
Self-assembly of ciprofloxacin and a tripeptide into an antimicrobial nanostructured hydrogel
10
Self-assembly of ciprofloxacin and a tripeptide into an antimicrobial nanostructured hydrogel Silvia Marchesan a, * , Yue Qu b , Lynne J. Waddington a , Christopher D. Easton a , Veronica Glattauer a , Trevor J. Lithgow b , Keith M. McLean a , John S. Forsythe c , Patrick G. Hartley a a CSIRO Materials Science and Engineering, Bayview Avenue, Clayton, Victoria 3053, Australia b Department of Biochemistry and Molecular Biology, Monash University, Victoria 3800, Australia c Department of Materials Engineering, Monash University, Victoria 3800, Australia article info Article history: Received 21 November 2012 Accepted 26 January 2013 Available online 17 February 2013 Keywords: Self-assembly Hydrogel Peptide Drug release abstract This work reports the self-assembly of a sparingly soluble antibiotic (ciprofloxacin) and a hydrophobic tripeptide ( D LeuePheePhe) into supramolecular nanostructures that yield a macroscopic hydrogel at physiological pH. Drug incorporation results in modified morphology and rheological properties of the self-assembled hydrogel. These changes can be correlated with intermolecular interactions between the drug and the peptide, as confirmed by spectroscopic analysis (fluorescence, circular dichroism, IR). The drug appears bound within the hydrogel by non-covalent interactions, and retains its activity over a prolonged release timescale. Antimicrobial activity of the ciprofloxacin-peptide self-assembled hydrogel was evaluated against Staphylococcus aureus, Escherichia coli, and a clinical strain of Klebsiella pneumoniae. Interestingly, the peptide hydrogel alone exhibited a mild anti-bacterial activity against Gram-negative bacteria. While toxic to bacteria, no major cytotoxicity was seen in haemolysis assays of human red blood cells or in mouse fibroblast cell cultures. This new approach of drug incorporation into the nanostructure of a simple tripeptide hydrogel by self-assembly may have important applications for cost-effective wound dressings and novel antimicrobial formulations. Crown Copyright Ó 2013 Published by Elsevier Ltd. All rights reserved. 1. Introduction Today there is an intense research effort directed towards the development of cost-effective, antimicrobial materials for topical applications such as wound dressings [1,2]. Nanomaterials offer the advantage of high surface area to volume ratio and the possibility to design their physical properties (such as porosity, mechanical strength etc.) to match natural tissue, and to selectively load drug molecules for their controlled release at the wound site [3]. In particular, the use of nanotechnology to develop innovative hydrogels is attractive for wound healing applications [4] since it combines the advantages described above with hydrogel properties known to accelerate the healing process, e.g. the moist and occlusive environment they provide, as well as their ability to allow for cell attachment and infiltration [1,2,5]. Tissue regeneration at the site of injury is often hampered by infection, which can be prevented or eradicated by the sustained release of relevant antimicrobials. Different approaches exist to prevent bacteria colonisation, such as the use of metal nano- particles [6], or the sustained release of antibiotics at a specific site of application [7]. In particular, the fluoroquinolone ciprofloxacin is one of the most effective antibiotics used clinically and has become the gold standard for a variety of topical applications, such as skin and eye infections. Drug formulations capable of sustained release are highly sought after, providing a means for drug concentration to be maintained for long periods of time above the minimum inhibitory concentration (MIC) for relevant pathogens. In order to address these requirements, formulations such as liposomes and gels are typically studied, as they offer good vehicles for the incorporation of this hydrophobic and sparingly soluble drug [7e10]. For delivery applications, hydrogels offer convenient drug de- livery matrices. They are typically composed of natural polymers (e.g. alginate), or crosslinked synthetic macromolecules (e.g. * Corresponding author. Present address: Center of Excellence for Nano- structured Materials, INSTM, Unit of Trieste, Dipartimento di Scienze Chimiche e Farmaceutiche, Università degli Studi di Trieste, Piazzale Europa 1, 34127 Trieste, Italy. E-mail addresses: [email protected], [email protected] (S. Marchesan). Contents lists available at SciVerse ScienceDirect Biomaterials journal homepage: www.elsevier.com/locate/biomaterials 0142-9612/$ e see front matter Crown Copyright Ó 2013 Published by Elsevier Ltd. All rights reserved. http://dx.doi.org/10.1016/j.biomaterials.2013.01.096 Biomaterials 34 (2013) 3678e3687
-
Upload
triestearchitettura -
Category
Documents
-
view
4 -
download
0
Transcript of Self-assembly of ciprofloxacin and a tripeptide into an antimicrobial nanostructured hydrogel

at SciVerse ScienceDirect
Biomaterials 34 (2013) 3678e3687
Contents lists available
Biomaterials
journal homepage: www.elsevier .com/locate/biomateria ls
Self-assembly of ciprofloxacin and a tripeptide into an antimicrobialnanostructured hydrogel
Silvia Marchesan a,*, Yue Qu b, Lynne J. Waddington a, Christopher D. Easton a, Veronica Glattauer a,Trevor J. Lithgowb, Keith M. McLean a, John S. Forsythe c, Patrick G. Hartley a
aCSIRO Materials Science and Engineering, Bayview Avenue, Clayton, Victoria 3053, AustraliabDepartment of Biochemistry and Molecular Biology, Monash University, Victoria 3800, AustraliacDepartment of Materials Engineering, Monash University, Victoria 3800, Australia
a r t i c l e i n f o
Article history:Received 21 November 2012Accepted 26 January 2013Available online 17 February 2013
Keywords:Self-assemblyHydrogelPeptideDrug release
* Corresponding author. Present address: Centestructured Materials, INSTM, Unit of Trieste, DipartimFarmaceutiche, Università degli Studi di Trieste, PiazItaly.
E-mail addresses: marchesan.silvia@gmail.(S. Marchesan).
0142-9612/$ e see front matter Crown Copyright � 2http://dx.doi.org/10.1016/j.biomaterials.2013.01.096
a b s t r a c t
This work reports the self-assembly of a sparingly soluble antibiotic (ciprofloxacin) and a hydrophobictripeptide (DLeuePheePhe) into supramolecular nanostructures that yield a macroscopic hydrogel atphysiological pH. Drug incorporation results in modified morphology and rheological properties of theself-assembled hydrogel. These changes can be correlated with intermolecular interactions between thedrug and the peptide, as confirmed by spectroscopic analysis (fluorescence, circular dichroism, IR). Thedrug appears bound within the hydrogel by non-covalent interactions, and retains its activity overa prolonged release timescale. Antimicrobial activity of the ciprofloxacin-peptide self-assembledhydrogel was evaluated against Staphylococcus aureus, Escherichia coli, and a clinical strain of Klebsiellapneumoniae. Interestingly, the peptide hydrogel alone exhibited a mild anti-bacterial activity againstGram-negative bacteria. While toxic to bacteria, no major cytotoxicity was seen in haemolysis assays ofhuman red blood cells or in mouse fibroblast cell cultures. This new approach of drug incorporation intothe nanostructure of a simple tripeptide hydrogel by self-assembly may have important applications forcost-effective wound dressings and novel antimicrobial formulations.
Crown Copyright � 2013 Published by Elsevier Ltd. All rights reserved.
1. Introduction
Today there is an intense research effort directed towards thedevelopment of cost-effective, antimicrobial materials for topicalapplications such as wound dressings [1,2]. Nanomaterials offer theadvantage of high surface area to volume ratio and the possibility todesign their physical properties (such as porosity, mechanicalstrength etc.) to match natural tissue, and to selectively load drugmolecules for their controlled release at the wound site [3]. Inparticular, the use of nanotechnology to develop innovativehydrogels is attractive for wound healing applications [4] since itcombines the advantages described abovewith hydrogel propertiesknown to accelerate the healing process, e.g. the moist and
r of Excellence for Nano-ento di Scienze Chimiche e
zale Europa 1, 34127 Trieste,
com, [email protected]
013 Published by Elsevier Ltd. All
occlusive environment they provide, as well as their ability to allowfor cell attachment and infiltration [1,2,5].
Tissue regeneration at the site of injury is often hampered byinfection, which can be prevented or eradicated by the sustainedrelease of relevant antimicrobials. Different approaches exist toprevent bacteria colonisation, such as the use of metal nano-particles [6], or the sustained release of antibiotics at a specific siteof application [7]. In particular, the fluoroquinolone ciprofloxacin isone of the most effective antibiotics used clinically and has becomethe gold standard for a variety of topical applications, such as skinand eye infections. Drug formulations capable of sustained releaseare highly sought after, providing ameans for drug concentration tobe maintained for long periods of time above the minimuminhibitory concentration (MIC) for relevant pathogens. In orderto address these requirements, formulations such as liposomesand gels are typically studied, as they offer good vehicles forthe incorporation of this hydrophobic and sparingly soluble drug[7e10].
For delivery applications, hydrogels offer convenient drug de-livery matrices. They are typically composed of natural polymers(e.g. alginate), or crosslinked synthetic macromolecules (e.g.
rights reserved.

