
Synthesis of morphology controllable porous Co 3 O 4 nanostructures with tunable textural properties...
10
Dalton Transactions PAPER Cite this: Dalton Trans., 2014, 43, 10248 Received 27th February 2014, Accepted 22nd April 2014 DOI: 10.1039/c4dt00608a www.rsc.org/dalton Synthesis of morphology controllable porous Co 3 O 4 nanostructures with tunable textural properties and their catalytic application† Mouni Roy, Sourav Ghosh and Milan Kanti Naskar* Porous cobalt oxide (Co 3 O 4 ) nanorod (50–100 nm) and nanosheet-like (70–100 nm) particles were syn- thesized by a facile hydrothermal method at 150 °C for 2–5 h and 12–24 h, respectively, using aqueous- based precursors like cobalt nitrate, urea and water in the absence of any templating agents followed by their calcination at 300 °C. Morphology and textural properties were tuned by changing the synthesis time at 150 °C. A 3D architecture of Co 3 O 4 was formed by the self-assembly of nanostructured (nanorod and nanosheet) particles. The BET surface area, pore volume and pore diameter of the sample prepared at 150 °C for 5 h were 112 m 2 g −1 , 0.5 cm 3 g −1 and 7.4 nm, respectively, and it exhibited the highest cata- lytic performance with a rate constant of 56.8 × 10 −3 min −1 for the degradation of Chicago Sky Blue 6B, a carcinogenic azo dye used in the textile, paper and food industries. Rod-like particles with a mesoporous structure rendered a better catalytic efficiency than sheet-like particles having both microporous and mesoporous structures. An interrelationship amongst the morphology, textural properties and the cata- lytic efficiency of Co 3 O 4 was established. 1. Introduction Transition metal oxides with varied morphologies have attracted intensive research interest because of their possible applications in materials science and engineering. Morpho- logical tuning of the materials is important because it influ- ences the different properties of the materials. Among the transition metal oxides, tricobalt tetraoxide (Co 3 O 4 ) finds wide- spread applications in chemical sensor, 1 lithium-ion bat- teries, 2 optical and magnetic materials, 3,4 and heterogeneous catalysts, 5 where their properties are strongly dependent on their size and shape. 6 In recent years, Co 3 O 4 with various mor- phologies such as nanorods, 7 nanowires, 8 nanotubes, 9 nano- belts, 10 nanocubes, 11 nanosheets, 12,13 etc. have been synthesized successfully. Porous transition metal oxides are of special research inter- est in finding potential applications in catalysis, sensors, elec- trode materials for batteries etc., 14,15 because of their large internal surface area, nanosized walls, and d electrons in an open shell. Mesoporous Co 3 O 4 has proved to be an excellent catalyst for many applications like CH 4 combustion, 16 oxygen evolution, 17 oxidation of toluene and methanol, 18 etc. The higher surface area of mesoporous Co 3 O 4 renders it catalyti- cally more active by facilitating the adsorption and diffusion of reactant molecules, which favours the catalytic performance of the oxides. It is reported that mesoporous Co 3 O 4 is generally synthesized by either hard templating route using KIT-6, SBA-15 and SBA-16 silica, 19,20 and carbon spheres 16 as hard templates or a soft templating process 21 using Pluronic block copolymer as the soft templating agent. The templating methods are tedious and complicated with extra processing time for the removal of the templates. However, there are a few reports for the synthesis of mesoporous Co 3 O 4 by the tem- plate-free method. Li et al. 22 synthesized mesoporous quasi- single-crystalline Co 3 O 4 nanowire arrays on different sub- strates, like transparent conductive glass, Si wafer and Cu foil. In recent times, Venugopal et al. 23 have reported the prepa- ration of self-assembled hollow mesoporous Co 3 O 4 using Li 2 O 2 as the oxidizing/precipitating agent. Wang et al. 24 have pre- pared different shaped Co 3 O 4 using a hydrothermal method. Zheng et al. 25 synthesized flower-like mesoporous Co 3 O 4 by a solvothermal method. In the present study, we have synthesized porous Co 3 O 4 nanostructures of tunable morphology and textural properties via a facile one-pot hydrothermal process at 150 °C by chan- ging the reaction times from 2 h–24 h in the presence of water- based precursors without using any templating agents fol- lowed by calcination at 300 °C. The synthesized particles of different morphologies have been used as catalysts for the † Electronic supplementary information (ESI) available. See DOI: 10.1039/ c4dt00608a Sol-Gel Division, CSIR-Central Glass and Ceramic Research Institute, Kolkata 700 032, India. E-mail: [email protected]; Fax: +91 33 24730957 10248 | Dalton Trans. , 2014, 43, 10248–10257 This journal is © The Royal Society of Chemistry 2014 Published on 22 April 2014. Downloaded by Central Glass & Ceramic Research Institute (CGCRI) on 14/07/2014 11:51:17. View Article Online View Journal | View Issue
Transcript of Synthesis of morphology controllable porous Co 3 O 4 nanostructures with tunable textural properties...

DaltonTransactions
PAPER
Cite this: Dalton Trans., 2014, 43,10248
Received 27th February 2014,Accepted 22nd April 2014
DOI: 10.1039/c4dt00608a
www.rsc.org/dalton
Synthesis of morphology controllable porousCo3O4 nanostructures with tunable texturalproperties and their catalytic application†
Mouni Roy, Sourav Ghosh and Milan Kanti Naskar*
Porous cobalt oxide (Co3O4) nanorod (50–100 nm) and nanosheet-like (70–100 nm) particles were syn-
thesized by a facile hydrothermal method at 150 °C for 2–5 h and 12–24 h, respectively, using aqueous-
based precursors like cobalt nitrate, urea and water in the absence of any templating agents followed by
their calcination at 300 °C. Morphology and textural properties were tuned by changing the synthesis
time at 150 °C. A 3D architecture of Co3O4 was formed by the self-assembly of nanostructured (nanorod
and nanosheet) particles. The BET surface area, pore volume and pore diameter of the sample prepared
at 150 °C for 5 h were 112 m2 g−1, 0.5 cm3 g−1 and 7.4 nm, respectively, and it exhibited the highest cata-
lytic performance with a rate constant of 56.8 × 10−3 min−1 for the degradation of Chicago Sky Blue 6B, a
carcinogenic azo dye used in the textile, paper and food industries. Rod-like particles with a mesoporous
structure rendered a better catalytic efficiency than sheet-like particles having both microporous and
mesoporous structures. An interrelationship amongst the morphology, textural properties and the cata-
lytic efficiency of Co3O4 was established.
1. Introduction
Transition metal oxides with varied morphologies haveattracted intensive research interest because of their possibleapplications in materials science and engineering. Morpho-logical tuning of the materials is important because it influ-ences the different properties of the materials. Among thetransition metal oxides, tricobalt tetraoxide (Co3O4) finds wide-spread applications in chemical sensor,1 lithium-ion bat-teries,2 optical and magnetic materials,3,4 and heterogeneouscatalysts,5 where their properties are strongly dependent ontheir size and shape.6 In recent years, Co3O4 with various mor-phologies such as nanorods,7 nanowires,8 nanotubes,9 nano-belts,10 nanocubes,11 nanosheets,12,13 etc. have beensynthesized successfully.
Porous transition metal oxides are of special research inter-est in finding potential applications in catalysis, sensors, elec-trode materials for batteries etc.,14,15 because of their largeinternal surface area, nanosized walls, and d electrons in anopen shell. Mesoporous Co3O4 has proved to be an excellentcatalyst for many applications like CH4 combustion,16 oxygenevolution,17 oxidation of toluene and methanol,18 etc. The
higher surface area of mesoporous Co3O4 renders it catalyti-cally more active by facilitating the adsorption and diffusion ofreactant molecules, which favours the catalytic performance ofthe oxides. It is reported that mesoporous Co3O4 is generallysynthesized by either hard templating route using KIT-6,SBA-15 and SBA-16 silica,19,20 and carbon spheres16 as hardtemplates or a soft templating process21 using Pluronic blockcopolymer as the soft templating agent. The templatingmethods are tedious and complicated with extra processingtime for the removal of the templates. However, there are a fewreports for the synthesis of mesoporous Co3O4 by the tem-plate-free method. Li et al.22 synthesized mesoporous quasi-single-crystalline Co3O4 nanowire arrays on different sub-strates, like transparent conductive glass, Si wafer and Cu foil.In recent times, Venugopal et al.23 have reported the prepa-ration of self-assembled hollow mesoporous Co3O4 using Li2O2
as the oxidizing/precipitating agent. Wang et al.24 have pre-pared different shaped Co3O4 using a hydrothermal method.Zheng et al.25 synthesized flower-like mesoporous Co3O4 by asolvothermal method.
