
Structural characterization and comparative modeling of PD-Ls 1–3, type 1 ribosome-inactivating...
12
Research paper Structural characterization and comparative modeling of PD-Ls 1e3, type 1 ribosome-inactivating proteins from summer leaves of Phytolacca dioica L. Antimo Di Maro a, * , Angela Chambery a , Vincenzo Carafa a , Susan Costantini b,c , Giovanni Colonna b , Augusto Parente a a Dipartimento di Scienze della Vita, Seconda Universita ` degli Studi di Napoli, Via Vivaldi 43, I-81100 Caserta, Italy b Dipartimento di Biochimica e Biofisica e CRISCEB (Centro di Ricerca Interdipartimentale per le Scienze Computazionali e Biotecnologiche), Seconda Universita ` degli Studi di Napoli, Via Costantinopoli 16, I-80138 Napoli, Italy c Istituto di Scienze dell’Alimentazione, CNR, Via Roma 52 A/C, I-83100 Avellino, Italy Received 25 June 2008; accepted 16 October 2008 Available online 26 October 2008 Abstract The amino acid sequence and glycan structure of PD-L1, PD-L2 and PD-L3, type 1 ribosome-inactivating proteins isolated from Phytolacca dioica L. leaves, were determined using a combined approach based on peptide mapping, Edman degradation and ESI-Q-TOF MS in precursor ion discovery mode. The comparative analysis of the 261 amino acid residue sequences showed that PD-L1 and PD-L2 have identical primary structure, as it is the case of PD-L3 and PD-L4. Furthermore, the primary structure of PD-Ls 1e2 and PD-Ls 3e4 have 81.6% identity (85.1% similarity). The ESI-Q-TOF MS analysis confirmed that PD-Ls 1e3 were glycosylated at different sites. In particular, PD-L1 contained three glycidic chains with the well known paucidomannosidic structure (Man) 3 (GlcNAc) 2 (Fuc) 1 (Xyl) 1 linked to Asn10, Asn43 and Asn255. PD-L2 was glycosylated at Asn10 and Asn43, and PD-L3 was glycosylated only at Asn10. PD-L4 was confirmed to be not glycosylated. Despite an overall high structural similarity, the comparative modeling of PD-L1, PD-L2, PD-L3 and PD-L4 has shown potential influences of the glycidic chains on their adenine polynucleotide glycosylase activity on different substrates. Ó 2008 Elsevier Masson SAS. All rights reserved. Keywords: Ribosome-inactivating proteins; Phytolacca dioica; Edman degradation; ESI-Q-TOF mass spectrometry; Comparative modeling 1. Introduction Ribosome-inactivating proteins (RIPs) are widely distrib- uted plant proteins present in various organs, in some cases, even in large amounts [1,2]. Moreover, although most of them await further confirmation, previous reports provide evidences of the presence of proteins of the RIP family also in fungi, bacteria and at least one alga [3]. RIPs are RNA N-b-glycosidases (EC 3.2.2.22) [4] and remove a site-specific single adenine residue (A 4324 in the case of rat liver ribosomes) from the highly conserved sarcin/ricin loop of the 28S rRNA, thus arresting protein synthesis [5]. As further results indicated that RIPs can depurinate DNA, RNA and poly(A), they were named polynucleotide:adenosine glycosidases (PAG; [6]), in turn renamed adenine poly- nucleotide glycosylases (APGs; [7]). RIPs are conventionally classified into three types: types 1, 2 and 3 on the basis of number and type of the constituting polypeptide chain(s) [8]. Type 1 RIPs are single-chain proteins endowed with glycosi- dase activity, while type 2 RIPs consist of a catalytic chain Abbreviations: RIPs, ribosome-inactivating proteins; PD-Ls, RIPs isolated from leaves of Phytolacca dioica L; PD-S2, RIP (major form) isolated from seeds of Phytolacca dioica L; CNBr, cyanogen bromide; Lys-C and Asp-N, endoproteinase Lys-C and Asp-N, respectively; PE-Cys, pyridylethylcysteine; Hse, homoserine; Hse>, homoserine lactone; hsDNA, herring sperm DNA; HPAEC-PAD, high performance anion-exchange chromatography with pulsed amperometric detection; Nbs2, 5,5 0 -[dithiobis (2-nitrobenzoic acid)]. For the amino acids the standard one or three-letter code has been used. * Corresponding author. Tel.: þ39 823 274535; fax: þ39 823 274571. E-mail address: [email protected] (A. Di Maro). 0300-9084/$ - see front matter Ó 2008 Elsevier Masson SAS. All rights reserved. doi:10.1016/j.biochi.2008.10.008 Available online at www.sciencedirect.com Biochimie 91 (2009) 352e363 www.elsevier.com/locate/biochi
Transcript of Structural characterization and comparative modeling of PD-Ls 1–3, type 1 ribosome-inactivating...

Available online at www.sciencedirect.com
Biochimie 91 (2009) 352e363www.elsevier.com/locate/biochi
Research paper
Structural characterization and comparative modeling of PD-Ls 1e3,type 1 ribosome-inactivating proteins from summer leaves of
Phytolacca dioica L.
Antimo Di Maro a,*, Angela Chambery a, Vincenzo Carafa a, Susan Costantini b,c,Giovanni Colonna b, Augusto Parente a
a Dipartimento di Scienze della Vita, Seconda Universita degli Studi di Napoli, Via Vivaldi 43, I-81100 Caserta, Italyb Dipartimento di Biochimica e Biofisica e CRISCEB (Centro di Ricerca Interdipartimentale per le Scienze Computazionali e Biotecnologiche), Seconda Universita
degli Studi di Napoli, Via Costantinopoli 16, I-80138 Napoli, Italyc Istituto di Scienze dell’Alimentazione, CNR, Via Roma 52 A/C, I-83100 Avellino, Italy
Received 25 June 2008; accepted 16 October 2008
Available online 26 October 2008
Abstract
The amino acid sequence and glycan structure of PD-L1, PD-L2 and PD-L3, type 1 ribosome-inactivating proteins isolated from Phytolaccadioica L. leaves, were determined using a combined approach based on peptide mapping, Edman degradation and ESI-Q-TOF MS in precursorion discovery mode. The comparative analysis of the 261 amino acid residue sequences showed that PD-L1 and PD-L2 have identical primarystructure, as it is the case of PD-L3 and PD-L4. Furthermore, the primary structure of PD-Ls 1e2 and PD-Ls 3e4 have 81.6% identity (85.1%similarity). The ESI-Q-TOF MS analysis confirmed that PD-Ls 1e3 were glycosylated at different sites. In particular, PD-L1 contained threeglycidic chains with the well known paucidomannosidic structure (Man)3 (GlcNAc)2 (Fuc)1 (Xyl)1 linked to Asn10, Asn43 and Asn255. PD-L2was glycosylated at Asn10 and Asn43, and PD-L3 was glycosylated only at Asn10. PD-L4 was confirmed to be not glycosylated. Despite anoverall high structural similarity, the comparative modeling of PD-L1, PD-L2, PD-L3 and PD-L4 has shown potential influences of the glycidicchains on their adenine polynucleotide glycosylase activity on different substrates.� 2008 Elsevier Masson SAS. All rights reserved.
Keywords: Ribosome-inactivating proteins; Phytolacca dioica; Edman degradation; ESI-Q-TOF mass spectrometry; Comparative modeling
1. Introduction
Ribosome-inactivating proteins (RIPs) are widely distrib-uted plant proteins present in various organs, in some cases,even in large amounts [1,2]. Moreover, although most of them
Abbreviations: RIPs, ribosome-inactivating proteins; PD-Ls, RIPs isolated
from leaves of Phytolacca dioica L; PD-S2, RIP (major form) isolated from
seeds of Phytolacca dioica L; CNBr, cyanogen bromide; Lys-C and Asp-N,
endoproteinase Lys-C and Asp-N, respectively; PE-Cys, pyridylethylcysteine;
Hse, homoserine; Hse>, homoserine lactone; hsDNA, herring sperm DNA;
HPAEC-PAD, high performance anion-exchange chromatography with pulsed
amperometric detection; Nbs2, 5,50-[dithiobis (2-nitrobenzoic acid)]. For the
amino acids the standard one or three-letter code has been used.
* Corresponding author. Tel.: þ39 823 274535; fax: þ39 823 274571.
E-mail address: [email protected] (A. Di Maro).
0300-9084/$ - see front matter � 2008 Elsevier Masson SAS. All rights reserved.
doi:10.1016/j.biochi.2008.10.008
await further confirmation, previous reports provide evidencesof the presence of proteins of the RIP family also in fungi,bacteria and at least one alga [3].
RIPs are RNA N-b-glycosidases (EC 3.2.2.22) [4] andremove a site-specific single adenine residue (A4324 in the caseof rat liver ribosomes) from the highly conserved sarcin/ricinloop of the 28S rRNA, thus arresting protein synthesis [5]. Asfurther results indicated that RIPs can depurinate DNA, RNAand poly(A), they were named polynucleotide:adenosineglycosidases (PAG; [6]), in turn renamed adenine poly-nucleotide glycosylases (APGs; [7]). RIPs are conventionallyclassified into three types: types 1, 2 and 3 on the basis ofnumber and type of the constituting polypeptide chain(s) [8].Type 1 RIPs are single-chain proteins endowed with glycosi-dase activity, while type 2 RIPs consist of a catalytic chain

353A. Di Maro et al. / Biochimie 91 (2009) 352e363
(A chain) linked to a sugar binding chain (B chain). Type 3RIPs, found in maize and barley, consist of an N-terminalactive chain linked to an unrelated C-terminal domain withunknown function [1,9].
