
The effect of low estrogen state on serotonin transporter function in mouse hippocampus: A...
11
Research Report The effect of low estrogen state on serotonin transporter function in mouse hippocampus: A behavioral and electrochemical study Paul P. Bertrand a , Udeni T. Paranavitane a,b , Carolina Chavez b , Andrea Gogos b,c , Margaret Jones d , Maarten van den Buuse b,c, * a Department of Physiology, University of Melbourne, Parkville VIC 3010, Australia b Behavioural Neuroscience Laboratory, Mental Health Research Institute, Parkville VIC 3052, Australia c Department of Pharmacology, University of Melbourne, Parkville VIC 3010, Australia d Prince Henry’s Institute of Medical Research, Clayton VIC 3168, Australia Accepted 11 October 2005 Available online 18 November 2005 Abstract Defects in serotonergic transmission, including serotonin transporter (SERT) function, have been implicated in depression, anxiety disorders and some aspects of schizophrenia. The sex steroid hormone estrogen is known to modulate functional SERT activity, but whether it is up- or down-regulated is unclear. The aim of the present study was to examine the effect of a low estrogen state in mice on the behavioral effect of drugs acting through the SERT, serotonin uptake kinetics and SERT density in the hippocampus. We compared control mice, ovariectomized (OVX) C57BL/6J mice and aromatase knockout (ArKO) mice that are unable to produce estrogen. Fluoxetine treatment, but not fenfluramine treatment, significantly increased prepulse inhibition (PPI), a measure of sensorimotor gating, in C57BL/6J mice. The effect of fluoxetine was greater in OVX compared to sham-operated mice. In ArKO and J129 wild-type mice, fluoxetine increased PPI to the same extent while fenfluramine increased PPI more in ArKO mice compared to controls. Measurement of the time-course for diffusion and reuptake of exogenous serotonin in the CA3 region of the hippocampus showed that, in OVX mice, the fluoxetine-induced slowing of signal decay after application of serotonin was enhanced when compared to sham-operated controls. Similarly, in ArKO mice, the effect of fluoxetine was enhanced, suggesting that SERT function was greater than in J129 wild-type controls. Measurement of SERT density by [ 3 H]- citalopram autoradiography, revealed an 18% decrease in hippocampus of OVX mice compared to intact controls. SERT density was also significantly reduced in nucleus accumbens (26%) but not in other regions, such as the raphe nuclei. Together, these results suggest that a low estrogen state increases SERT activity in the hippocampus despite an apparent reduction in SERT density. The behavioral consequences of these changes depend on the model of estrogen state used. D 2005 Elsevier B.V. All rights reserved. Theme: Neurotransmitters, modulators, transporters, and receptors Topic: Serotonin Keywords: Serotonin; Serotonin transporter; Estrogen; Hippocampus; Aromatase knockout (ArKO) 1. Introduction Defects in serotonergic transmission, including serotonin transporter (SERT) function, have been implicated in depression, anxiety disorders and some aspects of schizo- phrenia [18]. Levels of the sex steroid hormone, estrogen, have been correlated with the severity of these diseases and estrogen administration is known to change SERT expres- sion and function, but the literature is unclear on the 0006-8993/$ - see front matter D 2005 Elsevier B.V. All rights reserved. doi:10.1016/j.brainres.2005.10.018 Abbreviations: ANOVA, analysis of variance; ArKO, aromatase knockout mice; csf, cerebrospinal fluid; 5-HT, 5-hydroxytrytamine or serotonin; OVX, ovariectomized; PPI, prepulse inhibition; SEM, standard error of the mean; SERT, serotonin transporter * Corresponding author. Behavioural Neuroscience Laboratory, Mental Health Research Institute, 155 Oak Street, Parkville VIC 3052, Australia. Fax: +61 3 93875061. E-mail address: [email protected] (M. van den Buuse). Brain Research 1064 (2005) 10 – 20 www.elsevier.com/locate/brainres
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of The effect of low estrogen state on serotonin transporter function in mouse hippocampus: A...

www.elsevier.com/locate/brainres
Brain Research 1064
Research Report
The effect of low estrogen state on serotonin transporter function in mouse
hippocampus: A behavioral and electrochemical study
Paul P. Bertranda, Udeni T. Paranavitanea,b, Carolina Chavezb, Andrea Gogosb,c,
Margaret Jonesd, Maarten van den Buuseb,c,*
aDepartment of Physiology, University of Melbourne, Parkville VIC 3010, AustraliabBehavioural Neuroscience Laboratory, Mental Health Research Institute, Parkville VIC 3052, Australia
cDepartment of Pharmacology, University of Melbourne, Parkville VIC 3010, AustraliadPrince Henry’s Institute of Medical Research, Clayton VIC 3168, Australia
Accepted 11 October 2005
Available online 18 November 2005
Abstract
Defects in serotonergic transmission, including serotonin transporter (SERT) function, have been implicated in depression, anxiety
disorders and some aspects of schizophrenia. The sex steroid hormone estrogen is known to modulate functional SERT activity, but whether it
is up- or down-regulated is unclear. The aim of the present study was to examine the effect of a low estrogen state in mice on the behavioral
effect of drugs acting through the SERT, serotonin uptake kinetics and SERT density in the hippocampus. We compared control mice,
ovariectomized (OVX) C57BL/6J mice and aromatase knockout (ArKO) mice that are unable to produce estrogen. Fluoxetine treatment, but
not fenfluramine treatment, significantly increased prepulse inhibition (PPI), a measure of sensorimotor gating, in C57BL/6J mice. The effect
of fluoxetine was greater in OVX compared to sham-operated mice. In ArKO and J129 wild-type mice, fluoxetine increased PPI to the same
extent while fenfluramine increased PPI more in ArKO mice compared to controls. Measurement of the time-course for diffusion and
reuptake of exogenous serotonin in the CA3 region of the hippocampus showed that, in OVX mice, the fluoxetine-induced slowing of signal
decay after application of serotonin was enhanced when compared to sham-operated controls. Similarly, in ArKO mice, the effect of
fluoxetine was enhanced, suggesting that SERT function was greater than in J129 wild-type controls. Measurement of SERT density by [3H]-
citalopram autoradiography, revealed an 18% decrease in hippocampus of OVX mice compared to intact controls. SERT density was also
significantly reduced in nucleus accumbens (26%) but not in other regions, such as the raphe nuclei. Together, these results suggest that a low
estrogen state increases SERT activity in the hippocampus despite an apparent reduction in SERT density. The behavioral consequences of
these changes depend on the model of estrogen state used.
D 2005 Elsevier B.V. All rights reserved.
Theme: Neurotransmitters, modulators, transporters, and receptors
Topic: Serotonin
Keywords: Serotonin; Serotonin transporter; Estrogen; Hippocampus; Aromatase knockout (ArKO)
0006-8993/$ - see front matter D 2005 Elsevier B.V. All rights reserved.
doi:10.1016/j.brainres.2005.10.018
Abbreviations: ANOVA, analysis of variance; ArKO, aromatase
knockout mice; csf, cerebrospinal fluid; 5-HT, 5-hydroxytrytamine or
serotonin; OVX, ovariectomized; PPI, prepulse inhibition; SEM, standard
error of the mean; SERT, serotonin transporter
* Corresponding author. Behavioural Neuroscience Laboratory, Mental
Health Research Institute, 155 Oak Street, Parkville VIC 3052, Australia.
Fax: +61 3 93875061.
E-mail address: [email protected] (M. van den Buuse).
1. Introduction
Defects in serotonergic transmission, including serotonin
transporter (SERT) function, have been implicated in
depression, anxiety disorders and some aspects of schizo-
phrenia [18]. Levels of the sex steroid hormone, estrogen,
have been correlated with the severity of these diseases and
estrogen administration is known to change SERT expres-
sion and function, but the literature is unclear on the
(2005) 10 – 20

P.P. Bertrand et al. / Brain Research 1064 (2005) 10–20 11
direction of the change. For example, some neurochemical
studies have found that in female rats in a low estrogen state
there is a decreased SERT gene expression and binding in
comparison to estrogen treatment groups [23]. In the same
species, an in vivo microdialysis study yielded results
consistent with less activity of SERTwhen circulating levels
of estrogen were low as compared to when estrogen levels
were high [21]. Similarly, Lu et al. showed in ovariecto-
mized female macaques that SERT binding and 5-HT
uptake were reduced as compared to hormone treated
animals [19]. In contrast, two recent studies found that in
ovariectomized rats or monkeys there was an up-regulation
of SERT gene expression as compared to animals chroni-
cally treated with estrogen receptor modulators [28,39].