In the present study, we have synthesized porous Co3O4
nanostructures of tunable morphology and textural propertiesvia a facile one-pot hydrothermal process at 150 °C by chan-ging the reaction times from 2 h–24 h in the presence of water-based precursors without using any templating agents fol-lowed by calcination at 300 °C. The synthesized particles ofdifferent morphologies have been used as catalysts for the
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c4dt00608a
Sol-Gel Division, CSIR-Central Glass and Ceramic Research Institute,
Kolkata 700 032, India. E-mail: [email protected]; Fax: +91 33 24730957
10248 | Dalton Trans., 2014, 43, 10248–10257 This journal is © The Royal Society of Chemistry 2014
Publ
ishe
d on
22
Apr
il 20
14. D
ownl
oade
d by
Cen
tral
Gla
ss &
Cer
amic
Res
earc
h In
stitu
te (
CG
CR
I) o
n 14
/07/
2014
11:
51:1
7.
View Article OnlineView Journal | View Issue

degradation of Chicago Sky Blue 6B (CSB), a carcinogenic azodye used in the textile, paper, food and pharmaceutical indus-tries.26 Herein, to the best of our knowledge, we report for thefirst time a combined role of morphology, surface area, porevolume and pore geometry of Co3O4 particles on their catalyticefficiency, and their interrelationship.
2. Experimental2.1 Synthesis of Co3O4 nanostructures
In a typical preparation, cobalt nitrate hexahydrate, Co(NO3)2·6H2O (5 mmol) and urea, CO(NH2)2 (25 mmol) were dissolvedin 50 mL of deionized (DI) water under stirring. Then theabove solution was transferred into a 100 mL Teflon-linedautoclave, followed by a hydrothermal treatment at 150 °C for2 h, 5 h, 12 h and 24 h. After the reaction, the samples werecollected by centrifugation, washed with DI water, and dried at60 °C for 4 h. The dried as-prepared samples were calcined at300 °C with a heating rate of 1 °C min−1 and a dwell time of2 h each to obtain Co3O4 nanostructures.
2.2 Characterizations
X-ray diffraction (XRD) studies of the calcined powders wereperformed by a Philips X’Pert Pro PW 3050/60 powder diffracto-meter using Ni-filtered Cu-Kα radiation (λ = 0.15418 nm) oper-ated at 40 kV and 30 mA. The thermal behaviours of theuncalcined (as-prepared) particles were studied by thermogra-vimetry (TG) and differential thermal analysis (DTA; NetzschSTA 449C, Germany) from room temperature to 600 °C in anair atmosphere at a heating rate of 10 °C min−1. The character-istic vibration bands of the products were confirmed by FTIR(Nicolet 5PC, Nicolet Analytical Instruments, Madison, WI)with KBr pellets at a resolution of 4 cm−1. The Raman spec-trum was recorded with a RENISHAW spectrometer with514 nm radiation from an argon laser at room temperature.Nitrogen adsorption–desorption measurements were con-ducted at 77 K with a Quantachrome (ASIQ MP) instrument.The samples were outgassed in a vacuum at 250 °C for 4 hprior to the measurement. The surface area was obtainedusing the Brunauer–Emmet–Teller (BET) method within therelative pressure (P/P0) range of 0.05–0.20 and the pore size dis-tribution was calculated by the Barret–Joyner–Halenda (BJH)method. The nitrogen adsorption volume at a relative pressure(P/P0) of 0.99 was used to determine the pore volume. The mor-phology of the particles was examined by field emission scan-ning electron microscopy, FESEM with a Zeiss, Supra™ 35VPinstrument operating with an accelerating voltage of 10 kV,and transmission electron microscopy, TEM using a Tecnai G230ST (FEI) instrument operating at 300 kV. The UV visiblespectra were recorded using a UV-visible-NIR spectrophoto-meter (UV-3101PC, Shimadzu).
2.3 Catalytic test
In a typical catalytic test, 10 mg of the sample was mixed with4 mL of 10−4 M Chicago Sky Blue 6B (CSB) dye followed by the
addition of 0.44 mL hydrogen peroxide, H2O2 (30%). The finaldye (40 mL) concentration was adjusted to 10−5 M. The reac-tion mixture was monitored after a fixed interval of time usinga UV visible spectrophotometer. The decrease in absorptionintensity at λmax = 618 nm was observed. The kinetic equationsare:
Ct=C0 ¼ At=A0 ð1ÞlnðCt=C0Þ ¼ �kt ð2Þ
where Ct is the dye concentration at time t, C0 is the initial dyeconcentration, At and A0 are absorption intensity at λmax =618 nm at time t and 0 respectively, and k is the rate constantof the reaction. The degradation efficiency of the catalyst wascalculated as 1 − Ct/C0. The structure of Chicago sky blue dyeis as
3. Results and discussion3.1 Characterization of Co3O4 nanostructure
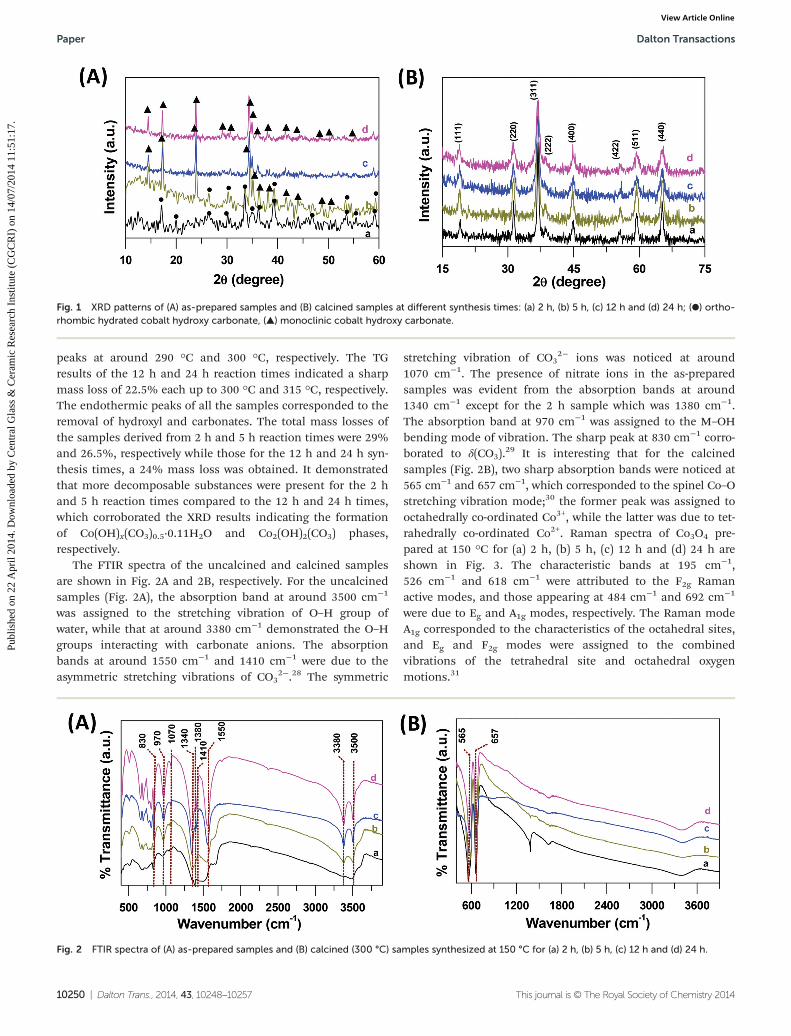
Fig. 1A shows the XRD patterns of the as-prepared samplesindicating the different forms of cobalt hydroxy carbonate. Thesample prepared at 150 °C for a 2 h reaction time resulted inorthorhombic hydrated cobalt hydroxy carbonate, Co(OH)x-(CO3)0.5·0.11H2O (JCPDS file no. 48-0083). Monoclinic cobalthydroxy carbonate, Co2(OH)2(CO3) (JCPDS file no. 29-1416)phase appeared in the presence of orthorhombic hydratedcobalt hydroxy carbonate phase for a 5 h reaction time.However, a complete transformation to monoclinic cobalthydroxy carbonate occurred for 12 h and 24 h synthesis timesat 150 °C. After calcination at 300 °C, cubic Co3O4 phase(JCPDS file no. 43-1003) was formed for all the samples pre-pared at 150 °C for 2 h, 5 h, 12 h and 24 h (Fig. 1B). It is to benoted that the presence of Co2+ in the raw materials is octa-hedrally coordinated by lattice oxygen, and it is in the meta-stable form. The Co3O4 has a spinel structure containing Co3+
in an octahedral coordination and Co2+ in a tetrahedral coordi-nation,27 which is thermodynamically stable. Therefore, in cal-cination of the as-prepared hydrated cobalt hydroxy carbonateand cobalt hydroxy carbonate in an air atmosphere, thethermodynamically stable Co3O4 spinel was formed.
