
Gastrointestinal tolerability of nimesulide, a selective cyclooxygenase-2 inhibitor, in experimental...
13
Inflammopharmacology, Vol. 8, No. 2, pp. 175– 187 (2000) VSP 2000. Gastrointestinal tolerability of nimesulide, a selective cyclooxygenase-2 inhibitor, in experimental animals and man GUDMUNDUR SIGTHORSSON, BJARNI THJODLEIFSSON, TAHER MAHMUD and INGVAR BJARNASON * Department of Medicine, Guy’s, King’s, St Thomas’ Medical School, Bessemer Road, London SE5 9PJ, UK Received 4 December 1999; revised 10 January 2000; accepted 10 January 2000 Abstract—These studies assessed the gastrointestinal tolerability and cyclooxygenase (COX) selec- tivity of nimesulide in animals and man. Nimesulide (15 and 30 mg/kg) had no significant ‘topical’ toxicity in animals as assessed by in vivo mitochondrial morphologcal studies and no increase in small intestinal permeability or inflammation in rats. Similarly, these doses did not affect mucosal prostaglandins significantly and there were no ulcers. At the very high does of 60 mg/kg, nimesulide had some ‘topical’ toxicity and lost some of its COX-2 selectivity, but this was not associated with inflammation or ulcers. Indomethacin (10 mg / kg, equivalent to 15 mg / kg of nimesulide) altered all of the above parameters and led to small bowel ulcers. This suggests that the gastrointestinal tolerability of nimesulide is not only due to its COX-2 selectivity, but that it is also due to its lack of ‘topi- cal’ toxicity. Thirty six healthy subjects participated in a randomised, double-blind, double-dummy crossover study (14 day treatment with a washout) of the effect of nimesulide (100 mg bid) and naproxen (500 mg bid) on the gastroduodenal mucosa and small intestine (absorption– permeability and inflammation). An assessment was also made on their COX selectivity. Nimesulide caused sig- nificantly less gastric (p = 0.0001) and duodenal (p = 0.0086) damage than naproxen. Naproxen increased intestinal permeability significantly (p = 0.004) and caused intestinal inflammation while nimesulide (p> 0.40) did not. Nimesulide had no significant (p> 0.1) effect on COX-1 dependant platelet aggregation, reduced serum thromboxane B 2 levels moderately by 24– 34% (p< 0.005) and decreased prostanoid generation in gastric biopsies by 9–30% (p> 0.5). By comparison, naproxen had significantly (p< 0.0001) greater effects on these functions: abolished platelet aggregation to arachidonic acid, suppressed serum thromboxane B 2 levels by 98% and gastric prostaglandin produc- tion by 76–82%. Nimesulide inhibited lipopolysaccharide-induced PGE 2 formation in whole blood to a significantly (p< 0.01) greater extent than naproxen. This study shows that nimesulide has a preferential selectivity for COX-2 over COX-1 in vivo at full therapeutic doses. This selectivity is associated with minimal gastrointestinal damage in the short term. * To whom correspondence should be addressed. E-mail: [email protected]
-
Upload
independent -
Category
Documents
-
view
5 -
download
0
Transcript of Gastrointestinal tolerability of nimesulide, a selective cyclooxygenase-2 inhibitor, in experimental...

Inflammopharmacology, Vol. 8, No. 2, pp. 175–187 (2000) VSP 2000.
