
Ironless transducer for measuring the mechanical properties of porous materials

ARTHRITIS & RHEUMATISMVol. 44, No. 10, October 2001, pp 2296–2310© 2001, American College of RheumatologyPublished by Wiley-Liss, Inc.
Synergistic Induction of Matrix Metalloproteinase 1 byInterleukin-1� and Oncostatin M in Human Chondrocytes
Involves Signal Transducer and Activator ofTranscription and Activator Protein 1 Transcription Factors
via a Novel Mechanism
J. B. Catterall,1 S. Carrere,1 P. J. T. Koshy,1 B. A. Degnan,1 W. D. Shingleton,1
C. E. Brinckerhoff,2 J. Rutter,2 T. E. Cawston,1 and A. D. Rowan1
Objective. To investigate the mechanism ofinterleukin-1� (IL-1�) and oncostatin M (OSM) syner-gistic regulation of matrix metalloproteinase 1(MMP-1) in human chondrocytes.
Methods. Using an immortalized human chon-drocyte cell line (T/C28a4), we investigated regulation ofthe MMP-1 gene. Northern blotting and flow cytometricanalysis were used to assess changes in receptor,MMP-1, and c-fos expression. Transient transfectionsusing MMP-1 promoter/luciferase constructs, electro-phoretic mobility shift assay, and site-directed mu-tagenesis were used to investigate MMP-1 promoteractivation.
Results. We found no alteration in the expressionof receptors used by these cytokines after stimulationwith IL-1�/OSM. Using MMP-1 promoter/luciferasereporter constructs, we found that the proximal (�517/�63) region of the MMP-1 promoter was sufficient tosupport a synergistic activation. A role for activatedsignal transducers and activators of transcription(STAT-3) was demonstrated, although no binding ofSTAT-3 to the MMP-1 promoter was found. However,
constitutive binding of activator protein 1 (AP-1) wasdetected, and changes in c-fos expression could modu-late promoter activity.
Conclusion. Since no changes in receptor expres-sion were observed, receptor modulation cannot accountfor the IL-1�/OSM synergy observed. Instead, the inter-play of various intracellular signaling pathways is amore likely explanation. STAT activation is required,but STAT proteins do not interact directly with theMMP-1 promoter. We propose that activated STATsstimulate c-fos expression, and changes in expression ofthe AP-1 components regulate MMP-1 expression. Wehighlight a new mechanism for MMP-1 regulation inhuman chondrocytes that could provide potential newtherapeutic targets.
Rheumatoid arthritis (RA) is a disease in whichthe end stage is characterized by loss of joint functiondue to degradation of articular cartilage. Proteoglycansand collagen are the 2 major components of this extra-cellular matrix; they provide both the resistance tocompressive forces and its tensile strength. A family ofclosely related enzymes, the matrix metalloproteinases(MMPs), which collectively can degrade all the compo-nents of the extracellular matrix of articular cartilage,have been strongly implicated in the disease process (1).Recent evidence now suggests that proteoglycan turn-over is mediated by ADAM-TS4 and/or ADAM-TS5(2,3). However, proteoglycan can be replaced relativelyrapidly once the proinflammatory cytokine stimulus,such as interleukin-1� (IL-1�), has been removed (4). Inmarked contrast, collagen is released much less readily,but when degradation of collagen does occur, the struc-
Supported by the Arthritis Research Campaign.1J. B. Catterall, PhD, S. Carrere, PhD, P. J. T. Koshy, PhD,
B. A. Degnan, PhD, W. D. Shingleton, PhD, T. E. Cawston, PhD,A. D. Rowan, PhD: University of Newcastle upon Tyne, Newcastleupon Tyne, UK; 2C. E. Brinckerhoff, PhD, J. Rutter, PhD: DartmouthMedical School, Hanover, New Hampshire.
Address correspondence and reprint requests to J. B. Catter-all, PhD, Department of Rheumatology, School of Clinical MedicalSciences, Cookson Building, The Medical School, University of New-castle upon Tyne, Framlington Place, Newcastle upon Tyne NE2 4HH,UK. E-mail: [email protected]
Submitted for publication November 10, 2000; accepted inrevised form May 24, 2001.
2296

tural integrity of the tissue is irreversibly lost (5).Collagen degradation therefore represents a key controlpoint in cartilage turnover.
The collagenases (MMP-1, MMP-8, and MMP-13) (6–8) specifically cleave triple-helical collagen togive characteristic three-quarter and one-quarter frag-ments, a unique ability that has made these MMPs andtheir regulators important targets for therapeutic inter-vention in rheumatic diseases. The relative contributionsof each collagenase in the degradation of cartilagecollagen within the rheumatoid joint remain unclear,although MMP-1 and MMP-13 have been most stronglyimplicated (9–12). MMP-1 has been shown to be presentin rheumatoid synovial fluid (13), can be localized to RAsynovial tissue (including sites of cartilage erosion)(9,14), and is up-regulated by IL-1� (15), which is alsopresent in RA synovial fluid (16).
These findings clearly suggest a role for MMP-1in the breakdown of cartilage collagen in arthritic tis-sues. We have demonstrated that IL-1� can synergizewith the IL-6 family cytokine oncostatin M (OSM) topromote dramatic cartilage collagen degradation thatwas associated with MMP-1 (17,18). The presence ofOSM in RA synovial fluid (18,19) has been shown, andrecently it was demonstrated that IL-6 can also synergizewith IL-1� in a manner similar to that of OSM inpromoting cartilage breakdown when a soluble IL-6receptor (sIL-6R) is present (20). Elevated levels of IL-6and sIL-6R have also been documented in RA (21–24).
OSM and IL-6 have been shown to induce tissueinhibitor of metalloproteinases 1 (TIMP-1) production(25,26), implying that they should be chondroprotective.However, when these cytokines are combined with IL-1�, they act in a proinflammatory manner to promotedramatic cartilage destruction (17,18,20). OSM mediatesits effects on chondrocytes via the OSM-specific recep-tor (OSMR�) (27) in conjunction with glycoprotein 130(gp130) (20), a common cell surface signaling subunitfor IL-6 family cytokines, which transduces signals pre-dominantly through the janus-activated kinase/signaltransducer and activator of transcription (JAK/STAT)pathway (28,29). An OSM response element (OSMRE)(30) in the human TIMP-1 and MMP-1 promoters hasbeen described (31), and consists of a bipartite activatorprotein 1 (AP-1) and STAT binding element (SBE), inwhich both sites must be occupied for maximum tran-scriptional activation (31).
AP-1 components are expressed in high levels inRA (32–34) compared with those in osteoarthritis (OA)(32,35), and are important in the transcriptional activa-tion of several MMPs, including MMP-1 (36). The
importance of AP-1 complexes in cartilage breakdown,in particular c-fos, was demonstrated when oligonucleo-tides that mimic an AP-1 binding site inhibited jointdestruction in a collagen-induced arthritis model (37).Activation of AP-1 was also shown to occur before theonset of arthritis in this model (35). Active STATs havebeen detected in inflamed synovial tissues (38,39), andRA synovial fluid can cause STAT activation (40). IL-1signal transduction is complex, via the type 1 IL-1receptor (IL-1RI) in association with an IL-1 receptoraccessory protein (IL-1R–Acp) (41,42), with severalsignaling cascades implicated, including p38 mitogen-activated protein kinase (p38 MAPK), protein kinases Aand C (PKA, PKC), and nuclear factor �B (NF-�B)(43,44).
We previously proposed that IL-1� and OSM(18) (as well as IL-1� and IL-6) (20) represent possibleimportant mechanisms by which cartilage collagen de-struction can be effected. We have also implicatedMMP-1 in this process (18,20). In the present study, weinitially investigated whether combinations of IL-1� andOSM up-regulated the expression of relevant cell sur-face receptors on chondrocytes. We then assessed thecontributions AP-1 and STAT complexes have on theinduction of MMP-1 in chondrocytes stimulated with acombination of IL-1� and OSM.
