
THE ATOMIC WIGHT OF RUBIDIUM THROUGH ...
99
THE ATOMIC WIGHT OF RUBIDIUM THROUGH DETERMINATIONS OF THE RUBIDIUM CHLORIDE—S1IATER RATIO BY THE STANDARD SOIUTION METHOD A Thesis submitted to The Department of Chemistry in partial fulfillment of The Degree of Master of Arts by Norman William Frederick Phillips 0 The University of British Columbia October 1935
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of THE ATOMIC WIGHT OF RUBIDIUM THROUGH ...

THE ATOMIC WIGHT OF RUBIDIUM THROUGH DETERMINATIONS
OF THE RUBIDIUM CHLORIDE—S1IATER RATIO
BY THE STANDARD SOIUTION METHOD
A Thesis
submitted to
The Department of Chemistry
i n p a r t i a l f u l f i l l m e n t of
The Degree of Master of Arts
by
Norman William Frederick P h i l l i p s
0
The University of B r i t i s h Columbia
October 1935

Acknowledgements
The author wishes to take this opportunity to
express his thanks to the following: to Dr. E. H. Archibald
for kindly advice and d i r e c t i o n , and to his friend J. Gilbert
Hooley for general collaboration, without whose services
th i s investigation would have been impossible; to Dr.
R.H. Clark, head of the Department of Chemistry, f o r
general cooperation; to Prof. G.P. Baxter for advice on
examination of a l k a l i metals i n the copper az?cj to Dr.
Arthur E. Scott for a copy of one of his papers prior to
publication; to Dr. C.R. Johnson for many reprints of
his published work; to Messrs. E.A. DSLisle, J. Hooley,
Eraser and Eraser, J. Eullerton^ J. Fyfe, H. McMahon, Dr.
M.J. Marshall, Mr. W.E. P h i l l i p s , Dr. W.F. Seyer and Dr.
W. Ure, for assistance i n construction and advice on the
design of apparatus; to Dr. H. Warren and Prof. G i l l i e s
for assistance in cutting quartz and porcelain; and to
Miss M. Perry for assistance i n the typing of this manuscript.

THE ATOMIC WEIGHT OF RUBIDIUM THROUGH DETERMINATIONS
OF THE RUBIDIUM CHLORIDE—SILYER EAT10
BY THE STANDARD SOLUTION METHOD
CONTENTS page
1. Introduction 1
2. Review of e a r l i e r investigations 3
3. P u r i f i c a t i o n of reagents 4
4*. The choice of the method of analysis 38
5. Balance, weights and weighings • 46
6. The nephelometer 48
7. The standard solutions 49
8. The analysis 52
9. The results of the analysis of rubidium chloride 59
10. The analysis of potassium chloride 63
11. Recalculation of the atomic weight of rubidium 65
from Archibald's data
12. The n e u t r a l i t y of fused rubidium chloride 69
13. Discussion of results 71
14. Bibliography 80

LIST OF FIGURES
facing page 1. Apparatus for the sublimation of phosphoric 11
oxide
£* The e l e c t r i c muffle furnace 18
3 . D e t a i l of element for e l e c t r i c muffle 19
4 . Apparatus f o r the generation of pure hydrogen 21
and the fusion of s i l v e r
5. The hydrogen generator 22
6. Filament for removal of oxygen 23
7 . R e c r y s t a l l i z a t i o n scheme fo r rubidium 29
dichloriodide
8. Centrifugal apparatus 30
9 . R e c r y s t a l l i z a t i o n scheme for rubidium 32
hydrogen tartrate
10. S i l v e r dissolving apparatus ' 52
11 . Apparatus for the fusion of rubidium 53
chloride i n an atmosphere of nitrogen
13 , Precipitant delivering device 5S

THE ATOMIC WEIGHT OF RUBIDIUM THROUGH DETERMINATIONS
OF THE RUBIDIUM CHLORIDE SILVER RATIO
BY THE STANDARD SOLUTION METHOD
She l a s t three decades have seen great advances i n the
technique of the determination of atomic weights. These
developments have centred c h i e f l y around the physical methods
as advanced by F.W. Aston and K.T. Bainbridge, and the chem
i c a l methods greatly improved by T.W, Richards, G.P. Baxter,
and 0. HBnigschmid with their many collaborators, and more
recently by C.R. Johnson. Many changes i n the values of these
constants have been thus effected, a l l but a dozen or so of
the 85 atomic weight values now appearing i n the International
Table having undergone correction since 1910. Recent years
have seen excellent c o r r e l a t i o n between atomic weights deter
mined by the physical and by the chemical methods.
Previous to the present research the atomic weight
of rubidium had not been determined by a chemical method for
nearly t h i r t y years. Since this constant had not been checked
by the mass spectrograph with the utmost precision i t was
thought that confirmation of the International value by the
most precise chemical methods available today would be advis
able.
Very l u c k i l y Dr. E.H. Archibald had on hand about
200 grams of rubidium s a l t s purchased at a time when the cost
of this material was not so prohibitive as at present. Due to
these reasons, combined with the prospect of the tr a i n i n g to

(2)
be acquired i n precise chemical measurement, the author was
able to accept with a l a c r i t y the opportunity offered to
determine anew the atomic weight of rubidium.

(3)
REVIEW OF EARLIER INVESTIGATIONS
Previous work on the atomic weight of rubidium has 1 2 "been completely reviewed by Archibald and by Clarke so i t
w i l l niat be necessary to do likewise here. A l l investigations
prior to that of Archibald embodied very i n f e r i o r a n a l y t i c a l
methods as well as s l i g h t l y questionable methods of p u r i f i c a
tion of materials. The most recent investigation p r i o r to
the present one, that of E.H, Archibald i n 1904, was
moderately extensive. P u r i f i c a t i o n was accomplished by
r e c r y s t a l l i z a t i o n of the di#chloriodide, followed either by
r e c r y s t a l l i z a t i o n of the hydrogen t a r t r a t e or by p r e c i p i t a t i o n
of the chloride. By spectroscopic observations the former
means was shown to be an e f f i c i e n t method for the elimination
of potassium and the l a t t e r methods likewise for caesium.
However, i n the l i g h t of present day research the a n a l y t i c a l
method may have been s l i g h t l y at f a u l t . This will, be discuss
ed subsequently, (see page ̂ 4) f . )
1. Trans. Ghem. S o c , 85, 777 (1904) 2. Nat. Acad. S c i . , Vol. 16, III mem. p. 120 (1922) 3. l o c . c i t . , pages 776-790

(4)
PURIFICATION OF REAGENTS
For the p u r i f i c a t i o n of the majority of the reagents
used i n thi s work much valuable information was drawn from
'Jp re par at ion of Pure Inorganic Substances"'''. However, f o r
the sake of c l a r i t y , and also to d e t a i l any technical or other
variations used i n these preparations, a f a i r l y complete out
l i n e of the methpds used w i l l be given. Since the methods
discussed i n the aforementioned work were followed wholly or
p a r t i a l l y i n the majority of p u r i f i c a t i o n s , reference to i t
w i l l not be made i n each s p e c i f i c case, but general acknow
ledgement i s made here.
The designation PI or PII af t e r a reagent s i g n i f i e s :
(1) (when such designation appears i n a t i t l e ) that the reagent
i s i n a state of purity as can be attained i n the manner
indicated below said t i t l e ;
(2) (when such designation appears i n the body of the a r t i c l e )
that the reagent i s i n a state of purity as can be attained
by the method given under the t i t l e containing t h i s designation,
A l l the water used i n t h i s research was water PI.
Owing to the frequent use of t h i s reagent i t w i l l always be
referred to merely as "water". The spe c i a l connotation how
ever, should not be forgotten.
Concerning general precautions, r i g i d l y observed i n
1. E.H, Archibald: "The Preparation of Pure Inorganic Substances", John Wiley and Sons, Inc., New York, 1932.

(5)
the present research, a few quotations from the work of 1
Eiohards and Willard are very applicable to the case at hand,
" A l l the s o l i d , l i q u i d and gaseous materials used i n
"this research were pur i f i e d with greatest care. The
"most insidious sources of impurity i n work of this kind
''are dust and the various gases sometimes contained i n the
''air of the laboratory, and the most e f f i c i e n t mehtods of
''purification may S a i l to give a pure product unless
"careful attention i s given to t h i s f a c t . " "The a i r of the room was, therefore, kept as pure as
"possible."
"Vessels containing pure material were always kept
"covered and under b e l l jars when not i n use. When i t
"was necessary to work under the hood, a large clean
"glass plate was suspended above the apparatus, to protect
''it. from p a r t i c l e s of dust f a l l i n g from above. A l l a the •
"heating was conducted e l e c t r i c a l l y i n order to avoid the
"deletrious effects of the products of combustion."
"The value of e l e c t r i c a l heating, both as to cleanliness "and wide range of application, was emphasized i n the ''present research."
1. J. Am. Ghem. S o c , 32, 4 (1910) a. i n a few eases i n the e a r l i e r part of the present work
gas was resorted to.

(6)
Water PI
The water used i n the p u r i f i c a t i o n and the analysis
was obtained by d i s t i l l i n g the ordinary d i s t i l l e d water of 1
the laboratory i n a special s t i l l , from a solution 0.001
molar i n potassium permanganate and 0.005 molar i n sodium
hydroxide, the f i r s t 10$ to 15$ of the d i s t i l l a t e being discarded. The water thus prepared had a s p e c i f i c conductivity
_c _n _ i of about 1 x 10 ohm cm. , and was nephelometrieally free
from chlorides. By evaporating large quantities of the water
i n a platinum dish, i t was found that the water contained not
more than 0.34 p.p.m. non—volatile matter at 120° and not
more than 0.14 p.p.m. non—volatile matter at 500°.
Hydrochloric Acid PI
Baker's 0..P. hydrochloric a c i d was diluted with
water to a concentration of 22$ 2. To each l i t e r of t h i s
mixture 2 gm. of potassium permanganate were added. It was
then d i s t i l l e d from an a l l Pyrex s t i l l , heated either by
gas or by e l e c t r i c i t y . The f i r s t 20$ of the d i s t i l l a t e was
discarded as was the la s t 20$. The middle f r a c t i o n was
collected i n a Pyrex f l a s k and was stored i n a Winchester,
which had for months pre¥ious contained concentrated hydrochlor
ic acid. Any impurities a r i s i n g from storage i n a bottle of this
nature would be soluble nonr-volatile ones. Such would i n t r o
duce no additional impurities i n the procedures i n which th i s
acid was employed. 1, Hooley and Phillips.} J. Chem. Ed., 11, 376 (1934) 2. Scott and Johnson| J. Pays. Chem., 33, 1979 (1929)

(V)
Hydrochloric Acid P H Hydrochloric acid PI was d i s t i l l e d i n an e l e c t r i c a l l y
heated fused s i l i c a r e t o r t . As "before, the middle f r a c t i o n
(60%) only was collected for use; this time however, the acad
was d i s t i l l e d just before using and the d i s t i l l a t e received i n
a fused s i l i c a vessel.
N i t r i c Acid PI
Baker's C.P. n i t r i c acid was d i s t i l l e d i n an
e l e c t r i c a l l y heated a l l Pyrex s t i l l . The f i r s t 20% d i s t i l l -
over was discarded as was the last 20% of the o r i g i n a l
amount taken. The d i s t i l l a t e was caught i n a Pyrex f l a s k
and was stored i n a Winchester which had f o r months previous
contained n i t r i c acid. The remarks i n the section hydrochloric
acid PI, with reference to impurities from this source, also
apply here. This acid was found to be nephelometrically free
from chlorides.
N i t r i c Acid PII
N i t r i c Acid PI was r e d i s t i l l e d i n an e l e c t r i c a l l y
heated fused s i l i c a retort shortly before use. The i n i t i a l
(20%) and the f i n a l (20%) fractions were discarded. The d i s
t i l l a t e was caught i n a Pyrex f l a s k and stored i n a stoppered
f l a s k of the same material. This acid v/as also free from chlorides
Sulphuric Acid PI
Baker's C P . sulphuric acid was d i s t i l l e d i n a Pyrex
retort heated by a meker burner. The f i r s t 20% of the d i s t i l l -
was discarded and a residue amounting to 10% of the t o t a l

(8)
quantity taken allowed to remain i n the r e t o r t . Small glass
tubes 0.1 cm. or less i n diameter and about 15 cm. long were found
to be conducive to even b o i l i n g of the l i q u i d . The acid was
received and stored as i n hydrochloric acid PI and the remarks
apertaining to the impurities introduced i n this- case also
apply here.
Sulphuric Acid PII
Sulphuric acid PI was d i s t i l l e d immediately before
use i n an e l e c t r i c a l l y heated fused s i l i c a r e t o r t , the f i r s t
and l a s t portions of the d i s t i l l a t e being discarded as i n the
preparation of sulphuric a c i d PI. The d i s t i l l a t e was c o l l e c t
ed i n a fused s i l i c a vessel.
No test for selenium or arsenic could be obtained with either acid PI or PII. Ammonium Hydroxide PI
1.5 l i t e r s of Baker's C P . ammonium hydroxide were
placed i n a Pyrex retort and 0*6 l i t e r s of water i n a receiving
f l a s k of the same material. The retort was then heeded u n t i l
about one l i t e r of the l i q u i d remained i n the r e t o r t . This
yielded a product of strength about 14$. It was d i s t i l l e d
just p r i o r to use and i t s non-volatile^was scarcely measurable.
Sodium Hydroxide PI
A 40$>, solution of B r i t i s h Drug Houses' A.R. sodium
hydroxide was electrolyzed for 4 hours with a current of 0.5
amperes i n a 500 cc. platinum dish. The dish acted as cathode
while platinum f o i l was used as the anode.

(9)
Although the o r i g i n a l material was shown to contain
as much as 0.007% iron and aluminum oxides, the r e s u l t i n g
product could not have contained more than 0.0001% of these
contaminants.
Formic Acid PI
Elmer and Amend C.P. 907o formic acid was d i s t i l l e d
just before use i n an e l e c t r i c a l l y heated Pyrex s t i l l . Of the
i n i t i a l quantity taken the f i r s t 20c/o and the l a s t 20% were
discarded. The acid was received i n a Pyrex f l a s k .
Ammon'fcum Carbonate Solution PI
Equal weights of water and Baker's C.P. ammonium
carbonate were placed i n a fJyrex retort and slowly heated with
small gas flame. The d i s t i l l a t e was collected i n a i?yz?ex
f l a s k and the l a s t 25% of the liquor was l e f t i n the r e t o r t .
Barium Hydroxide PI
Merck's C.P. barium hydroxide was dissolved by heat-*
ing with water. It was then immediately f i l t e r e d by suction
through a Whatman #42 f i l t e r supported by a platinum eone on
a steam funnel. The material was then r e e r y s t a l l i z e d 5 times,
i n platinum vessels. After each c r y s t a l l i z a t i o n the preeipitat
was centrifuged i n platinum Munroe crucibles f o r 10 minutes
at 2000 r.p.m. Not once a f t e r the preliminary f i l t r a t i o n did
the material come into contact with anything but platinum
ware. A 10 minute photographic exposure of the spectrum of
t h i s material when v o l a t i l i z e d i n the copper a r c a showed i t
a. see page 32.

