
Structural studies of shikimate 5-dehydrogenase from Mycobacterium tuberculosis
Upload
independentCategory
view
0download
0
Journal of Molecular Structure (Theochem), 160 (1987) l-15 Elsevier Science Publishers B.V.. Amsterdam - Printed in The Netherlands
STRUCTURAL STUDIES OF MESOGEN BENZALAZINE DERIVATIVES
Part I. Semiempirical MO calculations (CNDO/B, MNDO) on intramolecular H-bond and vicinal interactions
JOSE ELGUERO’, CARLOS JAIMEb, MERCEDES MARCOS’, ENRIQUE MELENDEZC, FRANCISCO SANCHEZ-FERRANDOb and JOSE LUIS SERRANOC
aZnstituto de Quimica Mt?dica, C.S.Z.C., Juan de la Cierva 3, 28006 Madrid (Spain) bDepartamento de Quimica Orghnica, Facultad de Ciencias, Universidad Autbnoma de Barcelona, Bellaterra (Barcelona) (Spain) CDepartamento de Q&mica Orghnica, Facultad de Ciencias, Universidad de Zaragoza, 50009 Zaragoza (Spain)
(Received 18 March 1986)
ABSTRACT
The molecular geometry of benzalazine derivatives was studied by spectroscopic and semiempirical MO (MNDO, CNDO/P) methods. The existence of a strong intramolecular H-bond (7-10 kcal mol”) was determined by ‘H NMR spectroscopy in molecules having a hydroxy group in the ortho position with reference to the diazine central bridge. A study of the H-bonds using the MNDO method proved to be unsatisfactory, whereas the results obtained by the CNDO/S method were in excellent agreement with experimental values. Both theoretical calculations (CNDO/S) and experimental evidence (UV and ‘H NMR) show that the existence of a first H-bond weakened the force of a second in benza- lazines.
INTRODUCTION
The benzalazines and ar,a!‘-dimethylbenzalazines are classical compounds in the study of liquid crystals [l, 21. The introduction of a hydroxy group in position 2 of the aromatic ring gives rise to the formation of an intramolec- ular H-bond between the hydroxylic hydrogen and the nearest nitrogen of the azine group, through a 6-member chelation ring. At- the same time an in- crease in the molecular planarity is observed. This H-bond modifies the
0166-1280/87/$03.50 o 1987 Elsevier Science Publishers B.V.

x -X’ Y Y’ 2 2’ I n-C8H170 H H CH, CH,
2 n-C8H170
3 n-C8H170
4 n-C8H170
5 n-C8H-,70
6 n-C8H170
7 n-C8H170
8 H
9 H
IO H
I I H
OH
OH
OH
OH
03
H
Ii
OH
OH
H
/ ,
H CH 3 CH3
OH CH3 CH3
03 CH3
H
OH H H
H H H
H H H
II CH3 CH3
OH CH3 CH3
OH H H
H Ii H
2
mesogenic behaviour of these compounds [ 3-51 to a considerable degree, producing some electronic shifts which play an important role in such modi- fication. Some authors attribute these shifts to the important contribution of quinoid canonical forms to the resonance hybrid [6].
Another decisive factor in the mesomorphism of a compound is the exis- tence of terminal chains. These groups should be in the direction of the main molecular axis and increasing the molecular length. The length of these chains has a considerable influence on the type of mesophase shown by the com- pound [7].
In this paper we report the study of the structural and electronic properties of eleven benzalazine derivatives (Sketch 2) using spectroscopic techniques (UV, ‘H NMR, IR). As ab initio calculations on molecules of this size are be- yond our computational facilities we have employed two of the most used
2’ Y
X
& 0 / v
0 X’
N-N
Y’
z
semiempirical methods, MNDO [8] and C!NDO/B [lo] to study the influence of the molecular structure on the mesogenic behaviour of these molecules. We have also studied the influence of the molecular structure on the forma- tion and strength of intramolecular H-bonds. The relative validity of the two semiempirical methods used to calculate the energy stabilization of the mol- ecules brought about by the H-bond was also studied.

3
Seven of these compounds, l-7, belong to seven series of mesogen com- pounds previously described [ 3, 5, 91, whereas the other four B-11, corre- spond to simplified structures which are necessary for the theoretical study of the H-bond.
RESULTS AND DISCUSSION
The mesogen compounds having an octyloxy group as terminal chain were selected among the components of each series [3, 5, 91 for spectroscopic studies. These compounds correspond to an intermediate position within the series* and show an adequate degree of solubility in non-polar solvents.
The main ‘H NMR and UV spectroscopic data for the eleven compounds studied are collected in Tables 1 and 2 respectively.
