
Role of Region C in Regulation of the Heat Shock Gene-Specific Sigma Factor of Escherichia coli, s...
10
JOURNAL OF BACTERIOLOGY, 0021-9193/99/$04.0010 June 1999, p. 3552–3561 Vol. 181, No. 11 Copyright © 1999, American Society for Microbiology. All Rights Reserved. Role of Region C in Regulation of the Heat Shock Gene-Specific Sigma Factor of Escherichia coli, s 32 FLORENCE ARSE ` NE, 1 TOSHIFUMI TOMOYASU, 1 AXEL MOGK, 1 CHRISTIANE SCHIRRA, 2 AGNES SCHULZE-SPECKING, 1 AND BERND BUKAU 1 * Institut fu ¨r Biochemie und Molekularbiologie, Universita ¨t Freiburg, D-79104 Freiburg, 1 and ZMBH, Universita ¨t Heidelberg, INF 282, D-69120 Heidelberg, 2 Germany Received 28 January 1999/Accepted 17 March 1999 Expression of heat shock genes is controlled in Escherichia coli by the antagonistic action of the s 32 subunit of RNA polymerase and the DnaK chaperone system, which inactivates s 32 by stress-dependent association and mediates s 32 degradation by the FtsH protease. A stretch of 23 residues (R122 to Q144) conserved among s 32 homologs, termed region C, was proposed to play a role in s 32 degradation, and peptide analysis identified two potential DnaK binding sites central and peripheral to region C. Region C is thus a prime candidate for mediating stress control of s 32 , a hypothesis that we tested in the present study. A peptide comprising the central DnaK binding site was an excellent substrate for FtsH, while a peptide comprising the peripheral DnaK binding site was a poor substrate. Replacement of a single hydrophobic residue in each DnaK binding site by negatively charged residues (I123D and F137E) strongly decreased the binding of the peptides to DnaK and the degradation by FtsH. However, introduction of these and additional region C alterations into the s 32 protein did not affect s 32 degradation in vivo and in vitro or DnaK binding in vitro. These findings do not support a role for region C in s 32 control by DnaK and FtsH. Instead, the s 32 mutants had reduced affinities for RNA polymerase and decreased transcriptional activities in vitro and in vivo. Furthermore, cysteines inserted into region C allowed cysteine-specific cross-linking of s 32 to RNA polymerase. Region C thus confers on s 32 a competitive advantage over other s factors to bind RNA polymerase and thereby contributes to the rapidity of the heat shock response. The major heat shock proteins (HSPs) of Escherichia coli are molecular chaperones and proteases that constitute a cytosolic system for folding, repair and degradation of proteins (5, 6, 11). Their synthesis is induced as part of the cellular heat shock response after exposure to a large variety of stress conditions which appear to have in common the ability to cause protein misfolding (4, 7, 10, 16, 40). When induced by upshift of the cells to a nonlethal temperature (e.g., 42°C), the heat shock response is transient and consists of a rapid induction phase followed by a shutoff phase starting approximately 5 to 10 min. after upshift. Expression of HSPs is positively controlled at the transcrip- tional level by the heat shock promoter-specific s 32 subunit of RNA polymerase, encoded by rpoH (4, 11, 42). Stress-depen- dent changes in heat shock gene expression are mediated by the antagonistic action of s 32 and negative modulators which act upon s 32 (34–36). These modulators are the DnaK chap- erone and its DnaJ and GrpE cochaperones, which inactivate s 32 by direct association and mediate its degradation by pro- teases (8, 9, 20, 21, 34, 35, 38). Degradation of s 32 is mediated mainly by FtsH (HflB), an ATP-dependent metalloprotease associated with the inner membrane (14, 37, 39, 40). FtsH degrades free s 32 but not RNA polymerase-bound s 32 , indi- cating that protease and RNA polymerase compete for binding to s 32 (40). The role of the chaperones in s 32 degradation is poorly understood. Inactivation of s 32 occurs by association of DnaK and DnaJ with the free form of s 32 , thereby preventing its binding to RNA polymerase (8, 9, 20, 22). There is increas- ing evidence that the sequestration of the DnaK chaperone system through binding to misfolded proteins is a direct deter- minant of the induction of the heat shock response (4, 7, 37, 40). Conversely, the shutoff of the heat shock response is as- sumed to result from HSP-mediated repair and degradation of misfolded proteins, which frees the DnaK chaperone system to inactivate s 32 and to promote its degradation. Furthermore, a competition may exist in vivo between s 32 and other sigma factors including s 70 for association with RNA polymerase. This competition is subject to stress-dependent changes and, consequently, leads to alterations in transcriptional activity of s 32 (2). A central open question is the identity of the binding sites within s 32 for DnaK, DnaJ, FtsH, and the core of RNA poly- merase and the functional interplay between these sites. Pre- vious work showed that the in vivo half-life of fusions between N-terminal fragments of s 32 and b-galactosidase increased when a stretch of 23 residues (R122 to Q144), located between conserved regions 2 and 3 of s 32 and termed region C (Fig. 1), is deleted or replaced by other residues (27). Within region C, a segment of 9 amino acids between residues 132 and 140 of s 32 (QRKLFFNLR) is almost entirely conserved within s 32 homologs but not other sigma factors; it was therefore termed the RpoH box (28). This specific conservation strongly suggests a regulatory role for the RpoH box. Consistent with this as- sumption were the results of a study in which a s 32 -derived peptide library was screened for DnaK binding sites. A high- affinity DnaK binding site exists within the RpoH box in the center of region C, and a second binding site was found close to the RpoH box at the periphery of region C (between resi- dues L118 and K125) (25). Based on this peptide analysis, the RpoH box, and possibly the peripheral DnaK binding motif, is a prime candidate for a regulatory site within the s 32 protein which allows binding of DnaK and possibly also degradation by FtsH (25). * Corresponding author. Mailing address: Institut fu ¨r Biochemie und Molekularbiologie, Universita ¨t Freiburg, Hermann-Herder Str. 7, D-79104 Freiburg, Germany. Phone: 49-761 203 52 22. Fax: 49-761 203 52 57. E-mail: [email protected]. 3552 on August 5, 2015 by guest http://jb.asm.org/ Downloaded from
-
Upload
uni-heidelberg -
Category
Documents
-
view
1 -
download
0
Transcript of Role of Region C in Regulation of the Heat Shock Gene-Specific Sigma Factor of Escherichia coli, s...

JOURNAL OF BACTERIOLOGY,0021-9193/99/$04.0010
June 1999, p. 3552–3561 Vol. 181, No. 11
Copyright © 1999, American Society for Microbiology. All Rights Reserved.
Role of Region C in Regulation of the Heat Shock Gene-SpecificSigma Factor of Escherichia coli, s32
FLORENCE ARSENE,1 TOSHIFUMI TOMOYASU,1 AXEL MOGK,1 CHRISTIANE SCHIRRA,2
AGNES SCHULZE-SPECKING,1 AND BERND BUKAU1*
Institut fur Biochemie und Molekularbiologie, Universitat Freiburg, D-79104 Freiburg,1 and ZMBH,Universitat Heidelberg, INF 282, D-69120 Heidelberg,2 Germany
Received 28 January 1999/Accepted 17 March 1999
Expression of heat shock genes is controlled in Escherichia coli by the antagonistic action of the s32 subunitof RNA polymerase and the DnaK chaperone system, which inactivates s32 by stress-dependent associationand mediates s32 degradation by the FtsH protease. A stretch of 23 residues (R122 to Q144) conserved amongs32 homologs, termed region C, was proposed to play a role in s32 degradation, and peptide analysis identifiedtwo potential DnaK binding sites central and peripheral to region C. Region C is thus a prime candidate formediating stress control of s32, a hypothesis that we tested in the present study. A peptide comprising thecentral DnaK binding site was an excellent substrate for FtsH, while a peptide comprising the peripheral DnaKbinding site was a poor substrate. Replacement of a single hydrophobic residue in each DnaK binding site bynegatively charged residues (I123D and F137E) strongly decreased the binding of the peptides to DnaK and thedegradation by FtsH. However, introduction of these and additional region C alterations into the s32 proteindid not affect s32 degradation in vivo and in vitro or DnaK binding in vitro. These findings do not support arole for region C in s32 control by DnaK and FtsH. Instead, the s32 mutants had reduced affinities for RNApolymerase and decreased transcriptional activities in vitro and in vivo. Furthermore, cysteines inserted intoregion C allowed cysteine-specific cross-linking of s32 to RNA polymerase. Region C thus confers on s32 acompetitive advantage over other s factors to bind RNA polymerase and thereby contributes to the rapidity ofthe heat shock response.
