
A Truncated Matched Filter Method for Interrupted Sampling ...

Biochemical and Biophysical Research Communications 308 (2003) 660–667
www.elsevier.com/locate/ybbrc
BBRC
Impairment of superoxide dismutase activation by N-terminallytruncated prion protein (PrP) in PrP-deficient neuronal cell line
Akikazu Sakudo,a,1 Deug-chan Lee,a,1 Keiichi Saeki,a Yuko Nakamura,a Keiichi Inoue,b
Yoshitsugu Matsumoto,a Shigeyoshi Itohara,b and Takashi Onoderaa,*
a Department of Molecular Immunology, School of Agricultural and Life Sciences, University of Tokyo, Bunkyo-ku, Tokyo 113-8657, Japanb Laboratory for Behavioral Genetics, Brain Science Institute, RIKEN, Wako, Saitama 351-0198, Japan
Received 12 July 2003
Abstract
Previous studies have reported a neuroprotective role for cellular prion protein (PrPC) against apoptosis induced by serum
deprivation in an immortalized prion protein gene (Prnp)-deficient neuronal cell line, but the mechanisms remain unclear. In this
study, to investigate the mechanisms by which PrPC prevents apoptosis, the authors compared apoptosis of Prnp�=� cells with that
of Prnp�=� cells expressing the wild-type PrPC or PrPC lacking N-terminal octapeptide repeat region under serum-free conditions.
Re-introduction of Prnp rescued cells from apoptosis, upregulated superoxide dismutase (SOD) activity, enhanced superoxide anion
elimination, and inhibited caspase-3/9 activation. On the other hand, N-terminally truncated PrPC enhanced apoptosis accompanied
by potentiation of superoxide production and caspase-3/9 activation due to inhibition of SOD. These results suggest that PrPC
protects Prnp�=� cells from apoptosis via superoxide- and caspase-3/9-dependent pathways by upregulating SOD activity. Fur-
thermore, the octapeptide repeat region of PrPC plays an essential role in regulating apoptosis and SOD activity.
� 2003 Elsevier Inc. All rights reserved.
Keywords: Antioxidant; Apoptosis; Caspase; Oxidative stress; Prion disease; Prion protein; PrP-deficient cell line; Superoxide dismutase
Prion diseases such as Creutzfeldt–Jakob disease(CJD), scrapie, and bovine spongiform encephalopathy
are disorders characterized by the intracerebral accu-
mulation of abnormal prion proteins (PrPSc) [1]. Cellu-
lar prion proteins (PrPC), which are constitutively
expressed on the cell surface of neural and nonneural
cells, are identical in amino acid sequence to PrPSc [2].
Mice lacking the expression of PrPC cannot be infected
with prion agents [3,4] and neurons lacking PrPC ex-pression cannot be killed by the toxicity of PrPSc [5,6].
Therefore, the expression of PrPC is essential for the
induction of prion diseases, and the conversion of PrPC
to PrPSc is closely associated with the main features of
prion diseases. Six lines of prion protein (PrP) gene
(Prnp) knockout mice, designated as Zur I [3], Zur II [7],
Npu [8], Ngsk [9], Rcm0 [10], and Rikn [11], have been
generated. Some lines do not display any anatomical or
* Corresponding author. Fax. +81-3-5841-8020.
E-mail address: [email protected] (T. Onodera).1 These two authors contributed equally to the work.
0006-291X/$ - see front matter � 2003 Elsevier Inc. All rights reserved.
doi:10.1016/S0006-291X(03)01459-1
developmental defects [3]. Although altered circadianrhythms [12] and electrophysiological abnormality [13]
have been reported, the latter remains controversial
[14,15]. Other lines of PrP-null mice exhibit a late-onset
ataxia, but this is possibly due to abnormal splicing by
the destruction of splicing acceptor [7,10]. Therefore,
little is known about the function of PrPC.
Recently, a line of PrP-deficient immortalized hippo-
campal neuronal cells derived from Rikn Prnp�=� micehas been established [16]. Moreover, it has been reported
that removal of serum from the cell culture causes ap-
optosis in Prnp�=� cells, but not in Prnpþ=þ cells.
Transfection of PrP gene suppresses apoptosis of
Prnp�=� cells under serum-free conditions [16]. However,
the mechanisms by which PrPC prevents apoptosis re-
main unclear. These recent studies suggest that PrPC may
itself work as an apoptosis suppressor and/or regulatorof apoptosis suppressors and that the loss of functions
causes neuronal cell death. Several apoptotic pathways
are activated by various stimuli. One of the pathways is
regulated by expression of the proto-oncogene bcl-2

A. Sakudo et al. / Biochemical and Biophysical Research Communications 308 (2003) 660–667 661
family, whose members include bcl-2 and bcl-x [17]. Afamily of cysteine proteases, termed caspases, which have
been shown to cleave a number of intracellular targets
including poly(ADP-ribose) polymerase (PARP), are
also involved in the initiation/execution of apoptosis [18].
Several reports concerning the relationship between
PrPC and superoxide dismutase (SOD) using brain or
primary cultures of neuron have provided inconsistent
results [19–22]. Additionally, the regulation of SOD ac-tivity by PrPC may not be a mechanism in an indepen-
dent cell but rather reflect a systemic process of
heterogeneous cell population in the brain. Recently,
rabbit kidney epithelial RK13 cells or human neuro-
blastoma SH-SY5Y cells overexpressing PrPC have been
resistant to hydrogen peroxide, 3-morpholinosydnoni-
mine (SIN-1) or copper induced toxicity possibly due to
increase of antioxidant enzyme activity [23,24]. However,since these cell lines have endogenous Prnp, the effect of
endogenous PrPC could not be excluded from the ex-
planation. Therefore, the antioxidative property of PrPC
was analyzed by investigating the mechanisms of apop-
tosis in the presence or absence of PrPC in Prnp�=� cells.