TABLE 1
‘H NMR chemical shifts for the hydroxyiic hygrogen in compounds 2-6, 9 and 10, and H-bond energy calculated by Schaefer’s equation
Compound Chemical shifts (6) Energy (kcal mol-I)
2 14.32 10.1 3 13.76 13.76 9.6 9.6 4 13.60 11.80 9.4 7.6 5 11.71 11.71 7.5 7.5 6 12.07 7.9 9 13.15 13.15 9.0 9.0
10 11.34 11.34 7.2 7.2
TABLE 2
UV spectroscopic data (A,, in nm) as registered in n-pentane for compounds l-11
Compound A max (log e)
1 2 3 4 5 6 7 8 9 10 11
395.5 sh 389 sh 383 sh 367.5 sh 346 sh
371 sh 377 sh
315(4.50) 349( 4.45) 379(4.50) 373(4.65) 368( 4.70) 352.5(4.65) 332(4.65) 292.5(4.35) 367.5(4.30) 359( 4.45) 309( 4.55)
367 sh 360 sh 354 sh 342 sh
*Compounds with terminal chains C-n-C&H,,+, where n = l-10, 12, 14, 16, 18 were described in each series.
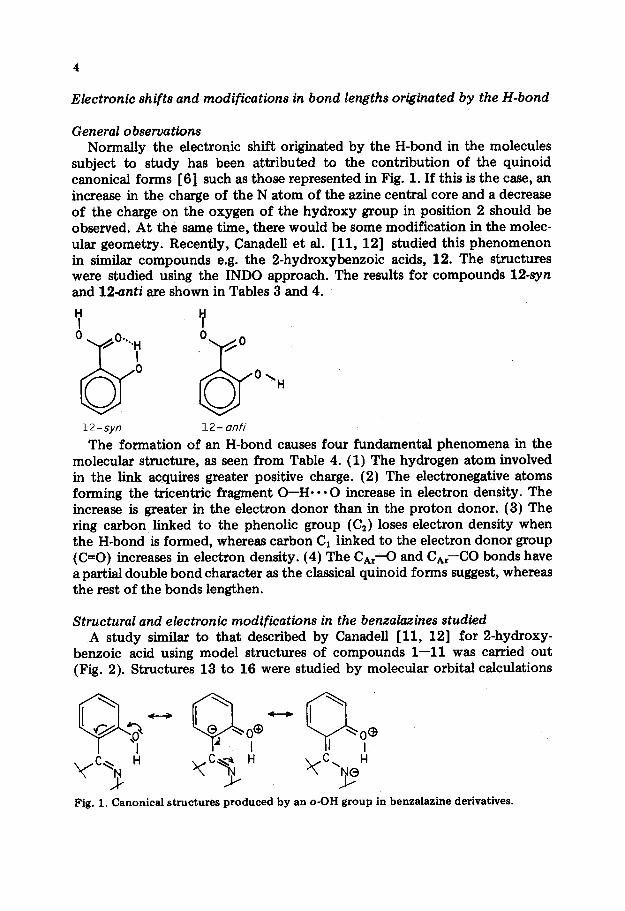
Electronic shifts and modifications in bond lengths originated by the H-bond
General observations Normally the electronic shift originated by the H-bond in the molecules
subject to study has been attributed to the contribution of the quinoid canonical forms [6] such as those represented in Fig. 1. If this is the case, an increase in the charge of the N atom of the azine central core and a decrease of the charge on the oxygen of the hydroxy group in position 2 should be observed. At the same time, there would be some modification in the molec- ular geometry. Recently, Canadell et al. [ 11, 121 studied this phenomenon in similar compounds e.g. the 2-hydroxybenzoic acids, 12. The structures were studied using the INDO approach. The results for compounds 12-syn and la-anti are shown in Tables 3 and 4.
7 ,O’YH ,O ,o r, 5 b
0 0
O\ H
12-syn 12-anti
The formation of an H-bond causes four fundamental phenomena in the molecular structure, as seen from Table 4. (1) The hydrogen atom involved in the link acquires greater positive charge. (2) The electronegative atoms forming the tricentric fragment O-H l ** 0 increase in electron density. The increase is greater in the electron donor than in the proton donor. (3) The ring carbon linked to the phenolic group (C,) loses electron density when the H-bond is formed, whereas carbon C1 linked to the electron donor group (PO) increases in electron density. (4) The CA,-0 and C,,-CO bonds have a partial double bond character as the classical quinoid forms suggest, whereas the rest of the bonds lengthen.
