
Genome-Wide Analysis of Sterol-Lipid Storage and Trafficking in Saccharomyces cerevisiae
14
EUKARYOTIC CELL, Feb. 2008, p. 401–414 Vol. 7, No. 2 1535-9778/08/$08.000 doi:10.1128/EC.00386-07 Copyright © 2008, American Society for Microbiology. All Rights Reserved. Genome-Wide Analysis of Sterol-Lipid Storage and Trafficking in Saccharomyces cerevisiae Weihua Fei, 1 †‡ Gabriel Alfaro, 2 † Baby-Periyanayaki Muthusamy, 3 Zachary Klaassen, 2 Todd R. Graham, 3 Hongyuan Yang, 1 and Christopher T. Beh 2 * Department of Biochemistry, National University of Singapore, Republic of Singapore 1 ; Department of Molecular Biology and Biochemistry, Simon Fraser University, Burnaby, British Columbia, Canada 2 ; and Department of Biological Sciences, Vanderbilt University, Nashville, Tennessee 3 Received 19 October 2007/Accepted 14 December 2007 The pandemic of lipid-related disease necessitates a determination of how cholesterol and other lipids are transported and stored within cells. The first step in this determination is the identification of the genes involved in these transport and storage processes. Using genome-wide screens, we identified 56 yeast (Saccha- romyces cerevisiae) genes involved in sterol-lipid biosynthesis, intracellular trafficking, and/or neutral-lipid storage. Direct biochemical and cytological examination of mutant cells revealed an unanticipated link between secretory protein glycosylation and triacylglycerol (TAG)/steryl ester (SE) synthesis for the storage of lipids. Together with the analysis of other deletion mutants, these results suggested at least two distinct events for the biogenesis of lipid storage particles: a step affecting neutral-lipid synthesis, generating the lipid core of storage particles, and another step for particle assembly. In addition to the lipid storage mutants, we identified mutations that affect the localization of unesterified sterols, which are normally concentrated in the plasma membrane. These findings implicated phospholipase C and the protein phosphatase Ptc1p in the regulation of sterol distribution within cells. This study identified novel sterol-related genes that define several distinct processes maintaining sterol homeostasis. Both cholesterol biosynthesis and storage are controlled in response to levels and localization of regulatory pools of sterols (33, 37, 54, 65). In response to high cholesterol levels in the endoplasmic reticulum (ER) membrane, the enzyme acyl coen- zyme A (CoA):sterol O-acyltransferase (ASAT) initiates sterol esterification and storage by covalently coupling fatty acids to cholesterol. Through an active process, the esterified cholesterol is amalgamated with other neutral lipids into lipid storage drop- lets that are released from the ER membrane (42, 88). The traf- ficking of unesterified sterols also affects the sterol distribution in regulatory pools. Although cholesterol is synthesized in the ER, the highest level of unesterified cholesterol is found in the plasma membrane (33) and maintenance of normal sterol levels requires the efficient transport of cholesterol from the ER membrane to the plasma membrane. The maintenance of cholesterol levels in the plasma membrane is affected by sorting from endosomal compartments and recycling back to the cell surface (33, 54), and feedback regulation of cholesterol on its own biosynthesis and storage also controls levels of cellular sterols (16, 68). These findings suggest that the maintenance of cellular cholesterol ho- meostasis requires the regulatory integration of cholesterol syn- thesis, storage, and transport pathways. As in mammalian cells, the budding yeast Saccharomyces cerevisiae synthesizes its own cholesterol-like lipids but, under normal aerobic conditions, yeast does not internalize exoge- nous sterol lipids. Apart from this difference, other elements of sterol homeostasis, including lipid storage and transport path- ways, appear to be conserved (70). In yeast, ASAT is encoded by two homologous genes, ARE1 and ARE2, which together generate steryl esters for lipid storage droplets (84). Lipid droplets are also comprised of triacylglycerols, which in yeast are produced by the acyl-CoA:diacylglycerol acyltransferase 2 (DGAT2) homologue encoded by DGA1 and by the phospho- lipid:diacylglycerol acyltransferase (PDAT) homologue en- coded by LRO1 (46). The genes encoding sterol and diacyl- glycerol acyltransferases are not essential, and a viable strain has been constructed that lacks all genes required for neutral- lipid biosynthesis (61, 66). These findings indicate that lipid storage is itself not required for yeast growth under normal culture conditions. A likely explanation for why neutral-lipid/ sterol storage is dispensable for yeast viability is that it repre- sents only one of several independent mechanisms that con- tribute to the maintenance of lipid and sterol homeostasis. This leads to the prediction that sterol regulatory pathways are functionally redundant and that growth defects occur only when several of these pathways are disrupted in concert. In the case of sterol storage and other sterol regulatory pathways, functional redundancy has been successfully ex- ploited to identify novel sterol-associated genes in yeast. ARV1, which affects the distribution of unesterified sterols, was orig- inally identified as a deletion mutation that is lethal in combi- nation with deletions of both ARE1 and ARE2 (72). This find- ing suggests that both sterol storage and trafficking make overlapping contributions to sterol homeostasis. ECM22 and UPC2 encode transcription factors that control another aspect of sterol homeostasis through the coordinate regulation of * Corresponding author. Mailing address: Department of Molecular Biology and Biochemistry, 8888 University Drive, Simon Fraser Uni- versity, Burnaby, British Columbia, Canada V5A 1S6. Phone: (778) 782-6801. Fax: (778) 782-5583. E-mail: [email protected]. ‡ Present address: School of Biomolecular Sciences and Biotechnol- ogy, University of New South Wales, Sydney, Australia. † W.F. and G.A. contributed equally to this work. Published ahead of print on 21 December 2007. 401 on January 22, 2015 by guest http://ec.asm.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Genome-Wide Analysis of Sterol-Lipid Storage and Trafficking in Saccharomyces cerevisiae

EUKARYOTIC CELL, Feb. 2008, p. 401–414 Vol. 7, No. 21535-9778/08/$08.00�0 doi:10.1128/EC.00386-07Copyright © 2008, American Society for Microbiology. All Rights Reserved.
Genome-Wide Analysis of Sterol-Lipid Storage and Trafficking inSaccharomyces cerevisiae�
Weihua Fei,1†‡ Gabriel Alfaro,2† Baby-Periyanayaki Muthusamy,3 Zachary Klaassen,2Todd R. Graham,3 Hongyuan Yang,1 and Christopher T. Beh2*
Department of Biochemistry, National University of Singapore, Republic of Singapore1; Department of Molecular Biology andBiochemistry, Simon Fraser University, Burnaby, British Columbia, Canada2; and Department of Biological Sciences,
Vanderbilt University, Nashville, Tennessee3
Received 19 October 2007/Accepted 14 December 2007
The pandemic of lipid-related disease necessitates a determination of how cholesterol and other lipids aretransported and stored within cells. The first step in this determination is the identification of the genesinvolved in these transport and storage processes. Using genome-wide screens, we identified 56 yeast (Saccha-romyces cerevisiae) genes involved in sterol-lipid biosynthesis, intracellular trafficking, and/or neutral-lipidstorage. Direct biochemical and cytological examination of mutant cells revealed an unanticipated link betweensecretory protein glycosylation and triacylglycerol (TAG)/steryl ester (SE) synthesis for the storage of lipids.Together with the analysis of other deletion mutants, these results suggested at least two distinct events for thebiogenesis of lipid storage particles: a step affecting neutral-lipid synthesis, generating the lipid core of storageparticles, and another step for particle assembly. In addition to the lipid storage mutants, we identifiedmutations that affect the localization of unesterified sterols, which are normally concentrated in the plasmamembrane. These findings implicated phospholipase C and the protein phosphatase Ptc1p in the regulation ofsterol distribution within cells. This study identified novel sterol-related genes that define several distinctprocesses maintaining sterol homeostasis.
Both cholesterol biosynthesis and storage are controlled inresponse to levels and localization of regulatory pools of sterols(33, 37, 54, 65). In response to high cholesterol levels in theendoplasmic reticulum (ER) membrane, the enzyme acyl coen-zyme A (CoA):sterol O-acyltransferase (ASAT) initiates sterolesterification and storage by covalently coupling fatty acids tocholesterol. Through an active process, the esterified cholesterolis amalgamated with other neutral lipids into lipid storage drop-lets that are released from the ER membrane (42, 88). The traf-ficking of unesterified sterols also affects the sterol distribution inregulatory pools. Although cholesterol is synthesized in the ER,the highest level of unesterified cholesterol is found in the plasmamembrane (33) and maintenance of normal sterol levels requiresthe efficient transport of cholesterol from the ER membrane tothe plasma membrane. The maintenance of cholesterol levelsin the plasma membrane is affected by sorting from endosomalcompartments and recycling back to the cell surface (33, 54), andfeedback regulation of cholesterol on its own biosynthesis andstorage also controls levels of cellular sterols (16, 68). Thesefindings suggest that the maintenance of cellular cholesterol ho-meostasis requires the regulatory integration of cholesterol syn-thesis, storage, and transport pathways.
As in mammalian cells, the budding yeast Saccharomycescerevisiae synthesizes its own cholesterol-like lipids but, under
normal aerobic conditions, yeast does not internalize exoge-nous sterol lipids. Apart from this difference, other elements ofsterol homeostasis, including lipid storage and transport path-ways, appear to be conserved (70). In yeast, ASAT is encodedby two homologous genes, ARE1 and ARE2, which togethergenerate steryl esters for lipid storage droplets (84). Lipiddroplets are also comprised of triacylglycerols, which in yeastare produced by the acyl-CoA:diacylglycerol acyltransferase 2(DGAT2) homologue encoded by DGA1 and by the phospho-lipid:diacylglycerol acyltransferase (PDAT) homologue en-coded by LRO1 (46). The genes encoding sterol and diacyl-glycerol acyltransferases are not essential, and a viable strainhas been constructed that lacks all genes required for neutral-lipid biosynthesis (61, 66). These findings indicate that lipidstorage is itself not required for yeast growth under normalculture conditions. A likely explanation for why neutral-lipid/sterol storage is dispensable for yeast viability is that it repre-sents only one of several independent mechanisms that con-tribute to the maintenance of lipid and sterol homeostasis. Thisleads to the prediction that sterol regulatory pathways arefunctionally redundant and that growth defects occur onlywhen several of these pathways are disrupted in concert.
In the case of sterol storage and other sterol regulatorypathways, functional redundancy has been successfully ex-ploited to identify novel sterol-associated genes in yeast. ARV1,which affects the distribution of unesterified sterols, was orig-inally identified as a deletion mutation that is lethal in combi-nation with deletions of both ARE1 and ARE2 (72). This find-ing suggests that both sterol storage and trafficking makeoverlapping contributions to sterol homeostasis. ECM22 andUPC2 encode transcription factors that control another aspectof sterol homeostasis through the coordinate regulation of
* Corresponding author. Mailing address: Department of MolecularBiology and Biochemistry, 8888 University Drive, Simon Fraser Uni-versity, Burnaby, British Columbia, Canada V5A 1S6. Phone: (778)782-6801. Fax: (778) 782-5583. E-mail: [email protected].
‡ Present address: School of Biomolecular Sciences and Biotechnol-ogy, University of New South Wales, Sydney, Australia.
† W.F. and G.A. contributed equally to this work.� Published ahead of print on 21 December 2007.
401
on January 22, 2015 by guesthttp://ec.asm
.org/D
ownloaded from

several sterol biosynthesis genes (79). Although the combineddeletion of ECM22 and UPC2 is not lethal, upc2� ecm22� cellsare inviable with the additional perturbation of sterols causedby the deletion of ERG2, which encodes the otherwise nones-sential enzyme C-8 sterol isomerase (79). Together these re-sults affirm that the disruption of just one sterol regulatorypathway is not detrimental unless there are additional defectsin sterol homeostasis.
In this study, we carried out a functional genomics screen toidentify yeast deletion mutants that cannot tolerate drug-in-duced disruptions in sterol homeostasis. This screen success-fully identified 56 known and novel genes that are required formaintenance of sterol homeostasis. The identified deletionmutants were analyzed by cellular and biochemical approachesto establish their specific roles in sterol-lipid biosynthesis, traf-ficking, and/or storage. In this study, we defined distinct stepsrequired for lipid storage droplet biogenesis and established alink between ASAT/DGAT lipid esterification and secretoryprotein glycosylation. Our findings provide insights into mech-anisms affecting sterol transport, synthesis, and neutral-lipidstorage, which together maintain sterol homeostasis and arepotentially linked to human lipid disorders.
MATERIALS AND METHODS
Strains and microbial and genetic techniques. Culture media and geneticmanipulations were as described previously (1). To select for the kan-MX4 gene,yeast were grown on yeast rich medium (YPD) containing 200 �g/ml Geneticinsulfate (G418) (Gibco BRL Life Technologies, Inc., Rockville, MD). YPD solidmedium containing nystatin (Sigma Chemicals, Inc., St. Louis, MO) or lovastatin(a gift of Merck & Co., Inc., NJ) was prepared as previously described (8).
Functional genomics screens were conducted using the nonessential kan-MX4-marked homozygous diploid deletion collection (isogenic derivates of BY4743),and subsequent analysis involved the MATa nonessential haploid deletion straincollection (isogenic derivates of BY4741) (81). The genotypes of other yeastmutant strains not obtained from the deletion mutant collections are listed inTable 1. Strains bearing multiple gene disruptions were generated through stan-dard genetic crosses.
Cloning and recombinant techniques. DNA cloning techniques and bacterialtransformations were performed by standard procedures (60) (Table 2). Restric-tion enzymes were obtained from New England Biolabs (Beverly, MA). Oligo-nucleotide primers for PCR were purchased from Operon Biotechnologies, Inc.(Huntsville, AL), and the yeast genomic DNA template for amplification wasisolated from BY4741. All oligonucleotide primers used for PCR amplificationsare listed in Table 3.
