
Sterol methyltransferase2: purification, properties, and inhibition
17
Sterol methyltransferase2: purification, properties, and inhibition Wenxu Zhou and W. David Nes * Department of Chemistry and Biochemistry, Texas Tech University, Lubbock, TX 79409-1061, USA Received 18 July 2003, and in revised form 26 August 2003 Abstract Expression of the Arabidopsis sterol methyltransferase2 (SMT2) cDNA in Escherichia coli yields a native protein, when purified to homogeneity, has the predicted molecular mass ca. 40 kDa on SDS–PAGE and recognizes native sterols synthesized by Ara- bidopsis with a D 24ð25Þ –bond (cycloartenol; K m 35 lM and k cat 0.001 s 1 ) and D 24ð28Þ -bond (24(28)-methylenelophenol; K m 28lM and k cat 0.01 s 1 ). Cycloartenol was converted to a single olefinic product-24(28)-methylenecycloartanol whereas 24(28)-methylenel- ophenol was converted to a mixture of three stereochemically related products with the D 24ð28Þ Z-ethylidene, D 24ð28Þ E-ethylidene, and D 25ð27Þ -24b-ethyl side chains. Structural determinants essential to activity were the nucleophilic features at C-3 and C-24. The double bond position in the sterol substrate influenced catalytic efficiency according to the order: side chain, D 24ð24Þ < D 24ð28Þ and nucleus, D 7 < D 8 < D 5 ¼ 9; 19-cyclopropane. The 14a-methyl group was harmful to catalysis, reducing the suitability of cycloartenol as a substrate. On the basis of substrate activity and product distribution, SMT action was probed further using substrate (26,27-de- hydrozymosterol: 26,27-DHZ) and intermediate (25-azacycloartenol: 25-AC) analogs of the SMT-catalyzed reactions. 26,27-DHZ was C-methylated to 26-homocholesta-8(9), 23(24)E, 26(26 0 )-trienol as well as 26-homocholesta-8(9),26(26 0 )-3b,24b-dienol by SMT2, K m of 15 lM, k cat of 0.001 s 1 . In addition, 26,27-DHZ acted as a mechanism-based irreversible inhibitor that results in the specific covalent modification of SMT2, exhibiting K i of 49 lM, k inact of 0.009 s 1 and partition ratio of 0.11. Substrate protection with zymosterol, 24(28)-methylenelophenol against 26,27-DHZ and similar inhibition of the first and second C 1 -transfer activities by the reversible inhibitor 25-AC of K i 20 nM suggested the analogs interacted at the same active site. [28E- 2 H]- and [28Z- 2 H]24(28)- methylenelanosterols were paired with AdoMet and differences of 2 H-incorporation in the enzyme-generated 24-ethyl olefins supported an antimechanism. The results suggest plant SMT2 has a position-specific substrate specificity for D 24ð25Þ -sterols and contains a single active center to catalyze the consecutive C 1 -transfer activities by substrate reaction channels similar to the fungal SMT1. Ó 2003 Elsevier Inc. All rights reserved. Keywords: Sterol methyltransferase; Arabidopsis thaliana; 26,27-Dehydrozymosterol; Cycloartenol; Sitosterol; Stereochemistry; AdoMet-dependent methyltransferase enzyme Sterol methyltransferases (SMTs) 1 synthesized by plants catalyze the conversion of cycloartenol (1), the ubiquitous C 30 intermediate of the phytosterol pathway, to a variety of 24-alkyl sterol olefins [1]. The crucial role of SMTs in the origin of phytosterol side chain diversity and function [2,3] has stimulated considerable interest in the number of SMT enzymes involved with campesterol (24-methyl) and sitosterol (24-ethyl) synthesis and the details of these related enzymatic transformations (Fig. 1). Extensive labeling and isolation studies have supported a C-methylation pathway in which SMT cat- alyzing the first C 1 -transfer activity converts cycloartenol (1) to 24(28)-methylenecycloartanol (2) and the exom- ethylene product is released from the enzyme [1,2]. The latter intermediate is processed to campesterol or it is transformed to 24(28)-methylenelophenol (6), a putative substrate for the second C 1 -transfer reaction to generate sitosterol. Recent genetic engineering experiments to modify the level of SMT expression in Arabidopsis tha- liana to change the ratio of campesterol to sitosterol or to * Corresponding author. Fax: 1-806-742-0135. E-mail address: [email protected] (W. David Nes). 1 Abbreviations used: SMT, sterol methyltransferase; 26,27-DHZ, 26,27-dehydrozymosterol; 25-AC, 25-azacycloartanol; SMT1, sterol methyl transferase catalyzing first C 1 -transfer reaction; SMT2, sterol methyl transferase catalyzing the second C 1 -transfer reaction; IPTG, isopropyl-b-D-thiogalactoside. 0003-9861/$ - see front matter Ó 2003 Elsevier Inc. All rights reserved. doi:10.1016/j.abb.2003.08.029 Archives of Biochemistry and Biophysics 420 (2003) 18–34 ABB www.elsevier.com/locate/yabbi
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Sterol methyltransferase2: purification, properties, and inhibition

Archives of Biochemistry and Biophysics 420 (2003) 18–34
ABBwww.elsevier.com/locate/yabbi
Sterol methyltransferase2: purification, properties, and inhibition
Wenxu Zhou and W. David Nes*
Department of Chemistry and Biochemistry, Texas Tech University, Lubbock, TX 79409-1061, USA
Received 18 July 2003, and in revised form 26 August 2003
Abstract
Expression of the Arabidopsis sterol methyltransferase2 (SMT2) cDNA in Escherichia coli yields a native protein, when purified
to homogeneity, has the predicted molecular mass ca. 40 kDa on SDS–PAGE and recognizes native sterols synthesized by Ara-
bidopsis with a D24ð25Þ–bond (cycloartenol; Km 35lM and kcat 0.001 s�1) and D24ð28Þ-bond (24(28)-methylenelophenol; Km 28lM and
kcat 0.01 s�1). Cycloartenol was converted to a single olefinic product-24(28)-methylenecycloartanol whereas 24(28)-methylenel-
ophenol was converted to a mixture of three stereochemically related products with the D24ð28ÞZ-ethylidene, D24ð28ÞE-ethylidene, andD25ð27Þ-24b-ethyl side chains. Structural determinants essential to activity were the nucleophilic features at C-3 and C-24. The double
bond position in the sterol substrate influenced catalytic efficiency according to the order: side chain, D24ð24Þ < D24ð28Þ and nucleus,
D7 < D8 < D5 ¼ 9; 19-cyclopropane. The 14a-methyl group was harmful to catalysis, reducing the suitability of cycloartenol as a
substrate. On the basis of substrate activity and product distribution, SMT action was probed further using substrate (26,27-de-
hydrozymosterol: 26,27-DHZ) and intermediate (25-azacycloartenol: 25-AC) analogs of the SMT-catalyzed reactions. 26,27-DHZ
was C-methylated to 26-homocholesta-8(9), 23(24)E, 26(260)-trienol as well as 26-homocholesta-8(9),26(260)-3b,24b-dienol by
SMT2, Km of 15lM, kcat of 0.001 s�1. In addition, 26,27-DHZ acted as a mechanism-based irreversible inhibitor that results in the
specific covalent modification of SMT2, exhibiting Ki of 49 lM, kinact of 0.009 s�1 and partition ratio of 0.11. Substrate protection
with zymosterol, 24(28)-methylenelophenol against 26,27-DHZ and similar inhibition of the first and second C1-transfer activities by
the reversible inhibitor 25-AC of Ki 20 nM suggested the analogs interacted at the same active site. [28E-2H]- and [28Z-2H]24(28)-
methylenelanosterols were paired with AdoMet and differences of 2H-incorporation in the enzyme-generated 24-ethyl olefins
supported an antimechanism. The results suggest plant SMT2 has a position-specific substrate specificity for D24ð25Þ-sterols and
contains a single active center to catalyze the consecutive C1-transfer activities by substrate reaction channels similar to the fungal
SMT1.
� 2003 Elsevier Inc. All rights reserved.
Keywords: Sterol methyltransferase; Arabidopsis thaliana; 26,27-Dehydrozymosterol; Cycloartenol; Sitosterol; Stereochemistry; AdoMet-dependent
methyltransferase enzyme
Sterol methyltransferases (SMTs)1 synthesized byplants catalyze the conversion of cycloartenol (1), the
ubiquitous C30 intermediate of the phytosterol pathway,
to a variety of 24-alkyl sterol olefins [1]. The crucial role
of SMTs in the origin of phytosterol side chain diversity
and function [2,3] has stimulated considerable interest in
* Corresponding author. Fax: 1-806-742-0135.
E-mail address: [email protected] (W. David Nes).1 Abbreviations used: SMT, sterol methyltransferase; 26,27-DHZ,
26,27-dehydrozymosterol; 25-AC, 25-azacycloartanol; SMT1, sterol
methyl transferase catalyzing first C1-transfer reaction; SMT2, sterol
methyl transferase catalyzing the second C1-transfer reaction; IPTG,
isopropyl-b-DD-thiogalactoside.
0003-9861/$ - see front matter � 2003 Elsevier Inc. All rights reserved.
doi:10.1016/j.abb.2003.08.029
the number of SMT enzymes involved with campesterol(24-methyl) and sitosterol (24-ethyl) synthesis and the
details of these related enzymatic transformations
(Fig. 1). Extensive labeling and isolation studies have
supported a C-methylation pathway in which SMT cat-
alyzing the first C1-transfer activity converts cycloartenol
(1) to 24(28)-methylenecycloartanol (2) and the exom-
ethylene product is released from the enzyme [1,2]. The
latter intermediate is processed to campesterol or it istransformed to 24(28)-methylenelophenol (6), a putative
substrate for the second C1-transfer reaction to generate
sitosterol. Recent genetic engineering experiments to
modify the level of SMT expression in Arabidopsis tha-
liana to change the ratio of campesterol to sitosterol or to

