
Randomized, placebo-controlled single-ascending-dose study to evaluate the safety, tolerability and...
8
Randomized, Placebo-Controlled, Single-Ascending-Dose Study of BMS-791325, a Hepatitis C Virus (HCV) NS5B Polymerase Inhibitor, in HCV Genotype 1 Infection Karen D. Sims, a Julie Lemm, b Timothy Eley, a Menping Liu, b Anna Berglind, c * Diane Sherman, a Eric Lawitz, d Apinya B. Vutikullird, e Pablo Tebas, f Min Gao, b Claudio Pasquinelli, a Dennis M. Grasela a Bristol-Myers Squibb Research and Development, Hopewell, New Jersey, USA a ; Bristol-Myers Squibb Research and Development, Wallingford, Connecticut, USA b ; Bristol- Myers Squibb Research and Development, Princeton, New Jersey, USA c ; The Texas Liver Institute, University of Texas Health Science Center, San Antonio, Texas, USA d ; WCCT Global LLC, Cypress, California, USA e ; University of Pennsylvania, Philadelphia, Pennsylvania, USA f BMS-791325 is a nonnucleoside inhibitor of hepatitis C virus (HCV) NS5B polymerase with low-nanomolar potency against ge- notypes 1a (50% effective concentration [EC 50 ], 3 nM) and 1b (EC 50 , 7 nM) in vitro. BMS-791325 safety, pharmacokinetics, and antiviral activity were evaluated in a double-blind, placebo-controlled, single-ascending-dose study in 24 patients (interferon naive and experienced) with chronic HCV genotype 1 infection, randomized (5:1) to receive a single dose of BMS-791325 (100, 300, 600, or 900 mg) or placebo. The prevalence and phenotype of HCV variants at baseline and specific posttreatment time points were assessed. Antiviral activity was observed in all cohorts, with a mean HCV RNA decline of 2.5 log 10 copies/ml ob- served 24 h after a single 300-mg dose. Mean plasma half-life among cohorts was 7 to 9 h; individual 24-hour levels exceeded the protein-adjusted EC 90 for genotype 1 at all doses. BMS-791325 was generally well tolerated, with no serious adverse events or discontinuations. Enrichment for resistance variants was not observed at 100 to 600 mg. At 900 mg, variants (P495L/S) associ- ated with BMS-791325 resistance in vitro were transiently observed in one patient, concurrent with an observed HCV RNA de- cline of 3.4 log 10 IU/ml, but were replaced with wild type by 48 h. Single doses of BMS-791325 were well tolerated; demonstrated rapid, substantial, and exposure-related antiviral activity; displayed dose-related increases in exposure; and showed viral kinetic and pharmacokinetic profiles supportive of once- or twice-daily dosing. These results support its further development in combi- nation with other direct-acting antivirals for HCV genotype 1 infection. (This trial has been registered at ClinicalTrials.gov un- der registration no. NCT00664625.) I t is estimated that between 130 and 170 million people, 2 to 3% of the world population, are chronically infected with hepatitis C virus (HCV) (1). The World Health Organization estimates that HCV infection results in approximately 350,000 deaths each year and is associated with long-term complications such as cirrhosis, liver failure, and hepatocellular carcinoma (2). Up until 2011, the standard of care for treatment of chronic HCV was based on the subcutaneous administration of pegylated alfa interferons (alfa-2a or alfa-2b) for periods of up to 48 weeks in combination with oral ribavirin (IFN-/RBV) (3, 4). However, the efficacy of IFN-/ RBV varies according to both viral and host genotype, specifically with respect to polymorphisms in IL28B (5–7), and is associated with a significant burden of treatment-limiting adverse events (AEs), which result in a high rate of on-treatment dose reductions and discontinuations (8). The poor tolerability of IFN-/RBV also results in its contraindication in approximately 17% of HCV-in- fected patients (9). For the more treatment-refractory genotype 1 infections that predominate in Europe, Japan, and the United States, a newer standard of care has emerged that combines one of the recently approved HCV NS3 protease inhibitors telaprevir and boceprevir with IFN-/RBV for part of the overall treatment (10). Although significantly improving posttreatment sustained viro- logic response rates, the use of either protease inhibitor also sig- nificantly adds to the AE burden by elevating rates of some IFN- /RBV-associated events, such as anemia, and by introducing new signature events, such as on-treatment rash (11). The goal of further improving sustained virologic response while reducing treatment-related toxicities has directed a major and ongoing research effort into the development of new direct- acting HCV antivirals with better tolerability profiles that can be used in all-oral, IFN-/RBV-free combinations to provide shorter and less toxic therapies with better posttreatment outcomes. All- oral regimens would also be expected to extend therapeutic ben- efit to those unable to receive or tolerate IFN-/RBV-based treat- ment, as well as provide an alternative option for those who have previously failed to achieve a sustained response. The HCV NS5B polymerase is an essential enzyme for HCV replication and has been validated as a target for anti-HCV ther- apy in clinical trials using both active-site and allosteric inhibitors (12–14). BMS-791325 (Fig. 1) is an orally bioavailable allosteric nonnucleoside inhibitor of NS5B that binds to the polymerase thumb 1 domain and inhibits HCV replication in genotype 1a and 1b subgenomic replicons at 50% effective concentrations (EC 50 s) of 3 and 7 nM, respectively, as described in the accompanying paper (15, 16). BMS-791325 is metabolized by cytochrome P450 Received 28 November 2013 Returned for modification 2 February 2014 Accepted 5 April 2014 Published ahead of print 14 April 2014 Address correspondence to Karen D. Sims, [email protected]. * Present address: Anna Berglind, AstraZeneca Research and Development, Mölndal, Sweden. Copyright © 2014, American Society for Microbiology. All Rights Reserved. doi:10.1128/AAC.02579-13 3496 aac.asm.org Antimicrobial Agents and Chemotherapy p. 3496 –3503 June 2014 Volume 58 Number 6 on April 23, 2016 by guest http://aac.asm.org/ Downloaded from
Transcript of Randomized, placebo-controlled single-ascending-dose study to evaluate the safety, tolerability and...

