
Kinetic characterization of the substrate specificity and mechanism of mushroom tyrosinase
10
Kinetic characterization of the substrate specificity and mechanism of mushroom tyrosinase Juan C. Espı ´n 1 , Ramo ´ n Varo ´n 2 , Lorena G. Fenoll 1 , M. Angeles Gilabert 1 , Pedro A. Garcı ´a-Ruı ´z 3 , Jose ´ Tudela 1 and Francisco Garcı ´a-Ca ´ novas 1 1 Grupo investigacio ´n Enzimologı ´a, Departamento de Bioquı ´mica y Biologı ´a Molecular-A, Facultad de Biologı ´a, Universidad de Murcia, Spain; 2 Departamento de Quı ´mica-Fı ´sica, Escuela Universitaria Polite ´cnica de Albacete, Universidad de Castilla-La Mancha, Spain; 3 Departamento de Quı ´mica Orga ´nica, Facultad de Quı ´mica, Universidad de Murcia, Spain This paper reports a quantitative study of the effect of ring substituents in the 1-position of the aromatic ring on the rate of monophenol hydroxylation and o-diphenol oxidation catalyzed by tyrosinase. A possible correlation between the electron density of the carbon atom supporting the oxygen from the monophenolic hydroxyl group and the V M max values for each monophenol was found. In the case of o-diphenols the same effect was observed but the size of the side-chain became very important. NMR studies on the monophenols justified the sequence of the V M max values obtained. As regards the o-diphenols, on the other hand, only a fair correlation between NMR and V D max values was observed due to the effect of the molecular size of the ring substituent. From these data, it can be concluded that the redox step k 33 is not the rate-determining step of the reaction mechanism. Thus, the monophenols are converted into diphenols, but the order of specificities towards monophenols is different to that of o-diphenols. The rate-limiting step of the monophenolase activity could be the nucleophilic attack k 5 1 of the oxygen atom of the hydroxyl group on the copper atoms of the active site of the enzyme. This step could also be similar to or have a lower rate of attack than the electrophilic attack (k 5 2 ) of the oxygen atom of the active site of oxytyrosinase on the C-3 of the monophenolic ring. However, the rate-limiting step in the diphenolase activity of tyrosinase could be related to both the nucleophilic power of the oxygen atom belonging to the hydroxyl group at the carbon atom in the 3-position k 32 and to the size of the substituent side-chain. On the basis of the results obtained, kinetic and structural models describing the monophenolase and diphenolase reaction mechanisms for tyrosinase are proposed. Keywords: diphenolase; enzyme kinetics; 3-methyl-2-benzothiazolinone hydrazone (MBTH); monophenolase; mushroom. The enzyme tyrosinase or polyphenol oxidase, PPO (mono- phenol, o-diphenol:oxygen oxidoreductase, EC 1.14.18.1) is of central importance in vertebrate melanin pigmentation. Enzy- matic browning in vegetables and fruits is caused by the activity of tyrosinase in plant tissues. Tyrosinase plays an important role in fruit and vegetable processing and during the storage of processed foods. This enzyme catalyzes the hydroxylation of monophenols (monophenolase activity) and the oxidation of o-diphenols to o-quinones (diphenolase activity). These o-quinones evolve nonenzymatically to yield several unstable intermediates, which polymerize to render melanins [1,2]. The active site of tyrosinase consists of two copper atoms and three states: ‘met’, ‘deoxy’, and ‘oxy’ [3–10]. Structural models for the active site of these three forms of tyrosinase have been proposed [11–15]. Recent advances on structural homology [16] and on the active-site conformation [17,18] have been reported. Nevertheless, the complete spatial structure of the active site is still unknown. The conversion of p-monophenols into o-diphenols is an interesting problem in industry [19], although the reaction is efficiently catalyzed by tyrosinase [1,2]. This has led to several approaches to mimic the active site of PPO and several binuclear Cu (I) complexes have been synthesized, whose catalytic efficiencies are lower than that of tyrosinase [19,20]. These synthetic complexes with Cu (I) show high catalytic activities on phenolic substrates with electron-donating groups in their side-chains [19,20]. This activity is equal to that Eur. J. Biochem. 267, 1270–1279 (2000) q FEBS 2000 Correspondence to F. Garcı ´a-Ca ´novas, Grupo Investigacio ´n Enzimologı ´a, Departamento de Bioquı ´mica y Biologı ´a Molecular-A, Facultad de Biologı ´a, Universidad de Murcia, E-30080 Murcia, Spain. Fax: 1 34 968363963, E-mail: [email protected] Abbreviations: d 3 , chemical displacement value at C-3; d 4 , chemical displacement value at C-4; D, o-diphenol; 2 H 2 O, deuterium oxide (heavy water); [D] 0 , initial o-diphenol concentration; [D] ss , o-diphenol concentration in the steady-state; [D] 1 , o-diphenol concentration at the final assay time; DHPAA, 3,4-dihydroxyphenyl acetic acid; DHPPA, 3,4-dihydroxyphenyl propionic acid; DMF, N, N 0 -dimethylformamide; : max , molar absorptivity at l max ; : i , molar absorptivity at l i ; E d , deoxy-tyrosinase (reduced form of tyrosinase with Cu 1 -Cu 1 in the active site); E m , met-tyrosinase (with Cu 21 -Cu 21 in the active site); E o , oxy-tyrosinase with Cu 21 -O 2 22 -Cu 21 in the active site; 4HA, 4-hydroxyanisole; k D cat , catalytic constant of tyrosinase towards o-diphenols; k M cat , catalytic constant of tyrosinase towards monophenols; K 1 = k 21 k 1 ; K 2 = k 22 k 2 ; K 6 = k 26 k 6 ; K 4 = k 24 k 4 ; K D m , apparent Michaelis constant of tyrosinase towards o-diphenols; K M m , apparent Michaelis constant of tyrosinase towards monophenols; l max , wavelength at the maximum of absorbance; l i , wavelength at the isosbestic point; M, monophenol; [M] 0 , initial monophenol concentration; MBTH, 3-methyl-2- benzothiazolinone hydrazone; PHPAA, p-hydroxyphenyl acetic acid; PHPPA, p-hydroxyphenyl propionic acid; [S] 0 , initial substrate concentration; TBC, 4-tert-butylcatechol; V D max , maximum steady-state rate of tyrosinase towards o-diphenols; V M max , maximum steady-state rate of tyrosinase towards monophenols; V ss , steady-state rate. (Received 26 July 1999, revised 25 October 1999, accepted 15 November 1999)
-
Upload
upcbarcelona -
Category
Documents
-
view
3 -
download
0
Transcript of Kinetic characterization of the substrate specificity and mechanism of mushroom tyrosinase

Kinetic characterization of the substrate specificity and mechanism ofmushroom tyrosinase
Juan C. EspõÂn1, Ramo n Varo n2, Lorena G. Fenoll1, M. Angeles Gilabert1, Pedro A. GarcõÂa-RuõÂz3, Jose Tudela1 andFrancisco GarcõÂa-Ca novas1
1Grupo investigacioÂn EnzimologõÂa, Departamento de BioquõÂmica y BiologõÂa Molecular-A, Facultad de BiologõÂa, Universidad de Murcia, Spain;2Departamento de QuõÂmica-FõÂsica, Escuela Universitaria PoliteÂcnica de Albacete, Universidad de Castilla-La Mancha, Spain;3Departamento de QuõÂmica OrgaÂnica, Facultad de QuõÂmica, Universidad de Murcia, Spain
This paper reports a quantitative study of the effect of ring substituents in the 1-position of the aromatic ring on
the rate of monophenol hydroxylation and o-diphenol oxidation catalyzed by tyrosinase. A possible correlation
between the electron density of the carbon atom supporting the oxygen from the monophenolic hydroxyl group
and the V Mmax values for each monophenol was found. In the case of o-diphenols the same effect was observed but
the size of the side-chain became very important. NMR studies on the monophenols justified the sequence of the
V Mmax values obtained. As regards the o-diphenols, on the other hand, only a fair correlation between NMR and
V Dmax values was observed due to the effect of the molecular size of the ring substituent. From these data, it can be
concluded that the redox step �k33� is not the rate-determining step of the reaction mechanism. Thus, the
monophenols are converted into diphenols, but the order of specificities towards monophenols is different to that
of o-diphenols. The rate-limiting step of the monophenolase activity could be the nucleophilic attack �k51� of the
oxygen atom of the hydroxyl group on the copper atoms of the active site of the enzyme. This step could also be
similar to or have a lower rate of attack than the electrophilic attack (k52) of the oxygen atom of the active site of
oxytyrosinase on the C-3 of the monophenolic ring. However, the rate-limiting step in the diphenolase activity of
tyrosinase could be related to both the nucleophilic power of the oxygen atom belonging to the hydroxyl group at
the carbon atom in the 3-position �k32� and to the size of the substituent side-chain. On the basis of the results
obtained, kinetic and structural models describing the monophenolase and diphenolase reaction mechanisms for
tyrosinase are proposed.
