
The crystal structure of horse deoxyhaemoglobin trapped in the high-affinity (R) state
14
J. Mol. Biol. (1996) 264, 743–756 The Crystal Structure of Horse Deoxyhaemoglobin Trapped in the High-affinity (R) State Julie Wilson 1 , Kathryn Phillips 2 and Ben Luisi 2 * Co-operative oxygen binding by the vertebrate haemoglobins arises from 1 Department of Chemistry an equilibrium between a quaternary structure with low affinity (T), University of York favoured in the absence of ligand, and a high affinity form (R) adopted by Heslington, York YO1 5DD the fully ligated protein. While R state haemoglobin has an oxygen affinity UK close to that of isolated subunits, the affinity of the T state is roughly 2 Department of Biochemistry 300-fold lower. The mechanism by which the T state restrains ligand University of Cambridge binding, and the pathway of the quaternary transition, have been largely Tennis Court Road revealed by detailed crystallographic analyses of a number of haemoglobin Cambridge, CB2 1QW, UK molecules in the equilibrium states, as well as intermediate forms of the T state including partially ligated species. The ligation intermediates of the R state, however, have not been as well characterized structurally. We report here the crystal structure of one such intermediate species, namely, horse deoxyhaemoglobin in the R state, at 1.8 Å resolution. While ligand binding in the T state may result in unfavourable stereochemistry in and around the haem–ligand complex, the more plastic R structure appears to accommodate equally well both liganded and ligand-free haem. Loss of ligand at the R state haem results in movements of the haem and shifts of the FG corners, which form characteristic intersubunit contacts that distinguish the quaternary states. The shifts are comparable in magnitude to the corresponding movements associated with de-ligation in the T state, although they differ in direction. These and other differences illustrate how the structural changes in the haem pocket are communicated to the subunit interfaces and how the movements that can occur in the R state may be impeded in the T state. 7 1996 Academic Press Limited Keywords: haemoglobin; co-operativity; R state; allosteric intermediates; X-ray crystallography *Corresponding author Introduction Well before the structure of haemoglobin (Hb) was elucidated, it was appreciated that ligand must affect the protein’s shape since crystals of oxy-hae- moglobin disintegrate under reducing conditions and deoxy haemoglobin (deoxyHb) crystals lose order when exposed to oxygen. Free in solution, deoxyHb strongly favours the T state, but ligation or oxidation of the haem groups (to the Met form) alters the allosteric equilibrium to favour the R state. The relationship between the T quaternary structure and its oxygen affinity have been corroborated by direct measurements from crystals and gels (Mozzarelli et al ., 1991; Shibayama & Saigo, 1995). Ligand binding in the T state appears to generate unfavourable stereochemistry in the haem pocket, and this strain is communicated to the subunit interfaces to favour the quaternary tran- sition (Abraham et al ., 1992; Anderson, 1973; Perutz et al ., 1987; Liddington et al ., 1988, 1992; Luisi et al ., 1990; Paoli et al ., 1996; Schumacher et al ., 1995). Here, we describe a species of deoxyHb trapped in the R state and examine how loss of ligand affects the haem pocket, the tertiary structure, and the subunit contacts. The deoxyHb R state crystals were prepared by treating horse haemoglobin with the cross-linking agent bis(N-maleimidoethyl) ether (BME; Figure 1). Crystals of the Met form of this cross-linked haemoglobin retain their high crys- talline order on chemical reduction to the deoxy Abbreviations used: BME, bis(N-maleimidoethyl) ether; BME-Hb, haemoglobin treated with bis(N-maleimidoethyl) ether; MetHb, oxidized Fe 3+ haemoglobin, with water as a distal ligand; deoxyHb, Fe 2+ haemoglobin without ligand; pMB, p-hydroxymercuribenzoate. 0022–2836/96/490743–14 $25.00/0 7 1996 Academic Press Limited
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of The crystal structure of horse deoxyhaemoglobin trapped in the high-affinity (R) state

J. Mol. Biol. (1996) 264, 743–756
The Crystal Structure of Horse DeoxyhaemoglobinTrapped in the High-affinity (R) State
Julie Wilson 1, Kathryn Phillips 2 and Ben Luisi 2*
Co-operative oxygen binding by the vertebrate haemoglobins arises from1Department of Chemistryan equilibrium between a quaternary structure with low affinity (T),University of Yorkfavoured in the absence of ligand, and a high affinity form (R) adopted byHeslington, York YO1 5DDthe fully ligated protein. While R state haemoglobin has an oxygen affinityUKclose to that of isolated subunits, the affinity of the T state is roughly2Department of Biochemistry 300-fold lower. The mechanism by which the T state restrains ligand
University of Cambridge binding, and the pathway of the quaternary transition, have been largelyTennis Court Road revealed by detailed crystallographic analyses of a number of haemoglobinCambridge, CB2 1QW, UK molecules in the equilibrium states, as well as intermediate forms of the
T state including partially ligated species. The ligation intermediates of theR state, however, have not been as well characterized structurally. Wereport here the crystal structure of one such intermediate species, namely,horse deoxyhaemoglobin in the R state, at 1.8 Å resolution. While ligandbinding in the T state may result in unfavourable stereochemistry in andaround the haem–ligand complex, the more plastic R structure appears toaccommodate equally well both liganded and ligand-free haem. Loss ofligand at the R state haem results in movements of the haem and shiftsof the FG corners, which form characteristic intersubunit contacts thatdistinguish the quaternary states. The shifts are comparable in magnitudeto the corresponding movements associated with de-ligation in the T state,although they differ in direction. These and other differences illustrate howthe structural changes in the haem pocket are communicated to the subunitinterfaces and how the movements that can occur in the R state may beimpeded in the T state.
7 1996 Academic Press Limited
Keywords: haemoglobin; co-operativity; R state; allosteric intermediates;X-ray crystallography*Corresponding author
Introduction
Well before the structure of haemoglobin (Hb)was elucidated, it was appreciated that ligand mustaffect the protein’s shape since crystals of oxy-hae-moglobin disintegrate under reducing conditionsand deoxy haemoglobin (deoxyHb) crystals loseorder when exposed to oxygen. Free in solution,deoxyHb strongly favours the T state, but ligationor oxidation of the haem groups (to the Met form)alters the allosteric equilibrium to favour the Rstate. The relationship between the T quaternary
structure and its oxygen affinity have beencorroborated by direct measurements from crystalsand gels (Mozzarelli et al., 1991; Shibayama &Saigo, 1995). Ligand binding in the T state appearsto generate unfavourable stereochemistry in thehaem pocket, and this strain is communicated to thesubunit interfaces to favour the quaternary tran-sition (Abraham et al., 1992; Anderson, 1973; Perutzet al., 1987; Liddington et al., 1988, 1992; Luisi et al.,1990; Paoli et al., 1996; Schumacher et al., 1995).
Here, we describe a species of deoxyHb trappedin the R state and examine how loss of ligand affectsthe haem pocket, the tertiary structure, and thesubunit contacts. The deoxyHb R state crystals wereprepared by treating horse haemoglobin with thecross-linking agent bis(N-maleimidoethyl) ether(BME; Figure 1). Crystals of the Met form of thiscross-linked haemoglobin retain their high crys-talline order on chemical reduction to the deoxy
Abbreviations used: BME, bis(N-maleimidoethyl)ether; BME-Hb, haemoglobin treated withbis(N-maleimidoethyl) ether; MetHb, oxidized Fe3+
haemoglobin, with water as a distal ligand; deoxyHb,Fe2+ haemoglobin without ligand; pMB,p-hydroxymercuribenzoate.
0022–2836/96/490743–14 $25.00/0 7 1996 Academic Press Limited

Haemoglobin Allosteric Intermediate Structure744
Figure 1. Chemical structure of the bi-functionalreagent bis(N-maleimidoethyl) ether (BME).
coefficients Fobs(BME) − Fobs(Met) and using phasescalculated from the MetHb model.)
