
Development of Polymer-based Gels for Multimodal Medical ...
Upload
independentCategory
view
3download
0
ARTICLE
Charles W. Wolgemuth Æ Alexander Mogilner
George Oster
The hydration dynamics of polyelectrolyte gels with applications to cellmotility and drug delivery
Received: 12 May 2003 / Revised: 20 June 2003 / Accepted: 8 July 2003 / Published online: 23 October 2003� EBSA 2003
Abstract We combine the physics of gels with thehydrodynamics of two-phase fluids to construct a set ofequations that describe the hydration dynamics ofpolyelectrolyte gels. We use the model to address threeproblems. First, we express the effective diffusion con-stants for neutral and charged spherically distributedgels in terms of microscopic parameters. Second, we usethe model to describe the locomotion of nematodesperm cells. Finally, we describe the swelling dynamicsof polyelectrolyte gels used for drug release.
Keywords Diffusion constants Æ Drug release ÆHydration dynamics Æ Nematode sperm cells ÆPolyelectrolyte gels
Introduction
Gelatinous materials are ubiquitous in nature, from thecell walls of bacteria (Burge et al. 1977) to the human eye(Elliott and Hodson 1998). They find practical applica-tions in fields as diverse as micromachines and drugdelivery (Cornejo-Bravo et al. 1995; Siegel et al. 1988).The theory of gel swelling has a long history, beginningwith the early work of Flory and Katchalsky (Flory1953; Katchalsky and Michaeli 1955). More recently,Tanaka and colleagues have addressed the mechanicalaspects of gel swelling by treating a neutral gel as a linear
elastic solid immersed in a viscous fluid (Tanaka andFillmore 1979; Tanaka et al. 1973). Although they ne-glected the motion of the fluid solvent, the model rea-sonably explained swelling of a gel to a nearbyequilibrium volume fraction. Subsequent studies relaxedthe constraint of linear elasticity by appealing to a freeenergy for the polymer mesh and defining the divergenceof the stress as the functional derivative of this energy(Durning and Morman 1993; Maskawa et al. 1999;Onuki 1989; Sekimoto et al. 1989; Yamaue et al. 2000).Most of these works still neglected the fluid flow thatmust accompany swelling. Wang et al. (1997) added influid flow by application of two-phase flow theory;however, they considered only the regime of smallpolymer volume fractions and small gradients in thevolume fraction. Durning and Morman (1993) also usedcontinuity equations to describe the flow of solvent andsolution in the gel, but used a diffusion approximationwith a constant diffusion coefficient to obtain the fluidmotion. This assumption does not treat the dynamics ofthe fluid and the gel on an equal footing, and so it onlyroughly approximates the viscous drag coupling betweenthe fluid and the polymer. They also proposed a freeenergy picture that did not include charged gels. Finally,there is a sizeable literature treating the swelling ofpolyelectrolyte gels in the context of cartilage mechanics.Several authors have used multiphasic fluid models inthis setting that include osmotic effects (Gu et al. 1998;Lai et al. 1991; Lanir 1996). Lanir (1996) derived aconstitutive relation for polyelectrolyte gels; however, hedid not treat the dynamics of the gel. Lai et al. (1991)derived dynamic equations similar to those we derivehere using a triphasic theory that also accounts for theflow of salt ions. This treatment, though rigorous in itsderivation of the dynamic equations, treats the free en-ergy as a phenomenological function and derives a stressthat is only valid for small deformations. Moreover, theenergy is treated as a function of strain rather than afunctional of position.
In this paper, we draw on the work cited above tounite gel mechanics with two-phase fluid dynamics. The
C. W. Wolgemuth Æ G. Oster (&)Departments of Molecular & Cellular Biology and ESPM,University of California, Berkeley, CA 94720-3112, USAE-mail: [email protected]: +1-510-6427428
A. MogilnerDepartment of Mathematics and Center for Genetics andDevelopment, University of California, Davis, CA 95616, USA
Present address: C. W. WolgemuthDepartment of Physiology, University of Connecticut HealthCenter, Farmington, CT 06030, USA
Eur Biophys J (2004) 33: 146–158DOI 10.1007/s00249-003-0344-5

resulting model provides a dynamic description of gelhydration that is suitable for addressing several situa-tions of biological interest. We derive the hydrationdynamics of a polyelectrolyte gel immersed in a coun-terion fluid by combining a Flory-type free energyfunctional with two-phase fluid dynamics. This modelenables us to compute the stresses that act within the geland to describe the motion of the gel and of the solvent.We apply this model to provide a novel description forthe crawling of nematode sperm, and to describe theswelling dynamics of medically important gels from thedry state to equilibrium. Most of the conservation andconstitutive equations of the model have appeared in theliterature before; however, our derivation of the model issimpler than others, and is more suitable for the bio-logical applications we address.
Methods
A model for polyelectrolyte gels
A polyelectrolyte gel is a charged, cross-linked polymer networkimmersed in a fluid. Let / denote the volume fraction of thepolymer, so that in a unit volume of the gel a fraction of thespace, /, is occupied by the polymer and the remaining space isfluid, (1)/). The total density of a unit volume of the system isq=/qp+(1)/)qf, with qp and qf the density of the polymer andthe fluid, respectively. The positions of material points of thepolymer are given by the vector X, and we define the initialposition of those points at time t=0 by xinit. The vectoru=X)xinit defines the displacement of polymer subunits from theinitial position. Each of the components obeys a continuityequation:
@ /qpð Þ@t ¼ �rX � /qput
� �
@ ð1�/Þqfð Þ@t ¼ �rX � ð1� /Þqfvð Þ
ð1Þ
where ut ” ¶u/¶t is the polymer velocity, v is the fluid velocity, andrX is the gradient operator with respect to the positions X. UsingEq. (1), and assuming that the fluid and polymer are incompress-ible (qf=constant, qp=constant), gives the conservation equation:
rX � /2ut þ 1� /ð Þ2vð Þ ¼ 0 ð2ÞAs the polymer moves, the polymer volume fraction will change
from its initial value, /init ” /(t=0). The following geometricalrelationship between /, /init, and u is derived in Appendix A:
//init
¼ det I þ @u@xinit
� �ð3Þ
where I is the identity matrix. In this paper, we define the swellingratio as the ratio of the initial polymer volume fraction to its finalvalue: /init//. Equations (2) and (3) describe the kinematics of thetwo-phase polymer–fluid system.
We shall treat only situations where the Reynolds number isvery low, so that the inertia of the polymer and the fluid are neg-ligible compared to the drag forces between them. Therefore, todescribe the motion of the gel, we construct a force balance on eachphase, with the constraint that the viscous drag between the solidand fluid phases are equal and opposite. For the fluid phase wewrite a Stokes-like equation (Happel and Brenner 1986):
f v� utð Þ|fflfflfflfflffl{zfflfflfflfflffl}
Drag force betweenfluid and gel
¼ grX �rX vþrX vT
2
� �
|fflfflfflfflfflfflfflfflfflfflfflfflfflfflfflfflfflfflffl{zfflfflfflfflfflfflfflfflfflfflfflfflfflfflfflfflfflfflffl}Fluid shear stress
� rX p|ffl{zffl}Fluid
pressure gradient
ð4Þ
Here f is the drag coefficient between the polymer and the fluid, g isthe fluid viscosity, and p is the fluid pressure. This equation ex-presses the balance between the force per unit volume acting on thefluid component and the drag force per unit volume that acts be-tween the fluid and polymer. The first term on the right side is thefluid shear, and the second term is the hydrostatic pressure, whichwe will later relate to the gel osmotic pressure. When the fluid shearterm is negligible, it is equivalent to Darcy�s law.
