rats. (C) GSSG level was significantly reduced in SOD1+GFP rats compared to GFP and SOD1+CAT rats. (D) GSH:GSSG ratio was significantly higher in SOD1+GFP rats than GFP and SOD1+GFP rats. Asterisk indicates a significant (p < 0.05) difference by ANOVA. GFP rats (n=13) (black bars); SOD1+GFP rats (n=11) (red bars); SOD1+CAT rats (n=10) (blue bars). Error bars represent S.E.M.

121
Figure 4-8. Decreased glutathione peroxidase (GPx) and glutathione reductase (GR) activities are observed in rats with overexpression of SOD1+GFP (A) GPx activity was measured in hippocampal samples from GFP, SOD1+GFP and SOD1+CAT rats. GPx activity was significantly reduced in SOD1+GFP rats relative to GFP and SOD1+CAT rats. (B) GR activity was significantly reduced in SOD1+GFP rats than GFP rats. In addition, there is a tendency of GR reduction in SPD1+GFP rats than SOD1+CAT rats. Asterisk indicates a significant (p < 0.05) difference by ANOVA. GFP rats (n=14) (black bars); SOD1+GFP rats (n=12) (red bars); SOD1+CAT rats (n=10) (blue bars). Error bars represent S.E.M.

122
CHAPTER 5
GENERAL DISCUSSION
Summary
Aging is associated with changes in physical performance and cognitive
impairment including deficits in learning and memory (Foster, 1999, 2012), which are
thought to be due to overproduction of free radicals (Harman, 1992). The free-radical
theory of aging indicates that the overproduction of endogenous ROS, including
superoxide, hydrogen peroxide, and hydroxyl radicals result in a pattern of cumulative
damage due to modification of proteins, nucleic acids, and polyunsaturated fatty acids
of the lipid membrane (Leutner et al., 2001a; Liu et al., 2002; Oliver et al., 1987; Smith
et al., 1991). Antioxidant therapy for cognitive function through supplement or
antioxidant enriched diet have little effect on normal aging possibly due in part to poor
penetration of the CNS through the blood brain barrier and (Bowry et al., 1992;
Grodstein et al., 2003; Maxwell et al., 2005). Alternately, antioxidant enzymes are the
major factors for defending ROS in the brain. Studies employing transgenic mice
indicate overexpression of SOD1 improves synaptic plasticity and memory during aging
(Kamsler et al., 2007; Kamsler and Segal, 2003b). Upregulation of SOD2, on the other
hand, has no obvious effect on synaptic plasticity or memory (Hu et al., 2007a). It is
unclear whether the improvement is due to a lifetime of overexpression, decreasing the
accumulation of oxidized molecules, or if increasing antioxidant enzymes in older
animals could reduce oxidative damage and improve cognitive function. In our
preliminary studies, we used lentivirus to modestly increase SOD1 expression in the
CA1 region of young and aged F344BN F1 rats. We tested the rats in various
behavioral tasks and found that modest overexpression of SOD1 in CA1 improved

123
learning in the spatial version of water maze in aged rats but had no effect in young rats
(Lee, 2008). Overexpression of SOD1 also did not affect rats’ performance in cue
discrimination, inhibitory avoidance or novel object recognition. However, lentivirus
mediated expression in the hippocampus was generally limited and variable. We
therefore, increased the transduction area using AAV in order to test the hypothesis that
overexpression of antioxidant enzymes (SOD1, SOD2, CAT and SOD1+CAT) in whole
hippocampi will delay age-related memory decline by reducing oxidative damage. This
study examined memory-related behavioral performance of the young (4-month) (19-
month) rats one month and four months post viral injection. The results from this study
indicated that overexpression of antioxidant enzymes reduced oxidative damage;
however, memory function was not related to the level of oxidative damage.
Overexpression of antioxidant enzymes did not affect learning and memory in young
rats. Further, increased expression of SOD2 and CAT did not affect learning and
memory in aged rats. Surprisingly, overexpression of SOD1, initiated in advanced age,
impaired learning. Moreover, increased expression of SOD1+CAT provided protection
from impairments associated with overexpression of SOD1 alone and appears to guard
against cognitive impairments in advanced age. An examination of protein expression
using western blots, revealed that aged rats with overexpression of SOD1 had
increased expression of GPx1. It was concluded that the learning impairment observed
in aged SOD1 rats might be due to overproduction of H2O2.
Previous studies from our laboratory suggest that a more oxidized intracellular
redox state mediates a decrease in NMDAR function, which could underlie impaired
cognition (Bodhinathan et al., 2010a, b). Therefore, we designed a serious of

124
experiments to examine the hypothesis that overexpression of antioxidant enzymes in
the hippocampi of aged rats would affect NMDAR function via regulating redox state.
Again, we used AAV to deliver SOD1+GFP or SOD1+CAT to the hippocampus of aged
(17-month) F344 rats and examined memory-related behavioral performance one-
month post injection. The results confirmed that SOD1 overexpression was associated
with impaired performance on the spatial version of the water maze. Importantly, SOD1
overexpression was associated with a decrease in NMDAR-mediated fEPSP.
Biochemical assays indicated that free glutathione, oxidized glutathione and total
glutathione decreased in aged rats with overexpression of SOD1. In addition, both
glutathione peroxidase and glutathione reductase activities decreased in aged rats with
overexpression of SOD1, which might result from altered redox state of the enzymes.
In conclusion, viral vector gene delivery provides a novel approach to test the
hypothesis that increased expression of antioxidant enzymes, specifically in
hippocampal neurons, will provide protection from age-related cognitive decline. The
results indicate that oxidative stress is a likely component of aging; however, it is
unclear whether increased production of ROS or the accumulation of oxidative damage
is the primary cause of functional decline. The results provide support for the idea that
altered redox sensitive signaling rather than the accumulation of damage may be of
greater significance in the emergence of age-related learning and memory.
Discussion
In this series of studies, we asked an important question in the cognitive aging and
memory field. Does ROS/ oxidative damage cause memory impairment during normal
aging? If so, can we rescue the memory deficits by overexpressing antioxidant enzymes
in the hippocampi using viral vectors? Surprisingly, enhanced overexpression of SOD1

125
in hippocampi using AAV did not help with memory in aged rats; instead, it impaired
memory. This result led us to examine the importance of tightly controlled antioxidant
system across life span.
One possibility is that SOD1 overexpression is toxic. Mutant SOD1 was
discovered to be the primary cause of 15 to 20 % of familial ALS cases (Rosen et al.,
1993). One possible mechanism proposed for ALS emphasizes reduced SOD activity
arising from instable or imperfectly folded enzymes, which could cause toxic oxidative
stress through an imbalance in oxidative defenses (Deng et al., 1993; Orrell et al., 1995).
However, increased expression of wild-type SOD1 in mice with ALS does not slow
disease progression or diminish the pathological changes in motor neurons (Brown,
1998). The failure of increased SOD1 activity to ameliorate the disease (Brown, 1998),
in addition to the fact that some SOD1 mutants retain high SOD activity (Ratovitski et al.,
1999), and targeted deletion of SOD1 in mice does not induce ALS-like symptoms
(Reaume et al., 1996) challenges the idea that mutant toxicity is linked to oxidative
stress arising from O2·-. Rather, mutant SOD1 is proposed to cause disease by the
acquisition of toxic properties through misfolded mutant SOD1 aggregates (Johnston et
al., 2000; Wang et al., 2003b). A recent study using conformation-specific antibody that
detects misfolded SOD1 (C4F6) found that oxidized wild-type SOD1 and mutant SOD1
share a conformation epitope that is not present in normal wild-type SOD1 but
commonly detected in motor neurons in the lumbosacral spinal cord in a subset of
human sporadic ALS (SALS) cases. The authors suggest that wild-type SOD1 can be
pathogenic in SALS most likely by dysregulating kinase signaling pathways (Bosco et
al., 2010).

126
In our current study, the memory impairment observed in aged rats is most likely
not due to SOD1 toxicity for several reasons. First, the memory deficit was limited to
aged SOD1 animals, indicating that it was not simply due to SOD1 overexpression.
Second, one would expect that toxicity should increase over time. On the contrary, we
observed that the memory deficits decreased following four months of SOD1
overexpression in our oldest rats. However, it would be interesting for future studies to
examine the effects of overexpression of wild-type SOD1 under severe oxidative stress
conditions such as in animal models of neurodegenerative diseases.
H2O2 Produced by Overexpression of SOD1 Affects Synaptic Plasticity and Memory in an Age-Dependent Manner
We emphasize the importance of H2O2 as a redox state regulator and suggest that
overexpression of SOD1, which converts O2·- into H2O2 in the hippocampus was
detrimental to learning and memory in aged rats. Several pieces of evidence point to an
increase in H2O2 in aged rats that overexpress SOD1. First, the memory impairment
was only observed in aged rats with overexpression of SOD1, and the impairment could
be rescued by co-overexpression of CAT in the same animal. CAT converts H2O2 to
water, suggesting that the rescue was due to a decrease in an elevated level of H2O2.
Similarly, GPx1 expression, examined by Western blot analysis, was increased in aged,
but not young, rats with overexpression of SOD1. Previous work in other tissues
indicates that elevated H2O2 induces an increase in GPx level (Hussain et al., 2004;
Noack et al., 1998; Rohrdanz et al., 2001; Wijeratne et al., 2005). Finally, the increased
GPx1 expression was not observed in aged rats with overexpression of SOD1+CAT.
The results support the idea that SOD1 overexpression was associated with an
elevated level of H2O2 in aged, but not young rats.

127
Young (5-month old) rats with one-month overexpression of SOD1 did not exhibit
memory deficits; however, as the rats aged (9-month old), following four months of
overexpression of SOD1, a mild memory deficit started to emerge. In a study using
superoxide sensitive probes to detect O2·- across life span of mammals and birds, an
age-related increase in O2·--dependent chemiluminescence was observed in the brains
of C56/BL6 mice, Wistar rats, and pigeons, with no change of SOD1 and SOD2 activity
(Sasaki et al., 2008). SOD1 converts O2·- to H2O2, suggesting that overexpression of
SOD1 may have increased H2O2 in the hippocampus as O2·- increased with advancing
age.
GSH is the major redox buffer for cellular H2O2. We observed that GSH, GSSG
and total GSH levels were decreased in SOD1+GFP rats, consistent with the idea that
the intracellular environment was in a more oxidized state, possibly due to an increased
level of H2O2. Although normally a reduction of GSSG indicates a more reduced
environment, the combination of decreased GSSG, in addition to loss of free GSH and
total GSH, in SOD1+GFP rats suggests a serious loss of GSH. A question remains
concerning the mechanism for the decrease in GSH. In this case, an increase in H2O2
may have induced oxidative inactivation of GSH redox cycling proteins such as GPx
and GR. We observed decreased activity of GPx and GR in aged rats with
overexpression of SOD1+GFP. Decreased GSH redox cycling function may have
contributed to the decreased level of GSH, GSSG and eventually insufficient antioxidant
buffering system.
Finally, NMDAR activity is required to control many physiological conditions
including neuronal development, synaptic plasticity and memory. NMDAR function is

128
known to be redox sensitive. H2O2 induces the formation of disulfide bonds between
pairs of cysteine residues in proteins, and thus change the structure and function of
these proteins, including proteins involved in multiple signaling cascades including Ca2+
signaling (Foster, 2007). Indeed, application of the cysteine specific reducing agent,
dithiothreitol, increased NMDAR but not AMPAR synaptic response and the magnitude
of LTP in hippocampal slices from older rats (Bodhinathan et al., 2010a). The
membrane-impermeable reducing agent, L-GSH, failed to increase the NMDAR
response when applied on the hippocampal slice; however, an increase of NMDAR
response was observed by intracellular delivery of L-GSH through the intracellular
recoding pipette. The results indicate that the intracellular redox state, rather than the
disulfide bonds on the extracellular cysteine residues, mediates the suppression of
NMDAR function and impaired LTP in aged rats. In our present study using rats with
overexpression of SOD1, the production of H2O2 is from the intracellular space;
however, because H2O2 is membrane permeable, we cannot rule out the possibility that
the newly synthesized H2O2 also affect extracellular cysteine residues. Regardless, the
fact that NMDAR, but not AMPAR synaptic responses were reduced in aged rats with
overexpression of SOD1+GFP provides further support for the idea that H2O2 produced
by SOD1 overexpression resulted in an oxidized redox environment in older animals.
Taken together, the weight of evidence indicates that SOD1 overexpression in aged
animals resulted in an increase in the level of H2O2.
Interestingly, aged (20-month old) rats with one-month overexpression of SOD1
showed severe memory deficits; however, the memory deficit became moderate with
advancing age (24-month old), following four-months overexpression of SOD1. In the