BME is highly reactive with thiol groups, and itis expected that this compound should have reactedwith Cys F9b. Indeed, since the crystalline BME-Hbis protected against reaction with mercurialcompounds, such as p-hydroxymercuribenzoate(pMB), the BME must be present but disordered.The compound could also be present at other sitesat low occupancy. The allosteric modulator,2,3-diphosphoglycerate, has roughly the sameeffect on the oxygen binding affinity of BME-Hband the control (Table 1), suggesting that theresidues of the binding site have not beenappreciably modified by the cross-linker (e.g. ValNH1b, His NH2b, His H21b and Lys EF6b). We didobserve weak density in the vicinity of Lys A12bthat contacts a neighbouring haemoglobin moleculein the crystal, and it is possible that thismodification may stabilize the crystal lattice. BMEappears to cause insignificant structural pertur-bation and may trap the R state by stabilizing thecrystal lattice.
Quaternary structural effects
Baldwin & Chothia (1979) have analysed the Rand T states and found a convenient referenceframe to describe the structural changes associatedwith the quaternary transition. The transition canbe thought of as a rotation and translation of twodimers, with each dimer composed of one a andone b subunit. The subunit interface within each abdimer (i.e. a1b1 and a2b2) changes comparativelylittle with the quaternary transition, while theinterface between the two dimers (i.e. a1b2 and a2b1)undergoes a repacking, and it is here that the maincommunication pathway between subunit interfaceand the haem pocket must lie.
In both R and T quaternary states the principalinterfacial contacts are pseudo-equivalent: thenon-helical segment joining the F and G helices (theFG corner) of the a1 subunit packs against the Chelix of the b2 subunit and the FG corner of the b2
subunit packs against the C helix of the a1 subunit.(The a2/b1 interface is the same by the moleculardyad symmetry.) However, closer inspection showsthat the contacts are entirely non-equivalent in thenature of their movements with the quaternarytransition. The a1 FG/b2 C interface acts as a pivotabout which the dimers rotate relative to eachother. The a1 C/b2 FG interface, on the other hand,undergoes much larger changes with the quater-nary transition as the b2FG corner, in particular HisFG4, switches between adjacent notches on the a1Chelix.
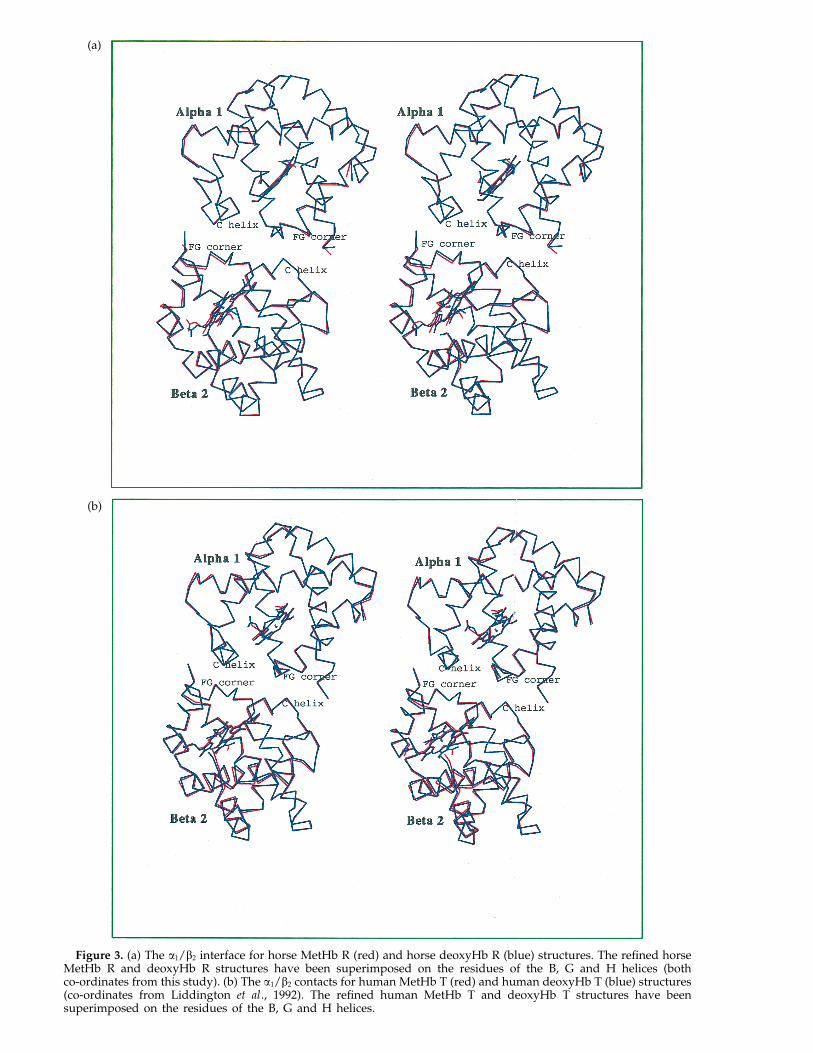
Loss of ligand in the R state results in smallstructural changes at the a1/b2 interface. InFigure 3(a), a comparison has been made of thea1/b2 interface of refined horse MetHb R and horsedeoxyHb R structures, which have been superim-posed in the reference frame of the B, G and Hhelices of the a1/b1 interface. The small movements
form; the structure of this trapped allostericintermediate is reported here at a resolution of1.8 A. Some aspects of this work have already beenpublished (Perutz et al., 1987; Luisi et al., 1990), butwe provide a more detailed comparison of theresponse of the globin and the haem to ligandbinding in the R and T states, and we discuss howthe quaternary state affects the communicationbetween the haem pocket and the intersubunitcontacts.
Results and Discussion
Crystals of horse Met BME-Hb retained highcrystalline order when reduced with sodiumdithionite. On the other hand, control crystals ofhorse MetHb, not reacted with BME disintegratedafter treatment with the reducing agent, showingthat the BME stabilizes the R state crystals in theabsence of ligand. The electron density maps ofBME-Hb confirm that both a and b subunits arevirtually free of ligand (Figure 2(a) and (b)), yet theprotein remains in the R quaternary state. As foundin the human deoxy T state (and myoglobin), awater molecule also occupies the distal haempocket of the a subunit of BME-Hb (Figure 2(a)) andis hydrogen bonded to histidine E7 (distance 2.9 A);it is too far from the haem Fe atom to be a ligand(3.5 A). As the water molecule is present in the asubunit of both the T and R states, its displacementmay not contribute to the free energy ofco-operativity. There is no water molecule in the bpocket in either the T or the R states.
Unlike some chemical modifications that havebeen used to constrain haemoglobin to onequaternary state, BME has only a small effect on theprotein’s oxygen binding characteristics, notablyincluding the co-operativity (Table 1). The modifiedhaemoglobin is therefore capable of switchingquaternary states.
Earlier studies suggested that the cross-linkingagent binds Cys F9b and His FG4b (Moffat, 1971;Moffat et al., 1971), although electron density mapsprepared from our refined model show no evidenceof the compound at either of these residues. (Thiswas also true of difference maps synthesized with

Haemoglobin Allosteric Intermediate Structure 745
(a)
(b)
Figure 2. The electron density of the deoxy R state (a) a haem and (b) b haem. Both maps are synthesized from2Fobserved − Fcalculated coefficients. The upper limit for the occupancy of ligand at the a haem is 10% and at the b haem,5%. A water molecule occupies the distal haem pocket of the a subunit and is hydrogen bonded to histidine E7.

Haemoglobin Allosteric Intermediate Structure746
Table 1. Oxygen binding propertiesAllosteric modulator
Hill’s Hill’sMolecule coefficient p50
a coefficient p50a
Horse BME-Hb 2.2 4.0 2.35 8.5Horse control Hb 2.4 5.1 2.70 11.0Human Hb 2.9 5.5 2.9 16.5
Measured with material from dissolved BME-Hb and controlMetHb crystals.
a Partial oxygen pressure (p50) at half-saturation of ligandbinding sites, in mm of Hg. The columns indicated withallosteric modulator are in the presence of 2,3-diphosphoglycer-ate. The differences in affinity between the BME-Hb and controlsamples is probably due to the large amount of MetHb presentin the BME-Hb sample (12% for BME-Hb versus 6% for thecontrol horse haemoglobin).
may affect the apparent differences: the greater thedistance of objects from the superimposing refer-ence, the greater the contribution of potential errorsto their observed features.