S. Marchesan et al. / Biomaterials 34 (2013) 3678e3687 3679
polyethylene glycol, polyvinyl alcohol), which offer bio-compatibility and desirable physical properties. However, there arealso significant limitations, such as undesired burst release ofdrugs, polymer shrinkage post-crosslinking, or even toxicity ofunreacted synthetic polymer precursors [11].
Peptide self-assembled systems in which non-covalent in-teractions are responsible for the physical assembly of peptidemolecules offer a viable alternative to generate hydrogels [12].Gelation can be triggered in a number of ways, for instance bychanges in pH [13], ionic strength [14], temperature [15], enzyme-catalysis [16], or a combination of different stimuli [17]. Thesesystems are attractive biomaterials in that they biodegrade intobenign catabolic products, their processing involves mild condi-tions that are compatible with physiological systems, and lowconcentrations (in the millimolar range) are required for gelation[12]. Moreover, some of these systems facilitate wound healing,allowing cell infiltration and retention of viability [18,19]. Materialssuch as these can also be used to entrap bioactive molecules andhydrophobic drugs for controlled release [18,20,21]. However, themajority of peptides capable of self-assembly into hydrogels consistof rather long molecules (>10 amino acids) [22], for which solid-phase synthetic preparation is expensive and difficult to scale-up.
In contrast, ultrashort peptides (i.e. made of 2 or 3 amino acids)are attractive candidates for hydrogels, as they can be readily pre-pared in solution-phase, making scale-up convenient [23]. Onlya few systems of this kind have been described in the literature ofwhich the majority exploit the presence of synthetic end-cappinggroups (e.g. Fmoc, naphthalene, etc.) to increase molecular hydro-phobicity and to assist self-assembly [24e26]. However, concernsexist for the potential in vivo toxicity of certain aromatic syntheticgroups, since examples of reduced viability of cells cultured onthese gels have been reported [27,28]. In addition, small amounts oforganic solvents are often required to aid peptide dissolution and toassist with secondary structure stability [27,29,30].
For biological applications, alternative ultrashort gelling agentsdevoid of synthetic end-capping groups, and that do not requireany organic solvent, are needed. We recently reported, to ourknowledge, the first hydrogel systems that satisfy these re-quirements, and that are based on uncapped tripeptides capable ofhydrogel formation under physiological conditions (DValePheePhe, DPheePheeVal, and DLeuePheePhe) [31,32]. Moreover, thesesystems display a D-amino acid at their N-terminus that could beadvantageous for prolonged stability in human tissue. These ul-trashort gelling peptides are composed of hydrophobic moleculeswhere aromatic interactions between the phenylalanine rings arepivotal to self-assembly. As a result, they could be a useful vehiclefor the sustained delivery of hydrophobic drugs that contain aro-matic groups, which may favourably interact with the peptide atthe nanostructural level. Moreover, these hydrogels are formedfrom the combination of two precursor solutions at acidic andalkaline pH respectively. Therefore, drugs that are sparingly solubleat physiological pH, but that present ionisable groups, could bedissolved in either of the precursor solutions (prior to entrapmentinto the hydrogel) at levels that are higher than their solubility limitat neutral pH.
In order to test this hypothesis, we chose the self-assemblingpeptide DLeuePheePhe and the antibiotic ciprofloxacin (CIP) asa model hydrophobic drug that is sparingly soluble at physiologicalpH. To the best of our knowledge, this is the first report of self-assembly of a drug and a tripeptide into a hydrogel with distinc-tive nanostructure that allows for controlled release of the drug.These materials were used to challenge cultures of Staphylococcusaureus, Escherichia coli, and a clinical isolate of Klebsiella pneumo-niae [34]. Sustained drug release from the hydrogels was reflectedby antibiotic concentrations of the surrounding solutions and by
agar-diffusion assays determining efficacy at bacterial killing. Theformulations were also validated for non-cytotoxicity to mamma-lian cells.
2. Materials and methods
2.1. General materials and methods
PheeWang resin, O-Benzotriazole-N,N,N,N0-tetramethyl-uronium-hexafluoro-phosphate (HBTU), and Fmoc protected L-phenylalanine were purchased from GLBiochem Ltd (China). Fmoc-protected D-leucine and D-valine were purchased fromMimotopes (Australia). All solvents purchased were of analytical grade from Merck(Australia). Piperidine, trifluoroacetic acid (TFA), diisopropyl ethyl amine (DIPEA)were from Acros (Australia). Sodium dihydrogen phosphate, disodium hydrogenphosphate, and sodium hydroxide were from BDH AnalaR (Australia). Ciprofloxacinwas purchased from Biochemika (SigmaeAldrich, Australia). L-phenylalanine (Phe),D-leucine (DLeu), and Thioflavin T were from Sigma (Australia). High purity Milli-Q-water (MQ water) with a resistivity greater than 18 MU cmwas obtained from an in-line Millipore RiOs/Origin system. Silicon wafers (M.M.R.C. Pty Ltd., Australia) werecleaned by ultrasonication (1 h) in a surfactant solution of 2% ethanol with 2% RBS 35(Pierce) followed by rinsing with copious amounts of MQ water and dried withnitrogen, then cleaned for 1 h in a UV/ozone Procleaner� (Bioforce Nanosciences,US). The HPLC (Agilent Infinity, Australia) was equipped with a quaternary pump(G1311B), analytical C-18 column (Luna, 5 microns, 100 Å, 150 � 4.60 mm, Phe-nomenex, Australia), autosampler (G1329B), diode array detector (G4212B), ther-mostat (G1330B) and thermostatted column compartment (G1316A). The gradientconsisted of acetonitrile (AcN)/water with 0.1% TFA with the following program:t ¼ 0e3 min. 5% AcN; t ¼ 18 min. 95% AcN; t ¼ 18e20 min. 95% AcN; tR ¼ 8 min forciprofloxacin; tR ¼ 11 min for DLeuePheePhe.
2.2. Peptide synthesis
The tripeptides were synthesised and purified according to standard solid-phase-peptide-synthesis procedures as reported previously [31,32].
2.3. Preparation of peptide hydrogel
The tripeptide DLeuePheePhe gelled immediately following a pH trigger [32].Briefly, the peptide (4.0mg) was added to a solution of sodium phosphate 0.1 M at pH11.8 (buffer A, 0.3 ml), and dissolution was achieved with sonication (5 min).Gelation was triggered by subsequent addition of an equal volume of 0.1 M sodiumphosphate at pH 5.7 (buffer B, 0.3 ml), resulting in a final pH of 7.4.
2.4. Preparation of CIP-loaded peptide hydrogel
CIP was dissolved in buffer A (2 or 4 mg ml�1 to obtain a final gel with 0.1% w/vor 0.2% w/v CIP, respectively) before adding the peptide. The resulting solution wasthen used following the procedure described above (Section 2.3). Gelation wasallowed to occur for 2 h, after which any residues of supernatant solution that mayhave been present (<2% total volume, containing <1% of total CIP) were gentlyaspirated with a pipette.
2.5. Fluorescence measurements
The interaction between CIP and the gelling peptide DLeuePheePhe wasobserved by fluorescence spectroscopy. Fluorescence measurements were madeusing a FlexStation 3 spectrofluorimeter (Molecular Devices). In order to avoid CIPprecipitation in the control samples (without the peptide), all measurements werecarried out on samples with a final CIP concentration of 0.5 mg ml�1 (i.e. 0.05% w/v)and increasing amounts of the gelling peptide (0, 3.9mM, 7.9mM,11.8mM, or 15.7mM
corresponding to 0%, 25%, 50%, 75%, or 100% of the peptide amount used in ourrelease studies), following the procedure described in section 2.3. Briefly, 100 ml ofeach precursor solutions were added onto each well of a black 96-well plate.Emission spectra were recorded after 2 h from 380 nm to 500 nmwith an excitationwavelength of 340 nm. At this timepoint, only samples with 50%, 75%, or 100%peptide formed hydrogels. Higher amounts of peptide could not be tested as theywould lead to peptide precipitation. Experiments were repeated twice in triplicates.Spectra were corrected by subtraction of the corresponding buffer signal (i.e. 0.1 M
sodium phosphate buffer, 100 ml at pH 11.8, and 100 ml at pH 5.7).
2.6. Circular dichroism (CD) analysis
The secondary structure of the peptides was analysed using a 0.1 cm quartz cellon a Jasco J815 Spectropolarimeter, with 1 s integrations, 1 accumulation and a stepsize of 1 nm with a band width of 1 nm over a range of wavelengths from 200 to270 nm. Peptide samples freshly prepared in buffer A were diluted with buffer Bdirectly in the CD cell, and spectrawere recorded after at 15e30 min. Measurements

S. Marchesan et al. / Biomaterials 34 (2013) 3678e36873680
were repeated at least 5 times and their average was plotted. Control samples withonly drug and buffer (no peptide) did not display any signal.