MATERIALS AND METHODS
Reagents. Chemicals were obtained from the followingsuppliers: IL-1� was a generous gift from Glaxo Wellcome(Stevenage, UK); OSM (45), leukemia inhibitory factor (LIF)(46), and antibodies to gp130 and OSMR� were from Profes-sor J. Heath (University of Birmingham, Birmingham, UK);cardiotrophin 1 (CT-1) was from Dr. C. Richards (McMasterUniversity, Hamilton, Ontario, Canada); and IL-6 and sIL-6Rwere purchased from R&D Systems (Abingdon, UK). All wererecombinant human in origin, stored at �70°C, and dilutedinto culture medium immediately prior to use. Human (unlessotherwise stated) complementary DNA (cDNA) for MMP-1and TIMP-1 was obtained from Dr. G. Wells (British Biotech,Oxford, UK); LIF receptor (LIFR) (46), gp130, OSMR� (27),and rat GAPDH cDNA (47) were obtained as referenced. Thefollowing MMP-1 promoter fragments were subcloned intopGL3-Basic vector (Promega, Madison, WI): �4372/�63,�3300/�63, �2900/�63, �1600/�63, and �517/�63 (48). The�711/�45 c-fos promoter/luciferase plasmid was as previouslydescribed (49). Expression vectors for c-fos (50) and theprotease inhibitor of activated STAT-3 (PIAS-3) (51) weregenerously provided by Dr. I. Verma (Salk Institute forBiological Studies, San Diego, CA) and Professor K. Shuai(University of California, Los Angeles), respectively. All otherchemicals and biochemicals were commercially availableanalytical-grade reagents obtained from Fisher Scientific UK
SYNERGISTIC INDUCTION OF MMP-1 BY IL-1� AND OSM 2297

(Loughborough, UK), BDH Chemicals (Poole, UK), or Sigma-Aldrich (Poole, UK).
Tissue culture. We used an immortalized human chon-drocyte cell line, T/C28a4, as described previously (52). Cellswere maintained in Dulbecco’s modified Eagle’s mediumsupplemented with 10% bovine fetal calf serum (FCS), 100IU/ml of penicillin, 100 �g/ml of streptomycin, 40 units/ml ofnystatin, and 10 �g/ml of ascorbate (C28a medium) usingstandard tissue culture procedures as described previously(18). Cytotoxicity was assessed using lactate dehydrogenase(LDH) in the CytoTox96 nonradioactive cytotoxicity assay(Promega).
Flow cytometric analysis. Flow cytometric analysis wasperformed as described previously (20). Briefly, cells weremaintained in 1% acid-treated FCS (ATFCS) with or withoutcytokine stimulation for 24–72 hours. Cells were harvested in0.5 mM EDTA before incubation with appropriate antibody orantiserum for 1 hour at 4oC. Antibody binding was detectedusing a flow cytometer with CellQuest software (Becton Dick-inson, Oxford, UK). Isotype control antibodies were used todetermine nonspecific binding. Optimal labeling concentra-tions of all antibodies and sera were determined using a panelof control cell lines known to express 1 of the various recep-tors: HeLa (�gp130, �OSMR�, �LIFR), A549 (�gp130,�OSMR�, �LIFR), Raji cells (�gp130, �OSMR�, �LIFR),SKW6.4 (�gp130, �OSMR�, �LIFR), and parental B cell–activating factor (�gp130, �OSMR�, �LIFR).
Northern blot analysis. Northern blotting was per-formed as described previously (18). Briefly, 95% confluentT/C28a4 cells were maintained in FCS-free medium with orwithout cytokine stimulation. Total RNA was isolated using anRNeasy kit (Qiagen, Crawley, UK). Samples were electropho-resed, transferred to GeneScreen Plus membrane (NEN LifeScience Products, Hounslow, UK), probed with random-primed 32P-labeled cDNA for 16 hours before detection eitherby using autoradiographic film or by exposure to a phosphorscreen (Molecular Dynamics, Chesham, UK), and visualizedwith the Storm 860 PhosphorImager (Molecular Dynamics).Radiolabeled bands were quantified by densitometry (Image-Master 1D image analysis software; Amersham PharmaciaBiotech, Little Chalfont, UK). Values were normalized toGAPDH expression.
Polyacrylamide gel electrophoresis and Western blot-ting. T/C28a4 cells were stimulated in ATFCS medium beforesolubilization in 200 �l of sample buffer (0.1M Tris HCl, pH6.8, 0.35M sodium dodecyl sulfate [SDS], 20% [weight/volume]glycerol, 2% (volume/volume) saturated bromphenol blue, and5% (v/v) �-mercaptoethanol). Samples were electrophoresedon a 10% mini SDS-polyacrylamide gel prior to transfer to aProtran nitrocellulose membrane (Schleicher & Schuell, Das-sel, Germany) by electroblotting in 20 mM Tris HCl, 0.6Mglycine, and 20% (v/v) methanol. Membranes were blocked in5% (w/v) milk proteins in Tris buffered saline before detectionof c-fos using a goat anti-human c-fos polyclonal antibody(c-fos[4]-G; Santa Cruz Biotechnology, Santa Cruz, CA) andhorseradish peroxidase–conjugated swine anti-goat IgG(Dako, Ely, UK). Protein bands were visualized with 25 ml of1.25 mM Na-luminol (Sigma) in 0.1M Tris HCl, pH 8.0, 250 mlof 41 mM iodophenol in DMSO, and 7.5 ml of 30% (v/v)hydrogen peroxide solution by exposure to autoradiographyHyperfilm (Amersham Pharmacia Biotech).
Transient transfection and reporter gene assay. Amethod modified from that described previously (31,53) wasused for transient transfection and reporter gene assay. Briefly,10-cm tissue culture dishes of T/C28a4 cells at 70–80% con-fluence were transfected with 8 �g of plasmid and 32 �l oflipofectamine (Gibco, Paisley, UK) for 5 hours in 2 ml ofFCS-free medium. Cells were left to recover overnight in C28amedium before splitting into 2 � 6 well plates and then left torecover for 24 hours in ATFCS medium. Transfected cellswere then stimulated overnight in ATFCS medium beforeharvesting, and luciferase activity was determined using 5�reporter lysis buffer and a luciferase assay (Promega) in awhite-walled 96-well plate (Wallac, Milton Keynes, UK) on a1450 MicroBeta TriLux liquid scintillation and luminescencecounter (Wallac). Luciferase results were normalized to theamount of plasmid per well, as determined by Hirt’s assay (54).In cotransfection experiments, equal amounts of plasmid wereused with the appropriate control vector.
Site-directed mutagenesis. Mutations were performedin the �517/�63 MMP-1 promoter construct using a 2-stagepolymerase chain reaction (PCR) process to replace TT withAA in the SBE. The reverse primer was also engineered in aHind III site by replacing CTT with AAG for subcloning. In theprimers listed below, the location of the SBE site is underlined,and mutated basepairs are shown in lowercase. The pointmutations used for this construct differ from those used for themutated SBE (mSBE)–OSMRE oligonucleotide (31) since thismutation also removes an Ets binding site, while the mutationsin this study only removed the STAT site as determined byMatInspector V2.2 software (55). In the first stage, 2 separatePCR reactions were performed using the following primer pairs:
Figure 1. Expression of matrix metalloproteinase 1 (MMP-1) andtissue inhibitor of metalloproteinases 1 (TIMP-1) messenger RNAfollowing stimulation of T/C28a4 chondrocytes with interleukin-1�(IL-1�) and/or oncostatin M (OSM). Confluent T/C28a4 chondrocyteswere serum starved for 24 hours before cytokine stimulation. Cellswere stimulated with IL-1� (1 ng/ml) and OSM (10 ng/ml) for 24 hoursas follows: 1) unstimulated, 2) IL-1�, 3) OSM, and 4) IL-1�/OSM.Total RNA was harvested and probed by Northern blot using comple-mentary DNA for human MMP-1 and TIMP-1. GAPDH was used tocorrect for RNA loading.
2298 CATTERALL ET AL

forward primer 5�-GCAATAGGGTACCAGGCAGCT and mu-tant reverse 5�-GCCCTTCCAGAttGCCAGAGGCTGTC-3�,mutant forward 5�-GACAGCCTCTGGCaaTCTGGAAGGGCand reverse primer 5�-GGCCTTTGTAAGCTTTCTCAGTG.From each of the first-stage reactions, 1 �l of each solution wascombined into a second PCR using the forward and reverseprimers. The resulting 593-bp product was gel purified usingthe QIAEX II gel extraction kit (Qiagen), restriction wasdigested with Hind III and Asp 718 (Roche Molecular Bio-chemicals, Lewes, UK), and was ligated into pGL3-Basicvector (Promega), which was prepared using the same en-zymes. The mutated construct was verified by automatedfluorescent dideoxy sequencing.
Electrophoretic mobility shift assay (EMSA). Crudenuclear extracts were prepared by modifying previous methods(31,53). All extraction buffers contained complete proteaseinhibitors with EDTA (Roche Molecular Biochemicals). Cellswere maintained for 24 hours in ATFCS medium beforestimulation. Cells were initially treated on ice with 20 mM
HEPES, pH 7.9, 1 mM dithiothreitol, and 0.2% (v/v) TritonX-100 for 5 minutes and pelleted at 21,000g at 4oC for 2minutes. The cell pellets were incubated with 100 �l of 20 mMHEPES, pH 7.9, 1 mM dithiothreitol, 0.4M NaCl, and 20%(v/v) glycerol on ice for another 40 minutes. Samples werecentrifuged again (5 minutes, 4oC at 21,000g) and the pelletswere discarded. Supernatants were aliquoted, snap-frozen inliquid nitrogen, and stored at �80oC until required. Proteinconcentration was then determined using the BCA assay(Pierce, Chester, UK).
