
Down regulation of serotonin-S 2 receptor sites in rat brain by chronic treatment with the...
11
Psychopharmacology (1986) 88:434-444 Psychopharmacology © Springer-Verlag 1986 Down regulation of serotonin-S2 receptor sites in rat brain by chronic treatment with the serotonin-S2 antagonists: ritanserin and setoperone J.E. Leysen a, p. Van Gompel 1, W. Gommeren 1, R. Woestenborghs 2, and P.A.J. Janssen a 1 Department of Biochemical Pharmacology and 2 Department of Drug Metabolism, Janssen Pharmaceutica, B-2340 Beerse, Belgium Abstract. Ritanserin is a potent and selective serotonin-Sz antagonist which slowly dissociates from the receptor sites, while setoperone has potent serotonin and moderate dopa- mine antagonistic properties and dissociates rapidly from the receptor sites. Acute administration of ritanserin (1-10 mg/kg) produced a non-competitive inhibition of 3H- ketanserin binding, measured ex vivo in washed frontal cor- tex membranes, which lasted for 12 h. This is in accordance with the slow dissociation of the drug from the receptor sites. Setoperone (1-10 mg/kg orally) also produced a par- tially non-competitive inhibition of 3H-ketanserin binding in washed membranes, which is unlike its rapid dissociation. In contrast, there was no inhibition of dopamine receptor binding in washed striatal membranes. Chronic oral admin- istration of 10 mg/kg, day of the drugs significantly reduced the Bm,xvalues of 3H-ketanserin, without changing the KD value when drug-free periods were longer than 1 day. The maximum reduction following 25 days' treatment with 14 mg/kg ritanserin was 50% at 1 day drug-free; the Bin, x values gradually returned to the control value in about 12 days. The receptor half-life was calculated to be 3.5 days and the receptor synthesis rate 4 fmoles/mg tissue, day. Ri- tanserin treatment did not alter radioligand binding to sero- tonin-S1, ~1-, 1~2- and fl-adrenergic, dopamine-D2, benzo- diazepine and substance P sites. Chronic treatment with setoperone at 10 mg/kg-day, orally, significantly reduced the Bma x value of 3H-ketanserin binding in frontal cortex but treatment with 1 mg/kg.day did not. In contrast, a dose-dependent increase in the number of striatal dopa- mine-D2 sites was observed, in accordance with the moder- ate dopamine-antagonistic properties of setoperone. Dopa- mine-D2 receptor up regulation up to 150% of control values, was maintained at the same level for 9 days, it started to decline 12 days after stopping drug treatment. Following chronic treatment and drug withdrawal for more than I day, ritanserin and setoperone levels in whole brain homogenates were below detection level (< 1 ng/g). The similar reduction in the Bma x values of 3H-ketanserin bind- ing following chronic treatment with the rapidly dissociat- ing setoperone and the slowly dissociating ritanserin, the absence of effect on the KD value, the slow reappearance of the receptor sites and the opposite effect on serotonin-S2 and dopamine-D2 receptors with setoperone suggest that real serotonin-S2 receptor down regulation occurs following antagonist treatment. The findings illustrate the difference Offprint requests to: J.E. Leysen in receptor regulation between the serotonergic and the do- paminergic system. The specific serotonin-Sz receptor down regulation produced by serotonin antagonists is probably achieved via drug interference with intracellular processes. In view of the hypothesis that supersensitive serotonin-S2 receptor sites may be involved in the etiology of certain mood disorders, acute blockade of these receptors followed by receptor down regulation may be beneficial for the treat- ment of such diseases. Key words: Serotonin-S2 receptors Radioligand binding - Receptor down regulation - Chronic drug treatment - Serotonin antagonist - Ritanserin - Setoperone In radioligand binding studies serotonin receptor sites have been classified as 5-hydroxytryptamine-1 (5HT1) or sero- tonin-S1 sites labelled with high affinity by 3H-serotonin and 5-hydroxytryptamine-2 (5HT2), or serotonin-S2 sites, which were first detected using 3H-spiperone in frontal cor- tex tissue (Leysen et al. 1978; Peroutka and Snyder 1979). The serotonin-S2 receptor sites have been further character- ized using the more selective serotonin antagonist 3H-ketan- serin (Leysen et al. 1982). They are found to be particularly concentrated in the frontal cortex and were detected in the striatum, mesolimbic areas and on blood platelets. Sero- tonin-S2 receptor sites were shown to have a role in sero- tonin agonist-induced behavioural excitation and discrimin- ative stimulus effects in rodents and they mediate serotonin- induced vasoconstriction and platelet function (for review see Leysen et al. 1984 and Leysen 1985). Recently, it was shown that inositol phospholipid turnover forms part of the signal transducing system coupled to the serotonin-Sz receptors (de Chaffoy de Courcelles et al. 1985). Serotonin-S2 receptor sites, like fl-adrenergic receptor sites, were found to be down regulated in rat brain following chronic treatment with the antidepressant amitriptyline (Peroutka and Snyder 1980). The finding was interpreted according to the receptor adaptation theory, that chronic deprival of receptor stimulation would result in receptor up regulation whereas persistent receptor stimulation would cause receptor down regulation. The receptor down regula- tion by amitriptyline was ascribed to the inhibition of amine uptake. The 3-week time course required for the establish- ment of the effect has been related to the time course for the development of therapeutic activity with the anfidepres-
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Down regulation of serotonin-S 2 receptor sites in rat brain by chronic treatment with the...

Psychopharmacology (1986) 88:434-444 Psychopharmacology © Springer-Verlag 1986
Down regulation of serotonin-S2 receptor sites in rat brain by chronic treatment with the serotonin-S2 antagonists: ritanserin and setoperone J.E. Leysen a, p. Van Gompel 1, W. Gommeren 1, R. Woestenborghs 2, and P.A.J. Janssen a 1 Department of Biochemical Pharmacology and 2 Department of Drug Metabolism, Janssen Pharmaceutica, B-2340 Beerse, Belgium
Abstract. Ritanserin is a potent and selective serotonin-Sz antagonist which slowly dissociates from the receptor sites, while setoperone has potent serotonin and moderate dopa- mine antagonistic properties and dissociates rapidly from the receptor sites. Acute administration of ritanserin (1-10 mg/kg) produced a non-competitive inhibition of 3H- ketanserin binding, measured ex vivo in washed frontal cor- tex membranes, which lasted for 12 h. This is in accordance with the slow dissociation of the drug from the receptor sites. Setoperone (1-10 mg/kg orally) also produced a par- tially non-competitive inhibition of 3H-ketanserin binding in washed membranes, which is unlike its rapid dissociation. In contrast, there was no inhibition of dopamine receptor binding in washed striatal membranes. Chronic oral admin- istration of 10 mg/kg, day of the drugs significantly reduced the Bm,xvalues of 3H-ketanserin, without changing the KD value when drug-free periods were longer than 1 day. The maximum reduction following 25 days' treatment with 14 mg/kg ritanserin was 50% at 1 day drug-free; the Bin, x values gradually returned to the control value in about 12 days. The receptor half-life was calculated to be 3.5 days and the receptor synthesis rate 4 fmoles/mg tissue, day. Ri- tanserin treatment did not alter radioligand binding to sero- tonin-S1, ~1-, 1~2- and fl-adrenergic, dopamine-D2, benzo- diazepine and substance P sites. Chronic treatment with setoperone at 10 mg/kg-day, orally, significantly reduced the Bma x value of 3H-ketanserin binding in frontal cortex but treatment with 1 mg/kg.day did not. In contrast, a dose-dependent increase in the number of striatal dopa- mine-D2 sites was observed, in accordance with the moder- ate dopamine-antagonistic properties of setoperone. Dopa- mine-D2 receptor up regulation up to 150% of control values, was maintained at the same level for 9 days, it started to decline 12 days after stopping drug treatment. Following chronic treatment and drug withdrawal for more than I day, ritanserin and setoperone levels in whole brain homogenates were below detection level (< 1 ng/g). The similar reduction in the Bma x values of 3H-ketanserin bind- ing following chronic treatment with the rapidly dissociat- ing setoperone and the slowly dissociating ritanserin, the absence of effect on the KD value, the slow reappearance of the receptor sites and the opposite effect on serotonin-S2 and dopamine-D2 receptors with setoperone suggest that real serotonin-S2 receptor down regulation occurs following antagonist treatment. The findings illustrate the difference
Offprint requests to: J.E. Leysen
in receptor regulation between the serotonergic and the do- paminergic system. The specific serotonin-Sz receptor down regulation produced by serotonin antagonists is probably achieved via drug interference with intracellular processes. In view of the hypothesis that supersensitive serotonin-S2 receptor sites may be involved in the etiology of certain mood disorders, acute blockade of these receptors followed by receptor down regulation may be beneficial for the treat- ment of such diseases.
Key words: Serotonin-S2 receptors Radioligand binding - Receptor down regulation - Chronic drug treatment - Serotonin antagonist - Ritanserin - Setoperone
In radioligand binding studies serotonin receptor sites have been classified as 5-hydroxytryptamine-1 (5HT1) or sero- tonin-S1 sites labelled with high affinity by 3H-serotonin and 5-hydroxytryptamine-2 (5HT2), or serotonin-S2 sites, which were first detected using 3H-spiperone in frontal cor- tex tissue (Leysen et al. 1978; Peroutka and Snyder 1979). The serotonin-S2 receptor sites have been further character- ized using the more selective serotonin antagonist 3H-ketan- serin (Leysen et al. 1982). They are found to be particularly concentrated in the frontal cortex and were detected in the striatum, mesolimbic areas and on blood platelets. Sero- tonin-S2 receptor sites were shown to have a role in sero- tonin agonist-induced behavioural excitation and discrimin- ative stimulus effects in rodents and they mediate serotonin- induced vasoconstriction and platelet function (for review see Leysen et al. 1984 and Leysen 1985). Recently, it was shown that inositol phospholipid turnover forms part of the signal transducing system coupled to the serotonin-Sz receptors (de Chaffoy de Courcelles et al. 1985).
Serotonin-S2 receptor sites, like fl-adrenergic receptor sites, were found to be down regulated in rat brain following chronic treatment with the antidepressant amitriptyline (Peroutka and Snyder 1980). The finding was interpreted according to the receptor adaptation theory, that chronic deprival of receptor stimulation would result in receptor up regulation whereas persistent receptor stimulation would cause receptor down regulation. The receptor down regula- tion by amitriptyline was ascribed to the inhibition of amine uptake. The 3-week time course required for the establish- ment of the effect has been related to the time course for the development of therapeutic activity with the anfidepres-

sant drugs. The theory was put forward, that supersensitive serotonin-S2 and fl-adrenergic receptor sites may be in- volved in the etiology of depressive illness (Peroutka and Snyder 1980; Vetulani et al. 1976).
