
Functionalisation of recombinant spider silk with conjugated polyelectrolytes
Upload
independentCategory
view
0download
0
ORIGINAL PAPER
The complete mitochondrial genome of the Antarctic sea spiderAmmothea carolinensis (Chelicerata; Pycnogonida)
Antonio Carapelli • Giulia Torricelli •
Francesco Nardi • Francesco Frati
Received: 9 October 2012 / Revised: 16 December 2012 / Accepted: 10 January 2013
� Springer-Verlag Berlin Heidelberg 2013
Abstract Mitochondria are responsible for the oxidative
phosphorylation process. Accordingly, putatively adaptive
changes in their genomic features have been variously
associated with major eco-physiological shifts in animal
evolution, including increased metabolic rates and heat
adaptation. Antarctic pycnogonids offer an interesting
system to test whether the selective pressure for heat pro-
duction and increased aerobic metabolism may be driving
genomic changes like: (a) unusual compositional biases at
the nucleotide and amino acid level, possibly related to
cold adaptation; (b) an accelerated rate of mutations/
genomic rearrangements, possibly related to the mutagenic
effects of oxygen intermediates. The complete mitochon-
drial genome (mtDNA) of the Antarctic sea spider
Ammothea carolinensis Leach, 1814 (Arthropoda: Pycno-
gonida), the type species for the genus Ammothea, has been
determined and is here compared to known genomes from
Antarctic and temperate species. We describe a marked
heterogeneity in base composition skewness parameters as
well as a strong signature of purifying selection toward an
increase in thymines at second codon positions, possibly
associated with an increased stability of hydrophobic inter-
membrane domains. We further observe a fairly high rate
of genomic changes, including a possible hot spot of
recombination at the level of tRNA-Q. Nevertheless, these
features do not seem to be restricted to the two Antarctic
pycnogonids analyzed, as to suggest a causal relationship
between cold adaptation and genomic changes, and are
better interpreted as basal features shared by the entire
group. The relevance of the newly determined sequence for
the phylogeny of pycnogonids, including its base compo-
sition and genomic rearrangements, is further discussed.
Keywords Pycnogonida �Mitochondrial DNA (mtDNA) �Molecular evolution � Antarctica � Nucleotide bias
Introduction
The mitochondrial genome of animals is almost invariably
a circular double-stranded molecule of 11–20 kb in size. It
includes the genes encoding for 13 protein products
involved in the oxidative phosphorylation (OXPHOS)
process, 22 transfer RNA (trnX), and two ribosomal rRNAs
(rrnS and rrnL), as well as a noncoding region (known as
AT-rich region in arthropods) where the signals for the
initiation of replication and transcription are located. Its
high-nucleotide substitution rate and variable gene order
combined with conservative gene content (Castellana et al.
2011), as well as the strict matrilinear inheritance and lack
of introns, made mtDNA one of the most popular markers
in phylogenetic studies, only paralleled by nuclear rRNA
markers. In addition, mitochondrial sequences are widely
used to identify unknown species (i.e., cytochrome oxidase
c subunit I in barcoding) and for population genetic studies
(Min and Hickey 2007), while the arrangement of genes
(gene order) within the organelle’s genome has been used
to infer deep phylogenetic relationships. The recent avail-
ability of large nuclear datasets for phylogenomic studies
(Regier et al. 2010; Rota-Stabelli et al. 2010; Rehm et al.
2011; Giribet and Edgecombe 2012), together with the
recognition that highly divergent rates of molecular
Electronic supplementary material The online version of thisarticle (doi:10.1007/s00300-013-1288-6) contains supplementarymaterial, which is available to authorized users.
A. Carapelli (&) � G. Torricelli � F. Nardi � F. Frati
Department of Life Sciences, University of Siena,
Via Aldo Moro 2, 53100 Siena, Italy
e-mail: [email protected]
123
Polar Biol
DOI 10.1007/s00300-013-1288-6

evolution (in primary sequence as well as gene order) may
affect specific taxa (Gissi et al. 2000; Hassanin 2006) with
undesirable outcomes on the analysis, has lowered the
interest in mitochondrial genomes as a phylogenetic mar-
ker, although attempts to improve data analysis are ongoing
(Carapelli et al. 2007; Gissi et al. 2008; Talavera and Vila
2011; Nardi et al. 2012a). Nevertheless, the accumulation
of over 2800 complete animal mitochondrial genomes from
a variety of different invertebrate and vertebrata taxa has
opened to the possibility of performing comparative studies
on the molecular features, functionality, and evolution of
the mitochondrial genome on a large scale, so far impos-
sible for other genomic compartments (nuclear and plas-
tidial). Key transitions of animal evolution (i.e., origin of
multicellularity and bilaterality) are associated with sub-
stantial modifications of the mitochondrial DNA architec-
ture (Lavrov 2011). Some of these changes represent
interesting features to be investigated in order to under-
stand the molecular evolution of the organelle genome. In
this respect, the mechanisms that lead to gene rearrange-
ments have been hypothesized following the discovery of
peculiar mtDNA organizations (Lavrov et al. 2002; San
Mauro et al. 2006; Juhling et al. 2012; Nardi et al. 2012b).
In addition, some posttranscriptional modifications (non-
templated nucleotide addition and insertion, which com-
pensate for defects in gene sequence and restore the
functionality of tRNAs), once believed to be exclusive of
plant mitochondrial and plastidial DNAs, have been
observed also in animals (Lavrov et al. 2002; Segovia et al.
2011). Eventually, sex-linked mtDNA genomes have been
discovered in some bivalves highlighting a transmission
route (Doubly uniparental inheritance) other than orthodox
matrilinear inheritance (Passamonti et al. 2011).
Six complete or almost complete mtDNAs have been
sequenced for Pycnogonida (Podsiadlowski and Braband
2006; Park et al. 2007; Masta et al. 2010; Dietz et al. 2011),
an enigmatic group of arthropods whose fossil record dates
back to the Upper-Cambrian (&490 Myr ago) (Waloszek
and Dunlop 2002) and that is characterized by a number of
peculiar morphological features that complicate the iden-
tification of their phylogenetic relationships (Budd and
Telford 2005; Maxmen et al. 2005; Edgecombe 2010).
