
In Silico inhibitors for Plasmodium falciparum lactate dehydrogenase

Eur. J. Biochem. 86, 519-530 (1978)
The Effect of Fatty Acids on the Regulation of Pyruvate Dehydrogenase in Perfused Rat Liver Roland SCHOLZ, Merle S. OLSON, Andreas J. SCHWAB, Ursula SCHWABE, Christiane NOELL, and Wolfgang BRAUN
Institut fur Physiologische Chemie, Physikalische Biochemie und Zellbiologie der Universitat Munchen
(Received October 10, 1977)
The effect of fatty acids on the rate of pyruvate decarboxylation was studied in perfused livers from fed rats. The production of 14C02 from infused [l-14C]pyruvate was employed as a monitor of the flux through the pyruvate dehydrogenase reaction. A correction for other decarboxylation reactions was made using kinetic analyses. Fatty acid (octanoate or oleate) infusion caused a stimulation of pyruvate decarboxylation at pyruvate concentrations in the perfusate below 1 mM (up to 3-fold at 0.05 mM pyruvate) but decreased the rate to one-third of control rates at pyruvate concentrations near 5 mM. These effects were half-maximal at fatty acid concentrations below 0.1 mM. Infusion of 3-hydroxybutyrate also caused a marked stimulation of pyruvate decarboxy- lation at low pyruvate concentrations.
The data suggest that the mechanism by which fatty acids stimulate the flux through the pyruvate dehydrogenase reaction in perfused liver at low (limiting) pyruvate concentrations involves an acceleration of pyruvate transport into the mitochondrial compartment due to an exchange with acetoacetate. Furthermore, it is proposed that a relationship exists between ketogenesis and the regulation of pyruvate oxidation at pyruvate concentrations approximating conditions in vivo.
The pyruvate dehydrogenase multienzyme com- plex from mammalian tissues is regulated by two different mechanisms. Early studies indicated that this enzyme is inhibited by products of the reaction, NADH and acetyl-CoA, each of which is a competi- tive inhibitor with respect to the corresponding sub- strate, i.e. NAD' and coenzyme A [4-61. Later it was found that pyruvate dehydrogenase is inactivated upon phosphorylation by a specific protein kinase, whereas the phosphorylated enzyme complex may be reactivated by a phosphoprotein phosphatase [7 - 91. Among the wide variety of effector molecules with a proposed involvement in the differential regulation of this kinase-phosphatase-mediated interconversion system are NADH and NAD' [ 10 - 131, monovalent and divalent metal cations [ 14- 161, ATP and ADP [17-221, acetyl-CoA and coenzyme A [ZO- 13,221, and pyruvate [17,23,24].
The oxidation of pyruvate in various tissues has been shown to be inhibited by fatty acids. The initial
Enzymes. Hydroxybutyrate dehydrogenase or D-3-hydroxy- butyrate:NAD oxidoreductase (EC 1.1 .I .30); malic enzyme or malate dehydrogenase (decarboxylating) or L-malate: NADP oxido- reductase (decarboxylating) (EC 1.1.1.40); multienzyme complex of pyruvate dehydrogenase or pyruvate: lipoate oxidoreductase (acceptor-acetylating) (EC 1.2.3.1); phosphoenolpyruvate carboxy- kinase or phosphoenoylpyruvate carboxylase or GTP : oxaloacetate carboxy-lyase (transphosphorylating) (4.1:1.32).
explanation for this effect was based upon the fact that the inhibitory products of the pyruvate dehydro- genase reaction, i.e. NADH and acetyl-CoA, are also produced during P-oxidation of fatty acids [4 - 61. However, the observation that addition of fatty acids to intact organs [25 - 271 or mitochondrial prepara- tions [ l l , 17,19,20] resulted in a conversion of active pyruvate dehydrogenase into its inactive form indi- cated that the fatty acid effect was more than a simple, direct feedback inhibition of the enzyme by NADH and acetyl-CoA. Initial proposals for the mechanism of the fatty-acid-mediated inactivation of pyruvate dehydrogenase have centered around the suggestion that the addition of long-chain fatty acids to tissues may cause an increased ATP/ADP ratio in the mito- chondrial compartment [20] which would accentuate the kinase-mediated inactivation, i.e. ATP is a sub- strate of the kinase while ADP is a competitive inhibitor [23]. This hypothesis was supported by experiments with isolated mitochondria in which the interconversion of pyruvate dehydrogenase between its active and inactive forms is sensitive to changes in the intramitochondrial ATP/ADP ratio [13,19, 20,221. On the other hand, several experiments sug- gested that the ATP/ADP ratio may not be primarily responsible for the fatty acid effect, but that the oxida- tion-reduction state of the mitochondrial pyridine

520 Regulation of Pyruvate Dehydrogenase
nucleotide system was likely involved [19,28]. This suggestion was pursued by Batenburg and Olson [l 1 ] and Hansford [22] who demonstrated that in liver mitochondria the conversion of active pyruvate de- hydrogenase into its inactive form was a consequence of increased NADH/NAD + and acetyl-CoA/CoASH ratios upon the addition of fatty acids. Moreover, this alternate hypothesis for the fatty acid effect on pyruvate dehydrogenase was supported by the findings that purified pyruvate dehydrogenase kinase is acti- vated by NADH and acetyl-CoA and is inhibited by NAD' and coenzyme A, while the phosphatase is inhibited by NADH [lo, 131.
Our present notion of the regulation of pyruvate oxidation in intact cells is derived mainly from mea- surements of enzyme activities in tissue homogenates or in isolated mitochondria. However, these data probably reflect only the capacity or potential for the regulation of the pyruvate dehydrogenase complex, whereas very little is known about the actual flux rates through the pyruvate dehydrogenase reaction in a given metabolic state. Moreover, the contribution of interconversion or feedback inhibition of pyruvate dehydrogenase to the overall regulation of pyruvate oxidation, i.e. fine or coarse control of this process, cannot be elucidated without measuring flux rates.
In view of the uncertainties in extrapolating data from isolated enzymes, mitochondria or tissue homo- genates to the condition in vivo, the following questions arise regarding the fatty acid effect on pyruvate dehydrogenase. First, is hepatic pyruvate oxidation inhibited by fatty acids under all metabolic conditions? Second, which of the regulatory mechanisms studied in experiments in vitro may be responsible for the fatty acid effect in vivo? The present study was initiated to investigate these questions by measuring the rate of pyruvate decarboxylation in perfused livers from fed rats.
It was the initial objective of this study to develop an assay system by which the flux through the pyr- uvate dehydrogenase reaction could be monitored continuously in a perfused organ such as the liver (Scholz, Braun, Josephi, Kiihn and Schwab, unpub- lished results). The production of I4CO2 from [1-14C]- pyruvate was used as a parameter of the flux through the pyruvate dehydrogenase reaction. As was shown previously [l], a quantitative evaluation of the rates can be achieved employing the specific activity of lactate in the effluent perfusate and a kinetic analysis to correct for additional decarboxylation processes. The following data indicated that the pyruvate dehy- drogenase complex in perfused rat liver is capable of interconversion between its active and inactive forms by the kinase/phosphatase system (Scholz and Braun, unpublished results). First, the pyruvate concentra- tion for half-maximal rates of pyruvate decarboxyla- tion was approximately 1 mM which is much higher
than the K, of the isolated enzyme complex, i.e. 10-20 pM pyruvate [23]. Second, a titration of pyru- vate decarboxylation in the presence of dichloroace- tate, an inhibition of pyruvate dehydrogenase kinase [29], resulted in an apparent K, of approximately 20 pM pyruvate similar to the value observed with the isolated enzyme. These results imply that the depen- dence of pyruvate decarboxylation on the concentra- tion of pyruvate (in the absence of dichloroacetate) was due to more than one process, i.e. pyruvate acting as the substrate for the active enzyme complex and, mainly, as an inhibitor of the inactivating en- zyme (Ki about 1 mM [23]).