We model the polymer mesh as a deformable elastic substanceand write its equation of motion as a balance between the viscousdrag force and elastic force:
f 2ut � 2vð Þ ¼ 2f p ð5Þ
where f is the same drag coefficient as in Eq. (4), and fp is the forceper unit total volume that acts on the polymer. Adding Eqs. (4) and(5), we find that:
rx � rT ¼ rx � rp �rxp þ grx �rxvþrxvT
2
� �¼ 0 ð6Þ
where rT=rp)pI+gðrx2vþrx2vTÞ=2 is the total stress and wehave used that the force per unit volume that acts on the polymer isrelated to the polymer stress, rp, by 2f p ¼ r � rp. Equations (4)and (5) are coupled because the hydrostatic pressure that acts onthe fluid is a function of the stress on the polymer. The coupling isdictated by the volume conservation equation. To complete theequations of motion we require an expression for the force per unitvolume acting on the polymer, f p.
In the literature, the polymer force per volume has been definedeither as the divergence of a polymer stress, or the functionalderivative of the free energy with respect to position (Onuki 1989).We shall follow the latter strategy:
2f p ¼ rx � rp ¼ � //init
dFdX
ð7Þ
where rp is the polymer stress tensor, F is the free energy, and d/dXis the functional derivative with respect to X. The free energy of thegel is the sum of four effects: the gel swells due to (1) the entropicdiffusion of its constituent chains and (2) their counterions; swellingis countered by (3) the elastic forces in the chains and (4) inter-chain attractive forces:
F ¼ Fmix þ Fion|fflfflfflfflfflfflffl{zfflfflfflfflfflfflffl}Expansion
þ Fel þ Fint|fflfflfflfflffl{zfflfflfflfflffl}Contraction
ð8Þ
Figure 1 illustrates these effects in a cartoon. In this model, wedo not directly account for the electrostatic repulsion of fixedcharges on the polymer; however, when the ion diffusion is fastcompared to the dynamics of the polymer and the bath ionicstrength is low, the electrostatic effects do not contribute to thetotal free energy (see Appendices B and C). In Appendix B we giveexplicit expressions for each of these terms. The expression for rp
that results from taking the functional derivative of Eq. (8) is de-rived in Appendices B and C. [These model equations are similar tothose derived for two-phase fluid flow without elastic forces derivedby He and Dembo (1997) and also the triphasic theory developedby Lai et al. (1991).] The model gel is described by Eqs. (1, 2, 3, 4,5, 6, 7, 8) with appropriate boundary conditions. The model vari-ables are listed in Table 1.
Results
Applications of the model
Diffusive swelling of gels
One of the earliest dynamic models for a gel was for-mulated by Tanaka and colleagues (Tanaka et al. 1973).
147

They assumed that the fluid remained stationary andthat the gel polymer behaved as a linear elastic solid. Forthe swelling of a spherically symmetric gel, theseassumptions led to a diffusion equation with a diffusioncoefficient as the ratio of the elastic modulus to the dragcoefficient between the fluid and the gel. This model fitwell the experiments on the swelling of neutral gels nearequilibrium. Since then, a number of dynamic modelshave been proposed that introduce a nonlinear elasticconstitutive relation for the gel polymer or incorporatesolvent coupling. All of these have been shown to reduceto a diffusion equation in the limit that the relativechange in volume of the gel is small. Likewise, our modelalso reproduces this result (see Appendix D). This dif-fusive behavior arises for small changes in the gel vol-ume as the motion of solvent becomes negligible anddeformations of the polymer remain in the linear elasticregime. These were the assumptions used to derive theoriginal diffusive model (Tanaka et al. 1973).
However, we can make further progress: since themodel utilizes a microscopic constitutive relation withexperimentally measurable parameters, the diffusioncoefficient for the gel can be estimated as a function ofmicroscopic model parameters. Using the linearizedmodel equations given in Appendices D and E withreasonable values for the gel parameters (see Table 2),we compute the diffusion coefficients for neutral gelswith a 5% and 2.5% equilibrium polymer volume frac-tion: D5=6.8·10)7 cm2/s and D2.5=4.6·10)7 cm2/s,respectively. These values are comparable to the diffu-sion constants observed experimentally for gels. Forthese volume fractions, the ratio D5/D2.5=1.45 has beenexperimentally measured for neutral gels (Tanaka et al.1973). The model also elucidates the dependence of thediffusion constant on the free counterion charge.Figure 2 shows how the swelling rate increases with theeffective free ion charge per monomer, a. As the gelpolymer becomes more charged, the diffusion constantincreases. Qualitatively, this effect is due to the contri-bution of the counterions to the osmotic pressure at thegel boundary, so that polyelectrolyte gels swell muchmore rapidly than neutral gels.
Crawling of nematode sperm
Many cell types can glide across a surface while main-taining their overall shape using a motile appendagecalled a lamellipod (see Fig. 3a) (Bray 2001). The spermof the nematode Ascaris suum has attracted muchattention recently because it represents a ‘‘strippeddown’’ version of a motile cell (Bottino et al. 2002;Roberts and Stewart 2000). The sperm cell is tens ofmicrons long and wide and a few microns high. It ad-vances steadily at a rate of �0.1 lm/s. A polymer net-work permeates the ventral surface consisting of flexiblefilaments composed of major sperm protein (MSP). Thefilaments are positively charged, having polymerizedfrom highly basic dimers. Strong hydrophobic interac-tions between the polymers aggregate them into higherorder rope-like complexes. The cell body that containsthe nucleus and mitochondria rides atop the cytoskeletalnetwork at the rear of the cell (see Fig. 3). Mitochon-drial activity maintains a spatial pH gradient across thelamellipod such that the cytoplasm at the rear is acidicwith respect to the front of the cell. There is evidencethat the local pH controls motility by affecting celladhesion, protrusion, and contraction (Bottino et al.2002).
The strength of adhesion of the lamellipod to thesurface increases with decreasing pH. Thus the cell at-taches to the surface strongly at the front and weaklytowards the rear. At the leading edge of the cell MSPdimers assemble onto the tips of existing polymers,pushing the leading edge forward (Bottino et al. 2002;Mogilner and Oster 1996). The rate of protrusion isindirectly regulated by the pH gradient and decreaseswith cell length. MSP filaments depolymerize in the lowpH environment at the rear of the cell and MSP dimers
Table 1 Model variables
Symbol Definition Units
/ Volume fraction of gel Dimensionlessu Gel displacement cmt Time sv Fluid velocity cm/sx Position cmp Hydrostatic pressure dyn/cm2
r Gel stress dyn/cm2
F Free energy dyn cmCb Counterion concentration Mh Stress due to changes in volume Dimensionless
Fig. 1 Cartoon illustration of the terms in the free energyexpression (Eq. 1). The counterion swelling pressure (left panel)arises from the diffusion of the mobile counterions (+) to the fixedfiber charges ()) that give rise to the Donnan potential at the gelsurface. The contractile elastic pressure (right top) arises from thetransverse thermal fluctuations of the gel fibers that tend to pull thecrosslinks together. The fiber interaction (right bottom), which maybe electrostatic or hydrophobic, also tends to deswell the gel byreducing the chain entropy
148

are recycled from the rear to the front by simple diffu-sion. The model of Bottino et al. (2002) is based on theassumption that at high pH the MSP polymers aggre-gate into bundles storing elastic energy and generatingthe force of protrusion. Then, in the low pH environ-ment under the cell body, the adhesions between thefilaments weaken and the complexes dissociate. Thisreleases the elastic energy stored in the gel so that it canentropically contract and pull the rear of the cell for-ward.