129
Sasaki study, the increase in O2·- level from birth until 20-month of age began to decline
with more advanced age in the brains of C56/BL6 mice (Sasaki et al., 2008). Similarly,
in tg-SOD1 mice, overexpression of SOD1 had an age-dependent effect on memory.
SOD1 overexpression impaired or had no effect on learning in young mice and
enhanced spatial memory in older mice (Kamsler et al., 2007; Kamsler and Segal,
2003b). The authors suggest that aged mice adapt to elevated H2O2. The results of the
present study, indicating no effect of memory in young SOD1 rats, severe memory
impairment in old SOD1 rats and moderate memory impairment in more advanced aged
SOD1 rats providing new evidences for the hypothesis that ROS generation increased
with age, and elevated ROS might be beneficial or less detrimental to animals at a more
advanced age.
In fact, one of the main driving forces for the current study was previous work
indicating that overexpression of SOD1 in aged mice was beneficial. Although
overexpression of SOD1 in young transgenic mice was found to be associated with
impaired LTP and impaired memory (Kamsler and Segal, 2003b), overexpression of
SOD1 in aged transgenic mice was associated with enhanced LTP and memory
compared to their age matched wild-type littermates (Kamsler et al., 2007; Kamsler and
Segal, 2003b). The authors proposed that H2O2 regulates synaptic plasticity in a
complex manner that depends on an interaction between the pre-existing redox state
and a transient, tetanic stimulation-induced rise in H2O2. According to their hypothesis,
young SOD1 transgenic mice that overproduce H2O2, contain an intracellular redox
environment with higher level of H2O2. The intracellular proteins therefore are
desensitized to the small rise of H2O2 that follows titanic stimulation, which cannot pass

130
the threshold necessary for producing LTP. The aged SOD1 transgenic mice have a
genetic desensitization of H2O2 caused by the overexpression of SOD1, accompanied
by age-dependent mitochondrial leakage (Beckman and Ames, 1998) that produces
more H2O2, resulting a state that is again sensitive to acute change of H2O2, promoting
LTP (Kamsler et al., 2007; Kamsler and Segal, 2003b). This hypothesis may be limited
to explain the age-dependent effects of overexpression of SOD1 on LTP induction in
hippocampal slices. However, if we consider that training and testing in the water maze
involves neural activity and activation of LTP mechanisms, our results could fit into this
hypothesis with a few exceptions. First of all, 4-month SOD1 rats had normal memory,
which is different from young SOD1 transgenic mice that were impaired in LTP and
memory. The different results might be explained by differences in the duration, extent,
or location of SOD1 overexpression. For our studies, SOD1 expression was increased
4-fold for one month in neurons of the hippocampus. In the case of transgenic mice,
SOD1 expression was increased 6-fold during development until 2-month of age in most
of the cells, if not all, including cells in the brain (Epstein et al., 1987). Again it should
be noted that a moderate memory impairment started to show up in our 9-month old
SOD1 rats with 4-month overexpression of SOD1, suggesting increased H2O2 at this
stage. In addition, our 20-month old SOD1 rats were memory impaired, which was
different from old SOD1 transgenic mice that exhibited enhanced LTP and memory.
Thus, age is an important variable. Consider the medium lifespan of SOD1 transgenic
mice is shorter than F344BN F1 (30 months versus 34 month)(Perez et al., 2009b;
Turturro et al., 1999), the 2-year old transgenic mouse was in a more advanced age
compared to a 20-month old F344BN F1 rats. Finally, at 24 months, the SOD1 rats with

131
4-month overexpression of SOD1 were not significantly different from the GFP rats at
the same age. It appears that the SOD1 rats may have adapted to the increase in
SOD1, exhibiting similar memory function compared to the GFP rats at this older age.
Future studies may want to examine the correlation of ROS generation and
overexpression of SOD1 in middle age rats, when ROS generation starts to exceed the
antioxidant capacity and in very old rats when ROS generation starts to slow down and
the endogenous level of ROS at this stage may not be sufficient for normal physiological
function.
Co-Overexpression of SOD1 and CAT in the Hippocampi is More Beneficial Than Overexpression of SOD1 or CAT Alone
Overexpression of SOD1 has also been reported to have several deleterious
effects, such as neuromuscular abnormalities and premature thymic involution,
suggesting that upregulation of SOD1 can lead to a toxic buildup H2O2, which can be
eliminated by the additional increase in CAT activity (Peled-Kamar et al., 1995; Peled-
Kamar et al., 1997). In the present study, we demonstrated that relative to animals that
overexpress SOD1, co-overexpression of SOD1 and CAT in hippocampi rescued
memory deficits observed in 20-month old F344BN F1 rats with overexpression for one
month, improved memory in 24-month old F344BN F1 rats with 4-months
overexpression, and improved memory in 18-month old F344 rats with one-month
overexpression.
If a high level of H2O2 during aging impairs memory, can removing H2O2 by
overexpression of CAT alone be beneficial? In our present study, we demonstrated that
overexpression of CAT alone in hippocampi has little effect on memory. The result may
be due to the fact that CAT activity can be inhibited by O2·- (Kono and Fridovich, 1982;

132
Shimizu et al., 1984). Therefore, co-overexpression of SOD1 and CAT could interact to
be more beneficial than overexpression of SOD1 or CAT alone by keeping CAT in an
active form through the increased elimination of O2·- and permitting CAT to remove
excess H2O2. Future studies may want to determine the protective effects of
overexpression of SOD1, CAT and co-overexpression of SOD1 and CAT against ROS-
mediated tissue injury including ischemia and reperfusion injury, hypoxic lung injury,
brain trauma, and neurotoxic drugs.
Mitochondrial ROS Has Minimal Effect on Synaptic Plasticity and Memory
Mitochondrial generation of O2·- during normal aerobic metabolism has been proposed
to be an important link between oxidants and aging (Balaban et al., 2005). However, in
a study of overexpression the mitochondrial SOD (SOD2) in transgenic mice, there was
no effect on LTP induction or spatial learning in the water maze (Hu et al., 2007a). The
results from our current study using AAV to upregulate SOD2 expression in hippocampi
were consistent with the SOD2 transgenic mice study. Overexpression of SOD2 in
hippocampi of young or aged rats had no effect on memory. The lack of influence of
SOD2 overexpression on synaptic plasticity and memory may be due to the
phospholipid bilayer structure of the mitochondrial membranes that does not allow free
diffusion of O2·- generated within mitochondria (Zeevalk et al., 2005). One study points
to a mitochondria membrane protein, voltage-dependent anion channels (VDAC), in
controlling the release of O2·- from mitochondria to the cytosol (Han et al., 2003).
Together with our results and the results from SOD2 transgenic mice, mitochondrial
ROS does not appear to contribute in a major way to synaptic plasticity and memory
under normal physiological conditions. In contrast, mitochondrial ROS has been

133
reported to play major role in memory dysfunction in neuropathological conditions, such
as Alzheimer’s disease (Dumont et al., 2009; Ma et al., 2011; Massaad et al., 2009).
Reactive Oxygen Species (ROS) and Reactive Nitrogen Species (RNS) as Signal Molecules in Synaptic Plasticity
One idea related to SOD1 and H2O2 influences on memory is that H2O2 may act
as a second messenger that is critical for memory. Second messenger molecules are
generated at the time of receptor activation, are short-lived, and act specifically on
effectors to transiently alter their activity (Forman et al., 2004). Indeed, ROS can be
generated at the time of receptor activation and are short-lived, but the specificity of
their action has been difficult to assess. Clearly, a reactive species such as OH cannot
have any specificity, as it reacts at nearly the rate of diffusion with almost any molecule.
H2O2 and O2·-, on the other hand, are less reactive. H2O2, in particular, has gained
credence as a second message as it is a highly diffusible molecule that can be
produced endogenously via receptor-mediated mechanisms in many cell types
(Lambeth, 2002) and is enzymatically metabolized. The targets of H2O2 include thiol
proteins, but these proteins have yet to be fully defined.
Targets for ROS modification include protein kinases, protein phosphatases, and
transcription factors that are involved in long-lasting LTP induction and long-term
memory formation. For example, LTP involves activation of extracellular signal-
regulated kinase (ERK), protein kinase A (PKA), protein kinase C (PKC), and
neurogranin (NG) (a PKC substrate that binds to and sequesters calmodulin (CaM)).
Neurogranin promotes the release of CaM, and thereby promotes Ca2+/CaM signaling
via the activation of CaMKII (Huang et al., 2000; Kishida et al., 2005; Klann et al., 1993;
Li et al., 1999). In contrast, ROS can inhibit protein phosphatases such as calcineurin

134
(protein-phosphatase 2B; PP2B), which has opposite effects to CaMKII and PKC
(Winder and Sweatt, 2001). ROS can also directly activate transcription factors such as
the redox-sensitive transcription factor NF-KB, or indirectly influence transcription factors
via the activity of ERK, PKA and CaMKII (Brigelius-Flohe et al., 2004). Similar to ROS,
NO and RNS can act as signaling molecules and are known to target thiol proteins and
metalloproteins (proteins contain a mental ion cofactor). RNS are capable of chemically
modifying critical thiols on numerous proteins and forming S-nitrosothiols, leading to
disruption and/or regulation of the target protein. Many proteins have been reported to
be at least partially regulated by S-nitrosylation, including NMDARs (Lipton et al., 1993).
Taken together, ROS/RNS signaling is critical for a number of cellular processes,
including synaptic plasticity and memory formation. Thus, indiscriminate suppression of
ROS/RNS may not always be an appropriate therapeutic method for treating
oxidative/nitrosative stress. The current study provides evidence for the complexity of
the ROS system and suggests the futility of modifying a single enzyme. Future studies
may want to examine possible differences in ROS/RNS signaling cascades in memory
impaired and memory unimpaired animals, and determine the proper ratio of ROS/RNS
and antioxidant enzymes as a potential therapeutic avenue.
LTP During Aging
LTP is thought to mediate learning and memory. Furthermore, LTP declines with
advancing age and depends on NMDAR function, which in turn is sensitive to the redox
environment. Therefore, in the current study we examined LTP induction in aged rats
with overexpression of SOD1+GFP, SOD1+CAT and GFP. The results indicated that
the LTP level was not changed in any treatment group and appear to contradict the

135
results in the study using SOD1 transgenic mice, in which overexpression of SOD1 in
aged transgenic mice results in larger LTP than that found in wild-type mice (Kamsler
and Segal, 2003b). The different results might be due to the different protocols used to
induce LTP in these two studies. In Kamsler’s study, LTP was induced by theta burst
stimulation (TBS) (10 trains of 4 pulses at 100 Hz separated by 200 msec intertrain
intervals, at the same intensity as the test stimulation). TBS is patterned after innate
rhythms in the hippocampus and is a relatively mild stimulation that is less likely to
saturate the LTP generating mechanism (Morgan and Teyler, 2001). In contrast, in our
study, LTP was induced by four episodes of high-frequency stimulation (HFS; 100 Hz, 1
s with 1 s interval). Compared to the 40 pulses using the TBS pattern, the 400 pulses
employed in the current study is relatively strong. It has been suggested that aging is
not associated with a difference in the magnitude of LTP, but an increase in the
threshold to induce LTP (Foster, 1999, 2012). Thus, age-related differences are not
observed when relatively strong induction stimulation is employed. In a previous study
examining redox influences on LTP in aged animals, a single burst of 100 Hz for 1 s
was able to distinguish between slices bathed in the reducing agent DTT and control
slices (Bodhinathan et al., 2010a). Therefore, the reason we were not able to see the
difference of LTP between treatment groups might be due to the failure to use a LTP
induction protocol that is near the threshold for LTP induction. Future studies may want
to work on finding the right LTP induction protocols that are suitable to distinguish the
fine synaptic plasticity difference in middle age animals.
Global Versus Local Redox State
The GSH: GSSG ratio is used to describe the global state of oxidized and reduced
species in the cell and is thought to play a major role in maintaining the cellular redox

136
state (Schafer and Buettner, 2001; Schafer et al., 2003). However, it has been argued
that since signal transduction reactions are not global but localized processes, there is
not a direct connection between the global GSH:GSSG ratio and any specific redox
signaling reaction (Forman et al., 2004). The importance of localized events is well
demonstrated by the sudden increase of cAMP which occurs after stimulation and
subsequent activation of the protein kinase A (PKA), even though enough
phosphodiesterase is present in the cell to decrease cAMP to its pre-stimulation level.
Thus cAMP activates PKA only within a short distance allowed by the
phosphodiesterase. Similarly, in our study, it seems that the global reduction of the GSH:
GSSG ratio should not be used to define the local redox state. In other word, the local
concentration of H2O2 may be more important in affecting its target molecules.
In this study, we observed abnormal level of GSH and GSSG and decreased
activities of glutathione peroxidase (GPx) and glutathione reductase (GR) in rats with
overexpression of SOD1+GFP. The combination of decreased GSSG in addition to loss
of free GSH and total GSH in SOD1+GFP rats suggests a serious loss of GSH, possibly
due to deficits in GSH redox cycling. Glutathione levels are maintained by the GSH
redox cycle consisting of glutathione peroxidase and glutathione reductase.
One possibility is that GPx and GR function was modified by the altered redox
environment. GPx’s are a family of peroxidases that contain a rare amino acid,
selenocysteine, which is essential for peroxidase activity and require GSH as a co-
substrate (Rocher et al., 1992; Tappel, 1984). GPx catalyzes the cellular reduction of
inorganic H2O2, lipid peroxide or organic hydroperoxide to water or corresponding
alcohols (Prabhakar et al., 2005). Selenium (Sec) from the active site of GPx is in the