Gastrointestinal tolerability of nimesulide, a selectivecyclooxygenase-2 inhibitor, in experimental animals andman
GUDMUNDUR SIGTHORSSON, BJARNI THJODLEIFSSON,TAHER MAHMUD and INGVAR BJARNASON∗
Department of Medicine, Guy’s, King’s, St Thomas’ Medical School,Bessemer Road, London SE5 9PJ, UK
Received 4 December 1999; revised 10 January 2000; accepted 10 January 2000
Abstract—These studies assessed the gastrointestinal tolerability and cyclooxygenase (COX) selec-tivity of nimesulide in animals and man. Nimesulide (15 and 30 mg/kg) had no significant ‘topical’toxicity in animals as assessed byin vivo mitochondrial morphologcal studies and no increase insmall intestinal permeability or inflammation in rats. Similarly, these doses did not affect mucosalprostaglandins significantly and there were no ulcers. At the very high does of 60 mg/kg, nimesulidehad some ‘topical’ toxicity and lost some of its COX-2 selectivity, but this was not associated withinflammation or ulcers. Indomethacin (10 mg/kg, equivalent to 15 mg/kg of nimesulide) altered all ofthe above parameters and led to small bowel ulcers. This suggests that the gastrointestinal tolerabilityof nimesulide is not only due to its COX-2 selectivity, but that it is also due to its lack of ‘topi-cal’ toxicity. Thirty six healthy subjects participated in a randomised, double-blind, double-dummycrossover study (14 day treatment with a washout) of the effect of nimesulide (100 mg bid) andnaproxen (500 mg bid) on the gastroduodenal mucosa and small intestine (absorption–permeabilityand inflammation). An assessment was also made on their COX selectivity. Nimesulide caused sig-nificantly less gastric (p = 0.0001) and duodenal (p = 0.0086) damage than naproxen. Naproxenincreased intestinal permeability significantly (p = 0.004) and caused intestinal inflammation whilenimesulide (p > 0.40) did not. Nimesulide had no significant (p > 0.1) effect on COX-1 dependantplatelet aggregation, reduced serum thromboxane B2 levels moderately by 24–34% (p < 0.005) anddecreased prostanoid generation in gastric biopsies by 9–30% (p > 0.5). By comparison, naproxenhad significantly (p < 0.0001) greater effects on these functions: abolished platelet aggregation toarachidonic acid, suppressed serum thromboxane B2 levels by 98% and gastric prostaglandin produc-tion by 76–82%. Nimesulide inhibited lipopolysaccharide-induced PGE2 formation in whole bloodto a significantly (p < 0.01) greater extent than naproxen.
This study shows that nimesulide has a preferential selectivity for COX-2 over COX-1in vivo atfull therapeutic doses. This selectivity is associated with minimal gastrointestinal damage in the shortterm.
∗To whom correspondence should be addressed. E-mail: [email protected]

176 G. Sigthorssonet al.
Key words: Intestinal inflammation; NSAID toxicity; cyclooxygenase.
1. INTRODUCTION
Nonsteroidal anti-inflammatory drugs (NSAIDs) are the most prescribed of theanti-rheumatic drugs. Their side effect on the gastrointestinal tract is of concernand includes gastroduodenal ulcers, with their attentant complications, as well assmall intestinal inflammation, which is associated with blood loss, protein loss andoccasionally strictures (Bjarnasonet al., 1993).
While it is generally accepted that the pathogenesis of NSAID enteropathy iscomplex it is now clear that there is an important initiating ‘topical’ aspect ofthe damage as well as a systemic one. With this in mind we have devised apathogenic framework for the damage to explain the pathogenesis of NSAID-induced damage to the gastrointestinal tract. Figure 1 shows the outline of thisframework. Essentially it is suggested that the ‘topical’ action involves an effecton mitochondria to uncouple oxidative phosphorylation or inhibit the respiratorychain leading to alterations in cellular ATP levels and consequentially increasedintestinal permeability (Somasundaramet al., 1995, 1997; Mahmudet al., 1996).Increased intestinal permeability, it is suggested, allows luminal aggressors accessto the mucosa thereby causing inflammation, as most of these are neutrophilchemoattractants. Concomitant, systemically or locally mediated, NSAID-inducedinhibition of cyclooxygenase (COX), under these circumstances, is instrumental inconverting this inflammation to ulcers, presumably because they not only inhibita compensatory increase in mucosal prostaglandin generation, but actually reduceprostaglandin levels below normal values. The damaging effect of the lack ofadequate mucosal prostaglandin levels is probably mediated by microcirculatoryvasoconstriction, reduced blood flow and ischemia (Nygardet al., 1994; Anthonyet al., 1995).
