
Synaptosomal Na, K-ATPase during forebrain ischemia in Mongolian gerbils
Upload
independentCategory
view
0download
0
SiN
CH*2
R
ob(1vtrueer(stacbpcrnrkmpotw
t
8
0CA
Archives of Biochemistry and BiophysicsVol. 366, No. 2, June 15, pp. 215–223, 1999Article ID abbi.1999.1198, available online at http://www.idealibrary.com on
timulation of Ouabain Binding to Na,K-ATPasen 40% Dimethyl Sulfoxide by a Factor from
a,K-ATPase Preparations
arlos F. L. Fontes,* Fabio E. Veiga Lopes,* Helena M. Scofano,*ector Barrabin,*,1 and Jens G. Nørby†
Departamento de Bioquımica, ICB, CCS, Bloco H2, Universidade Federal do Rio de Janeiro, Cidade Universitaria,1941-590 Rio de Janeiro, Brasil; and †Department of Biophysics, University of Aarhus, DK-8000 Århus C, Denmark
eceived June 29, 1998, and in revised form March 10, 1999
PkwsMharsrecm
ssb4fmowwplmf(fAsg
In 40% dimethyl sulfoxide (Me2SO) high-affinityuabain (O) binding to Na,K-ATPase (E) is promotedy Mg21 in the absence of inorganic phosphate (Pi)Fontes et al., Biochim. Biophys. Acta 1104, 215–225,995). Furthermore, in Me2SO the EO complex reactsery slowly with Pi and this ouabain binding canherefore be measured by the degree of inhibition ofapid phosphoenzyme formation. Here we found that,nexpectedly, the ouabain binding decreased with thenzyme concentration in the Me2SO assay medium. Wextracted the enzyme preparation with Me2SO or chlo-oform/methanol and demonstrated that the extracteddepleted) enzyme bound ouabain poorly. Addition ofuch extracts to assays with low enzyme concentra-ion or depleted enzyme fully restored the high-ffinity ouabain binding. Dialysis experiments indi-ated that the active principle had a molecular massetween 3.5 and 12 kDa. It was highly resistant toroteolysis. It was suggested that the active principleould either be a low-molecular-weight, proteolysis-esistant-peptide (e.g., a proteolipid) or a factor with aonproteinaceous nature. A polyclonal antibodyaised against the C-terminal 10 amino acids of the ratidney g-subunit was able to recognize this low-olecular-weight peptide present in the extracts. The
reviously depleted enzyme displayed lower amountsf the g-proteolipid in comparison to the native un-reated enzyme, as demonstrated by immunoreactionith the antibody. © 1999 Academic Press
Key Words: ouabain binding; dimethyl sulfoxide; pro-eolipid; ATPase; phosphorylation.
1
tTo whom correspondence should be addressed. Fax: 155 21 270
647. E-mail: [email protected].
003-9861/99 $30.00opyright © 1999 by Academic Pressll rights of reproduction in any form reserved.
It is firmly established that the cation transport AT-ase, Na,K-ATPase (EC 3.6.1.37), consists of a 110-Da subunit (a) and a glycosylated 56-kDa subunit (b)ith a protein mass equal to 35 kDa (see reviews on the
tructure of Na,K-ATPase by Lingrel et al. (1) andercer (2)). The a-subunit has long been known to
ave binding sites for both the physiological ligandsnd for the specific inhibitor ouabain, whereas, untilecently, the b-subunit was considered to be exclu-ively of structural importance and critical for the cor-ect expression of the enzyme (2). New evidence, how-ver, suggests that b also plays a functional role inertain intermediary steps of the reaction and pumpingechanism (3).A third subunit of unknown function named g, a
mall proteolipid with a molecular mass of 6.5 kDa (inome studies up to 12 kDa), has been demonstrated toe closely associated with the a- and b-subunits (2,–11). In evaluating the importance of the g-subunit
or the Na,K-pump it is of interest that several low-olecular-mass proteolipids have been associated with
ther ATPases. The best known (and the only one forhich its function is described) is phospholamban,hich plays a role as a regulator of the cardiac sarco-lasmic reticulum Ca-ATPase (12, 13). Other proteo-ipids in this category are purified from the sarcoplas-
ic reticulum of skeletal muscle (sarcolipin (14, 15)) orrom cardiac sarcolemmal vesicles (phospholemman16)) or they are found in connection with H-ATPasesrom different yeasts (17–19) or with bacterial Na-TPase (20). Of these, phospholemman has been
hown to have a certain sequence similarity with the-subunit of Na,K-ATPase (10, 16).The present investigation provides evidence for a func-
ional role of a low-molecular-weight factor in Na,K-AT-
215

Pobppepoadtatbtfaapmriga
E
utszab
R
tp
vw1sf
w7ftci
ps
Tp2Amfiafim
sat
OwtmwmSthpod
awraACoswrsuab
tawM
R
iMwtrstdmpcPMp
s
216 FONTES ET AL.
ase in the ouabain-binding process. The topology of theuabain-binding site is very complex (1, 11, 21). Theinding depends on the structural integrity of severalarts of the Na,KATPase assembly (21–24), and the exactosition and nature of the residues and subunits of thenzyme involved (4, 6, 16) is not solved. Our study wasrimarily based on the observation that the binding ofuabain to Na,K-ATPase in a medium of decreased waterctivity (40% (v/v) Me2SO) showed an unexpected depen-ence on the enzyme concentration: It decreased whenhe concentration of the Na,K-ATPase preparation in thessay was lowered. In this article we demonstrate thathe probable cause for this phenomenon is the extractiony Me2SO of a ouabain-binding-promoting factor fromhe Na,K-ATPase preparation. Enzyme depleted of thisactor binds ouabain with a significantly decreased ratend affinity, a deficiency which is reversed by addition ofMe2SO or chloroform/methanol extract of the enzyme
reparation. The factor present in these extracts has aolecular mass between 3.5 and 12 kDa and is highly
esistant to proteolysis. Also, a small peptide components recognized by the polyclonal antibody gC33 as being the-subunit of the pig kidney medulla Na,K-ATPase prep-ration.