Keywords: diphenolase; enzyme kinetics; 3-methyl-2-benzothiazolinone hydrazone (MBTH); monophenolase;
mushroom.
The enzyme tyrosinase or polyphenol oxidase, PPO (mono-phenol, o-diphenol:oxygen oxidoreductase, EC 1.14.18.1) is ofcentral importance in vertebrate melanin pigmentation. Enzy-matic browning in vegetables and fruits is caused by theactivity of tyrosinase in plant tissues. Tyrosinase plays animportant role in fruit and vegetable processing and during thestorage of processed foods. This enzyme catalyzes thehydroxylation of monophenols (monophenolase activity) andthe oxidation of o-diphenols to o-quinones (diphenolaseactivity). These o-quinones evolve nonenzymatically to yieldseveral unstable intermediates, which polymerize to rendermelanins [1,2]. The active site of tyrosinase consists of twocopper atoms and three states: `met', `deoxy', and `oxy' [3±10].Structural models for the active site of these three forms oftyrosinase have been proposed [11±15]. Recent advances onstructural homology [16] and on the active-site conformation[17,18] have been reported. Nevertheless, the completespatial structure of the active site is still unknown.
The conversion of p-monophenols into o-diphenols is aninteresting problem in industry [19], although the reaction isefficiently catalyzed by tyrosinase [1,2]. This has led to severalapproaches to mimic the active site of PPO and severalbinuclear Cu (I) complexes have been synthesized, whosecatalytic efficiencies are lower than that of tyrosinase [19,20].These synthetic complexes with Cu (I) show high catalyticactivities on phenolic substrates with electron-donating groupsin their side-chains [19,20]. This activity is equal to that
Eur. J. Biochem. 267, 1270±1279 (2000) q FEBS 2000
Correspondence to F. GarcõÂa-CaÂnovas, Grupo InvestigacioÂn EnzimologõÂa,
Departamento de BioquõÂmica y BiologõÂa Molecular-A, Facultad de
BiologõÂa, Universidad de Murcia, E-30080 Murcia, Spain.
Fax: 1 34 968363963, E-mail: [email protected]
Abbreviations: d3, chemical displacement value at C-3; d4, chemical
displacement value at C-4; D, o-diphenol; 2H2O, deuterium oxide (heavy
water); [D]0, initial o-diphenol concentration; [D]ss, o-diphenol
concentration in the steady-state; [D]1, o-diphenol concentration at the
final assay time; DHPAA, 3,4-dihydroxyphenyl acetic acid; DHPPA,
3,4-dihydroxyphenyl propionic acid; DMF, N, N 0-dimethylformamide;
:max, molar absorptivity at lmax; :i , molar absorptivity at li ;
Ed, deoxy-tyrosinase (reduced form of tyrosinase with Cu1-Cu1 in the
active site); Em, met-tyrosinase (with Cu21-Cu21 in the active site); E o,
oxy-tyrosinase with Cu21-O222-Cu21 in the active site; 4HA,
4-hydroxyanisole; k Dcat, catalytic constant of tyrosinase towards o-diphenols;
k Mcat , catalytic constant of tyrosinase towards monophenols; K1 = k21k1;
K2 = k22k2; K6 = k26k6; K4 = k24k4; K Dm , apparent Michaelis constant of
tyrosinase towards o-diphenols; K Mm , apparent Michaelis constant of
tyrosinase towards monophenols; lmax, wavelength at the maximum of
absorbance; li , wavelength at the isosbestic point; M, monophenol;
[M]0, initial monophenol concentration; MBTH, 3-methyl-2-
benzothiazolinone hydrazone; PHPAA, p-hydroxyphenyl acetic acid;
PHPPA, p-hydroxyphenyl propionic acid; [S]0, initial substrate
concentration; TBC, 4-tert-butylcatechol; V Dmax , maximum steady-state rate
of tyrosinase towards o-diphenols; V Mmax , maximum steady-state rate of
tyrosinase towards monophenols; Vss , steady-state rate.
(Received 26 July 1999, revised 25 October 1999, accepted
15 November 1999)

q FEBS 2000 Substrates and reaction mechanism of tyrosinase (Eur. J. Biochem. 267) 1271
reported in this paper with tyrosinase as enzymatic catalyst. Onthe other hand, several binuclear Cu (II) complexes have alsobeen synthesized [21], as this is the oxidation state of Cu inmet- and oxy-tyrosinase [10]. The incorporation of oxygen inthese synthetic complexes occurs through a chain of free radicalreactions, initiated by hydrogen abstraction from the phenolicsubstrate [21]. Thus, these studies have shown that theoxidation reaction by means of free radicals is not very specific[21]. The oxidation reaction of phenols catalysed by tyrosinaseis very specific and does not take place with free radicals[2,15]. The spatial docking of phenols within the active site oftyrosinase appears to be very important for the enzymaticgeneration of phenolic products with a specific hydroxylation.
The presence of three copper states in the active site oftyrosinase has led to a structural model for the reactionmechanism involved in the hydroxylation of monophenols andoxidation of the resulting o-diphenols being proposed [13]. Theoccurrence of a lag period in the monophenolase activity oftyrosinase is related to the presence of a dead-end complexEmM, as has been demonstrated by kinetic studies [22,23]. Thereaction mechanism of the enzyme has recently been reviewedtaking into consideration both kinetic and structural aspects[24].
Despite the well-established structural [13±18] and kinetic[22,23] mechanisms for the hydroxylation of monophenolicsubstrates to o-quinones catalyzed by tyrosinase, only a fewstudies have investigated the rate-limiting step in the reactionmechanism [7,23].
The disappearance constants of the Eo form in the presenceof monophenols and o-diphenols were found to be higher thanthe catalytic constant of the enzyme towards these substrates[7]. These authors concluded that the disappearance of the Eo
form is not the limiting step in the turnover and suggested thatmonophenol hydroxylation is the limiting step.
Another approach taken by our group was in agreement withthe above conclusion [23] by determining the experimentalparameter `R', which is the quantitative relationship betweenthe o-diphenol accumulated in the steady state ([D]ss) and theinitial monophenol concentration ([M]0), ([D] ss / [M]0). Theanalytical expression for this parameter is:
R � 1
2´
k5KDo
k7KMo
�See also �23��:
As the values of R were low and the order of magnitude of theMichaelis constants for several monophenol/o-diphenol pairswere the same, it could be deduced that k5 , k7. This indicatedthat the limiting step of the turnover might be the one governedby k5 (monophenol hydroxylation), in accordance with thehypothesis proposed by Makino and Mason [7].
Some studies have pointed to the lack of reliable and precisequantitative studies of the effect of the substituent side-chainson the hydroxylating rate of monophenols [2,15]. The lack ofsuch studies is mainly due to the high reactivity of enzyme-generated o-quinones preventing the system from reaching thesteady state, meaning that reliable and precise Vmax and Km
values could not be obtained.Recently, we have studied kinetically the oxidation of
various monophenols and o-diphenols with different substituentside-chains in C-1 catalysed by tyrosinases from severalsources [25±34].
The purpose of the study described in this paper is toobtain a sound kinetic and structural characterization of thereaction mechanism of tyrosinase. Several monophenols ando-diphenols are used to study the effect of different
substituents at C-1 on the enzymatic reactivity of the hydroxylgroups at C-4 in the case of monophenols, and C-3 and C-4 inthe case of o-diphenols. NMR studies are carried out in order toestablish the chemical reactivity of the OH groups of thephenolic compounds considered. The chemical displacementvalues for C-3 and C-4 of the benzene ring (d3 and d4,respectively) of these phenols is determined. A possiblecorrelation between the enzymatic and chemical reactivitiesfor the monophenols and o-diphenols is studied. Furthermore,we discuss the rate-limiting step of both (monophenolase anddiphenolase) reaction mechanisms.
