
Adult Urinary Catheter Insertion and Management policy for ...

Smm
GNa
b
c
ARRAA
KLHCCL
1
kis(tm
wcto
D1
0h
Electrochimica Acta 105 (2013) 99– 109
Contents lists available at SciVerse ScienceDirect
Electrochimica Acta
jo u r n al hom ep age: www.elsev ier .com/ locate /e lec tac ta
urfactant-assisted mild hydrothermal synthesis to nanostructuredixed orthophosphates LiMnyFe1−yPO4/C lithium insertion cathodeaterials
. Meligranaa, F. Di Lupoa, S. Ferrarib, M. Destroa, S. Bodoardoa,
. Garinoc, C. Gerbaldia,c,∗
Department of Applied Science and Technology – DISAT, Institute of Chemistry, Politecnico di Torino, C.so Duca degli Abruzzi 24, 10129 Torino, ItalyDepartment of Chemistry, Physical Chemistry Section, University of Pavia, Via Taramelli 16, 27100 Pavia, ItalyCenter for Space Human Robotics @Polito, Istituto Italiano di Tecnologia, Corso Trento 21, 10129 Torino, Italy
a r t i c l e i n f o
rticle history:eceived 20 December 2012eceived in revised form 24 April 2013ccepted 26 April 2013vailable online xxx
eywords:ithium iron manganese phosphateydrothermal synthesisathode
a b s t r a c t
Nanostructured mixed phospho-olivine LiMnyFe1−yPO4/C composites having different ratio of manganeseand iron (0 < y < 1) are prepared by an easy, quick and cost effective hydrothermal synthesis in the presenceof a cationic surfactant and their electrochemical behaviour as cathodes for Li-ion battery is demon-strated. A detailed study of the influence of Fe substitution with Mn on the structure, morphology andelectrochemical performances of the resulting samples is performed by means of X-ray powder diffrac-tion and Rietveld refinement of the diffraction patterns, inductively coupled plasma atomic emissionspectroscopy, X-ray photoelectron spectroscopy, N2 physisorption at 77 K, scanning and transmissionelectron microscopy, cyclic voltammetry and constant current charge–discharge cycling. The best per-forming LiMnyFe1−yPO4/C composite sample is able to deliver a very stable cycling behaviour for several
ycling stabilityithium battery
hundred cycles with good specific capacity values, high Coulombic efficiency and rate capability and isalso demonstrated to work in a real Li-ion battery configuration versus a graphite anode. These improvedelectrochemical properties are ascribed to the kind of synthesis adopted that leads to nanostructuredparticles, homogeneously coated by a thin layer of carbon, which exerts its positive effect by improv-ing electronic conductivity and suppressing Mn dissolution from the olivine structure during prolongedcycling.
. Introduction
Secondary Li-ion batteries are nowadays dominating the mar-et of portable electronics and rapidly expanding their applicationn the field of electricity powered transportation [1]. Despite this,erious barriers remain for their wider use in large scale devicese.g., batteries for electric vehicles), such as the elevated cost andoxicity of the Li-ion battery materials, especially Co-based cathode
aterials.In the last years, lithium transition-metal phosphates (LiMPO4,
here M = Fe, Mn) have attracted much attention due to their good
yclic performance, excellent chemical and thermal stability, lowoxicity and cost effectiveness. The three-dimensional frameworkf the olivine structure is stabilized by the strong covalent bonds∗ Corresponding author at: Department of Applied Science and Technology –ISAT, Institute of Chemistry, Politecnico di Torino, C.so Duca degli Abruzzi 24,0129 Torino, Italy. Tel.: +39 011 090 4638; fax: +39 011 090 4699.
E-mail addresses: [email protected], [email protected] (C. Gerbaldi).
013-4686/$ – see front matter © 2013 Elsevier Ltd. All rights reserved.ttp://dx.doi.org/10.1016/j.electacta.2013.04.153
© 2013 Elsevier Ltd. All rights reserved.
between oxygen and phosphorous, resulting in high structural sta-bility and safety [2–8]. Among others, LiFePO4 is now recognizedas the most promising cathode materials for automotive appli-cation. Nevertheless, the Fe2+/Fe3+ redox potential in LiFePO4 isabout 3.45 V vs. Li/Li+, thus setting a relatively low overall cellworking potential, particularly when anode materials with highworking potential are considered as the counter electrode in orderto realize a cell with high intrinsic safety. In this respect, the pro-gressive replacement of Fe by Mn in the LiFePO4 framework opensup new directions in optimizing the potential where lithium inter-calation/deintercalation takes place.
Olivine-type LiMnPO4 offers a redox potential exceeding 4.0 Vvs. Li/Li+ [9], which means a higher overall energy density comparedto that of LiFePO4, but early references reported that the kineticbehaviour of LiMnPO4 is too poor to show any reversible capacity[2]. Opinions about the suitability of LiMnPO4 as practical cath-
ode material are still controversial; for instance, LiMnPO4 preparedby polyol method [10–12] showed an interesting reversible capac-ity. Whereas, reports by several authors [13–19] demonstratedthe poor electrochemical performance of LiMnPO4 as cathode
100 G. Meligrana et al. / Electrochimica Acta 105 (2013) 99– 109
Table 1Precursors concentrations in the different samples prepared; the heat-treatment was carried out at 615 ◦C in flowing N2.
Sample y in LiMnyFe1−yPO4 FeSO4·7H2O (mmol) MnSO4·H2O (mmol) H3PO4(mmol) LiOH (mmol) CTAB (mmol)
LFM-0 0.00 1.24 0.00 1.24 3.72 1.37LFM-5 0.05 1.18 0.06 1.24 3.72 1.37LFM-25 0.25 0.93 0.31 1.24 3.72 1.37
2
3
4
mtDetM
irptapcvYaLui
ramao[cPtL
wphsitcaraa
2
2
MLblsr[
LFM-50 0.50 0.62 0.6LFM-75 0.75 0.31 0.9LFM-100 1.00 0.00 1.2
aterial for rechargeable Li-ion batteries. These authors revealedhat fundamental differences exist between LiFePO4 and LiMnPO4.elithiation of sub-micron-sized crystals of LiMnPO4 at ambi-nt temperature produced nonstoichiometric LiyMnPO4 phaseshat heated at temperatures close to 200 ◦C decomposed to form
n2P2O7, thus releasing highly flammable O2.For Mn substitution, the LiMnyFe1−yPO4 solid solution has been
ntensively studied. Yamada et al. [15,20] demonstrated that theeversible insertion of Li+ ions in mixed lithium manganese ironhosphate would generate higher energy density with respecto lithium iron phosphate and, also, that higher capacity can bechieved at higher Mn content. However, LiMnyFe1−yPO4 sam-les prepared by conventional synthetic routes showed decreasedapacity at higher Mn/Fe ratios. Thus, it can be concluded that theariation of y can be used to optimize the properties of this material.amada and Chung [15], in particular, optimized LiMnyFe1−yPO4s a possible 4.0 V cathode material. In their pioneering work,iMn0.6Fe0.4PO4, showed a high capacity and a good cyclability forp to 20 cycles. Hence, lithium bi-metal phosphates seem to be
nteresting cathode materials.The traditional synthesis method of lithium ion battery mate-
ials has been restricted in direct solid state reaction of precursort high temperature for a long time [21]. But, in recent years, wetethods have been more widely selected, as they can offer more
dvantages such as a better homogeneity, more regular morphol-gy, sub-micron sized particles, and larger specific surface area9,14,22–26]. Considerable improvements in the performances ofathode materials have been accomplished with these methods.articularly, carbon coating of the particle surface and nanostruc-uration have been adopted to achieve a higher capacity of bothiFePO4 [4,22,26] and LiMnPO4 [27,28].
In this paper we investigate the effect of Fe-site substitutionith Mn in LiFePO4. To the purpose, different nanostructured sam-les of LiMnyFe1−yPO4 (where 0 < y < 1) were prepared by a mildydrothermal method in the presence of a cationic surfactant. Thetructural–morphological changes in the cathode material due toncreasing dopant amount were thoroughly investigated. Besides,he effects of the substitution on the electrochemical activity,apacity and cycle performance were analysed at ambient temper-ture in laboratory-scale lithium cells. Finally, preliminary resultsegarding the capability of the best performing sample to work in
real Li-ion battery configuration versus a graphite anode will belso presented.
. Experimental
.1. Synthesis
Starting materials were FeSO4·7H2O (Aldrich, purity 99%),nSO4·H2O (Aldrich, purity 99%), H3PO4 (Aldrich, purity > 85%),
iOH (Aldrich, purity > 98%) and hexadecyltrimethylammoniumromide (Aldrich, C19H42BrN, CTAB). The carbon-containing mixed
ithium manganese iron phosphate samples were prepared bylight modification of a direct mild hydrothermal synthesis alreadyeported for the preparation of nanostructured LiFePO4/C powders22,26]. Following the receipt and procedure reported in ref. [26],
1.24 3.72 1.371.24 3.72 1.371.24 3.72 1.37
the precursor mixtures were prepared by dissolving stoichiometricamounts of reactants in doubly distilled water. The stoichiometricamounts of the precursor mixtures are given in Table 1.
