
The cell coat of the developing olfactory epithelium in the chick

lable at ScienceDirect
Biomaterials 35 (2014) 9199e9207
Contents lists avai
Biomaterials
journal homepage: www.elsevier .com/locate/biomater ia ls
The impact of nanoparticles on the mucosal translocationand transport of GLP-1 across the intestinal epithelium
Francisca Araújo a, b, c, Neha Shrestha c, Mohammed-Ali Shahbazi c, Pedro Fonte d, e,Ermei M. M€akil€a c, f, Jarno J. Salonen f, Jouni T. Hirvonen c, Pedro L. Granja a, b,H�elder A. Santos c, *, Bruno Sarmento a, d, **
a INEB e Instituto de Engenharia Biom�edica, University of Porto, Rua do Campo Alegre, 823, 4150-180 Porto, Portugalb ICBAS e Instituto Ciencias Biom�edicas Abel Salazar, University of Porto, Rua de Jorge Viterbo Ferreira, 4050-313 Porto, Portugalc Division of Pharmaceutical Chemistry and Technology, Faculty of Pharmacy, University of Helsinki, FI-00014 Helsinki, Finlandd INFACTS e Instituto de Investigaç~ao e Formaç~ao Avançada em Ciencias e Tecnologias da Saúde, Instituto Superior de Ciencias da Saúde-Norte,Department of Pharmaceutical Sciences, CESPU, Rua Central de Gandra, 1317, 4585-116 Gandra, Portugale REQUIMTE, Department of Chemical Sciences - Applied Chemistry Lab, Faculty of Pharmacy, University of Porto, Rua de Jorge Viterbo Ferreira,4050-313 Porto, Portugalf Laboratory of Industrial Physics, Department of Physics and Astronomy, FI-20014 University of Turku, Finland
a r t i c l e i n f o
Article history:Received 18 June 2014Accepted 19 July 2014Available online 7 August 2014
Keywords:Glucagon like peptide-1Oral delivery systemsNanoparticlesChitosanTriple co-cultureDiabetes
* Corresponding author. Tel.: þ358 2941 59661; fax** Corresponding author. INEB e Instituto de EngenhPorto, Rua do Campo Alegre, 823, 4150-180 Porto, Porfax: þ351 226094567.
E-mail addresses: [email protected] (H.A. Saup.pt (B. Sarmento).
http://dx.doi.org/10.1016/j.biomaterials.2014.07.0260142-9612/© 2014 Elsevier Ltd. All rights reserved.
a b s t r a c t
Glucagon like peptide-1 (GLP-1) is an incretin hormone that is in the pipeline for type 2 diabetes mellitus(T2DM) therapy. However, oral administration of GLP-1 is hindered by the harsh conditions of thegastrointestinal tract and poor bioavailability. In this study, three nanosystems composed by threedifferent biomaterials (poly(lactide-co-glycolide) polymer (PLGA), Witepsol E85 lipid (solid lipid nano-particles, SLN) and porous silicon (PSi) were developed and loaded with GLP-1 to study their perme-ability in vitro. All the nanoparticles presented a size of approximately 200 nm. The nanoparticles'interaction with the mucus and the intestinal cells were enhanced after coating with chitosan (CS). PSinanosystems presented the best association efficiency (AE) and loading degree (LD), even though a highAE was also observed for PLGA nanoparticles and SLN. Among all the nanosystems, PLGA and PSi werethe only nanoparticles able to sustain the release of GLP-1 in biological fluids when coated with CS. Thischaracteristic was also maintained when the nanosystems were in contact with the intestinal Caco-2 andHT29-MTX cell monolayers. The CS-coated PSi nanoparticles showed the highest GLP-1 permeationacross the intestinal in vitromodels. In conclusion, PLGA þ CS and PSi þ CS are promising nanocarriers forthe oral delivery of GLP-1.
© 2014 Elsevier Ltd. All rights reserved.
1. Introduction
Type 2 diabetes mellitus (T2DM) is the most prevalent meta-bolic disorder worldwide, making it the fourth leading cause ofdeath in the developed countries [1]. Currently, incretin hormonesare in the pipeline for the treatment of T2DM due to their ability topotentiate insulin secretion after meal in a glucose-dependentmanner. This incretin effect represents approximately 50e70% of
: þ358 2941 59138.aria Biom�edica, University oftugal. Tel.: þ351 226074900;
ntos), bruno.sarmento@ineb.
the total insulin secreted following oral glucose administration innormoglycemics [2,3]. Among all the incretin hormones, the 30amino acids peptide glucagon like peptide-1 (GLP-1) is the moststudied. Besides insulin production, GLP-1 also stimulates theneogenesis and proliferation of pancreatic b cells, which slows theprogression of T2DM, and also suppresses the glucagon release [4].Moreover, its insulinotropic effect is lost when glucose concentra-tion is below 77mg/dL, thus overcoming hypoglycemiawhich is themajor side effect of the currently available therapy [5,6].
Nevertheless, GLP-1 is currently administrated by parenteralroutes, which are not well accepted by patients, and also do notmimic the endogenous pathway of GLP-1 secretion [7,8]. Aiming toovercome the known disadvantages of this delivery route, attemptsto deliver GLP-1 orally have been made, specially using alternativedelivery systems to protect it from the harsh conditions of the

F. Araújo et al. / Biomaterials 35 (2014) 9199e92079200
gastrointestinal (GI) tract and prolong its release and therapeuticeffect [9e11]. However, these studies are still very preliminary andnoneof these formulationshaveprogressed towards further studies.
In this work, three different kinds of nanosystems were devel-oped in order to deliver GLP-1 orally. These nanosystems were pre-pared from three different types of materials with knownbiocompatibilityandbiodegradabilityproperties: (i) poly(lactide-co-glycolide) polymer (PLGA), (ii) lipid Witepsol E85, and (iii) poroussilicon (PSi) [12e15]. These are well known biomaterials that havebeen extensively studied in the drug delivery field. They have theability to help drugs to overcome the GI barriers and sustain and/orcontrol the drug release, thus improving the therapeutic efficiency ofthe drug and reducing unwanted side effects. Moreover, the surfacechemistry can be tailored conferring them specific properties, whichis another advantage of these materials [12,14,16e18].
Nonetheless, the successful protection of the cargos from the GIenvironment merely is not enough. The nanoparticles still have toface the protective mucus layer present in the intestine, whichmakes it difficult for the nanoparticles to approach the epithelialcells [19]. Thus, modifying the surface of the particles withmucoadhesive biomaterials, such as chitosan (CS), will promotemucus and cellular adhesion leading to the retention of the deliverysystems at the site of absorption for longer periods of time, therebyincreasing the oral bioavailability. Moreover, CS is also described asbeing able to increase the permeability of the drugs by transientlyopening the cellular tight junctions [20e22].
The aim of this work was to compare three different developednanosystems with different properties, and select the one withmost advantageous characteristics to the nanoparticles for oraldelivery. Moreover, the effect of CS adsorption on the surface ofthese nanoparticles was also studied. For that, an extensive char-acterization of the nanoparticles was made, as well as the study ofthe GLP-1 release in biological fluids performed. The interactionstudies of the nanoparticles with intestinal cell lines were alsoperformed together with the evaluation of GLP-1 permeationacross intestinal cellular monolayers.