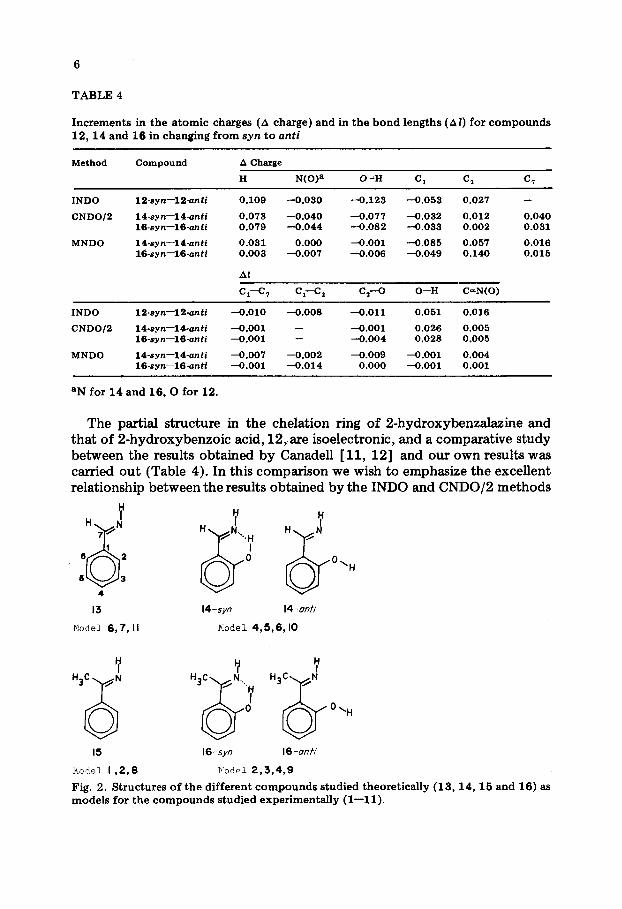
Structural and electronic modifications in the benzalazines studied A study similar to that described by Canadell [ 11, 121 for 2-hydroxy-
benzoic acid using model structures of compounds l-11 was carried out (Fig. 2). Structures 13 to 16 were studied by molecular orbital calculations
Fig. 1. Canonical structures produced by an o-OH group in benzalazine derivatives.

5
TABLE 3
Bond lengths, dihedral angle 2-l-7-N and energetic values for compounds 12-16 as calculated by INDO, CNDO/P and MNDO
Method Compound Bondlengths W2-l-l-N
Cl-c, C,-N(O)a q-C, C2-CI O-H
INDO ll-syn 1.433 1.289 1.413 1.356 1.087 la-anti 1.443 1.273 1.405 1.367 1.036
MNDO 13 1.482 1.290 1.417 - - 0.53 14-s&' 1.479 1.291 1.421 1.349 0.946 1.90 14-m& 1.480 1.287 1.429 1.358 0.947 104.45 15 1.498 1.297 1.419 - - 0.17 16-s&' 1.496 1.293 1.416 1.358 0.947 80.20 16-anti” 1.497 1.292 1.430 1.358 0.948 -81.97
CNDOj2 13 1.449 1.291 - - - 0.0 14-synb 1.445 1.296 - 1.366 1.058 0.0 14-w& 1.446 1.291 - 1.367 1.032 0.7 15 1.462 1.304 - -- 0.0 16-s&' 1.456 1.308 - 1.362 1.061 0.0 16-o&J 1.457 1.303 - 1.366 1.033 0.0
Atomic charzzes
N(Oja O-H Hc C1 C, C, Hf
INDO ll-syn -0.427 -0.394 0.265 -0.159 0.290 ll-anti -0.397 -0.271 0.156 -0.106 0.263
MNDO 13 -0.239 - - -0.086 0.094 42.33 14-synb 4.288 -0.252 0.228 -0.184 0.195 0.122 -4.80 14~ant+ -0.288 -0.251 0.177 -0.099 0.138 0.106 -6.08 15 -0.233 - - -0.065 0.022 0.055 38.24 16-eynb -0.220 -0.254 0.200 a.130 0.001 0.047 -12.91 16-ontib -0.213 -0.248 0.197 -0.081 0.141 0.067 -13.39
CND0/2 13 -0.178 - - 0.017 0.011 0.123 -66.56183Hd 14-synb -0.214 -0.304 0.206 -0.061 0.201 0.161 -85.04509H 14-ant+ -0.174 -0.227 0.133 -0.029 0.189 0.131 -85.02766H 15 -0.209 - - 0.007 0.011 0.145 -75.26405H 16-synb -0.248 -0.308 0.211 -0.071 0.201 0.182 -93.74898H 16-antib -0.204 -0.226 0.132 -0.038 0.199 0.151 -93.72960H
aN for 13-16 and 0 for 12.bTerms syn and anti refer to the conformation around C,- C,-O-H. CRefers to the hydroxylic hydrogen whenever it exists. dMNDO: heat of for- mation (kcal mol-’ ); CNDO/B : total energy (hartree).
using MNDO [8] and CNDO/B [lo] approaches. Table 3 contains the most significant results.