(10)
to be free from the most minute traces of strontium, calcium,
magnesium^aluminum, iron, potassium, lithium, caesium and
sodium.
Potassium Permanganate PI'
B r i t i s h Drug Houses' A.B. potassium permanganate was
r e c r y s t a l l i z e d 3 times from water i n Pyrex beakers. Heating
was done by a bunsen burner and the crystals were ca r e f u l l y
but not c e n t r i f u g a l l y drained.
Iodine PI
Mallinkrodt's O.P. resublimed iodine was • pulverized
with 10% of i t s weight of B r i t i s h Drug Houses' A.B. potassium
iodide i n a porcelain mortar. A suitable quantity of t h i s
mixture was next placed at one end of a long Pyrex tube. It
was then d i s t i l l e d to the centre of the tube, considerable
iodine being l e f t behind with the residue of potassium
iodide. The part of the tube containing the p a r t i a l l y
p u r i f i e d product was sealed o f f . The iodine was then at
one end of a.tube about two thirds as long as the o r i g i n a l .
It was then d i s t i l l e d to the extreme end of t h i s tube, which
portion was sealed o f f . This product was thence d i s t i l l e d
into a fused s i l i c a r e t o r t , an appreciable residue being
l e f t behind aft e r t h i s , as well as the preceding d i s t i l l a t i o n s .
T a r taric Acid PI
This was prepared by the r e c r y s t a l l i z a t i o n of
Baker's O.P. t a r t a r i c a c i d . B e c r y s t a l l i z a t i o n and f r a c t i o n
ation were carried out so as to give the product a purity


(11)
equivalent t o s i x r e c r y s t a l l i z a t i o n s of primary f r a c t i o n s . A l l
evaporations were done in platinum dishes and the mixtures
s t i r r e d and the c r y s t a l s transferred with a platinum spatula.
In a l l r e c r y s t a l l i z a t i o n s except the f i r s t the crystals were
drained by centrifuging i n Munroe crucibles for 10 minutes at
1500 r.p.m. In t h i s p u r i f i c a t i o n a l l possible precautions
were taken to avoid the atmospheric and other impurities. The
room i n which the operations were performed was free from a l l
fumes, and was used, during the duration o f this p u r i f i c a t i o n ,
for no other work. Solutions were evaporated on a copper
covered e l e c t r i c hot plate 30 cm. above which was suspended
a glass plate. Solutions and crystals were always kept under
glass to avoid contamination from dust. The product when
ignited i n a platinum dish gave no weighable residue.
Phosphoric Oxide PI
The chief impurities present i n phosphoric oxide are
the lower oxides PgOg and PgO^. For p u r i f i c a t i o n -Baker's C.P.
phosphoric oxide was resublimed i n a current of oxygen.
.-Wliitaker.'s modification of Finch and Peto's method
was used. Commercial tank oxygen was dried by passage through
wash bottles of concentrated sulphuric acid PI, a tube of s t i c k
potash and a tube of unpurified phosphoric oxide. The apparatus
employed was constructed of iron pipe as shown i n figure 1.
The contrivance was f i r s t heated e l e c t r i c a l l y but this resulted
1. J. Chem. S o c , 127, 2219 (1925) 2. i b i d . , 121, 692 (1922)

(IE)
i n melting the pipe flue to a "hot spot". The p u r i f i c a t i o n was
completed "by heating the tube with gas flames. TMs. was not
ent i r e l y s a t i s f a c t o r y owing to the large amount of heat required
to raise the iron apparatus to the correct temperature. An
apparatus made of high melting point glass would no doubt be
more conveniently heated.
The product, on being tested by Whitaker's method^
was found to be free from lower oxides. To make a test a
small quantity of the p u r i f i e d material was dissolved i n
water and boiled with mercuric chloride solution. No t u r b i d i t y
resulted. This test i s said to be very s e n s i t i v e .
Invert Sugar Solution PI
#360 gm. of commercial refined cane sugar were
dissolved i n 1 l i t e r of b o i l i n g water i n a 4 l i t e r Pyrex
beaker. The solution was allowed to cool and af t e r 4 days
the supernatant l i q u i d decanted and the residue of crystals
washed with water. The sugar was then dissolved to a satur
ated solution by agitat i o n with water. The splution was
f i l t e r e d , and made Vfo to 0.5% i n hydrochloric acid PI, and
warmed to 60°, at which temperature i t was kept hr., and
then allowed to cool. The res u l t i n g pale straw coloured l i q u i d
was stored i n a 2 l i t e r Pyrex beaker.
Calcium. Oxide PI
Baker's C P . calcium n i t r a t e was r e c r y s t a l l i z e d
twice i n Pyrex vessels. To induce c r y s t a l l i z a t i o n the
1. l o c . c i t .

(IS)
solutions were innoculated below 40° with very small crystals
of calcium n i t r a t e tetrahydrate. The calcium'nitrate crystals
were dissolved i n water and from th i s solution calcium
carbonate was precipitated with ammonium carbonate solution
PI. After f i l t e r i n g and washing the precipitate was dissolved
i n n i t r i c acid PI and the res u l t i n g solution eleetrolyzed with
a current of 1.2 amperes and a potential of 2.8 volts f o r 4
hours using platinum electrodes. From t h i s s o l u t i o n calcium
carbonate was again precipitated with ammonium carbonate
solution PI. The precipitate after thorough washing was dried o
for 18 hours at 120 . The dried calcium carbonate, contained
in a fused s i l i c a dish, was heated for s i x hours at a temp
erature of over, 800°. The product was kept i n a Pyrex
beaker i n a desiccator over calcium chloride. Analysis
showed i t to be en t i r e l y free from carbonate and iron .
Calcium Nitrate PI
This was prepared i n a similar manner to calcium
oxide PI, the f i n a l p r e c i p i t a t i o n with ammonium carbonate
and the subsequent i g n i t i o n of course being omitted.
Chlorine PI
Hydrochloric acid PI or P H was dropped onto
potassium permanganate PI i n an a l l glass apparatus. The
issuing gas was washes twice with water and passed through a
glass spray b a f f l e .
Iodine Monochloride PI Chlorine PI was passed into iodine PI contained i n

(14)
a fused s i l i c a retort u n t i l the brown l i q u i d f i r s t formed
began changing to a yellow s o l i d . The brown l i q u i d was then o
d i s t i l l e d at constant temperature of 101 into a fused s i l i c a
f l a s k or beaker.
Potassium Chloride PI
The potassium chloride used was prepared from .
Baker's C P . potassium carbonate and hydrochloric acid PI.
It was then c r y s t a l l i z e d four times i n fused s i l i c a and platinum vessels, with centrifugal draining on platinum,
o dried at 150 for an hour, pulverized i n an agate mortar,
fused i n a platinum crucible i n an e l e c t r i c muffle furnace,
and again pulverized i n a similar manner.
Hydrogen PI . .
This preparation w i l l fee described i n connection
with the fusion of s i l v e r .
S i l v e r PI "Perhaps as much attention has been paid to the
1 preparation of pure s i l v e r as to any other substance" .
Needless to say this attention was continued i n the present
case. The author f e e l s that the care given to this prepar
ation was as great as i s possible to a t t a i n . Today the majority of investigators preparing pure
s i l v e r follow, with or without variations, the c l a s s i c work E
of Richards and Wells . This work, with procedures of more 1. E.H. Archibald, l o c . c i t . , page 57 2. Pub. Car. Inst. Ho. £8 Page 16 (1905)

(15)
recent investigators, has been summarized by Archibald - 1-. The
actual sequence of operations employed i n the present research 2
follows that outlined by Baxter and Ishmaru . The procedure
given by them follows closely the methods used for the l a s t IX
two decades by the Harvard Laboratories . In outline, the processes employed were: double
p r e c i p i t a t i o n of the chloride, followed i n each case by r e
duction with invert sugar solution made alk a l i n e with sodium
hydroxide, fusion of the metal on a lime boat i n a blast
flame, solution i n n i t r i c acid and repeated c r y s t a l l i z a t i o n
of s i l v e r n i t r a t e , p r e c i p i t a t i o n of the metal with ammonium
formate, fusion on pure lime i n an e l e c t r i c a l l y heated
muffle, e l e c t r o l y s i s through a concentrated s i l v e r n i t r a t e
solution made from the same s i l v e r , fusion on pure lime i n
an atmosphere of pure dry e l e c t r o l y t i c hydrogen, etching with
n i t r i c acid and drying i n vacuum at 600°.
Two samples were p u r i f i e d independently. Sample 1
was prepared from 300 gm. Eimer and Amend s i l v e r f o i l 999 f i n e .
The s t a r t i n g point for sample 2 was 400 gm. Baker's O.P.
s i l v e r n i t r a t e . Sample 1 was dissolved i n dilute n i t r i c acdd
i n a Pyrex f l a s k and the r e s u l t i n g solution decanted from a
sl i g h t residue of carbon. Sample 2 was dissolved i n water to
make a dil u t e solution, and from th i s stage onward the t r e a t
ment of both samples was si m i l a r . 1. l o c . c i t . 2. J. Am. Ghem. S o c , 51, 1730 (1929) 3. for example*. Baxter: J. Am. Ghem. S o c , 44, 577 (1922)

(16)
Using hydrochloric acid PI and Pyrex glass vessels
s i l v e r chloride was precipitated from as dilute solutions of
the s i l v e r n i t r a t e srs was mechanically convenient. A thorough
washing with water followed. About 40 l i t e r s of wash water
were used for each sample.
This s i l v e r chloride was reduced to s i l v e r by warm
ing to 60° T- 80° with a solution containing equal volumes of
10 molal sodium hydroxide PI and invert sugar solution PI.
In practice i t was found convenient to take 300 cc. of the
above solution and 150 gms. of s i l v e r chloride i n a 500 cc.
platinum dish, i n which case the reduction was found to be
complete i n about 1 hour. Completeness of reduction was
shown when the residue dissolved e n t i r e l y when warmed with
an excess of dilute n i t r i c acid.
The reduced s i l v e r was washed thoroughly with water
and then dissolved i n n i t r i c acid PI a f t e r which the precip
i t a t i o n as chloride and the reduction to metal were completed.
As before, the p r e c i p i t a t i o n was done i n Pyrex glass beakers
and the reduction i n platinum dishes.
The reduced s i l v e r , contained i n a covered Pyrex
beaker was then dried i n an e l e c t r i c a l l y heated oven for 18 o
hours at ISO . The dried powder was then fused i n the flame
of a blast lamp whose nozzle had been carefully cleaned. The
container was a dish l i n e d with calcium oxide made as follows:
A suitable quantity of calcium oxide PI was pulverized i n
a porcelain mortal with 5$ to 10$ of i t s weight of calcium

(17)
n i t r a t e PI. The mixture was then moulded i n a 150 cc.
porcelain evaporating dish, to a depth of 0.6 em., with
the aid of a s l i g h t l y smaller dish. The mould and
support were then dried at 100° f o r a few hours and
f i n a l l y heated i n a "blast flame for 1 hour.
The p e l l e t s (20 gm.—35 gm.) r e s u l t i n g from the fusion, were,
i n succession, scrubbed, f i l e d , thoroughly etched with dilute
n i t r i c acid PI, washed with ammonium hydroxide PI, and then
with water.
The next stage consisted of solution of the s i l v e r
i n n i t r i c acid PI and r e c r y s t a l l i z a t i o n of the s i l v e r n i t r a t e
formed i n Pyrex beakers. The material was r e c r y s t a l l i z e d four
times. Of course, a l l evaporation was done under glass with
e l e c t r i c a l heating. The crystals were c a r e f u l l y , bpt not
c e n t r i f u g a l l y drained.
A suitable volume of s i l v e r n i t r a t e solution from
the crystals obtained above was placed i n a platinum dish with
double i t s equivalent of formic acid PI. It was then n e u t r a l
ized with ammonium hydroxide PI whereupon reduction commenced.
The solution was kept ammoniacal and warmed towards the end of
the reduction. The l a t t e r , besides hastening the reaction,
coagulated the s i l v e r and made decantation much easier. The
reduced s i l v e r was washed with water u n t i l no test f o r ammonia
was obtained by means of Hessler's reagent. The wet powder was
then dried i n a porcelain dish for at least 6 hours at 120°
A l l the s i l v e r n i t r a t e solution was thus treated.