The major heat shock proteins (HSPs) of Escherichia coli aremolecular chaperones and proteases that constitute a cytosolicsystem for folding, repair and degradation of proteins (5, 6,11). Their synthesis is induced as part of the cellular heat shockresponse after exposure to a large variety of stress conditionswhich appear to have in common the ability to cause proteinmisfolding (4, 7, 10, 16, 40). When induced by upshift of thecells to a nonlethal temperature (e.g., 42°C), the heat shockresponse is transient and consists of a rapid induction phasefollowed by a shutoff phase starting approximately 5 to 10 min.after upshift.
Expression of HSPs is positively controlled at the transcrip-tional level by the heat shock promoter-specific s32 subunit ofRNA polymerase, encoded by rpoH (4, 11, 42). Stress-depen-dent changes in heat shock gene expression are mediated bythe antagonistic action of s32 and negative modulators whichact upon s32 (34–36). These modulators are the DnaK chap-erone and its DnaJ and GrpE cochaperones, which inactivates32 by direct association and mediate its degradation by pro-teases (8, 9, 20, 21, 34, 35, 38). Degradation of s32 is mediatedmainly by FtsH (HflB), an ATP-dependent metalloproteaseassociated with the inner membrane (14, 37, 39, 40). FtsHdegrades free s32 but not RNA polymerase-bound s32, indi-cating that protease and RNA polymerase compete for bindingto s32 (40). The role of the chaperones in s32 degradation ispoorly understood. Inactivation of s32 occurs by association ofDnaK and DnaJ with the free form of s32, thereby preventingits binding to RNA polymerase (8, 9, 20, 22). There is increas-ing evidence that the sequestration of the DnaK chaperone
system through binding to misfolded proteins is a direct deter-minant of the induction of the heat shock response (4, 7, 37,40). Conversely, the shutoff of the heat shock response is as-sumed to result from HSP-mediated repair and degradation ofmisfolded proteins, which frees the DnaK chaperone system toinactivate s32 and to promote its degradation. Furthermore, acompetition may exist in vivo between s32 and other sigmafactors including s70 for association with RNA polymerase.This competition is subject to stress-dependent changes and,consequently, leads to alterations in transcriptional activity ofs32 (2).
A central open question is the identity of the binding siteswithin s32 for DnaK, DnaJ, FtsH, and the core of RNA poly-merase and the functional interplay between these sites. Pre-vious work showed that the in vivo half-life of fusions betweenN-terminal fragments of s32 and b-galactosidase increasedwhen a stretch of 23 residues (R122 to Q144), located betweenconserved regions 2 and 3 of s32 and termed region C (Fig. 1),is deleted or replaced by other residues (27). Within region C,a segment of 9 amino acids between residues 132 and 140 ofs32 (QRKLFFNLR) is almost entirely conserved within s32
homologs but not other sigma factors; it was therefore termedthe RpoH box (28). This specific conservation strongly suggestsa regulatory role for the RpoH box. Consistent with this as-sumption were the results of a study in which a s32-derivedpeptide library was screened for DnaK binding sites. A high-affinity DnaK binding site exists within the RpoH box in thecenter of region C, and a second binding site was found closeto the RpoH box at the periphery of region C (between resi-dues L118 and K125) (25). Based on this peptide analysis, theRpoH box, and possibly the peripheral DnaK binding motif, isa prime candidate for a regulatory site within the s32 proteinwhich allows binding of DnaK and possibly also degradation byFtsH (25).
* Corresponding author. Mailing address: Institut fur Biochemieund Molekularbiologie, Universitat Freiburg, Hermann-Herder Str. 7,D-79104 Freiburg, Germany. Phone: 49-761 203 52 22. Fax: 49-761 20352 57. E-mail: [email protected].
3552
on August 5, 2015 by guest
http://jb.asm.org/
Dow
nloaded from

The aim of the present study is to experimentally investigatethe regulatory role of region C, in particular of the two DnaKbinding motifs. In contrast to our expectations, we did not findevidence for a role of region C in chaperone binding anddegradation by FtsH. Instead, we found that region C wasinvolved in high-affinity binding of s32 to RNA polymerase,thereby providing to s32 a competitive advantage over othersigma factors in association with RNA polymerase.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions. Cells of strains BB2019[GW1000 recA441 sulA11 D(argF-lac)U169 supC(Ts) rpoH165(Am) pDMI,1] (8)and BB7089 (C600 thr-1 leuB6 thi-1 lacY supE44 rfbD1 fhuA21 lacIq PA1/lacO-1dnaKJ)(40) were grown aerobically at 30 or 42°C in Luria broth or M9 minimal mediumsupplemented with 0.2% glucose (M9-Glu) or 0.2% maltose (M9-Mal) as thecarbon source, thiamine (20 mg/ml), and appropriate amino acids (50 mg/ml).The growth media were also supplemented with 100 mM isopropyl-b-D-thioga-lactopyranoside (IPTG) for BB7089 and kanamycin (40 mg/ml) and ampicillin(100 mg/ml) when required. The wild-type rpoH gene cloned into plasmidpUHE21-2fdD12 (9) was used as template for mutagenesis by the method ofKunkel et al. (17). pBAD30 (rpoH) (wild type or mutant) was obtained by cloningthe EcoRI-HindIII fragment from pUHE21-2fdD12 (rpoH) into pBAD30 (12).For production of hexahistidine-tagged s32 variants, wild-type and mutant allelesof rpoH were subcloned by inserting the EcoRI-PstI fragment into pUHE211-1(rpoH) (carboxy-terminal histidine fusion, used for all mutant s32) or theMluI-HindIII fragment into pUHE212-1 (amino-terminal histidine fusion, usedfor s32-F137E) (8, 9). For C-terminal fusion of s32 to the intein-chitin bindingdomain, we amplified by PCR a 0.3-kb fragment corresponding to the codingsequence of the C-terminal 80 residues of s32, using primers P1 (59-GGAATTCTGCAGGATAAATCATCTAAC) and P2 (59-GGGGTACCCTTGGCAAAGCACGCTTCAATGGCAGCACGC) and pUHE21-2fdD12 (rpoH) as the tem-plate. The P1 sequence comprises internal coding sequences of rpoH containingan additional EcoRI site at the 59 end. The 39 end of P2 is complementary to the39-end coding sequence of rpoH (from the codon for amino acid 276 to the stopcodon). The 59 end of P2 is complementary to the sequence coding for intein (theN-terminal 6 amino acids) and contains a KpnI site. The sequence complemen-tary to the stop codon of rpoH (CAT) was changed to CAC, allowing us togenetically fuse s32 with the first cysteine of intein. The EcoRI-PstI fragmentfrom pCYB1 (New England Biolabs) was cloned into pUHE21-2fdD12 by usingas the linker a small DNA fragment containing a multiple-cloning site (XhoI,BglII, and HindIII). The 0.3-kb fragment obtained by PCR was cloned in thispUHE21-2fdD12 derivative (EcoRI-KpnI). Finally, the 59 end of the rpoH gene(EcoRI-PstI) (wild-type or mutant alleles) was cloned into the resulting plasmid.
Half-life determination. For determination of s32 stability in strain BB2019,the cells were grown at 30°C in M9-Glu without methionine or IPTG to anoptical density at 600 nm of 0.4. A 1-ml volume of culture was labeled for 2 minwith 70 mCi of [35S]methionine, and this was followed by chase with unlabeledmethionine (final concentration, 200 mg/ml). Aliquots of 100 ml were collected atthe indicated times, mixed with 10% (vol/vol) trichloroacetic acid (TCA), andincubated for 15 min on ice. After centrifugation for 15 min at 16.000 3 g, thepellets were washed with acetone and resuspended in 50 mM Tris-HCl (pH8.0)–1% sodium dodecyl sulfate (SDS)–1 mM EDTA. Half of each sample wassubjected to SDS-polyacrylamide gel electrophoresis (PAGE), and the other halfwas used for immunoprecipitation with s32-specific rabbit antiserum as describedpreviously (41). The immunoprecipitated material was separated by SDS-PAGEand quantified by phosphorimaging with MacBas software (Fuji Film Co.). For
half-life determination of s32 in strain BB7089, the cells were grown in M9-Malwithout methionine but with 100 mM IPTG to an optical density at 600 nm of 0.4.Expression of rpoH was induced by addition of arabinose (100 mM) for 5 min.Then [35S]methionine to label the cells and glucose (0.4%) to stop furtherexpression of rpoH were added, and the chase and immunoprecipitation stepswere performed as described above.