The effect of a specific mitochondrial uncoupler on the small bowel is toincrease intestinal permeability and cause low grade inflammation which, as it isasymptomatic, is unlikely to be clinically relevant. Selective inhibition of COX-1 onthe other hand does no harm as judged from the fact that COX-1 knockout mice donot appear to suffer any adverse effect and do not spontaneously develop intestinaldamage. One possibility of rendering conventional NSAIDs less toxic is to abolishthe ‘topical’ effect or render the agents more specific for COX-2 inhibition. Theformer is problematic as the uncoupling action of NSAIDs is dependant on theirproperties as weak acids and extreme lipid solubility. The same physicochemicalproperties underlie their COX inhibitory action. Indeed the acidic carboxylicgroup of conventional NSAIDs (oxicams and phenylbutazone have different acidicmoieties) binds by ionic interaction with an arginine group within the COX enzymesthereby causing steric hindrance to the access of arachidonic acid to the active siteand thereby causes inhibition of the enzymes. Most of the attempts to counteractthe ‘topical’ damaging effect have been unsuccessful in the long term, apart from

Gastrointestinal tolerability of nimesulide 177
Fig
ure
1.A
path
ogen
icfr
amew
ork
for
the
deve
lopm
ent
ofN
SA
ID-i
nduc
eden
tero
path
y.It
issu
gges
ted
that
NS
AID
sha
vea
dual
bioc
hem
ical
actio
nto
caus
eda
mag
e,na
mel
ya
non-
pros
tagl
andi
nm
edia
ted
‘topi
cal’
effe
ct,w
hich
invo
lves
unco
uplin
gof
mito
chon
dria
loxi
dativ
eph
osph
oryl
atio
n,an
di
nhib
ition
ofC
OX
-1.
Itis
sugg
este
dth
atth
em
itoch
ondr
iale
ffect
lead
sto
incr
ease
din
test
inal
perm
eabi
lity
and
alo
wgr
ade
infla
mm
ator
yre
actio
nw
hich
isdr
iven
byth
elu
min
alag
gres
sive
fact
ors
gain
ing
acce
ssto
the
muc
osa.
A‘n
orm
al’r
espo
nse
tosu
chin
jury
isan
incr
ease
inm
ucos
alpr
osta
glan
dins
whi
chpl
aya
par
tof
the
heal
ing
ofth
eda
mag
ein
part
byin
crea
sing
muc
osal
bloo
dflo
wan
dtis
sue
oxyg
enat
ion.
How
ever
,the
conc
omita
ntin
hibi
tion
ofC
OX
-1pr
even
tsth
is
incr
ease
inpr
osta
glan
dins
.T
heco
nseq
uenc
eis
,w
esu
gges
t,th
atth
elo
wgr
ade
infla
mm
atio
nis
conv
erte
dto
seve
rein
flam
mat
ion
with
ulce
rs.
The
prec
ise
role
ofvi
llus-
shor
teni
ngan
dva
scul
aris
chem
ia,b
oth
ofw
hich
appe
arto
beea
rlypa
thog
enic
even
tsin
the
ente
ropa
thy
requ
ires
furt
her
stud
y.

178 G. Sigthorssonet al.
the development of nabumetone. Nabumetone is a non-acidic pro-NSAID whichoutperforms most conventional NSAIDs when it comes to short or long term safety.The reason is almost certainly due to its lack of ‘topical’ toxicity as well as the factthat its active metabolite 6-methoxy-2-naphthylacetic acid is not excreted in bile.
Focus has therefore moved to the COX-2 specific (selective, preferential) in-hibitors. The idea is to inhibit COX-2 without affecting COX-1 significantly andindeed this approach seems to hold the greatest promise for improving the gastroin-testinal safety of anti-inflammatory analgesics. Early studies using meloxicam (anoxicam promoted as COX-2 selective), celecoxib and rofecoxib in man appear ex-ceptionally encouraging. However it is not clear if this enhanced tolerability is dueto COX selectivity, decreased ‘topical’ toxicity or both.
Nimesulide (4-nitro-2-phenoxymethanesulfonanilide) is a anti-inflammatory anal-gesic which was available for clinical use before the COX story broke. It has manyof the physicochemical andin vitro characteristics of selective COX-2 inhibitors.We have assessed the gastrointestinal tolerability of nimesulide in the rat (Sig-thorssonet al., 1998) and in man to assess the basis for and confirm its enhancedtolerability.
2. ANIMAL EXPERIMENTS
The following experiments assessed these keyin vivo pathophysiological steps inthe damage following:
i) Indomethacin (10 mg/kg) by gastric gavage which is the prototype model forNSAID-induced small intestinal damage.
ii) Nimesulide (15 mg/kg) by gastric gavage.
iii) Nimesulide (30 mg/kg) by gastric gavage.
iv) Nimesulide (60 mg/kg) by gastric gavage.
iv) The vehicle (DMSO less than 10%) only as control observations.