M A T E R I A L S A N D M E T H O D S
Reagents
4-Hydroxyanisole (4HA) and 4-ethoxyphenol were pur-chased from Aldrich (Spain); catechol, 4-methylcatecholand 4-tert-butylcatechol (TBC) were from Fluka (Spain),while 3,4-dihydroxyphenyl propionic acid (DHPPA), 3,4-dihydroxyphenyl acetic acid (DHPAA), l-dopa, dopamethyl ester, l-a-methyldopa, dopamine, l-isoproterenol,p-hydroxyphenyl propionic acid (PHPPA), p-hydroxyphenylacetic acid (PHPAA), tyramine, l-tyrosine, l-a-methyltyrosineand 3-methyl-2-benzothiazolinone hydrazone (MBTH) werefrom Sigma. All other chemicals were of analytical grade andsupplied by Merck. Stock solutions of the phenolic substrateswere prepared in 0.15 mm phosphoric acid to prevent auto-oxidation. The acidic character of MBTH required the use of50 mm sodium phosphate buffer in the assay medium. Todissolve the MBTH±quinone adducts, 2% (v/v) N,N 0-dimethyl-formamide (DMF) was added to the assay medium [25±35].Milli-Q system (Millipore Corp.) ultrapure water was usedthroughout this research.
Enzyme source
Mushroom tyrosinase (8300 U´mg21) was purchased fromSigma and purified by Duckworth and Coleman's procedure[36]. The enzyme concentration was calculated taking the valueof Mr as 120 000. Protein content was determined by Bradford'smethod [37] using bovine serum albumin as standard.
Spectrophotometric assays
Kinetic assays were carried out by measuring the appearance ofthe product in the reaction medium in an UV-visible Perkin±Elmer Lambda-2 spectrophotometer, on-line interfaced with aPC Intel-486 DX-66 microcomputer. Temperature was kept at25 8C using a Haake D1G circulating water-bath with a heater/cooler and checked using a Cole±Parmer digital thermometerwith a precision of ^ 0.1 8C. Reference cuvettes contained allthe components except the substrate, with a final volume of1 mL.
The monophenolase and diphenolase activities of mushroomtyrosinase were determined spectrophotometrically by usingMBTH, which is a potent nucleophile through its amino groupthat attacks the enzyme-generated o-quinones [35]. This assaymethod is highly sensitive, reliable and precise [25±35]. MBTHtraps the enzyme-generated o-quinones to render a solubleMBTH±quinone adduct with high molar absorptivity.MBTH±quinone adducts were soluble and stable at acidic pH[26±35]. At pH 6.8 (optimum pH for mushroom tyrosinase),these adducts were soluble and evolved showing an isosbesticpoint [26±35]. The stability of the MBTH±quinone adducts andthe rapid assays provide a reliable method for determining the

1272 J. C. EspõÂn et al. (Eur. J. Biochem. 267) q FEBS 2000
monophenolase and diphenolase activities of tyrosinase fromseveral sources [25±35].
Diphenolase activity on catechol and 4-methylcatechol wasdetermined by measuring the disappearance of NADH [38] inrapid kinetic assays. The respective MBTH±quinone adductswere not very soluble and so MBTH was not used as a coupledreagent [26,28]. The o-diphenol TBC yielded a highly stableo-quinone that did not react with MBTH [39]. Thediphenolase activity of tyrosinase on TBC was thereforedetermined by measuring the appearance of the enzyme-generated o-quinone in rapid kinetic assays [40]. The propertiesof the detectable species with the different assay methods arereported in Table 1.
Kinetic data analysis
K Mm , V M
max, K Dm and V D
max values of different monophenols ando-diphenols were calculated from triplicate measurements ofthe steady-state rate, Vss, for each initial substrate concen-tration, [S]0. The reciprocals of the variances of Vss were usedas weighting factors in the nonlinear regression fitting of Vss vs.[S]0 data to the Michaelis equation [41,42]. The fitting wascarried out by using Marquardt's algorithm [43] implementedin the sigma plot 2.01 program for windowsTM [44]. Initialestimations of K M
m , V Mmax , K D
m and V Dmax were obtained from
the Hanes±Woolf equation, a linear transformation of theMichaelis equation [41].
NMR assays13C NMR spectra of the different monophenols and o-diphenolsconsidered here were obtained in a Varian Unity spectrometerof 300 MHz. The spectra were obtained at the optimum pH formushroom tyrosinase (6.8) and using 2H2O as solvent for thesubstrates. d values were measured relative to those fortetramethylsilane (d � 0). The maximum wide line acceptedin the NMR spectra was 0.06 Hz. Therefore, the maximumaccepted error for each peak was ^ 0.03 p.p.m.
The dependence of d values in 13C for a carbon atom on itselectron density is known [45,46]. It has been previouslydemonstrated that there is a linear correlation between d in 13C
and the constant s, and such a correlation has been used in bothreaction rate and mechanistic studies [47]. d values in 13C havebeen used to corroborate or to calculate better s values [48,49].When several nucleophiles have the same nucleophilic atomand similar structural characteristics in the proximity of thenucleophilic site, the attack rate constants for the samesubstrate are usually well correlated with the basic characterand electron density of the nucleophiles [50±52]. Moreover,the electron-donating capacity of the oxygen atom fromdifferent phenolic compounds (nucleophilic power) has beencorrelated with the experimental d values in 13C for thecarbon atom that supports the hydroxyl group [53]. Takinginto account all the above, low values of dD
3 and dD4 in 13C
for o-diphenols and dM4 for monophenols indicate high
nucleophilic power.
R E S U LT S A N D D I S C U S S I O N
Diphenolase activity of tyrosinase
Kinetic assays. V Dmax values were very dependent on the
o-diphenols used (Table 2), following the sequence:catechol . 4-methylcatechol . TBC . DHPAA . DHPPA. dopamine . l-dopa . l-a-methyldopa . dopa methyl ester. l-isoproterenol. K D
m values did not follow the same sequence(Table 2).
NMR assays. The sequence of the V Dmax values (Table 2)
obtained for o-diphenols could be related to their differentnucleophilic powers (Table 3) and with the size of theirrespective side-chain substituents (Tables 2 and 3).
There are other chemical properties that also vary with theelectron density in the aromatic ring, especially in one-electronoxidation. However, it has not been demonstrated that freeradicals are present in the reactions catalysed by tyrosinase[2,15].
Structural reaction mechanism. Based on the results of previousstudies [22] and on the V D
max (Table 2) and NMR (Table 3)values obtained, structural (Scheme 1) and kinetic (Scheme 2)mechanisms are proposed. The Em form of the enzyme binds
Table 1. Properties of the detectable species from several substrates at the optimum pHs of the PPOs assayed. Conditions were for the calculation of
:max: 50 mm AB (pH 4.3), [MBTH]sat ranged from 0.2 to 3 mm, 2% DMF, and 1 mm mushroom tyrosinase. The substrate concentration was 10 mm for each
substrate/tyrosinase pair. The same conditions were used for calculation of :i but with sodium phosphate buffer 50 mm pH 6.8; [MBTH]sat ranged from 2
to 7 mm for the calculation of [MBTH]sat.
Substrate Assay method Detectable species lmax li
[MBTH]sat
(mm)2
:max
(m21´cm21)
:I
(m21´cm21)
4HA MBTH MBTH-adduct 492 464 2.5 �^ 0.1 31300 �^ 800 19200 �^ 400
4-Ethoxyphenol MBTH MBTH-adduct 488 458 2.5 �^ 0.1 35500 �^ 900 23900 �^ 500
DHPPA MBTH MBTH-adduct 500 466 4.0 �^ 0.2 40000 �^ 1000 20000 �^ 500
DHPAA MBTH MBTH-adduct 500 471 4.0 �^ 0.2 42600 �^ 1200 23000 �^ 500
TBC Formation of o-quinone o-Quinone 400 1150 �^ 50c
Dopamine MBTH MBTH-adduct 503 476 2.0 �^ 0.1 42500 �^ 1200 20700 �^ 400
Catechol Disappearance of NADH NADH 340 6300 �^ 400
4-Methyl catechol Disappearance of NADH NADH 340 6300 �^ 400
l-Dopa MBTH MBTH-adduct 507 484 5.0 �^ 0.2 38000 �^ 1000 22300 �^ 500
l-a-Methyl dopa MBTH MBTH-adduct 503 493 6.0 �^ 0.3 52000 �^ 2000 21400 �^ 500
Dopa methyl ester MBTH MBTH-adduct 503 476 4.0 �^ 0.2 52000 �^ 2000 23600 �^ 600
l-Isoproterenol MBTH MBTH-adduct 497 506 7.0 �^ 0.3 31500 �^ 1000 30000 �^ 800
a 50 mm sodium phosphate buffer (pH 6.8), 2% DMF, different MBTH concentrations, 5Km of substrate concentration and 6 nm tyrosinase. b [MBTH]sat used
in the assay conditions for determining activity of mushroom tyrosinase (optimum pH 6.8). c [40].

