
Efficiency of hydrogen recovery from reformate with a polymer electrolyte hydrogen pump

ARTICLE IN PRESS
0378-4371/$ - se
doi:10.1016/j.ph
�CorrespondE-mail addr
Physica A 365 (2006) 307–316
www.elsevier.com/locate/physa
The effect of oxygen–oxygen repulsion in the productionof water from hydrogen–oxygen reaction on the surface:
A computer simulation study
W. Ahmada,�, M. Parvezb, Musa Kaleem Baloachc, M. Khalidd
aPhysics Research Division, Pakistan Institute of Nuclear Science & Technology, P.O. Nilore, Islamabad, PakistanbComputer Division, Pakistan Institute of Nuclear Science & Technology, P.O. Nilore, Islamabad, Pakistan
cChemistry Department, Gomal University, D.I. Khan, PakistandPhysics Department, Gomal University, D.I. Khan, Pakistan
Received 19 April 2005; received in revised form 4 August 2005
Available online 21 October 2005
Abstract
The formation of water from hydrogen–oxygen reaction on a metal surface is of immense importance due to the
technological reasons. This reaction has been studied via a thermal mechanism on a Pt single crystal surface where the two
molecules, H2 and O2, have been adsorbed dissociatively in atomic form. The reaction takes place between the adsorbed
atoms through an intermediate OH radical. We have studied this reaction via a thermal (Langmuir–Hinshelwood
mechanism) as well as a non-thermal mechanism (precursor mechanism) by the Monte Carlo computer simulations. In this
study, we have applied a novel approach based upon the experimental observations that the dissociated oxygen atoms do
not sit next to one another on a catalytic surface. Some interesting results like the shifting of the phase transition points,
the broadening of the reaction window width and the elimination of the second-order phase transition in the non-thermal
reaction mechanism are obtained by considering various possibilities of the reaction scheme. The phase diagrams as well as
the snapshots of the surface covered with the reacting species are presented.
r 2005 Elsevier B.V. All rights reserved.
Keywords: Monte Carlo simulations; Catalytic surface reactions; Phase diagrams
1. Introduction
The hydrogen–oxygen (H2–O2) reaction is thought to be the simplest to study in the formation of water onmetal surfaces. The detailed knowledge of such a simple reaction would surely increase the possibility ofunderstanding more complex but technologically important reactions. Hydrogen burning fuel cells,electrolysis of water and photon-assisted electrochemical processes are a few areas, which demand the deepinsight to such reactions. In addition, the production of heavy water and the management of tritium in nuclear
e front matter r 2005 Elsevier B.V. All rights reserved.
ysa.2005.08.084
ing author.
ess: [email protected] (W. Ahmad).

ARTICLE IN PRESSW. Ahmad et al. / Physica A 365 (2006) 307–316308
reactors may utilize catalyzed exchange between hydrogen and water [1]. So the interaction of hydrogen,oxygen and water with a catalytic surface is of great interest.
It is observed that relatively less attention has been paid to the H2–O2 reaction compared with the CO–O2
reaction on the catalytic surfaces. The phenomenon of H2–O2 re-combination on metal surfaces was firstdiscussed by Faraday [2] who identified it as a property of a clean metal surface. Then Langmuir [3] made veryuseful kinetic studies in understanding this catalytic reaction. He deduced a mechanism for the H2–O2 reactionon a platinum (Pt) surface. It is generally agreed that Pt is the most active catalyst for this type of reaction atroom temperature or below. The reaction proceeds either via a thermal (Langmuir–Hinshelwood) or via anon-thermal (precursor) mechanism. In the thermal mechanism, both reactants adsorb (chemisorb) on thesurface before the reaction while in the non-thermal mechanism, one component is weakly held (physisorb)and reacts with a strongly adsorbed component. Christmann [4] gave a comprehensive review of theadsorption of hydrogen on metal surfaces.
The catalytic oxidation of hydrogen, which corresponds to the reaction of H2 and O2 through intermediateOH radical, has been the subject of numerous investigations [5–21]. On a Pt (1 1 1) single crystal surface, thereaction was found to take place even at 120K [14] below the desorption temperature of water (170K). Thedimer H2 and O2 are chemisorbed dissociatively in an atomic form and hence a mechanism comprising thereaction of the adsorbed H and O atoms through an intermediate OH radical has been suggested. However,the experimental data indicate that such a simple mechanism does not hold true in some cases [18].
