
Kinetic characterization of the substrate specificity and mechanism of mushroom tyrosinase
Upload
independentCategory
view
0download
0
ARCHIVES OF BIOCHEMISTRY AND BIOPHYSICS Vol. 297, No. 2, September, pp. 221-227, 1992
Proteolysis with Trypsin of Mammalian Tyrosinase lsoforms from B16 Mouse Melanoma’
Paloma Valverde, Jo,&-Carlos Garcia-Borr6n,2 Francisco Solano, and Josh-Antonio Lozano Departamento de Bioquimica y Biologia Molecular, Facultad de Medicina, Universidad de Murcia, 30100 Espinardo, Murk, Spain
Received January 15, 1992, and in revised form May 13, 1992
In spite of the central role of tyrosinase in mammalian pigmentation, few data are available on its structure and structure-function relationships based on direct analysis of the protein. A number of reasons have been invoked to account for this situation, including the problems for its purification and its resistance to proteases. However, no study on the effects of proteases on purified tyrosinase has been reported.
We have purified the melanosomal and cytosolic ty- rosinases from B16 mouse melanoma and analyzed their susceptibility to trypsin digestion. Both isoforms are sensitive to trypsin, and display similar peptide maps and kinetics of proteolysis, suggesting that they are products of the same gene. The peptide maps and the kinetics of appearance of the fragments were consistent with the sequential removal of N-terminal peptides, leading to a core of 55.3 kDa for the melanosomal form and 48.6 kDa for the cytosolic enzyme. This core was apparently re- sistant to further proteolysis and catalytically inactive. The difference in molecular weight for the core of the cytosolic and melanosomal forms is the same as that cal- culated for the native isoforms. The kinetics of enzyme inactivation indicate that the tyrosine hydroxylase and Dopa oxidase activities of tyrosinase are lost at the same rate, and should therefore display similar if not identical structural requirements. The results are discussed in terms of the relationship of both isoforms and of the pu- tative protein sequences deduced from the cDNA clones proposed for tyrosinase. 0 1992 Academic PWSS, Inc.
Mammalian tyrosinase (monophenol monooxygenase, EC 1.14.18.1) is a copper containing glycoprotein cata-
i This work was supported by Grant PB90/0269 from the DGICYT. Paloma Valverde is the recipient of a fellowship from the Instituto de Foment0 de la Comunidad Autonoma de Murcia.
* To whom correspondence should be addressed at Departamento de Bioquimica, Facultad de Medicina, Universidad de Murcia, Apt. 4021, 30100 Murcia, Spain. Fax: 34-68-834307.
0003.9861/92 $5.00 Copyright 0 1992 by Academic Press, Inc. All rights of reproduction in any form reserved.
lyzing the rate-limiting step of melanogenesis, the hy- droxylation of tyrosine to L-Dopa (1,2). Tyrosinase also catalyzes L-Dopa oxidation to dopaquinone and probably the conversion of 5,6-dihydroxyindole to indole-5,6-qui- none (3).
Within mammalian melanocytes, melanin synthesis is confined to specialized organelles, the melanosomes, but tyrosinase is present in a variety of isoenzymatic forms (4-6) with different subcellular location and electropho- retie mobility. Since the enzyme undergoes post-trans- lational modifications, including glycosylation (5, 6), it has been assumed that the different isoforms correspond to different degrees of maturation. The de novo micro- somal enzyme would be the precursor of the mature me- lanosomal form, whose molecular weight would be in- creased by the addition of carbohydrate moieties and maybe by interaction with other melanosomal compo- nents. The slightly heterogeneous glycosylation pattern might account for the microheterogeneity of the melano- somal enzyme (7). The soluble, cytosolic form of the en- zyme could arise by proteolytic cleavage of the melano- somal enzyme (8-ll), very possibly near its carboxyl terminus (12-14). However, it has been noted that this precursor-product relationship has never been conclu- sively demonstrated (15).
Several laboratories have recently cloned different genes that may encode proteins with tyrosinase activity in mouse (16-20) and human cells (21). So far, only the clone described by Muller et al. (20) has been unequivo- cally shown to code for a protein with tyrosine hydroxylase activity. However, these genes are considerably homolo- gous and an unambiguous identification of the correct(s)
3Abbreviations used: BSA, bovine serum albumin; L-Dopa, L-3,4- dihydroxyphenylalanine; EDTA, ethylenediaminetetraacetic acid; PAGE, polyacrylamide gel electrophoresis; PMSF, phenylmethylsulfonyl fluoride; SDS, sodium dodecyl sulfate; Temed, N,N,N’,N’-tetrameth- ylethylenediamine; TPCK, tosylphenylchloromethyl ketone; Tris, tris(hydroxymethyl)aminomethane.