The DTA and TG analysis of the as-prepared (uncalcined)samples is shown in Fig. S1 (ESI†). For a 2 h reaction time(Fig. S1a†), the DTA curve shows endothermic peaks at around165 °C, 260 °C and 285 °C accompanying a sharp mass loss of27% up to 300 °C in the TG curve, while the sample obtainedfor a 5 h reaction time shows endothermic peaks at around260 °C and 290 °C with a sharp mass loss of 24.5% up to300 °C (Fig. S1b†). Interestingly, for 12 h (Fig. S1c†) and 24 h(Fig. S1d†) reaction times, DTA curves exhibited endothermic
Dalton Transactions Paper
This journal is © The Royal Society of Chemistry 2014 Dalton Trans., 2014, 43, 10248–10257 | 10249
Publ
ishe
d on
22
Apr
il 20
14. D
ownl
oade
d by
Cen
tral
Gla
ss &
Cer
amic
Res
earc
h In
stitu
te (
CG
CR
I) o
n 14
/07/
2014
11:
51:1
7.
View Article Online
peaks at around 290 °C and 300 °C, respectively. The TGresults of the 12 h and 24 h reaction times indicated a sharpmass loss of 22.5% each up to 300 °C and 315 °C, respectively.The endothermic peaks of all the samples corresponded to theremoval of hydroxyl and carbonates. The total mass losses ofthe samples derived from 2 h and 5 h reaction times were 29%and 26.5%, respectively while those for the 12 h and 24 h syn-thesis times, a 24% mass loss was obtained. It demonstratedthat more decomposable substances were present for the 2 hand 5 h reaction times compared to the 12 h and 24 h times,which corroborated the XRD results indicating the formationof Co(OH)x(CO3)0.5·0.11H2O and Co2(OH)2(CO3) phases,respectively.
The FTIR spectra of the uncalcined and calcined samplesare shown in Fig. 2A and 2B, respectively. For the uncalcinedsamples (Fig. 2A), the absorption band at around 3500 cm−1
was assigned to the stretching vibration of O–H group ofwater, while that at around 3380 cm−1 demonstrated the O–Hgroups interacting with carbonate anions. The absorptionbands at around 1550 cm−1 and 1410 cm−1 were due to theasymmetric stretching vibrations of CO3
2−.28 The symmetric
stretching vibration of CO32− ions was noticed at around
1070 cm−1. The presence of nitrate ions in the as-preparedsamples was evident from the absorption bands at around1340 cm−1 except for the 2 h sample which was 1380 cm−1.The absorption band at 970 cm−1 was assigned to the M–OHbending mode of vibration. The sharp peak at 830 cm−1 corro-borated to δ(CO3).
29 It is interesting that for the calcinedsamples (Fig. 2B), two sharp absorption bands were noticed at565 cm−1 and 657 cm−1, which corresponded to the spinel Co–Ostretching vibration mode;30 the former peak was assigned tooctahedrally co-ordinated Co3+, while the latter was due to tet-rahedrally co-ordinated Co2+. Raman spectra of Co3O4 pre-pared at 150 °C for (a) 2 h, (b) 5 h, (c) 12 h and (d) 24 h areshown in Fig. 3. The characteristic bands at 195 cm−1,526 cm−1 and 618 cm−1 were attributed to the F2g Ramanactive modes, and those appearing at 484 cm−1 and 692 cm−1
were due to Eg and A1g modes, respectively. The Raman modeA1g corresponded to the characteristics of the octahedral sites,and Eg and F2g modes were assigned to the combinedvibrations of the tetrahedral site and octahedral oxygenmotions.31
Fig. 1 XRD patterns of (A) as-prepared samples and (B) calcined samples at different synthesis times: (a) 2 h, (b) 5 h, (c) 12 h and (d) 24 h; (●) ortho-rhombic hydrated cobalt hydroxy carbonate, (▲) monoclinic cobalt hydroxy carbonate.
Fig. 2 FTIR spectra of (A) as-prepared samples and (B) calcined (300 °C) samples synthesized at 150 °C for (a) 2 h, (b) 5 h, (c) 12 h and (d) 24 h.
Paper Dalton Transactions
10250 | Dalton Trans., 2014, 43, 10248–10257 This journal is © The Royal Society of Chemistry 2014
Publ
ishe
d on
22
Apr
il 20
14. D
ownl
oade
d by
Cen
tral
Gla
ss &
Cer
amic
Res
earc
h In
stitu
te (
CG
CR
I) o
n 14
/07/
2014
11:
51:1
7.