The drugs were administered in a volume of 1.0 ml. Extrapolated from maximumrecommended human doses of indomethacin the 15 mg/kg dose of nimesulide isroughly equivalent to the 10 mg/kg indomethacin dose. In relation to their anti-inflammatory activity in the carrageenan-induced rat paw oedema, and other anti-inflammatory models (Tofanettiet al., 1989; Velo 1991), the oral dose producing50% inhibition (ED50) of swelling nimesulide is quite comparable to that ofindomethacin (1.99 mg/kg and 2.95 mg/kg, respectively) (Swingleet al., 1976).
2.1. In vivo assessment of mitochondria
The in vivo effect of the above treatments on mitochondrial morphology wasassessed by electron microscopy as there are characteristic if not pathognomicchanges following administration of uncouplers of oxidative phosphorylation orrespiratory chain inhibitors. Four rats from each group were studied 2 h after
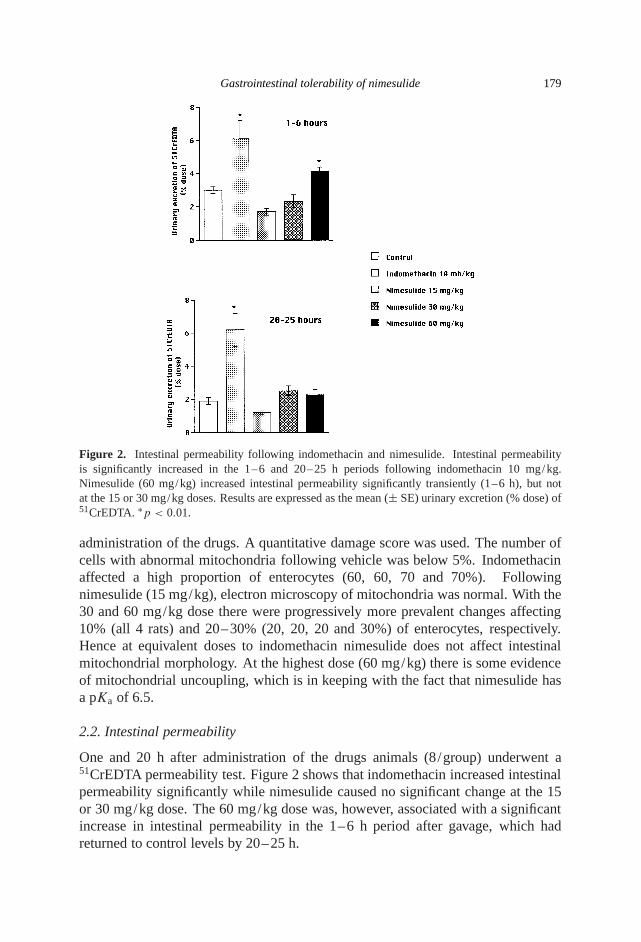
Gastrointestinal tolerability of nimesulide 179
Figure 2. Intestinal permeability following indomethacin and nimesulide. Intestinal permeabilityis significantly increased in the 1–6 and 20–25 h periods following indomethacin 10 mg/kg.Nimesulide (60 mg/kg) increased intestinal permeability significantly transiently (1–6 h), but notat the 15 or 30 mg/kg doses. Results are expressed as the mean (± SE) urinary excretion (% dose) of51CrEDTA. ∗p < 0.01.
administration of the drugs. A quantitative damage score was used. The number ofcells with abnormal mitochondria following vehicle was below 5%. Indomethacinaffected a high proportion of enterocytes (60, 60, 70 and 70%). Followingnimesulide (15 mg/kg), electron microscopy of mitochondria was normal. With the30 and 60 mg/kg dose there were progressively more prevalent changes affecting10% (all 4 rats) and 20–30% (20, 20, 20 and 30%) of enterocytes, respectively.Hence at equivalent doses to indomethacin nimesulide does not affect intestinalmitochondrial morphology. At the highest dose (60 mg/kg) there is some evidenceof mitochondrial uncoupling, which is in keeping with the fact that nimesulide hasa pKa of 6.5.
2.2. Intestinal permeability
One and 20 h after administration of the drugs animals (8/group) underwent a51CrEDTA permeability test. Figure 2 shows that indomethacin increased intestinalpermeability significantly while nimesulide caused no significant change at the 15or 30 mg/kg dose. The 60 mg/kg dose was, however, associated with a significantincrease in intestinal permeability in the 1–6 h period after gavage, which hadreturned to control levels by 20–25 h.