Randomized, Placebo-Controlled, Single-Ascending-Dose Study ofBMS-791325, a Hepatitis C Virus (HCV) NS5B Polymerase Inhibitor,in HCV Genotype 1 Infection
Karen D. Sims,a Julie Lemm,b Timothy Eley,a Menping Liu,b Anna Berglind,c* Diane Sherman,a Eric Lawitz,d Apinya B. Vutikullird,e
Pablo Tebas,f Min Gao,b Claudio Pasquinelli,a Dennis M. Graselaa
Bristol-Myers Squibb Research and Development, Hopewell, New Jersey, USAa; Bristol-Myers Squibb Research and Development, Wallingford, Connecticut, USAb; Bristol-Myers Squibb Research and Development, Princeton, New Jersey, USAc; The Texas Liver Institute, University of Texas Health Science Center, San Antonio, Texas, USAd;WCCT Global LLC, Cypress, California, USAe; University of Pennsylvania, Philadelphia, Pennsylvania, USAf
BMS-791325 is a nonnucleoside inhibitor of hepatitis C virus (HCV) NS5B polymerase with low-nanomolar potency against ge-notypes 1a (50% effective concentration [EC50], 3 nM) and 1b (EC50, 7 nM) in vitro. BMS-791325 safety, pharmacokinetics, andantiviral activity were evaluated in a double-blind, placebo-controlled, single-ascending-dose study in 24 patients (interferonnaive and experienced) with chronic HCV genotype 1 infection, randomized (5:1) to receive a single dose of BMS-791325 (100,300, 600, or 900 mg) or placebo. The prevalence and phenotype of HCV variants at baseline and specific posttreatment timepoints were assessed. Antiviral activity was observed in all cohorts, with a mean HCV RNA decline of �2.5 log10 copies/ml ob-served 24 h after a single 300-mg dose. Mean plasma half-life among cohorts was 7 to 9 h; individual 24-hour levels exceeded theprotein-adjusted EC90 for genotype 1 at all doses. BMS-791325 was generally well tolerated, with no serious adverse events ordiscontinuations. Enrichment for resistance variants was not observed at 100 to 600 mg. At 900 mg, variants (P495L/S) associ-ated with BMS-791325 resistance in vitro were transiently observed in one patient, concurrent with an observed HCV RNA de-cline of 3.4 log10 IU/ml, but were replaced with wild type by 48 h. Single doses of BMS-791325 were well tolerated; demonstratedrapid, substantial, and exposure-related antiviral activity; displayed dose-related increases in exposure; and showed viral kineticand pharmacokinetic profiles supportive of once- or twice-daily dosing. These results support its further development in combi-nation with other direct-acting antivirals for HCV genotype 1 infection. (This trial has been registered at ClinicalTrials.gov un-der registration no. NCT00664625.)
It is estimated that between 130 and 170 million people, 2 to 3%of the world population, are chronically infected with hepatitis
C virus (HCV) (1). The World Health Organization estimates thatHCV infection results in approximately 350,000 deaths each yearand is associated with long-term complications such as cirrhosis,liver failure, and hepatocellular carcinoma (2). Up until 2011, thestandard of care for treatment of chronic HCV was based on thesubcutaneous administration of pegylated alfa interferons (alfa-2aor alfa-2b) for periods of up to 48 weeks in combination with oralribavirin (IFN-�/RBV) (3, 4). However, the efficacy of IFN-�/RBV varies according to both viral and host genotype, specificallywith respect to polymorphisms in IL28B (5–7), and is associatedwith a significant burden of treatment-limiting adverse events(AEs), which result in a high rate of on-treatment dose reductionsand discontinuations (8). The poor tolerability of IFN-�/RBV alsoresults in its contraindication in approximately 17% of HCV-in-fected patients (9). For the more treatment-refractory genotype 1infections that predominate in Europe, Japan, and the UnitedStates, a newer standard of care has emerged that combines one ofthe recently approved HCV NS3 protease inhibitors telaprevir andboceprevir with IFN-�/RBV for part of the overall treatment (10).Although significantly improving posttreatment sustained viro-logic response rates, the use of either protease inhibitor also sig-nificantly adds to the AE burden by elevating rates of some IFN-�/RBV-associated events, such as anemia, and by introducing newsignature events, such as on-treatment rash (11).
The goal of further improving sustained virologic responsewhile reducing treatment-related toxicities has directed a major
and ongoing research effort into the development of new direct-acting HCV antivirals with better tolerability profiles that can beused in all-oral, IFN-�/RBV-free combinations to provide shorterand less toxic therapies with better posttreatment outcomes. All-oral regimens would also be expected to extend therapeutic ben-efit to those unable to receive or tolerate IFN-�/RBV-based treat-ment, as well as provide an alternative option for those who havepreviously failed to achieve a sustained response.
The HCV NS5B polymerase is an essential enzyme for HCVreplication and has been validated as a target for anti-HCV ther-apy in clinical trials using both active-site and allosteric inhibitors(12–14). BMS-791325 (Fig. 1) is an orally bioavailable allostericnonnucleoside inhibitor of NS5B that binds to the polymerasethumb 1 domain and inhibits HCV replication in genotype 1a and1b subgenomic replicons at 50% effective concentrations (EC50s)of 3 and 7 nM, respectively, as described in the accompanyingpaper (15, 16). BMS-791325 is metabolized by cytochrome P450
Received 28 November 2013 Returned for modification 2 February 2014Accepted 5 April 2014
Published ahead of print 14 April 2014
Address correspondence to Karen D. Sims, [email protected].
* Present address: Anna Berglind, AstraZeneca Research and Development,Mölndal, Sweden.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
doi:10.1128/AAC.02579-13
3496 aac.asm.org Antimicrobial Agents and Chemotherapy p. 3496 –3503 June 2014 Volume 58 Number 6
on April 23, 2016 by guest
http://aac.asm.org/
Dow
nloaded from

3A4 to an active N-desmethyl metabolite (BMS-794712) that isequipotent with the parent compound.
Here, we report the results of a proof-of-concept, single-as-cending-dose study to evaluate the safety, tolerability, pharmaco-kinetics, and antiviral activity of BMS-791325 in patients withchronic HCV genotype 1 infection, together with population se-quencing, clonal analysis, and phenotyping of baseline and emer-gent (postdose) NS5B sequence variants.
MATERIALS AND METHODSStudy design. This was a double-blind, placebo-controlled, randomized,single-ascending-dose study (study AI443-002; clinicaltrials.gov registra-tion no. NCT00664625) for genotype 1 HCV infection, comprising foursequential dosing cohorts of 100 mg, 300 mg, 600 mg, and 900 mg. Sixpatients in each cohort were randomized 5:1 to receive a single dose oforally administered BMS-791325 or placebo. Dose escalation betweencohorts occurred only after safety data from the previous cohort throughpostdose day 4 were reviewed by the sponsor and investigators. Withineach open dose cohort, randomization to receive BMS-791325 or placebowas made via an interactive voice response telephone system according toa computer-generated scheme prepared by the study sponsor. Initially,the 100-mg, 300-mg, and 900-mg cohorts were recruited sequentially.Subsequently, the 600-mg dosing cohort was enrolled using a higher-strength formulation (described below) to reduce capsule burden.