Oligonucleotides were annealed in equimolar amountsand 50-pM ends were labeled with �-32P–dCTP using Klenowend labeling. Binding reactions were performed under thefollowing conditions: 2 �l of 5� binding buffer (0.25M TrisHCl, pH 8.0, 5 mM EDTA, 5 mM dithiothreitol, 0.4M NaCl,and 25% [v/v] glycerol), 1 �l of poly(dI-dC) (2 �g/�l; Amer-sham Pharmacia Biotech), 3 �l of nuclear extract (5–20 �g ofprotein), 3 �l of labeled probe (1,500–3,000 cycles per second/�l), and 1 �l of water. This was incubated at room temperature
Figure 2. Cell surface expression of OSM receptor components glycoprotein 130 (gp130),leukemia inhibitory factor receptor (LIFR), and OSM–specific receptor (OSMR�) in T/C28a4chondrocytes following stimulation with IL-1� and/or OSM. Confluent T/C28a4 chondrocytes weregrown in acid-treated fetal calf serum medium for 24 hours prior to cytokine stimulation. Cells werestimulated with IL-1� (1 ng/ml) and OSM (10 ng/ml) for 24 hours. Cells were harvested using 0.5mM EDTA before labeling with either a specific or isotype-control antibody. Antibody binding wasassessed using flow cytometric analysis with fluoroscein isothiocyanate or R-phycoerythrin labeling.See Figure 1 for other definitions.
SYNERGISTIC INDUCTION OF MMP-1 BY IL-1� AND OSM 2299

for 20 minutes before separation on a 0.5� Tris–borate–EDTA (40 mM Tris–borate, pH 8.3, 1 mM EDTA) nondena-turing 5% (w/v) polyacrylamide (37.5:1) gel containing 3%(v/v) glycerol. Gels were dried and exposed to either radiog-raphy film or a phosphor screen.
The oligonucleotides (sense/antisense), with the AP-1and SBE underlined and altered bases in lowercase, were asfollows: OSMRE 5�-TCTAGATGAGTCAGACACCTCT-GGCTTTCTGGAAGGG, 5�-TCGACCCTTCCAGAAAGC-CAGAGGTGTCTGACTCATC; mSBE-OSMRE 5�-TCGA-GATGAGTCAGACACCTCTGGCTTTCTatcAGGG, 5�-TCGACCCTgatAGAAAGCCAGAGCTGTCTGACTCA-TC; mutated AP-1—OSMRE 5�-TCGAGtaGAGTCA-GACACCTCTGGCTTTCTGGAAGGG, 5�-TCGACCC-TTCCAGAAAGCCAGAGGTGTCTGACTCtaC (31); AP-15�-AAGTGATGAGTCAGAAT, 5�-AGCATTGTGACT-CATCA; SBE 5�-AAGTGTTTCTGGAACT, 5�-ACGAGT-TCCAGAAACA; high-affinity sis-inducible element (hSIE)5�-GTCGACATTTCCCGTAAATC, 5�-TCGACGATTTAC-GGGAAATG (53). In the competition EMSA, 1 �l of waterwas replaced with unlabeled competitor DNA (50–200-foldexcess). In the supershift assays, the water was replaced with 2�l of supershift concentration antibody that was added withthe labeled probe and incubated at room temperature for 1hour prior to gel electrophoresis. Two STAT-3 antibodies (200�g/�l SantaCruz C-20 or K-15) and 1 STAT-1 antibody(G16930, N-terminus; Transduction Laboratories, Lexington,KY) were used.
Statistical analysis. The difference between samplegroup means was tested for statistical significance using Stu-dent’s unpaired t-test. P values less than 0.01 were consideredsignificant.
RESULTS
IL-1� and OSM synergistically up-regulateMMP-1 expression in chondrocytes. The combination ofIL-1� and OSM caused a synergistic induction of
MMP-1 messenger RNA (mRNA) in the transformedhuman chondrocyte line T/C28a4 (Figure 1), as has beendescribed previously (18,20). Stimulation with OSMalone produced a small but transient increase in MMP-1mRNA and caused a marked induction of TIMP-1expression, as has been described previously (18,53)(Figure 1). No synergy was observed with the TIMP-1mRNA after IL-1�/OSM stimulation. We recently ob-served similar results in primary human articular chon-drocytes (20) and have therefore used the T/C28a4chondrocytes as a model cell line to study the synergisticeffects of IL-1� and OSM on the induction of MMP-1.Treatment with nontoxic levels of cycloheximide, asassessed by LDH assay, abolished the induction ofMMP-1 mRNA following stimulation with IL-1� andIL-1�/OSM (data not shown).
Mechanism of IL-1� and OSM synergy is notthrough up-regulation of cell surface receptors. Flowcytometric analyses of stimulated T/C28a4 chondrocytesrevealed no modulation in the surface expression ofgp130 and OSMR� (Figure 2). No LIFR expression wasevident, as found previously (20), and stimulation alsofailed to induce any detectable surface LIFR (Figure 2).This pattern of surface receptor expression was alsoobserved at 48 and 72 hours poststimulation (data notshown). We were also unable to detect any binding ofbiotinylated LIF, although we could detect the bindingof biotinylated OSM to the cell surface (data notshown).
Changes in cell surface receptor mRNA expres-sion were assessed using Northern blot analysis. OSMalone, and in combination with IL-1�, appeared to
Figure 3. Messenger RNA expression of OSM receptor components in T/C28a4 chondrocytesfollowing stimulation with IL-1� and/or OSM. Confluent T/C28a4 chondrocytes were grown inacid-treated fetal calf serum medium for 24 hours prior to cytokine stimulation. Cells werestimulated with IL-1� (1 ng/ml) and OSM (10 ng/ml) for up to 24 hours as follows: 1)unstimulated, 2) IL-1�, 3) OSM, and 4) IL-1�/OSM. Total RNA was harvested and probed byNorthern blot using complementary DNA for human glycoprotein 130 (gp130), OSM-specificreceptor (OSMR�), and leukemia inhibitory factor receptor (LIFR). GAPDH was used to correctfor RNA loading. See Figure 1 for other definitions.
2300 CATTERALL ET AL

stimulate a small (�2-fold) increase in gp130 andOSMR� mRNA (Figure 3), although these modestincreases were not accompanied by increased proteinexpression at the cell surface (Figure 2). Despite the lackof surface LIFR protein, we detected low, constitutivemultiple transcripts of LIFR mRNA that were unaf-
fected by any treatment. We were unable to detect anyexpression of I IL-1RI by either flow cytometry orNorthern blot analysis, suggesting very low expressionlevels. However, using reverse transcriptase–PCR (RT-PCR), we could detect mRNA for both IL-1RI and IL-1receptor accessory protein (IL-1R–Acp), neither ofwhich appeared to be modulated by cytokine (data notshown). Expression of the IL-1 decoy receptor (IL-1RII)was also assessed but no signal was detectable by RT-PCR (data not shown), as described previously (56).
The apparent lack of modulation of cell surfacegp130 or OSMR� on T/C28a4 chondrocytes after stim-ulation indicates that the synergistic increase in MMP-1expression after IL-1�/OSM stimulation was is not dueto increased surface expression of relevant receptorsubunits. Therefore, we propose that the synergisticinduction occurs downstream of these receptors.
Proximal region of MMP-1 promoter is sufficientfor synergistic induction by IL-1�/OSM. The synergisticeffect of IL-1� and OSM on the transcriptional activa-tion of the MMP-1 promoter was investigated furtherusing MMP-1 promoter/luciferase reporter gene con-structs transiently transfected into T/C28a4 chondro-cytes. No activation was observed for IL-1� alone, whilea modest induction was seen with OSM alone, and therewas a marked synergistic induction following IL-1�/OSM stimulation (Figure 4A). The lack of IL-1� stim-ulation has been previously observed with these con-structs (48). Although the largest construct (�4372/�63) was stimulated to a greater extent, the �517/�63construct showed a similar response to both OSM andIL-1�/OSM in terms of fold induction relative to basalactivity (Figure 4A). Three other MMP-1 promoterconstructs (�3300/�63, �2900/�63, and �1600/�63)(48) were tested and showed similar results (data notshown). The data indicate that elements that lie withinthe proximal region of the MMP-1 promoter (�517/�63) are sufficient to support the IL-1�/OSM synergis-tic activation.