2.7. FT-IR analysis
FT-IR spectra were collected on a Nicolet 6700 FT-IR spectrometer in ATR mode.Freshly prepared samples were left to settle for 2 h in a glass vial. A portion of the gelwas then transferred on a clean piece of silicon wafer (1 cm � 1 cm), gently spreadby pressing a coverslip on top, and dried under vacuum for 24 h. Dried samples onthe silicon wafers were placed directly to the ATR crystal, facing down. Scans werebetween 1800 and 600 cm�1 with 80 accumulations at a resolution of 0.4 cm�1.
2.8. Rheometry analysis
Dynamic sweep rheological analysis was conducted on an Ares rheometer (TAInstruments, USA) with 25 mm aluminium parallel plate geometry. A Peltier tem-perature controller was connected to the rheometer to maintain a temperature of25 �C. Hydrogels were analysed after 24 h. A gap of 300 mm was set. Strain sweepswere recorded using a frequency of 10 rad s�1. Frequency sweeps were recordedusing a controlled strain of 1%. Average measurements (n ¼ 3) have been plottedwith Excel.
2.9. Thioflavin T confocal fluorescence
Hydrogels were prepared as described above and after 2 h approximately 20 mlwere immediately placed on wells of a “m-Slide Angiogenesis” uncoated (Ibidi,Germany, through DKSH Australia). 20 ml of a solution of Thioflavin T (200 mM in50 mM GlycineeNaOH pH 7.5, 0.2 mm-filtered) were placed on top. After 15 min, theslides were imaged on a Leica SP5 microscope (63� water immersion objective, NA1.2, excitation 458 nm, emission 468e600 nm). Samples treated and stained with anidentical protocol but without the peptide, were used as control and did not revealany of the fluorescent structures described in the text (data not shown). The videowas obtained with Imaris software from a z-stack of a peptide gel with 0.2% w/v CIPafter 48 h of self-assembly.
2.10. Cryo-transmission electron microscopy (cryo-TEM) analysis
A laboratory-built humidity-controlled vitrification systemwas used to preparethe hydrogels for imaging in a thin layer of vitrified ice using cryo-TEM. Humiditywas kept close to 80% for all experiments, and ambient temperature was 22 �C. 200-Mesh copper grids coated with perforated carbon film (Lacey carbon film: ProSci-Tech, Qld, Australia) were used for all experiments. Grids were glow discharged innitrogen for 5 s immediately before use. Hydrogels were prepared as describedabove and analysed after 2 h. Hydrogels were agitated gently with the pipette tip inorder to liquefy them slightly if possible. Approximately 4 ml aliquots of sample werepipetted onto each grid prior to plunging. In the case of samples which could not beliquefied adequately, the gel was smeared gently onto the grid. After 30 s adsorptiontime the grid was blotted manually using Whatman 541 filter paper, for approx-imately 6 s. Blotting time was optimised for each sample. The grid was then plungedinto liquid ethane cooled by liquid nitrogen. Frozen grids were stored in liquid ni-trogen until required. The samples were examined using a Gatan 626 cryoholder(Gatan, Pleasanton, CA, USA) and Tecnai 12 Transmission Electron Microscope (FEI,Eindhoven, The Netherlands) at an operating voltage of 120 KV. At all times low doseprocedures were followed, using an electron dose of 8e10 electrons �2 for allimaging. Images were recorded using an FEI Eagle 4kx4k CCD camera (FEI, Eind-hoven, The Netherlands).
2.11. Negative staining TEM analysis
Carbon-coated 300-mesh copper grids were glow-discharged in nitrogen torender the carbon film hydrophilic. Hydrogels were prepared as described aboveand analysed after 2 h. A 4 ml aliquot of sample was pipetted onto each grid. After30 s adsorption time the excess was drawn off using Whatman 541 filter paper,followed by staining with 2% aqueous potassium phosphotungstate at pH 7.2, for10 s. Grids were air-dried until needed. The samples were examined using a Tecnai12 Transmission Electron Microscope (FEI, Eindhoven, The Netherlands) at anoperating voltage of 120 KV. Images were recorded using aMegaview III CCD cameraand AnalySIS camera control software (Olympus). Each grid was systematicallyexamined and imaged to reflect a representative view of the sample. This wasimportant as samples of this type can be very non-homogeneous on the grid; theyhad the tendency not to disperse evenly and the amyloid fibrils occured in distinctpatches.
2.12. AFM analysis
An Asylum Research MFP-3D atomic force microscope (Santa Barbara, CA, USA)was used to measure surface topography in tapping mode with ultrasharp siliconnitride tips (NSC15 noncontact silicon cantilevers, MikroMasch, Spain). The tips usedin this study had a typical force constant of 40 N/m and a resonant frequency of
320 kHz. Typical scan settings involved the use of an applied piezo deflection voltageof 0.6e0.7 V at a scan rate of 0.7 Hz. All images were processed (1st order flatteningalgorithm)using IgorPro software. Freshlypreparedhydrogel sampleswerepreparedas described above and left to settle for 2 h. A portion of each samplewas then spreadonto a clean square of siliconwafer (1 cm� 1 cm) by gently pressing a glass coverslipon top. Samples were then dried in a vacuum oven at room temperature for 24 h.
2.13. In vitro release studies
Self-assembled peptide gelswith 0.1%w/v, 0.2%w/vorwithout CIPwere prepareddirectly in a glass vial (diameter ¼ 1.8 cm, height ¼ 6.5 cm, 10 ml total volume), fol-lowing the procedure described above in Section 2.3 (final gel volumeof 0.6ml). After2 h, 10 ml of PBS buffer pH 7.4 were very gently added from the side of the vial,carefully avoiding disruption (and subsequent floating) of the gel disc. Vials wereincubated at 37 �C, 50 rpm, 70% humidity, and at selected time points 0.5 ml sampleswere taken and replaced with 0.5 ml of fresh buffer. Samples were analysed by HPLCand absorbance was monitored over time. HPLC signal integration was used toquantify the peptide (Abs at 215 nm) and CIP (Abs at 273 nm). Fresh solutions ofpeptide and CIPwere used as standards. Each experiment was repeated at least twicein triplicates, and mean and standard deviation values were calculated in Excel.
2.14. Quantitative determination of CIP released from the gel by broth micro-dilutionassay
Concentration of ciprofloxacin released from the gel system over a time coursewas also determined by a modification of broth micro-dilution assay from CLSI (M2-A8) [33]. Bacterial suspensions of S. aureus ATCC 29213, E. coli ATCC 25922,K. pneumoniae B5055 [34] were prepared in Mueller-Hinton broth (MHB) to reacha density of 1 � 106 CFU/ml. Ciprofloxacin solutions were sampled from the gel-releasing vials at 4 h, 8 h, 24 h, 48 h, day 4, and day 6 and prepared as serial doubledilutions (100 ml) in 96-well microplates with MHB. 100 ml of the bacterial suspen-sions were plated into microwells containing ciprofloxacin and the plates wereincubated at 35 �C overnight. Minimum Inhibitory Concentration (MIC) of cipro-floxacin against of S. aureusATCC 29213, E. coliATCC 25922,K. pneumoniaeB5055 [34]were also examined using broth micro-dilution assay and commercial ciprofloxacinpowder. This was to provide the baseline to determine the concentration of cipro-floxacin in the gel solution. Concentrationof ciprofloxacin ingredients releasedby thegel system at each time points were calculated, by using the formula:Concentration ¼ MIC/2�dilution factor. MIC was obtained as mentioned before. Thedilution factorwas obtained by counting the number of wellswithout visible growth.
2.15. Determination of anti-bacterial efficacy by micro-gel well diffusion assay
Micro-gel well diffusion assay was performed to examine the potential anti-bacterial activities of ciprofloxacin-gel system, by following the method describedby Magaldi et al. [35] Inocula of either S. aureus ATCC 29213, E. coli ATCC 25922, orK. pneumoniae B5055 [34] were prepared by growing cells to a log phase (5e6 h).The suspensions were adjusted with 0.85% NaCl to final optical density (OD) read-ings of 0.1 and 10 with a spectrophotometer at 600 nm. These two OD readingsrepresented bacterial cultures used for conventional antibiotic susceptibility testsand bacterial populations of higher resistance to antibiotics (i.e. biofilms) respec-tively [36,37]. 20 ml of MuellereHinton agar were melted, cooled to 55 �C and theninoculated with 1 ml of the bacterial suspension. The inoculated agar was pouredinto the Petri dish plate, and allowed to cool down on a levelled surface. Once themedium had solidified, 2 wells, each of 6 mm in diameter, were cut out of the agarwith a biopsy puncture. 70 ml of the gel with 0.2% w/v or without ciprofloxacin wereplaced intowells in the plate. The plateswere incubated in a humid chamber for 20 hat 35 �C. The plates with inhibition zone of bacterial growth were imaged.
2.16. Haemolysis assay
Haemolysis assay was performed as described by Ciornei et al. [38] Blood wascentrifuged at 800� g for 10 min, and then the supernatant (plasma) and ‘buffy coat’(white blood cells) were removed. Erythrocytes were rinsed three times and thenresuspended in phosphate-buffered saline (pH 7.4). Next, 400 ml of solutionsobtained from the in vitro release study for hydrogels with 0.2% w/v ciprofloxacin atdifferent time points (4 h, 8 h, 24 h, 48 h, day 4, and day 6 respectively) were used toreplace the PBS in the erythrocyte suspension and were followed by incubation of2 h at 37 �C with gentle end-over-end rotation. Triton X-100 at 2% (SigmaeAldrich)served as a positive control, and DMSO at 2% served as a negative control. Afterincubation, the samples were centrifuged at 800� g for 10 min. The release ofhaemoglobin was monitored by measuring the absorbance of the supernatant at540 nm and was expressed as a percentage of the value for Triton X-100-inducedhaemolysis.
