
Sites of limited proteolysis in the pyruvate decarboxylase component of the pyruvate dehydrogenase...
12
Sites of limited proteolysis in the pyruvate decarboxylase component of the pyruvate dehydrogenase multienzyme complex of Bacillus stearothermophilus and their role in catalysis Hitesh J. Chauhan, Gonzalo J. Domingo, Hyo-Il Jung and Richard N. Perham Cambridge Centre for Molecular Recognition, Department of Biochemistry, University of Cambridge, UK The E1 component (pyruvate decarboxylase) of the pyruvate dehydrogenase complex of Bacillus stearothermophilus is a heterotetramer (a 2 b 2 ) of E1a and E1b polypeptide chains. The domain structure of the E1a and E1b chains, and the protein–protein interactions involved in assembly, have been studied by means of limited proteolysis. It appears that there may be two conformers of E1a in the E1 heterotetramer, one being more susceptible to proteolysis than the other. A highly conserved region in E1a, part of a surface loop at the entrance to the active site, is the most susceptible to cleavage in E1 (a 2 b 2 ). As a result, the oxidative decarboxylation of pyruvate catalysed by E1 in the presence of dichlorophenol indophenol as an artificial electron acceptor is markedly enhanced, but the reductive acetylation of a free lipoyl domain is unchanged. The parameters of the interaction between cleaved E1 and the peripheral subunit-binding domain of the dihydrolipoyl acetyltransferase E2 component are identical to those of the wild-type E1. However, a pyruvate dehydrogenase complex assembled in vitro with cleaved E1p exhibits a markedly lower overall catalytic activity than that assembled with untreated E1. This implies that active site coupling between the E1 and E2 components has been impaired. This has important implications for the way in which a tethered lipoyl domain can interact with E1 in the assembled complex. Keywords: pyruvate decarboxylase; pyruvate dehydrogenase; multienzyme complex; limited proteolysis; enzyme catalysis. 2-Oxo acid dehydrogenase multienzyme complexes consist of multiple copies of three component enzymes that bring about the oxidative decarboxylation of 2-oxo acids and the transfer of the resulting acyl group to coenzyme A. In the pyruvate dehydrogenase (PDH) complexes of Gram-negative bacteria, such as Escherichia coli, these enzymes are normally found as three different types of polypeptide chain, corresponding to the pyruvate decarboxylase (E1p; EC 1.2.4.1), dihydrolipoyl acetyl- transferase (E2p; EC 2.3.1.12) and dihydrolipoyl dehydrogenase (E3; EC 1.8.1.4) components, and the E2p core consists of 24 copies of the E2p chain arranged with octahedral symmetry. On the other hand, the PDH complexes of Gram-positive bacteria, such as Bacillus stearothermophilus, and of mammals contain four different types of polypeptide chain: E1a and E1b (which form E1p, a 2 b 2 ), E2p and E3; and the E2p core consists of 60 copies of the E2p chain arranged with icosahedral symmetry (reviewed [1–3]). The B. stearothermophilus E2p chain consists of three domains separated by flexible linker regions: an N-terminal lipoyl domain that hosts the specific lysine residue (Lys42) to which the lipoyl group is attached; a peripheral subunit-binding domain (PSBD) [4], responsible for the tight but noncovalent binding of E1p and E3; and a C-terminal acetyltransferase domain responsible for assembly of the 60-mer icosahedral inner core [5] and catalytic transfer of the acetyl group to coenzyme A [1,6]. The E3 component, a homodimeric FAD- dependent enzyme, regenerates the oxidized form of lipoic acid, with the concomitant reduction of NAD. As in all icosahedral PDH complexes, the E1p of the B. stearothermophilus PDH complex is an a 2 b 2 heterotetramer of M r 152 000 [7]. The E1a chain (41 kDa) houses the conserved thiamin diphosphate-binding site [8,9], whereas the E1b chain (35 kDa) is responsible for the tight noncovalent binding of E1p to E2p [10–12], chiefly by interaction with the PSBD [13]. Reductive acetylation of the lipoyl group is the rate-limiting step of the overall PDH complex reaction [14–16]. Free lipoic acid or lipoamide can function as substrates for the E2p and E3 of the E. coli PDH complex [17], but E1p requires the lipoyl group to be attached to a folded lipoyl domain, for which k cat /K m is raised by a factor of 10 4 [18]. Moreover, E1 is highly specific for the lipoyl domain of its cognate E2 chain for both the PDH and 2-oxoglutarate dehydrogenase (OGDH) complexes [16,18,19]. The specificity of this interaction [20,21] forms the molecular basis of an elegant system of substrate Eur. J. Biochem. 267, 7158–7169 (2000) q FEBS 2000 Correspondence to R. N. Perham, Cambridge Centre for Molecular Recognition, Department of Biochemistry, University of Cambridge, 80 Tennis Court Road, Cambridge CB2 1GA, UK. Abbreviations: DCPIP, 2,6-dichlorophenolindophenol; E1, 2-oxo acid decarboxylase; E1a, alpha subunit of E1p; E1b, beta subunit of E1p; E1p, pyruvate decarboxylase of PDH complex; E2p, dihydrolipoyl acetyltransferase; E3, dihydrolipoyl dehydrogenase; FPLC, fast protein liquid chromatography; IPTG, isopropyl thio-b-d-galactoside; LplA, lipoate protein ligase A; OGDH, 2-oxoglutarate dehydrogenase; PDH, pyruvate dehydrogenase; PSBD, peripheral subunit-binding domain; RU, resonance units; SPR, surface plasmon resonance; TEMED, N, N, N 0 , N 0 -tetramethylethylenediamine; ThDP, thiamin diphosphate. Enzymes: pyruvate decarboxylase of pyruvate dehydrogense complex (EC 1.2.4.1); dihydrolipoyl acetyltransferase (EC 2.3.1.12); dihydrolipoyl dehydrogenase (EC 1.8.1.4). (Received 18 August 2000, accepted 12 October 2000)
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Sites of limited proteolysis in the pyruvate decarboxylase component of the pyruvate dehydrogenase...

Sites of limited proteolysis in the pyruvate decarboxylase componentof the pyruvate dehydrogenase multienzyme complex ofBacillus stearothermophilus and their role in catalysis
Hitesh J. Chauhan, Gonzalo J. Domingo, Hyo-Il Jung and Richard N. Perham
Cambridge Centre for Molecular Recognition, Department of Biochemistry, University of Cambridge, UK
The E1 component (pyruvate decarboxylase) of the pyruvate dehydrogenase complex of Bacillus
stearothermophilus is a heterotetramer (a2b2) of E1a and E1b polypeptide chains. The domain structure of
the E1a and E1b chains, and the protein±protein interactions involved in assembly, have been studied by means
of limited proteolysis. It appears that there may be two conformers of E1a in the E1 heterotetramer, one being
more susceptible to proteolysis than the other. A highly conserved region in E1a, part of a surface loop at the
entrance to the active site, is the most susceptible to cleavage in E1 (a2b2). As a result, the oxidative
decarboxylation of pyruvate catalysed by E1 in the presence of dichlorophenol indophenol as an artificial
electron acceptor is markedly enhanced, but the reductive acetylation of a free lipoyl domain is unchanged. The
parameters of the interaction between cleaved E1 and the peripheral subunit-binding domain of the dihydrolipoyl
acetyltransferase E2 component are identical to those of the wild-type E1. However, a pyruvate dehydrogenase
complex assembled in vitro with cleaved E1p exhibits a markedly lower overall catalytic activity than that
assembled with untreated E1. This implies that active site coupling between the E1 and E2 components has been
impaired. This has important implications for the way in which a tethered lipoyl domain can interact with E1 in
the assembled complex.
Keywords: pyruvate decarboxylase; pyruvate dehydrogenase; multienzyme complex; limited proteolysis; enzyme
catalysis.
2-Oxo acid dehydrogenase multienzyme complexes consist ofmultiple copies of three component enzymes that bring aboutthe oxidative decarboxylation of 2-oxo acids and the transfer ofthe resulting acyl group to coenzyme A. In the pyruvatedehydrogenase (PDH) complexes of Gram-negative bacteria,such as Escherichia coli, these enzymes are normally found asthree different types of polypeptide chain, corresponding to thepyruvate decarboxylase (E1p; EC 1.2.4.1), dihydrolipoyl acetyl-transferase (E2p; EC 2.3.1.12) and dihydrolipoyl dehydrogenase(E3; EC 1.8.1.4) components, and the E2p core consists of 24copies of the E2p chain arranged with octahedral symmetry. Onthe other hand, the PDH complexes of Gram-positive bacteria,such as Bacillus stearothermophilus, and of mammals containfour different types of polypeptide chain: E1a and E1b (which
form E1p, a2b2), E2p and E3; and the E2p core consists of 60copies of the E2p chain arranged with icosahedral symmetry(reviewed [1±3]).