Based on the Langmuir–Hinshelwood (LH) mechanism, Albano [5] studied this reaction system throughcomputer simulations. He found that the system exhibits a discontinuous phase transition at YH ¼ 2/3 (whereYH is the normalized partial pressure of hydrogen in the gas phase), separating the O+OH+vacanciespoisoned (saturated) state from another H+vacancies poisoned state. However, the desorption of the two Hatoms (in the form of H2 molecule), with probability P ¼ 1, produces a steady reactive state (SRS) forYH ¼ 0.70. This reactive state with a continuous production of water molecules continues until YH ¼ 1.0.Khan et al. [8] have shown that for Po1, a SRS, which is separated from the poisoned states by twoirreversible transitions, is obtained for this system. The position of the transition points depends upon thevalue of P. The observations of Albano [5] for P ¼ 1 is, therefore, one extreme case. He has also introducedthe OH+OH-H2O+O reaction in the simple simulation procedure. This reaction step cannot be neglectedat low temperatures and even at low oxygen coverage in actual catalytic systems [13,21]. The addition of thisstep leads to a reactive window (i.e., the continuous production of H2O) with two irreversible phase transitionsat Y1 ¼ 0.4525 and Y2 ¼ 0.6263. However, this situation has proved controversial as Zhonghuti et al. [7] haveshown by means of the Monte Carlo simulation and the mean field theory that the OH–OH reaction cannotchange the qualitative critical behavior of the system.
Many authors have given the evidence that non-thermal processes are also important to understand thecatalytic reactions [22–31]. The transient non-thermal mobility caused by the inability to instantaneouslydissipate the energy gained by a particle after the formation of the surface bond seems to be a common processin nature. Meakin [28] studied a monomer–monomer reaction of the type A+B-AB to explore the effect ofEley–Rideal process (direct reaction between a gas phase B atom and an adsorbed A atom) on the phasediagram of the reaction system. For a square lattice, the moment YB 6¼0, the continuous production of ABstarts which continues to increase till YB ¼ 0.495, where the surface is poisoned with B atoms.
Harris et al. [11] have studied the catalytic reaction of H2 and O2 on a polycrystalline Pt with a quartzcrystal microbeam in their early work. They have analyzed the data by means of a mean field approach andhave shown that this particular reaction system is an example of a precursor mechanism. Harris and Kasemo[31] have given a detailed discussion on the precursor mechanism of surface reactions. This mechanisminvolves direct collisions between chemisorbed species and molecules or atoms that are trapped in theneighborhood of the surface but have not thermalized. They have also shown that a H2 molecule after strikingthe surface of a metal breaks apart, leaving H atoms bound to the surface with a high parallel kinetic energy.The precursor kinetics is generally different from those characteristics of LH or ER mechanisms [11,23].
Khan et al. [32] have studied a dimmer–dimer catalytic surface reaction of the type A2+2B2-2AB2
(mimicking the production of water) along the lines envisaged by Harris and Kasemo [31]. Through thisprecursor model, they have reproduced some experimental results of the real system. They assumed that a B2
molecule incident on a metal site broke apart and produced two mobile B atoms on the surface. This scheme

ARTICLE IN PRESSW. Ahmad et al. / Physica A 365 (2006) 307–316 309
has added some interesting features in the phase diagram of the dimmer–dimer reaction, which were not seenby considering the LH mechanism only [9]. The model showed a steady reactive region limited by a continuousand a discontinuous irreversible transition. The phase diagram was qualitatively similar to the ZGB modelproposed by Ziff et al. [33] based on the LH mechanism. However, the width of the reactive region increasedwhen the mobility of the precursor was increased. The continuous transition point disappeared when theprecursor motion was extended up to the third nearest neighborhood. Some experimentally known facts, suchas the occurrence of the first-order transition and the dependence of the reaction rate on the coverage of Aatoms were also observed in this model.
The experiments performed under ultra-high-vacuum conditions using scanning tunneling microscope(STM) on a Pt (1 1 1) surface showed that the oxygen molecule impinging upon the surface dissociates into twooxygen atoms [34]. It was observed that the separation between the two O atoms is twice the Pt lattice constantas is directly seen in Fig. 1(a) of Ref. [34]. The possibility that the two oxygen atoms occupy adjacent sites onthe surface is very little. Keeping this experimental observation in view, we have introduced a novel approachin our simulation procedure that after dissociation, the two O atoms are not allowed to sit next to one anotheron adjacent lattice sites, i.e., if one oxygen atom is occupying a surface site, the other O atom may go to secondor third neighborhood for adsorption or reaction.