To construct a yeast plasmid that would rescue CNB1 mutant defects, theprimer combination of CBP263 and CBP264 was used to amplify the CNB1 geneby PCR. The amplified 1.1-kb fragment included all promoter and terminatorsequences for wild-type expression and was cloned into the EcoRI-XhoI sites of
TABLE 1. Yeast strains used in this study
Strain Genotype Source or referencea
BY4741 MATa ura3�0 leu2�0 his3�1 met15�0BY4743 MATa/MAT� ura3�0/ura3�0 leu2�0/leu2�0 his3�1/his3�1
met15�0/MET15 lys2�0/LYS2CBY2342 BY4741/pRS416CBY2344 BY4741 cnb1�::kan-MX4/pRS416CBY2346 BY4741 cax4�::kan-MX4/pRS416CBY2400 BY4741/pCB419CBY2404 BY4741 cax4�::kan-MX4/pCB419CBY2408 BY4741 cdc50�::kan-MX4/pRS416CBY2439 BY4741 cdc50�::kan-MX4/pKT1265CBY2443 BY4741/pCB456CBY2445 BY4741 cnb1�::kan-MX4/pCB456CBY2446 BY4741/pPTC1-1CBY2448 BY4741 ptc1�::kan-MX4/pRS416CBY2450 BY4741 ptc1�::kan-MX4/pPTC1-1CBY2464 BY4741 plc1�::kan-MX4/pRS416CBY2485 BY4741/pKT1265CRY1 MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 B. FullerJGY149 MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 cmd1-6 41JRY4130 MATa sec18 ura3-52 his4-619 6MMY09 MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 cna1::LEU2
cna2::URA341
MMY41-10B MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 CAN2�C 41MMY71 MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 cmk1�1::HIS3
cmk2::TRP141
RSY12 MAT� ura3-52 leu2-3,112 sec53-6 R. SchekmanRSY255 MAT� ura3-52 leu2-3,112 R. SchekmanW303-1A MATa ade2 his3 leu2 trp1 ura3 can1 J. NickelsYJN62 MATa ade2 his3 leu2 trp1 ura3 can1 lcb2ts J. NickelsYJN63 MATa ade2 his3 leu2 trp1 ura3 can1 lcb1-100ts J. Nickels
a Unless otherwise stated, all yeast strains were created as part of this study.
TABLE 2. Plasmids used in this study
Plasmid Markers Source or referencea
pCB419 CAX4 URA3 CENpCB456 CNB1 URA3 CENpCB523 VMA21 TRP1 CENpCB526 PLC1 TRP1 CENpKT10-GAL-HA PGAL-HA URA3 2 �m 39pRS416 URA3 CENpKT1265 CDC50 URA3 CEN 39pPTC1-1 PTC1 URA3 CEN J. ShawYCplac111 LEU2 CENYCplac111-CWH8 CAX4 LEU2 CEN
a Unless otherwise stated, all plasmids were created as part of this study.
402 FEI ET AL. EUKARYOT. CELL
on January 22, 2015 by guesthttp://ec.asm
.org/D
ownloaded from
pRS416 (64) to generate the plasmid pCB456. One set of primers used to amplifythe CAX4 gene were CBP267 and CBP268, and the amplified 1.9-kb fragmentwas cloned into the BamHI site of pRS416 to generate pCB419. The YHP1 andYHP2 primers were used to generate the CAX4 gene, after which the amplifiedfragment was cloned into the HindIII and BamHI restriction sites of YCplac111to generate the plasmid YCplac111-CWH8. The primer combination used toamplify VMA21 was CBP287 and CBP288, which produced a 0.6-kb fragmentthat was cloned into the EcoRI site of pRS416 to generate pCB523. Usingprimers CBP276 and CBP277, a 3.1-kb PLC1 fragment was amplified and clonedinto the EcoRI site of pRS416, producing pCB526.
Lovastatin and nystatin functional genomic screen. To screen the homozygousdeletion collection for sterol-sensitive mutants, a pin replicator was used totransfer equivalent inocula from strains arrayed and grown on solid medium into200 �l of sterile water. The resuspended cells were further diluted 100-fold insterile water into individual wells of microtiter plates. Using a pin replicator,strains were spotted and arrayed onto YPD solid rich medium and onto YPDcontaining 5 U/ml nystatin or 20 U/ml nystatin. Strains cultured on YPD solidmedium or YPD containing 5 U/ml nystatin were incubated for 1, 1 to 2, and 3days at 37, 30, and 23°C, respectively. Strains cultured on YPD solid mediumcontaining 20 U/ml nystatin were incubated at 37, 30, or 23°C for 2, 3, and 4 days,respectively. Resistance to nystatin was recorded only if the mutant grew in thepresence of 20 U/ml nystatin, whereas sensitivity was recorded only for strainsthat grew poorly on medium containing 5 U/ml nystatin. Growth defects wereassessed relative to the wild-type control (BY4743) and in comparison to growthof each respective deletion strain on YPD without nystatin. If the growth of aspecific deletion strain was affected by nystatin when cultured at two or more ofthe temperatures tested, the strain was picked and retested on nystatin-contain-ing medium to confirm the results. Once confirmed, these deletion strains werethen arrayed on solid medium and equivalent inocula were transferred to wellsof microtiter plates and diluted 100-fold in sterile water. Using a pin replicator,these strains were spotted onto YPD solid rich medium and YPD mediumcontaining 150 �g/ml lovastatin and incubated at 37, 30, and 23°C for 2, 3, and4 days, respectively. Relative to the wild-type control (BY4743), deletion mutantsthat were susceptible to lovastatin for at least two of the three culture temper-atures tested were retested (no lovastatin-resistant deletion mutants were iden-tified). The confirmed list of nystatin/lovastatin-affected homozygous deletionmutants includes all of the deletions shown in Table 4.
Filipin/sterol and Nile red fluorescence microscopy. To examine sterol-lipiddistribution, yeast cells were fixed and treated with filipin complex as previouslydescribed (6). For filipin and FM4-64 colocalization, 5.0 units of log-phase cellsat an optical density at 600 nm grown in synthetic complete medium at 30°C werepelleted and cultured at 30°C with 32 �M FXM4-64 (Molecular Probes/Invitro-gen, Carlsbad, CA) (78) for either 5 or 25 min. After the timed FXM4-64 uptake,cells were washed once with water, pelleted, and diluted to an optical density at600 nm of 0.7 units/ml with fresh medium. Cells were then fixed for 10 minfollowing the addition of formaldehyde to a final concentration of 3.75%. Thesecells were treated with filipin complex as described previously (6).
Lipid storage droplets were visualized by fluorescence microscopy after treat-ment with the lipophilic dye Nile red (Sigma Chemicals, St. Louis, MO). Cells
TABLE 4. Lipid droplet defects
Strain typeaAvg no. ofdroplets/
cellbVariability
(SD) P valueb Relative stainingintensityc
No.counted
(n)
Wild type 2.7 1.7 NA � 363cax4� 0.3 1.2 �0.0001 � 144rad51� 0.5 2.1 �0.0001 �/� 100lge1� 1.2 1.6 �0.0001 � 100hof1� 1.3 1.5 �0.0001 �/� 100rpl31a� 1.4 1.2 �0.0001 �/� 100rmd7� 1.5 1.1 �0.0001 � 200rim8� 1.6 1.7 �0.0001 � 100tkl1� 1.6 1.8 �0.0001 �/� 100srv2� 1.9 2.0 �0.0001 � 100stp1� 1.9 1.5 �0.0001 � 100van1� 2.1 1.9 0.002 � 100yel045c� 2.2 1.7 0.02 � 63zap1� 2.2 1.9 0.0051 � 100ydl133w� 2.2 1.6 0.006 �� 100yjl175w� 2.2 2.1 0.02 � 100tup1� 2.3 1.9 0.04 � 77tfp1� 2.3 1.4 0.03 � 100nut1� 2.5 1.3 � 100sif2� 2.6 2.1 � 100rim9� 2.6 1.6 � 100vma2� 2.6 1.6 � 200trp1� 2.7 1.6 � 100mck1� 2.7 1.7 � 100lbd7� 2.7 2.0 � 100yaf9� 2.8 1.8 � 100cnb1� 2.8 1.4 � 100erg6� 2.8 1.8 �� 100plc1� 2.9 1.4 � 100bck1� 2.9 2.8 � 92vps28� 3.0 1.5 � 100ram1� 3.1 1.9 � 100soh1� 3.1 1.9 � 100fyv4� 3.1 1.9 � 100sac3� 3.1 2.6 �� 100drs2� 3.1 2.0 0.04 � 100mdm39� 3.2 2.0 0.03 � 100bts1� 3.2 2.2 0.03 � 100erg3� 3.2 1.9 0.002 � 235cin4� 3.5 2.0 0.0004 � 100vma21� 3.5 2.3 �0.0001 � 300taf14� 3.5 2.1 0.0003 � 100cdc50� 3.5 1.9 �0.0001 ��� 445ptc1� 3.5 2.0 �0.0001 � 100rlr1� 3.6 3.1 0.0005 � 100srb5� 3.7 1.7 �0.0001 � 100sin4� 3.9 2.5 �0.0001 � 100pop2� 4.1 2.4 �0.0001 �� 100bud27� 4.1 3.1 �0.0001 � 100rsc2� 4.1 2.4 �0.0001 � 100vps69� 4.2 3.1 �0.0001 �� 100arv1� 4.2 3.3 �0.0001 �� 171ume6� 4.2 2.1 �0.0001 ���� 100vma9� 4.3 2.6 �0.0001 � 100vps65� 4.4 2.8 �0.0001 � 92bub1� 4.9 2.8 �0.0001 � 100pho88� 4.9 3.3 �0.0001 � 100
a All deletion mutants identified by sterol functional/chemical genomic arraysare listed. Deletion mutants that were further analyzed for lipid storage particledefects are in boldface.
b Significant differences compared to wild type, as indicated by particularly lowP values, are shown in boldface. P values and statistical significances comparedto wild-type cells were determined by unpaired t test computed at a 95% confi-dence interval. P values representing trivial differences are not shown.
c Compared to the wild-type control (�), the observed range of Nile redfluorescence intensity is indicated by a minimum (�) to a maximum (����).
TABLE 3. Oligonucleotide primers used in this study
Primer Correspondinggene Sequence
CBP267 CAX4 5� TGCGGATCCAGTTCACCCGCGGCG 3�
CBP268 CAX4 5� CTAGGATCCGGGTATACTAGCTCCATGTAAGGAGT 3�
CBP276 PLC1 5� CTAGAATTCGTCCATGAAGATTCCGCACG 3�
CBP277 PLC1 5� CTAGAATTCCCAAAGGATCCTAATTCAGTAATGCT 3�
CBP287 VMA21 5� CTTCTTTAATATGGAATACCTTGGCTGG 3�
CBP288 VMA21 5� ATCGTTATATATACTCATATATTTGAAAGAAAC 3�
YHP1 CAX4 5� CCCAAGCTTGGGAGATTCGCATGTAATT 3�
YHP2 CAX4 5� GCGGATCCCACAAACGCTCAAGATCGC 3�
VOL. 7, 2008 YEAST STEROL GENOMICS 403
on January 22, 2015 by guesthttp://ec.asm
.org/D
ownloaded from

from mid-logarithmic-phase-grown cultures were centrifuged, and the cell pelletwas resuspended with water before the addition of Nile red to a final concen-tration of 2 �g/ml. Nile red-stained cells were washed once with water beforevisualization. Osmotically susceptible mutant strains were fixed with 3.75% form-aldehyde for 15 min and washed in an equal volume of water before and afterNile red addition.
For all fluorescence microscopy, samples were mounted on poly-lysine-coatedslides, sealed under coverslips with nail polish, and imaged on a Leica DMRA2microscope microscope (Leica Microsystems, Wetzlar, Germany) equipped witha Orca-ER charge-coupled device digital camera (Hamamatsu Photonics,Hamamatsu City, Japan). Filipin and FM4-64 fluorescence was observed with aUV and fluorescein isothiocyanate (FITC) filter set using neutral-density filtersto preserve fluorescence. For each experimental trial shown, equal exposuretimes were used to compare cellular fluorescence. Image analysis was performedusing Improvision (Lexington, MA) Open Lab image analysis software.
In vivo assay for oleate incorporation into steryl esters and triacylglycerol.The incorporation of [3H]oleate into steryl ester and triacylglycerol was used asa measurement of sterol and diacylglycerol esterification rates as describedpreviously (46). Cells (5 ml) were grown in YPD liquid medium to mid-logarith-mic phase and then incubated at 30°C for 30 min with 5 �Ci of [3H]oleate. Toremove residual [3H]oleate, cells were washed twice with 0.5% Tergitol, washedonce with water, and then lyophilized. Dried cell pellets were resuspended in 50�l of lyticase solution (1,700 U/ml in 10% glycerol, 0.02% sodium azide), chilledfor 1 h at �70°C, and then incubated at 30°C for 15 min. Lipids were extractedby hexane and analyzed by thin-layer chromatography (TLC). The plates weredeveloped in hexane-diethyl ether-acetic acid (70:30:1) and stained with iodinevapor. Incorporation of label into lipids was determined after scintillation count-ing and normalization to a [14C]cholesterol internal standard and cell dry weight.For each assay, at least three independent strains of each genotype were used.Statistical analysis was performed using the paired t test.