Bottino et al. (2002) developed a 2-D finite elementmodel of the moving sperm cell, where the MSP gel wastreated as elastic solid and the fluid part of the cyto-plasm was neglected. Here we consider the sperm cellcytoskeleton as a hydrated polyelectrolyte gel governedby the equations derived in this paper. This allows us toadvance the model of cell crawling by explicitly takinginto account the movement of the liquid fraction of the
cytoplasm and including the effects of ion osmoticpressure. This allows us to compute the forces of the gelcontraction microscopically, rather than relying on a
Table 2 Model parametersSymbol Definition Value and units
/init Initial polymer volume fraction 0.025, 0.1f Drag coefficient between the polymer
and the fluid2·1011 dyn s/cm4 (Tanaka andFillmore 1979)
g Fluid viscosity 100 dyn s/cm2
Vm Volume of a monomer 0.1–1 nm3 (English et al. 1996)l Gel shear modulus 200 dyn/cm2 (Tanaka and
Fillmore 1979)/0 Material parameter setting unstressed
volume fraction�0.1
kBT Thermal energy �4.1 pN nmNA Avogadro�s number 6.02·1023Nx Number of monomers between crosslinks �10–100 (English et al. 1996)v Flory interaction parameter 0.6 (English et al. 1996)D Diffusion coefficient of the gel �10)7 cm2/s (computed here)a Number of charges per monomer �0.05 (assumed)Cb Bath ion concentration �0.07 M (assumed)j Permeability of gel/solvent interface �10)9 cm3/dyn s
Fig. 2 Model prediction of the swelling of a polyelectrolyte gelcompared to a neutral gel. a is the effective number of counterionsper gel monomer. The circles are data points from Tanaka et al.(1973). The curves for a=0.2 and 0.4 were computed from thelinearized model equations given in Appendices D and E
Fig. 3 a Side view of a crawling nematode sperm showing the gelstrip model. (b) Asymptotically stable stationary distributions ofthe polymer volume fraction, stress, and velocity and of the fluidvelocity from the rear to the front of the cell. (c) Trajectories of thefront and rear ends of the 1-D cytogel strip. Over a few hundredseconds the cell length regulates to a constant value
149

phenomenological contractile stress constitutive rela-tion. Following Bottino et al. (2002), this model assumesrubber-like elasticity for the MSP polymer: individualfilaments appear to be fairly flexible; there are no directmeasure of their persistence length but negativelystained filaments are curved and bent on the scale of tensof nanometers. We neglect chemical reactions in ourequations of mass balance because of the very high(�4 nM) concentration of MSP in the cytoplasm(Roberts and Stewart 2000). There are experimentalindications (Bottino et al. 2002; Roberts and Stewart2000) that a small number of enzymes activate MSPdimers into a polymerizable state, and that this is therate-limiting factor for MSP fiber assembly. Also, dif-fusion of MSP dimers is fast enough to recycle the di-mers from the rear to the front of the cell. Because ofthese factors, equations of chemical kinetics and diffu-sion uncouple from the equations for MSP gel, and ourmodel for the gel is self-consistent. The situation is morecomplicated for actin gels, and future work will incor-porate semi-flexible polymer energies and reaction-dif-fusion kinetics into the gel dynamic model to explore thequantitative differences between rubber-like and semi-flexible polymers and the role of MSP turnover. Here wereport the results of a 1-D simulation of a proximal-distal transect of the cytoskeletal gel strip at the center ofthe lamellipod. A 2-D simulation that takes into accountprocesses at the cell sides is much more involvednumerically; this work is in progress and will be reportedelsewhere; however, the 1-D model illustrates all of thebasic features driving the cell locomotion.
We model the dynamics of the 1-D cytoskeletal stripof gel solving the model equations (Eqs. 1, 2, 3, 4, 5, 6, 7)on a moving boundary domain. The quantitative detailsare presented in Appendix H. The rates of movement ofthe cell leading and trailing boundaries are given by theboundary velocities plus the rate of polymerization atthe front and depolymerization at the rear, respectively.We assume that the depolymerization rate is constant.We make the following assumptions consequent on theproximal to distal pH gradient. (1) The polymerization(protrusion) rate at the front is a decreasing function ofthe cell length. (2) There is an effective viscous dragbetween the cytoskeleton and the substratum; the cor-responding viscous drag coefficient increases signifi-cantly anterior to the cell body. (3) We model theunbundling of MSP filaments at the rear by increasingthe number of MSP dimers between adjacent crosslinksunder the cell body. We complement the model equa-tions by the conditions of zero gel stress at the bound-aries and no fluid flow through the leading edgeboundary. This amounts to assuming that the majorfluid flow into the basal gel strip comes from the cyto-plasm ‘‘above’’ the ventral layer we are modeling. Fromthe viewpoint of the 1-D model this enters as a distrib-uted fluid source.
Figure 3c shows the trajectories of the leading andtrailing edges of the cell. After initial transients decay,the steady mode of locomotion evolves such that the
cell achieves a constant length of tens of microns,similar to the observed length. It advances at the rateof few tenths of micron per second, in agreement withexperimental observations (Roberts and Stewart 2000).In the coordinate system moving with the cell, constantspatial distributions of densities, velocities, and forcesinside the cell also evolve. Figure 3b shows asymptot-ically stable spatial distributions of the polymer volumefraction, stress, gel and liquid velocities. The highlycrosslinked, unstressed gel has a steady-state polymervolume fraction �0.03. At the rear of the lamellipod,just in front of the cell body, the number of crosslinksdrops precipitously. Longer flexible polymer chainsbetween the adjacent crosslinks have a tendency to coil,like rubber strands (see Appendix H), so that thepolymer volume fraction, /0, increases. External andinternal viscous drag retards the polymer volume in-crease. Thus a contractile gel stress develops thatreaches its maximum at the center of the cell. Thepredicted total contractile force is tens of nanonewtons,comparable to that estimated experimentally for someother crawling cells (Oliver et al. 1995). This contractilestress does not affect the front of the cell because theadhesion to the substratum is strong at the front.However, because the adhesion is weak at the rear, thiscontractile stress pulls the rear of the cell forward. Thecontractile stress tends to compress the gel at the rearof the cell, where the polymer volume fraction reachesa new steady value of �0.045, relieving stress at therear.
The rate of advancement of the leading edge is equalto the polymerization rate. The steady state velocity of�0.1 lm/s at the cell rear is a consequence of the gelcontraction velocity that just balances the constantpolymerization rate at the front. The balance of forces issuch that if the lamellipod increases in length the pro-trusion velocity decreases, while at smaller length it in-creases, so the cell length is stable. The boundarycondition at the front ensures that the fluid velocity iszero there. However, in the middle of the cell, the largecontractile stress creates a significant hydrostatic pres-sure which drives the fluid phase of the cytoplasm bothforwards and backwards. At the very rear of the cell, thewater escapes upward into the region of the cell aroundthe cell body, where the cytoskeletal gel is less dense.Consequently, the cytoplasm near the dorsal surface ofthe cell should convect towards the front where new gelis polymerized. Computations show that the movementof the fluid is slow (a few nanometers per second) incomparison with gel movements. This justifies the cor-responding assumption made by Bottino et al. (2002).Thus, the model illustrates how stable rapid migration isachieved, so that the cell length and velocity regulate toconstant values.