Pycnogonids have been considered, in turn, as the sister
taxon to the chelicerates (Dunlop and Arango 2005) or
basal to all extant arthropods (Giribet et al. 2001). Unfor-
tunately, due to the heterogeneity of nucleotide composi-
tion compared to other arthropods, mitogenomic data failed
to provide a robust phylogenetic hypothesis and all phy-
logenetic reconstructions based on mtDNA recovered a
highly problematic placement of the group as branching
from within the Chelicerate group (mostly as sister group
to Acarida) that would imply a terrestrial origin of the
group (Hassanin 2006; Podsiadlowski and Braband 2006;
Park et al. 2007; Masta et al. 2010). Nevertheless, some
interesting molecular features that may in turn be deployed
as phylogenetic markers at a lower taxonomic level have
been described from the mitochondrial genomes of pyc-
nogonids. An extensive description of some of these
characters has been proposed by Masta et al. (2010),
although others are still being analyzed in more detail.
In this work, we describe the newly determined complete
mitochondrial genome of the Antarctic species Ammothea
carolinensis, provide a description of major molecular fea-
tures of the genome, and compare this with other published
pycnogonid mtDNAs.
Materials and methods
One living specimen of A. carolinensis was sampled off the
Antarctic coast in the vicinity of the Italian Research Station
Mario Zucchelli (Victoria Land) (74�410S, 164�070E), during
the XXI Italian-Antarctic expedition (2005/2006). The
specimen was identified by Dr. L. Dietz and colleagues from
the University of Bochum (Germany) and a small part of a leg
was frozen and preserved at -80 �C for the molecular anal-
yses. The remaining of the DNA extraction and of the ani-
mal’s body are preserved at the Department of Life Sciences
(University of Siena). Total genomic DNA was purified using
the Wizard SV genomic DNA purification system (Promega)
and used in all subsequent amplifications. Partial cox1 and
cob sequences were initially produced using primer pairs
C1-J-1718 (50-GGAGGATTTGGAAATTGATTAGTTCC-
30)/C1-N-2191 (50-CCCGGTAAAATTAAAATATAAAC
TTC-30) and CB-J-10933 (50-TATGTACTACCATGAGG
ACAAATATC)/CB-N-11367 (50-ATTACACCTCCTAA
TTTATTAGGAAT-30 (Simon et al. 1994) and standard
amplification procedures. Four specific primers were then
designed on these sequences to amplify the rest of the genome
in two segments of about 9 and 6 Kb, respectively: couples
ACA-cox1-1753J (50-GCTCATTCAGGATCCTCAGTAG
ATTTAAC-30)/ACA-cox1-1773N (50-GCCCCTAAAATT
GAAGATACTCCCGC-30) (fragment 1) and ACA-cob-
10682J (50-TTCTCAGTAAATAATGCCACCTTAAAT
CG-30)/ACA-cob-10906N (50-GTTAGCTGGAATGAAG
TTTTCTGGGTC-30) (fragment 2). Long-PCR conditions
were the following: fragment 1) 94 �C for 1 min, 60� for
1 min, and 68� for 12 min, 35 cycles; fragment 2) 94 �C
for 1 min, 50� for 1 min, and 68� for 8 min, 35 cycles.
Amplifications were performed in a 25 ll reaction volume
composed of 10.75 ll of sterilized distilled water, 2.5 ll
of LA PCR Buffer II (Takara), 2.5 ll of 25 mM MgCl2,
4 ll of dNTPs mix, 1.25 ll of each primer (10 lM),
2.5 ll of DNA template, and 0.25 ll (1.25 U) of TaKaRa
LA Taq polymerase (Takara). Long-PCR fragments were
purified using the Kit Wizard SV Gel and PCR Clean-up
Polar Biol
123

System (Promega) and completely sequenced on both
strands using a primer walking approach (primer list
available on request). The final sequence assembly was
produced using Sequencher 4.4.2 (Gene Codes) and
deposited in GenBank under accession number
GU065293.
Protein-coding, rRNA, and tRNA genes were identified
based on their amino acid translation or secondary structure
features, respectively. The AT-rich region was identified as
the single noncoding region of significant length and
according to its peculiar structure and base composition.
Start and stop codons of protein-encoding genes (PCGs)
were identified and gene boundaries were inspected for the
presence of gene overlaps. Secondary structures of tRNAs
were manually reconstructed and rendered using RnaViz
2.0 (de Rijk and de Wachter 1997). Basic sequence sta-
tistics and codon usage were calculated using PAUP* 4b10
(Swofford 2002) and codonw (Peden 1999), respectively.
Strand asymmetry was described using the formulas:
AT-skew = [A %-T %]/[A %?T %] and CG-skew =
[C %-G %]/[C %?G %] (Perna and Kocher 1995; Hass-
anin et al. 2005). The graphical representation of the
percentage of A?C and G?T nucleotides across the whole
J-strand was calculated using the program MacVector�
7.2.3 (Accelrys). The presence of repeated sequences
within the AT-rich region was assessed using the mreps
software (Kolpakov et al. 2003) and associated secondary
structure motifs were reconstructed using the software
Mfold (Zuker 2003) under default settings.
Results
Gene order
The gene order of six of the seven published mtDNA
genomes of pycnogonids is similar—although not identi-
cal—to that of Limulus polyphemus (Lavrov et al. 2000),
presumably the ancestral state for arthropods. In these
genomes, nevertheless, trnQ is never observed in its
putative original position between trnI and trnM (Masta
et al. 2010), suggesting either a ‘‘hot spot’’ for independent
rearrangements or the occurrence of an ancestral translo-
cation followed by independent repositioning in at least
three different lineages (Achelia ? Ammothea ? Tanysty-
lum, Colossendeis, and Nymphon). Notably, the actual
position of trnQ is unknown for two of the three incom-
plete genomes (Ammothea hilgendorfi and Nymphon
unguiculatum, erroneously reported as Colossendeis sp. in
Masta et al. 2010; Dietz et al. 2011), although this infor-
mation could be deduced based on the gene order of the
congeneric species A. carolinensis (Fig. 1) and Nymphon
gracile, where trnQ is located on the N-strand between the
AT-rich region and rrnS and between rrnS and trnN,
respectively. Furthermore, in Colossendeis megalonyx,
trnQ is not observed on the original position, yet it has not
been found in the sequenced fragment of the genome
(Dietz et al. 2011). Interestingly, in the taxa where this
gene has been sequenced, trnQ is always positioned at the
50-end of the rrnS gene, with the only exception of the
highly rearranged N. gracile (where it is found at the 30-end
of rrnS). This latter evidence suggests an ancestral trans-
location of trnQ at the base of the pycnogonid lineage,
either at the 30- or 50-end of rrnS, followed by additional
independent relocation in some lineages. The two Nym-
phon species further share the translocations of trnA, trnN,
and trnSgcu and the switch of position between trnE and
trnR. The observation that in all pycnogonid genomes, the
30-end of cox2 directly abuts the 50-end of cox1 without any
intergenic spacers while the two tRNA genes encoding
Leucine (trnLuag and trnLuaa) are located between rrnL
and nad1 unambiguously supports the placement of the
group outside the pancrustacean lineage (Boore et al.