XPERIMENTAL PROCEDURES
Enzyme preparations. Na,K-ATPase with a Vmax at 37°C of 15–25nits.mg protein21 was prepared from pig kidney outer medulla usinghe procedures of Jørgensen (25) as modified by Jensen et al. (26). Thetock preparations were stored at 280°C in a buffer (17.6 mM imida-ole, 0.625 mM EDTA, 250 mM sucrose, titrated to pH 7.4 with HCl)nd at a protein concentration of 4–5 mg.ml21. Protein was determinedy the method of Lowry (27) with a standard of bovine serum albumin.Antibody. gC33 polyclonal antibody was a kind donation of Dr.hoda Blostein.Preparation of extracts containing ouabain-binding-promoting fac-
or (OBPF).2 Extracts from the Na,K-ATPase preparations wererepared by two procedures:
(A) Enzyme stock solution was mixed at room temperature with 2ol of the Me2SO “standard medium” (see below), allowed to standith occasional shaking for 5 min, and then centrifuged for 10 min at48,000g to pellet the protein-containing membrane fragments. Theupernatant contained an ouabain-binding-promoting factor hence-orth called “OBPF-dmso.”
(B) Enzyme stock (250 ml) was diluted 16 times at room temperatureith a mixture (volume %) of 46% methanol, 46% chloroform, and 8%50 mM NH4HCO3 (5, 19), allowed to stand for 5 min, and then centri-uged for 3 min at 570g. The homogeneous supernatant, which con-ained a ouabain-binding-promoting factor that we here name “OBPF-hme,” was dried in a stream of N2, and the dry matter was resuspendedn 300 ml of the Me2SO standard medium (see below).
Ouabain inhibition of the rapid phosphorylation by inorganic phos-hate, Pi. The Me2SO standard medium for these experiments con-isted of 40% Me2SO (v/v), 5 mM MgCl2, 0.5 mM KCl, and 5 mM
2 Abbreviations used: OBPF, oubain-binding-promoting factor; EP,
fhosphoenzyme; PVDF, polyvinylidene fluoride; PS, phosphatidyl-erine; PC, phosphatidylcholine; SBT1, soya bean trypsin inhibitor.
ris–HCl, pH 7.0, temperature was 27°C. When ouabain wasresent, the enzyme was preincubated in the standard medium with0 mM ouabain for 5 min and then 20 mM radioactive 32Pi was added.fter 1 min, the amount of acid-stable phosphoenzyme, EP, waseasured by precipitation with a mixture of HClO4, Pi, and PPi andltering as described (28). The concentrations of enzyme, ouabain,nd 32Pi (when different from those just mentioned) are given in thegures. The results shown are generally the average of four experi-ents (SD is less than 0.1 nmol.mg21).Ouabain binding by incubation with radioactive ouabain. Mea-
urements of ouabain binding, including determinations of blanksnd unspecific radioactivity in the [3H]ouabain, were performed inhe Me2SO standard medium (no Pi) as described (29).
Proteolysis experiments with the ouabain-binding-promoting factorBPF-dmso. Several experiments with three different proteasesere performed, the protocol being dependent on the specific pro-
ease used. The OBPF-dmso was treated at 37°C in standard Me2SOedium, pH 7.4, for 1 h with trypsin (100 mg.ml21 of extract) or 24 hith chymotrypsin (50 mg.ml21 of extract) or proteinase-K (50g.ml21 of extract). The reactions were stopped with a 20-fold excessBTI for trypsin or 30 mM phenylmethylsulfonyl fluoride for chymo-rypsin. In the specific case of proteinase-K the long-term assay (24) leads to autodigestion of the proteolytic enzyme as observed in aarallel control. The effect of the treatment was tested by the abilityf the samples to stimulate ouabain inhibition of EP formation asescribed above.Western blot analysis of OBPF-chme using a g-subunit-specific
ntibody. OBPF-chme and native and previously depleted enzymeere submitted to a Tricine-based SDS–PAGE using a 16.5% sepa-
ating gel and a 4% stacking gel as described in (30). The proteinmount in each lane of the electrophoresis was equalized to 6 mg/slot.fter the run, the gel was spliced: one-half was stained by colloidaloomassie blue and the other half was submitted to electroblottingn a semidry (Bio-Rad) apparatus, using PVDF membranes as de-cribed in (31). The PVDF membranes were incubated for 1 h at 37°Cith an 1:5000 dilution of gC33 antiserum. This rabbit antiserum was
aised against the C-terminal 10 amino acids of the rat gammaubunit and it is able to detect the proteolipid in 0.5 mg/lane ofnpurified kidney microsomes of rat, pig, dog, or mouse. The specificntibody incorporation was revealed using an ECL-direct, anti-rab-it, luminescence kit using a procedure slightly modified from (32).Chemicals. The chemicals were analytical grade. 32Pi was ob-
ained from the Brazilian Institute of Atomic Energy and purifiedccording to Kessler et al. (33), ouabain was from Sigma, [3H]ouabainas purchased from NEN, and Me2SO (spectroscopic grade) fromerck.