Measurements of steady-state levels of unesterified sterol and neutral lipids.Lipid extractions were performed as described by Zhang et al. (86), and thequantification of neutral lipids and unesterified sterols were assayed by themethods of Zweytick et al. (88) with modifications. Log-phase cells were grownin rich medium and pelleted and then resuspended and pelleted twice in 0.5%Nonidet P-40 and once in distilled water before lyophilization. The dried cellpellets were resuspended in 50 �l of lyticase (1,700 U/ml in 10% glycerol; SigmaChemicals), incubated at 37°C for 15 min, and then freeze/thaw lysed at �70°Cfor 1 h and then at 37°C for 15 min. Lipids were extracted with hexane, blown drywith N2, and dissolved in 100 �l of chloroform-methanol (2:1 [vol/vol]). Sampleswere applied to Silica gel 60 F254 plates (Merck), and chromatograms weredeveloped in hexane-diethyl ether-acetic acid (85:15:1) with cholesterol, triolein,and cholesteryl ester (Sigma Chemicals) as the standard. Quantitative analysis ofunesterified sterol was carried out by densitometric scanning at 275 nm with aCAMAG TLC scanner. For quantitation of steryl ester and TAG, plates weredipped into methanolic MnCl2 solution (0.63 g MnCl2 · 4H2O, 60 ml water, 60 mlmethanol, and 4 ml concentrated sulfuric acid), dried, and heated at 120°C for 15min. Densitometric scanning was performed at 500 nm.
Immunoblots. Yeast extracts for Western blots were prepared as described byOhashi et al. (47). Prior to loading for sodium dodecyl sulfate-polyacrylamide gelelectrophoresis (SDS-PAGE), samples were incubated in sample buffer at 60°Cfor 10 min before loading. Transfer and immunoblot wash conditions were aspreviously described (7). Polyclonal antibodies were raised in rabbits againstglutathione S-transferase (GST)-fused Are1p (amino acids 12 to 191) and againstGST-fused Lro1p (amino acids 440 to 661). Rabbit anti-Are1p polyserum wasused at a 1:500 dilution; rabbit anti-Lro1p polyserum was used at 1:1,000. Rabbitanti-Vti1p was a gift from Wanjin Hong (Institute of Molecular and Cell Biology,Singapore) and was used at a dilution of 1:3,000. Bands were visualized with a1:3,000 dilution of horseradish peroxidase-conjugated antirabbit secondary an-tibody, followed by chemiluminescent detection (Pierce Chemical Co., Rockford,IL). Endoglycosidase H (endo H) removal of N-linked glycosylation was per-formed on protein extracts as described by the manufacturer (Sigma Chemicals,Inc.). Following deglycosylation, equivalent amounts of protein (20 �g) wereloaded per lane for SDS-PAGE and, after transfer, immunoblots were probedwith anti-Are1p and anti-Lro1p antibodies and detected by chemiluminescenceas described above.
Transmission electron microscopy. Samples were prepared for electron mi-croscopy as described previously (55). In brief, cells were fixed and embeddedafter treatment with osmium-thiocarbohydrazide and dehydration. Thin sectionswere stained with uranyl acetate and lead citrate prior to viewing on a PhilipsCM12 transmission electron microscope.
RESULTS
Identification of deletion mutants susceptible to sterol-lipidperturbation. Sterol lipids are essential for viability in almostall eukaryotic cells. The overall regulation of sterols, however,involves the control of sterol synthesis, as well as sterol trans-port and storage pathways. The individual pathways appear tobe dispensable for normal cell growth only because each path-way compensates for the others. We predicted that yeast mu-tations that disrupt any of these pathways might result in cellsthat are sensitized to further sterol perturbations and would beunable to compensate for imbalances in sterol homeostasis.With this in mind, a yeast functional/chemical genomics ap-proach was applied to identify nonessential genes representingeach of the general pathways that conspire to maintain sterolhomeostasis.
Ergosterol is a cholesterol-like sterol that is the bulk productof sterol biosynthesis in yeast. To identify potential candidatesterol-regulatory genes, we screened the �4,700 diploid ho-mozygous deletion strains (81) for sterol-related defects. Thismutant collection represents deletions corresponding to al-most all individual nonessential genes in the yeast genome(81). We screened the homozygous diploid collection, as op-posed to haploid deletions, to reduce false mutant identifica-tions due to nonspecific spontaneous recessive mutations. Forthe initial screening of the deletion collection, we analyzedmutant growth in the presence of the ergosterol-binding anti-biotic, nystatin (80, 82). Although the mechanism of nystatintoxicity is complex, it exerts its effects by direct binding toplasma membrane ergosterol and nystatin has been success-fully used to select viable mutants defective in ergosterol bio-synthesis (38). In this broad-based screen (see Materials andMethods), we identified 262 nystatin-susceptible mutants and95 nystatin-resistant mutants. Some of the same deletion mu-tants (e.g., ARV1, ERG3, CDC50, CNB1, DRS2, NUT1,PHO88, RIM9, SIF2, UME6, ZAP1, etc.) were independentlyidentified in previous genomic studies that examined the ef-fects of various sterol-affecting drugs on yeast growth (21, 34,49, 50). However, to eliminate from consideration those dele-tion mutants that did not specifically affect sterols, we per-formed a secondary screen using lovastatin. In yeast (andmammals), lovastatin reduces total amounts of sterols by in-hibiting the rate-limiting enzyme in sterol synthesis, 3-hydroxy-3-methyl-glutaryl-CoA reductase (5, 74). In this regard, nysta-tin and lovastatin have different inhibitory mechanisms but,because both drugs disrupt normal sterol regulation, bothwould affect deletion strains defective in sterol homeostasis.Thus, the 357 deletion strains affected by nystatin were testedfor lovastatin sensitivity (none were lovastatin resistant). Fifty-seven of the nystatin-susceptible deletion strains were lova-statin sensitive, whereas only 5 nystatin-resistant strains werealso lovastatin sensitive. Six of the mutants had been previouslyreported to exhibit nonspecific multidrug sensitivities (49), andthey were not analyzed further unless independent evidencesuggested otherwise (see below). Therefore, the 56 deletionmutations that affected sterol regulation or homeostasis arelisted in Table 4; 8 of these mutants had mutations that cor-responded to genes with established links to lipid synthesis,regulation, or transport (ARV1, BTS1, CDC50, DRS2, ERG3,ERG6, PLC1, and RAM1), whereas 3 corresponded to novel
404 FEI ET AL. EUKARYOT. CELL
on January 22, 2015 by guesthttp://ec.asm
.org/D
ownloaded from

genes (YEL045C, YDL133W, and YJL175W). The remainder ofmutants represented known genes that have not been previ-ously reported to have a role in sterol or lipid function. Ourstudy is complementary to a previous genomic approach thatsurveyed all nonessential deletion mutants for those that af-fected sterol uptake during anaerobic growth conditions (56).Except for the tkl1� and rlr1� mutants, the subset of mutantsidentified by these different approaches had no overlap. Todetermine how the deletion mutations we identified affect ste-rols, each of the 56 deletion mutants was analyzed for specificcellular defects in sterol storage, sterol synthesis, or in theintracellular membrane distribution of sterols.
(i) Mutant defects in neutral-lipid storage disrupt sterolhomeostasis. Esterified sterols within lipid droplets represent amajor pool of cellular sterols in most eukaryotes, includingyeast (42, 88). To determine if lipid storage was affected by anyof the deletion mutations we identified, mutant cells wereexamined after incubation with Nile red, a fluorescent dye thatstains neutral-lipid storage droplets. Nile red has been success-fully used both in yeast (84) and in Caenorhabditis elegansgenomic screens to examine lipid storage defects (3). Haploiddeletion mutants, representing each of the 56 sterol-relatedhomozygous deletion strains identified in the genomic screens,were individually cultured in rich medium to the logarithmicphase and stained with Nile red. The number, size, and inten-sity of Nile red-stained lipid droplets were determined by fluo-rescence microscopy and image analysis (Table 4). In wild-typecells, an average of 2.7 Nile red droplets was observed byfluorescence microscopy in a single optical section. Althoughmany of the deletion strains had relatively modest but repro-ducible deviations from the wild-type control in droplet num-ber or Nile red staining intensity, seven deletion mutants hadseverely reduced numbers of lipid droplets (fewer than halfthat of wild type) and 2 strains (ume6� and cdc50�) had asignificant increase in both intensity and droplet number (Ta-ble 4). These findings suggested that at least some of thedeletion mutants originally identified were susceptible to ste-rol-specific inhibitors because of lipid storage defects. As apragmatic approach, we conducted detailed analyses on justthose mutants having the greatest effects on the number andfluorescence intensity of Nile red-stained lipid droplets.
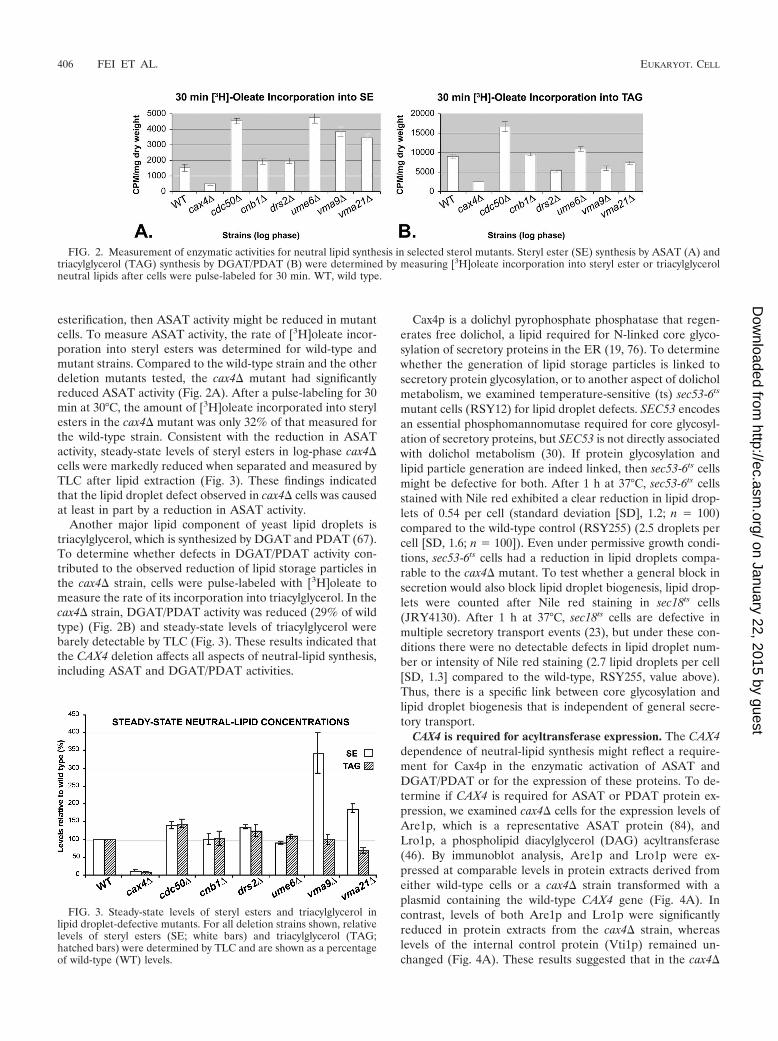
As shown in Fig. 1, the deletion of CAX4 drastically reducedlipid storage droplets. CAX4 encodes dolichyl pyrophosphatephosphatase (19), and the corresponding deletion mutant exhib-ited the greatest reduction in lipid droplet numbers of thoseanalyzed. Compared to wild-type cells, there were 9.1-fold-fewerlipid droplets in cax4� cells (Table 4 and Fig. 1). Of all thedeletion mutants with reduced numbers of lipid droplets, thedeletion of CAX4 had the greatest impact. To confirm thatthe observed Nile red staining defects were a direct result of thespecified deletion and were not due to another unlinked randommutation, we transformed the cax4� strain with a low-copy plas-mid containing its respective wild-type gene. The cax4� straintransformed with the CAX4 gene, but not the vector alone con-trol, fully rescued the lipid droplet defect phenotype (Fig. 1).These results implicated CAX4 as being required for lipid dropletbiogenesis.
In contrast to cax4� cells, deletion of either UME6 orCDC50 caused enhanced Nile red fluorescence and a prolifer-ation of lipid droplets relative to wild-type cells (Table 4). In
this regard, the ume6� and cdc50� mutants were unique inthat, in addition to the many lipid droplets, the intensity of Nilered fluorescence was significantly greater than those observedin the other mutants. For these reasons, the sterol defects inume6� and cdc50� cells were analyzed in detail. CDC50 en-codes a protein that is involved in cell polarization and alsoregulates the cellular localization of Drs2p, a lipid translocase(45, 58). The finding that the number of lipid droplets in-creased in cdc50� cells was particularly noteworthy because itwas recently shown that CDC50 genetically interacts with ste-rol biosynthetic genes (31). In cdc50� cells, wild-type CDC50expressed from a plasmid rescued the lipid droplet prolifera-tion and the observed increase in Nile red staining (Fig. 1).These findings indicated that the lipid droplet defect waslinked to the CDC50 locus. Significant increases in both lipiddroplet number and intensity were also observed in ume6�cells (Table 4). UME6 encodes a transcriptional regulator thatinduces early meiotic genes (77), but it also has a role as aregulator of specific mitotic genes (18, 69, 71). All told, ourresults established that several yeast genes, in addition to thosedirectly involved in neutral-lipid biosynthesis, are required forlipid storage particle biogenesis.