On the basis of these experimental results, experi- ments were performed to ascertain whether fatty acid infusion effects the rate of pyruvate decarboxylation in perfused rat liver and which regulatory mechanisms might be involved. It was found that addition of fatty acids changed the rates of pyruvate decarboxylation. However, the changes were modified both in extent and direction by the metabolic conditions of the liver and were highly dependent upon the pyruvate con- centration. The data indicate that in intact tissue the regulation of pyruvate dehydrogenase by fatty acids is a multifactorial system. In addition to the known mechanisms (i.e. feedback inhibition of pyruvate dehydrogenase by reaction products and intercon- version of the.enzyme) it appears that the transport of pyruvate into the mitochondria plays a role in the regulation of pyruvate metabolism.
MATERIALS AND METHODS
Male albino rats of the Wistar strain (Thomae, Biberach) weighing 200-220 g were used in these studies. The animals received a standard laboratory chow diet and water ad libitum prior to surgical removal of the liver under pentobarbital anaesthesia.
The hemoglobin-free liver perfusion technique [30] employing a non-recirculating perfusion system has been described elsewhere [31]. The perfusion medium was Krebs/Henseleit bicarbonate buffer [32], pH 7.4, and 37 "C, saturated with oxygen/carbon dioxide (95j5) . The perfusion medium was pumped through a membrane oxygenator prior to entering the h e r via the portal vein. Following passage through the liver the perfusate flowed past an oxygen electrode before it was collected in 250-ml conical flasks at intervals of 15, 30 or 60 s. Samples for metabolite analyses were removed and the residual perfusate was used for I4CO2 determination.
14C02 was trapped in phenethylamine and the radioactivity was measured by liquid scintillation spectroscopy. Glucose, lactate and pyruvate in the perfusate were isolated by anion-exchange chromatog-

R . Scholz, M. S. Olson, A. J . Schwab, U. Schwabe, Ch. Noell, and W. Braun 521
raphy employing columns filled with Dowex 1x8 to determine their specific radioactivities. Glucose, lac- tate, pyruvate, 3-hydroxybutyrate and acetoacetate were analyzed by standard enzymatic procedures [33]. The oxygen concentration in the venous perfusate was monitored continuously using a teflon-shielded micro- platinum electrode. The arterial concentrations of oxygen and substrates infused were measured at the end of each perfusion experiment in the absence of the liver. Metabolic rates were calculated from arterio-venous concentration differences and the con- stant flow rate and were referred to the wet weight of the liver.
Various substrates and the radioactive tracer were infused into the perfusion fluid prior to entering the liver. When the effects of fatty acids on pyruvate decarboxylation were studied, a solution of sodium octanoate or an emulsion of oleate-albumin (ratio 2 : 1 on a molar basis) were infused. In most experiments octanoate was chosen as the fatty acid species to avoid the various problems of water solubility and detergent properties of longer chain length fatty acids.
[l-'4C]Pyruvate (CFA 85, batch 83) was purchased from Ainersham Buchler Co. (Braunschweig). All chemicals were of reagent grade and were purchased from Merck Co. (Darmstadt), and Carl Roth Co. (Karlsruhe). Acetoacetate was prepared from the ethylester [34]. All enzymes and coenzymes used in the metabolite assays were products of Boehringer Mannheim GmbH (Tutzing). Octanoyl-D-( +)-carni- tine was a kind gift of Dr Denis McGarry (Dallas).
RESULTS
Experimental Protocol
Two types of experiments were performed: (A) con- tinuous infusion of [I -14C]pyruvate and measurement of the rates of 14C02 production under the various metabolic conditions (e.g. Fig. 1); (B) infusion of [l-'4C]pyruvate in trace amounts for 8 min (to reach a near isotopic equilibration between extracellular and intracellular pyruvate pools) and measurement of the washout kinetics of I4CO2 production following termination of the tracer infusion (e.g. Fig. 2). In experiments of type A, the rate of 14C02 production was used as an indicator for pyruvate decarboxylation by pyruvate dehydrogenase uncorrected for other decarboxylation processes which may contribute to the total I4CO2 production. On the other hand, the kinetic analyses in experiments of type B allow a cor- rection for the contribution of these decarboxylation processes as recently formulated by Scholz et al. [l]. In experiments of type A and B the dilution of the radioactivity of pyruvate inside the cell was determined using the specific radioactivity of lactate in the effluent perfusate as an indicator. It was considered that the
radioactivity of intracellular pyruvate should be more accurately reflected by lactate rather than pyruvate in the effluent perfusate. These corrections made by kinetic analyses and measurements of specific radio- activities provide a basis for the quantitation of the actual flux rates through the pyruvate deh ydrogenass reaction.
Difleventiul Effect of Octanoate on Rates qf 14C02 Production from [l-14C]Pyruvate
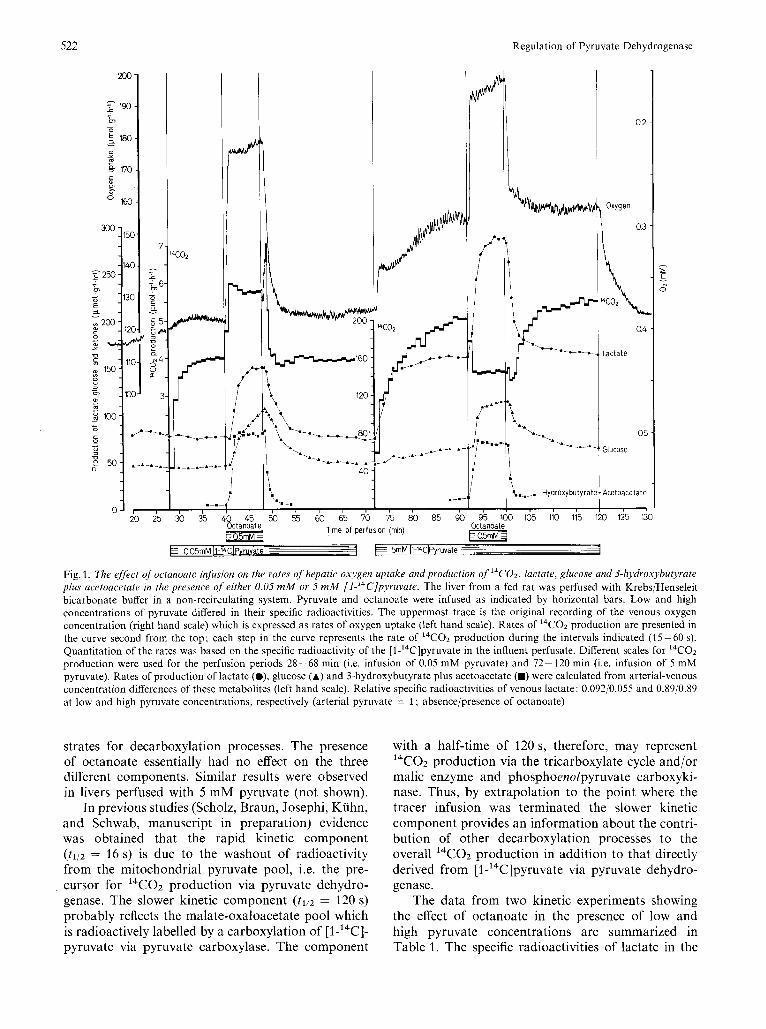
The experiment depicted in Fig. 1 illustrates the effect of octanoate on the rates of oxygen consumption, 14C02 production from [l -'4C]pyruvate, and the production of lactate, glucose and ketone bodies in the perfused liver from a fed rat. Octanoate was infused twice, first, at 0.05 mM and, second, at 5 mM pyruvate. Immediately following the initiation of octanoate infusion under both conditions, the rates of oxygen consumption and production of lactate and glucose were accelerated. The production of 3-hydroxybutyrate plus acetoacetate increased from a very low rate to rates approaching 80 pmol x g-' x h-'. The most interesting facet in this experiment was the observation that octanoate infusion resulted in a substantial increase in the rate of I4CO2 produc- tion at the low pyruvate concentration, while at 5 mM pyruvate octanoate decreased the rate of pyruvate decarboxylation. This apparent differential effect indicates that fatty acid addition to a complex meta- bolic system results in anything except an inhibition or inactivation of pyruvate dehydrogenase.