2. Material and methods
2.1. Materials and cell lines
GLP-1wasmanufactured by United Peptides, USA. PLGA 50:50was obtained fromPurac Biomaterials, Purasorb® PDLG 5004 and Witepsol E85 from Sasol, Germany.Polyvinyl alcohol (PVA), low molecular weight CS, fluorescein isothiocyanate (FITC),4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and glutaraldehydewere purchased from SigmaeAldrich®, USA. Culture flasks and Transwell® plateswere purchased from Corning Inc., USA. Dulbecco's Modified Eagle medium (DMEM),L-glutamine, non-essential amino acids (NEAA), Penicillin (100 IU/mL), Streptomycin(100mg/mL) and trypsineEDTAwere purchased fromHyClone, USA. Hank's balancedsalt solution (HBBS) and heat inactivated fetal bovine serum (FBS) were purchasedfrom Life Technologies Gibco®, USA. Human colon carcinoma (Caco-2) and Raji B celllines were obtained from American Type Culture Collection (ATCC), USA, and HT29-MTX cell linewas kindly provided by Dr. T. Lesuffleur (INSERMU178, Villejuif, France).
2.2. Preparation of PLGA nanoparticles and SLNs
Different formulations of polymeric and lipidic nanoparticles were preparedthrough modified solvent emulsificationeevaporation method, based on the water-in-oil-in-water (w/o/w) double emulsion technique [23,24]. The polymer/lipid wasdissolved in heated ethyl acetate in a concentration of 100 mg/mL. Then, 100 mL ofGLP-1 solution in MilliQ-water (2.5 mg/mL) was added to this solution and homo-genised for 30 s using a Vibra-Cell™ ultrasonic processor (Sonics®, Sonics and Ma-terials, Inc., USA). The primary emulsion (w/o) formed was then added into 4 mL ofthe surfactant solution, 2% of PVA in Milli-Q water, and homogenised again for 30 s.The second emulsion formed (w/o/w) was finally added to 7.5 mL of the samesurfactant solution and was left under magnetic stirring at 300 rpm for at least 3 hfor organic solvent evaporation.
CS solutionwas prepared by dissolving CS in deionizedwater containing 1% (v/v)acetic acid overnight undermagnetic stirringwith pH adjustment to 5.5, followed byfiltration through a paper filter Millipore #2 and stored at 4 �C. To coat the nano-particles with CS, the formulation was added into a solution of CS, at a ratio of 5:1(w/w) regarding the solid content of the solutions, and left overnight under
magnetic stirring, allowing surface layer deposition of the polymer on the surface ofthe particles.
Nanoparticles were washed three times in MilliQ-water and separated throughcentrifugation, using an ultra-centrifuge (Optima™ TL, Beckman Coulter, USA), at34,300 g for 20min for PLGA nanoparticles and 250,000 g for 150min for SLNs. Aftercentrifugation, the nanoparticles were re-dispersed in MilliQ-water and stored at4 �C for further analysis.
Drug-free nanoparticles were prepared according to this procedure but usingMilliQ-water instead of GLP-1 solution as internal aqueous solution. In order tovisualize the nanoparticles, FITC was also loaded into the nanoparticles according tothe procedure described above, but using FITC-solution instead of GLP-1 solution.
2.3. Preparation of the porous silicon (PSi) nanoparticles
Free-standing multilayer PSi films were anodized using single crystal Si wa-fers ⟨100⟩ of pþ-type with resistivity values of 0.01e0.02 Ucm in hydrofluoric acid(40%)eethanol mixture. The anodization profile consisted of successive low, highand zero current pulses applied on the Si wafer. Hydrocarbonization treatment forthe multilayer films consisted of exposing the particles to a 1:1 (v/v) flow of N2 andacetylene for 15 min at room temperature, followed by 15 min at 500 �C, and thencooling down to room temperature under N2 flush. The obtained thermally hydro-carbonized PSi (THCPSi) was then modified with 10-undecenoic acid for 16 h at120 �C to obtain partially carboxylic acid terminated UnTHCPSi. Wet ball milling wasused to reduce the size of the modified PSi films, while the surface oxidation wasminimized by using a 5 vol-% 10-undecenoic acide dodecane solution as the millingliquid. The final size separation and exchange of the suspension media were done bycentrifugation [13,25].
For theGLP-1 loading, a ratio of 1:5w/w (177mgofGLP-1:883mgofUnTHCPSi)wasused for GLP-1-loaded UnTHCPSi nanoparticles. The GLP-1 loading was performed bydispersing the UnTHCPSi nanoparticles in GLP-1 solution and stirring at room tem-perature for 90min at 300 rpm. After that, the suspensionwas centrifuged at 27,600 gfor 5 min to remove the excess free GLP-1. To coat the nanoparticles with CS throughphysical adsorption, the nanoparticleswerefirst centrifuged at 27,600 g for 5min, andthen mixed with a solution of CS at a ratio of 5:1 (w/w) regarding the solid content ofthe solutions, and left overnight under magnetic stirring at 300 rpm. For the nano-particlescoatedbyphysicaladsorptionwithCS, theGLP-1 loadingwasperformedpriorto the coating in order to prevent the blockage of the nanoparticles' pores by CS [26].
2.4. Nanoparticles characterization
After production, the nanoparticles were characterized for their average particlesize (Z-average), polydispersity index (PdI) and average zetaepotential by electro-phorethic light scattering using a Malvern Zetasizer Nano ZS instrument (MalvernInstruments Ltd, UK). For thesemeasurements, sampleswerediluted inMilliQ-water.
To characterize nanoparticles' morphology and confirm their size distribution,the nanoparticles were observed by transmission electron microscopy (TEM, Tec-nai™ F12, FEI Company, USA) using an acceleration voltage of 120 kV. For samplepreparation, nanoparticles were diluted in MilliQ-water, placed on copper coatedgrids, and contrasted with uranyl acetate. In each step, the excess of water wasremoved using a filter paper, and finally, grids were left at room temperature for afew min to dry before the imaging.
2.5. Association efficiency (AE) and loading degree (LD)
To determine the AE and LD of the developed nanosystems, the amount of GLP-1associated to the nanoparticles was calculated. This calculation was made by thedifference between the total amount of GLP-1 used to prepare the systems, and theamount of GLP-1 that remained in the aqueous phase after nanoparticles isolation bycentrifugation, according to the follow equations [27,28]:
AE�%�
¼ ðInitial mass of GLP-1Þ � ðmass of GLP-1 in supernatantÞTotal mass of GLP-1
� 100
LD %ð Þ ¼ Initial mass of GLP-1ð Þ � mass of GLP-1 in supernatantð ÞTotal mass of nanoparticles
� 100
The amount of GLP-1 was determined by HPLC (Agilent 1260, Agilent Technol-ogies, USA), using a C18 column (4.6 � 150 mm, 5 mm, Supelco Discovery® C18, USA).Themobile phase consisted of acetonitrile and 0.1% trifluoroacetic acid initially set atthe ratio of 30:70 (v/v), which linearly changed to 40:60 (v/v) gradient over 10 min.The flow rate was 1.0 mL/min and the injected volume of the sample was 75 mL. Thecolumn temperature was set at room temperature and the detection wavelength at240 nm. The total area under the curve (AUC) was used to quantify GLP-1.
2.6. In vitro release tests
The GLP-1 loaded nanoparticles (corresponding to 30 mg of GLP-1) were added to20 mL of pH 1.2 buffer (50 mM KCl) to simulate the gastric fluid. After 2 h, the nano-particles were centrifuged and introduced in a fasted state stimulated intestinal fluid(FaSSIF) (50 mM KH2PO4, 15 mM NaOH, 1.0% (w/v) pancreatin; pH 6.5) for more 4 h.

F. Araújo et al. / Biomaterials 35 (2014) 9199e9207 9201
Aliquots of 750 mLwere collected at specific time points (0.5,1, 2, 3, 4, 5 and6 h) duringthe dissolution experiments and the withdrawn volume was replaced with freshpre-heated buffer, keeping the volume constant. All the collected aliquots werecentrifuged at 27,600 g for 20 min and the supernatant was used for HPLC analysis inorder to quantify the GLP-1 released from the nanoparticles over time. All the testswere performed at 37 �C and 100 rpm under sink conditions.