Protein and peptide purification. Histidine-tagged s32 proteins were purifiedessentially as described previously (8), except that cell lysis was performed withthe French press (500 lb/in2) and buffer containing 100 mM NaCl, 20 mMTris-HCl (pH 7.9), 0.05% sodium deoxycholate, and 1 mM phenylmethylsulfonylfluoride. After elution from Ni21-nitrilotriacetic acid columns, the pooled frac-tions containing s32 were subjected to gel filtration on a Superose 12 column(Pharmacia) with a mobile buffer containing 40 mM HEPES-KOH (pH 8.0), 100mM KCl, 0.1 mM EDTA, 10% glycerol, and 1 mM b-mercaptoethanol. A finalpurification step was performed by using a MonoQ column (HR5/5; Pharmacia)and elution of the bound protein by a linear 100 to 1,000 mM KCl gradient.Untagged s32 and s32-F137E were purified as fusion proteins with self-cleavableintein-chitin binding domains on chitin affinity columns as specified by the sup-plier (New England Biolabs), except that a different running buffer was used (20mM Hepes-KOH, 500 mM NaCl, 0.5% Triton X-100) and an additional washingstep of the cell extract-loaded chitin column with 20 mM HEPES-KOH–500 mMNaCl–5 mM MgCl2–5 mM ATP was performed to elute DnaK bound to s32. Thechitin bound intein-s32 was eluted from the intein moiety with running buffercontaining 50 mM dithiothreitol (DTT), which induces self-splicing, and furtherpurified using a MonoQ column. RNA polymerase (holoenzyme and core), s70,DnaK, DnaJ, and FtsH were purified (purity of approximately 70% for s70 and.90% for the other proteins) as described previously (3, 23, 32, 39). Proteinconcentrations were routinely determined by a Bradford assay (Bio-Rad) withbovine serum albumin (BSA) as the standard and, for s32, calibrated by thebicinchoninic acid protein assay (Pierce). The peptides used were synthesized byR. Franck (ZMBH, University of Heidelberg, Heidelberg, Germany) and (fors32-E115-A131-C/I123D) by Jerini Bio Tools (Berlin, Germany). Peptide con-centrations were determined by measurement of the absorption at 280 nm.
3H labeling of proteins and in vitro degradation assays. To assay the degra-dation of s32, the proteins were labeled with N-succinimidyl-[2,3-3H]propionate(Amersham) as described previously (9), except that free N-succinimidyl-[2,3-3H]propionate was removed by dialysis against transcription buffer (20 mMTris-HCl [pH 8.0], 200 mM KCl, 5 mM MgCl2, 1 mM DTT, 5% glycerol).Degradation of labeled s32 by FtsH was assayed in a purified system adaptedfrom that of Tomoyasu et al. (39). Briefly, FtsH (1.2 mg) preincubated on ice for30 min with 50 mM Zn21–25 mM Tris-acetate (pH 8.0)–2.5 mM magnesiumacetate (final concentration) was mixed with s32 (1 mM, final concentration) ina final volume of 20 ml of reaction buffer (50 mM Tris-acetate [pH 8.0], 5 mMmagnesium acetate, 2 mM b-mercaptoethanol, 50 mM KCl, 5 mM ATP) andincubated at 42°C. Aliquots of 2 ml were withdrawn at the indicated times, mixedwith BSA (0.5 mg/ml) and EDTA (20 mM), and precipitated with TCA (10%,vol/vol). Radioactivity in the supernatant was determined in a scintillation cock-tail (Roth) with a Perkin-Elmer counter. To assay the degradation of peptides,the final volume of the reaction mixture was 60 ml and the concentration ofpeptide was 50 mM. At the indicated time points, aliquots of 18 ml were mixedwith 92 ml of 0.5% trifluoroacetic acid to stop the reaction. Products wereanalyzed by reverse-phase chromatography with a 5 to 80% acetonitrile gradientin 0.1% trifluoroacetic acid.
Analysis of protein interactions. Association of s32 with DnaK, DnaJ, andRNA polymerase core enzyme (RNAP core) was determined by gel filtrationwith a Superdex 200 column essentially as described previously (9). To determinethe association of s32 with RNAP core, 0.5 mM (in competition experiments) or1 mM s32 was incubated with 1.5 mM RNAP core for 10 min at 30°C in tran-scription buffer (20 ml, final volume). To determine the association of s32 withDnaK, 5 mM DnaK was incubated for 2 h at 30°C in transcription buffer (todiscourage oligomerization), mixed with 1 mM s32 in a final volume of 20 ml, andfurther incubated for 30 min at 30°C. These mixtures were shifted to ice, adjustedto 100 ml by addition of transcription buffer which for competition experimentscontained a 10- or 30-fold excess of unlabeled s70 or s32, respectively, and loadedon a Superdex 200 column at 4°C. Labeled s32 was detected in the elutionfractions by liquid scintillation counting.
Cross-linking experiments. To couple the cysteine-specific cross-linker N-(4-azido-2,3,5,6-tetrafluorobenzyl)-3-maleimidylpropionamide (TFPAM-3) to cys-teine-containing s32 mutant proteins, 50 ml of a 20 mM protein solution wasdialyzed against buffer A (20 mM HEPES-KCl [pH 8.0], 200 mM KCl, 5 mMEDTA), incubated for 1 h at 30°C in the dark with a 10-fold molar excess ofTFPAM-3, and then dialyzed against buffer B (20 mM HEPES-KOH [pH 8.0],200 mM KCl, 5 mM MgCl2) to remove free TFPAM-3. All other proteins weredialyzed against buffer B before use. For cross-linking, the proteins were mixedas indicated (150 pmol each) in 20 ml of buffer B and incubated at 30°C for 2 hin the dark. DnaJ (150 pmol) and ATP (5 mM) were added when indicated, andthe mixtures were incubated for a further 10 min on ice. Cross-linking wasinduced on ice by illumination under UV light (360 nm) for 5 min. After additionof sample buffer (18) and boiling for 5 min, the samples were subjected toelectrophoresis on SDS–10% polyacrylamide gels. The gels were silver stained orelectroblotted onto polyvinylidene difluoride membranes (Amersham) for im-munodetection. Immunoblots were developed with a Vistra ECF fluorescence
FIG. 1. Mutational alterations of s32. s32 is shown schematically, with thelocations of conserved regions 1 to 4 and details of region C including the RpoHbox indicated. Sequences of two potential DnaK binding sites identified bypeptide scanning are boxed, and mutational alterations of amino acid residuesintroduced into peptides and/or s32 proteins are indicated.
VOL. 181, 1999 ROLE OF REGION C IN REGULATION OF s32 3553
on August 5, 2015 by guest
http://jb.asm.org/
Dow
nloaded from

Western blotting kit (Amersham, Inc.), using Fluoroimager and MacBas soft-ware (Fuji Film Co.).
RESULTS
In vitro analysis of region C-derived peptides. To investigatewhether region C comprises binding sites for DnaK and FtsHthat are responsible for the control of s32 activity and stability,we designed amino acid alterations predicted to perturb thesesites (Fig. 1). For DnaK, this approach is straightforward sincetwo potential binding sites central and peripheral to region Chad been identified (25). Furthermore, the consensus sequencemotif recognized by DnaK and the sequence features whichprevent DnaK binding have been elucidated. The binding mo-tif consists of a hydrophobic core of up to five consecutivehydrophobic residues flanked by segments enriched in basicresidues (30), features which are compatible with the architec-ture of the substrate binding cavity of DnaK (29, 44). Intro-duction of negatively charged residues into the hydrophobiccore prevent DnaK binding (30). These findings provide arational basis for introducing alterations into the two potentialDnaK binding sites central and peripheral to region C. Recentresults concerning the substrate specificity of FtsH (40a) led usto speculate that this protease also recognizes hydrophobicstretches within protein sequences. We therefore changed hy-drophobic residues within the hydrophobic stretches located inregion C to negatively charged residues in order to perturbDnaK binding and degradation by FtsH.