180 G. Sigthorssonet al.
Fig
ure
3.In
test
inal
infla
mm
atio
nfo
llow
ing
indo
met
haci
nan
dni
mes
ulid
e.D
aily
faec
alex
cret
ion
ofG
MP
follo
win
gth
edr
ugs
(giv
enat
the
star
tof
day
1:ar
row
)de
mon
stra
ting
asi
gnifi
cant
infla
mm
ator
yre
actio
nfo
llow
ing
indo
met
haci
nbu
tnot
follo
win
gni
mes
ulid
e(1
5,30
or60
mg
/kg)
.R
esul
tsar
eex
pre
ssed
asm
ean±
SE
.∗D
iffer
ssi
gnifi
cant
ly(p<
0.01
)fr
omba
selin
e.

Gastrointestinal tolerability of nimesulide 181
Figure 4. Changes in intestinal prostanoid levels following indomethacin and nimesulide. Thevertical axis shows small intestinal prostanoid levels (positive) and the % decrease from control(negative). Indomethacin and nimesulide (30 and 60 mg/kg) decreased intestinal prostaglandin E,thromboxane B2 and 6-keto-prostaglandin F1α significantly (∗p < 0.01) by 71–96% and 65–74%,respectively, whilst the lower dose of nimesulide did not alter the prostanoid levels significantly(p > 0.25). Results are expressed as mean (± SE) % of control.
2.3. Intestinal inflammation
Granulocyte marker protein was determined from faecal samples obtained fromcollections made daily for three days prior to gavage with indomethacin, nimesulidedissolved or solvent. Figure 3 shows no significant inflammation following any ofthe nimesulide doses while indomethacin increased the granulocyte marker proteinby a factor of 15.
2.4. Intestinal prostanoid levels
Figure 4 shows the changes in small intestinal levels of prostaglandin E2, thrombox-ane B2 and 6-keto-prostaglandin F1α 5 h after administration of the drugs. There isa significant (p < 0.01) reduction in prostanoid levels following indomethacin andnimesulide starts losing some of its selectivity at doses exceeding 30 mg/kg.
2.5. Macroscopic damage
No small intestinal ulcers were seen 20 h or 7 days after administration of vehicleor nimesulide (15, 30 or 60 mg/kg doses). There were an average 38 (range25–49) pointed and 17 (range 9–22) longitudinal ulcers at 20 h and 7 (range 3–22)longitudinal, but no pointed ulcers 7 days after administration of indomethacin.

182 G. Sigthorssonet al.
2.6. Conclusion
In these studies indomethacin was associated with a ‘full house’ of pathophysiolog-ical abnormalities. It uncoupledin vivo, increased intestinal permeability, causedinflammation and decreased intestinal mucosal prostanoids. The combined effectsof these led to small intestinal ulcers. By comparison the 15 mg/kg dose of nime-sulide caused no pathophysiological abnormalities or ulcers. Overall, nimesulideuncouples only infrequently at the higher dosesin vivo. This was associated witha transient increase in intestinal permeability with the 60 mg/kg dose, but not in-flammation. The 30 and 60 mg/kg doses of nimesulide that are 15 and 30 timeshigher than that required for anti-inflammatory activity were associated with sig-nificant decreases in mucosal prostanoids (COX-1 dependant process) comparablewith indomethacin. However, this was not associated with any morphological dam-age. These results resemble in many aspects those obtained in the COX-1 knockoutanimals (Langenbachet al., 1995), namely, that COX-1 inhibition by itself does notlead to damage. The ‘topical’ action of NSAIDs appear to be essential to initiate thedamage, the role of mucosal prostanoids in the damage being primarily to do withthe modulation of the vascular or inflammatory response.
3. HUMAN STUDIES
Thirty six healthy Caucasian volunteers (24 males and 12 females, mean (± SD)age 48± 8 years, range 40–67 years) were studied in a randomised, double blind,double-dummy, crossover study, comparing the effect of nimesulide (100 mg twicea day) with that of naproxen (500 mg twice a day). The primary tolerability outcomemeasure for this trial was the change in the modified endoscopic Lanza score. Thesecondary outcomes were the Visual Analogue Scale and biopsy for gastroduodenaldamage (Aabakkenet al., 1990), the results of a combined absorption– permeabilitytest (Bjarnasonet al., 1989, 1995) and faecal calprotectin concentrations (Tibbleetal., 1999) which provide an objective quantitative measure of inflammation of thewhole gastrointestinal tract. Additionally the degree of COX-1 and 2 inhibitionduring the treatment period was assessed.