Fig. 1. Hypothetical phytosterol pathway in Arabidopsis thaliana.
W. Zhou, W. David Nes / Archives of Biochemistry and Biophysics 420 (2003) 18–34 19
change the ratio of phytosterol to cholesterol [4–6], and
experiments to establish the essentiality of sterol 24-al-
kylation determined by feeding high energy intermediate
(HEI) analogs of 24-alkylation catalysis to cultured plant
cells [7] all point to the requirement of sterol 24-alkyl-
ation genes for plant growth.
Completion of the Arabidopsis genome project and
related sequencing efforts revealed a set of at least 16cDNAs encoding SMT isoforms considered to catalyze
the first and second C1-transfer activities (Fig. 2). Com-
parison of the deduced amino acid sequences of these
enzymes and structure–activity tests using crude SMT
preparations have led one group of investigators to sug-
gest the evolution of two SMT families that act inde-
pendently on position-specific olefins. Thus SMT1
accepts D24ð25Þ-sterol substrates and generates D24ð28Þ-olefins (first C1-transfer activity) and SMT2 accepts
D24ð28Þ-sterol substrates and generates 24(28)Z-ethylidenesterols (second C1-transfer activity) [8–17]. The D24ð25Þ-and D24ð28Þ-substrates that bind productively to SMT1
and SMT2 enzymes are assumed to be conformationally
different, ‘‘bent’’ (e.g., cycloartenol) and ‘‘flat,’’ e.g.,
24(28)-methylenelophenol [14,18] thereby suggesting the
topography of their active sites are different. It has alsobeen hypothesized that the different sterol specificities
may result from unique sterol binding domains for the
D24ð25Þ—(e.g., Region 1 in Fig. 2) and D24ð28Þ-sterol (e.g.,
Region III in Fig. 2) acceptormolecules [8,17]. In terms of
the kinetic parameters for SMT action, studies of HEI
analogs of the C-methylation reaction and chemical
reasonableness suggest the second C1-transfer reaction in
sitosterol synthesis operate a step-wise mechanism
[14,18,19] whereas differential, inactivation and kinetic
isotope labeling studies suggest the first C1-transfer re-
action in ergosterol and campesterol synthesis operate a
non-stop mechanism [1,20]. Indeed, these apparent
mechanistic differences are further support that SMT1
and SMT2 enzymes can be separated into distinct classes.An alternative hypothesis based on our recent work
with the yeast SMT [21–26] is that a single plant SMT
can perform the consecutive C1-transfer reaction activ-
ities at the same active center and downregulation of
enzyme activity by the sterol membrane insert and
substrate channeling to one or more products is con-
trolled by the molecular shape and location of double
bonds and alkyl groups in the substrate as directed inthe steric–electric plug model [1,2]. Although it has been
tacitly assumed that the C-methylation reaction per-
formed by SMT2 synthesized in plants prefers D24ð28Þ-sterols and is product specific, no compelling evidence
for this proposal has been found. That is because only a
limited number of enzymes and sterol substrates have
been studied [1] and in cases where native enzyme
sources were tested the preparation likely containedendogenous sterol and contaminating enzymes that act
on sterols which can compromise the activity assays.
The low-abundance of SMTs in wild-type cells and the
lipophilic and insoluble nature of this membrane-
associated enzyme have greatly complicated its purifi-
cation and kinetic analysis therefore we considered an

Fig. 2. Alignment of ERG6 SMT family proteins. Key catalytic signature motifs are boxed; Region I is a wall of the sterol binding site and Region II
is the AdoMet binding site. Region III is a presumptive wall of the sterol binding site. Secondary structure elements reported as a-helices and b-sheetswere determined from homology building with related AdoMet-dependent proteins (Bujnicki and Nes, unpublished). The 16 SMTs, indicated by the
number on the left side are from: 1, Saccharomyces cerevisiae, CAA52308; 2, Candida albicans, AAC26626; 3, Schizosaccharomyces pombe,
CAB16897; 4, Neurospora crassa, CAB97289; 5, Pneumocystis carinii, AAK54439; 6, Glycine max, AAB04057; 7, Arabidopsis thaliana 1, AAG28462;
8, Ricinus communis, AAB62812; 9, Zea mays, AAC04265; 10, Oryza sativa sub sp. Japonica, AAC34988; 11, Triticum aestivum, AAB37769; and 12,
Nicotiana tabacum 1, AAC34951. 13, Oryza sativa, AAC34989; 14, Nicotiana tabacum 2, T03848; 15, Arabidopsis thaliana 2-1, CAA61966; 16,
Arabidopsis thaliana 2-2, AAB62809.
20 W. Zhou, W. David Nes / Archives of Biochemistry and Biophysics 420 (2003) 18–34
alternative strategy for generating a single enzyme using
recombinant DNA technology.
To test our hypothesis regarding the properties of
SMT2-type enzymes in relation to SMT1-type en-
zymes, we constructed a cDNA of the native A. tha-
liana SMT2 and expressed the resulting protein in E.
coli. Using the cloned SMT2 and sets of 2H-labeled
and non-labeled sterols paired with AdoMet, we
identified the binding determinants that underlie reg-
iochemistry and stereochemistry of the reaction and
determined the second C1-transfer reaction can
proceed by an ionic pathway involving the C-24
isofucosterol cation. Another aspect of the C-methyl-
ation process is the subject of the paper, namely, the
mode of interaction of the mechanism-based inactiva-
tor, 26,27-dehydrozymosterol (26,27-DHZ), previouslyfound to covalently attach to the SMT from Saccha-
romyces cerevisiae [26]. Our results also reveal the
properties and mechanism of plant SMT catalysis
share many similarities to that described for yeast
SMT action, suggesting the topography of the active
center of SMTs contain a highly conserved region for

W. Zhou, W. David Nes / Archives of Biochemistry and Biophysics 420 (2003) 18–34 21
the sterol binding domain consistent with a commonevolutionary origin for this class of enzyme.
Experimental procedures
Materials
The sources of reagents, authentic substrates and sterolanalogs isolated from nature or prepared synthetically,
[methyl-3H3]AdoMet (10–15Ci/mMol), [methyl-2H3]
AdoMet (99% atom enrichment) were as described in our
earlier publications [15,16,22,26,27]. [28E-2H]- and [28Z-2H]24(28)-methylenelanosterols (95% atom enrichment)
were prepared according to Arigoni [20] with modifica-
tions as specified [29]. 26,27-Dehydrozymosterol and
26,27-dehydrocycloartenol were prepared from the par-ent substrate [26]. 24(28)-Methylenecycloartanol isolated
from c-oryzanol was hydrogenated to give the epimeric
mixture of 24a=24b-methylcycloartanol and the respec-
tive diastereoisomers were purified by HPLC [28]. The
structures of sterols assayed in the present study are
shown in Figs. 1 and 3. Bradford protein assay kit, and
protein electrophoresis supplies were purchased from
Bio-Rad. IPTG was purchased from Research ProductsInternational Corp. Reagents and chemicals used in
growing bacteria, enzyme purification, and assays were
purchased fromSigma andFisher unless otherwise noted.
Instrumentation
Proton NMR spectra were recorded in deuterochlo-
roform at 300MHz using a Bruker AF300 spectrometer.Chemical shifts (d, ppm) are referenced to TMS (0.0).
Radioactivity measurements were obtained on a Beck-
Fig. 3. Structures of sterol su
man LS 6500 liquid scintillation counter with 5mL ofScintiVerse BD cocktail and were automatically quench
corrected. Mass spectra were recorded on a Hewlett-
Packard 6890 GC interfaced to a 5973 mass spectro-
meter [27,28].
Capillary gas liquid chromatography (GLC) was
performed using a DB-5 column (J&W, Folsom, CA),
30m� 250 lm, film thickness 0.25 lm, flow rate He
1.2mL/min, injector temperature 250 �C, initial columntemperature 170 �C, hold for 1min, ramp at 20 �C/min to
280 �C. Retention times in GLC on a 3% SE-30 packed
column operated isothermally at 245 �C with flame ion-
ization detection (retention time relative to cholesterol,
RRTc). Retention times of sterols in high-performance
liquid chromatography (HPLC), using a Whatman C18
reversed-phase 25-cm column eluted with acetonitrile–
isopropanol (9/1, v/v) and ultraviolet wavelength detec-tion set at 205 nm and motilities of compound are
relative to the retention time of cholesterol (ac). The FastProtein Liquid Chromatography (FPLC) system was
from Pharmacia (Pharmacia, NJ). The system included
two P-500 pumps, a UV-MII detector, a FRAC 100
fraction collector and LCC-501 plus controller.
Gel electrophoresis
SDS–PAGE was carried out using the standard buffer
system according to Laemmli in a 12% polyacrylamide
vertical slab gel preceded by a 4% stacking gel at a
constant voltage of 150V (Bio-Rad 1000/500 power
supply) for about 45min run in a Modular Mini-PRO-
TEAN II electrophoresis system (Bio-Rad). Gels were
stained with 2.0% (w/v) Coomassie brilliant blue R-250in methanol: acetic acid:water (40:10:50). Molecular
weight markers were purchased from Bio-Rad.
bstrates and inhibitors.