SERT is crucial for terminating the serotonin signal and for
restricting its activity to the synaptic cleft. If estrogen
induces alterations in SERT function, it would have major
consequences for serotonergic activity in the brain and,
hence, neurochemical and behavioral mechanisms that
depend on serotonergic transmission.
The aim of the present project was to use functional
rather than neurochemical measures to look at the effect of
estrogen on SERT function. We used two main experimental
approaches, prepulse inhibition (PPI) of acoustic startle and
electrochemical detection of serotonin reuptake by SERT,
and two animal models of low estrogen state, ovariecto-
mized (OVX) mice and aromatase knockout mice (ArKO).
ArKO mice have been generated by targeted disruption of
the aromatase gene (CYP19) [8] which renders these
animals unable to produce estrogen. These functional
measures were compared to SERT density measurements
in hippocampus and other brain regions of OVX mice.
The first experimental approach used PPI of acoustic
startle to examine sensorimotor gating [11]. A startle reflex
is reduced when a strong stimulus is preceded by a weak
non-startling prestimulus [3]. PPI is controlled by several
central pathways including serotonergic input to the
hippocampus [15,34]. Serotonergic projections to the
hippocampus arise from the raphe nuclei [1,22,36,37] and
PPI is disrupted when these nuclei are selectively lesioned
[17]. Some psychiatric disorders, such as schizophrenia, are
characterized by deficient sensorimotor gating. Because PPI
depends on hippocampal serotonin function, this behavioral
test is a useful way to assess SERT function in the intact
animal. The second experimental approach involved assess-
ing 5-HT uptake kinetics with in vitro electrochemistry. The
clearance of exogenously applied 5-HT can be measured in
hippocampal slices using carbon fiber electrodes and high
speed chronoamperometry [4] and studies have shown that
serotonin clearance is faster in the dorsal raphe nucleus than
in the CA3 region [25]. Similarly, the long-term effects of
SERT inhibitors and the graded destruction of serotonergic
neurons cause a reduction in serotonin clearance [2,26].
Thus, comparisons between animals with altered SERT
activity may be detected as differences in the rate of
serotonin clearance. SERT density was assessed using [3H]-
citalopram autoradiography on sections of different regions
of the brain. The results suggest that a low estrogen state
increases SERT activity in the hippocampus, despite an
apparent reduction in SERT density. The behavioral con-
sequences of these changes depend on the model of estrogen
state used.
2. Experimental procedures
2.1. Animals
Female C57BL/6J mice (weight range 21–27 g), female
ArKO mice (weight range 23–30 g) and wild-type
littermates (weight range 21–27 g) were used. The ArKO
mice and their wild-type littermates were obtained from Dr.
Margaret Jones at Prince Henry’s Institute for Medical
Research (Clayton, Victoria, Australia). Heterozygous males
and females were bred to produce wild-type and homozy-
gous-null offspring. Mice were genotyped at weaning at
Prince Henry’s Institute for Medical Research using a PCR
based strategy [29]. While the genetic background of these
mice is a mix of C57BL/6J and J129 [8], the proportion of
J129 in the cohort of animals used for the present study was
found to be approximately 90% vs. the component of
C57BL/6J (M. Jones, personal communication). Therefore,
for the purpose of this study, the wild-type controls of ArKO
mice will be referred to as J129 wild-type controls.
Mice were housed at the Mental Health Research
Institute (Parkville, Victoria, Australia) in groups of 2–4
in standard mouse cages. C57BL/6J mice had free access to
standard pellet food and tap water. ArKO and J129 wild-
type mice were maintained on a soy-free mouse chow (Glen
Forest Stockfeeders, Australia) and tap water. The mice
were maintained on a 12/12 h light/dark cycle (lights on at
06:30, lights off at 18:30) at a constant temperature of 22
-C. The University of Melbourne Animal Experimentation
Ethics Committee approved all surgical techniques, treat-
ments and experimental protocols.
2.2. Surgery
A total of 15 C57BL/6J mice were ovariectomized at
approximately 12 weeks of age. The animals were anesthe-
tized (isoflurane/oxygen breathing mixture) and placed on a
heat pad maintained at 37 -C. A longitudinal midline incision
was made through the skin and blunt-tipped scissors were
used to separate the connective tissue and to cut through the
peritoneal wall. The ovaries were located and removed
bilaterally and the incision surgically stapled closed. In
sham-operated animals, all steps except removal of ovaries
were carried out. All mice were given a 5 mg/kg subcutane-
ous injection of the non-steroidal, anti-inflammatory analge-
sic carprofen (ZenecarpR, 50 mg/ml, Heriot AgVet, VIC,
Australia) to reduce pain and discomfort. Behavioural
experiments commenced 1–2 weeks after surgery.

P.P. Bertrand et al. / Brain Research 1064 (2005) 10–2012
2.3. Drug treatment protocol
Each subgroup of C57BL/6J mice was randomly
injected with saline and 3 doses of one drug dissolved
in sterile saline. Using a randomized cross-over protocol,
every mouse from each group received all three drug
doses plus saline, with 3 or 4 days allowed between each
experiment to allow for the clearance of drugs. The first
group consisted of 7 OVX and 7 sham-operated mice and
received 3, 10 and 30 mg/kg of the selective SERT
inhibitor, fluoxetine ([T]-N-Methyl-g-[4-(trifluoromethyl)-
phenoxy] benzeneproanamine, Sigma-Aldrich, St. Louis,
Missouri, USA). The second group consisted of 8 OVX
and 8 sham-operated mice and received saline, 3, 10 or
30 mg/kg of the 5-HT releasing drug, fenfluramine (T-N-Ethyl-a-methyl-m-(trifluoromethyl) phenethylamine hydro-
chloride, Sigma-Aldrich). Fluoxetine or fenfluramine was
administered intraperitoneally, 20 min prior to the
commencement of the PPI session. Because of limited
availability of ArKO mice, these animals and their J129
wild-type littermates received only saline, 30 mg/kg of
fluoxetine or 30 mg/kg of fenfluramine in a randomized,
cross-over protocol.
2.4. Prepulse inhibition
Prepulse inhibition (PPI) experiments used a four-unit
automated SR-LAB startle system (San Diego Instruments,
CA, USA) that delivered startle stimuli and recorded
responses to a computer in an adjacent room. Mice were
individually placed into a transparent cylinder on a platform
with a piezoelectric transducer to detect motion. Animals
acclimatized for 5 min before the start of the PPI session.
During the session, animals were delivered a constant
background white noise of 70 decibels (dB) intensity
through speakers in the ceiling of the box.
The PPI session was approximately 50 min in duration
and consisted of 100 trials (pulses) [35,38]. It commenced
and ended with a group of 10 40 ms 115 dB startle-pulse
alone trials. Between these first and last groups of startle-
pulse alone trials, 80 more trials were delivered with a
variable inter-trial interval of 25 s average. These 80 trials
randomly consisted of 20 startle-pulse alone trials and 50
prepulse + startle-pulse trials. Prepulse trials consisted of
an 115 dB startle-pulse stimulus preceded 100 ms before
by a 20 ms prepulse of either 2, 4, 8, 12 or 16 dB (PP2,
PP4, PP8, PP12, PP16) over the 70 dB background (i.e.,
72, 74, 78, 82, 86 dB prepulses). In addition, there were
10 Fno stim_ trials to detect non-specific movement
artifacts. Drug effects on startle amplitude were assessed
using the 40 startle-pulse trials that were recorded
throughout the session. The percentage of PPI was
calculated as the difference of the magnitude of the startle
response to pulse-alone trials minus that to prepulse trials,
divided by the response to pulse-alone trials, expressed as
percentage [35,38].