The electronic shifts and the loss of planarity introduced in the molecule by the different substituents (H, CHB, OH) are of great interest in the study of mesogenic properties. The small variations in the bond lengths are less important. In order to reduce the calculation time CNDO/B was used, the ring structure was fixed according to standard geometry* and only the bonds involved in the chelation ring were optimized. As the MNDO minimization scheme is quicker, all the variables were allowed to optimize under this method.
*C-C = 1.470 A, C-C-C = 120”, C-H = 1.10 A, C-C-C-C = 0” and H-C-C-H = 0”.
6
TABLE 4
Increments in the atomic charges (A charge) and in the bond lengths (Al) for compounds 12,14 and 16 in changing from syn to anti
Method
INDO
CNDOl2
MNDO
Compound
la-syn-12-anti
14-syn-14-anti 16-syn-16-anti
14-syn-14-anti 16-syn-16-anti
A Charge
H NW8 O-H C, CZ C,
0.109 -0.030 -0.123 -0.053 0.027 -
0.073 -0.040 -0.077 -0.032 0.012 0.040 0.079 -0.044 -0.082 -0.033 0.002 0.031
0.031 0.000 -0.001 -0.085 0.057 0.016 0.003 -0.007 -0.006 -0.049 0.140 0.015
AZ
Cl-c, C,-C* C*-o O-I-I C=N(O)
INDO 12-syn-la-anti -0.010 -0.008 -0.011 0.051 0.016 CNDO/P 14-syn-14-anti -0.001 - -0.001 0.026 0.005
16-syn-16santi -0.001 - -0.004 0.028 0.005
MNDO 14-syn-14-anti -0.007 -0.002 -0.009 -0.001 0.004 16~syn-16-anti -0.001 -0.014 0.000 -0.001 0.001
aN for 14 and 16,O for 12.
The partial structure in the chelation ring of 2-hydroxybenzalazine and that of 2-hydroxybenzoic acid, 12, are isoelectronic, and a comparative study between the results obtained by Canadell [ 11, 121 and our own results was carried out (Table 4). In this comparison we wish to emphasize the excellent relationship between the results obtained by the INDO and CNDO/B methods
13 I4-syn 14-unfi
Model 6,7,11 Model 4,5,6,10
I5 IS-syn l6-unfi
Kodel I ,2,6 Model 2,3,4,9
Fig. 2. Structures of the different compounds studied theoretically (13,14,15 and 16) as models for the compounds studied experimentally (l-l 1).

7
for the four fundamental points previously mentioned. The structural modi- fications introduced by the OH group observed by CNDO/Z are generally equal, although slightly lower, to those found using the INDO method. This can be attributed to two fundamental factors: (i) the different behaviour of nitrogen and oxygen as electron donors in the chelation bond; (ii) the absence of complete optimization in CNDO/B, a point which has great influence on the small modifications observed in the corresponding bond lengths (C-C,, G-0).
All the molecules are planar ( W2_-1.-,_-N = 0.0”) when the CNDO/B method is used, and the total energies of the syn and anti conformers as calculated by CNDO/B reflect the stability of the H-bonds (syn conformers being more stable than anti by about 11-12 kcal mol-I). The results obtained using the MNDO approach disagree with those obtained by previous methods. When 13 and 1Csyn structures were compared, the results seem to support the contribution of the quinonic canonical forms (slight shortening of C1-C, and C,-O bonds and an important increase in the negative charge on the iminic N). These differences, however, cannot be attributed to the existence of H-bonds.
According to MNDO calculations the most stable conformation for 14-unti has the imine group outside the plane of the aromatic ring (Table 3) and with a heat of formation (&) of 1.28 kcal mol-’ lower than that of 14-syn in spite of the expected stabilization of the latter by H-bonds.
These results suggest to us that the modifications introduced by the OH group in 14 (MNDO results) were originated by the u and s bond orbitals and that the influence of the H-bond is minimal. It can be seen in Table 4 that: (1) The increase in the positive charge of the H atom forming the H-bond in syn conformers is much lower using MNDO than that observed using both INDO and CNDO/B. (2) the electronegative atom forming the tricentric frag- ment (0-H. l l O(N)) undergoes no increase in electron density according to MNDO calculations. (3) The net charges of the C1 and Cz carbon vary. These variations are of the same sign in all methods but have a higher absolute value from MNDO. (4) The OH bond length remains practically unchanged in 14-syn and 16anti according to MNDO. However, the other methods (INDO and CNDO/B) show a significant increase in the length of this bond.