Swelling and hydration dynamics in drug delivery
Gel swelling has many applications in the field of drugdelivery. One method is to chemically bind the drug to
150
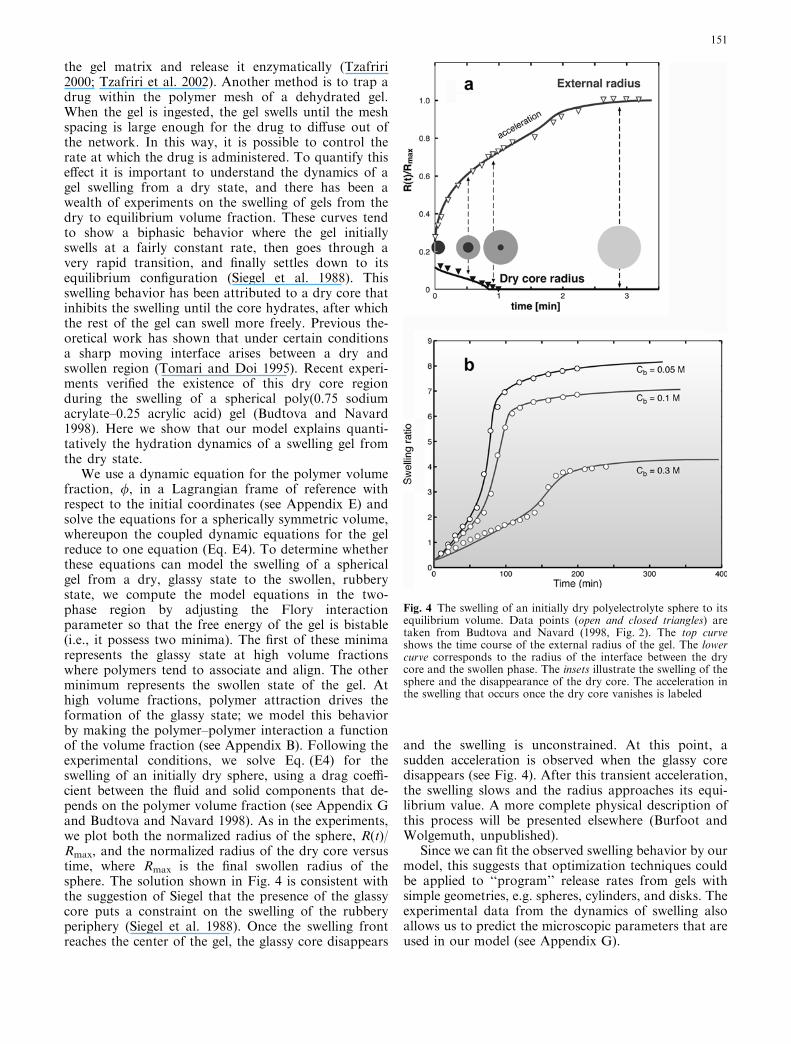
the gel matrix and release it enzymatically (Tzafriri2000; Tzafriri et al. 2002). Another method is to trap adrug within the polymer mesh of a dehydrated gel.When the gel is ingested, the gel swells until the meshspacing is large enough for the drug to diffuse out ofthe network. In this way, it is possible to control therate at which the drug is administered. To quantify thiseffect it is important to understand the dynamics of agel swelling from a dry state, and there has been awealth of experiments on the swelling of gels from thedry to equilibrium volume fraction. These curves tendto show a biphasic behavior where the gel initiallyswells at a fairly constant rate, then goes through avery rapid transition, and finally settles down to itsequilibrium configuration (Siegel et al. 1988). Thisswelling behavior has been attributed to a dry core thatinhibits the swelling until the core hydrates, after whichthe rest of the gel can swell more freely. Previous the-oretical work has shown that under certain conditionsa sharp moving interface arises between a dry andswollen region (Tomari and Doi 1995). Recent experi-ments verified the existence of this dry core regionduring the swelling of a spherical poly(0.75 sodiumacrylate–0.25 acrylic acid) gel (Budtova and Navard1998). Here we show that our model explains quanti-tatively the hydration dynamics of a swelling gel fromthe dry state.
We use a dynamic equation for the polymer volumefraction, /, in a Lagrangian frame of reference withrespect to the initial coordinates (see Appendix E) andsolve the equations for a spherically symmetric volume,whereupon the coupled dynamic equations for the gelreduce to one equation (Eq. E4). To determine whetherthese equations can model the swelling of a sphericalgel from a dry, glassy state to the swollen, rubberystate, we compute the model equations in the two-phase region by adjusting the Flory interactionparameter so that the free energy of the gel is bistable(i.e., it possess two minima). The first of these minimarepresents the glassy state at high volume fractionswhere polymers tend to associate and align. The otherminimum represents the swollen state of the gel. Athigh volume fractions, polymer attraction drives theformation of the glassy state; we model this behaviorby making the polymer–polymer interaction a functionof the volume fraction (see Appendix B). Following theexperimental conditions, we solve Eq. (E4) for theswelling of an initially dry sphere, using a drag coeffi-cient between the fluid and solid components that de-pends on the polymer volume fraction (see Appendix Gand Budtova and Navard 1998). As in the experiments,we plot both the normalized radius of the sphere, R(t)/Rmax, and the normalized radius of the dry core versustime, where Rmax is the final swollen radius of thesphere. The solution shown in Fig. 4 is consistent withthe suggestion of Siegel that the presence of the glassycore puts a constraint on the swelling of the rubberyperiphery (Siegel et al. 1988). Once the swelling frontreaches the center of the gel, the glassy core disappears
and the swelling is unconstrained. At this point, asudden acceleration is observed when the glassy coredisappears (see Fig. 4). After this transient acceleration,the swelling slows and the radius approaches its equi-librium value. A more complete physical description ofthis process will be presented elsewhere (Burfoot andWolgemuth, unpublished).
Since we can fit the observed swelling behavior by ourmodel, this suggests that optimization techniques couldbe applied to ‘‘program’’ release rates from gels withsimple geometries, e.g. spheres, cylinders, and disks. Theexperimental data from the dynamics of swelling alsoallows us to predict the microscopic parameters that areused in our model (see Appendix G).
Fig. 4 The swelling of an initially dry polyelectrolyte sphere to itsequilibrium volume. Data points (open and closed triangles) aretaken from Budtova and Navard (1998, Fig. 2). The top curveshows the time course of the external radius of the gel. The lowercurve corresponds to the radius of the interface between the drycore and the swollen phase. The insets illustrate the swelling of thesphere and the disappearance of the dry core. The acceleration inthe swelling that occurs once the dry core vanishes is labeled
151

Conclusion
We have assembled a model for the dynamics of poly-electrolyte gels from previously published componentsthat treat the gel as an elastic solid immersed in fluid. Wedescribe the dynamics by conservation equations forpolymer and fluid complemented by Stokes-like stressbalance equations for the polymer and fluid. The principalnovelty of the model is that it combines two-phase fluidflow, functional derivatives and a free energy derived fromFlory theory that contains microscopically measurableparameters. Though none of these separate concepts isnew, this model fully integrates them into a fairly simpletheory, generating predictions that can be tested againstexperimental data.
The model allows us to derive an effective diffusionequation for the swelling of neutral and charged gels thatgeneralizes previous work, and accounts for the fluidmovements that accompany gel swelling and de-swelling,factors that have been largely neglected heretofore. Thisenables us to express the effective diffusion coefficient ofthe gel in terms of microscopic, rather than phenomeno-logical macroscopic, parameters, revealing how swellingaccelerates as the gel charge increases. The main novelapplication of the model is the quantitative description ofthe lamellipodia of the nematode sperm cell. The modelexplains the nature of contractile stress that, when cou-pled to graded adhesion and depolymerization, drives thecell forward. The model reproduces some important fea-tures of steady cell movement. Finally, we model theisotropic swelling of charged gels. Solving themodel in thetwo-phase region accounts for the glassy state of the drygel. The model captures the qualitative features of theexperimental data on the swelling of spherical gels em-ployed for controlled drug release, and explains the ob-served biphasic swelling dynamics. This theory describessemi-quantitatively the swelling of a gel from the dry stateall the way to equilibrium.
Future elaborations of the model can deal with spe-cific geometries where shear stresses are important andswelling is driven or accompanied by chemical kinetics.This should be useful in a number of fields, includingbiomedical applications and cell mechanics.
Acknowledgements C.W. was supported by NSF grant 99-142.A.M. was supported by NSF grant DMS-1097746. G.O. wassupported by NSF grant DMS-9972826. Tom Roberts and MurrayStewart provided us with important biological input on the Ascarismodel. Dan Burfoot provided useful input on the drug deliverymodel. The authors especially thank Rami Tzafriri and HansWeinberger and the referees for their valuable comments and forsteering us to references we had missed.
