
A genetically modulated, intrinsic cingulate circuit supports human nicotine addiction
Upload
independentCategory
view
3download
0
Tyrosinase exacerbates dopamine toxicity but is not geneticallyassociated with Parkinson’s disease
Elisa Greggio,* Elisabetta Bergantino,* Donald Carter,� Rili Ahmad,� Gertrude-Emilia Costin,�Vincent J. Hearing,� Jordi Clarimon,� Andrew Singleton,� Johanna Eerola,§ Olli Hellstrom,¶Pentti J. Tienari,§ David W. Miller,� Alexandra Beilina,� Luigi Bubacco* and Mark R. Cookson�
*Department of Biology, University of Padova, Padova, Italy
�Laboratory of Neurogenetics, National Institute on Aging, Bethesda, Maryland, USA
�Pigment Cell Biology Section, Laboratory of Cell Biology, National Cancer Institute, Bethesda, Maryland, USA
§Department of Neurology, Helsinki University Central Hospital and University of Helsinki, Biomedicum-Helsinki, Neuroscience
Programme, Helsinki, Finland
¶Department of Neurology, Seinajoki Central Hospital, Seinajoki, Finland
Abstract
Tyrosinase is a key enzyme in the synthesis of melanin in skin
and hair and has also been proposed to contribute to the for-
mation of neuromelanin (NM). The presence of NM, which is
biochemically similar to melanin in peripheral tissues, identifies
groups of neurons susceptible in Parkinson’s disease (PD).
Whether tyrosinase is beneficial or detrimental to neurons is
unclear; whilst the enzyme activity of tyrosinase generates
dopamine-quinones and other oxidizing compounds, NM may
form a sink for such radical species. In the present study, we
demonstrated that tyrosinase is expressed at low levels in the
human brain. We found that mRNA, protein and enzyme
activity are all present but at barely detectable levels. In cell
culture systems, expression of tyrosinase increases neuronal
susceptibility to oxidizing conditions, including dopamine itself.
We related these in vitro observations to the human disease by
assessing whether there was any genetic association between
the gene encoding tyrosinase and idiopathic PD. We found
neither genotypic or haplotypic association with three poly-
morphic markers of the gene. This argues against a strong
genetic association between tyrosinase and PD, although the
observed contribution to cellular toxicity suggests that a
biochemical association is likely.
Keywords: dopamine, genetics, neuromelanin, Parkinson’s
disease, tyrosinase.
J. Neurochem. (2005) 93, 246–256.
Parkinson’s disease (PD) is characterized, in part, by theprogressive loss of select groups of neurons from variousbrain regions. Much attention has focused on the dopamin-ergic neurons of the substantia nigra, as loss of these cells isassociated with the movement-related symptoms of PD. Lossof projections from the substantia nigra results in decreaseddopamine in the striatum. Replacing this striatal dopamine,e.g. with the dopamine precursor L-DOPA, is currently themainstay of treatment for PD. This has led to the suggestionthat dopamine may contribute to the toxicity seen in thisdisease. However, it is clear that the presence or absence ofdopamine in neurons does not completely explain thepatterns of cell loss in PD. For example, there are non-dopaminergic neuronal groups that are affected during thedisease, such as norepinephrine-containing neurons in thelocus ceruleus (Zarow et al. 2003). Furthermore, there are
several neuronal populations that express dopamine, such asthe neurons of the ventral tegmental area, which arerelatively resistant to the processes of cell loss in PD (Hirsch1994).
There are many differences between these groups ofneurons that might explain their differential vulnerability inPD. An interesting observation is that the presence of
Received September 15, 2004; revised manuscript received November29, 2004; accepted December 1, 2004.Address correspondence and reprint requests to Elisa Greggio,
Department of Biology, University of Padova, Via Ugo Bassi 58b,35121, Padova, Italy. E-mail: [email protected] used: IP, immunoprecipitation; MOI, multiplicity of
infection; NM, neuromelanin; PD, Parkinson’s disease; pMEL, pig-mented melanoma cell lines.
Journal of Neurochemistry, 2005, 93, 246–256 doi:10.1111/j.1471-4159.2005.03019.x
246 � 2005 International Society for Neurochemistry, J. Neurochem. (2005) 93, 246–256No claim to original US government works

neuromelanin (NM) marks vulnerable neurons in both humanPD and experimental parkinsonism induced by the toxinMPTP. NM expression is especially high in primates andtends to accumulate during aging (Zecca et al. 2002).Chemically, NM is a stable polymer containing lipids, ironand quinone-like radicals (Zecca et al. 2001, 2003). Themethod of formation of NM is somewhat controversial.Dopamine and other catechols can be oxidized, through bothnon-enzymatic and enzymatic means, to form dopamine-quinones. One enzyme that could contribute to NM formationis tyrosinase (EC 1.14.18.1), a glycoprotein whose majorcatalytic activity is the hydroxylation of tyrosine andoxidation of L-DOPA, which occurs in the process of melaninformation (Sanchez-Ferrer et al. 1995). Therefore, tyrosinaseis the major enzyme required for the synthesis of melanin inskin and hair and may contribute to NM formation.
In addition, tyrosinase can oxidize the catechol ring ofdopamine to the highly reactive species dopamine-quinone(Miranda and Botti 1983). There are several potential targetsfor these dopamine-quinones in neurons. Within the cytosol,dopamine-quinone can react with the sulfhydryl group ofcysteine residues to form protein adducts, thus leading toirreversible alteration or inhibition of protein function(Asanuma et al. 2003). For example, in the presence oftyrosinase dopamine covalently modifies and inactivatestyrosine hydroxylase (Xu et al. 1998). Dopamine oxidationalso inhibits dopamine transporter (Whitehead et al. 2001),glutamate transport (Berman and Hastings 1997) andmitochondrial respiration (Berman and Hastings 1999).Cytosolic quinones can also enter the nucleus and producecovalent DNA modifications (Stokes et al. 1996; Cavalieriet al. 2002). Importantly for the pathophysiology of PD,dopamine-quinone can react with a-synuclein and stabilize atoxic intermediate in the process of fibril formation (Conwayet al. 2001). Mutations in the gene for a-synuclein areassociated with autosomal dominant PD and protein aggre-gation is likely to be causal for disease. These observationssuggest a hypothesis where tyrosinase could act on dopamineor other catechols and promote neuronal damage by mech-anisms including the exaggeration of a-synuclein toxicity.
Although dopamine-quinones are likely to be toxic, theformation of NM itself may be neuroprotective. NM iscapable of scavenging dopamine-quinone, thus decreasingtheir concentration within the cell. Furthermore, as NM formswithin bilamellar autophagic vacuoles/lysosomes within cells(Sulzer and Edwards 2000), trapped quinones are physicallysequestered from cytosolic targets. It has been proposed thatNM can be released into the cytoplasm and in the extracel-lular space from dying neurons in PD thus leading to asubsequent aggravation of neurodegeneration (Whiteheadet al. 2001; Wilms et al. 2003; Zecca et al. 2003).