Interest in RIPs is 3-fold: (i) their substrate is structurallycomplex as they act on native prokaryotic and eukaryoticribosomes, by removing a specific adenine [10]; (ii) they havepotential use in crop plant biotechnology with the aim ofincreasing resistance to fungal and virus pathogens [1], and(iii) if properly targeted they could be used as therapeuticagents [11,12].
The genus Phytolacca is traditionally known as suitablesource of several highly conserved RIPs, firstly discovered onthe basis of their antiviral properties [1]. Indeed, severalmembers of this genus have been found to contain type 1 RIPs,such as PAP forms from Phytolacca americana [13e15],dodecandrin from Phytolacca dodecandra [16], insularin fromPhytolacca insularis [17], heterotepalins from Phytolaccaheterotepala [18], PD-Ss and PD-Ls from seeds and leaves ofPhytolacca dioica, respectively [19,20]. The ability of PAP,isolated from Phytolacca americana leaves, to inhibit proteinsynthesis by enzymatically inactivating ribosomes wasinitially reported in 1973 [21].
Furthermore, for several RIPs from the Phytolaccaceaefamily a differential seasonal expression has been demon-strated: (i) PAP and PAP-II from P. americana, isolated fromspring and summer leaves, respectively [22] and (ii) RIPs fromP. dioica, expressed all over the year [23].
In particular, P. dioica leaves of adult plants contain at leastfour type 1 RIPs (PD-Ls), named PD-L1, PD-L2, PD-L3 andPD-L4 [20]. On the basis of their preliminary biochemicalcharacterization it was demonstrated that: (i) they havemolecular weights in the range 28e32 kDa and pI � 8.5; (ii)they are all glycosylated proteins with the exception of PD-L4;(iii) PD-L1 shares the same N-terminal sequence with PD-L2,as well as PD-L3 with PD-L4; (iv) they have different enzy-matic activity on substrates such as DNA, rRNA, poly(A) [20];and (v) some of them are endowed with a DNase activity onsupercoiled DNA [24] and antiviral activity against tobaccomosaic virus (TMV; unpublished data).
In the present work, we report the complete primarystructure, the glycosylation pattern and the comparative three-dimensional modeling of PD-L1, PD-L2 and PD-L3, isolatedfrom P. dioica summer leaves, with the aim to unravel thestructural basis responsible of the different biologicalactivities.
2. Materials and methods
2.1. Materials
Phytolacca dioica L. leaves were harvested at the end ofJune from a single plant in the garden of the BiologicalSciences of the Second University of Naples (Caserta). TheHPLC system was a Waters BREEZE� apparatus (Milan,Italy); HPLC-grade solvents and reagents were obtained fromCarlo Erba (Milan, Italy). Endoproteinase Lys-C and Asp-N
(sequencing grade) were purchased from Promega (Monza,Italy). Herring sperm DNA (hsDNA) and cyanogen bromidewere obtained from Sigma-Aldrich/Fluka (Milan, Italy).Reagents for automated Edman degradation were supplied byApplera (Monza, Italy). Trypsin and monosaccharides (fucose,Fuc; glucosamine, GlcN; mannose, Man; xylose, Xyl) werefrom Sigma (Milan, Italy), while trifluoroacetic acid (TFA)was from Pierce (Rockford, IL, USA).
Solvents were: solvent A, 0.1% TFA; solvent B, acetonitrilecontaining 0.1% TFA; solvent C, 5% CH3CN, containing 0.1%formic acid; solvent D, 2% CH3CN in Milli-Q water; solventE, 95% CH3CN in Milli-Q water.
2.2. Purification of native PD-Ls 1e4
Native PD-Ls 1e4 were purified from P. dioica summerleaves as described elsewhere using a general protocol for thepreparation of basic RIPs [20].
2.3. Determination of free sulfhydryl groups
Determination of SH groups was performed with Nbs2 [25]on the protein denatured in the presence of 6 M Gdn.Cl.
2.4. Reduction and S-pyridylethylation
Prior to enzymatic digestion, Edman degradation or afterCNBr cleavage (see later) native proteins were S-pyridylethylatedwith 4-vinylpyridine as previously reported [26]. Modifiedproteins were desalted by RP-HPLC using a C4 column(0.46 � 15 cm; Alltech, Italy), by eluting with a linear gradient ofsolvent A and solvent B, from 5 to 65%, over 60 min, at a flow rateof 1 mL/min, monitoring at 214 nm.
2.5. Analytical procedures
Experimental procedures for automated Edman degradation,chemical (with CNBr) or enzymatic (with endoproteinases Lys-C and Asp-N) cleavages on protein aliquots were performed aspreviously described [26,27]. Tryptic hydrolysis was performedin 0.1 M TriseCl, 20 mM CaCl2, pH 8.5, containing 10%acetonitrile. The enzyme was added in three steps with a finalenzyme-to-substrate ratio of 1:50 (w/w) and the reaction wascarried out at 37 �C for 24 h.
2.6. Peptide separation
Separation of Lys-C, Asp-N and tryptic peptides by RP-HPLC was obtained on a Waters Breeze instrument, usinga C18 Symmetry column (0.46 � 15 cm, 5 mm particle size;Waters, Milford, MA, USA) at a flow-rate of 1 mL/min.Peptide elution was obtained using a linear gradient of solventA and solvent B from 5 to 55% of solvent B over 150 min.Peptides were monitored at 214 nm. Insoluble material afterendoproteinase digestion, when present, was solubilized withformic acid and analyzed as described.

354 A. Di Maro et al. / Biochimie 91 (2009) 352e363
Separation of the CNBr digest was carried out on a C4column (0.46 � 25 cm; Vydac, Hesperia, CA, USA), from 5 to40% of solvent B over 80 min and from 40 to 70% over 70 min.Sequence analyses were performed by automated Edmandegradation as previously reported [26], using a Procisesequencer, Model 491C (Applied Biosystems, Foster City, CA).
2.7. Hydrolysis of glycosylated PD-Ls and HPAEC-PADanalysis
The monosaccharide composition was obtained using aBio-LC� (Dionex Corp., Sunnyvale, CA, USA) equipped witha CarboPac� PA10 column (2 � 250 mm, Dionex Milan,Italy) and a guard column Amino Trap� (2 � 50 mm, DionexMilan, Italy). Monosaccharides were separated with 18 mMNaOH at a flow rate of 0.25 mL/min and detected witha pulsed amperometric detector (PAD). Protocols employedwere as suggested by the manufacturer (Technical Note 40 byDionex Corp., Sunnyvale, CA, USA). The column wasregenerated with 0.2 M NaOH after every analysis. Data wereanalyzed with the on-line chromatography software, Chro-meleon� Chromatography Workstation Heat Block (DionexCorp., Sunnyvale, CA, USA).
The acid hydrolysis of glycosylated PD-Ls (w30 mg) wascarried out in 2 M TFA (100 mL). Protein samples were heatedat 100 �C for 4 h, dried in a SpeedVac concentrator (SavantInstruments Inc., Holbrook, NY, USA), solubilized in H2O,and then analyzed by high performance anion-exchangechromatography with pulsed amperometric detection(HPAEC-PAD).
2.8. Mass spectrometry of native proteins
The relative molecular masses (Mr) of PD-Ls were deter-mined using a Q-TOF Micro mass spectrometer equipped witha CapLC system (Waters, Manchester, UK) as previouslyreported [28]. The capillary source voltage and the conevoltage were set at 3000 and 43 V, respectively. The sourcetemperature was kept at 80 �C and nitrogen was used asa drying gas (flow rate about 50 L/h). RP-HPLC purifiedproteins at a concentration of about 1 pmol/mL were infusedinto the system at a flow rate of 5 mL/min. The acquisition anddeconvolution of data were performed by Mass Lynx software(Waters, Manchester, UK).
2.9. Identification of glycopeptides by LC-MS/MS in theprecursor ion discovery mode
Tryptic peptides were separated by means of a modularCapLC system connected to the Z-spray source of a Q-TOFMicro (Waters, Manchester, UK) equipped with the LockSprayinterface as reported [29e31]. The MS/MS data were pro-cessed using MaxEnt3 in MassLynx 4.0 software (Waters,Manchester, UK) for de-isotoping and deconvolution. Carbo-hydrate structure reconstruction was manually performed withthe assistance of Carbotools of the Biolynx application inMassLynx 4.0 software.
2.10. Biological activity assay
Adenine polynucleotide glycosylase activity was deter-mined on hsDNA, E. coli rRNA (16S þ 23S) and poly(A)substrates as described elsewhere [20,32].
2.11. Molecular modeling
The three-dimensional model of PD-Ls 1e2 protein chainwas created following the comparative modeling strategyalready used with success by our group [32e36]. In brief, thesearches for sequence similarity within databases wereperformed with the BLAST program [37]. These searchesevidenced that an high identity (87%) exists for the PD-Ls1e2 protein chain with the homologous protein PAP-S1(Pokeweed Antiviral Protein from seeds) from Phytolaccaamericana, so that the comparative modeling strategy can beapplied with success by using the 3D structure of PAP-S1complexed with adenine (PDB code: 1J1R) [38].
The MODELLER module implemented in the Quantamolecular simulation package (Accelrys, San Diego, CA) wasused to build 10 full-atom models of PD-Ls 1e2 protein chainby setting 4.0 A as RMS deviation among initial models andby full optimization of models, i.e. multiple cycles of refiningwith conjugate gradients minimization and moleculardynamics with simulated annealing. The best model amongthose obtained was chosen by evaluating the stereo chemicalquality of the models with the PROCHECK program [39] anda scoring function with ProsaII program [40]. The amino acidside chains were built by using SCWRL3 program [41], thatinvolves a backbone-dependent rotamer library [42], an energyfunction based on the log probabilities of these rotamers anda simple repulsive steric energy term. The total contribution ofhelices to protein stability was evaluated by considering fiveterms according to [43,44].