We previously suggested that OSM was uniqueamong the family of gp130 binding cytokines in its abilityto synergize with IL-1� to promote cartilage destruction(18). However, we have recently found that IL-6 in thepresence of sIL-6R can also synergize with IL-1� tocause cartilage breakdown via an induction of MMP-1(20). The effect of this combination was thereforeassessed using the same MMP-1 promoter constructs.IL-6 alone or in combination with IL-1� was unable toactivate the constructs. However, when sIL-6R wasadded with IL-6 there was a significant stimulation
Figure 4. Transcriptional activation of the human MMP-1 promoterin T/C28a4 chondrocytes following stimulation with IL-1� and/orOSM. Confluent T/C28a4 cells were transiently transfected with 2MMP-1/luciferase constructs (�517/�63 or �4372/�63). ThepGL3Basic vector was used as a negative vector control. Transfectedcells were split and grown in acid-treated fetal calf serum medium for24 hours before stimulating with IL-1� (1 ng/ml) and/or OSM (10ng/ml) (A), or IL-1� (1 ng/ml), IL-6 (50 ng/ml), and/or soluble IL-6receptor (sIL-6R) (200 ng/ml) (B) for another 24 hours. Cells werelysed and luciferase activity was assayed. Plasmid incorporation wasdetermined using Hirt’s assay and used to correct for transfectionefficiency. Experiments were repeated 3 times using 2 differentplasmid batches. All treatments were performed in triplicate. Valuesare the mean and SD. ● � P � 0.01 versus control. See Figure 1 fordefinitions.
SYNERGISTIC INDUCTION OF MMP-1 BY IL-1� AND OSM 2301

(Figure 4B). Furthermore, as with OSM, there was amarked synergistic response with IL-6 and sIL-6R incombination with IL-1�. The addition of sIL-6R aloneor with IL-1� led to significant promoter activationbecause T/C28a4 cells endogenously produce IL-6 (20).
Binding of AP-1, but not STAT proteins, to theMMP-1 promoter in T/C28a4 chondrocytes. The tran-sient transfection data (Figure 4A) suggested that theproximal region (�517/�63) of the MMP-1 promoter wasimportant for synergistic stimulation by IL-1�/OSM. Thisregion contains an OSMRE that is thought to play a role inOSM stimulation of MMP-1 (31). The relative roles of theAP-1 and SBE motifs were investigated using EMSAs.
Using oligonucleotides representing the OSMRE(31) with nuclear extracts prepared from stimulatedchondrocytes, 1 main and 2 other protein/DNA com-plexes were clearly visible (Figure 5A). There was noapparent change in the binding of the main complexafter treatment with IL-1�, OSM, or IL-1�/OSM at anyof the time points assessed. The main complex wasidentified as AP-1 binding by competition EMSA (Fig-
ure 5B), since an oligonucleotide specific for AP-1 wasable to compete for binding, while an OSMRE oligo-nucleotide with an mAP-1 site could not. The bandimmediately below the AP-1 complex appeared to showspecificity for the OSMRE, although this specificity didnot appear to involve either the AP-1 or SBE site(Figure 5B). No specific binding of proteins to the SBEbinding site within the OSMRE could be determined,since all bands could be equally competed with eitherthe OSMRE or the OSMRE with a mutated SBE.Competition with the SBE site alone was only able tocompete the top band, which appeared to be nonspecificsince all oligonucleotides used were able to compete thiscomplex. These experiments clearly show constitutivebinding of AP-1 complexes in T/C28a4 chondrocytes,which was still evident following IL-1� and/or OSMstimulation, but no STAT binding.
To further assess STAT binding in stimulated chon-drocytes, we used oligonucleotides representing theMMP-1 SBE (31) in EMSAs. No STAT binding wasevident using these oligonucleotides (results not shown),
Figure 5. Assessment of the binding of nuclear extracts from stimulated T/C28a4 chondrocytes tothe MMP-1 OSM response element (OSMRE). Confluent T/C28a4 chondrocytes were grown inacid-treated fetal calf serum medium for 24 hours prior to cytokine stimulation. A, Nuclear extractswere prepared from cells stimulated with IL-1� (1 ng/ml) and/or OSM (10 ng/ml) for up to 24 hoursand used in an electrophoretic mobility shift assay (EMSA) with OSMRE oligonucleotides. Theassay was repeated twice using different nuclear extracts. B, Nuclear extracts were prepared after15 minutes of stimulation with OSM and used in a competition EMSA using excess (70–200-fold)unlabeled competitor oligonucleotides. The assay was repeated twice using different nuclearextracts. mSBE � mutated signal transducers and activators of transcription (STAT) bindingelement; mAP1 � mutated activator protein 1; fp � free probe (see Figure 1 for other definitions).
2302 CATTERALL ET AL

suggesting that STAT proteins were not binding to theOSMRE within the MMP-1 promoter. This was furtherdemonstrated using a �517/�63 MMP-1 promoter con-struct in which the SBE had been destroyed by site-directed mutagenesis. In transient transfection assays, theactivation profile of this construct showed no differencefrom the wild-type construct (Figure 6), indicating that thissite was unimportant for OSM and IL-1�/OSM stimulationof the MMP-1 promoter in T/C28a4 chondrocytes. UsingMatInspector version 2.2 software (55), no further SBEcould be detected within the �517/�63 MMP-1 construct.
OSM causes STAT activation in T/C28a4 chon-drocytes. To determine whether STAT proteins wereactivated in T/C28a4 chondrocytes following cytokine stim-ulation, an oligonucleotide representing the STAT bindinghSIE from the c-fos promoter was used in EMSAs. STATbinding was evident after a 15-minute stimulation withOSM, reaching maximal binding after 30 minutes. Inter-estingly, STAT activation and binding persisted until 24hours poststimulation (Figure 7A). Maximum activation
was seen at 10 ng/ml OSM, with no significant increase athigher concentrations (Figure 7B). Specificity of STATbinding was evident following competition with excessunlabeled hSIE, whereas competition with excess OSMREfailed to compete the binding, further demonstrating theinability of the MMP-1 SBE to bind STAT proteins(Figure 7C). Supershift EMSA, with 2 different antibodiesfrom those in STAT-3, clearly identified STAT-3 binding(Figure 7C). Supershift assays were performed usingSTAT-1 antibodies but, due to the low amounts of acti-vated STAT-1 present, bands were only just detectable(results not shown). IL-1� stimulation did not cause STATactivation, while the combination of IL-1� and OSMinduced the same profile observed for OSM alone (resultsnot shown).
We also assessed other IL-6 cytokines for theirability to activate STAT proteins. When T/C28a4 chon-drocytes were stimulated with OSM, IL-6, LIF, or CT-1,only OSM was able to activate STATs (data not shown).However, since we have shown that IL-6 can mediate
Figure 6. Signal transducers and activators of transcription (STAT) binding element (SBE) in theMMP-1 promoter not required for OSM or IL-1�/OSM activation. Confluent T/C28a4 cells weretransiently transfected with either the wild-type (�517/�63) or a �517/�63 construct containinga mutated SBE (mSBE �517/�63) luciferase construct. Transfected cells were split and grown inacid-treated fetal calf serum medium for 24 hours before stimulation with IL-1� (1 ng/ml) and/orOSM (10 ng/ml) for another 24 hours. Cells were lysed and luciferase activity was assayed. Plasmidincorporation was determined using Hirt’s assay and used to correct for transfection efficiency.The experiment was repeated 3 times using 2 different plasmid batches. All treatments wereperformed in triplicate. Values are the mean and SD. See Figure 1 for other definitions.
SYNERGISTIC INDUCTION OF MMP-1 BY IL-1� AND OSM 2303

effects on chondrocytes when in the presence of sIL-6R(Figure 4B), the ability of this combination to causeSTAT activation was also investigated. An identicalSTAT activation profile to that produced by OSM wasobserved for IL-6 when in combination with sIL-6R(Figure 7D).
Protein inhibitor of activated STAT-3 (PIAS3)can inhibit OSM-induced transcriptional activation ofthe MMP-1 promoter. Since we demonstrated that thepredominantly activated STAT protein was STAT-3(Figure 7), we further investigated the role of STAT-3 inthe OSM-mediated transcriptional activation of theMMP-1 promoter. A PIAS-3 expression plasmid wastransiently cotransfected into T/C28a4 chondrocyteswith the proximal (�517/�63) MMP-1 promoter con-struct, and the results showed that inactivation ofSTAT-3 completely abolished the OSM-induced activa-tion of the MMP-1 promoter (Figure 8). This resultfurther confirmed an indirect role for activated STAT-3in the transcriptional activation of the MMP-1 promoterfollowing OSM stimulation of human chondrocytes.