These considerations show that tyrosinase has the potentialto damage neurons by producing dopamine-quinones or toprotect cells by enhancing NM synthesis. In the present
study, we have expressed tyrosinase within human neurob-lastoma cells or primary midbrain cultures and examined theeffects on toxicity induced by oxidative stress or exposure tomutant a-synuclein. We also considered tyrosinase as acandidate gene for risk of developing PD in humans.However, although several groups have suggested thattyrosinase mRNA is expressed in brain (Xu et al. 1997;Tief et al. 1998), some negative results have been reported(Gimenez et al. 2003). Studies of tyrosinase activity orprotein expression in human brain are limited. Higashi et al.(2000) showed tyrosinase immunoreactivity in human brainusing an anti-mushroom/human tyrosinase antibody, whilstother studies have failed to detect tyrosinase immunoreac-tivity (Ikemoto et al. 1998). Therefore, we first examined theexpression of tyrosinase mRNA and protein in samples fromhuman brain.
Materials and methods
RNA extraction, cDNA synthesis and RT-PCR
Post-mortem human brain tissues came from six individuals 24–
48 h after death and were stored at )80�C. Five brain regions
corresponding to cortex, caudate nucleus, globus pallidus, substantia
nigra and putamen were selected from each individual. Total RNA
was extracted using TRIZOL and approximately 50–100 mg of
brain tissue was homogenized using a Polytron PT2100 (Kinema-
tica, Lucerne, Switzerland). RNA integrity and concentration were
tested by denaturing agarose gel electrophoresis and spectropho-
tometer. Total RNA (1–3 lg) was denatured for 5 min at 75�C and
then reverse transcribed at 42�C for 1 h in a 20 lL reaction volume
in buffer containing 50 mM Tris-HCl (pH 8.3), 75 mM KCl, 3 mM
MgCl2, 10 mM dithiothreitol, 250 ng of oligo(dT) primers, 10 U of
AMV-RT, 1 mM of each dNTPs and 20 U of placental ribonuclease
inhibitor. cDNA (1 lL; 5–100 ng) was added in a 50-lL reaction to
amplify a 228-bp fragment using HT-F1 and HT-R1 primers (see
Table 1) with 35 (Fig. 1b) or 40 cycles (Fig. 1c) of denaturing at
95�C for 30 s, annealing at 61�C for 30 s, extension at 72�C 30 s
and the final extension performed at 72�C for 10 min. Dilutions
(50-fold) of reactions were re-amplified (20 cycles) with nested
primers HT-F2 and HT-R2 (174 bp) to confirm the specificity of the
RT-PCR. The full-length transcript was amplified under the same
conditions as above but with 3 min of extension and fully sequenced
by using the BigDye terminator kit on an ABI 3100 genetic analyser
(Applied Biosystems, Foster City, CA, USA). Primers sequences are
shown in Table 1.
Cloning and cell lines
Full-length human tyrosinase cDNA (coding sequence and
3¢untranslated region) was cloned into pcDNA3 vector (Invitrogen,
Carlsbad, CA, USA) using the EcoRI restriction site and the insert
was fully sequenced using the BigDye terminator kit on an ABI
3100 sequencer (Applied Biosystems). Transient transfections of
M17 human neuroblastoma cell lines were performed as described
by Miller et al. (2003). Human pigmented melanoma cell lines
(pMEL) expressing tyrosinase were kindly provided by David
Weinreich (National Cancer Institute, Bethesda, MD, USA).
Tyrosinase exacerbates dopamine toxicity 247
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 93, 246–256No claim to original US government works

Western blotting and immunoprecipitation
Western blotting from whole cell lysate and tissue samples was
performed as previously described (Miller et al. 2003). The primary
antibodies used were monoclonal anti-tyrosinase (clone T311;
1 : 200; Chemicon, Temencula, CA, USA) and rabbit antiserum
anti-tyrosinase aPEP7h (Virador et al. 2001) (1 : 1000). Blots were
developed with peroxidase-labeled secondary antibodies (1 : 5000;
Jackson Immunochemicals, West Grove, PA, USA) and captured on a
Storm phosphorimager (Amersham Biosciences, Uppsala, Sweden).
For immunoprecipitation (IP) experiments, �2 · 106 of M17 or
pMEL cells or �200 mg of striatal tissue were solubilized in
phosphate-buffered saline, pH 7.4, 1% NP40, phenylmethylsulfonyl
fluoride, 1 mM and protease inhibitor cocktail (Sigma, Poole, UK).
Cell (�0.5 mg of total proteins) or tissue (�5 mg of total proteins)
lysates were pre-cleared by incubation with 50 lL of pre-washed
Protein A agarose slurry (Amersham Biosciences, Uppsala, Sweden)
for 2 h on ice. IP was then performed by incubating the pre-cleared
lysate with 1 lL of polyclonal anti-tyrosinase (aPEP7h) overnight at4�C with continuous shaking, followed by addition of the beads and
further incubation for 2 h at 4�C. Beads were washed five times with
1 mL of phosphate-buffered saline, pH 7.4, containing 0.05%
NP40, phenylmethylsulfonyl fluoride, 1 mM and protease inhibitors
(1 : 100) and 50 lL of 2· Laemmli buffer containing 5% (v/v)
b-mercaptoethanol was added to the beads and incubated for 10 min
at 95�C. Supernatant fluids were processed for sodium dodecyl
sulfate–polyacrylamide gel electrophoresis and western blotting
with monoclonal anti-tyrosinase (clone T311, 1 : 200).
Some samples were also deglycosylated. Samples were denatured
(50 mM sodium phosphate, pH 7.0, 0.2% sodium dodecyl sulfate,
0.1% b-mercaptoethanol) at 100�C for 5 min, cooled and then
mixed with 1% NP40. Deglycosylation was then carried out
using the GLYKO kit (a mixture of PNGaseF, SialidaseA
and O-Glycanase) according to the manufacturer’s instructions
(Prozyme, San Leandro, CA, USA). Samples were digested at 37�Covernight and analysed by sodium dodecyl sulfate–polyacrylamide
gel electrophoresis and western blotting as above.