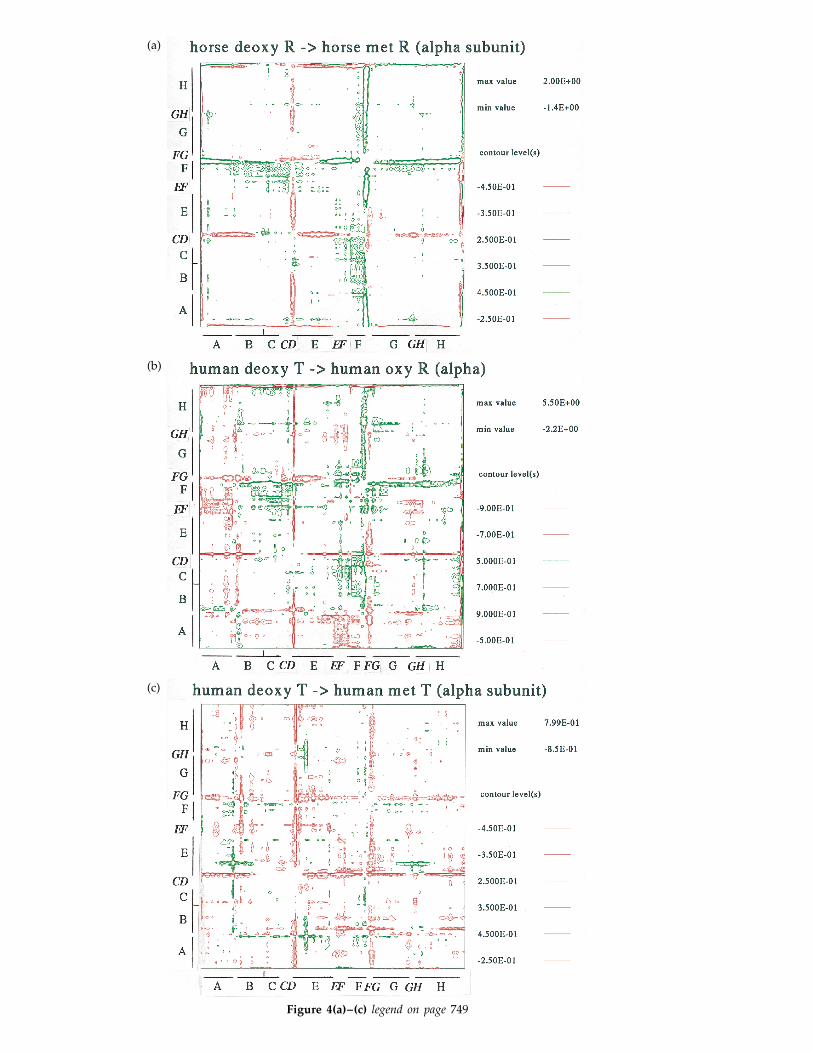
The distance matrix, which is the set of all thepair-wise separations of residues, provides astructural comparison that requires no superposi-tions, thereby eliminating errors associated with thefitting procedure. Differences in the matrix betweentwo states highlight structural changes in aninvariant co-ordinate frame. Large displacements ofsecondary structure appear as off-diagonal blocksin such matrices.
By comparing Figure 4(a) and (d), it is apparentthat the nature of the tertiary structural changesassociated with ligand binding in the R state differssignificantly for the a and b subunits. The excess ofpositive movements indicates that the b subunit isexpanding with ligand binding in the R state(Figure 4(d)), whereas the a subunit shows nocorresponding expansion (Figure 4(a)). The loss ofligand in the R state results principally in two shiftsin the a subunit: one occurs at the end of the Fhelix/FG corner with respect to A, B, EF, G, GH andH; the second principal shift is of the CD cornerwith respect to A, B, E, G and H. In the b subunit,the movements are more distributed. The end of theF helix and the FG corner have a smaller shift withrespect to the B helix in the b subunit comparedwith a. Also, the b CD corner moves little withrespect to the E helix, unlike the case for the asubunit. The FG corner and C helix (in the vicinityof the CD corner) play important roles in theco-operative mechanism, for they repack with thequaternary transition.
The same analysis may be extended to the T stateto examine the structural changes associated withoxidation. For the a subunit, there are shifts of theCD corner and smaller changes in the F helix (Fig-ure 4(c); Liddington et al., 1992), but the movementsof the FG corner are in a different direction fromthose occurring in the R state (compare Figure 4(a)and (c)). This perhaps indicates that mechanicalproperties of the corner in the two quaternary statesare different. For comparison, the changes associ-ated with the quaternary transition for the asubunit are shown in Figure 4(b).
For the T state b subunit (Figure 4(f)), mostinternal movements with oxidation occur in the Ehelix, whereas movements of the F helix and FGcorner are restricted compared with the R statechanges (Figure 4(d)). Again, as found for the asubunit, some of the b FG corner movements differfrom those found in the R state. Generally, the othertertiary structural changes associated with thequaternary transition appear to be a composite ofthe individual changes within the two states(Figure 4(b) and (e)).
The haem pockets
A comparison of the effects of oxidation on thehaem environs in the T and R states is shown in
at the a1/b2 interface associated with deligation arein the same direction as the R : T transition.Compared with the Met R species, the a FG cornerof the deoxyHb R state is displaced by roughly0.2 A with respect to the b C helix in a small slidingmovement. At the b FG/a CD contact, His FG4b inthe deoxyHb R state moves roughly 0.4 A relativeto the C helix of the a subunit. These small changesdo not greatly change the packing of the interfacesbut may in principle affect the stability of thedeoxyHb R intermediate in relation to the deoxyHbT state.
An analysis of the tertiary structural changes
The loss of ligand results in small tertiarystructural changes in BME haemoglobin. The a1b1
superposition in Figure 3(a) shows the tertiarystructural changes associated with oxidation in theR state; the corresponding changes with oxidationin the T state are shown in Figure 3(b). In both theT and R states, movements occur in the FG cornerof a and b subunits alike with respect to theinvariant interface.
In the R state a subunit, movements of the CDcorner occur with ligand binding but in exposedregions that do not make interfacial contacts.Movements in the b CD corner of the R state,however, do occur at an interfacial contact site. Thisinterfacial movement might be correlated withtranslational motion of the b haem groups,discussed further below. In the T state, only asmaller movement of the b CD corner occurs,probably as a consequence of restrictions imposedby the inter-subunit packing.
As the tertiary structural changes associated withligand binding in either the T or R state are smalland occur at some distance from the relativelyinvariant portions of the protein (i.e. the a1/b1
interface), it is necessary to choose a referenceframe carefully to describe the small changesaccurately. For instance, co-ordinate superpositionsare commonly used to evaluate structural changes,but the errors associated with fitting procedures
(a)
(b)
Figure 3. (a) The a1/b2 interface for horse MetHb R (red) and horse deoxyHb R (blue) structures. The refined horseMetHb R and deoxyHb R structures have been superimposed on the residues of the B, G and H helices (bothco-ordinates from this study). (b) The a1/b2 contacts for human MetHb T (red) and human deoxyHb T (blue) structures(co-ordinates from Liddington et al., 1992). The refined human MetHb T and deoxyHb T structures have beensuperimposed on the residues of the B, G and H helices.