1998).
Gene initiation and termination, intergenic spacers
Mitochondrial PCGs are notorious for the use of nonstan-
dard initiation and truncated stop codons (Boore 2006;
Carapelli et al. 2008). In A. carolinensis, nine genes start
Fig. 1 The mitochondrial genome organization of A. carolinensis.
Genes for proteins and rRNAs are indicated with standard abbrevi-
ations, whereas those for tRNAs are designated by a single letter for
the corresponding amino acid. Arrows indicate the direction of coding
regions. In black are genes encoded on the J-strand, in gray those with
opposite polarity
Polar Biol
123
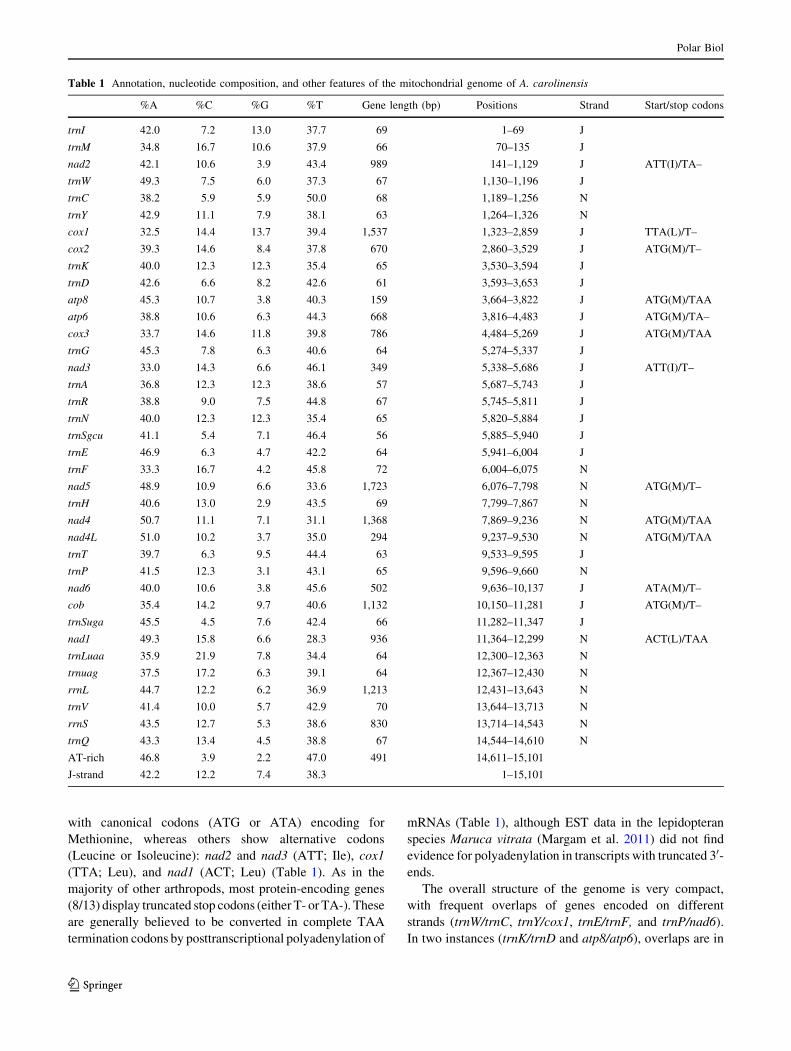
with canonical codons (ATG or ATA) encoding for
Methionine, whereas others show alternative codons
(Leucine or Isoleucine): nad2 and nad3 (ATT; Ile), cox1
(TTA; Leu), and nad1 (ACT; Leu) (Table 1). As in the
majority of other arthropods, most protein-encoding genes
(8/13) display truncated stop codons (either T- or TA-). These
are generally believed to be converted in complete TAA
termination codons by posttranscriptional polyadenylation of
mRNAs (Table 1), although EST data in the lepidopteran
species Maruca vitrata (Margam et al. 2011) did not find
evidence for polyadenylation in transcripts with truncated 30-ends.
The overall structure of the genome is very compact,
with frequent overlaps of genes encoded on different
strands (trnW/trnC, trnY/cox1, trnE/trnF, and trnP/nad6).