The results obtained when the molecular structures of imines derived from acetophenone and 2-hydroxyacetophenone (15 and 16, respectively) are considered are very different from those of the compounds previously de- scribed (Table 3). The structure of 16-syn is not completely planar in its MNDO energetic minimum. The phenyl and the imine group planes form an angle of 80.2”. The absence of conjugation between both groups did not allow us to observe the OH group effect on bond lengths.
Compound 16 is a good example for observing the underestimation of the H-bond energies (Table 3) and the overestimation of the non-bonding inter- actions for the short distances [8] by the MNDO method.
Our results coincide with and complement the work by Kolloman and

8
Allen [13] on the study of H-bonds using ab initio and semiempirical calcu- lations. A good agreement with ab initio calculations was obtained using CNDO/Z and INDO, whereas using EHT, MINDO/l and NPPO the results were unsatisfactory. MNDO can be included in the latter group.
Study of the intramolecular H-bond strength
General observations Information about H-bond strength can be obtained from spectroscopic
data. Schaefer proposes two different ways of obtaining this information: (i) from the chemical shift of the hydroxylic hydrogen in ‘H NMR [ 141; (ii) from the absorption corresponding to the rotation of the OH group ob- served in the far IR [ 151.
Both methods are interrelated by the following equation:
A6 = 0.017 Aot = E - 0.4 f 0.2
A6 corresponds to the difference between the observed chemical shift for the OH group originating in the H-bond and the OH of phenol. Aot corre- sponds to the difference (in cm-‘) between the torsional frequency of the phenolic OH and that of the OH of the compound under study (phenol OH torsional frequency in CSz is 310 cm-l) [ 151.
Schaefer recommends non-polar solvents for these experiments, but due to solubility problems we found it necessary to use CDC&. Phenol has a chemical shift of 4.59 ppm in this solvent at similar concentrations to those used for the compounds studied. The energy of the H-bond in the compounds studied, obtained by Schaefer’s equation, is included in Table 1. An attempt to check this value by using IR spectroscopy was made, but was impossible because the rotational frequency zone for the OH group (700-900 cm-‘) is masked by the bands of the aromatic ring.
H-bond strength in ketazines and benzalazines The H-bond strength in compounds of this type is quite large (7-10 kcal
mol-‘) (Table 1). These bonds are especially strong in the ketazines (AE = 2 kcal mol-’ higher in ketazines than in benzalazines). This could be caused by two factors: (i) the existence of a methyl group in ketazines which in- creases the electronic density of the azine N atom; c.f. N charges (CNDO/B) in pairs 13-15 or 16syn-16-syn (Table 3); (ii) the methyl group introduces steric hindrance with the aromatic hydrogen in the ortho position, pushing the azine N atom towards the OH group (Fig. 3). The non-bonded distances H/N in 13 and CHJN in 15 are 2.68 and 2.53 a respectively. These values were obtained by complete optimization using MNDO. The H-bond forma- tion is favoured in factor (i) by electrostatic attraction, and in factor (ii) by decrease in the H/N distance [ 161.

9
Fig. 3. Schematic representation of the steric hindrance between the methyl group and the o-hydrogen atom, which causes the azine N and the OH group to become closer.
H-bond strength in molecules with one or two OHgroups It is highly significant that the H-bond in compounds with one bond and
one hydroxyl in position 2 (2 and 6) has greater energy than those in com- pounds with two hydroxyl groups (3,4,5) (Table l), indicating that the for- mation of the second H-bond decreases the energy of the first. (E(2-3) = 0.5 kcal mol-* f 0.1; E(6-5) = 0.4 kcalmol-’ f 0.1; E(2-4) = 0.76 kcal mol-’ f 0.1; E(6-4) = 0.3 kcalmolS1 f 0.1.) The greater strength of the first H-bond could be explained by the contribution of the doubly canonic forms to the resonance hybrid (Fig. 4). The two negative charges on adjacent N atoms destabilize the system.
UV spectroscopy confirms the previous results (Table 2) qualitatively. The formation of a chelate bond causes a bathochromic shift greater in ketazines than in benzalazines (A3 - X1 = 64 nm and X5 - h, = 36 nm). The first chelate bond causes a higher bathochromic shift than the second in both compounds (hz - A1 = 34 nm, hJ - AZ = 30 nm, and X6 - X, = 20.5 nm, A5 - A6 = 15.5 nm).
MNDO calculation (Table 3) shows that 14-anti is more stable than 14-syn and that 16-syn exists in a conformation in which the phenyl and the imino group planes form an angle of 80.2”. These results confirm that MNDO is unsuitable for the study of the H-bond effects [ 171. On the other hand, CNDO/B (Table 3) gives completely planar structures and more negative energy values for syn-conformers than for anti-conformers. AE(anti - syn) is 10.9 kcal mole1 for compound 14 and 12.2 for compound 16. These values agree with the experimentally abtained H-bond strength for 5 and 2 (7.5 and 10.0 kcal mole’, respectively). The computational results also indicate that the H-bond is stronger in ketazines than in benzalazines.