ESULTS
In an aqueous milieu ouabain interacts slowly andncompletely with Na,K-ATPase in the presence of
g21 alone, whereas in the presence of Mg21 1 Pi,here the phosphoenzyme intermediate E2P is formed,
he system has a high affinity for ouabain and theeaction with ouabain is fast (34). However, as demon-trated by us previously (29), in a 40% Me2SO mediumhe interaction of Na,K-ATPase with ouabain and Pi isistinctly different from what happens in an aqueousilieu: Ouabain reacts rapidly with the enzyme in the
resence of Mg21 alone to form the enzyme–ouabainomplex, EO. The free enzyme also reacts rapidly withi to form EP, but EO is only slowly phosphorylated byg21 1 P and EP is only slowly ligated with ouabain to
iorm EOP.
eibftPceo
D
tiodtWoicpseibfli
tddbsccp
Fwmoatt
FEPsdPw
FOvtp
217STIMULATION OF OUABAIN BINDING TO Na,K-ATPase
Therefore, in 40% Me2SO ouabain will bind to thenzyme during incubation of the enzyme with ouabainn the absence of Pi, and the extent of this ouabaininding can be measured as an inhibition of the EPormation during the subsequent rapid phosphoryla-ion experiment by Pi. It has already been shown that
i binds with high affinity to the enzyme in a Me2SO-ontaining medium (28, 29, 35). These properties arexploited for developing a faster method to estimateuabain binding (29) and used in the following.
emonstration of Ouabain-Binding-Promoting Factorin Na,K-ATPase Preparations: PhosphorylationExperiments
The present study was initiated by the observationhat the extent of inhibition of phosphorylation by pre-ncubation with ouabain (indicative of the extent ofuabain-binding, see above) was significantly depen-ent on the concentration of the enzyme preparation inhe standard Me2SO preincubation medium (Fig. 1).
ith about 12 mg enzyme.ml21 there was no effect ofuabain on the inhibition of the phosphorylation by Pi,.e., no ouabain binding, whereas with higher enzymeoncentrations ouabain would bind and inhibit thehosphorylation of up to 60% of the enzyme. One pos-ible explanation for this unexpected finding is that thenzyme preparation contains a factor, which is solublen the reaction medium and which promotes ouabaininding. With the very low enzyme concentrations, theactor is efficiently extracted from the enzyme and thiseaves an insufficient amount to achieve optimal bind-
IG. 1. The dependence on enzyme concentration of the reactionith ouabain. The enzyme was preincubated in the standard Me2SOedium (which has 5 mM MgCl2 and no phosphate) with 20 mM
uabain for 5 min and then 10 mM 32Pi was added. After 1 min, themount of phosphoenzyme, EP, was determined. Symbols representwo different enzyme preparations. For other details, see Experimen-al Procedures.
ng of ouabain under the circumstances used.pv
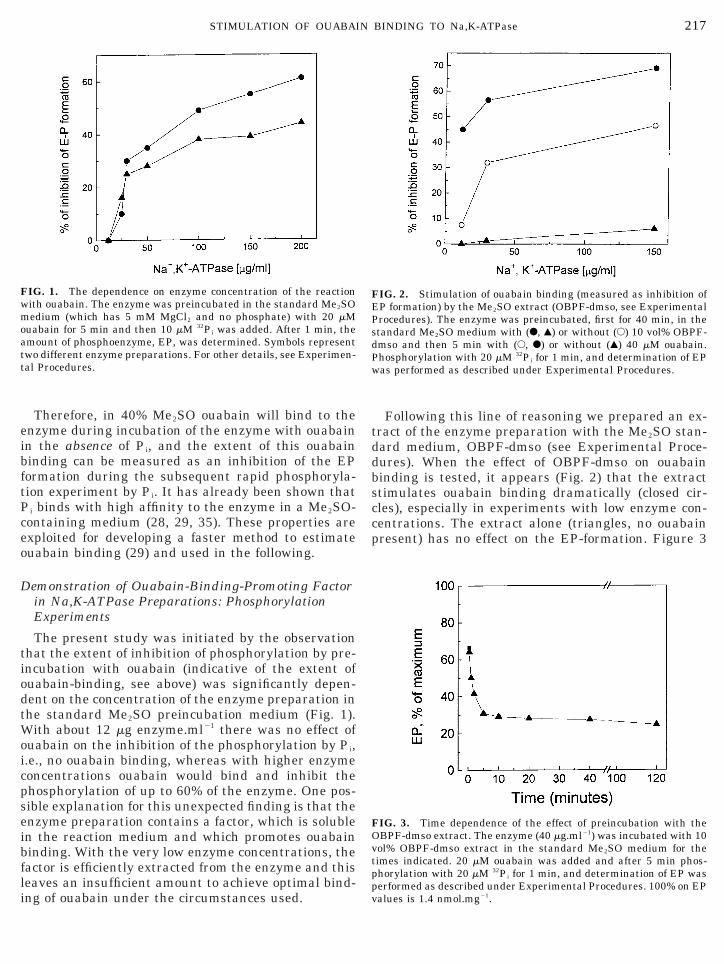
Following this line of reasoning we prepared an ex-ract of the enzyme preparation with the Me2SO stan-ard medium, OBPF-dmso (see Experimental Proce-ures). When the effect of OBPF-dmso on ouabaininding is tested, it appears (Fig. 2) that the extracttimulates ouabain binding dramatically (closed cir-les), especially in experiments with low enzyme con-entrations. The extract alone (triangles, no ouabainresent) has no effect on the EP-formation. Figure 3
IG. 2. Stimulation of ouabain binding (measured as inhibition ofP formation) by the Me2SO extract (OBPF-dmso, see Experimentalrocedures). The enzyme was preincubated, first for 40 min, in thetandard Me2SO medium with (F, Œ) or without (E) 10 vol% OBPF-mso and then 5 min with (E, F) or without (Œ) 40 mM ouabain.hosphorylation with 20 mM 32Pi for 1 min, and determination of EPas performed as described under Experimental Procedures.