Following the original observations, serotonin-S2 and fl-adrenergic receptor down regulation was observed with various purported antidepressant drugs, with different bio- chemical profiles (imipramine, desimipramine, iprindole, mianserin) (Blackshear and Sanders-Bush 1982; Gandolfi et al. 1984; Hall et al. 1984; Snyder and Peroutka 1982). However, the explanation of the mechanism of the sero- tonin-S2 receptor down regulation has been challenged by different findings. Chronic treatment with selective and po- tent serotonin uptake inhibitors not belonging to the class of tricyclic antidepressants (zimelidine, alaproclate, citalo- pram) did not reduce serotonin-Sz receptor numbers. Le- sioning or depletion of serotonergic neurones appeared not to abolish the serotonin-S2 receptor down regulation pro- duced by tricyclic antidepressants (Gandolfi et al. 1984; Hall et al. 1984; Hyttel et al. 1984). Moreover, drugs known as potent serotonin antagonists such as cyproheptadine, pi- zotifen, mianserin and ketanserin have been reported to produce a decrease in serotonin-S2 receptor numbers (Blackshear et al. 1983; Gandolfi et al. 1985). These obser- vations indicate that serotonin-S2 receptor regulation is un- like the current theory, but a likely explanation for the anomaly has not been found.
In this study we investigated serotonin-Sz receptor alter- ations in brain tissue following acute and chronic treatment of rats with the new serotonin antagonists ritanserin and setoperone (Janssen 1985). In vivo, ritanserin is a particu- larly selective and long-acting serotonin antagonist (Col- paert etal. 1985; Leysen etal. 1985; Niemegeers etal. 1984). Clinical studies have revealed therapeutic activity in dysthymic disorders (Reyntjens et al. 1984). Setoperone is primarily a serotonin antagonist, but in addition the drug reveals dopamine antagonistic properties (Niemegeers et al. 1984). Accordingly, neuroleptic-like activity has been ob- served (Ceulemans et al. 1985). Because of the dual action of setoperone, chronic treatment with this drug allowed investigation of both serotonin-Sz and dopamine-D2 recep- tor alterations.
Materials and methods
Drug treatment. Male Wistar rats, body weight 150g at the beginning of drug treatment, were used. For acute treat- ment an oral bolus dose (1 ml per 100 g body weight) of ritanserin or setoperone was given. For chronic treatment drugs were either given orally or were mixed with powdered food (see below). When drugs were mixed with the food, rats were kept individually in adapted cages where spread- ing of food was minimal. Weekly, body weight and food consumption were measured, from which drug intake in mg per kg of body weight was calculated. Chronic drug treatment (7, 25 or 28 days) was followed by drug-free peri- ods from 1 to 18 days, after which animals were sacrificed. Control rats were given solvent or powdered food without drug and were kept under exactly the same conditions as the drug-treated animals.
Receptor binding and neurotransmitter uptake assays. Fol- lowing the treatment period, rats were killed by decapita- tion, brains were immediately removed from the skull and
435
various brain areas were dissected. Tissues were either pro- cessed immediately or frozen in liquid nitrogen and kept in a biofreezer at - 8 0 ° C until assayed. Tissue storage never exceeded 2 weeks. Small brain areas (frontal cortex for serotonin-S2 receptor binding, striatum for dopamine- D2 receptor binding) were pooled (three rats) to provide sufficient tissue for assaying ligand saturation binding curves.
To assay drug inhibition of radioligand receptor binding or neurotransmitter uptake, naive female Wistar rats (150 g) and male Pirbright guinea pigs (200 g) were used. Tissues were dissected as above. For preparation of subcel- lular fractions, fresh tissue was homogenized in sucrose 0.25 M (10 v/w: v=volume, w = w e t weight tissue). The nu- clear fraction and cell debris were removed by centrifuga- tion at 10 min x 577 g. The pellet was rehomogenized in 5 v/w sucrose and again centrifuged. To obtain a (M + L) (heavy + ligh mitochondrial) fraction the supernatants were pooled and centrifuged at 10 minx23600g . To obtain a (M + L + P) fraction (P = microsomal fraction), the superna- tants after removal of the nuclear fraction were pooled and diluted 3-fold in Tris-HC1 buffer 50 mM, pH 7.6. The sus- pension was centrifuged at 20 minx 29900 g. For prepara- tion of a total particulate fraction the tissue was homoge- nized in Tris-HC1 50 mM, pH 7.6, 40 v/w and centrifuged at 20 minx 29 900 g. Pellets were washed twice by resuspen- sion in buffer and centrifugation. Final pellets were sus- pended in the assay buffer in a dilution as indicated in Table 1. Centrifugations were run at 0 ° C using a refriger- ated Sorvall RC-5B superspeed centrifuge and an SS34 ro- tor.
Tissue preparations and assay conditions for the various radioligand binding and neurotransmitter uptake models are summarized in Table 1. Further details are given in the legends to tables and figures. Following incubation, the mixtures were filtered under vacuum over Whatman GF/B glass fibre filters to separate bound from free radioactivity. Filters were rapidly rinsed with 2 x 5 ml ice-cold buffer and transferred to counting vials. Radioactivity was extracted by vigorous shaking with 7 ml Instagel. Vials were counted in a liquid scintillation counter equipped with an external standard and an in-built calculation device to express data in DPM referring to a standard quench curve for tritium.
Measurement of drug receptor dissociation rate. The dissoci- ation rate of unlabelled ritanserin and setoperone from var- ious receptor sites in vitro was measured using a recently described filter method (Leysen and Gommeren 1984; Ley- sen et al. 1985).
Determination of ritanserin and setoperone tissue residue lev- els. Brain tissue levels of ritanserin were selectively mea- sured by a high-performance liquid chromatographic (HPLC) method. Brain samples (range: 0.18-1.10 g) were homogenized (Ultra-Turrax) in 1 ml of a 0.05 M sodium borate decahydrate (borate) buffer, spiked with 100 ng of a suitable internal standard (R 56724) and extracted twice with 4-ml aliquots of heptane-isoamyl alcohol (98.5:1.5, v/v). The combined organic layers were back-extracted with 3 ml 0.05 M sulphuric acid and, after alkalinization of the aqueous phase with 0.15 ml concentrated ammonia, the compounds were re-extracted twice into 2-ml aliquots of the organic phase. The extracts were then evaporated to dryness under a stream of nitrogen in a water bath at 55 ° C.
436
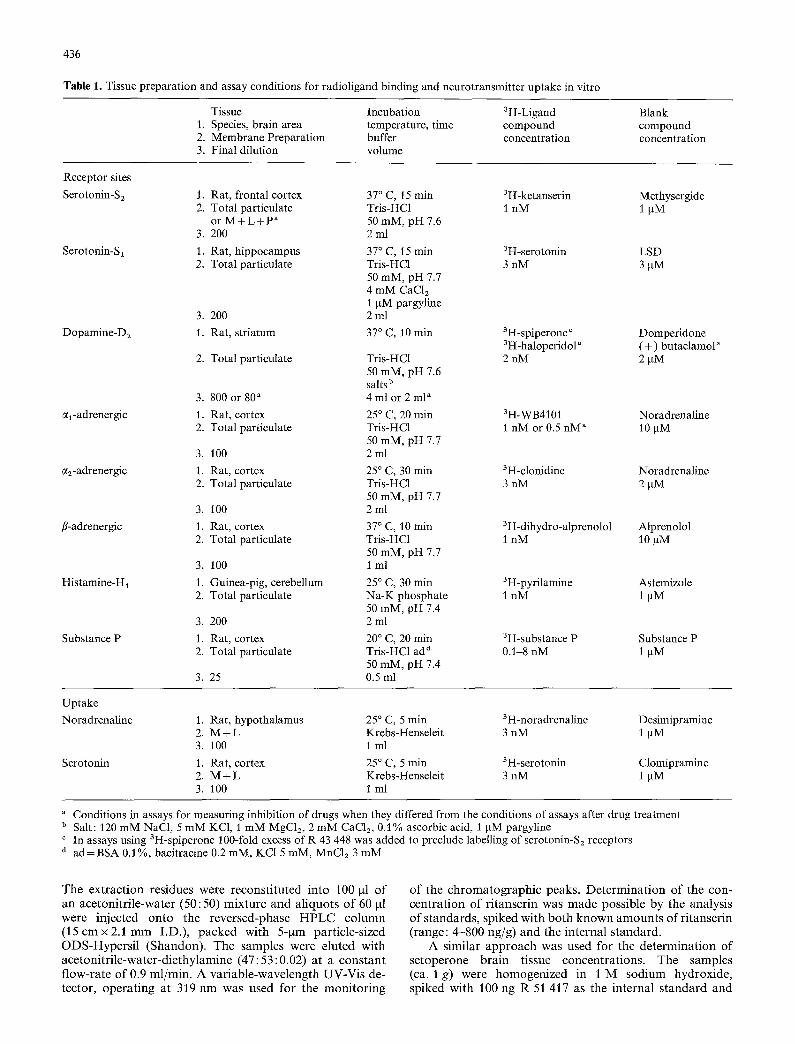
Table 1. Tissue preparation and assay conditions for radioligand binding and neurotransmitter uptake in vitro
Tissue Incubation 3H-Ligand Blank 1. Species, brain area temperature, time compound compound 2. Membrane Preparation buffer concentration concentration 3. Final dilution volume
Receptor sites
Serotonin-S2 t. Rat, frontal cortex 37 ° C, 15 min 3H-ketanserin Methysergide 2. Total particulate Tris-HC1 1 nM 1 gM
or M + L + p a 50 mM, pH 7.6 3. 200 2 ml
Serotonin-S1 1. Rat, hippocampus 37 ° C, 15 min 3H-serotonin LSD 2. Total particulate Tris-HC1 3 nM 3 gM
50 raM, pH 7.7 4 mM CaC12 1 gM pargyline
3. 200 2 ml
Dopamine-D2 1. Rat, striatum 37 ° C, 10 min 3H-spiperoneC Domperidone 3H-haloperidola (+) butaclamol a
2. Total particulate Tris-HC1 2 nM 2 gM 50 mM, pH 7.6 salts b
3. 800 or 80 a 4 ml or 2 ml a
cq-adrenergic 1. Rat, cortex 25 ° C, 20 rain 3H-WB4101 Noradrenaline 2. Total particulate Tris-HC1 1 nM or 0.5 nM" 10 gM
50 mM, pH 7.7 3. 100 2ml
~2-adrenergic 1. Rat, cortex 25 ° C, 30 min 3H-clonidine Noradrenaline 2. Total particulate Tris-HC1 3 nM 2 gM
50 mM, pH 7.7 3. 100 2ml
fl-adrenergic 1. Rat, cortex 37 ° C, 10 min 3H-dihydro-alprenolol Alprenolol 2. Total particulate Tris-HC1 1 nM 10 gM
50 mM, pH 7.7 3. 100 1 ml
Histamine-H1 1. Guinea-pig, cerebellum 25 ° C, 30 rain 3H-pyrilamine Astemizole 2. Total particulate Na-K phosphate 1 nM 1 ~M
50 mM, pH 7.4 3. 200 2 ml
Substance P 1. Rat, cortex 20 ° C, 20 rain 3H-substance P Substance P 2. Total particulate Tris-HC1 ad a 0.1-8 nM 1 gM
50 mM, pH 7.4 3. 25 0.5 rnl
Uptake
Noradrenaline
Serotonin
1. Rat, hypothalamus 25 ° C, 5 min 3H-noradrenaline Desimipramine 2. M + L Krebs-Henseleit 3 nM 1 gM 3. 100 1 ml
1. Rat, cortex 25 ° C, 5 rain 3H-serotonin Clomipramine 2. M + L Krebs-Henseleit 3 nM 1 gM 3. 100 1 ml
a Conditions in assays for measuring inhibition of drugs when they differed from the conditions of assays after drug treatment b Salt: 120 mM NaC1, 5 mM KC1, 1 mM MgC12, 2 mM CaC1/, 0.1% ascorbic acid, 1 gM pargyline c In assays using 3H-spiperone 100-fold excess of R 43 448 was added to preclude labelling of serotonin-S2 receptors a ad=BSA 0.1%, bacitracine 0.2 mM, KC1 5 mM, MnC12 3 mM
The extraction residues were reconstituted into 100 gl of an acetonitrile-water (50: 50) mixture and aliquots of 60 gl were injected onto the reversed-phase HPLC column (15 cmx2 .1 mm I.D.), packed with 5-gm particle-sized ODS-Hypersil (Shandon). The samples were eluted with acetonitrile-water-diethylamine (47:53 : 0.02) at a constant flow-rate of 0.9 ml/min. A variable-wavelength UV-Vis de- tector, operating at 319 nm was used for the monitor ing
of the chromatographic peaks. Determinat ion of the con- centration of ritanserin was made possible by the analysis of standards, spiked with both known amounts of ritanserin (range: 4-800 ng/g) and the internal standard.