The mixture, whose pH was adjusted between 7.2 and 7.5, wasvigorously stirred for 1 min and, then, quickly transferred in a250 mL airtight Teflon bottle and heated at 120 ◦C for 5 h. The bottlewas then cooled to ambient temperature and the resulting precip-itate washed with 500 mL of doubly distilled water, filtered anddried at about 55 ◦C overnight. The dried powders were introducedinto a vertical quartz tube equipped with a sintered glass filter (DISARaffaele e F.lli s.a.s., Milano – Italy). This set-up was indigenouslymade to assure a regular vertical diffusion of the inert gas throughthe glass filter and, consequently, a homogeneous diffusion throughthe powder, which is placed over the surface. The quartz tube waspositioned inside a vertical tubular furnace (Carbolite, UK) with atemperature control of ±1 ◦C (the heated length and diameter ofthe quartz reactor are 300 and 35 mm, respectively). The flow rateof the nitrogen used as carrier gas, directed from the bottom to thetop of the tube, was fixed at about 100 ml min−1. The samples wereheat-treated at 615 ◦C (heating rate of 2.0 ◦C min−1) under pure N2flux for 12 h in order to obtain the crystalline phase along with thecarbonisation of the organic surfactant, so obtaining a carbon filmthat homogeneously covers the grains [22].
By this process different samples having increasing amountsof Fe substitution by Mn were obtained, hereafter named LFM-y,where 0 < y < 100 indicates the degree of Fe substitution by Mn (seeTable 1). The LFM-0 sample was obtained without manganese andused as reference.
2.2. Analysis and characterization
The X-Ray powder diffraction (XRPD) profiles were obtainedby a Philips Xpert MPD powder diffractometer, equipped with CuK� radiation (V = 40 kV, i = 30 mA) and a curved graphite secondarymonochromator. The diffraction data were collected in the 10◦–80◦
2�-range with an acquisition step of 0.02◦ and a time per step of10 s. The structural and profile refinements were performed on thebasis of the Rietveld method with the TOPAS 3.0 software [29].The pattern refinements were carried out starting from the latticeparameters and the atomic positions obtained by Streltsov et al.[30].
Quantitative elemental analysis was carried out by inductivelycoupled plasma-atomic emission spectroscopy (ICP-AES) with aVarian Liberty 100 instrument. For analysis, the samples weredigested in hot concentrated HCl:HNO3 = 3:1 mixture. Specific sur-face areas (SSA) were determined using the Brunauer, Emmet,Teller (BET) method on an ASAP 2010 Micromeritics instrument.Prior to adsorption, approximately 50.0 mg of solid were placed inthe cell and evacuated at about 250 ◦C for 3 h.
Morphological characterization of the samples was performedemploying a FEI Quanta Inspect 200LV scanning electron micro-scope (SEM, max magnification of 1.5 × 105) equipped with an
Everhardt Thornley secondary electron detector (ET-SED). Prior toanalysis, all the samples were coated with a thin Cr layer (around10 nm thick) to minimize the effect of the electron beam irradia-tion. The samples were submitted to a high-resolution transmission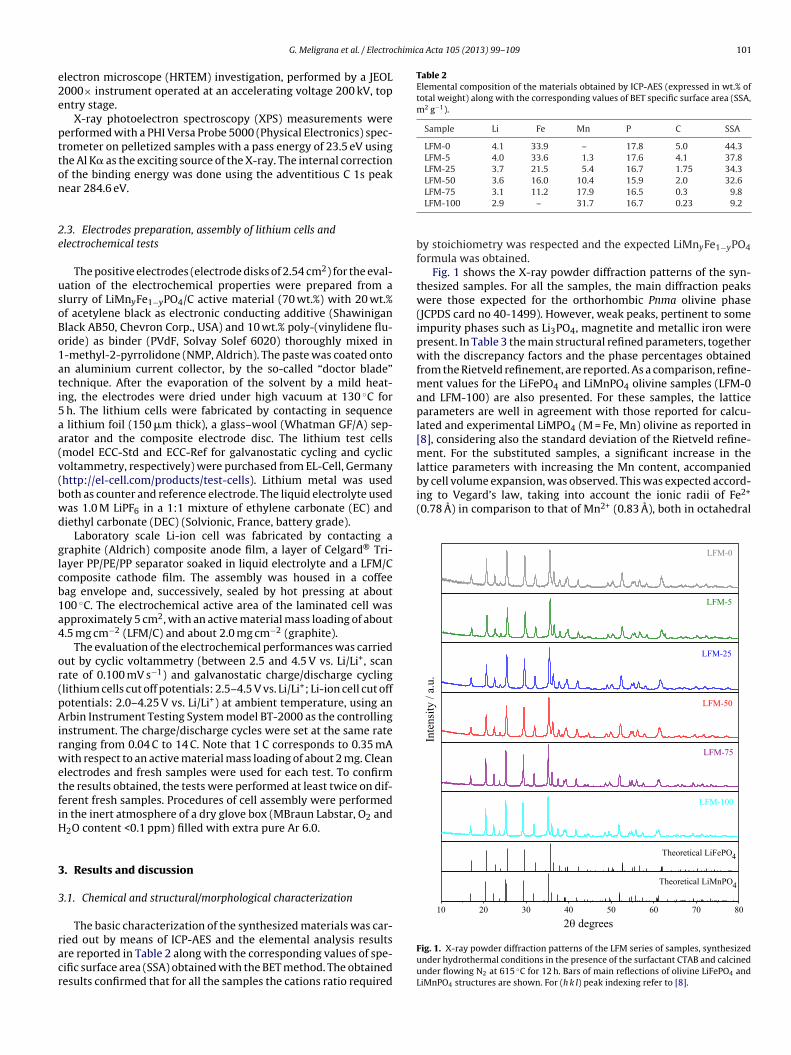
chimica Acta 105 (2013) 99– 109 101
e2e
ptton
2e
usoBo1ati5aa(v(bwd
glcb1a4
or(pAirwetfiH
3
3
racr
Table 2Elemental composition of the materials obtained by ICP-AES (expressed in wt.% oftotal weight) along with the corresponding values of BET specific surface area (SSA,m2 g−1).
Sample Li Fe Mn P C SSA
LFM-0 4.1 33.9 – 17.8 5.0 44.3LFM-5 4.0 33.6 1.3 17.6 4.1 37.8LFM-25 3.7 21.5 5.4 16.7 1.75 34.3LFM-50 3.6 16.0 10.4 15.9 2.0 32.6
by cell volume expansion, was observed. This was expected accord-ing to Vegard’s law, taking into account the ionic radii of Fe2+
(0.78 A) in comparison to that of Mn2+ (0.83 A), both in octahedral
10 20 30 40 50 60 70 80
Theoretical LiMnPO4
Theoretical LiFePO4
2θ degrees
LFM-100
Inte
nsi
ty /
a.u
.
LFM-75
LFM-50
LFM-25
LFM-5
LFM-0
G. Meligrana et al. / Electro
lectron microscope (HRTEM) investigation, performed by a JEOL000× instrument operated at an accelerating voltage 200 kV, topntry stage.
X-ray photoelectron spectroscopy (XPS) measurements wereerformed with a PHI Versa Probe 5000 (Physical Electronics) spec-rometer on pelletized samples with a pass energy of 23.5 eV usinghe Al K� as the exciting source of the X-ray. The internal correctionf the binding energy was done using the adventitious C 1s peakear 284.6 eV.
.3. Electrodes preparation, assembly of lithium cells andlectrochemical tests
The positive electrodes (electrode disks of 2.54 cm2) for the eval-ation of the electrochemical properties were prepared from alurry of LiMnyFe1−yPO4/C active material (70 wt.%) with 20 wt.%f acetylene black as electronic conducting additive (Shawiniganlack AB50, Chevron Corp., USA) and 10 wt.% poly-(vinylidene flu-ride) as binder (PVdF, Solvay Solef 6020) thoroughly mixed in-methyl-2-pyrrolidone (NMP, Aldrich). The paste was coated onton aluminium current collector, by the so-called “doctor blade”echnique. After the evaporation of the solvent by a mild heat-ng, the electrodes were dried under high vacuum at 130 ◦C for
h. The lithium cells were fabricated by contacting in sequence lithium foil (150 �m thick), a glass–wool (Whatman GF/A) sep-rator and the composite electrode disc. The lithium test cellsmodel ECC-Std and ECC-Ref for galvanostatic cycling and cyclicoltammetry, respectively) were purchased from EL-Cell, Germanyhttp://el-cell.com/products/test-cells). Lithium metal was usedoth as counter and reference electrode. The liquid electrolyte usedas 1.0 M LiPF6 in a 1:1 mixture of ethylene carbonate (EC) andiethyl carbonate (DEC) (Solvionic, France, battery grade).