221

222 VALVERDE ET AL.
clone(s) has been impossible due to the lack of direct in- formation on the structure of tyrosinase. It has even been suggested that mammalian melanocytes could contain more than one gene product with tyrosinase activity (15), and there is strong evidence indicating that the protein encoded by the clone described by Shibahara et al. (16) could also be catalytically competent (22). The situation is further complicated by the possibility of alternative splicing events of the primary transcripts (20, 23). The lack of direct data on the structure of mammalian tyros- inase is explained by a variety of factors including its N- terminal blockage and the problems in its purification. It is also widely accepted that mammalian tyrosinase is ex- tremely resistant to proteolytic degradation, since the melanosomal enzyme can be solubilized by limited pro- teolytic digestion with trypsin, yielding a form of slightly lower molecular weight and full enzymatic activity. How- ever, as far as we know, no study on the effect of proteases on purified tyrosinase isoforms has been reported.
We have examined the effect of trypsin on the activity and the electrophoretic pattern of two purified isoforms of B16 mouse melanoma tyrosinase. We have found that both the cytosolic and the melanosomal forms are sen- sitive to trypsin, and that proteolysis leads to a loss of tyrosine hydroxylase and Dopa oxidase activities.
MATERIALS AND METHODS
Reagents. The radioactive substrate L-[3,5-3H]tyrosine (sp act 58.8 Ci/mmol) was obtained from New England Nuclear (Boston, MA). L- Tyrosine, bovine serum albumin, hydroxyapatite type I suspension in 1 mM phosphate buffer, pH 6.8, L-Dopa, and TPCK-treated trypsin (Type XIII) were from Sigma (St. Louis, MO). Trichloroacetic acid, glycine, Tris, and Brij 35 were from Merck (Darmstadt, Germany). Acrylamide, bisacrylamide, Temed, and sodium persulfate were obtained from Bio-Rad (Richmond, CA). All other reagents were from Probus (Spain).
Animals and melanomas. B16/FlO mouse melanoma cells were orig- inally a kind gift from Dr. V. Hearing (NIH, Bethesda, MD). Tumors were maintained by serial transplantation into C57/Bl X DBA hybrid mice, as described (24).
Purification of tyrosinase isoforms. Freshly excised tumors were washed twice in ice-cold 10 mM phosphate buffer, pH 6.8,0.25 M sucrose, and 0.1 mM EDTA. The tumors were homogenized in a Polytron ho- mogenizer, in the same buffer supplemented with 0.1 mM PMSF, using a ratio of tumor weight (in g) to buffer volume (in ml) of 1:3. The ho- mogenate was centrifuged at 700g for 20 min in a Sorvall SS-34 rotor and the pellet was reextracted with a volume of buffer (in ml) equal to the weight of the starting material (in g) and centrifuged under the same conditions. A melanosomal pellet was obtained by centrifugation of the combined supernatants at ll,OOOg, 30 min. This pellet was solubilized by addition of 1% Brij 35 in 10 mM phosphate buffer, pH 6.8, using a volume (ml) twice the original weight of tumors. The suspension was gently shaken for 30 min and centrifuged at 105,OOOg, 1 h, in a Beckman 60 Ti rotor. The supernatant was used as a source of crude melanosomal tyrosinase. The cytosolic fraction was obtained by centrifugation of the ll,OOOg supernatant at 105,OOOg, 1 h.
To purify melanosomal tyrosinases crude extracts were brought to 33% saturation with ammonium sulfate, incubated for 3 h at 4’C with gentle shaking, and centrifuged at ll,OOOg, 30 min. The supernatants were brought to 75% saturation and kept at 4’C overnight. After cen- trifugation at ll,OOOg for 1 h the supernatants were discarded and the
pellets were redissolved in a volume (in ml) of 10 mM phosphate buffer, pH 6.8, containing 0.1% Brij 35 equal to the weight of starting material (in g) and extensively dialyzed against this buffer. To the dialyzed sample, hydroxyapatite suspension was added in a 1:l (v/v) ratio. The mixture was incubated 30 min and then centrifuged at 3OOOg, 10 min. The hy- droxyapatite pellet was washed with 1 vol of phosphate buffer and re- centrifuged. The combined supernatants were concentrated in an Amicon ultrafiltration cell and further purified by gel filtration chromatography on an Ultrogel AcA 34 column (52 X 2.6 cm) equilibrated and eluted at a rate of 12 ml/h with 50 mM phosphate buffer, pH 6.8, containing 0.1% Brij. The fractions with the highest Dopa oxidase specific activity were pooled, concentrated, and purified to apparent homogeneity by prepar- ative electrophoresis as described below.