22 W. Zhou, W. David Nes / Archives of Biochemistry and Biophysics 420 (2003) 18–34
SMT assays and product detection
The standard assay was performed in 600 lL of total
volume, 10 lg pure SMT (for kcat determination) or 2–
3mg of total protein (for routine kinetic studies), in
buffer A (50mM Tris buffer, 2mM MgCl2, 2mM 2-
mercaptoethanol and 20% (v/v) glycerol, pH 7.5), 50 lMsterol substrate, 50 lM [methyl-3H3]AdoMet at 0.6 lCiand Tween 80 (1%, v/v) to produce 104–106 dpm ofproduct in 45min at 35 �C. The incubation mixture was
terminated with 500 lL of a solution of 10% methanolic
KOH. The alkylated sterol product was extracted three
times with 2.0mL each hexane (Fisher). The resulting
organic layer was then transferred to a 7-mL scintilla-
tion vial dried and taken for liquid scintillation counting
to determine conversion rate. For kinetic analysis,
standard assays were performed at ten substrate con-centrations from 5 to 150 lM sterol with the AdoMet
concentration held at saturation of 100 lM. Velocity
measurements were made under initial conditions where
reaction of the varied substrate did not exceed 10%. All
assays were performed at least twice with duplicate
measurements, standard errors were typically in the
range of �5%. Increasing amounts of protein up to
3.0mg/mL protein produced a maximum velocity of660 pmol/min/mg for 24(28)-methylene lophenol, which
then fell off as the concentration of protein in the sample
was increased. The pH for the activity assay was deter-
mined over the range 5.5–10 as described previously [15]
and the optimal activity was found to be in the pH range
7.5–8.0. The production of radiolabeled product using
the soluble enzyme was linear with respect to time for up
to 3 h at a protein concentration of 3.0mg/mL, and allsubsequent experiments were carried out well within
these linear assay conditions. The rates of product for-
mation as a function of substrate concentration with
24(28)-methylenelophenol or cycloartenol gave rise to
typical hyperbolic saturation curves. Temperature de-
pendence studies on the SMT in the range 15–40 �Cindicated maximum activity between 32–40 and and
35 �C was chosen for activity assay measurements. Noapparent difference in catalytic competence of cycloar-
tenol or 24(28)-methylenelophenol was noted in the
conditions studied for activity assay. Based on internal
standardization with cholesterol, substrates and en-
zyme-generated products equivalent to 1 lM/assay can
be detected by GLC and HPLC methods. The product
distribution generated by the SMT was determined at
saturating levels of substrate and with sufficiently largepreparations (5mg/mL of protein) to insure accuracy by
GC-MS peak integration by the total ion current chro-
matogram [28]. Enzyme-generated products were char-
acterized by GC–MS and in select cases 1H and13CNMR. Reference sterol specimens were generated in
earlier studies [15,16,22,28]. SMT eluting in Mono Q
fractions was estimated by the A280 using an extinction
coefficient of 64,160M�1 cm�1. Bio-Rad determinationof the pure SMT overestimates the concentration of the
enzyme by about 1.5 [25].
Molecular weight determinations
A 500 lL sample of pure SMT (0.8mg) in buffer A and
emulphogen (polyoxoethylene 10 tridecylether, Sigma,
10%, v/v) (at 0.4%, volume of detergent/volume of buffer)was loaded onto a Hiprep 26/60 Sephacryl S-300 high
resolution gel filtration column (Pharmacia), calibrated
against a mixture of standards (Bio-Rad; thyroglobulin
(670,000Da), gamma globulin (158,000Da), ovalbumin
(44,000Da), myoglobin (17,000Da), and vitamin B-12
(1300Da)), and eluted isocratically with 100mM NaCl
containing 0.4% (v/v) emulphogen. Fractions eluted
from the FPLC column were assayed for activity andanalyzed by SDS–PAGE for SMT.
Data analysis
The initial velocity data were determined using the
computer program Sigmaplot 2001 plus the enzyme ki-
netics module software package. Measurement of
KmðappÞ and VmaxðappÞ for sterol employed a concentrationrange of 5–200 lM. Data were fitted to the equation
v ¼ Vmax � S=ðKm þ SÞ; ð1Þusing a non-linear least-squares approach. Kinetic con-
stants possessed �SE standard errors of 5% of the ex-
perimental measurement and R2 values were between
0.95 and 0.97.
Kinetic data from standard activity assays of product
inhibition and dead-end inhibition experiments were
analyzed with the software package in analogous fash-
ion to the initial velocity data analysis. The experimentaldata were analyzed to investigate whether competitive
(Eq. (2)), non-competitive (Eq. (3)) or uncompetitive
(Eq. (4)) inhibition was observed, the data were fitted to
the respective equations based on the algorithms defined
by Cleland as outlined by Copeland [29] using nonlinear
least squares analysis
v ¼ Vm=½1þ ðKm=SÞ � ð1þ I=KiÞ�; ð2Þ
v ¼ Vm=½ð1þ I=KiÞ � ð1þ Km=SÞ�; ð3Þ
v ¼ Vm=ð1þ I=Ki þ Km=SÞ: ð4ÞThe data for individual experiments with each in-
hibitor versus varied substrate were fit to all three in-hibitor models. Kinetic constants �standard errors, SE,
are shown as relevant. Vmax is the maximum velocity, Km
is the Michaelis constant for the varied substrate, S is
the concentration of sterol or AdoMet substrate, I is theconcentration of the inhibitor, and Ki is the dissociation
constant (assuming dissociation to free enzyme where
AdoMet binds prior to sterol). Choice of kinetic fit was

W. Zhou, W. David Nes / Archives of Biochemistry and Biophysics 420 (2003) 18–34 23
based on a combination of visual inspection and com-parison of SE values and residual for all three inhibition
types applied to the data sets.
Isoelectric focusing
Isoelectric focusing (IEF) was performed using a
prepoured PHAST gel on a Pharmacia PHAST system
at 16 �C. The isoelectric point of the protein was deter-mined by comparison to a standard calibration curve
using standards (IEF mix range 3–10, Pharmacia) ac-
cording to the manufacturer�s protocols. The theoreticalisoelectric point of the SMT enzyme was derived from
the inferred protein sequence using Vector NTI soft-
ware. The isoelectric points (pI) were calculated using
the pKa values for all ionizable amino acid side chains
and the N- and C-termini. The number and distributionof charged residues were determined by counting histi-
dine, lysine, and arginine as +1 charge and aspartic acid
and glutamic acid as )1 charge.
Protein sequencing
Approximately 5 lg of the purified SMT protein was
subjected to acid hydrolysis and subsequent amino acidanalysis at the Texas Tech University Biotechnology
Core Facility. In preparation for N-terminal sequence
analysis, SMT protein recovered from the Mono Q
column was separated on a SDS–PAGE. The protein
was transferred to a PVDF membrane. The protein on
the PVDF membrane was visualized by Coomassie
brilliant blue R-250 staining. The band corresponding to
ca 40 kDa was excised for protein sequencing. The se-quence of these samples was determined by Edman
degradation (10 cycles) performed on a Porton Instru-
ment 2020 automated sequencer coupled to a Beckman
Gold HPLC, which was eluted with a starting solvent of
water and 3% THF and 1% each of triethylamine and
sodium acetate, which served as ion-pairing agents.
Acetonitrile was then graded into this solvent mixture to
a final concentration of 90% aqueous acetonitrile.
Enzyme treatment and modification experiments
Aliquots of the 100,000g supernatant (calculated to
contain 0.06 lMSMT generally in 1mL) in buffer A were
incubated for up to 15min at 35 �Cwith 50 lMof [methyl-3H3]AdoMet and 5–20 lM of 26,27-DHZ or 26,27-de-
hydrocycloartanol, the cyclopropyl analog of the sub-strate for the first C1-transfer reaction. At the times
indicated, the samples were put into a dry ice ethanol pre-
cooled test tube to prevent catalysis. The samples were
thawed on ice and the non-reacted inhibitor removed
from the incubation mixture by Centricon (YM10, Am-
icon) filtration and washing with buffer A (0.5ml� 3).
Following treatment, the enzymatic reactionwas initiated
by addition of a saturating concentration of zymosterol(100 lM) and [methyl-3H3]AdoMet (100 lM).
The covalent nature of binding was established
by incubation of 100 lM 26,27-DHZ, 0.62 lM SMT,
and 100 lM [methyl-3H3]AdoMet. After dialysis and
Centricon (YM10) filtration to concentrate the protein
and remove any unbound inhibitor, the protein sample
was boiled in SDS loading buffer for 5min prior to
analysis by chromatographic separation on 12% SDS–PAGE gels. The SDS–PAGE was run at 150V for about
1 h until the dye reached the front edge of the gel. Gel
was removed from the cassette carefully and was washed
three times with 200ml of distilled water for 20min
each. The gel was stained with Gel-Code (Pierce) stain
solution for 20min. Stained gel was washed with water
(200mL). The gel was saturated with fluorescent en-
hancer (Fluoro-Hance, Research Product International)for 20min. Gel was dried by a gel drier at 80 �C for 2 h
and subjected to radiofluorography (Kodak XAR-5
X-ray film) for 4 weeks at )80 �C. The chemically
modified SMT band was identified based on the chro-
matographic mobility of Arabidopsis SMT2 enzyme in
SDS–PAGE. Thermal denaturation of the SMT activity
was carried out at 80 �C by monitoring loss of SMT
activity of aliquots when returned to 35 �C for activityassay. Complete inactivation was achieved within 5min.
The thermally inactivated sample was treated with the
cyclopropyl analog and prepared for SDS–PAGE and
fluorograpghy as before [30]. Unmodified controls
were included in all experiments. The mechanism-based
inactivation experiments were based on the protocols
discussed by Silverman [31].
Results
Gene cloning
The Arabidopsis SMT2 has been cloned and its se-
quence shown to be similar to other SMTs as shown in
Fig. 2. We originally amplified the full length cDNA ofSMT from an A. thaliana cDNA library (constructed
from two-week-old leaves ofA. thaliana ecotype 24) using
primers designed based on the published sequence
GenBank X89867 from the SMT genome [30], sense pri-
mer 50-ATGGACTCT TTAACACTCTTC-30 and anti-
senseprimer50-TCAAGAACTCTCTCCTCC GGTGA-30.The 1088 bp amplified fragment with a sticky A end was
subcloned into a PCR II cloning vector (Invitrogen). TheSMTsequencewas verified byDNAsequencing (ABI-310
automatic sequencer). In the earlier study, a fusion pro-
tein of SMTwas generated with aHis-tag and it proved to
be weakly active [32]. For this study, we desired the native
protein construct with the expectation that the resulting
purified protein will be highly active thereby allowing for
detailed kinetic analyses to be pursued. The native SMT2