In two instances (trnK/trnD and atp8/atp6), overlaps are in
Table 1 Annotation, nucleotide composition, and other features of the mitochondrial genome of A. carolinensis
%A %C %G %T Gene length (bp) Positions Strand Start/stop codons
trnI 42.0 7.2 13.0 37.7 69 1–69 J
trnM 34.8 16.7 10.6 37.9 66 70–135 J
nad2 42.1 10.6 3.9 43.4 989 141–1,129 J ATT(I)/TA–
trnW 49.3 7.5 6.0 37.3 67 1,130–1,196 J
trnC 38.2 5.9 5.9 50.0 68 1,189–1,256 N
trnY 42.9 11.1 7.9 38.1 63 1,264–1,326 N
cox1 32.5 14.4 13.7 39.4 1,537 1,323–2,859 J TTA(L)/T–
cox2 39.3 14.6 8.4 37.8 670 2,860–3,529 J ATG(M)/T–
trnK 40.0 12.3 12.3 35.4 65 3,530–3,594 J
trnD 42.6 6.6 8.2 42.6 61 3,593–3,653 J
atp8 45.3 10.7 3.8 40.3 159 3,664–3,822 J ATG(M)/TAA
atp6 38.8 10.6 6.3 44.3 668 3,816–4,483 J ATG(M)/TA–
cox3 33.7 14.6 11.8 39.8 786 4,484–5,269 J ATG(M)/TAA
trnG 45.3 7.8 6.3 40.6 64 5,274–5,337 J
nad3 33.0 14.3 6.6 46.1 349 5,338–5,686 J ATT(I)/T–
trnA 36.8 12.3 12.3 38.6 57 5,687–5,743 J
trnR 38.8 9.0 7.5 44.8 67 5,745–5,811 J
trnN 40.0 12.3 12.3 35.4 65 5,820–5,884 J
trnSgcu 41.1 5.4 7.1 46.4 56 5,885–5,940 J
trnE 46.9 6.3 4.7 42.2 64 5,941–6,004 J
trnF 33.3 16.7 4.2 45.8 72 6,004–6,075 N
nad5 48.9 10.9 6.6 33.6 1,723 6,076–7,798 N ATG(M)/T–
trnH 40.6 13.0 2.9 43.5 69 7,799–7,867 N
nad4 50.7 11.1 7.1 31.1 1,368 7,869–9,236 N ATG(M)/TAA
nad4L 51.0 10.2 3.7 35.0 294 9,237–9,530 N ATG(M)/TAA
trnT 39.7 6.3 9.5 44.4 63 9,533–9,595 J
trnP 41.5 12.3 3.1 43.1 65 9,596–9,660 N
nad6 40.0 10.6 3.8 45.6 502 9,636–10,137 J ATA(M)/T–
cob 35.4 14.2 9.7 40.6 1,132 10,150–11,281 J ATG(M)/T–
trnSuga 45.5 4.5 7.6 42.4 66 11,282–11,347 J
nad1 49.3 15.8 6.6 28.3 936 11,364–12,299 N ACT(L)/TAA
trnLuaa 35.9 21.9 7.8 34.4 64 12,300–12,363 N
trnuag 37.5 17.2 6.3 39.1 64 12,367–12,430 N
rrnL 44.7 12.2 6.2 36.9 1,213 12,431–13,643 N
trnV 41.4 10.0 5.7 42.9 70 13,644–13,713 N
rrnS 43.5 12.7 5.3 38.6 830 13,714–14,543 N
trnQ 43.3 13.4 4.5 38.8 67 14,544–14,610 N
AT-rich 46.8 3.9 2.2 47.0 491 14,611–15,101
J-strand 42.2 12.2 7.4 38.3 1–15,101
Polar Biol
123

turn observed between genes encoded on the same strand.
Whereas the overlap between atp8 and atp6 is a common
feature in arthropod and vertebrate mitochondrial genomes
(Ojala et al. 1981; Krzywinski et al. 2006; Podsiadlowski
et al. 2006), and polycistronic transcripts of 2 or 3 genes
(usually atp8/atp6, but also atp8/atp6/cox3, nad4L/nad4,
and nad6/cob) are sporadically observed in arthropod
mtDNA transcriptomes (Margam et al. 2011), the possi-
bility of alternative splicing may be considered for the pair
trnK/trnD.
Nucleotide composition and codon usage
The nucleotide composition of the A. carolinensis mtDNA
is very similar to A. hilgendorfi, with the highest value of
A?T (80.4 %) and the lowest of G-content (7.4 % on the
J-strand) ever detected for pycnogonid mtDNAs (Table 1).
Total A?C richness of the J-strand, a common feature
of arthropod mtDNAs, is not uniformly distributed across
the genome. While A?C % and G?T % are almost equal
in genes encoded on the J-strand, A?C % is invariably
higher than G?T % (calculated on the J-strand) in genes
encoded on the N-strand (either protein or RNA encoding)
(Fig. 2 and Supplementary material 1). This latter trend,
nevertheless, differs across codon positions, with G�C at
first and third positions and T�A at second positions
(Fig. 2). Skewness parameters calculated for all sea spi-
ders’ mtDNAs (J-strand) are very heterogeneous (Masta
et al. 2010), with negative values of both AT- and
CG-skew observed in Tanystylum orbiculare and N. ungui-
culatum and positive values in A. carolinensis (Table 2),
A. hilgendorfi, and N. gracile. In addition, Achelia bituber-
culata displays a negative AT-skew and a positive CG-skew
(Masta et al. 2010). The A. carolinensis J-strand is extremely
poor in G nucleotides (7.4 %), and it is not surprising that
the highest CG-skew values are observed at third codon
positions of PCGs (both orientations) (Supplementary
material 1).
Base composition asymmetries in the other five com-
plete pycnogonid mtDNAs (excluding N. gracile due to its
highly rearranged gene order) are in line with that observed
in A. carolinensis. The occurrence of a high number of Ts
at second codon positions of both J- and N-oriented genes
is also shared by all pycnogonids with the same gene order,
with AT-skew values ranging from -0.40 (C. megalonyx)
to -0.45 (A. carolinensis).
Strand and nucleotide bias significantly affect the codon
usage of protein-encoding genes in the mtDNA of A. car-
olinensis. In this respect, PCGs encoded on either J- and
N-strand show a remarkably higher frequency of NNA
(44 %) and NNU (40 %) triplets versus NNC (12 %) and
NNG (4 %) ones, in line with the observed A?T nucleo-
tide bias. Additionally, more than half (52 %) of the triplets
from fourfold degenerate codon families of J-strand genes
are represented by NNA triplets, whereas most (57 %) of
those oriented on the opposite strand are NNU. Remark-
ably, due to second codon positions, specific bias of
J-strand PCGs, NUNs, rather than NANs, are the most
employed codons (1,110 vs. 424, respectively).
AT-rich region
The AT-rich region of A. carolinensis (491 bp) is charac-
terized by the presence of four copies of a repeated
sequence (R1–R3 and PR), with three being complete and
identical (R1–R3 = 125 bp, corresponding to positions
14,611–14,735; 14,736–14,860; and 14,861–14,985 in the
annotated genome) and one incomplete and slightly dif-
ferent in primary sequence (PR = 42 bp, positions
14,986–15,027) (Fig. 3). Each complete repeat starts with a
Fig. 2 Graphical representation of AT- and CG-skew values in
different sets of J- and N-oriented protein-encoding sequences
(calculated on the J-strand): all = all positions of PCGs; first, second,
and third positions only
Table 2 Skew values
calculated for different sets of J-
and N-oriented nucleotides
All sites J-strand N-strand
PCGs only Third position only PCGs only Third position only
AT-skew 0.050 -0.060 0.032 -0.220 -0.138
CG-skew 0.241 0.197 0.603 -0.291 -0.645
Polar Biol
123

(AT)5 dinucleotide. At the 30-end of R3, there is a partial
repeat (PR) corresponding to the first 40 bases of the
complete repeats plus an additional ‘‘AT’’ resulting in a
(AT)6 sequence. This repeated region corresponds to the
majority (417/491 bases) of the noncoding sequence and is
followed by a 74-long stretch of non-repeated bases at the
30-end of PR. The whole AT-rich region can be folded into
a 8-stems secondary structure, mostly composed by paired
sequences of the aforementioned repeats (Fig. 3). While
the initial duplication may have originated through differ-
ent processes, including incorrect termination during rep-
lication, the maintenance and amplification of repeats is
probably due to replication slippage (Li et al. 2012). The
primary sequence of A. carolinensis AT-rich region does
not show significant similarity with the three other known
AT-rich regions, from A. bituberculata, N. gracile, and
T. orbiculare, whereas repeated stretches of sequences that
can be folded into secondary structure motifs were also
found in the congeneric A. bituberculata (Park et al. 2007).