(18)
For the next operation a small e l e c t r i c muffle
furnace was especially constructed. .The complete directions
are included here for the convenience of other workers desir—
ing a small, clean, convenient and inexpensive muffle furnace.
The author could f i n d no similar descriptions i n the l i t e r a t u r e .
Materials
22 metres #18 gauge Ghromel A wire
7 kilograms kaolin
G.8 kilograms p u r i f i e d asbestos
1 Y i t r e o s i l muffle with exterior dimensions 8 x 8 x 18 cm.
Probable cost of materials ... $10. Probable cost of similar furmace from supply house, not less than $150.
The wire used i n the construction of this furnace i s quite
resistant to oxidation. It w i l l stand a continuous temperature
of 1000° for an i n d e f i n i t e period, and for moderate intervals
temperatures up to 1200°. Due to i t s low temperature c o e f f i c
ient of resistance, furnaces made from i t may be brought up to
temperature very r a p i d l y . In thi s respect i t s superiority
over platinum i& evident, for the furnace described attained
a temperature of over 1100° i n 50 minutes.
Figure 2 shows the general assembly.
The wire c o i l s were made by winding the wire mechan
i c a l l y on a 0.75 cm. rod i n a lathe. 10 turns per cm. were used
and the wire was fed through the tool post through an oak


(19)
block d r i l l e d with a 0.38 cm. bole. The close wound c o i l s
were sprung apart to about twice th e i r former length and
divided into pieces such that each would carry 7 amperes at
110 vo l t s i n free a i r . Two of these were required. These
were further divided into two portions.
The supporting elements for the c o i l s and the•
insulating case were conveniently prepared fnom a mixture
of kaolin and asbestos. This was conveniently made i n about
1 kilogram l o t s . 7/8 kilogram of f i n e l y powdered kaoiin were
mixed with water to form a f a i r l y s t i f f paste, then about
l/8 kilogram of a good grade of asbestos (previously moistened)
was thoroughly mixed i n . The material then had a consistency
somewhat l i k e that of p l a s t i c i n e , and could be moulded any
shape desired. After forming i t was dried slowly and baked
above 500°, Four elements, 2 — 6.5 cm wide, 2— 8 cm. wide,
of the design shown i n figure 3 were made. The insulating
and supporting case (shown i n figure 2) was i n t e r n a l l y
reinforced with iron wire.
In assembling, each pair of elements were connected i n series and fastened to the binding posts as i l l u s t r a t e d ( f i g u r e 2).
The temperature was conveniently controlled with an external resistance.
The dried s i l v e r powder obtained by the formate
reduction was transferred by means of a platinum spatula to
porcelain crucibles lined with calcium oxide PI. The prepar—

(20)
ation of the calcium oxide l i n i n g has been described (page 16).
However, i n this case the .ignition was accomplished i n the
e l e c t r i c muffle furnace instead of the bla s t . A f t e r being
completely melted i n the muffle the p e l l e t s (10 gm. - 20 gm.)
were transferred with platinum foreepfc to a Pyrex beaker,
where they were thoroughly etched with n i t r i c acid PI and
completely washed with water.
The s i l v e r buttons were next subject to e l e c t r o l y t i c
p u r i f i c a t i o n . The e l e c t r o l y t i c solution, made by dissolving
several buttons i n n i t r i c acid PI, was placed i n a fused s i l i c a
dish. The p a r t i a l l y p u r i f i e d s i l v e r was made the anode and a
pure s i l v e r c r y s t a l the cathode. Oare was taken that the
platinum lead wires did not come into contact with the e l e c t r o
l y t e since platinum electrodes cause the formation of the
compound s i l v e r peroxynitrate (AggO^'AgETOg) which distributes
i t s e l f as a dark powder on the surface of the e l e c t r o l y t e .
The e l e c t r o l y z i n g current never exceeded 0.1 ampere and was
generally about 0.08 ampere. The apparatus was covered by
a 20 em. clock glass and a b e l l — j a r , i n a special room free
from fumes and dust, which was, during the course of the
e l e c t r o l y s i s , used only for this work. The buttons^were trans
ferred to the e l e c t r o l y t i c c e l l with platinum forceps. The
deposited crystals were placed s i m i l a r l y into water contained
i n a Pyrex beaker. After a preliminary r i n s i n g the crystals
were heated to b o i l i n g i n a fused s i l i c a beaker with Z ̂ 200 cc.
portions of water. After drying the s i l v e r was kept i n a


(21)
b e l l — jar_ u n t i l required for the next. operation.
The next step was the fusion of the .crystals i n
p u r i f i e d hydrogen. To t h i s purpose, an apparatus, shown
diagramatically i n figure 4, was made. Soda—lime glass was
employed i n the construction, unless otherwise indicated. A l l
joints were sealed, with the exception of the entrance to the
fusion tube. The parts of the apparatus are as follows:
A, the hydrogen generator, i s shown i n d e t a i l i n figure
5. Considerable d i f f i c u l t y was had i n fashioning this
large piece of apparatus with the f a c i l i t i e s at hand.
Several types of construction were t r i e d , but only this
proved succesful.
Band D are Emmerling towers containing concentrated sulphuric acid P I .
(3 i s a deoxidizing filament shown i n d e t a i l i n figure 6«
The f i r s t h a l f dozen or so units cracked on heating.
However t h i s f i n a l l y modified design was highly satisfacte
E, a tube 2.4 cm. x 40 cm. containing sodium hydroxide,
f r e s h l y fused i n s i l v e r trays.
3F, a tube 2.5 cm. x 30 cm. containing phosphoric oxide PI.
G are reservoirs ( t o t a l capacity 3 l i t e r s ) f or p u r i f i e d
hydrogen.
H, a tube 2.5 cm. x 25 cm. f i l l e d as E.
I, a tube 2.5 cm. x 41 cm. f i l l e d with Baker's C.P. phosphoric oxide.
J, a transparent fused s i l i c a tube 2.7 cm. x 45 cm.


(22) connected to the rest of the apparatus by graded seals.
K, a 700 watt tube furnace made i n this laboratory
especially for this fusion. Although d i f f e r i n g i n
external form, i n many detail s of construction this
furnace resembles thafc.i described on page 18.
~" Ii» M* £. £ S and Q are stopcocks c a r e f u l l y reground and
; lubricated with a s p e c i a l low vapour pressure stopcock
grease.
R and S are closed end mano m e' t ; e rs containing as a manometrie f l u i d concentrated sulphuric a c i d .
• The pump used was a Cenco Hyvac two stage rotary
o i l pump guaranteed to proefuce a vacuum of 0.001 cm. It was
protected from water vapour by H and I. The whole-assembly
when suitably tested was found to be free from leaks. The
hydrogen generator was f i l l e d to the indicated l e v e l with a
15$ aqueous solution of Baker's C P . sodium hydroxide.
To generate hydrogen one proceeded as follows:
(1) Open I, M,-U, 0, P and Q.
(2) Start pump and evacuate to 1—2 cm.
(3) Close L, M, U, and 0 leaving P and Q open and continue the evacuation of the remainder of the apparatus u n t i l the l i m i t of the pump i s reached.
(4) Close 0 and P.
( 5) Open L to vacuum u n t i l the e l e c t r o l y t e r i s e s to the top of the antifoam tube. Close 1.
(6) Close e l e c t r o l y s i s c i r c u i t adjusting current to 1
ampere and open $ slowly. At the same time close the


(23)
c i r c u i t of the deoxidizing filament, heating the platinum to red heat.
(7) < Continue e l e o t r o l y s i s , opening 1 car e f u l l y to vacuum
when necessary to readjust the l e v e l of the e l e c t r o l y t e .
(8) When H indicates a pressure of about 80 cm. repeat ( l )
to (7) i n c l u s i v e . Generation may be continued u n t i l a
pressure up to 2 atmospheres i s reached.
The hydrogen prepared could not have contained "as, a
maximum, any more than 0.0002% of oxygen by volume.
For fusion, the e l e c t r o l y t i c crystals were t r a n s f e r r
ed with platinum forceps to porcelain boats lined with calcium
oxide PI (see page 20). The boat and contents were then
placed i n the fusion tube. This was enabled by breaking the
hydrogen delivery tube at the place indicated i n figure 4.
Thus the removal of the fused s i l i c a stopper was f a c i l i t a t e d
and the precious charge pushed into the furnace witha long
wire* The stopper was replaced and the delivery tube joint
re—fused. A l l stopcocks were closed except Q and the pump
started. The temperature of the furnace was gradually raised
to 1000°. Before the s i l v e r melted however Q was closed and
P opened for amoment u n t i l S showed a pressure of 20 cm. The
s i l v e r was kept i n a state of quiescent fusion f o r 10 minutes
and then Q was opened and the tube evacuated. A new 20 cm.
pressure atmosphere of hydrogen was admitted through P and
allowed to remain 10 minutes. Evacuation and admission of
hydrogen again followed.* a f t e r which the s i l v e r was allowed to

(24)
oool slowly i n this f i n a l atmosphere. The furnace was charged,
and recharged u n t i l a l l the crystals had received t h i s treatment.
The p e l l e t s were transferred with platinum forceps
from the calcium oxide boat to a fused s i l i c a beaker, where
they were etched with dilute n i t r i c acid PI and washed
copiously with water. After drying the pell e t s were placed
on a s i l v e r plaque and cut with a s t e e l c h i s e l whose complete
surface had just been ground and polished. By this means the
5 — 10 gram p e l l e t s were reduced to 1 — 3 gram pieces. These
were very deeply etched i n a fused s i l i c a beaker with n i t r i c
acid PI, followed by washing with b o i l i n g water (see page 21).
The well washed p e l l e t s were transferred with
platinum forceps to a fused s i l i c a boat. This was introduced
into the same tube as was used for the fusion to hydrogen,
and i n the same manner. With a l l stopcocks except Q closed,
and the pump operating, the furnace was heated to 600° and
held at about th i s temperature f o r at least 1 hour. With
the pump s t i l l operating, the s i l v e r was allowed to slowly
cool. The completely p u r i f i e d s i l v e r was f i n a l l y transferred
with platinum forceps to 35 cc. weighing bottles, where i t
was kept for use over potassium hydroxide, freshly fused i n
s i l v e r trays.
Rubidium Chloride PI
Discussion
The following are quoted from "Preparation of Pure

( E5)
Inorganic Substances": 1
"The impurities most l i k e l y to be present i n C P .
"rubidium s a l t s are compounds of lithium, sodium,
"potassium, and caesium } the companions of rubidium
" i n the periodic table."
"As rubidium stands midway between potassium and
"caesium, not only as regards i t s atomic weight,
"but also i n respect to i t s properties, including
"the s o l u b i l i t i e s of i t s compounds, i t w i l l be much
"more d i f f i c u l t to prepare a pure rubidium s a l t than
"the corresponding compound of either potassium or
"caesium. The rubidium material w i l l have to be
"converted into one s a l t for the purpose of getting
" r i d of the potassium, and into another i n order
"that the caesium may be eliminated, and i t must be
"remembered that the process by which the porportion
"of caesium i n the material i s lowered w i l l raise the
"percentage of potassium, and on the other hand, the
"treatment that w i l l leave behind any potassium w i l l
"cause the proportion of caesium to increase".
In support of this l a s t statement, after r e c r y s t a l i z i n g as
dichloriodide rubidium material which was i n i t i a l l y
spectroscopically free from caesium, Archibald was able to 2
obtain a test for t h i s l a t t e r metal . Experimental 1. loo", c i t . page 46. 2. Trans. Ghem. S o c , 85, 779 (1904)

(26)
confirmation was not given that potassium impurities are ff
concentrated i n the r e c r y s t a l l i z a t i o n of rubidium acid
t a r t r a t e . It i s not obvious that they should be. The
author does not believe that the la s t quotation cited i s
true as a general p r i n c i p l e . To mention one example to the
contrary, Richards and Honigschmid 1 found that
r e c r y s t a l l i z a t i o n of the n i t r a t e i s a good method for removing
strontium impurities from calcium material, although calcium
n i t r a t e i s more than twice as soluble as strontium n i t r a t e .
In the author 1s opinion, at the present time no absolute
rule may be l a i d down re l a t i n g s o l u b i l i t y to ef f i c a c y of
r e c r y s t a l l i z a t i o n . Each i n d i v i d u a l case must be judged
according to i t s merits and subject to experimental t e s t .
The author thinks i t quite probable that when s t a r t i n g with
rubidium material containing not more than several percent
of potassium and caesium, that p u r i f i c a t i o n might be attained
by r e c r y s t a l l i z a t i o n of a single s a l t , such as the hydrogen
t a r t r a t e , sulphate of n i t r a t e . It must be remembered, of
course, that this i s only conjecture. Experimental
v e r i f i c a t i o n i s necessary. Archibald, f o r his atomic weight determinations
2 i n 1904 p u r i f i e d h i s rubidium material by r e c r y s t a l l i z a t i o n
as the dichloriodide, followed either by repeated
precipitations with hydrogen chloride, or by r e c r y s t a l l i z a t i o n
as the hydrogen t a r t r a t e . It is possible that better and 1. . J. Am. Ohem.Soc., 32, 1577 ( 1910) 2. Trans. Ohem.Soc, 85,776 ( 1904)

(27) more e f f i c i e n t methods of p u r i f i c a t i o n exist, The works of
B a l l 1 ^ Hobinsonf Davies 3, Missender 4, Gedroiz 5, Moser and
R i e t s c h e l 6 , Ato and Wada"'', Ereundler and Menager^, and of
Strecker and Diaz on the separation of rubidium, amid the
properties of i t s compounds i n a n a l y t i c a l reactions, were
considered. These works offered no procedures, which, as
f a r as the author could ascertain, were s e l f evidently
superior to those of Archibald. Time and material were not
available to test the ef f i c a c y and s u i t a b i l i t y of these other
methods, for preparing highly pure rubidium material. There
i s no doubt however, that although more convenient processes
may be found, the methods to be outlined are capable of
producing material i n a state of purity, at least high enough
to meet the exacting demands of modern atomic weight
determination.
Experimental
The s t a r t i n g materials where C.P. rubidium s a l t s ,
c h i e f l y the carbonate, with smaller amounts of the sulphate,
chloride and bromide. A l l but a small portion of these
chemicals were obtained from the B r i t i s h Drug Houses- These
compounds were dissolved i n water i n Pyrex beakers and the i r
T. J. Ghem. S o c , 95, 2126 30 (1909) 2. J. Ind. Eng.Ghem., 10, 50 (1918) 3. J. Ghem. S o c , 123, 2976 (1923) 4. Ghem.Hews, 124, 362 (1922) 5. Chemical Analysis of the S o i l , Series A, Ho.3, 1 258 (1923)
Commissariat of Agriculture, Petrograd, Russia. 6. Mbnatsch.A46, 9 22 (1935) 7. Soi. Papers Inst.Phys.Ghem.Research, 4, 263 93 (1926) 8. Compt. Rend., 182, 1158 (1926) 9. Z. anal. Ghem., 67, 321 41 (1925)

( SO)
solutions f i l t e r e d through #42 Whatman f i l t e r papers. Into the
bromide solution contained a fused s i l i c a dish was passed,
through a delivery tube of the same material, chlorine PI.
After chlorination for an hour or two the solution was evaporated
on an e l e c t r i c hot pLate to remove free bromine. The c h l o r i n a t -
ation and evaporation were repeated u n t i l the bromide had been
completely removed. The carbonate solution was treated with
a s l i g h t exoess of hydrochloric acid PII, To the sulphate
solution was added a solution of Baker's O.P. barium chloride
u n t i l equivalence had been attatined. The mixture was f i l t e r e d
through a #42 Whatman f i l t e r paper and the f i l t r a t e treated
with a solu t i o n of Baker's C P . ammonium carbonate and
f i l t e r e d as before. The solution was evaporated and ignited
i n a platinum dish and again f i l t e r e d .
To remove a l l foreign metals but caesium r e c r y s t a l l i z
ation of the dichloriodide must be very e f f i c i e n t . The potassium
s a l t i s much more unstable than that of rubidium and is: ten
times as soluble^. Neither lithium or sodium form the t r i h a l i d e . be ,
The caesium s a l t on the other hand can^ only about 1/40 as
soluble. The author estimates that the s o l u b i l i t y of rubidium
d i c l l o r i o d i d e i s about 80 gm.-J: 100 cc. at 20°, whereas E.H.
Archibald's estimate of the s o l u b i l i t y of caesium dichloriodide 2
at room temperature i s 2 gm./ 100 cc. Obviously i f enough caesium impurity i s present i n the s a l t i t s concentration i s quite l i k e l y . 1. Trans. Ghem. Soo., 85, 779 (1904) — 2. private communication.