24 W. Zhou, W. David Nes / Archives of Biochemistry and Biophysics 420 (2003) 18–34
was generated by amplifying the SMT cDNA from thePCR II vector using primers at the NcoI sites attached to
each terminus, the upstream primer 50-CATGCC AT
GGACTCTTTAACACTCTTC-30 and the downstream
primer 50-CATGCCATGGTCAAGAACTCTCCTCC
GGGTTA-30. The PCR primers were synthesized from
the start and stop codons and the resulting PCR DNA
product possessed NcoI sites on both 50 and 30. After di-
gestion with NcoI, the cDNA was subcloned into theE. coli expression vector pET15b (Promega) and then
transformed into competent BL21 (DE3) cells by heat
shock [24]. Sequencing confirmed no errors had been
introduced by the polymerase reactions.
Purification of sterol methyl transferase
A single colony of E. coli BL21(DE3) host cellsharboring A. thaliana SMT2 cDNA was inoculated into
250mL of Luria–Bertani (LB) medium containing am-
picillin (50 lg/mL) and grown for 8 h at 30 �C to reach
an optical density A600 of 0.7 (midlogarithmic phase
growth). From the starter culture, 25mL aliquots were
distributed to eight 2.7-L Fernbach flasks containing
1L each LB medium containing ampicillin (50 lg/mL).
The cells were incubated for 2 h at 30 �C to a densityA600, after which 0.5mL IPTG (0.4mM) was added to
each flask and incubated at 30 �C for another 3 h. The
induced SMT2 cultures were harvested by centrifuga-
tion at 10,000g for 10min and either used directly or
frozen in a 50mL Falcon tube with 15mL liquid ni-
trogen and stored at )80 �C. Generally, from eight
Fernbach flasks about 24 g fresh wt of cell pellet is
generated.The SMT2 was purified from the cell pellet using
anion exchange chromatography as shown in Table 1.
For routine purification of the enzyme, a portion of the
cell paste (5 g) was resuspended in 25mL buffer B
(50mM Tris–HCl, 2mM MgCl2, 2mM b-mercap-
toethanol, 1mM EDTA, and 5% glycerol (v/v) at pH
7.5) and lysed by passage through a French pressure cell
at 20,000 psi. Insoluble protein and cell debris wereremoved by centrifugation (4 �C, 100,000g, 1 h) and the
supernatant (25mL) assayed with 24(28)-methylene-
Table 1
Sterol methyl transferase purification from E. colia
Fractionation step Total protein (mg) Total acti
Cell lysate 359 18,387
100,000g supernatant 291 16,421
DEAE cellulose I 123 11,367
DEAE cellulose II 13 9497
Mono Qb 1.4 5764
a The data are based on the disruption of 5.0 g of fresh weight of bacteri
defined as pmol/min with 24(28)-methylenelophenol as substrate. The ass
concentrations were estimated by the method of Bradford.bRepresents pure SMT after the sample was chromatographed by a seco
lophenol for activity or processed for further analysis.All purification procedures were carried out at 4 �C. Inorder to reduce contaminants, the 100,000g supernatant
was applied to a column (2.5 cm� 5 cm) of DEAE cel-
lulose (referred to as DEAE cellulose I) pre-equilibrated
with buffer B. The column was eluted with buffer B (no
salt). To the first 25mL eluted from the column,
emulphogen detergent was added to final concentration
of 0.4% (v/v) and stirred on ice for 30min. This materialwas loaded onto a second DEAE cellulose column
(DEAE cellulose II) containing buffer B and emulpho-
gen detergent (0.4%, v/v) to minimize aggregation of the
protein. The column was developed in a gradient from 0
to 500mM NaCL in buffer B and detergent (0.4%).
Fractions every 15min (30mL) for 90min were moni-
tored for SMT activity and by SDS–PAGE gel elec-
trophoresis as described (20). Fractions correspondingto 250mM salt containing SMT activity were com-
bined, desalted by washing with fresh buffer A
(3� 5mL) and concentrated (Amicon ultra filtration
YM-10 membrane) to a final volume of 10mL. The
average yield to this point was about 13mg of total
protein and the level of SMT purity was about 85%.
Protein content during these purification steps was es-
timated by the method of Bradford [33]. The solutionfrom DEAE cellulose II was loaded onto a Pharmacia
Mono Q HR 10/10 column, a strong anion exchange
column, previously equilibrated with buffer B contain-
ing 0.4% emulphogen. The column was eluted with a
200mL linear gradient from 0 to 300mM NaCl in the
same buffer while monitoring the effluent as before
(Fig. 4). A set of fractions (2mL each) eluting between
140 and 280mM NaCl possessed a homogenous proteinby SDS–PAGE (Fig. 5). Analysis of the pure SMT by
gel permeation chromatography revealed a 160 kDa
protein (data not shown), suggesting a subunit organi-
zation that is tetrameric. Edman sequencing experi-
ments of the pure protein indicated the first 10 amino
acids at the N-terminus of the protein is MDSLTLF
FTG, as predicted by the complementary sequence
(Fig. 2). The pI of the pure protein was determined tobe 5.8, which is in good agreement with the predicted
value for the monomer.
vity (units) Specific activity (U/mg) Recovery (%)
51 100
56 81
92 34
731 4
4117 0.4
a cells (1.5L of culture) harboring Arabidopsis SMT cDNA. A unit is
ay conditions are described under Experimental procedures. Protein
nd FPLC as described under Experimental procedures.

Fig. 4. Elution profile of Arabidopsis SMT2 chromatographed on Mono Q HR. Protein as measured by absorption at 280 nm (–––), salt elution
gradients (- - -) and SMT activity (d- -dÞ are plotted. Description of chromatographic methods and assay conditions are provided under Experi-
mental procedures.
Fig. 5. SDS–PAGE of Arabidopsis SMT following the purification scheme described in Table 1. Migration of protein standards (molecular mass in
kDa) is indicated. The gel was stained with Coomassie brilliant blue. Lane 1, cell lysate; lane 2, 100,000g supernatant; lane 3, DEAE I fraction; lane 4,
DEAE II fraction; lane 5, Mono Q fraction, and lane 6, labeling of SMT by [methyl-3H3]AdoMet in the presence of 26,27-dehydrozymosterol-
radiofluorogram of SDS–PAGE.
W. Zhou, W. David Nes / Archives of Biochemistry and Biophysics 420 (2003) 18–34 25
SMT2 enzyme properties
Expression of the Arabidopsis cDNA plasmid in E.
coli resulted in the production of soluble native SMT
(>9mg/L of culture). This source of plant SMT was
used to provide preliminary characterization of the first
and second C1-transfer activities. 24(28)-Methylene-
lophenol was assumed to be the preferred substrate for
SMT action based on its natural occurrence in Ara-
bidopsis and the preliminary findings of Bouvier-Nave
et al. [34] who compared the substrate acceptability ofthe erg6-microsome-bound SMT2 against 24(28)-meth-
ylenelophenol, cycloartenol, lanosterol, obtusifoliol and
zymosterol and determined that 24(28)-methylene-
lophenol was the optimal substrate for the enzyme. We
re-examined the sterol specificity of SMT2 using the
native protein expressed in E. coli and a series of
potential sterol acceptor molecules modified in the
geometry and electronics of the substrate. These studies
suggested that the enzyme can C-methylate either
D24ð25Þ- or D24ð28Þ-sterol acceptor molecules (Table 2).Based on the catalytic competence of 19 compounds
tested against saturating concentrations of the co-sub-
strate (AdoMet, 100 lM) (Table 2), the affinity for
D24- sterols was found to be similar with an average Km
of 30 lM. However, the rates of product formation
differ according to variations in the structure of the
substrate. In buffer A, the native substrates 24(28)-
methylenelophenol and AdoMet were recognized by thepure SMT with equal affinity Km ca. 30 lM and
catalytic efficiency kcat of 0.01 s�1, values very similar
to the zymosterol and AdoMet bound to the yeast
SMT [24].
Three domains on the sterol substrate were evalu-
ated for their relevance to binding/catalysis (Fig. 1).
Testing pairs of substrates an all-or-none- response was