2.7. Cell culturing
Caco-2 (passage #31�40) and HT29-MTX (passage #15e30) cells grewseparately in culture flasks in a complete medium consisting of DMEM supple-mented with 10% (v/v) FBS, 1% (v/v) L-glutamine, 1% (v/v) NEAA, and 1% (v/v)antibioticeantimitotic mixture (final concentration of 100 U/ml Penicillin and100 U/ml Streptomycin). Cells were sub-cultured once a week using 0.25%trypsineEDTA (1�) to detach the cells from the flasks and seeded at a density of0.5 � 106 cells per 75 cm2
flask. The culture medium was replaced every otherday. Cells were maintained in an incubator (BB 16 gas incubator, Heraeus In-struments GmbH) at 37 �C and 5% CO2 and 95% relative humidity. Raji B cells(passage #26e35) were cultured in flasks with DMEM supplemented and at thesame conditions as described above.
2.8. Cell viability studies
For the viability tests, 100 mL of Caco-2 and HT29-MTX cells at 0.5 �106 cells/mLwere seeded separately in 96-well plates and were allowed to attach for 24 h. Afterthat, the medium was aspirated and the wells were washed twice with 100 mL offresh pre-heated HBSSeHEPES buffer (pH 7.4). After washing, 100 mL of the nano-particle solutions corresponding to GLP-1 amounts of 1.5, 3.75, 7.5 and 15 mg/mL ofGLP-1 were added to each well and the plates were incubated for a period of 3 and12 h at 37 �C. Afterwards, the plates were equilibrated at room temperature forabout 30 min and then washed twice with 100 mL of HBSS�HEPES (pH 7.4). About50 mL of the reagent assay CellTiter-Glo® (Promega Corporation, USA) were added to50 mL of HBSS�HEPES (pH 7.4) in each well; negative (HBSS buffer) and positive (1%Triton X-100) control wells were also used and treated similarly as described above.The plate was mixed for 10 min on an orbital shaker at room temperature, protectedfrom the light. Finally, the luminescence was measured using Varioskan Flash platereader (Thermo Fisher Scientific Inc., USA). The assay is based on the amount of ATPproduced by metabolically active cells, which is directly proportional to the numberof living cells present in the culture [29]. All data sets were compared with anegative control of HBSSeHEPES (pH 7.4), which was considered as 100% viability.
2.9. Celleparticle interactions
To verify the interactions between cells andnanoparticles, Caco-2:HT29-MTX co-cultures in the proportion of 90:10were seeded in Lab-Tek 8-chamber slides (ThermoFisher Scientific Inc., USA) and allowed to attach for 24 h. Afterwards, cells werewashed twice with fresh pre-heated HBSS�HEPES buffer (pH 7.4). Then, FITC-loadednanoparticleswere added to the cells and incubated at 37 �C for 3 h. After incubation,cells were washed twice with fresh pre-heated HBSS�HEPES buffer (pH 7.4). Then,the plasma membrane was stained by adding 200 mL of CellMask™ Orange (Invi-trogen, USA) and incubated for 3 min at 37 �C. The excess of staining solution waswashed twice with fresh HBSS�HEPES buffer and cells were fixed using 2.5%glutaraldehyde for 20 min. The localization of the FITC-loaded nanoparticles wasthen observedusing a Leica SP5 confocalmicroscope (LeicaMicrosystems, Germany).
2.10. Permeability experiments
For the permeability experiments, 7 � 104 cells/cm2 of Caco-2 and HT29-MTXcells in a ratio of 90:10 were seeded in 12-Transwell® cell culture inserts and wereallowed to grow and differentiate for 14 days with medium replacement everyother day. Then, 1.0 � 105 Raji B cells were added to the basolateral chamber for 7days more, in order to induce the phenotype change of Caco-2 cells into M cellsand to obtain a triple co-culture model [30]. The permeability experiments acrossthe cell monolayers were performed in the apical-to-basolateral direction inHBSS�HEPES at pH 6.5 (apical compartment) and pH 7.4 (basolateral compart-ment) at 37 �C using an orbital shaker (100 rpm). After removing the cell culturemedium, 0.5 mL of pure GLP-1 or nanoparticles in the concentration of 2 mg/mLwere pipetted into the apical side of the inserts. At different time points (0.5, 1, 2and 3 h), 0.75 mL samples were taken from the basolateral side of the inserts andthe same volume of fresh HBSS�HEPES (pH 7.4) buffer was added to replace thewithdrawn volume. Sample concentrations were quantified by EIA GLP-1 Kit(SigmaeAldrich®, USA) according to the manufacturer's instructions. The integrityof the cell monolayers was checked before and after the permeability experimentby measuring the transepithelial electric resistance (TEER) using Millicell®-Elec-trical Resistance System (Millipore, USA).
2.11. Morphological characterization of the coeculture monolayers
After the drug permeation studies, the nanoparticles suspension was carefullyremoved and cells were fixed with 2.5% glutaraldehyde for 20 min at room tem-perature. The wells were then washed twice with sodium cacodylate buffer (NaCac)
for 3 min. Afterwards, the cells were post-fixed with 1% osmium tetroxide in 0.1 M
NaCac buffer (pH 7.4) and then dehydrated and embedded in epoxy resin. Ultrathinsections (60 nm) were cut perpendicular to the insert, post-stained with uranylacetate and examined with TEM using an acceleration voltage of 120 kV.
2.12. Statistical analysis
All the experiments were performed in triplicate and are represented asmean ± standard deviation (SD). A one-way analysis of variance (ANOVA) withBonferroni post hoc-test (GraphPadPrism, GraphPad software Inc., CA, USA) wasused to analyse the data. The level of significancewas set at probabilities of *p < 0.05,**p < 0.01, and ***p < 0.001.
3. Results and discussion
3.1. Characterization of the nanoparticles
Nanoparticles were developed and characterized on the basis oftheir particle size, PdI and zeta-potential, as shown in Table 1. All ofthe uncoated nanoparticles presented average sizes of approxi-mately 200 nm with low PdI values (�0.12), suggesting the mon-odispersity of the nanosystems. TEM images supported theseresults in terms of mean size and their homogenous distribution(Fig. 1 and Fig. S1). From the TEM images, it can also be observedthat SLN and PLGA nanoparticles have spherical shape, in contrastwith the irregular shape of the UnTHCPSi nanoparticles. The shapeof the nanoparticles has also been described as having an impact ontheir interaction at the cellular level [31], wherein usually round-shaped nanoparticles are taken up more easily than irregularones with sharped edges [32]. Regarding the surface charge, it wasobserved that all of the uncoated nanoparticles are negativelycharged due to the nature of the biomaterials used. However, thenanoparticles with negative charge would lead to electrostaticrepulsionwith the negatively charged mucus layer on the intestinalwall (�50 mV), thereby inhibiting the nanoparticles to move closerto the intestinal epithelial wall [18]. Thus, to improve the interac-tion of the nanosystems with the intestinal epithelia, the nano-particles were then coated with CS, a positively chargedmucoadhesive polymer. The inheritance of the positive charge fromCS to the nanoparticles enhances the interactions with the nega-tively charged mucus and cells, which can lead to the improvementin the permeability and/or cellular internalization of the cells[18,33]. Moreover, CS is described as being an efficient intestinalabsorption enhancer since it is able to transiently open the tightjunctions between the epithelial cells [21,34]. The successful sur-face modification of the nanoparticles with CS was observed by thesignificant change in the zeta-potential and by the significant in-crease in the particle size, as shown in Table 1. These changes are inaccordance with other studies, in which the modification of thenanoparticles with CS were also performed [18,33].