Laboratory scale Li-ion cell was fabricated by contacting araphite (Aldrich) composite anode film, a layer of Celgard® Tri-ayer PP/PE/PP separator soaked in liquid electrolyte and a LFM/Composite cathode film. The assembly was housed in a coffeeag envelope and, successively, sealed by hot pressing at about00 ◦C. The electrochemical active area of the laminated cell waspproximately 5 cm2, with an active material mass loading of about.5 mg cm−2 (LFM/C) and about 2.0 mg cm−2 (graphite).
The evaluation of the electrochemical performances was carriedut by cyclic voltammetry (between 2.5 and 4.5 V vs. Li/Li+, scanate of 0.100 mV s−1) and galvanostatic charge/discharge cyclinglithium cells cut off potentials: 2.5–4.5 V vs. Li/Li+; Li-ion cell cut offotentials: 2.0–4.25 V vs. Li/Li+) at ambient temperature, using anrbin Instrument Testing System model BT-2000 as the controlling
nstrument. The charge/discharge cycles were set at the same rateanging from 0.04 C to 14 C. Note that 1 C corresponds to 0.35 mAith respect to an active material mass loading of about 2 mg. Clean
lectrodes and fresh samples were used for each test. To confirmhe results obtained, the tests were performed at least twice on dif-erent fresh samples. Procedures of cell assembly were performedn the inert atmosphere of a dry glove box (MBraun Labstar, O2 and
2O content <0.1 ppm) filled with extra pure Ar 6.0.
. Results and discussion
.1. Chemical and structural/morphological characterization
The basic characterization of the synthesized materials was car-
ied out by means of ICP-AES and the elemental analysis resultsre reported in Table 2 along with the corresponding values of spe-ific surface area (SSA) obtained with the BET method. The obtainedesults confirmed that for all the samples the cations ratio requiredLFM-75 3.1 11.2 17.9 16.5 0.3 9.8LFM-100 2.9 – 31.7 16.7 0.23 9.2
by stoichiometry was respected and the expected LiMnyFe1−yPO4formula was obtained.
Fig. 1 shows the X-ray powder diffraction patterns of the syn-thesized samples. For all the samples, the main diffraction peakswere those expected for the orthorhombic Pnma olivine phase(JCPDS card no 40-1499). However, weak peaks, pertinent to someimpurity phases such as Li3PO4, magnetite and metallic iron werepresent. In Table 3 the main structural refined parameters, togetherwith the discrepancy factors and the phase percentages obtainedfrom the Rietveld refinement, are reported. As a comparison, refine-ment values for the LiFePO4 and LiMnPO4 olivine samples (LFM-0and LFM-100) are also presented. For these samples, the latticeparameters are well in agreement with those reported for calcu-lated and experimental LiMPO4 (M = Fe, Mn) olivine as reported in[8], considering also the standard deviation of the Rietveld refine-ment. For the substituted samples, a significant increase in thelattice parameters with increasing the Mn content, accompanied
Fig. 1. X-ray powder diffraction patterns of the LFM series of samples, synthesizedunder hydrothermal conditions in the presence of the surfactant CTAB and calcinedunder flowing N2 at 615 ◦C for 12 h. Bars of main reflections of olivine LiFePO4 andLiMnPO4 structures are shown. For (h k l) peak indexing refer to [8].

102 G. Meligrana et al. / Electrochimica Acta 105 (2013) 99– 109
Table 3Structural parameters, compositions and discrepancy factors for the LFM series of samples, obtained from Rietveld refinement.
Sample LFM-0 LFM-5 LFM-25 LFM-50 LFM-75 LFM-100
a/Å 10.3268(3) 10.3358(3) 10.3670(3) 10.4004(4) 10.4301(3) 10.4508(4)b/Å 6.0019(2) 6.0094(2) 6.03270(2) 6.0585(2) 6.0799(2) 6.1040(2)c/Å 4.6994(1) 4.7039(2) 4.7158(2) 4.7285(2) 4.7470(2) 4.7513(2)V/Å3 291.27(1) 292.17(2) 294.93(2) 297.95(2) 301.03(2) 303.09(2)B(Li)/Å2 1.31(35) 0.95(32) 2.0(36) 1.75(36) 1.07(5) 0.72(50)B(M)/Å2 1.5(1) 0.91(4) 0.87(4) 0.45(5) 0.76(1) 0.77(50)N: LiM
0.982(2) 0.018(2) 0.972(3) 0.028(3) 0.968(3) 0.032(3) 0.963(1) 0.036(3) 0.962(4) 0.038(4) 0.984(3) 0.016(3)
Rwp/GoF 7.18/1.99 7.61/1.45 7.62/2.00 7.97/2.29 12.94/1.18 13.84/1.36Li3PO4 (wt.%) 1.57(15) 5.01(20) 2.64(2) 1.32(2) – 4.17(40)Fe O (wt.%) – 5.07(7) 2.33(9) 1.46(7) – –
cistittriiso3tfbftsctsseapobca[
iFLoctngtu4sr5oaog
3 4
Fe (wt.%) 2.72(3) – –
Mn2P2O7 (wt.%) – – –
oordination [8]. This consideration, together with the absence ofmpurity phases containing manganese, suggests that the iron sub-titution was effective with the exception of the LFM-75 sample,hat contains Mn2P2O7, which was most probably formed dur-ng thermal decomposition of the precursors [31]. To estimatehe influence of the impurity phases on the real stoichiometry ofhe Mn-substituted LiFePO4, the olivine-type stoichiometry can beecalculated on the basis of the impurity phase percentage reportedn Table 3. Even for the LFM-5 sample with a non-negligiblempurity amount (10% in total), the nominal and recalculated Fetoichiometry agrees quite well. In fact, the recalculated amountf the Fe/Mn ratio is 18.68, 2.66, 0.77 and 0.35 instead of 19.00,.00, 1.0 and 0.33 expected from the synthesis stoichiometry forhe LFM-5, LFM-25, LFM-50 and LFM-75 samples, respectively. Aurther investigation was performed concerning the cation distri-ution in the olivine lattice: slightly negative B factors were foundor the lithium site, thus indicating a lack of electronic density onhe same site. For this reason, occupancies were refined using auitable constraint on the total cation amount. The transition metalations appear to distribute both on Fe and Li sites for the substi-uted as well as for the not substituted olivine. This result is noto surprising, as the presence of the anti-site defect in the olivinetructure has been extensively discussed in the literature, see forxample [8,32,33] and references therein. In our case, the LFM-0nd LFM-100 samples with about 2% of anti-site defect appear toossess a lower degree of disorder compared to the substitutednes (LFM-75 has about 4% of anti-site defect). This difference cane likely ascribed to the simultaneous presence of two differentations in the olivine structure. Anyway, the overall amount ofnti-site defect is in complete agreement with previous reports34].