2.17. Fibroblast cell culture live/dead assay
Peptide self-assembled hydrogels without drug were prepared as describedabove (Section 2.3) by using 10 ml of each precursor solution which were filtered

S. Marchesan et al. / Biomaterials 34 (2013) 3678e3687 3681
(0.2 mm) and added to each microwell of a m-slide Angiogenesis uncoated (Ibidi) ina laminar-flow cabinet (final gel volume of 20 ml, 15.7 mM peptide). Controls con-sisted of empty microwells. Hydrogels were left to settle at room temperature for1 h. 30 ml of a solution of antibiotic-antimycotic (GIBCO)was gently added on top andincubated for 1 h at room temperature. The solution was then replaced with freshMEM/glutamax (Invitrogen) medium containing 10% foetal bovine serum with 1%non-essential amino acids and incubated for 1 h at room temperature. The solutionwas then replaced with media containing L929 fibroblasts (ATCC cell line CCL-1,Rockville, MD, USA) at 2000 cells per well. These were cultured at 37 �C in a hu-midified incubator for up to 3 days, exchanging the media with fresh (25 ml) every24 h. A few drops of sterile water were carefully placed between the microwells toreduce evaporation. Live/dead assays were performed at 24 h, 48 h, or 72 h byreplacing 25 ml of the media with 25 ml of freshly prepared live/dead solution con-taining calcein (live) and ethidium-homodimer (dead), (Molecular Probes, Invi-trogen) prepared according to the manufacturers’ instructions. Following 25 minincubation, cells were imaged on an inverted widefield fluorescence microscope(Nikon Eclipse TE2000-U). Each condition was repeated twice in triplicates. Rep-resentative images after 72 h incubation are shown.
2.18. MTS assay
Microwells containing hydrogels and cells, cells only (control), hydrogel only orempty wells (reagent blanks) were prepared as described in Section 2.17 andincubated for 24 h, 48 h, or 72 h. 25 ml of media were replaced with 25 ml of an MTSsolution containing 248 mM phenazine methosulfate (Sigma) and 680 mM 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (Promega) in media.Absorbance at 490 nm was read by placing 5 ml samples on a Nanodrop� ND-1000Spectrophotomer (BioLab). Each measurement was repeated at least three times persample, and each condition was repeated twice in triplicates. Absorbance at 490 nmof the blanks was subtracted to the absorbance at 490 nm of the samples andcontrols. Data are represented as means þ standard deviation (SD). Statisticalanalysis was performed by means of repeated measures ANOVA (RMANOVA) tests,with time as within subjects factor, and with condition as between subjects factor(at two levels: control and gel treatment). Follow-up RMANOVA tests were per-formed by splitting the data by condition.
3. Results and discussion
3.1. Ciprofloxacin interaction with the peptide within the hydrogel
The ciprofloxacin (CIP) loaded hydrogel was prepared by con-current encapsulation of the drug during peptide self-assembly(Fig. 1). The neutral form of CIP (pI ¼ 7.4) [39] is sparingly solublein water at physiological pH (0.07 mg ml�1 at 20 �C and0.11 mg ml�1 at 37 �C) [40]. However, ionisation under basic oracidic conditions leads readily to drug dissolution. CIP is highlysoluble at a pH > 10, thus, it could be dissolved in 0.1 M sodiumphosphate at pH 11.8 at a concentration of 4 mg ml�1 [39]. In ourexperiments, HPLC analysis confirmed CIP hydrolysis under theseconditions did not occur (data not shown), with the pH beingimmediately neutralised by addition of a second buffer. This pHchange triggered immediate gelation of the peptide as well asentrapment of the drug into the hydrogel matrix. Drug encapsu-lation was successful at levels of clinical relevance for gels (eg. 0.1%and 0.2% w/v) displaying high drug loading capacity (30% w/wrelative to tripeptide mass) compared to, for example, polymerhydrogels.
Fig. 1. Structures of ciprofloxacin (CIP) and peptide DLeuePheePhe
However, further increase in drug concentration led to systeminstability and hydrogel disruption. To monitor whether CIP inter-acted with the peptide at the molecular level, we employed fluo-rescence, FT-IR, circular dichroism (CD) spectroscopy andrheometric comparative analyses on peptide hydrogels in thepresence (0.1% and 0.2% w/v) and absence of drug.
3.1.1. Fluorescence studies on the hydrogelFluorescence was used to verify whether molecular interactions
occured between CIP and the peptide, since only the drug is fluo-rescent at the gelation pH of 7.4 [41]. Interestingly, CIP fluorescenceintensity was significantly increased when incorporated within thegel (Fig. 2A). In contrast, no significant difference was observedbetween samples with CIP only (control), and samples with CIP-peptide mixtures that were below the threshold concentrationrequired for gelation under our experimental conditions (25%peptide, i.e. 3.9 mM).
This particular behaviour for CIP-loaded hydrogels rules out aninteraction such as complex formation, which leads to partialquenching of CIP fluorescence, as observed in previous works onCIP and proteins or CIP and single amino acids (including phenyl-alanine and leucine) [42,43]. We propose instead that the inter-action with the gelling peptide DLeuePheePhe occurs as part ofself-assembly, possibly via aromatic interactions. It is plausible thatthe hydrophobicity of drug and peptide (which have similar HPLCretention times, see Section 2.1) may drive the molecules togetherin the core of the self-assembled hydrogel fibres. This hypothesis isconsistent with one recent report of drug fluorescence amplifica-tion when encapsulated within a peptide self-assembled hydrogel,as a result of molecular interactions [18].
3.1.2. Circular dichroism (CD) analysis of the hydrogel with andwithout ciprofloxacin
Circular dichroism (CD) analysis was used to assess the effect ofthe drug on the secondary structure of the peptide (Fig. 2B). Thetripeptide DLeuePheePhe gel displays the characteristic signatureof these systems, i.e. an intensemaximum atw226 nm indicative ofp stacking of phenylalanine aromatic rings, and a minimum in theregion 210e220 nm, due to anti-parallel beta-sheets [31,32].Interestingly, in the presence of CIP, the maximum displayeda blueshift of 2e3 nm, and a notable decrease in intensity, sug-gesting the drug interfered with phenylalanine p stacking. On thecontrary, the minimum showed a redshift of w5 nm and anincrease in intensity, suggesting enhanced beta-sheet con-formation. Finally, and only in the presence of the drug, a minimumin the region 230e270 nmwas registered, which can be attributedto phenylalanine residues with tertiary structure.
Taken together, this data confirmed that the major features ofthe secondary conformation of the peptide hydrogel were retainedin the presence of CIP, although some changes did occur. In par-ticular, it is plausible that CIP interfered with peptide p stacking via
, which self-assemble into a hydrogel following a pH trigger.
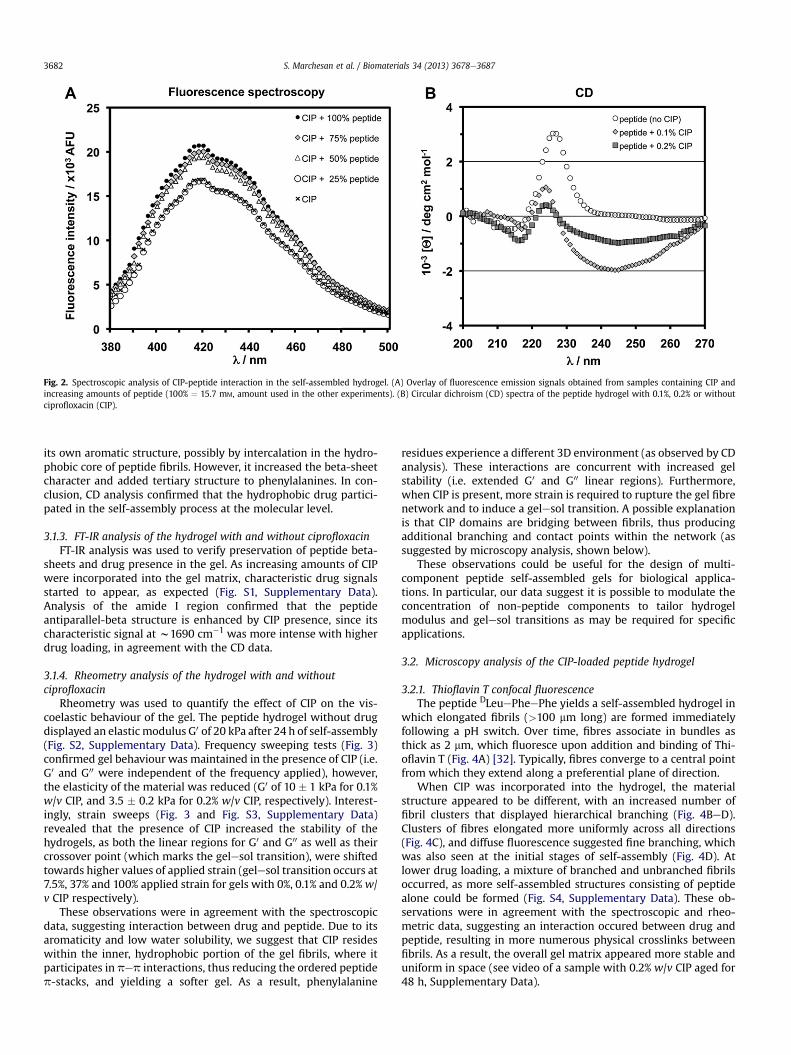
Fig. 2. Spectroscopic analysis of CIP-peptide interaction in the self-assembled hydrogel. (A) Overlay of fluorescence emission signals obtained from samples containing CIP andincreasing amounts of peptide (100% ¼ 15.7 mM, amount used in the other experiments). (B) Circular dichroism (CD) spectra of the peptide hydrogel with 0.1%, 0.2% or withoutciprofloxacin (CIP).