Secondary structures were assigned by the DSSP program[45]. Search for structural classification was performed onCATH database [46]. Molecular superimposition, RMSDvalues and figures were obtained with the InsightII package(Accelrys, Inc., San Diego, CA, USA).
The glycidic chains were built using the Biopolymermodule in InsightII (Accelrys, Inc., San Diego, CA).Hydrogen atoms were added with the ‘‘Modify/Add Hydro-gens tool’’. These structures were optimized according to theoptimization protocol of the Builder module in InsightII,starting with the steepest descent method and switching to theconjugate gradient algorithm only when the energy gradientreached the default threshold value.
2.12. Molecular simulations
Taking into account the glycosylation patterns found forPD-L1 and PD-L2, N-glycosylated structures were simulatedlinking three and two oligosaccharides, to the identical proteinchains, modeled by homology, creating N-glycosidic bondswith Asn10, Asn43 and Asn255 or with Asn10 and Asn43 byusing the Builder module of Insight II. The N-glycosylated

Table 1
Amino acid sequences of: (i) S-pyridylethylated protein, (ii) cyanogen
bromide (CB), (iii) endoproteinase Asp-N (D), (iv) endoproteinase Lys-C
(LC), and (v) trypsin (T) peptides used to assemble the amino acid sequence of
PD-L2a T1 peptide from PD-L1 tryptic digestion is also reported.
Protein/peptide Sequence Notesb
S-modified
PD-L2 1 INTITYDAGX TTINKYATFM
ESLRNEAKDP
SLQCYGIPML PNXSS
45
CB-1 66 LRRNNLYVM 74
CB-2 249 GLLNYVNGTC QTT 261 C-terminal
peptide
CB-3 21 ESLRNEAKDP SLQCYGIPMc 39
CB-4 1 INTITYDAGX TTINKYATFMc 20
CB-5 40 LPNXSSTIKY LLVKLQGASQ
KTITLMc65
CB-6 173 VSEAARFKYI ENQVKTNFNR
DFSPNDKILD LEENW.207 / (248)
CB-7 75 GYSDPFNGNC RYHIFNDITG
TERTNVENTL CSSSSSRDAK.114 / (172)
355A. Di Maro et al. / Biochimie 91 (2009) 352e363
structure of PD-L3 was simulated linking one glycidic chain toAsn10 of the PD-L4 structure [47].
Three sugar-protein systems were minimized for 500 stepsunder conjugate gradient algorithm and, then, subjected tomolecular dynamics simulations using Consistent ValenceForce Field (CVFF) to assign potentials and charges andmoving only the loop regions supporting the glycosylatedresidues according to the already used procedure [48]. A totalof 300-ps long MD simulations were performed at 300 K witha time step of 1 fs, setting the dielectric constant to 1 and usingthe Discover module in InsightII. Conformations were savedevery 1000 steps (i.e. every 1 ps). Data recorded during the last100 ps were used for further analyses.
In the average models, the docking of adenine into itsbinding pocket was performed using as starting reference thepositions of the adenine in the crystallographic template (PDBcode: 1J1R) [38]. The PD-Ls/adenine complexes were opti-mized according to the optimization protocol of the Buildermodule in InsightII in order to allow a better accommodationof the adenine in the binding groove and to decrease structuralconflicts due to steric hindrance.
The ‘‘ProteineProtein Interaction Server’’ [49] was used toidentify the amino acids in the adenine binding site. Thesolvent accessibility of these amino acids was evaluated withthe program NACCESS [50] by calculating the atomicaccessible surface defined by rolling a probe of 1.40 A aroundthe van der Waals surface of the protein models. H-bonds werecalculated with the Hbplus program [51].
D-1 1 INTITY 6
D-2 147 DIGKISGQSS FT 158
2.13. On-line bioinformatics tool D-3 78 DPFNGNCRYH IFN 90D-4 202 DLEENWGKIS TAIH 215
D-5 232 DGTKWIVLRV 241
D-6 247 DMGLLNYVNG TCQTT 261 C-terminal
peptide
D-7 216 DATNGALPKP LELKNA 231
D-8 91 DITGTERTNV ENTLCSSSSS R 211
D-9 7 DAGXTTINKY ATFMESLRNE AK 28
D-10 159 DKTEAKFLLV AIQMVSEAAR
FKYIENQVKT.188 / (192)
For multiple alignment of RIP amino acid sequences,a search for sequence similarities was performed with theBLAST program available on-line (http://www.ncbi.nlm.nih.gov/BLAST). Sequences were aligned using the Clustal Wsoftware in the default set-up. The alignment was thenanalyzed using the BOXSHADE software (http://bioweb.pasteur.fr/seqanal/interfaces/boxshade.html).
D-11 112 DAKPINYNSL YSTLEKKAEV
NSRSQVQLGI QILSS
146
2.14. UniProt Knowledgebase accession numbers
D-12 29 DPSLQCYGIP MLPNXSSTIKYLLVKLQGAS QKTITLMLR
77
LC-1 230 NADGTK 235
LC-2 236 WIVLRVDEIK 245
LC-3 151 ISGQSSFTDK 160
LC-4 188 TNFNRDFSPN DK 199
LC-5 210 ISTAIHDATN GALPK 224
The primary structures of PD-Ls 1e2 and PD-Ls 3e4 havebeen deposited in the UniProtKB (accession code P84853 forPD-Ls 1e2 and P84854 for PD-Ls 3e4).
3. Results and discussion
LC-6d 129 AEVNSRSQVQ LGIQILSSDI GK 150
T1 (236e261) 236 WIVLRVDEIK PDMGLLNYVX 261
3.1. Determination of the primary structure of PD-L2GTCQTT
T2 (236e261) 236 WIVLRVDEIK PDMGLLNYVN
GTCQTT
261
X, non-assigned amino acid residue.a For some CB and D peptides only the sequenced amino acid residues have
been reported.b (XXX), full length of the peptide fragment.c For CB peptides, the C-terminal Hse/Hse> is reported for methionine (M).d The sequence of the two peptides were assigned on the basis of their
different amount in the mixture.
The primary structure of PD-L2 was determined using thestrategy previously reported [26]. Briefly, the followingexperimental steps were employed: (i) PD-L2 purification andassessment of its homogeneity by SDSePAGE and ESI-Q-TOF MS; (ii) protein hydrolysis by chemical and enzymaticcleavages; and (iii) sequence determination by Edman degra-dation and alignment of CNBr peptides with overlappingendoproteinase Lys-C and Asp-N peptides.
CNBr, endoproteinase Asp-N and Lys-C peptides wereseparated by RP-HPLC (data not shown). Automated Edmandegradation of these peptides and of S-pyridylethylated PD-L2provided most of its amino acid sequence (Table 1). Theamino acid residues at positions 10 and 43 were not assignedby Edman degradation and are reported with X in Table 1. Asthe protein was S-pyridylethylated, this finding suggested thepresence of glycosylated asparaginyl residues at the two
356 A. Di Maro et al. / Biochimie 91 (2009) 352e363
positions. Therefore, we determined the carbohydratecomposition of PD-L2 by HPAEC-PAD analyses, whichrevealed the presence of four types of monosaccharide:mannose, glucosamine (derived from 2M TFA hydrolysis ofN-acetyl-glucosamine), fucose and xylose in the ratio 3:2:1:1.Then, the S-pyridylethylated protein was digested with trypsinand the resulting glycosylated peptides detected and charac-terized by ESI-Q-TOF MS/MS analysis in PID mode [29,31].
Two glycosylated tryptic peptides with m/z 1405.44([M þ 2H]2þ, precursor ion 2808.87) and m/z 1152.08([M þ 3H]3þ, precursor ion 3453.24) were found in PD-L2(Table 2). The glycopeptides identified in the PID experimentswere deconvoluted on the basis of fragment ion charge states,and their spectra were analyzed to gain insights into glycanchain structure and composition.
As an example, the MS/MS spectrum of the doubly chargedion at m/z 1405.44 is shown in the m/z 1600e2850 range,where the larger glycopeptide fragments are observed (Fig. 1S,supplementary material). The molecular ion at m/z 1638.72[M þ Hþ], corresponding to the deglycosylated tryptic peptide1e15 (theoretical Mr 1637.83), was clearly detected. Thecharacteristic ion (peptide þ HexNAc) was also found at m/z1841.76, with a rather high intensity (Fig. 1S). The clear-cutfragmentation pattern and the composition of the glycan chainobtained by HPAEC-PAD, allowed the determination of thespecific carbohydrate structure: (Man)3 (GlcNAc)2 (Fuc)1
(Xyl)1 (Fig. 1S). This glycan structure represents a standardplant paucidomannosidic N-glycosylation pattern, alreadyfound in other type 1 RIPs.
This glycan structure represents a standard plant paucido-mannosidic N-glycosylation pattern, already found in othertype 1 RIPs [4,31].
The same glycosylation pattern was found in tryptic peptide29e48 (Table 2). Thus, the assignment of the sequence posi-tions 10 and 43 as asparaginyl residues carrying glycidicchains, allowed us to complete the amino acid sequence ofPD-L2 which is reported in Fig. 1A, compared to the PD-S2sequence.
Furthermore, the four cysteinyl residues, located in positions34, 84, 105 and 258, were very likely engaged in disulfidebridges. This conclusion was based on the negative reaction ofnative PD-L2 with Nbs2, showing no free sulfhydryl groups. Asdemonstrated for other Phytolaccaceae, we confidently
Table 2
Glycosylated tryptic peptides detected in PD-Ls 1e3 by ESI-Q-TOF MS.