Cytosolic tyrosinase was purified from the 105,OOOg supernatant by ammonium sulfate precipitation, hydroxyapatite chromatography, ion- exchange chromatography, gel filtration, and preparative electrophoresis. Ammonium sulfate precipitation, hydroxyapatite, and gel filtration chromatographies were performed as for the melanosomal enzyme, ex- cept that the buffers were not supplemented with Brij 35. Ion-exchange chromatography was carried out by incubating at 4°C for 2 h the pooled supernatants from the hydroxyapatite treatment with 1 vol of CM- Sephadex previously washed and equilibrated with 10 mM phosphate buffer, pH 6.0. The suspension was filtered and the gel washed with 1 vol of buffer. The combined filtrates were concentrated and further pu- rified by gel filtration chromatography as described for the melanosomal enzyme.
Electrophoreticprocedures. Preparative electrophoresis was carried out at 5°C in a Protean electrophoresis cell from Bio-Rad, using 3-mm- thick slab gels prepared according to Laemmli (25). Two milliliters of partially purified tyrosinase, with a protein content of 4-5 mg, was mixed with 1 ml of sample buffer (0.18 M Tris-HCl, pH 6.8, 15% glycerol, 0.075% bromophenol blue, 9% SDS) and applied directly on top of a 3% stacking gel. Using higher protein loads, the purity of the electroeluted tyrosinase was lower. The gels were run at 45 mA until the front entered the 9% separating gel, and then the intensity of current was increased to 65 mA. To identify the tyrosinase band, gels were incubated at room temperature in 200 mM phosphate, pH 6.0, for 30 min, and then with 0.3 mg/ml L-Dopa in 10 mM phosphate buffer, pH 6.8, until the red- brown activity band was evident. The band was cut, washed with 10 mM phosphate buffer, pH 6.8, and divided into 2-mm cubes. These were electroeluted overnight at 4°C in a Bio-Rad 422 electroelution cell using the same buffer conditions as above and a current of 10 mA/tube. The electroeluted samples were extensively dialyzed against 10 mM phosphate buffer, pH 6.8, containing 0.1% Brij 35 in the case of the melanosomal enzyme. The dialyzed samples were centrifuged at 105,OOOg, 1 h, to re- move a small amount of melanin formed during activity stain and elec- troeluted with the protein. Figure 1 shows the electrophoretic homo- geneity of the final preparations.
Analytical SDS-PAGE was performed using the discontinuous method of Laemmli (25). The concentration of acrylamide was usually 12% for the separating gel and 3% for the stacking gel. Samples were routinely treated for 30 min at room temperature with sample buffer (0.18 M Tris- HCl, pH 6.8, 15% glycerol, 0.075% bromophenol blue, 9% SDS, 7.5% 2-mercaptoethanol), using a 2:l ratio (v:v) of sample to buffer. When indicated, mercaptoethanol was omitted from the sample buffer. Elec- trophoreses were run at a constant current of 20 mA. Molecular weight standards from Pharmacia (Uppsala, Sweden) were included in each run.
Tyrosinase activity determinations. Tyrosine hydroxylase activity determinations were carried out by the method of Pomerantz (26) mod- ified as described (27). When the activity of trypsin digestion mixtures was determined, the pH of the samples was brought to 6.8 by addition of HCl to avoid the rapid oxidation of the cofactor L-Dopa at basic pH.
Dopa oxidase activity was spectrophotometrically determined by re- cording the increase in absorbance at 475 nm due to dopachrome for- mation (e4r5 = 3700 M-’ cm-‘), according to Mason (28).

PROTEOLYSIS WITH TRYPSIN OF MAMMALIAN TYROSINASE 223
0 b
-92.5
- 31.0
FIG. 1. Electrophoretic homogeneity of purified cytosolic (a) and me- lanosomal (b) tyrosinases. 10% PAGE-SDS gels were overloaded with 10 pg of protein to detect possible minor contaminants. Samples were treated with P-mercaptoethanol prior to electrophoresis.