3 4
G 2 D 3 _ E 4
3) g _ E 3 ~ F 4
EL
E g — F 5 . G 4
E G ~ %
3
I3 *1
r 3 - — A — - v 2
3 — - £ 1
I \ 5i
The r e c r y s t a l l i z a t i o n of rubidium dichloriodide

fj^6/re 7- -See -.ores-...
i

(29)
The chloride solutions, then, were evaporated i n
fused s i l i c a beakers to a concentration of about 35% and made
0.3'"molar i n hydrochloric acid P H . A s l i g h t excess of
iodine monochloride PI was added and the r e s u l t i n g dichlor—
iodide subjected to systematic r e c r y s t a l l i z a t i o n . It is most
convenient to follow t h i s process by means of a schematic
diagram (figure 7). The l e t t e r s A , A etc. represent a unit
of material which is treated i n a s p e c i f i c manner indicated
i n the diagram, vftien a horozintal l i n e emerges from the
right hand side of a l e t t e r , and a v e r t i c a l l i n e from the,
lower side, i t i s indicated that a r e c r y s t a l l i z a t i o n has been
performed on the material. F i g u r a t i v e l y the precipitate has
been transferred i n the di r e c t i o n of the v e r t i c a l l i n e and the
mother liquor i n the di r e c t i o n of the horizontal. When a l i n e
approaches a l e t t e r from above, and a horizontal one from the
l e f t , t h i s s i g n i f i e s that the unit of material denoted by the
l e t t e r i n question i s composed of the precipitate from above
and the mother liquor from the l e f t . For example, A^ represents
the i n i t i a l dichloriodide; this i s r e c r y s t a l l i z e d , giving
c r y s t a l s to and mother liquor to Bg; B-̂ s i m i l a r l y gives
crystals to and mother liquor to Cgj B^ i s • r e c r y s t a l l i z e d
by evaporation giving crystals to Gg and mother liquor to C|p;
Gg, composed of mother liquor from B-|_ and crystals from Bg,
i s r e c r y s t a l l i z e d giving crystals to Dg and mother liquor to
Dg; and so on. The r e c r y s t a l l i z a t i o n was done i n fused s i l i c a
beakers and dishes. The solutions, owing to their marked

(30)
creeping tendency, were evaporated e n t i r e l y i n the beakers.
Pyrex hot plates, made according to the directions of
Smith and Tait 1 were found to be very clean and
convenient, and were used s o l e l y . Since the evaporation had
to be done i n the fume closet, due to the noxious fumes, a
large glass plate was supported by hollow glass rods over the
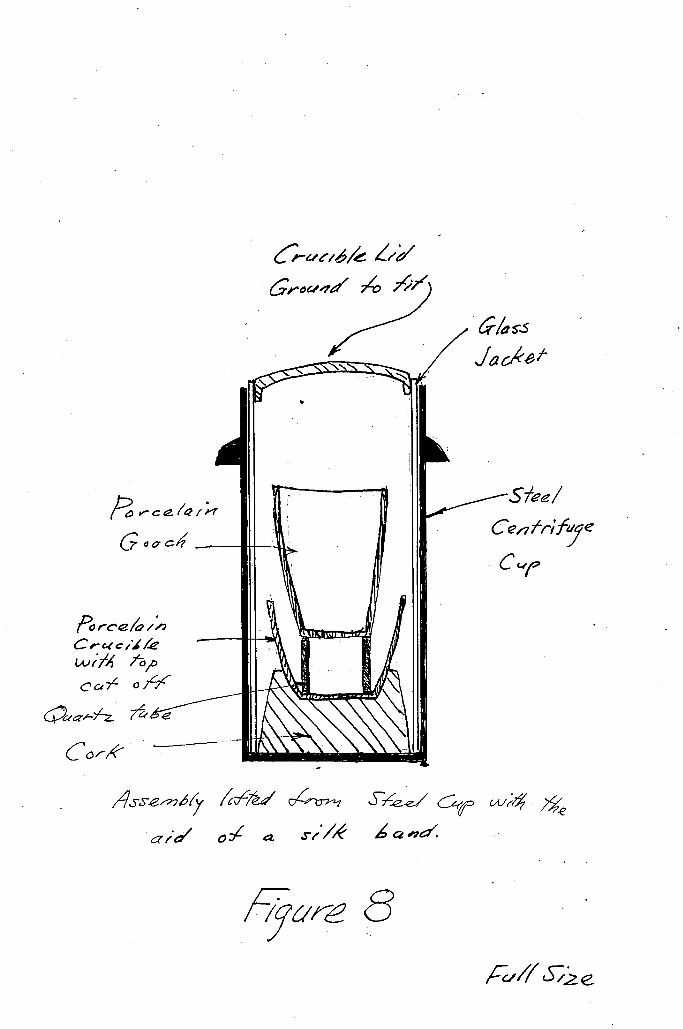
apparatus. After each r e c r y s t a l l i z a t i o n , there were 34 i n
a l l , the crystals were drained on the centrifuge at 1500 r.p.m.
for at least 10 minutes i n a s p e c i a l device which completely
prevented contamination (see figure 8), To counteract
decomposition, i t was necessary, from time to time, to make
small additions of iodine monochloride PI and hydrochloric
acid PII to the mother l i q u o r s . From the i n i t i a l 317 gm.
of dichloriodide were obtained the product K. , 1 9 , M and f> 1 ^ 3 >^ weighing 163 grams.
' ' ' - 2 3 The researches of Johnson and A l l e n , Archibald ,
Ato and Wada , and of Noyes, Halton and Williams indicate
that the hydrogen tartrate ion affords an e f f i c i e n t means of
separating rubidium and caesium compounds* The l a s t two
groups of investigators have shown, that, for p r e c i p i t a t i n g
rubidium completely, without contamination from caesium when
present i n moderate quantities, there i s no reagent superior
to the hydrogen tar t r a t e , with the possible exception of the
1. Proc. Roy. Soc. Ed i n . , 54, 88 ( 1933^-34) 2. Amer. J. S c i . and Arts, 35, ( i i ) , 94 (1863) 3. Trans..Ohem. S o c , 85, 781 (19.04) 4. S c i . Papers Inst. Pays. Ohem. Res*, 4, 263 (19 26) 5. IToyes and Bray: "A System of Qualitative Analysis for the
Rare Elements", The MaeMillan Company, New York, 1927, p.477.

(31)
6—chloro—5-nitrotoluenemetasulphonate\ The other investigators
have demonstrated the hydrogen tartrate's e f f i c a c y i n p u r i f i
cation by r e c r y s t a l l i z a t i o n . The method however i s not with
out i t s disadvantages; the low s o l u b i l i t y of the s a l t makes i t s
r e c r y s t a l l i z a t i o n somewhat inconvenient, and conversion of
other s a l t s to the hydrogen tartrate is sometimes tedious,
R e c r y s t a l l i z a t i o n of rubidium sulphate, a s a l t which is 6
tdimes as insoluble as the corresponding one of caesium and
does not possess the aforementioned f a u l t s , might be a better
procedure. However, since time and material were not available
to v e r i f y such a suggestion, i t was obvious that the only
policy was to adhere to a t r i e d and proven method.
An experiment was f i r s t performed to see i f i t were
possible to convert rubidium chloride d i r e c t l y to the hydrogen
tartrate^ by means t a r t a r i c acid. It was not. By treating
a saturated solution of rubidium chloride with a saturated
solution of t a r t a r i c acid, cooling to 0°, f i l t e r i n g , i g n i t i n g
the precipitate, treating the residue with hydrochloric acidj
evaporating and weighing, i t was found that only 35$ of the
chloride had been converted to the hydrogen t a r t r a t e . So
conversion was attained by n e u t r a l i z i n g the hydroxide with
t a r t a r i c acid.
Hence the pu r i f i e d dichloriodide was converted to
the sulphate. Fractions , Lg and were heated in a fused
s i l i c a dish on a Pyrex hot—plate u n t i l the extra halogens were
1. Davies: J. Ghem. Soc., 123, 2976 (1923)

Al : BB — — G 3
B l " " G S — D 3
°1 " D E — E 3
D x _ Eg p g
% — *2 %
P l S g
f i g u r e 9. R e c r y s t a l l i z a t i o n scheme for rubidium
hydrogen tartrate

(32)
expelled, and then evaporated with an equivalent amount of
sulphuric acid PH.. F r a c t i o n ^ ^ was converted to the sulphate
"by evaporating i t to dryness with a s l i g h t excess of sulphuric
ac i d P H . The l a t t e r i s adjudged the most convenient method.
In both cases the r e s u l t i n g residues of rubidium sulphate,
contained i n a fused s i l i c a beaker were heated to about 500°
at the mouth of an e l e c t r i c muffle (see page 18). After
cooling more sulphuric acid was added and'the heating repeated.
This treatment was continued u n t i l every trace of hydrogen
chloride was expelled. To the resmdaes dissolved i n warm
water i n a large platinum dish, a warm solution of barium
hydroxide PI was added u n t i l further ..addition gave no precipitate*
The excess barium was then precipitated with a small portion
of the rubidium sulphate solution which had been held i n
reserve. Tests with barium chloride solution, made on a
few drops of the mixture which had been f i l t e r e d through a
platinum mat, showed an excess sulphate to be present. By
centrifuging the mixture i n platinum, decanting the supernatant
l i q u i d from the prec i p i t a t e , and f i l t e r i n g through a platinum'
Munroe crucible the barium' sulphate was completely separated.
To the clear solution of carbonate and hydroxide contained in
a platinum dish was added a warm solution containing double the
equivalent of t a r t a r i c acid PI. The rubidium hydrogen
tartrate precipitate formed, was subjected to systematic
r e c r y s t a l l i z a t i o n in fused s i l i c a and platinum ware, the course
of which may be conveniently followed by r e f e r r i n g to figure 9.

(33)
Since the evaporation of the large quantities of solution
produced i n this procedure would have been somewhat slow on
the Pyrex hot—plates, t h i s was conveniently done with the
e l e c t r i c muffle furnace (see page 18) by turning i t on end and •
placing the fused s i l i c a beakers, or the platinum dishes, as
the case may have been, over the mouth of the furnace. Of
course, following each r e c r y s t a l l i z a t i o n , the cr y s t a l s ,
completely enclosed i n platinum, were centrifuged f o r at least
10 minutes at 2000 r.p.m. For further p u r i f i c a t i o n f r a c t i o n
F^ was ignited i n a platinum dish at the door of the e l e c t r i c
muffle, the residue thoroughly leached, the leachings f i l t e r e d
through a platinum mat, and the hydrogen tartscfete again
precipitated by the addition of a solution of t a r t a r i c acid
PI, followed by rigorous centrifuging. Fraction Gg, which
was very small, was not treated i n th i s manner. Fraction Gg
together with the reprecipitated f r a c t i o n F^ were then
ignited as just described. After treatment of the residue
with an excess of hydrochloric acid PH, the r e s u l t i n g solution
was f i l t e r e d through a platinum mat from a sl i g h t residue of
carbon. By slow evaporation of th i s solution i n platinum the
bulk of the chloride was separated as crystals and centrifuged
s i m i l a r l y to the hydrogen t a r t r a t e . Prior to analyses portions
of t h i s material were transferred with a platinum spatula to
a Munroe c r u c i b l e . Afew cc. of hot water were poured on the
crystals and the solution caught i n a platinum dish. After
s l i g h t evaporation the resulting crystals were centrifuged as

(54)
before, dried i n an e l e c t r i c oven.at 140° for several hoars
i n the same covered Munroe crucible in which they were
centrifuged and placed i n a desiccator over potassium hydroxide,
f r e s h l y fused i n s i l v e r trays. The product was then ready for
preliminary weighing.
The Purity of the Rubidium Chloride
That the e f f i c a c y of a c r y s t a l l i z a t i o n may be many
times increased by centrifugal drainage, has been demonstrated
by Richards"'". This method of treatment was made a spe c i a l
feature of t h i s research. Fortunately, at our disposal was
an;' :International Instrument Company's Type SB centrifuge, with
which cen t r i f u g a l v e l o c i t i e s as high as 3500 r.p.m. were
attainable. Its e f f i c i e n c y could not be improved upon.
For spectroscopic examination an Adam Hilger D~7
•Wavelength Spectrometer with metal camera was available, and
was found to be quite s a t i s f a c t o r y . Professor Gregory P.
Baxter, i n private communication, kindly gave the author some
advice on the detection of impurities. To quote from his
l e t t e r ,
" In examining s a l t s of the a l k a l i metals I use electrodes " of commercial copper about i inch i n diameter shaped l i k e "the diagram.
views of upper electrode
lower electrode the same without o
90 apart. cavity. •
1 * Chem. Soc., 27," 104 (1905)

(35)
"The wedge i s directed towards the spectroscope. Hew "electrodes are used each time,
"I use a quartz spectrograph, hut for Os, Rb and K "a glass instrument should be s a t i s f a c t o r y .
"The material to be examined i s placed om the lower
"electrode with platinum pincers before the arc i s struck,
"I use 110 v. D. G. current with enough resistance "so that the arc i s nht too intense.
"Photographic examination seems to me to be the best. "The eye i s not sensitive enough to detect traces of "impurity/
"Perhaps I should add that I use a condensing lens.
"Graphite electrodes are not satisfactory for the
" a l k a l i s because the carbon bands are located i n the regions
"to be examined. For most elements graphite serves well,
"although even the very best graphite i s not quite pure.
"Copper has only traces of impurity In my experience."
Information regarding the most sensitive l i n e s of
the impurities to be detected was obtained "Visual l i n e s for 1
Spectrum Analysis" . A ten minute exposure of a sample taken at the end
of the dichloriodide p u r i f i c a t i o n , revealed as the only
impurity a trace of sodium less than 0.001% (whose presence
could exert no measurable influence on the atomic weight); the 1. D. M. Smith, "Visual Lines for Spectrum Analysis", Adam
Hilger, Ltd;, London, 1928.

(36)
f i n a l product was found i n a similar manner to be even more gree from t h i s containmnant.
The author beleives, that, i n these two factors,
centrifugal drainage amd spectroscopic examination , th i s
p u r i f i c a t i o n i s superior to any others heretofore conducted*
He does not wish to imply, however, that previous work
yielded a productesufficiently contaminated to aff e c t a
determination of i t s equivalent weight.
Possible Modification of the Method of P u r i f i c a t i o n
Having conducted a p u r i f i c a t i o n of rubidium material
by the method outlinec/herein the author would suggest that the
following modification of the dichloriodide—tartrate method ,
wouihd be less time consuming and more convenient tharn^ the
present one, without any s a c r i f i c e of purity of the product*
The o r i g i n a l material should be f i r s t treated with sodium
hydrogen t a r t r a t e . The resultant precipitate should be
r e c r y s t a l l i z e d f o r removal of caesium. This product should
then be ignited and converted to the chloride with hydrochloric
acid, and then to the dichloriodide with iodine monochloride.
R e c r y s t a l l i z a t i o n of t h i s s a l t to remove a l l other impurities
w i l l be followed by conversion to the chloride by the usual
method.
Rubidium Nitrate PI
Fractions YgV Sg J (~ 2 and ^ g from the rubidium dichloriodide r e c r y s t a l l i z a t i o n (see figure 7 ) , which had a

(37)
purity equivalent to at least 6 r e c r y s t a l l i z a t i o n s of primary
fra c t i o n s , were evaporated i n a fused s i l i c a beaker with
n i t r i c acid P H on a Pyrex hot—plate u n t i l the solution
f a i l e d to y i e l d a nephelometric test for chloride. F i n a l l y ,
two..recrystallizations with centrifugal draining i n platinum
were followed by fusion of the product i n a platinum crucible
and desiccation over fused potassium hydroxide.