137
same column of elements in the periodic table as sulfur, and thus shares many
chemical properties with it. Therefore, this selenocysteine residue resembles a cysteine
residue in terms of chemical properties but with a higher reactivity (Huber and Criddle,
1967; Stadtman, 1980). The selenocysteine active site of GPx is highly sensitive to
oxidation modification. NO-mediated oxidation of the GPx selenocyeteine (oxidization of
Sec45 to form a selenenyl sulfide (Se-S) with a free thiol) has been shown to inactivate
the enzyme (Asahi et al., 1997). Similar to GPx, GR is another GSH related enzyme.
GR reduces disulfide glutathione (GSSG) to the sulfhydryl form of glutathione (GSH). Its
enzyme activity has been reported to be reversibly inactivated under oxidative stress
induced by myocardial ischemia-reperfusion injury (Yim and Ko, 1999), cardiac arrest
(Sharma et al., 2007), catecholamine (Remiao et al., 2000), and by S-nitrosoglutathione,
an endogenous NO donor (Becker et al., 1995). In conclusion, due to the fact that GPx
and GR are inactivated by a variety of oxidative modifications, it is reasonable to
hypothesize that the reduced activities of GPs and GR observed in rats with
overexpression of SOD1+GFP were at least partially due to chronic exposure to high
level of H2O2. See a model for this hypothesis (Fig. 5-1)

138
Figure 5-1. A model for altered redox state in aged rats with overexpression of SOD1.
Overexpression of SOD1 in aged rats results in an elevated level of H2O2 that cause oxidative modification on the structure and function of GSH-related enzymes, such as GPx and GR. Reduced activities of GPx and GR further influence normal GSH cycling, leading to decreased level of GSH and GSSG. The gray stars indicate oxidative modification

139
APPENDIX
ADDITIONAL FIGURES
Figure A-1. Control images for hippocampal immunofluorescence. (A) Hippocampi from a 25-month old rat injected with AAV-SOD1-myc showed intense myc (green) staining after incubating with myc-tag mouse monoclonal primary antibody and goat-anti mouse Alexa 488 secondary antibody. (B) No primary antibody negative control. Little to no staining was observed in hippocampi incubated with secondary antibody only. (C) Mouse IgG2a was used as an isotype

140
negative control. Little to no staining was shown in hippocampi. There was some non-specific background signal staining which was mainly observed in fiber tracts (e.g. corpus callosum, fimbria) appearing outside the hippocampi. All the images were merged with nuclear DAPI staining (Blue). All the images were taken with exposure time of 250 ms (green channel) and 700 ms (blue channel). Calibration bars represent 500 µm.

141
Figure A-2. SOD1 and myc expression are co-localized in the hippocampus of a rat with
overexpression of SOD1. Immunofluorescence shows a SOD1 staining in red (left), a myc staining in green (center), and a merged image of SOD1 and myc in yellow (right). White boxes in lower left indicate enhanced images of a small part of CA3 area labeled with dotted white box.

142
Figure A-3. Expression of corresponding antioxidant enzymes are higher in the hippocampus of rats with injection of AAV-SOD1, AAV-SOD2, or AAV-CAT. Immunofluorescence of endogenous expression of (A) SOD1 (red) and (E) CAT (red) in AAV-GFP rats. (C) Endogenous expression of SOD2 (green) in AAV-SOD1+CAT rats. (B, D, F) Immunofluorescence of overexpression of SOD1 (red), SOD2 (green) and CAT (red) in AAV-SOD1 rats (B), AAV-SOD2 rats (D) and AAV-CAT rats (F).

143
LIST OF REFERENCES
Abele, R., Lampinen, M., Keinanen, K., and Madden, D.R. (1998). Disulfide bonding and cysteine accessibility in the alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor subunit GluRD. Implications for redox modulation of glutamate receptors. J Biol Chem 273, 25132-25138.
Adams, S., Green, P., Claxton, R., Simcox, S., Williams, M.V., Walsh, K., and Leeuwenburgh, C. (2001). Reactive carbonyl formation by oxidative and non-oxidative pathways. Front Biosci 6, A17-24.
Aenlle, K.K., Kumar, A., Cui, L., Jackson, T.C., and Foster, T.C. (2009). Estrogen effects on cognition and hippocampal transcription in middle-aged mice. Neurobiol Aging 30, 932-945.
Afzal, M., Matsugo, S., Sasai, M., Xu, B., Aoyama, K., and Takeuchi, T. (2003). Method to overcome photoreaction, a serious drawback to the use of dichlorofluorescin in evaluation of reactive oxygen species. Biochem Biophys Res Commun 304, 619-624.
Aizenman, E., Hartnett, K.A., and Reynolds, I.J. (1990). Oxygen free radicals regulate NMDA receptor function via a redox modulatory site. Neuron 5, 841-846.
Aizenman, E., Lipton, S.A., and Loring, R.H. (1989). Selective modulation of NMDA responses by reduction and oxidation. Neuron 2, 1257-1263.
Akli, S., Caillaud, C., Vigne, E., Stratford-Perricaudet, L.D., Poenaru, L., Perricaudet, M., Kahn, A., and Peschanski, M.R. (1993). Transfer of a foreign gene into the brain using adenovirus vectors. Nat Genet 3, 224-228.
Al-Mehdi, A.B., Shuman, H., and Fisher, A.B. (1997). Intracellular generation of reactive oxygen species during nonhypoxic lung ischemia. Am J Physiol 272, L294-300.
Alisky, J.M., Hughes, S.M., Sauter, S.L., Jolly, D., Dubensky, T.W., Jr., Staber, P.D., Chiorini, J.A., and Davidson, B.L. (2000). Transduction of murine cerebellar neurons with recombinant FIV and AAV5 vectors. Neuroreport 11, 2669-2673.
Ames, B.N. (1989). Endogenous DNA damage as related to cancer and aging. Mutat Res 214, 41-46.
Ames, B.N., and Gold, L.S. (1991). Endogenous mutagens and the causes of aging and cancer. Mutat Res 250, 3-16.
Asahi, M., Fujii, J., Takao, T., Kuzuya, T., Hori, M., Shimonishi, Y., and Taniguchi, N. (1997). The oxidation of selenocysteine is involved in the inactivation of glutathione peroxidase by nitric oxide donor. J Biol Chem 272, 19152-19157.

144
Auerbach, J.M., and Segal, M. (1997). Peroxide modulation of slow onset potentiation in rat hippocampus. J Neurosci 17, 8695-8701.
Austad, S.N. (2005). Diverse aging rates in metazoans: targets for functional genomics. Mech Ageing Dev 126, 43-49.
Balaban, R.S., Nemoto, S., and Finkel, T. (2005). Mitochondria, oxidants, and aging. Cell 120, 483-495.
Ball, M.J. (1977). Neuronal loss, neurofibrillary tangles and granulovacuolar degeneration in the hippocampus with ageing and dementia. A quantitative study. Acta Neuropathol 37, 111-118.
Barford, D. (2004). The role of cysteine residues as redox-sensitive regulatory switches. Curr Opin Struct Biol 14, 679-686.
Barnes, C.A. (1979). Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psychol 93, 74-104.
Barnes, C.A., Rao, G., and Shen, J. (1997). Age-related decrease in the N-methyl-D-aspartateR-mediated excitatory postsynaptic potential in hippocampal region CA1. Neurobiol Aging 18, 445-452.
Barsoum, J., Brown, R., McKee, M., and Boyce, F.M. (1997). Efficient transduction of mammalian cells by a recombinant baculovirus having the vesicular stomatitis virus G glycoprotein. Hum Gene Ther 8, 2011-2018.
Bartke, A., and Brown-Borg, H. (2004). Life extension in the dwarf mouse. Curr Top Dev Biol 63, 189-225.
Bartlett, J.S., Samulski, R.J., and McCown, T.J. (1998). Selective and rapid uptake of adeno-associated virus type 2 in brain. Hum Gene Ther 9, 1181-1186.
Becker, K., Gui, M., and Schirmer, R.H. (1995). Inhibition of human glutathione reductase by S-nitrosoglutathione. Eur J Biochem 234, 472-478.
Beckman, K.B., and Ames, B.N. (1998). The free radical theory of aging matures. Physiol Rev 78, 547-581.
Benov, L., Sztejnberg, L., and Fridovich, I. (1998). Critical evaluation of the use of hydroethidine as a measure of superoxide anion radical. Free Radic Biol Med 25, 826-831.
Berlett, B.S., and Stadtman, E.R. (1997). Protein oxidation in aging, disease, and oxidative stress. J Biol Chem 272, 20313-20316.

145
Bernard, C.L., Hirsch, J.C., Khazipov, R., Ben-Ari, Y., and Gozlan, H. (1997). Redox modulation of synaptic responses and plasticity in rat CA1 hippocampal neurons. Exp Brain Res 113, 343-352.
Bessis, N., GarciaCozar, F.J., and Boissier, M.C. (2004). Immune responses to gene therapy vectors: influence on vector function and effector mechanisms. Gene Ther 11 Suppl 1, S10-17.
Bishop, N.A., Lu, T., and Yankner, B.A. (2010). Neural mechanisms of ageing and cognitive decline. Nature 464, 529-535.
Bizon, J.L., LaSarge, C.L., Montgomery, K.S., McDermott, A.N., Setlow, B., and Griffith, W.H. (2009). Spatial reference and working memory across the lifespan of male Fischer 344 rats. Neurobiol Aging 30, 646-655.
Blalock, E.M., Chen, K.C., Sharrow, K., Herman, J.P., Porter, N.M., Foster, T.C., and Landfield, P.W. (2003). Gene microarrays in hippocampal aging: statistical profiling identifies novel processes correlated with cognitive impairment. J Neurosci 23, 3807-3819.
Bliss, T.V., and Collingridge, G.L. (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31-39.
Block, M.L., Wu, X., Pei, Z., Li, G., Wang, T., Qin, L., Wilson, B., Yang, J., Hong, J.S., and Veronesi, B. (2004). Nanometer size diesel exhaust particles are selectively toxic to dopaminergic neurons: the role of microglia, phagocytosis, and NADPH oxidase. Faseb J 18, 1618-1620.
Blomer, U., Naldini, L., Kafri, T., Trono, D., Verma, I.M., and Gage, F.H. (1997). Highly efficient and sustained gene transfer in adult neurons with a lentivirus vector. J Virol 71, 6641-6649.
Bodhinathan, K., Kumar, A., and Foster, T.C. (2010a). Intracellular redox state alters NMDA receptor response during aging through Ca2+/calmodulin-dependent protein kinase II. J Neurosci 30, 1914-1924.
Bodhinathan, K., Kumar, A., and Foster, T.C. (2010b). Redox sensitive calcium stores underlie enhanced after hyperpolarization of aged neurons: role for ryanodine receptor mediated calcium signaling. J Neurophysiol 104, 2586-2593.
Bokoch, G.M., and Knaus, U.G. (2003). NADPH oxidases: not just for leukocytes anymore! Trends Biochem Sci 28, 502-508.
Bosco, D.A., Morfini, G., Karabacak, N.M., Song, Y., Gros-Louis, F., Pasinelli, P., Goolsby, H., Fontaine, B.A., Lemay, N., McKenna-Yasek, D., et al. (2010). Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci 13, 1396-1403.

146
Boveris, A. (1984). Determination of the production of superoxide radicals and hydrogen peroxide in mitochondria. Methods Enzymol 105, 429-435.
Boveris, A., and Chance, B. (1973). The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J 134, 707-716.
Bowry, V.W., Ingold, K.U., and Stocker, R. (1992). Vitamin E in human low-density lipoprotein. When and how this antioxidant becomes a pro-oxidant. Biochem J 288 ( Pt 2), 341-344.
Brigelius-Flohe, R., Banning, A., Kny, M., and Bol, G.F. (2004). Redox events in interleukin-1 signaling. Arch Biochem Biophys 423, 66-73.
Brody, H. (1955). Organization of the cerebral cortex. III. A study of aging in the human cerebral cortex. J Comp Neurol 102, 511-516.
Brown, D.I., and Griendling, K.K. (2009). Nox proteins in signal transduction. Free Radic Biol Med 47, 1239-1253.
Brown, R.H., Jr. (1998). SOD1 aggregates in ALS: cause, correlate or consequence? Nat Med 4, 1362-1364.
Buffenstein, R., and Jarvis, J.U. (2002). The naked mole rat--a new record for the oldest living rodent. Sci Aging Knowledge Environ 2002, pe7.
Burger, C., Gorbatyuk, O.S., Velardo, M.J., Peden, C.S., Williams, P., Zolotukhin, S., Reier, P.J., Mandel, R.J., and Muzyczka, N. (2004). Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol Ther 10, 302-317.
Burgess, N. (2006). Spatial memory: how egocentric and allocentric combine. Trends Cogn Sci 10, 551-557.
Burke, S.N., and Barnes, C.A. (2010). Senescent synapses and hippocampal circuit dynamics. Trends Neurosci 33, 153-161.
Burns, J.C., Friedmann, T., Driever, W., Burrascano, M., and Yee, J.K. (1993). Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc Natl Acad Sci U S A 90, 8033-8037.
Butterfield, D.A., Hensley, K., Cole, P., Subramaniam, R., Aksenov, M., Aksenova, M., Bummer, P.M., Haley, B.E., and Carney, J.M. (1997). Oxidatively induced structural alteration of glutamine synthetase assessed by analysis of spin label incorporation kinetics: relevance to Alzheimer's disease. J Neurochem 68, 2451-2457.