Table 1 Oligonucleotides used for the amplification of tyrosinase
cDNA
Name Sequence (5¢ fi 3¢)
HT-F1 TTTCATCCAAAGATCTGGGC
HT-F2 TTCAGACCCAGACTCTTTTCAA
HT-R1 TGAGGAGTGGCTGCTTTTCT
HT-R2 GCTGCTTTCTCTTGTGACGA
HT-5F AATGCTCCTGGCTGTTTTGT
HT-3R AGTGAGGTCAGGCTTTTTGG (a)
(b)
(c)
Fig. 1 Tyrosinase mRNA is expressed in human brain. (a) Approxi-
mately 50 ng of cDNA from human striatum was used to amplify a
1634-bp fragment (arrow) corresponding to the full-length transcript of
human tyrosinase using primers HT-5F and HT-3R (Table 1). (b)
cDNAs from cortex (Co), caudate nucleus (Ca), globus pallidus (Pa),
substantia nigra (SN) and putamen (Pu) were also amplified with
primers HT-F1 and HT-R1 to give a 228-bp PCR fragment (arrow,
upper panel). Subsequently, 50-fold dilutions of these reactions were
used for nested PCR with primers HT-F2 and HT-R2 to confirm the
specificity of the amplified products. The fragments obtained showed
the expected size of 174 bp (arrowhead, lower panel). (c) cDNAs from
the same brain regions as (b) were amplified with primers HT-F1 and
HT-R1 from six different control autopsied brains, using 40 cycles of
PCR to increase the sensitivity of detection.
248 E. Greggio et al.
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 93, 246–256No claim to original US government works

Enzyme assays
Tyrosinase enzyme activity was measured as described previously
(Hearing and Ekel 1976). Briefly, 30 lL of each sample to be tested
was added to 10 lLphosphate buffer containing 0.25 mMofDOPAas
cofactor and 10 lL 0.25 mCi/mmol [U-14C]-tyrosine (NEN, Boston,
MA, USA). The reactions (performed in triplicate) were mixed,
incubated at 37�C for either 3 or 22 h and 40 lL of each sample was
transferred to filter paper discs (3MM; Whatman, Clifton, NJ, USA)
and air-dried. Each filter was then washed three times in 0.1 N HCl,
twice in 95% ethanol, once in acetone and air-dried again. Radioactive
melanin retained by the filterswas determined in a scintillation counter
(Beckmann, Fullerton, CA, USA). Tyrosinase activity is defined as
pmol [14C]-tyrosine incorporated into melanin/lg protein/h at 37�C.
Primary cell cultures and viral transduction
Primary cell cultures were prepared from post-natal mouse midbrain
using methods described elsewhere (Petrucelli et al. 2002). Tyrosin-ase, a-synuclein wild-type and a-synuclein A53T control cDNAs
were cloned into pHSVPrpUC and packaged into recombinant viral
particles using 5dl1.2 helper virus and the 2-2 packaging cell line.
Recombinant viruseswere titred onM17 cells. Primary cells from3- to
4-week cultures were transduced with viral particles at a multiplicity
of infection (MOI) of 10. After 48 h, toxicity was assessed by staining
for tyrosine hydroxylase and microtubule-associated protein 2, again
as described previously (Petrucelli et al. 2002). For estimations of cell
numbers, six random microscope fields (using a 20· objective) were
counted in each of three sister cultures, for a total of 18 fields per
condition. For specific gene staining we used monoclonal anti-
tyrosinase (1 : 50; Chemicon, Temecula, CA, USA), monoclonal
anti-a-synuclein (1 : 200;Mc42; Translabs, Lexington, UK) or rabbit
anti-LacZ (1 : 500; Molecular Probes, Eugene, OR, USA).
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
assays
We expressed tyrosinase in M17 cells using HSV viruses at an MOI
of 1 for 24 h before 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetra-
zolium bromide assay for cell viability, with LacZ as a control for
transduction. Cells were exposed to dopamine (10–160 lM), H2O2
(300–1000 lM), paraquat (125– 2000 lM) or lactacystin (1–25 lM)for a further 24 h and viability assessed using the 3-(4,5-dimeth-
ylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. For each
experiment, eight wells per concentration were used and each
experiment was performed in duplicate. Statistical differences
between cell lines were assessed using two-way ANOVA.
Confocal microscopy
M17 cells were grown in 12-well plates with coverslips and
transduced with an MOI of 1 of tyrosinase-HSV. Slides were fixed
as described for the primary cultures and stained with primary
monoclonal anti-tyrosinase (1 : 50) and secondary AlexaFluor 568
(1 : 200; Molecular Probes). Slides were examined with a confocal
microscope (LSM510; Zeiss, Thornwood, NY, USA). Black
granules were identified using differential interference contrast on
the same microscope system.
Genetic association methods
We performed a case-control association analysis using 147 sporadic
late-onset PD patients (mean age 67.2, range 38–88, years, 41%
women) and 135 neurologically normal healthy control subjects
(mean age 65.8, range 37–80, years, 64% women). This population
from Finland has been described elsewhere (Clarimon et al. 2004)and is sufficiently large to detect the protective effects of
polymorphisms in the inducible nitric oxide synthase gene (Hague
et al. 2004). Affected individuals were diagnosed according to PD
Society Brain Bank criteria (Hughes et al. 1992). Patients with
dementia or patients who reported first-degree relatives with
parkinsonism were excluded. Written informed consent was
obtained from all participants. DNA was purified from peripheral
blood using standard phenol–chloroform extraction.
Three different polymorphisms were analysed. A biallelic
polymorphism consisting of a C to A exchange located at the
CCATT box in the TYR promoter region (nucleotide )199,rs1799989) was genotyped as previously described (Oetting et al.1991). A non-synonymous polymorphism Ser to Tyr at codon 192
(rs1042602) was genotyped as described previously (Giebel and
Spritz 1990). Finally, an A to G polymorphism located within intron
4 of the TYR gene was also analysed by using Taqman� Assays-by-
DesignSM SNP Genotyping (Applied Biosystems). Thermal cycling
and end-point PCR analysis were performed on an ABI PRISM�
7900HT Sequence Detection System. Primer and probe sequences
are available upon request.
Differences in allele and genotype distributions were analysed
using the v2 test. Haplotype frequencies were estimated with an
expectation-maximization algorithm, implemented in Arlequin
ver.2.000 program (Schneider et al. 2000). The D¢ statistic was
calculated with the EMLD program. Comparison of TYR haplotype
frequencies between cases and controls was performed using
Fisher’s exact test.
Results
Tyrosinase expression in human brain
As the presence of tyrosinase in human brain is controver-sial, we first determined: (i) the full-length transcriptsequence and (ii) the mRNA expression of tyrosinase indifferent regions of the adult human brain. By using aforward primer annealing between the 5¢untranslated regionand the first start codon of the mRNA (HT-5F) and a reverseprimer annealing to the 3¢untranslated region (HT-3R)(Table 1) we amplified the expected 1634-bp full-lengthtyrosinase transcript shown as a single band in Fig. 1(a).After cloning and sequencing, the amplification productshowed 100% alignment with the tyrosinase mRNAsequence (NM_000372). We next investigated the expressionof tyrosinase mRNA in different brain regions related to thedopamine circuit (cerebral cortex, substantia nigra, caudatenucleus, globus pallidus and putamen) from six post-mortembrains (Table 2). In these experiments, we used a nested PCRstrategy to maximize product yield. Figure 1(b) shows theexpression of tyrosinase in all the brain areas analysed (fromindividual 1). Although this is not a quantitative assay, theamplification product intensities are relatively weak after 35cycles of PCR, suggesting a very low expression of the
Tyrosinase exacerbates dopamine toxicity 249
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 93, 246–256No claim to original US government works

mRNA in all of the regions examined. RT-PCR experimentswere also performed for all six individuals with 40 cycles ofPCR in order to increase the intensity of the signals (Fig. 1a).The intensities of the amplified bands varied among theindividuals and the regions analysed, supporting the viewthat tyrosinase is likely to be expressed at similar but lowlevels in these regions.