IG. 3. Time dependence of the effect of preincubation with theBPF-dmso extract. The enzyme (40 mg.ml21) was incubated with 10ol% OBPF-dmso extract in the standard Me2SO medium for theimes indicated. 20 mM ouabain was added and after 5 min phos-horylation with 20 mM 32Pi for 1 min, and determination of EP was
erformed as described under Experimental Procedures. 100% on EPalues is 1.4 nmol.mg21.
soetn
et5Mmtm
1dpwfc
3
cptwrzoctibcd
D
ht
Fs(vc
FOmmmwp
FadOiftmt
218 FONTES ET AL.
hows that the extract reacts with the enzyme with a t½
f a few minutes, and Fig. 4 indicates that it has noffect on the affinity of the enzyme for Pi, corroboratinghe observation on Fig. 2 that the extract as such didot affect EP formation.An active factor was also prepared by extraction of the
nzyme preparation with a chloroform–methanol mix-ure (OBPF-chme; see Experimental Procedures). In Fig.
it is seen that this extract, like the one obtained ine2SO, reacts with the enzyme in a dose-dependentanner. Half-maximal effect in these experiments is ob-
ained with addition of about 30 ml extract to 500 ml ofedium with 40 mg enzyme protein.ml21 (OBPF-chme) or
IG. 4. Formation of EP from Pi during 1 min phosphorylation intandard Me2SO-medium (in the absence of ouabain). The enzyme40 mg.ml21) was preincubated for 40 min with (F) or without (E) 10ol% OBPF-chme extract. For further details, see Experimental Pro-edures.
IG. 5. Dose–response curve for the effect of the OBPF-dmso andBPF-chme extracts. The enzyme (12 mg.ml21 in OBPF-dmso and 40g.ml21 in OBPF-chme) was preincubated in the standard Me2SOedium (0.5 ml) with different concentrations of the extracts for 20in followed by addition of 20 mM ouabain. After 5 min, 20 mM 32Pi
oas added and phosphorylation performed as described under Ex-erimental Procedures.
2 mg enzyme protein.ml21 (OBPF-dmso). The OBPF-mso and OBPF-chme extracts also are both resistant toroteases, and in electrophoresis both have one main,ide band at the bottom of the gels (less than 10 kDa) as
ound by Navarre et al. (19) for the g-peptide from Sac-haromyces cerevisiae.
.2. Depletion of the Enzyme Preparation of theOuabain Binding Promoting Factor
Since the extracts appear to contain an active prin-iple for ouabain binding, it should be possible to de-lete the enzyme of this principle by repeated extrac-ions with the Me2SO standard medium (extractionith the chloroform-methanol mixture denatures the
emaining enzyme). From Fig. 6 it is obvious that en-yme thus depleted (triangles) reacts poorly withuabain and that addition of extract (OBPF-chme,losed circles) to depleted enzyme restores (or morehan restores at low enzyme concentrations) the capac-ty to bind ouabain. The occurrence of some ouabaininding at high protein concentrations (depleted curve)ould be an indication that the enzyme is not totallyepleted of the factor.
emonstration of the Ouabain-Binding-PromotingFactor: Ouabain-Binding Experiments
In the previous experiments (Figs. 1, 2, and 6) weave demonstrated that when the protein concentra-ion was low, e.g., 12 mg.ml21, incubation with 20 mM
IG. 6. Ouabain binding (measured as inhibition of EP formation)s a function of protein concentration for native enzyme (E), enzymeepleted of the OBPF-dmso (�), and depleted enzyme repleted withBPF-dmso (F). The enzyme was depleted of OBPF-dmso by dilution
n the standard Me2SO medium to 12 mg protein.ml21 and centri-uged to pellet the protein (28,000g for 30 min). After three suchreatments, the protein was resuspended in the standard Me2SOedium to 1.2–1.4 mg.ml21 and used for the experiments. For de-
ails, see Experimental Procedures.
uabain for 5 min resulted in no or very low ouabain

bpte7met
itatced1oMdeedtfea(t
P
l
wm(wip
FTme e wu
FTmotmec
219STIMULATION OF OUABAIN BINDING TO Na,K-ATPase
inding. With higher enzyme concentrations or in ex-eriments where OBPF was added, the 5-min incuba-ion with ouabain led to binding to 40–50% of thenzyme. From the ouabain-binding experiments in Fig.it is apparent that the relatively low binding after 5in is mainly due to a low rate of binding. Higher
nzyme concentrations or addition of OBPF increaseshe rate about 10 times.