Appendix
Appendix A: elasticity and functional derivatives
To define the stress tensor for the polymer mesh,Tanaka et al. (1973) originally assumed that the gel
could be treated as an elastic solid with an elastic energygiven by:
Fel ¼Z
l uik �1
3ujjdik
� �2
þ 1
2Ku2
jj
" #
d3x ðA1Þ
Here the components of the strain tensor areuik ¼ @ui=@Xk þ @uk=@Xið Þ=2, l is the shear modulus,and K is the bulk modulus. The first term in the integralis the energy associated with shear deformations of thesolid and the second term is the energy associated withchanges in volume. Moreover, they assumed that thesolvent was stationary and set the viscous drag forceacting on the gel polymer equal to the force obtainedfrom the functional derivative of this energy (Tanakaet al. 1973). Here we introduce a more completedescription of the free energy of the polyelectrolyte gelby taking into account not only the elastic energy but themixing, counterion, and polymer interaction energies aswell (English et al. 1996).
The energy we will use is dependent on twoparameters, the metric determinant,
ffiffiffigp ¼ /init
/ , and the
trace of the metric, Tr gij� �
¼ @Xi@xj
@Xi@xj. As such, the
energetic cost for deformations of the gel can be writ-ten as:
F ¼ZZZ
VH /ð Þ þ /
/init
! Tr gij� �� �� �
d3X ðA2Þ
with Q and ! functions that describe the energetic costfor volume and volume/shear deformations, respec-tively, and the integral being taken over the volume ofthe gel, V. This appendix defines notation and showshow functional derivatives are calculated and used toderive the forces per volume that act on the polymermesh.
We begin by defining the position of points in thepolymer mesh through the vector X. The initial positionof the polymer mesh at time t=0 is xinit=x(t=0). Fromthis, we can define the displacement vector for thesematerial points as u=X)xinit. The volume fraction ofthe polymer is obtained from this by:
/init
/¼ det
@X@xinit
� �¼ det I þ @u
@xinit
� �¼ ffiffiffi
gp ðA3Þ
with /init the initial polymer volume fraction and I theidentity matrix. Using this notation, we now discuss howto derive the force from a given energy function. Tobetter elucidate the method, we begin by consideringonly deformations that change the volume. The varia-tion in the energy is taken with respect to the initialposition vector, xinit, over the initial volume, V0, as thelimits of this integration are not altered by the variation:
dF ¼ZZZ
V0
@
@//init
/H /ð Þ
� �d/d3xinit ðA4Þ
It can be shown (Onuki 1989) that:
152

d/ ¼ �/X
l;m
@xinit;m@Xl
� �@
@xinit;mdXl
� �¼ �/
X
l
@
@XldXl
� �
ðA5Þ
Substituting this expression into Eq. (A4) andtransforming back to the displaced coordinates, we ob-tain:
dF ¼ZZZ
V� /2
/init
� �@
@//init
/H /ð Þ
� �X
l
@
@XldXl
� �d3X
ðA6Þ
This is then integrated by parts to give:
dF ¼ �ZZ
@V
X
l;m
nmPplmdXl dAþV
X
l;m
@
@XmPp
lm
� �dXl d
3X
ðA7Þ
where n is the unit outward normal vector to thesurface element dA, ¶V is the surface of the gel, and theisotropic polymer stress due to changes in volume isdefined as:
Pplm ¼ /
@H@/�H
� �dlm ðA8Þ
This also gives a force per volume:
2f p ¼ �rX �Pp ðA9Þ
where rX is the gradient over the displaced coordinates,X.
In a similar fashion, we can find the stress from thetrace term:
rtr;ij ¼ �/
NxVm
X
l
@Xi
@xl
@Xj
@xlðA10Þ
and we have used that:
! ¼ /init
NxVmTr gij� �
ðA11Þ
Combining these two terms, the total stress on thepolymer is:
rpij ¼ Pp
ij þ rsh;ij ðA12Þ
Appendix B: components of the gel free energy
The total free energy for the gel component can bebroken up into six independent components arising fromthe elasticity of the polymer, the entropic mixing due tothe fluid, interactions between polymer chains, thepressure due to the counter ions, Coulombic interac-tions, and a term that depends on gradients in the vol-ume fraction as:
F ¼ Fel þ Fmix þ Fint þ Fion þ Fcoulomb þ Fgrad ðB1Þ
We choose an elastic free energy based on rubberelasticity (Flory 1953):
Fel ¼kBT
2NxVm
ZZZ
V/ Tr gij
� �� 3� ln
/0
/
� �� �d3X ðB2Þ
where / is the polymer volume fraction, gij is the metricbetween the initial coordinates, x, and the final coordi-nates, X, kB is Boltzmann�s constant, T the absolutetemperature, Nx the number of monomers betweencrosslinks, /0 is a material parameter that sets the un-stressed volume fraction, Vm is the equivalent volumeoccupied by one monomer, and V is the volume of thegel. From these definitions, the number of chains pervolume is //NxVm. For the isotropic case:
gij ¼/init
/
� �2=3
I ðB3Þ
and:
Fel ¼kBT
2NxVm
ZZZ
V/
/0
/
� �� 1� 1
3ln
/0
/
� �� �d3X ðB4Þ
We choose the entropic mixing free energy defined byFlory theory (Flory 1953):
Fmix ¼kBTVm
ZZZ
V1� /ð Þln 1� /ð Þd3X ðB5Þ
and the solvent–polymer interaction energy representingtwo-body interactions is given by:
Fint ¼kBTVm
ZZZ
Vv/ 1� /ð Þd3X ðB6Þ
where v is the Flory parameter that measures thestrength of interaction between the polymer chains(Flory 1953). It is common to assume that v is inde-pendent of volume. However, in principle, it can be afunction of /; here we assume a dependence:
v ¼ v0 þ v1/2 ðB7Þ
to approximate the glass–rubber transition for swellinggels.
The contribution to the energy from the counterionsis obtained by assuming an ideal solution of chargeswith each species of charge labeled by the subscript i(English et al. 1996):
Fion ¼ �kBTNA
ZZZ
V
X
i
Ciln Cið Þð1� /Þ d3X ðB8Þ
where Ci is the molar concentration of the ith mobilecounterion and NA is Avogadro�s number. The factor of(1)/) in the integral is because the ions only reside in thefluid component of the mixture. Likewise, Ci is theconcentration with respect to the fluid volume, dV f, notthe total volume, dVT.
The contribution to the free energy from Coulombicinteractions is:
153

Fcoulomb ¼ZZZ
V
X
i
zieUselfCi d3X þ
ZZZ
VzFeUselfCF d
3X
ðB9Þ
where zi is the valence of the ith mobile counterion, eis the electronic charge, /self is the self-consistentpotential created by the polymer charges and themobile ions, and CF is the fixed charge density of thepolymer.
For cases where large differences in the volumefraction might exist over small lengths, such as volume-phase transitions in gels (Sekimoto et al. 1989; Tanaka1997), it is common to include a term that defines theenergy required to maintain the sharp gradient. As inLandau–Ginzburg theory and Cahn–Hilliard theory(Cahn and Hilliard 1958), we assume the form of theenergy to be:
Fgrad ¼ZZZ
V
c2r/r/ d3X ðB10Þ
where c is a material parameter that has units of energy/length. This type of energy has already been suggestedfor gels (Tanaka 1997).