3.2. Association efficiency (AE) and loading degree (LD)
UnTHCPSi nanoparticles, coated and uncoated with CS, pre-sented the highest AE with 85 ± 1% of the initial peptide associatedto the nanoparticles (Table 1). For PLGA nanoparticles, the AE ofGLP-1 was 67 ± 11% which was in the same range of values as CScoating (PLGA þ CS) with 60 ± 1% of AE (p > 0.05). In turn, SLN hadsignificantly higher AE than CS-coated SLN nanoparticles(SLN þ CS), with values of 73 ± 1% and 57 ± 6%, respectively. Thesereductions in AE for the CS-coated nanoparticles may be due toan additional step of mixing of the nanoparticles with the CSovernight, which could have led to some release of the drug [33].Taking into account that GLP-1 is a hydrophilic peptide, the AEobtained with these systems seemed to be higher than the usualvalues obtained for other hydrophilic peptides such as insulin[14,18,35,36]. This could be explained by the production method

F. Araújo et al. / Biomaterials 35 (2014) 9199e92079202
used here and the use of ethyl acetate as organic solvent, which isknown to enhance the rate of encapsulation of hydrophilic mole-cules [37].
Regarding the LD, UnTHCPSi nanoparticles also have the highestLD of 17%, which is 100 times higher than that of PLGA and SLNnanoparticles. Regarding the CS-coated nanoparticles it wasobserved that the LD values were slightly lower with no significantchanges when compared with the uncoated nanoparticles, exceptin UnTHCPSi where LD remained the same (Table 1).
Such differences in the AE and LD of PLGA and SLN in compar-ison with UnTHCPSi are explained with the methods used in theirproduction. With the UnTHCPSi nanoparticles, the payload will beretained in the pores of the nanoparticles, generally by physicaladsorption. The electrostatic interactions towards the payload willbe the main factors contributing for its incorporation and retentionin the nanoparticles [38,39]. However, for the PLGA and SLNnanosystems, the payload needs to be entrapped into the nano-particles. During the production of these nanoparticles, severaldifferent stages of the double emulsionmethod are involved, whichincreases the probability of loss of the peptide [38,40].
3.3. Cell viability studies
The in vitro cell viability experiments were performed using twodifferent intestinal cell lines, Caco-2 and HT29-MTX. As Caco-2 cellsresemble enterocytes that represent approximately 90% of the totalepithelial cells of the intestine and HT29-MTX mimic the gobletcells that are approximately 10% of intestinal cells [30], these celllines are suitable in vitro models to mimic the intestinal epithelia[30,41,42]. Cells viability was measured after exposure of thenanoparticles to the cells at different concentrations and incubationperiods. The aim of using different concentrations is to understandif there is any concentration dependent toxicity and to evaluate thelowest safe concentration of the nanoparticles that can be admin-istrated. Two different incubation periods, 3 and 12 h, were testedbecause they represent the average and maximum transit time inthe intestinal tract, respectively [43]. The results are presented inFig. 2.
For Caco-2 cells, it can be observed that the viability of the cellswas about 100% for all concentrations tested after 3 h of exposure tothe nanoparticles (Fig. 2A). After 12 h, the cell viability decreased ingeneral for all formulations; nevertheless, all of the cells presentedcell viabilities above 80%, with the exception of the UnTHCPSinanoparticles, which presented lower cell viabilities after 12 h(Fig. 2B). Regarding HT29-MTX cells, after 3 h incubation, the cellviability for all the formulations was very similar to the control,approximately 100%, with the exception of SLN þ CS (Fig. 2C). After12 h, no significant differences to the control were found for allsamples (Fig. 2D). With HT29-MTX cells, the nanoparticles showedless toxicity compared to the Caco-2 cells at the same time point.This may be explained by the fact that the HT29-MTX cells aremucus-producing cells, and the mucus layer protects the cells frominteracting strongly with the nanoparticles, minimizing the cyto-toxicity associated with them [30,42].
Table 1Size, PdI, z-potential, association efficiency (AE) and GLP-1 loading degree (LD) of the duncoated nanoparticles were compared with their corresponding CS-coated nanoparticl
Size (nm) PDI
PLGA 182.9 ± 5.3 0.08 ± 0.02PLGA þ CS 198.4 ± 5.7* 0.11 ± 0.01SLN 217.4 ± 2.0 0.11 ± 0.02SLN þ CS 223.6 ± 1.1* 0.11 ± 0.01UnTHCPSi 196.8 ± 6.0 0.12 ± 0.02UnTHCPSi þ CS 363.0 ± 8.0* 0.23 ± 0.02
3.4. In vitro release tests
The in vitro release tests intended to predict the release profilesof GLP-1 in conditions similar to the GI tract (stomach and smallintestine). Firstly, the nanoparticles were added to pH 1.2 buffer,which mimics the gastric environment. Then, after 2 h, the releasemedium was changed to FaSSIF (pH 6.5), which simulates thetransit of the nanoparticles to the small intestine. The releaseprofiles of GLP-1 from the uncoated and CS-coated nanoparticlesare shown in Fig. 3A and B, respectively.
PLGA nanoparticles did not release significant amounts of GLP-1at pH 1.2 for 2 h. At such low pH, the charge of GLP-1 (isoelectricpoint pI¼ 5.4) is highly positive, and thus, the interactions with thenegatively charged PLGA will be very strong resulting in lowerpercentages of the peptide released [20,28,44e46]. Moreover, thePLGA nanoparticles preserved their physical integrity avoiding thenanoparticle erosion, which also contributed to the reduced GLP-1release [28,45,46]. When added to the FaSSIF, a significant releaseof 22 ± 1.3% was observed, reaching 28 ± 1.6% of GLP-1 release after6 h. This could be attributed to the increase in the pH, which resultsin a change of the charge of the peptide, thereby causing a reverseinteraction with the nanoparticles and leading to the release of thepeptide [28,45]. The biphasic release pattern of drugs from thePLGA nanoparticles has been frequently reported in the literature[17,35,47,48]. Regarding the PLGAþ CS nanoparticles, a similar GLP-1 release profile was observed as for the PLGA nanoparticles but in amore sustained pattern. The absence of release at pH 1.2 was fol-lowed with a considerable release of 14.1 ± 0.4% in the FaSSIF thatreached 21% after 6 h. The decreased release of PLGA þ CS incomparison with uncoated PLGA nanoparticles in FaSSIF can beexplained by the interaction of the non-encapsulated GLP-1. ThisGLP-1�CS interactions probably led to a better release control ofGLP-1 from the nanoparticles [18]. The results obtained are inagreement with other reports inwhich CS has been described to beable to decrease the burst release effect of the encapsulated drugs[8,49].
For UnTHCPSi and UnTHCPSi þ CS nanoparticles, burst releasesof 24.2 ± 5.2% and 11.8 ± 6.3% were, respectively, observed at pH1.2 in the first 30 min followed by a constant release plateau. InFaSSIF, a rapid release was again observed for both nanoparticles,followed by a sustained GLP-1 release for the next 4 h. At 6 h, thetotal percentage of GLP-1 released from the UnTHCPSi andUnTHCPSiþ CS were 43.3 ± 2.7% and 35.3± 6.5%, respectively. Onceagain, the CS-coated UnTHCPSi nanoparticles provided less releasethan the uncoated ones, as was also seen for the PLGA nano-particles. These results confirm that the coating of the nano-particles with CS sustained the release of GLP-1.