Manual search for plausible regulatory sequences, includ-
ing the T-stretches commonly observed in other vertebrate
and arthropod lineages (Clayton 1982; Saito et al. 2005),
was also inconclusive. In addition, sequence length among
AT-rich regions is substantially variable in pycnogonid
mtDNAs, ranging from 191 bp in N. gracile to 977 in
A. bituberculata.
tRNAs
The complete mitochondrial genome of A. carolinensis was
searched for the presence of tRNA genes using the soft-
ware ARWEN (Laslett and Canback 2008). The complete
set of 22 mt-tRNA genes expected for Metazoa was found
and their putative secondary structure reconstructed (Sup-
plementary material 2). Paired stems of 2–5 bp are present
in DHU and TWC arms. Only three out of 22 tRNA genes
(trnN, trnC, and trnI) have a complete set of Watson and
Crick bonds, whereas all the others are characterized by
non-Watson and Crick bonds (G�U = 28) and/or mis-
matches (10).
Most of the tRNA genes can be folded into the canonical
cloverleaf structure, although two of them, trnA and
trnSgcu, have a truncated DHU arm (Podsiadlowski and
Braband 2006; Park et al. 2007; Masta et al. 2010), simi-
larly to what observed in all other pycnogonids with the
exception of C. megalonyx (Dietz et al. 2011). The two
tRNA genes trnK and trnD, both encoded on the J-strand,
overlap by 2 bp (Table 1; Supplementary material 2).
Notably, the anticodon for trnK is UUU as in all other
pycnogonids except for C. megalonyx, where the canonical
CUU 3-nucleotide sequence is, in turn, used (Masta et al.
2010; Dietz et al. 2011).
Discussion
Heterogeneity in nucleotide composition, also observed in
A. carolinensis (i.e., a higher content of A and T vs. C and
G), is a remarkable molecular feature of most arthropod
mtDNAs (Hassanin et al. 2005) and is thought to be a
consequence of different mutation and selection pressures
acting on the two strands. In addition, specific point
mutations occurring when the molecule is in a single strand
state, during the peculiar asymmetrical replication and/or
Fig. 3 Secondary structure of
the AT-rich region of
A. carolinensis (J-orientation)
with boundaries of complete
(R1–3) and partial (PR) repeats
highlighted
Polar Biol
123

transcription processes of the mtDNA, lead to deviations
from the second parity rule (PR2) (Frank and Lobry 1999)
and to a gradient of asymmetry in base composition
between J- and N-strands (Faith and Pollock 2003).
Accordingly, arthropods’ mitochondrial genomes fre-
quently have a J-strand with positive AT- and CG-skew
values (Hassanin et al. 2005; Wei et al. 2010), given that
alternative (asymmetric) mutation pressures may work on
opposite strands depending on the polarity of the origin of
replication, and therefore on the duration of time spent by
the N-strand into a single-stranded state (DssH) (Reyes
et al. 1998). This molecular bias usually leads to hetero-
geneity in nucleotide composition between J- and
N-strands, which are A?C- and G?T-rich, respectively. In
protein-encoding genes, a higher frequency of Ts in second
codon positions in genes encoded on the J- and N-strand
has already been observed for b-sheet structures of proteins
in humans and prokaryotes (Chiusano et al. 2000). This
may be associated, in mitochondrial genes, with the
necessity of assembling inter-membrane hydrophobic pro-
teins (Bradshaw et al. 2005) where 24 of 26 triplets
encoding for hydrophobic residues have T or C at second
position (Naylor et al. 1995). This observation would
suggest that the same selective pressure may be acting
similarly on the two strands, promoting the introduction of
a high number of T bases in second codon positions.
Notably, this pressure opposes, in genes encoded on the
J-strand, to the one introduced by deaminations, while is
concordant for genes encoded on the N-strand. To sum-
marize, given that the almost entire mitochondrial genome
is transcribed into mRNA or RNAs, its nucleotide content
is highly influenced by the chemical nature of the tran-
scripts. Pyrimidine richness may be for the most part driven
by the necessity to have T at second codon positions in
order to synthesize hydrophobic proteins, although the
overall nucleotide content frequently has a positive
AT-skews due to the double number of first and third
codon positions (with respect to second) and by the
occurrence of hydrophilic RNAs molecules, notoriously
purine-rich (Bradshaw et al. 2005).
The structure of tRNASgcu, lacking the DHU arm, but
with all Watson and Crick pairings in the remaining sec-
ondary structures, is in line with previous comparative
analyses that suggested that this tRNA may have lost the
DHU arm early before the radiation of Metazoa, as it has
never been detected in the mtDNA of multicellular organ-
isms (Wolstenholme 1992; Masta and Boore 2008). The
nucleotide overlap between tRNA genes oriented on the
same strand, observed in A. carolinensis, suggests that
alternative splicing of the immature mRNA or some other
mechanism may be needed in order to produce two complete
molecules. Notoriously, some tRNA genes undergo post-
transcriptional molecular manipulation (polyadenylation) to
restore their correct secondary structure (Yokobori and
Paabo 1995; Lavrov et al. 2000; Boore 2006; Segovia et al.
2011). Occurrence of similar overlaps in tRNA genes is very
frequent in several vertebrates and invertebrates, and some of
them (i.e., trnW/trnC and trnE/trnF) are almost invariably
present in Arthropoda (Juhling et al. 2012).
Conclusions
Our data support previous hypotheses on the molecular
evolution of arthropod mtDNAs (Hassanin et al. 2005;
Castellana et al. 2011). In mtDNAs of pycnogonids, a
strong purifying selection is seemingly acting on second
codon positions in order to maintain the functionality
(=hydrophobicity) of membrane proteins. Conversely,
changes at synonymous sites are characterized by the
common and widespread strand bias associated with the
peculiar asymmetrical replication system of the mtDNA.