We measured a possible difference in ouabain-bind-ng constants between native enzyme and enzyme ex-racted with Me2SO (depleted enzyme) at equilibriumnd low protein concentration (Figs. 8 and 9). Sinceheir rate of binding was low, it was necessary to in-ubate for up to 18 h. to reach equilibrium. The nativenzyme (Fig. 8) had one population of sites with aissociation constant of 59 6 6 nM, which is close to the12 nM determined earlier under similar conditions inur laboratory (29). When the enzyme is extracted withe2SO to deplete it of OBPF (see Experimental Proce-
ures), the affinity for ouabain of a fraction of thenzyme decreases by a factor of 100 (Fig. 9). The pres-nce in the depleted enzyme of two components withifferent ouabain affinities may suggest that the ex-raction of OBPF was not complete, as already inferredrom Fig. 6. The addition of OBPF to the depletednzyme was able to increase the apparent ouabainffinity of the low-affinity component by a factor of 10Fig. 9). These results, together with Fig. 7, indicatehat the addition of OBPF improved ouabain binding.
roperties of the Ouabain-Binding-Promoting Factor:Dialysis and Proteolysis
In an attempt to achieve an estimate of the molecu-
IG. 7. [3H]Ouabain binding to Na,K-ATPase in 40% Me2SO as a fuhe enzyme was incubated in the Me2SO standard medium with 5 measured as described (29). (A) Experiments performed with enzyme
nzyme concentration was 12 mg.ml21, but in one series (F) the enzymnder the binding assay.
ar mass of OBPF-dmso a series of dialysis experiments0p
as performed. Membranes permitting molecules with, 3.5 kDa or m , 12-14 kDa, respectively, were used
Table I). The activity of the OBPF-containing extractsas tested by their ability to increase the ouabain
nhibition of EP formation as described above. It ap-ears from the table that freshly prepared extract (row
tion of time and enzyme concentration and the effect of OBPF-dmso.[3H]ouabain for the times shown and the amount of bound ouabaincentrations of 12 (E) or 120 mg.ml21 (F). (B) In both experiments theas incubated with 50 ml.ml21 of OBPF-dmso for 40 min prior to and
IG. 8. [3H]Ouabain binding isotherms of native Na,K-ATPase.he enzyme, 12 mg.ml21, was incubated in the standard Me2SOedium at room temperature with [3H]ouabain for up to 18 h to
btain equilibrium. Bound ouabain was determined by a filteringechnique as described (29). Free ouabain was calculated as totalinus bound. The different symbols represent three independent
xperiments. The continuous lines are the nonlinear regressionurves, assuming one binding site per enzyme: Bmax 5 0.79, 0,73, and
21
ncMcon
.73 nmol.mg and Kdiss 5 62, 52, and 62 nM. (Inset) The Scatchardlots of the binding data.

2(ot1mm
mctrt
pOrpt
E
l
Fwb[dw(efoCblwA55B0
(((
(
dda1frm
(((
(
uptew
220 FONTES ET AL.
) or extract that had been standing for 24 h in the coldrow 3) was equally active in promoting the effect ofuabain. The active factor was apparently retained byhe 3.5-kDa cutoff membranes but not by the 12- to4-kDa cut-off membranes (row 4), suggesting that theolecular mass of the active principle, m, is 3.5 kDa ,, 12–14 kDa.An extensive number of proteolysis experiments (forore details, see Experimental Procedures) were also
arried out to identify the possible involvement of a pro-ein or peptide in the promotion of ouabain binding. Theesults given in Table II clearly indicate that neither the
IG. 9. [3H]Ouabain binding isotherms of depleted Na,K-ATPaseith or without OBPF. The depleted enzyme, 12 mg.ml21, was incu-ated in the standard Me2SO medium at room temperature with
3H]ouabain for up to 18 h to obtain equilibrium. Bound ouabain wasetermined by a filtering technique as described (29). Free ouabainas calculated as total minus bound. (A) The direct binding curves.
B) The Scatchard plots. The open symbols represent three differentxperiments with depleted enzyme; the closed symbols are two dif-erent experiments with depleted enzyme incubated with 50 ml.ml21
f OBPF-chme for 40 min prior to and during the binding assay.ontinuous lines are nonlinear fitting curves, with two independentinding sites, obtained for each individual experiment. The dottedine represents the mean curve for the native enzyme (see Fig. 8)ith Bmax 5 0.75 6 0.04 nmol.mg21 and Kdiss 5 59 6 6 nM. DepletedTPase (open symbols): Bmax1 5 0.47, 0.56, and 0.19 nmol.mg21, Kdiss1
7.5, 5.8, and 7.4 mM; Bmax2 5 0.31, 0.22, and 0.43 nmol.mg21, Kdiss2
96, 69, and 140 nM. Depleted ATPase plus OBPF (closed symbols):max1 5 0.61 and 0.52 nmol.mg21, Kdiss1 5 0.56 and 1.15 mM, Bmax2 5.20 and 0.42 nmol.mg21 and Kdiss2 5 11 or 16 nM.
reatment with trypsin nor those with chymotrypsin orai
roteinase-K were able to destroy the active principle inBPF-dmso (compare row 4 with rows 2 and 3). This
esistance to proteolysis seems to indicate either a non-roteinaceous nature or the presence of a protein withhe characteristics of a small proteolipid (9).
ffect of Phospholipids on OBPF-chme Activation ofOuabain Inhibition of Phosphorylation by Pi
To test the possibility of the OBPF being a phospho-ipid that could be extracted by the procedures de-
TABLE I
Range of Molecular Mass of the Ouabain-Promoting Factor(OBPF-dmso) as Determined by Dialysis
Conditions% inhibition ofE-P formation
1) 40 mM ouabain 40.5 6 2.12) ouabain 1 extract (freshly prepared) 70 6 0.33) ouabain 1 extract (after 24 h) 72.5 6 1.6
3.5-kDamembrane
12- to 14-kDamembrane
4) ouabain 1 24 hdialyzed extract 67 6 0.8 37.3 6 2.0
Note. A sample of the OBPF-dmso was dialyzed against the stan-ard Me2SO medium for 24 h in a cold room, and the remains in theialysis bag were tested for OBPF activity (with 40 mg enzyme ml21)s described in the text. The dialysis membranes were 3.5-kDa and2- to 14-kDa “cutoffs” from Spectrapore. The results are averages ofour experiments 6 SE. Control tests with Dextran Blue and methyled showed that the membranes were not disrupted by the Me2SOedium used.