2.5. Chronoamperometry
At least 3 days after the last behavioral experiments, mice
were deeply anesthetized with pentobarbitone sodium
(Nembutal, Rhone Merieux Australia Pty Ltd., QLD,
Australia) and decapitated. The brain was rapidly removed,
cut in the midsagittal plane and immersed in ice-cold
artificial cerebrospinal fluid (aCSF, composition in mM;
NaCl 126, NaHCO3 25, d-Glucose 11, KCl 2.5, CaCl2 2.4,
MgCl2 1.2, NaH2PO4 1.2) that was bubbled with 95% O2/
5% CO2. The initial solution also contained a high
magnesium concentration (7 mM). A 300 Am transverse
section of the brain was cut using a vibroslicer (Model
MA752, Campden Instruments Ltd., Leicestershire, UK)
then placed in high magnesium aCSF at room temperature
(22.0 T 3 -C) and gently heated to 37 -C for 1 h. One brain
slice was placed in an organ bath of 3 ml volume and held in
place with a metal mesh. The slice was superfused at 3 ml/
min with 35 -C aCSF of normal magnesium concentration
and allowed to equilibrate for 15–20 min. All electrochem-
ical experiments were conducted at 35 -C.A single 7 Am diameter carbon fiber was inserted into a
borosilicate glass micropipette (Harvard Apparatus, SDR
Clinical Technology, NSW, Australia) and pulled with a
micropipette puller (Narishige, Tokyo, Japan). A pellet of
Woods metal (Amac Alloys, VIC, Australia) was inserted
into the open end of the micropipette and copper wire (0.8
mm diameter, Oz wire, Australia) was inserted behind it.
The Woods metal was then gently heated to establish a
stable electrical connection between the copper and carbon
fiber. The exposed carbon fiber was trimmed to 100–200
Am in length. To improve the specificity of the electrodes,
the tips were cleaned in 5% acetone, then coated with
NafionR 117 solution (15 min, 3% solution, Fluka Chemika,
Switzerland). Nafion is a perfluorosulfonated ion exchange
resin that excludes negatively charged molecules such as
monoamine metabolites and ascorbic acid [10].
Serotonin was dissolved in distilled water (5 AM, 5-
hydroxytryptamine creatinine sulfate, Sigma-Aldrich) and
placed in a pipette which was attached to a pressure ejection
system (Picospritzer III, Parker Instrumentation, VIC, Aus-
tralia). This concentration of 5-HT is high enough to saturate
all of the SERT high affinity binding sites in the area local to
the pipette. The same solution was used to calibrate the
carbon fiber electrode at the beginning of each experiment
and to examine clearance of 5-HT from the hippocampus.
An oxidizing potential of +400 mV was applied to the
carbon fiber for 15–60 s with an electrochemistry amplifier
(NPI electronics VA-10, ALA Scientific instruments, NY,
USA) and 5-HT was pressure ejected onto the exposed
carbon fiber (50–150 ms pulse duration, 10 p.s.i.). The
electrode was returned to a holding potential of 0 mV
between applications of 5-HT. Serotonin oxidation signals
were digitized between 1 and 5 kHz (Digidata 1200)
recorded to a personal computer and analyzed with
Axoscope 9 (All from Axon Instruments, CA, USA).

P.P. Bertrand et al. / Brain Research 1064 (2005) 10–20 13
The hippocampus was visualized at 40� magnification
with a dissecting microscope. The carbon fiber electrode and
the 5-HTcontaining pipette were aligned tip to tip in the CA3
region of the hippocampus using mechanical micromanipu-
lators. This area was chosen because the noradrenaline
transporter is not a confounding factor in this area [5]. The
position of the electrode and micropipette was adjusted until
the greatest amplitude oxidation current to application of
serotonin was found—this amplitude corresponded to be-
tween 1 and 5 AM 5-HT. Control 5-HT oxidation currents
were recorded once every 5min for 25–35min (6–8 repeats).
Fluoxetine (1 nM) was superfused onto the bath and allowed
to equilibrate for 15–20 min before 5-HT oxidation currents
were again recorded every 5 min (6–8 repeats). In control
experiments, this concentration of fluoxetine was the
minimum needed to cause a clear reduction in 5-HT
clearance. Fluoxetine was allowed to wash out of the bath
for 15–20 min before recovery recordings were made (6–
8 repeats).
2.6. SERT density
For this experiment, another group of C57BL/6 mice was
ovariectomized or sham-operated as described above. There
were no ArKO mice available for SERT density measure-
ments. Four weeks after surgery, animals were killed by
decapitation and their brains were removed and frozen over
dry ice. Sections of the brain were cut at 14 Am on a cryostat
(Leica CM18-50, Leica Microsystems Nussloch GmbH,
Germany) at �20 -C and thaw-mounted onto gelatinized
microscope slides. Regions selected were at the level of
nucleus accumbens (bregma 1.18 to 0.86), dorsal hippo-
campus (bregma �1.82 mm to �2.18 mm) and raphe nuclei
(bregma �4.36 mm to �4.72 mm) [9]. Brain sections were
stored at �80 -C until use.
Sections were allowed to thaw, washed for 15 min in
Tris–HCl buffer (120 mM NaCl (Asia Pacific Specialty
Chemicals; Seven Hills, NSW, Australia), 50 mM Tris–
HCl (Sigma-Aldrich), pH 7.4) at room temperature and
dried with a stream of cool air. For specific total binding,
Tris–HCl buffer was supplemented with 2 nM [3H]-
citalopram (84.0 Ci/mmol; Amersham Bioscience; Castle
Hill, NSW, Australia). Non-specific binding was deter-
mined by adding 10 AM of the selective SERT inhibitor,
fluoxetine (Sigma-Aldrich) in addition to 2 nM [3H]-
citalopram. Fifty microliters of the 2 nM solution was
dispensed on top of each tissue section and allowed to
incubate for 1 h. Sections were then washed three times (1,
10 and 10 min, respectively) in ice-cold Tris–HCl buffer.
Once dry, sections were partially fixed with 4% parafor-
maldehyde vapor overnight. Fixed sections were apposed to
a BAS-TR2025 phosphoimaging plate (Fuji Imaging
Plates, Berthold Australia Pty. Ltd., Bundoora, Victoria,
Australia) for 10 days at room temperature in a cassette
together with autoradiographic [3H]-micro-scales (Amer-
sham Bioscience, Buckinghamshire, England).
Autoradiographic images on plates were scanned from
the phosphoimaging plate and images were retrieved for
analysis using AIS image analysis software (Analytical
Imaging Station, Imaging Research Inc., Ontario, Canada).
A standard curve was calculated according to the standard
tritium microscales, allowing conversion of photo-stimulat-
ed luminescence (psl) to desintegration units per minute per
milligram (dpm/mg) estimated tissue equivalent (ETE).
Images were then quantified against the standard curve.
The density of specific SERT binding was calculated by
subtracting the density from the non-specific samples from
that of the total binding samples. The numbers were then
converted from dpm/mg ETE to fmol/mg ETE using the
specific activity of the ligand and a decay factor.
2.7. Data analysis
All data were expressed as mean T standard error of the
mean (SEM). A one-way analysis of variance (ANOVA)
(SYSTAT 9, SPSS, Illinois, USA) followed by Bonferroni-
corrected t test was used to analyze body weight and uterus
weight data and SERT density data.
PPI data were analyzed using two- and three-way
ANOVA with repeated measures where appropriate
(SYSTAT). Data for startle amplitude and PPI from sham-
operated and OVX mice or J129 wild-type and ArKO mice
were compared with group (sham-operated and OVX or
J129 wild-type and ArKO) as a between-group factor and
dose (of either fenfluramine or fluoxetine in C57BL/6J) or
treatment (saline, fenfluramine or fluoxetine in ArKO mice)
as within-animal factor. An additional within-animal factor
was prepulse intensity for PPI analysis. Because the main
effect of prepulse intensity was always significant, it will not
be presented here in detail. Where appropriate, pair-wise
ANOVAs were used to further analyze the effect of
treatment or dose between the groups. Differences were
considered significant if P < 0.05.
Differences in electrochemical signals between control
and fluoxetine periods for a single brain slice were analyzed
by comparing the amplitude of the oxidation current and the
times for 50% and 90% decay to occur. Decay times were
calculated by taking the peak amplitude of the average trace
(3–6 repeats) for the control or fluoxetine period, and
finding the times at which the amplitude had fallen to one-
half peak amplitude and to one-tenth peak amplitude. The
absolute time course of the 5-HT induced current was
dependent on the placement of the electrode and the pipette.