Table 2
Substrate acceptability of SMT2a
Substrates Nucleus condition Structure
number
KmðappÞðlM)
VmaxðappÞ(pmol/min/mg)
Vmax=Km
(%)
24(28)-Methylenelophenol D7 6 30 660 22.0 (100)
Fecosterol D8 12 16 97 6.1 (28)
24(28)-Methylenecholesterol D5 13 36 72 2.0 (9)
Obtusifoliol D8 4 38 59 1.6 (7)
24(28)-Methylenecycloartanol 9,19-Ccyclopropane 2 40 15 0.4 (2)
24(28)-Methylenelanosterol D8 14 35 17 0.5 (2)
24(28)-Methylenecholest-7-enol D7 24 35 430 12.3 (56)
4a-Methylzymosterol D8 25 30 627 20.9 (95)
4a-Methylfecosterol D8 5 30 120 4.0 (25)
Cycloartenol 9,19-Cyclopropane 1 35 130 3.7 (17)
Zymosterol D8 15 28 512 18.3 (83)
Desmosterol D5 16 38 76 2.0 (9)
Dehydropollinastanol 9,19-Cyclopropane 17 23 89 3.9 (18)
Cholest-7,24-dienol D7 18 30 627 20.9 (95)
31-Norcycloartenol 9,19-Cyclopropane 19 30 125 4.2 (19)
26,27-Dehydrozymosterol D8 20 15 65 4.3 (20)
Cycloartanol 9,19-Cyclopropane 21 NAb NA NA
3-Desoxycycloartenol 9,19-Cyclopropane 22 NA NA NA
Cyclolaudenol 9,19-Cyclopropane 23 NA NA NA
aActivity assay performed with 100,000g as described under Experimental procedures.bNo activity.
26 W. Zhou, W. David Nes / Archives of Biochemistry and Biophysics 420 (2003) 18–34
apparent in the productive binding for cycloartenol 1
and 3-desoxycycloartenol 22 (Domain A) where 1 and
not 22 was transformed by SMT2. These results suggest
the C-3 hydroxyl group in the nucleus is essential to
activity. Methyl groups at C-4 which can interfere with
the hydrogen bonding ability of the 3-hydroxyl group
had little affect on affinity as shown by the activity assay
comparisons of 1 versus 17 and 19. In similar fashion, bycomparing the activities of cycloartenol 1 to cycloarta-
nol 21 (Domain C), it was found the D24-bond in the side
chain is essential to activity. Regiospecificity for the D24
versus D25-bond was established by comparing the ac-
tivities for cycloartenol, 24(28)-methylenecycloartanol
and cyclolaudenol 23. Structural specificity in favor of
the D24ð25Þ-bond was evident by comparing the activity
results of a series of pairs of D24ð25Þ- and D24ð28Þ-sterolacceptor molecules; 1/2, 25/5, 15/12, and 18/24. These
observations are consistent with the steric–electric plug
model where nucleophilic structures at either pole of the
molecule at C-3 and C-24 are compulsory for catalysis
and that the D24ð25Þ-sterol is the preferred substrate for
SMT catalysis [13].
Domain B of the sterol contains structural elements
which can affect the shape of the nucleus and the pla-narity of the a-face (back side of the molecule) [13]. By
comparing the pair of substrates 15/17 and 5/4, the
harmful affect of the 14a-methyl group to activity was
evident. By comparing the series of compounds 15/18/16
and 24/12/13 where the location of the double bond in
the nucleus is changed, we found the D7-bond was more
effective than the D8-bond and the D5-bond was less
effective in generating substrate acceptability. Fromthese data, the optimal substrate of SMT2 action is not
necessarily 24(28)-methylene lophenol but a sterol that
contains a critical side chain feature distinct from that
found in 24(28)-methylenelophenol. The preferred sub-
strate may contain a 3b-hydroxyl group, D7-nucleus and
D24ð25Þ-side chain rather than a D24ð28Þ-side chain. A 14a-methyl group is harmful to activity whereas the methyl
groups at C-4 are of little effect on productive binding.
Thus the expected preferred substrate for SMT2 actionwill contain a position-specific olefin side chain similar
to SMT1 action. It follows, the relevant 14a-desmethyl
D24ð25Þ-substrate is not generated under normal physio-
logical conditions in Arabidopsis thereby permitting the
most closely related substrate suited for SMT2 action to
be 24(28)-methylenelophenol which is synthesized by
Arabidopsis.
GC–MS analysis of the enzyme-generated productsfollowing activity assay with cycloartenol indicated
24(28)-methylenecycloartanol (25% yield), assay with
24(28)-methylenecycloartanol indicated 24(28)Z-ethy-
lidenecycloartanol (4% yield) and assay with 24(28)-
methylenelophenol indicated a set of three products
(Fig. 6) with side chains corresponding to 24(28)Z-eth-
ylidene, 24(28)E-ethylidene and 24b-ethyl (65% yield)
(Fig. 7). The identity of the seven compounds eluting inGC was based on the chromatographic and spectral
characteristics of authentic specimens available to us
[28]. In the case of the results with 24(28)-methylenecy-
cloartanol, we considered the initial experiments might
be misleading due to a problem with our limits of de-
tection in the activity assay because SMT2 was found to
accept the D24ð28Þ -substrate, 24(28)-methylenelophenol.
The possibility the SMT2 enzyme can mimic the soy-bean SMT1 [35] in generating a product set of three
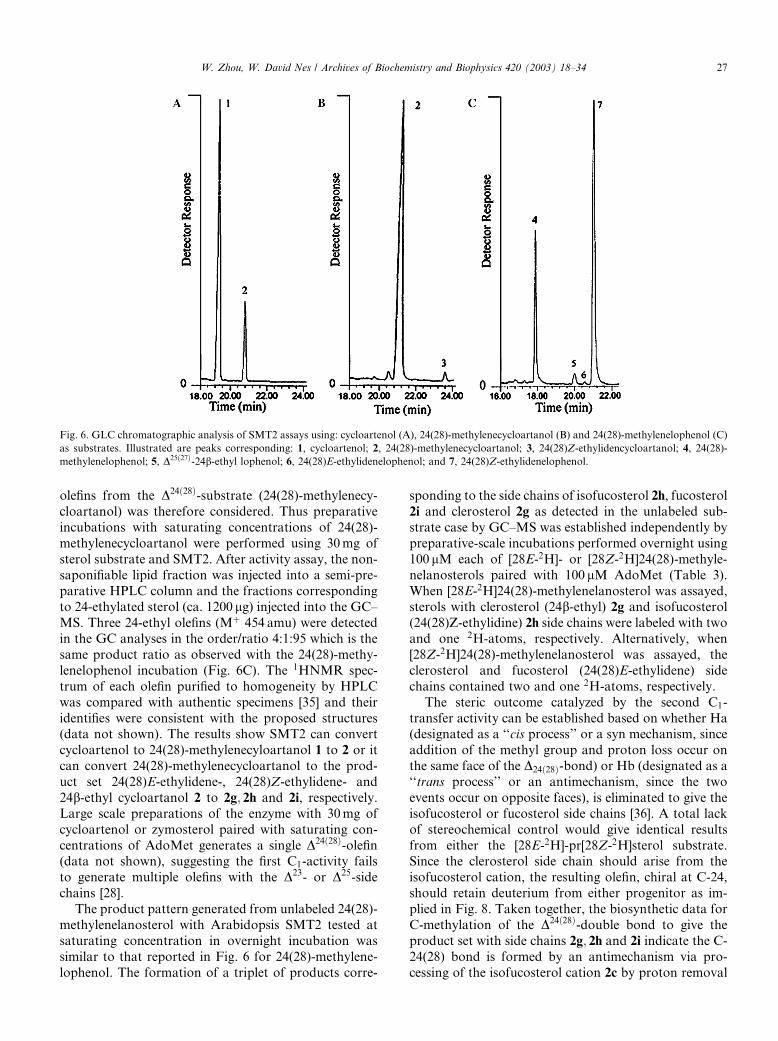
Fig. 6. GLC chromatographic analysis of SMT2 assays using: cycloartenol (A), 24(28)-methylenecycloartanol (B) and 24(28)-methylenelophenol (C)
as substrates. Illustrated are peaks corresponding: 1, cycloartenol; 2, 24(28)-methylenecycloartanol; 3, 24(28)Z-ethylidencycloartanol; 4, 24(28)-
methylenelophenol; 5, D25ð27Þ-24b-ethyl lophenol; 6, 24(28)E-ethylidenelophenol; and 7, 24(28)Z-ethylidenelophenol.
W. Zhou, W. David Nes / Archives of Biochemistry and Biophysics 420 (2003) 18–34 27
olefins from the D24ð28Þ-substrate (24(28)-methylenecy-
cloartanol) was therefore considered. Thus preparative
incubations with saturating concentrations of 24(28)-
methylenecycloartanol were performed using 30mg of
sterol substrate and SMT2. After activity assay, the non-
saponifiable lipid fraction was injected into a semi-pre-
parative HPLC column and the fractions correspondingto 24-ethylated sterol (ca. 1200 lg) injected into the GC–
MS. Three 24-ethyl olefins (Mþ 454 amu) were detected
in the GC analyses in the order/ratio 4:1:95 which is the
same product ratio as observed with the 24(28)-methy-
lenelophenol incubation (Fig. 6C). The 1HNMR spec-
trum of each olefin purified to homogeneity by HPLC
was compared with authentic specimens [35] and their
identifies were consistent with the proposed structures(data not shown). The results show SMT2 can convert
cycloartenol to 24(28)-methylenecyloartanol 1 to 2 or it
can convert 24(28)-methylenecycloartanol to the prod-
uct set 24(28)E-ethylidene-, 24(28)Z-ethylidene- and
24b-ethyl cycloartanol 2 to 2g; 2h and 2i, respectively.
Large scale preparations of the enzyme with 30mg of
cycloartenol or zymosterol paired with saturating con-
centrations of AdoMet generates a single D24ð28Þ-olefin(data not shown), suggesting the first C1-activity fails
to generate multiple olefins with the D23- or D25-side
chains [28].
The product pattern generated from unlabeled 24(28)-
methylenelanosterol with Arabidopsis SMT2 tested at
saturating concentration in overnight incubation was
similar to that reported in Fig. 6 for 24(28)-methylene-
lophenol. The formation of a triplet of products corre-
sponding to the side chains of isofucosterol 2h, fucosterol
2i and clerosterol 2g as detected in the unlabeled sub-
strate case by GC–MS was established independently by
preparative-scale incubations performed overnight using
100 lM each of [28E-2H]- or [28Z-2H]24(28)-methyle-
nelanosterols paired with 100 lM AdoMet (Table 3).
When [28E-2H]24(28)-methylenelanosterol was assayed,sterols with clerosterol (24b-ethyl) 2g and isofucosterol
(24(28)Z-ethylidine) 2h side chains were labeled with two
and one 2H-atoms, respectively. Alternatively, when
[28Z-2H]24(28)-methylenelanosterol was assayed, the
clerosterol and fucosterol (24(28)E-ethylidene) side
chains contained two and one 2H-atoms, respectively.
The steric outcome catalyzed by the second C1-
transfer activity can be established based on whether Ha(designated as a ‘‘cis process’’ or a syn mechanism, since
addition of the methyl group and proton loss occur on
the same face of the D24ð28Þ-bond) or Hb (designated as a
‘‘trans process’’ or an antimechanism, since the two
events occur on opposite faces), is eliminated to give the
isofucosterol or fucosterol side chains [36]. A total lack
of stereochemical control would give identical results
from either the [28E-2H]-pr[28Z-2H]sterol substrate.Since the clerosterol side chain should arise from the
isofucosterol cation, the resulting olefin, chiral at C-24,
should retain deuterium from either progenitor as im-
plied in Fig. 8. Taken together, the biosynthetic data for
C-methylation of the D24ð28Þ-double bond to give the
product set with side chains 2g; 2h and 2i indicate the C-
24(28) bond is formed by an antimechanism via pro-
cessing of the isofucosterol cation 2c by proton removal

ig. 7. Mass spectra of 24(28)-methylenelophenol and enzyme generated products of SMT2 incubated with 24(28)-methylenelophenol as shown in
ig. 6 and described under Experimental procedures.
28 W. Zhou, W. David Nes / Archives of Biochemistry and Biophysics 420 (2003) 18–34
F
F