Neutral-lipid synthesis is susceptible to defects in secretoryprotein glycosylation. A core component of lipid storage par-ticles is esterified ergosterol and the enzyme ASAT catalyzesthe coupling of sterols with fatty acids (83, 84). If cax4� mu-tations inhibit lipid droplet formation by blocking sterol trans-
FIG. 1. Examples of deletion mutants with defects in lipid dropletsand sterol lipid storage. Nile red-stained lipid droplets were visualizedby fluorescence microscopy in cax4� (CBY2346), cdc50� (CBY2408),and isogenic wild-type (WT; CBY2342) cells. (Left panels) Strainswere transformed with the vector control, and log-phase cells werestained with Nile red. (Right panels) Wild-type CDC50 and CAX4genes rescued the corresponding lipid droplet defects in the cax4�[CAX4] (CBY2404) and cdc50� [CDC50] (CBY2439) cells. The cax4�cells are an example of mutants that reduced the number of lipiddroplets, whereas an excess of lipid droplets were observed in thecdc50� example. In the cax4� mutant, few lipid droplets were everobserved, although a cytoplasmic background fluorescence was some-times seen. Arrows indicate increased lipid droplet size and stainingintensity. The scale bar for all panels is 10 �m.
VOL. 7, 2008 YEAST STEROL GENOMICS 405
on January 22, 2015 by guesthttp://ec.asm
.org/D
ownloaded from
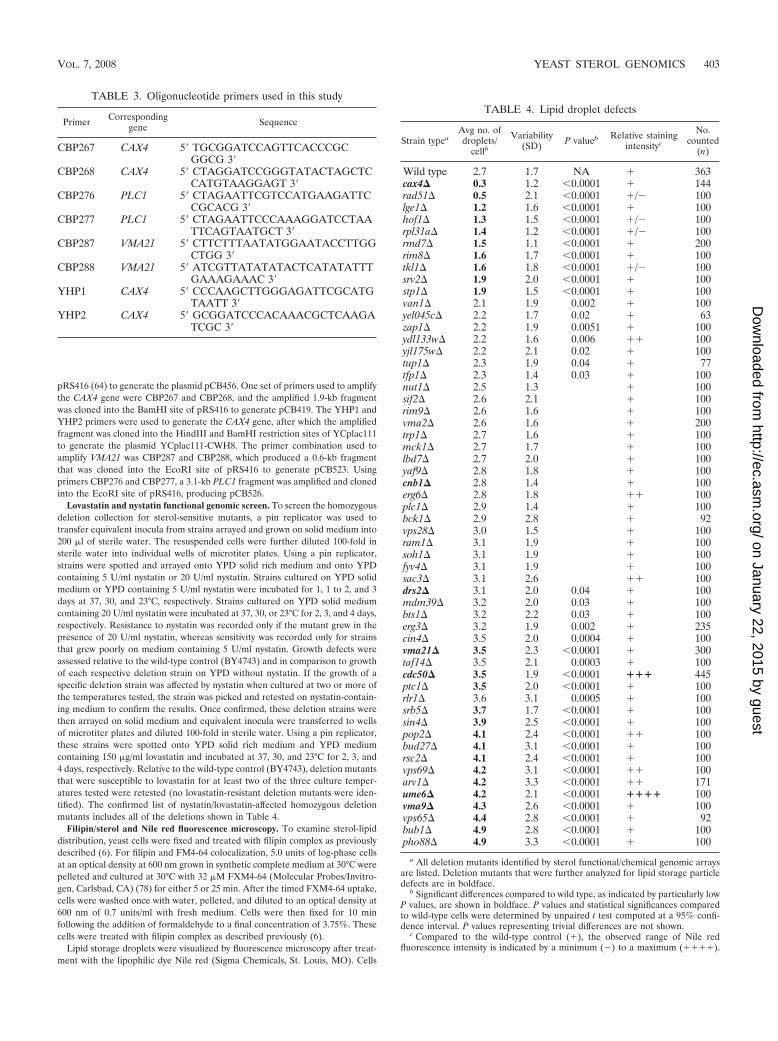
esterification, then ASAT activity might be reduced in mutantcells. To measure ASAT activity, the rate of [3H]oleate incor-poration into steryl esters was determined for wild-type andmutant strains. Compared to the wild-type strain and the otherdeletion mutants tested, the cax4� mutant had significantlyreduced ASAT activity (Fig. 2A). After a pulse-labeling for 30min at 30°C, the amount of [3H]oleate incorporated into sterylesters in the cax4� mutant was only 32% of that measured forthe wild-type strain. Consistent with the reduction in ASATactivity, steady-state levels of steryl esters in log-phase cax4�cells were markedly reduced when separated and measured byTLC after lipid extraction (Fig. 3). These findings indicatedthat the lipid droplet defect observed in cax4� cells was causedat least in part by a reduction in ASAT activity.
Another major lipid component of yeast lipid droplets istriacylglycerol, which is synthesized by DGAT and PDAT (67).To determine whether defects in DGAT/PDAT activity con-tributed to the observed reduction of lipid storage particles inthe cax4� strain, cells were pulse-labeled with [3H]oleate tomeasure the rate of its incorporation into triacylglycerol. In thecax4� strain, DGAT/PDAT activity was reduced (29% of wildtype) (Fig. 2B) and steady-state levels of triacylglycerol werebarely detectable by TLC (Fig. 3). These results indicated thatthe CAX4 deletion affects all aspects of neutral-lipid synthesis,including ASAT and DGAT/PDAT activities.
Cax4p is a dolichyl pyrophosphate phosphatase that regen-erates free dolichol, a lipid required for N-linked core glyco-sylation of secretory proteins in the ER (19, 76). To determinewhether the generation of lipid storage particles is linked tosecretory protein glycosylation, or to another aspect of dolicholmetabolism, we examined temperature-sensitive (ts) sec53-6ts
mutant cells (RSY12) for lipid droplet defects. SEC53 encodesan essential phosphomannomutase required for core glycosyl-ation of secretory proteins, but SEC53 is not directly associatedwith dolichol metabolism (30). If protein glycosylation andlipid particle generation are indeed linked, then sec53-6ts cellsmight be defective for both. After 1 h at 37°C, sec53-6ts cellsstained with Nile red exhibited a clear reduction in lipid drop-lets of 0.54 per cell (standard deviation [SD], 1.2; n 100)compared to the wild-type control (RSY255) (2.5 droplets percell [SD, 1.6; n 100]). Even under permissive growth condi-tions, sec53-6ts cells had a reduction in lipid droplets compa-rable to the cax4� mutant. To test whether a general block insecretion would also block lipid droplet biogenesis, lipid drop-lets were counted after Nile red staining in sec18ts cells(JRY4130). After 1 h at 37°C, sec18ts cells are defective inmultiple secretory transport events (23), but under these con-ditions there were no detectable defects in lipid droplet num-ber or intensity of Nile red staining (2.7 lipid droplets per cell[SD, 1.3] compared to the wild-type, RSY255, value above).Thus, there is a specific link between core glycosylation andlipid droplet biogenesis that is independent of general secre-tory transport.
CAX4 is required for acyltransferase expression. The CAX4dependence of neutral-lipid synthesis might reflect a require-ment for Cax4p in the enzymatic activation of ASAT andDGAT/PDAT or for the expression of these proteins. To de-termine if CAX4 is required for ASAT or PDAT protein ex-pression, we examined cax4� cells for the expression levels ofAre1p, which is a representative ASAT protein (84), andLro1p, a phospholipid diacylglycerol (DAG) acyltransferase(46). By immunoblot analysis, Are1p and Lro1p were ex-pressed at comparable levels in protein extracts derived fromeither wild-type cells or a cax4� strain transformed with aplasmid containing the wild-type CAX4 gene (Fig. 4A). Incontrast, levels of both Are1p and Lro1p were significantlyreduced in protein extracts from the cax4� strain, whereaslevels of the internal control protein (Vti1p) remained un-changed (Fig. 4A). These results suggested that in the cax4�
FIG. 2. Measurement of enzymatic activities for neutral lipid synthesis in selected sterol mutants. Steryl ester (SE) synthesis by ASAT (A) andtriacylglycerol (TAG) synthesis by DGAT/PDAT (B) were determined by measuring [3H]oleate incorporation into steryl ester or triacylglycerolneutral lipids after cells were pulse-labeled for 30 min. WT, wild type.
FIG. 3. Steady-state levels of steryl esters and triacylglycerol inlipid droplet-defective mutants. For all deletion strains shown, relativelevels of steryl esters (SE; white bars) and triacylglycerol (TAG;hatched bars) were determined by TLC and are shown as a percentageof wild-type (WT) levels.
406 FEI ET AL. EUKARYOT. CELL
on January 22, 2015 by guesthttp://ec.asm
.org/D
ownloaded from

mutant, inhibition of neutral-lipid synthesis was a result of aglobal reduction in ASAT and DGAT/PDAT protein expres-sion. Thus, CAX4 defines a mechanism that links secretoryprotein glycosylation with neutral-lipid acyltransferase expres-sion.
A trivial explanation for these results is that the variousneutral-lipid acyltransferases are all glycosylated, and their sta-bility is sensitive to even small perturbations in glycosylation.To test whether Are1p or Lro1p is glycosylated and whethertheir glycosylation state is affected by CAX4, protein extractsfrom wild-type and cax4� cells were treated with endo H toremove N-linked oligosaccharides. The molecular weight ofAre1p was unchanged whether in cax4� cells or in wild-typecells treated with endo H, which suggested that Are1p is not anN-linked glycoprotein (Fig. 4B). This result also suggested thatCAX4 does not affect Are1p stability through glycosylation.Although the molecular weight of Lro1p was reduced by endoH treatment, indicating that Lro1p is a glycoprotein, only theglycosylated form of Lro1p was detected in cax4� cells (Fig.4B). Given these results, CAX4 does not appear to affect neu-tral-lipid acyltransferase expression through their glycosyla-tion.
In cax4� cells and in cells where N-linked glycosylation hasbeen otherwise compromised, sphingolipid composition is sig-nificantly altered (52). Specifically, in cax4� cells there is aconsiderable reduction in inositolphosphorylceramides (IPCs),which represent a major class of sphingolipids (52). To test
whether the effect of CAX4 on sphingolipid composition has abearing on neutral lipid storage, lipid droplets in lcb1-100ts
(YJN63), lcb2ts (YJN64), and wild-type (W303-1A) cells werevisualized using Nile red fluorescence microscopy. The lcb1-100ts and lcb2ts mutants are defective in the first commitmentstep for the biosynthesis of all sphingolipids (43). After cul-tures were incubated at 37°C for 3 h, both the number andfluorescence intensity of Nile red-stained lipid droplets mark-edly increased in lcb1-100ts (average number of droplets percell, 9.5; n 101) and lcb2ts cells (average number of dropletsper cell, 7.9; n 251) as compared to the congenic wild-typecontrol (average number of droplets per cell, 5.2; n 228).When cultured at 23°C, all strains had a comparable number oflipid droplets (4.4 to 4.8 per cell). These findings indicated thatthe inhibition of all sphingolipid biosynthesis results in a con-comitant proliferation in lipid droplets. These results were,however, opposite to those observed in cax4� cells, in whichreduced IPC levels correlated with an absence of neutral lipidsand lipid droplets.
Sterol homeostasis is disrupted in mutants that accumulatelipid storage particles. In the genomic screen, several but notall deletion mutants corresponding to vacuolar H�-ATPasesubunits (e.g., vma2�, vma9�, vma21�, and tfp1�) were sus-ceptible to sterol inhibitors. To determine whether the vacuo-lar H�-ATPase is required for lipid storage, we inspected thesedeletion mutants for defects in neutral-lipid synthesis. In par-ticular, a significant increase in the number of lipid dropletswas observed in vma9� cells (Table 4). VMA9 encodes subunite of the V0 vacuolar H�-ATPase (59). Deletion of VMA9resulted in significant increases in ASAT activity (2.5-fold) andsteryl ester levels (3.4-fold) compared to wild-type cells (Fig.2A and Fig. 3). These increases in steryl esters were specificsince no change in triacylglycerol synthesis or levels was de-tected in vma9� cells (Fig. 2B and Fig. 3). The results sug-gested that other vacuolar H�-ATPase deletion mutants mightalso have specific effects on steryl ester storage. In vma21�cells, a modest increase in lipid droplet number and increasesin steryl ester synthesis and levels were detected (Fig. 2A and3). VMA21 encodes an ER-localized protein required for theassembly of the vacuolar H�-ATPase complex (25, 36). Noneof the other vacuolar H�-ATPase deletion mutants had signif-icant effects on lipid droplets, as determined by Nile red stain-ing (Table 4). These results indicated that steryl ester storageis not dependent on vacuolar H�-ATPase function per se.However, in vma9� cells, and to a lesser degree vma21� cells,steryl ester storage and triacylglycerol storage are uncoupled.The proliferation of lipid droplets is consistent with the in-creased steryl ester synthesis measured in these particular vac-uolar H�-ATPase mutants.
In contrast to vma9� cells, the profusion of lipid droplets inume6� and cdc50� cells was coupled with a striking increase inNile red fluorescence intensity (Table 4). Cdc50p regulates andphysically interacts with the Drs2p P-type ATPase aminophos-pholipid translocase (10, 22, 45), which generates phospholipidasymmetry in membranes (53, 58). This suggested that the lipiddroplet proliferation in cdc50� cells might be a consequence ofdefects in Drs2p phospholipid translocase activity. Consistentwith this possibility, drs2� was identified in our sterol genomicscreen and others have reported sterol defects in drs2� cells(56). However, we observed a very minor increase in lipid
FIG. 4. Immunoblot analysis of Are1p and Lro1p expression in thecax4� mutant. (A) As shown by anti-Are1p immunoblotting, Are1plevels were equivalent in the wild-type control (WT; BY4741) and thecax4� mutant rescued with a CAX4-containing plasmid (Yplac111-CWH8). Are1p levels, however, were clearly reduced in the cax4�mutant. As shown by anti-Lro1p immunoblotting, Lro1p levels werealso reduced in the cax4� mutant compared to the wild-type strain orthe cax4� transformant strain containing an episomal copy of CAX4.As an internal control for loading, samples were probed by immuno-blotting for Vti1p, which is a Golgi v-SNARE that has no directassociation with neutral lipid synthesis. (B.) Treatment of proteinextracts with endo H demonstrated that Are1p was not N-linked gly-cosylated, whereas Lro1p was an N-linked glycosylated protein. With-out added endo H (�), the molecular mass of Are1p was consistentwith its predicted unmodified molecular mass (72 kDa) and Lro1p was75 kDa. With the addition of endo H (�), the position of Are1p on thegel was unchanged but the migration of Lro1p indicated a reduction inmolecular mass. Equal amounts of protein were added to each lane.