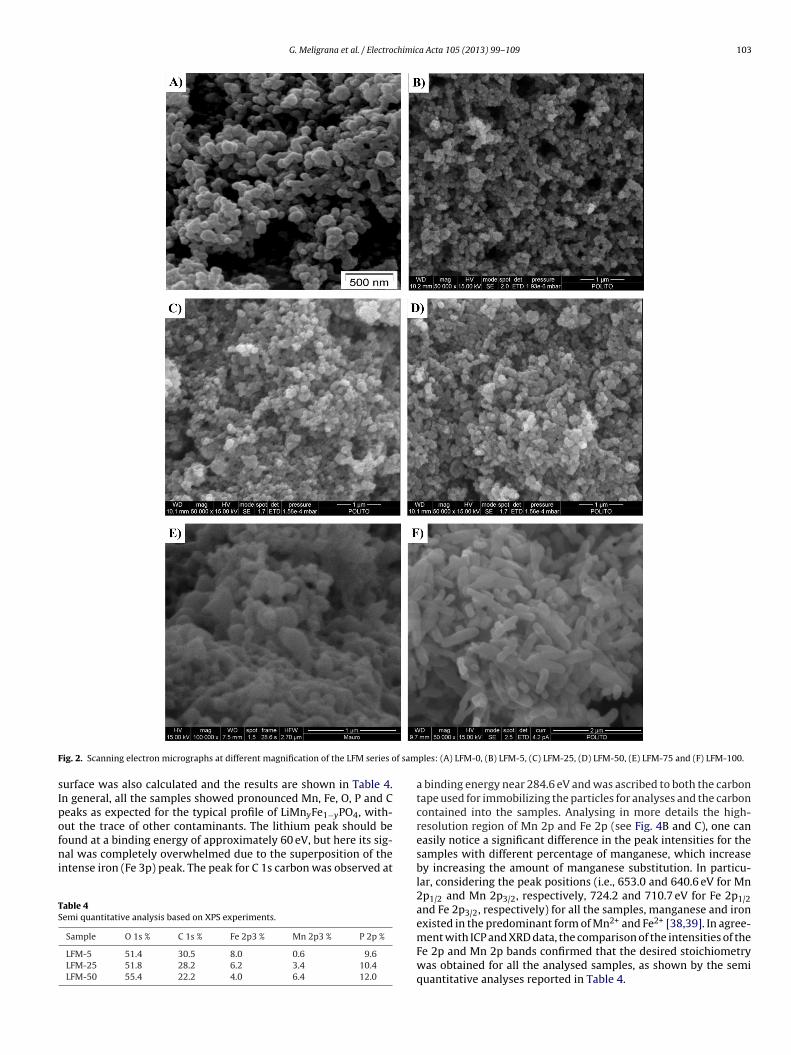
The morphology of the LFM series of samples was character-zed by SEM analysis. The resulting micrographs are shown inig. 2(A)–(F). The SEM micrographs of samples LFM-5, LFM-25 andFM-50 (see images B–D, respectively) pointed out the absencef noticeable morphological differences, both among them andompared to sample LFM-0 (not Mn substituted, image A). Allhese samples demonstrated sub-micrometric agglomerates andanometric grain sizes, with nanostructured particles very homo-eneous both in size and shape. No signs of coalescence amonghe grains to form big aggregates were distinguishable. In partic-lar, the grain size of LFM-0 could be roughly estimated around0–50 nm, while the grain size of the Mn-substituted samples waslightly higher, in good agreement with the Rietveld refinementesults for which, depending on the sample, values in the range0–60 nm were found. On the other hand, by increasing the amount
f Mn substitution to a higher extent (i.e., above 50%), notice-ble differences in the morphology of the samples were clearlybserved and, along with the presence of spherical particles, elon-ated structures appeared to be also present. In fact, LFM-75 (see0.68(3) – –– 7.02(21) –
micrograph E in Fig. 2) mainly showed particles with prevalentspherical morphology, but having increased dimensions that couldreach 200–300 nm, along with the presence of some elongated par-ticles. Considering the LFM-100 sample (image F), not only thedimensions but also the morphology was different. Actually, itmainly revealed regular elongated particles, homogeneous in shapeand with size in the micrometric range.
It has to be recalled here that particle morphology can play animportant role in the corresponding active material electrochem-ical behaviour; this has been recently demonstrated for LiMnPO4obtained with different shapes (plates, wedges and prisms) havingdifferent crystallographic orientations [35,36]. In fact, as lithiumions are constrained to move only parallel to b direction, only theac plane is active for Li+ extraction and insertion, thus allowinga fast Li+ ion diffusion rate and short diffusion length [37]. Thus,samples LFM-75 and LFM-100 are expected to show different elec-trochemical behaviors with respect to the other samples havinglower degree of Mn substitution.
BET specific surface area measurements confirmed the positiveinfluence of the use of the organic surfactant as dispersing agent inthis particular synthesis procedure, thus leading to powders withelevated SSA values [22]. It was possible to observe a slight pro-gressive decrease in SSA with the increment of the Mn percentage.This effect was found to be compatible with the observed increasein both cell volumes and grain dimensions (see structural charac-terization). Overall, it is important to remark that SSA values inthe range of 32–38 m2 g−1 were obtained for the Mn-substitutedsamples, which are significant values for bulk crystalline materials.Thus, the effectiveness of the CTAB surfactant in modulating bothparticle size and morphology was here confirmed [22,26] also forthe preparation of Mn-based phosphates.
The coating of the active material grains by a homogeneous layerof carbon was confirmed by HRTEM analysis. Some typical micro-graphs are shown in Fig. 3 for sample LFM-5, representative also forsamples LFM-25 and LFM-50 which showed similar features, withonly a slight progressive decrease in the thickness of the C layer, inagreement with the elemental analysis results. In fact, the carboncontent reduces from 4% in LFM-5 to about 2% in LFM-25 and LFM-50 (see Table 2). In general, the samples appeared well crystallizedand having all the grains covered with a uniform, very homoge-neous layer of carbon, approximately 3–5 nm thick. In particular,from the right hand sided micrograph it is possible to note thatsuch a layer of carbon presented also a certain degree of graphiti-sation. These characteristics of homogeneity and graphitisation ofthe carbon layer should positively affect the electronic conductivityof the samples and, correspondingly, their rate capability.
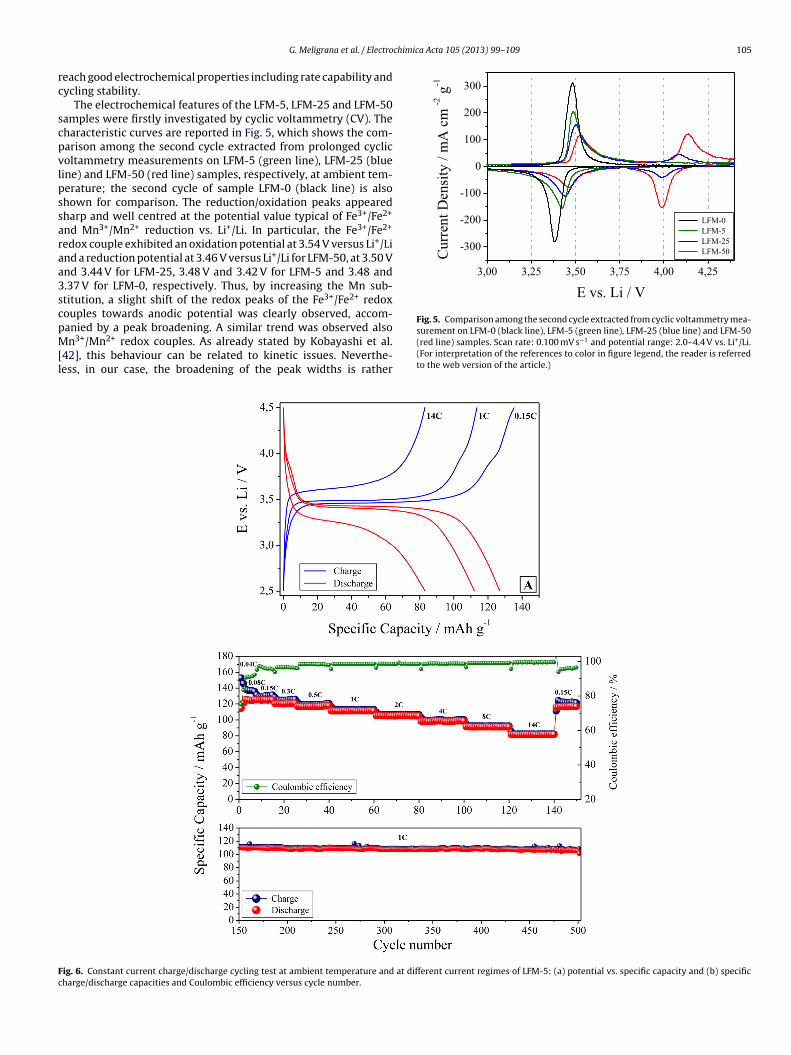
The surface chemical compositions and element states were alsoinvestigated by means of XPS analysis. Fig. 4 gives the wide XPSsurvey profile for the three samples with content of manganeseranging from 0 to 50. The atomic percentage of elements on the
G. Meligrana et al. / Electrochimica Acta 105 (2013) 99– 109 103
F f sam
sIpofni
TS
ig. 2. Scanning electron micrographs at different magnification of the LFM series o
urface was also calculated and the results are shown in Table 4.n general, all the samples showed pronounced Mn, Fe, O, P and Ceaks as expected for the typical profile of LiMnyFe1−yPO4, with-ut the trace of other contaminants. The lithium peak should be
ound at a binding energy of approximately 60 eV, but here its sig-al was completely overwhelmed due to the superposition of thentense iron (Fe 3p) peak. The peak for C 1s carbon was observed at
able 4emi quantitative analysis based on XPS experiments.