Up-regulation of c-fos and MMP-1 promoteractivity. We showed STAT binding to the hSIE, which isthought to be required for the activation of the c-fospromoter (49,57). Because c-fos is an AP-1 transcriptionfactor that could potentially play a role in the up-regulation of MMP-1 (58), we investigated whether itwas involved in the activation of MMP-1 in chondrocytes
Figure 7. Activation of signal transducers and activators of transcrip-tion (STAT) proteins in T/C28a4 chondrocytes by OSM. ConfluentT/C28a4 chondrocytes were grown in acid-treated fetal calf serummedium for 24 hours prior to stimulation. Labeled high-affinitysis-inducible element (hSIE) oligonucleotide was used in electro-phoretic mobility shift assay with nuclear extracts prepared fromT/C28a4 chondrocytes as follows: A, Cells stimulated with OSM (10ng/ml) for up to 24 hours. This was repeated 3 times using differentnuclear extracts. B, Cells stimulated with OSM (0–40 ng/ml) for 15minutes. This was repeated twice using different nuclear extracts. C,Cells stimulated with OSM (10 ng/ml) for 15 minutes. Antibodies toSTAT3 and competitor hSIE or OSM response element (OSMRE)oligonucleotides were added to the binding reactions to determineband specificity. This was repeated twice using different nuclearextracts. D, Cells stimulated with OSM (10 ng/ml), IL-6 (50 ng/ml),and/or soluble IL-6 receptor (sIL-6R) (200 ng/ml) for 15 minutes. Thiswas repeated twice using different nuclear extracts. fp � free probe;ns � nonspecific bands (see Figure 1 for other definitions).
Figure 8. Inhibition of OSM-stimulated MMP-1 promoter activationby protease inhibitor of activated signal transducers and activators oftranscription 3 (PIAS-3). Confluent T/C28a4 cells were transientlytransfected with the �517/�63 MMP-1 promoter/luciferase constructand either a plasmid cytomegalovirus (pCMV5) control vector or aPIAS-3 expression vector. Transfected cells were split and grown inacid-treated fetal calf serum medium for 24 hours before stimulatingwith OSM (10 ng/ml) for another 24 hours. Cells were lysed and theluciferase activity was assayed. Plasmid incorporation was determinedusing Hirt’s assay and used to correct for transfection efficiency. Theexperiment was repeated 3 times using 2 different plasmid batches. Alltreatments were performed in triplicate. Values are the mean and SD.See Figure 1 for other definitions.
2304 CATTERALL ET AL
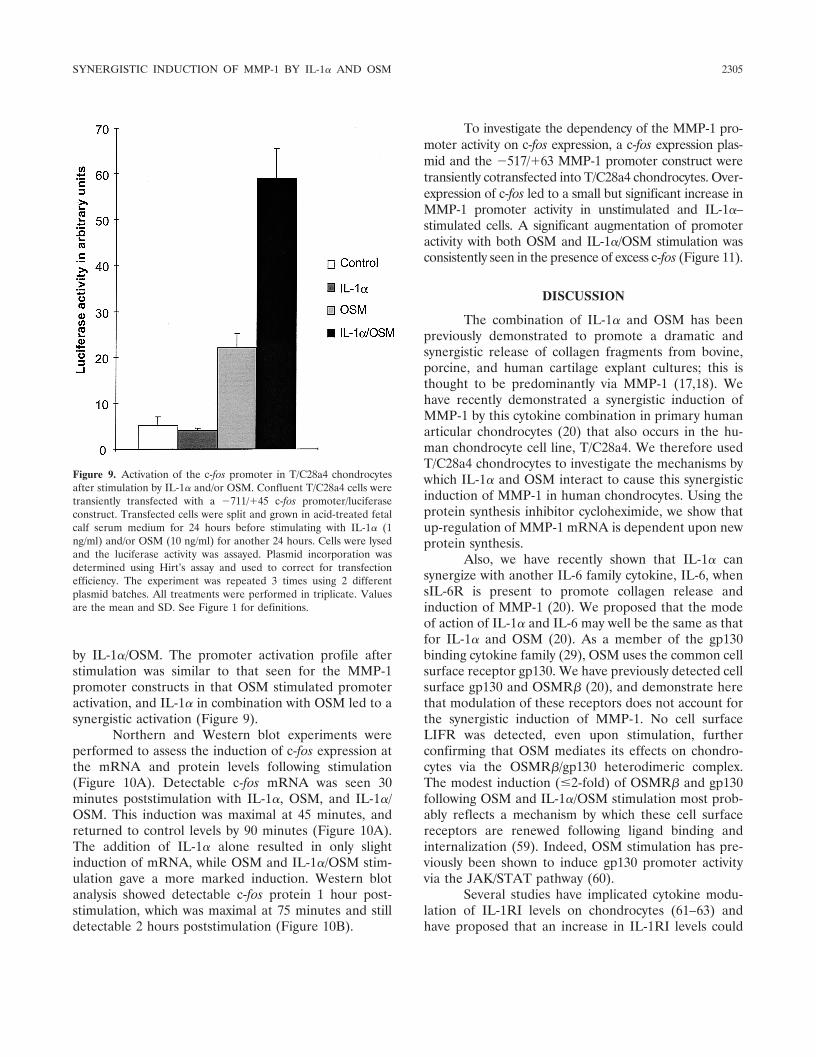
by IL-1�/OSM. The promoter activation profile afterstimulation was similar to that seen for the MMP-1promoter constructs in that OSM stimulated promoteractivation, and IL-1� in combination with OSM led to asynergistic activation (Figure 9).
Northern and Western blot experiments wereperformed to assess the induction of c-fos expression atthe mRNA and protein levels following stimulation(Figure 10A). Detectable c-fos mRNA was seen 30minutes poststimulation with IL-1�, OSM, and IL-1�/OSM. This induction was maximal at 45 minutes, andreturned to control levels by 90 minutes (Figure 10A).The addition of IL-1� alone resulted in only slightinduction of mRNA, while OSM and IL-1�/OSM stim-ulation gave a more marked induction. Western blotanalysis showed detectable c-fos protein 1 hour post-stimulation, which was maximal at 75 minutes and stilldetectable 2 hours poststimulation (Figure 10B).
To investigate the dependency of the MMP-1 pro-moter activity on c-fos expression, a c-fos expression plas-mid and the �517/�63 MMP-1 promoter construct weretransiently cotransfected into T/C28a4 chondrocytes. Over-expression of c-fos led to a small but significant increase inMMP-1 promoter activity in unstimulated and IL-1�–stimulated cells. A significant augmentation of promoteractivity with both OSM and IL-1�/OSM stimulation wasconsistently seen in the presence of excess c-fos (Figure 11).
DISCUSSION
The combination of IL-1� and OSM has beenpreviously demonstrated to promote a dramatic andsynergistic release of collagen fragments from bovine,porcine, and human cartilage explant cultures; this isthought to be predominantly via MMP-1 (17,18). Wehave recently demonstrated a synergistic induction ofMMP-1 by this cytokine combination in primary humanarticular chondrocytes (20) that also occurs in the hu-man chondrocyte cell line, T/C28a4. We therefore usedT/C28a4 chondrocytes to investigate the mechanisms bywhich IL-1� and OSM interact to cause this synergisticinduction of MMP-1 in human chondrocytes. Using theprotein synthesis inhibitor cycloheximide, we show thatup-regulation of MMP-1 mRNA is dependent upon newprotein synthesis.
Also, we have recently shown that IL-1� cansynergize with another IL-6 family cytokine, IL-6, whensIL-6R is present to promote collagen release andinduction of MMP-1 (20). We proposed that the modeof action of IL-1� and IL-6 may well be the same as thatfor IL-1� and OSM (20). As a member of the gp130binding cytokine family (29), OSM uses the common cellsurface receptor gp130. We have previously detected cellsurface gp130 and OSMR� (20), and demonstrate herethat modulation of these receptors does not account forthe synergistic induction of MMP-1. No cell surfaceLIFR was detected, even upon stimulation, furtherconfirming that OSM mediates its effects on chondro-cytes via the OSMR�/gp130 heterodimeric complex.The modest induction (�2-fold) of OSMR� and gp130following OSM and IL-1�/OSM stimulation most prob-ably reflects a mechanism by which these cell surfacereceptors are renewed following ligand binding andinternalization (59). Indeed, OSM stimulation has pre-viously been shown to induce gp130 promoter activityvia the JAK/STAT pathway (60).
Several studies have implicated cytokine modu-lation of IL-1RI levels on chondrocytes (61–63) andhave proposed that an increase in IL-1RI levels could
Figure 9. Activation of the c-fos promoter in T/C28a4 chondrocytesafter stimulation by IL-1� and/or OSM. Confluent T/C28a4 cells weretransiently transfected with a �711/�45 c-fos promoter/luciferaseconstruct. Transfected cells were split and grown in acid-treated fetalcalf serum medium for 24 hours before stimulating with IL-1� (1ng/ml) and/or OSM (10 ng/ml) for another 24 hours. Cells were lysedand the luciferase activity was assayed. Plasmid incorporation wasdetermined using Hirt’s assay and used to correct for transfectionefficiency. The experiment was repeated 3 times using 2 differentplasmid batches. All treatments were performed in triplicate. Valuesare the mean and SD. See Figure 1 for definitions.