(38)
THE CHOICE OF THE METHOD, OF ANALYSIS
The work of Richards, Baxter and H5nigschmid and
thei r numerous collaborators have during the l a s t twenty —
f i v e years established the so called t i t r a t i o n method as the
outstanding procedure now i n use i n the determination of
exact atomic weights. The general method consists i n estimat
ing the stoichiometrical r a t i o between a pure compound
furnishing chlorine or bromine ions, and pure s i l v e r , with
the use of the nephelometer.
''This method was f i r s t used by Richards and Weils
"in 1905 tp determine the NaCl:Ag r a t i o , from which the atomic
"weight of sodium can be calculated, by taking suitable
"values of the antecedent atomic weights of chlorine and s i l v e i
"The method has been subsequently applied to the analysis of
"other chlorides and bromides, to determine the atomic weights
"of many elements. In these analyses, the procedure described
by Richards and Wells has been used without essential
modification, i n determining the end — point of the nephelo— TI . metric t i t r a t i o n s (the equal—opalescence end—point) -f
"At the presenttime the accepted values of a large
"number of atomic weights rest almost entire l y upon ratios .ti
determined by the nephelometric method. This i s in part due
'to the convenience and s i m p l i c i t y of the method; i t has been
preferred by investigatos who have used modern methods to IT
prepare pure compounds for atomic weight analysis^ Further— % . J. Am. Ohem. S o c , 27, 502 (1905)

( 39)
"more, the 'propable error' of atomic weights calculated from "ratios determined nephelometrically is rather generally lower "than the 'probable error' of corresponding values calculated "from ratios found by other chemical methods."
lb question regarding the general applicability of the method was made until in 1931 G.R, Johnson outlined the situation as follows. To quote:
, "Curiously enough, in view of the wide application of "the nephelometric method in atomic weight work, the general "reliance placed on 'nephelometric' results, there have been "few attempts to demonstrate the unqualified applicability of "the method in the case of particular analyses. Richards and "Wells, in a series of tests which showed the applicability of "the method to the NaCl:Ag titration, established the essential "soundness of the equal—opalescence end—point. Nevertheless, "there are certain features of this end—point which make i t "seem desirable to extend the experiments of Richards and Wells, "more particularly to titrations in which multivalent ions are "present in the analytical solutions.
"In the titrations under considerations, an acid "solution containing the chloride or bromide ions from a weigh— "ed quantity of pure compound is precipitated with almost the "theoretical amount of pure silver, weighed, and dissolved in "ni t r i c acid. The titration consists in adjusting to equality "the silver and halide ions in the resulting supernatant so—
1. C.R. Johnson: J. Phys. Ghem., 35, 540 (1931)

(40))
'lution, by the use of the standard s i l v e r and halide solutions.
'In the equal—opalescence.method*.this adjustment i s based on
'nephelometric tests, which presumably, show the r e l a t i v e
'amounts of s i l v e r and halides ions i n the solution.
"The end—point of each t i t r a t i o n , and hence the
"calculated atomic weight, depends upon the comparison of two
" c o l l o i d a l suspensions of s i l v e r chloride. These suspensions
"are formed i n equal samples of the supernatant a n a l y t i c a l
"solution, In two matched test tubes, mnder conditions as
'nearly i d e n t i c a l as possible. S i l v e r n i t r a t e i s added i n
"excess to one tube. An equivalent excess of suitable a l k a l i
"halide i s added to the other tube, so that according to the
" ' s o l u b i l i t y product" p r i n c i p l e , one suspension represents
" p r a c t i c a l l y a l l of the s i l v e r ions, the other p r a c t i c a l l y
" a l l of the halide ions present i n the supernatant a n a l y t i c a l
" l i q u i d . At the stoichiometrical point, the numbers of the
" s i l v e r and halide ions i n the supernatant l i q u i d are,
"supposedly, equal. I f , from a solution at the stoichiometrical
"point, the above procedure results i n the production of two
"suspensions possessing equal l i g h t r e f l e c t i n g power during
"the time required for the nephelometric observations, the
"desired correspondence between the end—point and the
"stoichiometrical point i s attained. In this case the opal
escences of the two tubes are equal, and the two parts of the
"divided f i e l d seen through the nephelometer eyepiece are the
"same, when the r a t i o of the exposed lengths of the tubes i s

(41)
"1.00
"In every equal-opalescence t i t r a t i o n , i n spite of
"the attempt to compare s i l v e r chloride suspensions under
"exactly s i m i l a r conditions, there must be at least one very
"marked difference i n the two s o l s . One, s t a b i l i z e d by the
"adsorption of excess 01"" ions, i s negative; the other, stab-
" i l i z e d by the adsorption of excess Ag ions is positive .
"Experiments may be cited to show that under certain special
"conditions t h i s d i s s i m i l a r i t y introduces no error into the 2
'nephelometric readings. For example, Richards and Wells
"demonstrated that the 'extra' ions present i n the a n a l y t i c a l
"solutions had no ef f e c t on the end—point of the HaCl:Ag
'.titration. Scott and Johnson tested f i v e saturated
"solutions of s i l v e r chloride containing varying amounts of
' h i t r i c acid and found no deviation from equality of s i l v e r and
"chloride ions which would effect even the most accurate
'ktomic weight analyses.
'For the present purpose, i t seems desirable to
'emphasize the fact that the ions involved i n the above mention—
'fed tests were a l l univalent. In other atomic weight deter—
'minations, 'extra' ions of the most widely.varied character,
'fai—, t r i — , and tetravalent, derived from the compounds
'undergoing analysis, have been present i n the test solutions
'examined i n the nephelometer. The effect of the adsorption of 'these ions upon the quantity, state of di v i s i o n , structure, '1. lottermosert J, prakt. Chem., (2) 72, 39 (1905); 73,374 (1906) <E. J. Am. Chem. SOc.; 27, 505 (1§05) !6. J . Pays. Chem., 33, 1981 (1929)

U s )
"colour, ana s t a b i l i t y of the sols ( i . e . upon their l i g h t
" r e f l e c t i n g power) cannot certainly be stated. However, i t
" i s known that the coagulating power of some ions i s hundreds
"of times greater than that of other ions. Furthermore, any
" g i v e n ion i s more l i k e l y to a f f e c t a s o l of opposite charge
"than one having the same charge 1. Only rarely, then would one
"expect the action of any p a r t i c u l a r ion to be equal i n the two
"nephelometer tubes, — i n most cases i t s e f f e c t upon the
"properties of the two oppositely ohagged c o l l o i d a l suspensions
"should be d i f f e r e n t .
"The good general agreement between atomic weights
"determined nephelometrlcally and corresponding values deter-'
"ed by other methods insures that the effect under consideration
"must be small. Nevertheless, one would be u n j u s t i f i e d i n
"assuming that the effect was n e g l i g i b l e . The nephelometer, as
Richards and 'Wells have often pointed out, i s an extremely
"sensitive instrument. It seems very l i k e l y that the unbal—
"anced action of c e r t a i n ions (mainly multivalent ions) may
"cause differences i n the l i g h t r e f l e c t i n g power of the sols
"large enough to produce serious constant errors i n the neph— ir 3 elometric observations." .
However, in a very thorough series of experiments
involving extra ions of diverse valence and charge, he conclu
s i v e l y • shewed that "when tests are made with the necessary
"1. Schulze: J. prakt. Chem., (2) 25, 431 (1882); 27, 320 (1883$" 2. Am.' Chem. J., 31, 239,241.242 (1904)
J* Am. Chem. Soc., 27, 486 (1905) 3. J . Phys. Chem., 35, 540 (1931)

(43)
"precautions, the accuracy claimed for the nephelometric method
"of analysis by investigators mho have used i t i n atomic weight
"work may be attained, i n so far as any effects due to the presence
"of extra compounds come into consideration"^, thus removing
the doubt which may have been f e l t regarding the accuracy of
the equal—opalescence method.
In a l a t e r a r t i c l e though, Johnson and low reported
that the effect of the. potassium ion i n concentrations as low
as 0.06 molar was so great that equal—opalescence ra t i o s of
the excess s i l v e r and excess chloride suspensions i n solutions
containing equivalent amounts of s i l v e r and chloride varied
between 1.35 and 1.65. This i s at variance with the work of 3
Scott and Hurley who report values of the equal—opalescence
r a t i o within a few percent of unity even sfc concentrations of
potassium n i t r a t e as high as 0.6 molar. This discrepancy has
not been explained.
In place of the equal—opalescence end—point Johnson
proposed what he termed the standard solution end—point. In His
own words,
"The proposed standard solution method d i f f e r s from
"the equal—opalescence method only i n the determination of the
"end—point of the reaction under investigation. That i s , one
"precipitates an acid solution containing the chloride (or
"bromide) ions from a weighed quantity of the pure compound i n 1. J. Phys. Ohem., 35,2848 (1931) 8. i b i d . , 36, 2393 (1952) 3. J. Am. Ohem. S o c , 56, 333 (1934)

(44)
'"She usual manner, using with in a few tenths of a milligram
"of the t h e o r e t i c a l amount of pure s i l v e r , weighed, and
"dissolved i n n i t r i c a c i d . At this stage, without necessarily
"making further additions to the system, one determines the
"end—point "by measuring the absolute amounts of s i l v e r and
"halide ions i n the supernatant l i q u i d . This i s done by
"comparing test portions of the supernatant l i q u i d with
"standard solutions having p r a c t i c a l l y the same composition
"as the supernatant l i q u i d i t s e l f , with the aid of the
"nephelometer .""*"
This procedure seems to have attained the ideal 2
of Richards , who l a i d great stress on the necessity of
p r e c i p i t a t i n g nephelometric suspensions under conditions as
nearly i d e n t i c a l as possible. In the words of Dr. Richards,
"In my opinion, i f even moderately accurate a n a l y t i c a l
"results are to be had with the nephelometer, the one essential un-
"point to be heeded i s t h i s : the known solutions to be
"estimated must be treated i n exactly the same way as the
"known standard solutions, which serve as the basis of com—
"paEison. If this precaution i s adhered to, the changes of
"temperature, the presence of electr o l y t e s , the concentration
"of the solutions and a l l other variables, affecting each
"precipitate i n l i k e manner, are eliminated from the compari—
"son." Certainly the effects of one of these factors, namely 1. J. Phys. Chem., 35, 830 (1931) 2, Anu Chem. J., 35, 511 (1906) .

(45)
the presence of el e c t r o l y t e s , has not u n t i l recently, been
eliminated from nephelometric atomic weight determinations,
even aril the Harvard Laboratories. Johnson's idea is one of
such s i m p l i c i t y that one wonders why i t was not thought of
previously.
Subsequently, employing t h i s method, Johnson
determined the atomic weight of sodium 1 and more recently,
of potassium 2, obtaining values which do not d i f f e r • s i g n i f i c a n t
l y from the present International values 3. Anyone reading
Johnson's s e r i e s 4 of papers cannot help but agree that his work
offers a model of thoroughness and accuracy which i s not very
often attained. There seems to be no doubt that he has advanced
the t i t r a t i o n method to a degree of precision not hitherto reached,
His results have also shown, however, that the equal—opalescence
method i s s t i l l one of quite high accuracy as well as being
one of s i m p l i c i t y .
Although thus i t has been demonstrated that, i n
practice, the equal—opalescence method is an accurate procedure,
i t was decided to use the s l i g h t l y more precise standard
solution method. 1. J. Phys. Ghem., 36, 1942 (1932); 37, 923 (1933) 2. i b i d . , 36, 2390 (1932); 39, 781 (1935) 3. Johnson obtained 22.994 and 39.100 for the atomic weights
of sodium and potassium respectively whereas the present International values are 22.997 and 39.096.
4. J. Phys. Ohem.* 33, 1921 (1929) i b i d . , 35, 540 (1931) }£}•§•» ||» 830 i l 9 3 1 j j . Am. Chem. S o c , 55, 2258 (1933) i b i d : ; 35! 2581 1931 1M4.. 55, 2262 (1933) ibid.', 36 1942 (1932, i b i d . , 36 2390 (19320) i b i d . , 37, 923 (1933). . . i b i d . 39, 781 1935 i b i d . 39, 791 (1935) .

(46)
BALANCE* WEIGHTS. AND WEIGHINGS
The balance used was a No. 10 Troemner with which
we'ighings could be duplicated to 0.02 mg. The beam was d i v i d
ed into 5 large divisions*, the f i v e mg. r i d e r used was placed
when necessary on the unit divisions of the beam and the
weight to be added or subtracted was interpolated from the
determinations of zero points with loads d i f f e r i n g by 1 mg.
These were always c a r e f u l l y checked;, on the average they
agreed to about 0.04 divisions of the-pointer scale.
Two sets of lacquered brass weights were used. One
was always used for taring; the other was c a r e f u l l y standard
ized according to the method suggested by Richards"1". Weigh
ings were always made by the methted of substitution using
counterpoises of glass or platinum similar to the objects
being weighed.
For the purpose of correcting weighings to vacuo the formula,
¥.= m 1 -4- d_ / l _ 1 \ /
L K M J where Mis the mass of the object being weighed, m the mass of corresponding weights, d t h e i r density, d the density of
w a the medium i n which the weighing i s being conducted and d g
the density of the substance being weighed, was employed. It
was thus necessary to know the densities of rubidium chloride. 1. J, Am. Chem. S o c , 22, 144 (1900)

( 4 7 )
a i r , brass weights and s i l v e r . The l a t t e r constant has been
determined many times; for the present purposes i t was taken
to be 1 0 . 4 9 grams/cc Archibald and Setterberg have
determined the s p e c i f i c g ravities of the f i r s t mentioned s a l t
obtaining the values 2 . 7 5 3 and 2 . 8 0 7 respectively. Either
value was accurate enough for the purpose at hand although
the former was used. The density of the weights was taken 3
to be 8 . 4 grams/cc. Within the narrow range of atmospheric
conditions i n which the weighings were made the density of
the dry a i r was 0 . 0 0 1 1 7 grams/cc. with a maximum error of 2 $ .
Owing to the conditions under which the rubidium chloride was
bottled no psychrometrie correction to the a i r density was
necessary to obtain the true mass of this s a l t ; i n the case
of s i l v e r the factor i — i i s so small that even large d s d w
variations i n a i r density are negligible so that the
psychrometrie correction may be omitted. Substituting the
values for the different densities i n the formula, we obtain,
within the necessary l i m i t s of accuracy.
For rubidium chloride ^ ^ Q ^ m( 1 , 0 0 0 2 8 )
For s i l v e r M A g = m( 0.99997)-
, Eor potassium chloride the density was taken to be 1 . 9 9 grams/ cc. giving, : , • •
' M E C 1 ~ m ( 1 . 0 0 0 4 6 ) 1 . Trans.Ohem.Soc, 8 5 , 7 8 5 ( 1 9 0 4 ) ~ ~ ' ~ " 2 . Oefvers. Stockh. Acad. Forh., 3 9 , 2 3 ( 1 8 8 2 ) 3 . The method of c a l i b r a t i o n of the weights assumes that they
are a l l of equal density. This, of course, i s not true, but when making weighings under atmospheric conditions corresponding to those under which the standardization of the weights was made, no error i s involved by assuming a l l the weights i n the set have a density equal to that of the standard weight (usually a 1 0 gram brass weight).