The aim of this work is to study the formation of water on the basis of the LH as well as the precursormechanisms through the Monte Carlo simulations with the novel approach of repulsion of oxygen–oxygen atomson the surface. The paper is structured as follows: in the next section, the reaction mechanism and the simulationprocedure are discussed. The results are discussed in Section 3. Finally, the conclusions are inferred in Section 4.
2. Reaction model and simulation procedure
An infinite reservoir filled with H2 and O2 molecules is assumed having concentrations YH and YO,respectively. The total concentration is normalized to unity, i.e., YH+YO ¼ 1. The reservoir is in contact witha square surface of 64� 64 lattice size. It has been observed that an increase in the lattice size changes thecritical pressures slightly but the overall qualitative nature of the phase diagram is not affected [8,9]. Theperiodic boundary conditions are applied to mimic the infinite lattice.
The first part of the model for this reaction scheme, based upon the well-known LH thermal mechanism,can be written in the form of the following equations:
O2g þ 2x-Oc þOc; (1)
H2g þ 2x-Hc þHc; (2)
Hc þOc-OHc þ x; (3)
Hc þOHc-H2Og þ 2x; (4)
where x refers to a vacant site on the surface and superscripts g and c stand for the gas phase and thechemisorbed adatoms, respectively. The desorption of the species from the surface and the diffusion on thesurface has not been considered in this model. In this study, we have introduced the repulsion of the species onthe surface based upon the experimental findings [34].
The simulation starts with a clean surface, i.e., the entire lattice sites are empty. The selection of any one ofthe two molecules (H2 or O2) is made at random with relative probabilities YH and YO, respectively. If thestriking molecule is H2, then it requires two sites to be vacant in order to produce two chemisorbed atoms(step-1). If the striking molecule is O2, then it also requires two vacant sites to get adsorbed (step-2). In oursimulation, there is only one variable, i.e., the feed concentration (partial pressure) of hydrogen (YH). Theequilibrium coverages are measured as a function of YH. In order to locate the critical points, the programruns up to 50,000 Monte Carlo (MC) cycles. If the program completes 50,000 MC cycles without the latticegetting poisoned, the particular point is said to be within the SRS. Then in order to get the coveragescorresponding to a SRS, the initial 10,000 MC cycles are disregarded to achieve the equilibrium and theaverages are taken over the subsequent 40,000 MC cycles. The values of the coverages are obtained after every
ARTICLE IN PRESS
0.0 0.2 0.4 0.6 0.8 1.0
0.0
0.2
0.4
0.6
0.8
1.0
H
O
OH
H2O
0.0 0.2 0.4 0.6 0.8 1.0
0.0
0.2
0.4
0.6
0.8
1.0
H
O
OH
H2O
Sur
face
cov
erag
e an
d pr
oduc
tion
rate
Sur
face
cov
erag
e an
d pr
oduc
tion
rate
Feed concentration
Feed concentration
(a)
(b)
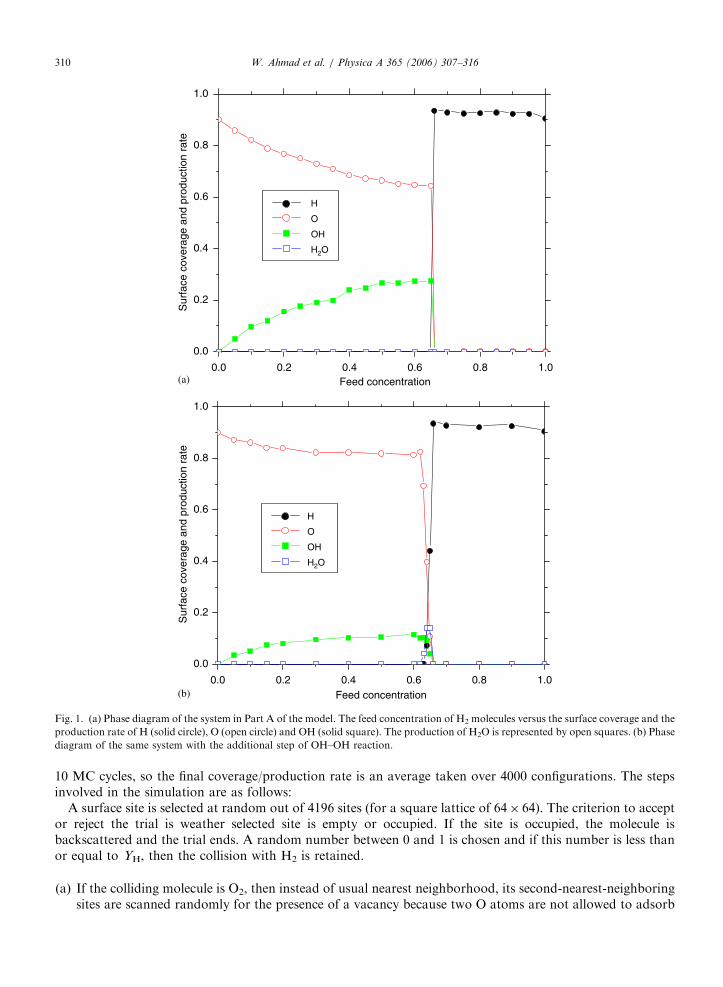
Fig. 1. (a) Phase diagram of the system in Part A of the model. The feed concentration of H2 molecules versus the surface coverage and the
production rate of H (solid circle), O (open circle) and OH (solid square). The production of H2O is represented by open squares. (b) Phase
diagram of the same system with the additional step of OH–OH reaction.
W. Ahmad et al. / Physica A 365 (2006) 307–316310
10 MC cycles, so the final coverage/production rate is an average taken over 4000 configurations. The stepsinvolved in the simulation are as follows:
A surface site is selected at random out of 4196 sites (for a square lattice of 64� 64). The criterion to acceptor reject the trial is weather selected site is empty or occupied. If the site is occupied, the molecule isbackscattered and the trial ends. A random number between 0 and 1 is chosen and if this number is less thanor equal to YH, then the collision with H2 is retained.
(a)
If the colliding molecule is O2, then instead of usual nearest neighborhood, its second-nearest-neighboringsites are scanned randomly for the presence of a vacancy because two O atoms are not allowed to adsorb
ARTICLE IN PRESSW. Ahmad et al. / Physica A 365 (2006) 307–316 311
next to one another. The trial ends if no vacancy is found there because O2 needs two vacant sites foradsorption. In case a second vacant site is available, then the four nearest-neighboring sites of each of thetwo selected sites are scanned. The trial ends if any O atom is found in any of their neighboring sites. If noO atom is found, then step-1 takes place to produce two chemisorbed O atoms.
(b)
If the colliding molecule is H2, then four nearest neighbors of the selected empty site are scanned randomlyfor the presence of another vacancy. The trial ends if no vacancy is found there because it also needs twovacant sites for adsorption. In case a second vacant site is available, then the H2 molecule is adsorbed inatomic form at these adjacent vacant sites as per step-2.After adsorption, step-3 takes place if H and O are sitting adjacent to one another. Being a lighter particle, itis more probable that H leaves its place and combines with O to form OH. So this OH occupies the site whereO was sitting and the other site where H was sitting becomes empty. Then the four nearest neighbors of OHare scanned to check the presence of H atoms. If an H atom is found, then H2O is formed, which desorbs fromthe surface leaving behind two vacant sites.
In part B of this model, we introduced the following additional step:
OHc þOHc-H2Og þOc þ x: (5)
Here, if an OH finds another OH sitting in its neighborhood, water is formed and one site is vacatedwhereas the O atom occupies the other site. This O atom then looks for a H atom in its four adjacent sites toform an OH for the reaction’s step-3.
In addition to the usual adsorption of atoms at second-nearest neighborhood, we consider the followingpossibilities in our simulations:
(i)
the two atoms may sit in the third-nearest-neighboring sites, (ii) the two atoms may sit in the second-nearest and third-nearest-neighboring sites with equal probability.Finally, H2 molecule was considered as a precursor where the following four steps were added instead ofstep-1 mentioned earlier:
H2g þ x-Hp þHp þ x; (6)
Hp þOc-OHc; (7)
Hp þOHc-H2Og þ x; (8)
Hp þ x-Hc: (9)
The difference in the two approaches is that here H2 needs only one vacant site for the process instead oftwo adjacent vacant sites for adsorption. So if the colliding molecule is H2, then after collision with thisrandomly chosen site two precursor atoms (HP) are produced via step-6, which move into the environment ofimpact (R). We have considered three different ranges of this environment: (i) first-nearest neighborhood(R ¼ 1), (ii) second-nearest neighborhood (R ¼
ffiffiffi
2p
and (iii) third-nearest neighbor (R ¼ 2). Each environmentconsists of a specific pattern for the set of sites around the striking site. For example, the first environmentconsists of four nearest-neighboring sites. The second environment contains four-nearest and four second-nearest-neighboring sites whereas the third environment contains all the eight sites of the second environmentand additional four third-nearest-neighboring sites.