Trypsin digestion. Tyrosinase isoforms were routinely treated with trypsin at pH 7.5 to 8.0 (adjusted with NaOH) and 37°C. Standard reaction mixtures contained 0.3 mg/ml of purified tyrosinase and trypsin ranging from 0.01 to 0.2 mg/ml in a final volume of 150 ~1. The reaction was stopped by addition of PMSF to a final concentration of 1 mM.
Protein determination. The protein content was determined by a modified Lowry method (29), using BSA as standard.
RESULTS AND DISCUSSION
Tyrosinase is considered to be protease resistant mainly because trypsin treatment of melanosomes leads to the solubilization of active enzyme, and because some forms of lower electrophoretic mobility obtained by solubiliza- tion of melanosomes with nonionic detergents can be transformed into active forms of higher mobility by pro- tease treatment (4-6, 8-11). However, no quantitative data have been reported on the recovery of tyrosinase activity after proteolysis.
On the other hand, we have previously shown that the cytosolic isoform of mouse melanoma tyrosinase is in- activated by cathepsin D (ll), a protease found in high levels in several tumors, including Bl6 mouse melanoma (30, 31). Moreover, the half-life of tyrosinase in murine melanoma is short as a result of active degradation of the enzyme (32). These observations suggested that tyrosinase could be sensitive to certain proteases that could be used to obtain information on the structure-function relation- ship of the mammalian enzyme.
The purified melanosomal form of B16 mouse mela- noma tyrosinase was found to be sensitive to trypsin. Fig- ure 2 shows that the extent of the proteolytic digestion at a fixed time was dependent on the concentration of trypsin and became evident at trypsin concentrations as
(w:w) resulted in the extensive digestion of the protein as judged by the disappearance of more than 80% of the 70.9-kDa band corresponding to intact tyrosinase.
The kinetics of proteolysis of the cytosolic and melano- somal isoforms were followed at a trypsin concentration of 0.02 mg/ml. The pattern of digestion fragments ob- tained (Figs. 3A and 3C) and the rate of disappearance of the native proteins (Fig. 3D) were very similar. The apparent molecular weights of the digestion fragments are shown in Table I. The differences in molecular weights obtained for the homologous fragments of each protein were very similar, and identical to the one observed for the native isoforms. This suggests that the sequences and tertiary structures of both isoforms are highly related, as expected if both isoforms display a precursor-product re- lationship. The melanosomal form appears to be bound to the melanosomal membrane through a single mem- brane spanning helix located at the C-terminus of the protein (19,20). Proteolytic digestion of the enzyme near the N-terminal side of this membrane-spanning fragment would lead to a soluble core with a difference in molecular weight with the melanosomal form in the range of the one observed in this study.
The pattern of degradation fragments observed in SDS- PAGE was different in the presence and absence of mer- captoethanol (Fig. 3B). In the absence of the reducing agent, the mobility of intact tyrosinase and its degradation products was higher and the protein bands were less in- tensely stained, suggesting that the protein is not fully denatured by SDS if intramolecular disulfide bridges are not reduced. The larger degradation fragment could only be detected in samples electrophoresed under reducing conditions. This result strongly suggests that the peptide
20 - .
I 0.0s
I ai
I I 015 0.2
Trypsin sonc*ntmtion (mg / ml ]
FIG. 2. Effect of trypsin concentration on the degradation of melano- somal tyrosinase. Purified melanosomal tyrosinase was incubated for 1 h at pH 8.0 and 37°C in the presence of varying amounts of trypsin. Proteolysis was stopped with PMSF and the reaction mixtures analyzed by SDS-PAGE in the presence of mercaptoethanol. The percentage of native tyrosinase remaining after trypsin treatment was calculated from the area of the tyrosinase bands determined by densitometric analysis
low as 0.02 mg/ml. Trypsin:tyrosinase ratios around 1:l of the gel.