VOL. 7, 2008 YEAST STEROL GENOMICS 407
on January 22, 2015 by guesthttp://ec.asm
.org/D
ownloaded from

droplet numbers (albeit statistically significant [P 0.04]) indrs2� cells stained with Nile red (Table 4), and insignificantlipid defects were detected in biochemical assays (Fig. 2 and 3[see below]). Because the S. cerevisiae genome contains fourother potential aminophospholipid translocases that, in somecases, have functional overlap with DRS2 (27), we testedwhether these P-type ATPases (DNF1 to DNF3, NEO1) af-fected lipid droplets. As observed by Nile red staining, noappreciable changes in the number of lipid droplets were ob-served in neo1-1 (ZHY628-15B), neo1-2 (ZHY628-34A),dnf1� dnf2� dnf3� (PFY3273A), or dnf1� dnf2� dnf3� drs2-31ts (ZHY410-3A) mutants, regardless of temperature (unpub-lished data). These results suggested that CDC50 affects lipiddroplets through a mechanism that is independent of the P-type aminophospholipid translocases.
To determine how lipid droplets are affected in cdc50� cells,lipid esterification activities and neutral lipid levels were ana-lyzed. To test if increased ASAT and DGAT/PDAT activitiescaused the increase in lipid droplets, cdc50� cultures werepulse-labeled with [3H]oleate to measure the rate of sterylester and triacylglycerol synthesis. Compared to the wild-typestrain, the cdc50� strain had a 3-fold increase in ASAT activityand a 1.9-fold increase in DGAT activity (Fig. 2A and B).However, these increases in enzyme activities manifested onlya modest 1.4-fold increase in both steady-state steryl ester andtriacylglycerol levels (Fig. 3). Thus, the proliferation of lipiddroplets in cdc50� cells as observed with Nile red is not en-tirely attributable to increases in neutral-lipid levels.
In ume6� cells, neutral-lipid levels were also unaffected (Fig.3) despite the observed increase in the number and fluores-cence intensity of Nile red-stained lipid droplets (Table 4).Similar to cdc50� cells, in ume6� cells ASAT activity wasmarkedly induced (3-fold) relative to the wild-type control,while DGAT activity was elevated only by 1.3-fold (Fig. 2).Despite the induction of ASAT activity in ume6� cells, nomeaningful changes in steady-state steryl ester levels were de-tected and triacylglycerol levels were normal. Based on theseresults, the lipid droplet defects observed by fluorescence mi-croscopy in ume6� cells are not attributable to changes inneutral-lipid levels. Based on these findings, the biogenesis oflipid storage particles is affected by two distinct classes ofmutants: one group that affects neutral-lipid synthesis (e.g.,cax4� and vma9�), and another that is independent of lipidsynthesis (e.g., cdc50� and ume6�).
CDC50 and UME6 deletions disrupt lipid storage particleultrastructure. In addition to the enzymes that synthesize neu-tral lipids, lipid storage also involves lipases that hydrolyze andrelease lipids from storage and structural factors for storageparticle assembly (42). Because steady-state levels of neutrallipids were not grossly affected in cdc50� and ume6� cells, weexamined lipid storage particles in these mutants for structuraldefects by using electron microscopy (Fig. 5). Consistent withNile red staining, the number of lipid droplets in cdc50� cellswas greater than wild type when viewed by electron micros-copy. In wild-type cells, we observed 0.9 lipid droplet per cellsection (n 186), whereas in cdc50� cells there were 1.8droplets per cell section (n 84). In ume6� cells, the numberof lipid droplets observed by electron microscopy was alsogreater than wild type (2.1 droplets per cell section; n 40).Because optical sections of Nile red-stained cells represent a
greater thickness through the cell than thin sections for elec-tron microscopy, the average number of lipid droplets countedon electron micrographs was fewer than those observed byfluorescence microscopy. When observed by electron micros-copy, the lipid droplet cortex in wild-type cells was surroundedby a discrete darkly stained “shell” (Fig. 5). In ume6� cells, thelipid droplet shell was exaggerated and clearly thicker than inwild-type cells (Fig. 5). In cdc50� cells, however, lipid dropletsappeared less well defined and had no distinct border (Fig. 5).These results not only confirmed previous results showing anincrease in Nile red-stained lipid droplets in cdc50� andume6� cells, but indicated lipid storage particle assembly wasdefective in both mutants. These findings also indicated thatCDC50 and UME6 have dramatically different effects on lipiddroplet ultrastructure.
(ii) Sterol homeostasis is disrupted by mutations affectingthe intracellular distribution of unesterified sterols. One ofthe deletion mutants identified (arv1�) has an established de-fect in ergosterol localization (6, 72). To determine if any ofother deletion mutations we identified disrupt the normal er-gosterol distribution, cells were fixed and treated with filipincomplex to visualize unesterified sterol lipids. Filipin is a spe-cific fluorescent probe for unesterified sterol localization inboth mammalian cells and yeast (6, 62). In wild-type yeast,filipin fluorescence is observed at the plasma membrane (6)and this pattern of localization is consistent with previous stud-ies that showed that ergosterol, the most abundant yeast sterol,is concentrated in the plasma membrane (87). In addition tothe plasma membrane staining, 15.7% of wild-type cells exhib-ited small filipin-fluorescent cytoplasmic spots. In 8.5% ofwild-type cells, membrane strands were also observed (Table 5and Fig. 6). In terms of morphology and localization, the mem-brane strands are consistent with peripheral ER. This could
FIG. 5. Ultrastructure of ume6� and cdc50� lipid droplet defects.Wild-type (WT; left panels), ume6� (middle panels), and cdc50�(right panels) cells were examined by electron microscopy. In wild-typecells, lipid droplets are distinguished by their light gray/white appear-ance encircled by a thin, dark, electron-dense border (black arrows).The discrete border surrounding the cortex of lipid droplets is exag-gerated in ume6� cells (white arrows) and absent in cdc50� cells. Thelarge electron-dense structures are vacuoles, and, consistent with pre-vious reports (39), vacuolar fragmentation was observed in cdc50�cells. The scale bar is 2 �m.
408 FEI ET AL. EUKARYOT. CELL
on January 22, 2015 by guesthttp://ec.asm
.org/D
ownloaded from
not be confirmed because of technical limitations of usingfilipin, which prevented costaining with ER markers. Nonethe-less, these results affirmed that the plasma membrane is theprimary repository of unesterified sterols in S. cerevisiae, butadditional filipin-stained structures were also evident.
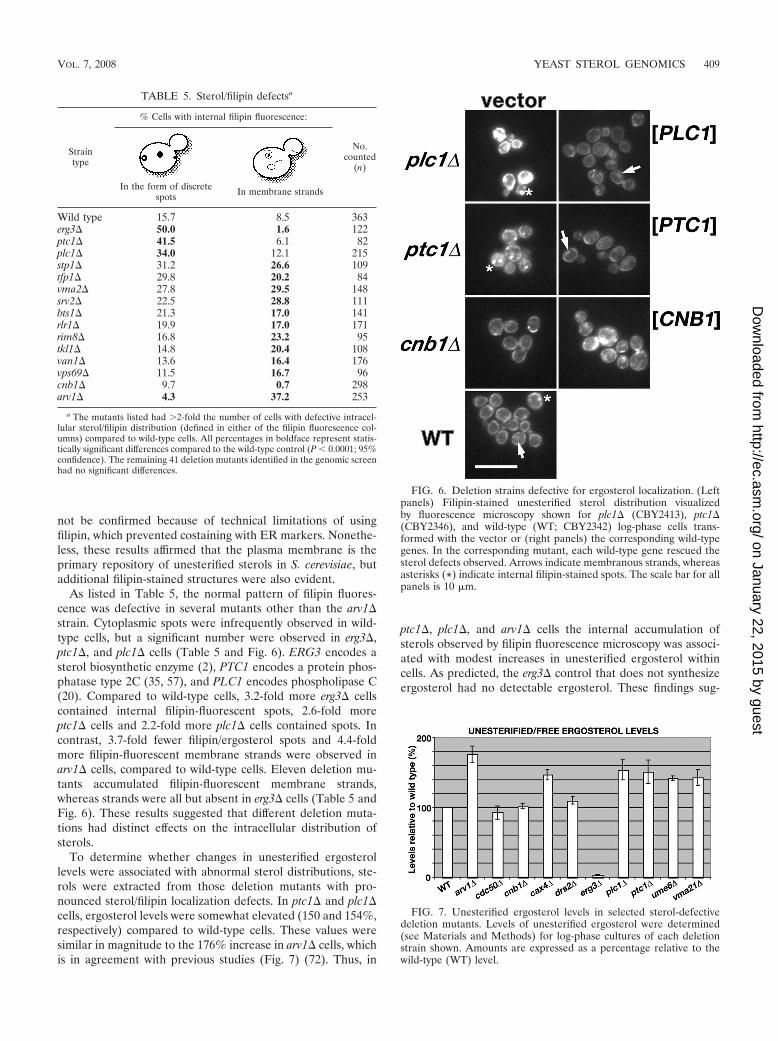
As listed in Table 5, the normal pattern of filipin fluores-cence was defective in several mutants other than the arv1�strain. Cytoplasmic spots were infrequently observed in wild-type cells, but a significant number were observed in erg3�,ptc1�, and plc1� cells (Table 5 and Fig. 6). ERG3 encodes asterol biosynthetic enzyme (2), PTC1 encodes a protein phos-phatase type 2C (35, 57), and PLC1 encodes phospholipase C(20). Compared to wild-type cells, 3.2-fold more erg3� cellscontained internal filipin-fluorescent spots, 2.6-fold moreptc1� cells and 2.2-fold more plc1� cells contained spots. Incontrast, 3.7-fold fewer filipin/ergosterol spots and 4.4-foldmore filipin-fluorescent membrane strands were observed inarv1� cells, compared to wild-type cells. Eleven deletion mu-tants accumulated filipin-fluorescent membrane strands,whereas strands were all but absent in erg3� cells (Table 5 andFig. 6). These results suggested that different deletion muta-tions had distinct effects on the intracellular distribution ofsterols.
To determine whether changes in unesterified ergosterollevels were associated with abnormal sterol distributions, ste-rols were extracted from those deletion mutants with pro-nounced sterol/filipin localization defects. In ptc1� and plc1�cells, ergosterol levels were somewhat elevated (150 and 154%,respectively) compared to wild-type cells. These values weresimilar in magnitude to the 176% increase in arv1� cells, whichis in agreement with previous studies (Fig. 7) (72). Thus, in
ptc1�, plc1�, and arv1� cells the internal accumulation ofsterols observed by filipin fluorescence microscopy was associ-ated with modest increases in unesterified ergosterol withincells. As predicted, the erg3� control that does not synthesizeergosterol had no detectable ergosterol. These findings sug-
FIG. 6. Deletion strains defective for ergosterol localization. (Leftpanels) Filipin-stained unesterified sterol distribution visualizedby fluorescence microscopy shown for plc1� (CBY2413), ptc1�(CBY2346), and wild-type (WT; CBY2342) log-phase cells trans-formed with the vector or (right panels) the corresponding wild-typegenes. In the corresponding mutant, each wild-type gene rescued thesterol defects observed. Arrows indicate membranous strands, whereasasterisks (*) indicate internal filipin-stained spots. The scale bar for allpanels is 10 �m.
TABLE 5. Sterol/filipin defectsa
Straintype
% Cells with internal filipin fluorescence:
No.counted
(n)
In the form of discretespots In membrane strands
Wild type 15.7 8.5 363erg3� 50.0 1.6 122ptc1� 41.5 6.1 82plc1� 34.0 12.1 215stp1� 31.2 26.6 109tfp1� 29.8 20.2 84vma2� 27.8 29.5 148srv2� 22.5 28.8 111bts1� 21.3 17.0 141rlr1� 19.9 17.0 171rim8� 16.8 23.2 95tkl1� 14.8 20.4 108van1� 13.6 16.4 176vps69� 11.5 16.7 96cnb1� 9.7 0.7 298arv1� 4.3 37.2 253
a The mutants listed had 2-fold the number of cells with defective intracel-lular sterol/filipin distribution (defined in either of the filipin fluorescence col-umns) compared to wild-type cells. All percentages in boldface represent statis-tically significant differences compared to the wild-type control (P � 0.0001; 95%confidence). The remaining 41 deletion mutants identified in the genomic screenhad no significant differences.
FIG. 7. Unesterified ergosterol levels in selected sterol-defectivedeletion mutants. Levels of unesterified ergosterol were determined(see Materials and Methods) for log-phase cultures of each deletionstrain shown. Amounts are expressed as a percentage relative to thewild-type (WT) level.
VOL. 7, 2008 YEAST STEROL GENOMICS 409
on January 22, 2015 by guesthttp://ec.asm
.org/D
ownloaded from

gested that like ARV1, PTC1 and PLC1 play a role in steroltrafficking within yeast cells.
We also investigated whether any of the lipid storage muta-tions affected unesterified ergosterol levels. Compared to thewild-type control, the level of unesterified ergosterol in cdc50�cells was unchanged and the ergosterol levels in cax4� andume6� strains were only a little higher (Fig. 7). These findingsindicated that in the mutants with lipid droplet mutations,increased concentrations of unesterified ergosterol were notnecessarily coupled with defects in sterol storage.