The above results support the contention that tyrosinasemRNA is expressed at low levels in human brain butwhether tyrosinase protein is present is similarly controver-sial. Initially we analysed the expression of tyrosinase bywestern blot using either monoclonal (T311) or rabbitpolyclonal (aPEP7h) antibodies to tyrosinase. The mono-clonal antibody was specific and sensitive for the detectionof endogenous tyrosinase in melanoma cell lines (pMEL)and of overexpressed tyrosinase in M17 neuroblastoma cellsbut we could not detect any specific immunoreactivity inbrain samples (data not shown). We therefore performed IPexperiments to increase both the sensitivity and specificity ofdetection. We used aPEP7h antibody, which binds to the C-terminal tail of the enzyme, for IP and the monoclonalantibody, which recognizes an epitope between amino acids1 and 433, for immunoblotting. This combination ofantibodies increased the sensitivity of detection of overex-pressed tyrosinase in M17 cells (Fig. 2a). Using the same IPconditions, we also detected a weak band of 70 kDacorresponding to tyrosinase from striatal extracts (Fig. 2b).However, this was not consistently present in all samples.An extra-band of about 90 kDa was also visible and couldrepresent a high molecular weight complex that we alsoobserved occasionally in M17 overexpressing tyrosinase(Fig. 2b). Tyrosinase is synthesized as a 60-kDa polypeptideand subsequently undergoes glycosylation in the endo-plasmatic reticulum to produce a mature 70–75-kDa protein.After IP, we subjected samples to deglycosylation and bothbrain and pMEL samples lost immunoreactivity around70 kDa (Fig. 2b). However, the size of tyrosinase afterdeglycosylation is similar to IgG heavy chains and thuscannot be clearly distinguished from the latter. Interestingly,the processing of tyrosinase seems to take place in the samefashion in M17 as in pMEL cells, suggesting that tyrosinaseis processed in the neuroblastoma cell model (and probablyin the brain) as it occurs in melanocytes (Fig. 2c).
Table 2 Clinical profiles of study subjects
Case Gender Age (years) Cause of death
1 Male 54 Heart attack
2 Male 54 Larynx and esophagus neoplasia
3 Male 44 Cirrhosis of the liver
4 Female 71 Acute pancreatitis
5 Male 20 Car accident
6 Male 82 Heart attack
(a)
(b)
(c)
Fig. 2 Immunoprecipitation and deglycosylation of tyrosinase. (a)
Tyrosinase overexpressed in M17 cells was immunoprecipitated from
0.5 mg of total protein extract using the polyclonal antibody aPEP7h
and blotted with monoclonal antibody to tyrosinase T311 (arrow).
Equivalent amounts of lysates and supernatant fluids (10 lg) were
loaded in the indicated lanes and 10 lL of denatured beads was
loaded for the immunoprecipitated sample (IP). Sample from dena-
tured beads alone is shown in the first lane (** and * are non-specific
bands). Markers on the right of the blots are in kDa. (b) Tyrosinase
was also immunoprecipitated from human striatum and pigmented
melanoma cell line (pMEL) cell extracts. After denaturation, the IP
supernatant fluids were analysed by western blotting (10 lL of dena-
tured beads) and demonstrated a 70-kDa band (arrow). The tyrosin-
ase band at 70 kDa was not visible after deglycosylation of an equal
amount of denatured beads, although the presence of several non-
specific bands from the IP (**; likely to be IgG heavy chain) in the same
region of the gel prevented us from demonstrating the expected
50-kDa band (see below). An extra-band of about 90 kDa is also
visible in both M17 cells expressing tyrosinase and brain extract
(arrowhead). This band also disappeared after deglycosylation sug-
gesting that it could represent a higher glycosylation form. (c) Lysates
of transfected M17 before (arrow) and after (arrowhead) deglycosy-
lation show the same pattern as pMEL cells expressing endogenous
tyrosinase (10 lg of protein extract loaded in each lane).
250 E. Greggio et al.
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 93, 246–256No claim to original US government works
As further evidence of the presence of tyrosinase in thebrain, we measured enzyme activity. Tyrosinase activity wasdetected in both human substantia nigra and caudate nucleus(Fig. 3). In substantia nigra, the activity after 3 h ofincubation was twice that of the caudate nucleus (Fig. 3).After 22 h we observed a decrease in the specific activity insubstantia nigra and a threefold decrease in caudate nucleus.This argues that the conversion of [14C]-tyrosine to melaninwas not due to a spontaneous process, as the rate of initialactivity decreased over time following enzyme kinetics.