A similar approach was used for the determination of setoperone brain tissue concentrations. The samples (ca. I g) were homogenized in I M sodium hydroxide, spiked with 100 ng R 51 417 as the internal standard and
437
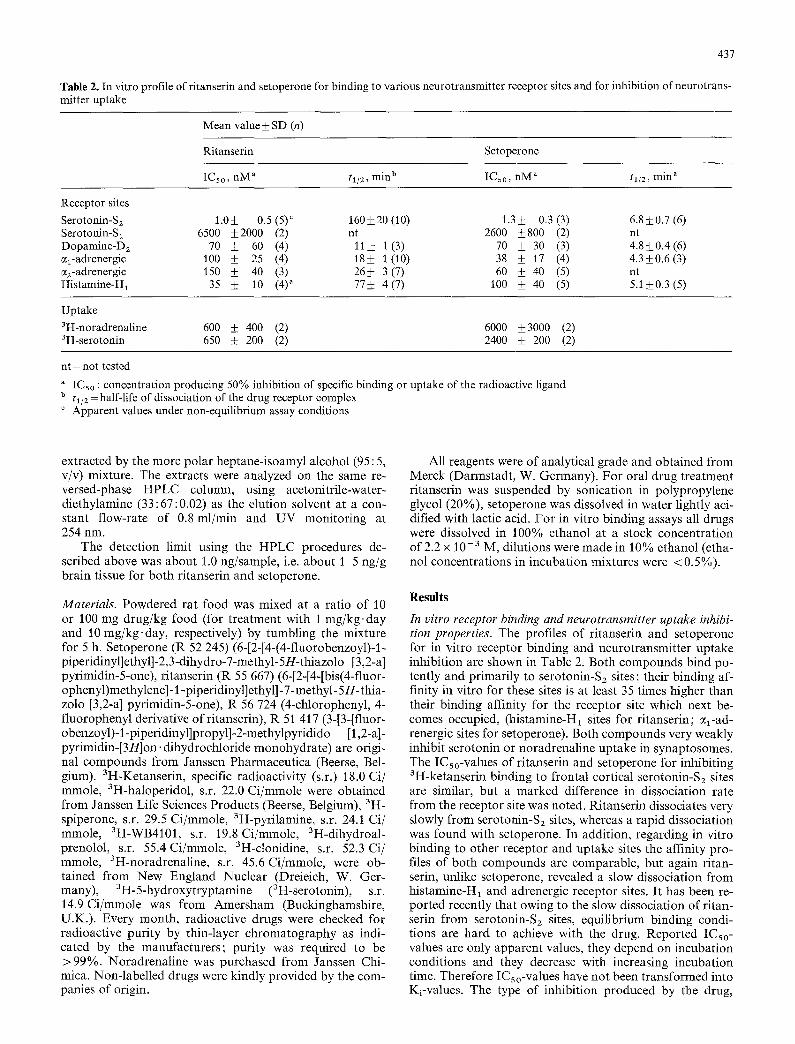
Table 2. In vitro profile of ritanserin and setoperone for binding to various neurotransmitter receptor sites and for inhibition of neurotrans- mitter uptake
Mean value _+ SD (n)
Ritanserin Setoperone
ICs0, nM a tl/2, m i n b IC50, nM a tl/z, rain a
Receptor sites Serotonin-Sz 1.0+_ 0.5 (5) c 160+_20 (10) 1.3_+ 0.3 (3) 6.8_+0.7 (6) Serotonin-S~ 6500 +_2000 (2) nt 2600 +_800 (2) nt Dopamine-D 2 70 ± 60 (4) 11+ 1(3) 70 _+ 30 (3) 4.8-+0.4(6) cq-adrenergic 100 ± 25 (4) 18_+ 1 (10) 38 ± 17 (4) 4.3±0.6(3) ~z-adrenergic 150 + 40 (3) 26_+ 3 (7) 60 ± 40 (5) nt Histamine-H1 35 _+ 10 (4) a 77+ 4 (7) 100 +_ 40 (5) 5.1 -+0.3 (5)
Uptake 3H-noradrenaline 600 + 400 (2) 6000 _+3000 (2) aH-serotonin 650 -t- 200 (2) 2400 _+ 200 (2)
nt = not tested
a ICso : concentration producing 50% inhibition of specific binding or uptake of the radioactive ligand b tin = half-life of dissociation of the drug receptor complex ° Apparent values under non-equilibrium assay conditions
extracted by the more polar heptane-isoamyl alcohol (95 : 5, v/v) mixture. The extracts were analyzed on the same re- versed-phase HPLC column, using acetonitrile-water- diethylamine (33:67:0.02) as the elution solvent at a con- stant flow-rate of 0.8 ml/min and UV monitoring at 254 nm.
The detection limit using the HPLC procedures de- scribed above was about 1.0 rig/sample, i.e. about 1-5 ng/g brain tissue for both ritanserin and setoperone.
Materials. Powdered rat food was mixed at a ratio of 10 or 100 mg drug/kg food (for treatment with 1 mg/kg.day and 10 mg/kg-day, respectively) by tumbling the mixture for 5 h. Setoperone (R 52 245) (6-[2-[4-(4-fluorobenzoyl)-l- piperidinyl]ethyl]-2,3-dihydro-7-methyl- 5H-thiazolo [3,2-a] pyrimidin-5-one), ritanserin (R 55 667) (6-[2-[4-[bis(4-fluor- ophenyl)methylene]-l-piperidinyl]ethyl]-7-methyl-5H-thia- zolo [3,2-a] pyrimidin-5-one), R 56 724 (4-chlorophenyl, 4- fluorophenyl derivative of ritansefin), R 51 417 (3-[3-[fluor- obenzoyl)-l-piperidinyl]propyl]-2-methylpyridido [1,2-a]- pyrimidin-[3H]on.dihydrochloride monohydrate) are origi- nal compounds from Janssen Pharmaceutica (Beerse, Bel- gium). 3H-Ketanserin, specific radioactivity (s.r.) 18.0 Ci/ mmole, 3H-haloperidol, s.r. 22.0 Ci/mmole were obtained from Janssen Life Sciences Products (Beerse, Belgium), 3H- spiperone, s.r. 29.5 Ci/mmole, 3H-pyrilamine, s.r. 24.1 Ci/ mmole, 3H-WB4101, s.r. 19.8 Ci/mmole, 3H-dihydroal- prenolol, s.r. 55.4 Ci/mmole, 3H-clonidine, s.r. 52.3 Ci/ mmole, 3H-noradrenaline, s.r. 45.6 Ci/mmole, were ob- tained from New England Nuclear (Dreieich, W. Ger- many), 3H-5-hydroxytryptamine (3H-serotonin), s.r. 14.9 Ci/mmole was from Amersham (Buckinghamshire, U.K.). Every month, radioactive drugs were checked for radioactive purity by thin-layer chromatography as indi- cated by the manufacturers; purity was required to be >99%. Noradrenaline was purchased from Janssen Chi- mica. Non-labelled drugs were kindly provided by the com- panies of origin.
All reagents were of analytical grade and obtained from Merck (Darmstadt, W. Germany). For oral drug treatment ritanserin was suspended by sonication in polypropylene glycol (20%), setoperone was dissolved in water lightly aci- dified with lactic acid. For in vitro binding assays all drugs were dissolved in 100% ethanol at a stock concentration of 2.2 x 10 .3 M, dilutions were made in 10% ethanol (etha- nol concentrations in incubation mixtures were < 0.5%).