147
Butterfield, D.A., and Sultana, R. (2007). Redox proteomics identification of oxidatively modified brain proteins in Alzheimer's disease and mild cognitive impairment: insights into the progression of this dementing disorder. J Alzheimers Dis 12, 61-72.
Capecchi, M.R. (2005). Gene targeting in mice: functional analysis of the mammalian genome for the twenty-first century. Nat Rev Genet 6, 507-512.
Carter, C.S., Leeuwenburgh, C., Daniels, M., and Foster, T.C. (2009). Influence of calorie restriction on measures of age-related cognitive decline: role of increased physical activity. J Gerontol A Biol Sci Med Sci 64, 850-859.
Catala, A. (2010a). A synopsis of the process of lipid peroxidation since the discovery of the essential fatty acids. Biochemical and biophysical research communications 399, 318-323.
Catala, A. (2010b). A synopsis of the process of lipid peroxidation since the discovery of the essential fatty acids. Biochem Biophys Res Commun 399, 318-323.
Chang, L.Y., Slot, J.W., Geuze, H.J., and Crapo, J.D. (1988). Molecular immunocytochemistry of the CuZn superoxide dismutase in rat hepatocytes. J Cell Biol 107, 2169-2179.
Choi, Y.B., and Lipton, S.A. (2000). Redox modulation of the NMDA receptor. Cell Mol Life Sci 57, 1535-1541.
Cini, M., and Moretti, A. (1995). Studies on lipid peroxidation and protein oxidation in the aging brain. Neurobiol Aging 16, 53-57.
Coil, D.A., and Miller, A.D. (2004). Phosphatidylserine is not the cell surface receptor for vesicular stomatitis virus. J Virol 78, 10920-10926.
Coleman, P.D., and Flood, D.G. (1987). Neuron numbers and dendritic extent in normal aging and Alzheimer's disease. Neurobiol Aging 8, 521-545.
Colman, R.J., and Anderson, R.M. (2011). Nonhuman primate calorie restriction. Antioxid Redox Signal 14, 229-239.
Commins, S., Cunningham, L., Harvey, D., and Walsh, D. (2003). Massed but not spaced training impairs spatial memory. Behav Brain Res 139, 215-223.
Consiglio, A., Gritti, A., Dolcetta, D., Follenzi, A., Bordignon, C., Gage, F.H., Vescovi, A.L., and Naldini, L. (2004). Robust in vivo gene transfer into adult mammalian neural stem cells by lentiviral vectors. Proc Natl Acad Sci U S A 101, 14835-14840.
Cui, L., Hofer, T., Rani, A., Leeuwenburgh, C., and Foster, T.C. (2009). Comparison of lifelong and late life exercise on oxidative stress in the cerebellum. Neurobiol Aging 30, 903-909.

148
Dash, P.K., Mach, S.A., Blum, S., and Moore, A.N. (2002). Intrahippocampal wortmannin infusion enhances long-term spatial and contextual memories. Learn Mem 9, 167-177.
Davies, K.J. (2001). Degradation of oxidized proteins by the 20S proteasome. Biochimie 83, 301-310.
Davis, H.P., Small, S.A., Stern, Y., Mayeux, R., Feldstein, S.N., and Keller, F.R. (2003). Acquisition, recall, and forgetting of verbal information in long-term memory by young, middle-aged, and elderly individuals. Cortex 39, 1063-1091.
Dawn-Linsley, M., Ekinci, F.J., Ortiz, D., Rogers, E., and Shea, T.B. (2005). Monitoring thiobarbituric acid-reactive substances (TBARs) as an assay for oxidative damage in neuronal cultures and central nervous system. J Neurosci Methods 141, 219-222.
de Haan, J.B., Cristiano, F., Iannello, R., Bladier, C., Kelner, M.J., and Kola, I. (1996). Elevation in the ratio of Cu/Zn-superoxide dismutase to glutathione peroxidase activity induces features of cellular senescence and this effect is mediated by hydrogen peroxide. Hum Mol Genet 5, 283-292.
Dei, R., Takeda, A., Niwa, H., Li, M., Nakagomi, Y., Watanabe, M., Inagaki, T., Washimi, Y., Yasuda, Y., Horie, K., et al. (2002). Lipid peroxidation and advanced glycation end products in the brain in normal aging and in Alzheimer's disease. Acta Neuropathol 104, 113-122.
Deng, H.X., Hentati, A., Tainer, J.A., Iqbal, Z., Cayabyab, A., Hung, W.Y., Getzoff, E.D., Hu, P., Herzfeldt, B., Roos, R.P., et al. (1993). Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science 261, 1047-1051.
Dexter, D., Carter, C., Agid, F., Agid, Y., Lees, A.J., Jenner, P., and Marsden, C.D. (1986). Lipid peroxidation as cause of nigral cell death in Parkinson's disease. Lancet 2, 639-640.
Dikalov, S.I., Dikalova, A.E., Bikineyeva, A.T., Schmidt, H.H., Harrison, D.G., and Griendling, K.K. (2008). Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic Biol Med 45, 1340-1351.
Driscoll, I., Howard, S.R., Stone, J.C., Monfils, M.H., Tomanek, B., Brooks, W.M., and Sutherland, R.J. (2006). The aging hippocampus: a multi-level analysis in the rat. Neuroscience 139, 1173-1185.
Dull, T., Zufferey, R., Kelly, M., Mandel, R.J., Nguyen, M., Trono, D., and Naldini, L. (1998). A third-generation lentivirus vector with a conditional packaging system. J Virol 72, 8463-8471.

149
Dumont, M., Wille, E., Stack, C., Calingasan, N.Y., Beal, M.F., and Lin, M.T. (2009). Reduction of oxidative stress, amyloid deposition, and memory deficit by manganese superoxide dismutase overexpression in a transgenic mouse model of Alzheimer's disease. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 23, 2459-2466.
Epstein, C.J., Avraham, K.B., Lovett, M., Smith, S., Elroy-Stein, O., Rotman, G., Bry, C., and Groner, Y. (1987). Transgenic mice with increased Cu/Zn-superoxide dismutase activity: animal model of dosage effects in Down syndrome. Proc Natl Acad Sci U S A 84, 8044-8048.
Figtree, G.A., Liu, C.C., Bibert, S., Hamilton, E.J., Garcia, A., White, C.N., Chia, K.K., Cornelius, F., Geering, K., and Rasmussen, H.H. (2009). Reversible oxidative modification: a key mechanism of Na+-K+ pump regulation. Circ Res 105, 185-193.
Fischer, A.C., Beck, S.E., Smith, C.I., Laube, B.L., Askin, F.B., Guggino, S.E., Adams, R.J., Flotte, T.R., and Guggino, W.B. (2003). Successful transgene expression with serial doses of aerosolized rAAV2 vectors in rhesus macaques. Mol Ther 8, 918-926.
Flannery, J.G., Zolotukhin, S., Vaquero, M.I., LaVail, M.M., Muzyczka, N., and Hauswirth, W.W. (1997). Efficient photoreceptor-targeted gene expression in vivo by recombinant adeno-associated virus. Proc Natl Acad Sci U S A 94, 6916-6921.
Forman, H.J., Fukuto, J.M., and Torres, M. (2004). Redox signaling: thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am J Physiol Cell Physiol 287, C246-256.
Forman, H.J., Torres, M., and Fukuto, J. (2002). Redox signaling. Mol Cell Biochem 234-235, 49-62.
Forster, M.J., Dubey, A., Dawson, K.M., Stutts, W.A., Lal, H., and Sohal, R.S. (1996). Age-related losses of cognitive function and motor skills in mice are associated with oxidative protein damage in the brain. Proc Natl Acad Sci U S A 93, 4765-4769.
Foster, T.C. (1999). Involvement of hippocampal synaptic plasticity in age-related memory decline. Brain Res Brain Res Rev 30, 236-249.
Foster, T.C. (2002). Regulation of synaptic plasticity in memory and memory decline with aging. Prog Brain Res 138, 283-303.
Foster, T.C. (2007). Calcium homeostasis and modulation of synaptic plasticity in the aged brain. Aging Cell 6, 319-325.
Foster, T.C. (2012). Dissecting the age-related decline on spatial learning and memory tasks in rodent models: N-methyl-D-aspartate receptors and voltage-dependent Ca(2+) channels in senescent synaptic plasticity. Prog Neurobiol.

150
Foster, T.C., and Kumar, A. (2002). Calcium dysregulation in the aging brain. Neuroscientist 8, 297-301.
Foster, T.C., and Norris, C.M. (1997). Age-associated changes in Ca(2+)-dependent processes: relation to hippocampal synaptic plasticity. Hippocampus 7, 602-612.
Foster, T.C., Rani, A., Kumar, A., Cui, L., and Semple-Rowland, S.L. (2008). Viral vector-mediated delivery of estrogen receptor-alpha to the hippocampus improves spatial learning in estrogen receptor-alpha knockout mice. Mol Ther 16, 1587-1593.
Foster, T.C., Sharrow, K.M., Kumar, A., and Masse, J. (2003). Interaction of age and chronic estradiol replacement on memory and markers of brain aging. Neurobiol Aging 24, 839-852.
Foster, T.C., Sharrow, K.M., Masse, J.R., Norris, C.M., and Kumar, A. (2001). Calcineurin links Ca2+ dysregulation with brain aging. J Neurosci 21, 4066-4073.
Fouquet, C., Petit, G.H., Auffret, A., Gaillard, E., Rovira, C., Mariani, J., and Rondi-Reig, L. (2011). Early detection of age-related memory deficits in individual mice. Neurobiol Aging 32, 1881-1895.
Frick, K.M., Baxter, M.G., Markowska, A.L., Olton, D.S., and Price, D.L. (1995). Age-related spatial reference and working memory deficits assessed in the water maze. Neurobiol Aging 16, 149-160.
Fridovich, I. (1983a). Superoxide dismutases: regularities and irregularities. Harvey Lect 79, 51-75.
Fridovich, I. (1983b). Superoxide radical: an endogenous toxicant. Annu Rev Pharmacol Toxicol 23, 239-257.
Friedberg, E.C. (1995). Out of the shadows and into the light: the emergence of DNA repair. Trends Biochem Sci 20, 381.
Fullerton, H.J., Ditelberg, J.S., Chen, S.F., Sarco, D.P., Chan, P.H., Epstein, C.J., and Ferriero, D.M. (1998). Copper/zinc superoxide dismutase transgenic brain accumulates hydrogen peroxide after perinatal hypoxia ischemia. Ann Neurol 44, 357-364.
Gahtan, E., Auerbach, J.M., Groner, Y., and Segal, M. (1998). Reversible impairment of long-term potentiation in transgenic Cu/Zn-SOD mice. Eur J Neurosci 10, 538-544.
Gajewski, E., Rao, G., Nackerdien, Z., and Dizdaroglu, M. (1990). Modification of DNA bases in mammalian chromatin by radiation-generated free radicals. Biochemistry 29, 7876-7882.

151
Gallagher, M., Landfield, P.W., McEwen, B., Meaney, M.J., Rapp, P.R., Sapolsky, R., and West, M.J. (1996). Hippocampal neurodegeneration in aging. Science 274, 484-485.
Gazzaley, A.H., Thakker, M.M., Hof, P.R., and Morrison, J.H. (1997). Preserved number of entorhinal cortex layer II neurons in aged macaque monkeys. Neurobiol Aging 18, 549-553.
Gemma, C., Mesches, M.H., Sepesi, B., Choo, K., Holmes, D.B., and Bickford, P.C. (2002). Diets enriched in foods with high antioxidant activity reverse age-induced decreases in cerebellar beta-adrenergic function and increases in proinflammatory cytokines. J Neurosci 22, 6114-6120.
Genoux, D., Haditsch, U., Knobloch, M., Michalon, A., Storm, D., and Mansuy, I.M. (2002). Protein phosphatase 1 is a molecular constraint on learning and memory. Nature 418, 970-975.
Geraerts, M., Eggermont, K., Hernandez-Acosta, P., Garcia-Verdugo, J.M., Baekelandt, V., and Debyser, Z. (2006). Lentiviral vectors mediate efficient and stable gene transfer in adult neural stem cells in vivo. Hum Gene Ther 17, 635-650.
Gerlai, R. (2001). Behavioral tests of hippocampal function: simple paradigms complex problems. Behav Brain Res 125, 269-277.
Gilbert, H.F. (1990). Molecular and cellular aspects of thiol-disulfide exchange. Adv Enzymol Relat Areas Mol Biol 63, 69-172.
Gilmer, L.K., Ansari, M.A., Roberts, K.N., and Scheff, S.W. (2010). Age-related changes in mitochondrial respiration and oxidative damage in the cerebral cortex of the Fischer 344 rat. Mech Ageing Dev 131, 133-143.
Goncalves, M.A. (2005). Adeno-associated virus: from defective virus to effective vector. Virol J 2, 43.
Gonzalez, A., Granados, M.P., Pariente, J.A., and Salido, G.M. (2006). H2O2 mobilizes Ca2+ from agonist- and thapsigargin-sensitive and insensitive intracellular stores and stimulates glutamate secretion in rat hippocampal astrocytes. Neurochem Res 31, 741-750.
Gorbunova, V., Bozzella, M.J., and Seluanov, A. (2008). Rodents for comparative aging studies: from mice to beavers. Age (Dordr) 30, 111-119.
Gozlan, H., Khazipov, R., Diabira, D., and Ben-Ari, Y. (1995). In CA1 hippocampal neurons, the redox state of NMDA receptors determines LTP expressed by NMDA but not by AMPA receptors. J Neurophysiol 73, 2612-2617.