Toxic effects of tyrosinase overexpression
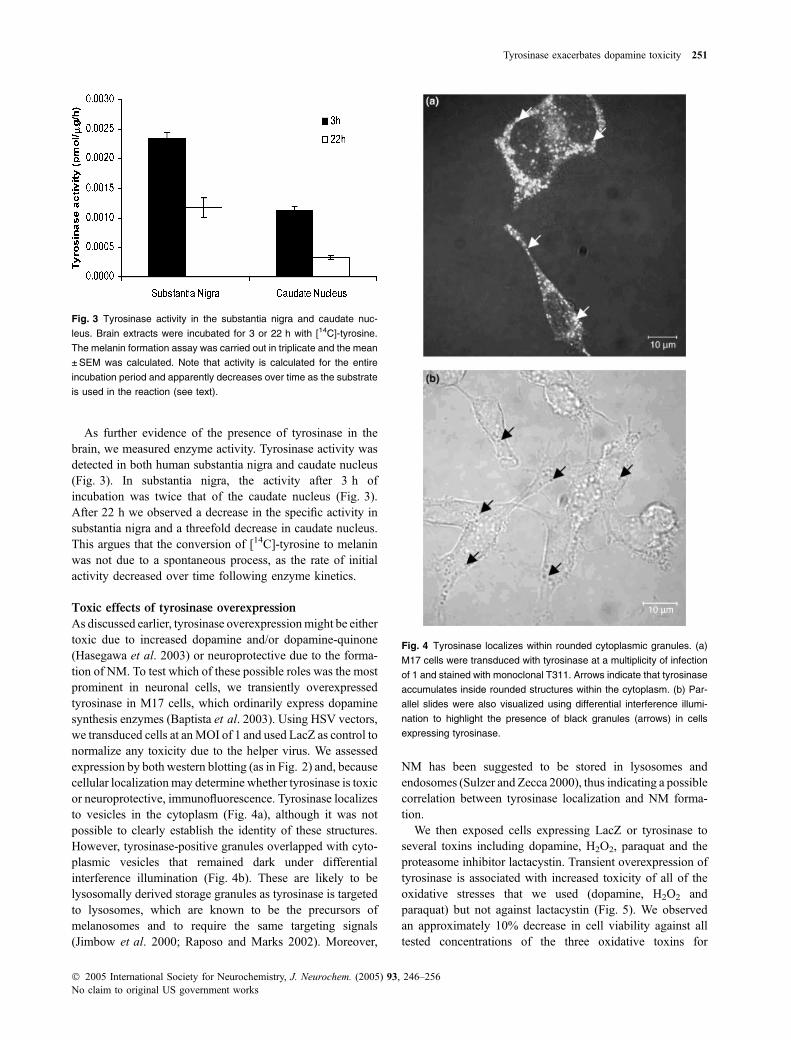
As discussed earlier, tyrosinase overexpressionmight be eithertoxic due to increased dopamine and/or dopamine-quinone(Hasegawa et al. 2003) or neuroprotective due to the forma-tion of NM. To test which of these possible roles was the mostprominent in neuronal cells, we transiently overexpressedtyrosinase in M17 cells, which ordinarily express dopaminesynthesis enzymes (Baptista et al. 2003). Using HSV vectors,we transduced cells at anMOI of 1 and used LacZ as control tonormalize any toxicity due to the helper virus. We assessedexpression by both western blotting (as in Fig. 2) and, becausecellular localization may determine whether tyrosinase is toxicor neuroprotective, immunofluorescence. Tyrosinase localizesto vesicles in the cytoplasm (Fig. 4a), although it was notpossible to clearly establish the identity of these structures.However, tyrosinase-positive granules overlapped with cyto-plasmic vesicles that remained dark under differentialinterference illumination (Fig. 4b). These are likely to belysosomally derived storage granules as tyrosinase is targetedto lysosomes, which are known to be the precursors ofmelanosomes and to require the same targeting signals(Jimbow et al. 2000; Raposo and Marks 2002). Moreover,
NM has been suggested to be stored in lysosomes andendosomes (Sulzer and Zecca 2000), thus indicating a possiblecorrelation between tyrosinase localization and NM forma-tion.
We then exposed cells expressing LacZ or tyrosinase toseveral toxins including dopamine, H2O2, paraquat and theproteasome inhibitor lactacystin. Transient overexpression oftyrosinase is associated with increased toxicity of all of theoxidative stresses that we used (dopamine, H2O2 andparaquat) but not against lactacystin (Fig. 5). We observedan approximately 10% decrease in cell viability against alltested concentrations of the three oxidative toxins for
Fig. 3 Tyrosinase activity in the substantia nigra and caudate nuc-
leus. Brain extracts were incubated for 3 or 22 h with [14C]-tyrosine.
The melanin formation assay was carried out in triplicate and the mean
±SEM was calculated. Note that activity is calculated for the entire
incubation period and apparently decreases over time as the substrate
is used in the reaction (see text).
Fig. 4 Tyrosinase localizes within rounded cytoplasmic granules. (a)
M17 cells were transduced with tyrosinase at a multiplicity of infection
of 1 and stained with monoclonal T311. Arrows indicate that tyrosinase
accumulates inside rounded structures within the cytoplasm. (b) Par-
allel slides were also visualized using differential interference illumi-
nation to highlight the presence of black granules (arrows) in cells
expressing tyrosinase.
Tyrosinase exacerbates dopamine toxicity 251
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 93, 246–256No claim to original US government works

tyrosinase compared with LacZ (p < 0.01 for differencesbetween cell lines for dopamine, H2O2 and paraquat).
We also transduced primary mouse midbrain neurons withtyrosinase or LacZ as a control. After transduction at an MOIof 10, more than 95% of tyrosine hydroxylase-positiveneurons expressed the respective proteins. After 48 h(Fig. 6a), we observed a toxic effect of tyrosinase com-pared with LacZ in tyrosine hydroxylase-positive cells(p < 0.001). We also examined the effect of the combinedoverexpression of tyrosinase with wild-type or A53T a-synuclein (Fig. 6b) to evaluate whether tyrosinase activitymight enhance a-synuclein toxicity via quinone-adductformation (Conway et al. 2001). Although tyrosinase wastoxic in the presence of wild-type a-synuclein (Fig. 6b), thiseffect was similar to that of expressing tyrosinase alone(Fig. 6a) or in the presence of additional LacZ virus tocontrol for the transduction of cells with two viral vectorseach at an MOI of 10 (Fig. 6b). In both cases, the effect oftyrosinase was significant (p < 0.01). As previously reported(Petrucelli et al. 2002), exposure to A53T mutant a-synuc-lein was toxic to these cells but exposure to wild-typea-synuclein was not. Furthermore, the toxicity of A53Ta-synuclein was increased by additional expression of
tyrosinase (Fig. 6b), suggesting that tyrosinase can enhancethe toxic effects of mutant a-synuclein.
Assessment of TYR gene polymorphisms in Finnish
Parkinson’s disease
The above results show that tyrosinase is expressed at lowlevels in human brain and enhances oxidative toxicity. Ifdopamine-derived radicals were associated with the patho-
Fig. 5 Tyrosinase enhances toxicity in the presence of oxidative tox-
ins. We expressed tyrosinase in M17 cells using HSV vectors before
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay for cell viability, with LacZ as a control for transduction. Cells
expressing tyrosinase showed decreased cell viability with respect to
control when treated for 24 h with dopamine (a), paraquat (b) and H2O2
(c) (p < 0.01). No enhancement of toxicity was observed after treat-
ment with the proteasome inhibitor lactasystin (d). Data are shown from
one set of experiments expressed as a percentage of cells transduced
with virus alone (n ¼ 8 wells per dose, error bars indicate SEM).
Fig. 6 Effect of overexpression of tyrosinase and a-synuclein on
survival of dopaminergic neurons. (a) Tyrosinase expression reduces
neuronal viability compared with LacZ control (***p < 0.01). Primary
midbrain cultures were transduced with tyrosinase for 48 h and cell
viability was determined by counting the number of dopaminergic
neurons stained with anti-tyrosine hydroxylase (TH) (n ¼ 6 fields;
experiments were carried out in triplicate). The data are expressed as
a percentage of control cells (LacZ), error bars indicate SEM. (b)
Additive effect of tyrosinase and A53T mutant a-synuclein. Primary
cells were cotransduced with tyrosinase and LacZ, wild-type (wt)
a-synuclein or mutant a-synuclein for 48 h. As in (a), there was a
strong effect of tyrosinase when coexpressed in the presence of LacZ
(***p < 0.001) or wt a-synuclein (**p < 0.01). There was an additional
loss of TH-positive neurons after coexpression of tyrosinase and A53T
a-synuclein compared with A53T a-synuclein alone (*p < 0.05).