Results
In vitro receptor binding and neurotransmitter uptake inhibi- tion properties. The profiles of ritanserin and setoperone for in vitro receptor binding and neurotransmitter uptake inhibition are shown in Table 2. Both compounds bind po- tently and primarily to serotonin-S2 sites; their binding af- finity in vitro for these sites is at least 35 times higher than their binding affinity for the receptor site which next be- comes occupied, (histamine-H1 sites for ritanserin; ~l-ad- renergic sites for setoperone). Both compounds very weakly inhibit serotonin or noradrenaline uptake in synaptosomes. The ICso-values of ritanserin and setoperone for inhibiting 3H-ketanserin binding to frontal cortical serotonin-S2 sites are similar, but a marked difference in dissociation rate from the receptor site was noted. Ritanserin dissociates very slowly from serotonin-S2 sites, whereas a rapid dissociation was found with setoperone. In addition, regarding in vitro binding to other receptor and uptake sites the affinity pro- files of both compounds are comparable, but again ritan- serin, unlike setoperone, revealed a slow dissociation from histamine-H1 and adrenergic receptor sites. It has been re- ported recently that owing to the slow dissociation of ritan- serin from serotonin-S2 sites, equilibrium binding condi- tions are hard to achieve with the drug. Reported ICso- values are only apparent values, they depend on incubation conditions and they decrease with increasing incubation time. Therefore ICs o-values have not been transformed into Ki-values. The type of inhibition produced by the drug,
438
Control (n: 10) Setoperone (n = 5 ) 10-9M
Setoperone (n= 5) 0.24 10 -9 M (preinc.)
mean values *- SD
KD(nM) Bmax f motes/rag tissue
0.38-* 0.08 23.0-* 2.0 1.8 + 0.3*** 26.0-* 3.0
1.9-'0.6 **~ 21.0+ 2.0
~ , *~*p<0.001
0.20-
,,=
m ~-~ 0 . 1 6 - ~//~TROL
Y_~'~ 0.12 ~k~.
uJuJ SETOPERONE 10-9M "~
006- / ~,
0.0/-,
0 i ' , , , ~ " ~ , 0 (103 0.06 0.09 0.12
3H-KETANSERIN SPECIFICALLY BOUND
(p moles/m[)
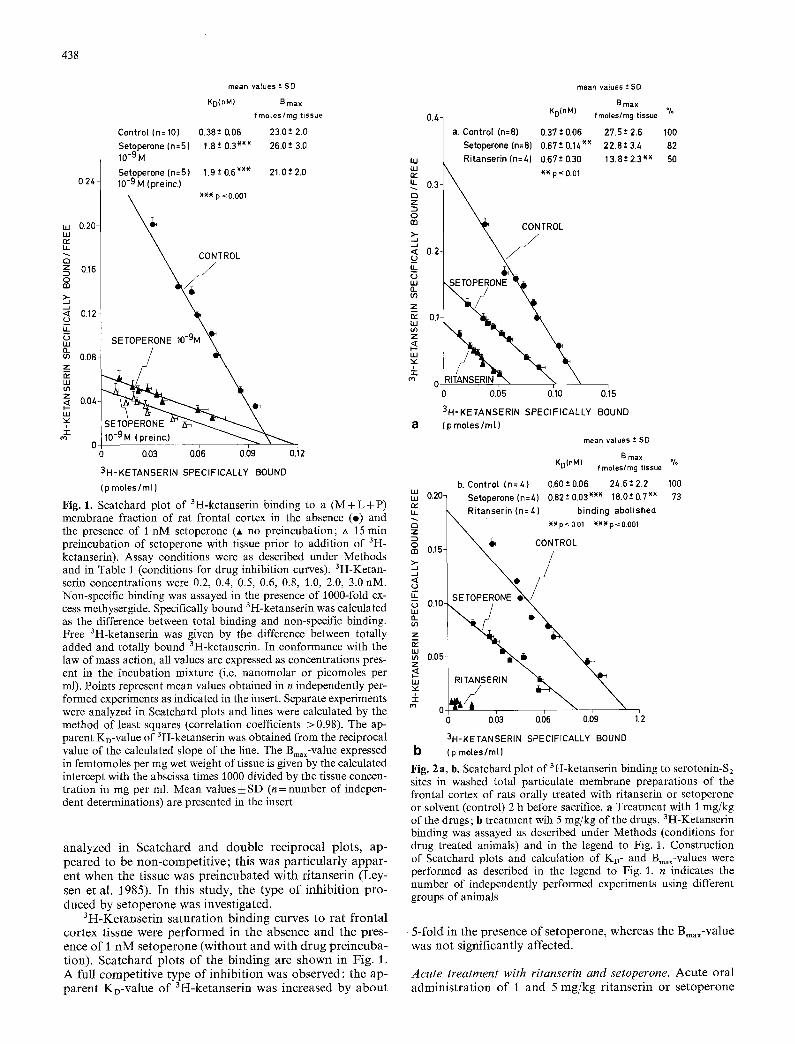
Fig. 1. Scatchard plot of 3H-ketanserin binding to a (M + L +P) membrane fraction of rat frontal cortex in the absence (o) and the presence of 1 nM setoperone (A no preincubation; zx 15 rain preincubation of setoperone with tissue prior to addition of 3H- ketanserin). Assay conditions were as described under Methods and in Table 1 (conditions for drug inhibition curves). 3H-Ketan- serin concentrations were 0.2, 0.4, 0,5, 0.6, 0.8, 1.0, 2.0, 3.0 nM. Non-specific binding was assayed in the presence of t000-fold ex- cess methysergide. Specifically bound 3H-ketanserin was calculated as the difference between total binding and non-specific binding. Free 3H-ketanserin was given by the difference between totally added and totally bound 3H-ketanserin. In conformance with the law of mass action, all values are expressed as concentrations pres- ent in the incubation mixture (i.e. nanomolar or picomoles per ml). Points represent mean values obtained in n independently per- formed experiments as indicated in the insert. Separate experiments were analyzed in Scatchard plots and lines were calculated by the method of least squares (correlation coefficients > 0.98). The ap- parent KD-value of 3H-ketanserin was obtained from the reciprocal value of the calculated slope of the line. The Bin,x-value expressed in femtomoles per mg wet weight of tissue is given by the calculated intercept with the abscissa times 1000 divided by the tissue concen- tration in mg per ml. Mean values _+ SD (n = number of indepen- dent determinations) are presented in the insert
analyzed in Scatchard and double reciprocal plots, ap- peared to be non-competit ive; this was particularly appar- ent when the tissue was preincubated with ritanserin (Ley- sen et al. 1985). In this study, the type of inhibit ion pro- duced by setoperone was investigated.
3H-Ketanserin saturation binding curves to rat frontal cortex tissue were performed in the absence and the pres- ence of 1 nM setoperone (without and with drug preincuba- tion). Scatchard plots of the binding are shown in Fig. 1. A full competitive type of inhibit ion was observed: the ap- parent KD-value of 3H-ketanserin was increased by about
mean values -+ SD
B ma x 0.4 KD(nM) fmoles/mg tissue %
a. Control (n=8) 0.37-*0.06 27.5-*2.6 100 Setoperone(n=8) 0.67"-0.14 ~*~ 22.8-'3./, 82
uJ Ritanserin (n=/,) 0.67*-0.30 13.8-+2.3 ~* 50 rytLI ~ ** p < 0.01 u_ 0 . 3 - Q z O m .J -~ >- ' ~ , ~ T R O L
< 0.2- o E
n. 0.1-
¢~ 0 i i 0.05 0.10 0.15
3H-KETANSERIN SPECIFICALLY BOUND a (p moles/m[)
mean values -* SD
B max IJ l L4 t O~
~DIn"u f moles/mg tissue
b. Control (n=/,) 0.60*-0.06 24.6*-2.2 100 m m 0.20- Setoperone(n=4) 0.62-*0.03*** 18.0+0.7 *~ 73 r~ tL ~ Ritanserin In: Z,) binding abol ished
Z ~ \ ~ ** p< 0.01 *** p< 0.001
-~ ~ CONTROL m ° 0.15
..a <
u_ 0.1C
LM 0_ if) z
LU m 0.05- Z
uJ
4- ¢ o 0 - i
0 0.03 0.06 0.09 1.2
3H-KETANSERIN SPECIFICALLY BOUND b (p mo[es/ml)
Fig. 2 a, b. Scatchard plot of 3H-ketanserin binding to serotonin-S2 sites in washed total particulate membrane preparations of the frontal cortex of rats orally treated with ritanserin or setoperone or solvent (control) 2 h before sacrifice, a Treatment with 1 mg/kg of the drugs; b treatment wih 5 mg/kg of the drugs. 3H-Ketanserin binding was assayed as described under Methods (conditions for drug treated animals) and in the legend to Fig. 1. Construction of Scatchard plots and calculation of KD- and Bin.x-values were performed as described in the legend to Fig. 1. n indicates the number of independently performed experiments using different groups of animals
5-fold in the presence of setoperone, whereas the Bin,x-value was not significantly affected.
Acute treatment with ritanserin and setoperone. Acute oral administrat ion of I and 5 mg/kg ritanserin or setoperone
439
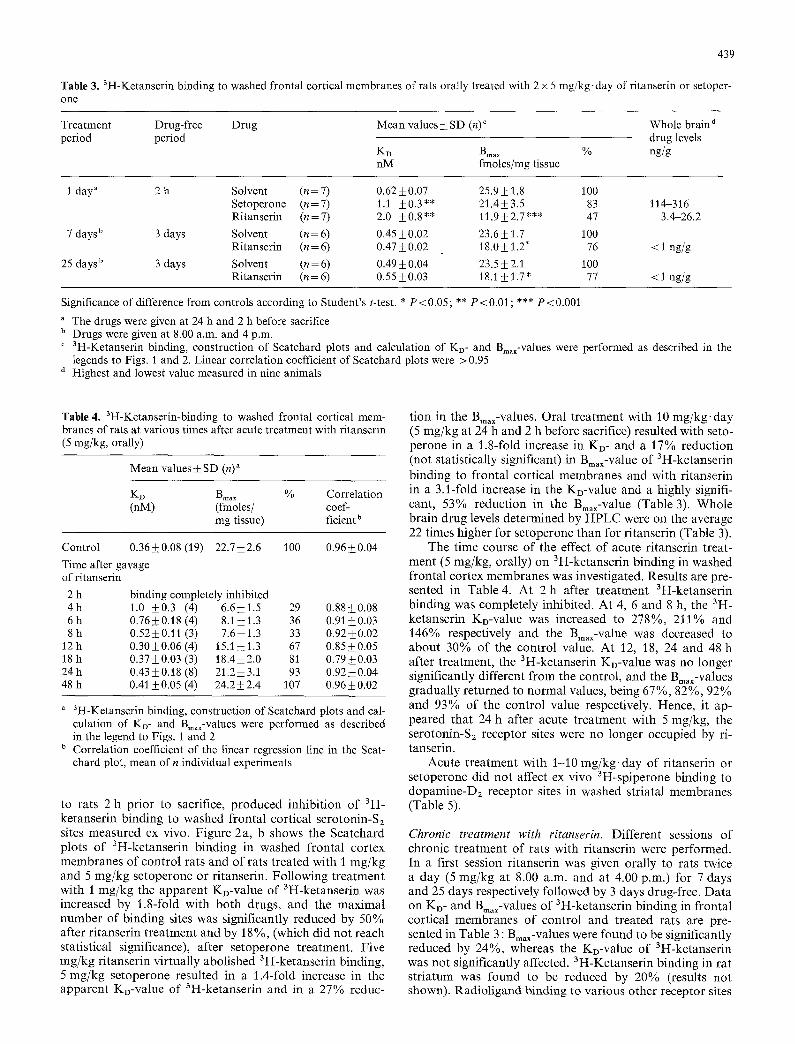
Table 3. 3H-Ketanserin binding to washed frontal cortical membranes of rats orally treated with 2 x 5 mg/kg, day of ritanserin or setoper- one
Treatment Drug-free Drug Mean values +- SD (n) ~ Whole brain d period period drug levels
KD B~. x % ng/g nM fmoles/mg tissue
1 day" 2 h Solvent (n=7) 0.62_+0.07 25.9_+ 1.8 100 Setoperone (n=7) 1.t _+0.3** 21.4_+3.5 83 114-316 Ritanserin (n=7) 2.0 +_0.8** 11.9_+2.7"** 47 3.4-26.2
7 days b 3 days Solvent (n=6) 0.45-+0.02 23.6+_1.7 100 Ritanserin (n = 6) 0.47 +_ 0.02 18.0 -+ 1.2" 76 < 1 ng/g