(a)
(b)
(c)
Figure 4(a)–(c) legend on page 749
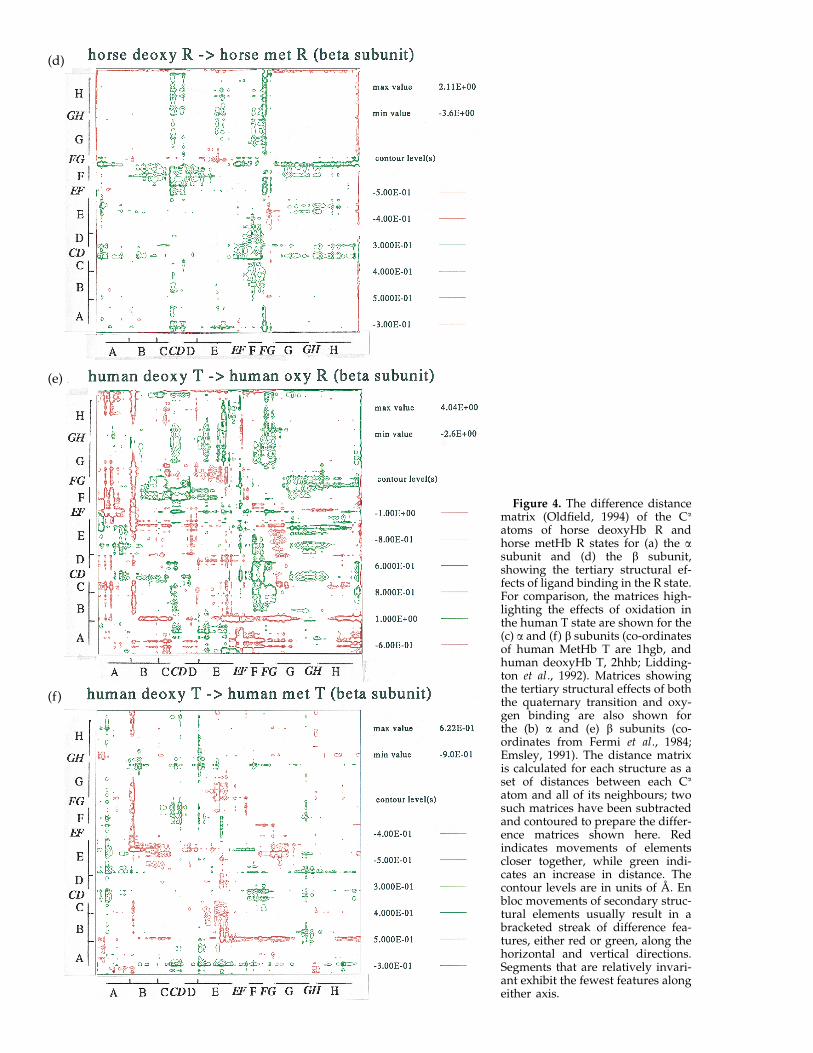
Figure 4. The difference distancematrix (Oldfield, 1994) of the Ca
atoms of horse deoxyHb R andhorse metHb R states for (a) the asubunit and (d) the b subunit,showing the tertiary structural ef-fects of ligand binding in the R state.For comparison, the matrices high-lighting the effects of oxidation inthe human T state are shown for the(c) a and (f) b subunits (co-ordinatesof human MetHb T are 1hgb, andhuman deoxyHb T, 2hhb; Lidding-ton et al., 1992). Matrices showingthe tertiary structural effects of boththe quaternary transition and oxy-gen binding are also shown forthe (b) a and (e) b subunits (co-ordinates from Fermi et al., 1984;Emsley, 1991). The distance matrixis calculated for each structure as aset of distances between each Ca
atom and all of its neighbours; twosuch matrices have been subtractedand contoured to prepare the differ-ence matrices shown here. Redindicates movements of elementscloser together, while green indi-cates an increase in distance. Thecontour levels are in units of A. Enbloc movements of secondary struc-tural elements usually result in abracketed streak of difference fea-tures, either red or green, along thehorizontal and vertical directions.Segments that are relatively invari-ant exhibit the fewest features alongeither axis.
(d)
(e)
(f)
Haemoglobin Allosteric Intermediate Structure750
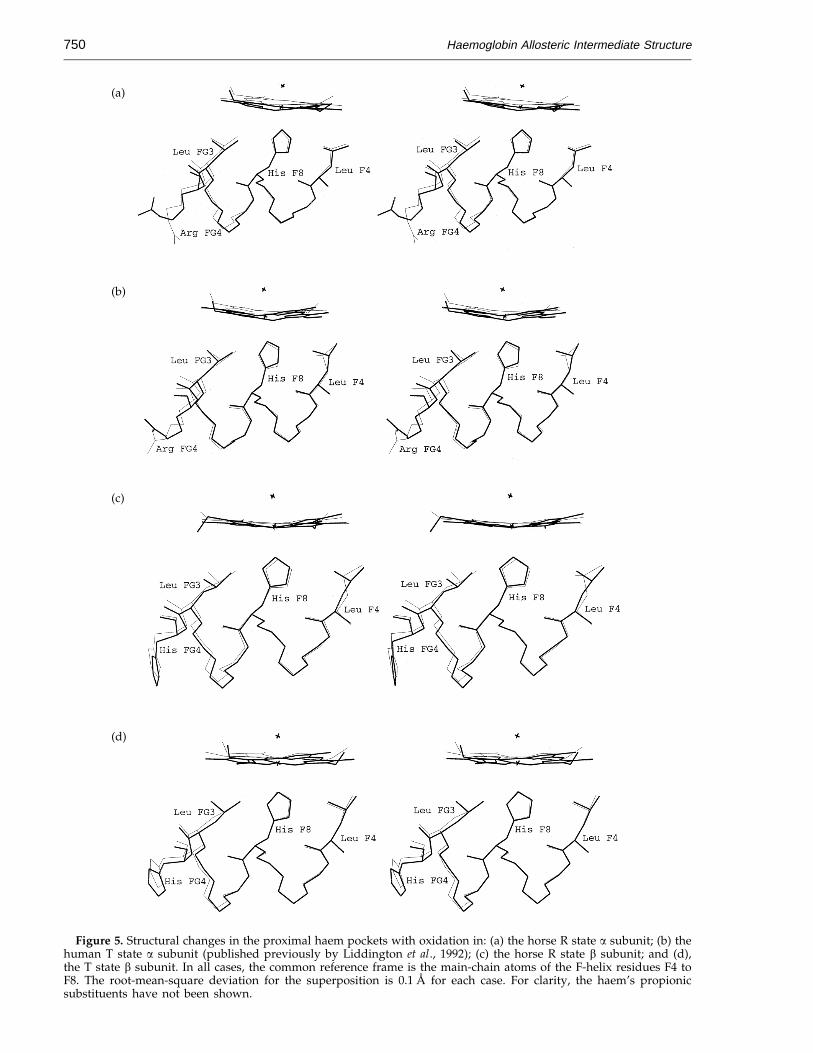
Figure 5. Structural changes in the proximal haem pockets with oxidation in: (a) the horse R state a subunit; (b) thehuman T state a subunit (published previously by Liddington et al., 1992); (c) the horse R state b subunit; and (d),the T state b subunit. In all cases, the common reference frame is the main-chain atoms of the F-helix residues F4 toF8. The root-mean-square deviation for the superposition is 0.1 A for each case. For clarity, the haem’s propionicsubstituents have not been shown.
(a)
(b)
(c)
(d)

Haemoglobin Allosteric Intermediate Structure 751
Figure 5. As before, the R state structures are fromhorse haemoglobin, while the T state structures arefrom human haemoglobin. The T state a subunitappears to permit limited movement of the haem(Figure 5(b)), corresponding to the restrictedmovements of the side-chains in the FG corner,such as Leu FG3 (Perutz et al., 1987; Liddingtonet al., 1988, 1992).
In deoxyHb the tension on the Fe–His F8 bond ofthe b subunit may be less than that of the a subunit(judging from the Fe–His distances of the b subunitin the deoxyHb R and deoxyHb T states in Table 2and from stretching frequencies observed fromresonance Raman spectroscopy (Nagai & Kitigawa,1980)). However, a second mechanism operates forthe b subunit to regulate ligand affinity: the distalresidues sterically occlude the ligand, and the Tstate globin restricts the movements required tofree the space (Perutz, 1970; Perutz et al., 1987;Nagai et al., 1987; Olson et al., 1988).