S. Marchesan et al. / Biomaterials 34 (2013) 3678e36873682
its own aromatic structure, possibly by intercalation in the hydro-phobic core of peptide fibrils. However, it increased the beta-sheetcharacter and added tertiary structure to phenylalanines. In con-clusion, CD analysis confirmed that the hydrophobic drug partici-pated in the self-assembly process at the molecular level.
3.1.3. FT-IR analysis of the hydrogel with and without ciprofloxacinFT-IR analysis was used to verify preservation of peptide beta-
sheets and drug presence in the gel. As increasing amounts of CIPwere incorporated into the gel matrix, characteristic drug signalsstarted to appear, as expected (Fig. S1, Supplementary Data).Analysis of the amide I region confirmed that the peptideantiparallel-beta structure is enhanced by CIP presence, since itscharacteristic signal at w1690 cm�1 was more intense with higherdrug loading, in agreement with the CD data.
3.1.4. Rheometry analysis of the hydrogel with and withoutciprofloxacin
Rheometry was used to quantify the effect of CIP on the vis-coelastic behaviour of the gel. The peptide hydrogel without drugdisplayed an elastic modulus G0 of 20 kPa after 24 h of self-assembly(Fig. S2, Supplementary Data). Frequency sweeping tests (Fig. 3)confirmed gel behaviour was maintained in the presence of CIP (i.e.G0 and G00 were independent of the frequency applied), however,the elasticity of the material was reduced (G0 of 10 � 1 kPa for 0.1%w/v CIP, and 3.5 � 0.2 kPa for 0.2% w/v CIP, respectively). Interest-ingly, strain sweeps (Fig. 3 and Fig. S3, Supplementary Data)revealed that the presence of CIP increased the stability of thehydrogels, as both the linear regions for G0 and G00 as well as theircrossover point (which marks the gelesol transition), were shiftedtowards higher values of applied strain (gelesol transition occurs at7.5%, 37% and 100% applied strain for gels with 0%, 0.1% and 0.2%w/v CIP respectively).
These observations were in agreement with the spectroscopicdata, suggesting interaction between drug and peptide. Due to itsaromaticity and low water solubility, we suggest that CIP resideswithin the inner, hydrophobic portion of the gel fibrils, where itparticipates in pep interactions, thus reducing the ordered peptidep-stacks, and yielding a softer gel. As a result, phenylalanine
residues experience a different 3D environment (as observed by CDanalysis). These interactions are concurrent with increased gelstability (i.e. extended G0 and G00 linear regions). Furthermore,when CIP is present, more strain is required to rupture the gel fibrenetwork and to induce a gelesol transition. A possible explanationis that CIP domains are bridging between fibrils, thus producingadditional branching and contact points within the network (assuggested by microscopy analysis, shown below).
These observations could be useful for the design of multi-component peptide self-assembled gels for biological applica-tions. In particular, our data suggest it is possible to modulate theconcentration of non-peptide components to tailor hydrogelmodulus and gelesol transitions as may be required for specificapplications.
3.2. Microscopy analysis of the CIP-loaded peptide hydrogel
3.2.1. Thioflavin T confocal fluorescenceThe peptide DLeuePheePhe yields a self-assembled hydrogel in
which elongated fibrils (>100 mm long) are formed immediatelyfollowing a pH switch. Over time, fibres associate in bundles asthick as 2 mm, which fluoresce upon addition and binding of Thi-oflavin T (Fig. 4A) [32]. Typically, fibres converge to a central pointfrom which they extend along a preferential plane of direction.
When CIP was incorporated into the hydrogel, the materialstructure appeared to be different, with an increased number offibril clusters that displayed hierarchical branching (Fig. 4BeD).Clusters of fibres elongated more uniformly across all directions(Fig. 4C), and diffuse fluorescence suggested fine branching, whichwas also seen at the initial stages of self-assembly (Fig. 4D). Atlower drug loading, a mixture of branched and unbranched fibrilsoccurred, as more self-assembled structures consisting of peptidealone could be formed (Fig. S4, Supplementary Data). These ob-servations were in agreement with the spectroscopic and rheo-metric data, suggesting an interaction occured between drug andpeptide, resulting in more numerous physical crosslinks betweenfibrils. As a result, the overall gel matrix appeared more stable anduniform in space (see video of a sample with 0.2% w/v CIP aged for48 h, Supplementary Data).
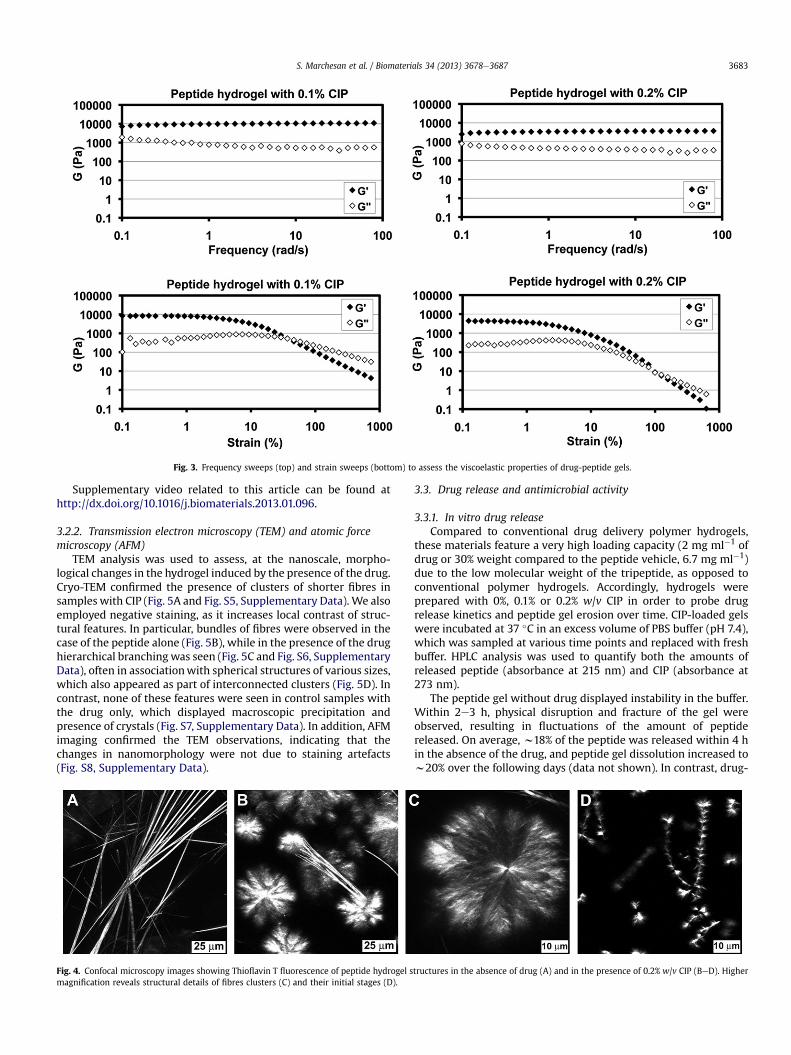
Fig. 3. Frequency sweeps (top) and strain sweeps (bottom) to assess the viscoelastic properties of drug-peptide gels.
S. Marchesan et al. / Biomaterials 34 (2013) 3678e3687 3683
Supplementary video related to this article can be found athttp://dx.doi.org/10.1016/j.biomaterials.2013.01.096.