q FEBS 2000 Substrates and reaction mechanism of tyrosinase (Eur. J. Biochem. 267) 1273
one molecule of o-diphenol (D) with no covalent interactions(EmD). The nucleophilic attack of the oxygen atom belongingto the hydroxyl groups at C-4 (EmD, k31
) and C-3 (EmD 0, k32)
can then take place with the production of 2H1. Next,oxidoreduction (EmD 00, k33
) should take place to yieldo-quinone (Q) with further protonation and productionof H2O. This would lead to the formation of the Ed form of theenzyme. Ed binds molecular oxygen (O2) in a fast step, inwhich the copper atoms are oxidized from Cu1 to Cu21. The O2
is reduced, yielding peroxide and generating the Eo formof the enzyme. This enzymatic form binds another molecule ofo-diphenol with no covalent interactions (EoD). After suchbinding, the nucleophilic attacks of the oxygen atoms belongingto the OH groups at C-4 (EoD, k71
) and C-3 (EoD 0, k72) take
place with the production of 2H1. The subsequent processwould involve oxidoreduction (EoD 00, k73
) to yield onemolecule of o-quinone. The peroxide group would oxidize thecopper atoms from Cu1 to Cu21 with the uptake of 3H1 andthe production of one molecule of H2O to render the Em form.This should close the catalytic cycle (Scheme 1).
The enzyme-generated o-quinones suffer the nucleophilicattack of the nucleophile MBTH to form the chromophoricMBTH±quinone adduct (Scheme 2). It should be noted that theregeneration of o-diphenol in the medium by means ofnonenzymatic reactions is not kinetically significant for thediphenolase activity because of the high initial concentration ofo-diphenol ([D]0 q [E]0) and the negligible consumption ofthis substrate during the assay ([Q]1 p [D]0, [D]1 < [D]0).
Kinetic reaction mechanism. The splitting of k3 and k7
(Scheme 1) has not been carried out for the kinetic analysisas it yields complex equations for V D
max and K Dm . Thus, a
simplified kinetic reaction mechanism has been proposed(Scheme 2). Assuming a rapid equilibrium, the analyticalexpressions for V D
max and k Dm in this reaction mechanism are
the following:
VDmax �
k3k7�E�0k3 1 k7
�1�
KDm �
k3k7
k3 1 k7
K6
k7
1K2
k3
� �� k3K6 1 k7K2
k3 1 k7
�2�
where K2 � k22/k2 and K6 � k26/k6. The V Dmax expression
(Eqn 1) only involves rate constants that governs transforma-tion steps in the reaction mechanism. The K D
m expression(Eqn 1) involves kinetic constants that govern both thetransformation and binding steps (Scheme 2). A simplifiedcase occur when K2 < K6 because K D
m < K2 < K6 (Eqn 2).The V D
max expression is directly dependent on the product of therate constants, k3 and k7 (Eqn 1) but not K D
m (Eqn 2). Therefore,any increase of the values of k3 and k7 will result in anincreased V D
max (Eqn 1), but not K Dm (Eqn 2).
Rate limiting step: electronic effects. The experimental datashow that d D
4 , d D3 (Table 3). For this reason, it has been
proposed (Scheme 1) that the oxygen atom belonging to theOH group at C-4 probably carries out the first nucleophilicattack (k31
; k71). This attack would be very fast and would not
therefore be the rate-limiting step in the diphenolase reactionmechanism. Then, a second nucleophilic attack would becarried out by the OH group at C-3 (k32
; k72). The oxygen atom
from this OH group at C-3 would be a poorer nucleophile thanthat at C-4, and so the V D
max values for o-diphenols (Table 2)could be arranged according to the electron density in C-3(Table 3).
Furthermore, the preference of PPO for reacting witho-diphenols with similar d D
3 values might also becombined with the contribution made by the low d D
4
values; this was the case for the catecholamines assayed(Table 3, from dopamine to l-isoproterenol).
Rate limiting step: steric effects. The o-diphenols with a lowmolecular size substituent side-chain showed high value ofV D
max (Tables 2 and 3). This was the case for catechol withhigher dD
3 values than other o-diphenols but no substituentside-chain. The absence of steric hindrance in the action of PPOon catechol led to this substrate being the best o-diphenolassayed. Note that 4-methylcatechol with a d D
3 value lower thanfor catechol is positioned at second place. The same wasobserved when DHPAA was compared with DHPPA, whendopamine was compared with l-dopa and l-a-methyldopa, andas dopa methyl ester with l-isoproterenol.
On the other hand, o-diphenols with lower d D3 (better
nucleophiles) than those for other o-diphenols were not very
Table 2. Kinetic constants for the diphenolase activity of mushroom tyrosinase. Conditions were: sodium phosphate buffer 50 mm pH 6.8, DMF 2%,
saturating MBTH concentration and 3.8 nm mushroom tyrosinase.
o-Diphenol V Dmax (mm min21) k D
cat (s21) K Dm (mm) k D
cat /K Dm (s21´mm21)
Catechola 200.1 �^ 4.6 877.6 �^ 20.2 0.3 �^ 0.02 2925 �^ 301
4-Methylcatechola 191.93 �^ 19.1.0 841.6 �^ 84.1 2.36 �^ 0.4 356.6 �^ 96
TBCb 146.35 �^ 6.7 641.8 �^ 29.5 2.8 �^ 0.3 229 �^ 36
DHPAA 144.0 �^ 10.1 631.6 �^ 30.5 5.1 �^ 0.2 123.8 �^ 11
DHPPA 126.2 �^ 6.1 553.5 �^ 25.6 1.89 �^ 0.1 294 �^ 29
Dopamine 100.1 �^ 5.2 439.0 �^ 17.6 2.2 �^ 0.1 200 �^ 17
l-Dopa 24.5 �^ 1.0 107.4 �^ 3.1 0.8 �^ 0.03 134 �^ 9
l-a-Methyldopa 10.1 �^ 0.5 44.3 �^ 1.8 6.8 �^ 0.2 6.5 �^ 0.5
Dopa methyl ester 8.1 �̂ 0.4 35.5 �^ 1.4 1.0 �^ 0.05 35.5 �^ 3
l-Isoproterenol 6.7 �^ 0.3 29.4 �^ 1.3 7.1 �^ 0.3 4 �^ 0.3
a Diphenolase activity of tyrosinase on catechol and 4-methylcatechol was determined by measuring the disappearance of NADH in rapid kinetic assays [38].b Diphenolase activity of tyrosinase on 4-tert-butylcatechol was determined by measuring the appearance of the enzymatic-generated o-quinone in rapid
kinetic assays. This o-quinone is highly stable and did not react with MBTH [39].

1274 J. C. EspõÂn et al. (Eur. J. Biochem. 267) q FEBS 2000
Sch
eme
1.
Pro
po
sed
stru
ctu
ral
rea
ctio
nm
ech
an
ism
for
the
mo
nop
hen
ola
sean
dth
ed
iph
enola
seact
ivit
ies
of
tyro
sin
ase
.T
he
num
eric
nota
tion
of
the
rate
const
ants
isth
atpre
vio
usl
yuse
d[1
6]
wit
hth
esp
litt
ing
of
k 1,
k 3,
k 5an
dk 7
rate
con
stan
ts.

q FEBS 2000 Substrates and reaction mechanism of tyrosinase (Eur. J. Biochem. 267) 1275
good substrates (low V Dmax) because of the high molecular size
of their side-chain substituent (Tables 2 and 3). This was thecase for TBC which, despite its d D
3 value (the lowest value inTable 3), was placed in third position as regards its V D
max value;moreover, it has a high K D
m value (Table 2). The same wasobserved when l-a-methyldopa was compared with dopamineand l-dopa, and l-isoproterenol compared with dopa methylester.
The K Dm values did not show the same dependence on
molecular size as the V Dmax values (Table 2). Perhaps, the
molecular size of the side-chain has different effects onthe binding rate constants (K2, K6) and on thetransformation rate constants (k3, k7). That would lead todifferent effects on the values of K D
m (Eqn 2) and V Dmax (Eqn 1).
The substrates studied here do not belong to a homologousseries. However, it can be observed that by lowering thecatalytic constant K D
m increases, with the exception of DHPAA,l-dopa and dopa methyl ester (Table 2). DHPAA has acarboxylic group close to the aromatic ring. This group couldaccess the active site of the enzyme in the opposite positionto the OH group at C-4, possibly acting as competitiveinhibitor and increasing the value of K D
m . In the case of l-dopaand dopa methyl ester, the low K D
m value could be a result of theCO and NH +
3 groups establishing some critical interactionincreasing the affinity of the enzyme towards thesesubstrates. In the case of l-a-methyl-dopa, the presence ofthe a-methyl group could cause some steric hindrance, whichwould explain the low affinity (high K D
m ) observed.The high nucleophilicity of the oxygen of the phenolic
hydroxyl groups could favour the formation of a hydrogen bondbetween the hydrogen of the phenolic hydroxyl and thedeprotonated nitrogen of a histidine residue of the active siteof tyrosinase. This would increase the values of k2 and k6 anddecrease the values of K2 and K6 and, therefore K D
m (Eqn 2).This is proposed from previous experimental data on the
dependence of K Dm on the pH that showed a pKa value near to
that histidine [54±57]. This suggestion should be investigatedin further structural studies. The histidine residue could belocated in the active site, but not in the binuclear copper center[18].