For the SLN nanoparticles, a significant burst release of68.0 ± 5.5% in the first 30 min was observed at pH 1.2. After thistime point, and since the GLP-1 released was in solution, the per-centage of GLP-1 was decreased over time due to its degradation,as observed by HPLC (data not shown). Even in FaSSIF, the per-centage of GLP-1 did not increase, being the GLP-1 released re-sidual when compared to the degraded GLP-1. This phenomenon
ifferent nanoparticles. Results are presented as mean ± SD (n � 3). The values ofes, *p < 0.05.
z-potential (mv) AE (%) GLP-1 LD (%)
�20.3 ± 1.0 67.0 ± 10.9 0.17 ± 0.03þ17.5 ± 1.2* 60.0 ± 0.8 0.15 ± 0.01�7.5 ± 0.3 73.3 ± 0.6 0.18 ± 0.01
þ13.4 ± 0.3* 57.0 ± 5.6* 0.14 ± 0.01�30.1 ± 2.8 85.0 ± 0.6 17.00 ± 0.05þ19.2 ± 0.4* 85.0 ± 0.5 17.00 ± 0.03

Fig. 1. TEM images of (A) PLGA nanoparticles, (B) SLN nanoparticles and (C) UnTHCPSi nanoparticles.
F. Araújo et al. / Biomaterials 35 (2014) 9199e9207 9203
was not observed in the other systems due to the controlled andsustained release of GLP-1, and thus, the amount of GLP-1 degradedis rather residual compared to the amount of GLP-1 that is released.The burst release followed by a sustained release is common in SLNsystems since hydrophilic peptides tend to accumulate at the o/winterface during preparation, thus remaining at the surface of thenanocarrier and in this case, the nanoparticles were not able tocontrol the release of the peptides [40]. Also of relevance is the factthat SLN were not as much negatively charged as the othernanoparticles tested, thus the interaction with the positivelycharged GLP-1 was not as strong as observed for the other nano-systems, resulting in a higher percentage of released GLP-1. Incontrast, SLN þ CS nanoparticles promoted a more sustainedrelease profile, with around 40% of the peptide released in the firsthour, after which the GLP-1 started to decrease. GLP-1 is releasedfrom the SLN by diffusion and also due to the degradation of thelipid matrix. When the nanosystems are unstable, their structuremay be compromised, and thus, this gives rise to a burst release ofthe drug. When the nanoparticles are coated with CS, not only thelipid matrix has an important role on the release process but alsoCS acts as a “layer” that the payload has to cross to be released fromthe nanoparticles. Therefore, the release of SLN þ CS was lowerthan the release of uncoated SLN. This is also supported by previ-ous studies that showed that under the same conditions, uncoatedSLN nanoparticles suffered 80% more degradation at 4 h than thecoated nanoparticles [40,50]. Thus, CS decreases the burst releaseeffect of the encapsulated peptides after administration and alsoincreases the stability of the nanoparticles and macromolecules[8,24,49].
Overall, the PLGA is the systemwhich could efficiently retain theGLP-1 from the harsh environment of the simulated conditions ofthe stomach without peptide release at pH 1.2, with sustained GLP-1 release thereafter. Despite the release at pH 1.2, the UnTHCPSisystems had a very similar behaviour to the PLGA systems, as thetotal percentage of GLP-1 available in FaSSIF at the end of the 6 hwas in the same range (15e20%). Regarding the SLN nanoparticles,they do not retain the peptide at low pH, causing a burst release(more than 50% of the loaded peptide) in the first 30 min at pH 1.2.Although CS coating more efficiently controlled the release fromSLN, it still showed a significantly higher GLP-1 release at pH 1.2 incomparison to the PLGA þ CS and UnTHCPSi þ CS nanoparticles.The differences between the uncoated and coated nanoparticlessuggested that CS improves the protection of the GLP-1 and itsrelease, providing a controlled drug delivery system.
3.5. Interaction of the nanoparticles with Caco-2:HT29-MTXcoeculture cells
As stated above, Caco-2 and HT29-MTX cells represent the twomost abundant cells in the intestinal epithelia. Thus, Caco-2:HT29-MTX co-culture in a 90:10 proportion is a reliable model to predictthe behaviour of the nanoparticles when in close contact with the
intestinal mucosa [30]. The interaction between the nanoparticlesand the Caco-2:HT29-MTX co-culture cells were observed withconfocal microscopy. In order to visualize the nanoparticles, theywere loaded with FITC, rendering them the green colour for easydetection. The cell membranes were stained with red using Cell-Mask™ Orange.
As it can be seen in Fig. 4 and Fig. S2, no interaction was seenbetween the cells and the uncoated nanoparticles. Regarding theCS-coated nanoparticles, it was observed that even after multiplewashes, the nanoparticles were still present in contact with thecells. In fact, therewere some placeswhere it was possible to observesome yellow colour, resulting from the overlap of the green and redlabelling, revealing a very close contact with the nanoparticles co-localized with the cell membranes. This result was expected due tothe presence of the mucoadhesive CS on the surface of the nano-particles which potentiates stronger electrostatic interactions withthe mucus present in the cellular co-culture, and also because it ispositively charged gainingmuchmore affinity to the cells'membrane[20,21].
3.6. Cell coeculture monolayers and permeability studies
To study the permeability of GLP-1 loaded into the threedifferent CS-coated nanosystems, a triple co-culture model wasused. This model comprises the most important features presentin the intestine, namely the presence of enterocytes (Caco-2 cells), the mucus-producing goblet cells (HT29-MTX) in phys-iological proportions (90:10) and the induction of M cells, asdescribed elsewhere [30]. M cells may have an important rolein intestinal absorption of drugs since they are specialized forantigen and microorganisms uptake, providing a possiblegateway for the absorption of proteins, as well as for nano-particles [51]. Together with Caco-2 and HT29-MTX cells, theyform a monolayer where cells are joined to each other by tightjunctions, mimicking the intestinal epithelia [42].