3.2.2. Transmission electron microscopy (TEM) and atomic forcemicroscopy (AFM)
TEM analysis was used to assess, at the nanoscale, morpho-logical changes in the hydrogel induced by the presence of the drug.Cryo-TEM confirmed the presence of clusters of shorter fibres insamples with CIP (Fig. 5A and Fig. S5, Supplementary Data). We alsoemployed negative staining, as it increases local contrast of struc-tural features. In particular, bundles of fibres were observed in thecase of the peptide alone (Fig. 5B), while in the presence of the drughierarchical branching was seen (Fig. 5C and Fig. S6, SupplementaryData), often in associationwith spherical structures of various sizes,which also appeared as part of interconnected clusters (Fig. 5D). Incontrast, none of these features were seen in control samples withthe drug only, which displayed macroscopic precipitation andpresence of crystals (Fig. S7, Supplementary Data). In addition, AFMimaging confirmed the TEM observations, indicating that thechanges in nanomorphology were not due to staining artefacts(Fig. S8, Supplementary Data).
Fig. 4. Confocal microscopy images showing Thioflavin T fluorescence of peptide hydrogel smagnification reveals structural details of fibres clusters (C) and their initial stages (D).
3.3. Drug release and antimicrobial activity
3.3.1. In vitro drug releaseCompared to conventional drug delivery polymer hydrogels,
these materials feature a very high loading capacity (2 mg ml�1 ofdrug or 30% weight compared to the peptide vehicle, 6.7 mg ml�1)due to the low molecular weight of the tripeptide, as opposed toconventional polymer hydrogels. Accordingly, hydrogels wereprepared with 0%, 0.1% or 0.2% w/v CIP in order to probe drugrelease kinetics and peptide gel erosion over time. CIP-loaded gelswere incubated at 37 �C in an excess volume of PBS buffer (pH 7.4),which was sampled at various time points and replaced with freshbuffer. HPLC analysis was used to quantify both the amounts ofreleased peptide (absorbance at 215 nm) and CIP (absorbance at273 nm).
The peptide gel without drug displayed instability in the buffer.Within 2e3 h, physical disruption and fracture of the gel wereobserved, resulting in fluctuations of the amount of peptidereleased. On average, w18% of the peptide was released within 4 hin the absence of the drug, and peptide gel dissolution increased tow20% over the following days (data not shown). In contrast, drug-
tructures in the absence of drug (A) and in the presence of 0.2% w/v CIP (BeD). Higher
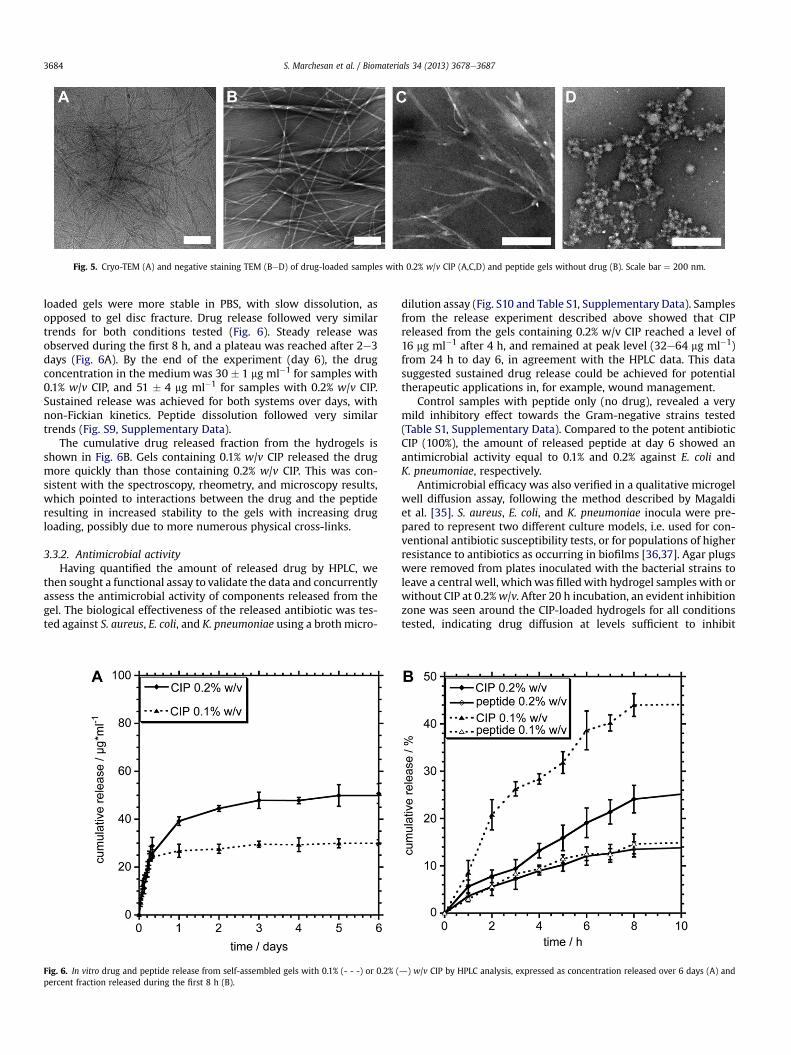
Fig. 5. Cryo-TEM (A) and negative staining TEM (BeD) of drug-loaded samples with 0.2% w/v CIP (A,C,D) and peptide gels without drug (B). Scale bar ¼ 200 nm.
S. Marchesan et al. / Biomaterials 34 (2013) 3678e36873684
loaded gels were more stable in PBS, with slow dissolution, asopposed to gel disc fracture. Drug release followed very similartrends for both conditions tested (Fig. 6). Steady release wasobserved during the first 8 h, and a plateau was reached after 2e3days (Fig. 6A). By the end of the experiment (day 6), the drugconcentration in the mediumwas 30 � 1 mg ml�1 for samples with0.1% w/v CIP, and 51 � 4 mg ml�1 for samples with 0.2% w/v CIP.Sustained release was achieved for both systems over days, withnon-Fickian kinetics. Peptide dissolution followed very similartrends (Fig. S9, Supplementary Data).
The cumulative drug released fraction from the hydrogels isshown in Fig. 6B. Gels containing 0.1% w/v CIP released the drugmore quickly than those containing 0.2% w/v CIP. This was con-sistent with the spectroscopy, rheometry, and microscopy results,which pointed to interactions between the drug and the peptideresulting in increased stability to the gels with increasing drugloading, possibly due to more numerous physical cross-links.
3.3.2. Antimicrobial activityHaving quantified the amount of released drug by HPLC, we
then sought a functional assay to validate the data and concurrentlyassess the antimicrobial activity of components released from thegel. The biological effectiveness of the released antibiotic was tes-ted against S. aureus, E. coli, and K. pneumoniae using a brothmicro-
Fig. 6. In vitro drug and peptide release from self-assembled gels with 0.1% (- - -) or 0.2% (percent fraction released during the first 8 h (B).
dilution assay (Fig. S10 and Table S1, Supplementary Data). Samplesfrom the release experiment described above showed that CIPreleased from the gels containing 0.2% w/v CIP reached a level of16 mg ml�1 after 4 h, and remained at peak level (32e64 mg ml�1)from 24 h to day 6, in agreement with the HPLC data. This datasuggested sustained drug release could be achieved for potentialtherapeutic applications in, for example, wound management.
Control samples with peptide only (no drug), revealed a verymild inhibitory effect towards the Gram-negative strains tested(Table S1, Supplementary Data). Compared to the potent antibioticCIP (100%), the amount of released peptide at day 6 showed anantimicrobial activity equal to 0.1% and 0.2% against E. coli andK. pneumoniae, respectively.
Antimicrobial efficacy was also verified in a qualitative microgelwell diffusion assay, following the method described by Magaldiet al. [35]. S. aureus, E. coli, and K. pneumoniae inocula were pre-pared to represent two different culture models, i.e. used for con-ventional antibiotic susceptibility tests, or for populations of higherresistance to antibiotics as occurring in biofilms [36,37]. Agar plugswere removed from plates inoculated with the bacterial strains toleave a central well, whichwas filled with hydrogel samples with orwithout CIP at 0.2%w/v. After 20 h incubation, an evident inhibitionzone was seen around the CIP-loaded hydrogels for all conditionstested, indicating drug diffusion at levels sufficient to inhibit
d) w/v CIP by HPLC analysis, expressed as concentration released over 6 days (A) and
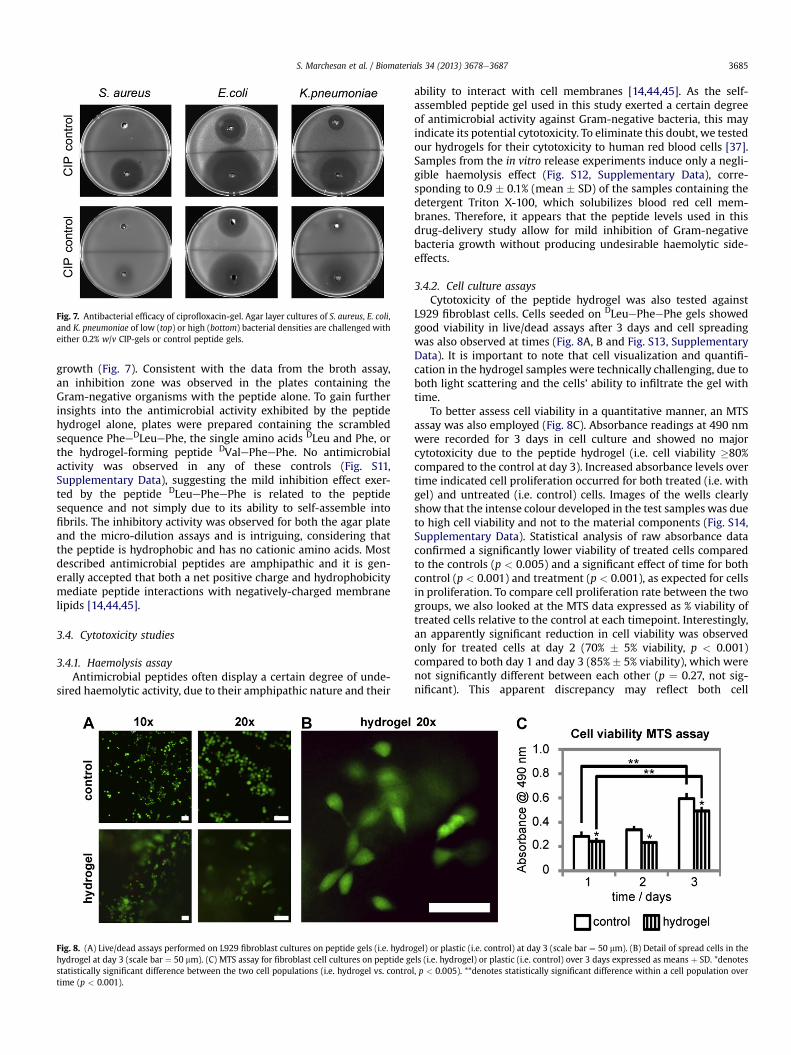
Fig. 7. Antibacterial efficacy of ciprofloxacin-gel. Agar layer cultures of S. aureus, E. coli,and K. pneumoniae of low (top) or high (bottom) bacterial densities are challenged witheither 0.2% w/v CIP-gels or control peptide gels.