Sample O 1s % C 1s % Fe 2p3 % Mn 2p3 % P 2p %
LFM-5 51.4 30.5 8.0 0.6 9.6LFM-25 51.8 28.2 6.2 3.4 10.4LFM-50 55.4 22.2 4.0 6.4 12.0
ples: (A) LFM-0, (B) LFM-5, (C) LFM-25, (D) LFM-50, (E) LFM-75 and (F) LFM-100.
a binding energy near 284.6 eV and was ascribed to both the carbontape used for immobilizing the particles for analyses and the carboncontained into the samples. Analysing in more details the high-resolution region of Mn 2p and Fe 2p (see Fig. 4B and C), one caneasily notice a significant difference in the peak intensities for thesamples with different percentage of manganese, which increaseby increasing the amount of manganese substitution. In particu-lar, considering the peak positions (i.e., 653.0 and 640.6 eV for Mn2p1/2 and Mn 2p3/2, respectively, 724.2 and 710.7 eV for Fe 2p1/2and Fe 2p3/2, respectively) for all the samples, manganese and ironexisted in the predominant form of Mn2+ and Fe2+ [38,39]. In agree-
ment with ICP and XRD data, the comparison of the intensities of theFe 2p and Mn 2p bands confirmed that the desired stoichiometrywas obtained for all the analysed samples, as shown by the semiquantitative analyses reported in Table 4.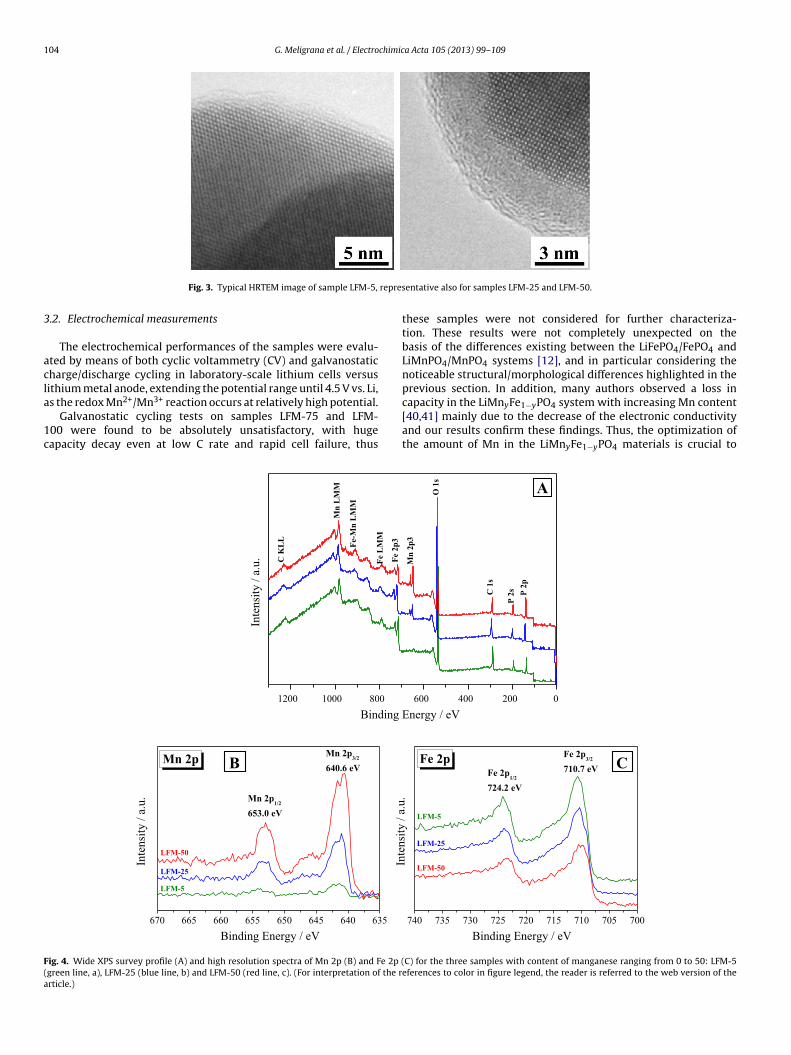
104 G. Meligrana et al. / Electrochimica Acta 105 (2013) 99– 109
repres
3
acla
1c
F(a
Fig. 3. Typical HRTEM image of sample LFM-5,
.2. Electrochemical measurements
The electrochemical performances of the samples were evalu-ted by means of both cyclic voltammetry (CV) and galvanostaticharge/discharge cycling in laboratory-scale lithium cells versusithium metal anode, extending the potential range until 4.5 V vs. Li,
s the redox Mn2+/Mn3+ reaction occurs at relatively high potential.Galvanostatic cycling tests on samples LFM-75 and LFM-00 were found to be absolutely unsatisfactory, with hugeapacity decay even at low C rate and rapid cell failure, thus
1200 1000 800
Fe
2p
3
Fe
LM
M
Fe-
Mn
LM
M
Mn
LM
M
C K
LL
Inte
nsi
ty /
a.u
.
Binding
670 665 660 655 650 645 640 635
Mn 2p1/2
653.0 eV
Inte
nsi
ty /
a.u
.
Mn 2p3/2
640.6 eV
LFM-5
LFM-25
LFM-50
Mn 2p
Binding Energy / eV
B
ig. 4. Wide XPS survey profile (A) and high resolution spectra of Mn 2p (B) and Fe 2p (green line, a), LFM-25 (blue line, b) and LFM-50 (red line, c). (For interpretation of the rrticle.)
entative also for samples LFM-25 and LFM-50.
these samples were not considered for further characteriza-tion. These results were not completely unexpected on thebasis of the differences existing between the LiFePO4/FePO4 andLiMnPO4/MnPO4 systems [12], and in particular considering thenoticeable structural/morphological differences highlighted in theprevious section. In addition, many authors observed a loss in
capacity in the LiMnyFe1−yPO4 system with increasing Mn content[40,41] mainly due to the decrease of the electronic conductivityand our results confirm these findings. Thus, the optimization ofthe amount of Mn in the LiMnyFe1−yPO4 materials is crucial to600 400 200 0
P 2
p
P 2
sC 1
s
O 1
s
Mn
2p
3
Energy / eV
740 735 730 725 720 715 710 705 700
Fe 2p1/2
724.2 eV
Fe 2p3/2
710.7 eV
Binding Energy / eV
Inte
nsi
ty /
a.u
.
Fe 2p
LFM-5
LFM-25
LFM-50
C
A
C) for the three samples with content of manganese ranging from 0 to 50: LFM-5eferences to color in figure legend, the reader is referred to the web version of the
chimica Acta 105 (2013) 99– 109 105
rc
scpvlpssaraa3scpM[l
3,00 3,25 3,50 3,75 4,00 4,25
-300
-200
-100
0
100
200
300
LFM-0
LFM-5
LFM-25
LFM-50Cu
rren
t D
ensi
ty /
mA
cm
-2 g
-1
E vs. Li / V
Fig. 5. Comparison among the second cycle extracted from cyclic voltammetry mea-
Fc
G. Meligrana et al. / Electro
each good electrochemical properties including rate capability andycling stability.
The electrochemical features of the LFM-5, LFM-25 and LFM-50amples were firstly investigated by cyclic voltammetry (CV). Theharacteristic curves are reported in Fig. 5, which shows the com-arison among the second cycle extracted from prolonged cyclicoltammetry measurements on LFM-5 (green line), LFM-25 (blueine) and LFM-50 (red line) samples, respectively, at ambient tem-erature; the second cycle of sample LFM-0 (black line) is alsohown for comparison. The reduction/oxidation peaks appearedharp and well centred at the potential value typical of Fe3+/Fe2+
nd Mn3+/Mn2+ reduction vs. Li+/Li. In particular, the Fe3+/Fe2+
edox couple exhibited an oxidation potential at 3.54 V versus Li+/Lind a reduction potential at 3.46 V versus Li+/Li for LFM-50, at 3.50 Vnd 3.44 V for LFM-25, 3.48 V and 3.42 V for LFM-5 and 3.48 and.37 V for LFM-0, respectively. Thus, by increasing the Mn sub-titution, a slight shift of the redox peaks of the Fe3+/Fe2+ redoxouples towards anodic potential was clearly observed, accom-
anied by a peak broadening. A similar trend was observed alson3+/Mn2+ redox couples. As already stated by Kobayashi et al.42], this behaviour can be related to kinetic issues. Neverthe-ess, in our case, the broadening of the peak widths is rather
surement on LFM-0 (black line), LFM-5 (green line), LFM-25 (blue line) and LFM-50(red line) samples. Scan rate: 0.100 mV s−1 and potential range: 2.0–4.4 V vs. Li+/Li.(For interpretation of the references to color in figure legend, the reader is referredto the web version of the article.)
ig. 6. Constant current charge/discharge cycling test at ambient temperature and at different current regimes of LFM-5: (a) potential vs. specific capacity and (b) specificharge/discharge capacities and Coulombic efficiency versus cycle number.