( 4 8 )
THE NEPHELOMETER
* In this type of atomic weight determination no less an important instrument than the balance is the nephelometer. To serve as such a Elett colorimeter was adapted. Eor a uniform source of parallel light a General Electric Point-o-Iite lamp was used in conjunction with a 5 in. Ramsden ocular. This is the ideal light for
1 nephelometry recommended "by Yoe . The complete apparatus was suitably housed to exclude stray light from the eyes.
1. J.H.Yoe: "Photometric Chemical Analysis, Volume II Nephelometry". John Wiley & Sons, Inc. New York 1929, Page.14,

(49) THE STANDARD SOLUTIONS
PRECIPITANTS
(a) For Silver Ion
Potassium chloride PI equivalent to 0.50000 gms of silver was weighed out and dissolved in water and diluted to 500.0 ml. in an accurately calibrated flask. It was necessary to weigh out 0.34554 gram of potassium chloride in vacuo, or 0.34539 gram in ai r . This yielded a solution containing the equivalent of 1^000 gEam/liter of silver. (b) For Chloride Ion
A pellet of silver PI weighing 1.16633 grams in vacuo was dissolved in 4 ml. of 6.4 molar n i t r i c acid PII in a special apparatus (see figure 9). It was made up with water to a total weight of 501.34 grams (in a i r ) . From this solution 285.89 grams ( in air) were removed and the remainder diluted to 500.0 ml. in an accurately calibrated flask. This yielded a solution containing silver nitrate equivalent to 1.002 gram/liter of silver. STANDARDS
These were made by accurate dilution of the precipitants plus appropriate additions of n i t r i c acid and of rubidium nitrate. The volumetric apparatus used was calibrated to give an accuracy of 1 part in 1000 or better. The solutions made up were as follows:

(50) Nephelometric Standards' (a) Silver Standard I
contained, Silver nitrate at a concentration equivalent to
0.500 mg./liter of silver N i t r i c acid PII at a concentration of 0.100 molar Rubidium nitrate PI at a concentration of 0.0250 molar
(b) Silver Standard II contained,
Silver nitrate at a concentration equivalent to 0.700 mg./liter of silver
Nitric acid PII at a concentration of 0.100 molar Rubidium nitrate PI at a concentration of 0.0250 molar
(c) Chloride Standard I contained,
Potassium chloride at a concentration equivalent to 0.500 mg./liter of silver
N i t r i c acid PII at a concentration of 0.100 molar Rubidium nitrate PI at a concentration of 0.0250 molar
( d) Chloride Standard II contained,
Potassium chloride at a concentration equivalent to 0.700 mg./liter of silver
N i t r i c acid PII at a concentration of 0.100 molar Rubidium nitrate PI at a concentration of 0.0250 molar

(51) Adjusting Standards
(a) S i l v e r Nitrate Adjusting Solution
contained,
S i l v e r n i t r a t e at a concentration equivalent to
0.100 gram/ l i t e r of s i l v e r
(b) Rubidium Chloride Adjusting Solution
contained,
Rubidium chloride at a concentration equivalent to
0.100 gram/ l i t e r of s i l v e r (on the basis off an assumed equivalent weight of
rubidium of 85.45±0.05)

FctlfSize

(52)
T H E A N A L Y S I S
As mentioned before, the analyses were to b© made by,the conventional t i t r a t i o n method,* f i r s t devised by Richards and Wells . However, i n th e i r work and subsequent investigations at the Harvard Laboratories the end point was followed by the equal opalescence method, whereas i n the present work the standard solution method of O.R.Johnson2 was employed.
Eollowing t h i s procedure a 2 or 3 gram pe l l e t of
s i l v e r was weighed to 0.01 mg. It was then dissolved i n such
a volume of 6.4 molar n i t r i c acid, that when diluted to 0.025
molar as regards s i l v e r n i t r a t e the n i t r i c acid concentration
would be 0.100 molar. The p e l l e t was dissolved i n a special
apparatus (see figure 10) from which oxides of nitrogen and
acid spray could escape only by means of a b a f f l e and water
trap,
A platinum boat of known weight was placed i n a
weighing bottle provided with a well f i t t i n g glass stopper.
Another weighing battle was prepared having nearly the same
weight as the f i r s t and containing a quantity of platinum very
nearly equal i n weight to the platinum boat. After
desiccation, the two bottles were allowed to remain in the
balance case, usually for about an hour but sometimes
overnight, c h i e f l y for allowing the effects of humidity on the
surface of the bottles to adjust themselves, following which
1. J.Am.Ghem.Soc., 27 v 502 (1905) 2. J. Phys.Ghem.* 35,830 (1931)


(53) the boat containing the bottle was carefully weighed using
the other bottle and i t s platinum as a counterpoise.
On the basis of an assumed atomic weight for rubidium an amount of the chloride E or 3 mg. greater than the weight equivalent to the s i l v e r p e l l e t was weighed into the platinum boat.
For the fusion i n an atmosphere of nitrogen which
was to follow, an apparatus shown diagrammatically in
figure I I was constructed. The component parts, which were,
i n the main, constructed of lime soda glass are as follows,
A and P are tubes 2 cm. x EO cm. packed with glass-wool
B and C are 400 cc. wash bottles containing i n each 150cc
of-C.B.. ammonium hydroxide (14$)
D i s a comnustion tube, 1.8 cm. x E6 cm. of Corning
No. 172 glass, joined to the remainder of the apparatus
by means of graded seals. It i s f i l l e d f or about EE cm.
of i t s length with closely rolled. 60 mesh copper gauze.
F i s a water trap, capacity about 400 cc.
G and E are washing bottles containing 6 molar sulphuric
ac i d .
I,J,K,I,£ and R are Emmerling towers, of which I,J and Q
contain a 40$ solution of C.P. potassium hydroxide, K
contains a 10$ solu t i o n of C.P. s i l v e r n i t r a t e and I and
R contain a concentrated solution of sulphuric acid PI,
a l l i n quantities not i n excess of that which could cause
bubbling of the l i q u i d s above the surface of the beads.
M and S are tubes 2.5 cm. x 40 cm. containing freshly

(54).
fused O.P. sodium hydroxide (see page 21)
E i s an e l e c t r i o furnace, made in th i s laboratory according
to methods si m i l a r to those described on pages 18 and 22.
During operation i t was maintained at a temperature of about
700° as indicated by a Chrome1 Alumel thermocouple.
E[ and T are tubes 2.5 cm. x 40 cm. containing phosphoric
oxide PI.
0 i s a z i g zag of glass tubing to allow a rotational motion
of U, V, W, X the Richards' b o t t l i n g apparatus. This was 1
according to Richards and W i l l a r d r s modification of the 2
o r i g i n a l apparatus devised by Richards and Parker . The component parts are,
U_, a soft glass tube with a niche for the stopper of the weighing bot t l e ;
V, the combustion or fusion tube; t h i s was o r i g i n a l l y
of a supposedly hard glass; however, after a couple of
fusions i t became very severely dil a t e d and was replaced
by a transparent fused s i l i c a tube, which was en t i r e l y
s a t i s f a c t o r y ;
¥ a tube 2.5 cm. x 40 cm. containing C P . phosphoric oxide;
X a long Pyrex glass rod ourating through a hole i n the
fusion tube which was made a i r t i g h t by means of an outer
sheath of rubber over the j o i n t .
I i s a wash bottle containing CP.concentrated sulphuric
acid,
1. Carnegie Inst, of Wash., Publication Ho.125, 20 (1910) 2. Proc.Amer. Acad., 32, 55 (1896)


(55)
The boat was then placed i n the fusion tube, the
weighing bottle and i t s stopper pushed into t h e i r positions,
and the tubes connected. A current of dry nitrogen was then
passed through the apparatus for about 30 minutes, i n order
to sweep out any moist a i r , and while this stream of gas was
s t i l l passing, the s a l t was heated with the flame from two or
three meker burners u n t i l i t had melted. After i t had been
fused for a few minutes i t was allowed to cool, and the
apparatus then swept out with a stream of dry a i r . Yilhen the.
a i r had completely displaced the nitrogen, by means of the
long Pyrex rod, the boat was pushed into the bottle, and the
stopper into i t , hence the bottle and contents to the
desiccator and then to the balance case.
After a weighing by substitution as before, the
boat containing the chloride was lowered into a 3 l i t e r
stoppered Pyrex Erlenmeyer flask, previously weighed to 1 gm,
and containing about 100 cc. of water, and the f l a s k slowly
rotated u n t i l the s a l t had dissolved. The beat was then
removed with a Pyrex rod and washed with water u n t i l the
washings, when examined nephelometrically, showed no
indications of the chlorine ion. The solution containing
the dissolved s i l v e r p e l l e t was then poured into a special
precipitant delivering device ( figure 12) along with a
suitable number of washings from the b i l v e r dissolving
apparatus (figure 10). Addition of the s i l v e r n i t r a t e
solution, at the rate of about 6 ml. per minute, to the
chloride solution kept i n motion, began immediately. This,

(56)
and subsequent operations involving s i l v e r chloride were done
i n the l i g h t of a Series OA Wratten Safelight. Of course,
t h i s excepts the small portions examined subsequently i n the
nephelometer.
As, during the fusion a trace of s a l t was
v o l a t i l i z e d and varying amounts of water were l o s t , exact
equivalence on the basis of the assumed atomic weight was not
i n general attained at t h i s point; so the requisite amount
of an adjusting solution of rubidium chloride or of s i l v e r
n i t r a t e , whichever was necessary, was then added to attain
equivalence.
By weighing the system to 1 gram, and adding the
necessary amount of water, the a n a l y t i c a l solution was
adjusted to 0.0250 molar i n rubidium n i t r a t e .
After a thorough shaking the f l a s k and contents
were packed i n i c e . From the cooled solution, after the
passage of at least 24 hours, a 50 ml* sample was pipetted off,
f i l t e r e d through a platinum mat, and allowed to come to the
temperature of the standard solutions.
Following Johnson's method, the supernatant l i q u i d
that had been pipetted o f f and had assumed the temperature of
the laboratory, was then analyzed for s i l v e r and chloride ion.
This was accomplished by nephelometric tests which consisted
i n comparing 20.00 ml. portions of the sample with standard
solutions (see page 49) of equal volume. In analyzing f o r
chloride, the standard and test solutions were precipitated
with two 2.00 ml. portions of the chloride ion precipitant

(57)
(see page 49); i n analyzing fo s i l v e r the standard and test
solutions were precipitated with 2.00 ml. portions of the
s i l v e r ion precipitant (see page 49). In the preparation of
the suspensions recourse was had to a special apparatus devised
by Scott and Hurley 1 and shown by these authors to produce
. unif orm^turbidities. The device consists essentially of
two Pyrex glass reservoirs, capacities about 5 and 50 ml.,
joined by a c a p i l l a r y Y-tube, the arms of which are such a
size that when 2 ml. of the pr e c i p i t a t i n g agent are placed In
reservoir and 20 ml. of either the standard of the unknown
are placed i n the other, and the device i s allowed to drain,
the solutions w i l l mix uniformly u n t i l the last drop has run
into a beaker placed below to receive the suspension.-
The suspensionshaving been prepared as indicated,
the cups and plungers of the nephelometer, previous rinsed
with water, were rinsed with the suspension with which they
were to come i n contact. Comparisons were then made of the
turbidities, of the several suspensions, the chloride Ion
unknown with the chloride ion standard, etc. In using the
nephelometer the precautions given b̂ r Yoe2 were observed.
If the analysis showed an excess of either s i l v e r
or chloride ion, suitable additions of the adjusting standard
solutions of rubidium chloride or of s i l v e r nitrate were made
(see page 51). The fl a s k and contents during this time were 1. J. Am. Chem. S o c , 56, 333 (1934) 2. J. H. Yoe: "Photometric Chemical Analysis, Volume II,
Hephelometry!? John Wiley and Sons, New York, 1929 p. 40 f*

(58)
of course maintained at 0° with ice. On the following day, a sample was withdrawn, analyzed as before, and i f necessary, further additions of rubidium chloride or silver nitrate adjusting solutions made. This procedure was continued until equivalence was attained, withiii about 0.1 mg.

( 59)
THE RESULTS OF THE ANALYSIS OF RUBIDIUM OHLORIDE
The results of the analysis of the purified rubidium chloride are shown in detail in Table I. The calculated values of the atomic weight of rubidium are based on the present accepted values for the atomic weights of silver and chlorine, 107.880 and 35.457 respectively.
To. illustrate completely the method of calculation the detailed computations for one of the analyses (Analysis
No. 7) are given below.
Weight of silver in vacuo 2.17978 grams Weight of rubidium chloride in vacuo 2.44580 grams Volume of analytical solution 0.810 l i t e r s Weight of silver added by means of adjusting standard
solution to correct end — point 0.00192 grams
FINAL ANALYSIS at end-point:
Concentration of standard solutions 0.50 mg. Ag/liter or the equivalent in chloride
Nephelometric Ratios: x : silver standard :: 1.20 : 1.00 x : chloride standard :: 1.10 : 1.00
Hence, Chloride ion concentration — 1.10 x 0.50 = 0.55 mg.
Ag/ l i t e r .

(60)
Silver ion concentration = 1.SO x 0.50=0.60 mg. Ag/liter.
And excess concentration of silver = 0.60 — 0.55 = 0.05 mg. Agfliter.
Therefore, total excess of silver — 0.810 x 0.05'— 0.04 mg Hence the weight of silver equivalent to E.44580 grams of
rubidium chloride = 2.17978 +0.000192 -0.00004 =2.18166 grams
And the ratio RbCl:Ag=1.12107 Whence the atomic weight of rubidium = 85.484
By a suitable choice of standards, nephelometric ratios were kept as close to unity as possible, the value 1.20 being a maximum.
From the last two columns of Table I can be obtained the averages, -
Average (including No. 2) S a t i o RbCl:Ag= 1.121110 db0.000034
Atomic weight of rubidium 85.488"± 0.003 Average (excluding No. 2) Ratio RbCl:Ag =1.12105
Zt 0.00002 Atomic weight of rubidium 85.482±0.002
Convention suggests the adoption of the latter value.