S. Marchesan et al. / Biomaterials 34 (2013) 3678e3687 3685
growth (Fig. 7). Consistent with the data from the broth assay,an inhibition zone was observed in the plates containing theGram-negative organisms with the peptide alone. To gain furtherinsights into the antimicrobial activity exhibited by the peptidehydrogel alone, plates were prepared containing the scrambledsequence PheeDLeuePhe, the single amino acids DLeu and Phe, orthe hydrogel-forming peptide DValePheePhe. No antimicrobialactivity was observed in any of these controls (Fig. S11,Supplementary Data), suggesting the mild inhibition effect exer-ted by the peptide DLeuePheePhe is related to the peptidesequence and not simply due to its ability to self-assemble intofibrils. The inhibitory activity was observed for both the agar plateand the micro-dilution assays and is intriguing, considering thatthe peptide is hydrophobic and has no cationic amino acids. Mostdescribed antimicrobial peptides are amphipathic and it is gen-erally accepted that both a net positive charge and hydrophobicitymediate peptide interactions with negatively-charged membranelipids [14,44,45].
3.4. Cytotoxicity studies
3.4.1. Haemolysis assayAntimicrobial peptides often display a certain degree of unde-
sired haemolytic activity, due to their amphipathic nature and their
Fig. 8. (A) Live/dead assays performed on L929 fibroblast cultures on peptide gels (i.e. hydrohydrogel at day 3 (scale bar ¼ 50 mm). (C) MTS assay for fibroblast cell cultures on peptide gestatistically significant difference between the two cell populations (i.e. hydrogel vs. controtime (p < 0.001).
ability to interact with cell membranes [14,44,45]. As the self-assembled peptide gel used in this study exerted a certain degreeof antimicrobial activity against Gram-negative bacteria, this mayindicate its potential cytotoxicity. To eliminate this doubt, we testedour hydrogels for their cytotoxicity to human red blood cells [37].Samples from the in vitro release experiments induce only a negli-gible haemolysis effect (Fig. S12, Supplementary Data), corre-sponding to 0.9 � 0.1% (mean � SD) of the samples containing thedetergent Triton X-100, which solubilizes blood red cell mem-branes. Therefore, it appears that the peptide levels used in thisdrug-delivery study allow for mild inhibition of Gram-negativebacteria growth without producing undesirable haemolytic side-effects.
3.4.2. Cell culture assaysCytotoxicity of the peptide hydrogel was also tested against
L929 fibroblast cells. Cells seeded on DLeuePheePhe gels showedgood viability in live/dead assays after 3 days and cell spreadingwas also observed at times (Fig. 8A, B and Fig. S13, SupplementaryData). It is important to note that cell visualization and quantifi-cation in the hydrogel samples were technically challenging, due toboth light scattering and the cells’ ability to infiltrate the gel withtime.
To better assess cell viability in a quantitative manner, an MTSassay was also employed (Fig. 8C). Absorbance readings at 490 nmwere recorded for 3 days in cell culture and showed no majorcytotoxicity due to the peptide hydrogel (i.e. cell viability �80%compared to the control at day 3). Increased absorbance levels overtime indicated cell proliferation occurred for both treated (i.e. withgel) and untreated (i.e. control) cells. Images of the wells clearlyshow that the intense colour developed in the test samples was dueto high cell viability and not to the material components (Fig. S14,Supplementary Data). Statistical analysis of raw absorbance dataconfirmed a significantly lower viability of treated cells comparedto the controls (p < 0.005) and a significant effect of time for bothcontrol (p < 0.001) and treatment (p < 0.001), as expected for cellsin proliferation. To compare cell proliferation rate between the twogroups, we also looked at the MTS data expressed as % viability oftreated cells relative to the control at each timepoint. Interestingly,an apparently significant reduction in cell viability was observedonly for treated cells at day 2 (70% � 5% viability, p < 0.001)compared to both day 1 and day 3 (85% � 5% viability), which werenot significantly different between each other (p ¼ 0.27, not sig-nificant). This apparent discrepancy may reflect both cell
gel) or plastic (i.e. control) at day 3 (scale bar ¼ 50 mm). (B) Detail of spread cells in thels (i.e. hydrogel) or plastic (i.e. control) over 3 days expressed as means þ SD. *denotesl, p < 0.005). **denotes statistically significant difference within a cell population over

S. Marchesan et al. / Biomaterials 34 (2013) 3678e36873686
infiltration and slow dye diffusion inside the gel (leading to lowermeasurements on day 2 for treated cells), and this effect wascompensated by partial hydrogel dissolution observed at day 3.Consistency of viability data between day 1 and day 3 suggests themild cytotoxic effect observed here is not amplified over the periodof 3 days, and thus does not significantly affect proliferation relativeto the control. Importantly, the data highlight the challenges rela-ted to the MTS assay when applied to hydrogels, and the need fordata validation and/or use of complimentary methods to assesssignificance of cell viability data.
In conclusion, both the live/dead assay and the MTS assay con-firmed the majority of cells treated with the gels were viable over 3days. Importantly, cells maintained the ability to proliferate.
4. Conclusions
We report the self-assembly of ciprofloxacin (CIP) and the tri-peptide DLeuePheePhe in a hydrogel with high drug loadings (i.e.2 mgml�1 and 30%w/w relative to the mass of the peptide vehicle).The drug actively participates in the gel nanostructure, yieldingsofter gels with increased stability. Interestingly, the peptide gelitself revealed a mild antimicrobial activity against the Gram-negative strains tested with no major haemolytic effect. Fibro-blasts infiltrate into the gel after 24 h, and this has relevance in thedevelopment of, for example, wound management dressings. Webelieve this study will be useful for the design of cost-effectivenanomaterials where drug incorporation in the vehicle nano-structure could lead to prolonged release.
Acknowledgements
The authors acknowledge the Australian Research Council (ARC)for funding (Y.Q. is an ARC Super Science Fellow, T.J.L. is an ARCFederation Fellow) and the facilities of Monash Micro Imaging,Monash University, Australia, and in particular Stephen Firth, Dr.Judy Callaghan and Dr. Alex Fulcher for their scientific and technicalassistance. The authors also acknowledge the CSIRO e MonashUniversity Collaborative Research Support Scheme (CRSS) forfunding.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.biomaterials.2013.01.096.
References
[1] Vasconcelos A, Cavaco-Paulo A. Wound dressings for a proteolytic-rich envi-ronment. Appl Microbiol Biotechnol 2011;90:445e60.
[2] Sweeney R, Miraftab M, Collyer G. A critical review of modern and emergingabsorbent dressings used to treat exudingwounds. IntWound J 2012;9:601e12.
[3] Singh AV, Aditi AS, Gade WN, Vats T, Lenardi C, Milani P. Nanomaterials: newgeneration therapeutics in wound healing and tissue repair. Curr Nanosci2010;6:577e86.
[4] Martin JE, Patil AJ, Butler MF, Mann S. Guest-molecule-directed assembly ofmesostructured nanocomposite polymer/organoclay hydrogels. Adv FunctMater 2011;21:674e81.
[5] Kundu B, Kundu SC. Silk sericin/polyacrylamide in situ forming hydrogels fordermal reconstruction. Biomaterials 2012;33:7456e67.