In this study,weassessed roles ofPrPC andPrPC lacking
an octapeptide repeat region in the apoptosis of Prnp�=�
cells. The results suggested that antioxidative property ofPrPC was the mechanism for preventing apoptosis, and
that the octapeptide repeat region of PrPC essentially
contributed to regulation of SOD activity, superoxide
elimination, and inhibition of caspase-3/9 activation.
Materials and methods
Cell cultures and animals. Murine PrP-deficient cell line (HpL3-4)
[16] was maintained in Dulbecco’s modified Eagle’s medium (DMEM)
(Sigma, St. Louis, MO) supplemented with 10% fetal calf serum (FCS)
at 37 �C in a humidified 5% CO2 incubator. Occasionally, cells were
serum-deprived. For serum deprivation, cells were plated on 60-mm
culture dishes (5� 105 cells per dish) in a complete medium. Two dayslater, cells were washed twice with a serum-free medium and then
placed in a fresh serum-free medium for the indicated time. The male
Rikn Prnp�=� mice [11] and C57BL/6CrSlc (WT) mice purchased from
Nippon SLC were analyzed at 8 weeks of age.
Plasmid construction and gene transfer. In the gene transfer of Prnp,
the coding region of Prnp cDNA was amplified by polymerase chain
reaction (PCR) using the following primers:
Prnp:
50-GCGAATTCCGCCACCATGGCGAACCTTGGCTACTGGCTG-30
50-CACGAATTCCACCTCAATTGAAAGAGCTACAGGTGG-30
The EcoRI linker is underlined and the Kozak consensus sequence
(GCCACCATGG) [25] is boxed. The PCR products were cloned
into pT7Blue vector and subcloned into the multicloning site of
pMSCVpuro (Clontech, Palo Alto, CA). The resulting construct
pMSCVpuro-PrP was transiently transfected into the packaging cell
line PT67 by the Lipofectamine-mediated method (Life Technologies,
Gaithersburg, MD). Viral supernatants were harvested for the trans-
duction of HpL3-4 cells. These cells were selected for more than 10
days in a complete medium containing 20 lg/ml puromycin (WAKO,Osaka, Japan). Ten individual colonies were further selected. Of 10
stable Prnp-transfected lines, two clones (HpL3-4-PrPNo3 and HpL3-
4-PrPNo7) with abundant PrPC were used for this study. Additionally,
a plasmid containing a Prnp cDNA rendering an N-terminally trun-
cated PrP lacking the octapeptide repeat region [PrP(D53–94, Q52H)]was constructed using the restriction enzymes Eco81I and KpnI and a
DNA blunting kit (Takara Bio, Otsu, Japan). The Prnp and Prnp
deletion cDNAs were subcloned into the multicloning site of
pMSCVpuro-EGFP, which was constructed by insertion of the BglII–
HpaI fragment of pIRES2-EGFP (Clontech) into pMSCVpuro,
resulting in pMSCVpuro-PrP-EGFP and pMSCVpuro-PrP(D53–94,Q52H)-EGFP. HpL3-4 cells were transduced with pMSCVpuro-
EGFP, pMSCVpuro-PrP-EGFP or pMSCVpuro-PrP(D53–94, Q52H)-EGFP by a retrovirus-mediated method as described above. The
resulting stable transfectants were named HpL3-4-EM, HpL3-4-PrP,
and HpL3-4-D#1, respectively.SOD activity assay. A SOD assay kit-WST (Dojindo, Kumamoto,
Japan) was used for the quantification of SOD activity. This method is
a xanthine-based spectrophotometric assay. Each protein extract
(20 lg) was assayed and compared with 1U of bovine erythrocyte Cu/Zn-SOD (Sigma S2515) activity. The SOD activity was estimated using
the standard curve of SOD activity versus absorbance.
Western blot assay. To fractionate tissue samples, 10% (w/v) ho-
mogenate was prepared in PBS by sonication. After centrifugation at
600g for 15min at 4 �C, supernatants were ultracentrifuged at 200,000gfor 1 h, at 4 �C. The pellet was solubilized by 27-gauge syringe. Eachfraction was solubilized in radio-immunoprecipitation assay (RIPA)
buffer composed of 10mM Tris–HCl (pH 7.4) containing 1% sodium
deoxycholate, 1% Nonidet P-40, 0.1% sodium dodecyl sulfate (SDS),
and 0.15M sodium chloride supplemented with 2mM phenylmethyl-
sulfonyl fluoride (PMSF).
For detection of Bcl-2, Bcl-xL, and Cu/Zn-SOD, tissue homogen-
ates were directly lysed in RIPA buffer, while the membrane fraction of
tissue samples was used for detection of PrPC.
Cells were pelleted by centrifugation and washed with PBS before
lysis with RIPA buffer. After centrifugation at 10,000g for 10min at
4 �C, the protein concentration of the supernatant was measured usingthe Bio-Rad DC assay, and an equal quantity of protein (60lg forPrPC, 50lg for Bcl-2, 30 lg for Bcl-x, and 20 lg for Cu/Zn-SOD,cleaved caspase-3, cleaved caspase-9, and cleaved PARP) was dena-
tured in 2� SDS gel-loading buffer [90mM Tris–HCl (pH 6.8), 10%
mercaptoethanol, 2% SDS, 0.02% bromophenol blue, and 20% glyc-
erol] and separated on SDS/12% polyacrylamide gel before electrical
transfer onto polyvinyl difluoride (PVDF) membrane (Hybond-P;
Amersham–Pharmacia Biotech, Piscataway, NJ) for 60min at 15V.