As an experimental starting point, we used peptides com-prising either the RpoH box (including the central DnaK bind-ing site) or the peripheral DnaK binding site to test the effectsof sequence alterations in vitro. This approach is suitable sincepeptides can bind DnaK with high affinity in an ATP-depen-dent fashion (25, 31) and can be degraded efficiently by FtsHin the presence of ATP (40a). A 21-mer peptide comprising thewild-type sequence of the RpoH box (s32-Q132-Q151-C) hasvery high affinity for DnaK (Kd, 40 nM) and is rapidly degradedby FtsH in the presence of ATP (t1/2, 7 min) (Fig. 2). Toperturb the single DnaK binding motif within the RpoH box,we replaced F137, located in the center of the hydrophobiccore, by E (s32-Q132-Q151-C/F137E). This replacement in-creased the Kd of the s32-DnaK complex by 50-fold (Kd, 2 mM)and strongly reduced the efficiency of degradation by FtsH(t1/2, of 28 min) (Fig. 2). A 18-mer peptide comprising theDnaK binding site located peripheral to region C (s32-E115-A131-C) has high affinity for DnaK (Kd, 0.2 mM) (Fig. 2) but isonly slowly degraded by FtsH in the presence of ATP (t1/2, 60min) (data not shown). A replacement I123 by D, predicted toprevent DnaK binding to its binding site within this peptide(s32-E115-A131-C/I123D), caused a strong decrease in affinityfor DnaK (Kd, 3.4 mM) (Fig. 2) and no observable degradationby FtsH (data not shown).
These results show that at the peptide level, the RpoH boxcontains overlapping or identical recognition sites for DnaKand FtsH which are efficiently perturbed by the F137E ex-change and that the N-terminal end of region C contains abinding site for DnaK which is perturbed by the I123D ex-change.
In vivo activity of s32 mutant proteins with altered region C.The above results formed the basis for a rational design ofmutational alterations of region C within the s32 protein(Fig. 1). rpoH was mutated to introduce the F137E (rpoH-F137E) or I123D mutation (rpoH-I123D), or the rpoH-WRI121,122,123ART mutation. This mutation generates a mu-tant protein with increased similarity to s70. The mutant rpoHgenes were cloned into plasmid pUHE21-2fdD12 (9) such that
their expression is controlled by the IPTG-regulatable PA1/lacO-1
promoter. When produced to the levels used in the experi-ments described below, the three mutant s32 proteins wererecovered in the soluble fractions of cells growing at 30 and42°C (data not shown). The mutational alterations thereforedid not cause structural changes in s32 leading to aggregation,allowing further analysis of the in vivo activities of the mutantproteins.
We first tested the ability of plasmids containing the rpoHmutant alleles to complement the temperature-sensitive growthof rpoH165(Am) mutant cells (BB2019) in liquid culture andon agar plates. The rpoH-WRI121,122,123ART and rpoH-I123D alleles allowed IPTG-dependent complementation ofgrowth at 42°C, but the colonies were smaller than thoseformed by cells expressing wild-type rpoH. The rpoH-F137Emutant allele allowed only partial complementation of growthat 42°C, leading to formation of a reduced number of slow-growing colonies (data not shown).
We then determined in pulse experiments the ability of theplasmid-borne mutant alleles to restore the heat shock re-sponse in rpoH165(Am) cells after a shift from 30 to 42°C. Theinduction of expression of the rpoH-WRI121,122,123ART (datanot shown) and rpoH-I123D (Fig. 3) alleles by IPTG allowedthe induction of expression of heat shock genes at 30 and 42°C.However, the amplitude of the response was two- and fourfold
FIG. 2. DnaK binding and FtsH-mediated degradation of peptides derivedfrom region C. (A) Amino acid sequences of the peptides used. Mutated residuesare boxed. (B) Dissociation constants (Kd) of the peptide-DnaK complexes. TheKd values were determined by peptide titration with fluorescently labeled peptides32-Q132-Q144-C-IAANS as competitor as described previously (25). (C) Invitro degradation of peptides by FtsH. Degradation is shown as a percentage ofthe amount of peptide remaining.
3554 ARSENE ET AL. J. BACTERIOL.
on August 5, 2015 by guest
http://jb.asm.org/
Dow
nloaded from

lower for cells expressing rpoH-WRI121,122,123ART and rpoH-I123D, respectively, than for cells expressing wild-type rpoH.Furthermore, the induction of the heat shock response wasdelayed by 5 to 10 min in cells expressing rpoH-I123D. Theinduction of expression of the rpoH-F137E allele allowed anincrease in heat shock gene expression only after the temper-ature upshift to 42°C, and the induction of the heat shockresponse was delayed (10 min) compared to the response incells expressing wild-type rpoH. However, in cells producingeither one of the mutant proteins, a shutoff phase of the heatshock response was observed, suggesting that the DnaK-medi-ated inactivation of s32 is operative in vivo. These resultscorrelate well with the growth complementation profile of themutant allele. Together, these data indicate that the muta-tional alterations I123D and WRI121,122,123ART of s32
cause only partial regulatory defects of s32 whereas the F137Emutation causes stronger regulatory defects in vivo.
Proteolysis of s32 mutant proteins in vivo. We investigatedthe effects of the mutational alterations in region C on s32
stability by performing pulse-chase experiments followed byimmunoprecipitation of s32. The s32-F137E mutant proteinproduced from a plasmid was stabilized in rpoH165(Am) mu-tant cells at 30°C (t1/2, 30 min) (Fig. 4) and at 42°C (t1/2, 17 min[data not shown]). In comparison, wild-type s32 produced fromplasmids was rapidly degraded after prolonged growth at bothtemperatures (t1/2, ca. 1 min) (Fig. 4).
The above experiments were complicated by our finding thatthe in vivo activity of s32-F137E was low at 30°C (Fig. 3).Consequently, in the rpoH165(Am) mutant cells producings32-F137E, the synthesis of HSPs was reduced compared tothat in cells producing wild-type s32. This reduction includesthe synthesis of DnaK and DnaJ, which are limiting for activityand stability control of s32 (37, 40), and this may profoundlyperturb our stability determinations. We therefore testedwhether the observed stabilization of s32-F137E in rpoH165(Am) mutant cells is due to the low levels of DnaK and DnaJ.For this purpose, s32 stability experiments were performedwith a C600 strain derivative (BB7089) in which DnaK andDnaJ levels can be kept constant by replacement of the chro-mosomal dnaK dnaJ operon by an IPTG-regulatable artificialoperon (40). The concentration of IPTG was adjusted suchthat dnaK and dnaJ were not overexpressed compared to theirexpression in the parental C600 wild-type strain. Under ourexperimental conditions for immunoprecipitation, the chromo-somally encoded wild-type s32 was not detectable due to its lowabundance. Wild-type and mutant rpoH alleles were expressedfrom plasmids under the control of an arabinose-induciblepromoter (pBAD30) (12), which allows for tight repression inthe absence of inducer. This promoter was induced for only ashort time (5 min) with 100 mM arabinose, so that the synthesisof s32 produced from plasmids increased only slightly, justenough to detect labeled s32 by immunoprecipitation. At thisshort time after induction of s32 synthesis, the levels of HSPswere not increased detectably (data not shown). In these cells,only minor differences existed between the half-lives of wild-type s32 (1.3 6 1 min) and the three s32 mutant proteins (1.1 60.1 min for s32-WRI-ART, 2.8 6 2 min for s32-I123D, and1.6 6 1 min for s32-F137E). Figure 4 shows the data fors32-F137E. These results indicate that the mutations intro-duced into region C did not affect the half-life of s32 in vivoprovided that the levels of DnaK and DnaJ were kept constant.
Proteolysis of s32 mutant proteins in vitro. To further sub-stantiate these in vivo results, we investigated the half-life ofeach mutated s32 proteins in vitro by using purified histidine-tagged s32 and FtsH. The three s32 mutant proteins had wild-type-like elution profiles during gel filtration and ion-exchange
chromatography. Furthermore, they were indistinguishablefrom the wild type with respect to the proteolysis pattern ob-tained by partial proteinase K and trypsin digestion (data notshown). We thus have no indication for changes in their overalltertiary structures.
All three mutant proteins (s32-F137E, s32-I123D, and s32-WRI121,122,123ART) were degraded by FtsH in presence ofATP, with similar kinetics to those of wild-type s32 (Fig. 5 anddata not shown). The histidine tags fused to the s32 proteinswere not responsible for the degradation, since the authentics32-F137E mutant protein, purified after cleavage from anintein-chitine fusion, was degraded with the same efficiency asthe tagged derivative (data not shown). Thus, consistent withthe in vivo data, the alterations introduced within region C didnot affect the efficiency of s32 degradation by FtsH in vitro.
DnaK and DnaJ binding to s32 mutant proteins in vitro.Several in vitro approaches were used to test whether DnaKand DnaJ binding to s32 is impaired by the mutational alter-ations in region C. In one approach, we used gel filtration todetect complexes between s32 and the chaperones. 3H-labeledwild-type and mutant s32 proteins, all histidine tag fusions,were incubated with DnaK and subjected to gel filtration to
FIG. 3. Ability of rpoH mutant alleles to restore the heat shock response inrpoH165(Am) cells. Cells of strain BB2019 [rpoH165(Am)] which carry plasmids(pUHE21-2fdD12) expressing wild-type or mutant rpoH were grown in M9-Gluwithout methionine. At midexponential growth phase, expression of plasmid-borne rpoH alleles was induced by IPTG (500 mM) for 10 min. The cultures weresplit and further grown at 30 and 42°C. At the indicated times, aliquots (160 ml)were pulse-labeled with [35S]methionine (7.5 mCi) for 1 min and 40 ml of fivefold-concentrated sample buffer was added. Aliquots were subjected to SDS-PAGE(12% polyacrylamide) followed by development with a phosphorimager. Thepositions of GroEL, DnaK, and s32 are indicated.