224 VALVERDE ET AL.
A 0 15 30 60 120 BI 2
66.2- -66.2
-45.0
45D-
31.0- -21.5
21.5 -
D
20 40 60 00 100 120
Time I min )
FIG. 3. Time-dependent degradation of the cytosolic and melanosomal isoforms of tyrosinase by trypsin. Purified tyrosinase samples at a final concentration of approximately 0.3 mg/ml were incubated in the presence of trypsin (0.02 mg/ml final concentration) at pH 8.0 and 37°C. Aliquots were withdrawn at fixed times, shown in minutes on top of the corresponding lanes, and the samples containing 6 pg of protein were analyzed by SDS-PAGE. A, kinetics of proteolysis of the melanosomal enzyme. B, effect of P-mercaptoethanol on the electrophoretic pattern of a melanosomal tyrosinase sample digested with trypsin (0.02 mg/ml) for 1 h. The sample was electrophoresed in the presence (1) or absence (2) of 7.5% p- mercaptoethanol. C, kinetics of proteolysis of the cytosolic enzyme. D, comparison of the rates of disappearance of native cytosolic (0) and melanosomal(0) tyrosinase isoforms, treated for different times with 0.02 mg/ml trypsin, as determined by densitometric analysis of SDS-PAGE gels run in the presence of mercaptoethanol.
resulting from the first proteolytic cleavage remained The pattern obtained was consistent with a precursor- bound to the core of the molecule by at least one disulfide product relationship for the fragments, the final product bridge. being a highly protease-resistant 55.3-kDa core.
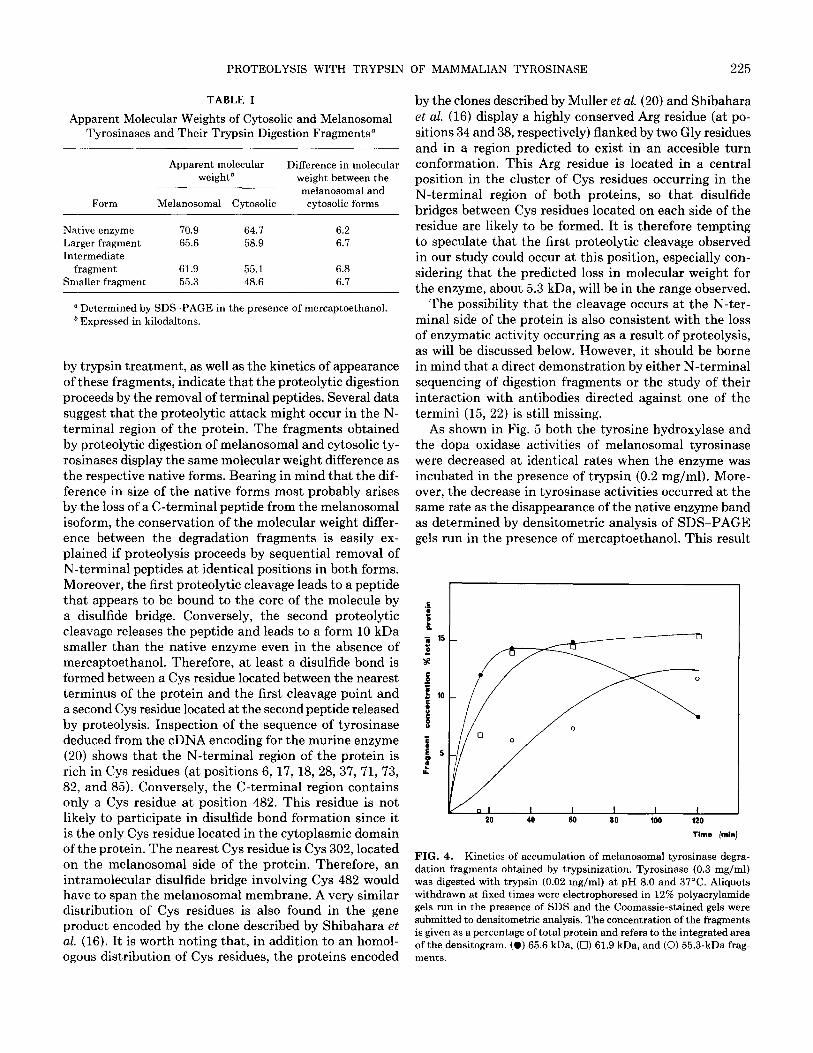
The kinetics of accumulation of the three proteolytic The small difference in molecular weight of native ty- fragments of melanosomal tyrosinase is shown in Fig. 4. rosinase and the different degradation fragments obtained
PROTEOLYSIS WITH TRYPSIN OF MAMMALIAN TYROSINASE 225
TABLE I
Apparent Molecular Weights of Cytosolic and Melanosomal Tyrosinases and Their Trypsin Digestion Fragments”
Apparent molecular Difference in molecular weightb weight between the
melanosomal and Form Melanosomal Cytosolic cytosolic forms
Native enzyme 70.9 64.7 6.2 Larger fragment 65.6 58.9 6.7 Intermediate
fragment 61.9 55.1 6.8 Smaller fragment 55.3 48.6 6.7
by the clones described by Muller et al. (20) and Shibahara et al. (16) display a highly conserved Arg residue (at po- sitions 34 and 38, respectively) flanked by two Gly residues and in a region predicted to exist in an accesible turn conformation. This Arg residue is located in a central position in the cluster of Cys residues occurring in the N-terminal region of both proteins, so that disulfide bridges between Cys residues located on each side of the residue are likely to be formed. It is therefore tempting to speculate that the first proteolytic cleavage observed in our study could occur at this position, especially con- sidering that the predicted loss in molecular weight for the enzyme, about 5.3 kDa, will be in the range observed.