Blots were treated by BLOCK-ACE (Dainippon Pharmaceutical,
Osaka, Japan) at 4 �C overnight and then incubated in PBS containing0.1% Tween 20 (PBS–T) and 10% BLOCK-ACE for 1 h at room
temperature with one of the following: 3F11 monoclonal anti-Bcl-2
(Pharmingen, San Diego, CA), polyclonal anti-Bcl-x (Pharmingen),
polyclonal anti-Cu/Zn-SOD (Stressgen, Victoria, BC), polyclonal
anti-cleaved caspase-3 (Cell Signaling Technology, Beverly, MA),
polyclonal anti-caspase-9 (Cell Signaling Technology), polyclonal anti-
cleaved PARP (Cell Signaling Technology), P8 polyclonal anti-PrP,
which recognizes amino acids 92–109 of mouse PrP (GGTHNQWN
KPSKPKTNLK), or 6H4 monoclonal anti-PrP, which recognizes
residues 144–152 of mouse PrP. After washing thrice with PBS–T, the
membrane was incubated in horseradish peroxidase-conjugated sec-
ondary antibody for 1 h at room temperature. After washing twice
with PBS–T, the probed proteins were detected using an enhanced
chemiluminescence detection kit (Amersham–Pharmacia Biotech).
Apoptosis assays. Internucleosomal DNA fragmentation was de-
tected after 24-h incubation in a serum-free medium by DNA lad-
dering assay [16]. Cells were lysed with lysis buffer [10mM Tris–HCl
(pH 7.4), 10mM EDTA (pH 8.0), and 0.5% Triton X-100], incubated
for 20min on ice, and centrifuged at 12,000g for 30min at 4 �C. Theaqueous phase was incubated with 400lg/ml DNase-free RNase A for1 h at 37 �C. Proteins were digested with 400lg/ml of proteinase K for1 h at 37 �C and then DNA was precipitated in a solution of 0.4M

Fig. 1. Re-introduction of Prnp did not change the expression of Bcl-2,
Bcl-xL, and Cu/Zn-SOD. Western blot analysis of protein extracts
using anti-PrP (P8), Bcl-2, Bcl-x, and Cu/Zn-SOD antibodies. The
expressions of Bcl-2, Bcl-xL, and Cu/Zn-SOD were not significantly
different between Prnp transfected HpL3-4 cells [HpL3-4-PrPNo3 (lane
2) and HpL3-4-PrPNo7 (lane 3)] and the parental HpL3-4 cell lines
(lane 1). Bcl-2, Bcl-xL, and Cu/Zn-SOD were similarly expressed in WT
and Prnp�=� (KO) mouse brain.
662 A. Sakudo et al. / Biochemical and Biophysical Research Communications 308 (2003) 660–667
NaCl in 50% isopropanol overnight at )20 �C. Precipitated DNAsamples were resuspended in TE buffer [10mM Tris–HCl (pH 7.4),
1mM EDTA (pH 8.0)] and electrophoresed through 2.0% agarose gel.
The gel was stained with ethidium bromide (EB) (0.5mg/ml) for 10min
and destained with milli-Q for 10min. DNA bands were visualized by
UV light-transilluminator and photographed.
Reactive oxygen species probes. Intracellular H2O2 and superoxide
anion levels were determined based on methods adapted from those
previously described [26,27] using 20,70-dichlorofluorescein diacetate
(DCFH-DA) (Lambda Fluoreszenztechnologie, Graz, Austria) and
Dihydroethidium (DH) (Molecular Probes, Eugene, OR), respectively.
DCFH-DA is incorporated into cells and hydrolyzed by intracellular
esterase, generating the nonfluorescent molecule DCFH. H2O2 oxi-
dizes DCFH to the fluorescent molecule DCF. DH permeates to viable
cells and is directly oxidized to EB by superoxide anion intracellularly
to yield fluorescence for measurement.
DCFH-DA or DH was added to cell cultures to a final concen-
tration of 10lM and incubated for 30min at 37 �C. The cells werecollected, washed twice, and then analyzed with flow cytometry.
Fluorescence was collected using an excitation of 488 nm and a 530/
30-nm band pass for DCF and a 585/42-nm band pass for EB.
Caspase activity assay. Equal quantity of protein (20lg for caspase-3/7 and 60lg for caspase-9) from cells before and after serum depri-
vation for the indicated time was incubated in reaction buffer [20mM
Hepes (pH 7.5); 10% glycerol; and 2mM DTT] containing 100lMAc-LEHD-methyl-coumaryl-7-amide (MCA) (specific substrate for
caspase-9) or Ac-DEVD-MCA (specific substrate for caspase-3/7)
obtained from Peptide Institute (Osaka, Japan). LEHD and DEVD
cleavage was assessed by monitoring fluorescence with excitation and
emission wavelengths of 355 and 460 nm using a Fluoroscan fluo-
rometer (Labsystems Oy, Helsinki, Finland).
Statistical analysis. Quantification of SOD activity, superoxide
anion production, and caspase activity was tested statistically by the
unpaired Student’s t test. p Values smaller than 0.05 were considered tobe statistically significant.
Results
Influence of PrP expression on the protein levels of Bcl-2,
Bcl-xL, and Cu/Zn-SOD
Elucidation of the mechanisms by which PrPC inhibits
apoptosis was approached by analysis of Prnp-trans-
fectedPrnp�=� cells byWestern blotting. Re-introductionof Prnp was confirmed by Western blotting (Fig. 1) and
flow cytometry (data not shown). There were no differ-
ences in the expression level of Bcl-2, Bcl-xL, and Cu/Zn-
SOD protein among HpL3-4, HpL3-4-PrPNo3, and
HpL3-4-PrPNo7 (Fig. 1). In a similar tendency, equal
expressions of Bcl-2, Bcl-xL, and Cu/Zn-SOD protein
were observed betweenwild-type (WT) andPrnp�=� (KO)
murine brain (Fig. 1). In the brain samples, Bcl-xL wasdetected as a doublet with a molecular mass of approxi-
mately 28 kDa. The higher band of this doublet may be a
phosphorylated form of the Bcl-xL protein [28].