Table 3
Mass spectra of SMT2 generated products
Substrates Product distribution
24b-ethyl sterol 24(28)E-ethylidene sterol 24(28)Z-ethylidene sterol
Mþ [Mþ–CH3] [Mþ–CH3–
H2O]
Mþ [Mþ–CH3] [Mþ–CH3–
H2O]
Mþ [Mþ–CH3] [Mþ–CH3–
H2O]
24(28)-Methylenelanosterol 454 439 421 454 439 421 454 439 421
½28E-2H�24(28)- methylenelanosterol 455 440 422 454 439 421 455 440 422
½28Z-2H�24(28)- methylenelanosterol 455 440 422 455 440 422 454 439 421
Fig. 8. Stereochemical model for the formation of phytosterol olefins by C1- and C2-activities performed by soybean SMT; SN, sterol nucleus.
W. Zhou, W. David Nes / Archives of Biochemistry and Biophysics 420 (2003) 18–34 29
on the opposite face from AdoMet attack. These pre-
dicted results serve to confirm the stepwise sequence il-lustrated in Fig. 8 in which the isofucosterol cation
either undergoes deprotonation at C-28 to generate the
isofucosterol side chain structure or rotation about
C24(28) by 120� occurs to position the Ha atom adja-
cent to the same active site base involved with depro-
tonation of Ha from the isofucosterol cation. In this
way, both the isofucosterol and fucosterol side chains
can be formed at the same active site involving the samebase. Because of the increase steric bulk evolving from
the 24-ethyl sterol side chain in the clerosterol cation,
some movement of the side chain and the active site
structure may occur in the activated complex to juxta-
pose an active site base normally out of position to de-
protonate C-27 to generate the clerosterol side chain.
Inhibition of SMT activity
The general proposal for the coupled methylation-
deprotonation reaction by plant SMTs first involves
C-methylation of the D24-double bond of the sterol ac-
ceptor via b-face attack of the methyl group from
AdoMet to give a HEI, which is converted to an olefin
after hydride transfer and C-28 deprotonation. The
process is repeated in the overall catalytic cycle to gen-
erate a doubly 24-alkylated sterol. The product analog
with the 24b-methyl stereochemistry and ammoniumanalog of the HEI with a nitrogen at C-25 therefore
were considered to be potential inhibitors of the enzy-
matic C-methylation of D24-substrates to 24-alkyl
products. To test this possibility, 24b-methylcycloarta-
nol 27 and 25-azacycloartenol 29, respectively, together
with related analogs 24a-methylcycloartanol 26, citro-
stadienol 6, sitosterol 10, and cholesterol 28 (Figs. 1 and
3) were prepared and assayed as described under Ex-perimental procedures.
The ammonium analog 29 of the HEI of the C-
methylation reaction was designed with the expectation
that at physiological pH it would be protonated and
thereby serve as a carbocation mimic of the reaction
intermediate. Hence, it was expected to bind many or-
ders of magnitude more tightly than the substrate, as
might be expected for a transition state analog [18]. Thedissociation constant for 25-azacycloartenol was similar
for both C1-transfer activities, Ki 20 nM, consistent with
the hypothesis and provided preliminary evidence the
two C1-transfer activities operated at the same active
center. The non-competitive nature of 25-azacycloarta-
nol toward the sterol substrates (Fig. 9) suggest the in-
hibitor might be bound to a different subsite in the active
center from that of the sterol substrate [37]. These data
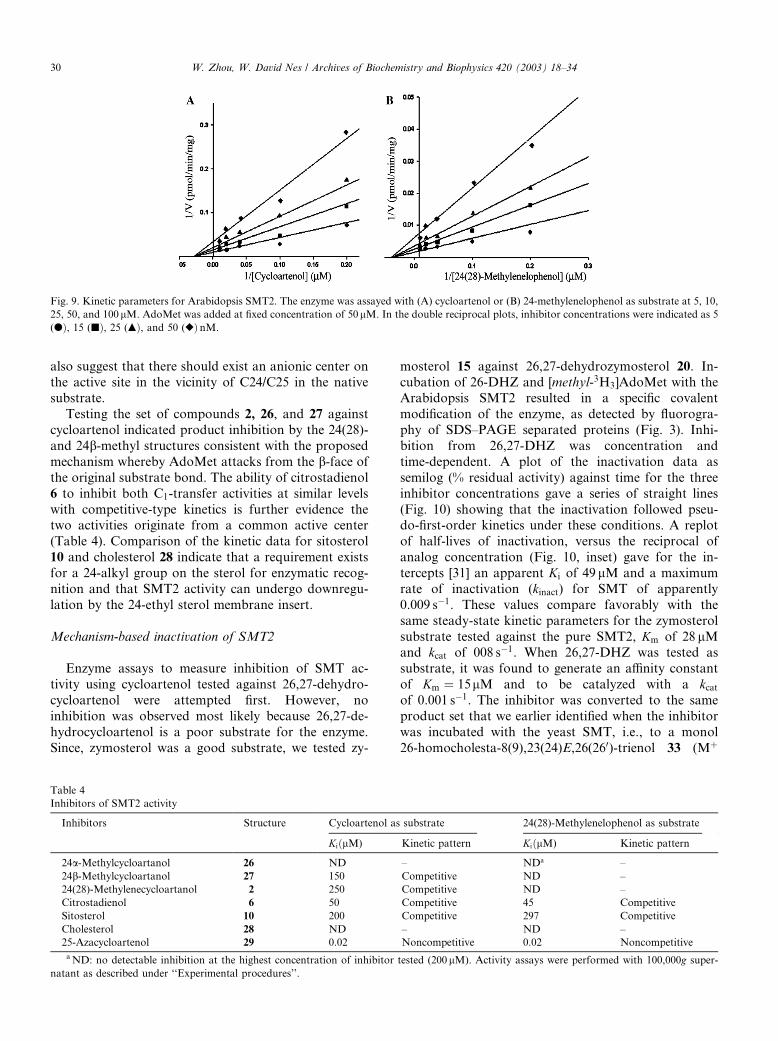
Fig. 9. Kinetic parameters for Arabidopsis SMT2. The enzyme was assayed with (A) cycloartenol or (B) 24-methylenelophenol as substrate at 5, 10,
25, 50, and 100lM. AdoMet was added at fixed concentration of 50lM. In the double reciprocal plots, inhibitor concentrations were indicated as 5
(dÞ, 15 (jÞ, 25 (mÞ, and 50 (rÞ nM.
30 W. Zhou, W. David Nes / Archives of Biochemistry and Biophysics 420 (2003) 18–34
also suggest that there should exist an anionic center on
the active site in the vicinity of C24/C25 in the nativesubstrate.
Testing the set of compounds 2, 26, and 27 against
cycloartenol indicated product inhibition by the 24(28)-
and 24b-methyl structures consistent with the proposed
mechanism whereby AdoMet attacks from the b-face ofthe original substrate bond. The ability of citrostadienol
6 to inhibit both C1-transfer activities at similar levels
with competitive-type kinetics is further evidence thetwo activities originate from a common active center
(Table 4). Comparison of the kinetic data for sitosterol
10 and cholesterol 28 indicate that a requirement exists
for a 24-alkyl group on the sterol for enzymatic recog-
nition and that SMT2 activity can undergo downregu-
lation by the 24-ethyl sterol membrane insert.
Mechanism-based inactivation of SMT2
Enzyme assays to measure inhibition of SMT ac-
tivity using cycloartenol tested against 26,27-dehydro-
cycloartenol were attempted first. However, no
inhibition was observed most likely because 26,27-de-
hydrocycloartenol is a poor substrate for the enzyme.
Since, zymosterol was a good substrate, we tested zy-
Table 4
Inhibitors of SMT2 activity
Inhibitors Structure Cycloartenol as
KiðlM)
24a-Methylcycloartanol 26 ND
24b-Methylcycloartanol 27 150
24(28)-Methylenecycloartanol 2 250
Citrostadienol 6 50
Sitosterol 10 200
Cholesterol 28 ND
25-Azacycloartenol 29 0.02
aND: no detectable inhibition at the highest concentration of inhibitor
natant as described under ‘‘Experimental procedures’’.
mosterol 15 against 26,27-dehydrozymosterol 20. In-
cubation of 26-DHZ and [methyl-3H3]AdoMet with theArabidopsis SMT2 resulted in a specific covalent
modification of the enzyme, as detected by fluorogra-
phy of SDS–PAGE separated proteins (Fig. 3). Inhi-
bition from 26,27-DHZ was concentration and
time-dependent. A plot of the inactivation data as
semilog (% residual activity) against time for the three
inhibitor concentrations gave a series of straight lines
(Fig. 10) showing that the inactivation followed pseu-do-first-order kinetics under these conditions. A replot
of half-lives of inactivation, versus the reciprocal of
analog concentration (Fig. 10, inset) gave for the in-
tercepts [31] an apparent Ki of 49 lM and a maximum
rate of inactivation (kinact) for SMT of apparently
0.009 s�1. These values compare favorably with the
same steady-state kinetic parameters for the zymosterol
substrate tested against the pure SMT2, Km of 28 lMand kcat of 008 s�1. When 26,27-DHZ was tested as
substrate, it was found to generate an affinity constant
of Km ¼ 15lM and to be catalyzed with a kcatof 0.001 s�1. The inhibitor was converted to the same
product set that we earlier identified when the inhibitor
was incubated with the yeast SMT, i.e., to a monol
26-homocholesta-8(9),23(24)E,26(260)-trienol 33 (Mþ
substrate 24(28)-Methylenelophenol as substrate
Kinetic pattern KiðlM) Kinetic pattern
– NDa –
Competitive ND –
Competitive ND –
Competitive 45 Competitive
Competitive 297 Competitive
– ND –
Noncompetitive 0.02 Noncompetitive
tested (200 lM). Activity assays were performed with 100,000g super-