Study conduct and ethics. The study was conducted at four sites in theUnited States. Patients underwent screening within 28 days prior to ad-ministration of study medication on day 1. Patients who received 100,300, or 900 mg BMS-791325 remained at their clinical centers for the first2 days postdose and were furloughed on day 3; two further outpatientvisits occurred on days 4 and 7. Due to gastrointestinal intolerance at the900-mg dose level (presumed due to the 18 capsules of 50-mg strengthrequired to deliver the dose), patients who subsequently received 600 mgBMS-791325 were administered 4 capsules of 150-mg strength and fur-loughed at day 7, with an outpatient visit at day 14.
The study was conducted according to local regulatory requirementsand in accordance with the principles of the Declaration of Helsinki andwith Good Clinical Practice, as defined by the International Conferenceon Harmonization and the ethical principles underlying the EuropeanUnion Directive 2001/20/EC and the United States Code of Federal Reg-ulations, Title 21, Part 50 (21CFR50). The protocol, amendments, andpatient informed consent were approved by the Institutional ReviewBoard of each participating site. Written informed consent was obtainedfrom all patients prior to study procedures.
Study objectives. The primary objective was to assess the safety andtolerability of a single oral dose of 100 to 900 mg BMS-791325 in patientswith chronic HCV genotype 1 infection. Secondary objectives included
assessment of the pharmacokinetics (PK) of BMS-791325 and its metab-olite BMS-794712, assessment of antiviral activity, and characterization ofresistance-associated NS5B amino acid substitutions following a singledose.
Patients. Eligible patients were men and women aged 18 to 60 yearswith chronic HCV genotype 1 infection, a screening plasma HCV RNAlevel of at least 100,000 IU/ml, and a body mass index between 18 and 35kg/m2. Patients were noncirrhotic (screening FibroTest score of �0.59with an aminotransferase/platelet ratio index of �2 or with absence ofcirrhosis documented by biopsy within the previous 12 months) andcould be either treatment naive or have previously received and discon-tinued alfa interferon, with or without RBV, at least 6 months beforeenrollment. Patients with previous exposure to HCV NS5A or NS5B in-hibitors were excluded, as were patients coinfected with human immuno-deficiency virus or hepatitis B virus or infected with other HCV genotypes.Pregnant or nursing women were also excluded, as were women of child-bearing age unwilling to use contraception from 1 month predosethrough 8 weeks postdose. Men were excluded if unwilling to practicebarrier contraception with female partners for at least 12 weeks postdose.
Dosing. Study drug was administered with water in the fasted state ashard gelatin capsules containing 50 mg (100-mg, 300-mg, and 900-mgdose cohorts) or 150 mg (600-mg cohort) BMS-791325 or placebo. Pa-tients continued to fast for 4 h postdose.
HCV RNA assessments. Plasma HCV RNA levels were determinedusing the Roche TaqMan HCV quantitative assay (lower limit of quanti-tation, 25 IU/ml) at screening, predose, and 2, 4, 6, 8, 12, 16, 24, 36, 48, 72,and 144 h postdose.
Safety assessments. Blood and urine samples for clinical laboratoryevaluations and vital sign measurements were collected at specified timepoints throughout the study. Information about AEs was collected byspontaneous report or elicited by open-ended questioning, examination,or evaluation. Event intensity was categorized by the investigator as mild,moderate, severe, or very severe based on level of incapacitation or inter-ference in everyday activities. A 12-lead electrocardiogram was recordedat the screening visit, on day �1, predose on day 1 and at 0.5, 1, 1.5, 2, 3,4, 6, 8, 12, 24, and 48 h postdose, and on days 7 and 14 postdose. Safetyassessments were based on AE reports and the results of vital sign mea-surements, electrocardiograms, physical examinations, and clinical labo-ratory tests.
Pharmacokinetics. Serial blood samples for PK analyses were ob-tained predose and at 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 16, 24, 48, and 72h postdose. Concentrations of BMS-791325 and the BMS-794712 metab-olite were determined from 100-�l aliquots of EDTA-treated plasma us-ing liquid-liquid extraction and a validated liquid chromatography assaywith tandem mass spectrometry at Tandem Labs (West Trenton, NJ,USA). Analyte concentrations were calculated by linear regression overconcentration ranges of 0.1 to 50.0 ng/ml using BMS-791325–D6 or BMS-794712-13CD3 internal standards. The mass spectrometer was operated inmultiple reaction monitoring mode under optimized conditions for thedetection of positive analyte/internal standard ions formed by electros-pray ionization. Assay precision (percent coefficient of variability) andaccuracy (percent bias) were 4.5 to 6.9% and 0.0 to 6.5%, respectively, forBMS-791325 and 3.8 to 6.8% and 0.0 to 6.0%, respectively, for the BMS-794712 metabolite.
Parameters derived from plasma concentration-versus-time data in-cluded maximum observed concentration (Cmax), time to maximum con-centration, terminal half-life (t1/2), concentration at 24 h postdose (C24),and area under the concentration-time curve extrapolated to infinite time(AUCinf). Dose proportionality was assessed for key BMS-791325 expo-sure parameters (Cmax, AUCinf, and C24) by generating point estimatesand 90% confidence intervals for the slope (�) of the log-transformed(linearized) dose-exposure relationship using a power model.
Pharmacodynamics. Antiviral activity was assessed by the magnitude(mean decline at 24 h and mean maximum decline) of change in log10
plasma HCV RNA levels from the day 1 predose baseline. Exposure-re-
FIG 1 Structure of BMS-791325 and its N-desmethyl metabolite BMS-794712.
BMS-791325 Single-Dose Efficacy and Safety
June 2014 Volume 58 Number 6 aac.asm.org 3497
on April 23, 2016 by guest
http://aac.asm.org/
Dow
nloaded from

sponse relationships for AUCinf, Cmax, and C24 were assessed graphicallyby plotting maximum change from baseline in log10 HCV RNA versusestimated PK parameters and by estimation of Pearson’s correlation co-efficients.
Resistance analyses. Serum samples were analyzed predose and at 12,24, 48, and 144 h postdose for NS5B sequence variants potentially associ-ated with BMS-791325 resistance, particularly codon 495 variants charac-teristically associated with loss of susceptibility to thumb 1 allosteric in-hibitors, including BMS-791325 (16, 17). Viral RNA was extracted usingthe QIAamp MiniElute virus vacuum kit, and first-strand cDNA synthesiswas performed using random hexamer primers and the Superscript IIIfirst-strand synthesis system (Invitrogen). The full NS5B coding regionwas sequenced from amplimers generated by sequential or nested PCRsusing genotype-specific degenerate primers. To determine the sensitivityfor minority populations, mixed wild-type and codon-495 proline-to-leucine (P495L) variant DNAs were sequenced at ratios of 100:0, 90:10,80:20, and 50:50. The P495L variant was readily detected at 20% preva-lence. Relevant amplimers were subcloned using a TOPO TA cloning kit(Invitrogen) and transfected into Escherichia coli for subsequent clonalsequence analysis of individual colonies.