252 E. Greggio et al.
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 93, 246–256No claim to original US government works

genesis of PD, we reasoned that tyrosinase would be acandidate for genetic association with PD. Therefore, weanalysed whether three known polymorphisms located in thepromoter (Oetting et al. 1991), exon 1 (Giebel and Spritz1990) and intron 4 contribute to the risk of sporadic PD in acase-control series. Although none of these SNPs have beenproven to have functional effects, we chose these two as theywere relatively frequent alleles in the population and becausethey spanned the length of the gene, thus increasing ourability to detect association. Allele and genotype distribu-tions for the three polymorphisms are shown in Table 3.Distributions of genotypes were at Hardy–Weinberg equilib-rium. No significant differences (p > 0.1) were found in theallele or genotype frequencies between cases and controls(Table 3). All pairs of loci displayed significant linkagedisequilibrium independent of diagnostic group (D¢ valuesranging from 0.45 to 0.99). Haplotypes for the threepolymorphisms were estimated using an expectation-max-imization algorithm method on the basis of genotypesobserved. Comparison of the haplotype distributions betweenstudy groups did not reveal any significant difference
(Fisher’s exact test, p ¼ 0.989; Table 4). Taken togetherthese results indicate that TYR does not play a major role inthe genetic predisposition to PD in this population.
Discussion
In the present study, we have collected data that support theexistence of tyrosinase in human brain and shown that theenzyme can enhance oxidative toxicity in vitro. The expres-sion of tyrosinase in brain is controversial (Xu et al. 1997;Tief et al. 1998). In this study, we have also shown that theTYR gene is expressed at low levels but ubiquitously inhuman brain and the transcript sequence is identical to thatexpressed in melanocytes. This suggests that the corres-ponding protein product in the brain contains the 18 aminoacid signal peptide for the targeting to melanosomes or, morelikely in the brain, lysosomes (Hasegawa et al. 2003). Wehave also demonstrated the presence of the protein product ofTYR gene by IP and activity assays. Tyrosinase appears in thebrain as a very weak band of 70 kDa similar to that seen inmelanoma cell lines or transfected neuroblastoma cell lines.Interestingly, both brain and M17 samples show an extra-band around 90 kDa. The finding of tyrosinase as a doubletis a common feature in most cell types (Berson et al. 2000;Halaban et al. 2001) and they were shown to representdifferent glycosylation forms (Olivares et al. 2003). Aftercomplete removal of oligosaccharide residues, both forms ofthe protein appeared to be successfully deglycosylated, thussupporting the specificity of the product detected in the brainextracts. Further experiments are warranted to unambigu-ously demonstrate tyrosinase in human brain. Subcellularfractionation might be useful if tyrosinase is restricted tolysosomal compartments but this would require substantialamounts of human brain samples. However, we were able todetect enzymatic activity in substantia nigra and caudate
Table 3 TYR genotypes and allele frequencies in Parkinson’s disease (PD) cases and controls
RS1799989 CC CA AA v2 p, d.f. ¼ 2 C allele v2 p, d.f. ¼ 1
PD cases (%) 96 (67.1) 44 (30.8) 3 (2.1) 1.339
p > 0.50
238 (82.6) 1.161
p > 0.25
Controls (%) 97 (73.5) 33 (25) 2 (1.5) 227 (86)
RS1042602 AA AC CC v2 p, d.f. ¼ 2 A allele v2 p, d.f. ¼ 1
PD cases (%) 6 (4.1) 50 (34.0) 91 (61.9) 0.391
p > 0.80
62 (21.1) 0.003
p > 0.90
Controls (%) 4 (3) 49 (36.6) 81 (60.4) 57 (21.3)
RS1827430 AA AG GG v2 p, d.f. ¼ 2 A allele v2 p, d.f. ¼ 1
PD cases (%) 57 (40.4) 66 (46.8) 18 (12.8) 0.584
p > 0.70
180 (63.8) 0.359
p > 0.50
Controls (%) 46 (35.9) 65 (50.8) 17 (13.3) 157 (61.3)
Table 4 TYR haplotype frequencies in cases and controls
PD Controls
CAA 0.21 0.19
CCA 0.37 0.37
CCG 0.25 0.28
ACG 0.11 0.09
Other* 0.06 0.07
Fisher’s exact test, p ¼ 0.989.
*Other haplotypes with frequencies lower than 5%.
The order of SNPs is: RS1799989, RS1042602 and RS1827430. PD,
Parkinson’s disease.
Tyrosinase exacerbates dopamine toxicity 253
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 93, 246–256No claim to original US government works

nucleus. Tyrosinase activity was found to be higher insubstantia nigra, a region rich in dopamine, the majortyrosinase substrate. However, the tyrosinase activity that wemeasured in the brain is approximately 100 000 times lowerthan in normal melanocytes (Jimenez et al. 1988), a valuethat is consistent with data from RT-PCR and IP suggestingthat tyrosinase is only expressed at low levels.
To assess the effect of tyrosinase overexpression we thentransferred the gene into M17 neuroblastoma cells andprimary neurons. We observed that M17 cells expressingtyrosinase showed decreased cell viability compared withcontrols if treated with dopamine. Tyrosinase also increasedthe toxicity of H2O2, an enhancer of the oxidative stressand inhibitor of dopamine transporter (Huang et al. 2003),and of the oxidizing agent paraquat, which can promotedopamine synthesis by an increase in tyrosine hydroxylaseactivity (McCormack et al. 2002). This effect was restric-ted to oxidative toxins as we did not observe increasedtoxicity in cells treated with the proteasome inhibitorlactacystin. Given that tyrosinase can convert dopamine todopamine-quinones, it is highly likely that generation ofthese oxidizing species is the major route to enhancedtoxicity.
The toxic effect of tyrosinase overexpressed in neurobla-stoma cells was further confirmed in primary neurons. Weobserved a reduced neuronal viability in cultures transducedwith tyrosinase compared with controls. Additionally, coex-pression of tyrosinase and a-synuclein A53T mutant leads tofurther reduction of cell viability. Several studies support thehypothesis that the interaction between a-synuclein andoxidized dopamine stabilizes toxic protofibrils (Conwayet al. 2001). Additionally, fibril formation is acceleratedrelative to wild-type by the A53T mutant (Conway et al.2000). Therefore, it is possible that dopamine-quinones aretoxic in these cells by reaction with a-synuclein.