To reduce this source of variability, the percent of control
change was calculated and used for all further analyses.
The close proximity of the electrode and 5-HT containing
pipette meant that the initial decay phase of the current (50%
decay time) was due to diffusion of 5-HT away from the
electrode while the effects of the SERT in the surrounding
tissue would be detected primarily at the 90% decay times.
Similarly, the amplitude of the current was primarily
controlled by the concentration of 5-HT in the pipette.
Table 2
Body weights, uterus weights, average startle amplitudes and average
percentage prepulse inhibition (PPI) of sham-operated and ovariectomized
(OVX) C57BL/6J mice treated with fenfluramine
Group Sham (n = 7) OVX (n = 7)
Body weight (g) 21.7 T 0.3 25.1 T 1.2*
Uterus weight (mg) 57.2 T 4.9 16.3 T 0.5*
Uterus/body weight ratio 2.64 T 0.24 0.65 T 0.21*
P.P. Bertrand et al. / Brain Research 1064 (2005) 10–2014
When near-maximal responses were recorded, as they were
in here, fluoxetine would not be expected to enhance them.
The electrochemical data were analyzed using an
ANOVA (SYSTAT). Comparisons were made for the effects
of fluoxetine in Sham vs. OVX mice and for the effects of
fluoxetine in J129 wild-type vs. ArKO mice. Differences
were considered significant if P < 0.05.
Average startle amplitude
Saline 107 T 13 146 T 24
Fenfluramine 3 mg/kg 132 T 13 189 T 38
Fenfluramine 10 mg/kg 160 T 14 282 T 80
Fenfluramine 30 mg/kg 122 T 20 221 T 32
Average %PPI
Saline 48.5 T 2.6 42.1 T 2.6
Fenfluramine 3 mg/kg 50.1 T 4.8 46.2 T 5.2
Fenfluramine 10 mg/kg 54.6 T 3.3 39.6 T 6.4
Fenfluramine 30 mg/kg 50.7 T 3.7 49.4 T 5.3
* P < 0.05 compared with sham-operated mice (t test). For further
statistical comparison, see text.
3. Results
Body weights and uterus weights at the time of sacrifice
were used to confirm the estrogen state of the animals used in
PPI and uptake kinetics experiments (Tables 1–3). Body
weight was significantly higher in one group of OVX mice
vs. their respective sham-operated controls (Tables 1 and 2).
Similarly, ArKO mice had a significantly higher body weight
compared to J129 wild-type mice (Table 3). Uterus weight
was markedly reduced in all OVX mice compared to sham-
operated C57BL/6J mice and, consequently, the uterus
weight/body weight ratio was significantly reduced (Tables
1 and 2). Uterus weight and uterus/body weight ratio were
similarly lower in ArKOmice than in J129 wild-type controls
(Table 3).
3.1. Effect of fluoxetine or fenfluramine on PPI in C57BL/6J
mice
Treatment with fluoxetine significantly increased startle
amplitudes (main effect of dose F(3,42) = 17.9, P < 0.001).
However, ovariectomy did not influence startle amplitude
(Table 1). Treatment with fluoxetine caused an increase of
PPI (main effect of dose F(3,42) = 4.2, P = 0.011) and this
effect was greater for lower prepulse intensities (interaction
of prepulse and dose F(12,168) = 7.4, P < 0.001). Although
Table 1
Body weights, uterus weights, average startle amplitudes and average
percentage prepulse inhibition (PPI) of sham-operated and ovariectomized
(OVX) C57BL/6J mice treated with fluoxetine
Group Sham (n = 7) OVX (n = 7)
Body weight (g) 25.5 T 0.6 24.5 T 0.6
Uterus weight (mg) 86.2 T 7.7 11.0 T 1.3*
Uterus/body weight ratio 3.38 T 0.29 0.45 T 0.05*
Average startle amplitude
Saline 140 T 16 138 T 17
Fluoxetine 3 mg/kg 130 T 16 149 T 19
Fluoxetine 10 mg/kg 192 T 22 180 T 21
Fluoxetine 30 mg/kg 208 T 30 215 T 16
Average %PPI
Saline 47.2 T 2.9 45.2 T 3.7
Fluoxetine 3 mg/kg 49.7 T 4.7 46.2 T 2.6
Fluoxetine 10 mg/kg 54.4 T 4.2 54.8 T 3.0
Fluoxetine 30 mg/kg 58.8 T 3.2 59.8 T 3.2
* P < 0.05 compared with sham-operated mice (t test). For further
statistical comparison, see text.
there was no main effect of Group (Table 1), the prepulse-
dependent effect of fluoxetine on PPI was significantly
greater in OVX mice than in sham-operated controls
(interaction of prepulse � dose � group F(12,168) =
10.1, P < 0.001) (Fig. 1). Treatment with fenfluramine
significantly increased startle amplitudes (main effect of
dose F(3,36) = 4.9, P = 0.006). However, as with fluoxetine
treatment, ovariectomy did not influence startle amplitude
(Table 2). Fenfluramine treatment had no effect on PPI in
either sham-operated or OVX mice (Fig. 2).
3.2. Effect of fluoxetine and fenfluramine on PPI in ArKO
mice
Analysis of combined data obtained after saline, fluox-
etine and fenfluramine treatment revealed a significant main
Table 3
Body weights, uterus weights, average startle amplitudes and average
percentage prepulse inhibition (PPI) of J129 wild-type controls and ArKO
mice
Group J129 wild-type (n = 8) ArKO (n = 8)
Body weight (g) 24.3 T 0.8 26.8 T 0.8*
Uterus weight (mg) 151.3 T 24.3 10.6 T 1.7*
Uterus/body weight ratio 6.38 T 1.13 0.39 T 0.06*
Average startle amplitude
Saline 174 T 47 222 T 52
Fluoxetine 30 mg/kg 198 T 36 338 T 51
Fenfluramine 30 mg/kg 168 T 46 270 T 54
Average %PPI
Saline 63.6 T 4.6 50.3 T 4.7
Fluoxetine 30 mg/kg 65.1 T 3.5 65.1 T 3.5
Fenfluramine 30 mg/kg 68.3 T 2.0 70.8 T 3.2
* P < 0.05 compared with wild-type J129 mice (t test). For further
statistical comparison, see text.
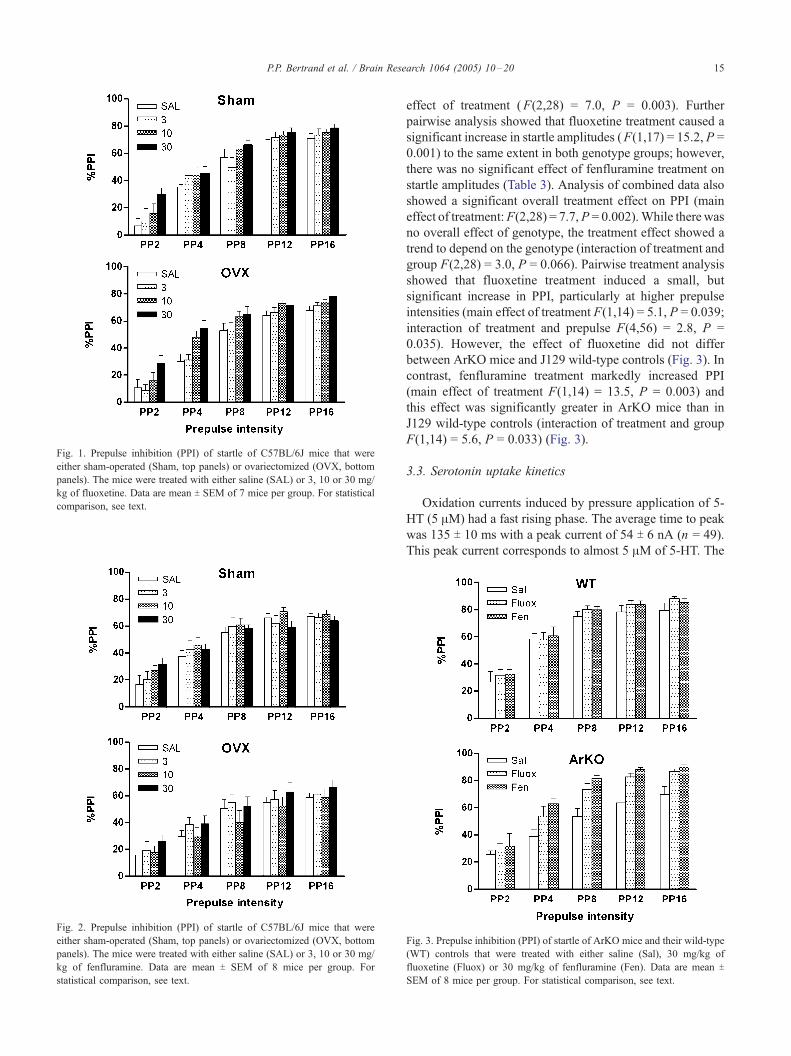
Fig. 2. Prepulse inhibition (PPI) of startle of C57BL/6J mice that were
either sham-operated (Sham, top panels) or ovariectomized (OVX, bottom
panels). The mice were treated with either saline (SAL) or 3, 10 or 30 mg/
kg of fenfluramine. Data are mean T SEM of 8 mice per group. For
statistical comparison, see text.