View Article Online
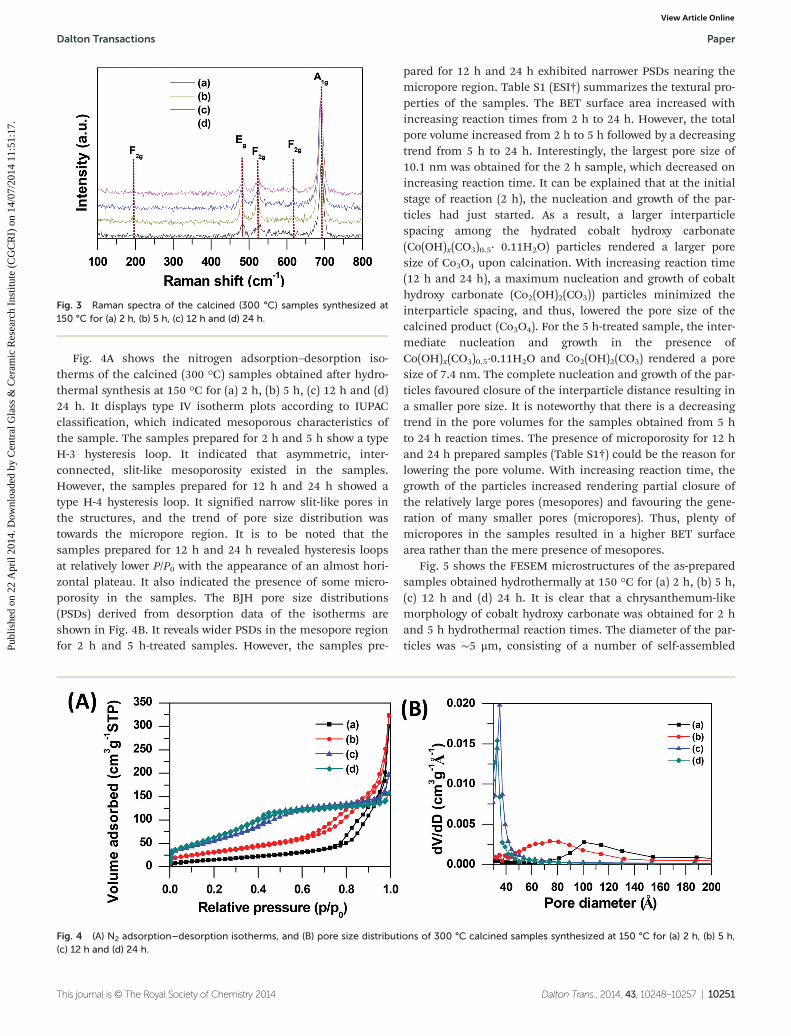
Fig. 4A shows the nitrogen adsorption–desorption iso-therms of the calcined (300 °C) samples obtained after hydro-thermal synthesis at 150 °C for (a) 2 h, (b) 5 h, (c) 12 h and (d)24 h. It displays type IV isotherm plots according to IUPACclassification, which indicated mesoporous characteristics ofthe sample. The samples prepared for 2 h and 5 h show a typeH-3 hysteresis loop. It indicated that asymmetric, inter-connected, slit-like mesoporosity existed in the samples.However, the samples prepared for 12 h and 24 h showed atype H-4 hysteresis loop. It signified narrow slit-like pores inthe structures, and the trend of pore size distribution wastowards the micropore region. It is to be noted that thesamples prepared for 12 h and 24 h revealed hysteresis loopsat relatively lower P/P0 with the appearance of an almost hori-zontal plateau. It also indicated the presence of some micro-porosity in the samples. The BJH pore size distributions(PSDs) derived from desorption data of the isotherms areshown in Fig. 4B. It reveals wider PSDs in the mesopore regionfor 2 h and 5 h-treated samples. However, the samples pre-
pared for 12 h and 24 h exhibited narrower PSDs nearing themicropore region. Table S1 (ESI†) summarizes the textural pro-perties of the samples. The BET surface area increased withincreasing reaction times from 2 h to 24 h. However, the totalpore volume increased from 2 h to 5 h followed by a decreasingtrend from 5 h to 24 h. Interestingly, the largest pore size of10.1 nm was obtained for the 2 h sample, which decreased onincreasing reaction time. It can be explained that at the initialstage of reaction (2 h), the nucleation and growth of the par-ticles had just started. As a result, a larger interparticlespacing among the hydrated cobalt hydroxy carbonate(Co(OH)x(CO3)0.5· 0.11H2O) particles rendered a larger poresize of Co3O4 upon calcination. With increasing reaction time(12 h and 24 h), a maximum nucleation and growth of cobalthydroxy carbonate (Co2(OH)2(CO3)) particles minimized theinterparticle spacing, and thus, lowered the pore size of thecalcined product (Co3O4). For the 5 h-treated sample, the inter-mediate nucleation and growth in the presence ofCo(OH)x(CO3)0.5·0.11H2O and Co2(OH)2(CO3) rendered a poresize of 7.4 nm. The complete nucleation and growth of the par-ticles favoured closure of the interparticle distance resulting ina smaller pore size. It is noteworthy that there is a decreasingtrend in the pore volumes for the samples obtained from 5 hto 24 h reaction times. The presence of microporosity for 12 hand 24 h prepared samples (Table S1†) could be the reason forlowering the pore volume. With increasing reaction time, thegrowth of the particles increased rendering partial closure ofthe relatively large pores (mesopores) and favouring the gene-ration of many smaller pores (micropores). Thus, plenty ofmicropores in the samples resulted in a higher BET surfacearea rather than the mere presence of mesopores.
Fig. 5 shows the FESEM microstructures of the as-preparedsamples obtained hydrothermally at 150 °C for (a) 2 h, (b) 5 h,(c) 12 h and (d) 24 h. It is clear that a chrysanthemum-likemorphology of cobalt hydroxy carbonate was obtained for 2 hand 5 h hydrothermal reaction times. The diameter of the par-ticles was ∼5 μm, consisting of a number of self-assembled
Fig. 3 Raman spectra of the calcined (300 °C) samples synthesized at150 °C for (a) 2 h, (b) 5 h, (c) 12 h and (d) 24 h.