25 days b 3 days Solvent (n = 6) 0.49 _+ 0.04 23.5 _+ 2.1 100 Ritanserin (n = 6) 0.55 _+ 0.03 18.1 _+ 1.7 * 77 < 1 ng/g
Significance of difference from controls according to Student's t-test. * P < 0.05; ** P < 0.01; *** P < 0.001
The drugs were given at 24 h and 2 h before sacrifice b Drugs were given at 8.00 a.m. and 4 p.m. c 3H_Ketanserin binding, construction of Scatchard plots and calculation of KD- and Bm~-values were performed as described in the
legends to Figs. 1 and 2. Linear correlation coefficient of Scatchard plots were > 0.95 d Highest and lowest value measured in nine animals
Table 4. 3H-Ketanserin-binding to washed frontal cortical mem- branes of rats at various times after acute treatment with ritanserin (5 mg/kg, orally)
Mean values_+ SD (n) a
KD Bm,x % Correlation (nM) (fmoles/ coef-
mg tissue) ficient b
Control 0.36_+0.08 (19) 22.7_+2.6 100 0.96+_0.04
Time after gavage of ritanserin
2 h binding completely inhibited 4h 1.0 +_0.3 (4) 6.6_+1.5 29 0.88_+0.08 6h 0.76_+0.18(4) 8.1_+1.3 36 0.91_+0.03 8 h 0.52+0.11 (3) 7.6_+1.3 33 0.92_+0.02
12 h 0.30_+0.06 (4) 15.1 _+ 1.3 67 0.85+_0.05 18 h 0.37_+0.03 (3) 18.4_+2.0 81 0.79+_0.03 24 h 0.43_+0.18 (8) 21.2_+3.1 93 0.92_+0.04 48 h 0.41 +0.05 (4) 24.2_+2.4 107 0.96_+0.02
a aH_Ketanserin binding, construction of Scatchard plots and cal- culation of KD- and Bm,~-values were performed as described in the legend to Figs. 1 and 2
b Correlation coefficient of the linear regression line in the Scat- chard plot, mean of n individual experiments
to rats 2 h pr ior to sacrifice, p roduced inhibi t ion of 3H- ketanserin binding to washed frontal cortical serotonin-S2 sites measured ex vivo. Figure 2a, b shows the Scatchard plots o f 3H-ketanserin binding in washed frontal cortex membranes of control rats and of rats t reated with I mg/kg and 5 mg/kg setoperone or ritanserin. Fol lowing t rea tment with 1 mg/kg the apparen t KD-value of 3H-ketanserin was increased by 1.8-fold with both drugs, and the maximal number of binding sites was significantly reduced by 50% after r i tanserin t rea tment and by 18 %, (which did not reach statistical significance), after setoperone treatment. Five mg/kg ri tanserin vir tual ly abolished 3H-ketanserin binding, 5 mg/kg setoperone resulted in a 1.4-fold increase in the apparent KD-value of 3H-ketanserin and in a 27% reduc-
tion in the Bmax-values. Oral t reatment with 10 mg/kg, day (5 mg/kg at 24 h and 2 h before sacrifice) resulted with seto- perone in a 1.8-fold increase in KD- and a 17% reduction (not statistically significant) in Bmax-value of 3H-ketanserin binding to frontal cortical membranes and with ri tanserin in a 3.1-fold increase in the KD-value and a highly signifi- cant, 53% reduct ion in the Bmax-value (Table 3). Whole brain drug levels determined by HPLC were on the average 22 times higher for setoperone than for r i tanserin (Table 3).
The time course of the effect of acute ri tanserin treat- ment (5 mg/kg, orally) on 3H-ketanserin binding in washed frontal cortex membranes was investigated. Results are pre- sented in Table 4. At 2 h after t reatment 3H-ketanserin binding was completely inhibited. At 4, 6 and 8 h, the 3H- ketanserin KD-value was increased to 278%, 2J1% and 146% respectively and the Bmax-value was decreased to about 30% of the control value. At 12, 18, 24 and 48 h after t reatment, the 3H-ketanserin KD-value was no longer significantly different from the control, and the Bma~-values gradual ly returned to normal values, being 67%, 82%, 92% and 93% of the control value respectively. Hence, it ap- peared that 24 h after acute t reatment with 5 mg/kg, the serotonin-S2 receptor sites were no longer occupied by ri- tanserin.
Acute t reatment with 1-10 m g / k g . d a y of r i tanserin or setoperone did not affect ex vivo 3H-spiperone binding to dopamine-D2 receptor sites in washed striatal membranes (Table 5).
Chronic treatment with ritanserin. Different sessions of chronic t reatment of rats with ri tanserin were performed. In a first session ri tanserin was given orally to rats twice a day (5 mg/kg at 8.00 a.m. and at 4.00 p.m.) for 7 days and 25 days respectively followed by 3 days drug-free. Da ta on KD- and Bmax-values of 3H-ketanserin binding in frontal cortical membranes of control and treated rats are pre- sented in Table 3 : Bmax-values were found to be significantly reduced by 24%, whereas the KD-value of 3H-ketanserin was not significantly affected. 3H-Ketanserin binding in rat s tr iatum was found to be reduced by 20% (results not shown). Rad io l igand binding to various other receptor sites

440
Table 5. 3H-Spiperone binding to dopamine-D2 sites in washed striatal membranes of rats acutely or chronically treated with ritan- serin or setoperone
Mean values _+ SD"
KD B,,,x % of (nM) (fmoles control
• r a g - 1 tissue)
Solvent (n = 33) 0.046 + 0210
Ritanserin
1. Acutely
I mg/kg at - 2 h; n=4 0.035_+0.002 5 mg/kg at - 2 h; n = 4 0.053__0.006 5 mg/kg at - 2 4 h 0.044_+0.010 and - 2 h; n=7
2. Chronically b
10 mg/kg.day; n=7 0.045_+0.009
Setoperone
1. Acutely
i mg/kg at - 2 h; n=7 0.042_+0.010 5 mg/kg at - 2 h; n=4 0.050_+0.007 5 mg/kg at - 24 h 0.050-+ 0.008 and - 2 h; n=7
2. Chronically °
i mg/kg-day; n=4 0.040_+0.006 10 mg/kg'day; n=7 0.044__0.010
Haloperidol
I mg/kg.day; n=3 0.059+_0.008
28.2-+3.1 100
27.8 _+ 2.4 99 27.6_+ 1.3 98 27.3 _+ 2.4 97
29.4_+3.4 106
27.6_+2.7 98 28.5_+0.9 101 30.5_+4.5 108
32.7_+3.3 l l5 43.2_+7.5*** 153
49.6___2.2*** 176
*** Significance of difference from control values according to Stu- dent's t-test. P<0.00I
a 3H_Spiperon e binding, construction of Scatchard plots and cal- culation of Ko- and Bmax-values were as described under legend to Fig. 5
b Rats treated with ritanserin for 25 days, followed by 3 days drug- free (cf legend to Fig. 3) Rats treated with setoperone or haloperidol for 28 days, followed by 5 days drug-free (cf legend to Figs. 4-5)
was unaffected, i.e. 3H-spiperone and 3H-haloperidol bind- ing to striatal dopamine-D2 sites, 3H-WB4J01 binding to ~l-adrenergic, 3H-clonidine binding to c~2-adrenergic and 3H-dihydroalprenolol binding to fl-adrenergic sites in corti- cal tissue, 3H-flunitrazepam binding to cortical benzodiaze- pine sites, 3H-serotonin binding to hippocampal serotonin- S, sites and 3H-substance P binding to cortical membranes.