In the presence of 0.05 mM pyruvate, immediately following the termination of octanoate infusion, there occurred a rapid increase (with peak maximum around 50 s) followed by a rapid decrease in the 14C02 production. This spike was invariably observed at low pyruvate concentrations (see also Fig. 6-8). In the presence of 5 mM pyruvate, on the other hand, the inhibition of 14C02 production by octanoate was reversed with a time lag of about 2 min in contrast to the rapid reversal of changes in other metabolic parameters. These phenomena may be indicative of differences in the time constants of several regulatory processes which are affecting this system.
Rates of'Suhstrate Flux through the Pyvuvate Dehydvogenase Reaction
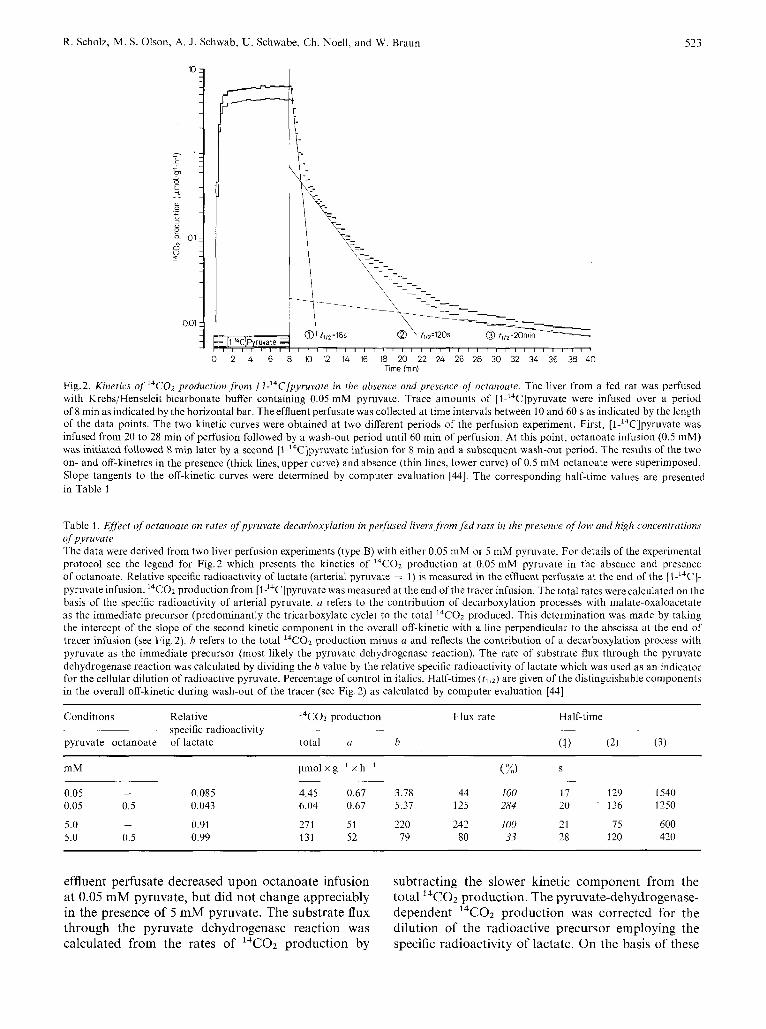
The kinetics of l4CO2 production from [1-'4C]- pyruvate following onset and termination of tracer infusion are shown in Fig.2. The liver was perfused with 0.05 mM pyruvate, first, in the absence and, second, in the presence of octanoate. The overall washout kinetics exhibit a superposition of at least 3 exponential kinetics with half-times of 16, 120 and 1200 s indicating different intracellular pools of sub-
7
Regulation of Pyruvate Dehydrogenase
.-.-/ 1 ‘,.-._. Hydroxybutyrate: I Acetoacetate
, , I # . . I t 4
50 55 $0 65 7 0 75 80 85 9b 95 160 165 110 115 120 125 1: g25;;2 &tar%te I rne of perfusion (rnin) g05mMi 005rnMll-W~Pyruvate =-- 1 E -~ 5rnM U-’CIPyruvate ---- .~
Fig. 1. The effect of octanoate infusion on the rates of hepatic oxygen uptake and production of I4CO2, lactate, glucose and 3-hydroxybutyrate plus acetoacetate in the presence of either 0.05 mM or 5 m M [l-’4C]pyruvate. The liver from a fed rat was perfused with KrebsjHenseleit bicarbonate buffer in a non-recirculating system. Pyruvate and octanoate were infused as indicated by horizontal bars. Low and high concentrations of pyruvate differed in their specific radioactivities. The uppermost trace is the original recording of the venous oxygen concentration (right hand scale) which is expressed as rates of oxygen uptake (left hand scale). Rates of 14C02 production are presented in the curve second from the top; each step in the curve represents the rate of 14C02 production during the intervals indicated (15-60 s). Quantitation of the rates was based on the specific radioactivity of the [l-’4C]pyruvate in the influent perfusate. Different scales for I4CO2 production were used for the perfusion periods 28-68 min (i.e. infusion of 0.05 mM pyruvate) and 72- 120 min (i.e. infusion of 5 mM pyruvate). Rates of production of lactate (O), glucose (A) and 3-hydroxybutyrate plus acetoacetate (m) were calculated from arterial-venous concentration differences of these metabolites (left hand scale). Relative specific radioactivities of venous lactate: 0.092j0.055 and 0.89/0.89 at low and high pyruvate concentrations, respectively (arterial pyruvate = 1 ; absencejpresence of octanoate)
strates for decarboxylation processes. The presence of octanoate essentially had no effect on the three different components. Similar results were observed in livers perfused with 5 mM pyruvate (not shown).
In previous studies (Scholz, Braun, Josephi, Kiihn, and Schwab, manuscript in preparation) evidence was obtained that the rapid kinetic component (t112 = 16 s) is due to the washout of radioactivity from the mitochondria1 pyruvate pool, i.e. the pre- cursor for 14C02 production via pyruvate dehydro- genase. The slower kinetic component (fl/2 = 120 s) probably reflects the malate-oxaloacetate pa01 which is radioactively labelled by a carboxylation of [1-14C]- pyruvate via pyruvate carboxylase. The component
with a half-time of 120 s, therefore, may represent 14C02 production via the tricarboxylate cycle and/or malic enzyme and phosphoenolpyruvate carboxyki- nase. Thus, by extrapolation to the point where the tracer infusion was terminated the slower kinetic component provides an information about the contri- bution of other decarboxylation processes to the overall 14C02 production in addition to that directly derived from [1 -14C]pyruvate via pyruvate dehydro- genase.