The permeability profiles of GLP-1 alone and from differentnanosystems are shown in Fig. 5. The highest GLP-1 permeation,when free in solution, may be explained with the high amount ofpeptide available during the time of the test, whereas a slower GLP-1 released from the nanoparticles was observed. Moreover, sincethese experiments were performed in HBSSeHEPES buffer tominimize the damage of the cellular monolayers and the buffer didnot present any surfactant as in FaSSIF, the GLP-1 release from thenanoparticles may have more sustained pattern than could bepredicted, thereby explaining the higher permeability of GLP-1 infree solution. Regarding the nanoparticles, the UnTHCPSi þ CSnanoparticles had the highest amount of GLP-1 permeated acrossthe intestinal cell monolayers followed by SLN þ CS nanoparticles(p> 0.05) and then by the PLGAþ CS nanoparticles (p< 0.05). Theseresults are in accordance with the data obtained for the in vitrorelease tests that showed that PLGA þ CS nanoparticles sustainedthe release of GLP-1 longer than the other nanoparticles. The
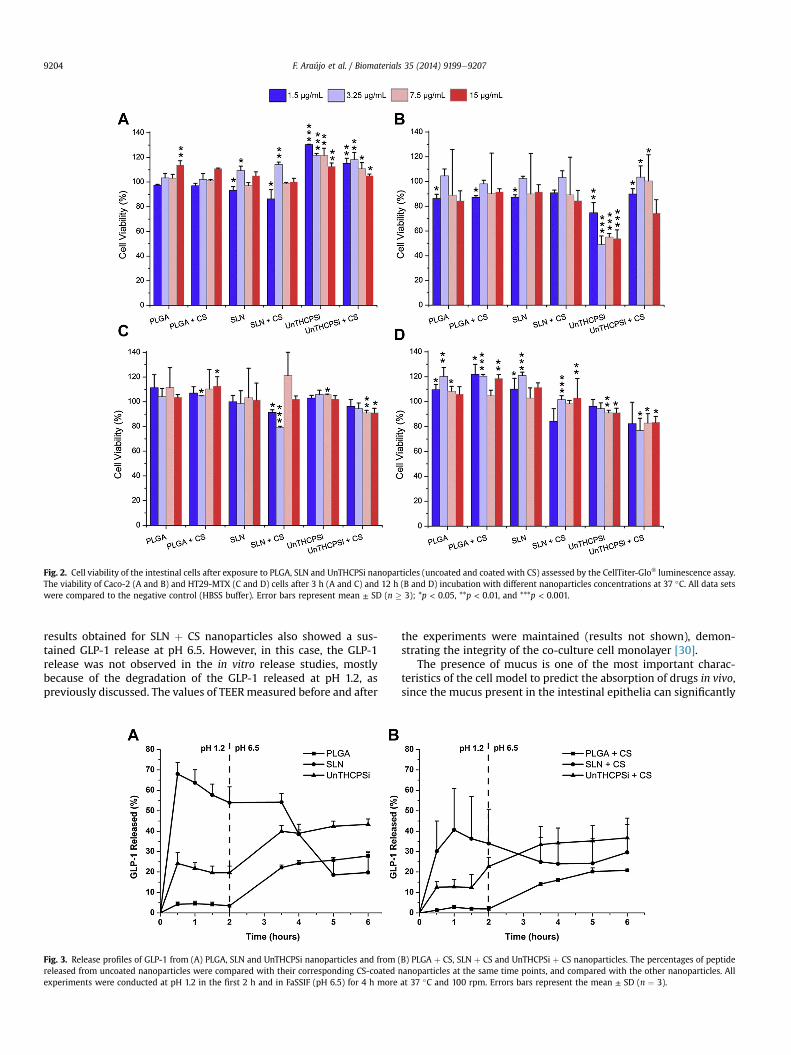
Fig. 2. Cell viability of the intestinal cells after exposure to PLGA, SLN and UnTHCPSi nanoparticles (uncoated and coated with CS) assessed by the CellTiter-Glo® luminescence assay.The viability of Caco-2 (A and B) and HT29-MTX (C and D) cells after 3 h (A and C) and 12 h (B and D) incubation with different nanoparticles concentrations at 37 �C. All data setswere compared to the negative control (HBSS buffer). Error bars represent mean ± SD (n � 3); *p < 0.05, **p < 0.01, and ***p < 0.001.
F. Araújo et al. / Biomaterials 35 (2014) 9199e92079204
results obtained for SLN þ CS nanoparticles also showed a sus-tained GLP-1 release at pH 6.5. However, in this case, the GLP-1release was not observed in the in vitro release studies, mostlybecause of the degradation of the GLP-1 released at pH 1.2, aspreviously discussed. The values of TEERmeasured before and after
Fig. 3. Release profiles of GLP-1 from (A) PLGA, SLN and UnTHCPSi nanoparticles and fromreleased from uncoated nanoparticles were compared with their corresponding CS-coatedexperiments were conducted at pH 1.2 in the first 2 h and in FaSSIF (pH 6.5) for 4 h more
the experiments were maintained (results not shown), demon-strating the integrity of the co-culture cell monolayer [30].
The presence of mucus is one of the most important charac-teristics of the cell model to predict the absorption of drugs in vivo,since the mucus present in the intestinal epithelia can significantly
(B) PLGA þ CS, SLN þ CS and UnTHCPSi þ CS nanoparticles. The percentages of peptidenanoparticles at the same time points, and compared with the other nanoparticles. Allat 37 �C and 100 rpm. Errors bars represent the mean ± SD (n ¼ 3).
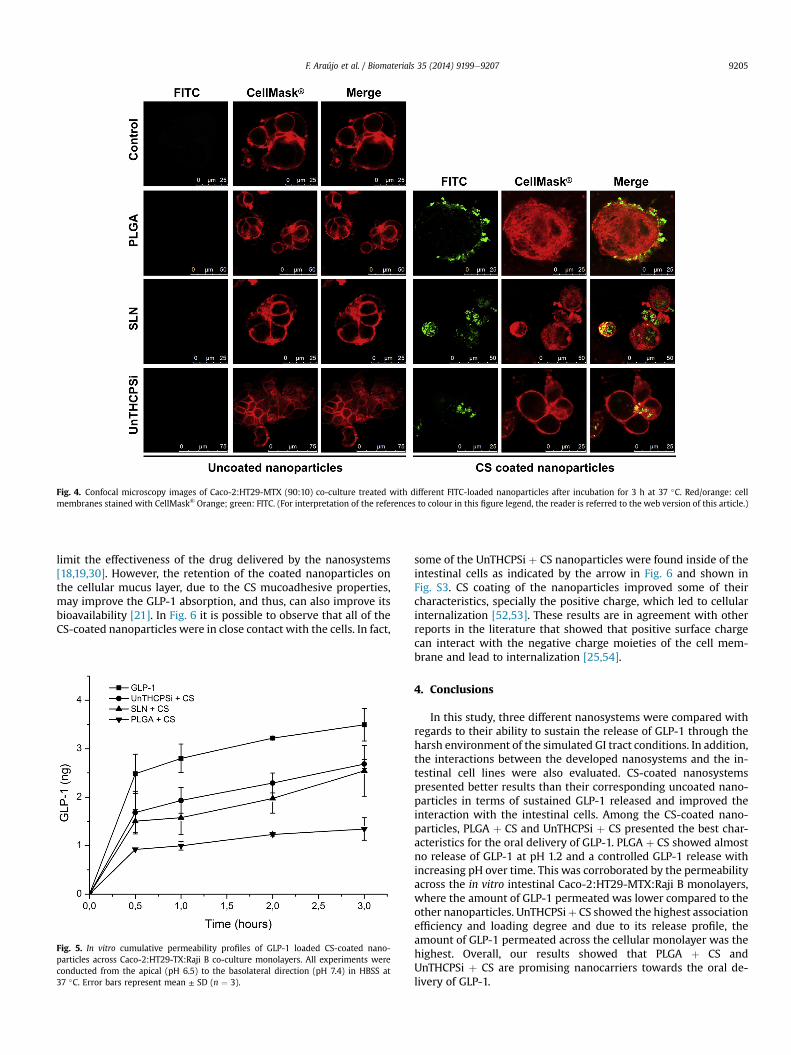
Fig. 4. Confocal microscopy images of Caco-2:HT29-MTX (90:10) co-culture treated with different FITC-loaded nanoparticles after incubation for 3 h at 37 �C. Red/orange: cellmembranes stained with CellMask® Orange; green: FITC. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
F. Araújo et al. / Biomaterials 35 (2014) 9199e9207 9205
limit the effectiveness of the drug delivered by the nanosystems[18,19,30]. However, the retention of the coated nanoparticles onthe cellular mucus layer, due to the CS mucoadhesive properties,may improve the GLP-1 absorption, and thus, can also improve itsbioavailability [21]. In Fig. 6 it is possible to observe that all of theCS-coated nanoparticles were in close contact with the cells. In fact,
Fig. 5. In vitro cumulative permeability profiles of GLP-1 loaded CS-coated nano-particles across Caco-2:HT29-TX:Raji B co-culture monolayers. All experiments wereconducted from the apical (pH 6.5) to the basolateral direction (pH 7.4) in HBSS at37 �C. Error bars represent mean ± SD (n ¼ 3).
some of the UnTHCPSi þ CS nanoparticles were found inside of theintestinal cells as indicated by the arrow in Fig. 6 and shown inFig. S3. CS coating of the nanoparticles improved some of theircharacteristics, specially the positive charge, which led to cellularinternalization [52,53]. These results are in agreement with otherreports in the literature that showed that positive surface chargecan interact with the negative charge moieties of the cell mem-brane and lead to internalization [25,54].