Correlation of PrPC production and SOD activity using
tissue and cell lines
For simple analysis of PrPC distribution, the precip-
itate after ultracentrifugation (200,000g for 1 h at 4 �C)
of the tissue homogenate (considered to be the mem-
brane fraction) and the supernatant (the soluble frac-
tion) were employed. PrPC was detected only in the
membrane fraction of WT mouse brain, whereas Cu/Zn-SOD was detected in the soluble fraction of WT and KO
mouse tissues (Fig. 2A). The SOD activity in the above-
mentioned fraction was then measured. The membrane
fraction of WT mouse brain, where PrPC is abundant,
showed elevated SOD activity as compared with the
membrane fraction of other tissues (Fig. 2B). The SOD
activity of the soluble fraction of Prnp�=� mouse brain
and testis indicated a slight but significant decreasewhen compared to WT mouse brain and testis, respec-
tively (Fig. 2C).
To further examine the relationship between expres-
sion of PrPC and SOD activity, PrP-deficient neuronal
cells were analyzed. The lysates of HpL3-4-PrPNo3 and
HpL3-4-PrPNo7 cells, which expressed PrPC (Fig. 1),
revealed enhanced SOD activity as compared with
HpL3-4 cells (data not shown). Flow cytometry analysisusing ROS probes demonstrated that HpL3-4PrPNo3
cells eliminated superoxide anion generation and en-
hanced H2O2 production as compared with HpL3-4 cells
under serum deprivation for 6 h (Figs. 3A and B), sug-
gesting that augmented SOD activity by PrPC effectively
converted superoxide anion into H2O2.
Role of the N-terminal octapeptide repeat region of
PrPC for inhibition of apoptosis and superoxide anion
elimination
Because the N-terminal octapeptide repeat region of
PrPC has been shown to bind to copper and is involvedin the enzymatic function [29], it was of interest to de-
termine whether the octapeptide repeat region of PrPC
affected the suppression of apoptotic neuronal cell death
under serum deprivation. This was examined in PrP-
negative cells (HpL3-4-EM), PrP-positive cells (HpL3-4-

Fig. 3. Re-introduction of Prnp eliminated superoxide anion and in-
creased hydrogen peroxide. Flow cytometric analysis of ROS in serum-
deprived cells. (A) The amount of intracellular superoxide anion was
determined by DH. (B) Hydrogen peroxide was detected by the
staining of DCFH-DA. HpL3-4 and HpL3-4PrPNo3 (PrPNo3) cells
were stained with the fluorescent probes as described in Materials and
methods. The histograms of cells under serum-deprivation for 6 h and
control (0 h) are indicated by filled and open areas, respectively.
Fig. 2. SOD activity in the wild-type and Prnp�=� mouse tissues. Brain
and testis homogenates from Prnp�=� and WT mice were fractionated
by ultracentrifugation. (A) PrPC and Cu/Zn-SOD in precipitate (Ppt)
and supernatant (Sup) were examined by Western blotting. PrPC was
detected in the wild-type (WT) mouse brain but not in Prnp�=� (KO)
mouse brain. PrPC was detected only in the membrane fraction of WT
mouse brain, while Cu/Zn-SOD was detected in the soluble fraction of
WT and KO mouse tissues. (B–C) SOD activity of the above-men-
tioned fraction of tissue was determined. SOD activity correlates with
the expression level of PrPC in the membrane fraction (B). In the
soluble fraction, KO mouse tissue showed reduced SOD activity as
compared with WT mouse tissue (C). Differences with p < 0:05 (*)
between WT and KO tissue and differences with p < 0:01 (**) versus
other fraction of tissue were significant.
A. Sakudo et al. / Biochemical and Biophysical Research Communications 308 (2003) 660–667 663
PrP), and cells transfected with a plasmid expressingPrnp cDNA rendering an N-terminally truncated PrP
lacking the octapeptide repeat region (HpL3-4-D#1).Western blotting showed that HpL3-4-PrP and HpL3-4-
D#1 expressed PrPC at the equivalent level on WT
mouse brain (Fig. 4A). When compared with empty
vector transfected cells (HpL3-4-EM), the cells ex-
pressing PrPC (HpL3-4-PrP) were markedly resistant to
apoptosis following serum deprivation for 24 h (Fig. 4B).The cells expressing N-terminally truncated PrPC
(HpL3-4-D#1), which lacks an octapeptide repeat re-gion, showed enhanced apoptosis when compared with
HpL3-4-EM (Fig. 4B), thus illustrating the importance
of this region for survival in serum-derived medium.
To examine whether the increased susceptibility of
HpL3-4-D#1 cells to serum deprivation was due to ab-errant activity of SOD, SOD activity and superoxide
anion production in the presence and absence of serum
were analyzed. HpL3-4-D#1 cells demonstrated signifi-cantly low activity of SOD and a significant increase in
superoxide anion production compared with HpL3-4-
EM cells under serum deprivation for 6 h (Figs. 4C and
D). However, HpL3-4-PrP cells showed significantly
high activity of SOD and a significant decrease in su-peroxide anion production compared with HpL3-4-EM
cells (Figs. 4C and D).