Fig. 10. Inactivation of Arabidopsis SMT2 with the mechanism-based inactivator, 26,27-dehydrozymosterol. Semilog plots of residual activity versus
time at 0 (dÞ, 5 (jÞ, 10 (mÞ, and 20 (r)lM concentrations of inhibitor. (Inset) Replot of enzyme half life (t1=2) for inactivation versus 1/[I].
W. Zhou, W. David Nes / Archives of Biochemistry and Biophysics 420 (2003) 18–34 31
396 amu) and a diol 26-homocholesta-8(9),26(260)-3b; 24b-dienol 32a (Mþ 414 amu) (26). Although, we
did not characterize the 26,27-DHZ enzyme-generated
products by 1HNMR due to a paucity of sample, we
performed a second activity assay with 26,27-DHZ and
[methyl-2H3]AdoMet to show the products contained
three 32 rather than a two mass 35 atom increasewhich is consistent with the proposed structures having
the methyl addition to C-26 rather than to C-24 of the
sterol side chain (Fig. 11) [26]. The partition ratio, a
measure of the production of product per inactivation
event, can be calculated from the ratio of kcat=kinact tobe 0.11, suggesting a high kill to turnover ratio for the
inhibitor. Co-incubation of 5 lM 26,27-DHZ with 50
and 100 lM either zymosterol or 24(28)-methylenel-ophenol afforded protection against inactivation gen-
erating 45% and 70% C-methylation activity,
Fig. 11. Proposed pathway for mechanism-based inactivation of SMT; ‘‘kill
implies the substrate is transformed to a C-methyl product and is released from
dehydrozymosterol. The methyl donor species represented in the scheme as
respectively, relative to the C-methylation activity of a
control incubation that contained saturating amounts
of substrate and coenzyme only. In separate experi-
ments using 26,27-DHZ as inhibitor tested against in-
creasing concentrations of either zymosterol and
24(28)-methylenelophenol, the Ki and kinetic patterns
were similar, 49 lM and competitive-type. These resultsare further evidence that the first and second C1-
transfer reactions occur at the same binding site. To
determine the stoichiometry of binding, 100 lM 26,27-
DHZ and 100 lM [methyl-3H3]AdoMet were incubated
with 0.62 lM of SMT2 in buffer B for 45min at 35 �C.Assay of an aliquot of the incubation mixture con-
firmed loss of >99% of the original SMT2 activity. The
incubation mixture was dialyzed for 48 h against bufferB and then subjected to repeated ultrafiltration using
an Amicon concentrator with YM-10 membrane to
’’ implies covalent attachment of inhibitor to enzyme and ‘‘turnover’’
the enzyme during the reaction progress. SN, sterol nucleus for 26,27-
CD3 originates with [2H3-methyl]AdoMet.

32 W. Zhou, W. David Nes / Archives of Biochemistry and Biophysics 420 (2003) 18–34
give a final tritium activity to protein which was cal-culated to correspond to 0.82� 0.15 equiv. of inhibitor
per mole of enzyme (native enzyme as the tetramer).
The above results are readily explained by a mecha-
nism-based inactivation process as shown in path a of
Fig. 11. Experiments to establish the mechanism of
inactivation and to determine the site of covalent
modification are in progress.
Discussion
The side chain structures of campesterol and sitos-
terol arise from the consecutive C-methylation of D24-
sterols. Generation of these structures can be catalyzed
by distinct SMTs designed for each chemical step and
tailored through evolution to recognize specific molec-ular shapes and functional groups within the substrate.
The rationale for the existence of two classes and the
chemical mechanism for catalysis is only poorly de-
scribed. As important as SMT action appears to be to
plant biochemistry few attempts have been made to in-
vestigate the pure enzyme to establish specificity and
reactivity of the C-methylation reaction. As a result, the
possibility for a single plant SMT to generate multipleproducts has gone unnoticed. Indeed, examination of
the phytosterol composition of Arabidopsis reveals the
plant synthesizes more compounds than is usually
framed in the pathway (e.g., fucosterol, the 24(28)E-
ethylidene isomer of isofucosterol 9) [5]. As in the case of
Arabidopsis which can synthesize several 24-ethyl(lid-
ene) sterols, at least 60 sterols have been isolated from
Zea mays and the type and amount of 24-alkyl sterolpresent in the plant was found to change with matura-
tion [28]. The change in phytosterol homeostasis ex-
pressed in corn and reported in many other plants
[38,39] suggest a more complex catalytic nature of SMT
enzymes than is usually recognized.
This is the first report of a SMT enzyme from plants
on purification and characterization of its properties.
The native SMT2 from Arabidopsis was expressed in E.
coli and purified to homogeneity. In characterizing the
properties of SMT2, the importance of sterically bulky
groups and ionic interactions between the substrate and
enzyme were determined. In past studies, the kinetic
constants of SMT have not been well defined because it is
technically demanding to characterize the in vitro prop-
erties of the enzyme. For example, the native substrates
for these enzymes are not commercially available and toobtain these compounds they must be isolated from na-
ture or prepared synthetically. Large amounts of micro-
somal protein are required to obtain evenminimal kinetic
constants. Thus the Vmax values reported seem to refer to
enzyme activities and therefore to corresponding varia-
tions in the amounts of the enzyme in different systems
rather than to fundamental kinetic parameters of the
enzyme reactions. The substrate concentration cannot beincreased arbitrarily because the solubility of sterol ac-
ceptor molecule is limited. In some cases, the lack of
sterol availability or knowledge of the three-dimensional
shape of the molecule for structure–activity tests have led
to misrepresentations about the substrate conformation
and position-specific olefin requirements of the enzyme.
Finally, SMT is a membrane-bound enzyme [40], as a
result its purification can be very difficult [20].In our studies with SMT2 we overcame many of the
problems discussed above. Preparative amounts of the
relevant sterols were isolated from plants or generated
chemically and purified by HPLC. The chiral nature of
the 24-alkyl groups and flat conformation of these com-
pounds was determined by a combination of high field1H- or 13CNMR (solution) and X-ray crystallography
(solid sate) [15,41]. E. coli was used as the expressionsystem rather than the yeast (erg6) expression system
because the bacteria can be induced to generate large
amounts of soluble SMT in a sterol-less background.
Hence, the SMT2 was purified from E. coli enabling the
estimation of actual enzyme concentrations, and thereby
the comparison of exacting kinetic parameters.
Assuming that 24(28)-methylenelophenol is the pre-
ferred substrate for the SMT2 [5], the Michaelis con-stant for 24(28)-methylenelophenol is in the range of
values for SMTs generally [1,2]. The subunit organiza-
tion of SMT2 is tetrameric, the pH optima occurs in the
range 7.0–8.0 and its activity is subject to downregula-
tion by a 24-alkyl sterol, in agreement with observations
for related SMTs [1,2]. Substrate (zymosterol) and
product inhibition patterns for plant SMT2 were similar
to those observed for the yeast SMT1 [26]. Like thefungal SMT1, the plant SMT2 recognizes a set of strict
features that relate to the nucleophilicity and three-di-
mensional shape of the molecule and is a slow enzyme
with a low turnover number for the preferred substrate,
ca. 0.01 s�1. A critical new finding related to substrate
acceptability is that the native Arabidopsis SMT2 cat-
alyzes sterol acceptors with the D24ð25Þ-bond with similar
efficiency to the Saccharomyces SMT1. For example,cholesta-7,24-dienol and 4a-methylzymosterol are,
within experimental variation, equivalent to 24(28)-
methylenelophenol as substrates for SMT2. However,
under normal physiological conditions these compounds
are not available for binding whereas 24(28)-methyle-
nelophenol is made available to the enzyme. Cycloar-
tenol might be a substrate, it has D24ð25Þ-bond, but it
binds poorly because of the 14a-methyl group whichperturbs association. Taken together, these results sug-
gest the structure of the enzyme–substrate ternary
complex is fundamentally the same for SMTs of plant
and fungal origin as directed in the steric–electric plug
model [1].
A series of analogs were tested to (i) establish the cat-
ionic process required in catalysis of D24-substrates, (ii)