4. Conclusions
In this study, three different nanosystems were compared withregards to their ability to sustain the release of GLP-1 through theharsh environment of the simulated GI tract conditions. In addition,the interactions between the developed nanosystems and the in-testinal cell lines were also evaluated. CS-coated nanosystemspresented better results than their corresponding uncoated nano-particles in terms of sustained GLP-1 released and improved theinteraction with the intestinal cells. Among the CS-coated nano-particles, PLGA þ CS and UnTHCPSi þ CS presented the best char-acteristics for the oral delivery of GLP-1. PLGA þ CS showed almostno release of GLP-1 at pH 1.2 and a controlled GLP-1 release withincreasing pH over time. This was corroborated by the permeabilityacross the in vitro intestinal Caco-2:HT29-MTX:Raji B monolayers,where the amount of GLP-1 permeated was lower compared to theother nanoparticles. UnTHCPSiþ CS showed the highest associationefficiency and loading degree and due to its release profile, theamount of GLP-1 permeated across the cellular monolayer was thehighest. Overall, our results showed that PLGA þ CS andUnTHCPSi þ CS are promising nanocarriers towards the oral de-livery of GLP-1.

Fig. 6. TEM images of flat embedded ultrathin sections after permeability showing control cells (A), PLGA þ CS nanoparticles (B), SLN þ CS nanoparticles (C), and UnTHCPSi þ CSnanoparticles (D).
F. Araújo et al. / Biomaterials 35 (2014) 9199e92079206
Acknowledgements
The authors would like to acknowledge Bishal Silwal for his helpand support. This work was financed by the European RegionalDevelopment Fund (ERDF) through the Programa OperacionalFactores de Competitividade e COMPETE, by the Portuguese fundsthrough Fundaç~ao para a Ciencia e a Tecnologia (FCT) in theframework of the project PEst-C/SAU/LA0002/2013, and co-financed by the North Portugal Regional Operational Programme(ON.2 e O Novo Norte) in the framework of the project SAESCTN-PIIC&DT/2011, under the National Strategic Reference Framework(NSRF). Francisca Araújo would like to thank to FCT for financialsupport (SFRH/BD/87016/2012). Dr. H�elder A. Santos acknowledgesfinancial support from the Academy of Finland (decision no.252215), the University of Helsinki Research Funds, the BiocentrumHelsinki, and the Finnish Center for International Mobility (grant noTM-13-9048).
Appendix A. Supplementary data
Supplementary data related to this article can http://dx.doi.org/10.1016/j.biomaterials.2014.07.003.
References
[1] Russell S. Incretin-based therapies for type 2 diabetes mellitus: a review ofdirect comparisons of efficacy, safety and patient satisfaction. Int J ClinPharmacol 2013;35:159e72.
[2] Campbell JE, Drucker DJ. Pharmacology, physiology, and mechanisms ofincretin hormone action. Cell Metab 2013;17:819e37.
[3] Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology2007;132:2131e57.
[4] Tahrani AA, Piya MK, Kennedy A, Barnett AH. Glycaemic control in type 2diabetes: targets and new therapies. Pharmacol Ther 2010;125:328e61.
[5] Nauck MA, Heimesaat MM, Behle K, Holst JJ, Nauck MS, Ritzel R, et al. Effects ofglucagon-like peptide 1 on counterregulatory hormone responses, cognitivefunctions, and insulin secretion during hyperinsulinemic, stepped hypogly-cemic clamp experiments in healthy volunteers. J Clin Endocrinol Metab2002;87:1239e46.
[6] Rekha MR, Sharma CP. Oral delivery of therapeutic protein/peptide fordiabetes-future perspectives. Int J Pharm 2013;440:48e62.
[7] Araújo F, Fonte P, Santos HA, Sarmento B. Oral delivery of glucagon-likepeptide-1 and analogs: alternatives for diabetes control? J Diabetes SciTechnol 2012;6:1486e97.
[8] Gupta S, Jain A, Chakraborty M, Sahni JK, Ali J, Dang S. Oral delivery of ther-apeutic proteins and peptides: a review on recent developments. Drug Deliv2013;20:237e46.
[9] Joseph JW, Kalitsky J, St-Pierre S, Brubaker PL. Oral delivery of glucagon-likepeptide-1 in a modified polymer preparation normalizes basal glycaemia indiabetic db/db mice. Diabetologia 2000;43:1319e28.
[10] Chae SY, Jin CH, Shin HJ, Youn YS, Lee S, Lee KC. Preparation, character-ization, and application of biotinylated and biotin-PEGylated glucagon-likepeptide-1 analogues for enhanced oral delivery. Bioconjug Chem 2008;19:334e41.
[11] Nguyen HN, Wey SP, Juang JH, Sonaje K, Ho YC, Chuang EY, et al. The glucose-lowering potential of exendin-4 orally delivered via a pH-sensitive nano-particle vehicle and effects on subsequent insulin secretion in vivo. Bio-materials 2011;32:2673e82.
[12] Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Preat V. PLGA-basednanoparticles: an overview of biomedical applications. J Control Release2012;161:505e22.
[13] Bimbo LM, Sarparanta M, Santos HA, Airaksinen AJ, M€akil€a E, Laaksonen T,et al. Biocompatibility of thermally hydrocarbonized porous silicon nano-particles and their biodistribution in rats. ACS Nano 2010;4:3023e32.
[14] Sarmento B, Martins S, Ferreira D, Souto EB. Oral insulin delivery by means ofsolid lipid nanoparticles. Int J Nanomed 2007;2:743e9.
[15] Shahbazi MA, Santos HA. Improving oral absorption via drug-loaded nano-carriers: absorption mechanisms, intestinal models and rational fabrication.Curr Drug Metab 2013;14:28e56.
[16] Salonen J, Kaukonen AM, Hirvonen J, Lehto VP. Mesoporous silicon in drugdelivery applications. J Pharm Sci 2008;97:632e53.
[17] Fredenberg S, Wahlgren M, Reslow M, Axelsson A. The mechanisms of drugrelease in poly(lactic-co-glycolic acid)-based drug delivery systems-a review.Int J Pharm 2011;415:34e52.
[18] Zhang X, Sun M, Zheng A, Cao D, Bi Y, Sun J. Preparation and characterizationof insulin-loaded bioadhesive PLGA nanoparticles for oral administration. EurJ Pharm Sci 2012;45:632e8.
[19] Ensign LM, Cone R, Hanes J. Oral drug delivery with polymeric nano-particles: the gastrointestinal mucus barriers. Adv Drug Deliv Rev 2012;64:557e70.
[20] Sarmento B, Ribeiro A, Veiga F, Ferreira D, Neufeld R. Oral bioavailability ofinsulin contained in polysaccharide nanoparticles. Biomacromolecules2007;8:3054e60.
[21] Andrade F, Antunes F, Nascimento AV, da Silva SB, das Neves J, Ferreira D, et al.Chitosan formulations as carriers for therapeutic proteins. Curr Drug DiscovTechnol 2011;8:157e72.
[22] Shahbazi MA, Hamidi M. The impact of preparation parameters on typicalattributes of chitosan-heparin nanohydrogels: particle size, loading efficiency,and drug release. Drug Dev Ind Pharm 2013;39:1774e82.