This hypothesis might explain the values obtained for thekinetic constants of the diphenols characterized here (Table 2),i.e. the decrease of k D
cat, the increase of K Dm and the resulting
decrease of k Dcat /K D
m . The only exceptions to this explanation areTBC, DHPAA, l-dopa and dopa methyl ester, because of the
Scheme 2. Kinetic reaction mechanism for the monophenolase and the diphenolase activities of tyrosinase coupled to nonenzymatic reactions
from o-quinone. The numeric notation of the rate constants is that previously used [16] with the splitting of k5 rate constant.
Table 3. dD3 and dD
4 values for C-3 and C-4, respectively, of the
aromatic ring of o-diphenols. Conditions were: saturating substrate
concentration in 2H2O at pH 6.8. d values were measured relative to
those for tetramethylsilane (d � 0). The maximum wide line accepted in
the NRM spectra was 0.06 Hz. Therefore, the maximum error accepted for
each peak of the spectrum was ^ 0.03 p.p.m.
o-Diphenol d D3 (p.p.m.) dD
4 (p.p.m.).
Catechol 146�.59 146�.59
4-Methylcatechola 146�.43 144�.06
DHPAA 146�.43 144�.96
DHPPA 146�.51 144�.61
TBC 146�.24 144�.09
Dopamine 146�.79 145�.58
l-Dopa 146�.92 146�.06
l-a-Methyldopa 146�.74 146�.19
Dopa methyl ester 146�.90 146�.18
l-Isoproterenol 146�.87 146�.82
a The values of d D3 and dD
4 are corresponding to the C-3 and C-4 atoms of
the benzene ring, whose support OH groups. The C-1 atom support the
side-chain substituent. Thus, the systematic name of 4-methylcatechol is
considered here as 1-methylcatechol.

1276 J. C. EspõÂn et al. (Eur. J. Biochem. 267) q FEBS 2000
reasons discussed above. Note that the values of k Dcat for the
best and the worst o-diphenols show a ratio of < 30. Thecorresponding ratio between their catalytic efficiencies,k D
cat / K Dm , is < 731, reflecting the effect discussed above on K D
m .
Monophenolase activity of tyrosinase
V Mmax values were also very dependent on the monophenol used
(Table 3), following the sequence: 4HA . 4-ethoxyphenol .PHPPA . PHPAA . tyramine . phenol . l-tyrosine . l-a-methyltyrosine. This order reveals that the monophenols with ahigh electron-donor side-chain oriented towards the benzenering were oxidized more quickly. This property has beenproposed previously by other authors [58] and is supported byour experimental data (Table 4). As in the case of diphenolaseactivity, the K M
m values did not follow the same sequence(Table 4).
NMR assays
dM4 values for each monophenol at the optimum pH for
mushroom tyrosinase were determined by NMR assays(Table 5). In fact, a good correlation was found between thedM
4 values and V Mmax values (Fig. 1), the lowest dM
4 valuescorresponding to the highest V M
max values.
Structural reaction mechanism. The melanogenesis pathwayfrom monophenol starts with the monophenolase activity oftyrosinase, which consists of two catalytic cycles overlappingthrough three common intermediates. The stoichiometry of thepathway implies that one molecule of tyrosinase mustaccomplish two turnovers in the hydroxylase cycle for eachone in the oxidase cycle (Schemes 1 and 2 [22]). The Em formof the enzyme does not act on monophenols but has affinitytowards monophenols and binds them (k11
) through a dead-endpathway. The resulting form is a dead-end complex EmM[16,17] which suffers deprotonation (k12
) to yield EmM 0. Theexistence of this dead-end complex may explain the existenceof a lag period prior to attainment of the steady-state [22,23],which is characteristic of the monophenolase activity oftyrosinase. The turnover of the steady-state of tyrosinase actingon monophenols involves several processes (Schemes 1 and 2).The Eo form of the enzyme binds one molecule of monophenolwith no covalent interactions (EoM). The nucleophilic attack(k51
) of the oxygen belonging to the OH group on the copperatoms of the active site of the enzyme should then take placewith the further production of H1 to render EoM 0. We have nodata to support this proposal as the pKa value of the phenolicOH could be different at the active site from that presented in
solution. One of two forms, phenol or phenolate, may attack theenzyme. Whatever the case, the electronic effects and therelative sequence of nucleophilic power for the substrateswould not change. After this attack, a hydroxylation reactioninvolving electrophilic attack (k51
) would yield the EmD 00 form.Oxidoreduction (k33
) would then lead to production of onemolecule of o-quinone (Q) to yield the Ed form of the enzymewith Cu1-Cu1 in the active site, accompanied by theconsumption of H1 and formation of H2O. This enzymaticform binds O2 to yield the Eo form with Cu21-O 22
2 -Cu21
in the active site. As the concentration of o-diphenol hasnot changed during the steady state, one more catalyticcycle is accomplished in the hydroxylase pathway andanother molecule of monophenol is consumed to yieldanother molecule of o-quinone. MTBH traps the enzyme-generated o-quinones resulting in production of the MBTH±quinone adduct, while one more molecule of o-diphenolaccumulates in the medium (Scheme 2). Because the o-diphenol level has increased and the enzyme has to maintainthe steady state, the Eo form binds o-diphenol through thediphenolase pathway. The o-diphenol level decreases in thislatter pathway but it is restored from the two enzyme-generatedmolecules of o-quinone.
To conclude, the monophenolase activity of tyrosinase(Schemes 1 and 2) starts with the single operation of thehydroxylase cycle and evolves towards the steady state, withtwo turnovers in the hydroxylase cycle for each one in theoxidase cycle, accompanied by a net decrease in the early EmMdue to the regeneration of o-diphenol in the nonenzymaticreactions from o-quinone 1 MBTH. This sequence of couplednonenzymatic reactions (o-quinone 1 MBTH) in the mono-phenolase system is therefore of great importance.
Kinetic reaction mechanism. The reaction mechanism oftyrosinase acting on monophenols is a bicyclic system(Scheme 1) involving many rate constants and resulting in com-plex expressions for V M
max and K Mm . To simplify the kinetic
analysis, the mechanism described in Scheme 1 can be reducedto that described in Scheme 2. The expression of the steady-state rate (V M
ss ) is:
V Mss �
b0�D�0�O2��E�0c0 1 c1�D�0 1 c2�O2�1 c3�M�0�O2�1 c4�D�0�O2� �3�
V Mmax �
b0R�E�0c3 1 c4R
�4�
Table 4. Kinetic constants for the monophenolase activity of mushroom tyrosinase. Conditions were as in Table 2.