152
Grodstein, F., Chen, J., and Willett, W.C. (2003). High-dose antioxidant supplements and cognitive function in community-dwelling elderly women. Am J Clin Nutr 77, 975-984.
Gulinello, M., Gertner, M., Mendoza, G., Schoenfeld, B.P., Oddo, S., LaFerla, F., Choi, C.H., McBride, S.M., and Faber, D.S. (2009). Validation of a 2-day water maze protocol in mice. Behav Brain Res 196, 220-227.
Gupta, S.P., Patel, S., Yadav, S., Singh, A.K., Singh, S., and Singh, M.P. (2010). Involvement of nitric oxide in maneb- and paraquat-induced Parkinson's disease phenotype in mouse: is there any link with lipid peroxidation? Neurochem Res 35, 1206-1213.
Haberman, R.P., McCown, T.J., and Samulski, R.J. (1998). Inducible long-term gene expression in brain with adeno-associated virus gene transfer. Gene Ther 5, 1604-1611.
Halliwell, B. (1992). Reactive oxygen species and the central nervous system. J Neurochem 59, 1609-1623.
Han, D., Antunes, F., Canali, R., Rettori, D., and Cadenas, E. (2003). Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J Biol Chem 278, 5557-5563.
Han, D., Williams, E., and Cadenas, E. (2001). Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem J 353, 411-416.
Harman, D. (1992). Role of free radicals in aging and disease. Ann N Y Acad Sci 673, 126-141.
Hasty, P., Campisi, J., Hoeijmakers, J., van Steeg, H., and Vijg, J. (2003). Aging and genome maintenance: lessons from the mouse? Science 299, 1355-1359.
Hasty, P., and Vijg, J. (2004). Accelerating aging by mouse reverse genetics: a rational approach to understanding longevity. Aging Cell 3, 55-65.
Head, E., Liu, J., Hagen, T.M., Muggenburg, B.A., Milgram, N.W., Ames, B.N., and Cotman, C.W. (2002). Oxidative damage increases with age in a canine model of human brain aging. J Neurochem 82, 375-381.
Hofer, T., Seo, A.Y., Prudencio, M., and Leeuwenburgh, C. (2006). A method to determine RNA and DNA oxidation simultaneously by HPLC-ECD: greater RNA than DNA oxidation in rat liver after doxorubicin administration. Biol Chem 387, 103-111.

153
Hogge, M., Adam, S., and Collette, F. (2008). Directed forgetting and aging: the role of retrieval processes, processing speed, and proactive interference. Neuropsychol Dev Cogn B Aging Neuropsychol Cogn 15, 471-491.
Hu, D., Cao, P., Thiels, E., Chu, C.T., Wu, G.Y., Oury, T.D., and Klann, E. (2007a). Hippocampal long-term potentiation, memory, and longevity in mice that overexpress mitochondrial superoxide dismutase. Neurobiol Learn Mem 87, 372-384.
Hu, D., Klann, E., and Thiels, E. (2007b). Superoxide dismutase and hippocampal function: age and isozyme matter. Antioxid Redox Signal 9, 201-210.
Hu, W., Feng, Z., Eveleigh, J., Iyer, G., Pan, J., Amin, S., Chung, F.L., and Tang, M.S. (2002). The major lipid peroxidation product, trans-4-hydroxy-2-nonenal, preferentially forms DNA adducts at codon 249 of human p53 gene, a unique mutational hotspot in hepatocellular carcinoma. Carcinogenesis 23, 1781-1789.
Huang, K.P., Huang, F.L., Li, J., Schuck, P., and McPhie, P. (2000). Calcium-sensitive interaction between calmodulin and modified forms of rat brain neurogranin/RC3. Biochemistry 39, 7291-7299.
Huber, R.E., and Criddle, R.S. (1967). Comparison of the chemical properties of selenocysteine and selenocystine with their sulfur analogs. Arch Biochem Biophys 122, 164-173.
Huppert, F.A., and Kopelman, M.D. (1989). Rates of forgetting in normal ageing: a comparison with dementia. Neuropsychologia 27, 849-860.
Hussain, S.P., Amstad, P., He, P., Robles, A., Lupold, S., Kaneko, I., Ichimiya, M., Sengupta, S., Mechanic, L., Okamura, S., et al. (2004). p53-induced up-regulation of MnSOD and GPx but not catalase increases oxidative stress and apoptosis. Cancer Res 64, 2350-2356.
Infanger, D.W., Sharma, R.V., and Davisson, R.L. (2006). NADPH oxidases of the brain: distribution, regulation, and function. Antioxid Redox Signal 8, 1583-1596.
Ingram, D.K. (1988). Complex maze learning in rodents as a model of age-related memory impairment. Neurobiol Aging 9, 475-485.
Ingram, D.K., Cutler, R.G., Weindruch, R., Renquist, D.M., Knapka, J.J., April, M., Belcher, C.T., Clark, M.A., Hatcherson, C.D., Marriott, B.M., et al. (1990). Dietary restriction and aging: the initiation of a primate study. J Gerontol 45, B148-163.
Jakobsson, J., Ericson, C., Jansson, M., Bjork, E., and Lundberg, C. (2003). Targeted transgene expression in rat brain using lentiviral vectors. J Neurosci Res 73, 876-885.

154
Jang, Y.C., Perez, V.I., Song, W., Lustgarten, M.S., Salmon, A.B., Mele, J., Qi, W., Liu, Y., Liang, H., Chaudhuri, A., et al. (2009). Overexpression of Mn superoxide dismutase does not increase life span in mice. The journals of gerontology 64, 1114-1125.
Johnson, T.E. (2003). Advantages and disadvantages of Caenorhabditis elegans for aging research. Exp Gerontol 38, 1329-1332.
Johnston, J.A., Dalton, M.J., Gurney, M.E., and Kopito, R.R. (2000). Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 97, 12571-12576.
Jones, M.L. (1992). Longevity of mammals in captivity--an update. In Vivo 6, 363-366.
Jones, M.W., Errington, M.L., French, P.J., Fine, A., Bliss, T.V., Garel, S., Charnay, P., Bozon, B., Laroche, S., and Davis, S. (2001). A requirement for the immediate early gene Zif268 in the expression of late LTP and long-term memories. Nat Neurosci 4, 289-296.
Jonker, C., Launer, L.J., Hooijer, C., and Lindeboom, J. (1996). Memory complaints and memory impairment in older individuals. J Am Geriatr Soc 44, 44-49.
Kaemmerer, E., Schutt, F., Krohne, T.U., Holz, F.G., and Kopitz, J. (2007). Effects of lipid peroxidation-related protein modifications on RPE lysosomal functions and POS phagocytosis. Invest Ophthalmol Vis Sci 48, 1342-1347.
Kametani, H., Bresnahan, E.L., Chachich, M.E., Spangler, E.L., and Ingram, D.K. (1989). Comparison of retention performance between young rats with fimbria-fornix lesions and aged rats in a 14-unit T-maze. Behav Brain Res 35, 253-263.
Kamsler, A., Avital, A., Greenberger, V., and Segal, M. (2007). Aged SOD overexpressing mice exhibit enhanced spatial memory while lacking hippocampal neurogenesis. Antioxidants & redox signaling 9, 181-189.
Kamsler, A., and Segal, M. (2003a). Hydrogen peroxide modulation of synaptic plasticity. J Neurosci 23, 269-276.
Kamsler, A., and Segal, M. (2003b). Paradoxical actions of hydrogen peroxide on long-term potentiation in transgenic superoxide dismutase-1 mice. J Neurosci 23, 10359-10367.
Kaplitt, M.G., Leone, P., Samulski, R.J., Xiao, X., Pfaff, D.W., O'Malley, K.L., and During, M.J. (1994). Long-term gene expression and phenotypic correction using adeno-associated virus vectors in the mammalian brain. Nat Genet 8, 148-154.

155
Keaney, M., and Gems, D. (2003). No increase in lifespan in Caenorhabditis elegans upon treatment with the superoxide dismutase mimetic EUK-8. Free Radic Biol Med 34, 277-282.
Keaney, M., Matthijssens, F., Sharpe, M., Vanfleteren, J., and Gems, D. (2004). Superoxide dismutase mimetics elevate superoxide dismutase activity in vivo but do not retard aging in the nematode Caenorhabditis elegans. Free Radic Biol Med 37, 239-250.
Keller, G.A., Warner, T.G., Steimer, K.S., and Hallewell, R.A. (1991). Cu,Zn superoxide dismutase is a peroxisomal enzyme in human fibroblasts and hepatoma cells. Proc Natl Acad Sci U S A 88, 7381-7385.
Kelner, M.J., Bagnell, R., Montoya, M., Estes, L., Uglik, S.F., and Cerutti, P. (1995). Transfection with human copper-zinc superoxide dismutase induces bidirectional alterations in other antioxidant enzymes, proteins, growth factor response, and paraquat resistance. Free Radic Biol Med 18, 497-506.
Keuker, J.I., Luiten, P.G., and Fuchs, E. (2003). Preservation of hippocampal neuron numbers in aged rhesus monkeys. Neurobiol Aging 24, 157-165.
Kiffin, R., Christian, C., Knecht, E., and Cuervo, A.M. (2004). Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell 15, 4829-4840.
Kilinc, A., Yalcin, A.S., Yalcin, D., Taga, Y., and Emerk, K. (1988). Increased erythrocyte susceptibility to lipid peroxidation in human Parkinson's disease. Neurosci Lett 87, 307-310.
Kishida, K.T., Pao, M., Holland, S.M., and Klann, E. (2005). NADPH oxidase is required for NMDA receptor-dependent activation of ERK in hippocampal area CA1. J Neurochem 94, 299-306.
Klann, E., Chen, S.J., and Sweatt, J.D. (1993). Mechanism of protein kinase C activation during the induction and maintenance of long-term potentiation probed using a selective peptide substrate. Proc Natl Acad Sci U S A 90, 8337-8341.
Klein, R.L., Meyer, E.M., Peel, A.L., Zolotukhin, S., Meyers, C., Muzyczka, N., and King, M.A. (1998). Neuron-specific transduction in the rat septohippocampal or nigrostriatal pathway by recombinant adeno-associated virus vectors. Exp Neurol 150, 183-194.
Knoferle, J., Koch, J.C., Ostendorf, T., Michel, U., Planchamp, V., Vutova, P., Tonges, L., Stadelmann, C., Bruck, W., Bahr, M., et al. (2010). Mechanisms of acute axonal degeneration in the optic nerve in vivo. Proc Natl Acad Sci U S A 107, 6064-6069.

156
Kogan, J.H., Frankland, P.W., Blendy, J.A., Coblentz, J., Marowitz, Z., Schutz, G., and Silva, A.J. (1997). Spaced training induces normal long-term memory in CREB mutant mice. Curr Biol 7, 1-11.
Kono, Y., and Fridovich, I. (1982). Superoxide radical inhibits catalase. J Biol Chem 257, 5751-5754.
Kral, V.A. (1962). Senescent forgetfulness: benign and malignant. Can Med Assoc J 86, 257-260.
Krebs, H.A. (1967). The redox state of nicotinamide adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Adv Enzyme Regul 5, 409-434.
Krebs, H.A., and Gascoyne, T. (1968). The redox state of the nicotinamide-adenine dinucleotides in rat liver homogenates. Biochem J 108, 513-520.
Krebs, H.A., and Veech, R.L. (1969). Equilibrium relations between pyridine nucleotides and adenine nucleotides and their roles in the regulation of metabolic processes. Adv Enzyme Regul 7, 397-413.
Kumar, A. (2011). Long-Term Potentiation at CA3-CA1 Hippocampal Synapses with Special Emphasis on Aging, Disease, and Stress. Front Aging Neurosci 3, 7.
Kumar, A., Bodhinathan, K., and Foster, T.C. (2009). Susceptibility to Calcium Dysregulation during Brain Aging. Front Aging Neurosci 1, 2.
Kumar, A., and Foster, T.C. (2005). Intracellular calcium stores contribute to increased susceptibility to LTD induction during aging. Brain Res 1031, 125-128.
Kumar, A., Thinschmidt, J.S., Foster, T.C., and King, M.A. (2007). Aging effects on the limits and stability of long-term synaptic potentiation and depression in rat hippocampal area CA1. J Neurophysiol 98, 594-601.
Lal, H., Pogacar, S., Daly, P.R., and Puri, S.K. (1973). Behavioral and neuropathological manifestations of nutritionally induced central nervous system "aging" in the rat. Prog Brain Res 40, 128-140.
Lambeth, J.D. (2002). Nox/Duox family of nicotinamide adenine dinucleotide (phosphate) oxidases. Curr Opin Hematol 9, 11-17.
Lambeth, J.D. (2004). NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 4, 181-189.
Lane, M.A. (2000). Nonhuman primate models in biogerontology. Exp Gerontol 35, 533-541.