Protein Sequence positiona m/z Charge state Experimental Mr
PD-L1 1e15 1405.35 2 2808.70
29e48 1152.12 3 3453.36
241e261 1196.39 3 3586.17
PD-L2 1e15 1405.44 2 2808.87
29e48 1152.08 3 3453.24
PD-L3 1e15 1389.24 2 2776.49
Protein, sequence position, m/z, charge state, experimental masses of the precurso
reported.a Containing N-glycosylation site.b Including the mass of the asparaginyl residue (114.04 uma).
assumed that the two disulfide bridges were located betweenCys34eCys258 and Cys84eCys105 [18,31,52].
The PD-L2 amino acid sequence accounts for a calculatedMr of 29,222.2, which is in good agreement with the experi-mental Mr obtained on native PD-L2 by ESI-Q-TOF MS(31,560.50), taking into account the presence of two disulfidebridges (�4 uma) and two glycosidic chains (þ2342 uma).
3.2. Determination of the primary structure of PD-L1
It was already known that PD-L1 shared with PD-L2 thesame N-terminal sequence up to residue 45 and that their Mr
differed by 1171 ([20] and this paper). Therefore, we decidedto complete its sequence by tryptic peptide mapping and ESI-Q-TOF MS analysis in PID mode having as reference the dataobtained for PD-L2.
The RP-HPLC tryptic peptide maps revealed a highreproducibility and the same retention times for almost allpeptides (Fig. 2). Differences were found for peptide T2(236e261), absent in PD-L1 and T1(236e261), eluted at a lowerretention time. The two peaks were collected and sequencedby Edman degradation (Table 1), which revealed that bothpeptides corresponded to the C-terminal region 236e261(236-WIVLRVDEIK PDMGLLNYVN GTCQTT-261). InPD-L2, the potential consensus N-glycosylation sequence(NGT, reported in bold) was found. On the contrary, thesequence position 255 was not assigned in PD-L1, stronglysuggesting the presence of a modified asparaginyl residue.Therefore, the tryptic digest of PD-L1 was analyzed by ESI-Q-TOF MS in PID mode. In this instance, three glycosylatedpeptides were detected, two of which were identical to thosefound in PD-L2 (sequence positions 1e15 and 29e48, Table2), while the third glycopeptide, showed a sequence corre-sponding to positions 236e261, and the same glycan moietyreported for the other two peptides (Table 2). Therefore, weconcluded that PD-L1 differed from PD-L2 for an additionalglycan moiety on Asn255 (see Scheme 1A).
The mass spectrometry analysis revealed also the presenceof a micro heterogeneity at position 20 in the two RIPs.Indeed, two tryptic peptides corresponding to sequence posi-tions 16e24, carrying a Met or a Leu/Ile residue (Fig. 2S,supplementary material), were clearly detected and charac-terized by tandem MS. The presence of protein isoforms have
Theoretical Mr of
deglycosylated peptidesbGlycidic
chain Mr
Theoretical Mr of
glycosylated peptide
D
1637.83 1171 2808.83 �0.13
2282.11 1171 3453.11 0.25
2415.15 1171 3586.15 0.02
1637.83 1171 2808.83 0.04
2282.11 1171 3453.11 0.13
1605.84 1171 2776.84 �0.35
r ions together with mass accuracy compared with theoretical mass values are

Fig. 1. (A) Amino acid sequence of PD-Ls 1e2 and PD-Ls 3e4 compared with PD-S2. ‘‘*’’ identical, ‘‘:’’ conserved and ‘‘.’’ semi-conserved amino acid residues.
The asparaginyl residues in a consensus sequence for N-glycosylation have been shaded. �, indicates conserved amino acid residues found in the active site of
RIPs. The amino acid sequence of PD-S2 was taken from [64]. PD-L1 has the same amino acid sequence of PD-L2 and Asn255 glycosylated, differently from PD-
L2. In the case of PD-L3, the amino acid sequence is the same of PD-L4, but Asn10 is glycosylated. (B) Identityesimilarity matrix of PD-Ls 1e2, PD-Ls 3e4 and
PD-S2.
357A. Di Maro et al. / Biochimie 91 (2009) 352e363
been widely reported for the RIP family, justifying theoccurrence of more than one Mr in the mass spectra.
3.3. Determination of the primary structure of PD-L3
A methodological approach similar to that applied for PD-Ls 1e2 was adopted for determining the primary structure ofPD-L3. In this case, we had as reference the already knownsequence of PD-L4 [32]. Indeed, PD-L3 shares the first 45 N-terminal amino acid residues with PD-L4. Furthermore,a mass difference of 1171 Da and the presence of carbohy-drates had been demonstrated for PD-L3 but not for PD-L4[20]. Therefore, a tryptic LC-peptide mapping of PD-L3 wasperformed and compared with that obtained from PD-L4. TheRP-HPLC pattern of PD-L3 and PD-L4 are reported in Fig. 3.Also in this case, almost all peptides eluted with the sameretention times in both chromatograms. However, peptideT4(1e15) was absent in the PD-L4 map, while in the PD-L3map T3(1e15) was present at a lower retention time. Edmandegradation analysis revealed that both peptides, T3(1e15)and T4(1e15), corresponded to the N-terminal region 1e15(1-VNTITFDVGNATINK-15) which contained a potentialconsensus sequence for N-glycosylation in PD-L4 (NAT,
reported in bold). Again, sequence position 10 was notassigned in PD-L3, due to the likely presence of a glycanchain on this residue. Tryptic digests of PD-L3 and PD-L4were then analyzed by ESI-Q-TOF MS in PID mode, whichshowed that PD-L3 contained only one glycosylated peptide,with m/z 1389.24 ([M þ 2H]2þ, precursor ion 2776.49; Table2), whose fragmentation revealed a glycidic chain structureidentical to that found in PD-L1 and PD-L2 (data not shown).
The PD-L3 sequence (Fig. 1A) accounts for a calculatedmolecular mass of 29,190.2 Da, which is in good agreementwith the value obtained by ESI-Q-TOF MS on the nativeprotein (30,357.7 Da), taking into account the presence of theglycan chain and two disulfide bridges (see earlier). Therefore,we concluded that PD-L3 differed from PD-L4 for a glycanmoiety on Asn10 (see Scheme 1B).
3.4. Sequence analysis
The amino acid sequences of PD-Ls 1e2 and PD-Ls 3e4were compared each other and with PD-S2, the major RIPform isolated from seeds of the same plant (Fig. 1A).
The primary structure of PD-Ls 1e2 and PD-Ls 3e4have 81.6% identity (85.1% similarity). When compared with
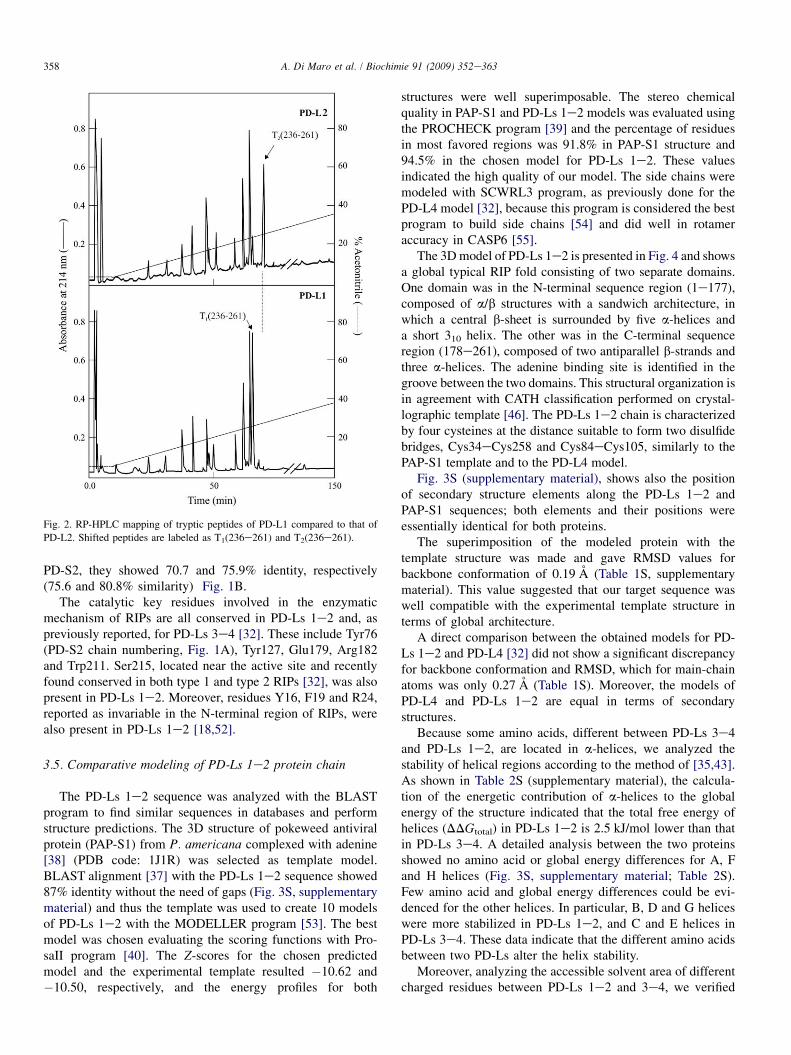
Fig. 2. RP-HPLC mapping of tryptic peptides of PD-L1 compared to that of
PD-L2. Shifted peptides are labeled as T1(236e261) and T2(236e261).
358 A. Di Maro et al. / Biochimie 91 (2009) 352e363
PD-S2, they showed 70.7 and 75.9% identity, respectively(75.6 and 80.8% similarity) Fig. 1B.
The catalytic key residues involved in the enzymaticmechanism of RIPs are all conserved in PD-Ls 1e2 and, aspreviously reported, for PD-Ls 3e4 [32]. These include Tyr76(PD-S2 chain numbering, Fig. 1A), Tyr127, Glu179, Arg182and Trp211. Ser215, located near the active site and recentlyfound conserved in both type 1 and type 2 RIPs [32], was alsopresent in PD-Ls 1e2. Moreover, residues Y16, F19 and R24,reported as invariable in the N-terminal region of RIPs, werealso present in PD-Ls 1e2 [18,52].