Monophenol V Dmax (mm min21) K M
cat (s21) K Mm (mm) k M
cat / K Mm (s21´mm21)
4HA 42.0 �^ 3.1 184.2 �^ 6.1 0.08 �^ 0.003 2300 �^ 162
4-Ethoxyphenol 30.1 �^ 2.2 132.0 �^ 4.4 0.17 �^ 0.005 776 �^ 48
PHPPA 15.2 �^ 1.1 66.7 �^ 2.7 0.44 �^ 0.01 152 �^ 10
PHPAA 10.1 �^ 0.7 44.3 �^ 1.5 1.91 �^ 0.08 23 �^ 2
Tyramine 5.9 �̂ 0.3 25.9 �^ 1.1 0.51 �^ 0.02 51 �^ 4
Phenol 2.9 �^ 0.1 12.7 �^ 0.6 0.70 �^ 0.04 18 �^ 2
l-Tyrosine 1.8 �^ 0.06 7.9 �^ 0.3 0.21 �^ 0.01 38 �^ 3
l-a-Methyl tyrosine 0.13 �^ 0.005 0.6 �^ 0.03 1.20 �^ 0.08 0.5 �^ 0.06
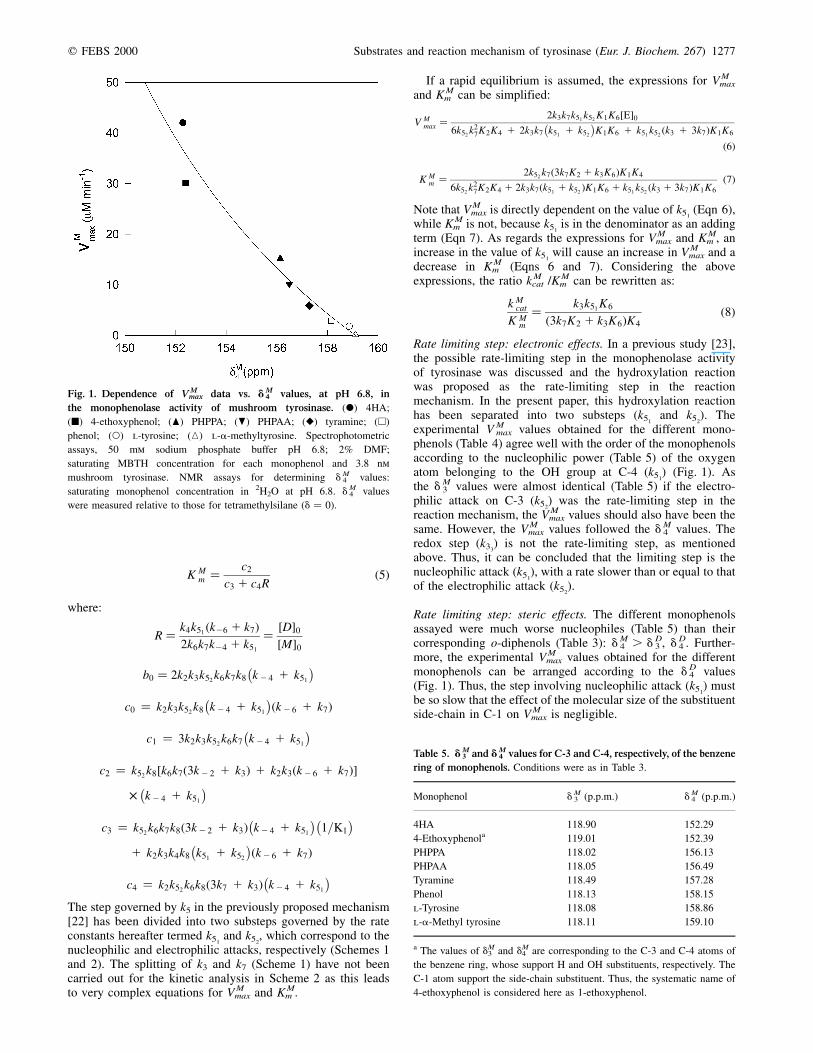
q FEBS 2000 Substrates and reaction mechanism of tyrosinase (Eur. J. Biochem. 267) 1277
K Mm �
c2
c3 1 c4R�5�
where:
R � k4k51�k26 1 k7�
2k6k7k24 1 k51
� �D�0�M�0
b0 � 2k2k3k52k6k7k8 k 2 4 1 k51
ÿ �c0 � k2k3k52
k8 k 2 4 1 k51
ÿ �k 2 6 1 k7� �
c1 � 3k2k3k52k6k7 k 2 4 1 k51
ÿ �c2 � k52
k8 k6k7 3k 2 2 1 k3� � 1 k2k3 k 2 6 1 k7� �� �� k 2 4 1 k51
ÿ �c3 � k52
k6k7k8 3k 2 2 1 k3� � k 2 4 1 k51
ÿ �1=K1
ÿ �1 k2k3k4k8 k51
1 k52
ÿ �k 2 6 1 k7� �
c4 � k2k52k6k8 3k7 1 k3� � k 2 4 1 k51
ÿ �The step governed by k5 in the previously proposed mechanism[22] has been divided into two substeps governed by the rateconstants hereafter termed k51
and k52, which correspond to the
nucleophilic and electrophilic attacks, respectively (Schemes 1and 2). The splitting of k3 and k7 (Scheme 1) have not beencarried out for the kinetic analysis in Scheme 2 as this leadsto very complex equations for V M
max and K Mm .
If a rapid equilibrium is assumed, the expressions for V Mmax
and K Mm can be simplified:
V Mmax �
2k3k7k51k52
K1K6 E� �06k52
k27K2K4 1 2k3k7 k51
1 k52
ÿ �K1K6 1 k51
k52k3 1 3k7� �K1K6
�6�
K Mm �
2k52k7�3k7K2 1 k3K6�K1K4
6k52k2
7K2K4 1 2k3k7�k511 k52
�K1K6 1 k51k52�k3 1 3k7�K1K6
�7�
Note that V Mmax is directly dependent on the value of k51
(Eqn 6),while K M
m is not, because k51is in the denominator as an adding
term (Eqn 7). As regards the expressions for V Mmax and K M
m , anincrease in the value of k51
will cause an increase in V Mmax and a
decrease in K Mm (Eqns 6 and 7). Considering the above
expressions, the ratio k Mcat /K M
m can be rewritten as:
k Mcat
K Mm
� k3k51K6
�3k7K2 1 k3K6�K4
�8�
Rate limiting step: electronic effects. In a previous study [23],the possible rate-limiting step in the monophenolase activityof tyrosinase was discussed and the hydroxylation reactionwas proposed as the rate-limiting step in the reactionmechanism. In the present paper, this hydroxylation reactionhas been separated into two substeps (k51
and k52). The
experimental V Mmax values obtained for the different mono-
phenols (Table 4) agree well with the order of the monophenolsaccording to the nucleophilic power (Table 5) of the oxygenatom belonging to the OH group at C-4 (k51
) (Fig. 1). Asthe dM
3 values were almost identical (Table 5) if the electro-philic attack on C-3 (k52
) was the rate-limiting step in thereaction mechanism, the V M
max values should also have been thesame. However, the V M
max values followed the dM4 values. The
redox step (k33) is not the rate-limiting step, as mentioned
above. Thus, it can be concluded that the limiting step is thenucleophilic attack (k51
), with a rate slower than or equal to thatof the electrophilic attack (k52
).
Rate limiting step: steric effects. The different monophenolsassayed were much worse nucleophiles (Table 5) than theircorresponding o-diphenols (Table 3): dM
4 . d D3 , d D
4 . Further-more, the experimental V M
max values obtained for the differentmonophenols can be arranged according to the d D
4 values(Fig. 1). Thus, the step involving nucleophilic attack (k51
) mustbe so slow that the effect of the molecular size of the substituentside-chain in C-1 on V M
max is negligible.
Fig. 1. Dependence of V Mmax data vs. dM
4 values, at pH 6.8, in
the monophenolase activity of mushroom tyrosinase. (X) 4HA;
(B) 4-ethoxyphenol; (O) PHPPA; (P) PHPAA; (V) tyramine; (A)
phenol; (W) l-tyrosine; (K) l-a-methyltyrosine. Spectrophotometric
assays, 50 mm sodium phosphate buffer pH 6.8; 2% DMF;
saturating MBTH concentration for each monophenol and 3.8 nm
mushroom tyrosinase. NMR assays for determining dM4 values:
saturating monophenol concentration in 2H2O at pH 6.8. dM4 values
were measured relative to those for tetramethylsilane (d � 0).
Table 5. dM3 and dM
4 values for C-3 and C-4, respectively, of the benzene
ring of monophenols. Conditions were as in Table 3.
Monophenol dM3 (p.p.m.) dM
4 (p.p.m.)
4HA 118�.90 152�.29
4-Ethoxyphenola 119�.01 152�.39
PHPPA 118�.02 156�.13
PHPAA 118�.05 156�.49
Tyramine 118�.49 157�.28
Phenol 118�.13 158�.15
l-Tyrosine 118�.08 158�.86
l-a-Methyl tyrosine 118�.11 159�.10
a The values of dM3 and dM
4 are corresponding to the C-3 and C-4 atoms of
the benzene ring, whose support H and OH substituents, respectively. The
C-1 atom support the side-chain substituent. Thus, the systematic name of
4-ethoxyphenol is considered here as 1-ethoxyphenol.

1278 J. C. EspõÂn et al. (Eur. J. Biochem. 267) q FEBS 2000
Other studies on the effects of changes in the side-chain onthe monophenol activity reached similar conclusions to thoseabove [13]. They found little effect when the side-chain on thearomatic ring was modified, except for a drastic decrease in theactivity of ptert-butylphenol compared with that of othersubstrates. They proposed that the bulky substituent was thereason for this drastic decrease in the V M
max value. However,we think that the results obtained by these authors could beexplained by the fact that this monophenol yields a highlystable o-quinone. No chemical reactions take place after theenzymatic reaction. The o-diphenol is not regenerated in themedium and the system cannot reach a true steady-state as wedemonstrated in a previous work [39].