SYNERGISTIC INDUCTION OF MMP-1 BY IL-1� AND OSM 2305

augment the IL-1� response, leading to increased pro-teinase induction (62). Our present study suggests thatstimulation of T/C28a4 chondrocytes with IL-1�, OSM,and IL-1�/OSM does not cause an increase in IL-1RIlevels during a period in which MMP-1 is synergisticallyinduced. Flow cytometric analysis failed to detect sur-face IL-1RI protein, although the presence of IL-1RImRNA was confirmed. We conclude that T/C28a4 sur-face levels of IL-1RI were relatively low, which has beenshown previously for articular chondrocytes (56), andwere below the limit of detection using flow cytometry.Increased IL-1RI has been observed in OA articularchondrocytes (56) and this may reflect a different dis-ease mechanism between RA and OA, although it hasbeen suggested that only a small proportion of receptorsis required for maximum collagenase production (56).The accessory protein, IL-1R–Acp, essential for IL-1RIsignal transduction (64), was constitutively expressedand unaffected following stimulation. We also failed todetect IL-1RII mRNA, which corroborates previousfindings (56), although mRNA for both receptor types
has been detected using in situ hybridization of murinechondrocytes, but this was dependent upon cartilagelocation (65).
Transient transfection studies in T/C28a4 chon-drocytes revealed that the proximal region of theMMP-1 promoter (�517/�63) was sufficient to supportthe synergistic activation by IL-1�/OSM, but the previ-ously reported SBE (31) did not appear to be involved inthis activation. Although this SBE is a match for theSTAT consensus sequence, other studies show that it isonly a weak STAT site (66). Indeed, the bovine MMP-1promoter lacks this motif, and a proximal bovineMMP-1 promoter fragment behaves similarly to thehuman counterpart when transiently transfected intohuman chondrocytes (Catterall JB, Rowan AD: unpub-lished observations). However, we were able to demon-strate rapid STAT activation in T/C28a4 chondrocytesfollowing OSM stimulation using hSIE and showed thatthis was predominantly STAT-3. Moreover, an indirectrole for STAT-3 was confirmed using a specific negativeregulator of STAT-3, PIAS-3 (51).
Figure 10. Expression of c-fos in T/C28a4 chondrocytes after OSM stimulation. A, Confluent T/C28a4 chondrocytes were serum starved for 24 hoursbefore cytokine stimulation. Cells were stimulated with IL-1� (1 ng/ml) and/or OSM (10 ng/ml) for up to 90 minutes. Total RNA was harvested andprobed for human c-fos. GAPDH was used to correct for RNA loading. B, Confluent T/C28a4 chondrocytes were grown in acid-treated fetal calfserum medium for 24 hours before stimulation with OSM (10 ng/ml) for up to 2 hours. Cells were solubilized in sodium dodecyl sulfate sample buffer,electrophoresed, and Western blotted with a c-fos antibody using a chemiluminescent detection system. This was repeated twice using different cellextracts. See Figure 1 for definitions.
2306 CATTERALL ET AL

In fibroblasts, OSM predominantly activatesSTAT-1 (31), which may be an important factor inbinding to OSMRE in the MMP-1 promoter in thesecells. Interestingly, no other gp130 binding cytokine,except IL-6 in the presence of sIL-6R, caused STATactivation under the culture conditions used, whichcorrelated with the synergistic activation of the proximalMMP-1 promoter (�517/�63) construct when used incombination with IL-1�. IL-6 and OSM can signalthrough common pathways (59), and these results sup-port a common signaling pathway for MMP-1 stimula-tion by these cytokines. This further confirms our previ-ous observations that only these 2 cytokines cansynergize with IL-1�, and the lack of any effect with theother IL-6 family members may be due to the lack ofLIFR on chondrocytes (20). IL-1� alone was unable tostimulate the promoter fragments used, as has beenpreviously reported (48). It is possible that IL-1� maycontribute to the synergistic induction of endogenousMMP-1 mRNA via posttranscriptional effects such asincreasing mRNA stability, which has been shown pre-viously (67). The lack of response on the �4372/�63 bppromoter fragment is probably because it is not the
full-length promoter and so may lack some elementsrequired by IL-1� for stimulation.
Although there was no apparent STAT bindingto the OSMRE oligonucleotide, we did detect constitu-tive AP-1/DNA binding using the �77 AP-1 site fromthe MMP-1 promoter. Deletion of this AP-1 site leads toreduction of the stimulated transcriptional activity tobasal levels (Carrere S: unpublished observations), fur-ther substantiating the importance of AP-1 interactionswith the MMP-1 promoter in chondrocytes. The AP-1complexes are well known to be important for theregulation of MMPs in general (36) and for the specificregulation of MMP-1 (67,68).
A role for changes in AP-1 after OSM stimula-tion has been demonstrated using the TIMP-1 promoter,in which an increase in bound c-fos containing AP-1complexes was observed (53). We were able to showc-fos promoter stimulation and a transient production ofboth mRNA and protein in chondrocytes after stimula-tion with IL-1� and OSM. In bovine articular chondro-cytes, IL-1� stimulates c-fos production, and this hasbeen linked to MMP-1 induction (69). Our data confirmthis, although we find that OSM is a much more potent
Figure 11. Effect of c-fos overexpression on MMP-1 promoter activity in T/C28a4 chondrocytes.Confluent T/C28a4 cells were transiently transfected with the �517/�63 MMP-1 promoter/luciferase construct and either a c-fos expression vector or a plasmid cytomegalovirus controlplasmid. Transfected cells were split and grown in acid-treated fetal calf serum medium for 24hours before stimulating with IL-1� (1 ng/ml) or OSM (10 ng/ml) for another 24 hours. Cells werelysed and the luciferase activity was assayed. Plasmid incorporation was determined using Hirt’sassay and used to correct for transfection efficiency. The experiment was repeated twice using 2different plasmid batches. All treatments were performed in triplicate. Values are the mean andSD. See Figure 1 for definitions.
SYNERGISTIC INDUCTION OF MMP-1 BY IL-1� AND OSM 2307

inducer of c-fos mRNA than is IL-1�. Overexpression ofc-fos led to consistently augmented activation of theMMP-1 promoter when stimulated with OSM, andoverexpression per se did lead to a small transcriptionalactivation of the promoter. This suggests that c-fos doesplay a role in MMP-1 promoter activation, and that c-fosavailability could be rate limiting. This implies thatup-regulation of other AP-1 transcription factors, suchas members of the Jun family, or phosphorylation ofAP-1 complexes is required for transcriptional activationto occur.
The results presented here suggest a new mech-anism by which IL-1� and OSM stimulate the synergisticproduction of MMP-1 in human chondrocytes. Thismechanism is different from that previously proposedfor OSM regulation of MMP-1 (31) since activatedSTAT proteins do not directly bind to the MMP-1promoter. We propose that STAT proteins, in particularSTAT-3, are activated following OSM stimulation butact indirectly, up-regulating other transcription factorssuch as c-fos, c-Jun, and jun B, as has been demonstratedpreviously (53,70–72). These could then form differentAP-1 complexes that may be further activated by MAPKpathways (38) and bind to the AP-1 sites in the proximalregion of the MMP-1 promoter. IL-1� stimulates otherpathways, including c-Jun N-terminal kinase (JNK),MAPK, and NF-�B (41), although findings in a recentstudy suggested that these are not involved in IL-1�up-regulation of MMP-1 (73). Reactive oxygen specieshave also been implicated in IL-1� up-regulation ofMMP-1 in articular chondrocytes (69).
The potential importance of signaling as a ther-apeutic target has already been demonstrated with thediscovery that one of the major treatments for RA,glucocorticoids, can interfere with the ability of bothNF-�B and AP-1 transcription factors to activate genes(74). Coupled with this finding, an understanding of theunderlying mechanisms involved in IL-1� and OSMsynergy may be of considerable importance in determin-ing the mode of action of some of the present therapies,as well as generating future therapeutic targets for RA.
ACKNOWLEDGMENTSWe thank Professor J. Heath, Professor K. Shuai,
Professor M. Goldring, Dr. I. Verma, and Dr. D. Leaman forthe generous supply of reagents, and Professor C. Richardsand Dr. E. Dawes for their technical advice.
REFERENCES1. Cawston TE. Matrix metalloproteinases and TIMPs: properties
and implications for the rheumatic diseases [review]. Mol MedToday 1998;4:130–7.
2. Abbaszade I, Liu RQ, Yang F, Rosenfeld SA, Ross OH, Link JR,et al. Cloning and characterization of ADAMTS11, an aggre-canase from the ADAMTS family. J Biol Chem 1999;274:23443–50.