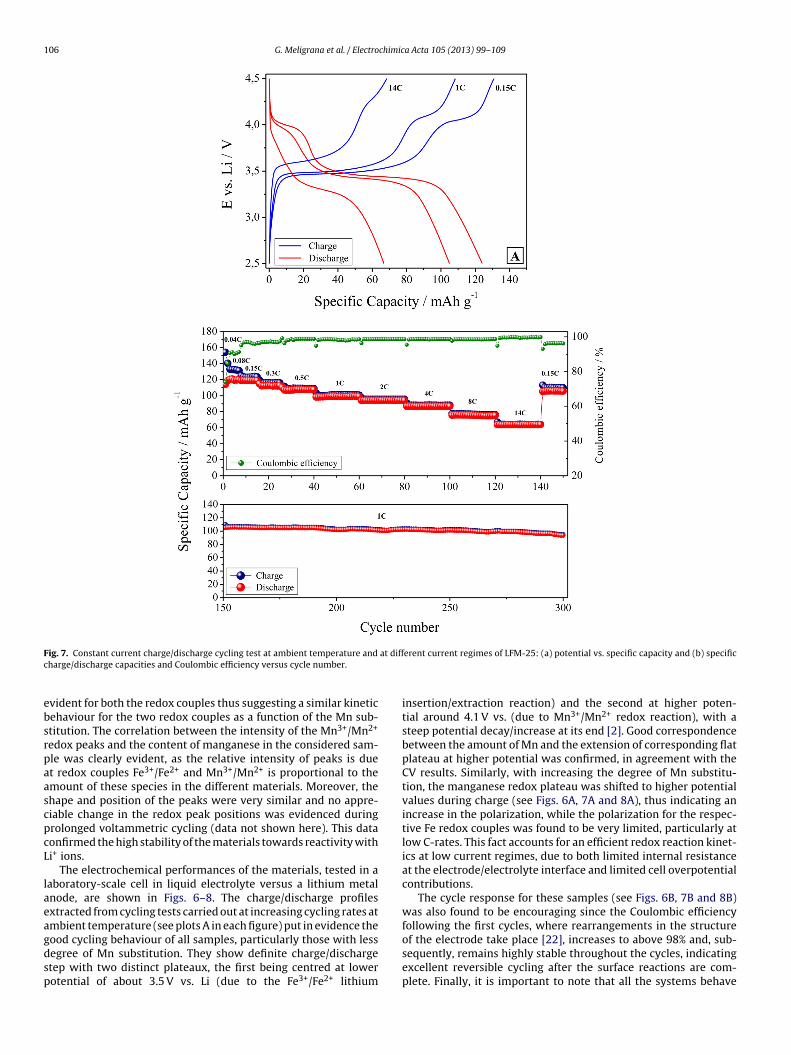
106 G. Meligrana et al. / Electrochimica Acta 105 (2013) 99– 109
F at diffc
ebsrpaascpcL
laeagdsp
ig. 7. Constant current charge/discharge cycling test at ambient temperature andharge/discharge capacities and Coulombic efficiency versus cycle number.
vident for both the redox couples thus suggesting a similar kineticehaviour for the two redox couples as a function of the Mn sub-titution. The correlation between the intensity of the Mn3+/Mn2+
edox peaks and the content of manganese in the considered sam-le was clearly evident, as the relative intensity of peaks is duet redox couples Fe3+/Fe2+ and Mn3+/Mn2+ is proportional to themount of these species in the different materials. Moreover, thehape and position of the peaks were very similar and no appre-iable change in the redox peak positions was evidenced duringrolonged voltammetric cycling (data not shown here). This dataonfirmed the high stability of the materials towards reactivity withi+ ions.
The electrochemical performances of the materials, tested in aaboratory-scale cell in liquid electrolyte versus a lithium metalnode, are shown in Figs. 6–8. The charge/discharge profilesxtracted from cycling tests carried out at increasing cycling rates atmbient temperature (see plots A in each figure) put in evidence the
ood cycling behaviour of all samples, particularly those with lessegree of Mn substitution. They show definite charge/dischargetep with two distinct plateaux, the first being centred at lowerotential of about 3.5 V vs. Li (due to the Fe3+/Fe2+ lithiumerent current regimes of LFM-25: (a) potential vs. specific capacity and (b) specific
insertion/extraction reaction) and the second at higher poten-tial around 4.1 V vs. (due to Mn3+/Mn2+ redox reaction), with asteep potential decay/increase at its end [2]. Good correspondencebetween the amount of Mn and the extension of corresponding flatplateau at higher potential was confirmed, in agreement with theCV results. Similarly, with increasing the degree of Mn substitu-tion, the manganese redox plateau was shifted to higher potentialvalues during charge (see Figs. 6A, 7A and 8A), thus indicating anincrease in the polarization, while the polarization for the respec-tive Fe redox couples was found to be very limited, particularly atlow C-rates. This fact accounts for an efficient redox reaction kinet-ics at low current regimes, due to both limited internal resistanceat the electrode/electrolyte interface and limited cell overpotentialcontributions.
The cycle response for these samples (see Figs. 6B, 7B and 8B)was also found to be encouraging since the Coulombic efficiencyfollowing the first cycles, where rearrangements in the structure
of the electrode take place [22], increases to above 98% and, sub-sequently, remains highly stable throughout the cycles, indicatingexcellent reversible cycling after the surface reactions are com-plete. Finally, it is important to note that all the systems behave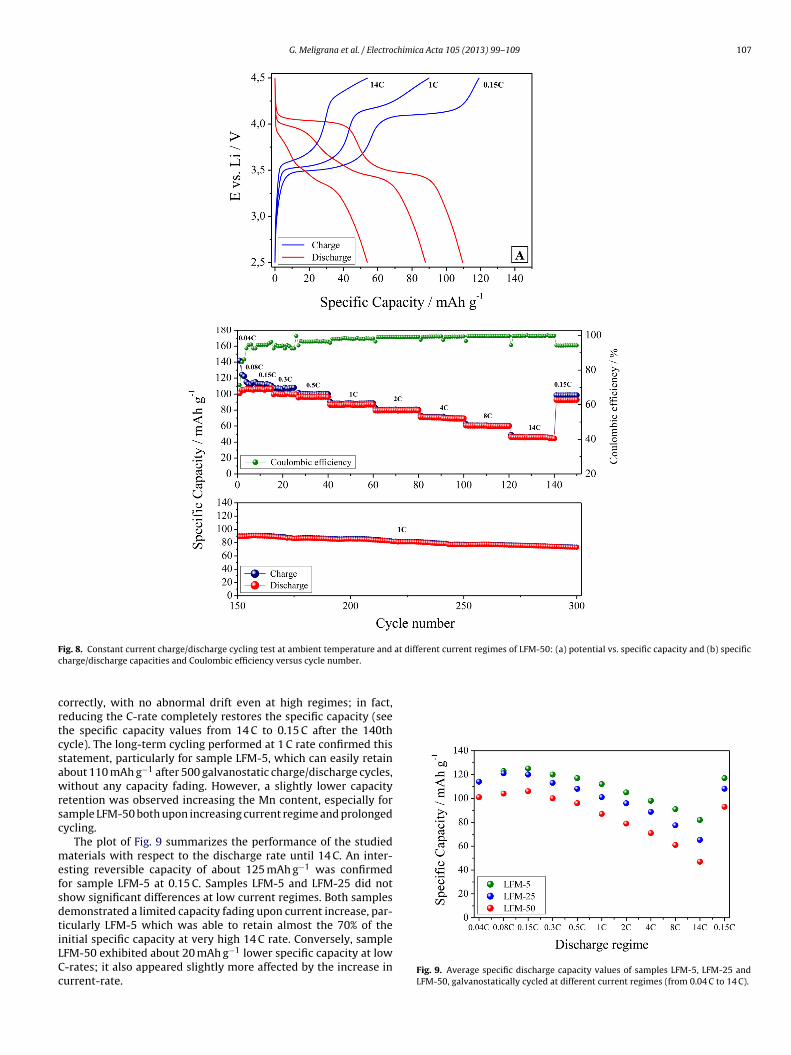
G. Meligrana et al. / Electrochimica Acta 105 (2013) 99– 109 107
F at different current regimes of LFM-50: (a) potential vs. specific capacity and (b) specificc
crtcsawrsc
mefsdtiLCc
ig. 8. Constant current charge/discharge cycling test at ambient temperature and
harge/discharge capacities and Coulombic efficiency versus cycle number.
orrectly, with no abnormal drift even at high regimes; in fact,educing the C-rate completely restores the specific capacity (seehe specific capacity values from 14 C to 0.15 C after the 140thycle). The long-term cycling performed at 1 C rate confirmed thistatement, particularly for sample LFM-5, which can easily retainbout 110 mAh g−1 after 500 galvanostatic charge/discharge cycles,ithout any capacity fading. However, a slightly lower capacity
etention was observed increasing the Mn content, especially forample LFM-50 both upon increasing current regime and prolongedycling.
The plot of Fig. 9 summarizes the performance of the studiedaterials with respect to the discharge rate until 14 C. An inter-
sting reversible capacity of about 125 mAh g−1 was confirmedor sample LFM-5 at 0.15 C. Samples LFM-5 and LFM-25 did nothow significant differences at low current regimes. Both samplesemonstrated a limited capacity fading upon current increase, par-icularly LFM-5 which was able to retain almost the 70% of the
nitial specific capacity at very high 14 C rate. Conversely, sampleFM-50 exhibited about 20 mAh g−1 lower specific capacity at low-rates; it also appeared slightly more affected by the increase inurrent-rate.Fig. 9. Average specific discharge capacity values of samples LFM-5, LFM-25 andLFM-50, galvanostatically cycled at different current regimes (from 0.04 C to 14 C).