In mammalian cells, the late endosome represents a sortingcompartment for internalized cholesterol (54). To determinewhether the cytoplasmic filipin-fluorescent spots observed inyeast correspond to an endosomal compartment, cells wereincubated with the endosome-specific dye FM4-64 (78) andthen fixed and stained with filipin. FM4-64 is a fluorescentlipophilic dye that is internalized from the yeast plasma mem-brane, through endosomal compartments, to the vacuole (78).Colocalization of filipin and FM4-64 fluorescence was detected25 min after FM4-64 internalization but not at earlier times ofendocytosis (Fig. 8). This finding suggested that in wild-typecells, filipin stains both ergosterol in the plasma membraneand, in a minority of cells, sterols in late endosomes.
To determine whether the excess filipin-stained spots ob-served in plc1� and ptc1� cells corresponded to endosomes,cells were incubated with FM4-64 prior to fixation and thenfilipin stained. After 25 min (and not before), the colocaliza-tion of FM4-64- and filipin-stained spots was significantly
greater in plc1� and ptc1� cells than in wild-type cells (Fig. 8).In wild-type cells (n 239), filipin fluorescence was detected in8% of FM4-64-stained late endosomes. In contrast, 41% oflate endosomes costained with filipin in plc1� cells (n 310)and 27% costained with filipin in ptc1� cells (n 255). Theseresults suggested that in the absence of PLC1 or PTC1 func-tion, unesterified sterols accumulated in late endosomes,though not exclusively.
DISCUSSION
In a genome-wide screen, we identified 56 mutants from theyeast nonessential deletion collection that were susceptible todrug-induced perturbations in sterol homeostasis. Sterol ho-meostasis is maintained through the interplay of several pro-cesses: sterol transport between membranes, the regulation ofsterol biosynthesis, and the storage of sterol esters in lipiddroplets/lipid storage particles (Fig. 9). Some of the mutantswe identified affected sterol homeostasis as a result of defectsin lipid droplet generation (Fig. 9). Direct examination ofspecific mutants defined at least two distinct events in lipidstorage particle biogenesis, namely neutral lipid synthesis andlipid droplet organelle assembly (Fig. 10). Further biochemicalanalysis of neutral-lipid synthesis in some of these mutantsrevealed an unanticipated link between neutral-lipid synthesisand secretory protein glycosylation. Yet another group of mu-tants we identified had mutations that affected sterol ho-meostasis through defects in the membrane localization ofunesterified sterols (Fig. 9). These results implicated phospho-lipase C (Plc1p) and protein phosphatase type 2C (Ptc1p) inthe intracellular trafficking of unesterifed sterols. These find-ings affirmed that multiple independent pathways contribute tothe maintenance of cellular sterol-lipid homeostasis.
Previous genomic studies have analyzed the pharmacologi-cal effects of sterol-targeting drugs on yeast deletion strains(21, 34, 49), but the causal basis for the drug sensitivity was notexplored. The nonessential deletion collection was also
FIG. 8. Colocalization of FM4-64 late endosome fluorescence andinternal filipin fluorescence. Wild-type (WT; BY4741), ptc1�(CBY2448), and plc1� (CBY2464) strains were incubated withFM4-64 in synthetic medium for 25 min at 30°C. Cells were fixed andstained with filipin, and the coincident staining of FM4-64-fluorescentendosomes (red) and filipin-stained membranes (false-colored green)was observed by fluorescence microscopy. In wild-type cells, internalfilipin-stained spots overlapped with FM4-64-fluorescent late endo-somes, as shown by arrows pointing to overlapping yellow spots in themerged image. Asterisks (*) indicate examples of filipin-stained spotsthat did not colocalize with FM4-64. The colocalizations detected didnot represent fluorescence bleed-through since FM4-64 was not de-tected by DAPI (4�,6�-diamidino-2-phenylindole) fluorescence and fili-pin was not detected by Texas red fluorescence channels (data notshown). The scale bar for all panels is 10 �m.
FIG. 9. Processes and genes contributing to the maintenance ofergosterol homeostasis. The control of sterol synthesis, sterol transportbetween membranes, and the storage of sterols as neutral lipid estersall contribute to sterol homeostasis. The functional redundancy be-tween these processes was the premise for the genomic screen thatidentified 56 sterol homeostasis genes. Of the 56 genes, those whosesterol-related function was previously established, as well as thosewhose specific role in sterol homeostasis was determined in this study,are shown.
410 FEI ET AL. EUKARYOT. CELL
on January 22, 2015 by guesthttp://ec.asm
.org/D
ownloaded from

screened for mutants that cannot grow in anaerobic cultures inorder to identify sterol uptake mutants (56). Oxygen is essen-tial for sterol synthesis, and under anaerobic conditions yeastmust import sterols from the medium to survive. Under theseanaerobic conditions, however, sterol-esterification-defectivemutants grow normally, which explains why lipid storage mu-tants were not represented in the list of 37 anaerobically sen-sitive mutants (56). In fact, only 2 of these 37 deletion mutants(tkl1� and rlr1�) were also identified by our approach. As such,our study complements previous genome-wide screens andidentifies novel sterol-associated genes.
As a confirmation of the efficacy of our approach, we iden-tified several deletion mutants that correspond to previouslyidentified sterol-related genes. Deletion mutations that affectisoprenoid and sterol biosynthesis were identified, includingbts1�, ram1�, erg3�, and erg6� (15), as well as arv1�, whichaffects the normal distribution of unesterified sterols (72).Many of the deletion mutations corresponded to general tran-scription factors that could affect the expression of genes re-quired for sterol biosynthesis and homeostasis. However, thedeletion of UME6, a transcriptional regulator in mitotic andmeiotic cells (18, 69, 71, 77), had pronounced and specificeffects on sterol-lipid storage. Thus, our unbiased analysis ofyeast nonessential gene deletions identified both predicted tar-gets as well as novel mutations not previously linked to sterolhomeostasis.
In our mutant identification, there were some genes withdirect and inferred connections to sterol homeostasis that werenot detected. Because the screen was performed under aerobicconditions, most anaerobic mutations that affect sterol uptakewere not identified (56), and of course essential genes or thosewith redundant/overlapping functions would also not be de-tected. We note that deletion mutations representing lipiddroplet-localized proteins were not detected. From genome-wide protein localization studies (4, 27), we compiled a list of29 potential lipid droplet proteins. Of the 29 correspondingdeletion mutants, only 2 mutants had lipid droplet defects asobserved by Nile red fluorescence microscopy. In tgl3� cells, anincrease in Nile red/lipid droplet fluorescence was detected
whereas ldb16/ycl005w� cells had decreased numbers of lipiddroplets (unpublished results); TGL3 encodes a TAG lipase(4) and LDB16 has a potential role in protein glycosylation(11). These results suggested that the vast majority of lipiddroplet proteins are either functionally redundant or not re-quired for lipid droplet formation/maintenance. Since proteinson lipid droplets have such limited effects, the implication isthat lipid droplet biogenesis is mainly regulated by nonresidentproteins.
Two distinct steps in lipid storage particle biogenesis. Fromthe studies of many different cell types, a general model forlipid droplet biogenesis has been established (14, reviewed inreference 42). This model posits that neutral lipid synthesisoccurs in specific ER microdomains wherein lipid dropletscoalesce between the bilayer leaflets. Lipid storage particlematuration results after the neutral lipid core is sheathed witha phospholipid monolayer and buds from the ER surface intothe cytoplasm. In Fig. 10, our findings are integrated into thismodel to depict the distinct events in yeast lipid droplet bio-genesis revealed by the defects in cax4�, cdc50�, ume6�,vma9�, and vma21� cells.
The first requisite step in lipid droplet biogenesis is thesynthesis of its bulk neutral lipid components by the acyltrans-ferases ASAT (Are1p and Are2p) and DGAT/PDAT (Dga1pand Lro1p). Remarkably, the expression of these enzymes wasdependent on CAX4, which otherwise has a secondary role insecretory protein N glycosylation (19, 76). Lipid droplet bio-genesis was also dependent on SEC53, which also affects N-linked glycosylation (30). However, direct glycosylation did notappear to play a role in regulating the expression or turnoverof the neutral lipid acyltransferases. Alternatively, acyltrans-ferase expression might be indirectly affected by signaling path-ways that respond to unfolded and/or unglycosylated secretoryproteins. However, none of the glycoprotein folding and/orquality control mutants we tested (i.e., ire1�, HAC1-238[S238A] [40], and cne1�) had significant lipid droplet defects(unpublished results). Another less-understood consequenceof N-glycosylation defects is a concomitant change in sphingo-lipid composition (52). Whereas in cax4� cells, the reduction
FIG. 10. A model for the initial steps in lipid storage particle biogenesis in yeast. (A) The requisite first step in lipid droplet formation in theER is the synthesis of neutral lipids, the core component of storage particles. (B) ASAT, encoded by ARE1 and ARE2, esterifies sterols with fattyacids (R), which produces a major lipid component of the emerging lipid droplet. The other neutral-lipid component is triacylglycerol, which issynthesized by DGAT/PDAT. Neutral-lipid synthesis is dependent on CAX4 and SEC53, whereas VMA9 negatively affected ASAT but notDGAT/PDAT activities. (C) After synthesis, neutral lipids either coalesce spontaneously within the membrane bilayer or are actively amalgamated.The deletion of UME6 or CDC50 disrupted lipid droplet morphology. (D) After release from the ER, neutral lipids are sheathed in a phospholipidmonolayer and resident lipid droplet proteins are associated with the periphery (14, 38). As in mammalian cells, mature lipid droplet organellesmight arise from the fusion of smaller particles and the release of stored lipid esters from formed lipid droplets is mediated by acyl-lipases.
VOL. 7, 2008 YEAST STEROL GENOMICS 411
on January 22, 2015 by guesthttp://ec.asm
.org/D
ownloaded from

in sphingolipid (IPC) levels was associated with fewer lipiddroplets, we found that the number of lipid droplets dramati-cally increased in mutants that block sphingolipid biosynthesis.These findings suggest a potential link between sphingolipids,N glycosylation, and neutral-lipid storage, but the mechanismis anything but straightforward.
Other mutants were defective in another aspect of yeast lipiddroplet biogenesis, the structural assembly of the lipid-storageparticle (Fig. 10). Both CDC50 and UME6 deletion mutationshad striking effects on lipid droplet structure. In addition to amarked increase in the number of lipid droplets and a modestincrease in neutral lipids, the lipid storage particles in cdc50�cells exhibited a distinctive morphological defect. Lipid drop-lets in cdc50� cells lacked the discrete electron-dense cortex,suggesting that CDC50 affects the addition of protein(s) ontolipid droplets. In contrast, UME6 had the opposite effect on thestructural assembly of lipid droplet organelles. In ume6� cells,the intensity of Nile red fluorescence was more intense and theelectron-dense shell surrounding the lipid storage particles wasmore prominent than in wild-type cells. Since Ume6p is aCys[6] zinc binuclear transcription factor that coordinates mi-totic and meiotic gene expression (69), these findings suggest arole for Ume6p in regulating the mitotic and meiotic prolifer-ation of lipid storage particles. UME6 might affect lipid dropletmaturation during meiosis and sporulation, when large in-creases in neutral-lipid content occur (28). Despite their dif-ferent effects, both UME6 and CDC50 define new processes inlipid storage particle assembly.
Potential regulators of intracellular ergosterol distribution.In wild-type yeast, sterol lipids are concentrated in the plasmamembrane (87), but some filipin/sterol fluorescence was ob-served in internal membranes. Based on the overlap of filipin/FM4-64 fluorescence, some of the internalized sterols corre-spond to the late endosome. Many deletion mutants affectedthe normal pattern of filipin/sterol staining, but plc1� andptc1� cells exhibited the most striking defects. In these dele-tion mutants, a significant increase in filipin fluorescence wasobserved, most of which corresponded to endosomes. Sinceunesterified and esterified sterol levels in plc1� and ptc1� cellswere only modestly higher than wild type, increases in internalfilipin fluorescence were not caused by larger amounts of ste-rols but rather by sterol redistribution. A simple explanationfor this redistribution is that endosomal sorting of sterols isdefective in PLC1 and PTC1 mutants. Of the many endosomalmutants represented in the deletion collection, however, nonewere identified in our screen, suggesting that the role of PLC1and PTC1 in sterol sorting is independent of establishedendosomal trafficking pathways.
Tentative connections have been reported that link PLC1and PTC1 to endosomal function. PTC1 encodes a PP2C phos-phatase that is best described as a negative regulator of theHOG mitogen-activated protein kinase pathway, which re-sponds to osmotic stress by increasing cellular glycerol concen-trations (85). Independent of this function, however, ptc1�genetically interacts with conditional alleles of the clathrinheavy chain gene (9), which affect Golgi and endocytic traf-ficking (51). Ptc1p purportedly binds the ASAT Are2p (26),although no lipid droplet defects were detected in our analysisof ptc1� cells. Plc1p, the phospholipase C homologue, hydro-lyzes phosphatidylinositol bisphosphate to produce DAG (20),
which in turn stimulates vacuolar membrane dynamics (29). Inplc1� cells, fragmented vacuoles accumulate, suggesting a de-fect in vacuolar/endosomal trafficking or vacuole fusion (29).Both Ptc1p and Plc1p also share a link to osmoregulation (32),and both PTC1 and PLC1 are linked to calcium signaling (29,63, 73). Regardless of whether sterol trafficking involves theseor a novel function, Ptc1p and Plc1p appear to play an impor-tant role in how sterols are distributed within cells.