Fig. 4 (A) N2 adsorption–desorption isotherms, and (B) pore size distributions of 300 °C calcined samples synthesized at 150 °C for (a) 2 h, (b) 5 h,(c) 12 h and (d) 24 h.
Dalton Transactions Paper
This journal is © The Royal Society of Chemistry 2014 Dalton Trans., 2014, 43, 10248–10257 | 10251
Publ
ishe
d on
22
Apr
il 20
14. D
ownl
oade
d by
Cen
tral
Gla
ss &
Cer
amic
Res
earc
h In
stitu
te (
CG
CR
I) o
n 14
/07/
2014
11:
51:1
7.
View Article Online
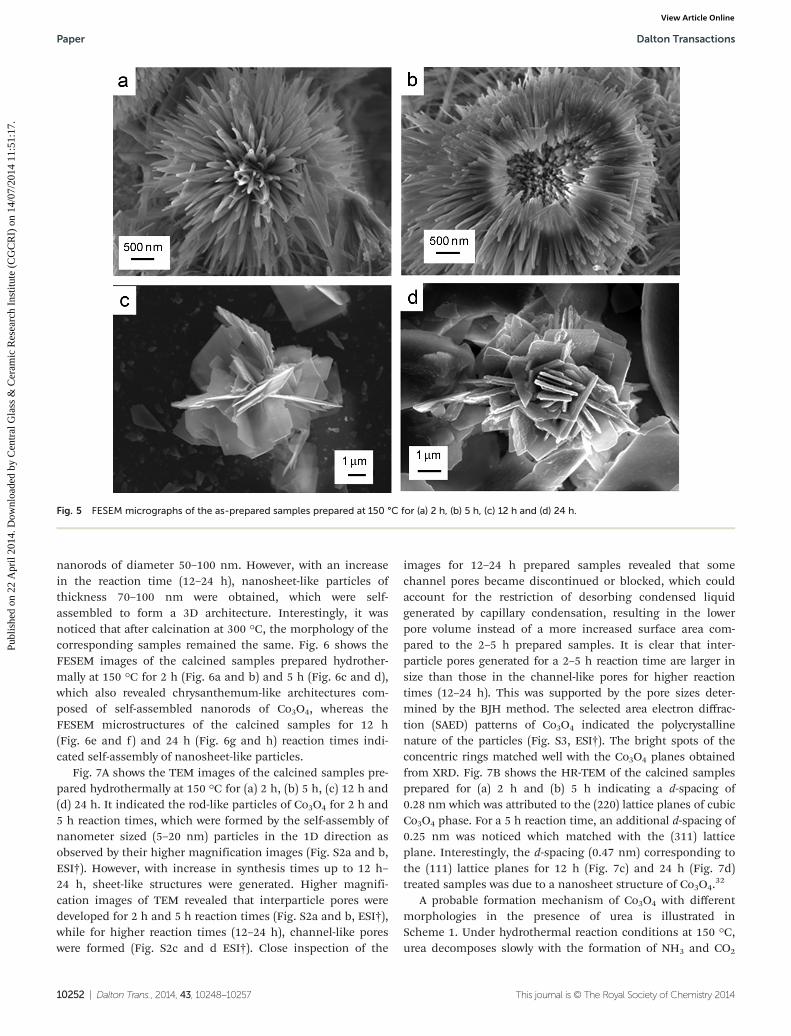
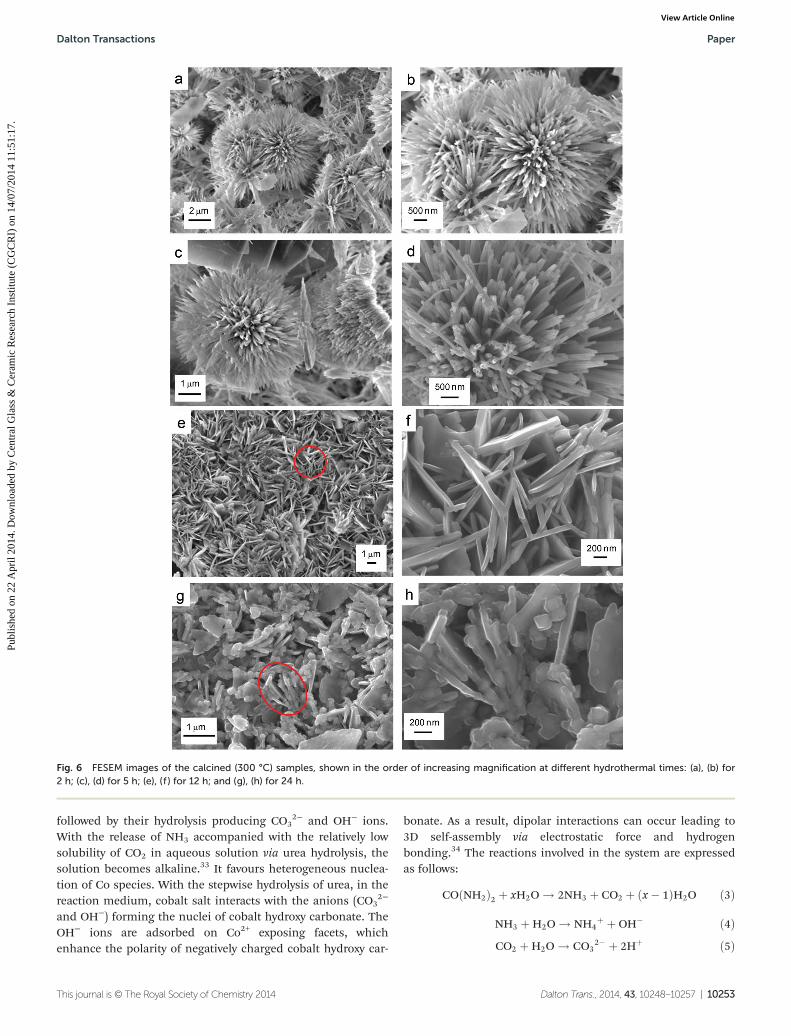
nanorods of diameter 50–100 nm. However, with an increasein the reaction time (12–24 h), nanosheet-like particles ofthickness 70–100 nm were obtained, which were self-assembled to form a 3D architecture. Interestingly, it wasnoticed that after calcination at 300 °C, the morphology of thecorresponding samples remained the same. Fig. 6 shows theFESEM images of the calcined samples prepared hydrother-mally at 150 °C for 2 h (Fig. 6a and b) and 5 h (Fig. 6c and d),which also revealed chrysanthemum-like architectures com-posed of self-assembled nanorods of Co3O4, whereas theFESEM microstructures of the calcined samples for 12 h(Fig. 6e and f) and 24 h (Fig. 6g and h) reaction times indi-cated self-assembly of nanosheet-like particles.
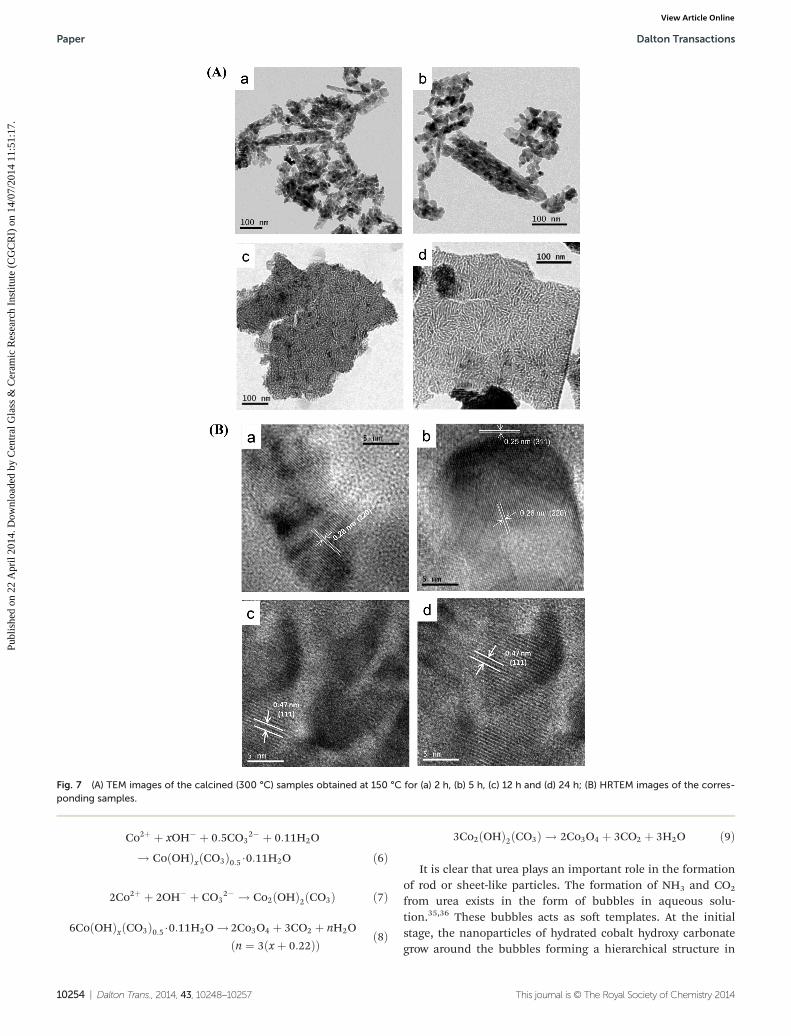
Fig. 7A shows the TEM images of the calcined samples pre-pared hydrothermally at 150 °C for (a) 2 h, (b) 5 h, (c) 12 h and(d) 24 h. It indicated the rod-like particles of Co3O4 for 2 h and5 h reaction times, which were formed by the self-assembly ofnanometer sized (5–20 nm) particles in the 1D direction asobserved by their higher magnification images (Fig. S2a and b,ESI†). However, with increase in synthesis times up to 12 h–24 h, sheet-like structures were generated. Higher magnifi-cation images of TEM revealed that interparticle pores weredeveloped for 2 h and 5 h reaction times (Fig. S2a and b, ESI†),while for higher reaction times (12–24 h), channel-like poreswere formed (Fig. S2c and d ESI†). Close inspection of the
images for 12–24 h prepared samples revealed that somechannel pores became discontinued or blocked, which couldaccount for the restriction of desorbing condensed liquidgenerated by capillary condensation, resulting in the lowerpore volume instead of a more increased surface area com-pared to the 2–5 h prepared samples. It is clear that inter-particle pores generated for a 2–5 h reaction time are larger insize than those in the channel-like pores for higher reactiontimes (12–24 h). This was supported by the pore sizes deter-mined by the BJH method. The selected area electron diffrac-tion (SAED) patterns of Co3O4 indicated the polycrystallinenature of the particles (Fig. S3, ESI†). The bright spots of theconcentric rings matched well with the Co3O4 planes obtainedfrom XRD. Fig. 7B shows the HR-TEM of the calcined samplesprepared for (a) 2 h and (b) 5 h indicating a d-spacing of0.28 nm which was attributed to the (220) lattice planes of cubicCo3O4 phase. For a 5 h reaction time, an additional d-spacing of0.25 nm was noticed which matched with the (311) latticeplane. Interestingly, the d-spacing (0.47 nm) corresponding tothe (111) lattice planes for 12 h (Fig. 7c) and 24 h (Fig. 7d)treated samples was due to a nanosheet structure of Co3O4.
32
A probable formation mechanism of Co3O4 with differentmorphologies in the presence of urea is illustrated inScheme 1. Under hydrothermal reaction conditions at 150 °C,urea decomposes slowly with the formation of NH3 and CO2
Fig. 5 FESEM micrographs of the as-prepared samples prepared at 150 °C for (a) 2 h, (b) 5 h, (c) 12 h and (d) 24 h.