Specific substitutions identified by sequence analysis were assessedphenotypically in genotype 1a or 1b replicon clones by site-directed mu-tagenesis using a Stratagene QuikChange mutagenesis kit according to themanufacturer’s directions. Mutant clones were verified by sequence anal-ysis and evaluated for replication efficiency and compound sensitivityusing transient replicon assays as described elsewhere (16).
To evaluate the phenotype of the entire patient-derived NS5B gene, ashuttle replicon system was developed by creating unique SpeI/ClaI (ge-notype 1a) or SpeI/SnaBI (genotype 1b) restriction sites flanking the NS5Bgene and reamplifying patient-derived NS5B amplicons using oligomerprimers bearing the relevant sites. Clones generated by standard tech-niques were verified by sequence analysis, and multiple clones per timepoint were evaluated for replication efficiency and compound sensitivityusing transient replication assays.
Statistical considerations. Five patients receiving BMS-791325 in adose cohort provided 80% probability of observing at least 1 occurrence ofany AE occurring at a 28% incidence in the overall population.
RESULTSPatients. A total of 198 patients were screened, of whom 24 wererandomized and treated between June 2008 and June 2009. Themajority of screened patients who were not randomized werescreening failures meeting one or more study exclusion criteria(153/174; 88%). A small number of patients withdrew consent orwere lost to follow-up after screening (6/174 [3%] each). Otherreasons for nonrandomization included administrative exclu-sions and failure to attend study day 1. Baseline demographics and
disease characteristics are shown in Table 1, and a patient dispo-sition diagram is shown in Fig. 2. Baseline characteristics weregenerally comparable across all treatment groups. All patientscompleted the study.
Safety and tolerability. BMS-791325 was generally well toler-ated. There were no deaths, serious AEs, discontinuations due toAEs, laboratory or physical examination findings reported as AEs,or clinically relevant changes in vital signs or electrocardiogramparameters. All AEs were of mild intensity except for two moder-ate gastrointestinal events in the 900-mg cohort. All AEs resolvedprior to study completion. There were 14 AEs in total, occurring in10 patients, of which eight events (in five patients) were consid-ered to be at least possibly related to treatment. Treatment-relatedevents in the placebo arm were mild nausea and headache (n � 1)and mild decreased appetite (n � 1). All treatment-related eventson BMS-791325 occurred in the 900-mg cohort: mild vomiting,diarrhea, and headache (n � 1); moderate nausea (n � 1); and
TABLE 1 Baseline demographics and disease characteristics
Characteristic
BMS-791325
Placebo (n � 4)100 mg (n � 5) 300 mg (n � 5) 600 mg (n � 5) 900 mg (n � 5)
Age, median (range), yr 44 (33–59) 43 (24–50) 47 (40–55) 46 (35–50) 49 (41–60)Male, n (%) 5 (100) 5 (100) 3 (60) 3 (60) 2 (50)
Race, n (%)White 4 (80) 5 (100) 5 (100) 5 (100) 4 (100)Black/African American 1 (20) 0 0 0 0
Wt, mean (range), kg 79 (60–91) 82 (61–117) 73 (61–94) 82 (63–91) 75 (67–78)HCV RNA, mean (SD), log10 IU/ml 5.90 (0.48) 5.99 (0.62) 5.86 (0.46) 5.75 (0.53) 5.97 (0.58)Genotype 1a/1b, n/n 5/0 4/1 4/1 3/2 4/0Prior IFN treatment, n (%) 0 (0) 2 (40) 1 (20) 1 (20) 3 (75)
FIG 2 Patient disposition.
Sims et al.
3498 aac.asm.org Antimicrobial Agents and Chemotherapy
on April 23, 2016 by guest
http://aac.asm.org/
Dow
nloaded from

moderate vomiting (n � 1). The high proportion of gastrointes-tinal events in the 900-mg cohort may have been due, at least inpart, to high capsule burden (18 capsules). Events not consideredto be related to treatment were ecchymosis (n � 1; 300 mg), uri-
nary tract infection (n � 1; 900 mg), pruritus (n � 1; 900 mg),application-site erythema and headache (n � 1; 900 mg), and painin extremity (n � 1; placebo). Overall, only nausea (n � 2), vom-iting (n � 2), and headache (n � 2) were reported for more thana single patient.
Pharmacokinetics. Two patients in the BMS-791325 900-mgcohort who experienced vomiting at approximately 2 h postdosewere excluded from the PK analyses. Figure 3 shows mean con-centration-time data for BMS-791325 and the BMS-794712 me-tabolite, and Table 2 summarizes key PK parameters. Peak BMS-791325 concentrations were attained between 2 and 4 h, andcompound t1/2 was 6.8 to 9.4 h. Exposure to BMS-791325 (Cmax
and AUCinf) was dose dependent and appeared slightly more thandose proportional, with point estimates and 90% confidence in-tervals for � of 1.1 (0.99 to 1.22) for Cmax and 1.18 (0.99 to 1.36)for AUCinf. All patients had 24-hour plasma concentrations ex-ceeding the protein-adjusted EC90 for HCV genotype 1. The PKprofile of the BMS-794712 metabolite was generally similar to thatof the parent compound, but the plasma exposure was approxi-mately 22% of the parent value. No assessment of dose propor-tionality was performed for the metabolite.
Antiviral response. Mean and individual changes from base-line in log10 HCV RNA following single doses of BMS-791325 orplacebo are shown in Fig. 4A through Fig. 4F. Antiviral activitywas dose related and appeared to plateau between 300 mg and 600mg, with the majority of the dose-dependent increase seen be-tween 100 mg and 300 mg. Mean maximum log10 HCV RNAdeclines (standard deviations and ranges) were �1.3 (0.54; range,�0.7 to �2.2) at 100 mg, �2.52 (0.55; �1.8 to �2.9) at 300 mg,�2.78 (0.42; �2.2 to �3.4) at 600 mg, and �2.55 (0.79; �1.3 to�3.4) at 900 mg. Mean and median times to maximum declineincreased slightly with dose from 100 mg (mean, 16.8 h; median,12 h) through 600 mg (mean and median, 24 h). Mean time tomaximum decline at 900 mg was 34.4 h (median, 36 h) but with alarge standard deviation (14.3 h). One patient in the 900-mg co-hort showed consistently poorer virologic response than did theothers (Fig. 4F), associated with low BMS-791325 levels at all sam-pling time points (data not shown). This patient was one of thetwo excluded from the PK analyses for early vomiting (2.3 h);however, the cause of the poor drug exposure is uncertain, as
FIG 3 Mean plasma concentration-time profile of BMS-791325 (A) andBMS-794712 metabolite (B) after single oral doses in HCV-infected patients.