Another potential regulator of sterols that was identified iscalcineurin. In the absence of the calcineurin regulatory sub-unit, encoded by CNB1, cells were lovastatin sensitive butnystatin resistant. However, no significant sterol biosynthesis,sterol transport, or sterol storage defects were detected in ouranalysis of cnb1� cells. This finding is noteworthy only becauseseveral of the other deletion mutations identified (i.e., bts1�,bck1�, cax4�, drs2�, ptc1�, van1�, vma2�, and vma21�) arelethal in combination with cnb1� (44, 45, 49, 63, 75). In both S.cerevisiae and Candida species, calcineurin has been previouslyimplicated in promoting resistance to sterol biosynthetic inhib-itors through an adaptive mechanism (12, 13, 17, 24, 48). Per-haps calcineurin plays a similar role in adaptation to the de-fects in sterol homeostasis caused by some nonessentialdeletion mutants.
We note that many of the sterol homeostasis genes that wereidentified by this yeast functional/chemical genomics approachare conserved in mammals. In particular, yeast genes that af-fect lipid droplet biogenesis might play a conserved role inhumans. For instance, the link between neutral-lipid synthesisand secretory protein glycosylation might be applicable to hu-man adipocytes, as in yeast. Thus, the study of sterol homeosta-sis in yeast might not only be pertinent to human cholesterolregulation, but also valuable for providing novel gene targetsfor treating obesity.
ACKNOWLEDGMENTS
We give special thanks to Trisha Davis, Wanjin Hong, Joe Nickels,Randy Schekman, Janet Shaw, and Kazuma Tanaka for strains, anti-bodies, and plasmid constructs. We gratefully acknowledge MichelLeroux, Keith Kozminski, and Nancy Hawkins for suggestions on themanuscript and Aaron Chiam for technical assistance on the analysisof lipid droplet resident proteins.
C.T.B. was supported by grants from by a Natural Science andEngineering Research Council (NSERC) of Canada, an SFU Presi-dent’s Research Grant, and by joint contributions for microscopyequipment from the Canadian Foundation for Innovation (CFI) andthe British Columbia Knowledge and Development Fund (BCKDF).H.Y. was supported by the Ministry of Education, National Medicaland Biomedical Research Councils of Singapore.
This study was initiated at the University of California, Berkeley.
REFERENCES
1. Adams, A., D. E. Gottsschling, C. A. Kaiser, and T. Stearns. 1997. Methodsin yeast genetics. Cold Spring Harbor Harbor Laboratory Press, Cold SpringHarbor, NY.
2. Arthington, B. A., L. G. Bennett, P. L. Skatrud, C. J. Guynn, R. J. Barbuch,C. E. Ulbright, and M. Bard. 1991. Cloning, disruption and sequence of thegene encoding yeast C-5 sterol desaturase. Gene 102:39–44.
3. Ashrafi, K., F. Y. Chang, J. L. Watts, A. G. Fraser, R. S. Kamath, J. Ahringer,and G. Ruvkun. 2003. Genome-wide RNAi analysis of Caenorhabditis elegans fatregulatory genes. Nature 421:268–271.
4. Athenstaedt, K., D. Zweytick, A. Jandrositz, S. D. Kohlwein, and G. Daum.1999. Identification and characterization of major lipid particle proteins ofthe yeast Saccharomyces cerevisiae. J. Bacteriol. 181:6441–6448.
5. Basson, M. E., M. Thorsness, and J. Rine. 1986. Saccharomyces cerevisiaecontains two functional genes encoding 3-hydroxy-3-methylglutaryl-coen-zyme A reductase. Proc. Natl. Acad. Sci. USA 83:5563–5567.
412 FEI ET AL. EUKARYOT. CELL
on January 22, 2015 by guesthttp://ec.asm
.org/D
ownloaded from

6. Beh, C. T., and J. Rine. 2004. A role for yeast oxysterol-binding proteinhomologs in endocytosis and in the maintenance of intracellular sterol-lipiddistribution. J. Cell Sci. 117:2983–2996.
7. Beh, C. T., V. Brizzio, and M. D. Rose. 1997. KAR5 encodes a novel pher-omone-inducible protein required for homotypic nuclear fusion. J. Cell Biol.139:1063–1076.
8. Beh, C. T., L. Cool, J. Phillips, and J. Rine. 2001. Overlapping functions ofthe yeast oxysterol-binding protein homologues. Genetics 157:1117–1140.
9. Bensen, E. S., G. Costaguta, and G. S. Payne. 2000. Synthetic genetic inter-actions with temperature-sensitive clathrin in Saccharomyces cerevisiae: rolesfor synaptojanin-like Inp53p and dynamin-related Vps1p in clathrin-depen-dent protein sorting at the trans-Golgi network. Genetics 154:83–97.
10. Chen, C. Y., M. F. Ingram, P. H. Rosal, and T. R. Graham. 1999. Role forDrs2p, a P-type ATPase and potential aminophospholipid translocase, inyeast late Golgi function. J. Cell Biol. 147:1223–1236.
11. Corbacho, I., I. Olivero, and L. M. Hernandez. 2005. A genome-wide screenfor Saccharomyces cerevisiae nonessential genes involved in mannosyl phos-phate transfer to mannoprotein-linked oligosaccharides. Fungal Gen. Biol.42:773–790.
12. Cowen, L. E., and S. Lindquist. 2005. Hsp60 potentiates the rapid evolutionof new traits: drug resistance in diverse fungi. Science 309:2185–2189.
13. Cruz, M. C., A. L. Goldstein, J. R. Blankenship, M. Del Poeta, D. Davis,M. E. Cardenas, J. R. Perfect, J. H. McCusker, and J. Heitman. 2002.Calcineurin is essential for survival during membrane stress in Candidaalbicans. EMBO J. 21:546–559.
14. Czabany, T., K. Athenstaedt, and G. Daum. 2007. Synthesis, storage anddegradation of neutral lipids in yeast. Biochim. Biophys. Acta 12:299–309.
15. Daum, G., N. D. Lees, M. Bard, and R. Dickson. 1998. Cell biology of lipidsof Saccharomyces cerevisiae. Yeast 14:1471–1510.
16. Du, X., Y. H. Pham, and A. J. Brown. 2004. Effects of 25-hydroxycholesterolon cholesterol esterification and sterol regulatory element-binding proteinprocessing are dissociable: implications for cholesterol movement to theregulatory pool in the endoplasmic reticulum. J. Biol. Chem. 279:47010–47016.
17. Edlind, T., L. Smith, K. Henry, S. Katiyar, and J. Nickels. 2002. Antifungalactivity in Saccharomyces cerevisiae is modulated by calcium signalling. Mol.Microbiol. 46:257–268.
18. Einerhand, A. W. C., W. Kos, W. C. Smart, A. J. Kal, H. K. Tabak, and T. G.Cooper. 1995. The upstream region of the FOX3 gene encoding peroxisomal3-oxoacyl-coenzyme A thiolase in Saccharomyces cerevisiae contains ABF1-and replication protein A-binding sites that participate in its regulation byglucose repression. Mol. Cell. Biol. 15:3405–3414.
19. Fernandez, F., J. S. Rush, D. A. Toke, G. S. Han, J. E. Quinn, G. M. Carman,J. Y. Choi, D. R. Voelker, M. Aebi, and C. J. Waechter. 2001. CWH8 geneencodes a dolichyl pyrophosphate phosphatase with a luminally orientedactive site in the endoplasmic reticulum of Saccharomyces cerevisiae. J. Biol.Chem. 276:41455–41464.
20. Flick, J. S., and J. Thorner. 1998. An essential function of a phosphoinositide-specific phospholipase C is relieved by inhibition of a cyclin-dependent proteinkinase in the yeast Saccharomyces cerevisiae. Genetics 148:33–47.
21. Giaever, G., A. M. Chu, L. Ni, C. Connelly, L. Riles, S. Veronneau, S. Dow, A.Lucau-Danila, K. Anderson, B. Andre, A. P. Arkin, A. Astromoff, M. El-Bakk-oury, R. Bangham, R. Benito, S. Brachat, S. Campanaro, M. Curtiss, K. Davis,A. Deutschbauer, K. D. Entian, P. Flaherty, F. Foury, D. J. Garfinkel, M.Gerstein, D. Gotte, U. Guldener, J. H. Hegemann, S. Hempel, Z. Herman, D. F.Jaramillo, D. E. Kelly, S. L. Kelly, P. Kotter, D. LaBonte, D. C. Lamb, N. Lan,H. Liang, H. Liao, L. Liu, C. Luo, M. Lussier, R. Mao, P. Menard, S. L. Ooi,J. L. Revuelta, C. J. Roberts, M. Rose, P. Ross-Macdonald, B. Scherens, G.Schimmack, B. Shafer, D. D. Shoemaker, S. Sookhai-Mahadeo, R. K. Storms,J. N. Strathern, G. Valle, M. Voet, G. Volckaert, C. Y. Wang, T. R. Ward, J.Wilhelmy, E. A. Winzeler, Y. Yang, G. Yen, E. Youngman, K. Yu, H. Bussey,J. D. Boeke, M. Snyder, P. Philippsen, R. W. Davis, and M. Johnston. 2002.Functional profiling of the Saccharomyces cerevisiae genome. Nature 418:387–391.
22. Graham, T. R. 2004. Flippases and vesicle-mediated protein transport.Trends Cell Biol. 14:670–677.
23. Graham, T. R., and S. D. Emr. 1991. Compartmental organization of Golgi-specific protein modification and vacuolar protein sorting events defined ina yeast sec18 (NSF) mutant. J. Cell Biol. 114:207–218.
24. Heitman, J. 2005. A fungal Achilles’ heel. Science 309:2175–2176.25. Hill, K. J., and T. H. Stevens. 1994. Vma21p is a yeast membrane protein
retained in the endoplasmic reticulum by a di-lysine motif and is required forthe assembly of the vacuolar H(�)-ATPase complex. Mol. Biol. Cell 5:1039–1050.
26. Ho, Y., A. Gruhler, A. Heilbut, G. D. Bader, L. Moore, S. L. Adams, A. Millar,P. Taylor, K. Bennett, K. Boutilier, L. Yang, C. Wolting, I. Donaldson, S.Schandorff, J. Shewnarane, M. Vo, J. Taggart, M. Goudreault, B. Muskat, C.Alfarano, D. Dewar, Z. Lin, K. Michalickova, A. R. Willems, H. Sassi, P. A.Nielsen, K. J. Rasmussen, J. R. Andersen, L. E. Johansen, L. H. Hansen, H.Jespersen, A. Podtelejnikov, E. Nielsen, J. Crawford, V. Poulsen, S. D. Sorensen,J. Matthiesen, R. C. Hendrickson, F. Gleeson, T. Pawson, M. F. Moran, D.Durocher, M. Mann, C. W. Hogue, D. Figeys, and M. Tyers. 2002. Systematic
identification of protein complexes in Saccharomyces cerevisiae by mass spec-trometry. Nature 415:180–183.
27. Hua, W. K., J. V. Falvo, L. C. Gerke, A. S. Carroll, R. W. Howson, J. S.Weissman, and E. K. O’Shea. 2003. Global analysis of protein localization inbudding yeast. Nature 425:671–672.
28. Illingworth, R. F., A. H. Rose, and A. Beckett. 1973. Changes in the lipidcomposition and fine structure of Saccharomyces cerevisiae during ascusformation. J. Bacteriol. 113:373–386.
29. Jun, Y., R. A. Fratti, and W. Wickner. 2004. Diacylglycerol and its formationby phospholipase C regulate Rab- and SNARE-dependent yeast vacuolefusion. J. Biol. Chem. 279:53186–53195.
30. Kepes, F., and R. Schekman. 1988. The yeast SEC53 gene encodes phos-phomannomutase. J. Biol. Chem. 263:9155–9161.
31. Kishimoto, T., T. Yamamoto, and K. Tanaka. 2005. Defects in structuralintegrity of ergosterol and the Cdc50p-Drs2p putative phospholipid translo-case cause accumulation of endocytic membranes, onto which actin patchesare assembled in yeast. Mol. Biol. Cell 16:5592–5609.
32. Lin, H., P. Nguyen, and A. Vancura. 2002. Phospholipase C interacts withSgd1p and is required for expression of GPD1 and osmoresistance in Sac-charomyces cerevisiae. Mol. Genet. Genomics 267:313–320.
33. Liscum, L., and N. J. Munn. 1999. Intracellular cholesterol transport. Bio-chim. Biophys. Acta 1438:19–37.
34. Lum, P. Y., C. D. Armour, S. B. Stepaniants, G. Cavet, M. K. Wolf, J. S.Butler, J. C. Hinshaw, P. Garnier, G. D. Prestwich, A. Leonardson, P.Garrett-Engele, C. M. Rush, M. Bard, G. Schimmack, J. W. Phillip, C. J.Roberts, and D. D. Shoemaker. 2004. Discovering modes of action for ther-apeutic compounds using a genome-wide screen of yeast heterozygotes. Cell116:121–137.
35. Maeda, T., S. M. Wurgler-Murphy, and H. Saito. 1994. A two-componentsystem that regulates an osmosensing MAP kinase cascade in yeast. Nature369:242–245.
36. Malkus, P., L. A. Graham, T. H. Stevens, and R. Schekman. 2004. Role ofVma21p in assembly and transport of the yeast vacuolar ATPase. Mol. Biol.Cell 15:5075–5091.
37. Maxfield, F. R., and A. K. Menon. 2006. Intracellular sterol transport anddistribution. Curr. Opin. Cell Biol. 18:379–385.
38. McCammon, M. T., M.-A. Hartmann, C. D. Bottema, and L. W. Parks. 1984.Sterol methylation in Saccharomyces cerevisiae. J. Bacteriol. 157:475–483.