With the quaternary transition, the b haem planerotates and translates with respect to the invarianta1/b1 interface (Baldwin & Chothia, 1979), and this isthought to clear a space for the ligand. Surprisingly,our modelling suggests that the steric occlusion bythe distal residues is only partially relieved in thedeoxyHb R species (Figure 6). This steric occlusionpresumably has little effect on the affinity, since re-placement of the Val by Ala in human haemoglobinleaves KR, the R state binding constant, unchanged(Nagai et al., 1987). The R state must therefore besufficiently flexible that little work is required forthe remaining movements of the haem that free thenecessary space for the ligand to bind.
How is flexibility of the haem environs in the bsubunit conferred to the R state but restricted in theT state? In the R state, haem oxidation is associatedwith translation and a slight tilt of the porphyrin.Crystallographic studies show that the b subunithaem tilts roughly equally well in both the T andR states in response to ligand binding, buttranslation of the porphyrin with respect to thea1/b1 interface occurs only in the R state (Luisi et al.,1990). The translation of the haem in the R state iscorrelated with the movements of the b CD corner,and the corresponding movement in the T state ismuch smaller. The apparently restricted movementof the b CD corner in the T state, through the effectsof interfacial contacts, might therefore be in partresponsible for controlling the b haem affinity.
Another restriction that is associated with the Tstate is in movement of Leu FG3 on the proximalside of the haem pocket of the b subunit. Themovements in the T state differ from those foundin the R state (Figure 5(a) and (b)). It is possible thatthis restriction in the T state prevents the same typeof haem movements that are permitted within theR state.
Details of the haem stereochemistry
How does the deoxy haem fit into the R state?Comparing the distance between the Fe atom and
the porphyrin plane in the deoxyHb R anddeoxyHb T states, we note that the Fe is roughly0.1 A closer to the haem plane in the deoxyHb Rstate (Table 2). This difference exactly matches thechange of the Fe displacement in model haemgroups designed to mimic the deoxyHb R anddeoxyHb T states (Fe2+ (TpivPP) (2-MeIm) and Fe2+
(Piv2C8) (1-MeIm), Table 2). The small butsignificant differences between the deoxy haem inthe two quaternary states suggest that the T stateprotein pulls the Fe atom towards the F helixcreating the so-called ‘‘tension at the haem’’described by Perutz (1970). Resonance Ramanspectroscopy measurements suggest that the strainenergy associated with the stretched Fe–His bond isvery small (roughly 0.03 kcal/mol for the a subunitand 0.004 kcal/mol for the b subunit (Nagai &Kitigawa, 1980)).
The haem stereochemistry of the deoxy Rhaemoglobin is similar to that of deoxymyoglobinand the non-co-operative R state b tetramer(Borgstahl et al., 1994), which may be consideredmodels for the unconstrained, free a and b subunits,respectively. Observations from resonance Ramanspectroscopy reveal comparatively little strain onthe Fe–His bond in the b tetramer compared withthe deoxyHb T state of the normal a/b tetramer(Kitigawa, 1992; Nagai & Kitigawa, 1980). Byinference, the globin places little mechanicaltension on the deoxyhaem in the R state incomparison with the free subunits, since they allhave similar haem stereochemistry. It cannot beproved from structural scrutiny alone that the haemis indeed stereochemically unstrained in thedeoxyHb R molecule, since strain energy can bedistributed throughout the structure in smalldeformations (Hopfield, 1973).
The thermodynamic preference for deoxyHb Tover the deoxyHb R state
The haem–ligand complex in the T state appearsto be associated with an unfavourable stereochem-istry, and this apparent strain can be relieved by thequaternary transition (Perutz et al., 1987; Lidding-ton et al., 1988, 1992; Luisi et al., 1990). Thestructures studied here suggest that there may beno corresponding strain in the deoxyHb R state todrive the quaternary transition in the oppositedirection, since the ligand-free haem can beaccommodated into the R state structure withapparently regular stereochemistry. The deoxyHbR structure studied here therefore confirms the lackof strain already suggested by the near-identity ofthe oxygen affinity of the R tetramer and the freesubunits. What then drives the relative stability ofthe deoxyHb T state over the deoxyHb Rintermediate?
Perutz has proposed that salt-bridges at thecarboxy termini account for the more favourablefree energy of the deoxyHb T state compared withthe deoxyHb R state, and that most of the freeenergy difference between these species (about
Haemoglobin Allosteric Intermediate Structure752
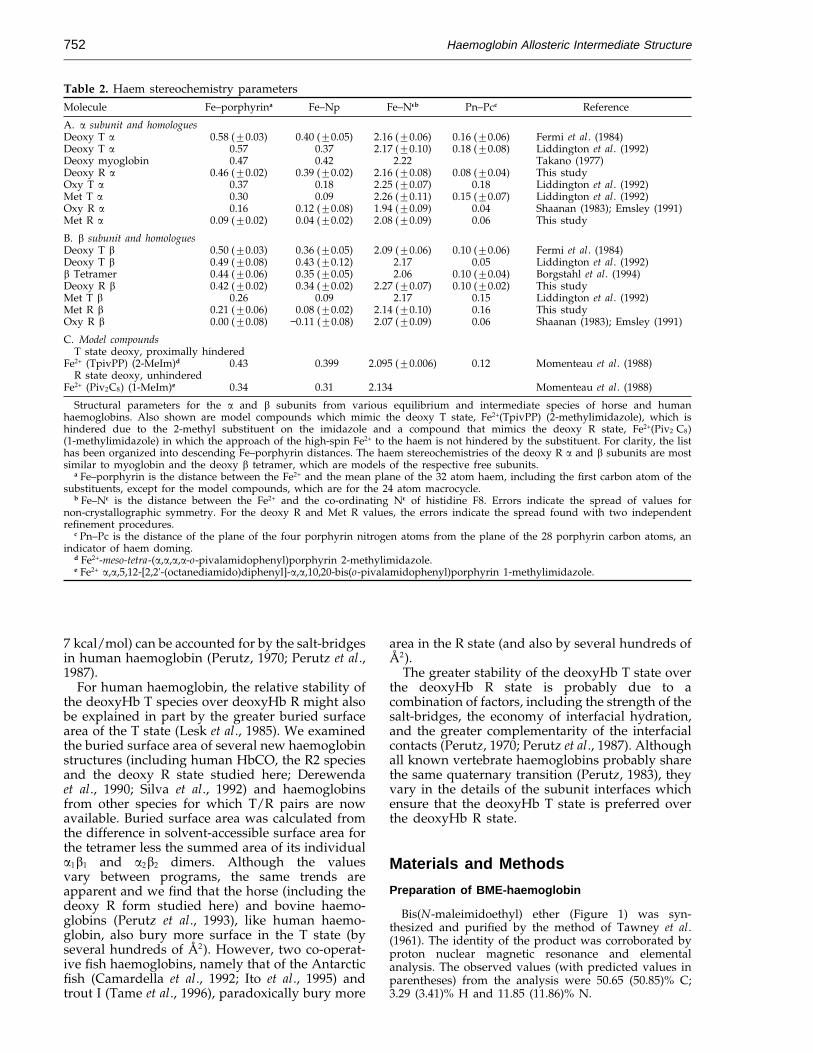
Table 2. Haem stereochemistry parametersMolecule Fe–porphyrina Fe–Np Fe–Neb Pn–Pcc Reference
A. a subunit and homologuesDeoxy T a 0.58 (20.03) 0.40 (20.05) 2.16 (20.06) 0.16 (20.06) Fermi et al. (1984)Deoxy T a 0.57 0.37 2.17 (20.10) 0.18 (20.08) Liddington et al. (1992)Deoxy myoglobin 0.47 0.42 2.22 Takano (1977)Deoxy R a 0.46 (20.02) 0.39 (20.02) 2.16 (20.08) 0.08 (20.04) This studyOxy T a 0.37 0.18 2.25 (20.07) 0.18 Liddington et al. (1992)Met T a 0.30 0.09 2.26 (20.11) 0.15 (20.07) Liddington et al. (1992)Oxy R a 0.16 0.12 (20.08) 1.94 (20.09) 0.04 Shaanan (1983); Emsley (1991)Met R a 0.09 (20.02) 0.04 (20.02) 2.08 (20.09) 0.06 This study
B. b subunit and homologuesDeoxy T b 0.50 (20.03) 0.36 (20.05) 2.09 (20.06) 0.10 (20.06) Fermi et al. (1984)Deoxy T b 0.49 (20.08) 0.43 (20.12) 2.17 0.05 Liddington et al. (1992)b Tetramer 0.44 (20.06) 0.35 (20.05) 2.06 0.10 (20.04) Borgstahl et al. (1994)Deoxy R b 0.42 (20.02) 0.34 (20.02) 2.27 (20.07) 0.10 (20.02) This studyMet T b 0.26 0.09 2.17 0.15 Liddington et al. (1992)Met R b 0.21 (20.06) 0.08 (20.02) 2.14 (20.10) 0.16 This studyOxy R b 0.00 (20.08) −0.11 (20.08) 2.07 (20.09) 0.06 Shaanan (1983); Emsley (1991)
C. Model compoundsT state deoxy, proximally hindered
Fe2+ (TpivPP) (2-MeIm)d 0.43 0.399 2.095 (20.006) 0.12 Momenteau et al. (1988)R state deoxy, unhindered
Fe2+ (Piv2C8) (1-MeIm)e 0.34 0.31 2.134 Momenteau et al. (1988)
Structural parameters for the a and b subunits from various equilibrium and intermediate species of horse and humanhaemoglobins. Also shown are model compounds which mimic the deoxy T state, Fe2+(TpivPP) (2-methylimidazole), which ishindered due to the 2-methyl substituent on the imidazole and a compound that mimics the deoxy R state, Fe2+(Piv2 C8)(1-methylimidazole) in which the approach of the high-spin Fe2+ to the haem is not hindered by the substituent. For clarity, the listhas been organized into descending Fe–porphyrin distances. The haem stereochemistries of the deoxy R a and b subunits are mostsimilar to myoglobin and the deoxy b tetramer, which are models of the respective free subunits.