a Determined by SDS-PAGE in the presence of mercaptoethanol. b Expressed in kilodaltons.
by trypsin treatment, as well as the kinetics of appearance of these fragments, indicate that the proteolytic digestion proceeds by the removal of terminal peptides. Several data suggest that the proteolytic attack might occur in the N- terminal region of the protein. The fragments obtained by proteolytic digestion of melanosomal and cytosolic ty- rosinases display the same molecular weight difference as the respective native forms. Bearing in mind that the dif- ference in size of the native forms most probably arises by the loss of a C-terminal peptide from the melanosomal isoform, the conservation of the molecular weight differ- ence between the degradation fragments is easily ex- plained if proteolysis proceeds by sequential removal of N-terminal peptides at identical positions in both forms. Moreover, the first proteolytic cleavage leads to a peptide that appears to be bound to the core of the molecule by a disulfide bridge. Conversely, the second proteolytic cleavage releases the peptide and leads to a form 10 kDa smaller than the native enzyme even in the absence of mercaptoethanol. Therefore, at least a disulfide bond is formed between a Cys residue located between the nearest terminus of the protein and the first cleavage point and a second Cys residue located at the second peptide released by proteolysis. Inspection of the sequence of tyrosinase deduced from the cDNA encoding for the murine enzyme (20) shows that the N-terminal region of the protein is rich in Cys residues (at positions 6, 17, 18, 28, 37, 71, 73, 82, and 85). Conversely, the C-terminal region contains only a Cys residue at position 482. This residue is not likely to participate in disulfide bond formation since it is the only Cys residue located in the cytoplasmic domain of the protein. The nearest Cys residue is Cys 302, located on the melanosomal side of the protein. Therefore, an intramolecular disulfide bridge involving Cys 482 would have to span the melanosomal membrane. A very similar distribution of Cys residues is also found in the gene product encoded by the clone described by Shibahara et al. (16). It is worth noting that, in addition to an homol- ogous distribution of Cys residues, the proteins encoded
The possibility that the cleavage occurs at the N-ter- minal side of the protein is also consistent with the loss of enzymatic activity occurring as a result of proteolysis, as will be discussed below. However, it should be borne in mind that a direct demonstration by either N-terminal sequencing of digestion fragments or the study of their interaction with antibodies directed against one of the termini (15, 22) is still missing.
As shown in Fig. 5 both the tyrosine hydroxylase and the dopa oxidase activities of melanosomal tyrosinase were decreased at identical rates when the enzyme was incubated in the presence of trypsin (0.2 mg/ml). More- over, the decrease in tyrosinase activities occurred at the same rate as the disappearance of the native enzyme band as determined by densitometric analysis of SDS-PAGE gels run in the presence of mercaptoethanol. This result
Time (min)
FIG. 4. Kinetics of accumulation of melanosomal tyrosinase degra- dation fragments obtained by trypsinization. Tyrosinase (0.3 mg/ml) was digested with trypsin (0.02 mg/ml) at pH 8.0 and 37°C. Aliquots withdrawn at fixed times were electrophoresed in 12% polyacrylamide gels run in the presence of SDS and the Coomassie-stained gels were submitted to densitometric analysis. The concentration of the fragments is given as a percentage of total protein and refers to the integrated area of the densitogram. (0) 65.6 kDa, (0) 61.9 kDa, and (0) 55.3-kDa frag- ments.