39. Misu, K., K. Fujimura-Kamada, T. Ueda, A. Nakano, H. Katoh, and K.Tanaka. 2003. Cdc50p, a conserved endosomal membrane protein, controlspolarized growth in Saccharomyces cerevisiae. Mol. Biol. Cell 14:730–747.
40. Mori, K., N. Ogawa, T. Kawahara, H. Yanagi, and T. Yura. 2000. mRNAsplicing-mediated C-terminal replacement of transcription factor Hac1p isrequired for efficient activation of the unfolded protein response. Proc. Natl.Acad. Sci. USA 97:4660–4665.
41. Moser, M. J., J. R. Geiser, and T. N. Davis. 1996. Ca2�-calmodulin promotessurvival of pheromone-induced growth arrest by activation of calcineurin andCa2�-calmodulin-dependent protein kinase. Mol. Cell. Biol. 16:4824–4831.
42. Murphy, D. J., and J. Vance. 1999. Mechanisms of lipid-body formation.Trends Biochem. Sci. 24:109–115.
43. Nagiec, M. M., J. A. Baltisberger, G. B. Wells, and R. L. Lester. 1994. TheLCB2 gene of Saccharomyces and the related LCB1 gene encode subunits ofserine palmitoyltransferase, the initial enzyme in sphingolipid synthesis.Proc. Natl. Acad. Sci. USA 91:7899–7902.
44. Nakamura, T., T. Ohmoto, D. Hirata, E. Tsuchiya, and T. Miyakawa. 1996.Genetic evidence for the functional redundancy of the calcineurin- andMpk1-mediated pathways in the regulation of cellular events important forgrowth in Saccharomyces cerevisiae. Mol. Gen. Genet. 251:211–219.
45. Natarajan, P., J. Wang, Z. Hua, and T. R. Graham. 2004. Drs2p-coupledaminophospholipid translocase activity in yeast Golgi membranes and rela-tionship to in vivo function. Proc. Natl. Acad. Sci. USA 101:10614–10619.
46. Oelkers, P., A. Tinkelenberg, N. Erdeniz, D. Cromley, J. T. Billheimer, andS. L. Sturley. 2000. A lecithin cholesterol acyltransferase-like gene mediatesdiacylglycerol esterification in yeast. J. Biol. Chem. 275:15609–15612.
47. Ohashi, M. A., J. Gibson, I. Gregor, and G. Schatz. 1982. Import of proteinsinto mitochondria. J. Biol. Chem. 257:13042–13047.
48. Onyewu, C., J. R. Blankenship, M. Del Poeta, and J. Heitman. 2003. Ergos-terol biosynthesis inhibitors become fungicidal when combined with cal-cineurin inhibitors against Candida albicans, Candida glabrata, and Candidakrusei. Antimicrob. Agents Chemother. 47:956–964.
49. Parsons, A. B., R. L. Brost, H. Ding, Z. Li, C. Zhang, B. Sheikh, G. W.Brown, P. M. Kane, T. R. Hughes, and C. Boone. 2004. Integration ofchemical-genetic and genetic interaction data links bioactive compounds tocellular target pathways. Nat. Biotechnol. 22:62–69.
50. Parsons, A. B., A. Lopez, I. E. Givoni, D. E. Williams, C. A. Gray, J. Porter,G. Chua, R. Sopko, R. L. Brost, C. H. Ho, J. Wang, T. Ketela, C. Brenner,J. A. Brill, G. E. Fernandez, T. C. Lorenz, G. S. Payne, S. Ishihara, Y. Ohya,B. Andrews, T. R. Hughes, B. J. Frey, T. R. Graham, R. J. Andersen, and C.Boone. 2006. Exploring the mode-of-action of bioactive compounds by chem-ical-genetic profiling in yeast. Cell 126:611–625.
51. Payne, G. S., D. Baker, E. van Tuinen, and R. Schekman. 1988. Protein
VOL. 7, 2008 YEAST STEROL GENOMICS 413
on January 22, 2015 by guesthttp://ec.asm
.org/D
ownloaded from

transport to the vacuole and receptor-mediated endocytosis by clathrin heavychain-deficient yeast. J. Cell Biol. 106:1453–1461.
52. Pittet, M., D. Uldry, M. Aebi, and A. Conzelmann. 2006. The N-glycosylationdefect of cwh8� yeast cells causes a distinct defect in sphingolipid biosyn-thesis. Glycobiology 16:155–164.
53. Pomorski, T., R. Lombardi, H. Riezman, P. F. Devaux, G. van Meer, andJ. C. Holthuis. 2003. Drs2p-related P-type ATPases Dnf1p and Dnf2p arerequired for phospholipid translocation across the yeast plasma membraneand serve a role in endocytosis. Mol. Biol. Cell 14:1240–1254.
54. Prinz, W. A. 2002. Cholesterol trafficking in the secretory and endocyticsystems. Semin. Cell Dev. Biol. 13:197–203.
55. Reider, S. E., L. M. Banta, K. Kohrer, J. M. McCaffery, and S. D. Emr. 1996.Multilamellar endosome-like compartment accumulates in the yeast vps28vacuolar protein sorting mutant. Mol. Biol. Cell 7:985–999.
56. Reiner, S., D. Micolod, G. Zellnig, and R. Schneiter. 2006. A genomewidescreen reveals a role of mitochondria in anaerobic uptake of sterols in yeast.Mol. Biol. Cell 17:90–103.
57. Robinson, M. K., W. H. van Zyl, E. M. Phizicky, and J. R. Broach. 1994.TPD1 of Saccharomyces cerevisiae encodes a protein phosphatase 2C-likeactivity implicated in tRNA splicing and cell separation. Mol. Cell. Biol.14:3634–3645.
58. Saito, K., K. Fujimura-Kamada, N. Furuta, U. Kato, M. Umeda, and K.Tanaka. 2004. Cdc50p, a protein required for polarized growth, associateswith the Drs2p P-type ATPase implicated in phospholipid translocation inSaccharomyces cerevisiae. Mol. Biol. Cell 15:3418–3432.
59. Sambade, M., and P. M. Kane. 2004. The yeast vacuolar proton-translocatingATPase contains a subunit homologous to the Manduca sexta and bovine esubunits that is essential for function. J. Biol. Chem. 279:17361–17365.
60. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: alaboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, ColdSpring Harbor, NY.
61. Sandager, L., M. H. Gustavsson, U. Stahl, A. Dahlqvist, E. Wiberg, A. Banas,M. Lenman, H. Ronne, and S. Stymne. 2002. Storage lipid synthesis isnon-essential in yeast. J. Biol. Chem. 277:6478–6482.
62. Severs, N. J. 1997. Cholesterol cytochemistry in cell biology and disease.Subcell. Biochem. 28:477–505.
63. Shitamukai, A., D. Hirata, S. Sonobe, and T. Miyakawa. 2004. Evidence forantagonistic regulation of cell growth by the calcineurin and high osmolarityglycerol pathways in Saccharomyces cerevisiae. J. Biol. Chem. 279:3651–3661.
64. Sikorski, R. S., and P. Hieter. 1989. A system of shuttle vectors and yeasthost strains designed for efficient manipulation of DNA in Saccharomycescerevisiae. Genetics 122:19–27.
65. Soccio, R. E., and J. L. Breslow. 2004. Intracellular cholesterol transport.Aterioscler. Thromb. Vasc. Biol. 24:1150–1160.
66. Sorger, D., K. Athenstaedt, C. Hrastnik, and G. Daum. 2004. A yeast strainlacking lipid particles bears a defect in ergosterol formation. J. Biol. Chem.279:31190–31196.
67. Sorger, D., and G. Daum. 2003. Triacylglycerol biosynthesis in yeast. Appl.Microbiol. Biotechnol. 61:289–299.
68. Steck, T. L., and Y. Lange. 2002. SCAP, an ER sensor that regulates cellcholesterol. Dev. Cell 3:306–308.
69. Strich, R., R. T. Surosky, C. Steber, E. Dubois, F. Messenguy, and R. E.Esposito. 1994. UME6 is a key regulator of nitrogen repression and meioticdevelopment. Genes Dev. 8:796–810.
70. Sturley, S. L. 2000. Conservation of eukaryotic sterol homeostasis: newinsights from studies in budding yeast. Biochim. Biophys. Acta 1529:155–163.
71. Sweet, D. H., Y. K. Jang, and G. B. Sancar. 1997. Role of UME6 in tran-scriptional regulation of a DNA repair gene in Saccharomyces cerevisiae.Mol. Cell. Biol. 17:6223–6235.
72. Tinkelenberg, A. H., Y. Liu, F. Alcantara, S. Khan, Z. Guo, M. Bard, andS. L. Sturley. 2000. Mutations in yeast ARV1 alter intracellular sterol distri-bution and are complemented by human ARV1. J. Biol. Chem. 275:40667–40670.
73. Tisi, R., F. Belotti, S. Wera, J. Winderickx, J. M. Thevelein, and E. Marte-gani. 2004. Evidence for inositol triphosphate as a second messenger forglucose-induced calcium signaling in budding yeast. Curr. Genet. 45:83–89.
74. Tolbert, J. A. 2003. Lovastatin and beyond: the history of the HMG-CoAreductase inhibitors. Nat. Rev. Drug Discov. 2:517–526.
75. Tong, A. H., M. Evangelista, A. B. Parsons, H. Xu, G. D. Bader, N. Page, M.Robinson, S. Raghibizadeh, C. W. Hogue, H. Bussey, B. Andrews, M. Tyers,and C. Boone. 2001. Systematic genetic analysis with ordered arrays of yeastdeletion mutants. Science 294:2364–2368.
76. van Berkel, M. A., M. Rieger, S. te Heesen, A. F. Ram, H. van den Ende, M.Aebi, and F. M. Klis. 1999. The Saccharomyces cerevisiae CWH8 gene isrequired for full levels of dolichol-linked oligosaccharides in the endoplasmicreticulum and for efficient N-glycosylation. Glycobiology 9:243–253.
77. Vershon, A. K., and M. Pierce. 2000. Transcriptional regulation of meiosis inyeast. Curr. Opin. Cell Biol. 12:334–339.
78. Vida, T. A., and S. D. Emr. 1995. A new vital stain for visualizing vacuolarmembrane dynamics and endocytosis in yeast. J. Cell Biol. 128:779–792.
79. Vik, A., and J. Rine. 2001. Upc2p and Ecm22p, dual regulators of sterolbiosynthesis in Saccharomyces cerevisiae. Mol. Cell. Biol. 21:6395–6405.
80. Walker-Caprioglio, H. M., J. M. MacKenzie, and L. W. Parks. 1989. Anti-bodies to nystatin demonstrate polyene sterol specificity and allow immuno-labeling of sterols in Saccharomyces cerevisiae. Antimicrob. Agents Che-mother. 33:2092–2095.
81. Winzeler, E. A., D. D. Shoemaker, A. Astromoff, H. Liang, K. Anderson, B.Andre, R. Bangham, R. Benito, J. D. Boeke, H. Bussey, A. M. Chu, C.Connelly, K. Davis, F. Dietrich, S. W. Dow, M. El Bakkoury, F. Foury, S. H.Friend, E. Gentalen, G. Giaever, J. H. Hegemann, T. Jones, M. Laub, H.Liao, N. Liebundguth, D. J. Lockhart, A. Lucau-Danila, M. Lussier, N.M’Rabet, P. Menard, M. Mittmann, C. Pai, C. Rebischung, J. L. Revuelta, L.Riles, C. J. Roberts, P. Ross-MacDonald, B. Scherens, M. Snyder, S.Sookhai-Mahadeo, R. K. Storms, S. Veronneau, M. Voet, G. Volckaert, T. R.Ward, R. Wysocki, G. S. Yen, K. Yu, K. Zimmermann, P. Philippsen, M.Johnston, and R. W. Davis. 1999. Functional characterization of the Sac-charomyces cerevisiae genome by gene deletion and parallel analysis. Science285:901–906.
82. Woods, R. A. 1971. Nystatin-resistant mutants of yeast: alterations in sterolcontent. J. Bacteriol. 108:69–73.
83. Xu, X. X., and I. Tabas. 1991. Sphingomyelinase enhances low densitylipoprotein uptake and ability to induce cholesteryl ester accumulation inmacrophages. J. Biol. Chem. 266:24849–24858.
84. Yang, H., M. Bard, D. A. Bruner, A. Gleeson, R. J. Deckelbaum, G. Aljinovic,T. M. Pohl, R. Rothstein, and S. L. Sturley. 1996. Sterol esterification inyeast: a two-gene process. Science 272:1353–1356.
85. Young, C., J. Mapes, J. Hanneman, S. Al-Zarban, and I. Ota. 2002. Role ofPtc2 type 2C Ser/Thr phosphatase in yeast high-osmolarity glycerol pathwayinactivation. Eukaryot. Cell 1:1032–1040.
86. Zhang, Q., H. K. Chieu, C. P. Low, S. Zhang, C. K. Heng, and H. Yang. 2003.Schizosaccharomyces pombe cells deficient in triacylglycerols synthesis un-dergo apoptosis upon entry into the stationary phase. J. Biol. Chem. 278:47145–47155.
87. Zinser, E., F. Paltauf, and G. Daum. 1993. Sterol composition of yeastorganelle membranes and subcellular distribution of enzymes involved insterol metabolism. J. Bacteriol. 175:2853–2858.
88. Zweytick, D., K. Athenstaedt, and G. Daum. 2000. Intracellular lipid particlesof eukaryotic cells. Biochim. Biophys. Acta 1469:101–120.
414 FEI ET AL. EUKARYOT. CELL
on January 22, 2015 by guesthttp://ec.asm
.org/D
ownloaded from




