3.5. Comparative modeling of PD-Ls 1e2 protein chain
The PD-Ls 1e2 sequence was analyzed with the BLASTprogram to find similar sequences in databases and performstructure predictions. The 3D structure of pokeweed antiviralprotein (PAP-S1) from P. americana complexed with adenine[38] (PDB code: 1J1R) was selected as template model.BLAST alignment [37] with the PD-Ls 1e2 sequence showed87% identity without the need of gaps (Fig. 3S, supplementarymaterial) and thus the template was used to create 10 modelsof PD-Ls 1e2 with the MODELLER program [53]. The bestmodel was chosen evaluating the scoring functions with Pro-saII program [40]. The Z-scores for the chosen predictedmodel and the experimental template resulted �10.62 and�10.50, respectively, and the energy profiles for both
structures were well superimposable. The stereo chemicalquality in PAP-S1 and PD-Ls 1e2 models was evaluated usingthe PROCHECK program [39] and the percentage of residuesin most favored regions was 91.8% in PAP-S1 structure and94.5% in the chosen model for PD-Ls 1e2. These valuesindicated the high quality of our model. The side chains weremodeled with SCWRL3 program, as previously done for thePD-L4 model [32], because this program is considered the bestprogram to build side chains [54] and did well in rotameraccuracy in CASP6 [55].
The 3D model of PD-Ls 1e2 is presented in Fig. 4 and showsa global typical RIP fold consisting of two separate domains.One domain was in the N-terminal sequence region (1e177),composed of a/b structures with a sandwich architecture, inwhich a central b-sheet is surrounded by five a-helices anda short 310 helix. The other was in the C-terminal sequenceregion (178e261), composed of two antiparallel b-strands andthree a-helices. The adenine binding site is identified in thegroove between the two domains. This structural organization isin agreement with CATH classification performed on crystal-lographic template [46]. The PD-Ls 1e2 chain is characterizedby four cysteines at the distance suitable to form two disulfidebridges, Cys34eCys258 and Cys84eCys105, similarly to thePAP-S1 template and to the PD-L4 model.
Fig. 3S (supplementary material), shows also the positionof secondary structure elements along the PD-Ls 1e2 andPAP-S1 sequences; both elements and their positions wereessentially identical for both proteins.
The superimposition of the modeled protein with thetemplate structure was made and gave RMSD values forbackbone conformation of 0.19 A (Table 1S, supplementarymaterial). This value suggested that our target sequence waswell compatible with the experimental template structure interms of global architecture.
A direct comparison between the obtained models for PD-Ls 1e2 and PD-L4 [32] did not show a significant discrepancyfor backbone conformation and RMSD, which for main-chainatoms was only 0.27 A (Table 1S). Moreover, the models ofPD-L4 and PD-Ls 1e2 are equal in terms of secondarystructures.
Because some amino acids, different between PD-Ls 3e4and PD-Ls 1e2, are located in a-helices, we analyzed thestability of helical regions according to the method of [35,43].As shown in Table 2S (supplementary material), the calcula-tion of the energetic contribution of a-helices to the globalenergy of the structure indicated that the total free energy ofhelices (DDGtotal) in PD-Ls 1e2 is 2.5 kJ/mol lower than thatin PD-Ls 3e4. A detailed analysis between the two proteinsshowed no amino acid or global energy differences for A, Fand H helices (Fig. 3S, supplementary material; Table 2S).Few amino acid and global energy differences could be evi-denced for the other helices. In particular, B, D and G heliceswere more stabilized in PD-Ls 1e2, and C and E helices inPD-Ls 3e4. These data indicate that the different amino acidsbetween two PD-Ls alter the helix stability.
Moreover, analyzing the accessible solvent area of differentcharged residues between PD-Ls 1e2 and 3e4, we verified
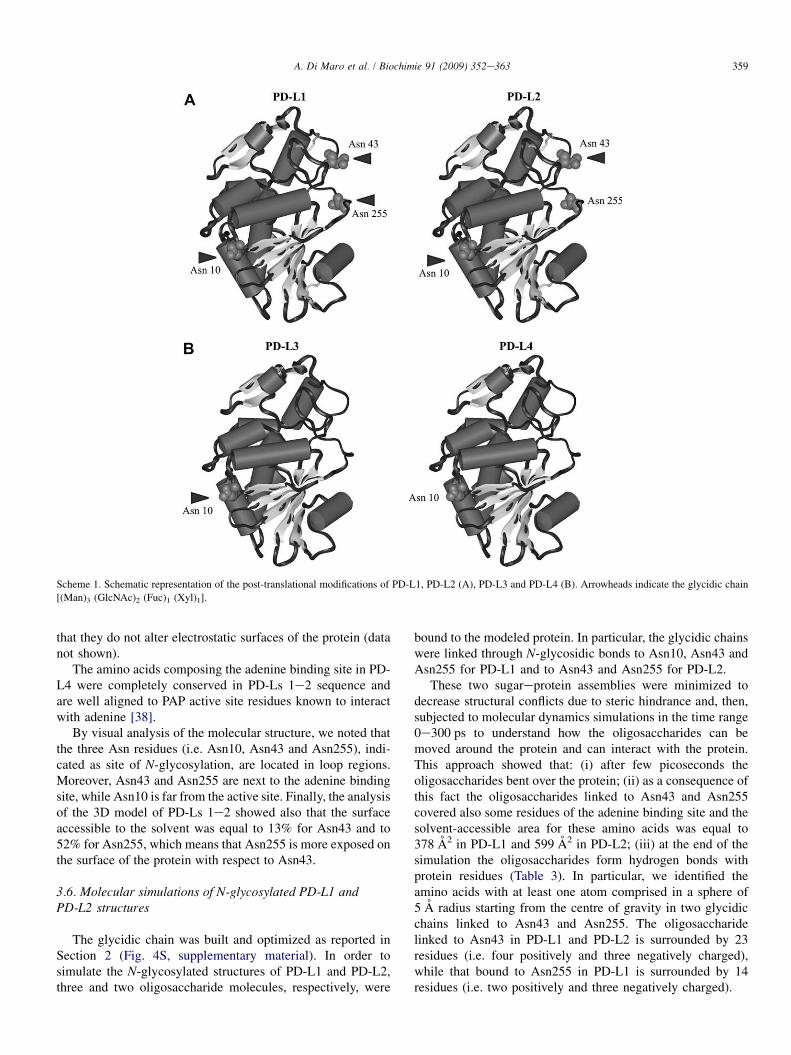
Scheme 1. Schematic representation of the post-translational modifications of PD-L1, PD-L2 (A), PD-L3 and PD-L4 (B). Arrowheads indicate the glycidic chain
[(Man)3 (GlcNAc)2 (Fuc)1 (Xyl)1].
359A. Di Maro et al. / Biochimie 91 (2009) 352e363
that they do not alter electrostatic surfaces of the protein (datanot shown).
The amino acids composing the adenine binding site in PD-L4 were completely conserved in PD-Ls 1e2 sequence andare well aligned to PAP active site residues known to interactwith adenine [38].
By visual analysis of the molecular structure, we noted thatthe three Asn residues (i.e. Asn10, Asn43 and Asn255), indi-cated as site of N-glycosylation, are located in loop regions.Moreover, Asn43 and Asn255 are next to the adenine bindingsite, while Asn10 is far from the active site. Finally, the analysisof the 3D model of PD-Ls 1e2 showed also that the surfaceaccessible to the solvent was equal to 13% for Asn43 and to52% for Asn255, which means that Asn255 is more exposed onthe surface of the protein with respect to Asn43.
3.6. Molecular simulations of N-glycosylated PD-L1 andPD-L2 structures
The glycidic chain was built and optimized as reported inSection 2 (Fig. 4S, supplementary material). In order tosimulate the N-glycosylated structures of PD-L1 and PD-L2,three and two oligosaccharide molecules, respectively, were
bound to the modeled protein. In particular, the glycidic chainswere linked through N-glycosidic bonds to Asn10, Asn43 andAsn255 for PD-L1 and to Asn43 and Asn255 for PD-L2.
These two sugareprotein assemblies were minimized todecrease structural conflicts due to steric hindrance and, then,subjected to molecular dynamics simulations in the time range0e300 ps to understand how the oligosaccharides can bemoved around the protein and can interact with the protein.This approach showed that: (i) after few picoseconds theoligosaccharides bent over the protein; (ii) as a consequence ofthis fact the oligosaccharides linked to Asn43 and Asn255covered also some residues of the adenine binding site and thesolvent-accessible area for these amino acids was equal to378 A2 in PD-L1 and 599 A2 in PD-L2; (iii) at the end of thesimulation the oligosaccharides form hydrogen bonds withprotein residues (Table 3). In particular, we identified theamino acids with at least one atom comprised in a sphere of5 A radius starting from the centre of gravity in two glycidicchains linked to Asn43 and Asn255. The oligosaccharidelinked to Asn43 in PD-L1 and PD-L2 is surrounded by 23residues (i.e. four positively and three negatively charged),while that bound to Asn255 in PD-L1 is surrounded by 14residues (i.e. two positively and three negatively charged).