VOL. 181, 1999 ROLE OF REGION C IN REGULATION OF s32 3555
on August 5, 2015 by guest
http://jb.asm.org/
Dow
nloaded from

separate DnaK-s32 complexes from free s32 (Fig. 6). Underthe conditions used, approximately 75% of wild-type 3H-la-beled s32 was recovered in complex with DnaK (eluting infractions 12 to 17). The 3H-labeled s32 mutant proteins (s32-F137E, s32-I123D, s32-WRI121,122,123ART) showed similarefficiencies of complex formation, even under chase conditionsin which the complexes were separated after the addition of a30-fold molar excess of unlabeled wild-type s32. Thus, themutations introduced into region C of s32 had no defect inbinding of DnaK. For the s32-F137E mutant protein and wild-type s32, we verified that these results are also valid for au-thentic proteins lacking histidine tags (data not shown). Fur-thermore, no defect in chaperone binding was observed whenthe s32 mutant proteins were incubated with DnaK togetherwith DnaJ in the presence of ATP (data not shown).
In a second approach, we tested the interaction of DnaKwith s32 proteins by a functional assay which relies on theability of substrates to stimulate the ATPase activity of DnaK(25, 31). In single-turnover ATPase assays in the presence ofDnaJ, wild-type s32 and the s32-F137E mutant protein stimu-lated ATP hydrolysis to similar extents (approximately 10-fold)(data not shown). The s32-I123D mutant protein stimulatedATP hydrolysis by DnaK efficiently, although to a slightly lowerlevel compared to the two other proteins. We do not considerthis difference to be significant with respect to the DnaK-s32
interaction.In a third approach, we used plasmon surface resonance
spectroscopy to analyze the ability of the s32 mutant proteinsto interact with DnaJ. This method was used previously to
detect the interaction of DnaJ with wild-type s32 (9). We didnot observe any difference between the wild type and the threes32 mutant proteins in affinity for DnaJ (data not shown).Taken together, these data indicate that none of the muta-tional alterations within region C affects the affinity of DnaKand DnaJ for s32.
RNAP binding of s32 mutant proteins in vitro. Our findingsthat the mutational alterations introduced into region C of s32
failed to show defects in the interaction with FtsH and DnaK-DnaJ led us to search for other roles for this region. Sinceseveral s32 mutant proteins analyzed in this study had defectsin activity, we investigated whether the mutated segments ofregion C are involved in the interaction of s32 with the RNAPcore enzyme. We determined the efficiency of association of3H-labeled s32 with RNAP by using gel filtration. We focusedon the s32-F137E and s32-I123D mutant proteins, since theyshowed functional defects in vivo. The relative amounts of thes32-I123D mutant protein recovered in association with RNAP(eluting in fractions 8 to 15) were similar to those of wild-types32. In contrast, the amounts of s32-F137E recovered in asso-ciation with RNAP were reduced two- to threefold comparedto those of wild-type s32 (data not shown). We then deter-mined the half-lives of the RNAP holoenzymes in chase ex-periments as outlined in Fig. 7A. The amounts of holoenzymescontaining 3H-labeled wild-type or mutant s32 were deter-mined by gel filtration at various time points after the additionof excess unlabeled wild-type s32. Differences in the stability ofthe mutant s32-core complexes were evident immediately afteraddition of the competitor, and 5 min after addition almost no
FIG. 4. In vivo stability of the s32-F137E mutant protein at 30°C. Wild-type s32 and the s32-F137E mutant protein were produced from plasmids containing rpoHand tested for their stabilities in both BB2019 cells expressing the dnaK and dnaJ genes from authentic s32-dependent heat shock promoters (left panel) and BB7089cells expressing the dnaK and dnaJ genes from the IPTG-regulated PA1/lacO-1 promoter. After pulse-labeling with [35S]methionine and a chase step, aliquots were takenat the indicated time points followed by immunoprecipitation of s32 (top). The bottom panels show quantification of the precipitated proteins relative to time zero.Mean values of the results of at least two experiments are given.
3556 ARSENE ET AL. J. BACTERIOL.
on August 5, 2015 by guest
http://jb.asm.org/
Dow
nloaded from

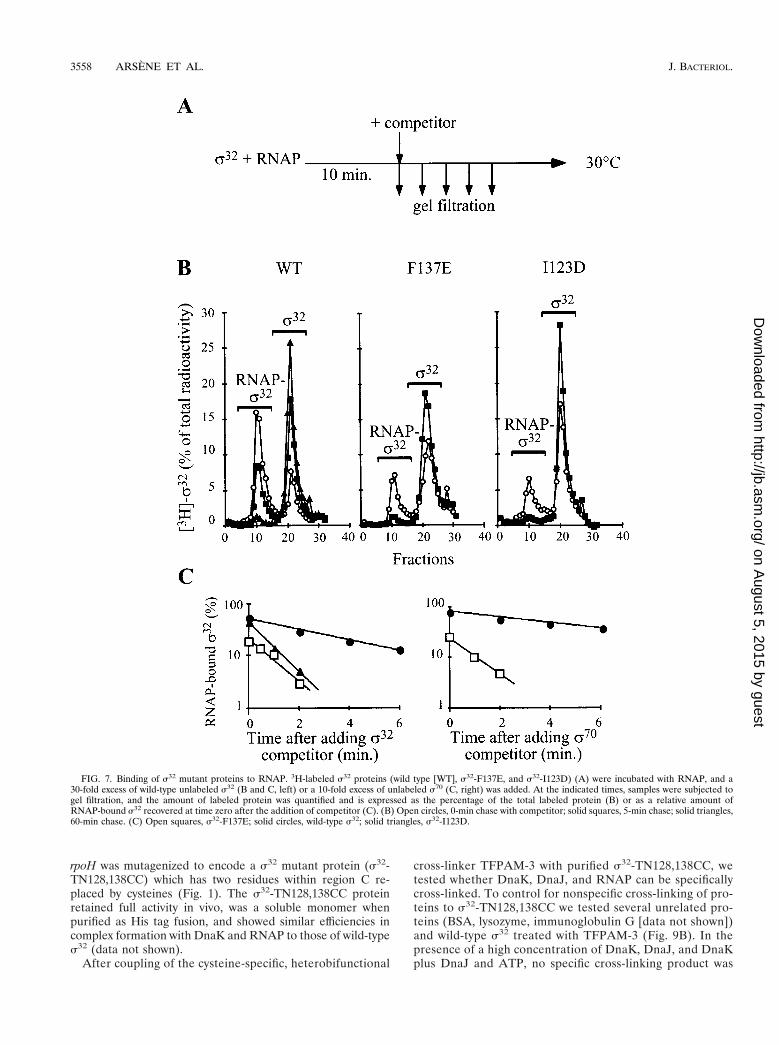
holoenzymes containing the s32-F137E and s32-I123D mutantproteins were recovered (Fig. 7B). We therefore chose shorterchase times to determine the half-lives of the holoenzymes(Fig. 7C, left panel). The half-lives of the holoenzymes con-taining 3H-labeled s32-F137E (0.7 6 0.07 min) and 3H-labeleds32-I123D (0.8 6 0.2 min) were five- and fourfold reduced,respectively, compared to the half-life of the holoenzyme con-
taining the wild-type 3H-labeled s32 (3.5 6 0.2 min). For the3H-labeled s32-F137E mutant protein, we performed addi-tional experiments with a 10-fold excess of s70 as competitorand found a 6-fold decrease in the half-life (6.4 6 0.6 min forthe holoenzyme containing 3H-labeled s32 and 1 6 0.1 min forthe holoenzyme containing 3H-labeled s32-F137E) (Fig. 7C,right panel). These results show that the mutational alterationsin region C decrease the affinity of s32 for RNAP by four- tosixfold.