Serum deprivation induced caspase-3/9 activation which
was inhibited by PrPC and enhanced by N-terminally
truncated PrPC lacking octapeptide repeat region
To further characterize the effects of PrPC and N-
terminally truncated PrPC lacking the octapeptide repeat
region on apoptosis, levels and activities of apoptosis
related proteins including caspase-3, caspase-9, and
PARP in HpL3-4-EM, HpL3-4-PrP, and HpL3-4-D#1cells were examined by Western blotting and caspaseactivity assay. Western blot analysis demonstrated that
active cleaved forms of caspase-3 (17 kDa) and caspase-
9 (37 and 39 kDa) were observed in HpL3-4-EM and
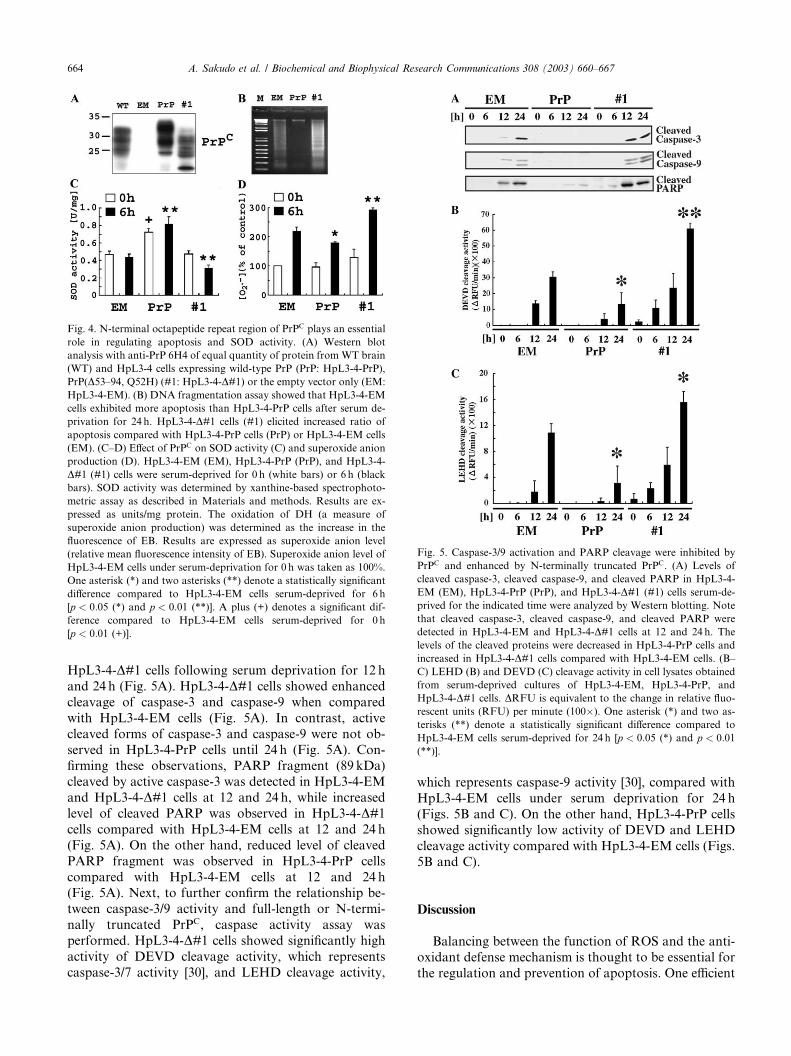
Fig. 4. N-terminal octapeptide repeat region of PrPC plays an essential
role in regulating apoptosis and SOD activity. (A) Western blot
analysis with anti-PrP 6H4 of equal quantity of protein fromWT brain
(WT) and HpL3-4 cells expressing wild-type PrP (PrP: HpL3-4-PrP),
PrP(D53–94, Q52H) (#1: HpL3-4-D#1) or the empty vector only (EM:HpL3-4-EM). (B) DNA fragmentation assay showed that HpL3-4-EM
cells exhibited more apoptosis than HpL3-4-PrP cells after serum de-
privation for 24 h. HpL3-4-D#1 cells (#1) elicited increased ratio ofapoptosis compared with HpL3-4-PrP cells (PrP) or HpL3-4-EM cells
(EM). (C–D) Effect of PrPC on SOD activity (C) and superoxide anion
production (D). HpL3-4-EM (EM), HpL3-4-PrP (PrP), and HpL3-4-
D#1 (#1) cells were serum-deprived for 0 h (white bars) or 6 h (blackbars). SOD activity was determined by xanthine-based spectrophoto-
metric assay as described in Materials and methods. Results are ex-
pressed as units/mg protein. The oxidation of DH (a measure of
superoxide anion production) was determined as the increase in the
fluorescence of EB. Results are expressed as superoxide anion level
(relative mean fluorescence intensity of EB). Superoxide anion level of
HpL3-4-EM cells under serum-deprivation for 0 h was taken as 100%.
One asterisk (*) and two asterisks (**) denote a statistically significant
difference compared to HpL3-4-EM cells serum-deprived for 6 h
[p < 0:05 (*) and p < 0:01 (**)]. A plus (+) denotes a significant dif-
ference compared to HpL3-4-EM cells serum-deprived for 0 h
[p < 0:01 (+)].
Fig. 5. Caspase-3/9 activation and PARP cleavage were inhibited by
PrPC and enhanced by N-terminally truncated PrPC. (A) Levels of
cleaved caspase-3, cleaved caspase-9, and cleaved PARP in HpL3-4-
EM (EM), HpL3-4-PrP (PrP), and HpL3-4-D#1 (#1) cells serum-de-prived for the indicated time were analyzed by Western blotting. Note
that cleaved caspase-3, cleaved caspase-9, and cleaved PARP were
detected in HpL3-4-EM and HpL3-4-D#1 cells at 12 and 24 h. Thelevels of the cleaved proteins were decreased in HpL3-4-PrP cells and
increased in HpL3-4-D#1 cells compared with HpL3-4-EM cells. (B–
C) LEHD (B) and DEVD (C) cleavage activity in cell lysates obtained
from serum-deprived cultures of HpL3-4-EM, HpL3-4-PrP, and
HpL3-4-D#1 cells. DRFU is equivalent to the change in relative fluo-rescent units (RFU) per minute (100�). One asterisk (*) and two as-terisks (**) denote a statistically significant difference compared to
HpL3-4-EM cells serum-deprived for 24 h [p < 0:05 (*) and p < 0:01
(**)].
664 A. Sakudo et al. / Biochemical and Biophysical Research Communications 308 (2003) 660–667
HpL3-4-D#1 cells following serum deprivation for 12 h
and 24 h (Fig. 5A). HpL3-4-D#1 cells showed enhancedcleavage of caspase-3 and caspase-9 when compared
with HpL3-4-EM cells (Fig. 5A). In contrast, active
cleaved forms of caspase-3 and caspase-9 were not ob-
served in HpL3-4-PrP cells until 24 h (Fig. 5A). Con-firming these observations, PARP fragment (89 kDa)
cleaved by active caspase-3 was detected in HpL3-4-EM
and HpL3-4-D#1 cells at 12 and 24 h, while increasedlevel of cleaved PARP was observed in HpL3-4-D#1cells compared with HpL3-4-EM cells at 12 and 24 h
(Fig. 5A). On the other hand, reduced level of cleaved
PARP fragment was observed in HpL3-4-PrP cells
compared with HpL3-4-EM cells at 12 and 24 h(Fig. 5A). Next, to further confirm the relationship be-
tween caspase-3/9 activity and full-length or N-termi-
nally truncated PrPC, caspase activity assay was
performed. HpL3-4-D#1 cells showed significantly highactivity of DEVD cleavage activity, which represents
caspase-3/7 activity [30], and LEHD cleavage activity,
which represents caspase-9 activity [30], compared with
HpL3-4-EM cells under serum deprivation for 24 h
(Figs. 5B and C). On the other hand, HpL3-4-PrP cells
showed significantly low activity of DEVD and LEHD
cleavage activity compared with HpL3-4-EM cells (Figs.