3. Tortorella MD, Burn TC, Pratta MA, Abbaszade I, Hollis JM, LiuR, et al. Purification and cloning of aggrecanase-1: a member ofthe ADAMTS family of proteins. Science 1999;284:1664–6.
4. Page Thomas DP, King B, Stephens T, Dingle JT. In vivo studiesof cartilage regeneration after damage induced by catabolin/interleukin-1. Ann Rheum Dis 1991;50:75–80.
5. Jubb RW, Fell HB. The breakdown of collagen by chondrocytes.J Pathol 1980;130:159–67.
6. Stricklin GP, Bauer EA, Jeffrey JJ, Eisen AZ. Human skincollagenase: isolation of precursor and active forms from bothfibroblast and organ cultures. Biochemistry 1977;16:1607–15.
7. Hasty KA, Pourmotabbed TF, Goldberg GI, Thompson JP,Spinella DG, Stevens RM, et al. Human neutrophil collagenase: adistinct gene product with homology to other matrix metallopro-teinases. J Biol Chem 1990;265:11421–4.
8. Freije JMP, Diez-Itza I, Balbin M, Sanchez LM, Blasco R, ToliviaJ, et al. Molecular cloning and expression of collagenase-3, a novelhuman matrix metalloproteinase produced by breast carcinomas.J Biol Chem 1994;269:16766–73.
9. Tetlow LC, Woolley DE. Comparative immunolocalization studiesof collagenase 1 and collagenase 3 production in the rheumatoidlesion, and by human chondrocytes and synoviocytes in vitro. Br JRheumatol 1998;37:64–70.
10. Lindy O, Konttinen YT, Sorsa T, Ding Y, Santavirta S, Ceponis A,et al. Matrix metalloproteinase 13 (collagenase 3) in humanrheumatoid synovium. Arthritis Rheum 1997;40:1391–9.
11. Mitchell PG, Magna HA, Reeves LM, Lopresti-Morrow LL,Yocum SA, Rosner PJ, et al. Cloning, expression, and type IIcollagenolytic activity of matrix metalloproteinase-13 from humanosteoarthritic cartilage. J Clin Invest 1996;97:761–8.
12. Fernandes JC, Martel-Pelletier J, Lascau-Coman V, Moldovan F,Jovanovic D, Raynauld JP, et al. Collagenase-1 and collagenase-3synthesis in normal and early experimental osteoarthritic caninecartilage: an immunohistochemical study. J Rheumatol 1998;25:1585–94.
13. Clark IM, Powell LK, Ramsey S, Hazleman BL, Cawston TE. Themeasurement of collagenase, tissue inhibitor of metalloproteinases(TIMP), and collagenase–TIMP complex in synovial fluids frompatients with osteoarthritis and rheumatoid arthritis. ArthritisRheum 1993;36:372–9.
14. Brinckerhoff CE. Joint destruction in arthritis: metalloproteinasesin the spotlight [editorial]. Arthritis Rheum 1991;34:1073–5.
15. Saklatvala J, Pilsworth LMC, Sarsfield SJ, Gavrilovic J, Heath JK.Pig catabolin is a form of interleukin 1: cartilage and bone resorb,fibroblasts make prostaglandin and collagenase, and thymocyteproliferation is augmented in response to one protein. Biochem J1984;224:461–6.
16. Westacott CI, Whicher JT, Barnes IC, Thompson D, Swan AJ,Dieppe PA. Synovial fluid concentration of five different cytokinesin rheumatic diseases. Ann Rheum Dis 1990;49:676–81.
17. Cawston TE, Ellis AJ, Humm G, Lean E, Ward D, Curry V.Interleukin-1 and oncostatin M in combination promote therelease of collagen fragments from bovine nasal cartilage inculture. Biochem Biophys Res Commun 1995;215:377–85.
18. Cawston TE, Curry VA, Summers CA, Clark IM, Riley GP, LifePF, et al. The role of oncostatin M in animal and humanconnective tissue collagen turnover and its localization within therheumatoid joint. Arthritis Rheum 1998;41:1760–71.
19. Okamoto H, Yamamura M, Morita Y, Harada S, Makino H, OtaZ. The synovial expression and serum levels of interleukin-6,interleukin-11, leukemia inhibitory factor, and oncostatin M inrheumatoid arthritis. Arthritis Rheum 1997;40:1096–105.
20. Rowan AD, Koshy PJT, Shingleton WD, Degnan BA, Heath JK,
2308 CATTERALL ET AL

Vernallis AB, et al. Synergistic effects of glycoprotein 130 bindingcytokines in combination with interleukin-1 on cartilage collagenbreakdown. Arthritis Rheum 2001;44:1620–32.
21. Robak T, Gladalska A, Stepien H, Robak E. Serum levels ofinterleukin-6 type cytokines and soluble interleukin-6 receptor inpatients with rheumatoid arthritis. Mediators Inflamm 1998;7:347–53.
22. Mihara M, Moriya Y, Kishimoto T, Ohsugi Y. Interleukin-6 (IL-6)induces the proliferation of synovial fibroblastic cells in thepresence of soluble IL-6 receptor. Br J Rheumatol 1995;34:321–5.
23. Brozik M, Rosztoczy I, Meretey K, Balint G, Gaal M, Balogh Z, etal. Interleukin 6 levels in synovial fluids of patients with differentarthritides: correlation with local IgM rheumatoid factor andsystemic acute phase protein production. J Rheumatol 1992;19:63–8.
24. Houssiau FA, Devogelaer J-P, van Damme J, de DeuxchaisnesCN, van Snick J. Interleukin-6 in synovial fluid and serum ofpatients with rheumatoid arthritis and other inflammatory arthrit-ides. Arthritis Rheum 1988;31:784–8.
25. Richards CD, Shoyab M, Brown TJ, Gauldie J. Selective regula-tion of metalloproteinase inhibitor (TIMP-1) by oncostatin M infibroblasts in culture. J Immunol 1993;150:5596–603.
26. Silacci P, Dayer JM, Desgeorges A, Peter R, Manueddu C, GuernePA. Interleukin (IL)-6 and its soluble receptor induce TIMP-1expression in synoviocytes and chondrocytes, and block IL-1-induced collagenolytic activity. J Biol Chem 1998;273:13625–9.
27. Mosley B, De Imus C, Friend D, Boiani N, Thoma B, Park LS, etal. Dual oncostatin M (OSM) receptors: cloning and characteriza-tion of an alternative signaling subunit conferring OSM-specificreceptor activation. J Biol Chem 1996;271:32635–43.
28. Ihle JN. STATs: signal transducers and activators of transcription.Cell 1996;84:331–4.
29. Taga T, Kishimoto T. Gp130 and the interleukin-6 family ofcytokines. Annu Rev Immunol 1997;15:797–819.
30. Bugno M, Graeve L, Gatsios P, Koj A, Heinrich PC, Travis J, et al.Identification of the interleukin-6/oncostatin M response elementin the rat tissue inhibitor of metalloproteinases-1 (TIMP-1) pro-moter. Nucleic Acids Res 1995;23:5041–7.
31. Korzus E, Nagase H, Rydell R, Travis J. The mitogen-activatedprotein kinase and JAK-STAT signaling pathways are required foran oncostatin M-responsive element-mediated activation of matrixmetalloproteinase 1 gene expression. J Biol Chem 1997;272:1188–96.
32. Asahara H, Fujisawa K, Kobata T, Hasunuma T, Maeda T,Asanuma M, et al. Direct evidence of high DNA binding activity oftranscription factor AP-1 in rheumatoid arthritis synovium. Arthri-tis Rheum 1997;40:912–8.
33. Kinne RW, Boehm S, Iftner T, Aigner T, Vornehm S, Weseloh G,et al. Synovial fibroblast-like cells strongly express jun-B and C-fosproto-oncogenes in rheumatoid and osteoarthritis. Scand J Rheu-matol Suppl 1995;101:121–5.
34. Trabandt A, Aicher WK, Gay RE, Sukhatme VP, Fassbender HG,Gay S. Spontaneous expression of immediately early responsegenes c-fos and egr-1 in collagenase-producing rheumatoid syno-vial fibroblasts. Rheumatol Int 1992;12:53–9.
35. Han Z, Boyle DL, Manning AM, Firestein GS. AP-1 and NF-kappaB regulation in rheumatoid arthritis and murine collagen-induced arthritis. Autoimmunity 1998;28:197–208.
36. Benbow U, Brinckerhoff CE. The AP-1 site and MMP generegulation: what is all the fuss about? Matrix Biol 1997;15:519–26.
37. Shiozawa S, Shimizu K, Tanaka K, Hino K. Studies on thecontribution of c-fos/AP-1 to arthritic joint destruction. J ClinInvest 1997;99:1210–6.
38. Firestein GS, Manning AM. Signal transduction and transcriptionfactors in rheumatic disease [review]. Arthritis Rheum 1999;42:609–21.