The B. stearothermophilus E2p chain consists of threedomains separated by flexible linker regions: an N-terminallipoyl domain that hosts the specific lysine residue (Lys42) towhich the lipoyl group is attached; a peripheral subunit-bindingdomain (PSBD) [4], responsible for the tight but noncovalentbinding of E1p and E3; and a C-terminal acetyltransferasedomain responsible for assembly of the 60-mer icosahedralinner core [5] and catalytic transfer of the acetyl group tocoenzyme A [1,6]. The E3 component, a homodimeric FAD-dependent enzyme, regenerates the oxidized form of lipoicacid, with the concomitant reduction of NAD.
As in all icosahedral PDH complexes, the E1p of theB. stearothermophilus PDH complex is an a2b2 heterotetramerof Mr 152 000 [7]. The E1a chain (41 kDa) houses theconserved thiamin diphosphate-binding site [8,9], whereas theE1b chain (35 kDa) is responsible for the tight noncovalentbinding of E1p to E2p [10±12], chiefly by interaction with thePSBD [13].
Reductive acetylation of the lipoyl group is the rate-limitingstep of the overall PDH complex reaction [14±16]. Free lipoicacid or lipoamide can function as substrates for the E2p andE3 of the E. coli PDH complex [17], but E1p requires thelipoyl group to be attached to a folded lipoyl domain, for whichkcat/Km is raised by a factor of 104 [18]. Moreover, E1 ishighly specific for the lipoyl domain of its cognate E2 chainfor both the PDH and 2-oxoglutarate dehydrogenase (OGDH)complexes [16,18,19]. The specificity of this interaction [20,21]forms the molecular basis of an elegant system of substrate
Eur. J. Biochem. 267, 7158±7169 (2000) q FEBS 2000
Correspondence to R. N. Perham, Cambridge Centre for Molecular
Recognition, Department of Biochemistry, University of Cambridge,
80 Tennis Court Road, Cambridge CB2 1GA, UK.
Abbreviations: DCPIP, 2,6-dichlorophenolindophenol; E1, 2-oxo acid
decarboxylase; E1a, alpha subunit of E1p; E1b, beta subunit of E1p;
E1p, pyruvate decarboxylase of PDH complex; E2p, dihydrolipoyl
acetyltransferase; E3, dihydrolipoyl dehydrogenase; FPLC, fast protein
liquid chromatography; IPTG, isopropyl thio-b-d-galactoside; LplA,
lipoate protein ligase A; OGDH, 2-oxoglutarate dehydrogenase; PDH,
pyruvate dehydrogenase; PSBD, peripheral subunit-binding domain;
RU, resonance units; SPR, surface plasmon resonance; TEMED,
N,N,N 0,N 0-tetramethylethylenediamine; ThDP, thiamin diphosphate.
Enzymes: pyruvate decarboxylase of pyruvate dehydrogense complex
(EC 1.2.4.1); dihydrolipoyl acetyltransferase (EC 2.3.1.12); dihydrolipoyl
dehydrogenase (EC 1.8.1.4).
(Received 18 August 2000, accepted 12 October 2000)

channelling in the complexes [1,2]. Crystal structures of thehomologous E1(a2b2) components of the branched-chain 2-oxoacid dehydrogenase complexes from Pseudomonas putida andhumans have been determined [22,23]. These structures showthe E1 heterotetramer to be tightly packed, with a b2 dimer heldbetween the jaws of a `vice' formed by the two a subunits.
Limited proteolysis has proved very useful in the study of thestructure, assembly and mechanism of the 2-oxo aciddehydrogenase complexes [1,6]. With the PDH and OGDHcomplexes from E. coli, trypsin excises the lipoyl domainswithout causing further disassembly [24±27]. Similarly, themain effect of trypsin on the B. stearothermophilus PDHcomplex is cleavage of the E2p chains, accompanied by lossof the peripheral E1p and E3 subunits from the residual E2pcore, and a slower cleavage of the E1a chains [28]. In contrast,treatment of the same complex with chymotrypsin cleaves onlythe lipoyl domains from the E2p chains, and disassembly doesnot ensue [29]. With mammalian PDH complexes, the E1aand E2 chains are again susceptible to limited proteolysis, butin the complex from ox kidney the E1a chains are found tobe the more susceptible [30,31]. Similarly, selective proteolysisof the mammalian OGDH complex with trypsin promotesdissociation of peripheral subunits and rapid inactivation as aresult of a single cleavage in its E1 (a2) component [32]. In allthese instances, the E3 and the E1b components remainuncleaved upon exposure to trypsin or chymotrypsin, hence lossof catalytic activity is associated with proteolytic cleavage ofthe E1a and/or E2 chains.
Studies of the proteolysis of the homodimeric (a2) E1p ofA. vinelandii has indicated that the N-terminal region could beinvolved in binding to the E2p core [33,34]; however, such anN-terminal sequence is absent from the heterotetrameric (a2b2)E1s, suggestive of a different mode of binding in theseenzymes. In the present paper, we describe the results of limiteddigestion of the E1p component of the PDH complex ofB. stearothermophilus with trypsin and chymotrypsin. Theresults are important in defining the domain structure of theE1p polypeptide chains, in separating the catalytic steps inreductive acetylation of the lipoyl domain, and in throwingnew light on the protein±protein interactions involved inassembly and catalysis in the intact complex.
M A T E R I A L S A N D M E T H O D S
Materials
Milli-QTM and deionized water was used for all experimentsunless otherwise stated. For most purposes, reagents ofanalytical grade or the purest available were employed. Allchemicals were obtained from Sigma Chemical Co. Ltd (Poole,UK) unless otherwise stated. Isopropyl thio-b-d-galactoside(IPTG) was from Melford Laboratories (Ipswich, UK).Ampicillin was from Beecham Research Laboratories(Brentford, UK). Kanamycin sulfate was from Calbiochem-Novabiochem Ltd (Nottingham, UK). Bacteriological agar andtryptone were from Unipath Ltd (Basingstoke, UK). Yeastextract was from Beta Lab (West Molesey, UK). Solid[2-14C]pyruvic acid, sodium salt (specific activity,15.9 mCi´mmol21) was from New England Nuclear (NENLife Science Products, Boston, MA, USA).
E. coli host strain TG1 recO was from Amersham Inter-national. Plasmids pKBstE1a and pKBstE1b expressing genesencoding the E1a and E1b subunits, respectively, of theB. stearothermophilus PDH complex have been describedpreviously [7]. E. coli strain BL21 (DE3) pLysS was from
Novagen Inc. Plasmids pETBstE2 and pBSTNAV/E3 forexpression of the B. stearothermophilus E2 and E3 genes,respectively, have been described previously [35]. E. colihost strain BL21 (DE3) [36] was from Novagen Inc. Theplasmid pET11d2D encodes the di-domain (lipoyl domainplus PSBD), which comprises the first 171 amino-acidresidues of the B. stearothermophilus E2p chain (S. J. Bowden& R. N. Perham, unpublished work). The plasmid pET11ThDDencodes a thrombin-cleavable di-domain (ThDD), with athrombin-cleavage site inserted in the linker region betweenthe lipoyl domain and the PSBD [20].
Purification of E1(a2b2), E1a and E1b
Cultures of E. coli strain TG1 recO cells transformed withplasmids pKBstE1a or pKBstE1b, were grown in Luria±Bertanimedium and induced with IPTG to over-express B. stearo-thermophilus E1p and its constituent subunits (E1a and E1b),as described elsewhere [7].
Purification of E2p and E3
The 60-mer apo-E2 component was purified from E. coli BL21(DE3) pLysS cells transformed with pETBstE2 and the E3component from E. coli TG1 recO cells transformed withpBSTNAV/E3 [35].
Purification of di-domain and lipoyl domain
Cultures of E. coli strain BL21 (DE3) cells transformed withthe plasmid pET11d2D were grown in 2 � TY medium andinduced with IPTG to over-express di-domain. The di-domainwas purified essentially as described elsewhere [37]. Culturesof E. coli strain BL21 (DE3) cells transformed with the plasmidpET11ThDD were also grown in 2 � TY medium and inducedwith IPTG to over-express ThDD. Purified ThDD was treatedwith thrombin in order to detach the lipoyl domain from thePSBD, and then subsequently with chymotrypsin to remove theflexible linker from the C-terminus (at Phe85) of the lipoyldomain [20].
Lipoylation of unlipoylated lipoyl domain
The apo-form of the lipoyl domain was lipoylated using E. colirecombinant lipoate-protein ligase A (LplA) obtained fromE. coli BL21 (DE3) cells transformed with the plasmid TM202[38]. The procedure was carried out essentially as describedelsewhere [39]. Mass spectrometry verified that the lipoyldomain was lipoylated (Mr 9458 Da for lipoylated protein). E2(Mr 46383 Da for lipoylated chain) [35] and di-domain (Mr
18573 Da for lipoylated protein) [39] were lipoylated similarly.
Proteolysis of the E1(a2b2) component
The E1p component (6 mg´mL21) was exposed to trypsin(TPCK-treated) or chymotrypsin (TLCK-treated) in 20 mmpotassium phosphate buffer, pH 7.0, on ice at a substrate toenzyme ratio of 200 : 1 (w/w), for various periods of time. Theproteolytic action was terminated by adding phenylmethane-sulfonyl fluoride (final concentration 1 mm). Whenever the useof chymotrypsin is mentioned, this refers to a-chymotrypsin.