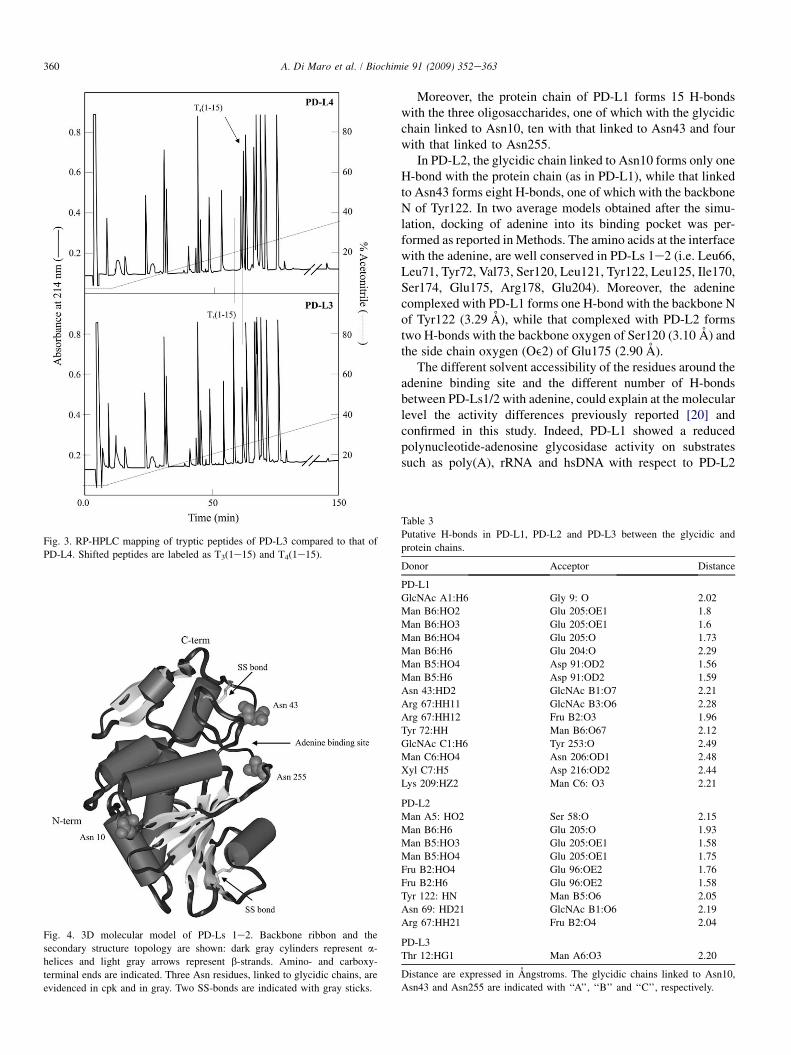
Fig. 3. RP-HPLC mapping of tryptic peptides of PD-L3 compared to that of
PD-L4. Shifted peptides are labeled as T3(1e15) and T4(1e15).
Fig. 4. 3D molecular model of PD-Ls 1e2. Backbone ribbon and the
secondary structure topology are shown: dark gray cylinders represent a-
helices and light gray arrows represent b-strands. Amino- and carboxy-
terminal ends are indicated. Three Asn residues, linked to glycidic chains, are
evidenced in cpk and in gray. Two SS-bonds are indicated with gray sticks.
360 A. Di Maro et al. / Biochimie 91 (2009) 352e363
Moreover, the protein chain of PD-L1 forms 15 H-bondswith the three oligosaccharides, one of which with the glycidicchain linked to Asn10, ten with that linked to Asn43 and fourwith that linked to Asn255.
In PD-L2, the glycidic chain linked to Asn10 forms only oneH-bond with the protein chain (as in PD-L1), while that linkedto Asn43 forms eight H-bonds, one of which with the backboneN of Tyr122. In two average models obtained after the simu-lation, docking of adenine into its binding pocket was per-formed as reported in Methods. The amino acids at the interfacewith the adenine, are well conserved in PD-Ls 1e2 (i.e. Leu66,Leu71, Tyr72, Val73, Ser120, Leu121, Tyr122, Leu125, Ile170,Ser174, Glu175, Arg178, Glu204). Moreover, the adeninecomplexed with PD-L1 forms one H-bond with the backbone Nof Tyr122 (3.29 A), while that complexed with PD-L2 formstwo H-bonds with the backbone oxygen of Ser120 (3.10 A) andthe side chain oxygen (Oe2) of Glu175 (2.90 A).
The different solvent accessibility of the residues around theadenine binding site and the different number of H-bondsbetween PD-Ls1/2 with adenine, could explain at the molecularlevel the activity differences previously reported [20] andconfirmed in this study. Indeed, PD-L1 showed a reducedpolynucleotide-adenosine glycosidase activity on substratessuch as poly(A), rRNA and hsDNA with respect to PD-L2
Table 3
Putative H-bonds in PD-L1, PD-L2 and PD-L3 between the glycidic and
protein chains.
Donor Acceptor Distance
PD-L1
GlcNAc A1:H6 Gly 9: O 2.02
Man B6:HO2 Glu 205:OE1 1.8
Man B6:HO3 Glu 205:OE1 1.6
Man B6:HO4 Glu 205:O 1.73
Man B6:H6 Glu 204:O 2.29
Man B5:HO4 Asp 91:OD2 1.56
Man B5:H6 Asp 91:OD2 1.59
Asn 43:HD2 GlcNAc B1:O7 2.21
Arg 67:HH11 GlcNAc B3:O6 2.28
Arg 67:HH12 Fru B2:O3 1.96
Tyr 72:HH Man B6:O67 2.12
GlcNAc C1:H6 Tyr 253:O 2.49
Man C6:HO4 Asn 206:OD1 2.48
Xyl C7:H5 Asp 216:OD2 2.44
Lys 209:HZ2 Man C6: O3 2.21
PD-L2
Man A5: HO2 Ser 58:O 2.15
Man B6:H6 Glu 205:O 1.93
Man B5:HO3 Glu 205:OE1 1.58
Man B5:HO4 Glu 205:OE1 1.75
Fru B2:HO4 Glu 96:OE2 1.76
Fru B2:H6 Glu 96:OE2 1.58
Tyr 122: HN Man B5:O6 2.05
Asn 69: HD21 GlcNAc B1:O6 2.19
Arg 67:HH21 Fru B2:O4 2.04
PD-L3
Thr 12:HG1 Man A6:O3 2.20
Distance are expressed in Angstroms. The glycidic chains linked to Asn10,
Asn43 and Asn255 are indicated with ‘‘A’’, ‘‘B’’ and ‘‘C’’, respectively.
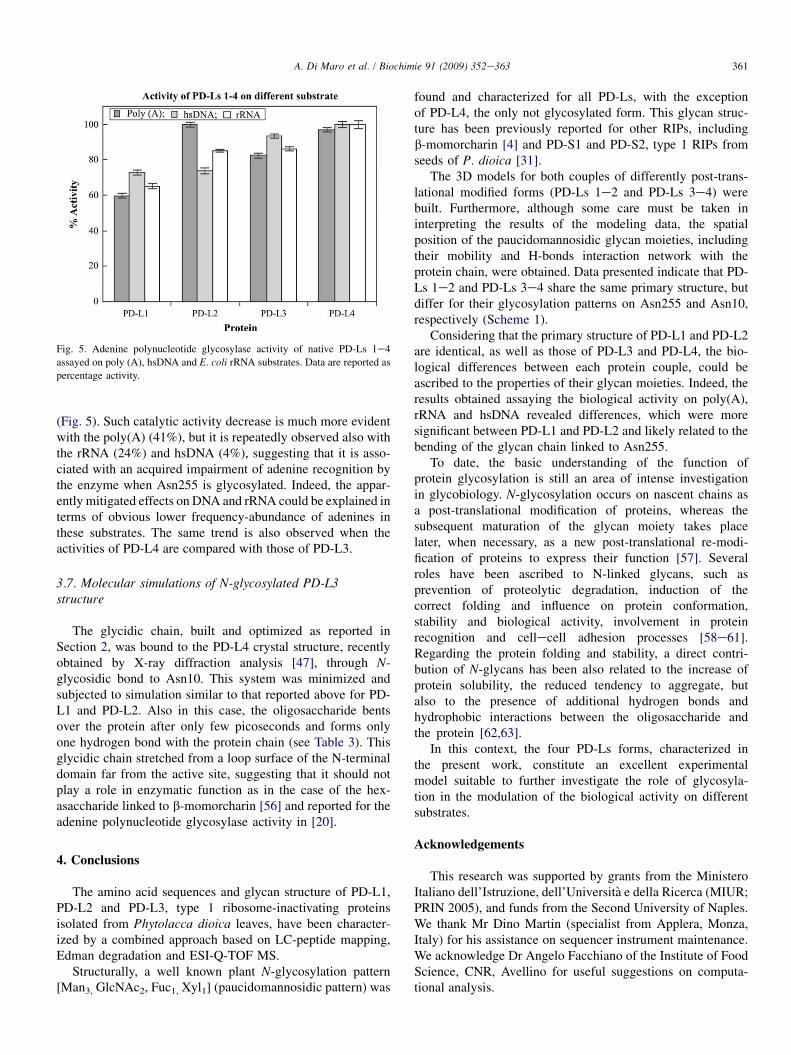
Fig. 5. Adenine polynucleotide glycosylase activity of native PD-Ls 1e4
assayed on poly (A), hsDNA and E. coli rRNA substrates. Data are reported as
percentage activity.
361A. Di Maro et al. / Biochimie 91 (2009) 352e363
(Fig. 5). Such catalytic activity decrease is much more evidentwith the poly(A) (41%), but it is repeatedly observed also withthe rRNA (24%) and hsDNA (4%), suggesting that it is asso-ciated with an acquired impairment of adenine recognition bythe enzyme when Asn255 is glycosylated. Indeed, the appar-ently mitigated effects on DNA and rRNA could be explained interms of obvious lower frequency-abundance of adenines inthese substrates. The same trend is also observed when theactivities of PD-L4 are compared with those of PD-L3.