Repeated stretches of sequences as those observed in the
A. carolinensis AT-rich region, usually including both
complete and incomplete units, are commonly found in
arthropod taxa (Nardi et al. 2001; Carapelli et al. 2006) and
are generally associated with imprecise initiation/termina-
tion mechanisms of mtDNA replication, and/or to regions
with abrupt shift of polarity between differently oriented
genes (Boore 2000; Carapelli et al. 2008).
The occurrence of truncated stop codons observed in
some A. carolinensis genes is though to be a common
feature shared by most animal lineages and probably con-
nected with the extreme compactness of the genome.
Recent studies, mostly based on Drosophila species, have
demonstrated that mitochondrially encoded genes are
transcribed in polycistronic mRNA sequences, which are
secondarily cleaved into separate mRNAs. According to
the tRNA punctuated model (Ojala et al. 1981), the poly-
cistronic mt-mRNA is cleaved at gene junctions, thanks to
oligonucleolytic enzymes that recognize the secondary
structure of tRNA genes which are interspersed between
PCGs. Enzymes responsible for the endonucleolytic
cleavage at the 50- and 30-ends of tRNA within the poly-
cistronic transcripts (RNase P and RNase Z, respectively)
have been identified (Rossmanith 2012). Nonetheless, the
lack of intervening tRNA genes between some genes fre-
quently leads to the synthesis of bicistronic units that are
alternatively translated. In addition, when a truncated stop
codon is present, proteins encoded in the nucleus are
necessary to synthesize enzymes that restore termination
codon functionality. This latter process is believed to be
accomplished through two fundamental steps: (1) a yet
unknown enzyme restores termination codons via oligoa-
denylation; (2) a poly A polymerase (mtPAP) polyadeny-
lates with about 50 nt the 30 terminus of the mRNA.
Polar Biol
123

Additionally, several enzymes, such as the bicoid stability
factor (BSF) of Drosophila and homologs of the mam-
malian RNA-binding protein (SLIRP), are also believed to
contribute to the stabilization of polyadenylated mRNAs
(Sterky et al. 2010; Bratic et al. 2011).
Extreme environmental conditions, as those experienced
by Antarctic organisms, are generally associated with the
occurrence of peculiar physiological, as well as morpho-
logical, features. Polar gigantism and peculiar pigmenta-
tion patterns are only some of these morphological features
exhibited by marine and terrestrial invertebrates as an
adaptation to the environmental conditions of the Antarctic
Continent (Chapelle and Peck 1999). Considering that the
key feature of Antarctic environment is the extremely low
temperature, and that the mitochondrion is the central
power plant of the cell, there has been quite a lot of
speculation that modifications in the respiratory chain may
arise that divert part of the energy accumulated in the
respiratory chain from ATP to heat production. Neverthe-
less, neither the species here under study, nor the other
pycnogonid Antarctic species (N. unguiculatum), seem to
display molecular features of the mitochondrial genome
and/or of the genes encoding for the respiratory chain that
may be regarded as an adaptation to cold environment.
It is generally thought that large-bodied organisms of
high-latitude marine species (polar gigantism) are mostly
due to high oxygen availability combined with low meta-
bolic rates (Chapelle and Peck 1999; Woods and Moran
2008), but recent works on Antarctic pycnogonids seem to
reject, at least in this group, the oxygen hypothesis (Woods
et al. 2009). Oxygen uptake is a fundamental process of
metazoan taxa that play a key role in energy production
(oxidative phosphorylation) in the mitochondrion inner
membrane. Higher eukaryotic aerobic organisms rely on
this mechanism to generate ATP, transferring electrons to
oxygen by the mitochondrial electron transport chain to
produce water. Reactive intermediates (ROS), which are
very deleterious for the cell because they induce damages
to the milieu and can cause mutations of the DNA, are
however generated during this process (Giustarini et al.
2009). Higher levels of oxygen may therefore increase the
frequency of mutations of mt- and, to a lesser extent,
nu-DNAs. Comparison of pycnogonid mtDNAs does not
provide clear evidence for higher frequencies of mutational
incidence occurring either in the two Antarctic species of
this group (A. carolinensis and N. unguiculatum), or in the
Sub-Antarctic one (C. megalonyx). Accelerated rates in
nucleotide substitution with respect to L. polyphemus (a
basal chelicerate species) and other arthropod groups seem
to be a common molecular feature shared by most cheli-
cerates that leads to long branches in phylogenetic com-
parisons (Podsiadlowski and Braband 2006). This process
is probably dating as early as the initial stages of
diversification of the whole group, rather than being a
consequence of a higher mutational frequency limited to a
specific subgroup.
Acknowledgments This work was funded by the Italian Program of
Research in Antarctica (PNRA). The logistic support of PNRA during
the collection of the material is acknowledged. Partial support was
also provided by the University of Siena. We also thank Dr. Laura
Bianciardi for her collaboration with the analysis of data.
References
Boore JL (2000) The duplication/random loss model for gene
rearrangement exemplified by mitochondrial genomes of deu-
terostome animals. In: Sankoff D, Nadeau J (eds) Comparative
genomics, computational biology (Series vol 1). Kluwer,
Dordrecht, pp 133–147
Boore JL (2006) The complete sequence of the mitochondrial genome
of Nautilus macromphalus (Mollusca: Cephalopoda). BMC
Genomics 7:182
Boore JL, Lavrov DV, Brown WM (1998) Gene translocation links
insects and crustaceans. Nature 392:667–668
Bradshaw PC, Rathi A, Samuels DC (2005) Mitochondrial-encoded
membrane protein transcripts are pyrimidine-rich while soluble
protein transcripts and ribosomal RNA are purine-rich. BMC
Genomics 6:136
Bratic A, Wredenberg A, Gronke S, Stewart JB, Mourier A,
Ruzzenente B, Kukat C et al (2011) The bicoid stability factor
controls polyadenylation and expression of specific mitochon-
drial mRNAs in Drosophila melanogaster. PLoS Genet
7(10):e1002324
Budd GE, Telford MJ (2005) Evolution: along came a sea spider.