Transcriptional activity of s32 mutant proteins in vitro. Thereduced affinity of the s32 mutant proteins for RNAP may haveconsequences for their activity in the transcription of heatshock genes, in particular in a situation of competition withother sigma factors. This possibility was tested by runoff tran-scription assays with the s32-dependent P2 heat shock pro-moter of the dnaK dnaJ operon as a template. The reactionswere performed in the presence or absence of s70 as compet-itor, with the s32-F137E and s32-I123D mutant proteins, sincethey had reduced activities in vivo and reduced ability to com-pete with s70 for RNAP binding in vitro. In the absence ofcompetitor, both s32 mutant proteins had strongly reducedactivities compared to wild-type s32 (Fig. 8). Moreover, theaddition of equimolar concentrations of s70 was sufficient tostrongly reduce the activities of both s32 mutant proteins,whereas the activity of wild-type s32 was affected only in thepresence of a fivefold excess of s70 (Fig. 8). The s32-F137E ands32-I123D mutant proteins thus had reduced activities in heatshock gene transcription, which were further reduced in thepresence of s70 as competitor.
Cross-linking of region C of s32 to RNAP. To obtain phys-ical evidence for a role of region C in the association of s32
with RNAP, we determined by cysteine-specific cross-linkingwhether region C is surface exposed within s32 and in proxim-ity to the RNAP in the holoenzyme. This approach was facil-itated by the fact that s32 lacks cysteine, which allowed us tospecifically engineer cysteines into region C. Plasmid-borne
FIG. 5. In vitro degradation of s32 mutant proteins by FtsH. 3H-labeled s32
proteins and 3H-labeled s70 (as control) were incubated with FtsH in the absenceor presence of 5 mM ATP, followed by TCA precipitation at the indicated timepoints. The curves represent the percentage of the radioactivity in the superna-tants which contain the proteolytic fragments. s32 and s32-I123D have C-termi-nal histidine tags, and s32-F137E has an N-terminal histidine tag. N-terminallyand C-terminally histidine tagged s32 did not differ in the kinetics of degradation(data not shown). Open squares, s32-F137E; solid circles, wild-type s32; solidtriangles, s32-I123D; open circles, s70; open triangles, wild-type s32 withoutATP.
FIG. 6. Binding of s32 mutant proteins to DnaK. 3H-labeled s32, s32-F137E, and s32-I123D proteins were incubated with DnaK and the reaction mixtures weresubjected to gel filtration either immediately (2 competitor) or after further incubation with a 30-fold excess of unlabeled wild-type s32 (1 competitor). Labeled proteinwas quantified in the elution fractions and is expressed as a percentage of total radioactivity. The peaks corresponding to the DnaK-s32 complex and free s32 areindicated. Solid circles, wild-type s32; open circles, s32-I123D; solid triangles, s32-F137E.
VOL. 181, 1999 ROLE OF REGION C IN REGULATION OF s32 3557
on August 5, 2015 by guest
http://jb.asm.org/
Dow
nloaded from
rpoH was mutagenized to encode a s32 mutant protein (s32-TN128,138CC) which has two residues within region C re-placed by cysteines (Fig. 1). The s32-TN128,138CC proteinretained full activity in vivo, was a soluble monomer whenpurified as His tag fusion, and showed similar efficiencies incomplex formation with DnaK and RNAP to those of wild-types32 (data not shown).
After coupling of the cysteine-specific, heterobifunctional
cross-linker TFPAM-3 with purified s32-TN128,138CC, wetested whether DnaK, DnaJ, and RNAP can be specificallycross-linked. To control for nonspecific cross-linking of pro-teins to s32-TN128,138CC we tested several unrelated pro-teins (BSA, lysozyme, immunoglobulin G [data not shown])and wild-type s32 treated with TFPAM-3 (Fig. 9B). In thepresence of a high concentration of DnaK, DnaJ, and DnaKplus DnaJ and ATP, no specific cross-linking product was
FIG. 7. Binding of s32 mutant proteins to RNAP. 3H-labeled s32 proteins (wild type [WT], s32-F137E, and s32-I123D) (A) were incubated with RNAP, and a30-fold excess of wild-type unlabeled s32 (B and C, left) or a 10-fold excess of unlabeled s70 (C, right) was added. At the indicated times, samples were subjected togel filtration, and the amount of labeled protein was quantified and is expressed as the percentage of the total labeled protein (B) or as a relative amount ofRNAP-bound s32 recovered at time zero after the addition of competitor (C). (B) Open circles, 0-min chase with competitor; solid squares, 5-min chase; solid triangles,60-min chase. (C) Open squares, s32-F137E; solid circles, wild-type s32; solid triangles, s32-I123D.
3558 ARSENE ET AL. J. BACTERIOL.
on August 5, 2015 by guest
http://jb.asm.org/
Dow
nloaded from

observed in silver-stained gels (Fig. 9A) or after immuno-staining (data not shown). In contrast, three low-abundancecross-linking products of more than 100 kDa (CL1, CL2,and CL3) formed in the presence of RNAP. These productswere not generated when wild-type s32 lacking cysteines was
used (Fig. 9B). Immunostaining revealed that the cross-linking products contain RNAP and s32-TN128,138CC (Fig.9). The amount of the lower-molecular-weight band (CL3)strongly decreased in the presence of an equimolar amountof wild-type s32 as competitor and thus shows specificity(Fig. 9A). The amounts of the other two cross-linking prod-ucts (CL1 and CL2) showed only a slight but detectablereduction upon addition of s32. These results are consistentwith a direct role of region C in the association of s32 withRNAP but not with DnaK and DnaJ.
DISCUSSION
The aim of this study was to establish whether region C playsa role in the regulation of s32. Such a role had been suggestedby (i) the high conservation of region C, in particular of thenonameric RpoH box, specifically among s32 homologs (28);(ii) demonstration that the in vivo stability of protein fusionsbetween s32 segments and b-galactosidase strongly increaseswhen region C is deleted or replaced by other residues (27);and (iii) identification at the peptide level of two high-affinitybinding sites for DnaK within the RpoH box and peripheral toregion C (25).
In view of the above evidence, it seemed plausible to postu-late that region C provides recognition sites for DnaK andFtsH which allow the regulation of s32 activity and stability. Toanalyze this possibility, we mutationally altered region C by
FIG. 8. In vitro transcriptional activity of s32 mutant proteins. Runoff tran-scription assays were performed in transcription buffer (20 mM Tris-HCl [pH8.0], 200 mM KCl, 5 mM MgCl2, 1 mM DTT, 5% glycerol) as described previ-ously (9), with a template consisting of a linear 360-bp DNA fragment (bluntends) containing the P2 promoter of dnaK. The transcription assay mixturescontained 120 nM RNAP, s70, wild-type s32, s32-F137E, or s32-I123D as indi-cated. 11, fivefold molar excess (600 nM) of the corresponding protein over theother proteins in the assay. Transcripts were analyzed by polyacrylamide-urea gelelectrophoresis (9) followed by autoradiography. The relative amounts of tran-scripts were quantified and are expressed as a percentage of the transcriptobtained with wild-type s32 in the absence of s70 (defined as 100%).
FIG. 9. Cysteine-specific cross-linking of s32-TN128,138CC with RNAP. RNAP, DnaK, wild-type s32, or s32-TN128,138CC proteins were mixed as indicated (1,150 pmol; 11, 450 pmol). After exposure to UV light, the samples were separated by SDS-PAGE followed by silver staining or immunostaining with s32- orRNAP-specific antiserum. Cross-linking products (CL1, CL2, and CL3), DnaK, DnaJ, s32, and subunits of RNAP (a, b, b9) are indicated.
VOL. 181, 1999 ROLE OF REGION C IN REGULATION OF s32 3559
on August 5, 2015 by guest
http://jb.asm.org/
Dow
nloaded from

rational design. Using peptides, McCarty et al. found that a31-mer peptide comprising the wild-type sequence of region Cis a high-affinity substrate for DnaK (Kd, ca. 80 nM) (25). Thisqualifies region C as recognition site for DnaK. We found thatthe 21-mer peptide comprising only the RpoH box is an excel-lent substrate for DnaK (Kd, 40 nM), in accordance with ourearlier findings (25), but also for FtsH (t1/2, 7 min). Replace-ment of a single hydrophobic residue in this 21-mer peptide,positioned in the hydrophobic core segment of the DnaK bind-ing motif, by a negatively charged residue (F137E) stronglydecreased the affinity for DnaK (Kd, 2 mM) and the efficiencyof degradation by FtsH (t1/2, 28 min). To our knowledge, thisis the first evidence that a protease and a chaperone recognizethe same sequence stretch within a substrate, possibly estab-lishing a competitive relationship allowing kinetic partitioningof the substrate between the chaperone and the protease.