5B and C).
Discussion
Balancing between the function of ROS and the anti-
oxidant defense mechanism is thought to be essential for
the regulation and prevention of apoptosis. One efficient

A. Sakudo et al. / Biochemical and Biophysical Research Communications 308 (2003) 660–667 665
anti-oxidant enzyme in neurons is SOD. This enzymeconverts superoxide anion radicals into H2O2. Accord-
ing to Brown et al. [19], the brains of Prnp�=� mice have
reduced the activity of cytosolic Cu/Zn-SOD. Further-
more, PrPC per se elicits SOD activities. The N-terminal
octapeptide repeat region has been shown to bind to
copper and is involved in the enzymatic function of PrPC
[29]. These studies suggest that PrPC plays a role in
cellular resistance to oxidative stress. However, a re-cently conflicting report has demonstrated that the
amount of brain copper and the activity of cuproen-
zymes, such as SOD and cytochrome C oxidase, do not
differ with the PrPC expression level [20]. More recent
papers demonstrated that PrPC overexpression increased
resistance to hydrogen peroxide, SIN-1 or copper in
induced toxicity in RK13 or SH-SY5Y cell lines possibly
due to increase of antioxidant enzyme activity [23,24].However, since RK13 and SH-SY5Y cells have endog-
enous Prnp, the effect of endogenous PrPC could not be
excluded from the explanation. Therefore, further
studies using PrP-deficient immortalized neuronal cells
are warranted to fully elucidate the relationship between
PrPC and SOD activity. The present study analyzed this
relationship using PrP-deficient neuronal cells; Prnp�=�
neuronal cells were apoptotic after serum deprivation,and re-introduction of Prnp upregulated SOD activity,
suggesting that PrPC inhibited apoptosis by upregula-
tion of SOD activity. Moreover, PrPC did not change
Bcl-2, Bcl-xL, and Cu/Zn SOD expression levels, sug-
gesting that PrPC inhibits apoptosis regardless of Bcl-2,
Bcl-xL and Cu/Zn SOD expression levels. Furthermore,
as transfection of SOD-1, which encodes Cu/Zn SOD,
but not treatment of Cu/Zn SOD inhibited apoptosis inPrnp�=� cells (data not shown), superoxide anion gen-
erated within an individual cells and acting inside cells is
an important event in apoptosis. PrPC expression in
Prnp�=� cells resulted in effective elimination of super-
oxide anion and H2O2 accumulation under serum de-
privation, suggesting that SOD activated by PrPC
efficiently converts superoxide anion into H2O2. These
data support the notion that PrPC acts to regulate SODactivity and superoxide anion elimination in an inde-
pendent cell.
The membrane fraction of WT mouse brain, where
PrPC is highly expressed, manifested a higher level of
SOD activity than those of other membrane fractions of
tissues such as WT and Prnp�=� mouse testis and
Prnp�=� mouse brain, suggesting indeed that PrPC per se
elicits SOD activity. In addition, the soluble fraction ofWT mouse brain and testis, where cytosolic Cu/Zn-SOD
exists, showed higher SOD activity compared with
Prnp�=� mouse brain and Prnp�=� mouse testis, respec-
tively, suggesting that low levels of PrPC are necessary
for the activation of cytosolic Cu/Zn-SOD because ex-
pression of PrPC in testis is low but sufficient for stabi-
lizing SOD activity. Furthermore, re-introduction of
Prnp upregulated SOD activity without changing Cu/Zn-SOD expression levels. It has been reported that copper
binding promotes PrPC endocytosis [31]. N-terminal
deletion mutants of chicken PrPC show diminished en-
docytosis [32]. These suggest that PrPC upregulates SOD
activity by incorporation of copper into Cu/Zn-SOD via
the N-terminal domain of PrPC. Zeng et al. [24] recently
reported that substitution of signal peptide sequence of
PrPC for the uncleaved signal sequence and transmem-brane sequence compromises cellular SOD activity. In-
terestingly, our studies indicated that PrPC lacking an
octapeptide repeat region accelerates apoptosis and in-
hibits SOD activity. As the N-terminal deletion mutant
of PrPC enhanced caspase-3/9-mediated pathway, it is
suggested that the toxicity of the deletion mutant of PrPC
functions in the upstream of caspase-3/9 cascade. It is
consistent with the recent report that overexpression ofPrPC suppresses cell death induced by Bax-overexpres-
sion, but PrPC lacking an octapeptide repeat region does
not enhance Bax-induced cell death in human primary
culture neurons [33]. Taken together with our studies, the
toxicity of PrPC lacking an octapeptide repeat region
functions in the upstream of caspase-3/9 cascade at least
in part by inhibition of SOD activity. As the N-termi-
nally truncated PrPC decreased SOD activity, these re-sults raise the possibility that some factors, which
interact with PrPC, may mediate copper transfer from
PrPC to Cu/Zn-SOD, and the existence of the octapep-
tide repeat region of PrPC may be indispensable for
copper transfer and regulation of SOD activity. Stress-
inducible protein 1 (STI1) is a candidate for involvement
in such an interaction with PrPC [34]. STI1, which me-
diates PrPC dependent neuroprotective signals, interactswith amino acids 113–128 of PrPC [34]. However, to test
the possibility, further study to investigate the functional
interaction of PrPC and copper with STI1 or unknown
factors would be necessary. Overall, the present study
supports the notion that PrPC aids the elimination of
superoxide anion in an independent cell and N-terminal
octapeptide repeat region of PrPC is essential for its
regulation of apoptosis and SOD activity.The present study showed that serum deprivation
caused apoptosis of Prnp�=� cells via intracellular su-
peroxide production and caspase-3/9 activation, and
PrPC suppressed apoptosis, at least in part, by upregu-
lation of SOD activity and inhibition of caspase-3/9
activation via N-terminal octapeptide repeat region of
PrPC. These results advocate the fact that superoxide
anion production and caspase-3/9 activation are criticalevents in the apoptosis of Prnp�=� cells under serum
deprivation, and the octapeptide repeat region of PrPC
essentially contributes to regulation of SOD activity,
superoxide elimination, and inhibition of caspase-3/9
activation. When animals are infected with prions, PrPSc
is converted from PrPC and accumulates. The conver-
sion of PrPC to PrPSc leads to PrPC deficiency [11].