Nature 437:1099–1102
Carapelli A, Vannini L, Nardi F, Boore JL, Beani L, Dallai R, Frati F
(2006) The mitochondrial genome of the entomophagous endopar-
asite Xenos vesparum (Insecta: Strepsiptera). Gene 376:248–259
Carapelli A, Lio P, Nardi F, van der Wath E, Frati F (2007)
Phylogenetic analysis of mitochondrial protein coding genes
confirms the reciprocal paraphyly of Hexapoda and Crustacea.
BMC Evol Biol 7(Suppl 2):S8
Carapelli A, Comandi S, Convey P, Nardi F, Frati F (2008) The
complete mitochondrial genome of the Antarctic springtail
Cryptopygus antarcticus (Hexapoda: Collembola). BMC
Genomics 9:315
Castellana S, Vicario S, Saccone C (2011) Evolutionary patterns of
the mitochondrial genome in Metazoa: exploring the role of
mutation and selection in mitochondrial protein coding genes.
Genome Biol Evol 3:1067–1079
Chapelle G, Peck LS (1999) Polar gigantism dictated by oxygen
availability. Nature 399:114–115
Chiusano ML, Alvarez-Valin F, Di Giulio M, D’Onofrio G,
Ammirato G, Colonna G, Bernardi G (2000) Second codon
positions of genes and the secondary structures of proteins.
Relationships and implications for the origin of the genetic code.
Gene 261:63–69
Clayton DA (1982) Replication of animal mitochondrial DNA. Cell
28:693–705
de Rijk P, de Wachter R (1997) RnaViz, a program for the
visualisation of RNA secondary structure. Nucleic Acids Res
25:4679–4684
Dietz L, Mayer C, Arango CP, Leese F (2011) The mitochondrial
genome of Colossendeis megalonyx supports a basal position of
Polar Biol
123

Colossendeidae within the Pycnogonida. Mol Phylogenet Evol
58:553–558
Dunlop JA, Arango CP (2005) Pycnogonid affinities: a review. J Zool
Syst Evol Res 43:8–21
Edgecombe GD (2010) Arthropod phylogeny: an overview from the
perspectives of morphology, molecular data and the fossil
record. Arthropod Struct Dev 39:74–87
Faith JJ, Pollock DD (2003) Likelihood analysis of asymmetrical
mutation bias gradients in vertebrate mitochondrial genomes.
Genetics 165:735–745
Frank AC, Lobry JR (1999) Asymmetric substitution patterns: a
review of possible underlying mutational or selective mecha-
nisms. Gene 238:65–77
Giribet G, Edgecombe GD (2012) Reevaluating the arthropod tree of
life. Annu Rev Entomol 57:167–186
Giribet G, Edgecombe GD, Wheeler WC (2001) Arthropod phylog-
eny based on eight molecular loci and morphology. Nature
413:157–161
Gissi C, Reyes A, Pesole G, Saccone C (2000) Lineage-specific
evolutionary rate in mammalian mtDNA. Mol Biol Evol
17:1022–1031
Gissi C, Iannelli F, Pesole G (2008) Evolution of the mitochondrial
genome of Metazoa as exemplified by comparison of congeneric
species. Heredity 101:301–320
Giustarini D, Dalle-Donne I, Tsikas D, Rossi R (2009) Oxidative
stress and human diseases: origin, link, measurement, mecha-
nisms, and biomarkers. Crit Rev Clin Lab Sci 46:241–281
Hassanin A (2006) Phylogeny of Arthropoda inferred from mito-
chondrial sequences: strategies for limiting the misleading
effects of multiple changes in pattern and rates of substitution.
Mol Phylogenet Evol 38:100–106
Hassanin A, Leger N, Deutsch J (2005) Evidence for multiple
reversals of asymmetric mutational constraints during the
evolution of the mitochondrial genome of Metazoa, and
consequences for phylogenetic inferences. Syst Biol 54:277–298
Juhling F, Putz J, Bernt M, Donath A, Middendorf M, Florentz C,
Stadler PF (2012) Improved systematic tRNA gene annotation
allows new insights into the evolution of mitochondrial tRNA
structures and into the mechanisms of mitochondrial genome
rearrangements. Nucleic Acids Res 40:2833–2845
Kolpakov R, Bana G, Kucherov G (2003) mreps: efficient and flexible
detection of tandem repeats in DNA. Nucleic Acids Res
31:3672–3678
Krzywinski J, Grushko OG, Besansky NJ (2006) Analysis of the
complete mitochondrial DNA from Anopheles funestus: an
improved dipteran mitochondrial genome annotation and a
temporal dimension of mosquito evolution. Mol Phylogenet
Evol 39:417–423
Laslett D, Canback B (2008) ARWEN, a program to detect tRNA
genes in metazoan mitochondrial nucleotide sequences. Bioin-
formatics 24:172–175
Lavrov DV (2011) Key transitions in animal evolution: a mitochon-
drial DNA perspective. In: Schierwater B, DeSalle R (eds) Key
transitions in animals evolution. Science Publishers & CRC
Press, Enfield, New Hampshire, pp 35–54
Lavrov DV, Boore JL, Brown WM (2000) The complete mitochon-
drial DNA sequence of the horseshoe crab Limulus polyphemus.
Mol Biol Evol 17:813–824
Lavrov DV, Boore JL, Brown WM (2002) Complete mtDNA
sequences of two millipedes suggest a new model for mitochon-
drial gene rearrangements: duplication and non-random loss.
Mol Biol Evol 19:163–169
Li H, Liu H, Shi A, Stys P, Zhou X, Cai W (2012) The complete
mitochondrial genome and novel gene arrangement of the
unique-headed bug Stenopirates sp. (Hemiptera: Enicocephali-
dae). PLoS ONE 7(1):e29419
Margam VM, Coates BS, Hellmich RL, Agunbiade T, Seufferheld
MJ, Sun W, Ba MN et al (2011) Mitochondrial genome sequence
and expression profiling for the legume pod borer Maruca vitrata(Lepidoptera: Crambidae). PLoS ONE 6(2):e16444
Masta SE, Boore JL (2008) Parallel evolution of truncated transfer
RNA genes in arachnid mitochondrial genomes. Mol Biol Evol
25:949–959
Masta SE, McCall A, Longhorn SJ (2010) Rare genomic changes and
mitochondrial sequences provide independent support for con-
gruent relationships among the sea spiders (Arthropoda, Pycno-
gonida). Mol Phylogenet Evol 57:59–70
Maxmen A, Browne WE, Martindale MQ, Giribet G (2005)
Neuroanatomy of sea spiders implies an appendicular origin of
the protocerebral segment. Nature 437:1144–1148
Min XJ, Hickey DA (2007) DNA barcodes provide a quick preview of
mitochondrial genome composition. PLoS ONE 2(3):e325
Nardi F, Carapelli A, Fanciulli PP, Dallai R, Frati F (2001) The
complete mitochondrial DNA sequence of the basal hexapod
Tetrodontophora bielanensis: evidence for heteroplasmy and
tRNA translocations. Mol Biol Evol 18:1293–1304
Nardi F, Carapelli A, Frati F (2012a) Internal consistency as a method
to assess the quality of dating estimates using multiple markers.