TABLE II
Resistance of the Ouabain-Promoting Factor(OBPF-dmso) to Proteolysis
Conditions% inhibition ofEP formation
1) 40 mM ouabain 40 6 0.72) ouabain 1 extract (freshly prepared) 71.3 6 1.03) ouabain 1 control extract
(after 24 h at 37°C) 72.6 6 0.6
Trypsin Chymotrypsin Proteinase-K
4) ouabain 1 digestedextract at 37°C 67 6 0.9 78 6 2.0 69 6 2.0
Note. The proteolysis experiments were performed as describednder Experimental Procedures, and the OBPF-activity of the sam-les was then tested by its ability to stimulate the ouabain inhibi-ion of EP formation (see text). The results are averages of sixxperiments 6 SE with each proteolytic enzyme. Controls performedith corresponding inhibited proteolysis medium (SBTI, PMSF, or
utodigestion treatment) in the absence of ouabain displayed nonhibition.
sttptdiwpf
I
stebc[cuMt
D
btiNephterMPcbpc
NPP
wsasbtfrimi
FFlsc
221STIMULATION OF OUABAIN BINDING TO Na,K-ATPase
cribed above, we performed a series of phosphoryla-ion by Pi measurements with addition of Me2SO ex-racts of a mixture of phosphatidylserine (PS) andhosphatidylcholine (PC) (Table III). The preincuba-ion of the native diluted enzyme (24 mg/ml), withifferent amounts of PS 1 PC extract, was not able toncrease the OBPF effects. The phospholipids testedere also unable to increase the ouabain inhibition ofhosphorylation by Pi and did not affected the EPormation (controls).
TABLE III
Effect of Phospholipids on Ouabain Inhibitionof Phosphorylation by Pi
No ouabainPlus 10 mM
ouabain
Plus 10 mMouabain andOBPF-CM
o PL added 0.68 6 0.07 0.44 6 0.02 (35) 0.27 6 0.03 (60)lus 5 mg PL 0.65 6 0.02 (4) 0.50 6 0.01 (27) 0.28 6 0.06 (59)lus 20 mg PL 0.65 6 0.02 (4) 0.43 6 0.04 (36) 0.29 6 0.04 (57)
Note. An aliquot containing 130 mg of PS 1 PC in equal proportionsas dried, ressuspended in 300 ml of standard Me2SO medium, and
onified in bath for 1 h. After addition of the phospholipids (in themounts indicated) to native enzyme (12 mg), the suspension wasonified for 20 min. Then the enzyme suspensions were preincubatedy extra 20 min with or without OBPF-chme (10% final vol) prior tohe addition of ouabain. All points with ouabain were preincubatedor 5 more min before the addition of Pi to start the phosphorylationeaction. Phosphoenzyme was measured as described under Exper-mental Procedures. The results are averages of EP levels (mmols.
g21) of four experiments 6 SE; values in parenthesis are the % ofnhibition respective to the control EP (no ouabain and no PL added).
IG. 10. Tricine–SDS–PAGE and Western blot analysis of OBPF-chor details, see Experimental Procedures. Lane 1, molecular weight
ane 4, depleted enzyme (6 mg). (B) Western blotting. For details, s33
pecific polyclonal antibody (gC ). Lane 1, native enzyme (6 mg); laoncentration of the samples (except for OBPF) was determined by met
munnoidentification of g-Subunit on OBPF
Western blot analysis (Fig. 10) identified a singletrong band of immunoreaction of the g antibody withhe OBPF-chme and with the native enzyme, withoutxhibiting cross-reaction of the antibody with a-or-subunits. The results also show that the g-subunitontent was enriched during the extraction processcompare rows 2 and 3 (A) and 1 and 2 (B)]. The rowontaining the depleted enzyme displayed less g-sub-nit content than the native enzyme, indicating thate2SO was able to partially extract the g-subunit from
he Na,K-ATPase.
ISCUSSION
In the present study, ouabain binding was measuredoth by the ouabain inhibition of enzyme phosphoryla-ion from Pi and by measurement of [3H]ouabain bind-ng (Figs. 7–9) (29). The efficiency of ouabain binding toa,K-ATPase decreased significantly with decreasing
nzyme concentration under the conditions of the phos-horylation assay (Fig. 1). The experiments reportedere have made it likely that this is because a factorhat promotes ouabain binding is removed from thenzyme-containing particles when the enzyme prepa-ation is diluted in the standard Me2SO medium (40%e2SO (v/v)) used. First, an extract of the Na,K-AT-ase preparation with the standard Me2SO mediumontains a factor that increases the rate of ouabaininding (Figs. 2, 6, and 7), and second, enzyme de-leted of the factor by repeated Me2SO extractionslearly binds ouabain with a lower rate (Fig. 6) and
. (A) Colloidal Coomassie blue staining of Tricine-based SDS–PAGE.rkers; lane 2, native Na,K-ATPase (6 mg); lane 3, OBPF-chme; andExperimental Procedures. The blots were probed with a g-subunit
memaee
ne 2, OBPF-chme; lane 3, depleted enzyme (6 mg). The proteinhod of Lowry (27).