Fig. 1. Prepulse inhibition (PPI) of startle of C57BL/6J mice that were
either sham-operated (Sham, top panels) or ovariectomized (OVX, bottom
panels). The mice were treated with either saline (SAL) or 3, 10 or 30 mg/
kg of fluoxetine. Data are mean T SEM of 7 mice per group. For statistical
comparison, see text.
P.P. Bertrand et al. / Brain Research 1064 (2005) 10–20 15
effect of treatment (F(2,28) = 7.0, P = 0.003). Further
pairwise analysis showed that fluoxetine treatment caused a
significant increase in startle amplitudes (F(1,17) = 15.2, P =
0.001) to the same extent in both genotype groups; however,
there was no significant effect of fenfluramine treatment on
startle amplitudes (Table 3). Analysis of combined data also
showed a significant overall treatment effect on PPI (main
effect of treatment:F(2,28) = 7.7,P = 0.002).While there was
no overall effect of genotype, the treatment effect showed a
trend to depend on the genotype (interaction of treatment and
group F(2,28) = 3.0, P = 0.066). Pairwise treatment analysis
showed that fluoxetine treatment induced a small, but
significant increase in PPI, particularly at higher prepulse
intensities (main effect of treatment F(1,14) = 5.1, P = 0.039;
interaction of treatment and prepulse F(4,56) = 2.8, P =
0.035). However, the effect of fluoxetine did not differ
between ArKO mice and J129 wild-type controls (Fig. 3). In
contrast, fenfluramine treatment markedly increased PPI
(main effect of treatment F(1,14) = 13.5, P = 0.003) and
this effect was significantly greater in ArKO mice than in
J129 wild-type controls (interaction of treatment and group
F(1,14) = 5.6, P = 0.033) (Fig. 3).
3.3. Serotonin uptake kinetics
Oxidation currents induced by pressure application of 5-
HT (5 AM) had a fast rising phase. The average time to peak
was 135 T 10 ms with a peak current of 54 T 6 nA (n = 49).
This peak current corresponds to almost 5 AM of 5-HT. The
Fig. 3. Prepulse inhibition (PPI) of startle of ArKO mice and their wild-type
(WT) controls that were treated with either saline (Sal), 30 mg/kg of
fluoxetine (Fluox) or 30 mg/kg of fenfluramine (Fen). Data are mean T
SEM of 8 mice per group. For statistical comparison, see text.
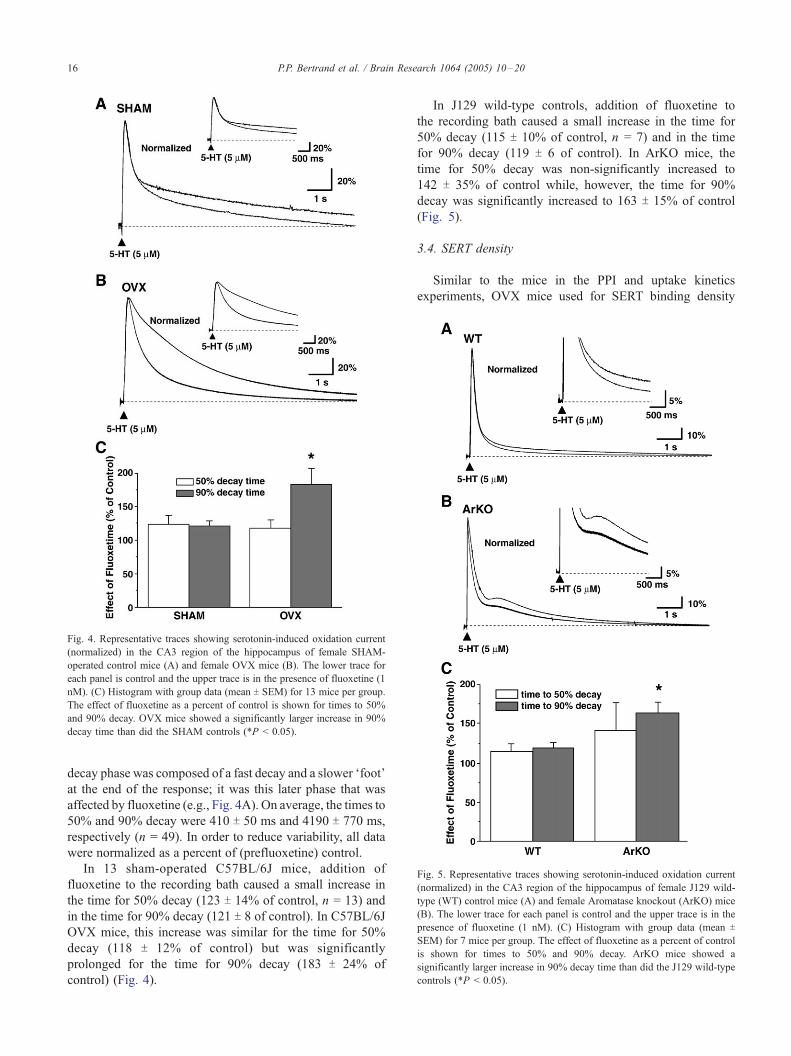
Fig. 5. Representative traces showing serotonin-induced oxidation current
(normalized) in the CA3 region of the hippocampus of female J129 wild-
type (WT) control mice (A) and female Aromatase knockout (ArKO) mice
(B). The lower trace for each panel is control and the upper trace is in the
presence of fluoxetine (1 nM). (C) Histogram with group data (mean T
SEM) for 7 mice per group. The effect of fluoxetine as a percent of control
is shown for times to 50% and 90% decay. ArKO mice showed a
significantly larger increase in 90% decay time than did the J129 wild-type
controls (*P < 0.05).
Fig. 4. Representative traces showing serotonin-induced oxidation current
(normalized) in the CA3 region of the hippocampus of female SHAM-
operated control mice (A) and female OVX mice (B). The lower trace for
each panel is control and the upper trace is in the presence of fluoxetine (1
nM). (C) Histogram with group data (mean T SEM) for 13 mice per group.
The effect of fluoxetine as a percent of control is shown for times to 50%
and 90% decay. OVX mice showed a significantly larger increase in 90%
decay time than did the SHAM controls (*P < 0.05).
P.P. Bertrand et al. / Brain Research 1064 (2005) 10–2016
decay phase was composed of a fast decay and a slower Ffoot_at the end of the response; it was this later phase that was
affected by fluoxetine (e.g., Fig. 4A). On average, the times to
50% and 90% decay were 410 T 50 ms and 4190 T 770 ms,
respectively (n = 49). In order to reduce variability, all data
were normalized as a percent of (prefluoxetine) control.
In 13 sham-operated C57BL/6J mice, addition of
fluoxetine to the recording bath caused a small increase in
the time for 50% decay (123 T 14% of control, n = 13) and
in the time for 90% decay (121 T 8 of control). In C57BL/6J
OVX mice, this increase was similar for the time for 50%
decay (118 T 12% of control) but was significantly
prolonged for the time for 90% decay (183 T 24% of
control) (Fig. 4).