Appendix C: the counterion swelling pressure of the gel
In Eq. (B8), the energy of the counterions is that of anideal solution of charges. We assume that the diffusionof the ions is much quicker than the motions of thesolvent and polymer. Therefore, we use the steady-stateconcentrations for the ions inside the gel. Because thecounterions are confined within the solvent, the con-centration is taken with respect to the solvent volume,dVf=(1)/)dVT, where dVT is a total unit volume of thegel. If we assume that there is an effective charge permonomer, a, on the polymer, the fixed charge density ofthe polymer is equal to aN/(dVf) where N=//Vm is thenumber of monomers per volume and Vm is the volumeof a monomer. From this, the fixed charge density of thepolymer is:
CF ¼a/
ð1� /ÞVmðC1Þ
Assuming a univalent salt in the solvent, theconcentrations of the positive and negative ions, C+
and C), respectively, can be related to the fixedcharge density via the Donnan equilibrium (Overbeek1956). Equating the chemical potentials across theboundary:
Cþ ¼1
24C2
b þ C2F
� �1=2�CF
C� ¼1
24C2
b þ C2F
� �1=2þCF
ðC2Þ
where Cb is the bulk molar concentration of positive(or negative) ions outside the gel. Therefore, thetotal molar ion concentration inside the gel,Cion=C++C), can be related to the fixed charge con-centration:
Cion ¼ 2Cb
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
1þ a/2 1� /ð ÞNAVmCb
� �2s
ðC3Þ
Note that in the limit of the neutral gel, Cion=2Cb
(2Cb is the total ion bath concentration), while in thelimit of a highly charged gel, the concentration is (1/Vm)(a//(1)/)). As /! 1, this concentration diverges.We expect that the effective fixed charge per monomerdecreases with volume fraction due to the condensationof charges onto the polymer as the gel dehydrates.Demanding that in the absence of solvent all the chargesare condensed onto the polymer sets a / ¼ 1ð Þ ¼ 0. Tosatisfy this constraint, we choose a=a0(1)/), with a0 aconstant.
Using Eqs. (C1) and (C2), we can find the Coulombiccontribution to the energy:
Fcoulomb ¼ZZZ
VeUselfðCþ �C�Þd3X þ
ZZZ
VzFeUselfCF d
3X
¼ZZZ
V�eUselfCF d
3X þZZZ
VzFeUselfCF d
3X
¼ 0 ðC4Þ
Therefore, the electrostatic contribution to the total freeenergy is zero. This is only valid at low ionic strengthsand when the diffusion of the ions is much quicker thanthe dynamics of the polymer.
The free energy for the gel can now be written com-pletely in terms of the polymer volume fraction and ashear term. Using the free energy given in Appendix Band Eq. (A8) and (A10) to calculate the polymer stress,we find:
Vm
kBT
� �Pp
ij ¼ hð/Þdij �/
NxVm
X
l
@Xi
@xl
@Xj
@xlðC5Þ
where:
hð/Þ ¼ �lnð1� /Þ � /� v/2 þ NAVm Cion � 2Cbð Þ
þ /2Nxþ c /r2/� 1
2r/ð Þ2
� �ðC6Þ
Experiments have shown that, at low volume frac-tions, the gel shear modulus is much less than the bulkmodulus (Tanaka and Fillmore 1979). If we neglectthe shear component [which is the same as using theisotropic form of the elastic free energy (Eq. B4)] andthe term that comes from the gradient squared piecein the energy, the polymer stress in Eq. (C5) reducesto:
154

Vm
kBT
� �Pp
ij ¼ hð/Þdij ðC7Þ
where:
hð/Þ ¼ �lnð1� /Þ � /� v/2 þ NAVm Cion � 2Cbð Þ
þ /0
Nx
1
2
//0
� �� /
/0
� �13
!
(C8)
is the dimensionless swelling pressure and we have as-sumed that the parameters v, /0, a0, Nx, and c areconstants. We have also subtracted 2NAVmCb to ac-count for the ion pressure outside the gel.
If we only assume a purely entropic energy as givenby Eq. (B5), then:
hð/Þ ¼ �lnð1� /Þ � / ðC9Þ
Plugging this into Eqs. (5) and (7), we find that:
f@u@t� v
� �¼ � kBT/
Vm 1� /ð Þ
� �r/ ðC10Þ
For small /, this should reproduce normal diffusion.Using that Cm�//Vm is the concentration of gelmonomer per fluid volume, we get:
f@u@t� v
� �¼ �kBTCmrCm ðC11Þ
For normal diffusion, the drag coefficient, fD, is de-fined as the drag per particle. In our representation, f isthe drag per total volume. The relation between these isf ¼ fDCm, which shows that these equations reproducepure diffusive behavior when only entropic effects areconsidered and we recover the Einstein relation for thediffusion coefficient, D ¼ kBT =fD:
Appendix D: linear approximation
The gel equations are a coupled, nonlinear set ofequations. To gain insight into the dynamics thatthey describe, we linearize the isotropic expansionequations about an initial polymer volume fraction,/init, setting /=/init(1)�), and using the rescalingsu! Lu, with L a relevant length scale. We useEqs. (3), (5), and (7), and expand to first order in � toobtain:
e � �ruut ¼ Ar2uþ Br� r� uð Þ
� � ðD1Þ
where:
A ¼ 1� /initð Þ 4~l=3þ /init @h=@/initð Þð Þ ðD2Þ
and:
B ¼ 1� /initð Þ ~l=3þ /init @h=@/initð Þð Þ ðD3Þ
with:
~l ¼ Vm=kBTð Þl ðD4Þ
h is the dimensionless swelling pressure that is derived inAppendix C, and @h=@/init the derivative of h with re-spect to / evaluated at /=/init. The latter is equivalentto the equation derived by Tanaka et al. (1973) withbulk modulus:
K ¼ kBT=Vmð Þ 1� /initð Þ/init @h=@/ð Þ ðD5Þ
This equation can now be used to calculate the rate ofchange in polymer volume fraction and the fluid velocityfor swelling of a gel near some initial configuration /init.This can then be compared with the work that has al-ready been done on spherical swelling which has ne-glected fluid velocity.
We begin by assuming that all variables are onlyfunctions of r, the radial coordinate, and that all vectorsare only in the radial direction, u ¼ uðrÞr and v ¼ vðrÞr.This allows the conservation equation to be integratedand the solvent velocity is obtained in terms of thepolymer velocity:
vðrÞ ¼ cðtÞ1� /ð Þr2 �
/ut
1� /ð Þ ðD6Þ
with c(t) a time-dependent constant. For a finite solutionat r=0, c(t)=0, giving (1)/)v+/ut=0, i.e. the center ofmass of the gel is stationary. Thus v>0 for hydratinggels; the fluid flows into the gel at a velocity equal to theratio of gel volume to fluid volume. Using this result, thefirst-order polymer velocity equation is:
ut � ~lþ /init
@h@/init
� �r2
r u ðD7Þ
with rr being the radial gradient. This gives a diffusivedynamics:
ut ¼ �Dr2r u ðD8Þ
with a diffusion coefficient:
D ¼ � kBTfVm
1� /initð Þ ~lþ /init
@h@/init
� �ðD9Þ
[The minus sign in the diffusion constant is due to the factthat as the gel swells, the displacement increases. InEq. (D9), D>0.] If we assume a neutral gel, a=0, andutilize reasonable values of /0=0.1, Vm=10)22 cm3,v=0.6, Nx=60 (English et al. 1996), l=200 dyn/cm2,and f=2·1011 dyn s/cm4 (Tanaka and Fillmore 1979),we find D=6.8·10)7 cm2/s with /init=0.05 andD=4.7·10)7 cm2/s with /init=0.025, which are compa-rable to the diffusion constants previously observed forgels with these polymer volume fractions and the ratioD5/D2.5=1.45 is what is experimentally observed (Tanakaet al. 1973). Using the continuity equation (Eq. 2) and thelinearized equations, we obtain the effective diffusionequation for the polymer volume fraction:
155

et ¼ Dr2r e ðD10Þ
When the gel/solvent interface is sufficiently perme-able, water can flow into the gel to satisfy any imposedboundary condition on the gel. Using a zero stressboundary condition sets the polymer volume fraction atthe interface to the unstressed volume fraction and thegel will relax to this unstressed state on a characteristictime scale given by s�L2/D. This time decreases as thecharge per monomer, a, is increased. Therefore, apolyelectrolyte gel swells faster than a neutral gel, as isobserved experimentally.