TABLE 2 Summary pharmacokinetic parameters for BMS-791325 and BMS-794712 metabolite
Parametera
BMS-791325 BMS-794712
100 mg(n � 5)
300 mg(n � 5)
600 mg(n � 5)
900 mg(n � 3)
100 mg(n � 5)
300 mg(n � 5)
600 mg(n � 5)
900 mg(n � 3)
Cmax, ng/ml, geometricmean (% CV)
1,432 (23) 4,782 (22) 10,199 (20) 16,545 (31) 243.1 (17) 694.1 (47) 1,832 (40) 2,546 (41)
AUCinf, ng · h/ml, geometricmean (% CV)
17,167 (42) 69,360 (23) 112,033 (27) 290,247 (27) 3,651 (40) 13,199 (38) 25,551 (45) 65,637 (31)
tmax, h, median (range) 2.00 (2.0–6.0) 4.00 (2.0–4.0) 4.00 (1.5–4.0) 3.00 (3.0–4.0) 4.0 (2.5–10) 4.0 (4.0–8.0) 4.0 (2.5–4.0) 8.0 (6.0–10)t1/2, h, mean (SD) 8.62 (2.25) 9.37 (1.94) 6.81 (0.91) 7.78 (0.29) 10.3 (2.8) 10.2 (2.1) 7.3 (1.0) 9.0 (0.3)C24, ng/ml, geometric mean
(% CV)161.7 (63) 863.4 (44) 1,149 (49) 5,141 (20) 46.3 (57) 203.7 (57) 318.3 (56) 1,481 (27)
CLT/F, ml/min, geometricmean (% CV)
97.1 (59) 72.1 (26) 89.3 (23) 51.7 (31)
Metabolic ratio, geometricmean (% CV)
0.217 (22) 0.194 (29) 0.233 (23) 0.231 (7)
a Abbreviations: AUCinf, area under the plasma concentration-time curve extrapolated to infinite time; C24, 24-hour plasma concentration; CLT/F, apparent total body clearance;Cmax, maximum plasma concentration; tmax, time to maximum plasma concentration; t1/2, terminal plasma half-life; CV, coefficient of variation.
BMS-791325 Single-Dose Efficacy and Safety
June 2014 Volume 58 Number 6 aac.asm.org 3499
on April 23, 2016 by guest
http://aac.asm.org/
Dow
nloaded from

pre-emesis BMS-791325 levels at 1.0 and 1.5 h postdose were alsolow, and the other excluded patient had a strong virologic re-sponse (maximum RNA decline, �2.86 log10) despite vomiting at1.8 h.
Pharmacodynamic relationships. Individual maximum de-clines in HCV RNA across all dosing cohorts were associated withAUCinf, Cmax, and C12 (Fig. 5), with Pearson’s correlation coeffi-cients of 0.76, 0.76, and 0.75, respectively. A similar association
FIG 4 Mean (A) and individual changes from baseline in HCV RNA following a single dose of placebo (B) or BMS-791325 at 100 mg (C), 300 mg (D), 600 mg(E), and 900 mg (F).
Sims et al.
3500 aac.asm.org Antimicrobial Agents and Chemotherapy
on April 23, 2016 by guest
http://aac.asm.org/
Dow
nloaded from
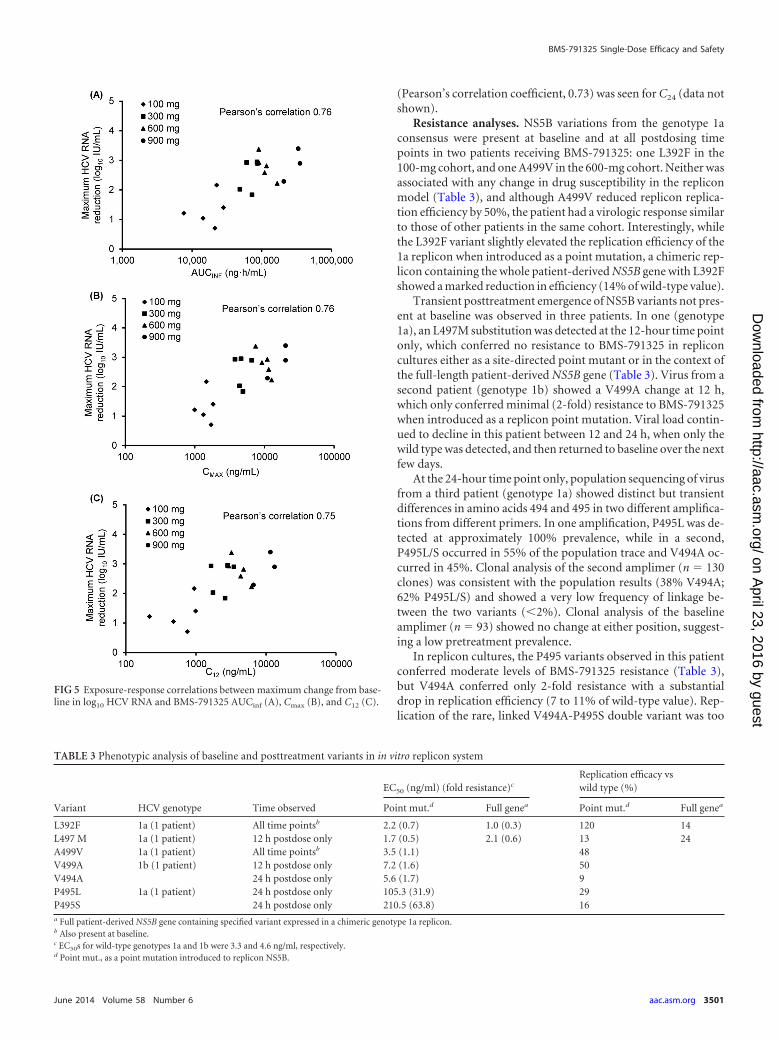
(Pearson’s correlation coefficient, 0.73) was seen for C24 (data notshown).
Resistance analyses. NS5B variations from the genotype 1aconsensus were present at baseline and at all postdosing timepoints in two patients receiving BMS-791325: one L392F in the100-mg cohort, and one A499V in the 600-mg cohort. Neither wasassociated with any change in drug susceptibility in the repliconmodel (Table 3), and although A499V reduced replicon replica-tion efficiency by 50%, the patient had a virologic response similarto those of other patients in the same cohort. Interestingly, whilethe L392F variant slightly elevated the replication efficiency of the1a replicon when introduced as a point mutation, a chimeric rep-licon containing the whole patient-derived NS5B gene with L392Fshowed a marked reduction in efficiency (14% of wild-type value).
Transient posttreatment emergence of NS5B variants not pres-ent at baseline was observed in three patients. In one (genotype1a), an L497M substitution was detected at the 12-hour time pointonly, which conferred no resistance to BMS-791325 in repliconcultures either as a site-directed point mutant or in the context ofthe full-length patient-derived NS5B gene (Table 3). Virus from asecond patient (genotype 1b) showed a V499A change at 12 h,which only conferred minimal (2-fold) resistance to BMS-791325when introduced as a replicon point mutation. Viral load contin-ued to decline in this patient between 12 and 24 h, when only thewild type was detected, and then returned to baseline over the nextfew days.