3.7. Molecular simulations of N-glycosylated PD-L3structure
The glycidic chain, built and optimized as reported inSection 2, was bound to the PD-L4 crystal structure, recentlyobtained by X-ray diffraction analysis [47], through N-glycosidic bond to Asn10. This system was minimized andsubjected to simulation similar to that reported above for PD-L1 and PD-L2. Also in this case, the oligosaccharide bentsover the protein after only few picoseconds and forms onlyone hydrogen bond with the protein chain (see Table 3). Thisglycidic chain stretched from a loop surface of the N-terminaldomain far from the active site, suggesting that it should notplay a role in enzymatic function as in the case of the hex-asaccharide linked to b-momorcharin [56] and reported for theadenine polynucleotide glycosylase activity in [20].
4. Conclusions
The amino acid sequences and glycan structure of PD-L1,PD-L2 and PD-L3, type 1 ribosome-inactivating proteinsisolated from Phytolacca dioica leaves, have been character-ized by a combined approach based on LC-peptide mapping,Edman degradation and ESI-Q-TOF MS.
Structurally, a well known plant N-glycosylation pattern[Man3, GlcNAc2, Fuc1, Xyl1] (paucidomannosidic pattern) was
found and characterized for all PD-Ls, with the exceptionof PD-L4, the only not glycosylated form. This glycan struc-ture has been previously reported for other RIPs, includingb-momorcharin [4] and PD-S1 and PD-S2, type 1 RIPs fromseeds of P. dioica [31].
The 3D models for both couples of differently post-trans-lational modified forms (PD-Ls 1e2 and PD-Ls 3e4) werebuilt. Furthermore, although some care must be taken ininterpreting the results of the modeling data, the spatialposition of the paucidomannosidic glycan moieties, includingtheir mobility and H-bonds interaction network with theprotein chain, were obtained. Data presented indicate that PD-Ls 1e2 and PD-Ls 3e4 share the same primary structure, butdiffer for their glycosylation patterns on Asn255 and Asn10,respectively (Scheme 1).
Considering that the primary structure of PD-L1 and PD-L2are identical, as well as those of PD-L3 and PD-L4, the bio-logical differences between each protein couple, could beascribed to the properties of their glycan moieties. Indeed, theresults obtained assaying the biological activity on poly(A),rRNA and hsDNA revealed differences, which were moresignificant between PD-L1 and PD-L2 and likely related to thebending of the glycan chain linked to Asn255.
To date, the basic understanding of the function ofprotein glycosylation is still an area of intense investigationin glycobiology. N-glycosylation occurs on nascent chains asa post-translational modification of proteins, whereas thesubsequent maturation of the glycan moiety takes placelater, when necessary, as a new post-translational re-modi-fication of proteins to express their function [57]. Severalroles have been ascribed to N-linked glycans, such asprevention of proteolytic degradation, induction of thecorrect folding and influence on protein conformation,stability and biological activity, involvement in proteinrecognition and cellecell adhesion processes [58e61].Regarding the protein folding and stability, a direct contri-bution of N-glycans has been also related to the increase ofprotein solubility, the reduced tendency to aggregate, butalso to the presence of additional hydrogen bonds andhydrophobic interactions between the oligosaccharide andthe protein [62,63].
In this context, the four PD-Ls forms, characterized inthe present work, constitute an excellent experimentalmodel suitable to further investigate the role of glycosyla-tion in the modulation of the biological activity on differentsubstrates.
Acknowledgements
This research was supported by grants from the MinisteroItaliano dell’Istruzione, dell’Universita e della Ricerca (MIUR;PRIN 2005), and funds from the Second University of Naples.We thank Mr Dino Martin (specialist from Applera, Monza,Italy) for his assistance on sequencer instrument maintenance.We acknowledge Dr Angelo Facchiano of the Institute of FoodScience, CNR, Avellino for useful suggestions on computa-tional analysis.

362 A. Di Maro et al. / Biochimie 91 (2009) 352e363
Appendix A. Supplementary material
Supplementary information for this manuscript can bedownloaded at doi: 10.1016/j.biochi.2008.10.008
References
[1] W.J. Peumans, Q. Hao, E.J. Van Damme, Ribosome-inactivating proteins
from plants: more than RNA N-glycosidases? FASEB J 15 (2001) 1493e
1506.
[2] T. Girbes, J.M. Ferreras, F.J. Arias, F. Stirpe, Description, distribution,
activity and phylogenetic relationship of ribosome-inactivating proteins
in plants, fungi and bacteria, Mini Rev. Med. Chem. 4 (2004) 461e476.
[3] R.S. Liu, J.H. Yang, W.Y. Liu, Isolation and enzymatic characterization of
lamjapin, the first ribosome-inactivating protein from cryptogamic algal
plant (Laminaria japonica A), Eur. J. Biochem. 269 (2002) 4746e4752.
[4] L. Barbieri, M.G. Battelli, F. Stirpe, Ribosome-inactivating proteins from
plants, Biochim. Biophys. Acta 1154 (1993) 237e282.
[5] Y. Endo, K. Tsurugi, RNA N-glycosidase activity of ricin A-chain.
Mechanism of action of the toxic lectin ricin on eukaryotic ribosomes,
J. Biol. Chem. 262 (1987) 8128e8130.
[6] L. Barbieri, P. Valbonesi, E. Bonora, P. Gorini, A. Bolognesi, F. Stirpe,
Polynucleotide: adenosine glycosidase activity of ribosome-inactivating
proteins: effect on DNA, RNA and poly(A), Nucleic Acids Res. 25
(1997) 518e522.
[7] A. Bolognesi, L. Polito, C. Lubelli, L. Barbieri, A. Parente, F. Stirpe,
Ribosome-inactivating and adenine polynucleotide glycosylase activities
in Mirabilis jalapa L. tissues, J. Biol. Chem. 277 (2002) 13709e13716.
[8] K. Nielsen, R.S. Boston, Ribosome-inactivating proteins: a plant
perspective, Annu. Rev. Plant Physiol, Plant Mol. Biol. 52 (2001)
785e816.
[9] S. Reinbothe, C. Reinbothe, J. Lehmann, W. Becker, K. Apel, B. Parthier,
JIP60, a methyl jasmonate-induced ribosome-inactivating protein
involved in plant stress reactions, Proc. Natl. Acad. Sci. USA 91 (1994)
7012e7016.
[10] B. Vester, L.H. Hansen, S. Douthwaite, The conformation of 23S rRNA
nucleotide A2058 determines its recognition by the ErmE methyl-
transferase, RNA 1 (1995) 501e509.
[11] F. Stirpe, Ribosome-inactivating proteins, Toxicon 44 (2004) 371e383.
[12] F. Stirpe, M.G. Battelli, Ribosome-inactivating proteins: progress and
problems, Cell. Mol. Life Sci. 63 (2006) 1850e1866.
[13] J.D. Irvin, Purification and partial characterization of the antiviral protein
from Phytolacca americana which inhibits eukaryotic protein synthesis,
Arch. Biochem. Biophys. 169 (1975) 522e528.
[14] J.D. Irvin, T. Kelly, J.D. Robertus, Purification and properties of a second
antiviral protein from Phytolacca americana which inactivates eukaryotic
ribosomes, Arch. Biochem. Biophys. 200 (1980) 418e425.
[15] S.W. Park, C.B. Lawrence, J.C. Linden, J.M. Vivanco, Isolation and
characterization of a novel ribosome-inactivating protein from root
cultures of pokeweed and its mechanism of secretion from roots, Plant
Physiol. 130 (2002) 164e178.
[16] S. Thomsen, H.S. Hansen, U. Nyman, Ribosome-inhibiting proteins from
in vitro cultures of Phytolacca dodecandra, Planta Med. 57 (1991) 232e
236.
[17] S.K. Song, Y. Choi, Y.H. Moon, S.G. Kim, Y.D. Choi, J.S. Lee, Systemic
induction of a Phytolacca insularis antiviral protein gene by mechanical
wounding, jasmonic acid, and abscisic acid, Plant Mol. Biol. 43 (2000)
439e450.
[18] A. Di Maro, A. Chambery, A. Daniele, P. Casoria, A. Parente, Isolation and
characterization of heterotepalins, type 1 ribosome-inactivating proteins
from Phytolacca heterotepala leaves, Phytochemistry 68 (2007) 767e776.
[19] A. Parente, P. De Luca, A. Bolognesi, L. Barbieri, M.G. Battelli,
A. Abbondanza, M.J. Sande, G.S. Gigliano, P.L. Tazzari, F. Stirpe,
Purification and partial characterization of single-chain ribosome-inac-
tivating proteins from the seeds of Phytolacca dioica L, Biochim. Bio-
phys. Acta 1216 (1993) 43e49.
[20] A. Di Maro, P. Valbonesi, A. Bolognesi, F. Stirpe, P. De Luca,
G. Siniscalco Gigliano, L. Gaudio, P. Delli Bovi, P. Ferranti, A. Malorni,
A. Parente, Isolation and characterization of four type-1 ribosome-inac-
tivating proteins, with polynucleotide:adenosine glycosidase activity,
from leaves of Phytolacca dioica L, Planta 208 (1999) 125e131.
[21] T.G. Obrig, J.D. Irvin, B. Hardesty, The effect of an antiviral peptide on
the ribosomal reactions of the peptide elongation enzymes, EF-I and EF-
II, Arch. Biochem. Biophys. 155 (1973) 278e289.
[22] L.L. Houston, S. Ramakrishnan, M.A. Hermodson, Seasonal variations in
different forms of pokeweed antiviral protein, a potent inactivator of
ribosomes, J. Biol. Chem. 258 (1983) 9601e9604.
[23] A. Parente, B. Conforto, A. Di Maro, A. Chambery, P. De Luca,
A. Bolognesi, M. Iriti, F. Faoro, Type 1 ribosome-inactivating proteins
from Phytolacca dioica L. leaves: differential seasonal and age expression,
and cellular localization, Planta (2008). doi:10.1007/s00425-008-0796-z.