PAGE
Proteins or peptides were analyzed by means of SDS/PAGE(10±20% acrylamide resolving gel, 4% acrylamide stacking
q FEBS 2000 Limited proteolysis of the E1 component (Eur. J. Biochem. 267) 7159

gel) using a discontinuous Tris/glycine buffer system [40] ora Pharmacia PhastSystem (Pharmacia Biotech Ltd, St Albans,UK), in which case either 12 or 20% ready-made (precast)gels (Phastgels, Pharmacia) were utilized. After electro-phoresis, gels were stained with Coomassie Brilliant BlueR-250 or electroblotted onto ProBlott poly(vinylidene difluoride)membranes for N-terminal sequence analysis. Some gels, afterdestaining, were subjected to densitometric analysis using anEpson GT9000 scanner, with the software packages Adobephotoshop and phoretix 1D gel analysis. NondenaturingPAGE utilizing 12 or 20% precast gels was carried out using aPharmacia Phastsystem.
RP-HPLC
A protein C4 reversed phase column with the appropriateprotein C4 guard column (Vydac, 300 AÊ , 5 mm, 4.6 mmi.d. � 250 mm, Hesperia, CA, USA) was equilibrated with 5%(HPLC grade) acetonitrile 1 0.1% trifluoroacetic acid. Theprotein/peptide sample (10±100 mg in 50 mL) was theninjected and the column washed with 5% acetonitrile 1 0.1%trifluoroacetic acid for 10 min at a flow rate of 0.5 mL´min21.After 10 min, a gradient starting at 5% acetonitrile 1 0.1%trifluoroacetic acid and ending at 100% acetonitrile 1 0.1%trifluoroacetic acid was applied over 50 min. Finally, thecolumn was washed with 100% acetonitrile 1 0.1% trifluoro-acetic acid for a further 10 min before re-equilibrating with 5%acetonitrile 1 0.1% trifluoroacetic acid. Fractions of 200 mLwere collected.
Mass spectrometry
Protein or peptide samples which were not in a suitable solventfor mass spectrometry were buffer exchanged into waterusing centrifugal filter devices (Millipore) with the appropriatecut-off before mixing with an equal volume of acetonitrile and1% (v/v) acetic acid. Positive electrospray ionization (ESI)mass spectrometry was performed either on a VG BioQquadrapole mass spectrometer (VG Fisons, VG BiotechAltrincham, Cheshire, UK) or a Quattro-LC triple quadrapolemass spectrometer (Micromass) using sperm whale myoglobinas the calibration standard. MALDI mass spectrometry wasperformed on a MALDI IV time-of-flight (TOF) instrument(KRATOS) in the Protein and Nucleic Acid Chemistry Facilityin the Department of Biochemistry.
Enzyme assays
DCPIP assay for oxidative decarboxylation activity of E1. Thecatalytic activity of wild-type and proteolysed E1 wasmeasured in 1-mL cuvettes at 30 8C using DCPIP as electronacceptor [41]. The linear decrease in the absorbance at 600 nmover the first 30 s was monitored [7]. The molar absorptioncoefficient at 600 nm for DCPIP is 22 000 m21´cm21. One unitof activity is the amount of enzyme required to reduce 1 mmolof DCPIP per min under the conditions of the assay.
Assay for reductive acetylation activity of E1. The reductiveacetylation of lipoyl domains by wild-type and proteolysed E1component was assayed using a modified version of the method
Fig. 1. Time-course of limited proteolysis of B. stearothermophilus E1p, as revealed by SDS/PAGE. E1p was exposed to trypsin (A) or chymotrypsin (B)
for times between 0 and 720 min, and overnight (Z, < 24 h). Digestion was carried out in 20 mm potassium phosphate, pH 7.0, on ice at an enzyme/substrate
ratio of 1 : 200 (w/w). In (A), the pattern of bands does not change up to 180 min, but two additional bands (at 14 and 8 kDa) appear at longer exposure
times. In (B), a similar pattern of bands occurs up to 180 min, apart from the almost complete absence of the 22-kDa fragment. An additional faint band
(8 kDa) appears if the digestion is prolonged. Lane M, molecular mass markers (42, 30, 20 and 14 kDa); lane L, molecular mass markers (17, 14, 10, 8 and
6 kDa).
7160 H. J. Chauhan et al. (Eur. J. Biochem. 267) q FEBS 2000

of Packman et al. [42], essentially as described elsewhere [39].The incorporation of 14C from sodium [2-14C]pyruvate into thelipoyl domain in the absence of coenzyme A was measuredafter precipitating the lipoyl domain with cold 10% (w/v)trichloroacetic acid at measured times. Duplicate samples werecounted twice for 1 min. Blank reactions were carried out withunlipoylated lipoyl domain or no lipoyl domain.
Assay for PDH complex activity. The PDH activities ofcomplexes reconstituted with wild-type and proteolysed E1component were measured in 1-mL cuvettes at 30 8C in aspectrophotometer using the NAD1-reduction methoddescribed elsewhere [13]. The molar ratio E1/E2/E3 used was0.5 : 1.0 : 1.0. The linear increase in the absorbance at 340 nmover the first 30 s was monitored. One unit of PDH activity isdefined as the amount of enzyme required to generate 1 mmolof NADH per min. The specific catalytic activity of thereconstituted PDH complex is expressed as units per mg of E2.
Surface plasmon resonance analysis
The interaction of immobilized, lipoylated di-domain withwild-type and proteolysed E1 component was investigatedusing surface plasmon resonance (SPR) detection (BIAcore,Pharmacia Biosensor AB). All experiments were performed at25 8C using a flow rate of 5 mL´min21. The di-domain wasimmobilized on a CM5 sensor chip using the surface thiolmethod [12]. The immobilization levels of di-domain variantswere adjusted from 50 to 150 resonance units (RU) to
minimize potential mass transport phenomena. To obtainkinetic parameters, the E1 was injected and allowed to interactwith the sensor surface for 3 min (association phase) atconcentrations ranging from 12.5 to 200 nm. The flow wascontinued for a further 5 min with the equilibrium buffer toobtain the dissociation phase. The sensor was regenerated asdescribed previously [12]. The association and dissociation ofsurface-bound complex was treated as a pseudo-first-orderreaction [12]. Where it was possible from the experimentaldata, values for kon (association rate constant), koff (dissociationrate constant) and Kd (equilibrium dissociation constant) weredetermined using the biaevaluation software supplied by themanufacturer.
R E S U L T S
Digestion of the E1p component with trypsin orchymotrypsin
The isolated E1p (a2b2) component (153 kDa) of the PDHcomplex of B. stearothermophilus was treated with eithertrypsin or chymotrypsin (1 : 200, w/w) under nondenaturingconditions (0 8C, pH 7) for up to 24 h. In both instances, theE1a component was cleaved into several fragments, whereasthe E1b component remained intact. With trypsin, the E1acomponent (41 kDa) was cleaved to form three stable
Fig. 2. Amino-acid sequence of the B. stearothermophilus E1a chain,
with proteolytic cleavage sites identified. Underlined R or K residues are
sites of limited proteolysis with trypsin and underlined Y or W residues are
sites of limited proteolysis with chymotrypsin (see Table 1). In all cases,
cleavage occurs on the C-terminal side of the residue indicated. The region
(residues 172±202) in italics is the ThDP-binding motif [9].
Fig. 3. Model of the E1a subunit of B. stearothermophilus PDH
complex. This structure is based on the structure of the E1a subunit in
the E1a2b2 of P. putida 2-oxoisovalerate dehydrogenase complex [22]. The
structure was calculated using the automated protein modeling server,
swissmodel. There is 31% sequence identity of the template (the P. putida
E1a subunit) with the target sequence (E1a subunit of B. stearothermo-
philus PDH complex). The bound cofactor, ThDP, is shown in ball and stick
conformation, and the Mg21 is shown as a grey sphere. All regions which
are cleaved by trypsin/chymotrypsin are in loops, and are depicted in
various colours: RL (residues 71±72, red), RF (residues 203±204, dark
green), KQ (residues 212±213, light green), YRSKE (residues 281±285,
blue), and WA (residues 290±291, yellow). The N-terminus is at the bottom
left, the C-terminus at the top right. The diagram was created using the
program molscript [43].
q FEBS 2000 Limited proteolysis of the E1 component (Eur. J. Biochem. 267) 7161

polypeptides of apparent sizes 30, 22 and 10 kDa, as judged bySDS/PAGE (Fig. 1A). With chymotrypsin, the two stableproducts formed from the E1a component were 30 and10 kDa in size, also as judged by SDS/PAGE (Fig. 1B). Thepattern of the bands remained the same throughout thedigestion, from 15 to 180 min; at longer times, up to < 24 h,the pattern was similar but, with trypsin in particular, additionalweaker bands (at 14 and 8 kDa) appeared. Densitometry of thegels revealed that exposure to chymotrypsin led to < 35% ofthe E1a chain being cut after just 15 min, with a further 10±20% being cut by 240 min. Moreover, after prolonged exposure(up to 720 min), only < 70% had been cleaved. Even after 24 hexposure to proteinase, the E1a band was not completelydegraded (with trypsin and chymotrypsin, respectively, < 5%and < 20% remained undigested).