Tyrosinase overexpressed in M17 cells clearly accumu-lates inside rounded cytoplasmic structures that probablycorrespond to lysosomes (Hasegawa et al. 2003), organellesthat are related to melanosomes. Lysosomes are thought to beinvolved in the uptake of excess cytoplasmic dopamine,which would otherwise undergo oxidation (Sulzer and Zecca2000). In particular, it was shown both in vitro (Sulzer et al.2000) and in human brain (Liang et al. 2004) that NMsynthesis is driven by excess cytosolic dopamine notaccumulated by synaptic vesicles and cytosolic dopaminecontent is correlated to the brain vesicular monoaminetransporter (VMAT2) expression. Within the lysosomes,tyrosinase can catalyse the conversion of imported dopamineand other cathecols into NM, thus detoxifying these species.However, the experimental system which we have used toexpress tyrosinase within neurons does not allow us todistinguish the effects of NM formation from the productionof dopamine-quinones. Therefore, we cannot assess whetherNM itself is beneficial or detrimental.
We also analysed three known polymorphisms within theTYR gene in a case-control series. The data obtained from thegenetic analysis did not identify polymorphisms thatincreased the risk of sporadic PD. Therefore, despite theenhancement of oxidative toxicity seen in vitro, we did notfind evidence to support the hypothesis that this is a majorrisk factor for the development of the human disease.Although it is possible that there are additional polymor-phisms that are associated with PD, it is unlikely that wewould have found no association in a sample of this size ifthere were a strong causal link, especially given that thep-values generated in this study were substantially differentfrom significance. This population has previously been usedin our laboratory to establish that there is a protective effectof variation in the gene for inducible nitric oxide synthase.However, further analyses in different populations may berequired in order to fully assess the role of this locus in thegenetic predisposition of sporadic PD.
In conclusion, from the data presented in this work it isreasonable to suppose that, under conditions such asincreased oxidative stress, tyrosinase may be harmful forcathecolaminergic neurons by increasing the concentration ofdopamine-derived radical species.
Acknowledgements
We are sincerely grateful to Prof. A. Fassina and Dr E. Pegoraro
(University of Padova) for providing the tissue samples, Dr D.
Weinreich (National Cancer Institute, NIH) for supplying the pMEL
and Prof. R.A. Spritz (University of Wisconsin) for the kind gift of
the tyrosinase cDNA.
References
Asanuma M., Miyazaki I. and Ogawa N. (2003) Dopamine- or L-DOPA-induced neurotoxicity: the role of dopamine quinone formation andtyrosinase in a model of Parkinson’s disease. Neurotox. Res. 5,165–176.
Baptista M. J., O’Farrell C., Daya S., Ahmad R., Miller D. W., HardyJ., Farrer M. J. and Cookson M. R. (2003) Co-ordinate tran-scriptional regulation of dopamine synthesis genes by alpha-synuclein in human neuroblastoma cell lines. J. Neurochem. 85,957–968.
Berman S. B. and Hastings T. G. (1997) Inhibition of glutamate transportin synaptosomes by dopamine oxidation and reactive oxygenspecies. J. Neurochem. 69, 1185–1195.
Berman S. B. and Hastings T. G. (1999) Dopamine oxidation altersmitochondrial respiration and induces permeability transition inbrain mitochondria: implications for Parkinson’s disease. J. Neur-ochem. 73, 1127–1137.
Berson J. F., Frank D. W., Calvo P. A., Bieler B. M. and Marks M.S. (2000) A common temperature-sensitive allelic form ofhuman tyrosinase is retained in the endoplasmic reticulum atthe nonpermissive temperature. J. Biol. Chem. 275, 12 281–12 289.
Cavalieri E. L., Li K. M., Balu N., Saeed M., Devanesan P., Higgin-botham S., Zhao J., Gross M. L. and Rogan E. G. (2002) Cate-chol ortho-quinones: the electrophilic compounds that form
254 E. Greggio et al.
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 93, 246–256No claim to original US government works

depurinating DNA adducts and could initiate cancer and otherdiseases. Carcinogenesis 23, 1071–1077.
Clarimon J., Eerola J., Hellstrom O., Tienari P. J. and Singleton A. (2004)Paraoxonase 1 (PON1) gene polymorphisms and Parkinson’sdisease in a Finnish population. Neurosci. Lett. 367, 168–170.
Conway K. A., Lee S. J., Rochet J. C., Ding T. T., Harper J. D., Will-iamson R. E. and Lansbury P. T. Jr (2000) Accelerated oligome-rization by Parkinson’s disease linked alpha-synuclein mutants.Ann. NY Acad. Sci. 920, 42–45.
Conway K. A., Rochet J. C., Bieganski R. M. and Lansbury P. T. Jr(2001) Kinetic stabilization of the alpha-synuclein protofibril by adopamine-alpha-synuclein adduct. Science 294, 1346–1349.
Giebel L. B. and Spritz R. A. (1990) RFLP for MboI in the humantyrosinase (TYR) gene detected by PCR. Nucl. Acids Res. 18,3103.
Gimenez E., Lavado A., Giraldo P. and Montoliu L. (2003) Tyrosinasegene expression is not detected in mouse brain outside the retinalpigment epithelium cells. Eur. J. Neurosci. 18, 2673–2676.
Hague S., Peuralinna T., Eerola J., Hellstrom O., Tienari P. J. and Sin-gleton A. B. (2004) Confirmation of the protective effect of iNOSin an independent cohort of Parkinson disease. Neurology 62, 635–636.
Halaban R., Cheng E., Svedine S., Aron R. and Hebert D. N. (2001)Proper folding and endoplasmic reticulum to golgi transport oftyrosinase are induced by its substrates, DOPA and tyrosine.J. Biol. Chem. 276, 11 933–11 938.
Hasegawa T., Matsuzaki M., Takeda A., Kikuchi A., Furukawa K.,Shibahara S. and Itoyama Y. (2003) Increased dopamine and itsmetabolites in SH-SY5Y neuroblastoma cells that express tyros-inase. J. Neurochem. 87, 470–475.
Hearing V. J. and Ekel T. M. (1976) Mammalian tyrosinase. A com-parison of tyrosine hydroxylation and melanin formation. Biochem.J. 157, 549–557.
Higashi Y., Asanuma M., Miyazaki I. and Ogawa N. (2000) Inhibition oftyrosinase reduces cell viability in catecholaminergic neuronalcells. J. Neurochem. 75, 1771–1774.
Hirsch E. C. (1994) Biochemistry of Parkinson’s disease with specialreference to the dopaminergic systems.Mol. Neurobiol. 9, 135–142.
Huang J., Bai Y. X., Han S. W., Ng S. S., Jing da D., Wong B. C., HuangC. F., Kung H. F. and Lin M. C. (2003) A human TERT C-terminalpolypeptide sensitizes HeLa cells to H2O2-induced senescencewithout affecting telomerase enzymatic activity. Biochem. Biophys.Res. Commun. 301, 627–632.
Hughes A. J., Daniel S. E., Kilford L. and Lees A. J. (1992) Accuracy ofclinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J. Neurol. Neurosurg. Psychiat.55, 181–184.