The molecular size of the substituent side-chain in C-1 couldaffect the K M
m values, as for the diphenolase activity. Thedecrease in the value of the catalytic constant implies anincrease in K M
m (Table 4), as in the case of o-diphenols(Table 2). l-Tyrosine is an exception because of the presence ofthe CO and NH +
3 groups. PHPAA shows a higher value of K Mm
than PHPPA. The explanation could be the same as describedpreviously for the o-diphenol DHPAA compared with DHPPA.Phenol is not the first in the series of monophenols, whichcould indicate that the binding constants participate in thepossible binding by hydrogen bridge with the phenolichydroxyl group, as proposed for diphenolase activity. Thissame effect can be observed in the ratio k M
cat / K Mm (Table 4).
This ratio k Mcat / K M
m (Eqn 8) is directly related to thenucleophilicity of the oxygen atom from the hydroxyl groupat C-4 (k51
) as well as to the affinity constants K4 � k24/k4. Thehigh nucleophilicity of the phenolic hydroxyl group couldfavour the formation of a hydrogen bond between the hydrogenof the phenolic hydroxyl and the deprotonated nitrogen of ahistidine residue of the active site of tyrosinase, as proposed forthe diphenolase activity. This could increase the value of k4 anddecrease the value of K4, increasing the value of the catalyticefficiency k M
cat / K Mm (Eqn 8). This occurs with all the mono-
phenols with the exception of l-tyrosine and PHPAA. (Notethat now the values of k M
cat for the best and the worstmonophenols show a ratio of < 307; Table 4.) This value ishigher than the corresponding value for o-diphenols < 30(Table 2), because of the involvement of binding constants aswell as transformation constants in the expression of k M
cat
(Eqn 6). Both types of constant are present in theexpression of K M
m (Eqn 7). For this reason, a ratio of < 4600is obtained for the corresponding catalytic efficiencies,k M
cat / K Mm (Table 4), which is higher than that for o-diphenols
< 731 (Table 2).
Conclusions
The kinetics of the catalytic activities of mushroom tyrosinaseon several o-diphenols (Table 2) and monophenols (Table 4)has been characterized. The electron densities of C-3 and C-4of the phenolic ring have also been determined by NMR(Tables 3 and 5; Fig. 1). From these data, a reaction mechanismhas been proposed (Schemes 1 and 2) and several conclusionshave been obtained:
The redox step (k33) is not limiting. This affirmation is based on
various observations. The k Dcat of the diphenols (Table 2) are
not ordered according to their nucleophilic power (k32)
(Table 3) and steric hindrances intervene. The monophenolsare oxidized after being transformed into diphenols. If the redoxstep were limiting, the order of the monophenols k M
cat (Table 4)would be the same as the order of the diphenol nucleophilicpower (Table 3); this is not the case (Table 4). Moreover, the
case of the phenol is of great interest as it does not contain aside-chain in C-1 but it is not among the first monophenols(Table 4). Therefore, in monophenols too, steric hindrances arenot limiting.
The electrophilic attack step (k52) is not limiting. This
affirmation is based on the fact that the side-chain in C-1does not influence on the electron density of C-3 (Table 5). Ifthis step were limiting, the k M
cat values would be the same for allthe monophenols, regardless of the substituting group in C-1;this does not occur (Table 4).
The nucleophilic attack step (k51) is of the same order or less
than the electrophilic attack step (k52). This is the conclusion
reached based on the above results.
A C K N O W L E D G E M E N T S
This paper was partially supported by the `ComisioÂn Interministerial de
Ciencia y TecnologõÂa', project number CICYT PB98-0403-CO2. J. C. E.
has a fellowship from the Programa Nacional de FormacioÂn del Personal
Investigador, Ministerio de EducacioÂn y Ciencia (Spain), reference AP93
34785457. F. G.-C. dedicates this paper to Professor Dr Jose Antonio
Lozano, who first introduced him to the topic and provided encouragement
to carry out research into tyrosinase.
R E F E R E N C E S
1. Robb, D.A. (1984) Tyrosinase. In Copper Proteins and Copper
Enzymes (Lontie, R., ed. ), Vol. 2, pp. 207±240. CRC Press, Boca
Raton.
2. Prota, G. (1992) Tyrosinase. In Melanins and Melanogenesis
(Jovanovich, H.B., ed.), pp. 34±62. Academic Press, San Diego.
3. Mason, H.S. (1956) Structure and functions of the phenolase complex.
Nature 177, 79±81.
4. Jolley, R.L. Jr, Evans, L.H. & Mason, H.S. (1972) Reversible
oxygenation of tyrosinase. Biochem. Biophys. Res. Commun. 46,
878±884.
5. Schoot-Uiterkamp, A.J. & Mason, H.S. (1973) Magnetic dipole-dipole
coupled Cu (II) pairs in nitric oxide-treated tyrosinase: structural
relationship between the active site of tyrosinase and hemocyanin.
Proc. Natl Acad. Sci. USA 70, 993±996.
6. Jolley, R.L. Jr, Evans, L.H., Makino, N. & Mason, H.S. (1974)
Oxytyrosinase. J. Biol. Chem. 249, 335±345.
7. Makino, N. & Mason, H.S. (1973) Reactivity of oxytyrosinase towards
substrates. J. Biol. Chem. 248, 5731±5735.
8. Makino, N., McMahill, P., Mason, H.S. & Moss, T.H. (1974) The
oxidation state of copper in resting tyrosinase. J. Biol. Chem. 249,
6062±6066.
9. Schoot-Uiterkamp, A.J.M., Evans, L.H., Jolley, R.L. & Mason, H.S.
(1976) Absorption and circular dichroism spectra of different forms
of mushroom tyrosinase. Biochim. Biophys. Acta 453, 200±204.
10. Lerch, K. (1981) Copper monooxygenases: tyrosinase and dopamine
beta-hydroxylase. In Metal Ions in Biological Systems (Sigel, H., ed.),
pp. 143±186. Marcel Dekker Inc., New York.
11. Himmelwright, R.S., Eickman, N.C., Lu Bien, C.D., Lerch, K. &
Solomon, E.I. (1980) Chemical and spectroscopic studies of the
binuclear copper active site of Neurospora tyrosinase: Comparison to
hemocyanins. J. Am. Chem. Soc. 102, 7339±7344.
12. Solomon, E.I. (1981) Binuclear copper active site: Hemocyanin,
tyrosinase and type 3 copper oxidases. In Copper Proteins (Spiro,
T.G., ed.), Vol. 3, pp. 41±108. Wiley-Interscience, New York.
13. Wilcox, D.E., Porras, A.G., Hwang, Y.T., Lerch, K., Winkler, M.E. &
Solomon, E.I. (1985) Substrate analogue binding to the coupled
binuclear copper active site in tyrosinase. J. Am. Chem. Soc. 107,
4015±4027.
14. Solomon, E.I. & Lowery, M.D. (1993) Electronic structure contri-
butions to function in bioinorganic chemistry. Science 259,
1575±1581.

q FEBS 2000 Substrates and reaction mechanism of tyrosinase (Eur. J. Biochem. 267) 1279
15. Solomon, E.I., Sundaram, U.M. & Machonkin, T.E. (1996) Multi-
copper oxidases and oxygenases. Chem. Rev. 96, 2563±2605.
16. Van Gelder, C.W.G., Flurkey, W.A. & Wichers, H.J. (1997)
Sequence and structural features of plant and fungal tyrosinases.
Phytochemistry 45, 1309±1323.
17. Klabunde, T., Eicken, C., Sacchettini, J.C. & Krebs, B. (1998) Crystal
structure of a plant catechol oxidase containing a dicopper center.
Nat. Struct. Biol. 5, 1084±1090.
18. Bubacco, L., Salgado, J., Tepper, A.W.J.W., Vijgenboom, E. & Canters,
G.W. (1999) 1H NMR spectroscopy of the binuclear Cu (II)
active site of Streptomyces antibioticus tyrosinase. FEBS Lett.
442, 215±220.
19. Chioccara, F., Di Gennaro, P., La Monica, G., Sebastiano, R. &
Rindone, B. (1991) Selective ortho-hydroxilation of phenols in
copper (I) complexes. Tetrahedron 47 (25), 4429±4434.
20. Sayre, L.M. & Nadkarni, D.V. (1994) Direct conversion of phenols to
o-quinones by copper (I) dioxygen. Questions regarding the
monophenolase activity of tyrosinase mimics. J. Am. Chem. Soc.
116, 3157±3158.
21. Kitajima, N., Koda, T., Iwata, Y. & Moro-oka, Y. (1990) Reaction
aspects of a micro-peroxo binuclear copper (II) complex. J. Am.
Chem. Soc. 112, 8833±8839.
22. RodrõÂguez-LoÂpez, J.N., Tudela, J., VaroÂn, R., GarcõÂa-Carmona, F. &
GarcõÂa-CaÂnovas, F. (1992) Analysis of a kinetic model for melanin
biosynthesis pathway. J. Biol. Chem. 267, 3801±3810.