During its motion in a particular environment, a precursor HP strikes a surface site, which is randomlychosen out of the total number of sites of the environment. If this site is occupied by a chemisorbed atom OC,then reaction step-7 takes place and the precursor ends its life with the formation of an OH radical. In asimilar way if the randomly picked site is occupied by a chemisorbed radical OHC, then reaction step-8 takesplace and the precursor ends its life. So the formation of H2O takes place, which desorbs from the surface andthe site occupied by OHC is vacated. Therefore, the collision of this precursor atom is taken at random so thatthe formation of OHC and H2O is equally probable. In case the precursor does not find any chemisorb reacting

ARTICLE IN PRESSW. Ahmad et al. / Physica A 365 (2006) 307–316312
species within a particular environment, then it takes one vacant site (randomly chosen) from the environmentto become chemisorbed (step-9) and the precursor ends its life. This chemisorbed atom scans its fourneighboring sites for the presence of OC or OHC in order to complete reaction step-3 and step-4, respectively.If an OC is found, then an OHC is formed and the site occupied by HC is vacated. If an OHC is found, thenH2O is formed, which desorbs from the surface and the sites occupied by HC and OHC are vacated. It shouldbe noted that after adsorption, the OC atoms also scan their respective nearest neighborhood in order tocomplete possible reaction step-3.
If one of the precursors does not end its life through any of the above-mentioned ways, then it adsorbs onthe surface through reaction step-9. If both the precursors of the H2 molecule do not end their lives, then theyreturn to the gas phase and hence the trial ends.
Fig. 2. (a) Snapshot of the O+OH+vacancies poisoned state. Here *, 0 and+represents H, O and OH, respectively. (b) Snapshot of the
H+vacancies poisoned state. (c) Snapshot of the steady reaction state (SRS).
ARTICLE IN PRESSW. Ahmad et al. / Physica A 365 (2006) 307–316 313
3. Results and discussion
The results of 2H2+O2-2H2O reaction on a square lattice, using the LH mechanism, without repulsion ofspecies on the surface are well known [5]. The system exhibits a discontinuous phase transition at YH ¼ 0.66(where YH is the partial pressure of hydrogen in the gas phase) separating an O+OH+vacancies poisonedstate from another H+vacancies poisoned state. Applying the same mechanism first, we discuss the simplecase when one H atom (after dissociation of a H2 molecule) occupies the site of impact and the other H atomoccupies the second-nearest-neighboring site. The phase diagram of the system, with and without OH–OHreaction is given in Figs. 1(a) and (b), respectively. The snapshots of the species sitting on the surface beforeand after the transition points as well as the steady reactive region are shown in Fig. 2. Fig. 2(a) represents anO+OH+vacancies poisoned state, i.e., before the first transition point; Fig. 2(b) is of a H+vacanciespoisoned state, i.e., after the second transition point; and Fig. 2(c) is of the steady reactive region where thecontinuous production of H2O exists, i.e. between the two transition points. The results with and withoutOH–OH reaction are exactly in accordance with the findings of Albano [5] where a first-order phase transitionoccurred at YH ¼ 0.66.
Interesting results were found when the other H atom was allowed to sit at the third-nearest-neighboringsite. Without OH–OH reaction, production of H2O started at Y1 ¼ 0.672 and terminated at Y2 ¼ 0.720.