The data from two kinetic experiments showing the effect of octanoate in the presence of low and high pyruvate concentrations are summarized in Table 1. The specific radioactivities of lactate in the
R. Scholz, M. S. Olson, A. J . Schwab, U. Schwabe, Ch. Noell, and W. Braun 523
i-I 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34 36 38 40
Time fmin)
Fig.2. Kinetics of 14C02 production ,from [l-'4C]pyvuvate in the absence and presence of octanoute. The liver from a fed rat was perfuscd with Krebs/Henseleit bicarbonate buffer containing 0.05 mM pyruvate. Trace amounts of [l-14C]pyruvate were infused over a period of 8 min as indicated by the horizoiital bar, The effluent perfusate was collected at time intervals between 10 and 60 s as indicated by the length of the data points. The two kinetic curves were obtained at two different periods of the perfusion experiment. First, [l-14C]pyruvate was infused from 20 to 28 min of perfusion followed by a wash-out period until 60 min of perfusion. At this point, octanoate infusion (0.5 mM) was initiated followed 8 min later by a second [l-14C]pyruvate infusion for 8 min and a subsequent wash-out period. The results of the two on- and off-kinetics in the presence (thick lines, upper curve) andabsence (thin lines, lower curve) of 0.5 mM octanoate were superimposed. Slope tangents to the off-kinetic curves were determined by computer evaluation [44]. The corresponding half-time values are presented in Table I
Table 1. Efect ojoctanoate on rates ofpyruvate decarboxylution in perfused livers,from,fed ruts in the presence of low und high concentrations of pyruvate The data were derived from two liver perfusion experiments (type B) with either 0.05 mM or 5 mM pyruvate. For details of the experimental protocol see the legend for Fig.2 which presents the kinetics of l4COZ production at 0.05 mM pyruvate in the absence and presence of octanoate. Relative specific radioactivity of lactate (arterial pyruvate = 1) is measured in the effluent perfusate at the end of the [I-'"C]- pyruvate infusion. 14C02 production from [1'4C]pyruvate was measured at the end of the tracer infusion. The total rates were calculated on the basis of the specific radioactivity of arterial pyruvate. a refers to the contribution of decarboxylation processes with malate-oxaloacetate as the immediate precursor (predominantly the tricarboxylate cycle) to the total 14COz produced. This determination was made by taking the intercept of the slope of the second kinetic component in the overall off-kinetic with a line perpendicular to the abscissa at the end of tracer infusion (see Fig, 2). h refers to the total 14C02 production minus u and reflects the contribution of a decarboxylation process with pyruvate as the immediate precursor (most likely the pyruvate dehydrogenase reaction). The rate of substrate flux through the pyruvate dehydrogenase reaction was calculated by dividing the b value by the relative specific radioactivity of lactate which was used as an indicator for the cellular dilution of radioactive pyruvate. Percentage of control in italics. Half-times (t l lZ) are given of the distinguishable components in the overall off-kinetic during wash-out of the tracer (see Fig. 2) as calculated by computer evaluation [44]
Conditions Relative ~ ~
l4co2 production ~~~ specific radioactivity ~~
pyruvate octanoate of lactate total a b
~~
Flux rate Half-time
S ~ -
mM pmol x g-' x h I ( 70) ~~ ~-
0 05 - 0 085 445 067 378 44 100 17 129 1540 0 05 0 5 0 043 604 067 5 37 125 284 20 136 1250
5 0 - 0 91 271 51 220 242 100 21 75 600 5 0 0 5 0 99 131 52 79 80 33 28 120 420
effluent perfusate decreased upon octanoate infusion at 0.05 mM pyruvate, but did not change appreciably in the presence of 5 mM pyruvate. The substrate flux through the pyruvate dehydrogenase reaction was calculated from the rates of 14C02 production by
subtracting the slower kinetic component from the total 14C02 production. The pyruvate-dehydrogenase- dependent I4CO2 production was corrected for the dilution of the radioactive precursor employing the specific radioactivity of lactate. On the basis of these

524
c
Regulation of Pyruvate Dehydrogenase
I I I I 8 I 3 8 I I I 8 8 8 I I I 8 I I S ,
j **
\
0 1 0 01 0.2 0.3 0.4 0.5 0.6 0.7 0.8
[Octanoate] (mM)
Fig. 4. Octanoate eJfi'ct on rates ofpyruvute decarho.xylation: depen- dence upon the octanoate concentrations. The octanoate concentra- tions of the influent perfusate were varied between 0.02 and 0.7 mM in 3 experiments of type B similar to that presented in Fig.2. The pyruvate concentrations were either 0.25 mM (0, A) or 2 mM (0)
calculations, octanoate infusion caused a nearly 3-fold increase in the flux rates when the liver was perfused with 0.05 mM pyruvate. At high pyruvate concen- tration, the flux through the pyruvate dehydrogenase reaction was calculated to decrease to one third following octanoate infusion. Thus, despite some uncertainties regarding the validity of lactate as an
indicator for the intracellular dilution of the radio- active precursor, the data of Table 1 quantitate the observations presented in Fig. 1.
Dependence of the Differential Efiect of Fatty Acids upon the Pyruvute Concentration
In a series of experiments similar to that shown in Fig. 2, the influent pyruvate concentration was varied over a range between 0.02 and 5 mM. The fatty acid added was either octanoate or oleate. The changes in flux through the pyruvate dehydrogenase reaction following addition of fatty acids were calculated according to the procedure described in Table 1. The data expressed as percentage of control are summarized in Fig.3. As can be clearly seen, the infusion of fatty acids at pyruvate concentrations below 0.5 mM resulted in a stimulation while the same type of experiment performed at pyruvate concen- trations greater than 1 mM resulted in a restriction of the flux. The percentage stimulation and inhibition were strongly dependent upon the pyruvate concen- tration, i.e. the stimulatory or inhibitory effects became larger with decreasing or increasing pyruvate concentration, respectively. Octanoate and oleate exhibited similar effects on pyruvate decarboxylation, although their metabolism differs in many respects.
Dependence upon the Concentration of Fatty Acids
When the rate of I4CO2 production from [1-I4C]- pyruvate was studied at low (0.25 mM) or high (2 mM) pyruvate concentrations while the concentration of the infused octanoate was varied in the range between 0.08 and 0.8 mM the dose response curves shown in Fig. 4 were obtained. Half-maximal inhibitory and stimulatory effects were observed at octanoate con- centrations below 0.1 mM. It should be noted that changes in the redox state, oxygen consumption and ketogenesis exhibited a similar dependency upon the octanoate concentration (data not shown).
Effect of3-Hydroxybutyrate and Acetoacetate Infusion
In order to study the effect of the mitochondrial redox state on pyruvate oxidation 3-hydroxybutyrate or acetoacetate were infused. The metabolic fate of these substrates should be merely conversion to aceto- acetate or 3-hydroxybutyrate via the 3-hydroxybuty- rate dehydrogenase thus elevating or lowering the mitochondrial NADH/NAD' ratio with little effect on the acetyl-CoA/CoASH ratio.