39. Wang F, Sengupta TK, Zhong Z, Ivashkiv LB. Regulation of the
balance of cytokine production and the signal transducer andactivator of transcription (STAT) transcription factor activity bycytokines and inflammatory synovial fluids. J Exp Med 1995;182:1825–31.
40. Sengupta TK, Wang F, Zhong Z, Darnell JE Jr, Ivashkiv LB.Cytokines and the Jak-STAT signaling pathway in inflammatoryarthritis [abstract]. Arthritis Rheum 1995;38 Suppl 9:S190.
41. O’Neill LA. Towards an understanding of the signal transductionpathways for interleukin 1. Biochim Biophys Acta 1995;1266:31–44.
42. Wesche H, Korherr C, Kracht M, Falk W, Resch K, Martin MU.The interleukin-1 receptor accessory protein (IL-1RAcP) is essen-tial for IL-1-induced activation of interleukin-1 receptor-associ-ated kinase (IRAK) and stress-activated protein kinases (SAPkinases). J Biol Chem 1997;272:7727–31.
43. O’Neill LA, Greene C. Signal transduction pathways activated bythe IL-1 receptor family: ancient signaling machinery in mammals,insects, and plants. J Leukoc Biol 1998;63:650–7.
44. Geng Y, Valbracht J, Lotz M. Activation of the MAP kinasesubgroups JNK and p38 is selectively associated with IL-1 or TNFstimulation of human articular chondrocytes [abstract]. ArthritisRheum 1996;39 Suppl 9:S130.
45. Staunton D, Hudson KR, Heath JK. The interactions of thecytokine-binding homology region and immunoglobulin-like do-mains of gp130 with oncostatin M: implications for receptorcomplex formation. Protein Eng 1998;11:1093–102.
46. Robinson RC, Grey LM, Staunton D, Vankelecom H, VernallisAB, Moreau JF, et al. The crystal structure and biological functionof leukemia inhibitory factor: implications for receptor binding.Cell 1994;77:1101–16.
47. Fort P, Marty L, Piechaczyk M, el Sabrouty S, Dani C, Jeanteur P,et al. Various rat adult tissues express only one major mRNAspecies from the glyceraldehyde-3-phosphate-dehydrogenase mul-tigenic family. Nucleic Acids Res 1985;13:1431–42.
48. Rutter JL, Benbow U, Coon CI, Brinckerhoff CE. Cell-typespecific regulation of human interstitial collagenase-1 gene expres-sion by interleukin-1� (IL-1�) in human fibroblasts and BC-8701breast cancer cells. J Cell Biochem 1997;66:322–36.
49. Leaman DW, Pisharody S, Flickinger TW, Commane MA,Schlessinger J, Kerr IM, et al. Roles of JAKs in activation ofSTATs and stimulation of c-fos gene expression by epidermalgrowth factor. Mol Cell Biol 1996;16:369–75.
50. Carrozza ML, Jacobs H, Acton D, Verma I, Berns A. Overexpres-sion of the FosB2 gene in thymocytes causes aberrant developmentof T cells and thymic epithelial cells. Oncogene 1997;14:1083–91.
51. Chung CD, Liao J, Liu B, Rao XP, Jay P, Berta P, et al. Specificinhibition of Stat3 signal transduction by PIAS3. Science 1997;278:1803–5.
52. Goldring MB, Birkhead JR, Suen LF, Yamin R, Mizuno S,Glowacki J, et al. Interleukin-1�-modulated gene expression inimmortalized human chondrocytes. J Clin Invest 1994;94:2307–16.
53. Botelho FM, Edwards DR, Richards CD. Oncostatin M stimulatesc-fos to bind a transcriptionally responsive AP-1 element withinthe tissue inhibitor of metalloproteinase-1 promoter. J Biol Chem1998;273:5211–8.
54. Hirt B. Selective extraction of polyoma DNA from infected mousecell cultures. J Mol Biol 1967;26:365–9.
55. Quandt K, Frech K, Karas H, Wingender E, Werner T. MatIndand MatInspector: new fast and versatile tools for detection ofconsensus matches in nucleotide sequence data. Nucleic Acids Res1995;23:4878–84.
56. Martel-Pelletier J, McCollum R, DiBattista J, Faure M-P, ChinJA, Fournier S, et al. The interleukin-1 receptor in normal andosteoarthritic human articular chondrocytes: identification as thetype 1 receptor and analysis of binding kinetics and biologicfunction. Arthritis Rheum 1992;35:530–40.
57. Hill CS, Treisman R. Differential activation of c-fos promoter
SYNERGISTIC INDUCTION OF MMP-1 BY IL-1� AND OSM 2309

elements by serum, lysophosphatidic acid, G-proteins andpolypeptide growth factors. EMBO J 1995;14:5037–47.
58. Tsuji M, Hirakawa K, Kato A, Fujii K. The possible role of c-fosexpression in rheumatoid cartilage destruction. J Rheumatol 2000;27:1606–21.
59. Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L.Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J 1998;334:297–314.
60. O’Brien CA, Manolagas SC. Isolation and characterization of thehuman gp130 promoter: regulation by STATS. J Biol Chem1997;272:15003–10.
61. Pronost S, Segond N, Macro M, Redini F, Penfornis H, JullienneA, et al. Modulation of interleukin-1 receptor expression bytransforming growth factor-beta in cultured rabbit articular chon-drocytes: analysis by reverse transcription-polymerase chain reac-tion. Osteoarthritis Cartilage 1995;3:147–55.
62. Harvey AK, Stack ST, Chandrasekhar S. Differential modulationof degradative and repair responses of interleukin-1 treated chon-drocytes by platelet-derived growth factor. Biochem J 1993;292:129–36.
63. Chandrasekhar S, Harvey AK. Induction of interleukin-1 receptorson chondrocytes by fibroblast growth factor: a possible mechanismfor modulation of interleukin-1 activity. J Cell Physiol 1989;138:236–46.
64. Korherr C, Hofmeister R, Wesche H, Falk W. A critical role forinterleukin-1 receptor accessory protein in interleukin-1 signaling.Eur J Immunol 1997;27:262–7.
65. Xu LX, Kukita T, Nakano Y, Yu H, Hotokebuchi T, Kuratani T,et al. Osteoclasts in normal and adjuvant arthritis bone tissuesexpress the mRNA for both type I and II interleukin-1 receptors.Lab Invest 1996;75:677–87.
66. Lamb P, Seidel HM, Haslam J, Milocco L, Kessler LV, Stein RB,et al. STAT protein complexes activated by interferon-gamma andgp130 signaling molecules differ in their sequence preferences and
transcriptional induction properties. Nucleic Acids Res 1995;23:3283–9.
67. Vincenti MP, Coon CI, Lee O, Brinckerhoff CE. Regulation ofcollagenase gene expression by IL-1� requires transcriptional andpost-transcriptional mechanisms. Nucleic Acids Res 1994;22:4818–27.
68. White LA, Brinckerhoff CE. Two activator protein-1 elements inthe matrix metalloproteinase-1 promoter have different effects ontranscription and bind Jun D, c-Fos, and Fra-2. Matrix Biol1995;14:715–25.
69. Lo YY, Conquer JA, Grinstein S, Cruz TF. Interleukin-1 betainduction of c-fos and collagenase expression in articular chondro-cytes: involvement of reactive oxygen species. J Cell Biochem1998;69:19–29.
70. Kojima H, Nakajima K, Hirano T. IL-6-inducible complexes on anIL-6 response element of the junB promoter contain Stat3 and 36kDa CRE-like site binding protein(s). Oncogene 1996;12:547–54.
71. Solis-Herruzo JA, Rippe RA, Schrum LW, de La Torre P, GarciaI, Jeffrey JJ, et al. Interleukin-6 increases rat metalloproteinase-13gene expression through stimulation of activator protein 1 tran-scription factor in cultured fibroblasts. J Biol Chem 1999;274:30919–26.
72. Sjin RM, Lord KA, Abdollahi A, Hoffman B, Liebermann DA.Interleukin-6 and leukemia inhibitory factor induction of JunB isregulated by distinct cell type-specific cis-acting elements. J BiolChem 1999;274:28697–707.
73. Mengshol JA, Vincenti MP, Coon CI, Barchowsky A, BrinckerhoffCE. Interleukin-1 induction of collagenase 3 (matrix metallopro-teinase 13) gene expression in chondrocytes requires p38, c-JunN-terminal kinase, and nuclear factor �B: differential regulation ofcollagenase 1 and collagenase 3. Arthritis Rheum 2000;43:801–11.
74. Gottlicher M, Heck S, Herrlich P. Transcriptional cross-talk, thesecond mode of steroid hormone receptor action. J Mol Med1998;76:480–9.
2310 CATTERALL ET AL
Copyright © 2022 FDOKUMEN



