F. Araújo et al. / Biomaterials 35 (2014) 9199e9207 9207
[23] Zhang N, Ping Q, Huang G, Xu W, Cheng Y, Han X. Lectin-modified solid lipidnanoparticles as carriers for oral administration of insulin. Int J Pharm2006;327:153e9.
[24] Garcı
a-Fuentes M, Torres D, Alonso MJ. Design of lipid nanoparticles for theoral delivery of hydrophilic macromolecules. Colloids Surf B Biointerfaces2003;27:159e68.
[25] Shahbazi MA, Almeida PA, M€akil€a EM, Kaasalainen MH, Salonen JJ,Hirvonen JT, et al. Augmented cellular trafficking and endosomal escape ofporous silicon nanoparticles via zwitterionic bilayer polymer surface engi-neering. Biomaterials 2014;35:7488e500.
[26] Shrestha N, Shahbazi MA, Araújo F, Zhang H, M€akil€a E, Kauppila J, et al.Chitosan-modified porous silicon microparticles for enhanced permeabilityof insulin across intestinal cell monolayers. Biomaterials 2014;35:7172e9.
[27] Li X, Guo S, Zhu C, Zhu Q, Gan Y, Rantanen J, et al. Intestinal mucosapermeability following oral insulin delivery using core shell corona nano-lipoparticles. Biomaterials 2013;34:9678e87.
[28] Sarmento B, Ribeiro A, Veiga F, Ferreira D. Development and characterizationof new insulin containing polysaccharide nanoparticles. Colloids Surf B Bio-interfaces 2006;53:193e202.
[29] Bimbo LM, M€akil€a E, Raula J, Laaksonen T, Laaksonen P, Strommer K, et al.Functional hydrophobin-coating of thermally hydrocarbonized porous siliconmicroparticles. Biomaterials 2011;32:9089e99.
[30] Araújo F, SarmentoB. Towards the characterizationof an invitro triple co-cultureintestine cell model for permeability studies. Int J Pharm 2013;458:128e34.
[31] Verma A, Stellacci F. Effect of surface properties on nanoparticle-cell in-teractions. Small 2010;6:12e21.
[32] Caldorera-Moore M, Guimard N, Shi L, Roy K. Designer nanoparticles: incor-porating size, shape and triggered release into nanoscale drug carriers. ExpertOpin Drug Deliv 2010;7:479e95.
[33] Wang Y, Li P, Kong L. Chitosan-modified PLGA nanoparticles with versatilesurface for improved drug delivery. AAPS PharmSciTech 2013;14:585e92.
[34] Chen MC, Mi FL, Liao ZX, Hsiao CW, Sonaje K, Chung MF, et al. Recent advancesin chitosan-based nanoparticles for oral delivery of macromolecules. AdvDrug Deliv Rev 2013;65:865e79.
[35] Emami J, Hamishehkar H, Najafabadi AR, Gilani K, Minaiyan M, Mahdavi H,et al. A novel approach to prepare insulin-loaded poly(lactic-co-glycolicacid) microcapsules and the protein stability study. J Pharm Sci 2009;98:1712e31.
[36] Fonte P, Nogueira T, Gehm C, Ferreira D, Sarmento B. Chitosan-coated solidlipid nanoparticles enhance the oral absorption of insulin. Drug Deliv TranslRes 2011;1:299e308.
[37] Cohen-Sela E, Chorny M, Koroukhov N, Danenberg HD, Golomb G. A newdouble emulsion solvent diffusion technique for encapsulating hydrophilicmolecules in PLGA nanoparticles. J Control Release 2009;133:90e5.
[38] Liu D, Bimbo LM, M€akil€a E, Villanova F, Kaasalainen M, Herranz-Blanco B, et al.Co-delivery of a hydrophobic small molecule and a hydrophilic peptide byporous silicon nanoparticles. J Control Release 2013;170:268e78.
[39] Kovalainen M, M€onk€are J, M€akil€a E, Salonen J, Lehto VP, Herzig KH, et al.Mesoporous silicon (PSi) for sustained peptide delivery: effect of PSi micro-particle surface chemistry on peptide YY3-36 release. Pharm Res 2012;29:837e46.
[40] Almeida AJ, Souto E. Solid lipid nanoparticles as a drug delivery system forpeptides and proteins. Adv Drug Deliv Rev 2007;59:478e90.
[41] Antunes F, Andrade F, Araujo F, Ferreira D, Sarmento B. Establishment of atriple co-culture in vitro cell models to study intestinal absorption of peptidedrugs. Eur J Pharm Biopharm 2013;83:427e35.
[42] Sarmento B, Andrade F, da Silva SB, Rodrigues F, das Neves J, Ferreira D. Cell-based in vitro models for predicting drug permeability. Expert Opin DrugMetab Toxicol 2012;8:607e21.
[43] Shah S, Shah P, Todkar J, Gagner M, Sonar S, Solav S. Prospective controlledstudy of effect of laparoscopic sleeve gastrectomy on small bowel transit timeand gastric emptying half-time in morbidly obese patients with type 2 dia-betes mellitus. Surg Obes Relat Dis 2010;6:152e7.
[44] Huotari A, Xu W, Monkare J, Kovalainen M, Herzig KH, Lehto VP, et al. Effect ofsurface chemistry of porous silicon microparticles on glucagon-like peptide-1(GLP-1) loading, release and biological activity. Int J Pharm 2013;454:67e73.
[45] Martins S, Sarmento B, Souto EB, Ferreira DC. Insulin-loaded alginate micro-spheres for oral delivery - effect of polysaccharide reinforcement on physi-cochemical properties and release profile. Carbohydr Polym 2007;69:725e31.
[46] Li J, Jiang G, Ding F. The effect of pH on the polymer degradation and drugrelease from PLGA-mPEG microparticles. J App Polym Sci 2008;109:475e82.
[47] Buske J, Konig C, Bassarab S, Lamprecht A, Muhlau S, Wagner KG. Influence ofPEG in PEG-PLGA microspheres on particle properties and protein release. EurJ Pharm Biopharm 2012;81:57e63.
[48] Giteau A, Venier-Julienne MC, Aubert-Pouessel A, Benoit JP. How to achievesustained and complete protein release from PLGA-based microparticles? Int JPharm 2008;350:14e26.
[49] Chakravarthi SS, Robinson DH. Enhanced cellular association of paclitaxeldelivered in chitosan-PLGA particles. Int J Pharm 2011;409:111e20.
[50] Garcia-Fuentes M, Torres D, Alonso MJ. New surface-modified lipid nano-particles as delivery vehicles for salmon calcitonin. Int J Pharm 2005;296:122e32.
[51] des Rieux A, Ragnarsson EG, Gullberg E, Preat V, Schneider YJ, Artursson P.Transport of nanoparticles across an in vitro model of the human intestinalfollicle associated epithelium. Eur J Pharm Sci 2005;25:455e65.
[52] Huhn D, Kantner K, Geidel C, Brandholt S, De Cock I, Soenen SJ, et al. Polymer-coated nanoparticles interacting with proteins and cells: focusing on the signof the net charge. ACS Nano 2013;7:3253e63.
[53] Lee SY, Huh MS, Lee S, Lee SJ, Chung H, Park JH, et al. Stability and cellularuptake of polymerized siRNA (poly-siRNA)/polyethylenimine (PEI) complexesfor efficient gene silencing. J Control Release 2010;141:339e46.
[54] Fuller JE, Zugates GT, Ferreira LS, Ow HS, Nguyen NN, Wiesner UB, et al.Intracellular delivery of core-shell fluorescent silica nanoparticles. Bio-materials 2008;29:1526e32.
Copyright © 2022 FDOKUMEN




