157
Lattal, K.M., Mullen, M.T., and Abel, T. (2003). Extinction, renewal, and spontaneous recovery of a spatial preference in the water maze. Behav Neurosci 117, 1017-1028.
LeBel, C.P., Ischiropoulos, H., and Bondy, S.C. (1992). Evaluation of the probe 2',7'-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol 5, 227-231.
LeDoux, J.E. (2000). Emotion circuits in the brain. Annu Rev Neurosci 23, 155-184.
Lee, W.H. (2008). Role of SOD1 in brain aging. In Society for Neuroscience 38th Anneak Meeting, Washington, DC.
Lee, W.H., Kumar, A., Rani, A., Herrera, J., Xu, J., Someya, S., and Foster, T.C. (2012). Influence of viral vector-mediated delivery of superoxide dismutase and catalase to the hippocampus on spatial learning and memory during aging. Antioxid Redox Signal 16, 339-350.
Leutner, S., Eckert, A., and Muller, W.E. (2001a). ROS generation, lipid peroxidation and antioxidant enzyme activities in the aging brain. J Neural Transm 108, 955-967.
Leutner, S., Eckert, A., and Muller, W.E. (2001b). ROS generation, lipid peroxidation and antioxidant enzyme activities in the aging brain. J Neural Transm 108, 955-967.
Levin, E.D., Brady, T.C., Hochrein, E.C., Oury, T.D., Jonsson, L.M., Marklund, S.L., and Crapo, J.D. (1998). Molecular manipulations of extracellular superoxide dismutase: functional importance for learning. Behav Genet 28, 381-390.
Levin, E.D., Christopher, N.C., Lateef, S., Elamir, B.M., Patel, M., Liang, L.P., and Crapo, J.D. (2002). Extracellular superoxide dismutase overexpression protects against aging-induced cognitive impairment in mice. Behav Genet 32, 119-125.
Levine, R.L., Williams, J.A., Stadtman, E.R., and Shacter, E. (1994). Carbonyl assays for determination of oxidatively modified proteins. Methods Enzymol 233, 346-357.
Li, J., Pak, J.H., Huang, F.L., and Huang, K.P. (1999). N-methyl-D-aspartate induces neurogranin/RC3 oxidation in rat brain slices. J Biol Chem 274, 1294-1300.
Li, Y., Huang, T.T., Carlson, E.J., Melov, S., Ursell, P.C., Olson, J.L., Noble, L.J., Yoshimura, M.P., Berger, C., Chan, P.H., et al. (1995). Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet 11, 376-381.
Linden, A., Gulden, M., Martin, H.J., Maser, E., and Seibert, H. (2008a). Peroxide-induced cell death and lipid peroxidation in C6 glioma cells. Toxicol In Vitro 22, 1371-1376.

158
Linden, A., Gulden, M., Martin, H.J., Maser, E., and Seibert, H. (2008b). Peroxide-induced cell death and lipid peroxidation in C6 glioma cells. Toxicol In Vitro 22, 1371-1376.
Liochev, S.I., and Fridovich, I. (2007). The effects of superoxide dismutase on H2O2 formation. Free Radic Biol Med 42, 1465-1469.
Lipman, R.D. (1997). Pathobiology of aging rodents: inbred and hybrid models. Exp Gerontol 32, 215-228.
Lipman, R.D., Chrisp, C.E., Hazzard, D.G., and Bronson, R.T. (1996). Pathologic characterization of brown Norway, brown Norway x Fischer 344, and Fischer 344 x brown Norway rats with relation to age. J Gerontol A Biol Sci Med Sci 51, B54-59.
Lipman, R.D., Dallal, G.E., and Bronson, R.T. (1999). Effects of genotype and diet on age-related lesions in ad libitum fed and calorie-restricted F344, BN, and BNF3F1 rats. J Gerontol A Biol Sci Med Sci 54, B478-491.
Lipton, S.A., Choi, Y.B., Pan, Z.H., Lei, S.Z., Chen, H.S., Sucher, N.J., Loscalzo, J., Singel, D.J., and StamLer, J.S. (1993). A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature 364, 626-632.
Lipton, S.A., Choi, Y.B., Takahashi, H., Zhang, D., Li, W., Godzik, A., and Bankston, L.A. (2002). Cysteine regulation of protein function--as exemplified by NMDA-receptor modulation. Trends Neurosci 25, 474-480.
Liu, H., Wang, H., Shenvi, S., Hagen, T.M., and Liu, R.M. (2004). Glutathione metabolism during aging and in Alzheimer disease. Ann N Y Acad Sci 1019, 346-349.
Liu, J., Atamna, H., Kuratsune, H., and Ames, B.N. (2002). Delaying brain mitochondrial decay and aging with mitochondrial antioxidants and metabolites. Ann N Y Acad Sci 959, 133-166.
Liu, R., Liu, I.Y., Bi, X., Thompson, R.F., Doctrow, S.R., Malfroy, B., and Baudry, M. (2003). Reversal of age-related learning deficits and brain oxidative stress in mice with superoxide dismutase/catalase mimetics. Proc Natl Acad Sci U S A 100, 8526-8531.
Lu, S.C. (1999). Regulation of hepatic glutathione synthesis: current concepts and controversies. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 13, 1169-1183.
Ma, T., Hoeffer, C.A., Wong, H., Massaad, C.A., Zhou, P., Iadecola, C., Murphy, M.P., Pautler, R.G., and Klann, E. (2011). Amyloid beta-induced impairments in hippocampal synaptic plasticity are rescued by decreasing mitochondrial superoxide. J Neurosci 31, 5589-5595.

159
Macdonald, S.W., Stigsdotter-Neely, A., Derwinger, A., and Backman, L. (2006). Rate of acquisition, adult age, and basic cognitive abilities predict forgetting: new views on a classic problem. J Exp Psychol Gen 135, 368-390.
Maeda, H., Yamamoto, K., Nomura, Y., Kohno, I., Hafsi, L., Ueda, N., Yoshida, S., Fukuda, M., Fukuyasu, Y., Yamauchi, Y., et al. (2005). A design of fluorescent probes for superoxide based on a nonredox mechanism. J Am Chem Soc 127, 68-69.
Malenka, R.C., and Nicoll, R.A. (1999). Long-term potentiation--a decade of progress? Science 285, 1870-1874.
Malinska, D., Kudin, A.P., Debska-Vielhaber, G., Vielhaber, S., and Kunz, W.S. (2009). Chapter 23 Quantification of superoxide production by mouse brain and skeletal muscle mitochondria. Methods Enzymol 456, 419-437.
Malleret, G., Alarcon, J.M., Martel, G., Takizawa, S., Vronskaya, S., Yin, D., Chen, I.Z., Kandel, E.R., and Shumyatsky, G.P. (2010). Bidirectional regulation of hippocampal long-term synaptic plasticity and its influence on opposing forms of memory. J Neurosci 30, 3813-3825.
Mandel, R.J., Rendahl, K.G., Spratt, S.K., Snyder, R.O., Cohen, L.K., and Leff, S.E. (1998). Characterization of intrastriatal recombinant adeno-associated virus-mediated gene transfer of human tyrosine hydroxylase and human GTP-cyclohydrolase I in a rat model of Parkinson's disease. J Neurosci 18, 4271-4284.
Maragos, W.F., Jakel, R., Chesnut, D., Pocernich, C.B., Butterfield, D.A., St Clair, D., and Cass, W.A. (2000). Methamphetamine toxicity is attenuated in mice that overexpress human manganese superoxide dismutase. Brain Res 878, 218-222.
Marchesi, E., Rota, C., Fann, Y.C., Chignell, C.F., and Mason, R.P. (1999). Photoreduction of the fluorescent dye 2'-7'-dichlorofluorescein: a spin trapping and direct electron spin resonance study with implications for oxidative stress measurements. Free Radic Biol Med 26, 148-161.
Markowska, A.L. (1999). Life-long diet restriction failed to retard cognitive aging in Fischer-344 rats. Neurobiol Aging 20, 177-189.
Markowska, A.L., and Savonenko, A. (2002). Retardation of cognitive aging by life-long diet restriction: implications for genetic variance. Neurobiol Aging 23, 75-86.
Marnett, L.J. (1999). Lipid peroxidation-DNA damage by malondialdehyde. Mutat Res 424, 83-95.
Martin, S.J., and Clark, R.E. (2007). The rodent hippocampus and spatial memory: from synapses to systems. Cell Mol Life Sci 64, 401-431.

160
Martins, D.B., Mazzanti, C.M., Franca, R.T., Pagnoncelli, M., Costa, M.M., de Souza, E.M., Goncalves, J., Spanevello, R., Schmatz, R., da Costa, P., et al. (2012). 17-beta estradiol in the acetylcholinesterase activity and lipid peroxidation in the brain and blood of ovariectomized adult and middle-aged rats. Life Sci 90, 351-359.
Massaad, C.A., Washington, T.M., Pautler, R.G., and Klann, E. (2009). Overexpression of SOD-2 reduces hippocampal superoxide and prevents memory deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A 106, 13576-13581.
Maxwell, C.J., Hicks, M.S., Hogan, D.B., Basran, J., and Ebly, E.M. (2005). Supplemental use of antioxidant vitamins and subsequent risk of cognitive decline and dementia. Dement Geriatr Cogn Disord 20, 45-51.
McCown, T.J. (2005). Adeno-associated virus (AAV) vectors in the CNS. Curr Gene Ther 5, 333-338.
McCown, T.J. (2011). Adeno-Associated Virus (AAV) Vectors in the CNS. Curr Gene Ther 11, 181-188.
McCown, T.J., Xiao, X., Li, J., Breese, G.R., and Samulski, R.J. (1996). Differential and persistent expression patterns of CNS gene transfer by an adeno-associated virus (AAV) vector. Brain Res 713, 99-107.
McLay, R.N., Freeman, S.M., Harlan, R.E., Kastin, A.J., and Zadina, J.E. (1999). Tests used to assess the cognitive abilities of aged rats: their relation to each other and to hippocampal morphology and neurotrophin expression. Gerontology 45, 143-155.
Melov, S., Ravenscroft, J., Malik, S., Gill, M.S., Walker, D.W., Clayton, P.E., Wallace, D.C., Malfroy, B., Doctrow, S.R., and Lithgow, G.J. (2000). Extension of life-span with superoxide dismutase/catalase mimetics. Science 289, 1567-1569.
Merrill, D.A., Chiba, A.A., and Tuszynski, M.H. (2001). Conservation of neuronal number and size in the entorhinal cortex of behaviorally characterized aged rats. J Comp Neurol 438, 445-456.
Merrill, D.A., Roberts, J.A., and Tuszynski, M.H. (2000). Conservation of neuron number and size in entorhinal cortex layers II, III, and V/VI of aged primates. J Comp Neurol 422, 396-401.
Miletic, H., Fischer, Y.H., Neumann, H., Hans, V., Stenzel, W., Giroglou, T., Hermann, M., Deckert, M., and Von Laer, D. (2004). Selective transduction of malignant glioma by lentiviral vectors pseudotyped with lymphocytic choriomeningitis virus glycoproteins. Hum Gene Ther 15, 1091-1100.
Milner, A.J., Cummings, D.M., Spencer, J.P., and Murphy, K.P. (2004). Bi-directional plasticity and age-dependent long-term depression at mouse CA3-CA1 hippocampal synapses. Neurosci Lett 367, 1-5.

161
Milner, B. (1972). Disorders of learning and memory after temporal lobe lesions in man. Clin Neurosurg 19, 421-446.
Milner, B., Squire, L.R., and Kandel, E.R. (1998). Cognitive neuroscience and the study of memory. Neuron 20, 445-468.
Mitchell, D.B., Brown, A.S., and Murphy, D.R. (1990). Dissociations between procedural and episodic memory: effects of time and aging. Psychol Aging 5, 264-276.
Miwa, S., St-Pierre, J., Partridge, L., and Brand, M.D. (2003). Superoxide and hydrogen peroxide production by Drosophila mitochondria. Free Radic Biol Med 35, 938-948.
Mockett, R.J., Bayne, A.C., Kwong, L.K., Orr, W.C., and Sohal, R.S. (2003). Ectopic expression of catalase in Drosophila mitochondria increases stress resistance but not longevity. Free Radic Biol Med 34, 207-217.
Montine, T.J., Neely, M.D., Quinn, J.F., Beal, M.F., Markesbery, W.R., Roberts, L.J., and Morrow, J.D. (2002). Lipid peroxidation in aging brain and Alzheimer's disease. Free Radic Biol Med 33, 620-626.
Morgan, S.L., and Teyler, T.J. (2001). Electrical stimuli patterned after the theta-rhythm induce multiple forms of LTP. J Neurophysiol 86, 1289-1296.
Morris, R. (1984). Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods 11, 47-60.
Morris, R.G., Garrud, P., Rawlins, J.N., and O'Keefe, J. (1982). Place navigation impaired in rats with hippocampal lesions. Nature 297, 681-683.
Morris, R.G., Schenk, F., Tweedie, F., and Jarrard, L.E. (1990). Ibotenate Lesions of Hippocampus and/or Subiculum: Dissociating Components of Allocentric Spatial Learning. Eur J Neurosci 2, 1016-1028.
Morrison, J.H., and Hof, P.R. (1997). Life and death of neurons in the aging brain. Science 278, 412-419.
Moser, E.I., Kropff, E., and Moser, M.B. (2008). Place cells, grid cells, and the brain's spatial representation system. Annu Rev Neurosci 31, 69-89.
Muller, F.L., Liu, Y., and Van Remmen, H. (2004). Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem 279, 49064-49073.
Murabito, J.M., Yuan, R., and Lunetta, K.L. (2012). The search for longevity and healthy aging genes: insights from epidemiological studies and samples of long-lived individuals. J Gerontol A Biol Sci Med Sci 67, 470-479.