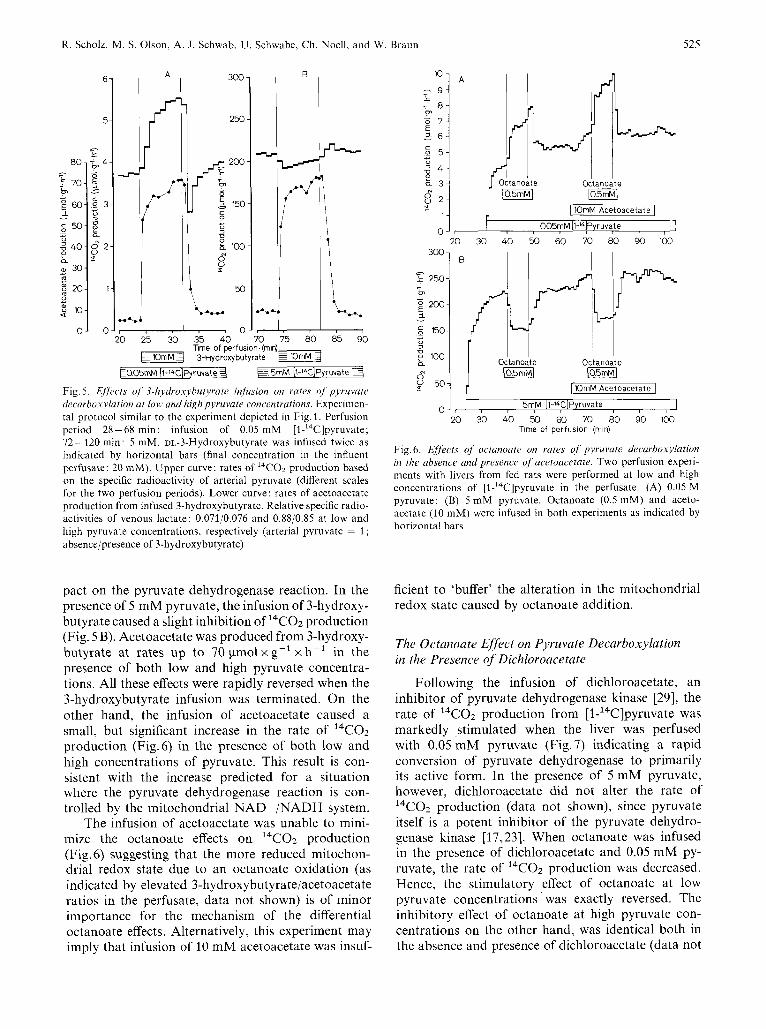
When 3-hydroxybutyrate was infused in the pres- ence of 0.05 mM pyruvate, the rate of I4CO2 produc- tion from [l-14C]pyruvate was nearly doubled (Fig. 5A). The results are opposite to those one would ex- pect if the mitochondrial redox state has a greater im-
R. Scholz. M. S. Olson, A. J. Schwab, U . Schwabe, Ch. Noell, and W. Braun 525
lo-
9 - i 7 8 - F" a 7 - E 2 6 -
5 5 -
z 4 - g 3 -
+ "
g 2 -
1 -
0 -
A
-
E005rnM \1-14C]Pyruvate 3 5rnM [1-l4C]Pyruvate 4
80-
f 70-
60- L U
50- = a : 4 0 - 9 2
a, 30-
- - - F " -
- 7
g r
9 20-
+ c m
0
'l
c
z 10-
0-
Fig. 5. E//kcts of 3-hydroxybutyrate infusion on rates oJ pjruvate decarbolcylation ut low and high pyruvate concentrations. Experimen- tal protocol similar to the experiment depicted in Fig. 1. Perfusion period 28-68 min: infusion of 0.05 mM [l-'4C]pyruvate; 72- 120 min: 5 mM. DL-3-Hydroxybutyrate was infused twice as indicated by horizontal bars (final concentration in the influent perfusate: 20 mM). Upper curve: rates of I4CO2 production based on the specific radioactivity of arterial pyruvate (different scales for the two perfusion periods). Lower curve: rates of acetoacetate production from infused 3-hydroxybutyrate. Relative specific radio- activities of venous lactate: 0.071/0.076 and 0.88/0.85 at low and high pyruvate concentrations, respectively (arterial pyruvate = 1 ; absence/presence of 3-hydroxybutyrate)
T~ 4
$ 3
1
0
pact on the pyruvate dehydrogenase reaction. In the presence of 5 mM pyruvate, the infusion of 3-hydroxy- butyrate caused a slight inhibition of 14C02 production (Fig. 5 B). Acetoacetate was produced from 3-hydroxy- butyrate at rates up to 70 pmolxg- 'xh- ' in the presence of both low and high pyruvate concentra- tions. All these effects were rapidly reversed when the 3-hydroxybutyrate infusion was terminated. On the other hand, the infusion of acetoacetate caused a small, but significant increase in the rate of 14C02 production (Fig.6) in the presence of both low and high concentrations of pyruvate. This result is con- sistent with the increase predicted for a situation where the pyruvate dehydrogenase reaction is con- trolled by the mitochondrial NAD + /NADH system.
The infusion of acetoacetate was unable to mini- mize the octanoate effects on l4CO2 production (Fig.6) suggesting that the more reduced mitochon- drial redox state due to an octanoate oxidation (as indicated by elevated 3-hydroxybutyrate/acetoacetate ratios in the perfusate, data not shown) is of minor importance for the mechanism of the differential octanoate effects. Alternatively, this experiment may imply that infusion of 10 mM acetoacetate was insuf-
Octanoate Octanoate
[lOrnM Acetoacetate 1
30 40 50 60 70 80 90 100
I 1 n" (05mMJ
Octanoate Octanoate
[IOmM Acetoacetate I 5mM [l-l4C]Pyruvate
20 30 40 50 60 70 80 90 100 Time of perfusion (min)
0 5 0 1 r ( , , , , , , , , I
Fig. 6. Effects of octunoate on rates of pyruvate decurboxylation in the absence and presence of acetoacetate. Two perfusion experi- ments with livers from fed rats were performed at low and high concentrations of [l-'4C]pyruvate in the perfusate. (A) 0.05 M pyruvate; (B) 5 mM pyruvate. Octanoate (0.5 mM) and aceto- acetate (10 mM) were infused in both experiments as indicated by horizontal bars
ficient to 'buffer' the alteration in the mitochondrial redox state caused by octanoate addition.
The Octanoate Effect on Pyruvate Decarboxylation in the Presence of Dichloroacetate
Following the infusion of dichloroacetate, an inhibitor of pyruvate dehydrogenase kinase [29], the rate of 14C02 production from [l-'4C]pyruvate was markedly stimulated when the liver was perfused with 0.05 mM pyruvate (Fig.7) indicating a rapid conversion of pyruvate dehydrogenase to primarily its active form. In the presence of 5 mM pyruvate, however, dichloroacetate did not alter the rate of 14 CO2 production (data not shown), since pyruvate itself is a potent inhibitor of the pyruvate dehydro- genase kinase [17,23]. When octanoate was infused in the presence of dichloroacetate and 0.05 mM py- ruvate, the rate of 14C02 production was decreased. Hence, the stimulatory effect of octanoate at low pyruvate concentrations was exactly reversed. The inhibitory effect of octanoate at high pyruvate con- centrations on the other hand, was identical both in the absence and presence of dichloroacetate (data not

526 Regulation of Pyruvate Dehydrogenase
1 I I I I
I I Octanoate
I 3rnM Dichloroacetate I 005rnM [1-"+C]Pyruvate
20 30 40 50 60 70 80 90 100 Trne of perfusion (rnin)
0 I , / , , , , , , , 1
Fig. 7. Effects of octanoate on rates of oxygen uptake (upper trace) and ''C02 production from [l-'4C]pyruvate in the absence and presence of dichloroacetate. This perfusion experiment was per- formed essentially as described in the legend to Fig. 1. The influent perfusate contained 0.05 mM [l-14C]pyruvate. Octanoate (0.5 mM) and dichloroacetate (3 mM) were infused as indicated by horizontal bars
shown). One may conclude, therefore, that fatty acids inhibit pyruvate oxidation only under conditions where pyruvate dehydrogenase is converted primarily into its active form.
Substitution of 3-hydroxybutyrate for octanoate in an experiment similar to that described in Fig.7 yielded exactly the same result, i.e. 3-hydroxybutyrate stimulated pyruvate decarboxylation in the absence of dichloroacetate but strongly inhibited I4CO2 pro- duction in its presence (data not shown).