To calculate the swelling radius versus time for thelinear approximation of the spherically symmetric gel,we use the result derived in Tanaka and Fillmore(1979):
DaðtÞ ¼ 6
p2
� �Da0
Xn�2exp
�Dn2ta2
� �ðD11Þ
where Da is the change in radius of the gel, Da0 is thetotal change in radius, D is the diffusion coefficientfound in Eq. (D9) and the sum is over n.
Appendix E: equations for the swelling gel
In this Appendix, we derive a dynamic equation for thepolymer volume fraction, /, in a Lagrangian frame ofreference with respect to the initial coordinates. Thecoupled dynamic equations for the gel can be reduced toone equation by neglecting shear. Symmetry conditionspermit this for certain geometries (e.g., spheres andslabs). For other geometries, the magnitude of the shearterm is a few orders of magnitude smaller than that ofthe volume deformation term for a wide range ofphysically relevant parameters (Tanaka and Fillmore1979). Moreover, we will see that neglecting sheargreatly simplifies the analysis, yet fits the experimentaldata closely.
For the purposes of our calculations, it is easier towork in a reference frame fixed on the initial positionof the gel, since this frame does not change in time. Inthis frame, the continuity equation for the polymervolume fraction does not contain convection, and,therefore:
@tjx/ ¼ �/rX � 2ut ðE1Þ
where @tjx represents the partial derivative with respectto time at fixed material point xinit. Derivatives withrespect to the initial position are given by:
@
@Xi¼ @xinit;j
@Xi
� �@
@xinit;jðE2Þ
and we can define the metric as:
gij ¼@Xk
@xinit;i
@Xk
@xinit;jðE3Þ
Substituting Eq. (5) into Eq. (2) and adding andsubtracting the divergence of the polymer velocity,yields:
rX �1
f/� 1ð Þ2f p þ 2ut
� �¼ 0 ðE4Þ
Plugging this into Eq. (E1) and using Eqs. (E2) and(E3) leads to a dynamic equation for /:
@tjx/ ¼ �/2
/init
@
@xinit;i
/init
f/1� /ð Þf p
i
� �ðE5Þ
where f pi is the ith component of the polymer force and
it has been used that the metric determinant:
g1=2 ¼ /init=/ ðE6Þ
For a spherically symmetric gel:
Rr
� �2@R@r¼ /init
/ðE7Þ
where R is the radial coordinate and r is the radius of theinitial state.
If shear is neglected, and we assume isotropy:
gij ¼ //init
� �2=d
dij ðE8Þ
where d is the dimension of space and dij is the Kro-encker delta function, then the polymer force can bewritten completely in terms of / and @/=@xinit. Thissimplifies the dynamics by giving a single nonlinear PDEfor / rather than the coupled set of equations (Eqs. 2, 3,4, 5), and greatly simplifies the problem.
For the drug delivery swelling problem, Eq. (E5) wassolved numerically on a spherically symmetric volumewith initial volume fraction /0=0.95 everywhere, exceptat the boundary point where we seed the low-energyswollen state with /=0.1. We used boundary conditions(see Eq. F1) with the drag coefficient discussed inAppendix G. The microscopic energy parameters werefound from fits to the experimental data (see AppendixG). Equation (E5) was solved on the fixed initial coor-dinates using a variable time step method (MATLABroutine ode15s) and a forward explicit numericalscheme.
Appendix F: boundary conditions for a swelling gel
Taking the functional derivative of the free energy pro-duces both an integral over the volume and surfaceterms [see, for example, Eq. (A8)]. The integrand of thevolume integral gives the force per volume that acts onthe gel. The integrand of the surface area integral pro-vides the natural boundary conditions on the stress. Incircumstances where there are no external forces acting,these boundary conditions apply. For the swelling gel,the natural boundary conditions are:
156

rp � n ¼ 0c/r/ � n ¼ 0
ðF1Þ
Tokita and Tanaka (1991) found that f / /1:5 when/<1; this is expected from Flory scaling theory since thedrag coefficient scales like 1/n2, where n is the lengthcorrelation for the gel. Scaling theory suggests thatn / /�3=4. We use this dependence for the drag coeffi-cient. We assume a volume fraction dependence on c,and find the best fits to the data when c / /�1:
Appendix G: parameter fits from swelling experiments
Our model depends on a number of microscopicparameters that describe the material and physicalproperties of gels. The data of Budtova and Navard(1998) allow us to fit a number of the microscopicparameters in this model and enable us to fit the dy-namic behavior of the swelling of dry gels (see Fig. 2 andFig. 3). In fig. 2 of Budtova and Navard (1998), theswelling curves for spherical poly(0.75 sodium acrylate–0.25 acrylic acid) gels of differing radii are plotted. Thebath ion concentration for this plot is Cb=0. Based onthe mole fraction of the crosslinking density to that ofthe monomer, an estimate of the number of monomersper crosslink for these gels is Nx=1600 (Budtova andNavard 1998). If we assume that the volume of amonomer is �0.1 nm3 (English et al. 1996), then the onlyparameters left to fit are v, a0, f, and /0. f sets the overalltime scale for the process, and is found by scaling thecomputer time (time step interval) to the experimentaltime, giving f(/=1)�1013 dyn s/cm4, which is compa-rable to experiment (Tokita and Tanaka 1991). v0, a0,and /0 were fit to provide a swelling ratio comparable tothat observed in the experiments (Budtova and Navard1998). v1 was set by fitting the experimental swellingdata to the numerical results. We find v=0.6+2.3/2,a0=1.0, and /0=0.5. These values are reasonablecompared to what has been measured previously (Eng-lish et al. 1996).
Appendix H: model equations for cell crawling
We model the dynamics of the 1-D cytoskeletal strip ofgel with the following four equations:
/init
/¼ 1þ @u
@xðH1Þ
@
@x/@u@tþ 1� /ð Þv
� �¼ 0 ðH2Þ
f v� @u@t
� �¼ � @p
@xðH3Þ
fex@u@tþ f
@u@t� v
� �¼ @r
p
@xðH4Þ
Here Eqs. (H1) and (H2) are 1-D conservation equa-tions for the polymer (Eq. 3) and the gel (Eq. 2),respectively. Equation (H3) is the 1-D force balance forthe liquid (Eq. 4). Finally, Eq. (H4) is the 1-D gel forcebalance (Eq. 5), where we have added the effective vis-cous drag between the cytoskeleton and the surface; fexis the corresponding viscous drag coefficient. The gelstress is:
rpð/Þ ¼ ðkBT=VsiteÞhð/Þ ðH5Þ
where the stress due to changes in volume, h(/) is givenby Eq. (C8). The shear terms are absent in 1-D in boththe polymer and liquid force balance equations. Wesolve the problem on the moving boundary domainr(t)<x<f(t), where r(t) and f(t) are the coordinates ofthe rear and front of the cell, respectively. The rates ofmovement of the cell edges are given by the gel velocitiesat the edges, plus the rate of polymerization at the frontand depolymerization at the rear, respectively. Thus theboundary conditions are:
drdt¼ Vd þ
@u@tjx¼rðtÞ;
dfdt¼ Vpðr; f Þ þ
@u@tjx¼f ðtÞ; Vpðr; f Þ
¼V 0p
f � r � l1ðH6Þ
Here Vd is the constant depolymerization rate. Thepolymerization (protrusion) rate at the front is thedecreasing function of the cell length, l(t)=f(t))r(t); l1 isa model parameter.