In a second experimental session rats were given ritan- serin mixed with food for a period of 25 days. Mean daily ritanserin intake amounted to 14___ 1 mg/kg. The ritanserin treatment period was followed by drug-free periods of 1, 3, 6, 12 and 18 days. Scatchard plots of 3H-ketanserin bind- ing to frontal cortical membranes of control and treated rats are shown in Fig. 3. The KD-value of 3H-ketanserin was slightly, but significantly increased in tissue of rats with a l -day drug-free period, in rats with longer drug-free peri- ods Ko-values were not significantly different from control values. Bma~-values were highly significantly reduced in tis- sues of chronically treated rats after I day drug-free ( - 5 0 % ) , 3 days drug-free ( - 3 4 % ) and 6 days drug-free ( - 1 8 % ) . In rats which had been drug-free for 12 and
uJ LU
U_
r~ Z
O m
>. .,J
kL G uJ Q_ oej
z
ILl
OA
-r ¢~
MEAN V A L U E S : SD ( n = 3 )
Ko BMAX % l nM tmoles /rag 0.3 C O N T R O L 0 , 3 7 ; 0 . 0 4 19 .7 ; 1.9 1 0 0
D A Y S AFTER T R E A T M E N T
1 0 . 5 1 : 0 . 0 7 )~ 9 . 8 : 0 . 6 )~*)~ 5 0 3 0 . 4 3 : 0 . 0 5 13.0 : 0.7 )~*)~ 66
0.2-
0.1-
iiiiiiiii iiiiiiii ii p < 0 . 0 5 X~<X p<0.001
2
0.025 0.05 0,075 0.1
3H KETANSERIN SPECIFICALLY BOUND (pmotes/ml)
Fig. 3. Scatchard plots of 3H-ketanserin binding to washed total particulate fractions of frontal cortex of rats chronically treated with ritanserin mixed with food (daily drug intake 14+ l mg/kg. day). Rats were treated with ritanserin for 25 days, followed by drug-free periods of J, 3, 6, 12 and 18 days as indicated on the graph. Control rats, kept under the same conditions as treated rats, were concomitantly assayed in each session; since interassay variations were not significant, the average of all control values was calculated (n = 15 for control values), 3H-Ketanserin binding, construction of Scatchard plots and calculation of KD- and Bma x- values were performed as described in the legend to Figs. I and 2. Ritanserin whole brain levels, determined by HPLC, were below detection level (< 5 ng/g) in all the tissues• Insert: Semilogarithmic plot of the rate of receptor reappearance according to Mauger et al. (1982) and Sladeczek and Bockaert (1983). Rss = steady state receptor concentration; R, = receptor concentration at time t; k= rate constant of receptor degradation• When receptor repopulation is given by receptor production (with rate constant r) and receptor degradation (with rate constant k), then
R _ r ~ = r - - k ( R t ) or t - ~ ( / - - e -kt)
when t ~ oe, than Rt approaches k ' which is equal to
r Rss =
Hence
Rt=Rss ( l - e kt)
and
In Rss = k t Rss-- R~
from k = 0.2 day- 1 and Rss = 19.7 fmoles/mg tissue r = 3.94 fmoles/ mg tissue-day; the receptor half-life is 3.5 days
LIJ LIJ re" LL
C~ Z
0 m
>- .-4
K G m &. m z
U.I m Z
u j
Ko(nM)
Control (n=7) 0.5± 0.2 Setoperone (n=/.) 0.4*- 0.1 1 mg/kg Setoperone (n=7) 0.6 t 0.1
0.20 \ lO mg/kg \ * p <0.05 \%
t \ \ o ,.OL
0.10- 10 mg~/kg ~ ~ ~
0.05-
mean va lues t SD
B m a x % f m e t e s . r a g -1 t i s s u e
20.5"-1.9 100 18.5 *- 1.6 90
16.1"-3.0 ~ 78
0 -
0 0.03 0.06 0.09 0.12 3H-KETANSERIN SPECIFICALLY BOUND (p moles/m[)
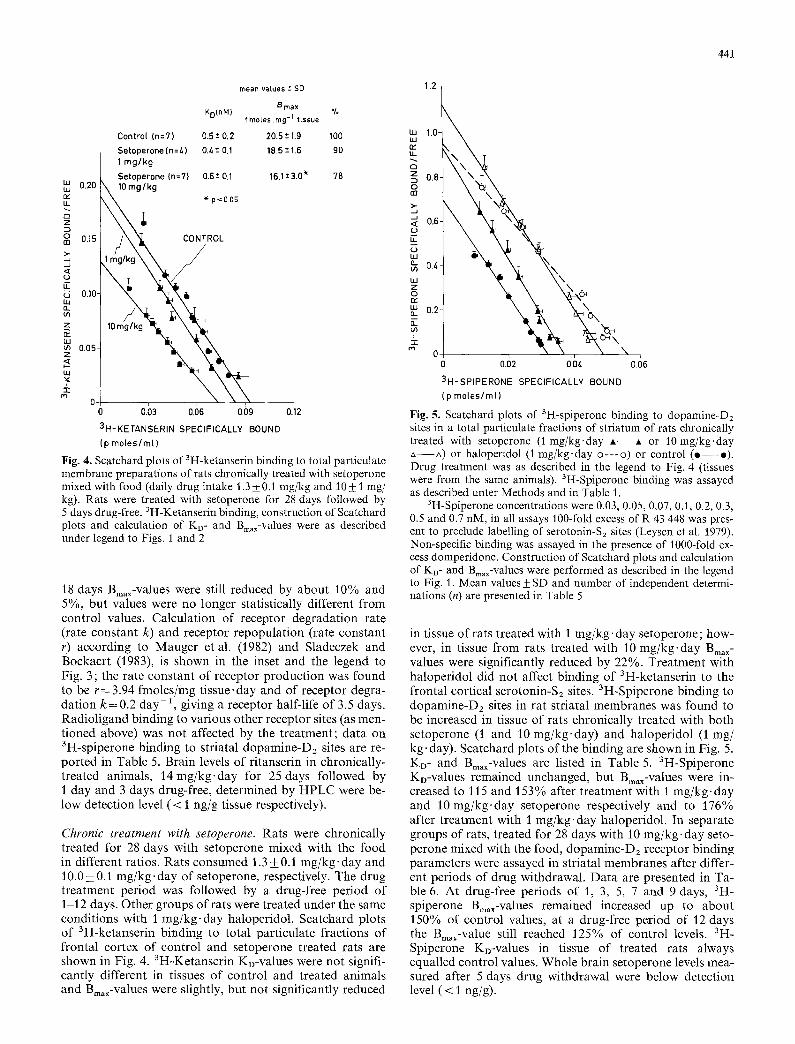
Fig. 4. Scatchard plots of 3H-ketanserin binding to total particulate membrane preparations of rats chronically treated with setoperone mixed with food (daily drug intake 1.3 4-0.1 mg/kg and 10_ 1 mg/ kg). Rats were treated with setoperone for 28 days followed by 5 days drug-free. 3H-Ketanserin binding, construction of Scatchard plots and calculation of Ko- and Bmax-values were as described under legend to Figs. 1 and 2
18 days Bmax-values were still reduced by about 10% and 5%, but values were no longer statistically different from control values. Calculation of receptor degradation rate (rate constant k) and receptor repopulation (rate constant r) according to Mauger et al. (1982) and Sladeczek and Bockaert (1983), is shown in the inset and the legend to Fig. 3 ; the rate constant of receptor production was found to be r = 3.94 fmoles/mg tissue, day and of receptor degra- dation k = 0.2 day-2, giving a receptor half-life of 3.5 days. Radioligand binding to various other receptor sites (as men- tioned above) was not affected by the treatment; data on 3H-spiperone binding to striatal dopamine-D2 sites are re- ported in Table 5. Brain levels of ritanserin in chronically- treated animals, 14mg/kg.day for 25 days followed by 1 day and 3 days drug-free, determined by HPLC were be- low detection level (< 1 ng/g tissue respectively).
Chronic treatment with setoperone. Rats were chronically treated for 28 days with setoperone mixed with the food in different ratios. Rats consumed 1.3 + 0.1 mg/kg-day and 10.0 + 0.1 mg/kg.day of setoperone, respectively. The drug treatment period was followed by a drug-free period of 1-12 days. Other groups of rats were treated under the same conditions with 1 mg/kg-day haloperidol. Scatchard plots of 3H-ketanserin binding to total particulate fractions of frontal cortex of control and setoperone treated rats are shown in Fig. 4. 3H-Ketanserin KD-values were not signifi- cantly different in tissues of control and treated animals and Bin,x-values were slightly, but not significantly reduced
441
1.2
W UJ
U_
0
O (13
...1
U_
W
1.O
0.8-
0.6-
0i 0. \
0 0.(72 \
0.04 o.~6
3H-SPIPERONE SPECIFICALLY BOUND (p moles/mr)
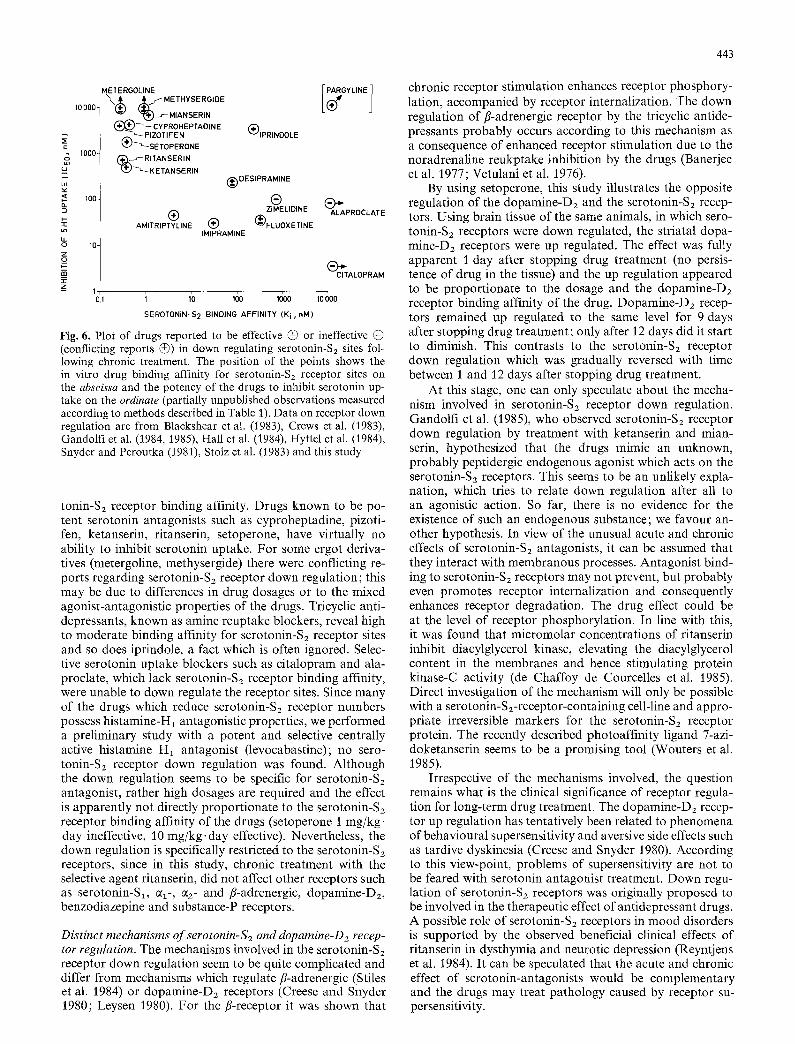
Fig. 5. Scatchard plots of 3H-spiperone binding to dopamine-D2 sites in a total particulate fractions of striatum of rats chronically treated with setoperone (1 mg/kg.day A- -A or 10 mg/kg.day A--A) or haloperidol (1 mg/kg.day o---o) or control ( o - -* ) . Drug treatment was as described in the legend to Fig. 4 (tissues were from the same animals). 3H-Spiperone binding was assayed as described unter Methods and in Table 1.
3H-Spiperone concentrations were 0.03, 0.05, 0.07, 0.1, 0.2, 0.3, 0.5 and 0.7 nM, in all assays 100-fold excess of R 43 448 was pres- ent to preclude labelling of serotonin-Sz sites (Leysen et al. 1979). Non-specific binding was assayed in the presence of t000-fold ex- cess domperidone. Construction of Scatchard plots and calculation of Ko- and Bmax-values were performed as described in the legend to Fig. 1. Mean values 4- SD and number of independent determi- nations (n) are presented in Table 5
in tissue of rats treated with I mg/kg, day setoperone; how- ever, in tissue from rats treated with 10 mg/kg-day Bin, x- values were significantly reduced by 22%. Treatment with haloperidol did not affect binding of 3H-ketanserin to the frontal cortical serotonin-S2 sites. 3H-Spiperone binding to dopamine-Dz sites in rat striatal membranes was found to be increased in tissue of rats chronically treated with both setoperone (1 and 10 mg/kg-day) and haloperidol (1 mg/ kg. day). Scatchard plots of the binding are shown in Fig. 5. KD- and Bmax-values are listed in Table 5. 3H-Spiperone KD-values remained unchanged, but Bmax-values were in- creased to 115 and 153% after treatment with 1 mg/kg.day and 10 mg/kg.day setoperone respectively and to 176% after treatment with 1 mg/kg.day haloperidol. In separate groups of rats, treated for 28 days with 10 mg/kg-day seto- perone mixed with the food, dopamine-D2 receptor binding parameters were assayed in striatal membranes after differ- ent periods of drug withdrawal. Data are presented in Ta- ble 6. At drug-free periods of 1, 3, 5, 7 and 9 days, aH- spiperone Bmax-values remained increased up to about 150% of control values, at a drug-free period of 12 days the Bmax-value still reached 125% of control levels. 3H- Spiperone KD-values in tissue of treated rats always equalled control values. Whole brain setoperone levels mea- sured after 5 days drug withdrawal were below detection level (< 1 ng/g).