Effects of L-Carnitine
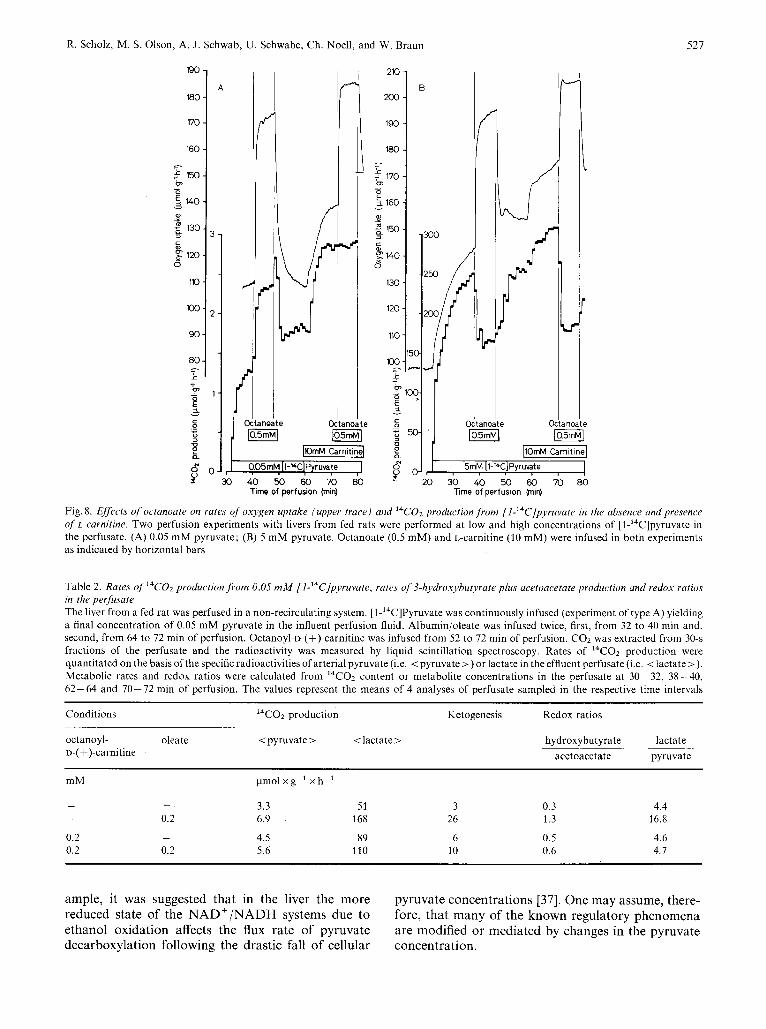
The infusion of L-carnitine increased the rates of oxygen consumption and 14C02 production from [l-'4C]pyruvate in the presence of both low and high pyruvate concentrations (Fig. 8). Moreover, no effect on pyruvate decarboxylation was observed when octanoate was infused in the presence of L-carnitine and 0.05 mM pyruvate, suggesting that the maximal stimulation due to fatty acids is already achieved by L-carnitine. On the other hand, the inhibitory effect of octanoate at 5 mM pyruvate was unaffected by L-carnitine.
Effects of Octanoyl-D-( +)-Carnitine
The question arises if the stimulatory effect of fatty acids on pyruvate decarboxylation at low pyru- vate concentrations is due to the mere presence of the fatty acid (i.e. detergent effect) or if the mechanism involves the activation or oxidation of the fatty acid. Octanoyl-D-( +)-carnitine was considered a suitable inhibitor [35] to distinguish between the two possi- bilities, since derivatives of D-( +)-carnitine are known to inhibit the transport of the activated form of long- chain-length fatty acids into the mitochondria, thus, preventing P-oxidation [36]. As shown in Table 2, infusion of oleate caused a large increase in the rate of ketogenesis and the 3-hydroxybutyrate/acetoacetate ratio accompanied by a 3-fold stimulation of pyruvate decarboxylation. These effects were diminished signif- icantly when oleate was infused in the presence of octanoyl-D-(4)-carnitine indicating that metabolism of the fatty acid is a prerequisite for its stimulatory effect on pyruvate decarboxylation.
DISCUSSION
Studies of the Regulation of Pyruvate Dehydrogenase under Conditions in vivo and in vitro
The initial objectives of the present study were to design experiments using the isolated perfused liver in which the various regulatory characteristics of the pyruvate dehydrogenase multienzyme complex could be demonstrated under conditions near in vivo. The significance of these objectives derives from the fact that most of our current understanding of the regula- tory properties of pyruvate dehydrogenase has been formulated on the basis of experiments performed using the purified enzyme complex and isolated mito- chondria. Only relatively few studies have been con- ducted using intact organs or cells and most of these investigations have been concerned with hormonal or nutritional effects on the interconversion of pyru- vate dehydrogenase between its active and inactive forms by measuring the activity state of the enzyme.
Moreover, most experiments in vitro concerned with the regulation of pyruvate metabolism were performed under highly artificial conditions with respect to the substrate concentrations employed. Pyruvate concentrations generally reported in the literature between 2 and 10 mM are at least one to two orders of magnitude higher than the concentra- tions in vivo. Pyruvate, however, not only serves as a substrate for pyruvate dehydrogenase (K, zz 0.02 mM) and pyruvate carboxylase (K, zz 0.4mM) but it acts also as an activator of the dehydrogenase by inhib- iting the inactivating kinase (Ki zz 1 mM). Under con- ditions in vivo, therefore, the flux through the pyruvate dehydrogenase reaction may be strongly influenced by changes in the pyruvate concentration. For ex-
R. Scholz, M. S. Olson, A. J. Schwab, U. Schwabe, Ch. Noell, and W. &dun 527
190 - A
180
no -
-
160 - =-. .$ 150- ?
5 140 - 2 130- 3 3 -
- - 0 1
c
g 120- 0
10 -
loo-
90 -
60 -
2 -
- - r
b - 1 -
!
210 -
m-
190 -
180 -
B
- i - 170 - I- m
I
Fig.8. Effects of octanoate on rates of oxygen uptake (upper trace) and l4Co2 production from [I-'4C]pyruvate in the absence and presence of L-carnitine. Two perfusion experiments with livers from fed rats were performed at low and high concentrations of [l-14C]pyruvate in the perfusate. (A) 0.05 mM pyruvate; (B) 5 mM pyruvate. Octanoate (0.5 mM) and L-carnitine (10 mM) were infused in both experiments as indicated by horizontal bars
Table 2. Rates of l4C02 production from 0.05 mM f l-14C]pyruvate, rates of 3-hydroxyhutyrute plus acetoacetate production and redox ratios in the perfusate The liver from a fed rat was perfused in a non-recirculating system. [l-'4C]Pyruvate was continuously infused (experiment of type A) yielding a final concentration of 0.05 mM pyruvate in the influent perfusion fluid. Albumin/oleate was infused twice, first, from 32 to 40 min and, second, from 64 to 72 min of perfusion. Octanoyl-o-(+)-carnitine was infused from 52 to 72 min of perfusion. COz was extracted from 30-s fractions of the perfusate and the radioactivity was measured by liquid scintillation spectroscopy. Rates of I4CO2 production were quantitated on the basis ofthe specific radioactivities of arterial pyruvate (i.e. < pyruvate >) or lactate in the effluent perfusate (i.e. <lactate >).
Conditions l4Co2 production Ketogenesis Redox ratios ~ ._ . . . ocranoyi- oleate < pyruvate > < lactate > D-( +)-carnitine
lactate ~~~~ -
hydroxybutyrate ~~
acetoacetate pyruvate
mM pmol x g-' x h-'
3.3 51 3 0.3 - 0.2 6.9 168 26 1.3
- 4.5 89 6 0.5 0.2
- -
- - _ _ ^ ^
4.4 16.8
4.6 . _ U.L 0.L 5.6 110 10 0.6 4.7
ample, it was suggested that in the liver the more reduced state of the NAD+/NADH systems due to ethanol oxidation affects the flux rate of pyruvate decarboxylation following the drastic fall of cellular
pyruvate concentrations [37]. One may assume, there- fore, that many of the known regulatory phenomena are modified or mediated by changes in the pyruvate concentration.

528 Regulation of Pyruvate Dehydrogenase
It is generally accepted that pyruvate oxidation is inhibited by fatty acids. However, this concept is derived mainly from experiments with isolated mito- chondria incubated with artificially high pyruvate concentrations. One of our objectives was to re-eva- luate the fatty acid effect on pyruvate decarboxylation at physiologically relevant pyruvate concentrations (i.e. below 0.1 mM).
Differential Effects of Fatty Acids on Pyruvate Metabolism : Fatty Acids Inhibit Pq’ruvate Decarboxylation at High Pyruvate Concentrations but Stimulate at Low Concentrations
Our studies with perfused rat livers confirm the present notion of the fatty acid effect on pyruvate oxidation : In the presence of artificially high pyruvate concentrations (i.e. 2 mM) the infusion of fatty acids caused a decrease in the rate of pyruvate decarboxyla- tion as measured by 14C02 production from [1-I4C]- pyruvate. However, our data also indicate that this effect is not a general characteristic, since the phenom- enon strongly depends on the experimental conditions, i.e. the pyruvate concentration. When livers were perfused with low pyruvate concentrations, a substan- tial increase in the decarboxylation rate due to fatty acid infusion was observed.