Identification of proteolytic cleavage sites
The products of digesting E1 with trypsin or chymotrypsin wereseparated by means of RP-HPLC. The supposed fragments ofE1a were identified by their mobility on SDS/PAGE (data notshown) and then subjected to N-terminal sequence analysis andmass spectrometry. The 30-kDa fragment generated withtrypsin was found to have the same N-terminal sequence(GVKTFQ-) as native E1a; the N-terminal sequence of the22-kDa fragment was found to be LGFYAP-, and that ofthe 10-kDa fragment to be SKELEN-(major) and ELENEW-(minor). With chymotrypsin, the 30-kDa fragment alsoexhibited the same N-terminal sequence (GVKTFQ-) asundigested E1a but the 10-kDa fragment revealed twoN-terminal sequences, RSKELE-and AKKDPL-. The 14-kDafragment that was generated after long periods of incubationwith trypsin was found to have the N-terminal sequenceLGFYAP-, and the 8-kDa fragment revealed three N-terminalsequences: GVKTFQ-, FAISTP-and QTVAKT-. All theseN-terminal sequences can be located in the amino acidsequence of E1a (Fig. 2).
The masses obtained for various of the fragments by massspectrometry (Table 1), enabled an accurate estimation of theirC-terminal extents. The 30-kDa tryptic fragment was observedto contain two species with slightly different molecular masses,consistent with their being N-terminal portions of E1agenerated by cleavages at positions Arg282 (major) andLys284 (minor). Similarly, the 10-kDa fragment containedtwo species, with molecular masses and N-terminal sequencesconsistent with their being the corresponding C-terminalportions generated by cleavages at Arg282 (major) andLys284 (minor). Conclusive mass spectra were not obtainedfor the 22-, 14- and 8-kDa fragments resulting from trypticdigestion of E1 (see Fig. 1). With chymotrypsin, the mass ofthe 30-kDa fragment was consistent with a cleavage of the E1achain at Tyr281. The 10-kDa fragment again exhibited twospecies, consistent with their being C-terminal portions of theE1a chain, generated by cleavages at Tyr281 (major) andTrp290 (minor).
A structural model of the E1a subunit of the B. stearo-thermophilus PDH complex with the proteolysis sites high-lighted is shown in Fig. 3.
Gel filtration of E1p after limited proteolysis
Samples of E1(a2b2), E1a and E1b were each run in turn on acalibrated Superdex 200 gel-filtration column equilibrated with20 mm potassium phosphate, pH 7.0. The E1a component, theE1b component, and E1a2b2 (heterotetramer) were eluted atdifferent volumes (data not shown), as described previously [7].Samples of proteolytic digests of E1a2b2 were also run onthe same Superdex 200 column. The elution volume of thetreated E1a2b2 was the same as that of the untreated E1a2b2,and SDS/PAGE revealed that all the fragments remainedassociated during gel filtration (Fig. 4). The proteolyticdigestion evidently caused no disassembly of the E1a2b2 andno major fragments became detached.
Table 1. Sizes of the proteolytic fragments of B. stearothermophilus E1p (a2b2) determined by means of electrospray mass spectrometry. The exact
masses of the corresponding peptides were calculated from the E1a sequence; their N-terminal sequences, as determined experimentally are also given. The
N-terminal sequence shown in parentheses is an inference based on the mass spectrum of the peptide and its possible position in the E1a sequence.
Protein/peptide fraction N-terminal sequence Estimated molecular mass (Da) Mass calculated from sequence (Da)
Undigested E1
E1a- GVKTFQ- 41 343 �^ 13 41 338 (res. 1±368)
E1b- AQMTMV- 35 339 �^ 12 35 329 (res. 1±324)
Undigested E1a GVKTFQ- 41 342 �^ 8 41 338 (res. 1±368)
Undigested E1b AQMTMV- 35 337 �^ 10 35 329 (res. 1±324)
Trypsin-treated E1
E1a- GVKTFQ- 41 356 �^ 8 41 338 (res. 1±368)
E1b- AQMTMV- 35 340 �^ 13 35 329 (res. 1±324)
30 k- GVKTFQ- 31 251 �^ 13 31 238 (res. 1±284)
30 k- GVKTFQ- 31 034 �^ 14 31 022 (res. 1±282)
10 k- SKELEN- 10 335 �^ 1 10 316 (res. 283±368)
10 k- (ELENEW-) 10 120 �^ 1 10 100 (res. 285±368)
Chymotrypsin-treated E1
E1a- GVKTFQ- 41 338 �^ 13 41 338 (res. 1±368)
E1b- AQMTMV- 35 338 �^ 10 35 329 (res. 1±324)
30 k- GVKTFQ- 30 872 �^ 10 30 866 (res. 1±281)
10 k- RSKELE- 10 495 �^ 6 10 472 (res. 282±368)
10 k- AKKDPL- 9318 �^ 3 9300 (res. 291±368)
7162 H. J. Chauhan et al. (Eur. J. Biochem. 267) q FEBS 2000

Interaction of proteolysed E1p with the E2p component
In the B. stearothermophilus PDH complex, the E1p com-ponent is bound to the E2p core mainly by tight interaction withthe PSBD of the E2p chains. This interaction can be studiedconveniently in vitro using E1p and a recombinant di-domain ofthe E2p chain comprising the lipoyl domain and PSBD joinedby the native linker region [11]. Samples of trypsin- orchymotrypsin-treated E1p (incubated for 600 min) that hadbeen subjected to gel filtration on a Superdex 200 column,and hence were free of any proteolytic enzymes (trypsin/chymotrypsin), were mixed with a sample of excess di-domainin 20 mm potassium phosphate, pH 7.0. After 30 min at 0 8C,the mixture was subjected to nondenaturing PAGE and gel
filtration. As judged by nondenaturing PAGE (Fig. 5A), thebinding of E1p to the PSBD of E2p, revealed by the decrease inthe electrophoretic mobility of the E1p [11], was unaffectedby the limited proteolysis with trypsin. Likewise, the trypsin-treated E1p component was now eluted earlier from theSuperdex 200 column, separated from the surplus unbounddi-domain, again consistent with its having bound tightly to thePSBD (Fig. 5B). From the size of the peak of the residual freedi-domain, it could be inferred that the trypsin-treated E1p wasbinding the PSBD in a 1 : 1 molar ratio, as described for nativeE1p (a2b2) [11]. Identical results were obtained withchymotrypsin-treated E1p (data not shown).
Surface plasmon resonance (SPR) detection was used todetermine the kinetic parameters for the interaction between
Fig. 4. Gel filtration of B. stearothermophilus
E1p (a2b2) before and after limited
proteolysis with trypsin or chymotrypsin.
Samples were analysed by means of SDS/PAGE.
(A) Proteolysis with trypsin for 720 min at
pH 7.0 and 0 8C. Lane 1, untreated E1; lane 2,
trypsin-treated E1 before gel filtration; lane 3,
trypsin-treated E1 after gel filtration; lane 4,
molecular mass markers (42, 30, 20 and 14 kDa);
lane 5, molecular mass markers (17, 14, 10, 8 and
6 kDa). The apparent molecular masses of the
major bands in lanes 2 and 3 are 41, 35, 30, 23,
22, 14, 10 and 8 kDa. (B) As in (A), except
that the proteolytic enzyme was chymotrypsin.
Lane 1, untreated E1; lane 2,
chymotrypsin-treated E1 before gel filtration;
lane 3, chymotrypsin-treated E1 after gel
filtration; lanes 4 and 5, molecular mass markers.
The apparent molecular masses of the bands in
lanes 2 and 3 are 41, 35, 30 and 10 kDa.
Fig. 5. Interaction of proteinase-treated E1p
of B. stearothermophilus with E2p di-domain.
A sample of trypsin-treated E1a2b2 was mixed
with E2p di-domain (at a molar ratio of E1 to
di-domain of 1 : 1.5) in 20 mm potassium
phosphate, pH 7.0. After 30 min, the mixture
was submitted to nondenaturing PAGE and gel
filtration. (A) Nondenaturing PAGE. Lane 1,
native E1p; lane 2, E2p di-domain; lane 3, native
E1p 1 di-domain; lane 4, trypsin-treated E1p;
lane 5, trypsin treated E1p 1 di-domain. The
shift in the position of the band of E1p (lower
electrophoretic mobility) indicates tight
association with the di-domain (lanes 3 1 5).