Ikemoto K., Nagatsu I., Ito S., King R. A., Nishimura A. and Nagatsu T.(1998) Does tyrosinase exist in neuromelanin-pigmented neuronsin the human substantia nigra? Neurosci. Lett. 253, 198–200.
Jimbow K., Park J. S., Kato F., Hirosaki K., Toyofuku K., Hua C. andYamashita T. (2000) Assembly, target-signaling and intracellulartransport of tyrosinase gene family proteins in the initial stage ofmelanosome biogenesis. Pigment Cell Res. 13, 222–229.
Jimenez M., Kameyama K., Maloy W. L., Tomita Y. and Hearing V. J.(1988) Mammalian tyrosinase: biosynthesis, processing, andmodulation by melanocyte-stimulating hormone. Proc. Natl Acad.Sci. USA 85, 3830–3834.
Liang C. L., Nelson O., Yazdani U., Pasbakhsh P. and German D. C.(2004) Inverse relationship between the contents of neuromelaninpigment and the vesicular monoamine transporter-2: human mid-brain dopamine neurons. J. Comp. Neurol. 473, 97–106.
McCormack A. L., Thiruchelvam M., Manning-Bog A. B., Thiffault C.,Langston J. W., Cory-Slechta D. A. and Di Monte D. A. (2002)
Environmental risk factors and Parkinson’s disease: selectivedegeneration of nigral dopaminergic neurons caused by the her-bicide paraquat. Neurobiol. Dis. 10, 119–127.
Miller D. W., Ahmad R., Hague S. et al. (2003) L166P mutant DJ-1,causative for recessive Parkinson’s disease, is degraded throughthe ubiquitin-proteasome system. J. Biol. Chem. 278, 36 588–36595.
Miranda M. and Botti D. (1983) Harding-passey mouse-melanomatyrosinase inactivation by reaction products and activation byL-epinephrine. Gen. Pharmacol. 14, 231–237.
Oetting W. S., Roed C. M., Mentink M. M. and King R. A. (1991) PCRdetection of a TaqI polymorphism at the CCAATT box of thehuman tyrosinase (TYR) gene. Nucl. Acids Res. 19, 5800.
Olivares C., Solano F. and Garcia-Borron J. C. (2003) Conformation-dependent post-translational glycosylation of tyrosinase. Require-ment of a specific interaction involving the CuB metal binding site.J. Biol. Chem. 278, 15 735–15 743.
Petrucelli L., O’Farrell C., Lockhart P. J. et al. (2002) Parkin protectsagainst the toxicity associated with mutant alpha-synuclein: pro-teasome dysfunction selectively affects catecholaminergic neurons.Neuron 36, 1007–1019.
Raposo G. and Marks M. S. (2002) The dark side of lysosome-relatedorganelles: specialization of the endocytic pathway for melano-some biogenesis. Traffic 3, 237–248.
Sanchez-Ferrer A., Rodriguez-Lopez J. N., Garcia-Canovas F. andGarcia-Carmona F. (1995) Tyrosinase: a comprehensive review ofits mechanism. Biochim. Biophys. Acta 1247, 1–11.
Schneider S., Kueffer J., Roessli D. and Excoffier L. (2000) Arlequinver.2000: a Software Environment for the Analysis of PopulationGenetics Data. Genetics Biometry Laboratory, University ofGeneva, Geneva, Switzerland.
Stokes A. H., Brown B. G., Lee C. K., Doolittle D. J. and Vrana K. E.(1996) Tyrosinase enhances the covalent modification of DNA bydopamine. Brain Res. Mol. Brain Res. 42, 167–170.
Sulzer D. and Edwards R. (2000) Vesicles: equal in neurotransmitterconcentration but not in volume. Neuron 28, 5–7.
Sulzer D. and Zecca L. (2000) Intraneuronal dopamine-quinone syn-thesis: a review. Neurotox. Res. 1, 181–195.
Sulzer D., Bogulavsky J., Larsen K. E. et al. (2000) Neuromelaninbiosynthesis is driven by excess cytosolic catecholamines notaccumulated by synaptic vesicles. Proc. Natl Acad. Sci. USA 97, 11869–11 874.
Tief K., Schmidt A. and Beermann F. (1998) New evidence for presenceof tyrosinase in substantia nigra, forebrain and midbrain. BrainRes. Mol. Brain Res. 53, 307–310.
Virador V., Matsunaga N., Matsunaga J. et al. (2001) Production ofmelanocyte-specific antibodies to human melanosomal proteins:expression patterns in normal human skin and in cutaneous pig-mented lesions. Pigment Cell Res. 14, 289–297.
Whitehead R. E., Ferrer J. V., Javitch J. A. and Justice J. B. (2001)Reaction of oxidized dopamine with endogenous cysteine residuesin the human dopamine transporter. J. Neurochem. 76, 1242–1251.
Wilms H., Rosenstiel P., Sievers J., Deuschl G., Zecca L. and Lucius R.(2003) Activation of microglia by human neuromelanin isNF-kappaB dependent and involves p38 mitogen-activated proteinkinase: implications for Parkinson’s disease. Faseb J. 17, 500–502.
Xu Y., Stokes A. H., Freeman W. M., Kumer S. C., Vogt B. A. and VranaK. E. (1997) Tyrosinase mRNA is expressed in human substantianigra. Brain Res. Mol. Brain Res. 45, 159–162.
Xu Y., Stokes A. H., Roskoski R. Jr and Vrana K. E. (1998) Dopamine,in the presence of tyrosinase, covalently modifies and inactivatestyrosine hydroxylase. J. Neurosci. Res. 54, 691–697.
Zarow C., Lyness S. A., Mortimer J. A. and Chui H. C. (2003) Neuronalloss is greater in the locus coeruleus than nucleus basalis and
Tyrosinase exacerbates dopamine toxicity 255
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 93, 246–256No claim to original US government works

substantia nigra in Alzheimer and Parkinson diseases. Arch. Neu-rol. 60, 337–341.
Zecca L., Tampellini D., Gerlach M., Riederer P., Fariello R. G. andSulzer D. (2001) Substantia nigra neuromelanin: structure, syn-thesis, and molecular behaviour. Mol. Pathol. 54, 414–418.
Zecca L., Fariello R., Riederer P., Sulzer D., Gatti A. and Tampellini D.(2002) The absolute concentration of nigral neuromelanin, assayed
by a new sensitive method, increases throughout the life and isdramatically decreased in Parkinson’s disease. FEBS Lett. 510,216–220.
Zecca L., Zucca F. A., Wilms H. and Sulzer D. (2003) Neuromelanin ofthe substantia nigra: a neuronal black hole with protective and toxiccharacteristics. Trends Neurosci. 26, 578–580.
256 E. Greggio et al.
� 2005 International Society for Neurochemistry, J. Neurochem. (2005) 93, 246–256No claim to original US government works
Copyright © 2022 FDOKUMEN




