a Fe–porphyrin is the distance between the Fe2+ and the mean plane of the 32 atom haem, including the first carbon atom of thesubstituents, except for the model compounds, which are for the 24 atom macrocycle.
b Fe–Ne is the distance between the Fe2+ and the co-ordinating Ne of histidine F8. Errors indicate the spread of values fornon-crystallographic symmetry. For the deoxy R and Met R values, the errors indicate the spread found with two independentrefinement procedures.
c Pn–Pc is the distance of the plane of the four porphyrin nitrogen atoms from the plane of the 28 porphyrin carbon atoms, anindicator of haem doming.
d Fe2+-meso-tetra-(a,a,a,a-o-pivalamidophenyl)porphyrin 2-methylimidazole.e Fe2+ a,a,5,12-[2,2'-(octanediamido)diphenyl]-a,a,10,20-bis(o-pivalamidophenyl)porphyrin 1-methylimidazole.
7 kcal/mol) can be accounted for by the salt-bridgesin human haemoglobin (Perutz, 1970; Perutz et al.,1987).
For human haemoglobin, the relative stability ofthe deoxyHb T species over deoxyHb R might alsobe explained in part by the greater buried surfacearea of the T state (Lesk et al., 1985). We examinedthe buried surface area of several new haemoglobinstructures (including human HbCO, the R2 speciesand the deoxy R state studied here; Derewendaet al., 1990; Silva et al., 1992) and haemoglobinsfrom other species for which T/R pairs are nowavailable. Buried surface area was calculated fromthe difference in solvent-accessible surface area forthe tetramer less the summed area of its individuala1b1 and a2b2 dimers. Although the valuesvary between programs, the same trends areapparent and we find that the horse (including thedeoxy R form studied here) and bovine haemo-globins (Perutz et al., 1993), like human haemo-globin, also bury more surface in the T state (byseveral hundreds of A2). However, two co-operat-ive fish haemoglobins, namely that of the Antarcticfish (Camardella et al., 1992; Ito et al., 1995) andtrout I (Tame et al., 1996), paradoxically bury more
area in the R state (and also by several hundreds ofA2).
The greater stability of the deoxyHb T state overthe deoxyHb R state is probably due to acombination of factors, including the strength of thesalt-bridges, the economy of interfacial hydration,and the greater complementarity of the interfacialcontacts (Perutz, 1970; Perutz et al., 1987). Althoughall known vertebrate haemoglobins probably sharethe same quaternary transition (Perutz, 1983), theyvary in the details of the subunit interfaces whichensure that the deoxyHb T state is preferred overthe deoxyHb R state.
Materials and Methods
Preparation of BME-haemoglobin
Bis(N-maleimidoethyl) ether (Figure 1) was syn-thesized and purified by the method of Tawney et al.(1961). The identity of the product was corroborated byproton nuclear magnetic resonance and elementalanalysis. The observed values (with predicted values inparentheses) from the analysis were 50.65 (50.85)% C;3.29 (3.41)% H and 11.85 (11.86)% N.
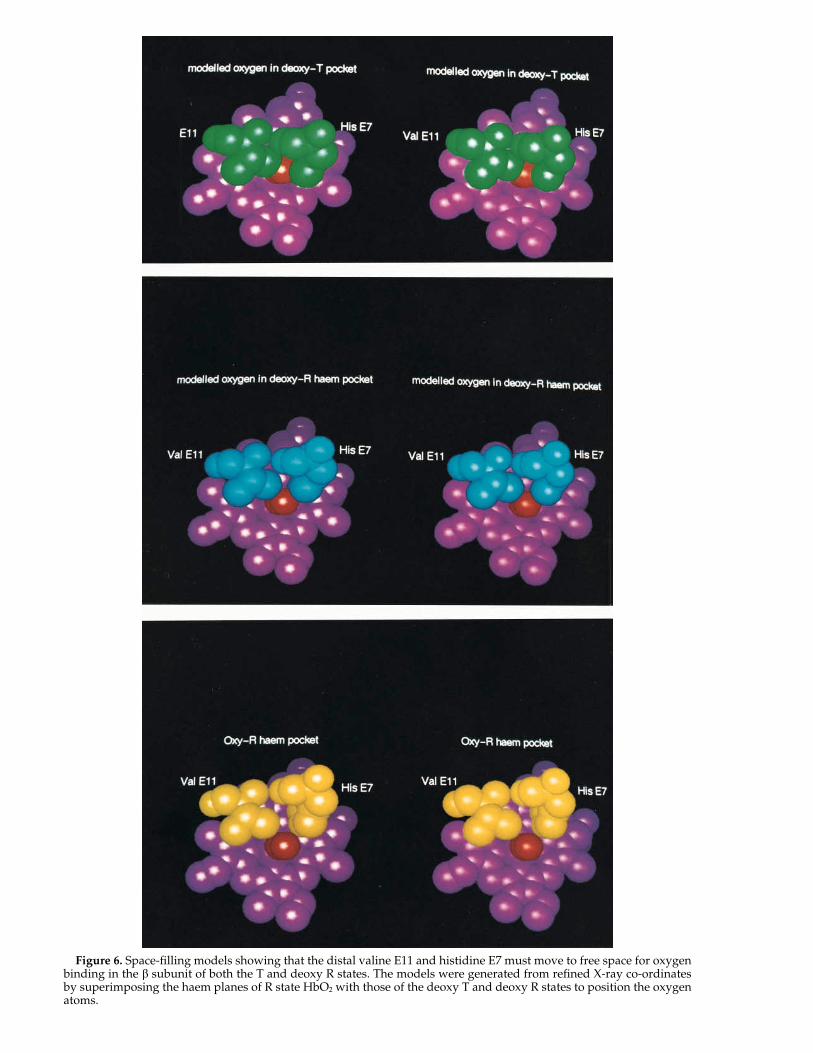
Figure 6. Space-filling models showing that the distal valine E11 and histidine E7 must move to free space for oxygenbinding in the b subunit of both the T and deoxy R states. The models were generated from refined X-ray co-ordinatesby superimposing the haem planes of R state HbO2 with those of the deoxy T and deoxy R states to position the oxygenatoms.