The trypsin-treated E1p migrates slightly
differently from the native E1p; probably as a
result of a change in the conformation and/or
charge state of the E1 after proteolysis. (B) Gel
filtration on a Superdex 200 column in 20 mm
potassium phosphate, pH 7.0. Solid line,
trypsin-treated E1p in the absence of di-domain;
dashed line, di-domain in the absence of E1p;
hatched areas, trypsin-treated E1p after
addition of di-domain. The shift in the peak of
trypsin-treated E1p to a position of higher
molecular mass in the presence of di-domain
indicates tight association, and the areas of the
peaks are consistent with a stoichiometry of
interaction of 1 : 1 (E1p:di-domain).
q FEBS 2000 Limited proteolysis of the E1 component (Eur. J. Biochem. 267) 7163
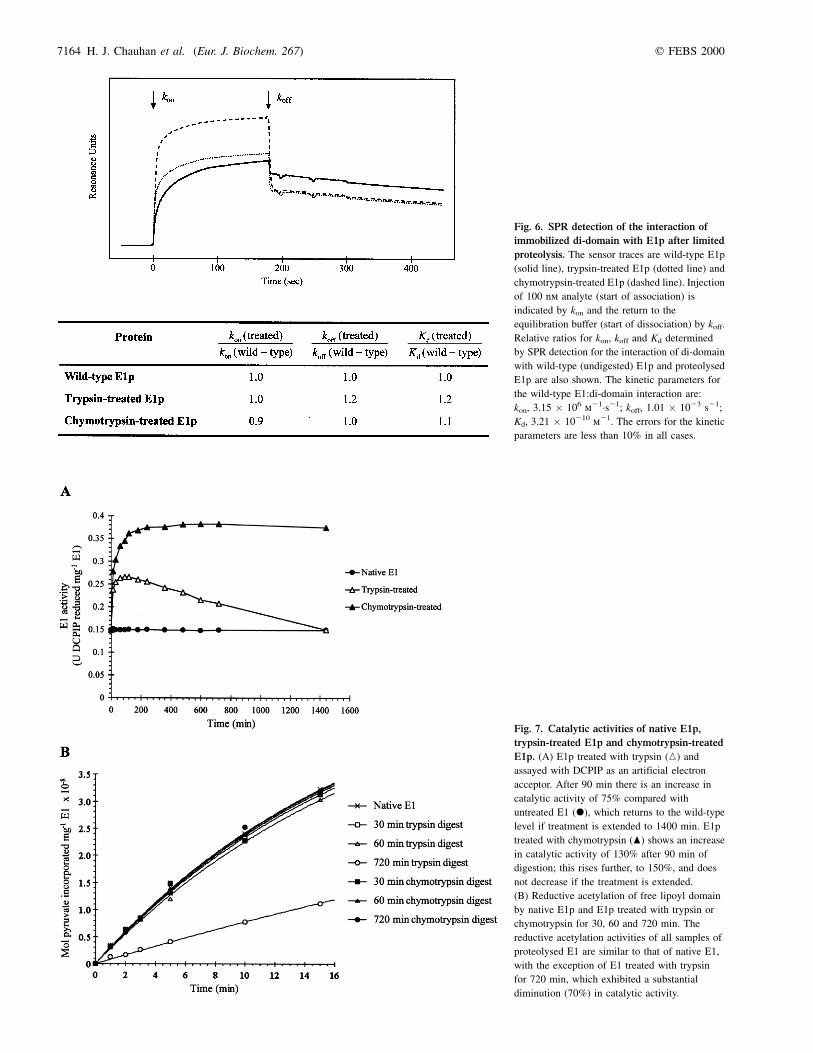
Fig. 6. SPR detection of the interaction of
immobilized di-domain with E1p after limited
proteolysis. The sensor traces are wild-type E1p
(solid line), trypsin-treated E1p (dotted line) and
chymotrypsin-treated E1p (dashed line). Injection
of 100 nm analyte (start of association) is
indicated by kon and the return to the
equilibration buffer (start of dissociation) by koff.
Relative ratios for kon, koff and Kd determined
by SPR detection for the interaction of di-domain
with wild-type (undigested) E1p and proteolysed
E1p are also shown. The kinetic parameters for
the wild-type E1:di-domain interaction are:
kon, 3.15 � 106 m21´s21; koff, 1.01 � 1023 s21;
Kd, 3.21 � 10210 m21. The errors for the kinetic
parameters are less than 10% in all cases.
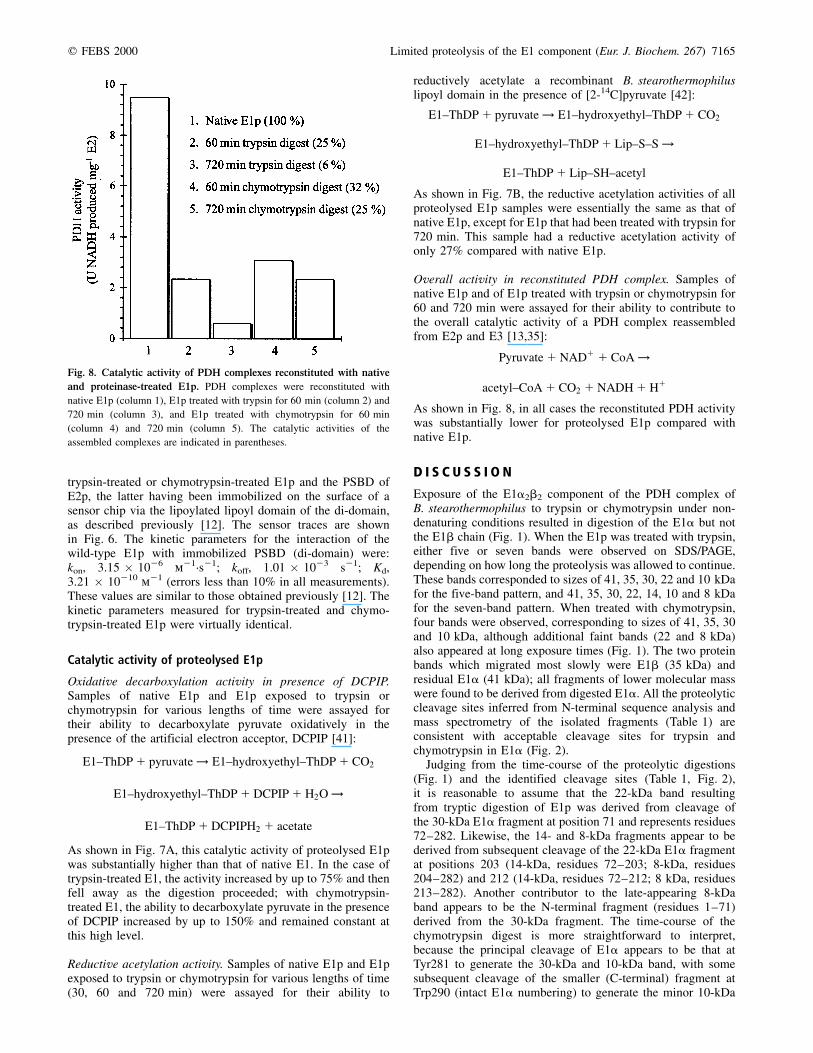
Fig. 7. Catalytic activities of native E1p,
trypsin-treated E1p and chymotrypsin-treated
E1p. (A) E1p treated with trypsin (K) and
assayed with DCPIP as an artificial electron
acceptor. After 90 min there is an increase in
catalytic activity of 75% compared with
untreated E1 (X), which returns to the wild-type
level if treatment is extended to 1400 min. E1p
treated with chymotrypsin (O) shows an increase
in catalytic activity of 130% after 90 min of
digestion; this rises further, to 150%, and does
not decrease if the treatment is extended.
(B) Reductive acetylation of free lipoyl domain
by native E1p and E1p treated with trypsin or
chymotrypsin for 30, 60 and 720 min. The
reductive acetylation activities of all samples of
proteolysed E1 are similar to that of native E1,
with the exception of E1 treated with trypsin
for 720 min, which exhibited a substantial
diminution (70%) in catalytic activity.
7164 H. J. Chauhan et al. (Eur. J. Biochem. 267) q FEBS 2000
trypsin-treated or chymotrypsin-treated E1p and the PSBD ofE2p, the latter having been immobilized on the surface of asensor chip via the lipoylated lipoyl domain of the di-domain,as described previously [12]. The sensor traces are shownin Fig. 6. The kinetic parameters for the interaction of thewild-type E1p with immobilized PSBD (di-domain) were:kon, 3.15 � 1026 m21´s21; koff, 1.01 � 1023 s21; Kd,3.21 � 10210 m21 (errors less than 10% in all measurements).These values are similar to those obtained previously [12]. Thekinetic parameters measured for trypsin-treated and chymo-trypsin-treated E1p were virtually identical.