Paper Dalton Transactions
10252 | Dalton Trans., 2014, 43, 10248–10257 This journal is © The Royal Society of Chemistry 2014
Publ
ishe
d on
22
Apr
il 20
14. D
ownl
oade
d by
Cen
tral
Gla
ss &
Cer
amic
Res
earc
h In
stitu
te (
CG
CR
I) o
n 14
/07/
2014
11:
51:1
7.
View Article Online
followed by their hydrolysis producing CO32− and OH− ions.
With the release of NH3 accompanied with the relatively lowsolubility of CO2 in aqueous solution via urea hydrolysis, thesolution becomes alkaline.33 It favours heterogeneous nuclea-tion of Co species. With the stepwise hydrolysis of urea, in thereaction medium, cobalt salt interacts with the anions (CO3
2−
and OH−) forming the nuclei of cobalt hydroxy carbonate. TheOH− ions are adsorbed on Co2+ exposing facets, whichenhance the polarity of negatively charged cobalt hydroxy car-
bonate. As a result, dipolar interactions can occur leading to3D self-assembly via electrostatic force and hydrogenbonding.34 The reactions involved in the system are expressedas follows:
COðNH2Þ2 þ xH2O ! 2NH3 þ CO2 þ ðx� 1ÞH2O ð3Þ
NH3 þH2O ! NH4þ þ OH� ð4Þ
CO2 þH2O ! CO32� þ 2Hþ ð5Þ
Fig. 6 FESEM images of the calcined (300 °C) samples, shown in the order of increasing magnification at different hydrothermal times: (a), (b) for2 h; (c), (d) for 5 h; (e), (f ) for 12 h; and (g), (h) for 24 h.
Dalton Transactions Paper
This journal is © The Royal Society of Chemistry 2014 Dalton Trans., 2014, 43, 10248–10257 | 10253
Publ
ishe
d on
22
Apr
il 20
14. D
ownl
oade
d by
Cen
tral
Gla
ss &
Cer
amic
Res
earc
h In
stitu
te (
CG
CR
I) o
n 14
/07/
2014
11:
51:1
7.
View Article Online
Co2þ þ xOH� þ 0:5CO32� þ 0:11H2O
! CoðOHÞxðCO3Þ0:5 �0:11H2O ð6Þ
2Co2þ þ 2OH� þ CO32� ! Co2ðOHÞ2ðCO3Þ ð7Þ
6CoðOHÞxðCO3Þ0:5 �0:11H2O ! 2Co3O4 þ 3CO2 þ nH2O
ðn ¼ 3ðxþ 0:22ÞÞ ð8Þ
3Co2ðOHÞ2ðCO3Þ ! 2Co3O4 þ 3CO2 þ 3H2O ð9Þ
It is clear that urea plays an important role in the formationof rod or sheet-like particles. The formation of NH3 and CO2
from urea exists in the form of bubbles in aqueous solu-tion.35,36 These bubbles acts as soft templates. At the initialstage, the nanoparticles of hydrated cobalt hydroxy carbonategrow around the bubbles forming a hierarchical structure in
Fig. 7 (A) TEM images of the calcined (300 °C) samples obtained at 150 °C for (a) 2 h, (b) 5 h, (c) 12 h and (d) 24 h; (B) HRTEM images of the corres-ponding samples.
Paper Dalton Transactions
10254 | Dalton Trans., 2014, 43, 10248–10257 This journal is © The Royal Society of Chemistry 2014
Publ
ishe
d on
22
Apr
il 20
14. D
ownl
oade
d by
Cen
tral
Gla
ss &
Cer
amic
Res
earc
h In
stitu
te (
CG
CR
I) o
n 14
/07/
2014
11:
51:1
7.
View Article Online
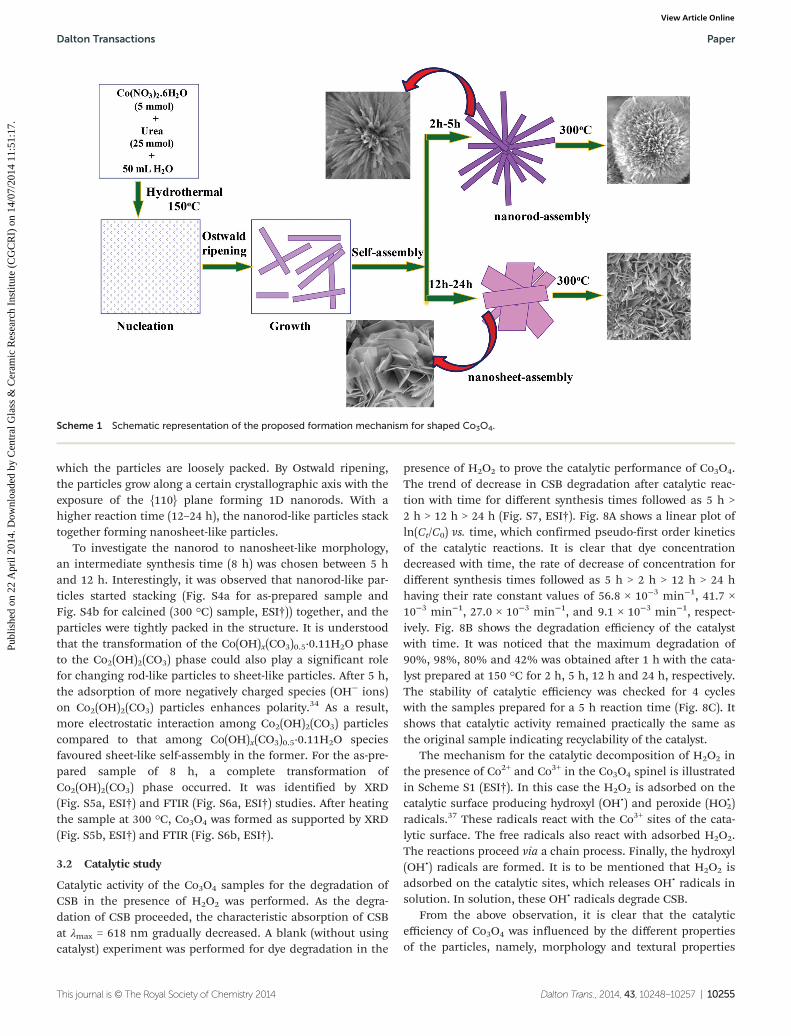
which the particles are loosely packed. By Ostwald ripening,the particles grow along a certain crystallographic axis with theexposure of the {110} plane forming 1D nanorods. With ahigher reaction time (12–24 h), the nanorod-like particles stacktogether forming nanosheet-like particles.
To investigate the nanorod to nanosheet-like morphology,an intermediate synthesis time (8 h) was chosen between 5 hand 12 h. Interestingly, it was observed that nanorod-like par-ticles started stacking (Fig. S4a for as-prepared sample andFig. S4b for calcined (300 °C) sample, ESI†)) together, and theparticles were tightly packed in the structure. It is understoodthat the transformation of the Co(OH)x(CO3)0.5·0.11H2O phaseto the Co2(OH)2(CO3) phase could also play a significant rolefor changing rod-like particles to sheet-like particles. After 5 h,the adsorption of more negatively charged species (OH− ions)on Co2(OH)2(CO3) particles enhances polarity.34 As a result,more electrostatic interaction among Co2(OH)2(CO3) particlescompared to that among Co(OH)x(CO3)0.5·0.11H2O speciesfavoured sheet-like self-assembly in the former. For the as-pre-pared sample of 8 h, a complete transformation ofCo2(OH)2(CO3) phase occurred. It was identified by XRD(Fig. S5a, ESI†) and FTIR (Fig. S6a, ESI†) studies. After heatingthe sample at 300 °C, Co3O4 was formed as supported by XRD(Fig. S5b, ESI†) and FTIR (Fig. S6b, ESI†).