Haemoglobin Allosteric Intermediate Structure754
Table 3. X-ray data for deoxy R and Met R horse haemoglobinsResolution Complete Rsym
a Rb
Molecule (A) Obs. Unique (%) (%) (%)
BME-deoxyHb 1.8 144,335 32,227 98.1 5.9 18.1MetHb 2.0 23,235 99.6 7.0 19.0
a Rsym = S=�I� − Ii =/S�I�, where �I� is the mean of the symmetry-related multiplymeasured reflections for each unique reflection.
b R = S=Fobs − Fcalc=/S=Fobs= where Fobs and Fcalc are the experimentally determined andcalculated structure factors, respectively.
Horse haemoglobin was prepared from freshly drawnblood obtained from the Equine Research Centre,Newmarket, UK and the ‘‘fast component’’ fraction waspurified by the method of Kilmartin (1969) usingAmberlite CG 50. The optimal reaction condition withBME was determined by evaluating the productswith isoelectric focussing and monitoring the loss ofreactivity with pMB, which reacts with CysF9b. Thehaemoglobin and cross-linking agent were mixedtogether in a 1.9:1.0 molar ratio of BME:Hb tetramer forone to two hours at 4°C under a carbon monoxideatmosphere in 10 mM sodium phosphate (pH 7.0). Thehaemoglobin was then immediately gel-filtered onSephadex G25 and purified on CM 52 equilibrated withCO-saturated buffer. A well-resolved fraction wasobtained of material which was unreactive to pMB, andthe yield was roughly 60%. Incubation of this material,hereafter referred to as BME-Hb, for 1.5 hours at roomtemperature in the presence of a 200:1 molar excess ofeither sodium dithionite or dithiothreitol followed by gelfiltration, did not free the sulphydryl groups for reactionwith pMB. Amino acid composition analysis byhigh-pressure liquid chromatography (following acidhydrolysis) indicated an anomalous peak, which elutedafter aspartic acid and at the usual position forcarboxymethyl cysteine.
The BME-Hb was stored under CO. To prepare the Metform for crystallizations, the bound CO was removed byphotolysis followed by oxidation with K3Fe(CN)6, gelfiltration, extensive dialysis against distilled water, andthen centrifugation to remove traces of precipitate.Crystals were prepared using the conditions described byPerutz (1968), and the most suitable crystals for datacollection grew from solutions A (pH approximately 7.5)and B (pH approximately 7.0).
Human BME-haemoglobin was prepared by a similarprocedure, but crystallization trials were unsuccessful.
X-ray data collection
The horse Met BME-Hb crystals were reduced under anitrogen atmosphere by exposure to 2 mM sodiumdithionite in degassed crystallization buffer containing1 mM bovine catalase for 12 hours. The crystals werethen mounted in glass capillaries under nitrogen.X-ray diffraction data were collected from threecrystals using a multiwire area detector at theUniversity of California, San Diego. The space group isC2 and cell dimensions are a = 108.18(20.08) A,b = 63.18(20.04) A, c = 54.58(0.03) A, b = 111.10(20.04)°(the reported values for horse MetHb are 108.24 A,63.13 A, 54.54 A and b = 110.85°; Ladner et al., 1977).
At the completion of X-ray data collection, the crystalswere dissolved in 0.1 M sodium phosphate (pH 6.4),which had been saturated with carbon monoxide, andoptical spectra were recorded. No haem oxidation was
detectable from any of the three crystals used to collectdata, and the upper limit to the Met content, estimatedfrom errors in measurement, was 10%. The sulphydrylgroups of the crystallized, dithionite-treated BME-Hbwere found to have remained blocked to the reactionwith pMB.
A 2.5 A data set was also collected from five crystalsusing a Watts-Hilger diffractometer, with cell dimensionsa = 108.17 (20.06) A, b = 63.19 (20.03) A, c = 54.56(20.03) A and b = 111.11 (20.03)°. Fourier differencemaps using the BME-Hb and MetHb structure factorsfrom diffractometer and area detector data exhibited thesame features.
The asymmetric unit contains one ab dimer (the axesof the molecular dyad and crystallographic 2-foldcoincide) and the horse MetHb co-ordinates published byLadner et al. (1977) were used as the initial model forrefinement of the deoxy BME-Hb. The horse MetHbco-ordinates were re-refined by a similar procedure forcomparison with the deoxy BME-Hb structure. The CCP4suite was used for many of the crystallographiccalculations (CCP4, 1994). The MetHb and deoxyHb Rstructures were initially refined using the procedure ofJack & Levitt (1978) and re-refined with PROLSQ(Konnert & Hendrickson, 1980) and O (Jones &Kjeldgaard, 1991). Difference maps suggested an an-isotropic movement of the Fe atom perpendicular to thehaem plane in the deoxy BME-Hb, and this was modelledsuccessfully using the program SHELXL-93 (Sheldrick,1990). The root-mean-square differences from ideality forBME-Hb and MetHb are, respectively, 0.024 and 0.029 Afor bond distances; 0.130 and 0.170 for chiral volumes;3.06 and 2.92° for planar torsion angles; and 0.018 and0.021 A for planes (Table 3).
Oxygen binding curves
Crystals of Met BME-Hb and control horse MetHbcrystals were dissolved in buffer and dialysed extensivelyagainst 1 mM Tris-HCl (pH 7.9), 10 mM EDTA, and thenreduced with a tenfold excess of sodium dithionite andfiltered on Sephadex G25. The material was then dialysedagainst 1 mM Tris-HCl (pH 7.9), 1 mM EDTA solutionsaturated with CO, centrifuged to remove traces ofprecipitate and stored frozen in liquid nitrogen. Oxygenbinding measurements were made from this materialafter photolysis. The conditions were 25°C in 50 mMbis-Tris (pH 7.2), 0.1 M NaCl, 20 mg catalase, 12.5 to15 mM haemoglobin. The concentration of Met duringthe measurements was 12% for horse BME-Hb, 6% forthe horse Hb control and <2% for the human control. Theresults are summarized in Table 1. The difference inaffinity between the BME-Hb and the control samples isprobably due to greater oxidation of the BME-Hb sample(compared with the MetHb species).

Haemoglobin Allosteric Intermediate Structure 755
AcknowledgementsWe thank Claude Poyart for measuring the haemo-
globin oxygen binding curves; Veronique Baudin andHenri Wajcman for proteolytic analysis of BME-haemo-globin; Chris Christopolous for determining the NMRspectrum of BME; and Ng H. Xuong and colleagues atUCSD for use of X-ray facilities. We are grateful toKiyoshi Nagai, Jeremy Tame, Cyrus Chothia, GiulioFermi and Max Perutz for extensive discussions andencouragement. This study was supported by theMedical Research Council. The co-ordinates of BME-Hband refined MetHb co-ordinates have been depositedwith the Protein Data Bank (no. 1IBE).
ReferencesAbraham, D. J., Peascoe, R. A., Randad, R. S. &
Panikker, J. (1992). X-ray diffraction study of di andtetra-ligated T-state haemoglobin from high saltcrystals. J. Biol. Chem. 227, 480–492.
Anderson, L. (1973). Intermediate structure of normalhuman haemoglobin: methaemoglobin in the deoxyquaternary conformation. J. Mol. Biol. 79, 495–506.
Baldwin, J. & Chothia, C. (1979). Haemoglobin: thestructural changes related to ligand binding and itsallosteric mechanism. J. Mol. Biol. 129, 175–220.
Borgstahl, G. E. O., Rogers, P. H. & Arnone, A. (1994). The1.9 A structure of deoxyb4 hemoglobin: analysis ofthe partitioning of quaternary-associated and ligand-induced changes in tertiary structure. J. Mol. Biol.236, 831–843.
Camardella, L., Caruso, C., D’Avino, R., di Prisco, G.,Rutigliano, B., Tamburrini, M., Fermi, G. & Perutz,M. F. (1992). Haemoglobin of the antarctic fishPagothenia bernacchii: amino acid sequence, oxygenequilibria and crystal structure of its carbonmonoxyderivative. J. Mol. Biol. 224, 449–460.