W. Zhou, W. David Nes / Archives of Biochemistry and Biophysics 420 (2003) 18–34 33
determine the number of sterol binding sites necessary forcatalysis, and (iii) deduce their relative importance to
regulate C-methylation activity. Several of the com-
pounds analyzed as reversible inhibitors, including ci-
trostadienol and sitosterol, as well as the mechanism-
based inactivator, 26,27-DHZ have been shown to be
mutually competitive inhibitors toward the first and sec-
ond C1-transfer reactions, and 25-azacycloarteol was
equally potent toward the position-specific olefins. Thestoichiometry of binding of 26,27-DHZ to the Arabid-
opsis SMT2 indicates a single binding site for sterol,
which is consistent with work on the yeast SMT [26].
Taken together these results suggest that both activities
arise at a common active center. However, the two
C1-transfer reactions do not proceed by identical mech-
anisms. In the case of the C-methylation of the D24ð25Þ-bond the reaction proceeds via a non-covalent pathwaywhereby methyl addition to the 24,25-double bond and
deprotonation of C-28 gives rise to a nucleophilic rear-
rangement in whichH-24migrates to C-25 on theRe-face
of the substrate double bond in concert with the initial
ionization (1, 32). In the case of the second C1-transfer
reaction, catalysis proceeds by a stepwise ionic pathway
involving a C-24 isofucosterol cation, suggesting that
distinct chemical mechanisms operate in the consecutiveC-methylation of theD24-bond to adoublyC-24 alkytated
sterol side chain. The ability for sitosterol to impair SMT
activity is consistent with a regulatory role for the mem-
brane insert to control phytosterol genesis [2].
The remarkable fidelity in mechanism catalyzed by
SMT1 and SMT2 enzymes and the existence of a com-
mon sterol binding site may reflect these proteins share
common ancestral genes that underwent diversificationand gene duplication coincidentally with evolution of
the membrane requirements. That SMTs are so similar
in much of their primary sequence to other AdoMet-
dependent methyltransferases in spite of taxonomic
differences from which they were derived [1], suggests
they arose by divergent evolution from a common an-
cestral AdoMet-dependent methyltransferase early in
evolution. The sequence homology showing a conservedRegion III (Fig. 2) is, we believe, a wall of the binding
site common to D24ð25Þ- and D24ð28Þ-sterols. In support of
this hypothesis, a leucine-screening program whereby
leucine is substituted for each of the highly conserved
residues in the yeast SMT1 and binding/catalysis mea-
sured [26] suggested that Glu195 is a critical residue for
SMT action (1 and unpublished observations).
It is interesting that the retention of an equatorial C-4methyl group on the substrate is not a serious impedi-
ment to catalysis but an axial methyl group at C-14 can
prevent sterol acceptability. Clearly, the hydrophobic
cleft of the fungal SMT1 is tailored to accept 14a-methyl
sterols [22] whereas the plant SMT2 is not, suggesting
the planarity of the back face of the sterol molecule
contributes to differences in catalytic efficiency. Modifi-
cation of the three-dimensional shape of sterol followinga change in the nucleus structure during phytosterol
transformation by the plant from 9,19-cyclopropane
(cycloartenol) to D8 (obtusifoliol) to D7 (24(28)-methy-
lenelophenol) generates increased sterol suitability and
regiospecificity for SMT2 catalysis. These changes in
sterol structure accompany recognition by the enzyme
and play a key role in channeling and controlling the
reaction pathway. Tight conformational control (i.e.,productive binding to generate the D24ð28Þ-structure) canpreclude alternate reaction pathways as observed with
the first C1-transfer reaction and implies proximity of
the anionic center with the nearest double bond of the
substrate during C-methylation.
The functional importance of molecular recognition
and catalytic control over SMT1 and SMT2 action raise
the intriguing possibility that genetic manipulation oftheir transcriptional regulation in situ and their active
site structure can lead to value-added traits, such as
insect resistance. A switching of the C-methylation ac-
tivities was reported to be influenced by development
[27,38,39], light [42] and ATP [2] thereby suggesting that
the proportion of C1/C2-activities and hence ratio of
substrates and products provide a branch point system
to affect phytosterol homeostasis. It follows, that syn-thesis of 24(28)-methylenelophenol during cycloartenol
conversion to sitosterol is made possible by the coordi-
nate regulation of SMT1 and SMT2. Further investi-
gations at the level of gene regulation may provide
additional information about the role of SMT1 and
SMT2 in plant sterol production and processing.
Acknowledgments
This work was supported by grants from the Welch
Foundation (D-1276), National Science Foundation
(MCB 0115401) and National Institutes of Health (GM
63477) to W.D.N.
References
[1] W.D. Nes, Phytochemistry 64 (2003) 75–95.
[2] W.D. Nes, Biochim. Biophys. Acta 1529 (2000) 63–88.
[3] H. Schaller, Prog. Lipid Res. 42 (2003) 163–175.
[4] S.D. Clouse, Plant Cell 14 (2002) 1995–2000.
[5] A.C. Diener, H. Li, W. Zhou, W.J. Whoriskey, W.D. Nes, G.R.
Fink, Plant Cell 12 (2000) 853–870.
[6] F.M. Carland, S. Fujioka, S. Takatsuto, S. Yoshida, T. Nelson,
Plant Cell 14 (2002) 2045–2058.
[7] W.D. Nes, G.G. Janssen, R.A. Norton, M. Kalinowska, F.G.
Crumley, B. Tal, A. Bergenstrahle, L. Jonsson, Biochem. Biophys.
Res. Commun. 177 (1991) 566–574.
[8] R.J. Grebenok, D.W. Galbraith, D.D. Penna, Plant Mol. Biol. 34
(1997) 891–896.
[9] K.L. Jensen-pergakes, M.A. Kennedy, N.D. Lees, R. Barbuch, C.
Koegel, M. Bard, Antimicrobiol. Agents Chemother. 42 (1998)
1160–1167.

34 W. Zhou, W. David Nes / Archives of Biochemistry and Biophysics 420 (2003) 18–34
[10] K.G. Hardwick, H.R.B. Pelham, Yeast 10 (1994) 265–269.
[11] J. Shi, R.A. Gonzales, M.K. Bhattacharyya, J. Biol. Chem. 271
(1996) 9384–9389.
[12] P. Bouvier-nave, T. Husselstein, P. Benveniste, Eur. J. Biochem.
256 (1998) 88–96.
[13] E.S. Kaneshiro, J.A. Rosenfeld, M. Basselin-eiweida, J.R.
Stringer, S.P. Keely, A.G. Smulian, J.-L. Giner, Mol. Microbiol.
44 (2002) 989–999.
[14] A. Rahier, M. Taton, P. Bouvier-nave, P. Schmitt, P. Benveniste,
F. Schuber, A.S. Arula, L. Cattel, C. Anding, P. Place, Lipids 21
(1986) 52–62.
[15] W.D. Nes, G.G. Janssen, A. Bergenstrahle, J. Biol. Chem. 266
(1991) 15202–15212.
[16] A.T. Mangla, W.D. Nes, Bioorg. Med. Chem. 8 (2000) 925–
936.
[17] E.S. Kaneshiro, Drug Resistance Updates 5 (2002) 259–268.
[18] A. Rahier, J.-C. Genot, F. Schuber, P. Benveniste, A.S. Narula, J.
Biol. Chem. 259 (1984) 15213–15215.
[19] A.C. Oehlschlager, R.H. Angus, A.M. Pierce, H.D. Pierce Jr., R.
Srinivasan, Biochemistry 23 (1984) 3582–3589.
[20] D. Arigoni, Ciba Found. Symp. 60 (1978) 243–258.
[21] W. Zhou, D. Guo, W.D. Nes, Tetrahedron Lett. 37 (1996) 1339–
1342.
[22] M. Venkatramesh, D. Guo, Z. Jia, W.D. Nes, Biochim. Biophys.
Acta 1299 (1996) 313–324.
[23] W.D. Nes, D. Guo, W. Zhou, Arch. Biochem. Biophys. 342 (1997)
68–81.
[24] W.D. Nes, B.S. McCourt, W. Zhou, J. Ma, L.A. Peek, M.
Brennan, Arch. Biochem. Biophys. 353 (1998) 297–311.
[25] W.D. Nes, B.S. McCourt, J.A. Marshall, J. Ma, A.L. Dennis, M.
Lopez, H. Li, L. He, J. Org. Chem. 64 (1999) 1535–1542.
[26] W.D. Nes, J.A. Marshall, Z. Jia, T.T. Jaradat, Z. Song,
P. Jayasimha, J. Biol. Chem. 277 (2002) 42549–42556.
[27] D. Guo, Z. Jia, W.D. Nes, J. Am. Chem. Soc. 118 (1996) 8507–
8508.
[28] D. Guo, M. Venkatramesh, W.D. Nes, Lipids 30 (1995) 203–219.
[29] R.A. Copeland, Enzymes: A Practical Introduction to Structure,
Mechanism and Data Analysis, second ed., Wiley, New York,
2000.
[30] J.A. Marshall, W.D. Nes, Bioorg. Med. Chem. Lett. 9 (1999)
1533–1536.
[31] R.B. Silverman, in: Mechanism-Based Enzyme Inactivation:
Chemistry and Enzymology, Vols. I and II, CRC Press, Boca
Raton, FL, 1988.
[32] Y. Tong, B.S. McCourt, D. Guo, A.T. Mangla, W.-X. Zhou,
M.D. Jenkins, W. Zhou, M. Lopez, W.D. Nes, Tetrahedron Lett.
38 (1997) 6115–6118.
[33] M.M. Bradford, Anal. Biochem. 72 (1976) 680–685.
[34] P. Bouvier-nave, T. Husselstein, T. Desprez, P. Benveniste, Eur. J.
Biochem. 246 (1997) 518–529.
[35] A.L. Dennis, W.D. Nes, Tetrahedron Lett. 43 (2002) 7017–7021.
[36] T. Okuzumi, Y. Kaji, H. Hamada, Y. Fujimoto, Tetrahedron
Lett. 41 (2000) 3623–3626.
[37] M.A. Ator, S.J. Schmidt, J.L. Adams, R.E. Dolle, Biochemistry
28 (1989) 9633–9640.
[38] W.D. Nes, Rec. Adv. Phytochem. 24 (1990) 283–327.
[39] M. Kalinowska, W.R. Nes, F.G. Crumley, W.D. Nes, Phyto-
chemistry 29 (1990) 3427–3434.
[40] M.A. Hartmann-Bouillon, P. Benveniste, Phytochemistry 17
(1978) 1037–1042.
[41] W.D. Nes, K. Koike, Z. Jia, Y. Sakamoto, T. Satou, T. Nikaido,
J.F. Griffin, J. Am. Chem. Soc. 120 (1998) 5970–5980.
[42] Y. Sauvaire, B. Tal, R. Heupel, R. England, P.K. Hanners, W.D.
Nes, W.D.J.B. Mudd, in: P.K. Stumpf, J.B. Mudd, W.D. Nes
(Eds.), The Metabolism, Structure and Function of Plant Lipids,
Plenum Press, New York, 1987, pp. 107–110.




