[6] Fullenkamp DE, Rivera JG, Gong Y, Lau KHA, He L, Varchney R, et al. Mussel-inspired silver-releasing antibacterial hydrogels. Biomaterials 2012;33:3783e91.
[7] Khoo X, Simons EJ, Chiang HH, Hickey JM, Sabharwal V, Pelton SI, et al. For-mulations for trans-timpanic antibiotic delivery. Biomaterials 2012;34:1281e8.
[8] Wong B, Boyer C, Steinbeck C, Peters D, Schmidt J, van Zanten R, et al. Designand in situ characterization of lipid containers with enhanced drug retention.Adv Mater 2011;23:2320e5.
[9] Hui A, Sheardown H, Jones L. Acetic and acrylic acid molecular imprintedmodel silicone hydrogel materials for ciprofloxacin-HCl delivery. Materials2012;5:85e107.
[10] Budai L, Hajdu M, Budai M, Grof P, Beni S, Noszal B, et al. Gels and liposomes inoptimized ocular drug delivery: studies on ciprofloxacin formulations. Int JPharm 2007;343:34e40.
[11] Hatefi A, Amsden B. Biodegradable injectable in situ forming drug deliverysystems. J Control Release 2002;80:9e28.
[12] Chung HJ, Park TG. Self-assembled and nanostructured hydrogels for drugdelivery and tissue engineering. Nano Today 2009;4:429e37.
[13] Jayawarna V, Ali M, Jowitt TA, Miller AE, Saiani A, Gough JE, et al. Nano-structured hydrogels for three-dimensional cell culture through self-assemblyof fluorenylmethoxycarbonyl-dipeptides. Adv Mater 2006;18:611e4.
[14] Veiga AS, Sinthuvanich C, Gaspar D, Franquelim HG, Castanho MARB,Schneider JP. Arginine-rich self-assembling peptides as potent antibacterialgels. Biomaterials 2012;33:8907e16.
[15] Castelletto V, Moulton CM, Cheng G, Hamley IW, Hicks MR, Rodger A, et al.Self-assembly of Fmoc-tetrapeptides based on the RGDS cell adhesion motif.Soft Matter 2011;7:11405e15.
[16] Toledano S, Williams RJ, Jayawarna V, Ulijn RV. Enzyme-triggered self-assembly of peptide hydrogels via reversed hydrolysis. J Am Chem Soc2006;128:1070e1.
[17] Liu Y, Tirrell JL, Tsao CY, Wu HC, Javvaji V, Kim E, et al. Biofabricating multi-functional soft matter with enzymes and stimuli-responsive materials. AdvFunct Mater 2012;22:3004e12.
[18] Altunbas A, Lee SJ, Rajasekaran SA, Schneider JP, Pochan DJ. Encapsulation ofcurcumin in self-assembling peptide hydrogels as injectable drug deliveryvehicles. Biomaterials 2011;32:5906e14.
[19] Luo Z, Zhang S. Designer nanomaterials using chiral self-assembling peptidesystems and their emerging benefit for society. Chem Soc Rev 2012;41:4736e54.
[20] Williams RJ, Hall TE, Glattauer V, White J, Pasic PJ, Sorensen AB, et al. Thein vivo performance of an enzyme-assisted self-assembled peptide/proteinhydrogel. Biomaterials 2011;32:5304e10.
[21] Webber MJ, Matson JB, Tamboli VK, Stupp SI. Controlled release of dex-amethasone from peptide nanofiber gels to modulate inflammatory response.Biomaterials 2012;33:6823e32.
[22] Kope�cek JI, Yang J. Peptide-directed self-assembly of hydrogels. Acta Biomater2009;5:805e16.
[23] Adams DJ. Dipeptide and tripeptide conjugates as low-molecular-weighthydrogelators. Macromol Biosci 2011;11:160e73.
[24] Smith AM, Williams RJ, Tang C, Coppo P, Collins RF, Turner ML, et al. Fmoc-diphenylalanine self assembles to a hydrogel via a novel architecture based onpi-pi interlocked beta-sheets. Adv Mater 2008;20:37e41.
[25] Chen L, Revel S, Morris K, Serpell LC, Adams DJ. Effect of molecular structureon the properties of naphthalene-dipeptide hydrogelators. Langmuir 2010;26:13466e71.
[26] Mishra A, Loo YH, Deng RS, Chuah YJ, Hee HT, Ying JY, et al. Ultrasmall naturalpeptides self-assemble to strong temperature-resistant helical fibers in scaf-folds suitable for tissue engineering. Nano Today 2011;6:438.
[27] Orbach R, Adler-Abramovich L, Zigerson S, Mironi-Harpaz I, Seliktar D, Gazit E.Self-assembled Fmoc-peptides as a platform for the formation of nano-structures and hydrogels. Biomacromolecules 2009;10:2646e51.
[28] Wang HM, Yang CH, Tan M, Wang L, Kong DL, Yang ZM. A structure-gelationability study in a short peptide-based ‘Super Hydrogelator’ system. SoftMatter 2011;7:3897e905.
[29] Panda JJ, Dua R, Mishra A, Mittra B, Chauhan VS. 3D cell growth and prolif-eration on a RGD functionalized nanofibrillar hydrogel based on a conforma-tionally restricted residue containing dipeptide. ACS Appl Mater Interfaces2010;2:2839e48.
[30] Mahler A, Reches M, Rechter M, Cohen S, Gazit E. Rigid, self-assembledhydrogel composed of a modified aromatic dipeptide. Adv Mater 2006;18:1365e70.
[31] Marchesan S, Easton CD, Kushkaki F, Waddington L, Hartley PG. Tripeptideself-assembled hydrogels: unexpected twists of chirality. Chem Commun2012;48:2195e7.
[32] Marchesan S, Waddington L, Easton CD, Winkler DA, Goodall L, Forsythe J,et al. Unzipping the role of chirality in nanoscale self-asssembly of tripeptidehydrogels. Nanoscale 2012;4:6752e60.
[33] Clinical and Laboratory Standards Institute/NCCLS Approved Standards forAerobic Bacteria. Performance standards for antimicrobial disk susceptibilitytests. CLSI document (M2-A8). 8th ed. Wayne, PA: CLSI; 2003.
[34] Clements A, Gaboriaud F, Duval JF, Farn JL, Jenney AW, Lithgow T, et al. Themajor surface-associated saccharides of Klebsiella pneumoniae contribute tohost cell association. PLoS One 2008;3:e3817.
[35] Magaldi S, Mata-Essayag S, De Capriles CH, Perez C, Colella MT, Olaizola C,et al. Well diffusion for antifungal susceptibility testing. Int J Infect Dis 2004;8:39e45.
[36] Qu Y, Daley AJ, Istivan TS, Rouch DA, Deighton MA. Densely adherent growthmode, rather than extracellular polymer substance matrix build-up ability,contributes to high resistance of Staphylococcus epidermidis biofilms to an-tibiotics. J Antimicrob Chemother 2010;65:1405e11.
[37] Mizunaga S, Kamiyama T, Fukuda Y, Takahata M, Mitsuyama J. Influence ofinoculum size of Staphylococcus aureus and Pseudomonas aeruginosa onin vitro activities and in vivo efficacy of fluoroquinolones and carbapenems.J Antimicrob Chemother 2005;56:91e6.
[38] Ciornei CD, Sigurdardottir T, Schmidtchen A, Bodelsson M. Antimicrobial andchemoattractant activity, lipopolysaccharide neutralization, cytotoxicity, and

S. Marchesan et al. / Biomaterials 34 (2013) 3678e3687 3687
inhibition by serum of analogs of human cathelicidin LL-37. AntimicrobAgents Chemother 2005;49:2845e50.
[39] Ross DL, Riley CM. Aqueous solubilities of some variously substituted quino-lone antimicrobials. Int J Pharm 1990;63:237e50.
[40] Caco AI, Varanda FT, Pratas de Melo MJ, Dias AMA, Dohrn R, Marrucho IM.Solubility of antibiotics in different solvents. Part II. Non-hydrochloride formsof tetracycline and ciprofloxacin. Ind Eng Chem Res 2008;47:8083e9.
[41] Yang R, Fu Y, Li LD, Liu JM. Medium effects on fluorescence of ciprofloxacinhydrochloride. Spectrochim Acta A Mol Biomol Spectrosc 2003;59:2723e32.
[42] Qin P, Su B, Liu R. Probing the binding of two fluoroquinolones to lysozyme:a combined spectroscopic and docking study. Mol Biosyst 2012;8:1222e9.
[43] AlizadethK,MobarrezM,GanjaliMR,Norouzi P, ChaichiMJ. Spectrofluorimetricstudy of the interaction of ciprofloxacin with amino acids in aqueous solutionfollowing solvatochromic studies. Spectrochim Acta A 2012;94:72e7.
[44] Fjell CD, Hiss JA, Hancock REW, Schneider G. Designing antimicrobial pep-tides: form follows function. Nat Rev Drug Discov 2012;11:37e51.
[45] Nguyen LT, Haney EF, Vogel HJ. The expanding scope of antimicrobial peptidestructures and their modes of action. Trends Biotechnol 2011;29:464e72.




