Catalytic activity of proteolysed E1p
Oxidative decarboxylation activity in presence of DCPIP.Samples of native E1p and E1p exposed to trypsin orchymotrypsin for various lengths of time were assayed fortheir ability to decarboxylate pyruvate oxidatively in thepresence of the artificial electron acceptor, DCPIP [41]:
E1±ThDP 1 pyruvate! E1±hydroxyethyl±ThDP 1 CO2
E1±hydroxyethyl±ThDP 1 DCPIP 1 H2O!
E1±ThDP 1 DCPIPH2 1 acetate
As shown in Fig. 7A, this catalytic activity of proteolysed E1pwas substantially higher than that of native E1. In the case oftrypsin-treated E1, the activity increased by up to 75% and thenfell away as the digestion proceeded; with chymotrypsin-treated E1, the ability to decarboxylate pyruvate in the presenceof DCPIP increased by up to 150% and remained constant atthis high level.
Reductive acetylation activity. Samples of native E1p and E1pexposed to trypsin or chymotrypsin for various lengths of time(30, 60 and 720 min) were assayed for their ability to
reductively acetylate a recombinant B. stearothermophiluslipoyl domain in the presence of [2-14C]pyruvate [42]:
E1±ThDP 1 pyruvate! E1±hydroxyethyl±ThDP 1 CO2
E1±hydroxyethyl±ThDP 1 Lip±S±S!
E1±ThDP 1 Lip±SH±acetyl
As shown in Fig. 7B, the reductive acetylation activities of allproteolysed E1p samples were essentially the same as that ofnative E1p, except for E1p that had been treated with trypsin for720 min. This sample had a reductive acetylation activity ofonly 27% compared with native E1p.
Overall activity in reconstituted PDH complex. Samples ofnative E1p and of E1p treated with trypsin or chymotrypsin for60 and 720 min were assayed for their ability to contribute tothe overall catalytic activity of a PDH complex reassembledfrom E2p and E3 [13,35]:
Pyruvate 1 NAD1 1 CoA!
acetyl±CoA 1 CO2 1 NADH 1 H1
As shown in Fig. 8, in all cases the reconstituted PDH activitywas substantially lower for proteolysed E1p compared withnative E1p.
D I S C U S S I O N
Exposure of the E1a2b2 component of the PDH complex ofB. stearothermophilus to trypsin or chymotrypsin under non-denaturing conditions resulted in digestion of the E1a but notthe E1b chain (Fig. 1). When the E1p was treated with trypsin,either five or seven bands were observed on SDS/PAGE,depending on how long the proteolysis was allowed to continue.These bands corresponded to sizes of 41, 35, 30, 22 and 10 kDafor the five-band pattern, and 41, 35, 30, 22, 14, 10 and 8 kDafor the seven-band pattern. When treated with chymotrypsin,four bands were observed, corresponding to sizes of 41, 35, 30and 10 kDa, although additional faint bands (22 and 8 kDa)also appeared at long exposure times (Fig. 1). The two proteinbands which migrated most slowly were E1b (35 kDa) andresidual E1a (41 kDa); all fragments of lower molecular masswere found to be derived from digested E1a. All the proteolyticcleavage sites inferred from N-terminal sequence analysis andmass spectrometry of the isolated fragments (Table 1) areconsistent with acceptable cleavage sites for trypsin andchymotrypsin in E1a (Fig. 2).
Judging from the time-course of the proteolytic digestions(Fig. 1) and the identified cleavage sites (Table 1, Fig. 2),it is reasonable to assume that the 22-kDa band resultingfrom tryptic digestion of E1p was derived from cleavage ofthe 30-kDa E1a fragment at position 71 and represents residues72±282. Likewise, the 14- and 8-kDa fragments appear to bederived from subsequent cleavage of the 22-kDa E1a fragmentat positions 203 (14-kDa, residues 72±203; 8-kDa, residues204±282) and 212 (14-kDa, residues 72±212; 8 kDa, residues213±282). Another contributor to the late-appearing 8-kDaband appears to be the N-terminal fragment (residues 1±71)derived from the 30-kDa fragment. The time-course of thechymotrypsin digest is more straightforward to interpret,because the principal cleavage of E1a appears to be that atTyr281 to generate the 30-kDa and 10-kDa band, with somesubsequent cleavage of the smaller (C-terminal) fragment atTrp290 (intact E1a numbering) to generate the minor 10-kDa
Fig. 8. Catalytic activity of PDH complexes reconstituted with native
and proteinase-treated E1p. PDH complexes were reconstituted with
native E1p (column 1), E1p treated with trypsin for 60 min (column 2) and
720 min (column 3), and E1p treated with chymotrypsin for 60 min
(column 4) and 720 min (column 5). The catalytic activities of the
assembled complexes are indicated in parentheses.
q FEBS 2000 Limited proteolysis of the E1 component (Eur. J. Biochem. 267) 7165

product. With both proteinases, it is clear that the initialproteolytic cleavage sites cluster in a short region of amino acidsequence, RYRSKE, located at residues 280±285 in the E1achain. Further tryptic cleavages at Arg71, Arg203 and Lys212can take place. As the E1b subunit is not degraded, it must beassumed that all the cleavage sites for trypsin and chymotrypsinin this polypeptide chain are in structured regions and/orprotected by the E1a subunits.
It is interesting that B. stearothermophilus E1p remainsrelatively resistant to chymotryptic digestion after the initialperiod of proteolysis (see Fig. 1). Densitometry measurementsrevealed that < 35% of the E1a component was cleaved after15 min of digestion, with 45±55% being cleaved after 240 min,and < 70% being cleaved after 720 min. This suggests that theE1a chains in the heterotetramer (a2b2) may exist in twodifferent conformations, one of which is more susceptible, andone of which is more resistant, to chymotryptic digestion. In thecase of tryptic digestion of E1 (Fig. 1), the appearance ofadditional bands, which may be due to secondary cleavagespromoted by rearrangements in the tertiary structure, make thedensitometric pattern too complex to interpret unequivocally;
even so, it is clear that some E1a chain remains intact afterlong periods of exposure to the proteinase. It is known thatthe tetrameric protein environment plays a significant rolein maintaining the active-site geometry [22], and thatlong-range nonbonded interactions are often influential inmaintaining structurally important features. The idea ofconformational nonequivalence resulting in active and inactive(or less active) conformers has been advanced previously foryeast pyruvate decarboxylase [44] and E1p from pigeon breastmuscle [45].
Proteolytic cleavage sites in globular proteins are generallyfound in relatively unstructured surface loop regions associatedwith higher crystallographic B factors [46,47]. Mapping thetrypsin and chymotrypsin cleavage sites on to the structuralmodel of the E1a subunit of the B. stearothermophilus PDHcomplex (Fig. 3) reveals them, as expected, to be in surfaceloops. Analysis of the B factors in the crystal structure of thehomologous E1 (a2b2) of the P. putida 2-oxoisovaleratedehydrogenase complex (Fig. 9), on which the E1a structuralmodel of the B. stearothermophilus PDH complex (Fig. 3)is based, indicates that the regions corresponding to residues
Fig. 9. Space-filling representations of the structural organization of heterotetrameric E1a2b2 of P. putida 2-oxoisovalerate dehydrogenase complex
[22]. (A) The a subunits are shown in shades of blue or lilac and the b subunits in shades of green. Each subunit makes extensive surface contacts with the
other three subunits, such that a b dimer is tightly held between the jaws of the two a subunits. The N- and C-termini of the subunits are indicated. The
diagrams were created using the program molscript [43]. (B) Molecular surface representation with regions cleaved by trypsin or chymotrypsin shown in
red: R71 (res. 71±72), R203 (res. 203±204), K212 (res. 212±213), Y281 (res. 280±283), K284 (res. 284±285) and W290 (res. 290±291). The diagrams were
created using the program protein explorer. In both (A) and (B), the right-hand image is that of the left-hand image rotated about a vertical axis by 908.
7166 H. J. Chauhan et al. (Eur. J. Biochem. 267) q FEBS 2000

281±283 (YRS) and 71±72 (RL) are more flexible than theregion corresponding to residues 203±204 (RF), which occursimmediately after the ThDP-binding region (Fig. 2). Cleavageat the RYRSKE site appears to render the Arg71 site, andsubsequently the Arg203 and Lys212 sites, more susceptible toproteolysis by trypsin. This may be the result of a confor-mational change in the E1a chain after cleavage at theconserved RYRSKE site (the YR being highly conservedresidues in sequence alignments).
Gel filtration and SDS/PAGE revealed that the E1 (a2b2)remained fully assembled after proteolysis with trypsin orchymotrypsin (Fig. 4). Moreover, the proteolysed E1 wascapable of interacting normally with the PSBD of theB. stearothermophilus E2p chain, as judged both by non-denaturing PAGE and gel filtration (Fig. 5) and by surfaceplasmon resonance (Fig. 6). Thus, the cleavage of the E1achain can take place without effect on the E1±E2 interaction,consistent with an earlier observation that the PSBD appearsto interact only with the E1b of the a2b2 heterotetramer[12].