w((ose
mAmmrIefA
crgftbtsr
numadkatbt
tgcNnpsemttounotrr
pphracagrtlkh
HisM
A
aDetRsCEPRum
R
1
1
1
222 FONTES ET AL.
ith a much lower affinity than the native enzymeFig. 9). The effect of the OBPF is “dose” dependentFig. 5), but we have no means of assessing the stoichi-metry of the factor to, for example, the a-subunit,ince we do not know the concentration of OBPF in thextracts.The active factor can also be extracted by chloroform/ethanol as it is used for isolation of proteolipids fromTPases (4, 5, 19). An estimate of the molecular mass,, by dialysis experiments (Table I) yielded 3.5 kDa ,, 12–14 kDa, and the factor was found to be very
esistant to proteolysis by various proteases (Table II).t will appear from the following that OBPF has sev-ral properties in common with the small proteolipidsound both in Na,K-ATPase preparations and in otherTPase preparations.The proteolipids of Na,K-ATPase were isolated and
haracterized by Reeves et al. (5). A solubilized prepa-ation was fractionated on Sepharose and the so-called-fraction was lyophilized and extracted with chloro-orm/methanol with NH4HCO3. The molecular mass ofhese proteolipids was estimated by electrophoresis toe around 12 kDa. Collins and Lesyk (9) observed thathe g-subunit is very resistant to proteolysis by tryp-in, carboxypeptidase, pepsin, and Staphylococcus au-eus V8 protease.
Mercer et al. (10) have used molecular biology tech-iques to characterize the cDNA that codes the g-sub-nit. They showed that the g-subunit has a molecularass of 6.5 kDa with a pronounced homology in the
mino acid sequence among different species. It wasemonstrated that a and g colocalize in the sheepidney and that g is found only where there is a. Theylso showed, using photoaffinity ouabain derivatives,hat “only the polypeptides that correspond to the la-eled (photoaffinity ouabain derivative) g-subunit ofhe Na,K-ATPase are immunoreactive” to g-antisera.
One possibility is that the factor (OBPF) studied inhe present work, due to the similarities, may be the-subunit. In fact, the OBPF described in this paperontains the g-subunit; furthermore, the depleteda,K-ATPase preparation contains less g-subunit thanative enzyme, suggesting that the proteolipid maylay a role in the modulation of the ouabain-bindingite on the Na,K-ATPase. It is probable that the pres-nce of this g-subunit is not an indispensable require-ent for ouabain binding but that it could influence
he kinetics of ouabain binding as observed here. Con-rary to what has been found for the H-ATPase prote-lipid from Saccharomyces cerevisiae (17), the g-sub-nit is not essential for Na,K-ATPase activity (8) and isot a required subunit for reconstituting the pump (36)r for its expression in yeast (37, 38). At the present,here is no consensus about the functional propertieselated to g. Other interesting possibilities regarding
egulation of Na,K-ATPase have recently been pro-1
osed. Dowd et al. (39) found two low-molecular-weighteptides that were protein kinase A substrates in beefeart Na,K-ATPase preparations. Therien et al. (40)ecently suggested that differences in the Na,K pumpsffinity ratio KNa/KK between tissues and isoformsould be “the result of modulation of the enzyme bynother associated protein.” Recent work (41), sug-ested that the g-proteolipid can be a tissue-specificegulator of some Na,K-ATPase’s properties. The au-hors used a specific antibody and identified significantevels of g-subunit expression only in membranes fromidney medulla tissues and not those from axolemma,eart, red blood cells, or kidney glomeruli.An alternative possibility is that OBPF is a lipid.owever we failed to observe any effect of phospholip-
ds when the enzyme was preincubated with suspen-ions of phosphatidylcholine and phosphatidylserine ine2SO medium (Table III).
CKNOWLEDGMENTS
We thank Monica M. Freire, U.F.R.J., for her excellent technicalssistance and Birthe Bjerring Jensen and Angielina Damgaard,epartment of Biophysics, Aarhus University, for preparing thenzyme. We also thank Dr. Rhoda Blostein for a kindly donation ofhe gC33, gamma subunit polyclonal antibody. We also thank Dr.obert W. Mercer for helping us obtain the antibody. This work wasupported by grants from the Conselho Nacional de Desenvolvimentoientıfico e Tecnologico (CNPq), Fundacao de Amparo a Pesquisa dostado do Rio de Janeiro (FAPERJ), Financiadora de Estudos erojetos (FINEP), PRONEX (7697.1000.00) and The Biomembraneesearch Centre, University of Aarhus. F.E.V.L. is recipient of anndergraduate fellowship from Conselho Nacional de Desenvolvi-ento Cientıfico e Tecnologico (CNPq).
EFERENCES
1. Lingrel, J. B., Orlowski, J., Shull, M. M., and Price, E. M. (1990)Prog. Nucleic Acids Res. Mol. Biol. 38, 37–89.
2. Mercer, R. W. (1993) Int. Rev. Cytol. 137C, 139–168.3. Lutsenko, S., and Kaplan, J. H. (1993) Biochemistry 32, 6737–
6743.4. Forbush, B., III, Kaplan, J. H., and Hoffman, J. F. (1978) Bio-
chemistry 17, 3667–3676.5. Reeves, A. S., Collins, J. H., and Schwartz, A. (1980) Biochem.
Biophys. Res. Commun. 95, 1591–1598.6. Collins, J. H., Lane, L. K., Ling, E., Schwartz, A., Zot, A., and
Forbush, B., III (1983) Curr. Top. Membr. Transplant. 19, 131–134.