226 VALVERDE ET AL.
Time [mln)
FIG. 5. Effect of proteolysis on the enzymatic activities of melano- somal tyrosinase. Melanosomal tyrosinase (0.3 mg/ml) was digested with trypsin (0.2 mg/ml final concentration) at pH 8.0 and 37’C. Aliquots were withdrawn at different times and their residual dopa oxidase (0) and tyrosine hydroxylase (m) activities were measured, as well as the amount of residual native tyrosinase (0) as determined by densitometric analysis of SDS-PAGE gels run in the presence of @-mercaptoethanol.
suggests that none of the observed digestion fragments is enzymatically active. This was further proven by activity staining with L-Dopa of gels run in the absence of mer- captoethanol. The band corresponding to native tyrosi- nase was progressively lost as proteolysis proceeded, but no other activity bands of higher mobility were detected.
The perfect match between the loss of the native band in reducing SDS-PAGE gels and the loss of enzyme ac- tivity indicates that inactivation occurs as a result of the first proteolytic event releasing a 65.6-kDa fragment and a 5.3-kDa peptide from the melanosomal form. However, the N terminus of the protein is predicted to be located far away in the primary structure of the protein from the putative copper-binding sites, located in the central region of the molecule (20, 33). Therefore, it is possible that the terminal peptide would contribute to the stabilization of the active conformation of the molecule, rather than be a part of the active site. It is worth noting that a similar situation is found in several amphibian tyrosinases that exist in a proenzymatic form activated by the proteolytic removal of a small terminal fragment (34-36). In any case, the suggestion that the N terminus of the protein is es- sential for its catalytic activity is consistent with the re- cent characterization by several groups of point mutations in the tyrosinase gene, leading to an enzymatically in- active molecule as a result of single amino acid changes within this region of the protein (37-40). On the other hand, the loss of tyrosine hydroxylase and Dopa oxidase activities occurred at the same rate, suggesting that the conformational requirements for both activities are very similar if not identical.
The finding that trypsin is able to inactivate purified tyrosinase is in clear contrast with the observation that trypsin treatment of melanosomes leads to the solubili-
zation of active enzyme and promotes the interconversion of several active forms in crude extracts. The high resis- tance to proteolytic degradation of tyrosinase in crude preparations has even been exploited for its purification by proteinase K digestion of crude melanosomal extracts, followed by ion-exchange chromatography (41). In this case, proteinase K digestion resulted in a loss of over 40% of the initial tyrosinase activity. Moreover, the molecular weight of the purified protein was lower than the one observed for the native enzyme. In our hands, tyrosinase in crude melanosomal preparations is also highly resistant to trypsin digestion, but the purified enzyme is efficiently degraded by the protease. So far, the more likely expla- nation for this observation is that tyrosinase in crude extracts could be partially protected by interaction with other melanosomal components. Supporting this view, preliminary experiments indicate that purified tyrosinase is partially protected by its substrate, L-tyrosine. Exper- iments to identify the melanosomal components respon- sible of tyrosinase protection are underway.
From the results reported above, it can be concluded that, as opposed to the current view, mammalian tyros- inase is sensitive to proteolytic digestion. Proteases appear to be useful probes for the study of the structure and function of the enzyme.
REFERENCES
1.
2.
3. 4.
5.
6.
7.
8.
9. 10.
11.
12.
13.
14.
15.
16.
Lerner, A. B., Fitzpatrick, T. B., Calkins, E., and Summerson, W. H. (1949) J. Biol. Chm. 178,185-195. Hearing, V. J., and Jimenez, M. (1987) Znt. J. Biochem. 19, 1141- 1147.
Korner, A., and Pawelek, J. (1982) Science 217, 1163-1165.
Hearing, V. J., Nicholson, J. M., Montague, P. H., Ekel, T. M., and Tomecki, K. J. (1978) Biochim. Biophys. Actu 522,327-339.
Hearing, V. J., Ekel, T. M., and Montague, P. M. (1981) Znt. J. Biochem. 13,99-103. Laskin, J. D., and Piccinini, L. A. (1986) J. Biol. Ckem. 261,16626- 16635. Ferrini, V., Mileo, A. M., and Hearing, V. J. (1987) Znt. J. Biochem. 19,227-234. Quevedo, W. C., Holstein, T. J., and Bienieki, T. C. (1975) Proc. Sot. Exp. Biol. Med. 150, 735-740. Iwata, K., and Takeuchi, T. (1977) J. Invest. Dermatol. 68.88-92.
Nishioka, K. (1977) FEBS Lett. 80, 225-228.
Martinez-Liarte, J. H., Solano, F., and Lozano, J. A. (1989) Cell B&hem. Funct. 7,21-26.