CNDO/B calculation on compound 17, a model for structures having two H-bonds, shows that the strength of the second H-bond is less than that of the first. The total energy difference between 17-syn-anti and 17-syn-syn is
Fig. 4. Doubly quinonic canonic form in 3,4, 5,9 and 10.

10
8.03 kcal mol-l favouring 17-synsyn and between 17-anti--anti and 17- syn-anti is 8.33 kcal mol-’ favouring 17-syn-anti. Consequently the second H-bond implies a stabilization of only 0.3 kcal mol-l (Fig. 5). This value is very similar to that obtained experimentally (0.5-0.4 kcal mol-‘, see Table 1).
Figure 5 contains all the bond lengths and atomic charges of 17 obtained by CNDO/B. The elongation of C=N, C=C and O-H bonds and the shorten- ing of C-C and C-O bonds due to H-bond formation can be seen in the anti conformer in this Figure. The increase in the N, 0, and H charges on forming the H-bond is clearly observed also. It is noteworthy that the molecule is planar in both halves but these are not coplanar. The torsional angle Wz-la_, is 108.3“ and 69.2” in conformers syn-syn and syn--anti respectively (Fig. 5).
Molecular structure study
Study of the molecular geometry of l-11 was carried out by two different methods i.e. MO calculations and by the usual spectroscopic techniques.
The CNDO/B program [lo] makes it possible to work with up to 35 atoms and has a very slow minimization process [ 181. For this reason it is not suit- able to calculate the whole molecular structure of the benzalazines. The MNDO method is one of the most commonly used because calculations can be quicker in spite of its well-known overvaluation of Hf in sterically-con- gested molecules and its underestimation of the H-bond energy. In order to study the molecular structure it is necessary to optimize the largest possible number of variables, and thus MNDO was the method of choice. From our observations and other reports [ 171 we found that MNDO does not take the H-bond into account. However, our hypothesis is that the method could be used to calculate the total energies of the molecule (provided the experi- mentally-obtained H-bond energies are used as correction values).
The charge shifts and deviation from planarity observed in the molecule are the fundamental variables which influence the study of mesogen com- pounds, whereas changes in bond lengths are of secondary importance. With this in mind, we established several approximations in order to simplify and reduce calculation time.
Instead of studying structures l-7, the analogous compounds (1’ to 7’) were studied. The octyloxy group was substituted by the methoxy group. In the same way, the aromatic rings were defined as standard geometries and were not optimized. Only the variables related to the azine group and the o&o-substituent were optimized. For each product, only two conformers having “C,” symmetry were studied (one planar and one non-planar). The non-planar conformer was that in which the dihedral angles were all mini- mized, whereas in the planar conformer some dihedral angles were fixed. The MNDO results for these 14 structures are shown in Table 5.
The one non-planar conformer always proved more stable than the planar conformers (Table 5). The Hf difference between both conformers is small (l-3 kcal mol-‘) for molecules having interactions resembling those of

11
If onfi-anti
W &&&C=270° E = -108.6435525530 hartrees
H H
17 syn-onfi
W C_N_N_C=69. 2’ E = -108.6568193230 hartrem
17 syn-syn
~~_~_~_~=108.3’ E = -108.6696182784 hartrees
Fig. 5. Total energy (E), bond lengths (in A), atomic charges and C-N-N-C torsional angles for compound 17 in conformers anti-anti, syn-anti and syn-syn.

12
TABLE 5
Heat of formation (Hf) for planar and non-planar conformers of compounds l;-7’ and 8-11 as calculated by MNDO
Compound*
1’ 2’ 3’ 4’ 5’ 6’ 7’ 8 9
10 11
Hi (kcal mol-‘) AHf
Planar Non-planar Hfplanar - Hfnon-planar
7.59 -1.90 9.49 -33.27 -43.38 10.11 -80.20 -92.37 12.17 -79.27 -85.91 6.64 -78.57 -79.62 1.05 -31.94 -35.30 3.36
12.48 11.15 1.33 87.15 77.47 9.58 -1.49 -14.36 12.87
0.33 -1.35 1.68 92.55 91.15 1.40
AHtb
9.49 0.01
-7.03 -10.36 -13.95
-4.54 1.33 9.68
-5.13 -12.72
1.40
sFor l’-7’ X = X’ = OCH,. bAHf corrected by using the experimentally-obtained H-bond energies (see Table 1). A$ = AHr- energy of H-bond.