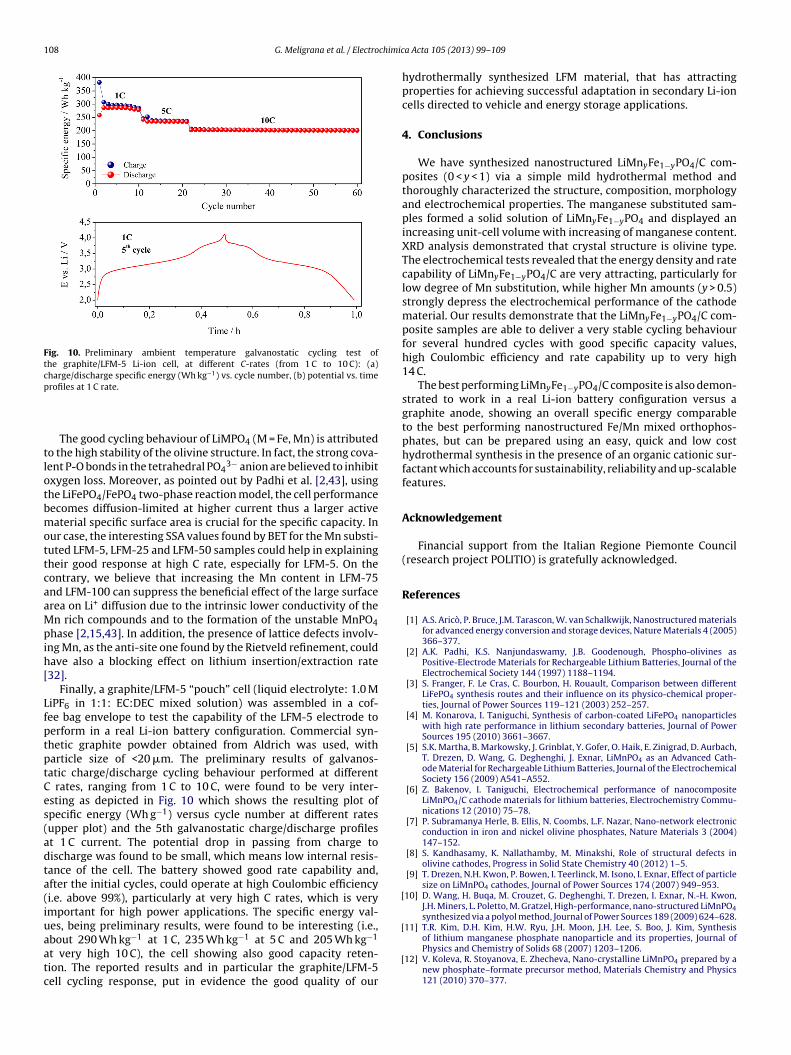
108 G. Meligrana et al. / Electrochimic
Fig. 10. Preliminary ambient temperature galvanostatic cycling test ofthe graphite/LFM-5 Li-ion cell, at different C-rates (from 1 C to 10 C): (a)charge/discharge specific energy (Wh kg−1) vs. cycle number, (b) potential vs. timep
tlotbmottcaaMpih[
LfptptCes(adta(iuaatc
[
[of lithium manganese phosphate nanoparticle and its properties, Journal of
rofiles at 1 C rate.
The good cycling behaviour of LiMPO4 (M = Fe, Mn) is attributedo the high stability of the olivine structure. In fact, the strong cova-ent P-O bonds in the tetrahedral PO4
3− anion are believed to inhibitxygen loss. Moreover, as pointed out by Padhi et al. [2,43], usinghe LiFePO4/FePO4 two-phase reaction model, the cell performanceecomes diffusion-limited at higher current thus a larger activeaterial specific surface area is crucial for the specific capacity. In
ur case, the interesting SSA values found by BET for the Mn substi-uted LFM-5, LFM-25 and LFM-50 samples could help in explainingheir good response at high C rate, especially for LFM-5. On theontrary, we believe that increasing the Mn content in LFM-75nd LFM-100 can suppress the beneficial effect of the large surfacerea on Li+ diffusion due to the intrinsic lower conductivity of then rich compounds and to the formation of the unstable MnPO4
hase [2,15,43]. In addition, the presence of lattice defects involv-ng Mn, as the anti-site one found by the Rietveld refinement, couldave also a blocking effect on lithium insertion/extraction rate32].
Finally, a graphite/LFM-5 “pouch” cell (liquid electrolyte: 1.0 MiPF6 in 1:1: EC:DEC mixed solution) was assembled in a cof-ee bag envelope to test the capability of the LFM-5 electrode toerform in a real Li-ion battery configuration. Commercial syn-hetic graphite powder obtained from Aldrich was used, witharticle size of <20 �m. The preliminary results of galvanos-atic charge/discharge cycling behaviour performed at different
rates, ranging from 1 C to 10 C, were found to be very inter-sting as depicted in Fig. 10 which shows the resulting plot ofpecific energy (Wh g−1) versus cycle number at different ratesupper plot) and the 5th galvanostatic charge/discharge profilest 1 C current. The potential drop in passing from charge toischarge was found to be small, which means low internal resis-ance of the cell. The battery showed good rate capability and,fter the initial cycles, could operate at high Coulombic efficiencyi.e. above 99%), particularly at very high C rates, which is verymportant for high power applications. The specific energy val-es, being preliminary results, were found to be interesting (i.e.,
−1 −1 −1
bout 290 Wh kg at 1 C, 235 Wh kg at 5 C and 205 Wh kgt very high 10 C), the cell showing also good capacity reten-ion. The reported results and in particular the graphite/LFM-5ell cycling response, put in evidence the good quality of our[
a Acta 105 (2013) 99– 109
hydrothermally synthesized LFM material, that has attractingproperties for achieving successful adaptation in secondary Li-ioncells directed to vehicle and energy storage applications.
4. Conclusions
We have synthesized nanostructured LiMnyFe1−yPO4/C com-posites (0 < y < 1) via a simple mild hydrothermal method andthoroughly characterized the structure, composition, morphologyand electrochemical properties. The manganese substituted sam-ples formed a solid solution of LiMnyFe1−yPO4 and displayed anincreasing unit-cell volume with increasing of manganese content.XRD analysis demonstrated that crystal structure is olivine type.The electrochemical tests revealed that the energy density and ratecapability of LiMnyFe1−yPO4/C are very attracting, particularly forlow degree of Mn substitution, while higher Mn amounts (y > 0.5)strongly depress the electrochemical performance of the cathodematerial. Our results demonstrate that the LiMnyFe1−yPO4/C com-posite samples are able to deliver a very stable cycling behaviourfor several hundred cycles with good specific capacity values,high Coulombic efficiency and rate capability up to very high14 C.
The best performing LiMnyFe1−yPO4/C composite is also demon-strated to work in a real Li-ion battery configuration versus agraphite anode, showing an overall specific energy comparableto the best performing nanostructured Fe/Mn mixed orthophos-phates, but can be prepared using an easy, quick and low costhydrothermal synthesis in the presence of an organic cationic sur-factant which accounts for sustainability, reliability and up-scalablefeatures.
Acknowledgement
Financial support from the Italian Regione Piemonte Council(research project POLITIO) is gratefully acknowledged.
References
[1] A.S. Aricò, P. Bruce, J.M. Tarascon, W. van Schalkwijk, Nanostructured materialsfor advanced energy conversion and storage devices, Nature Materials 4 (2005)366–377.
[2] A.K. Padhi, K.S. Nanjundaswamy, J.B. Goodenough, Phospho-olivines asPositive-Electrode Materials for Rechargeable Lithium Batteries, Journal of theElectrochemical Society 144 (1997) 1188–1194.
[3] S. Franger, F. Le Cras, C. Bourbon, H. Rouault, Comparison between differentLiFePO4 synthesis routes and their influence on its physico-chemical proper-ties, Journal of Power Sources 119–121 (2003) 252–257.
[4] M. Konarova, I. Taniguchi, Synthesis of carbon-coated LiFePO4 nanoparticleswith high rate performance in lithium secondary batteries, Journal of PowerSources 195 (2010) 3661–3667.
[5] S.K. Martha, B. Markowsky, J. Grinblat, Y. Gofer, O. Haik, E. Zinigrad, D. Aurbach,T. Drezen, D. Wang, G. Deghenghi, J. Exnar, LiMnPO4 as an Advanced Cath-ode Material for Rechargeable Lithium Batteries, Journal of the ElectrochemicalSociety 156 (2009) A541–A552.
[6] Z. Bakenov, I. Taniguchi, Electrochemical performance of nanocompositeLiMnPO4/C cathode materials for lithium batteries, Electrochemistry Commu-nications 12 (2010) 75–78.