At the 24-hour time point only, population sequencing of virusfrom a third patient (genotype 1a) showed distinct but transientdifferences in amino acids 494 and 495 in two different amplifica-tions from different primers. In one amplification, P495L was de-tected at approximately 100% prevalence, while in a second,P495L/S occurred in 55% of the population trace and V494A oc-curred in 45%. Clonal analysis of the second amplimer (n � 130clones) was consistent with the population results (38% V494A;62% P495L/S) and showed a very low frequency of linkage be-tween the two variants (�2%). Clonal analysis of the baselineamplimer (n � 93) showed no change at either position, suggest-ing a low pretreatment prevalence.
In replicon cultures, the P495 variants observed in this patientconferred moderate levels of BMS-791325 resistance (Table 3),but V494A conferred only 2-fold resistance with a substantialdrop in replication efficiency (7 to 11% of wild-type value). Rep-lication of the rare, linked V494A-P495S double variant was too
FIG 5 Exposure-response correlations between maximum change from base-line in log10 HCV RNA and BMS-791325 AUCinf (A), Cmax (B), and C12 (C).
TABLE 3 Phenotypic analysis of baseline and posttreatment variants in in vitro replicon system
Variant HCV genotype Time observed
EC50 (ng/ml) (fold resistance)c
Replication efficacy vswild type (%)
Point mut.d Full genea Point mut.d Full genea
L392F 1a (1 patient) All time pointsb 2.2 (0.7) 1.0 (0.3) 120 14L497 M 1a (1 patient) 12 h postdose only 1.7 (0.5) 2.1 (0.6) 13 24A499V 1a (1 patient) All time pointsb 3.5 (1.1) 48V499A 1b (1 patient) 12 h postdose only 7.2 (1.6) 50V494A 24 h postdose only 5.6 (1.7) 9P495L 1a (1 patient) 24 h postdose only 105.3 (31.9) 29P495S 24 h postdose only 210.5 (63.8) 16a Full patient-derived NS5B gene containing specified variant expressed in a chimeric genotype 1a replicon.b Also present at baseline.c EC50s for wild-type genotypes 1a and 1b were 3.3 and 4.6 ng/ml, respectively.d Point mut., as a point mutation introduced to replicon NS5B.
BMS-791325 Single-Dose Efficacy and Safety
June 2014 Volume 58 Number 6 aac.asm.org 3501
on April 23, 2016 by guest
http://aac.asm.org/
Dow
nloaded from

low to assess resistance accurately; similarly, a chimeric repliconcontaining the entire NS5B gene for this patient at 24 h had areplication efficiency that was too low to assess resistance. Thepatient’s baseline NS5B gene yielded a replicon with normal BMS-791325 susceptibility but low replication (35% of control). Ofinterest, this patient also displayed the highest maximum HCVRNA decline in the 900-mg cohort (�3.39 log10), which coincidedwith the transient emergence of resistance. Viremia in this patientreturned toward baseline levels at the same rate as it did for otherpatients in the cohort.
DISCUSSION
This single-ascending-dose study represents a proof of concept forthe use of BMS-791325 in the treatment of chronic HCV infec-tion. Single doses of this agent between 100 mg and 900 mg re-sulted in strong and rapid dose- and concentration-dependentreductions in viral RNA between 1 log10 and over 3 log10 IU/mlamong patients infected with HCV genotypes 1a and 1b. Mediantime to maximum antiviral response across all patients receivingdrug was 24 h postdose (range, 12 to 48 h), which was also themodal time (12/20 patients). At the lowest dose of 100 mg, mediantime to maximum response was shorter at 12 h postdose (3/5patients; range, 12 to 24 h). Maximum antiviral response was pos-itively correlated with plasma drug exposure.
The PK of BMS-791325 indicate that exposure is slightlygreater than dose proportional and are consistent with the expec-tation of once- or twice-daily dosing. Although t1/2 appears to bebelow 10 h, achievable exposures at 24 h greatly exceeded effectiveconcentrations established in vitro. Plasma concentrations at alldoses exceeded the protein-adjusted EC90 value in replicon cul-tures (52 ng/ml) within 1 h of dosing and remained above the EC90
at 24 h in all patients, which suggests that robust antiviral re-sponses can be expected for repeated administration even at dosescomparable to the lowest tested dose of 100 mg in the currentstudy. Additionally, preliminary exposure-response assessmentssuggest that single doses above 300 mg provide little additionalantiviral benefit. The time to maximum concentration and t1/2 ofBMS-794712—the active, equipotent metabolite of BMS-791325—were consistent with those of the parent compound. TheAUC of the active metabolite in plasma was approximately 20 to30% of that of BMS-791325 and may therefore contribute signif-icantly to the antiviral activity observed.
Single doses of BMS-791325 were generally well tolerated, withvomiting the only dose-associated event observed, and only at thehighest dose of 900 mg. Since the 900-mg cohort was dosed with18 capsules of 50 mg BMS-791325, it is likely that at least part ofthis gastrointestinal intolerance was excipient related. It is notablethat no gastrointestinal or other treatment-related events werereported in the 600-mg cohort, in which patients received onlyfour capsules of BMS-791325, 150 mg each. There were no indi-cations of drug-associated changes in laboratory parameters, vitalsigns, or cardiac function.
The highest observed maximum reduction in HCV RNA was3.4 log10 IU/ml and was achieved by two patients, one in the600-mg cohort (predose baseline, 6.6 log10 IU/ml) and one in the900-mg cohort (predose baseline, 6.5 log10 IU/ml). Both patientsachieved this reduction at 24 h postdose, but the patient in the900-mg cohort exhibited a 24-hour plasma BMS-791325 level fivetimes greater than that in the 600-mg patient (5,340 versus 1,090ng/ml). It is therefore of note that the only patient who demon-
strated transient enrichment for NS5B proline 495-associated re-sistance variants in this study was the 900-mg patient with the3.4-log10 reduction. Since selective pressure for the emergence ofviral drug resistance is a function of antiviral potency, drug con-centration, and residual replication (18), it is logical that resis-tance in this single-dose study would have been observed at thehighest plasma concentration of BMS-791325 associated with thehighest antiviral response, at the time that maximal response wasachieved. Poor fitness of these early variants under decaying drug-selective pressure was evidenced by a return to NS5B wild type by48 h postdose. Further in vivo data will be needed to establishwhether ongoing drug selection results in compensatory mu-tagenesis and the restoration of viral fitness.