162
Murakami, K., Kondo, T., Kawase, M., Li, Y., Sato, S., Chen, S.F., and Chan, P.H. (1998). Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J Neurosci 18, 205-213.
Muramatsu, S., Fujimoto, K., Ikeguchi, K., Shizuma, N., Kawasaki, K., Ono, F., Shen, Y., Wang, L., Mizukami, H., Kume, A., et al. (2002). Behavioral recovery in a primate model of Parkinson's disease by triple transduction of striatal cells with adeno-associated viral vectors expressing dopamine-synthesizing enzymes. Hum Gene Ther 13, 345-354.
Nadon, N.L. (2006). Exploiting the rodent model for studies on the pharmacology of lifespan extension. Aging Cell 5, 9-15.
Naldini, L., Blomer, U., Gage, F.H., Trono, D., and Verma, I.M. (1996a). Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc Natl Acad Sci U S A 93, 11382-11388.
Naldini, L., Blomer, U., Gallay, P., Ory, D., Mulligan, R., Gage, F.H., Verma, I.M., and Trono, D. (1996b). In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272, 263-267.
Navarro, A., Sanchez Del Pino, M.J., Gomez, C., Peralta, J.L., and Boveris, A. (2002). Behavioral dysfunction, brain oxidative stress, and impaired mitochondrial electron transfer in aging mice. Am J Physiol Regul Integr Comp Physiol 282, R985-992.
Newman, A.B., Glynn, N.W., Taylor, C.A., Sebastiani, P., Perls, T.T., Mayeux, R., Christensen, K., Zmuda, J.M., Barral, S., Lee, J.H., et al. (2011). Health and function of participants in the Long Life Family Study: A comparison with other cohorts. Aging (Milano) 3, 63-76.
Nicklin, S.A., Buening, H., Dishart, K.L., de Alwis, M., Girod, A., Hacker, U., Thrasher, A.J., Ali, R.R., Hallek, M., and Baker, A.H. (2001). Efficient and selective AAV2-mediated gene transfer directed to human vascular endothelial cells. Mol Ther 4, 174-181.
Nicolle, M.M., Gonzalez, J., Sugaya, K., Baskerville, K.A., Bryan, D., Lund, K., Gallagher, M., and McKinney, M. (2001). Signatures of hippocampal oxidative stress in aged spatial learning-impaired rodents. Neuroscience 107, 415-431.
Noack, H., Lindenau, J., Rothe, F., Asayama, K., and Wolf, G. (1998). Differential expression of superoxide dismutase isoforms in neuronal and glial compartments in the course of excitotoxically mediated neurodegeneration: relation to oxidative and nitrergic stress. Glia 23, 285-297.
O'Keefe, J., and Dostrovsky, J. (1971). The hippocampus as a spatial map. Preliminary evidence from unit activity in the freely-moving rat. Brain Res 34, 171-175.

163
Oliver, C.N., Ahn, B.W., Moerman, E.J., Goldstein, S., and Stadtman, E.R. (1987). Age-related changes in oxidized proteins. J Biol Chem 262, 5488-5491.
Olsen, A., Vantipalli, M.C., and Lithgow, G.J. (2006). Using Caenorhabditis elegans as a model for aging and age-related diseases. Ann N Y Acad Sci 1067, 120-128.
Opalach, K., Rangaraju, S., Madorsky, I., Leeuwenburgh, C., and Notterpek, L. (2010). Lifelong calorie restriction alleviates age-related oxidative damage in peripheral nerves. Rejuvenation Res 13, 65-74.
Orr, W.C., Mockett, R.J., Benes, J.J., and Sohal, R.S. (2003). Effects of overexpression of copper-zinc and manganese superoxide dismutases, catalase, and thioredoxin reductase genes on longevity in Drosophila melanogaster. J Biol Chem 278, 26418-26422.
Orr, W.C., and Sohal, R.S. (1993). Effects of Cu-Zn superoxide dismutase overexpression of life span and resistance to oxidative stress in transgenic Drosophila melanogaster. Arch Biochem Biophys 301, 34-40.
Orr, W.C., and Sohal, R.S. (1994). Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster. Science 263, 1128-1130.
Orrell, R., de Belleroche, J., Marklund, S., Bowe, F., and Hallewell, R. (1995). A novel SOD mutant and ALS. Nature 374, 504-505.
Packard, M.G., and McGaugh, J.L. (1992). Double dissociation of fornix and caudate nucleus lesions on acquisition of two water maze tasks: further evidence for multiple memory systems. Behav Neurosci 106, 439-446.
Pakkenberg, B., and Gundersen, H.J. (1997). Neocortical neuron number in humans: effect of sex and age. J Comp Neurol 384, 312-320.
Papaioannou, N., Tooten, P.C., van Ederen, A.M., Bohl, J.R., Rofina, J., Tsangaris, T., and Gruys, E. (2001). Immunohistochemical investigation of the brain of aged dogs. I. Detection of neurofibrillary tangles and of 4-hydroxynonenal protein, an oxidative damage product, in senile plaques. Amyloid 8, 11-21.
Park, D.C., Royal, D., Dudley, W., and Morrell, R. (1988). Forgetting of pictures over a long retention interval in young and older adults. Psychol Aging 3, 94-95.
Peled-Kamar, M., Lotem, J., Okon, E., Sachs, L., and Groner, Y. (1995). Thymic abnormalities and enhanced apoptosis of thymocytes and bone marrow cells in transgenic mice overexpressing Cu/Zn-superoxide dismutase: implications for Down syndrome. Embo J 14, 4985-4993.

164
Peled-Kamar, M., Lotem, J., Wirguin, I., Weiner, L., Hermalin, A., and Groner, Y. (1997). Oxidative stress mediates impairment of muscle function in transgenic mice with elevated level of wild-type Cu/Zn superoxide dismutase. Proc Natl Acad Sci U S A 94, 3883-3887.
Pellmar, T.C., Hollinden, G.E., and Sarvey, J.M. (1991). Free radicals accelerate the decay of long-term potentiation in field CA1 of guinea-pig hippocampus. Neuroscience 44, 353-359.
Perez, V.I., Bokov, A., Van Remmen, H., Mele, J., Ran, Q., Ikeno, Y., and Richardson, A. (2009a). Is the oxidative stress theory of aging dead? Biochim Biophys Acta 1790, 1005-1014.
Perez, V.I., Van Remmen, H., Bokov, A., Epstein, C.J., Vijg, J., and Richardson, A. (2009b). The overexpression of major antioxidant enzymes does not extend the lifespan of mice. Aging Cell 8, 73-75.
Peters, A., Leahu, D., Moss, M.B., and McNally, K.J. (1994). The effects of aging on area 46 of the frontal cortex of the rhesus monkey. Cereb Cortex 4, 621-635.
Phelan, J.P. (1992). Genetic variability and rodent models of human aging. Exp Gerontol 27, 147-159.
Prabhakar, R., Vreven, T., Morokuma, K., and Musaev, D.G. (2005). Elucidation of the mechanism of selenoprotein glutathione peroxidase (GPx)-catalyzed hydrogen peroxide reduction by two glutathione molecules: a density functional study. Biochemistry 44, 11864-11871.
Pratico, D. (2001). Lipid peroxidation in mouse models of atherosclerosis. Trends Cardiovasc Med 11, 112-116.
Raha, S., McEachern, G.E., Myint, A.T., and Robinson, B.H. (2000). Superoxides from mitochondrial complex III: the role of manganese superoxide dismutase. Free Radic Biol Med 29, 170-180.
Rapp, P.R., and Gallagher, M. (1996). Preserved neuron number in the hippocampus of aged rats with spatial learning deficits. Proc Natl Acad Sci U S A 93, 9926-9930.
Rasmussen, T., Schliemann, T., Sorensen, J.C., Zimmer, J., and West, M.J. (1996). Memory impaired aged rats: no loss of principal hippocampal and subicular neurons. Neurobiol Aging 17, 143-147.
Ratovitski, T., Corson, L.B., Strain, J., Wong, P., Cleveland, D.W., Culotta, V.C., and Borchelt, D.R. (1999). Variation in the biochemical/biophysical properties of mutant superoxide dismutase 1 enzymes and the rate of disease progression in familial amyotrophic lateral sclerosis kindreds. Hum Mol Genet 8, 1451-1460.

165
Reaume, A.G., Elliott, J.L., Hoffman, E.K., Kowall, N.W., Ferrante, R.J., Siwek, D.F., Wilcox, H.M., Flood, D.G., Beal, M.F., Brown, R.H., Jr., et al. (1996). Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet 13, 43-47.
Rebrin, I., Bayne, A.C., Mockett, R.J., Orr, W.C., and Sohal, R.S. (2004). Free aminothiols, glutathione redox state and protein mixed disulphides in aging Drosophila melanogaster. Biochem J 382, 131-136.
Reiser, J., Harmison, G., Kluepfel-Stahl, S., Brady, R.O., Karlsson, S., and Schubert, M. (1996). Transduction of nondividing cells using pseudotyped defective high-titer HIV type 1 particles. Proc Natl Acad Sci U S A 93, 15266-15271.
Remiao, F., Carmo, H., Carvalho, F.D., and Bastos, M.L. (2000). Inhibition of glutathione reductase by isoproterenol oxidation products. J Enzyme Inhib 15, 47-61.
Rempel-Clower, N.L., Zola, S.M., Squire, L.R., and Amaral, D.G. (1996). Three cases of enduring memory impairment after bilateral damage limited to the hippocampal formation. J Neurosci 16, 5233-5255.
Rendahl, K.G., Leff, S.E., Otten, G.R., Spratt, S.K., Bohl, D., Van Roey, M., Donahue, B.A., Cohen, L.K., Mandel, R.J., Danos, O., et al. (1998). Regulation of gene expression in vivo following transduction by two separate rAAV vectors. Nat Biotechnol 16, 757-761.
Reynolds, I.J., Rush, E.A., and Aizenman, E. (1990). Reduction of NMDA receptors with dithiothreitol increases [3H]-MK-801 binding and NMDA-induced Ca2+ fluxes. Br J Pharmacol 101, 178-182.
Reznick, A.Z., and Packer, L. (1994). Oxidative damage to proteins: spectrophotometric method for carbonyl assay. Methods Enzymol 233, 357-363.
Rikke, B.A., Murakami, S., and Johnson, T.E. (2000). Paralogy and orthology of tyrosine kinases that can extend the life span of Caenorhabditis elegans. Mol Biol Evol 17, 671-683.
Robillard, J.M., Gordon, G.R., Choi, H.B., Christie, B.R., and MacVicar, B.A. (2011). Glutathione restores the mechanism of synaptic plasticity in aged mice to that of the adult. PLoS One 6, e20676.
Rocher, C., Lalanne, J.L., and Chaudiere, J. (1992). Purification and properties of a recombinant sulfur analog of murine selenium-glutathione peroxidase. Eur J Biochem 205, 955-960.
Rohrdanz, E., Schmuck, G., Ohler, S., Tran-Thi, Q.H., and Kahl, R. (2001). Changes in antioxidant enzyme expression in response to hydrogen peroxide in rat astroglial cells. Arch Toxicol 75, 150-158.

166
Rosen, D.R., Siddique, T., Patterson, D., Figlewicz, D.A., Sapp, P., Hentati, A., Donaldson, D., Goto, J., O'Regan, J.P., Deng, H.X., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59-62.
Rota, C., Chignell, C.F., and Mason, R.P. (1999). Evidence for free radical formation during the oxidation of 2'-7'-dichlorofluorescin to the fluorescent dye 2'-7'-dichlorofluorescein by horseradish peroxidase: possible implications for oxidative stress measurements. Free Radic Biol Med 27, 873-881.
Rozing, M.P., Westendorp, R.G., de Craen, A.J., Frolich, M., de Goeij, M.C., Heijmans, B.T., Beekman, M., Wijsman, C.A., Mooijaart, S.P., Blauw, G.J., et al. (2010). Favorable glucose tolerance and lower prevalence of metabolic syndrome in offspring without diabetes mellitus of nonagenarian siblings: the Leiden longevity study. J Am Geriatr Soc 58, 564-569.
Russell, S.J., and Cosset, F.L. (1999). Modifying the host range properties of retroviral vectors. J Gene Med 1, 300-311.
Sampayo, J.N., Gill, M.S., and Lithgow, G.J. (2003a). Oxidative stress and aging--the use of superoxide dismutase/catalase mimetics to extend lifespan. Biochem Soc Trans 31, 1305-1307.
Sampayo, J.N., Olsen, A., and Lithgow, G.J. (2003b). Oxidative stress in Caenorhabditis elegans: protective effects of superoxide dismutase/catalase mimetics. Aging Cell 2, 319-326.
Sancar, A. (1996). DNA excision repair. Annu Rev Biochem 65, 43-81.
Sasaki, T., Unno, K., Tahara, S., Shimada, A., Chiba, Y., Hoshino, M., and Kaneko, T. (2008). Age-related increase of superoxide generation in the brains of mammals and birds. Aging Cell 7, 459-469.
Schafer, F.Q., and Buettner, G.R. (2001). Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med 30, 1191-1212.
Schafer, M., Schafer, C., Ewald, N., Piper, H.M., and Noll, T. (2003). Role of redox signaling in the autonomous proliferative response of endothelial cells to hypoxia. Circ Res 92, 1010-1015.
Scheibel, A.B. (1979). The hippocampus: organizational patterns in health and senescence. Mech Ageing Dev 9, 89-102.
Scheibel, M.E., Lindsay, R.D., Tomiyasu, U., and Scheibel, A.B. (1976). Progressive dendritic changes in the aging human limbic system. Exp Neurol 53, 420-430.