3.1. Endoscopy results
The results of the grading of gastric and duodenal mucosal damage on the Lanzascale are shown in Table 1. The difference in number of subjects developing Lanzascores of greater than 1 in the stomach (p = 0.0001) or duodenum (p = 0.0086)was significantly greater for naproxen.
Figure 5 shows the differences of the effects of the two drugs on the gastroduode-nal mucosa the Visual Analogue Scale was used in relation to gastric and duodenalhaemorrhagic and erosive lesions.

Gastrointestinal tolerability of nimesulide 183
Table 1.Gastric and duodenal damage scores (Lanza) after nimesulide and naproxen
Lanza Nimesulide Naproxenscore (n = 35) (n = 36)
Stomach: endoscopic findingsnormal 0 20 (57%) 4 (11%)1 erosion or submucosal haemorrhage 1 5 (14%) 2 (6%)2–10 erosion or submucosal haemorrhage 2 9 (26%) 9 (25%)>10 erosion or submucosal haemorrhage 3 1 (3%) 16 (44%)ulcer 4 0 (0%) 5 (14%)
Duodenum: endoscopic findingsnormal 0 28 (80%) 20 (56%)1 erosion or submucosal haemorrhage 1 4 (11%) 4 (11%)2–10 erosion or submucosal haemorrhage 2 2 (6%) 7 (19%)>10 erosion or submucosal haemorrhage 3 0 (0%) 3 (8%)ulcer 4 1 (3%) 2 (6%)
Significantly more subjects had a Lanza score of 0 and 1 in the stomach (p = 0.0001) andduodenum (p = 0.0086) following nimesulide as compared with naproxen.
Table 2.Intestinal absorption and permeability before and after nimesulide and naproxen
Test substance Nimesulide Naproxen
Baseline Day 10 Baseline Day 10
3-0-D-glucose 46.2± 2.9% 41.3± 2.2% 45.5± 2.2% 41.3± 2.4%D-xylose 28.2± 1.5% 26.6± 1.2% 29.1± 1.3% 27.0± 1.6%L-rhamnose 8.4± 0.5% 8.0± 0.4% 9.0± 0.7% 8.8± 0.7%Lactulose 0.227± 0.003% 0.211±0.021% 0.243± 0.038% 0.384±0.246%Lactulose/L-rhamnose 0.0282± 0.003 0.0271± 0.021 0.0268± 0.003 0.0433±0.009∗
The values represent mean (±SE) 5 h urinary excretion (% dose) of the ingested saccharides whilethe differential urinary excretion of lactulose/L-rhamnose is an index of intestinal permeability.∗ p < 0.05.
3.2. Intestinal absorption– permeability
Table 2 shows the results of the combined absorption– permeability test in theIcelandic volunteers. Neither treatment led to any significant alterations in theurinary excretion of 3-O-methyl-D-glucose, D-xylose, L-rhamnose or lactulose.However, naproxen increased intestinal permeability significantly (p = 0.003;Wilcoxon–Pratt test) while nimesulide did not (p = 0.486).
3.3. Intestinal inflammation
Baseline calprotectin concentrations did not differ significantly (p > 0.5) betweenthe nimesulide (6.1± 2.3 mg/ l) and naproxen (5.5± 1.2 mg/ l) groups. There was

184 G. Sigthorssonet al.
Figure 5. Damage scores in stomach and duodenum before and after nimesulide and naproxen. Gas-tric and duodenal haemorrhagic and erosive scores (VAS= Visual Analogue Scale) are consistentlygreater following naproxen.∗p < 0.05.
no significant increase in faecal calprotectin concentrations during treatment withnimesulide (6.9± 1.3, p > 0.8). However, naproxen caused a significant increase(12.1± 2.1; p < 0.0002) in faecal calprotectin concentrations.
4. PROSTANOID METABOLISM
COX-1 and COX-2 dependant functions were assessed to investigate whethernimesulide was in fact a selective COX-2 inhibitor.