Substitutions at proline 495 are signature drug resistance mu-tations for the thumb 1 NS5B inhibitors (17, 19–21) and have beenpreviously observed under BMS-791325 selection both in vitro inreplicon cultures (15, 16) and in vivo in virologic rebound samplesfrom patients receiving BMS-791325 in combination with IFN-�/RBV (22). Additional substitutions in NS5B, such as A421V andL392I, have been identified under sustained clinical or in vitroselective pressure of BMS-791325 (15, 16, 22), but the significanceof the transient V494A variant observed here in a single patientunder single-dose selection is difficult to assess. However, theemergence of resistance after a single dose of a potent allostericviral polymerase inhibitor is well established in the human immu-nodeficiency virus model (23) and emphasizes the importance oftreating HCV infection with combinations of active agents.
These data suggest that BMS-791325 has potent anti-HCV ac-tivity and holds significant promise in the treatment of HCV in-fection, and its further evaluation in combination with other an-tiviral agents is warranted. Phase 2 clinical studies of BMS-791325at doses of 75 mg and 150 mg twice daily in combination withIFN-�/RBV (study AI443-012; clinicaltrials.gov registration no.NCT01193361) (24) and in combination with the NS5A replica-tion complex inhibitor daclatasvir and the NS3 protease inhibitorasunaprevir (study AI443-014; clinicaltrials.gov registration no.NCT01455090) (25) have shown promising preliminary resultsfor the treatment of HCV genotype 1 infection.
ACKNOWLEDGMENTS
This study was wholly funded by Bristol-Myers Squibb. Editorial assis-tance was provided by Nick Fitch of Articulate Science, London, UnitedKingdom, with funding from Bristol-Myers Squibb. A. B. Vutikullird re-ports no conflicts of interest; K. Sims, J. Lemm, T. Eley, M. Liu, A. Ber-glind, D. Sherman, M. Gao, C. Pasquinelli, and D. M. Grasela are, or wereat the time that the work described was carried out, employees of Bristol-Myers Squibb and may be stockholders thereof; P. Tebas has receivedcompensation from the University of Pennsylvania for clinical study workdescribed here and has received consultancy fees from Merck and Gileadand adjudication committee fees from GlaxoSmithKline; E. Lawitz hasreceived speaker fees from Gilead, Kadmon, Merck, and Vertex and re-search or grant support from AbbVie, Achillion, Boehringer, Bristol-Myers Squibb, Gilead, GlaxoSmithKline, Idenix, Intercept, Janssen,Medtronic, Merck, Novartis, Presidio, Roche, Santaris, and Vertex andhas participated in advisory boards for AbbVie, Achillion, BioCryst, Bi-otica, Enanta, Idenix, Janssen, Merck, Novartis, Santaris, Theravance, andVertex.
REFERENCES1. Lavanchy D. 2009. The global burden of hepatitis C. Liver Int. 29:74 – 81.
http://dx.doi.org/10.1111/j.1478-3231.2008.01934.x.
Sims et al.
3502 aac.asm.org Antimicrobial Agents and Chemotherapy
on April 23, 2016 by guest
http://aac.asm.org/
Dow
nloaded from

2. World Health Organization. 2012. Prevention and control of viral hep-atitis infection: framework for global action. World Health Organization,Geneva, Switzerland. http://www.who.int/csr/disease/hepatitis/GHP_framework.pdf. Accessed 5 September 2013.
3. European Association for the Study of the Liver. 2011. EASL clinicalpractice guidelines: management of hepatitis C virus infection. J. Hepatol.55:245–264. http://dx.doi.org/10.1016/j.jhep.2011.02.023.
4. Ghany MG, Strader DB, Thomas DL, Seeff LB, American Associationfor the Study of Liver Diseases. 2009. Diagnosis, management, andtreatment of hepatitis C: an update. Hepatology 49:1335–1374. http://dx.doi.org/10.1002/hep.22759.
5. Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ,Heinzen EL, Qiu P, Bertelsen AH, Muir AJ, Sulkowski M, McHutchisonJG, Goldstein DB. 2009. Genetic variation in IL28B predicts hepatitis Ctreatment-induced viral clearance. Nature 461:399 – 401. http://dx.doi.org/10.1038/nature08309.
6. Rauch A, Kutalik Z, Descombes P, Cai T, Di Iulio J, Mueller T, BochudM, Battegay M, Bernasconi E, Borovicka J, Colombo S, Cerny A,Dufour JF, Furrer H, Gunthard HF, Heim M, Hirschel B, Malinverni R,Moradpour D, Mullhaupt B, Witteck A, Beckmann JS, Berg T, Berg-mann S, Negro F, Telenti A, Bochud PY, Swiss Hepatitis C CohortStudy, Swiss HIV Cohort Study. 2010. Genetic variation in IL28B isassociated with chronic hepatitis C and treatment failure: a genome-wideassociation study. Gastroenterology 138:1338 –1345. http://dx.doi.org/10.1053/j.gastro.2009.12.056.
7. Suppiah V, Moldovan M, Ahlenstiel G, Berg T, Weltman M, Abate ML,Bassendine M, Spengler U, Dore GJ, Powell E, Riordan S, Sheridan D,Smedile A, Fragomeli V, Muller T, Bahlo M, Stewart GJ, Booth DR,George J. 2009. IL28B is associated with response to chronic hepatitis Cinterferon-alpha and ribavirin therapy. Nat. Genet. 41:1100 –1104. http://dx.doi.org/10.1038/ng.447.
8. McHutchison JG, Lawitz EJ, Shiffman ML, Muir AJ, Galler GW, Mc-Cone J, Nyberg LM, Lee WM, Ghalib RH, Schiff ER, Galati JS, BaconBR, Davis MN, Mukhopadhyay P, Koury K, Noviello S, Pedicone LD,Brass CA, Albrecht JK, Sulkowski MS, IDEAL Study Team. 2009.Peginterferon alfa-2b or alfa-2a with ribavirin for treatment of hepatitis Cinfection. N. Engl. J. Med. 361:580 –593. http://dx.doi.org/10.1056/NEJMoa0808010.
9. Talal AH, Lafleur J, Hoop R, Pandya P, Martin P, Jacobson I, Han J,Korner EJ. 2013. Absolute and relative contraindications to pegylated-interferon or ribavirin in the US general patient population with chronichepatitis C: results from a US database of over 45 000 HCV-infected,evaluated patients. Aliment. Pharmacol. Ther. 37:473– 481. http://dx.doi.org/10.1111/apt.12200.
10. Ghany M, Nelson DR, Strader DB, Thomas DL, Seeff LB. 2011. An updateon treatment of genotype 1 chronic hepatitis c virus infection: 2011 practiceguidelines by the American Association for the Study of Liver Diseases. Hepa-tology 54:1433–1444. http://dx.doi.org/10.1002/hep.24641.