The identification of amino acid substitutions within regionC peptides that affect the recognition of DnaK and FtsH pro-vided a rational basis for specific mutagenesis of s32. It wassurprising that s32 mutant proteins carrying the I123D andF137E substitutions, as well as a third mutant protein carryingthe WRI121,122,123ART substitution which renders the s32
sequence more similar to s70, showed no defects in affinity forDnaK or in degradation by FtsH. With respect to DnaK, wild-type-like interactions with the s32 mutant proteins were foundin vitro by gel filtration, surface plasmon resonance spectros-copy, and a functional assay for substrate binding to DnaK.Furthermore, in cells producing s32-F137E, s32-I123D, or s32-WRI121,122,123ART, a DnaK-mediated shutoff of the heatshock response was still observed. However, in these cells theamplitude of the heat shock response was reduced, and in cellsproducing the s32-F137E and s32-I123D mutant proteins theinduction kinetics were slower than in cells expressing wild-type s32. These differences are assumed to result from reducedaffinities of the s32 mutant proteins for RNAP (see below).With respect to proteolytic susceptibility, the half-life of eachs32 mutant protein was almost normal in vivo at 30°C, providedthat the DnaK and DnaJ levels were adjusted, and in vitro inFtsH- and ATP-dependent degradation assays. It is importantto note that the amino acid substitutions in the three s32
mutant proteins do not appear to cause overall structuralchanges, as evidenced by the unaltered partial proteolysis pat-tern and solubility of the proteins. It is therefore unlikely thatthe ability of DnaK and FtsH to recognize the s32 mutantproteins is caused by exposure of novel recognition sites in-duced by unfolding. Together, our data indicate that region Cis not a regulatory site in s32 that is essential for binding ofDnaK and degradation by FtsH. Recently, it was proposed thatthe C termini of protein substrates including lcI and s32 aredeterminants for degradation by FtsH (1, 13), although recentexperiments from our laboratory question the importance ofthe C terminus of s32 for degradation (unpublished results).
In retrospect, the identification of DnaK and FtsH recogni-tion sites at the peptide level did not lead to elucidation ofessential DnaK and FtsH recognition sites within the s32 pro-tein. Several reasons may account for this failure. It is possiblethat region C plays a nonessential role in the DnaK- andFtsH-dependent regulation. Alternatively, and perhaps moreprobably, the segments within region C that act as recognitionsites for DnaK and FtsH at the peptide level may not beaccessible to interactions with these ligands in the context ofthe folded s32. For DnaK, this may be caused by helix forma-tion by the respective segments in the folded state of s32, aconformation that is incompatible with the architecture of thesubstrate binding cavity (29, 44). It should be emphasized thatthe approach of using peptides as first indicators for potential
chaperone and protease binding sites in protein substrates isnot devalued by our findings. However, as expected for foldedprotein substrates, peptides do not provide information on theaccessibility of such sites in the context of a folded protein.Validation of peptide data by transfer of corresponding muta-tions into the protein substrate is therefore an essential step ofthis approach. Here we investigated two of approximatelyseven major DnaK binding regions within the s32 sequence.Further experiments will be performed to investigate the reg-ulatory potential of the other regions.
Our experimental results provide evidence for a role ofregion C in the interaction of s32 with the core enzyme ofRNAP. This is consistent with a similar conclusion obtainedfrom an independent study of mutational alterations within theentire s32 protein (15). By glycerol gradient sedimentationanalysis, Joo et al. showed that a s32 mutant protein altered inregion C (F136L) has decreased affinity for RNAP and onlypartial transcriptional activity in vitro. However, this study didnot provide quantitative data on the dissociation rate of theRNAP holoenzyme and did not investigate the in vivo activityof the s32 mutant protein and the interaction with FtsH andDnaK. Our present work thus considerably extends the find-ings of Joo et al. We quantified the RNAP binding defects ofthe s32-F137E or s32-I123D mutant proteins by measuring thehalf-lives of the RNAP holoenzymes in the presence of com-petitor. For both proteins, a sixfold-decreased half-life wasobtained compared to that of the holoenzyme containing wild-type s32. These defects in core binding are sufficient to accountfor the reduced activities of the mutant proteins in vivo at 30and 42°C and in runoff transcription in vitro at 30°C. However,the in vivo activity of the s32-F137E mutant protein is moredramatically affected than that of the s32-I123D mutant pro-tein. The reason for this difference is unclear. We speculatethat the s32-F137E mutant protein has additional defects inpromoter recognition.
The ability of region C to increase the affinity of s32 forRNAP has physiological consequences. Region C may providecompetitive advantages of both s32 over other sigma factorsfor RNAP binding and RNAP over DnaK, DnaJ, and FtsH fors32 binding. Region C thereby may increase the efficiency andspeed by which the heat shock response is induced upon tem-perature upshift and the efficiency of heat shock gene tran-scription under steady-state conditions. In fact, only 10 to 30molecules of s32 that exist in a cell growing at 30°C (35) aresufficient to produce HSPs that account for at least 5% of thetotal cytosolic protein (11). The observed delay in the induc-tion of the heat shock response in cells producing the s32-F137E and s32-I123D mutant proteins is consistent with thisproposed role of region C.
Our cross-linking experiments provide the first indicationthat region C is in close proximity to the RNAP. It is possiblethat region C provides directly the physical contacts whichincrease the affinity of s32 for RNAP core. However, since thesequence of region C is absent in other sigma factors notbelonging to the s32 branch, it is clear that this region cannotbe involved in essential contacts to RNAP but that it seems tobe required to enhance affinity. Several regions within thepolypeptide chains of s32 and s70, including the conservedregions 2.1., 2.2., 3, and 4, have been implicated in core inter-action (15, 19, 24, 26, 33, 43). This multitude of potentialinteracting sites suggests a high structural complexity of thesigma factor-RNAP interaction which may be fully understoodonly upon elucidation of the atomic structures of the involvedproteins.
3560 ARSENE ET AL. J. BACTERIOL.
on August 5, 2015 by guest
http://jb.asm.org/
Dow
nloaded from

ACKNOWLEDGMENTS
We thank T. Laufen for assistance with the ATPase assays, S. Ru-diger for determination of the Kd of DnaK-peptide complexes, K. Paalfor analysis by plasmon surface resonance spectroscopy, and M. P.Mayer and T. Ogura for fruitful discussions and comments on themanuscript.
F.A. is a recipient of a Marie Curie training grant of the EEC. Thiswork was support by a grant of the Deutsche Forschungsgemeinschaftto B.B. and the Fonds der Chemischen Industrie.
REFERENCES
1. Blaszczak, A., C. Georgopoulos, and K. Liberek. 1999. On the mechanism ofFtsH-dependent degradation of the s32 transcriptional regulator of Esche-richia coli and the role of the DnaK chaperone machine. Mol. Microbiol.31:157–166.
2. Blaszczak, A., M. Zylicz, C. Georgopoulos, and K. Liberek. 1995. Bothambient temperature and the DnaK chaperone machine modulate the heatshock response in Escherichia coli by regulating the switch between s70 ands32 factors assembled with RNA polymerase. EMBO J. 14:5085–5093.
3. Buchberger, A., A. Valencia, R. McMacken, C. Sander, and B. Bukau. 1994.The chaperone function of DnaK requires the coupling of ATPase activitywith substrate binding through residue E171. EMBO J. 13:1687–1695.
4. Bukau, B. 1993. Regulation of the E. coli heat shock response. Mol. Micro-biol. 9:671–680.
5. Bukau, B. (ed.). 1999. Molecular chaperones and folding catalysts—regula-tion, cellular function and mechanisms. Harwood Academic Publishers, Am-sterdam, The Netherlands.
6. Burkholder, W. F., and M. E. Gottesman. 1999. Genetic evidence for theroles of molecular chaperones in Escherichia coli metabolism, p. 105–138. InB. Bukau (ed.), Molecular chaperones and folding catalysts—regulation,cellular function and mechanisms. Harwood Academic Publishers, Amster-dam, The Netherlands.
7. Craig, E. A., and C. A. Gross. 1991. Is hsp70 the cellular thermometer?Trends Biochem. Sci. 16:135–140.
8. Gamer, J., H. Bujard, and B. Bukau. 1992. Physical interaction between heatshock proteins DnaK, DnaJ, GrpE and the bacterial heat shock transcrip-tional factor s32. Cell 69:833–842.
9. Gamer, J., G. Multhaup, T. Tomoyasu, J. S. McCarty, S. Rudiger, H.-J.Schonfeld, C. Schirra, H. Bujard, and B. Bukau. 1996. A cycle of binding andrelease of the DnaK, DnaJ and GrpE chaperones regulates activity of the E.coli heat shock transcription factor s32. EMBO J. 15:607–617.