7. Lowndes, J. M., Hokin-Neaverson, M., and Ruoho, A. E. (1984)J. Biol. Chem. 259, 10533–10538.
8. Hardwicke, P. M. D., and Freytag, J. W. (1981) Biochem. Bio-phys. Res. Commun. 102, 250–257.
9. Collins, J. H., and Leszyk, J. (1987) Biochemistry 26, 8665–8668.0. Mercer, R. W., Biemesderfer, D., Bliss, D. P., Jr., Collins, J. H.,
and Forbush, B., III (1993) J. Cell Biol. 121, 579–586.1. Forbush, B., III (1983) Curr. Top. Membr. Transplant. 19, 167–
201.2. Tada, M., and Kadoma, M. (1989) BioEssays 10, 157–163.
3. James, P., Inui, M., Tada, M., Chiesi, M., and Carafoli, E. (1989)Nature 342, 90–92.

1
1
1
1
1
1
2
2
2
2
2
22
2
2
2
3
3
3
3
33
33
3
3
4
223STIMULATION OF OUABAIN BINDING TO Na,K-ATPase
4. MacLennan, D. H., Yip, C. C., Iles, G. H., and Seeman, P. (1972)Cold Spring Harbor Symp. Quant. Biol. 37, 469–478.
5. Wawryznow, A., Theibert, J. L., Murphy, C., Jona, I., Martonosi,A., and Collins, J. C. (1992) Arch. Biochem. Biophys. 298, 620–623.
6. Palmer, C. J., Scott, B. T., and Jones, L. R. (1991) J. Biol. Chem.266, 11126–11130.
7. Navarre, C., Catty, P., Leterme, S., Dietrich, F., and Goffeau, A.(1994) J. Biol. Chem. 269, 21262–21268.
8. Dufour, J-P., and Goffeau, A. (1978) J. Biol. Chem. 253, 7026–7032.
9. Navarre, C., Ghislain, M., Leterma, S., Ferroud, C., Dufour,J. P., and Goffeau, A. (1992) J. Biol. Chem. 267, 6425–6428.
0. Kakinuma, Y., Kakinuma, S., Kazuma, T., Konishi, K., Igarashi,K., and Yamato, I. (1993) Biochem. Biophys. Res. Commun. 195,1063–1069.
1. Palasis, M., Kuntzweiler, T. A., Arguello, J. M., and Lingrel, J. B.(1996) J. Biol. Chem. 271, 14176–14182.
2. Schwappach, B., Stuermer, W., Apell, H-J., and Karlish, S. J. D.(1994) J. Biol. Chem. 269, 21620–21626.
3. Or, E., Goldshleger, R., Tal, D. M., and Karlish, S. J. D. (1996)Biochemistry 35, 6853–6864.
4. Price, E. M., and Lingrel, J. B. (1988) Biochemistry 27, 8400–8408.
5. Jørgensen, P. L. (1974) Biochim. Biophys. Acta 356, 36–52.6. Jensen, J., Nørby, J. G., and Ottolenghi, P. (1984) J. Physiol.
(London) 346, 219–241.
7. Lowry, O. H., Rosebrough, N. J., Farr, A. L., and Randall, R. J.(1951) J. Biol. Chem. 193, 265–275.4
8. Barrabin, H., Fontes, C. F. L., Scofano, H. M., and Nørby, J. G.(1990) Biochim. Biophys. Acta 1023, 266–273.
9. Fontes, C. F. L., Scofano, H. M., Barrabin, H., and Nørby, J. G.(1995) Biochim. Biophys. Acta 1235, 43–51.
0. Schager, H., and Von Jagow, G. (1987) Anal. Biochem. 166,368–379.
1. Towbin, H., Staehelin, T., and Gordon, J. (1979) Proc. Natl.Acad. Sci USA 76, 4350–4354.
2. Harlow, E., and Lane, D. (1988) Antibodies: A Laboratory Man-ual, pp. 475–504, Cold Spring Harbor Laboratory Press, ColdSpring Harbor, NY.
3. Kessler, R. J., Vaughn, D. A., and Fanestil, D. D. (1986) Anal.Biochem. 158, 117–118.
4. Hansen, O. (1984) Pharmacol. Rev. 36, 143–163.5. De Moraes, V. L. G., and de Meis, L. (1987) FEBS Lett. 222,
163–166.6. Goldin, S. M. (1977) J. Biol. Chem. 252, 5630–5642.7. Horowitz, B., Eakle, K. A., Scheiner-Bobis, G., Randolph, G. R.,
Chen, C. Y., Hitzeman, R. A., and Farley, R. A. (1990) J. Biol.Chem. 265, 4189–4192.
8. Scheiner-Bobis, G., and Farley, R. A. (1994) Biochim. Biophys.Acta 1193, 226–234.
9. Dowd, F. J., Jr., Pitts, B. J. R., and Schwartz, A. (1976) Arch.Biochem. Biophys. 175, 321–331.
0. Therien, A. G., Nestor, B. N., Ball, W. R., and Blostein, R. (1996)J. Biol. Chem. 271, 7104–7112.
1. Therien, A. G., Goldshleger, R., Karlish, S. J. D., and Blostein, R.(1997) J. Biol. Chem. 272, 32628–32634.
Copyright © 2022 FDOKUMEN















![The effect of di]methylsulfoxide on the substrate site of Na +/K +ATPase studied through phosphorylation by inorganic phosphate and ouabain binding](https://static.fdokumen.com/doc/165x107/6323a6724d8439cb620d023f/the-effect-of-dimethylsulfoxide-on-the-substrate-site-of-na-k-atpase-studied.jpg)