11. Pawlotsky JM. 2011. The results of phase III clinical trial with telaprevirand boceprevir presented at the Liver Meeting 2010: a new standard of carefor hepatitis C virus genotype 1 infection, but with issues still pending.Gastroenterology 140:746 –754. http://dx.doi.org/10.1053/j.gastro.2011.01.028.
12. Kneteman NM, Howe AY, Gao T, Lewis J, Pevear D, Lund G, DouglasD, Mercer DF, Tyrrell DL, Immermann F, Chaudhary I, Speth J,Villano SA, O’Connell J, Collett M. 2009. HCV796: a selective nonstruc-tural protein 5B polymerase inhibitor with potent anti-hepatitis C virusactivity in vitro, in mice with chimeric human livers, and in humans in-fected with hepatitis C virus. Hepatology 49:745–752. http://dx.doi.org/10.1002/hep.22717.
13. Legrand-Abravanel F, Nicot F, Izopet J. 2010. New NS5B polymeraseinhibitors for hepatitis C. Expert Opin. Invest. Drugs 19:963–975. http://dx.doi.org/10.1517/13543784.2010.500285.
14. Pierra C, Amador A, Benzaria S, Cretton-Scott E, D’Amours M, Mao J,Mathieu S, Moussa A, Bridges EG, Standring DN, Sommadossi JP,
Storer R, Gosselin G. 2006. Synthesis and pharmacokinetics of valopic-itabine (NM283), an efficient prodrug of the potent anti-HCV agent 2=-C-methylcytidine. J. Med. Chem. 49:6614 – 6620. http://dx.doi.org/10.1021/jm0603623.
15. Lemm JA, Liu M, Gentles RG, Ding M, Voss S, Pelosi LA, Wang Y-K,Rigat KL, Mosure KW, Bender JA, Knipe JO, Colonno R, Meanwell NA,Kadow JF, Santone KS, Roberts SB, Gao M. 2014. Preclinical character-ization of BMS-791325, an allosteric inhibitor of hepatitis C virus NS5Bpolymerase. Antimicrob. Agents Chemother. 58:3485–3495. http://dx.doi.org/10.1128/AAC.02495-13.
16. Pelosi LA, Voss S, Liu M, Gao M, Lemm JA. 2012. Effect on hepatitis Cvirus replication of combinations of direct-acting antivirals, includingNS5A inhibitor daclatasvir. Antimicrob. Agents Chemother. 56:5230 –5239. http://dx.doi.org/10.1128/AAC.01209-12.
17. Tomei L, Altamura S, Bartholomew L, Biroccio A, Ceccacci A, Pacini L,Narjes F, Gennari N, Bisbocci M, Incitti I, Orsatti L, Harper S, Stans-field I, Rowley M, De Francesco R, Migliaccio G. 2003. Mechanism ofaction and antiviral activity of benzimidazole-based allosteric inhibitors ofthe hepatitis C virus RNA-dependent RNA polymerase. J. Virol. 77:13225–13231. http://dx.doi.org/10.1128/JVI.77.24.13225-13231.2003.
18. Coffin JM. 1995. HIV population dynamics in vivo: implications for ge-netic variation, pathogenesis, and therapy. Science 267:483– 489. http://dx.doi.org/10.1126/science.7824947.
19. Kukolj G, McGibbon GA, McKercher G, Marquis M, Lefebvre S, Thau-vette L, Gauthier J, Goulet S, Poupart MA, Beaulieu PL. 2005. Bindingsite characterization and resistance to a class of non-nucleoside inhibitorsof the hepatitis C virus NS5B polymerase. J. Biol. Chem. 280:39260 –39267. http://dx.doi.org/10.1074/jbc.M506407200.
20. Tomei L, Altamura S, Paonessa G, De Francesco R, Migliaccio G. 2005.HCV antiviral resistance: the impact of in vitro studies on the develop-ment of antiviral agents targeting the viral NS5B polymerase. Antivir.Chem. Chemother. 16:225–245.
21. Larrey D, Lohse AW, de Ledinghen V, Trepo C, Gerlach T, Zarski JP,Tran A, Mathurin P, Thimme R, Arastéh K, Trautwein C, Cerny A,Dikopoulos N, Schuchmann M, Heim MH, Gerken G, Stern JO, Wu K,Abdallah N, Girlich B, Scherer J, Berger F, Marquis M, Kukolj G,Böcher W, Steffgen J. 2012. Rapid and strong antiviral activity of thenon-nucleosidic NS5B polymerase inhibitor BI 207127 in combinationwith peginterferon alfa 2a and ribavirin. J. Hepatol. 57:39 – 46. http://dx.doi.org/10.1016/j.jhep.2012.02.015.
22. McPhee F, Falk P, Lemm J, Liu M, Kirk M, Hernandez D, Cooney E,Hughes EA, Gao M. 2012. Characterization of viral escape in HCV geno-type 1-infected patients treated with BMS-791325 and pegylated interfer-on-alfa and ribavirin. J. Hepatol. 56(Suppl 2):S473. (Abstract.) http://dx.doi.org/10.1016/S0168-8278(12)61206-9.
23. de Bethune MP. 2010. Non-nucleoside reverse transcriptase inhibitors(NNRTIs), their discovery, development, and use in the treatment ofHIV-1 infection: a review of the last 20 years (1989 –2009). Antiviral Res.85:75–90. http://dx.doi.org/10.1016/j.antiviral.2009.09.008.
24. Tatum HA, Thuluvath PJ, Lawitz E, Martorell C, DeMicco M, Cohen S,Rustgi V, Ravendhran N, Ghalib R, Hanson J, Zamparo J, Yang R,Hughes EA, Cooney E. 2012. A phase 2a study of BMS-791325, an NS5Bpolymerase inhibitor, with peginterferon alfa-2a and ribavirin in treat-ment-naive patients with genotype 1 chronic hepatitis C virus infection. J.Hepatol. 56(Suppl 2):S460. (Abstract.) http://dx.doi.org/10.1016/S0168-8278(12)61175-1.
25. Everson GT, Sims KD, Rodriguez-Torres M, Hézode C, Lawitz E,Bourlière M, Loustaud-Ratti V, Rustgi V, Schwartz H, Tatum H,Marcellin P, Pol S, Thuluvath PJ, Eley T, Wang X, Huang SP, McPheeF, Wind-Rotolo M, Chung E, Pasquinelli C, Grasela DM, Gardiner DF.2014. Efficacy of an interferon- and ribavirin-free regimen of daclatasvir,asunaprevir, and BMS-791325 in treatment-naive patients with HCV ge-notype 1 infection. Gastroenterology 146:420 – 429. http://dx.doi.org/10.1053/j.gastro.2013.10.057.
BMS-791325 Single-Dose Efficacy and Safety
June 2014 Volume 58 Number 6 aac.asm.org 3503
on April 23, 2016 by guest
http://aac.asm.org/
Dow
nloaded from




