After digestion with trypsin or chymotrypsin, the ability ofthe E1 component to decarboxylate pyruvate in the presence ofDCPIP was found to increase by up to 17 or 150%, respectively(Fig. 7A). This increase could be due to a change in theconformation of the E1, such that the active site of theproteolysed enzyme facilitates release of the product and/or ismore accessible to the DCPIP. A similar suggestion has beenadvanced previously in describing the proteolysis of E1p fromporcine heart muscle [48]. Certainly, the initial proteolyticcleavages occur in a region (RYRSKE) in a surface loop,apparently at the entrance to the active site (Fig. 3). Whendigestion with trypsin was continued beyond 120 min, however,the catalytic activity began to decrease from its peak value of175% (compared with native E1p), and gradually reverted tothe wild-type level after 24 h (Fig. 7A). This decrease inactivity was accompanied by the appearance of further cleavageproducts (Fig. 1). The digestion of E1p with chymotrypsinalso led to an increase in catalytic activity but this didnot diminish on prolonged exposure (Fig. 7A), nor didadditional proteolytic cleavages appear (Fig. 1). The generationof the 22- and 14- and 8-kDa fragments of E1a (which occursonly on tryptic digestion) is thus associated with the gradualloss of catalytic activity from its high point in the earlier phaseof digestion.
In contrast, the reductive acetylation activity of E1p afterdigestion with trypsin or chymotrypsin, measured with a freelipoyl domain as substrate, was found to be almost unchangedfrom that of native E1p (Fig. 7B). However, when E1p wasdigested with trypsin for a prolonged period (720 min), theactivity was found to decrease substantially (Fig. 7B). Thisdecrease in reductive acetylation activity again coincides withthe appearance of the 14- and 8-kDa E1a fragments, associatedwith cleavage at Arg71 and Arg203. The latter residueimmediately follows the ThDP-binding motif (Fig. 2) and asa consequence is close in space to the ThDP, and thus to theactive site (Fig. 3). On the other hand, the overall catalyticactivity of a PDH complex reconstituted with trypsin- orchymotrypsin-treated E1p was substantially lower than that of aPDH complex reconstituted with native E1p (Fig. 8). Inparticular, cleavage at Tyr281 with chymotrypsin (60 and720 min) caused the overall PDH activity to decrease by< 70%; similar results were observed with trypsin (60 min),and subsequent cleavage at the Arg203 site (as at 720 min) wasaccompanied by a further decrease in activity to only 6% ofwild-type.
There is no obvious difference between native E1p andproteolysed E1p in terms of their ability to bind to the PSBDof the E2p chain of the B. stearothermophilus PDH complex(see above). Likewise, the proteolysed E1p is indistinguishablefrom native E1p in the reductive acetylation of the free lipoyldomain (except after prolonged exposure to trypsin) (Fig. 7B),but it is substantially less effective than native E1p in makingan active PDH complex with native E2p (Fig. 8). In the intactPDH complex, the lipoyl domains are not totally free but aretethered by the long linker segment between the C-terminus ofthe lipoyl domain and the N-terminus of the PSBD in the E2pchain; this could restrict the trajectory of the lipoyl domain inits approach to the active site of the bound E1 component. Thisin turn might be adversely affected by the proteolytic cleavagesin E1a at the entrance to the E1 active site. The way the lipoyldomain can interact with E1p is crucially important to ourunderstanding of the assembled complex and deserves furtherexperimental investigation, particularly in view of the recentevidence of restricted motion of the lipoyl-lysine `swingingarm' in the lipoyl domain [49].
A C K N O W L E D G E M E N T S
We are grateful to the Biotechnology and Biological Sciences Research
Council for a research grant (to R. N. P.) and the award of a BBSRC
Special Research Studentship (to H. J. C.), and to the Cambridge Overseas
Trust and St John's College, Cambridge, for financial support of H.-I. J.
We would like to thank Mr M. Weldon for N-terminal sequence analysis
and Mr P. Sharratt for amino acid analysis. The core facilities of the
Cambridge Centre for Molecular Recognition are supported by the BBSRC
and The Wellcome Trust.
R E F E R E N C E S
1. Perham, R.N. (1991) Domains, motifs, and linkers in 2-oxo acid
dehydrogenase multienzyme complexes: a paradigm in the design of
a multifunctional enzyme. Biochemistry 30, 8501±8512.
2. Perham, R.N. (2000) Swinging arms and swinging domains in
multifunctional enzymes: catalytic machines for multistep reactions.
Annu. Rev. Biochem. 69, 963±1006.
3. de Kok, A., Hengeveld, A.F., Martin, A. & Westphal, A.H. (1998) The
pyruvate dehydrogenase multienzyme complex from Gram-negative
bacteria. Biochim. Biophys. Acta 1385, 353±366.
4. Kalia, Y.N., Brocklehurst, S.M., Hipps, D.S., Appella, E., Sakaguchi,
K. & Perham, R.N. (1993) The high-resolution structure of the
peripheral subunit-binding domain of dihydrolipoamide acetyltrans-
ferase from the pyruvate dehydrogenase complex of Bacillus
stearothermophilus. J. Mol. Biol. 230, 323±341.
5. Izard, T., ávarsson, A., Allen, M.D., Westphal, A.H., Perham, R.N., de
Kok, A. & Hol, W.G.J. (1999) Principles of quasi-equivalence and
Euclidean geometry govern the assembly of cubic and dodecahedral
cores of pyruvate dehydrogenase complexes. Proc. Natl Acad. Sci.
USA 96, 1240±1245.
6. Reed, L.J. & Hackert, M.L. (1990) Structure-function relationships in
dihydrolipoamide acyltransferases. J. Biol. Chem. 265, 8971±8974.
7. Lessard, I.A.D. & Perham, R.N. (1994) Expression in Escherichia coli
of genes encoding the E1a and E1b subunits of the pyruvate
dehydrogenase complex of Bacillus stearothermophilus and assembly
of a functional E1 component (a2b2) in vitro. J. Biol. Chem. 269,
10378±10383.
8. Stepp, L.R. & Reed, L.J. (1985) Active-site modification of
mammalian pyruvate dehydrogenase by pyridoxal 5 0-phosphate.
Biochemistry 24, 7187±7191.
9. Hawkins, C.F., Borges, A. & Perham, R.N. (1989) A common
structural motif in thiamin pyrophosphate-binding enzymes. FEBS
Lett. 255, 77±82.
10. Wynn, R.M., Chuang, J.L., Davie, J.R., Fisher, C.W., Hale, M.A., Cox,
q FEBS 2000 Limited proteolysis of the E1 component (Eur. J. Biochem. 267) 7167

R.P. & Chuang, D.T. (1992) Cloning and expression in Escherichia
coli of mature E1b subunit of bovine mitochondrial branched-chain
alpha-keto acid dehydrogenase complex ± mapping of the E1 beta
binding region on E2. J. Biol. Chem. 267, 1881±1887.
11. Lessard, I.A.D. & Perham, R.N. (1995) Interaction of component
enzymes with the peripheral subunit-binding domain of the pyruvate
dehydrogenase multienzyme complex of Bacillus stearothermo-
philus: stoichiometry and specificity in self assembly. Biochem. J.
306, 727±733.
12. Lessard, I.A.D., Fuller, C. & Perham, R.N. (1996) Competitive
interaction of component enzymes with the peripheral subunit-
binding domain of the pyruvate dehydrogenase multienzyme com-
plex of Bacillus stearothermophilus: Kinetic analysis using surface
plasmon resonance detection. Biochemistry 35, 16863±16870.
13. Domingo, G.J., Chauhan, H.J., Lessard, I.A.D., Fuller, C. & Perham,
R.N. (1999) Self-assembly and catalytic activity of the pyruvate
dehydrogenase multienzyme complex from Bacillus stearothermo-
philus. Eur. J. Biochem. 266, 1136±1146.
14. Danson, M.J., Fersht, A.R. & Perham, R.N. (1978) Rapid intra-
molecular coupling of active sites in the pyruvate dehydrogenase
complex of Escherichia coli: mechanism for rate enhancement in a
multimeric structure. Proc. Natl Acad. Sci. USA 75, 5386±5390.
15. Cate, R.L., Roche, T.E. & Davis, L.C. (1980) Rapid intersite transfer of
acetyl groups and movement of pyruvate dehydrogenase components
in the kidney pyruvate dehydrogenase complex. J. Biol. Chem. 255,
7556±7562.
16. Berg, A., Westphal, A.H., Bosma, H.J. & de Kok, A. (1998) Kinetics
and specificity of reductive acylation of wild-type and mutated lipoyl
domains of 2-oxo acid dehydrogenase complexes from Azotobacter
vinelandii. Eur. J. Biochem. 252, 45±50.
17. Reed, L.J., Koike, M., Levitch, M.E. & Leach, F.R. (1958) Studies on
the nature and reactions of protein-bound lipoic acid. J. Biol. Chem.