In J129 wild-type controls, addition of fluoxetine to
the recording bath caused a small increase in the time for
50% decay (115 T 10% of control, n = 7) and in the time
for 90% decay (119 T 6 of control). In ArKO mice, the
time for 50% decay was non-significantly increased to
142 T 35% of control while, however, the time for 90%
decay was significantly increased to 163 T 15% of control
(Fig. 5).
3.4. SERT density
Similar to the mice in the PPI and uptake kinetics
experiments, OVX mice used for SERT binding density
P.P. Bertrand et al. / Brain Research 1064 (2005) 10–20 17
had significantly lower uterus weights either as absolute
values (24.9 T 3.6 mg vs. 84.8 T 8.8 mg in controls) or
as ratio of body weight (1.0 T 0.2 mg/g vs. 3.7 T 0.4
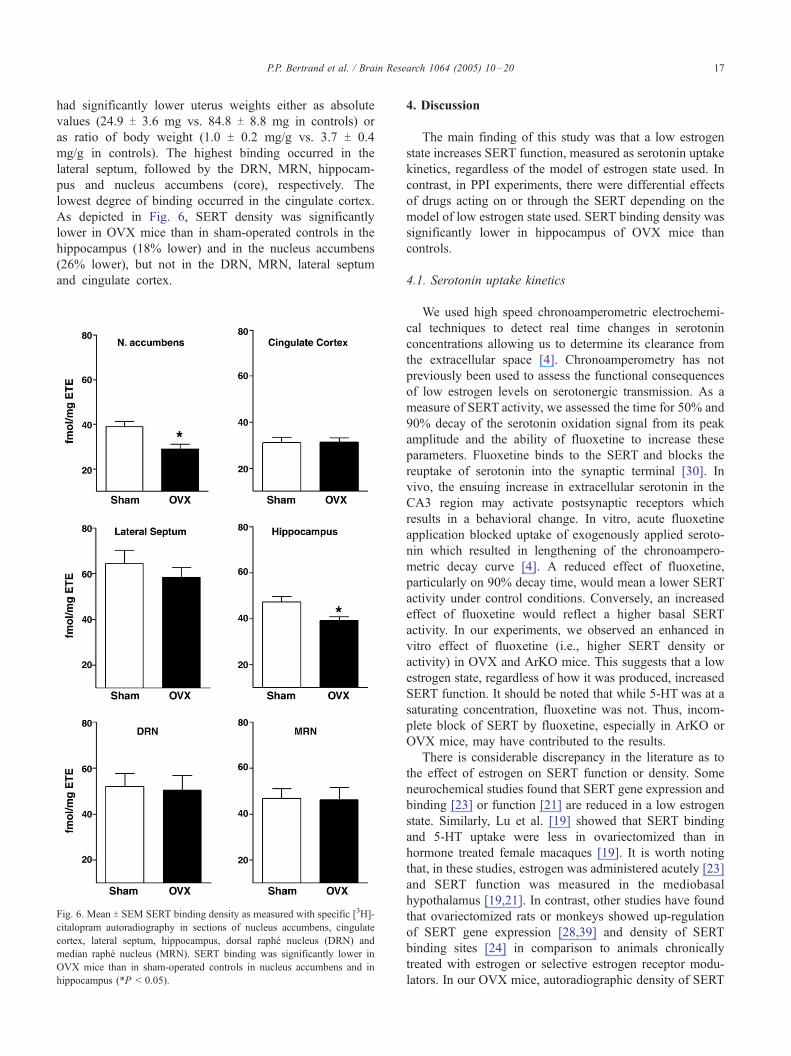
mg/g in controls). The highest binding occurred in the
lateral septum, followed by the DRN, MRN, hippocam-
pus and nucleus accumbens (core), respectively. The
lowest degree of binding occurred in the cingulate cortex.
As depicted in Fig. 6, SERT density was significantly
lower in OVX mice than in sham-operated controls in the
hippocampus (18% lower) and in the nucleus accumbens
(26% lower), but not in the DRN, MRN, lateral septum
and cingulate cortex.
Fig. 6. Mean T SEM SERT binding density as measured with specific [3H]-
citalopram autoradiography in sections of nucleus accumbens, cingulate
cortex, lateral septum, hippocampus, dorsal raphe nucleus (DRN) and
median raphe nucleus (MRN). SERT binding was significantly lower in
OVX mice than in sham-operated controls in nucleus accumbens and in
hippocampus (*P < 0.05).
4. Discussion
The main finding of this study was that a low estrogen
state increases SERT function, measured as serotonin uptake
kinetics, regardless of the model of estrogen state used. In
contrast, in PPI experiments, there were differential effects
of drugs acting on or through the SERT depending on the
model of low estrogen state used. SERT binding density was
significantly lower in hippocampus of OVX mice than
controls.
4.1. Serotonin uptake kinetics
We used high speed chronoamperometric electrochemi-
cal techniques to detect real time changes in serotonin
concentrations allowing us to determine its clearance from
the extracellular space [4]. Chronoamperometry has not
previously been used to assess the functional consequences
of low estrogen levels on serotonergic transmission. As a
measure of SERT activity, we assessed the time for 50% and
90% decay of the serotonin oxidation signal from its peak
amplitude and the ability of fluoxetine to increase these
parameters. Fluoxetine binds to the SERT and blocks the
reuptake of serotonin into the synaptic terminal [30]. In
vivo, the ensuing increase in extracellular serotonin in the
CA3 region may activate postsynaptic receptors which
results in a behavioral change. In vitro, acute fluoxetine
application blocked uptake of exogenously applied seroto-
nin which resulted in lengthening of the chronoampero-
metric decay curve [4]. A reduced effect of fluoxetine,
particularly on 90% decay time, would mean a lower SERT
activity under control conditions. Conversely, an increased
effect of fluoxetine would reflect a higher basal SERT
activity. In our experiments, we observed an enhanced in
vitro effect of fluoxetine (i.e., higher SERT density or
activity) in OVX and ArKO mice. This suggests that a low
estrogen state, regardless of how it was produced, increased
SERT function. It should be noted that while 5-HT was at a
saturating concentration, fluoxetine was not. Thus, incom-
plete block of SERT by fluoxetine, especially in ArKO or
OVX mice, may have contributed to the results.
There is considerable discrepancy in the literature as to
the effect of estrogen on SERT function or density. Some
neurochemical studies found that SERT gene expression and
binding [23] or function [21] are reduced in a low estrogen
state. Similarly, Lu et al. [19] showed that SERT binding
and 5-HT uptake were less in ovariectomized than in
hormone treated female macaques [19]. It is worth noting
that, in these studies, estrogen was administered acutely [23]
and SERT function was measured in the mediobasal
hypothalamus [19,21]. In contrast, other studies have found
that ovariectomized rats or monkeys showed up-regulation
of SERT gene expression [28,39] and density of SERT
binding sites [24] in comparison to animals chronically
treated with estrogen or selective estrogen receptor modu-
lators. In our OVX mice, autoradiographic density of SERT

P.P. Bertrand et al. / Brain Research 1064 (2005) 10–2018
was reduced in hippocampus and nucleus accumbens,
consistent with a reduction in SERT gene expression or
numbers in the low estrogen state. These data are at odds
with our chronoamperometry results (discussed above). It is
not clear how reduced SERT binding sites could cause a
relatively greater capacity for serotonin reuptake. Further
chronoamperometry experiments would be needed in
control and in OVX mice to determine whether these
conditions cause a change in the maximal velocity of SERT
transport or the affinity for 5-HT.
4.2. Prepulse inhibition
Administration of fluoxetine caused an increase in startle
amplitude and PPI. Ovariectomy enhanced the effect of
fluoxetine on PPI, but not startle amplitude. The effect of
fluoxetine on startle and PPI has not previously been tested
in mice. Data in rats suggest that an increase in extracellular
serotonin levels, as a result of fluoxetine treatment, does not
affect PPI [20]. The difference between this study and our
results could be because of species differences or due to the
fact that we used female mice vs. male rats.