CCP4. (1994). The CCP4 suite: programs for proteincrystallography. Acta Crystallog. sect. D, 50, 760–763.
Derewenda, Z., Dodson, G., Emsley, P., Harris, D., Nagai,K., Perutz, M. & Renaud, J.-P. (1990). Stereochem-istry of carbon monoxide binding to normal humanadult and Cowtown haemoglobins. J. Mol. Biol. 211,515–519.
Emsley, P. E. (1991). Crystallographic studies of ligandbinding to haemoglobin. PhD thesis, University ofYork.
Fermi, G., Perutz, M. F., Shaanan, B. & Fourme, R. (1984).The crystal structure of human deoxy haemoglobinat 1.74 A resolution. J. Mol. Biol. 175, 159–174.
Hopfield, J. J. (1973). Relation between structure,co-operativity and spectra in a model of hemoglobinaction. J. Mol. Biol. 77, 207–222.
Ito, N., Komiyama, N. H. & Fermi, G. (1995). Structure ofdeoxyhaemoglobin of the antartic fish Pagotheniabernacchii with an analysis of the structural basis ofthe Root effect by comparison of the liganded andunliganded haemoglobin structures. J. Mol. Biol. 250,648–658.
Jack, A. & Levitt, M. (1978). Refinement of largestructures by simultaneous minimization of energyand R-factor. Acta Crystallog. sect. A, 34, 931–935.
Jones, T. A. & Kjeldgaard, M. (1991). O: The Manual,Uppsala University, Sweden.
Kilmartin, J. V. (1969). Carbon dioxide binding byhemoglobin. PhD thesis, University of Cambridge.
Kitagawa, T. (1992). Investigation of higher orderstructures of proteins by ultraviolet resonance
Raman spectroscopy. Prog. Biophys. Mol. Biol. 58,1–18.
Konnert, J. H. & Hendrickson, W. (1980). A restrainedparameter thermal factor refinement procedure. ActaCrystallog. sect. A, 36, 344–350.
Ladner, R. C., Heidner, E. J. & Perutz, M. F. (1977). Thestructure of horse methaemoglobin at 2.0 A resol-ution. J. Mol. Biol. 114, 385–414.
Lesk, A. M., Janin, J., Wodak, S. & Chothia, C. (1985).Haemoglobin: the surface buried between the a1b1and a2b2 dimers in the deoxy and oxy structures.J. Mol. Biol. 183, 267–270.
Liddington, R., Derewenda, Z., Dodson, G. & Harris, D.(1988). Structure of the liganded T state ofhaemoglobin identifies the origin of cooperativeoxygen binding. Nature, 331, 725–728.
Liddington, R., Derewenda, Z., Dodson, E., Hubbard, R.& Dodson, G. (1992). High-resolution crystalstructures and comparisons of T-state deoxy-haemoglobin and two liganded T-state haemo-globins: T(a-oxy) haemoglobin and T (Met)haemoglobin. J. Mol. Biol. 228, 551–579.
Luisi, B. F., Liddington, R. C., Fermi, G. & Shibayama, N.(1990). Structure of deoxy-quaternary haemoglobinwith liganded beta subunits. J. Mol. Biol. 214, 7–14.
Moffat, J. K. (1971). Structure and functional properties ofchemically modified horse hemoglobin. II. J. Mol.Biol. 58, 79–88.
Moffat, J. K., Simon, S. R. & Konigsberg, W. H. (1971).Structure and functional properties of chemicallymodified horse hemoglobin. III. J. Mol. Biol. 58,89–101.
Momenteau, M., Scheidt, W. R., Eigenbrot, C. W. & Reed,C. A. (1988). A deoxymyoglobin model with asterically unhindered axial imidazole. J. Am. Chem.Soc. 110, 1207–1215.
Mozzarelli, A., Rivetti, C., Rossi, G. L., Henry, E. R. &Eaton, W. A. (1991). Crystals of haemoglobin withthe T quaternary structure bind oxygen noncoopera-tively with no Bohr effect. Nature, 51, 416–419.
Nagai, K. & Kitigawa, T. (1980). Differences inFe(II)-Ne(His-F8) stretching frequencies betweendeoxyhemoglobins in the two alternative quaternarystructures. Proc. Natl Acad. Sci. USA, 77, 2033–2037.
Nagai, K., Luisi, B., Shih, D., Miyazaki, G., Imai, K.,Poyart, C., De Young, A., Kwiatkowsky, L., Noble,R. W., Lin, S.-H. & Yu, N.-T. (1987). Distal residuesin the oxygen binding site of haemoglobin studiedby protein engineering. Nature, 329, 858–860.
Oldfield, T. J. (1992). SQUID: a program for the analysisand display of data from crystallography andmolecular dynamics. J. Mol. Graphics, 10, 247–252.
Olson, J. S., Mathews, A. J., Rohlfs, R. J., Springer, B. A.,Edeberg, K. D., Tame, J., Renaud, J. P. & Nagai, K.(1988). The role of the distal histidine in myoglobinand haemoglobin. Nature, 336, 265–266.
Paoli, M., Liddington, R., Tame, J., Wilkinson, A. &Dodson, G. (1996). Crystal structure of T statehaemoglobin with oxygen bound at all four haems.J. Mol. Biol. 256, 775–792.
Perutz, M. F. (1968). Preparation of haemoglobin crystals.J. Crystal Growth, 2, 54–56.
Perutz, M. (1970). Stereochemistry of cooperative effectsin haemoglobin. Nature, 228, 726–739.
Perutz, M. F. (1983). Species adaptation in a proteinmolecule. Mol. Biol. Evol. 1, 1–28.
Perutz, M. F., Fermi, G., Luisi, B. F., Shaanan, B. &Liddington, R. C. (1987). Stereochemistry of

Haemoglobin Allosteric Intermediate Structure756
cooperative mechanisms in hemoglobin. Acc. Chem.Res. 20, 309–321.
Perutz, M. F., Fermi, G., Poyart, C., Pagnier, J. &Kister, J. (1993). A novel allosteric mechanismin haemoglobin: structure of bovine deoxy-haemoglobin, absence of specific chloride-bindingsites and origin of the chloride-linked Bohr effect inbovine and human haemoglobin. J. Mol. Biol. 233,536–545.
Schumacher, M. A., Dixon, M. M., Kluger, R., Jones, R. T.& Brennan, R. G. (1995). Allosteric transitionintermediates modelled by crosslinked haemo-globin. Nature, 375, 84–87.
Shaanan, B. (1983). Structure of human oxyhaemoglobinat 2.1 A resolution. J. Mol. Biol. 171, 31–59.
Sheldrick, G. (1990). Phase annealing in Shelx-90: directmethods for larger structures. Acta Crystallog. sect. A,46, 467–473.
Shibayama, N. & Saigo, S. (1995). Fixation of thequaternary structures of human adult haemoglobinby encapsulation in transparent porous silica gels.J. Mol. Biol. 251, 203–209.
Silva, M. M., Rogers, P. H. & Arnone, A. (1992). A thirdquaternary structure of human haemoglobin A at1.7 A resolution. J. Biol. Chem. 267, 17248–17256.
Takano, T. (1977). Structure of myoglobin refined at 2.0 Aresolution. II. Structure of deoxymyoglobin fromsperm whale. J. Mol. Biol. 110, 569–584.
Tame, J. R. H., Wilson, J. C. & Weber, R. E. (1996). Thecrystal structure of trout Hb I in the deoxy andcarbonmonoxy forms. J. Mol. Biol. 259, 749–760.
Tawney, P. O., Snyder, R. H., Conger, R. P., Leibbrand,K. A., Stitler, C. H. & Williams, A. R. (1961). Thechemistry of maleimide and its derivatives. II.Maleimide and N-methylolmaleimide. J. Org. Chem.26, 15–21.
Edited by K. Nagai
(Received 4 April 1996; received in revised form 6 September 1996; accepted 18 September 1996)




