10. Goff, S. A., and A. L. Goldberg. 1985. Production of abnormal proteins in E.coli stimulates transcription of lon and other heat shock genes. Cell 4:587–595.
11. Gross, C. A. 1996. Function and regulation of the heat shock proteins, p.1382–1399. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin,K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, andH. E. Umbarger (ed.), Escherichia coli and Salmonella: cellular and molec-ular biology, 2nd ed. ASM Press, Washington, D.C.
12. Guzman, L.-M., D. Belin, M. J. Carson, and J. Beckwith. 1995. Tight regu-lation, modulation, and high-level expression by vectors containing the arab-inose PBad promoter. J. Bacteriol. 177:4121–4130.
13. Herman, C., D. Thevenet, P. Boulox, G. C. Walker, and R. D’Ari. 1998.Degradation of carboxy-terminal-tagged cytoplasmic proteins by the Esche-richia coli protease HflB (FtsH). Genes Dev. 12:1348–1355.
14. Herman, C., D. Thevenet, R. D’Ari, and P. Bouloc. 1995. Degradation ofsigma 32, the heat shock regulator in Escherichia coli, is governed by HflB.Proc. Natl. Acad. Sci. USA 92:3516–3520.
15. Joo, D. M., A. Nolte, R. Calendar, Y. N. Zhou, and D. J. Jin. 1998. Multipleregions on the Escherichia coli heat shock transcription factor s32 determinecore RNA polymerase binding specificity. J. Bacteriol. 180:1095–1102.
16. Kanemori, M., H. Mori, and T. Yura. 1994. Induction of heat shock proteinsby abnormal proteins results from stabilization and not increased synthesis ofsigma 32 in Escherichia coli. J. Bacteriol. 176:5648–5653.
17. Kunkel, T. A., K. Bebenek, and J. McClary. 1991. Efficient site-directedmutagenesis using uracil-containing DNA. Methods. Enzymol. 204:125–139.
18. Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly ofthe head of bacteriophage T4. Nature (London) 227:680–685.
19. Lesley, S. A., and R. R. Burgess. 1989. Characterization of the Escherichiacoli transcription factor sigma 70: localization of a region involved in theinteraction with core RNA polymerase. Biochemistry 28:7728–7734.
20. Liberek, K., T. P. Galitski, M. Zylicz, and C. Georgopoulos. 1992. The DnaKchaperone modulates the heat shock response of Escherichia coli by bindingto the s32 transcription factor. Proc. Natl. Acad. Sci. USA 89:3516–3520.
21. Liberek, K., and C. Georgopoulos. 1993. Autoregulation of the Escherichia
coli heat shock response by the DnaK and DnaJ heat shock proteins. Proc.Natl. Acad. Sci. USA 90:11019–11023.
22. Liberek, K., D. Wall, and C. Georgopoulos. 1995. The DnaJ chaperonecatalytically activates the DnaK chaperone to preferentially bind the sigma32 heat shock transcriptional regulator. Proc. Natl. Acad. Sci. USA 92:6224–6228.
23. Lowe, P. A., D. A. Hager, and R. R. Burgess. 1978. Purification and proper-ties of the s subunit of Escherichia coli DNA-dependent RNA polymerase.Biochemistry 18:1344–1352.
24. Malhotra, A., E. Severinova, and S. A. Darst. 1996. Crystal structure of a s70
subunit fragment from E. coli RNA polymerase. Cell 87:127–136.25. McCarty, J. S., S. Rudiger, H.-J. Schonfeld, J. Schneider-Mergener, K.
Nakahigashi, T. Yura, and B. Bukau. 1996. Regulatory region C of the E. coliheat shock transcription factor, s32, constitutes a DnaK binding site and isconserved among eubacteria. J. Mol. Biol. 256:829–837.
26. Nagai, H., and N. Shimamoto. 1998. Regions of the Escherichia coli primarysigma factor s70 that are involved in interaction with RNA polymerase coreenzyme. Genes Cells 2:725–734.
27. Nagai, H., H. Yuzawa, M. Kanemori, and T. Yura. 1994. A distinct segmentof the s32 polypeptide is involved in DnaK-mediated negative control of theheat shock response in Escherichia coli. Proc. Natl. Acad. Sci. USA 91:10280–10284.
28. Nakahigashi, K., H. Yanagi, and T. Yura. 1995. Isolation and sequenceanalysis of rpoH genes encoding sigma 32 homologs from gram negativebacteria: conserved mRNA and protein segments for heat shock regulation.Nucleic Acids Res. 23:4383–4390.
29. Rudiger, S., A. Buchberger, and B. Bukau. 1997. Interaction of Hsp70 chap-erones with substrates. Nat. Struct. Biol. 4:342–349.
30. Rudiger, S., L. Germeroth, J. Schneider-Mergener, and B. Bukau. 1997.Substrate specificity of the DnaK chaperone determined by screening cellu-lose-bound peptide libraries. EMBO J. 16:1501–1507.
31. Russell, R., R. Jordan, and R. McMacken. 1998. Kinetic characterization ofthe ATPase cycle of the DnaK molecular chaperone. Biochemistry 37:596–607.
32. Schonfeld, H.-J., D. Schmidt, H. Schroder, and B. Bukau. 1995. The DnaKchaperone system of Escherichia coli: quaternary structures and interactionsof the DnaK and GrpE components. J. Biol. Chem. 270:2183–2189.
33. Severinova, E., K. Severinov, D. Fenyo, M. Marr, E. N. Brody, J. W. Roberts,B. T. Chait, and S. A. Darst. 1996. Domain organization of the Escherichiacoli RNA polymerase s70 subunit. J. Mol. Biol. 263:637–647.
34. Straus, D., W. Walter, and C. Gross. 1990. DnaK, DnaJ, and GrpE heatshock proteins negatively regulate heat shock gene expression by controllingthe synthesis and stability of s32. Genes Dev. 4:2202–2209.
35. Straus, D. B., W. A. Walter, and C. A. Gross. 1987. The heat shock responseof E. coli is regulated by changes in the concentration of s32. Nature (Lon-don) 329:348–350.
36. Straus, D. B., W. A. Walter, and C. A. Gross. 1989. The activity of s32 isreduced under conditions of excess heat shock protein production in Esch-erichia coli. Genes Dev. 3:2003–2010.
37. Tatsuta, T., T. Tomoyasu, B. Bukau, M. Kitagawa, H. Mori, K. Karata, andT. Ogura. 1998. Heat shock regulation in the ftsH null mutant of Escherichiacoli: dissection of stability and activity control mechanisms of sigma32 in vivo.Mol. Microbiol. 30:583–593.
38. Tilly, K., N. McKittrick, M. Zylicz, and C. Georgopoulos. 1983. The DnaKprotein modulates the heat-shock response of Escherichia coli. Cell 34:641–646.
39. Tomoyasu, T., J. Gamer, B. Bukau, M. Kanemori, H. Mori, A. J. Rutman,A. B. Oppenheim, T. Yura, K. Yamanaka, H. Niki, S. Hiraga, and T. Ogura.1995. Escherichia coli FtsH is a membrane-bound, ATP-dependent proteasewhich degrades the heat-shock transcription factor sigma 32. EMBO J.14:2551–2560.
40. Tomoyasu, T., T. Ogura, T. Tatsuta, and B. Bukau. 1998. Levels of DnaKand DnaJ provide tight control of heat shock gene expression and proteinrepair in E. coli. Mol. Microbiol. 30:567–581.
40a.Tomoyasu, T., and B. Bukau. Unpublished data.41. Yano, R., H. Nagai, K. Shiba, and T. Yura. 1990. A mutation that enhances
synthesis of sigma 32 and suppresses temperature-sensitive growth of therpoH15 mutant of Escherichia coli. J. Bacteriol. 172:2124–2130.
42. Yura, T., H. Nagai, and H. Mori. 1993. Regulation of the heat-shock re-sponse in bacteria. Annu. Rev. Microbiol. 47:321–350.
43. Zhou, Y. N., W. A. Walter, and C. A. Gross. 1992. A mutant sigma 32 with asmall deletion in conserved region 3 of sigma has reduced affinity for coreRNA polymerase. J. Bacteriol. 174:5005–5012.
44. Zhu, X., X. Zhao, W. F. Burkholder, A. Gragerov, C. M. Ogata, M. Gottes-man, and W. A. Hendrickson. 1996. Structural analysis of substrate bindingby the molecular chaperone DnaK. Science 272:1606–1614.
VOL. 181, 1999 ROLE OF REGION C IN REGULATION OF s32 3561
on August 5, 2015 by guest
http://jb.asm.org/
Dow
nloaded from




