The mechanism of action of the ovariectomy-induced
enhancement of fluoxetine action on PPI remains unclear. It
has been shown that serotonin levels are decreased in the
hippocampus of OVX mice [14]. This could be caused by
two distinct mechanisms. First, reduced levels could be the
result of enhanced release. In this case, the effect of
fluoxetine in our experiments to increase PPI may be
enhanced in OVX mice because of an already increased
serotonin release in the low estrogen state. Second, the
reduced 5-HT levels in OVX mice could be due to increased
SERT activity as an up-regulation of SERT gene expression
[28,39] or density of SERT binding sites [24]. This is inline
with our chronoamperometry results which showed an
increased SERT function in OVX mice. In this case,
blockade of the SERT by fluoxetine would then cause a
relatively greater increase in extracellular serotonin levels in
OVX mice than in sham-operated controls and, consequent-
ly, a more marked behavioral effect, as was seen in our PPI
experiments.
It is harder to reconcile our findings with those that
show that ovariectomy causes a decrease in SERT density.
It was found that ovariectomy reduced SERT gene
expression in the brain, particularly in the dorsal raphe
nucleus and septum [23,33], as compared to acutely
ovariectomized rats injected with estrogen. This is consis-
tent with microdialysis experiments that showed that, in
diestrus, when circulating levels of estrogen are low, acute
treatment with fluoxetine caused a markedly smaller
increase in extracellular serotonin levels when compared
to estrus, when circulating levels of estrogen are high [21].
If, in our experiments, a loss of estrogen by ovariectomy
caused lower levels or activity of SERT, then it is difficult
to explain why there was an enhancement of fluoxetine
action on PPI. Thus, a decrease in SERT is consistent with
our autoradiography results, but not our chronoamperom-
etry or PPI results.
The administration of the serotonin releasing drug
fenfluramine, produced an increase in startle amplitude in
C57BL/6J mice, showing fenfluramine was active in the
brain at this dose. Nevertheless, fenfluramine treatment did
not significantly influence PPI in C57BL/6J mice. While the
effect of fenfluramine on PPI in mice has not previously
been examined, Dulawa and Geyer [6] reported that another
serotonin releasing drug, MDMA ((+)3,4-methylenedioxy-
N-methylamphetamine), had no effect on the startle
response in C57BL/6J mice but that PPI was decreased. It
may be that MDMA was having an effect on PPI that was
not related to its actions on the serotonergic system, but
rather by its action on central dopaminergic activity [6].
Fenfluramine is a substrate type releaser that initially binds
to the SERT and is transported into the nerve terminal [30].
There it promotes non-exocytotic release of serotonin by a
carrier-mediated exchange process [30]. It would appear that
these mechanisms are not altered in OVX C57BL/6J mice. It
is unclear why fluoxetine treatment increased PPI in sham-
operated and OVX C57BL/6 mice, whereas fenfluramine
treatment had no effect. Further experiments assessing
SERT gene expression and SERT binding in the brain are
needed to investigate this apparent discrepancy.
Fluoxetine administration caused an increase in startle
amplitudes and PPI in J129 wild-type controls and ArKO
mice; however, in contrast to the effect of fluoxetine in
C57BL/6J mice, there were no differences in the extent of
this effect between the groups. Treatment with fenfluramine,
which did not influence PPI in C57BL/6J mice, increased
PPI in J129 wild-type controls and ArKO mice. Moreover,
the effect of fenfluramine was greater in the ArKO mice
than in their controls.
4.3. Differences in models of estrogen state
There were two main differences between C57BL/6J
sham-operated and OVX mice on the one hand and J129
wild-type and ArKO mice on the other. First, the genetic
background may have played a role. Numerous studies have
shown mouse strain differences in behavior, including PPI
(for references, see [12,27]). This could explain why sham-
operated C57BL/6 mice differ from J129 wild-type mice
with respect to their responsiveness to fluoxetine and
fenfluramine. It would then be of particular interest to study
the effect of ovariectomy in J129 wild-type controls to see if
a similar enhancement of the effect of fenfluramine will
occur as seen in ArKO mice or if an enhancement of the
effect of fluoxetine would occur, similar to that seen in OVX
C57BL/6 mice. Furthermore, measurement of SERT density
would have to be done in ArKO mice.
The other main difference between the two models is the
way the low estrogen state is achieved. Ovariectomy will
remove one major source of circulating sex steroids;
however, this procedure will not inhibit production of

P.P. Bertrand et al. / Brain Research 1064 (2005) 10–20 19
estrogen elsewhere in the body, in particular the brain [32].
It is becoming clear that, in the brain, steroid hormones are
synthesized and may act locally to influence behavior in a
paracrine fashion [31]. Unlike ovariectomy, targeted dis-
ruption of the CYP19 gene in ArKO mice will also remove
components of this brain steroid production. This may
explain some of the differences in behavioral effects
between the low estrogen state achieved by ovariectomy
vs. disruption of the CYP19 gene.
It is important to note that ArKO mice not only show
reduced estrogen production but, as a consequence of the
mutation, also high circulating testosterone levels [8].
Estrogen is produced by conversion of testosterone by the
aromatase enzyme and blockade of this conversion may be
the cause of a Fbuild-up_ of testosterone [8]. It is possible
that high testosterone levels in the ArKO mice are
involved in the differential effects seen on the effect of
fluoxetine and fenfluramine on PPI [7], as we have shown
for the effect of the 5-HT1A receptor agonist, 8-OH-DPAT
[13]. Finally, as with other animal models using knockout
strategies, the deletion of the aromatase gene could have
effects throughout development and may trigger compen-
satory changes in responsiveness to drugs, such a
fluoxetine and fenfluramine.
Follow-up experiments should be aimed at other manip-
ulations of estrogen levels, particularly by following the
normal variation of these levels during the estrus cycle.
However, other hormone levels also change during this
cycle and therefore the present study was focused on just
estrogen.
In conclusion, we observed that low estrogen state,
induced either by ovariectomy or in ArKO mice,
increased SERT function as measured by chronoamper-
ometry in hippocampal slices. SERT binding density was
reduced in hippocampus of OVX mice, which is at odds
with the increased SERT kinetics. Interestingly, the
behavioral responses to drugs acting on or through the
SERT depended on how the low estrogen state was
induced. Considering that sensory motor gating is
disrupted in schizophrenia and other mental illnesses,
our experiments on the effect of low estrogen state may
provide further insight into the possible beneficial effects
of estrogen as adjuncts to these treatments [16].
Acknowledgments
This work was supported by the National Health and
Medical Research Council, Australia. Dr. M. van den
Buuse was supported by the Griffith Senior Research
Fellowship of the University of Melbourne. Dr. M. Jones
was supported by a U.S. Public Health Service Grant
(R37 AG-08174). The Mental Health Research Institute is
a Stanley Research Centre supported by the Stanley
Medical Research Institute, Bethesda, MD, USA (http://
www.stanleyresearch.org/programs/stanley_research.asp).
References
[1] A. Adell, P. Celada, M.T. Abellan, F. Artigas, Origin and functional
role of the extracellular serotonin in the midbrain raphe nuclei, Brain
Res. Rev. 39 (2002) 154–180.
[2] S. Benmansour, W.A. Owens, M. Cecchi, D.A. Morilak, A. Frazer,
Serotonin clearance in vivo is altered to a greater extent by
antidepressant-induced downregulation of the serotonin transporter
than by acute blockade of this transporter, J. Neurosci. 22 (2002)
6766–6772.
[3] D.L. Braff, M.A. Geyer, N.R. Swerdlow, Human studies of prepulse
inhibition of startle: normal subjects, patient groups, and pharmaco-
logical studies, Psychopharmacology 156 (2001) 234–258.
[4] L.C. Daws, G.M. Toney, D.J. Davis, G.A. Gerhardt, A. Frazer, In vivo
chronoamperometric measurements of the clearance of exogenously
applied serotonin in the rat dentate gyrus, J. Neurosci. Methods 78
(1997) 139–150.
[5] L.C. Daws, G.M. Toney, G.A. Gerhardt, F. A, In vivo chronoampero-
metric measures of extracellular serotonin clearance in rat dorsal
hippocampus: contribution of serotonin and norepinephrine trans-
porters, J. Pharmacol. Exp. Ther. 286 (1998) 967–976.
[6] S.C. Dulawa, M.A. Geyer, Effects of strain and serotonergic agents on
prepulse inhibition and habituation in mice, Neuropharmacology 39
(2000) 2170–2179.