442
Table 6. 3H-Spiperone binding to dopamine-D2 sites in washed striatal membranes of rats treated for 28 days with 10 mg/kg.day setoperone mixed with food, followed by different drug free periods
Mean values_+ SD"
Ko Bmax % (nM) (fmoles/mg tissue)
0.043_+0.009 24.2_+2.6 Control (n = 24)
Setoperone treated Drug-free period
1 day (n=4) 3 days (n = 4) 5 days (n = 4) 7 days (n = 4) 9 days (n = 4)
12 days (n = 4)
0.047_+0.004 37.3_+2.5 154 0.040_+0.003 36.9_+ 1.4 152 0.046_+0.007 34.3_+3.0 142 0.041 _ 0.004 37.9 + 2.7 157 0.042_+0.007 37.2_+ 1.3 154 0.035-+-0.006 30.4_+2.8 126
3H-Spiperone binding, construction of Scatchard plots and cal- culations of KD- and Bmax-values were as described under legend to Fig. 5
Discussion
Receptor binding properties and specificity of action of ritan- serin and setoperone. Ritanserin has previously been de- scribed to bind primarily and with high affinity to sero- tonin-Sz receptor sites and to dissociate slowly from these sites in vitro. Because of the latter property, the inhibition by ritanserin of 3H-ketanserin binding was found to be partially non-competitive. In ex vivo and in vivo receptor binding studies, the compound shows a pronounced selec- tivity towards serotonin-S2 receptors. Full occupation of frontal cortical serotonin-S2 sites was obtained with 0.63 mg/kg ritanserin given subcutaneously, whereas adren- ergic and dopaminergic receptor sites were not occupied by doses up to 160 mg/kg (Leysen et al. 1985; Niemegeers et al. 1984). Setoperone revealed in in vitro assays a receptor binding affinity profile which is rather similar to that of ritanserin, but in contrast to ritanserin, setoperone dissoci- ates rapidly from all the receptor sites (Table 2). According- ly, we could demonstrate a fully competitive inhibition in vitro between binding of setoperone and 3H-ketanserin to serotonin-S2 receptor sites (Fig. 1). In in vivo receptor bind- ing assays setoperone was found to readily occupy frontal cortical serotonin-S2 receptor sites at dosages between 0.01 and 0.63 mg/kg, but, unlike ritanserin, setoperone also oc- cupied striatal dopamine-D2 receptor sites at dosages be- tween 0.16 and 10 mg/kg (Leysen et al. 1986; Niemegeers et al. 1984). The difference between ritanserin and setoper- one as regards interference with dopamine receptor sites in vivo is probably to be ascribed to drug distribution and levels in the brain. In this study, we found that following acute administration of 5 mg/kg orally, whole brain levels of ritanserin were about 22 times lower than those of seto- perone. It can be assumed that the free ritanserin concentra- tion in the vicinity of striatal dopamine-D2 receptors is never sufficiently high to yield receptor occupation.
Acute drug effects on ex vivo receptor binding. Investigation of ex vivo receptor binding in washed brain membrane preparations following acute drug treatment revealed some unexpected findings. Both ritanserin and setoperone given acutely at i mg/kg, 5 mg/kg or 2 x 5 mg/kg per os produced a marked inhibition of 3H-ketanserin binding to serotonin-
$2 sites in frontal cortical membranes. In each case, the inhibition produced by ritanserin was more pronounced than the inhibition produced by setoperone (Fig. 2; Ta- ble 3). The drugs appeared not to be removed from the serotonin-S2 receptor sites by extensive membrane washing. This is not uncommon for a slowly dissociating drug such as ritanserin, but it is not in agreement with the rapid disso- ciation from serotonin-S2 receptors of setoperone. More- over, the inhibition by setoperone, measured ex vivo, was partially non-competitive (increased KD-, reduced Bma ~- value), in contrast to the fully competitive inhibition by setoperone of 3H-ketanserin binding upon in vitro co-incu- bation of the compounds (Figs. 1, 2). In contrast, setoper- one appeared to be completely removed by the membrane washing procedure from striatal dopamine-D2 receptor sites (no persistent inhibition of 3H-spiperone binding to striatal membranes, Table 5). This already demonstrates a differ- ence in events on serotonin-S2 and dopamine-D2 receptor sites. When drugs hit serotonin-S2 receptor sites in in vivo conditions, they become captured by the receptor, in such a way that they are difficult to wash out. The in vivo recep- tor blockade following acute treatment with ritanserin, is only slowly reversed over a period of 24 h. This time course of acute drug effect differs from the findings of Blackshear and Sanders-Bush (1982), who observed a reduction in sero- tonin-S2 receptor binding for several days following a single dose of mianserin. These authors suggested an acute down regulation of the receptor sites; our findings with setoper- one could partially support this view.
Down regulation of serotonin-S2 receptor sites by chronic serotonin antagonist treatment. Apparent serotonin-S2 re- ceptor down regulation was found following chronic ad- ministration of a high dose of either ritanserin or setoper- one. In several independently performed experiments it was consistently observed that 3H-ketanserin binding in washed frontal cortical membranes was fully non-competitively in- hibited, i.e. Ko-values were unchanged and Bmax-values were significantly reduced. The time course of receptor re- population following chronic ritanserin treatment spanned about 12 days. The derived serotonin-S2 receptor half-life of 3.5 days corresponds to the reported half-life of 2-3 days of the acetylcholine receptor (Dolly and Barnard 1984). The slow receptor reappearance following chronic treat- ment differs from the observation following acute ritanserin treatment, where 3H-ketanserin binding was found to be normalized after 24 h.
The fully non-competitive type of inhibition, the similar observation with the slowly dissociating ritanserin and the rapidly dissociating setoperone, the slow reappearance of receptor numbers and the fact that 1 day after stopping drug treatment the drug levels in the brain were below de- tection level (< 1 ng/g) suggest that the observed reduction in Bma x by chronic treatment with the serotonin antagonists represents a real receptor down regulation. Receptor down regulation as a consequence of receptor blockade is unlike the current theory. Yet, when re-evaluating reported find- ings, it appears to be a consistent observation following treatment with drugs having serotonin antagonistic proper- ties. This is illustrated in Fig. 6, where reported serotonin- $2 receptor down regulation has been related to the binding affinity of the drugs for serotonin-S2 receptor sites and to their potency to inhibit serotonin uptake. All the drugs, except pargyline, which reportedly caused serotonin-S2 re- ceptor down regulation, showed moderate to high sero-
443
=
Lt)
2: z
10000-
1000-
100-
10-
METERGOLINE "~..) ( ~ f ME TH YSE R GIDE
~ " 1" 1~ J-- MIA N SE RtN (~}~ ~'~ CY PROH EPTAD IN E
~ PIZOTIFEN (~ '~SE TOPERONE
%%",TANSE, ,N KETANSERIN
( ~.~GY LINE ]
(~IPRINDOLE
(~ DESIPRAMINE
® (~ ZIMELIDINE ALAPROCLATE
AMITRIPTYLINE (~ ('~FLUOXETINE IMIPRAMINE
CITALOPRAM
1 o.1 i io Ido Idoo ioooo
SEROTONiN-S 2 BINDING AFFINITY (Ki, r,M)
Fig. 6. Plot of drugs reported to be effective @ or ineffective @ (conflicting reports @) in down regulating serotonin-S2 sites fol- lowing chronic treatment. The position of the points shows the in vitro drug binding affinity for serotonin-Sz receptor sites on the abscissa and the potency of the drugs to inhibit serotonin up- take on the ordinate (partially unpublished observations measured according to methods described in Table 1). Data on receptor down regulation are from Blackshear et al. (1983), Crews et al. (1983), Gandolfi et al. (1984, 1985), Hall et al. (1984), Hyttel et al. (1984), Snyder and Peroutka (1981), Stolz et al. (1983) and this study
tonin-Sz receptor binding affinity. Drugs known to be po- tent serotonin antagonists such as cyproheptadine, pizoti- fen, ketanserin, ritanserin, setoperone, have virtually no ability to inhibit serotonin uptake. For some ergot deriva- tives (metergoline, methysergide) there were conflicting re- ports regarding serotonin-S2 receptor down regulation; this may be due to differences in drug dosages or to the mixed agonist-antagonistic properties of the drugs. Tricyclic anti- depressants, known as amine reuptake blockers, reveal high to moderate binding affinity for serotonin-S2 receptor sites and so does iprindole, a fact which is often ignored. Selec- tive serotonin uptake blockers such as citalopram and ala- proclate, which lack serotonin-S2 receptor binding affinity, were unable to down regulate the receptor sites. Since many of the drugs which reduce serotonin-S2 receptor numbers possess histamine-H 1 antagonistic properties, we performed a preliminary study with a potent and selective centrally active histamine H1 antagonist (levocabastine); no sero- tonin-S2 receptor down regulation was found. Although the down regulation seems to be specific for serotonin-S2 antagonist, rather high dosages are required and the effect is apparently not directly proportionate to the serotonin-S2 receptor binding affinity of the drugs (setoperone I mg/kg- day ineffective, 10 mg/kg, day effective). Nevertheless, the down regulation is specifically restricted to the serotonin-S2 receptors, since in this study, chronic treatment with the selective agent ritanserin, did not affect other receptors such as serotonin-S1, ~- , c~2- and /~-adrenergic, dopamine-D2, benzodiazepine and substance-P receptors.
Distinct mechanisms of serotonin-S2 and doparnine-D2 recep- tor regulation. The mechanisms involved in the serotonin-S2 receptor down regulation seem to be quite complicated and differ from mechanisms which regulate fl-adrenergic (Stiles et al. 1984) or dopamine-Dz receptors (Creese and Snyder 1980; Leysen 1980). For the fl-receptor it was shown that
chronic receptor stimulation enhances receptor phosphory- lation, accompanied by receptor internalization. The down regulation of fl-adrenergic receptor by the tricyclic antide- pressants probably occurs according to this mechanism as a consequence of enhanced receptor stimulation due to the noradrenaline reukptake inhibition by the drugs (Banerjee et al. 1977; Vetulani et al. 1976).
By using setoperone, this study illustrates the opposite regulation of the dopamine-D2 and the serotonin-S2 recep- tors. Using brain tissue of the same animals, in which sero- tonin-S2 receptors were down regulated, the striatal dopa- mine-D2 receptors were up regulated. The effect was fully apparent 1 day after stopping drug treatment (no persis- tence of drug in the tissue) and the up regulation appeared to be proportionate to the dosage and the dopamine-D2 receptor binding affinity of the drug. Dopamine-Dz recep- tors remained up regulated to the same level for 9 days after stopping drug treatment; only after 12 days did it start to diminish. This contrasts to the serotonin-S2 receptor down regulation which was gradually reversed with time between 1 and J 2 days after stopping drug treatment.