232, 143±148.
18. Graham, L.D., Packman, L.C. & Perham, R.N. (1989) Kinetics and
specificity of reductive acylation of lipoyl domains from 2-oxo
acid dehydrogenase multienzyme complexes. Biochemistry 28,
1574±1581.
19. Schulze, E., Westphal, A.H., Veeger, C. & de Kok, A. (1992)
Reconstitution of pyruvate dehydrogenase multienzyme complexes
based on chimeric core structures from Azotobacter vinelandii and
Escherichia coli. Eur. J. Biochem. 206, 427±435.
20. Wallis, N.G., Allen, M.D., Broadhurst, R.W., Lessard, I.A.D. &
Perham, R.N. (1996) Recognition of a surface loop of the lipoyl
domain underlies substrate channelling in the pyruvate dehydro-
genase multienzyme complex. J. Mol. Biol. 263, 463±474.
21. Howard, M.J., Chauhan, H.J., Domingo, G.J., Fuller, C. & Perham,
R.N. (2000) Protein±protein interaction revealed by NMR T2
relaxation experiments: the lipoyl domain and E1 component of the
pyruvate dehydrogenase multienzyme complex of Bacillus stearo-
thermophilus. J. Mol. Biol. 295, 1023±1037.
22. ávarsson, A., Seger, K., Turley, S., Sokatch, J.R. & Hol, W.G.J. (1999)
Crystal structure of 2-oxoisovalerate dehydrogenase and the archi-
tecture of 2-oxo acid dehydrogenase multienzyme complexes. Nat.
Struct. Biol. 6, 785±792.
23. ávarsson, A., Chuang, J.L., Wynn, R.M., Turley, S., Chuang, D.T. &
Hol, W.G.J. (2000) Crystal structure of human branched-chain a-keto
acid dehydrogenase and the molecular basis of multienzyme
complex deficiency in maple syrup urine disease. Structure Fold.
Des. 8, 277±291.
24. Bleile, D.M., Munk, P., Oliver, R.M. & Reed, L.J. (1979) Subunit
structure of dihydrolipoyl transacetylase component of pyruvate
dehydrogenase complex from Escherichia coli. Proc. Natl Acad. Sci.
USA 76, 4385±4389.
25. Hale, G. & Perham, R.N. (1979) Limited proteolysis of the pyruvate
dehydrogenase multienzyme complex of Escherichia coli. Eur. J.
Biochem. 94, 119±126.
26. Perham, R.N. & Roberts, G.C. (1981) Limited proteolysis and proton
NMR spectroscopy of the 2-oxoglutarate dehydrogenase multi-
enzyme complex of Escherichia coli. Biochem. J. 199, 733±740.
27. Packman, L.C., Hale, G. & Perham, R.N. (1984) Repeating functional
domains in the pyruvate dehydrogenase multienzyme complex of
Escherichia coli. EMBO J. 3, 1315±1319.
28. Perham, R.N. & Wilkie, A.O.M. (1980) Inner core and domain
structure of the pyruvate dehydrogenase multienzyme complex of
Bacillus stearothermophilus. Biochem. Int. 1, 470±477.
29. Duckworth, H.W., Jaenicke, R., Perham, R.N., Wilkie, A.O.M., Finch,
J.T. & Roberts, G.C.K. (1982) Limited proteolysis and proton NMR
spectroscopy of Bacillus stearothermophilus pyruvate dehydrogenase
multienzyme complex. Eur. J. Biochem. 124, 63±69.
30. Kresze, G.B. & Ronft, H. (1980) Bovine kidney pyruvate dehydro-
genase complex. Limited proteolysis and molecular structure of the
lipoate acetyltransferase component. Eur. J. Biochem. 112, 589±599.
31. Kresze, G.B., Ronft, H., Dietl, B. & Steber, L. (1981) Limited
proteolysis of 2-oxoglutarate dehydrogenase multienzyme complex
from bovine kidney. FEBS Lett. 127, 157±160.
32. McCartney, R.G., Rice, J.E., Sanderson, S.J., Bunik, V., Lindsay, H. &
Lindsay, J.G. (1998) Subunit interactions in the mammalian a-keto-
glutarate dehydrogenase complex. Evidence for direct association of
the a-ketoglutarate dehydrogenase and dihydrolipoamide dehydro-
genase components. J. Biol. Chem. 273, 24158±24164.
33. Hengeveld, A.F., Westphal, A.H. & de Kok, A. (1997) Expression and
characterization of the homodimeric E1 component of the Azoto-
bacter vinelandii pyruvate dehydrogenase complex. Eur. J. Biochem.
250, 260±268.
34. Hengeveld, A.F., Schoustra, S.E., Westphal, A.H. & de Kok, A. (1999)
Pyruvate dehydrogenase from Azotobacter vinelandii ± properties
of the N-terminally truncated enzyme. Eur. J. Biochem. 265,
1098±1107.
35. Lessard, I.A.D., Domingo, G.J., Borges, A. & Perham, R.N. (1998)
Expression of genes encoding the E2 and E3 components of the
Bacillus stearothermophilus pyruvate dehydrogenase complex and
the stoichiometry of subunit interaction in assembly in vitro. Eur. J.
Biochem. 258, 491±501.
36. Studier, F.W. & Moffatt, B.A. (1986) Use of bacteriophage T7 RNA
polymerase to direct selective high-level expression of cloned genes.
J. Mol. Biol. 189, 113±130.
37. Hipps, D.S. & Perham, R.N. (1992) Expression in Escherichia coli of a
sub-gene encoding the lipoyl and peripheral subunit-binding domains
of the dihydrolipoamide acetyltransferase component of the pyruvate
dehydrogenase complex of Bacillus stearothermophilus. Biochem. J.
283, 665±671.
38. Morris, T.W., Reed, K.E. & Cronan, J.E. (1994) Identification of
the gene encoding lipoate-protein ligase A of Escherichia coli.
Molecular cloning and characterization of the lplA gene and gene
product. J. Biol. Chem. 269, 16091±16100.
39. Jones, D.D., Horne, J.H., Reche, P.A. & Perham, R.N. (2000) Structural
determinants of post-translational modification and catalytic
specificity for the lipoyl domains of the pyruvate dehydrogenase
multienzyme complex of Escherichia coli. J. Mol. Biol. 295,
289±306.
40. Laemmli, U.K. (1970) Cleavage of structural proteins during the
assembly of the head of bacteriophage T4. Nature 227, 680±685.
41. KhaõÆlova, L.S., Bernkhardt, R. & Khiubner, G. (1977) Study of the
kinetic mechanism of pyruvate-2,6-dichlorophenolindophenol reduc-
tase activity of muscle pyruvate dehydrogenase. Biokhimiia (Mos-
cow) 42, 113±117.
42. Packman, L.C., Perham, R.N. & Roberts, G.C.K. (1984) Domain
structure and 1H-n.m.r. spectroscopy of the pyruvate dehydrogenase
complex of Bacillus stearothermophilus. Biochem. J. 217, 219±227.
43. Kraulis, P.J. (1991) MOLSCRIPT: a program to produce both detailed
and schematic plots of protein structures. J. Appl. Cryst. 24, 946±950.
44. Lu, G., Dobritzsch, D., Baumann, S., Schneider, G. & KoÈnig, S. (2000)
The structural basis of substrate activation in yeast pyruvate
decarboxylase. A crystallographic and kinetic study. Eur. J. Biochem.
267, 861±868.
45. Khailova, L.S. & Korochkina, L.J. (1982) Determination of the number
of active-centers in the pyruvate dehydrogenase component of the
7168 H. J. Chauhan et al. (Eur. J. Biochem. 267) q FEBS 2000

pyruvate dehydrogenase complex from pigeon breast muscle.
Biochem. Int. 5, 525±532.
46. Fontana, A., Fassina, G., Vita, C., Dalzoppo, D., Zamai, M. &
Zambonin, M. (1986) Correlation between sites of limited proteolysis
and segmental mobility in thermolysin. Biochemistry 25, 1847±1851.
47. Fontana, A., de Zambonin, M., Laureto, P.P., DeFillipis, V.,
Clementi, A. & Scaramella, E. (1997) Probing the conformational
state of apomyoglobin by limited proteolysis. J. Mol. Biol. 266,
223±230.
48. Koike, K., Urata, Y. & Goto, S. (1992) Proteinase-catalyzed
activation of porcine heart muscle pyruvate dehydrogenase
and identification of its cleavage site. Biochim. Biophys. Acta 1118,
223±230.
49. Jones, D.D., Stott, K.M., Howard, M.J. & Perham, R.N. (2000)
Restricted motion of the lipoyl-lysine swinging arm in the
pyruvate dehydrogenase complex of Escherichia coli. Biochemistry
39, 8448±8459.
q FEBS 2000 Limited proteolysis of the E1 component (Eur. J. Biochem. 267) 7169




