Mol Phylogenet Evol 62:874–879
Nardi F, Carapelli A, Frati F (2012b) Repeated regions in mitochon-
drial genomes: distribution, origin and evolutionary significance.
Mitochondrion 12:483–491
Naylor GJ, Collins TM, Brown WM (1995) Hydrophobicity and
phylogeny. Nature 373:565–566
Ojala D, Montoya J, Attardi G (1981) tRNA punctuation model of
RNA processing in human mitochondria. Nature 290:470–474Park SJ, Lee YS, Hwang UW (2007) The complete mitochondrial
genome of the sea spider Achelia bituberculata (Pycnogonida,
Ammotheidae): arthropod ground pattern of gene arrangement.
BMC Genomics 8:343
Passamonti M, Ricci A, Milani L, Ghiselli F (2011) Mitochondrial
genomes and Doubly Uniparental Inheritance: new insights from
Musculista senhousia sex-linked mitochondrial DNAs (Bivalvia
Mytilidae). BMC Genomics 12:442
Peden JF (1999) Analysis of codon usage. Dissertation. University of
Nottingham, UK
Perna NT, Kocher TD (1995) Patterns of nucleotide composition at
fourfold degenerate sites of animal mitochondrial genomes.
J Mol Evol 41:353–358
Podsiadlowski L, Braband A (2006) The complete mitochondrial
genome of the sea spider Nymphon gracile (Arthropoda:
Pycnogonida). BMC Genomics 7:284
Podsiadlowski L, Carapelli A, Nardi F, Dallai R, Koch M, Boore JL,
Frati F (2006) The mitochondrial genomes of Campodea fragilisand Campodea lubbocki (Hexapoda: Diplura): high genetic
divergence in a morphologically uniform taxon. Gene 381:49–61
Regier JC, Shultz JW, Zwick A, Hussey A, Ball B, Wetzer R, Martin
JW et al (2010) Arthropod relationships revealed by phyloge-
nomic analysis of nuclear protein-coding sequences. Nature
463:1079–1083
Rehm P, Borner J, Meusemann K, von Reumont BM, Simon S, Hadrys
H, Misof B et al (2011) Dating the arthropod tree based on large-
scale transcriptome data. Mol Phylogenet Evol 61:880–887
Reyes A, Gissi C, Pesole G, Saccone C (1998) Asymmetrical
directional mutation pressure in the mitochondrial genome of
mammals. Mol Biol Evol 15:957–966
Rossmanith W (2012) Of P and Z: mitochondrial tRNA processing
enzymes. Biochim Biophys Acta 1819:1017–1026
Rota-Stabelli O, Kayal E, Gleeson D, Daub J, Boore JL, Telford MJ,
Pisani D et al (2010) Ecdysozoan mitogenomics: evidence for a
common origin of the legged invertebrates, the Panarthropoda.
Genome Biol Evol 2:425–440
Polar Biol
123

Saito S, Tamura K, Aotsuka T (2005) Replication origin of
mitochondrial DNA in insects. Genetics 171:1695–1705
San Mauro D, Gower DJ, Zardoya R, Wilkinson M (2006) A hotspot
of gene order rearrangement by tandem duplication and random
loss in the vertebrate mitochondrial genome. Mol Biol Evol
23:227–234
Segovia R, Pett W, Trewick S, Lavrov DV (2011) Extensive and
evolutionarily persistent mitochondrial tRNA editing in the
velvet worms (Phylum Onychophora). Mol Biol Evol 28:2873–
2881
Simon C, Frati F, Beckenbach A, Crespi B, Liu H, Flook P (1994)
Evolution, weighting, and phylogenetic utility of mitochondrial
gene sequences and a compilation of conserved polymerase
chain reaction primers. Ann Entomol Soc Am 87:651–701
Sterky FH, Ruzzenente B, Gustafsson CM, Samuelsson T, Larsson
NG (2010) LRPPRC is a mitochondrial matrix protein that is
conserved in metazoans. Biochem Biophys Res Commun
398:759–764
Swofford DL (2002) PAUP*: phylogenetic analysis using parsimony
(* and other methods), version 4.0. Sinauer Associates,
Sunderland
Talavera G, Vila R (2011) What is the phylogenetic signal limit from
mitogenomes? The reconciliation between mitochondrial and
nuclear data in the Insecta class phylogeny. BMC Evol Biol
11:315
Waloszek D, Dunlop JA (2002) A larval sea spider (Arthropoda:
Pycnogonida) from the Upper Cambrian ‘Orsten’ of Sweden, and
the phylogenetic position of pycnogonids. Palaeontology
45:421–446
Wei SJ, Shi M, Chen XX, Sharkey MJ, van Achterberg C, Gong-Yin
Y, Jun-Hua H (2010) New views on strand asymmetry in insect
mitochondrial genomes. PLoS ONE 5(9):e12708
Wolstenholme DR (1992) Animal mitochondrial DNA: structure and
evolution. Int Rev Cytol 141:173–216
Woods HA, Moran AL (2008) Temperature–oxygen interactions in
Antarctic nudibranch egg masses. J Exp Biol 211:798–804
Woods HA, Moran AL, Arango CP, Mullen L, Shields C (2009)
Oxygen hypothesis of polar gigantism not supported by perfor-
mance of Antarctic pycnogonids in hypoxia. Proc R Soc B
276:1069–1075
Yokobori S, Paabo S (1995) Transfer RNA editing in land snail
mitochondria. Proc Natl Acad Sci USA 92:10432–10435
Zuker M (2003) Mfold web server for nucleic acid folding and
hybridization prediction. Nucleic Acids Res 31:3406–3415
Polar Biol
123
Copyright © 2022 FDOKUMEN




