[24] S. Aceto, A. Di Maro, B. Conforto, G.G. Siniscalco, A. Parente, P. Delli Bovi,
L. Gaudio, Nicking activity on pBR322 DNA of ribosome inactivating
proteins from Phytolacca dioica L. leaves, Biol. Chem. 386 (2005) 307e317.
[25] A. Parente, B. Merrifield, G. Geraci, G. D’Alessio, Molecular basis of
superreactivity of cysteine residues 31 and 32 of seminal ribonuclease,
Biochemistry 24 (1985) 1098e1104.
[26] A. Di Maro, P. Ferranti, M. Mastronicola, L. Polito, A. Bolognesi,
F. Stirpe, A. Malorni, A. Parente, Reliable sequence determination of
ribosome- inactivating proteins by combining electrospray mass spec-
trometry and Edman degradation, J. Mass Spectrom. 36 (2001) 38e46.
[27] E. Gross, The cyanogen bromide reaction, Methods Enzymol. 11 (1967)
238e257.
[28] A. Chambery, A. de Donato, A. Bolognesi, L. Polito, F. Stirpe, A. Parente,
Sequence determination of lychnin, a type 1 ribosome-inactivating protein
from Lychnis chalcedonica seeds, Biol. Chem. 387 (2006) 1261e1266.
[29] R.H. Bateman, R. Carruthers, J.B. Hoyes, C. Jones, J.I. Langridge,
A. Millar, J.P. Vissers, A novel precursor ion discovery method on
a hybrid quadrupole orthogonal acceleration time-of-flight (Q-TOF) mass
spectrometer for studying protein phosphorylation, J. Am. Soc. Mass
Spectrom. 13 (2002) 792e803.
[30] A. Chambery, V. Severino, A. D’Aniello, A. Parente, Precursor ion
discovery on a hybrid quadrupole-time-of-flight mass spectrometer for
gonadotropin-releasing hormone detection in complex biological
mixtures, Anal. Biochem. 374 (2008) 335e345.
[31] A. Chambery, A. Di Maro, A. Parente, Primary structure and glycan
moiety characterization of PD-Ss, type 1 ribosome-inactivating proteins
from Phytolacca dioica L. seeds, by precursor ion discovery on a Q-TOF
mass spectrometer, Phytochemistry 69 (2008) 1973e1982.
[32] A. Chambery, M. Pisante, A. Di Maro, E. Di Zazzo, S. Costantini,
G. Colonna, A. Parente, Invariant Ser211 is involved in the catalysis of
PD-L4, type I RIP from Phytolacca dioica leaves, Proteins Struct. Funct.
Bioinform 67 (2007) 209e218.
[33] A. Chambery, M. Pisante, A. Di Maro, E. Di Zazzo, M. Ruvo, S. Costantini,
G. Colonna, A. Parente, Invariant Ser211 is involved in the catalysis of PD-
L4, type I RIP from Phytolacca dioica leaves, Proteins 67 (2007) 209e218.
[34] S. Costantini, M. Rossi, G. Colonna, A.M. Facchiano, Modelling of
HLA-DQ2 and its interaction with gluten peptides to explain molecular
recognition in celiac disease, J. Mol. Graph. Model 23 (2005) 419e431.
[35] A.M. Facchiano, S. Costantini, A. Di Maro, D. Panichi, A. Chambery,
A. Parente, S. Di Gennaro, E. Poerio, Modeling the 3D structure of wheat
subtilisin/chymotrypsin inhibitor (WSCI). Probing the reactive site with
two susceptible proteinases by time-course analysis and molecular
dynamics simulations, Biol. Chem. 387 (2006) 931e940.
[36] R. Dosi, A. Di Maro, A. Chambery, G. Colonna, S. Costantini, G. Geraci,
A. Parente, Characterization and kinetics studies of water buffalo
(Bubalus bubalis) myoglobin, Comp. Biochem. Physiol. B Biochem.
Mol. Biol. 145 (2006) 230e238.
[37] S.F. Altschul, T.L. Madden, A.A. Schaffer, J. Zhang, Z. Zhang,
W. Miller, D.J. Lipman, Gapped BLAST and PSI-BLAST: a new
generation of protein database search programs, Nucleic Acids Res. 25
(1997) 3389e3402.
[38] E. Honjo, D. Dong, H. Motoshima, K. Watanabe, Genomic clones
encoding two isoforms of pokeweed antiviral protein in seeds (PAP-S1

363A. Di Maro et al. / Biochimie 91 (2009) 352e363
and S2) and the N-glycosidase activities of their recombinant proteins on
ribosomes and DNA in comparison with other isoforms, J. Biochem,
(Tokyo) 131, 2002, 225-231.
[39] R.A. Laskowski, M.W. MacArthur, D.S. Moss, J.M. Thornton, PRO-
CHECK: a program to check the stereochemical quality of protein
structures, J. Appl. Cryst. 26 (1993) 283e291.
[40] M.J. Sippl, Recognition of errors in three-dimensional structures of
proteins, Proteins 17 (1993) 355e362.
[41] A.A. Canutescu, A.A. Shelenkov, R.L. Dunbrack Jr., A graph-theory
algorithm for rapid protein side-chain prediction, Protein Sci. 12 (2003)
2001e2014.
[42] R.L. Dunbrack Jr., Rotamer libraries in the 21st century, Curr. Opin.
Struct. Biol. 12 (2002) 431e440.
[43] V. Munoz, L. Serrano, Elucidating the folding problem of helical
peptides using empirical parameters. II. Helix macrodipole effects and
rational modification of the helical content of natural peptides, J. Mol.
Biol. 245 (1995) 275e296.
[44] A.M. Facchiano, G. Colonna, R. Ragone, Helix stabilizing factors and
stabilization of thermophilic proteins: an X-ray based study, Protein Eng
11 (1998) 753e760.
[45] W. Kabsch, C. Sander, Dictionary of protein secondary structure: pattern
recognition of hydrogen-bonded and geometrical features, Biopolymers
22 (1983) 2577e2637.
[46] C.A. Orengo, A.D. Michie, S. Jones, D.T. Jones, M.B. Swindells,
J.M. Thornton, CATH: a hierarchic classification of protein domain
structures, Structure 5 (1997) 1093e1108.
[47] A. Ruggiero, A. Chambery, A. Di Maro, A. Parente, R. Berisio, Atomic
resolution (1.1 A) structure of the ribosome-inactivating protein PD-L4
from Phytolacca dioica L. leaves, Proteins 71 (2007) 8e15.
[48] A. Paladino, S. Costantini, G. Colonna, A.M. Facchiano, Molecular
modelling of miraculin: structural analyses and functional hypotheses,
Biochem. Biophys. Res. Commun. 367 (2008) 26e32.
[49] S. Jones, J.M. Thornton, Principles of protein-protein interactions, Proc.
Natl. Acad. Sci. USA 93 (1996) 13e20.
[50] S.J. Hubbard, S.F. Campbell, J.M. Thornton, Molecular recognition.
Conformational analysis of limited proteolytic sites and serine proteinase
protein inhibitors, J. Mol. Biol. 220 (1991) 507e530.
[51] I.K. McDonald, J.M. Thornton, Satisfying hydrogen bonding potential in
proteins, J. Mol. Biol. 238 (1994) 777e793.
[52] L. Barbieri, L. Polito, A. Bolognesi, M. Ciani, E. Pelosi, V. Farini,
A.K. Jha, N. Sharma, J.M. Vivanco, A. Chambery, A. Parente, F. Stirpe,
Ribosome-inactivating proteins in edible plants and purification and
characterization of a new ribosome-inactivating protein from Cucurbita
moschata, Biochim. Biophys. Acta 1760 (2006) 783e792.
[53] A. Sali, T.L. Blundell, Comparative protein modelling by satisfaction of
spatial restraints, J. Mol. Biol. 234 (1993) 779e815.
[54] B. Wallner, A. Elofsson, All are not equal: a benchmark of different
homology modeling programs, Protein Sci. 14 (2005) 1315e1327.
[55] M. Tress, I. Ezkurdia, O. Grana, G. Lopez, A. Valencia, Assessment of
predictions submitted for the CASP6 comparative modeling category,
Proteins 61 (Suppl. 7) (2005) 27e45.
[56] Y.-R. Yuan, Y.-N. He, J.-P. Xiong, Z.-X. Xia, Three-dimensional struc-
ture of b-momorcharin at 2.55 A resolution, Acta Crystallogr D55 (1999)
1144e1151.
[57] A. Ceriotti, M. Duranti, R. Bollini, Effects of N-glycosylation on the
folding and structure of plant proteins, J. Exp. Bot. 49 (1998)
1091e1103.
[58] A.D. Elbein, The role of N-linked oligosaccharides in glycoprotein
function, Trends Biotechnol. 9 (1991) 346e352.
[59] H. Lis, N. Sharon, Protein glycosylation. Structural and functional
aspects, Eur. J. Biochem. 218 (1993) 1e27.
[60] N. Sharon, H. Lis, Carbohydrates in cell recognition, Sci. Am. 268
(1993) 82e89.
[61] A. Varki, Biological roles of oligosaccharides: all of the theories are
correct, Glycobiology 3 (1993) 97e130.
[62] S.E. O’Connor, B. Imperiali, Modulation of protein structure and
function by asparagine-linked glycosylation, Chem. Biol. 3 (1996)
803e812.
[63] D.F. Wyss, G. Wagner, The structural role of sugars in glycoproteins,
Curr. Opin. Biotechnol. 7 (1996) 409e416.
[64] F. Del Vecchio Blanco, A. Bolognesi, A. Malorni, M.J. Sande, G. Savino,
A. Parente, Complete amino-acid sequence of PD-S2, a new ribosome-
inactivating protein from seeds of Phytolacca dioica L, Biochim. Bio-
phys. Acta 1338 (1997) 137e144.




