( 61)
TABLE I No. Fusion Silver Rubidium Chloride Silver
Atmosphere Sample Vacuum Vacuum ... Crams Grams
(1) Nitrogen. 2 2,41226 2.15397 (2), Nitrogen 2 2.77942 2.47489 ( 3 ) Nitrogen 2 2.90458 2.58899 (4) Nitrogen 2 2.51028 2.23922 (5) Laboratory 2 3.04508 2.73799 (6)
Sir 2.73799 (6) Nitrogen ' 1 2.25778 2.01719 in Nitrogen 1 2.44580 2.17978 (8) Nitrogen 1 2.59528 2.51011
Table I (continued) No. i n i t i a l Silver Added NEPHELOMETRIC ANALYSES AT END-POINT
Volume to Solution Mg. of Silver per lifcer as Liters Mg. Chloride 8 , Silver
(1) 0.800 -2.35 0.62 0.56 (2) 0.920 -0.64 0.58 0.43 ( 3 ) 1.040 • -L«91 0.55 0.40 (4) 0.830 -0.30 0.84 0.78 (5) 1.014 -21.57 0.66 0.60 (6) 0.750 -3.10 0.60 0.60 (7) 0.810 1.92 0.55 0.60 (8) 0.858 -4.02 0.60 0.60
a . The values in this column are the chloride concentrations in milligrams per l i t e r , multiplied by the factor Ag/Cl.

(62)
Table I (continued)
Wo. Deficiency of End Point Correction Total Correcti S i l v e r at to S i l v e r to S i l v e r End Point Mg. Mg.
Mg. per l i t e r
(1) 0.06 0.05 -2.30 (2) 0.15 0.14 -0.05 (3) 0.15 0.15 2.06 (4) 0.06 0.05 -0.25 (5) 0.06 0.06 -21.51 (6) 0.00 0.00 -3.10 (7) -0.05 -0.04 1.88 (8) 0.00 0.00 - 4,02
Table I (continued)
Ho. Ag Equivalent BbCl Ratio Atomic Weight to RbCl Vacuum RbCltAg of Rubidium Grams Grams
(1) 2.15167 . 2.41226 X * 1 S H I 85.488 (2) 2.47848 2.77942 1.12142 85.619 (3), 2.59105 2.90548 1,12101 85.477 (4) 2.2389 7 2,51028 1.12117 85.495 (5) 2.71648 3.04508 1.1209 7 85.473 (6) 2.01409 2.25778 1,12099 85.474 (7) 2.18166 2.44580 1.12107 85.484 (8) 2.31509 2.59528 1.12103 85.479

(63)
THE ANALYSIS OE POTASSIUM CHLORIDE
To check the technique and method, two determinations
were made of the atomic weight of potassium using as material
for analysis potassium chloride PI. The samples were fused
i n the e l e c t r i c muffle furnace (see page 18), and agter
cooling and desiccation were weighed out for analysis on a
watch glass. Johnson 1 has shown that potassium chloride fused
i n the moist a i r ( r e l a t i v e humidity about 60$) of the laboratory
hydrolyzes only to the extent of about 2 moles 0H~ in 10 6
moles of s a l t ; hence the method of fusion employed, here
w i l l introduce no errors i n the present analysis. The constancy
i n weight of the fused s a l t when exposed to the atmosphere
has also been demonstrated .
Aft e r the weighing of the salt the analysis follows
the method used f o r the rubidium chloride.
The r e s u l t s are shown i n d e t a i l i n Tahle II. The
atomic weights of potassium were calculated using the present
day accepted values for chlorine and i l s v e r of 55.457 and
107.880 respectively. The mean value, 39.100, i s reassuringly
close to 39,096, the accepted value today.
1. J. Phys.Chem., 39, 791 (1935) 2. Johnson: i b i d .

So. Fusion S i l v e r Atmosphere Sample
(64)
TABLE II
Potassium Chloride Vacuum Grams
Silve r Vacuum Grams
(1) Laboratory A i r 2 (2) . Laboratory A i r 2
4.37911
3.76620 6.33662
5.44976
Table II (continued) No. I n i t i a l S i l v e r Added NEPHELOMETRIC ANALYSES AT END POINT
Volume L i t e r s
to Solution Mg. S i l v e r per L i t e r as Chloride 8 , S i l v e r
(1)
(2)
2.40
2.17
0.00
0.00 0.69
0.39 0.63
0.70 a. See footnote.a, Table I page 61.
Table II (continued) No. Deficiency of End Point Correction Total Correction
S i l v e r at to S i l v e r End Point Mg.
Mg. per L i t e r
to S i l v e r Mg.
(1) (2)
0.06
0.31 0.14
0.67 0.14
— 0.67
Table II (continued) No. Ag Equivalent KC1 Ratio Atomic Weight
to KCl Vacuum KCl:Ag of Potassium Grams Grams
(1) (2)
6.33676
5.44909
4.37911 0.691067
3.76620 0.691161
39.095
39.105

(65)
REOALCULATION OF. THE, ATOMIC WEIGHT .OF RUBIDIUM
FROM ARCHIBALD1-S DATA
Using the present day accepted atomic weights of chlorine, "bromine and s i l v e r (35.457, 79.916 and 107"-.a80 respectively ) the atomic weight of rubidium has been completely recalculated from the data of E. H. Archibald 1. For convenience i n comparison and discussion the recalculated values are shown i n fable I I I . The table shows the rati o s RbCl:AgCl, RbCl:Ag, RbBr:AgBr and RbBr:Ag obtianed from the analysis of 14 weighed quantities of rubidium chloride and 7 similar quantities of the bromide, together with their corresponding atomic weights.
1. Trans. Chem. S o c , 85, 786 (1904); 85, 789 (1904)

(66)
TABLE III
No. Rubidium Chloride S i l v e r Chloride Ratio Atomic Wi Vacuum "Vacuum RbGl:AgCl of Rubii Grams Grams
(1) 1.99966 2.37070 0,84349 85.446 (2) 2.06480 2.44778 0.84354 i 3b.85.453 (3) 2.29368 2.71960 0.84339 85.432 (4) ' 1.09495 1.29 796 0.84360 85.459 (5) 2.14381 2.54118 0.84364 85.468 (6) 2.89700 3.43475 9.84344 85.439 (7) 2.19692 2.60452 0.84350 85.448 (8) 2.14543 2.54386 0.84338 85.431 (9) 2.12164 2.51557 0.84341 85.435 (10) . 2.25777 2.67685 0.84344 85.439 (11) 2.18057 2.58528 0.84346 85.442 (12) 2.32699 2.75878 0.84348 85.445 (13) 4.00035 4.74233 0.84354 85.453 (14) 2.43440 2.88613 0.84348 85.442
Average 85.445

(67)
Table III (continued)
S i l v e r Ratio Atomic Weight
Vacuum RbCl:Ag of Rubidium
Grams
(15) 1.99966 1.78454 1.12054 85.426 (16) 2.06480 1.84241 1.12070 85.444 (17) 2.29368 2V04710 1.12046 85.418 (18) 1*09495 0.97702 Cfi.12070 85.444 (19) 2.14381 1.91316 1.12056 85.429 (20) 2.89700 2.58550 1.12047 85.419 (21) 2.19692 1.96076 1.12044 85.416 (22) 2.14543 \ 1.91462 1.12055 85.428 ( 230 2.12164 1.89346 1.12052 85.428 (24) 2.25777 2.01515 l*i2040 85.412 ( 250 2.18057 1.94594 1.12057 85.430 (26) 2.32699 2.07668 1.12053 855426 (27) 4.00035 3.56998 IL • 13055 85.428 (28) 2.43440 1.12064 85.438
Average 85.427
No. Rubidium Chloride
Vacuum
Grams

(68) Table III (continued)
ffo. Rubidium Bromide S i l v e r Bromide Ratio Atomic Weight
"V Vacuum Vacuum RbBr:AgBr of Rubidium Grams Grams
(29) 2.68170 3.04578 0.88047 85.433 (30) 2.07280 2.35401 0.88054 85.446 (31) 2.10086 2.38589 0.88053 85.444 (32) 2.61044 2.96462 0.88055 85.444 (33) 3.84082 4.36215 0.88049 85.436 (34) 3.77852 4.29084 0.88061 85.457 (35) 4.34299 4.93210 0.88056 85.450
Average 85.444 Table III (continued)
No.- Rubidium Bromide S i l v e r Ratio Atomic Weight Vacuum Vacuum RbBr:Ag of Rubidium Grams Grams
(36) 2.68170 1.74930 1.53301 85.465 (37) 2.07280 1.35230 1.53280 85.442 (38) 2.10086 1.37061 1.53278 85.440 (39) 2.61044 1.70300 1.55285 85.448 (40) 3.84082 2.50590 1.53272 85.434 (41) 3.77852 2.46502 1.53287 85.450
(42) 4.34299 2.83340 1.53278 85.440
Average 85.446

(69)
THE NEUTRALITT.OF FUSED RUBIDIUM CHLORIDE
"By c r y s t a l l i z a t i o n i n platinum sodium chloride
"and potassium chloride may "be obtained free from a l l weigh—
"able impurities except water, platinum, and atmospheric gases,
"The platinum impurities are e a s i l y kept to negligible amounts,
"and the water and gases may be removed by fusion of the s a l t s ,
"bpt there i s a l i m i t set upon the purity of the material by
"the s l i g h t hydrolysis which occurs during f u s i o n . " 1 In two
series of experiments i n which the platinum ware was weighed 2
before and .after using C.R. Johnson shewed that the platinum
impurities when c a r e f u l l y c r y s t a l l i z e d i n platinum vessels
could not be greater than about 0.0003$. It did not, therefore
seem necessary to repeat a similar series of experiments i n the
case of rubidium chloride.
However the hydrolysis of rubidium chloride was a
point which demanded attention, especially since "It has also
been shown by Johnson and A l l e n that fusion of caesium or
"rubidium chloride i n a moist atmosphere was l i k e l y to cause
"decomposition; ©,s i n t h e i r analysis of caesium chloride,
"where the fusion took place i n the a i r of the laboratory a
"solution of the fused s a l t was always found to be s l i g h t l y
" a l kaline." Time was not available to conduct an extensive 1. C.R, Johnson: J. Phys. Chem., 39, 791 (1935) 2. J. Phys. Chem.,.37, 924 (1933); 782 (1935) 3. Archibalds Trans. Chem. S o c , 85, 782 (1904)

(70)
series of experiments l i k e those of O.K. Johnson on the
hydrolysis of potassium and sodium chlorides* However
s,amples of rubidium chloride fused i n the atmosphere of the
laboratory, when their solutions were tested with Indicators
and potentiometrically, f a i l e d to show a degree of hydrolysis
which would effect even the most accurate atomic weight
determinations• From the experiments performed an estimate of
the hydrolysis was hard to obtain but i t must have been less
than about 1 mole 0H~ per 10 6 moles of s a l t . One sample of
s a l t fused i n the laboratory a i r was analyzed. The equivalent
weight obtained for t h i s sample (Ho. 5) was s l i g h t l y less than
the mean of the values of the samples fused i n pure dry
nitrogen; hydrolysis would have produced an opposite eff e c t .
These experiments are conclusive enough to invalidate any
observations which may have been made :by Hohnson and Allen .
Although a rigorous estimate of the extent of hydrol
ysis on fusion of rubidium chloride has not been made, the
few experiments cited should j u s t i f y i t s fusion i n an atmosphere
of nitrogen rather than a p a r t i a l or t o t a l atmosphere of
hydrogen chloriMLe, which may eas i l y cause, i f not. s u f f i c i e n t
care i s taken, the fused sample to;become acid. That this
l a s t factor i s quite apparent In the"ease of sodium chloride
has been demonstrated by C.R. Johnson .
2. Amer. J . S c i . and Arts, ( i i ) , 35, 94 (1863) 3. J. Phys. Ghem., 37, 930 (1933)

(71)
DISCUSSION OF RESULTS
Comparison of the Results of E.H.Archibald with
those obtained i n the Present Investigation
The average value 85.482 for the atomic weight of
rubidium obtained i n t h i s research i s approximately 0.04 units
greater than that found by Archibald 1. Considering the
rigorous treatment employed i n the p u r i f i c a t i o n of rubidium
s a l t s for both researches i t seems unlikely that the
preparations employed could d i f f e r i n purity by an amount
large enough to account for even a small f r a c t i o n of the
observed difference in atomic weight. Any accounting for the
differences i n results then, i t would seem, should best be
based on differences i n the methods of analysis. This section
then, w i l l be devoted to a discussion of these differences and
t h e i r p l a u s i b i l i t y i n accounting for the differences i n
observed values.
The high accuracy of the t i t r a t i o n method, and
especially of the standard solution modification has already
been discussed (see page 45). The methods used i n 1904 have
been long since discarded.
Some inconsistencies i n Archibald's results w i l l
now be pointed out. In his method, by precipitating the
chloride from a weighed amount of rubidium chloride, using a
s l i g h t l y greater than equivalent amount of s i l v e r , weighed
and dissolved i n n i t r i c acid, and by weighing the s i l v e r
T~. Trans.Chem.boc., 8i?, Y O Y , I iyu4t; ~

(7E)
chloride formed and determining the slight excess of silver not required to precipitate the silver chloride, he was able to determine the ratios RbCl:Ag, Rb01:AgCl and also Ag:AgCl. The f i r s t two ratios are shown in Table III together with the corresponding atomic weights calculated on the basis of the 1935 values for silver and chlorine. Whereas the values of the atomic weight obtained from the ratio Rb01:AgCl agree very well with those obtained from the Analyses of the bromide, those obtained from the ratio RbOlrAg do not, via:
Average atomic weight of rubidium from the ratio RbCl: AgOl .... 85.445 BbBr:AgBr .... 85.444 RbBr: Ag .... 85.446
but from RbCl: Ag .... .85.427
This inconsistency is consistent with the value obtained for the Ag:AgOl ratio of 0.75274, This ratio, while agreeing closely with that of Stas, 0.75276, is higher than that obtained by Richards and Wells . The latter value, 0.75263, conforms with the 1935 values of the atomic weights of silver and chlorine. This apparent excess of silver in the silver
• 2 chloride seems to check with the observations of C.R.Johnson who has shown that silver chloride, when washed with pure water, retains an excess of silver over chlorine, the washings, T. J.Am.Chem.Soc., 27, 502 (1905) 2. ibid,, 55, 2258 (1933)
J.Phys.Chem., 55, 2241 (1931) ; 36, 1945 (1932)