23. Ros, J.R., RodrõÂguez-LoÂpez, J.N. & GarcõÂa-CaÂnovas, F. (1994)
Tyrosinase: kinetic analysis of the transient phase and the steady-
state. Biochim. Biophys. Acta 1204, 33±42.
24. SaÂnchez-Ferrer, A., RodrõÂguez-LoÂpez, J.N., GarcõÂa-CaÂnovas, F. &
GarcõÂa-Carmona, F. (1995) Tyrosinase: a comprehensive review of its
mechanism. Biochim. Biophys. Acta 1247, 1±11.
25. RodrõÂguez-LoÂpez, J.N., Escribano, J. & GarcõÂa-CaÂnovas, F. (1994) A
continuous spectrophotometric method for the determination of
monophenolase activity of tyrosinase using 3-methyl-2-benzothiali-
zone hydrazone. Anal. Biochem. 216, 205±212.
26. EspõÂn, J.C., Morales, M., VaroÂn, R., Tudela, J. & GarcõÂa-CaÂnovas, F.
(1995) A continuous spectrophotometric method for determining the
monophenolase and diphenolase activities of apple polyphenol
oxidase. Anal. Biochem. 231, 237±246.
27. EspõÂn, J.C., Morales, M., VaroÂn, R., Tudela, J. & GarcõÂa-CaÂnovas, F.
(1995) Monophenolase activity of polyphenol oxidase from verde-
doncella apple. J. Agric. Food Chem. 43, 2807±2812.
28. EspõÂn, J.C., Morales, M., VaroÂn, R., Tudela, J. & GarcõÂa-CaÂnovas, F.
(1996) A continuous spectrophotometric method for determining the
monophenolase and diphenolase activities of pear polyphenol
oxidase. J. Food Sci. 61, 1177±1201.
29. EspõÂn, J.C., Morales, M., VaroÂn, R., Tudela, J. & GarcõÂa-CaÂnovas, F.
(1997) Monophenolase activity of polyphenol oxidase from Blan-
quilla pear. Phytochemistry 44, 17±22.
30. EspõÂn, J.C., Morales, M., GarcõÂa-Ruiz, P.A., Tudela, J. & GarcõÂa-
CaÂnovas, F. (1997) Improvement of a continuous spectrophotometric
method for determining the monophenolase and diphenolase
activities of mushroom polyphenol oxidase. J. Agric. Food Chem.
45, 1084±1090.
31. EspõÂn, J.C., Trujano, M.F., Tudela, J. & GarcõÂa-CaÂnovas, F. (1997)
Monophenolase activity of polyphenol oxidase from Haas
avocado. J. Agric. Food Chem. 45, 1091±1096.
32. EspõÂn, J.C., Ochoa, M., Tudela, J. & GarcõÂa-CaÂnovas, F. (1997)
Monophenolase activity of strawberry polyphenol oxidase. Phyto-
chemistry 45, 667±670.
33. EspõÂn, J.C., VaroÂn, R., Tudela, J. & GarcõÂa-CaÂnovas, F. (1997) Kinetic
study of the oxidation of 4-hydroxyanisole catalized by tyrosinase.
Biochem. Mol. Biol. Int. 41, 1265±1276.
34. EspõÂn, J.C., GarcõÂa-Ruiz, P.A., Tudela, J. & GarcõÂa-CaÂnovas, F. (1998)
Study of the stereospecificity in mushroom tyrosinase. Biochem. J.
331, 547±551.
35. Winder, A.J. & Harris, H. (1991) New assays for the tyrosine
hydroxilase and dopa oxidase activities of tyrosinase. Eur. J.
Biochem. 198, 317±326.
36. Duckworth, H.W. & Coleman, J.E. (1970) Physicochemical and
kinetic properties of mushroom tyrosinase. J. Biol. Chem. 245,
1613±1625.
37. Bradford, M. (1976) A rapid and sensitive method for the quatification
of microgram quatities of proteins utilizing the principle of protein-
dye binding. Anal. Biochem. 72, 248±256.
38. Carlson, B.W. & Miller, L.L. (1985) Mechanism of the oxidation of
NADH by quinones. Energetics of one-electron and hydride
routes. J. Am. Chem. Soc. 107, 479±485.
39. Ros, J.R., RodrõÂguez-LoÂpez, J.N., VaroÂn, R. & GarcõÂa-CaÂnovas, F.
(1994) Kinetics study of the oxidation of 4-tert-butylphenol by
tyrosinase. Eur. J. Biochem. 222, 449±452.
40. Waite, J.H. (1976) Calculating extinction coefficients for enzymatically
produced o-quinones. Anal. Biochem. 75, 211±218.
41. Wilkinson, G.N. (1961) Statistical estimations in enzyme kinetics.
Biochem. J. 80, 324±332.
42. Endrenyi, L. (1981). Kinetic Data Analysis: Design and Analysis of
Enzyme and Pharmacokinetics Experiments. Plenum Press, New
York.
43. Marquardt, D. (1963) An algorithm for least-squares estimation of non-
linear parameters. J. Sci. Ind. Appl. Math. 11, 431±444.
44. Jandel Scientific (1994) Sigma Plot 2.01 for WindowsTM (Jandel
Scientific, ed.). Jandel Scientific, Corte Madera.
45. GuÈnther, H. (1980) Nuclear magnetic resonance of fluorine-19 and
carbon-13. In NMR Spectroscopy, pp. 364±374. John Wiley & Sons,
New York.
46. Farnun, D.G. (1975) Charge density-NMR chemical shift correlations
in organic ions. In Advances in Physical Organic Chemistry (Gold, V.
& Bethell, D., eds), Vol. 11, pp. 123±173. Academic Press, New
York.
47. Levy, G.C. & Nelson, G.L. (1976) Resonancia MagneÂtica Nuclear de
Carbono 13 Para QuõÂmicos OrgaÂnicos, pp. 100±107. Ediciones
Bellaterra S.A., Barcelona.
48. Marriot, S. & Topsom, R.D. (1985) A theorical scale of
substituent resonance parameters (soR). J. Chem. Soc. Perkin
Trans. II 1045±1047.
49. Bromilow, J., Brownlee, R.T.C., Lopez, V.O. & Taft, R.W. (1979) Para
substituent carbon-13 chemical shifts in substituted benzenes. 1.
Updating the s0R scale and analysis of aprotic solvent effects. J. Org.
Chem. 44, 4766±4770.
50. Hirsch, J.A. (1972) Concepts. In Theoretical Organic Chemistry, pp.
184±189. Allyn and Bacon Inc. Boston.
51. Bordell, F.G. & Hughes, D.L. (1984) SN2 Reactions of nitranions with
benzyl chlorides. J. Am. Chem. Soc. 106, 3234±3240.
52. Bordell, F.G. & Hughes, D.L. (1983) Steric and electronic effects in
SN2 reactions of 9-substituted fluorenyl and a-cyano carbanions with
benzyl chloride in dimethyl sulfoxide solution. J. Org. Chem. 48,
2206±2215.
53. Tomiyama, S., Sakai, S., Nihiyama, T. & Yasmada, F. (1993) Factors
influencing the antioxidant activities of phenols by an ab initio study.
Bull. Chem. Soc. Jpn. 66, 205±211.
54. Dietler, C. & Lerch, K. (1982) Tyrosinase. In Oxidases and Related
Redox Systems, pp. 305±317. Pergamon Press, Oxford.
55. GarcõÂa-CaÂnovas, F., Tudela, J., MartõÂnez Madrid, C., VaroÂn, R., GarcõÂa-
Carmona, F. & Lozano, J.A. (1987) Kinetic study on the suicide
inactivation of tyrosinase induced by catechol. Biochim. Biophys.
Acta 912, 417±423.
56. Tudela, J., GarcõÂa-CaÂnovas, F., VaroÂn, R., JimeÂnez, M., GarcõÂa-
Carmona, F. & Lozano, J.A. (1987) Kinetic characterization of
dopamine as a suicide substrate of tyrosinase. J. Enzyme Inhib. 2,
47±56.
57. GarcõÂa-Moreno, M., VaroÂn, R., SaÂnchez-Gracia, Angela Tudela, J. &
GarcõÂa-CaÂnovas, F. (1994) The effect of pH on the suicide
inactivation of frog epidermis tyrosinase. Biochim. Biophys. Acta
1205, 282±288.
58. Passi, S. & Nazzarro-Porro, M. (1981) Molecular basis of substrate and
inhibitory of tyrosinase: phenolic compounds. Br. J. Dermatol. 104,
659±665.




