Fig. 6(a). This difference increases up to 10 kcal mol-l when the H of the benzalazines is substituted by a methyl group (Fig. 6(b)). Moreover, a mol- ecule having eclipsed bonds like those shown in Fig. 6, appears excessively congested because MNDO predicts that molecules such as 7’ and 11 should have the aromatic ring and the azine group forming a dihedral angle of approximately 90” (Table 5).
The existence of two methyl groups in the CC and cy ’ position relative to the azine N atom produces considerable destabilization of the planar conformer (see l’-3’, 7’-9 in Table 5) and the existence of two ortho-hydroxy groups does not stabilize the planar conformers as might be expected from the intra- molecular H-bond (Table 5).
For MNDO, the introduction of an ortho-hydroxy group only causes an increase of steric congestion at the other side of the molecule, and AH, for compounds 2’, 3’ and 9 is lo-13 kcal mol-‘. However, if we correct the AH, values obtained by MNDO with the experimental values for the H-bond energy for the different molecules (Table 1) we obtain AH,* values (Table 5). From these values we can establish that compounds which have no H-bonds
Fig. 6. Two different cases of eclipsed interactions present in benzalazine derivatives: (a) 1,3-interaction type; (b) 1,4-interaction type.

13
will be non-planar (l', '7', 8 and 11) (AH,* > 0). Those which have two H- bonds will be planar (3’, 4’, 5’ and 10) (AH: < 0) and the conformation of those having only one H-bond will depend on whether they are ketazines (2’, AHf z 0) or benzalazines (6’, AH,* < 0).
These tentative conclusions about the molecular geometry obtained by MNDO calculations agree qualitatively with experimental results. It can thus be observed that molecules without hydroxy groups (benzalazines 7 and 11) show values of X,,, higher than those of the corresponding ketazines (1 and 8) despite the bathochromic effect of the methyl group (Table 2). This bathochromic effect can be attributed to the greater coplanarity of the benz- alazines compared with the ketazines, as a consequence of the steric repulsion between the a-methyl group and the ortho-hydrogen (Fig. 3). This repulsion prevents the aromatic ring from being coplanar with the central azine group. X-Ray diffraction data of analogous molecules also confirm these results. While anisaldazine [19] is practically planar, the 4,4’-dihydroxy-cup’-di- methylbenzalazine [20] has a much more irregular and far from planar structure.
The introduction of two hydroxy groups in position 2 causes a strong bathochromic shift, higher in ketazines (3 and 9) than in benzalazines (5 and 10). This can be attributed to the additional effect of the methyl group (- 5 nm) [ 211 because values for compound 4, with only one methyl in the central core, lie between those of the two previous cases. This helps to con- firm that formation of the two intramolecular H-bonds forces the molecules to adopt a coplanar disposition, and justify the increase in the conjugation and the strong bathochromic shift observed. X-Ray diffraction data of analo- gous molecules also confirm these results. The salicylaldazine [6] and the 4,4’-diethoxy-2,2’-dihydroxyap’-dimethyl benzalazine [ 221 have practically planar structures. Obviously, molecules with only one ortho-OH group are intermediate; the aromatic ring with the OH group tends to remain coplanar with the central azine group while the other aromatic ring tends to become twisted in order to minimize the steric repulsions.
Electronic and steric effect of the alkoxy chains
The appearance of mesogenic properties in a compound generally requires the presence of terminal chains. In the azines under consideration, these chains are linear alkoxy groups (0-n-&HZ,+ 1, n = l-10, 12, 14,16,18). The influence of these alkoxy groups on the mesomorphism of the com- pounds depends on two fundamental factors: (i) the length of the terminal chains which is a decisive parameter in the mesogenic properties of any com- pound [7, 231. This factor depends on the length of each aIkoxy group and being an individual factor has not been dealt with in this study; (ii) the par- ticipation of the alkoxy group in the ?r-electronic delocalization of the central core using the lone pair of the oxygen. This effect is common to all the alkoxy groups considered.

14
The alkoxy group can contribute to the existence of quinoid forms with a charge distribution similar to that shown in Fig. 1. The existence of these canonical forms causes strengthening of the intramolecular H-bond. CNDO/B calculations on molecules 18syn and l&anti, indicate that the energy differ- ence syn-anti is 12.6 kcal mol-‘. This value is slightly higher than that calcu- lated for 14 (10.9 kcal mol-I). Experimentally-obtained values agree with
18- syn 18-anti
calculated values. Compounds with para-alkoxy groups (3 and 5) have slightly stronger H-bonds than analogous molecules without (9 and 10) as deduced from ‘H NMR (Table 1). In UV spectroscopy (Table 2), the batho- chromic effect introduced by para-alkoxy groups is greater in compounds without ortho-OH groups (l-8, 7-ll), than in compounds with two chela- tion bonds (3-9, 5-10). This confirms that the contribution of the canoni- cal forms in which the alkoxy group takes part is greater in molecules with no chelation bonds. These observations show that molecules with terminal alkoxy groups (l-7) have a slightly more planar geometry than their ana- logues (8-11) although this effect is much more marked in molecules with- out chelation bonds.