Great care was taken in this study to avoid the danger of misinterpreting the experimental results because of the problems related to the intracellular dilution of radioactive substrates and rigorous identi- fication of radioactive products in metabolic systems. The stimulatory effect of fatty acids was clearly demonstrated by the total I4CO2 production from [l-14C]pyruvate. It became more evident when rates of 14C02 production were corrected, first, for the dilution of the radioactivity of the precursor inside the cell using the specific radioactivity of effluent lactate as an indicator and, second, for the contri- bution of other decarboxylation processes apart from the pyruvate dehydrogenase reaction employing kine- tic analyses (see Table 1). The kinetic data suggest that decarboxylations not dependent on pyruvate dehydrogenase did not contribute more than 20 % of the total 14C02 production and that these processes were not significantly altered by octanoate. Changes in the I4CO2 production from [ l-14C]pyruvate, there- fore, reflect mainly changes in the flux through the pyruvate dehydrogenase reaction.
Probably, the present study represents the first documented evidence in an intact metabolic system that fatty acid administration causes any other effect than an inhibition or inactivation of the pyruvate dehydrogenase complex although a recent paper of Taylor et al. [19] could imply a similar conclusion. It was reported that in adipose tissue the inhibitory
effect of fatty acids on pyruvate decarboxylation was almost undetectable when the pyruvate concentra- tions were lowered to 1 mM.
Mechanisms of the Stimulutory and Inhibitory Effects of Octanoate on Pyruvate Decarboxylation
Several experiments were performed during the course of the present study to elucidate the mechanism of the stimulatory and inhibitory effects of octanoate on pyruvate decarboxylation. First, acetoacetate was co-infused with octanoate in order to ‘buffer’ any redox changes due to octanoate oxidation. However, both effects of octanoate were unchanged in the presence of acetoacetate. Second, co-infusion of L-carnitine in an attempt to ‘buffer’ changes in the acetyl-CoA/CoASH ratio upon fatty acid addition was essentially without effect on the inhibition of 14C02 production due to octanoate. However, L-carni- tine itself stimulated I4CO2 production which could not be enhanced further by octanoate in the presence of low pyruvate concentrations. Third, when pyruvate dehydrogenase was converted nearly completely into its active form by dichloroacetate, octanoate infusion caused an inhibition of pyruvate decarboxylation in the presence of both low and high pyruvate concen- trations, thus, reversing the stimulatory effect of octanoate at 0.05 mM pyruvate. Fourth, 3-hydroxy- butyrate stimulated the rate of decarboxylation in the presence of low pyruvate concentrations similar to the effect of octanoate, despite the fact that infusion of 3-hydroxybutyrate resulted in a more reduced mito- chondrial redox state and, therefore, should have depressed pyruvate oxidation. However, in the pres- ence of high pyruvate concentrations octanoate and 3-hydroxybutyrate differed in that the latter com- pound had only a slight effect on pyruvate decarboxy- lation.
These experimental data are difficult to explain in terms of feedback inhibition of pyruvate dehydro- genase by NADH and acetyl-CoA or interconversion of the enzyme between its active and inactive forms. It appears unlikely on the basis of these data that changes in the mitochondrial NADH/NAD’ and acetyl-CoA/CoASH ratios are involved in the stimula- tory effect. Moreover, their involvement in the inhib- itory effect is unlikely, too, since acetoacetate and L-carnitine failed to minimize the octanoate effect at high pyruvate concentrations. On the other hand, changes in the mitochondrial ATP/ADP ratio (as a consequence of enhanced fatty acid oxidation or inhibition of adenine nucleotide transport [20,38]) could explain the inhibitory but not the stimulatory effect of octanoate. Thus, the present data failed to provide a clear cut explanation for the mechanism of the observed phenomena.

R. Scholz, M. S. Olson, A. J. Schwab, U . Schwabe, Ch. Noell, and W. Braun 529
Possible Role of Pyruvate Transport as a Rate-Controlling Factor in Pyruvate Metabolism
However, one observation which suggests a pos- sible mechanism of the stimulatory effect of fatty acids on pyruvate oxidation is that the infusion of 3-hydroxybutyrate was capable of enhancing the rate of pyruvate decarboxylation at low substrate concen- trations but, in contrast to the findings with octanoate, had almost no effect at high pyruvate concentrations (see Fig. 5). In liver, 3-hydroxybutyrate is mainly converted into acetoacetate which is also a primary product of octanoate oxidation. Acetoacetate leaves the mitochondrial compartment probably via a carrier- mediated transport [39]. On the other hand, pyruvate is transported into the mitochondria also via a mono- carboxylate translocase [39 - 431 which involves a pyruvate : hydroxyl exchange. Papa and Paradies [39] demonstrated recently that hydroxyl ions can be effec- tively substituted by acetoacetate.
It is our proposal that the stimulation of pyruvate decarboxylation by fatty acids or 3-hydroxybutyrate at low pyruvate concentrations is a result of an accelerated entry of pyruvate into the mitochondrial compartment in exchange for acetoacetate which is generated intramitochondrially at high rates under these conditions. Once translocated into the mito- chondria, pyruvate serves as the substrate of pyruvate dehydrogenase and as an activator by retarding the kinase-mediated inactivation of the multienzyme com- plex. At low pyruvate concentrations, this proposed transport-mediated effect of fatty acids must be exceedingly potent to overcome the known inhibitory actions of fatty acid oxidation on the inhibitionlinacti- vation of pyruvate dehydrogenase. At high pyruvate concentrations, only the inhibitory properties (likely mediated by the increased NADH/NAD + and acetyl- CoA/CoASH ratios or a decreased ATP/ADP ratio) are observed as pyruvate transport seems to be maximal.
The stimulation of pyruvate decarboxylation fol- lowing the addition of dichloroacetate in the presence of low pyruvate concentrations (see Fig. 7), however, is difficult to explain in view of our working hypothesis. It is envisioned that the small portion of pyruvate de- hydrogenase present in the active form is operating at maximal velocity due to its high substrate affinity ( K , < 0.02 mM pyruvate). Pyruvate transport across the mitochondrial membrane, therefore, would be sufficient to maintain the necessary substrate concen- tration and also to allow even a higher flux when the multienzyme complex is totally converted into its active form by dichloroacetate. Presuming that the stimulatory effect of octanoate is caused by an enhanced pyruvate transport due to acetoacetate egress, one has to assume that the mitochondrial pyruvate concentration increases considerably result-
ing in an activation of the enzyme via inhibition of the inactivating kinase (K, z 1 mM pyruvate). Further, it is proposed that when octanoate is added under conditions where pyruvate dehydrogenase is fully activated either by extremely high extracellular pyru- vate concentrations or by tlichloroacetate, the active form of the enzyme could be inhibited by acetyl-CoA and NADH. Thus, product:; of octanoate metabolism, i.e. (a) acetyl-CoA and NA.DH and (b) acetoacetate, exert opposing effects on pyruvate oxidation. Whether the effects of acetyl-CoA and NADH or that of aceto- acetate predominates, depends on the experimental condition.
Physiological Relevance j i ) r a Possible Interaction of Ketogenesis and Pyruvatc Metabolism
An accelerated conversion of pyruvate to acetyl- CoA during fatty acid oxidcation seems to be contrary to any principle of metabolic economy. It is possible, however, that the pyruva te : acetoacetate exchange system evolved as a regulatory process designed to facilitate pyruvate entry info the mitochondrial com- partment during periods of starvation, i.e. limited supply of carbohydrates and rapid oxidation of fatty acids associated with ketogenesis. An increase in mitochondrial pyruvate concentrations should stimu- late pyruvate carboxylatiori (K , of isolated pyruvate carboxylase zz 0.4 mM pyruvate [43]) with subsequent acceleration of gluconeogenesis. The question, how- ever, cannot be answered whether the increased flux through the pyruvate dehydrogenase reaction repre- sents a real regulatory effect of fatty acids or if the experiments with the isolated perfused rat liver provide only insight into a potential interaction which is suf- ficiently counteracted in the intact organism.