167
Schriner, S.E., Linford, N.J., Martin, G.M., Treuting, P., Ogburn, C.E., Emond, M., Coskun, P.E., Ladiges, W., Wolf, N., Van Remmen, H., et al. (2005). Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 308, 1909-1911.
Scoville, W.B., and Milner, B. (1957). Loss of recent memory after bilateral hippocampal lesions. J Neurol Neurosurg Psychiatry 20, 11-21.
Seeberg, E., Eide, L., and Bjoras, M. (1995). The base excision repair pathway. Trends Biochem Sci 20, 391-397.
Shacter, E. (2000a). Quantification and significance of protein oxidation in biological samples. Drug Metab Rev 32, 307-326.
Shacter, E. (2000b). Quantification and significance of protein oxidation in biological samples. Drug metabolism reviews 32, 307-326.
Sharma, A.B., Sun, J., Howard, L.L., Williams, A.G., Jr., and Mallet, R.T. (2007). Oxidative stress reversibly inactivates myocardial enzymes during cardiac arrest. Am J Physiol Heart Circ Physiol 292, H198-206.
Shen, Y., Muramatsu, S.I., Ikeguchi, K., Fujimoto, K.I., Fan, D.S., Ogawa, M., Mizukami, H., Urabe, M., Kume, A., Nagatsu, I., et al. (2000). Triple transduction with adeno-associated virus vectors expressing tyrosine hydroxylase, aromatic-L-amino-acid decarboxylase, and GTP cyclohydrolase I for gene therapy of Parkinson's disease. Hum Gene Ther 11, 1509-1519.
Shetty, P.K., Huang, F.L., and Huang, K.P. (2008). Ischemia-elicited oxidative modulation of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem 283, 5389-5401.
Shibutani, S., Takeshita, M., and Grollman, A.P. (1991). Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature 349, 431-434.
Shimizu, N., Kobayashi, K., and Hayashi, K. (1984). The reaction of superoxide radical with catalase. Mechanism of the inhibition of catalase by superoxide radical. J Biol Chem 259, 4414-4418.
Shringarpure, R., Grune, T., Mehlhase, J., and Davies, K.J. (2003). Ubiquitin conjugation is not required for the degradation of oxidized proteins by proteasome. J Biol Chem 278, 311-318.
Singer, T., Verhaeghen, P., Ghisletta, P., Lindenberger, U., and Baltes, P.B. (2003). The fate of cognition in very old age: six-year longitudinal findings in the Berlin Aging Study (BASE). Psychol Aging 18, 318-331.

168
Sinn, P.L., Sauter, S.L., and McCray, P.B., Jr. (2005). Gene therapy progress and prospects: development of improved lentiviral and retroviral vectors--design, biosafety, and production. Gene Ther 12, 1089-1098.
Smith, C.D., Carney, J.M., Starke-Reed, P.E., Oliver, C.N., Stadtman, E.R., Floyd, R.A., and Markesbery, W.R. (1991). Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc Natl Acad Sci U S A 88, 10540-10543.
Smith, C.V., Jones, D.P., Guenthner, T.M., Lash, L.H., and Lauterburg, B.H. (1996). Compartmentation of glutathione: implications for the study of toxicity and disease. Toxicol Appl Pharmacol 140, 1-12.
Soh, N. (2006). Recent advances in fluorescent probes for the detection of reactive oxygen species. Anal Bioanal Chem 386, 532-543.
Spangler, E.L., Waggie, K.S., Hengemihle, J., Roberts, D., Hess, B., and Ingram, D.K. (1994). Behavioral assessment of aging in male Fischer 344 and brown Norway rat strains and their F1 hybrid. Neurobiol Aging 15, 319-328.
Spreng, M., Rossier, J., and Schenk, F. (2002). Spaced training facilitates long-term retention of place navigation in adult but not in adolescent rats. Behav Brain Res 128, 103-108.
Sprott, R.L. (1991). Development of animal models of aging at the National Institute of Aging. Neurobiol Aging 12, 635-638.
Sprott, R.L., and Ramirez, I. (1997). Current Inbred and Hybrid Rat and Mouse Models for Gereontological Research. Ilar J 38, 104-109.
Stadtman, T.C. (1980). Selenium-dependent enzymes. Annu Rev Biochem 49, 93-110.
Sun, J., Folk, D., Bradley, T.J., and Tower, J. (2002). Induced overexpression of mitochondrial Mn-superoxide dismutase extends the life span of adult Drosophila melanogaster. Genetics 161, 661-672.
Tappel, A.L. (1984). Selenium--glutathione peroxidase: properties and synthesis. Curr Top Cell Regul 24, 87-97.
Thibault, O., Hadley, R., and Landfield, P.W. (2001). Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: relationship to impaired synaptic plasticity. J Neurosci 21, 9744-9756.
Thibault, O., and Landfield, P.W. (1996). Increase in single L-type calcium channels in hippocampal neurons during aging. Science 272, 1017-1020.

169
Thiels, E., Urban, N.N., Gonzalez-Burgos, G.R., Kanterewicz, B.I., Barrionuevo, G., Chu, C.T., Oury, T.D., and Klann, E. (2000). Impairment of long-term potentiation and associative memory in mice that overexpress extracellular superoxide dismutase. J Neurosci 20, 7631-7639.
Townsend, D.M., Tew, K.D., and Tapiero, H. (2003). The importance of glutathione in human disease. Biomed Pharmacother 57, 145-155.
Turrens, J.F., and Boveris, A. (1980). Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J 191, 421-427.
Turturro, A., Witt, W.W., Lewis, S., Hass, B.S., Lipman, R.D., and Hart, R.W. (1999). Growth curves and survival characteristics of the animals used in the Biomarkers of Aging Program. J Gerontol A Biol Sci Med Sci 54, B492-501.
van der Staay, F.J., and Blokland, A. (1996). Behavioral differences between outbred Wistar, inbred Fischer 344, brown Norway, and hybrid Fischer 344 x brown Norway rats. Physiol Behav 60, 97-109.
Van Luijtelaar, E.L., and Coenen, A.M. (1988). Circadian rhythmicity in absence epilepsy in rats. Epilepsy Res 2, 331-336.
Van Vliet, K.M., Blouin, V., Brument, N., Agbandje-McKenna, M., and Snyder, R.O. (2008). The role of the adeno-associated virus capsid in gene transfer. Methods Mol Biol 437, 51-91.
Vasquez, B.J., Martinez, J.L., Jr., Jensen, R.A., Messing, R.B., Rigter, H., and McGaugh, J.L. (1983). Learning and memory in young and aged Fischer 344 rats. Arch Gerontol Geriatr 2, 279-291.
Vorhees, C.V., and Williams, M.T. (2006). Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc 1, 848-858.
Wang, H., Liu, H., and Liu, R.M. (2003a). Gender difference in glutathione metabolism during aging in mice. Exp Gerontol 38, 507-517.
Wang, J., Slunt, H., Gonzales, V., Fromholt, D., Coonfield, M., Copeland, N.G., Jenkins, N.A., and Borchelt, D.R. (2003b). Copper-binding-site-null SOD1 causes ALS in transgenic mice: aggregates of non-native SOD1 delineate a common feature. Hum Mol Genet 12, 2753-2764.
Weimann, A., Belling, D., and Poulsen, H.E. (2002). Quantification of 8-oxo-guanine and guanine as the nucleobase, nucleoside and deoxynucleoside forms in human urine by high-performance liquid chromatography-electrospray tandem mass spectrometry. Nucleic Acids Res 30, E7.

170
West, M.J., Coleman, P.D., Flood, D.G., and Troncoso, J.C. (1994). Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer's disease. Lancet 344, 769-772.
Wijeratne, S.S., Cuppett, S.L., and Schlegel, V. (2005). Hydrogen peroxide induced oxidative stress damage and antioxidant enzyme response in Caco-2 human colon cells. J Agric Food Chem 53, 8768-8774.
Wilkniss, S.M., Jones, M.G., Korol, D.L., Gold, P.E., and Manning, C.A. (1997). Age-related differences in an ecologically based study of route learning. Psychol Aging 12, 372-375.
Winder, D.G., and Sweatt, J.D. (2001). Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nat Rev Neurosci 2, 461-474.
Winterbourn, C.C., and Metodiewa, D. (1999). Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic Biol Med 27, 322-328.
Wood, R.D. (1996). DNA repair in eukaryotes. Annu Rev Biochem 65, 135-167.
Wu, K., Meyers, C.A., Guerra, N.K., King, M.A., and Meyer, E.M. (2004a). The effects of rAAV2-mediated NGF gene delivery in adult and aged rats. Mol Ther 9, 262-269.
Wu, K., Meyers, C.A., Guerra, N.K., King, M.A., and Meyer, E.M. (2004b). The effects of rAAV2-mediated NGF gene delivery in adult and aged rats. Mol Ther 9, 262-269.
Wykle, M.L., Whitehouse, P.J., and Morris, D.L. (2005). Successful aging through the life span : intergenerational issues in health (New York, Springer Pub. Co.).
Xie, Y., Yang, H., Cunanan, C., Okamoto, K., Shibata, D., Pan, J., Barnes, D.E., Lindahl, T., McIlhatton, M., Fishel, R., et al. (2004). Deficiencies in mouse Myh and Ogg1 result in tumor predisposition and G to T mutations in codon 12 of the K-ras oncogene in lung tumors. Cancer Res 64, 3096-3102.
Yang, Y.J., Wu, P.F., Long, L.H., Yu, D.F., Wu, W.N., Hu, Z.L., Fu, H., Xie, N., Jin, Y., Ni, L., et al. (2010). Reversal of aging-associated hippocampal synaptic plasticity deficits by reductants via regulation of thiol redox and NMDA receptor function. Aging Cell 9, 709-721.
Yim, T.K., and Ko, K.M. (1999). Schisandrin B protects against myocardial ischemia-reperfusion injury by enhancing myocardial glutathione antioxidant status. Mol Cell Biochem 196, 151-156.
Yoritaka, A., Hattori, N., Uchida, K., Tanaka, M., Stadtman, E.R., and Mizuno, Y. (1996). Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci U S A 93, 2696-2701.

171
Zarkovic, K. (2003). 4-hydroxynonenal and neurodegenerative diseases. Mol Aspects Med 24, 293-303.
Zeevalk, G.D., Bernard, L.P., Song, C., Gluck, M., and Ehrhart, J. (2005). Mitochondrial inhibition and oxidative stress: reciprocating players in neurodegeneration. Antioxid Redox Signal 7, 1117-1139.
Zeier, Z., Madorsky, I., Xu, Y., Ogle, W.O., Notterpek, L., and Foster, T.C. (2011). Gene expression in the hippocampus: regionally specific effects of aging and caloric restriction. Mech Ageing Dev 132, 8-19.
Zola-Morgan, S., Squire, L.R., and Amaral, D.G. (1986). Human amnesia and the medial temporal region: enduring memory impairment following a bilateral lesion limited to field CA1 of the hippocampus. J Neurosci 6, 2950-2967.
Zufferey, R., Dull, T., Mandel, R.J., Bukovsky, A., Quiroz, D., Naldini, L., and Trono, D. (1998). Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J Virol 72, 9873-9880.
Zufferey, R., Nagy, D., Mandel, R.J., Naldini, L., and Trono, D. (1997). Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol 15, 871-875.

172
BIOGRAPHICAL SKETCH
Wei-Hua Lee was born in 1980 at Taipei, Taiwan. She graduated from National
Taiwan Normal University in 2003, where she received a Bachelor of Science degree in
biology. She came to the United States in 2004 to begin her journey of being a
biological researcher. Wei-Hua Lee enrolled the University of Iowa in 2004 and joined
the lab of Dr. Chun-Fang Wu for her Master of Science degree. After that she joined Dr.
Thomas Foster lab at University of Florida, where she was focusing on studying
memory deficits during normal aging, and eventually dedicated herself into
understanding the relationship between aging, reactive oxygen species, synaptic
plasticity and memory. Wei-Hua Lee was active in her academic career and had
participated in several national conferences. Her scientific discoveries had been
published in different journals. She received her Ph.D. from the University of Florida in
the summer of 2012. In the future she will devote herself to help the community and
human society as a scientist by her research.




