We assume that the pH gradient regulates theexternal drag coefficient so that fex=3f(1+0.8 -tanh((x)r)l2)/l3)), where l2 and l3 are model parame-ters. (The internal drag coefficient, f, is given inTable 2.) The external drag coefficient is small and al-most constant under the cell body but constant andlarge under the lamellipod. We also assume that thenumber of dimers between the adjacent crosslinks inthe gel increases significantly from 25 at the front to 75at the rear. We describe this effect quantitatively with afunction similar to drag coefficient spatial behavior:Nx=50)25 tanh((x)r)l2)/l3). Finally, we reason thatthe equilibrium volume fraction of the gel scales as/ / N1=2
x . Indeed, the equilibrium size of a long flexiblefilament is proportional to the square root of its con-tour length (Landau et al. 1980). Thus, the number ofMSP dimers in unit volume scales like the number ofdimers between the adjacent crosslinks divided by theequilibrium distance between these crosslinks, Nx=N 1=2
x .Therefore, we assume that:
/0 ¼ 0:005N 1=2x ðH7Þ
where the coefficient is set such that the equilibriumvolume fraction at the front is approximately 0.035. as isobserved experimentally. For simplicity, we keep othermodel parameters constant; they are given in Table 2. Insimulations, we used the length scale L=10 lm, the timescale:
157

fVsiteL2�
kBT � 0:01 s ðH8Þ
and the force scale:
kBTL2�
Vsite � 100 dyn ðH9Þ
We also used additional parameters: l1=5 lm,l2=12 lm, l3=(1/15) lm, Vd=0.2 lm/s, V 0
p¼ 0:4lm=s.We non-dimensionalized the model equations usingthese scales.
Then, we used Eqs. (H2) and (H3) to express the fluidvelocity in terms of the hydrostatic pressure gradient:
v ¼ �/1@p@x
ðH10Þ
(This equation is valid up to a constant, but thisconstant is almost zero due to the conditions at theleading edge, where v=0, and the pressure gradient isexponentially small.) From Eqs. (H3) and (H4) we find:
@p@x¼ @r
p
@x� 1ex
@u@t
ðH11Þ
Substituting into Eq. (H4), we obtain:
fex 1� /ð Þ þ fð Þ @u@t¼ 1� /ð Þ @r
@xðH12Þ
where:
r ¼ rð/Þ;/ ¼ /init
1þ @u@x
ðH13Þ
We solve the equations on a uniform grid using aforward explicit integration method. At each time step,we update the gel displacement, u, using the gel stressfrom the previous step. Then, we use Eqs. (H10) and(H11) to find the pressure gradient and liquid velocity.Then, using Eq. (H13) we compute the new values of thegel stress using zero stress boundary conditions. Finally,we use the boundary conditions (Eq. H6) to move thecell boundaries. We start from the initial conditions ofzero stress and velocities everywhere and compute untilthe asymptotically stable steady stress, density, andvelocity distributions evolve.
References
Bottino D, Mogilner A, Roberts T, Stewart M, Oster G (2002)How nematode sperm crawl J Cell Sci 115:367–384
Bray D (2001) Cell movements: from molecules to motility, 2ndedn. Garland, New York
Budtova T, Navard P (1998) Swelling kinetics of a polyelectrolytegel in water and salt solutions. Coexistence of swollen andcollapsed phases. Macromolecules 31:8845–8850
Burge RE, Fowler AG, Reaveley DA (1977) Structure of thepeptidogylcan of bacterial cell walls. I. J Mol Biol 117:927–953
Cahn JW, Hilliard JE (1958) Free energy of a nonuniform system.I. Interfacial free energy. J Chem Phys 28:258–267
Cornejo-Bravo J, Arias-Sanchez V, Alvarez-Anguiano A, Siegel R(1995) Kinetics of drug release from hydrophobic polybasicgels: effect of buffer acidity. J Controlled Release 33:223–229
Durning CJ, Morman KN Jr (1993) Nonlinear swelling of polymergels. J Chem Phys 98:4275–4293
Elliott G, Hodson S (1998) Cornea, and the swelling of polyelec-trolyte gels of biological interest. Rep Prog Phys 61:1325–1365
English AE, Mafe S, Manzanares JA, Xiahong Y, Grosberg AY,Tanaka T (1996) Equilibrium swelling properties of poly-ampholytic hydrogels. J Chem Phys 104:8713–8720
Flory P (1953) Principles of polymer chemistry. Cornell UniversityPress, Ithaca, NY
Gu WY, Lai WM, Mow VC (1998) A mixture theory for charged-hydrated soft tissues containing multi-electrolytes: passivetransport and swelling behaviors. Trans ASME J Biomech Eng120:169–180
Happel J, Brenner H (l986) Low Reynolds number hydrodynamics,vol 1. Nijhoff, The Hague
He X, Dembo M (1997) On the mechanics of the first cleavagedivision of the sea urchin egg. Exp Cell Res 233:252–273
Katchalsky A, Michaeli I (1955) Polyelectrolyte gels in salt solu-tions. J Polym Sci 15:69–86
Lai WM, Hou JS, Mow VC (1991) A triphasic theory for theswelling and deformation behaviors behaviors of articular car-tilage. J Biomech Eng 113:245–258
Landau LD, Lifshits EM, Pitaevskii LP (1980) Statistical physics,3rd edn. Pergamon Press, Oxford
Lanir Y (1996) Plausibility of structural constitutive equations forswelling tissues-implications of the C-N and S-E conditions.Trans ASME J Biomech Eng 118:10–16
Maskawa J, Takeuchi T, Maki K, Tsujii K, Tanaka T (1999)Theory and numerical calculation of pattern formation inshrinking gels. J Chem Phys 110:10993–10999
Mogilner A, Oster G (1996) Cell motility driven by actin poly-merization. Biophys J 71:3030–3045
Oliver T, Dembo M, Jacobson K (1995) Traction forces in loco-moting cells. Cell Motil Cytoskel 31:225–240
Onuki A (1989) Theory of pattern formation in gels: surface foldingin highly compressible elastic bodies. Phys Rev A 39:5932–5948
Overbeek JTG (1956) The Donnan equilibrium. Prog BiophysBiophys Chem 6:58–84
Roberts T, Stewart M (2000) Acting like actin: the dynamics of thenematode major sperm protein (MSP) cytoskeleton indicate apush-pull mechanism for amoeboid cell motility. J Cell Biol149:7–12
Sekimoto K, Suematsu N, Kawasaki K (1989) Spongelike domainstructure in a two-dimensional model gel undergoing volume-phase transition. Phys Rev A 39:4912–4914
Siegel R, Falamarzian M, Firestone B, Moxley B (1988) pH-con-trolled release from hydrophobic/polyelectrolyte copolymerhydrogels. J Controlled Release 8:179–182
Tanaka H (1997) Viscoelastic model of phase separation. Phys RevE 56:4451–4462
Tanaka T, Fillmore D (1979) Kinetics of swelling of gels. J ChemPhys 70:1214–1218
Tanaka T, Hocker L, Benedek G (1973) Spectrum of light scatteredfrom a viscoelastic gel. J Chem Phys 59:5151–5159
Tokita M, Tanaka T (1991) Friction coefficient of polymernetworks of gels. J Chem Phys 95:4613–4619
Tomari T, Doi M (1995) Hysteresis and incubation in the dynamicsof volume transition of spherical gels. Macromolecules28:8334–8343
Tzafriri A (2000) Mathematical modeling of diffusion-mediatedrelease from bulk degrading matrices. J Controlled Release63:69–79
Tzafriri A, Bercovier M, Parnas H (2002) Reaction diffusion modelof the enzymatic erosion of Insoluble fibrillar matrices. BiophysJ 83:776–793
Wang C, Li Y, Hu Z (1997) Swelling kinetics of polymer gels.Macromolecules 30:4727–4732
Yamaue T, Taniguchi T, Doi M (2000) Shrinking process of gels bystress-diffusion coupled dynamics. Prog Theor Phys Supp138:416–417
158
Copyright © 2022 FDOKUMEN




