666 A. Sakudo et al. / Biochemical and Biophysical Research Communications 308 (2003) 660–667
Our studies suggest that neuronal cells are susceptible tooxidative stress due to reduced SOD activity and cas-
pase-3/9 activation induced by PrPC deficiency. These
findings suggest that not only accumulation of PrPSc but
also deficiency of PrPC are crucial events in the
neurodegeneration of prion diseases.
Acknowledgments
The authors are grateful to Dr. Yoshio Yamakawa (Department of
Biochemistry and Cell Biology, National Institute of Infectious Dis-
eases) for the gift of anti-prion protein P8. Thanks are due to Dr. An-
thony Foong for reading the manuscript. This work was supported by
the Sasakawa Scientific Research Grant from The Japan Science Soci-
ety (to A.S.) and aGrant-in-Aid for Science Research from theMinistry
of Education, Science, Culture and Technology of Japan (to T.O.).
References
[1] S.B. Prusiner, Prions, Proc. Natl. Acad. Sci. USA 95 (1998)
13363–13383.
[2] P.E. Bendheim, H.R. Brown, R.D. Rudelli, L.J. Scala, N.L.
Goller, G.Y. Wen, R.J. Kascsak, N.R. Cashman, D.C. Bolton,
Nearly ubiquitous tissue distribution of the scrapie agent precur-
sor protein, Neurology 42 (1992) 149–156.
[3] H. Bueler, M. Fischer, Y. Lang, H. Bluethmann, H.P. Lipp, S.J.
DeArmond, S.B. Prusiner, M. Aguet, C. Weissmann, Normal
development and behaviour of mice lacking the neuronal cell-
surface PrP protein, Nature 356 (1992) 577–582.
[4] H. Bueler, A. Aguzzi, A. Sailer, R.A. Greiner, P. Autenried, M.
Aguet, C. Weissmann, Mice devoid of PrP are resistant to scrapie,
Cell 73 (1993) 1339–1347.
[5] S. Brandner, S. Isenmann, A. Raeber, M. Fischer, A. Sailer, Y.
Kobayashi, S. Marino, C. Weissmann, A. Aguzzi, Normal host
prion protein necessary for scrapie-induced neurotoxicity, Nature
379 (1996) 339–343.
[6] A. Giese, D.R. Brown, M.H. Groschup, C. Feldmann, I. Haist,
H.A. Kretzschmar, Role of microglia in neuronal cell death in
prion disease, Brain Pathol. 8 (1998) 449–457.
[7] D. Rossi, A. Cozzio, E. Flechsig, M.A. Klein, T. Rulicke, A.
Aguzzi, C. Weissmann, Onset of ataxia and Purkinje cell loss in
PrP null mice inversely correlated with Dpl level in brain, EMBO
J. 20 (2001) 694–702.
[8] J.C. Manson, A.R. Clarke, M.L. Hooper, L. Aitchison, I.
McConnell, J. Hope, 129/Ola mice carrying a null mutation in
PrP that abolishes mRNA production are developmentally
normal, Mol. Neurobiol. 8 (1994) 121–127.
[9] S. Sakaguchi, S. Katamine, N. Nishida, R. Moriuchi, K.
Shigematsu, T. Sugimoto, A. Nakatani, Y. Kataoka, T. Houtani,
S. Shirabe, H. Okada, S. Hasegawa, T. Miyamoto, T. Noda, Loss
of cerebellar Purkinje cells in aged mice homozygous for a
disrupted PrP gene, Nature 380 (1996) 528–531.
[10] R.C. Moore, I.Y. Lee, G.L. Silverman, P.M. Harrison, R. Strome,
C. Heinrich, A. Karunaratne, S.H. Pasternak, M.A. Chishti, Y.
Liang, P. Mastrangelo, K. Wang, A.F. Smit, S. Katamine, G.A.
Carlson, F.E. Cohen, S.B. Prusiner, D.W. Melton, P. Tremblay,
L.E. Hood, D. Westaway, Ataxia in prion protein (PrP)-deficient
mice is associated with upregulation of the novel PrP-like protein
doppel, J. Mol. Biol. 292 (1999) 797–817.
[11] T. Yokoyama, K.M. Kimura, Y. Ushiki, S. Yamada, A.
Morooka, T. Nakashiba, T. Sassa, S. Itohara, In vivo conversion
of cellular prion protein to pathogenic isoforms, as monitored by
conformation-specific antibodies, J. Biol. Chem. 276 (2001)
11265–11271.
[12] I. Tobler, S.E. Gaus, T. Deboer, P. Achermann, M. Fischer, T.
Rulicke, M. Moser, B. Oesch, P.A. McBride, J.C. Manson,
Altered circadian activity rhythms and sleep in mice devoid of
prion protein, Nature 380 (1996) 639–642.
[13] J. Collinge, M.A. Whittington, K.C. Sidle, C.J. Smith, M.S.