EXPERIMENTAL
Synthesis
Compounds 1 to 7 have been previously described [3,5,9]. The prepara- tion of compounds 8-11 was carried out using reported methods [3,5,9].
Techniques
‘H NMR spectra were obtained with a Bruker wpSO-SY instrument using CDCIJ solutions and TMS as internal reference. UV spectra were obtained with a Perkin-Elmer 200 instrument using n-pentane solutions. IR spectra were obtained with a Perkin-Elmer 283 instrument.

15
ACKNOWLEDGEMENTS
A fellowship from “Ministerio de Educacibn y Ciencia”, Spain (to C. J.) is gratefully acknowledged. This work was carried out under contract with the C.A.I.C.Y.T. (Spain).
REFERENCES
1 W. G. Shaw and G. Brown, J. Am. Chem. Sot., 81 (1959) 2532. 2 A. Roviello and A. Sirigu, Mol. Cryst. Liq: Cryst., 33 (1976) 19. 3 M. Marcos, E. Melendez and J. L. Serrano, Mol. Cryst. Liq. Cryst., 91 (1983) 157. 4 E. Melendez and J. L. Serrano, Mol. Cry&. Liq. Cryst., 91 (1983) 173. 5 J. L. Alabart, E. Melendez, B. Ros and J. L. Serrano, Mol. Cryst. Liq. Cryst., 107
(1984) 397. 6 G. Arcorito, M. Bonamico, A. Dominicano and A. Vaciago, J. Chem. Sot. B, (1969)
733. 7 G. W. Gray, Liquid Crystals and Plastic Crystals, Vol. I. Ellis Horwood, Chichester,
1974. 8 M. J. S. Dewar and W. Theil, J. Am. Chem. Sot., 99(15) (1977) 4907. 9 M. Artigas, M. Marcos, E. Melendez and J. L. Serrano, Mol. Cryst. Liq. Cryst., 130
(1985) 337. 10 J. A. Pople and D. L. Beveridge, Quantum Chemistry Program Exchange, Program
No. 389. 11 (a) E. Canadell, Doctoral Thesis, Universidad Autonoma de Madrid, Madrid (1976).
(b) J. Catalan, E. Canadell and J. I. Fernindez-Alonso. Progress in Theoretical Organic Chemistry, Vol. 2, Elsevier, Amsterdam, 1977.
12 E. Canadell, J. Catalan and J. I. Fernandez-Alonso, Adv. Mol. Relaxation and Inerac- tion Processes, 12 (1978) 265.
13 P. A. Kollman and L. C. Allem, Chem. Rev., 72(3) (1972) 283. 14 T. Schaefer, J. Phys. Chem., 79 (1975) 1888. 15 G. Fateley, G. L. Carlson and F. F. Bentley, J. Phys. Chem.,79(3) (1975) 199. 16 R. M. Silverstein, G. Claydon-Bassler and T. C. Monill, Spectrometric Identification of
Organic Compouncs, John Wiley, New York, 3rd edn., 1974, 77 pp. 17 (a) K. Ya. Burstein and A. N. Isaev. Theor. Chim. Acta, 54( 5) (1984) 397.
(b) K. Ya. Burstein and A. N. Isaev, Zh. Strukt. Khin., 25( 1) (1984) 25. 18 Adapted version of Rosenbrock’s method. (a) H. H. Rosenbrock, Comp. J., 3 (1960)
175. (b) M. Muchara and A. Mulawa, Comm. ACM, 16 (1983) 482.
19 J. L. Galigne and J. Falgueirettes, Acta Crystallogr., Sect. B, 25 (1968) 1523. 20 H. Garcia-Mina, F. Arrese, M. Martinez-Ripoll, S. Garcia-Blanc0 and J. L. Serrano,
Acta Crystallogr., Sect. B, 38 (1982) 2726. 21 E. Pretch, T. Clerc, J. Seibl and W. Simon, Tabellen zur Strukturaufhirung Organischer
verbindungen mit Spektroskopischen Methoden, Springer Verlag, Berlin, 1976. 22 J. Fayos, M. Martinez-Ripoll, M. Garcia-Mina, J. Gonzalez and F. Arrese, Acta Crys-
tallogr., Sect. B, 36 (1980) 1952. 23 G. W. Gray, Molecular Structure and the Properties of Liquid Crystals, Academic Press,
New York, 1962.
Copyright © 2022 FDOKUMEN




