0.0 0.2 0.4 0.6 0.8 1.0
0.0
0.2
0.4
0.6
0.8
1.0
H O OH H2O
Sur
face
cov
erag
e an
d pr
oduc
tion
rate
Feed concentration
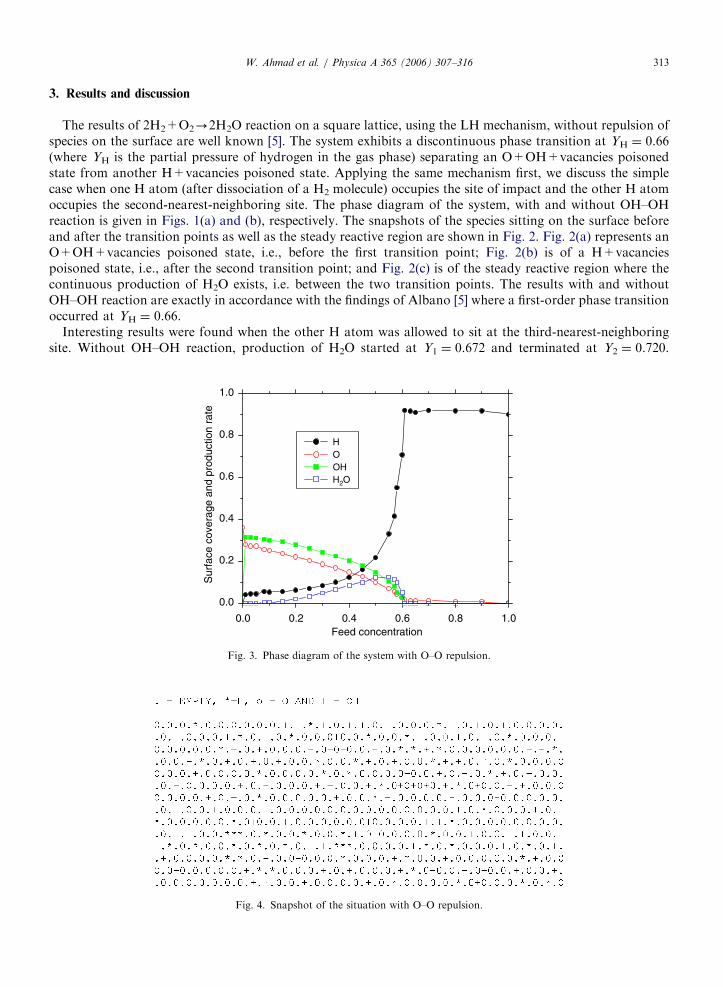
Fig. 3. Phase diagram of the system with O–O repulsion.
Fig. 4. Snapshot of the situation with O–O repulsion.
ARTICLE IN PRESSW. Ahmad et al. / Physica A 365 (2006) 307–316314
Considering OH–OH reaction, the production started a little earlier (Y1 ¼ 0.652) but the termination pointwas the same. The two transition points shift to lower values (Y1 ¼ 0.63 and Y2 ¼ 0.68) when the other Hatom was allowed to sit at second or third-nearest neighbor with equal probability.
Similar to this, no SRS was found when O atoms after dissociation of an O2 molecule were allowed to sit atthe next nearest neighborhood. A small reactive window of width ’ 0:05 with Y1 ¼ 0.62 and Y2 ¼ 0.67 wasobserved when the other O atom was allowed to sit at the third-nearest neighbor in both cases (with andwithout OH–OH reaction).
Introducing O–O repulsion, i.e., the two O atoms were not allowed to sit next to one another, theproduction of H2O started much earlier. The transition points were found to be at Y1 ¼ 0.15 and Y2 ¼ 0.65
0.0 0.2 0.4 0.6 0.8 1.0
0.0
0.2
0.4
0.6
0.8
1.0
H
O
OH
H2O
0.0 0.2 0.4 0.6 0.8 1.00.0
0.2
0.4
0.6
0.8
1.0
H
O
OH
H2O
Sur
face
cov
erag
e an
d pr
oduc
tion
rate
Sur
face
cov
erag
e an
d pr
oduc
tion
rate
Feed concentration
Feed concentration
(a)
(b)
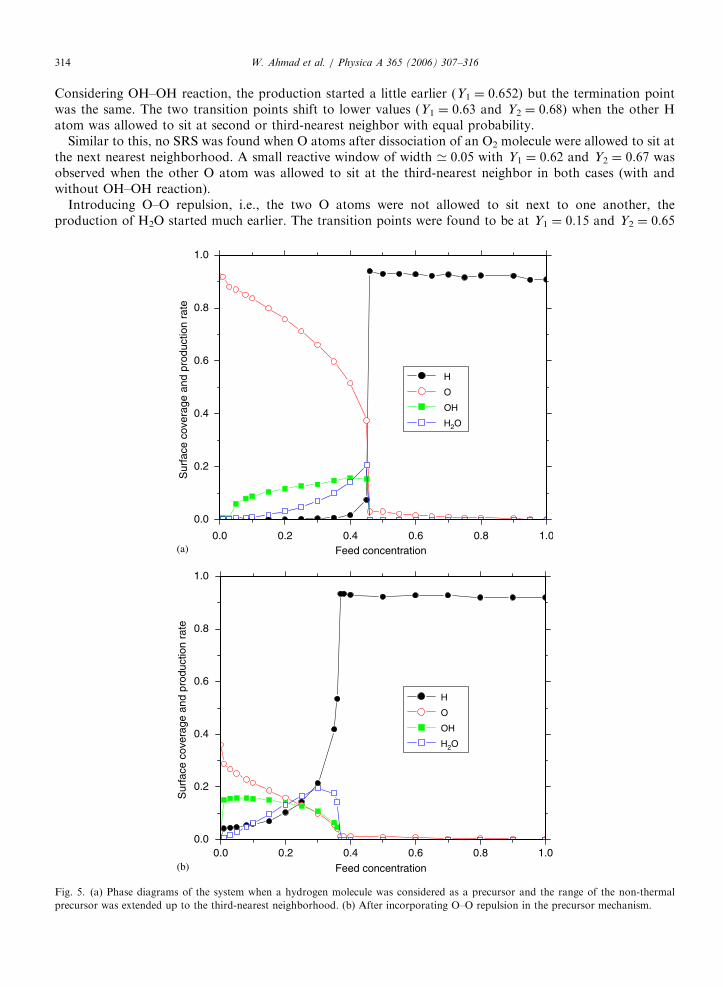
Fig. 5. (a) Phase diagrams of the system when a hydrogen molecule was considered as a precursor and the range of the non-thermal
precursor was extended up to the third-nearest neighborhood. (b) After incorporating O–O repulsion in the precursor mechanism.

ARTICLE IN PRESSW. Ahmad et al. / Physica A 365 (2006) 307–316 315
when one oxygen atom occupied the vacant site of impact and the other O atom occupied the second-nearest-neighboring site. Y1 shifted towards even lower value (Y1 ¼ 0.05) when the other O atom was allowed to sit atthe third-nearest neighbor. Here, the window width (production rate) was exceptionally large (�0:05). Onemore possibility was also considered, i.e., the other O atom was allowed to occupy the second or thirdneighboring site with equal