In conclusion, the present study has characterized a metabolic situation in the perfused liver in which fatty acid oxidation results in an acceleration of pyruvate decarboxylation. Neither this observation nor our interpretation shoiild suggest that the previ- ously reported mechanisms for the regulation of pyruvate dehydrogenase are inoperative. This study does accentuate the necessity to define carefully all potential modifiers or regulatory processes in a highly complex metabolic system before general statements are made concerning how a given enzyme may be regulated in vivo.
This work was supported by a grant from Deutsche Forschungs- genzeinschuft, SFB 51 “Mrdizinische Molrkulurhiologie und Bio- chemie”, D/7. M.S.O. was a recipient of an NIH Fogarty Internatio- nal Center Senior Fellowship. Computations were performed at the Leihniz-Rechenzentrum der Buyeri.i.chen Akudemie der Wissenschuf’ ten, Munich, We thank Dr Ennci Jorn for writing the computer programme for half-time evaluation. Parts of this work were presented elsewhere [I -31.

530 R. Scholz, M. S. Olson, A. J. Schwab, U. Schwabe, Ch. Noell, and W. Braun: Regulation of Pyruvate Dehydrogenase
REFERENCES
1. Scholz, R., Braun, W., Kuhn, A. & Zimmer, P. (1976) Hoppe-
2. Scholz, R., Braun, W. & Kuhn, A. (1976) 20th Int. Congr.
3. Olson, M. S. & Scholz, R. (1977) Fed. Proc. 36, 875. 4. Garland, P. B. & Randle, P. J. (1964) Biochem. J . 91, 6c-7c. 5. Bremer, J. (1969) Eur. J . Biochem. 8, 535-540. 6. Tsai, C. S., Burgett, M. W. & Reed, L. J . (1973) J . Biol. Chem.
7. Linn, T. C., Pettit, F. H. & Reed, L. J. (1969) Proc. Natl Acad.
8. Linn, T. C., Pettit, F. H., Hucho, F. & Reed, L. J. (1969) Proc.
9. Wieland, 0. & Siess, E. (1970) Proc. Nut1 Acad. Sci. U.S.A. 65,
10. Pettit, F. H., Pelley, J. W. & Reed, L. J. (1975) Biochem. Bio-
11. Batenburg, J. J. & Olson, M. S. (1976) J . Bid. Chem. 251,
12. Cooper, R. H., Randle, P. J. & Denton, R. M. (1975) Nature (Lond.), 257,808-809.
13. Kerbey, A. L., Rdndk, P. J . , Copper, R . H., Whitehouse, S., Pask, H. T. & Denton, R. M. (1976) Biochem. J. 154, 327- 348.
14. Pettit, F. H., Rocher, T. E. & Reed, L. J . (1972) Biochern. Biophys. Res. Commun. 49, 563 - 571.
15. Schuster, S. M. & Olson, M. S. (1974) J. Biol. Chem. 249,
16. Roche, T. E. & Reed, L. J. (1974) Biochem. Biophys. Res. Com-
17. Portenhauser, R. & Wieland, 0. (1972) Eur. J . Biochem. 31,
18. Martin, B. R., Denton, R. M., Pask, H. T. & Randle, P. J.
19. Taylor, S. I., Mukherjee, C. & Jungas, R. L. (1975) J . Biol.
20. Wieland, 0 . & Portenhauser, R. (1974) Eur. J . Biochem. 45,
21. Walajtys, E. I., Gottesman, D. P. & Williamson, J. R. (1974)
Seyler’s Z. Physiol. Chem. 357, 279-280.
Biochem., Abstr. p. 388.
248, 8348-8352.
Sci. U.S.A. 64, 234-241.
Natl Acad. Sci. U.S.A. 64, 227-234.
947- 954.
phys. Res. Commun. 65, 575- 582.
1 364 - 1370.
7159-7165.
mun. 59, 1341 - 1348.
308-314.
(1972) Biochem. 1. 129,763-713.
Chem. 250,2028 - 2035.
577- 588.
J . Biol. Chem. 249, 1857-1865.
22. Hansford, R. G. (1976) J . Bid. Chem. 251, 5483-5489. 23. Hucho, F., Randall, D. D., Roche, T. E., Burgett, M. W.,
Pelley, J. W. & Reed, L. J. (1972) Arch. Biochem. Biophys.
24. Chiang, P. K.&Sacktor, B.(1975)J. Bid. Chern.250,339-348. 25. Wieland, O., von Fnncke, H. & Loffler, G. (1971) FEBS Lett.
26. Patzelt, E., Loffler, G. & Wieland, 0. (1973) Eur. J . Biochem. 33, 117- 122.
27. Taylor, S. I., Mukherjee, E. & Jungas, R. L. (1973) J . Biol. Chem. 248,73 - 81.
28. Batenburg, J. J. & Olson, M. S. (1975) Biochem. Biophys. Res. Commun. 66,533-540.
29. Whitehouse, S., Cooper, R. H. & Randle, P. J. (1974) Biochem. J. 141,761 -774.
30. Scholz, R. & Bucher, Th. (1965) in Control of Energy Metah- olism (Chance, B., Estabroock, R. W. & Williamson, J. R., eds) pp. 393-414, Academic Press, New York.
31. Scholz, R., Hansen, W. & Thurman, R. G. (1973) Eur. f. Biochem. 38,64 - 72.
32. Krebs, H. A. & Henseleit, K. (1932) Hoppe-Seyler’s 2. Physiol. Chem. 210,33 - 66.
33. Bergmeyer, H. U., ed. (1974) Methods of Enzymatic Analyses 2nd edn, Verlag Chemie, Weinheim.
34. Hall, L. M. (1 962) Anal. Biochem. 3, 75 - 80. 35. McGarry, J . D. &Foster, D. W. (1974) Diabetes, 23,485-493. 36. Delisle, G. &Fritz, I. B. (1967) Proc. Nut1 Acad. Sci. U.S.A. 58,
790 ~ 797. 37. Scholz, R., Kaltstein, A,, Schwabe, U. & Thurman, R. G.
(1964) in Alcohol and Aldehyde Metabolizing Systems (Thur- man, R. G., Yonetani, T. , Williamson, J . R. & Chance, B., eds) pp. 315 - 325, Academic Press, New York.
38. Siess, E. A. & Wieland, 0. H. (1976) Biochem. J . 1 5 6 , 9 1 ~ 102. 39. Papa, S. & Paradies, G. (1974) Eur. J . Biochem. 49, 265-274. 40. Papa, S., Francavilla, A,, Paradies, G. & Meduri, B. (1971)
41. Halestrap, A. P. & Denton, R. M. (1974) Biochem. J . 138,
42. Halestrap, A. P. (1975) Biochem. J . 148, 85-96. 43. Scrutton, M. C., Olmsted, M. R. & Utter, M. F. (1969) Methods
44. Brdess, D. (1967) Computing, 2, 309-319.
151, 328 - 340.
15,295 - 298.
FEBS Lett. 12,285-288.
313- 316.
Enzymol. 13, 235 -249.
R. Scholz, A. J. Schwab, U. Schwabe, Ch. Noell, and W. Braun, Institut fur Physiologische Chemie, Physikalische Biochemie und Zellbiologie der Ludwig-Maximilians-UniversitPt Munchen, Goethestrasse 33, D-8000 Munchen 2, Federal Republic of Germany
M. S. Olson, Department of Biochemistry, University of ,Texas Health Science Center at San Antonio, San Antonio, Texas, U.S.A. 78284
Copyright © 2022 FDOKUMEN








![Conversion of Hyperpolarized [1-13C]Pyruvate in Breast ...](https://static.fdokumen.com/doc/165x107/6328a69be491bcb36c0bdd22/conversion-of-hyperpolarized-1-13cpyruvate-in-breast-.jpg)