3.2 Catalytic study
Catalytic activity of the Co3O4 samples for the degradation ofCSB in the presence of H2O2 was performed. As the degra-dation of CSB proceeded, the characteristic absorption of CSBat λmax = 618 nm gradually decreased. A blank (without usingcatalyst) experiment was performed for dye degradation in the
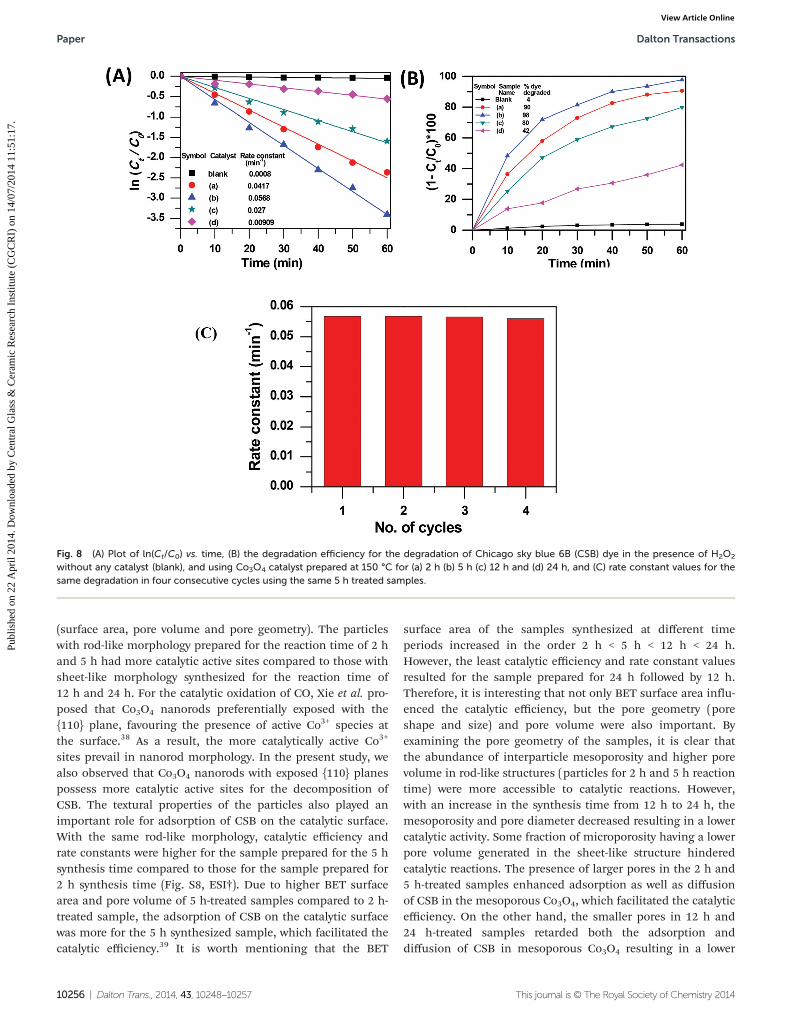
presence of H2O2 to prove the catalytic performance of Co3O4.The trend of decrease in CSB degradation after catalytic reac-tion with time for different synthesis times followed as 5 h >2 h > 12 h > 24 h (Fig. S7, ESI†). Fig. 8A shows a linear plot ofln(Ct/C0) vs. time, which confirmed pseudo-first order kineticsof the catalytic reactions. It is clear that dye concentrationdecreased with time, the rate of decrease of concentration fordifferent synthesis times followed as 5 h > 2 h > 12 h > 24 hhaving their rate constant values of 56.8 × 10−3 min−1, 41.7 ×10−3 min−1, 27.0 × 10−3 min−1, and 9.1 × 10−3 min−1, respect-ively. Fig. 8B shows the degradation efficiency of the catalystwith time. It was noticed that the maximum degradation of90%, 98%, 80% and 42% was obtained after 1 h with the cata-lyst prepared at 150 °C for 2 h, 5 h, 12 h and 24 h, respectively.The stability of catalytic efficiency was checked for 4 cycleswith the samples prepared for a 5 h reaction time (Fig. 8C). Itshows that catalytic activity remained practically the same asthe original sample indicating recyclability of the catalyst.
The mechanism for the catalytic decomposition of H2O2 inthe presence of Co2+ and Co3+ in the Co3O4 spinel is illustratedin Scheme S1 (ESI†). In this case the H2O2 is adsorbed on thecatalytic surface producing hydroxyl (OH•) and peroxide (HO•
2)radicals.37 These radicals react with the Co3+ sites of the cata-lytic surface. The free radicals also react with adsorbed H2O2.The reactions proceed via a chain process. Finally, the hydroxyl(OH•) radicals are formed. It is to be mentioned that H2O2 isadsorbed on the catalytic sites, which releases OH• radicals insolution. In solution, these OH• radicals degrade CSB.
From the above observation, it is clear that the catalyticefficiency of Co3O4 was influenced by the different propertiesof the particles, namely, morphology and textural properties
Scheme 1 Schematic representation of the proposed formation mechanism for shaped Co3O4.
Dalton Transactions Paper
This journal is © The Royal Society of Chemistry 2014 Dalton Trans., 2014, 43, 10248–10257 | 10255
Publ
ishe
d on
22
Apr
il 20
14. D
ownl
oade
d by
Cen
tral
Gla
ss &
Cer
amic
Res
earc
h In
stitu
te (
CG
CR
I) o
n 14
/07/
2014
11:
51:1
7.
View Article Online
(surface area, pore volume and pore geometry). The particleswith rod-like morphology prepared for the reaction time of 2 hand 5 h had more catalytic active sites compared to those withsheet-like morphology synthesized for the reaction time of12 h and 24 h. For the catalytic oxidation of CO, Xie et al. pro-posed that Co3O4 nanorods preferentially exposed with the{110} plane, favouring the presence of active Co3+ species atthe surface.38 As a result, the more catalytically active Co3+
sites prevail in nanorod morphology. In the present study, wealso observed that Co3O4 nanorods with exposed {110} planespossess more catalytic active sites for the decomposition ofCSB. The textural properties of the particles also played animportant role for adsorption of CSB on the catalytic surface.With the same rod-like morphology, catalytic efficiency andrate constants were higher for the sample prepared for the 5 hsynthesis time compared to those for the sample prepared for2 h synthesis time (Fig. S8, ESI†). Due to higher BET surfacearea and pore volume of 5 h-treated samples compared to 2 h-treated sample, the adsorption of CSB on the catalytic surfacewas more for the 5 h synthesized sample, which facilitated thecatalytic efficiency.39 It is worth mentioning that the BET
surface area of the samples synthesized at different timeperiods increased in the order 2 h < 5 h < 12 h < 24 h.However, the least catalytic efficiency and rate constant valuesresulted for the sample prepared for 24 h followed by 12 h.Therefore, it is interesting that not only BET surface area influ-enced the catalytic efficiency, but the pore geometry (poreshape and size) and pore volume were also important. Byexamining the pore geometry of the samples, it is clear thatthe abundance of interparticle mesoporosity and higher porevolume in rod-like structures (particles for 2 h and 5 h reactiontime) were more accessible to catalytic reactions. However,with an increase in the synthesis time from 12 h to 24 h, themesoporosity and pore diameter decreased resulting in a lowercatalytic activity. Some fraction of microporosity having a lowerpore volume generated in the sheet-like structure hinderedcatalytic reactions. The presence of larger pores in the 2 h and5 h-treated samples enhanced adsorption as well as diffusionof CSB in the mesoporous Co3O4, which facilitated the catalyticefficiency. On the other hand, the smaller pores in 12 h and24 h-treated samples retarded both the adsorption anddiffusion of CSB in mesoporous Co3O4 resulting in a lower
Fig. 8 (A) Plot of ln(Ct/C0) vs. time, (B) the degradation efficiency for the degradation of Chicago sky blue 6B (CSB) dye in the presence of H2O2
without any catalyst (blank), and using Co3O4 catalyst prepared at 150 °C for (a) 2 h (b) 5 h (c) 12 h and (d) 24 h, and (C) rate constant values for thesame degradation in four consecutive cycles using the same 5 h treated samples.