probability. No change in the results was found and the transition points remainedat Y1C0.15 and Y2C0.60. This means that the selection of any one of the two gases as feed concentration didnot change the nature of transition but the window width was much higher when O–O repulsion wasconsidered. A phase diagram of this system is given in Fig. 3 and the snapshots in Fig. 4.
A charge appeared when the hydrogen molecules were considered as precursors. The first-order phasetransition point was eliminated and the production of water started as soon as the feed concentration ofhydrogen increased from zero. The phase diagram of the case with the precursor range up to the third-nearestneighborhood is shown in Fig. 5(a) and after incorporating O–O repulsion, the diagram is shown in Fig. 5(b).In the latter case, the transition points are at Y1 ¼ 0.0 and Y2 ¼ 0.37. These findings are very similar to ourprevious studies with the precursor mechanism on the dimmer–dimer catalytic reaction [35–37] when the rangeof the precursor is extended up to the third nearest neighborhood. This is also in accordance with theexperimental observation that the first-order phase transition has never been observed in such reactions. So weconclude that the non-thermal precursor mechanism gives results closer to the real situation than theconventional LH thermal mechanism.
4. Conclusion
The application of the Monte Carlo computer simulations to study the production of H2O from H2–O2
reaction on a metal surface has produced some interesting results when a novel approach of O–O repulsion isintroduced in the usual LH procedure. The width of the reactive window, i.e., the production of water,increased to a larger extent. Some interesting results were found for the adsorption of the species at the third-nearest-neighboring site. Here, continuous production of water was observed bounded by two transitionpoints. Different possibilities of adsorption and reaction were applied to eliminate the second-order phasetransition point. Finally, this transition disappeared when H2 was considered as a precursor. The productionof H2O starts as soon as the feed concentration of H2 is non-zero. These findings are close to the experimentalobservations.