Wittbjer, A., Dahlbiick, B., Odh, G., Rosengren, A., Rosengren, E., and Rorsman, H. (1989) Acta Derm. Venereal. (Stockholm) 69, 125- 131.
Wittbjer, A., Odh, G., Rosengren, A., Rosengren, E., and Rorsman, H. (1990) Acta Derm. Venereal. (Stockholm) 70,291-294.
Wittbjer, A., Odh, G., Rosengren, A., Rosengren, E., and Rorsman, H. (1990) Pigment Cell Res. 3, 168-172.
Jimenez, M., Maloy, W. L., and Hearing, V. J. (1989) J. Biol. Ckem. 264,3397-3403. Shibahara, S., Tomita, Y., Sakakura, T., Nager, C., Chaudhuri, B., and Muller, R. (1986) Nucleic Acids Res. 14,2413-2427.

PROTEOLYSIS WITH TRYPSIN OF MAMMALIAN TYROSINASE 227
17. Yamamoto, H., Takeuchi, S., Kudo, T., Makino, K., Nakata, A.,
18.
19.
20.
Shinoda, T., and Takeuchi, T. (1987) JapanJ. Genet. 62,271-274.
Jackson, I. (1988) Proc. Natl. Acad. Sci. USA 85,4392-4396.
Kwon, B., Wakulchik, M., Haq, A., Halaban, R., and Kestler, D. (1988) Bioc~~. Biophys. Res. Commun. 153,1301-1309.
Muller, G., Ruppert, S., Schmid, E., and Schutz, G. (1988) EMBO J. 7, 2723-2730.
21.
22.
Kwon, B., Haq, A., Pomerantz, S., and Halaban, R. (1987) Proc. Natl. Acad. Sci. USA 84,7473-7477.
Jimenez, M., Tsukamoto, K., and Hearing, V. J. (1991) J. Btil. C/rem. 266,1147-1156.
23.
24.
25.
26.
Terao, M., Tabe, L., Garattini, E., Sartori, D., Studer, M., and Mintz, B. (1989) Biochem. Biophys. Res. Commun. 159,848-853.
Aroca, P., Garcia-Borron, J. C., Solano, F., and Lozano, J. A. (1990) Biochim. Biophys. Acta 1035, 266-275.
Laemmli, U. K. (1970) Nature 277,680-682.
Pomerantz, S. H. (1964) Biochem. Biophys. Res. Commun. 16,188- 194.
27. Jara, J. R., Solano, F., and Lozano, J. A. (1988) Pigment Cell Res. 1.332-339.
28. Mason, H. S. (1948) J. Biol. Chem. 172,83-90.
29. Hartree, E. (1972) Anal. Biochem. 48,422-427.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
Sloane, B. F., Dunn, J. R., and Honn, K. V. (1981) Science 212,
1151-1153.
Sloane, B. F., Honn, K. V., Sadler, J. G., Turner, W. A., Kimpson, J. J., and Taylor, S. D. (1982) Cancer Res. 42,980-986. JimCnez, M., Kameyama, K., Maloy, W., Tomita, Y., and Hearing, V. J. (1988) Proc. Natl. Acad. Sci. USA 85, 3830-3834. Hearing, V. H., and Jimenez, M. (1989) Pigment Cell Res. 2, 7% 85.
Barisas, B. G., and McGuire, J. S. (1974) J. Biol. Chem. 249,3151- 3156.
Penafiel, R., Galindo, J. D., Pedreiio, E., and Lozano, J. A. (1982) Biochem. J. 205,397-404.
Garcia-Borron, J. C., Escribano, J., Jimenez, M., and Iborra, J. L. (1982) Anal. Biochem. 125, 277-285.
Giebel, L., Strunk, K., King, R., Hanifin, J., and Spritz, R. (1990) Proc. Natl. Acad. Sci. USA 87, 3255-3258.
Jackson, I., and Bennet, D. (1990) Proc. Natl. Acad. Sci. USA 87, 7010-7014.
Shibahara, S., Okinaga, S., Tomita, Y., Takeda, A., Yamamoto, H., Sato, M., and Takeuchi, T. (1990) Eur. J. Biochem. 189,455-461. Takeda, A., Tomita, Y., Matsunaga, J., Tagami, H., and Shibahara, S. (1990) J. Biol. Chem. 265, 17792-17797. Yurkow, E. J., and Laskin, J. D. (1989) Arch. Biochem. Biophys. 275,122-129.
Copyright © 2022 FDOKUMEN




