Paper Dalton Transactions
10256 | Dalton Trans., 2014, 43, 10248–10257 This journal is © The Royal Society of Chemistry 2014
Publ
ishe
d on
22
Apr
il 20
14. D
ownl
oade
d by
Cen
tral
Gla
ss &
Cer
amic
Res
earc
h In
stitu
te (
CG
CR
I) o
n 14
/07/
2014
11:
51:1
7.
View Article Online

catalytic efficiency. Therefore, it can be demonstrated that manyfactors like morphology, surface area, pore volume and poregeometry of the particles are important for catalytic reactions.
4. Conclusions
In summary, we have demonstrated a facile hydrothermal syn-thesis of Co3O4 nanostructures with controllable morphologyand tunable textural properties via an aqueous based route inthe absence of templating agents. Catalytic performance forthe degradation of Chicago Sky Blue 6B in the presence ofH2O2 was studied with Co3O4 particles. The affects of mor-phology and textural properties on the catalytic behaviors ofCo3O4 were examined. It was revealed that nanorod-like par-ticles with a higher pore volume and mesoporosity obtainedfrom 2 h and 5 h treated samples were more active in catalysiscompared to those of nanosheet-like particles obtained from12 h and 24 h treated samples having a smaller pore volumealong with the presence of microporosity which hindered cata-lytic activity. By tuning the crystal morphology and surface pro-perties, Co3O4 and other transition metal oxides could beapplicable in many other fields.
Acknowledgements
The authors would like to thank the Director of this institutefor his kind permission to publish this paper. The authors(M. Roy and S. Ghosh) are thankful to CSIR for their fellow-ship. The Nano Structured Materials Division is thankfullyacknowledged for Raman spectra. The work was funded byDST-SERB Project, Government of India (no. GAP 0616).
References
1 W. Y. Li, L.-N. Xu and J. Chen, Adv. Funct. Mater., 2005, 15,851.
2 P. Polzot, S. Laruelle, S. Grugeon, L. Dupont andJ. M. Tarascon, Nature, 2000, 407, 496.
3 G. Wang, X. Shen, J. Horvat, B. Wang, H. Liu, D. Wexlerand J. Yao, J. Phys. Chem. C, 2009, 113, 4357.
4 M. Yada, Y. Inoue, M. Koikawa, T. Torikai and T. Watari,CrystEngComm, 2012, 14, 7374.
5 T.-L. Lai, Y.-L. Lai, C.-C. Lee, Y.-Y. Shu and C.-B. Wang,Catal. Today, 2008, 131, 105.
6 H. Sun, H. Ming Ang, M. O. Tade and S. Wang, J. Mater.Chem. A, 2013, 1, 14427.
7 X. Ke, J. Cao, M. Zheng, Y. Chen, J. Liu and G. Ji, Mater.Lett., 2007, 61, 3901.
8 Y. Li, B. Tan and Y. Wu, Nano Lett., 2008, 8, 265.9 X. W. Lou, D. Deng, J. Y. Lee, J. Feng and L. A. Archer, Adv.
Mater., 2008, 20, 258.10 Y. Wang, H. Xia, L. Lu and J. Lin, ACS Nano, 2010, 4, 1425.11 M. Wang, L. Zeng and Q. Chen, Dalton Trans., 2011, 40,
597.
12 L. Chen, J. Hu, R. Richards, S. Prikhodko andS. Kodambaka, Nanoscale, 2010, 2, 1657.
13 X. Wang, H. Guan, S. Chen, H. Li, T. Zhai, D. Tang,Y. Bando and D. Golberg, Chem. Commun., 2011, 47, 12280.
14 K. Nakajima, T. Fukui, H. Kato, M. Kitano, J. N. Kondo,S. Hayashi and M. Hara, Chem. Mater., 2010, 22, 3332.
15 J. P. Cheng, X. B. Zhang, Y. Ye, J. P. Tu, F. Liu, X. Y. Tao,H. J. Geise and T. G. Van, Microporous Mesoporous Mater.,2005, 81, 73.
16 C.-A. Wang, S. Li and L. An, Chem. Commun., 2013, 49,7427.
17 J. Rosen, G. S. Hutchings and F. Jiao, J. Am. Chem. Soc.,2013, 135, 4516.
18 Y. Xia, H. Dai, H. Jiang and L. Zhang, Catal. Commun.,2010, 11, 1171.
19 G. Wang, H. Liu, J. Horvat, B. Wang, S. Qiao, J. Park andH. Ahnidl, Chem. – Eur. J., 2010, 16, 11020.
20 J. Deng, L. Zhang, H. Dai, Y. Xia, H. Jiang, H. Zhang andH. He, J. Phys. Chem. C, 2010, 114, 2694.
21 N. Dahal, I. A. Ibarra and S. M. Humphrey, J. Mater. Chem.,2012, 22, 12675.
22 Y. Li, B. Tan and Y. Wu, J. Am. Chem. Soc., 2006, 128,14258.
23 N. Venugopal, D.-J. Lee, Y. J. Lee and Y.-K. Sun, J. Mater.Chem. A, 2013, 1, 13164.
24 H. Wang, L. Zhang, X. Tan, C. M. B. Holt, B. Zahiri,B. C. Olsen and D. Mitlin, J. Phys. Chem. C, 2011, 115,17599.
25 J. Zheng, J. Liu, D. Lv, Q. Kuang, Z. Jiang, Z. Xie, R. Huangand L. Zheng, J. Solid State Chem., 2010, 183, 600.
26 A. K. Mohammed and K. T. McKenzie, J. Environ. Sci., 2005,17, 869.
27 S. C. Petitto, E. M. Marsh, G. A. Carson and M. A. Langell,J. Mol. Catal. A: Chem., 2008, 281, 49.
28 J. Yang, H. Cheng and R. L. Frost, Spectrochim. Acta, Part A,2011, 78, 420.
29 R. Xu and H. C. Zeng, J. Phys. Chem. B, 2003, 107, 12643.30 H.-P. Cong and S.-H. Yu, Cryst. Growth Des., 2009, 9, 210.31 C. W. Na, H.-S. Woo, H.-J. Kim, U. Jeong, J.-H. Chung and
J.-H. Lee, CrystEngComm, 2012, 14, 3337.32 H. Liang, J. M. Raitano, L. Zhang and S.-W. Chan, Chem.
Commun., 2009, 7569.33 J. H. Pan, X. Zhang, A. J. Du, H. Bai, J. Ng and D. Sun, Phys.
Chem. Chem. Phys., 2012, 14, 7481.34 J. H. Pan, Q. Huang, Z. Y. Koh, D. Neo, X. Z. Wang and
Q. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 6292.35 J. Sun, G. Chen, J. Wu, H. Dong and G. Xiong, Appl. Catal.,
B, 2013, 132–133, 304.36 C. Zhou, Y. Zhao, T. Bian, L. Shang, H. Yu, L.-Z. Wu,
C.-H. Tung and T. Zhang, Chem. Commun., 2013, 49, 9872.37 S.-S. Lin and M. D. Gurol, Environ. Sci. Technol., 1998, 32,
1417.38 X. Xie, Y. Li, Z.-Q. Liu, M. Haruta and W. Shen, Nature,
2009, 458, 746.39 C. Yu, X. Dong, L. Guo, J. Li, F. Qin, L. Zhang, J. Shi and
D. Yan, J. Phys. Chem. C, 2008, 112, 13378.
Dalton Transactions Paper
This journal is © The Royal Society of Chemistry 2014 Dalton Trans., 2014, 43, 10248–10257 | 10257
Publ
ishe
d on
22
Apr
il 20
14. D
ownl
oade
d by
Cen
tral
Gla
ss &
Cer
amic
Res
earc
h In
stitu
te (
CG
CR
I) o
n 14
/07/
2014
11:
51:1
7.
View Article Online




