At this stage, one can only speculate about the mecha- nism involved in serotonin-S2 receptor down regulation. Gandolfi et al. (1985), who observed serotonin-S2 receptor down regulation by treatment with ketanserin and mian- serin, hypothesized that the drugs mimic an unknown, probably peptidergic endogenous agonist which acts on the serotonin-S2 receptors. This seems to be an unlikely expla- nation, which tries to relate down regulation after all to an agonistic action. So far, there is no evidence for the existence of such an endogenous substance; we favour an- other hypothesis. In view of the unusual acute and chronic effects of serotonin-S2 antagonists, it can be assumed that they interact with membranous processes. Antagonist bind- ing to serotonin-Sz receptors may not prevent, but probably even promotes receptor internalization and consequently enhances receptor degradation. The drug effect could be at the level of receptor phosphorylation. In line with this, it was found that micromolar concentrations of ritanserin inhibit diacylglycerol kinase, elevating the diacylglycerol content in the membranes and hence stimulating protein kinase-C activity (de Chaffoy de Courcelles et al. 1985). Direct investigation of the mechanism will only be possible with a serotonin-S2-receptor-containing cell-line and appro- priate irreversible markers for the serotonin-S2 receptor protein. The recently described photoaffinity ligand 7-azi- doketanserin seems to be a promising tool (Wouters et al. 1985).
Irrespective of the mechanisms involved, the question remains what is the clinical significance of receptor regula- tion for long-term drug treatment. The dopamine-Da recep- tor up regulation has tentatively been related to phenomena of behavioural supersensitivity and aversive side effects such as tardive dyskinesia (Creese and Snyder 1980). According to this view-point, problems of supersensitivity are not to be feared with serotonin antagonist treatment. Down regu- lation of serotonin-S2 receptors was originally proposed to be involved in the therapeutic effect of antidepressant drugs. A possible role of serotonin-S2 receptors in mood disorders is supported by the observed beneficial clinical effects of ritanserin in dysthymia and neurotic depression (Reyntjens et al. 1984). It can be speculated that the acute and chronic effect of serotonin-antagonists would be complementary and the drugs may treat pathology caused by receptor su- persensitivity.

444
Acknowledgement. Part of this study was supported by a grant from the Instituut voor Wetenschappelijk Onderzoek in Landbouw en Nijverheid. The help of Fred Lenaerts in accommodating the animals during chronic drug treatment is much appreciated. Thanks to Diane Verkuringen, Jef Van Mierlo and Lambert Leijs- sen for typing and art-work and to David Ashton for linguistic correction.
References
Banerjee SP, Kung LS, Riggi SJ, Chanda S (1977) Development of/?-adrenergic receptor subsensitivity by antidepressants. Na- ture (London) 268 : 455M56
Blackshear MA, Sanders-Bush E (1982) Serotoniu receptor sensi- tivity after acute and chronic treatment with mianserin. J Phar- macol Exp Ther 221 : 303-308
Blackshear MA, Friedman RL, Sanders-Bush E (1983) Acute and chronic effects of serotonin (5HT) antagonists on serotonin binding sites. Naunyn-Schmiedebergs Arch Pharmacol 324:125-129
Ceulemans DLS, Gelders YG, Hoppenbrouwers M-LJA, Reynt- jens ASM, Janssen PAJ (1985) Effect of serotonin antagonism in schizophrenia: a pilot study with setoperone. Psychopharma- cology 85:329-339
Colpaert FC, Meert TF, Niemegeers CJE, Janssen PAJ (1985) Be- havioral and 5-HT antagonist effects of ritanserin : a pure and selective antagonist of LSD discrimination in rat. Psychophar- macology 86:45-54
Creese I, Snyder SH (1980) Chronic neuroleptic treatment and dopamine receptor regulation. In: Cattabeni F, Racagni G, Spano PF, Costa E (eds) Long-Term Effects of Neuroleptics (Adv Biochem Psychopharmacol Vol 24), Raven Press, New York, pp 89-94
Crews FT, Scott JA, Shorstein NM (1983) Rapid down regulation of serotonin-S2 receptor binding during combined administra- tion of tricyclic antidepressant drugs and c~z antagonists. Neu- ropharmacology 22:1203-1209
de Chaffoy de Courcelles D, Leysen JE, De Clerck F, Van Belle H, Janssen PAJ (1985) Evidence that phospholipid turnover is the signal transducing system coupled to serotonin-S2 recep- tor sites. J Biol Chem 260:7603-7608
Dolly JO, Barnard EA (1984) Nicotinic acetylcholine receptors: an overview. Biochem Pharmacol 33 : 841-858
Gandolfi O, Barbaccia ML, Costa E (1984) Comparison of iprin- dole, imipramine and mianserin action on brain serotonergic and beta adrenergic receptors. J Pharmacol Exp Ther 229 : 782-786
Gandolfi O, Barbaccia ML, Costa E (1985) Different effects of serotonin antagonists on 3H-mianserin and 3H-ketanserin rec- ognition sites. Life Sci 36:713 721
Hall H, Ross SB, S/illemark M (1984) Effect of destruction of central noradrenergic and serotonergic nerve terminals by sys- temic neurotoxins on the long-term effects of antidepressants on fl-adrenoceptors and 5-HT2 binding sites in the rat cerebral cortex. J Neural Transm 59:9-23
Hyttel J, Fredericson Overo K, Arnt J (1984) Biochemical effects and drug levels in rats after long-term treatment with the specif- ic 5-HT-uptake inhibitor, citalopram. Psychopharmacology 83 : 20-27
Janssen PAJ (1985) Pharmacology of potent and selective S2-sero- tonergic antagonists. J Cardiovasc Pharmacol 7 [Suppl] 7:$2- $11
Leysen JE (1980) 3H-Apomorphine receptors in various rat brain regions: A study of specific and non-specific binding and the influence of chronic neuroleptic treatment. In: Cattabeni F, Racagni G, Spano PF, Costa E (eds) Long-Term Effects of Neuroleptics (Adv Biochem Psychopharmacol Vol 24), Raven Press, New York, pp 123-132
Leysen JE (1985) Characterization of serotonin receptor binding sites. In: Green AR (ed) Neuropharmacology of Serotonin, Oxford University Press, Oxford, pp 79 116
Leysen JE, Gommeren W (1984) The dissociation rate of unlabelled dopamine antagonists and agonists from the dopamine-D2 re- ceptor, application of an original filter method. J Recept Res 4: 81%845
Leysen JE, Niemegeers CJE, Tollenaere JP, Laduron PM (1978) Serotonergic component of neuroleptic receptors. Nature 272:168-171
Leysen JE, Gommeren W, Laduron PM (1979) Distinction between dopaminergic and serotonergic components of neuroleptic binding sites in limbic brain areas. Biochem Pharmacol 28 : 447M48
Leysen JE, Niemegeers CJE, Van Nueten JM, Laduron PM (1982) [3H]-Ketanserin (R 41 468), a selective 3H-ligand for serotonin2 receptor binding sites. Mol Pharmacol 21:301-314
Leysen JE, de Chaffoy de Courcelles D, De Clerck F, Niemegeers CJE, Van Nueten JM (1984) Serotonin-Sz receptor binding sites and functional correlates. Neuropharmacology 23:1493-1501
Leysen JE, Gommeren W, Van Gompel P, Wynants J, Janssen PFM, Laduron PM (1985) Receptor-binding properties in vitro and in vivo of ritanserin. A very potent and long-acting sero- tonin-S2 antagonist. Mol Pharmacol 27:600-611
Leysen JE, Van Gompel P, Gommeren W, Laduron PM (1986) Differential regulation of dopamine-D2 and serotonin-S2 recep- tors by chronic treatment with the serotonin-S2 antagonists, ritanserin and setoperone. In: Dahl SG, Gram LF, Paul SM, Potter WZ (eds) Clinical Pharmacology in Psychiatry IV. Selec- tivity in Psychotropic Drug Action - Promises or Problems? Springer-Verlag, Heidelberg (in press)
Mauger JP, Sladeczek F, Bockaert J (1982) Characteristics and metabolism of cq-adrenergic receptors in a nonfusing muscle cell line. J Biol Chem 257:875-879
Niemegeers CJE, Leysen JE, Laduron PM, Janssen PAJ (1984) Differential pharmacological and biochemical profiles of sero- tonin-S2 antagonists. In: Collegium Internationale Neuro-Psy- chopharmacologicum, 14th Congress, Book of Abstracts, p 889
Peroutka S J, Snyder SH (1979) Multiple serotonin receptors differ- ential binding of [3H] 5-hydroxytryptamine, [3H] lysergic acid diethylamide, and [3HI spiroperidol. Mol Pharmacol 16: 687-699
Peroutka S J, Snyder SH (1980) Regulation of serotonin2 (5-HT2) receptors labeled with [3H] spiroperidol by chronic treatment with the antidepressant amitriptyline. J Pharmacol Exp Ther 215:582-587
Reyntjens A, Waelkens J, Gelders Y, Ceulemans D, Janssen PAJ (1984) A novel approach for psychosomatic normalization: a placebo-controlled pilot trial of R 55 667, a potent indolamine antagonist. Collegium Internationale Neuro-Psychopharmaco- logicum, 14th Congress, p 566 (Book of Abstracts)
Sladeczek F, Bockaert J (1983) Turnover in vivo of alphal-adrener- gic receptors in rat submaxillary glands. Mol Pharmacol 23 : 282-288
Snyder SH, Peroutka SJ (1982) A possible role of serotonin recep- tors in antidepressant drug action. Pharmacopsychiatry 15:131-134
Stiles GL, Caron MG, Lefkowitz RJ (1984)/~-Adrenergic receptor biochemical mechanisms of physiological regulation. Physiol Rev 64:661-743
Stolz JF, Marsden CA, Middlemiss DN (1983) Effect of chronic antidepressant treatment and subsequent withdrawal on [3H] 5-hydroxytryptamine and [3H] spiperone binding in rat frontal cortex and serotonin receptor mediated behaviour. Psychophar- macology 80 : 150-155
Vetulani J, Starwarz RJ, Dingell JV, Sulser F (1976) A possible common mechanism of action of antidepressant treatments. Arch Pharmacol 239:109-114
Wouters W, Van Dun J, Leysen JE, Laduron PM (1985) Photoaf- finity probes for serotonin and histamine receptors: synthesis and characterization of two azide analogues of ketanserin. J Biol Chem 260 : 8423
Received July 8, 1985

![Bis[(1 S *,2 S *)- trans -1,2-bis(diphenylphosphinoxy)cyclohexane]chloridoruthenium(II) trifluoromethanesulfonate dichloromethane disolvate](https://static.fdokumen.com/doc/165x107/63360a7bcd4bf2402c0b5520/bis1-s-2-s-trans-12-bisdiphenylphosphinoxycyclohexanechloridorutheniumii.jpg)


