[7] P. Subramanya Herle, B. Ellis, N. Coombs, L.F. Nazar, Nano-network electronicconduction in iron and nickel olivine phosphates, Nature Materials 3 (2004)147–152.
[8] S. Kandhasamy, K. Nallathamby, M. Minakshi, Role of structural defects inolivine cathodes, Progress in Solid State Chemistry 40 (2012) 1–5.
[9] T. Drezen, N.H. Kwon, P. Bowen, I. Teerlinck, M. Isono, I. Exnar, Effect of particlesize on LiMnPO4 cathodes, Journal of Power Sources 174 (2007) 949–953.
10] D. Wang, H. Buqa, M. Crouzet, G. Deghenghi, T. Drezen, I. Exnar, N.-H. Kwon,J.H. Miners, L. Poletto, M. Gratzel, High-performance, nano-structured LiMnPO4
synthesized via a polyol method, Journal of Power Sources 189 (2009) 624–628.11] T.R. Kim, D.H. Kim, H.W. Ryu, J.H. Moon, J.H. Lee, S. Boo, J. Kim, Synthesis
Physics and Chemistry of Solids 68 (2007) 1203–1206.12] V. Koleva, R. Stoyanova, E. Zhecheva, Nano-crystalline LiMnPO4 prepared by a
new phosphate–formate precursor method, Materials Chemistry and Physics121 (2010) 370–377.

chimic
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
G. Meligrana et al. / Electro
13] R. Dominko, M. Bele, M. Gasberscek, M. Remskar, D. Hanzel, J.M. Goupil, S.Pejovnik, J. Jamnik, Porous olivine composites synthesized by sol–gel tech-nique, Journal of Power Sources 153 (2006) 274–280.
14] N.N. Bramnik, H. Ehrenberg, Precursor-based synthesis and electrochemi-cal performance of LiMnPO4, Journal of Alloys and Compounds 464 (2008)259–264.
15] A. Yamada, S.C. Chung, Crystal chemistry of the Olivine-type Li(MnyFe1−y)PO4
and (MnyFe1−y)PO4 as possible 4 V cathode materials for lithium batteries,Journal of the Electrochemical Society 148 (2001) A960–A967.
16] G. Chen, T.J. Richardson, Solid solution phases in the olivine-typeLiMnPO4/MnPO4 system, Journal of the Electrochemical Society 156 (2009)A756–A762.
17] S.-W. Kim, J. Kim, H. Gwon, K. Kang, Phase stability study of Li1-xMn PO4
(0 ≤ x ≤ 1) cathode for Li rechargeable battery, Journal of the ElectrochemicalSociety 156 (2009) A635–A638.
18] N.N. Bramnik, K. Nikolowsky, S.M. Trots, H. Ehrenberg, Thermal stability ofLiCoPO4 cathodes, Electrochemistry and Solid-State Letters 11 (2008) A89–A93.
19] G. Chen, T.J. Richardson, Thermal instability of olivine-type LiMnPO4 cathodes,Journal of Power Sources 195 (2010) 1221–1224.
20] A. Yamada, Y. Kudo, K.Y. Liu, Phase diagram of Lix(MnyFe1-y)PO4 (0 ≤ x ≤ 1),Journal of the Electrochemical Society 148 (2001) A1153–A1158.
21] A. Yamada, M. Hosoya, S.-Ch. Chung, Y. Kudo, K. Hinokuma, K.-Y. Liu, Y. Nishi,Olivine-type cathodes: achievements and problems, Journal of Power Sources119–121 (2003) 232–238.
22] G. Meligrana, C. Gerbaldi, A. Tuel, S. Bodoardo, N. Penazzi, Hydrothermal syn-thesis of high surface LiFePO4 powders as cathode for Li-ion cells, Journal ofPower Sources 160 (2006) 516–522.
23] G. Liang, P. Charest, M. Gauthier, A. Guerfi, C. Michot, K. Zaghib, Int. PatentAppl. A method for preparing a particulate cathode materials, and the materialobtained by said method (2008) WO/2008/067677.
24] S. Bodoardo, C. Gerbaldi, G. Meligrana, F. Di Lupo, N. Penazzi, D. Fontana, Int.Patent Appl., Hydrothermal Process for the production of LiFePO4 powder(2011) WO/2011/057646.
25] F. Brochu, A. Guerfi, J. Trottier, M. Kopec, A. Mauger, H. Groult, C.M. Julien, K.Zaghib, Structure and electrochemistry of scaling nano C–LiFePO4 synthesizedby hydrothermal route: complexing agent effect, Journal of Power Sources 214(2012) 1–6.
26] S. Bodoardo, C. Gerbaldi, G. Meligrana, A. Tuel, S. Enzo, N. Penazzi, Optimisationof some parameters for the preparation of nanostructured LiFePO4/C cathode,Ionics 15 (2009) 19–26.
27] G.H. Li, H. Azuma, M. Tohda, LiMnPO4 as the Cathode for Lithium Batteries,Electrochemistry and Solid-State Letters 5 (2002) A135–A137.
28] M. Yonemura, A. Yamada, Y. Takei, N. Sonoyama, R. Kanno, Comparative kineticstudy of olivine LixMPO4 (M = Fe, Mn), Journal of the Electrochemical Society151 (2004) A1352–A1356.
[
a Acta 105 (2013) 99– 109 109
29] Bruker AXS 2000: TOPAS V3. 0; general profile and structural analysis softwarefor powder diffraction data, in: User Manual, Bruker AXS, Karlsruhe, Germany,2000.
30] V.A. Streltsov, E.L. Belokoneva, V.G. Tsirelson, N.K. Hansen, Multipole analysisof the electron density in Triphylite, LiFePO4, using X-ray diffraction data, ActaCrystallographica B 49 (1993) 147–153.
31] R. von Hagen, H. Lorrmann, K.-C. Möller, S. Mathur, ElectrospunLiFe1−yMnyPO4/C nanofiber composites as self-supporting cathodes inLi-Ion batteries, Advanced Energy Materials 2 (2012) 553–559.
32] G.R. Gardiner, M.S. Islam, Anti-Site defects and ion migration in theLiFe0.5Mn0.5PO4 mixed-metal cathode material, Chemistry of Materials 22(2010) 1242–1248.
33] M. Bini, S. Ferrari, D. Capsoni, P. Mustarelli, G. Spina, F. Del Giallo, M. Lantieri, C.Leonelli, A. Rizzuti, V. Massarotti, Pair distribution function analysis and möss-bauer study of defects in microwave-hydrothermal LiFePO4, RSC Advances 2(2012) 250–258.
34] M.S. Whittingham, Lithium batteries and cathode materials, Chemical Reviews104 (2004) 4271–4301.
35] X.-L. Pan, C.-Y. Xu, L. Zhen, Synthesis of LiMnPO4 microspheres assembledby plates, wedges and prisms with different crystallographic orientationsand their electrochemical performance, CrystEngComm 14 (2012) 6412–6418.
36] Z. Qin, X. Zhou, Y. Xia, C. Tang, Z. Liu, Morphology controlled synthesis and mod-ification of high-performance LiMnPO4 cathode materials for Li-ion batteries,Journal of Materials Chemistry 22 (2012) 21144–21153.
37] G. Chen, X. Song, T.J. Richardson, Electron microscopy study of the LiFePO4
to FePO4 phase transition, Electrochemistry and Solid-State Letters 9 (2006)A295–A298.
38] T. Yamashita, P. Hayes, Analysis of XPS spectra of Fe2+ and Fe3+ ions in oxidematerials, Applied Surface Science 254 (2008) 2441–2449.
39] Y.-H. Rho, L.F. Nazar, L. Perry, D. Ryan, Surface chemistry of LiFePO4 stud-ied by mössbauer and X-ray photoelectron spectroscopy and its effect onelectrochemical properties, Journal of the Electrochemical Society 154 (2007)A283–A289.
40] Z.-H. Wang, L.-X. Yuan, W.-X. Zhang, Y.-H. Huang, LiFe0.8Mn0.2PO4/C cathodematerial with high energy density for lithium-ion batteries, Journal of Alloysand Compounds 532 (2012) 25–30.
41] B. Zhang, X. Wang, H. Li, X. Huang, Electrochemical performances ofLiFe1−xMnxPO4 with high Mn content, Journal of Power Sources 196 (2011)6992–6996.
42] G. Kobayashi, A. Yamada, S. Nishimura, R. Kanno, Y. Kobayashi, S. Seki, Y. Ohno,
H. Miyashiro, Shift of redox potential and kinetics in Lix(MnyFe1−y)PO4, Journalof Power Sources 189 (2009) 397–401.43] A.K. Padhi, K.S. Nanjundaswamy, C. Masquelier, S. Okada, J.B. Goodenough,Effect of structure on the Fe3+/Fe2+ redox couple in iron phosphates, Journalof the Electrochemical Society 144 (1997) 1609–1613.
Copyright © 2022 FDOKUMEN




