(75) of course, containing an excess of chloride over silver.
At the same time as Archibald analysed rubidium chloride "several analyses were made of a sample of very pure potassium chloride in order to test the method and also
1 the purity of the silver used". "The results .will form the subject of a future communication."2 (Trans.Roy.Soc. Can., Ser.II, Sec. i i i , p.47 (1904)). The values obtained in . this analysis which was performed in an identical manner to that of rubidium chloride, are at variance with those obtained 3 4 by Richards and Staehler , Richards and Mueller , and later by Konigschmid and Sachtleben^and Baxter and MaoNevin^. These latter values have acceptance today. Although Archibald's silver was not fused in a current of hydrogen, the author feels more inclined to put the discrepancy down to the method of analysis rather than to the purity of any of the compounds used.
It should be pointed out, however, that these discrepancies just outlined are not of the highest order, being in general slightly less than that between the accepted atomic weight of rubidium and the value obtained in this investigation.
For convenience in cr i t i c i z i n g Archibald's analytical procedure i t w i l l be outlined below: l7~ Trans. Ohem.Soc, 85, 785 (1904) 2. ibid. 3. Pub.Oarn.Inst.Wn.,No.69 p.7 (1907) 4. ibid., p.27. 5. Z.anorg.allgem.Ghem., 171, 1 11933) 6. J.Am.Chem.Soc , 55, 3185 (1935)

(74)
S i l v e r chloride was precipitated from a weighed
amount of fused rubidium chloride with a s l i g h t excess of
s i l v e r , weighed and dissolved in n i t r i c acid, and the whole
system vigorously shaken. After several hours the mother
liquor was f i l t e r e d o f f through an asbestos mat of a weighed
Gooch crucible and the precipitate washed twice with 2 150 c
portions of water. These washings were added to the main
f i l t r a t e . The precipitate was then washed with 2 300 cc.
portions of water and these washings kept separate. After
washing the precipitate onto the f i l t e r , i t was dried at 125°
desiccated and weighed. To obtain the weight of s i l v e r
chloride equivalent to rubidium chloride, corrections were
applied for the asbestos carried away i n the f i l t r a t e , the
loss of water on fusion of the s i l v e r chloride, and the
amount of s i l v e r chloride dissolved by the last 600 cc. of
wash water. The s o l u b i l i t y correction was made on the basis
of a nephelometric analysis of the l a t t e r 600 cc, of wash
water. In this procedure a solution containing a known
amount of s i l v e r n i t r a t e was compared with the unknown
solution, hydrochloric acid being added to the former and
s i l v e r n i t r a t e solution to the l a t t e r . To obtain the r a t i o
Rb01;Ag the o r i g i n a l weight of s i l v e r taken was corrected for
the amount of s i l v e r , as s i l v e r n i t r a t e , found in the
supernatant liquor from the precipitation. The l a t t e r
quantity was determined by preci p i t a t i n g the excess s i l v e r
n i t r a t e with hydrochloric acid, washing the precipitate with
a very dilute solution of the precipitant, drying and

(75)
weighing.
Some p o i n t s i n the f o r e g o i n g procedure which are
questionable are as f o l l o w s :
(.1) Vigorous Shaking with A n a l y s i s f o l l o w i n g soon a f t e r .
Systems c o n t a i n i n g s i l v e r c h l o r i d e and other e x t r a
ions when v i g o r o u s l y shaken i f not allowed a considerable
r e s t p e r i o d before a n a l y s i s show an excess o f c h l o r i d e over 1
s i l v e r amounting to s e v e r a l tenths of a m i l l i g r a m per l i t e r .
T h i s e f f e c t i s e l i m i n a t e d by gradual r e d u c t i o n of shaking to
a minimum over a p e r i o d of a few days.
( S) Washing of S i l v e r C h l o r i d e with Pure 'Water.
Johnson has shown that much washings c o n t a i n an
excess of c h l o r i d e over s i l v e r ions to the extent of s e v e r a l
tenths of a m i l l i g r a m per l i t e r l e a v i n g the p r e c i p i t a t e with
an excess o f s i l v e r . "This source of constant e r r o r . . . . . . . 3
would tend to make the c a l c u l a t e d atomic weight too low."
( 3) C o r r e c t i o n to be A p p l i e d to the O r i g i n a l Weights o f
S i l v e r C h l o r i d e and o f S i l v e r . (a) The Nephelometric A n a l y s i s
T h i s o b v i o u s l y v i o l a t e s the general p r i n c i p l e f i r s t
l a i d down by Richards, "In my o p i n i o n , i f even moderately
ac c u r a t e a n a l y t i c a l r e s u l t s are t o be had with the
nephelometer, the one e s s e n t i a l p o i n t to be heeded i s t h i s :
the unknown s o l u t i o n s to be estimated must be t r e a t e d i n
TI C.R.Johnson; J.Phys.Chem., 35, 2582 (1931) 2. J.Am.Chem.Soc, 55, 2258 (1935) 3. C*R.Johnson: i b i d . , 35, 2242 (1931)

(76) 1
exactly the same way as the known standard solutions....." p
The discussion of Johnson on the coagulating po?/er of
different ions and the r e s u l t i n g difference i n l i g h t
intensity of s i l v e r chloride suspensions i s to the point.
(b) No Correction due to the 2 150 cc. Portions of vYash
Water.
Presumably since the f i r s t E washings were
transferred to the supernatant l i q u i d (I.e.the i n i t i a l
f i l t r a t e ) they were intended to wash out occluded and other—
wise held excess s i l v e r n i t r a t e and so enable the o r i g i n a l
weight.of s i l v e r taken to be accurately corrected to obtain
a precise value for the RbCl:Ag r a t i o . To obtain the
expected results one must assume that these E 150 cc. portions
of wash water remove completely the excess s i l v e r nitrate and
at the same time dissolve no s i l v e r chloride. This i s
hardly possible. Any s i l v e r chloride dissolved, then, would
affect both the weight of the main s i l v e r chloride precipitate
and also the weight of the s i l v e r chloride precipitate from
the supernatant l i q u i d . In this connection, another point
which i s not obvious to the author i s , that i f the
experimenter considered the S 150 cc. portions of wash water
s u f f i c i e n t to remove the excess s i l v e r nitrate from the s i l v e r
chloride precipitate, why then did he wash with 600 cc.more.
If he did not.consider the 2 150 ce. portions s u f f i c i e n t for
t h i s , then the corrected weight of s i l v e r , intended to be
—iSfrShem.j., 30. o n i i y o b j ^ 2. J.Phys.Chem., 35, 541 (1931)

(77)
equivalent to the rubidium chloride, would be subject to error.
It is hard to estimate how the sum t o t a l of the a n a l y t i c a l errors outlined i n sections (1), (2), (3a) and (3b) would affect the f i n a l atomic weight, either in magnitude or i n sign.
•Comparison, of the Results of this Investigation
with those of F.W.Aston.
By photometry of i t s mass spectra F.W.Aston1 found ft K
the most probable r a t i o of the two istopes of rubidium, Rb 87
and Rb , to be 3.0^0.05. i'he packing f r a c t i o n of rubidium has not bean determined, butt i f i t i s assumed to be the same
8 6 as that of Kr ( i . e . — 8 . 2 parts in 10,000) the atomic weight on the physical scale ( 0 ^16.0000 ) can be calculated to be 85.46±0.03. 'When concerted to the chemical scale ( o 1 6 , 1 7 , 1 8
2 =• 16.0000)by means of Meeke and Childs conversion factor
(1.00022) based on measurements of the abundance of the oxygen
isotopes 0 1 7 and O 1 8 one obtains the value 85.43^0.03.
This is i n good agreement with the International value of 85.44.
However, there i s good reason to believe that this conversion
factor "may be i n error. In the words of F.W.A'ston, "a
general survey of the relations between the measurements of
the masses of the isotopes and the chemical atomic weights of
the elements suggests that this factor i s too high" (he was
discussing Meeke and Childs' value). T. Proc.Roy.Soc, A134. 575 (1932) 2. Z e i t s . f . Physik., 68, 362 (1931) 3. F.W.Aston: T,Mass Spectra and Isotopes" Edward Arnold and Co., London 1933. page 143.

(78)
It should he noted that the abundance ratios of
the two isotopes have not been determined with very high
precision, r e s u l t i n g i n a considerable probable error i n the
value for the atomic weight.
The existence of other isotopes of rubidium i s very
un l i k e l y . The fact that no element of odd atomic number (
(that of rubidium i s 57) is known to possess more than
2 isotopes has led Aston to enunciate the rule: "Elements of 1
odd atomic number never have more than 2 isotopes." Even
considering t h i s l a s t factor, lack of definite information
regarding the values of the packing fractions of the isotopes
combined with the aforementioned not very high order of
accuracy of the mass spectroscopic photometric determination
of the isOtopic r a t i o Bb 8 5:Rb 8 7 makes the disagreement between
the value for the atomic weight of rubidium obtained i n this
investigation and-that of E.W.Aston determined mass
spectrosoopically not very serious.
1. i b i d . , page 175.

(79)
SUMMARY
(1) Rubidium chloride has been prepared i n , as far as i s
• possible to ascertain, a state of purity i n excess of
that necessary to affect the value of the atomic weight
of rubidium as much as 0.001 unit.
(2) Two samples of s i l v e r have been prepared by methods
shown by investigators to produce a product of highest purity.
( 3) The aforementioned samples of rubidium chloride and
s i l v e r have been compared by means of G.R.Johnson's
standard solution modification of the conventional
t i t r a t i o n method of Richards and Wells.
(4) I f the values of the atomic weights of Silv e r and
chlorine are taken as 107.880 and 35.457 respectively
the value obtained for the atomic weight of rubidium
is 85.482.
(5) So far as a f f e c t i n g an atomic weight determination the
hydrolysis of rubidium chloride when fused i n the
laboratory a i r or dry nitrogen is negligible,
(6) Reasons for the discrepancy between the value obtained
for the atomic weight of rubidium i n this investigation
and the International value have been suggested.

(80)
BIBLIOGRAPHY
i l l Archibald, E.H.r Trans. Chem. S o c , 85, 776 (1904)
fS) Archibald, E.H.: Proc. Roy. S o c Can., II, S e c i i i
p. 47 (1904)
(3) Archibald, E.H.; "Preparation of Pure Inorganic Substances"
John Wiley and Sons, Inc. New York, 1931. (4) Aston, F.W.: P r o c Roy. S o c , A134* ,575 (1932)
(5) Aston , F.W.: "Mass Spectra and Isotopes", Edward Arnold
and Co., London, 1933.
(6) Ato and Wada: S c i . Papers Inst. Phys. Chem. Res.,
4, 263 (1926)
(7) B a l l : J. Chem. S o c , 95, 2126 (1909)
(8) Baxter, G.P.: J. fcmGhem* .Soc, 44, 577 (1922)
(9) Baxter and Ishmaru: J. Am. Chem. S o c , 51, 1730 (1929)
(10) Baxter and MacNevin: J. Am. Chem. S o c , 55, 3185 (1933) -
(11) Bray, see Noyes
(12) Childs, see Meoke
(13) Diaz, see Strecker
(14) LaIton, see Noyes
(15) Davies: J. Chem. S o c , 123, 2976 (1923)
(16) Finch and Peto: i b i d . , 121, 692 (1922)
(17) Freundler ani Menager: Compt. Rend., 182, 1158 (19 260
(18) Gedroiz: Chemical Analysis of the S o i l , Series A, No.3, (1923), Commissariat of Agriculture, Setrograd.
(19) HBnigschmid and Sachteleben; Z. anorg. allgem. Chem., 171, 1 (1933)

(81)
HBnigschmid, see also Richards
Hooley and P h i l l i p s : J» Ghem, Ed., 11, 576 (1934)
Johnson and Alle n : Am. J. S c i . Arts, 35 ( i i ) 94 (1963)
Johnson, C.R.: J. Phys. Ghem., 35, 540 (1931)
Johnson, O.R.: i b i d . , 55, 830 (1931)
Johnson , O.R.s i b i d . , 35, £237 (1931)
Johnson, C.R.: i b i d . , 35, 2581 (1931)
Johnson, G.R.: i b i d , , 36, 1942 (1932)
Johnson, G.R.t i b i d . , 37, 923 (1933)
Johnson, G.R.S i b i d . , 39, 781 (1935)
Johnson, G.R.: i b i d . , 39, 791 (1955)
Johnson and Hullet: ft, 5Am. ::Ghemw5Soc., 55, 2258 (1933)
Johnson and low: i b i d . , 55, 2262 (1953)
Johnson and Low: J . Phys. Chem., 36, 2390 (1938)
Johnson, also see Scott
Meoke and Childs: Z e i t s . f . Physik., 68, 362 (1931)
Missender: Ghem. News, 124 362 (19£2)
Moser and Rietschel: Monatsch., 46, 9 (1926)
Mueller, see Richards
Noyes and Bray (Daltcm and Williams): "A System of
Qualitative Analysis for the Rare Elements",
The MacMillan Co., New York 1927
(40) Parker, see Richards (41) P h i l l i p s , see Hooley (42) Richards, T.W.: Am. Chem. J., 35, 511 (1906)

(82)
Richards, T.W,: J. Am. Chem. S o c * 22, 144 (1900)
Richards and Parker: P r o c Am. Acad., 32, 55 (1896)
Richards and Mueller: Pub. Cam. Inst. Wn., Ho* 69
p. 27 (1907)
- Richards and Staehler: i b i d . , p. 7
Richards and Wells: J. Am. Chem. S o c , 27, 502 (1905)
Richards and Wells: Am, Chem. J., 31, 239 f , (1904)
Richards and Willard: Cam. Inst, of Wn., Pub. Ho. 125,
(1910)
Richards and Willard: J. Am. Chem. S o c , 32, 4 (1910) Rietscnel, see Moser
Robinson: J . Ind. Eng. Chem., 10, 50 (1918)
Richards, T.W.$ J. Am, Chem. Soc,, 27, 104 (1905)
Richards and Honigschmid: J. Am. Ghem. S o c , 32, 1577
(1910)
ticSoott and Johnson: J• Phys.. Chem., 33, 1979 (1929)
Smith, D.M.; "Visual l i n e s for Spectrum Analysis",
Adam Hilger l t d . , london, 1928.
Smith and T a i t : P r o c Roy. S o c Edin., 54, 88 (1933-34)
Setterberg: Oefvers. Stockh. Acad. Eorh., 39, 23 (1882)
Strecker and Diaz: Z. anal. Chem., 67, 321 (1925)
Wells, R.G.f Am. Chem. J., 99 (1906)
Wells,.E . G . : Am. Chem. J., 508 (1906)
Wells, see also Richards
Whitaker: J* Ohem. S o c , 127, 2219 (1925)
Yoe, J.H.: "Photometric Chemical Analysis, V o l . II': Hephelometry" John Wiley and Sons, Inc. london, 19 29.




