[7] G. Fink, B. Sumner, R. Rosie, H. Wilson, J. McQueen, Androgen
actions on central serotonin neurotransmission: relevance for mood,
mental state and memory, Behav. Brain Res. 105 (1999) 53–68.
[8] C.R. Fisher, K.H. Graves, A.F. Parlow, E.R. Simpson, Characteriza-
tion of mice deficient in aromatase (ArKO) because of targeted
disruption of the cyp19 gene, Proc. Natl. Acad. Sci. U. S. A. 95 (1998)
6965–6970.
[9] K.B.J. Franklin, G. Paxinos, The Mouse Brain in Stereotaxic
Coordinates, Academic Press, San Diego, 1996.
[10] G.A. Gerhardt, A.F. Oke, G. Nagy, B. Moghaddam, R.N. Adams,
Nafion-coated electrodes with high selectivity for CNS electrochem-
istry, Brain Res. 290 (1984) 390–395.
[11] M.A. Geyer, A. Markou, Animal models of psychiatric disorders, in:
F.E. Bloom, D.J. Kupfer (Eds.), Psychopharmacology: The Fourth
Generation of Progress, Raven Press, New York, 1995, pp. 787–798.
[12] M.A. Geyer, K.L. McIlwain, R. Paylor, Mouse genetic models for
prepulse inhibition: an early review, Mol. Psychiatry 7 (2002)
1039–1053.
[13] A. Gogos, M. Van den Buuse, Castration reduces the effect of
serotonin-1A receptor stimulation on prepulse inhibition in rats,
Behav. Neurosci. 117 (2003) 1407–1415.
[14] T. Heikkinen, J. Puolivali, L. Liu, A. Rissanen, H. Tanila, Effects of
ovariectomy and estrogen treatment on learning and hippocampal
neurotransmitters in mice, Horm. Behav. 41 (2002) 22–32.
[15] M. Koch, The neurobiology of startle, Prog. Neurobiol. 59 (1999)
107–128.
[16] J. Kulkarni, A. Riedel, A.R. de Castella, P.B. Fitzgerald, T.J. Rolfe, J.
Taffe, H. Burger, Estrogen—A potential treatment for schizophrenia,
Schizophr. Res. 48 (2001) 137–144.
[17] S. Kusljic, D.L. Copolov, M. Van den Buuse, Differential role of
serotonergic projections arising from the dorsal and median raphe
nuclei in locomotor hyperactivity and prepulse inhibition, Neuro-
psychopharmacology 28 (2003) 2138–2147.
[18] A.L. Lopez-Figueroa, C.S. Norton, M.O. Lopez-Figueroa, D. Armel-
lini-Dodel, S. Burke, H. Akil, J.F. Lopez, S.J. Watson, Serotonin 5-
HT1A, 5-HT1B, and 5-HT2A receptor mRNA expression in subjects
with major depression, bipolar disorder, and schizophrenia, Biol.
Psychiatry 55 (2004) 225–233.
[19] N.Z. Lu, A.J. Eshleman, A. Janowsky, C.L. Bethea, Ovarian
steroid regulation of serotonin reuptake transporter (SERT) binding,
distribution, and function in female macaques, Mol. Psychiatry
8 (2003) 353–360.
[20] D.L. Martinez, M.A. Geyer, Characterization of the disruptions of

P.P. Bertrand et al. / Brain Research 1064 (2005) 10–2020
prepulse inhibition and habituation of startle induced by alpha-
ethyltryptamine, Neuropsychopharmacology 16 (1997) 255–264.
[21] S. Maswood, W. Truitt, M. Hotema, M. Caldarola-Pastuszka, L.
Uphouse, Estrous cycle modulation of extracellular serotonin in
mediobasal hypothalamus: role of the serotonin transporter and
terminal autoreceptors, Brain Res. 831 (1999) 146–154.
[22] R. McQuade, T. Sharp, Functional mapping of dorsal and median
raphe 5-hydroxytryptamine pathways in forbrain of the rat using
microdialysis, J. Neurochem. 69 (1997) 791–796.
[23] J.K. McQueen, H. Wilson, G. Fink, Estradiol-17 beta increases
serotonin transporter (SERT) mRNA levels and the density of SERT-
binding sites in female rat brain, Mol. Brain Res. 45 (1997) 13–23.
[24] J.K. McQueen, H. Wilson, B.E. Sumner, G. Fink, Serotonin
transporter (SERT) mRNA and binding site densities in male rat brain
affected by sex steroids, Mol. Brain Res. 63 (1999) 241–247.
[25] S. Montanez, L.C. Daws, G.G. Gould, G.A. Gerhardt, A. Frazer,
Differential in vivo clearance of serotonin in rat dorsal raphe nucleus
and CA3 region, Brain Res. 955 (2002) 236–244.
[26] S. Montanez, L.C. Daws, G.G. Gould, A. Frazer, Serotonin (5-HT)
transporter (SERT) function after graded destruction of serotonergic
neurons, J. Neurochem. 87 (2003) 861–867.
[27] R. Paylor, J.N. Crawley, Inbred strain differences in prepulse
inhibition of the mouse startle response, Psychopharmacology 132
(1997) 169–180.
[28] M. Pecins-Thompson, N.A. Brown, C.L. Bethea, Regulation of
serotonin re-uptake transporter mRNA expression by ovarian steroids
in rhesus macaques, Mol. Brain Res. 53 (1998) 120–129.
[29] K.M. Robertson, L. O’Donnell, M.E.E. Jones, S.J. Meachem, W.C.
Boon, C.R. Fischer, K.H. Graves, R.I. McLachlan, E.R. Simpson,
Impairment of spermatogenesis in mice lacking a functional aromatase
(cyp 19) gene, Proc. Natl. Acad. Sci. U. S. A. 96 (1999) 7986–7991.
[30] R.B. Rothman, M.H. Baumann, Monoamine transporters and psy-
chostimulant drugs, Eur. J. Pharmacol. 479 (2003) 23–40.
[31] R. Rupprecht, F. di Michele, B. Hermann, A. Strohle, M. Lancel, E.
Romeo, F. Holsboer, Neuroactive steroids: molecular mechanisms of
action and implications for neuropsychopharmacology, Brain Res.
Rev. 37 (2001) 59–67.
[32] E.R. Simpson, Sources of estrogen and their importance, J. Steroid
Biochem. Mol. Biol. 86 (2003) 225–230.
[33] B.E. Sumner, K.E. Grant, R. Rosie, C. Hegele-Hartung, K.H.
Fritzemeier, G. Fink, Effects of tamoxifen on serotonin transporter
and 5-hydroxytryptamine2A receptor binding sites and mRNA levels
in the brain of ovariectomized rats with or without acute estradiol
replacement, Mol. Brain Res. 73 (1999) 119–128.
[34] N.R. Swerdlow, M.A. Geyer, D. Braff, Neural circuit regulation of
prepulse inhibition of startle in the rat: current knowledge and future
challenges, Psychopharmacology 156 (2001) 194–215.
[35] M. Van den Buuse, M. Morris, C. Chavez, S. Martin, J.H. Wang,
Effect of adrenalectomy and corticosterone replacement on prepulse
inhibition and locomotor activity in mice, Br. J. Pharmacol. 142
(2004) 543–550.
[36] R.P. Vertes, A PHA-L analysis of ascending projections of the dorsal
raphe nucleus in the rat, J. Comp. Neurol. 313 (1991) 643–668.
[37] R.P. Vertes, W.J. Fortin, A.M. Crane, Projections of the median raphe
nucleus in the rat, J. Comp. Neurol. 407 (1999) 555–582.
[38] J.H. Wang, J.L. Short, C. Ledent, A.J. Lawrence, M. van den Buuse,
Reduced startle habituation and prepulse inhibition in mice lacking the
adenosine A2A receptor, Behav. Brain Res. 143 (2003) 201–207.
[39] W. Zhou, N. Koldzic-Zivanovic, C.H. Clarke, R. de Beun, K.
Wassermann, P.S. Bury, K.A. Cunningham, M.L. Thomas, Selective
estrogen receptor modulator effects in the rat brain, Neuroendocrino-
logy 75 (2002) 24–33.




