Palmer, A.R. Clarke, J.G. Jefferys, Prion protein is necessary for
normal synaptic function, Nature 370 (1994) 295–297.
[14] J.W. Herms, H.A. Kretzchmar, S. Titz, B.U. Keller, Patch–clamp
analysis of synaptic transmission to cerebellar Purkinje cells of
prion protein knockout mice, Eur. J. Neurosci. 7 (1995) 2508–2512.
[15] P.M. Lledo, P. Tremblay, S.J. DeArmond, S.B. Prusiner, R.A.
Nicoll, Mice deficient for prion protein exhibit normal neuronal
excitability and synaptic transmission in the hippocampus, Proc.
Natl. Acad. Sci. USA 93 (1996) 2403–2407.
[16] C. Kuwahara, A.M. Takeuchi, T. Nishimura, K. Haraguchi, A.
Kubosaki, Y. Matsumoto, K. Saeki, T. Yokoyama, S. Itohara, T.
Onodera, Prions prevent neuronal cell-line death, Nature 400
(1999) 225–226.
[17] C. Borner, The Bcl-2 protein family: sensors and checkpoints for
life- or-death decisions, Mol. Immunol. 39 (2003) 615–647.
[18] V. Cryns, J. Yuan, Proteases to die for, Genes Dev. 12 (1998)
1551–1570.
[19] D.R. Brown, W.J. Schulz-Schaeffer, B. Schmidt, H.A. Kretzsch-
mar, Prion protein-deficient cells show altered response to
oxidative stress due to decreased SOD-1 activity, Exp. Neurol.
146 (1997) 104–112.
[20] D.J. Waggoner, B. Drisaldi, T.B. Bartnikas, R.L. Casareno, J.R.
Prohaska, J.D. Gitlin, D.A. Harris, Brain copper content and
cuproenzyme activity do not vary with prion protein expression
level, J. Biol. Chem. 275 (2000) 7455–7458.
[21] B.S. Wong, T. Pan, T. Liu, R. Li, P. Gambetti, M.S. Sy,
Differential contribution of superoxide dismutase activity by prion
protein in vivo, Biochem. Biophys. Res. Commun. 273 (2000)
136–139.
[22] F. Klamt, F. Dal-Pizzol, M.J. Conte da Frota, R. Walz, M.E.
Andrades, E.G. da Silva, R.R. Brentani, I. Izquierdo, J.C.
Fonseca Moreira, Imbalance of antioxidant defense in mice
lacking cellular prion protein, Free Radic. Biol. Med. 30 (2001)
1137–1144.
[23] W. Rachidi, D. Vilette, P. Guiraud, M. Arlotto, J. Riondel, H.
Laude, S. Lehmann, A. Favier, Expression of prion protein
increases cellular copper binding and antioxidant enzyme activ-
ities but not copper delivery, J. Biol. Chem. 278 (2003) 9064–9072.
[24] F. Zeng, N.T. Watt, A.R. Walmsley, N.M. Hooper, Tethering the
N-terminus of the prion protein compromises the cellular response
to oxidative stress, J. Neurochem. 84 (2003) 480–490.
[25] M. Kozak, At least six nucleotides preceding the AUG initiator
codon enhance translation in mammalian cells, J. Mol. Biol. 196
(1987) 947–950.
[26] P.K. Narayanan, E.H. Goodwin, B.E. Lehnert, Alpha particles
initiate biological production of superoxide anions and hydrogen
peroxide in human cells, Cancer Res. 57 (1997) 3963–3971.
[27] A. Imrich, Y.Y. Ning, L. Kobzik, Intracellular oxidant produc-
tion and cytokine responses in lung macrophages: evaluation of
fluorescent probes, J. Leukoc. Biol. 65 (1999) 499–507.
[28] A.L. Johnson, J.T. Bridgham, T. Jensen, Bcl-X(LONG) protein
expression and phosphorylation in granulosa cells, Endocrinology
140 (1999) 4521–4529.
[29] D.R. Brown, B.S. Wong, F. Hafiz, C. Clive, S.J. Haswell, I.M.
Jones, Normal prion protein has an activity like that of superoxide
dismutase, Biochem. J. 344 (1999) 1–5.
[30] N.A. Thornberry, T.A. Rano, E.P. Peterson, D.M. Rasper, T.
Timkey, M. Garcia-Calvo, V.M. Houtzager, P.A. Nordstrom, S.
Roy, J.P. Vaillancourt, K.T. Chapman, D.W. Nicholson, A
combinatorial approach defines specificities of members of the

A. Sakudo et al. / Biochemical and Biophysical Research Communications 308 (2003) 660–667 667
caspase family and granzyme B. Functional relationships estab-
lished for key mediators of apoptosis, J. Biol. Chem. 272 (1997)
17907–17911.
[31] P.C. Pauly, D.A. Harris, Copper stimulates endocytosis of the
prion protein, J. Biol. Chem. 273 (1998) 33107–33110.
[32] S.L. Shyng, K.L. Moulder, A. Lesko, D.A. Harris, The N-
terminal domain of a glycolipid-anchored prion protein is
essential for its endocytosis via clathrin-coated pits, J. Biol.
Chem. 270 (1995) 14793–14800.
[33] Y. Bounhar, Y. Zhang, C.G. Goodyer, A. LeBlanc, Prion protein
protects human neurons against Bax-mediated apoptosis, J. Biol.
Chem. 276 (2001) 39145–39149.
[34] S.M. Zanata, M.H. Lopes, A.F. Mercadante, G.N. Hajj, L.B.
Chiarini, R. Nomizo, A.R. Freitas, A.L. Cabral, K.S. Lee, M.A.
Juliano, E. de Oliveira, S.G. Jachieri, A. Burlingame, L. Huang,
R. Linden, R.R. Brentani, V.R. Martins, Stress-inducible protein
1 is a cell surface ligand for cellular prion that triggers neuropro-
tection, EMBO J. 21 (2002) 3307–3316.
Copyright © 2022 FDOKUMEN




