References
[1] M. Hammerbi, J.P. Butler, W.H. Stevens, Int. J. Hydrogen Energy 4 (1979) 85.
[2] M. Faraday, Experimental Researches in Electricity, vol.1, Richard and John E. Taylor, London, 1849, p. 561.
[3] I. Langmuir, Trans. Faraday Soc. 17 (1922) 621.
[4] K. Christmann, Bull. Soc. Chem. Belg. 88 (1979) 519.
[5] E.V. Albano, J. Phys. A 25 (1992) 2557.
[6] A. Maltz, E.V. Albano, Surf. Sci. 277 (1992) 414.
[7] H. Zhonghaui, Y. Lingfa, X. Houwen, Phys. Rev. E 58 (1998) 234.
[8] K.M. Khan, K. Yaldram, N. Ahmad, Qamar-ul-Haque, Physica A 268 (1999) 89.
[9] K.M. Khan, K. Yaldram, N. Ahmad, J. Chem. Phys. 109 (1998) 5054.
[10] K. Yaldram, K.M. Khan, N. Ahmad, M.A. Khan, J. Phys. A 26 (1993) 2663.
[11] J. Harris, B. Kesemo, E. Tarnqvist, Surf. Sci. 105 (1981) L288.
[12] L.K. Verheij, Surf. Sci. 371 (1997) 100.
[13] L.K. Verheij, M. Freitag, M.B. Hugenschmidt, I. Kempf, B. Poelsema, G. Comsa, Surf. Sci. 272 (1992) 276.
[14] G.B. Fisher, J.L. White, Surf. Sci. 139 (1984) 43.
[15] K.M. Ogle, J.M. White, Surf. Sci. 139 (1984) 43.
[16] J.L. Gland, G.B. Fisher, E.B. Kollin, J. Catal. 77 (1982) 263.
[17] L.K. Verheij, M.B. Hugenschmidt, Surf. Sci. 324 (1995) 185.
[18] S. Volkening, K. Bedurfug, K. Jacobi, J. Wintterlin, G. Ertl, Phys. Rev. Lett. 83 (1999) 2672.
[19] B. Hellsing, B. Kasemo, V.P. Zhdanov, J. Catal. 132 (1991) 210.
[20] T.A. Germer, W. Ho, Chem. Phys. Lett. 163 (1989) 449.
[21] A. Anton, D.C. Cadogan, Surf. Sci. 239 (1990) L548.
[22] R.P. Thorman, D. Anderson, S.L. Bernasek, Phys. Rev. Lett. 44 (1980) 743.

ARTICLE IN PRESSW. Ahmad et al. / Physica A 365 (2006) 307–316316
[23] N.O. Wolf, D.R. Burgess, D.K. Hoffman, Surf. Sci. 100 (1980) 453.
[24] S.P. Singh-Boparai, M. Bowker, D.A. King, Surf. Sci. 53 (1975) 55.
[25] G. Boato, P. Cantini, L. Mattera, J. Chem. Phys. 65 (1976) 544.
[26] G. Comsa, R. David, B.J. Schumacher, Surf. Sci. 95 (1980) L210.
[27] B. Jackson, M. Persson, J. Chem. Phys. 103 (1995) 6257.
[28] P. Meakin, J. Chem. Phys. 93 (1990) 2903.
[29] H. Brune, J. Wintterlin, R.J. Behm, G. Ertl, Phys. Rev. Lett. 68 (1992) 624.
[30] V.D. Pereyra, E.V. Albano, Appl. Phys. A 57 (1993) 291.
[31] J. Harris, B. Kasemo, Surf. Sci. 105 (1981) L281.
[32] K.M. Khan, E.V. Albano, R.A. Monetti, Surf. Sci. 481 (2001) 78.
[33] R.M. Ziff, E. Gulari, Y. Barshad, Phys. Rev. Lett. 56 (1986) 2553.
[34] J. Wintterlin, R. Schuster, G. Ertl, Phys. Rev. Lett. 77 (1996) 123.
[35] W. Ahmad, K.M. Khan, Nucleus 37 (2000) 125.
[36] K.M. Khan, W. Ahmad, K. Iqbal, Int. J. Mod. Phys. C 14 (2003) 141.
[37] M. Kahlid, A.U. Qaisrani, W. Ahmad, Chin. Phys. Lett. 6 (2005) 1553.
Copyright © 2022 FDOKUMEN




