4.1. COX-1 inhibition
i) Platelet aggregation: Nimesulide had no significant effect on arachidonic in-duced platelet aggregation (COX-1 dependant process), ADP-induced platelet

Gastrointestinal tolerability of nimesulide 185
aggregation (partially COX-1 dependant) or thrombin receptor activator peptide-induced platelet aggregation (COX-1 independent process). However, naproxenreduced all significantly (p = 0.001,p = 0.0098 andp = 0.024, respectively;Wilcoxon–Pratt Test).
ii) Gastric mucosal biopsies were obtained during endoscopy and prostanoid pro-duction was assessed: Prior to treatment there were no significant differencesin the production of PGE2 and 6-keto-PGF1α between the nimesulide (126±14and 228± 35 ng/mg protein) and naproxen (145± 25 and 258± 35 ng/mgprotein) treatment groups. Nimesulide did not decrease decreased mucosal pro-duction of PGE2 and 6-keto-PGF1α significantly (p > 0.05) while naproxendecreased the prostanoid production by 79% and 77%, respectively (p <
0.001).
iii) Nimesulide decreased serum TXB2 levels significantly (between 24 and 34%;p < 0.01) on days 3, 10 and 15. Naproxen was however significantly morepotent in this respect (p < 0.01) decreasing serum TXB2 levels by 96–99%.
4.2. COX-2 inhibition
COX-2 inhibition was assessed by anex in vivotechnique. Nimesulide decreasedlipopolysaccharide-induced PGE2 synthesis in whole blood by 91–93% (p <
0.0001; Wilcoxon–Pratt Test) on days 3, 7 and 10 while naproxen was significantlyless (p < 0.001) potent in this respect, decreasing PGE2 levels by 74–77%(p < 0.0001).
5. CONCLUSION
This study demonstrates that in the short-term nimesulide, administered at the max-imum recommended doses, causes minimal macroscopic damage to the gastroduo-denal mucosa in man. The changes in this study were certainly no greater than thatreported for the highly-specific inhibitors flosulide (discontinued), celecoxib androfecoxib when studied under similar conditions.
The animal studies showed that nimesulide was a poor uncoupler of oxidativephosphorylation. Here, we have shown that nimesulide, at full therapeutic doses inman, was not associated with any measurable ‘topical’ toxicity as assessed by anintestinal permeability test. In agreement with the proposed pathogenic framework(Fig. 1), this lack of an effect on intestinal permeability was translated into non-measurable effects on intestinal inflammation. By comparison, naproxen increasedintestinal permeability and caused inflammation which is so characteristic of acidic-NSAIDs (Sigthorssonet al., 1998; Tibbleet al., 1999).
The excellent gastrointestinal tolerability of nimesulide in this study could eitherbe explained on the basis of reduced ‘topical’ toxicity as assessed from boththe animal and human study and/or selectivity for COX-2 inhibition. The latterpossibility was assessed directly by three measures of COX-1 dependant processes

186 G. Sigthorssonet al.
and the degree of COX-2 inhibition. Naproxen inhibited COX-1 significantly andseverely, as expected, in all the assays. Nimesulide had no significant on thesemeasures except it was associated with a modest 24–34% decrease in serum TXB2
levels, which needs to be viewed in the context of the extreme inhibition (>95%)required to suppress platelet aggregation and alter haemostatic function (Svenssonand Samuelson, 1983).
Of particular note is that nimesulide did not affect gastric biopsy prostaglandingeneration which is important as, following ingestion of the drugs, the stomach (andsmall bowel) is exposed to the high concentrations of the drug where the maximalbiological action of the drug would be expected.
The degree of COX-2 inhibition, as assessed by the lipopolysaccharide stimulatedwhole blood assay, which is thought to relate to the therapeutic effects, wassignificantly greater following nimesulide than naproxen. The differences arehowever unlikely to be biologically or therapeutically important, as both drugs causeprofound inhibition of the COX-2 enzyme.
In summary we have shown in these two studies that nimesulide is almost devoidof the ‘topical’ toxicity of conventional NSAIDs, that at maximum therapeutic dosesnimesulide maintains a higher degree of selectivity for the COX-2 enzyme in manthan expected fromin vitro assay and it seems likely that these two factors underliethe gastrointestinal tolerability of the drug. Longer term studies are required tosee if the gastrointestinal tolerability are maintained, but the study endorses theidea that ‘selective’ COX-2 inhibition is associated with enhanced gastrointestinaltolerability.
REFERENCES
L. Aabakken, S. Larsen and M. Osnes (1990). Visual analogue scales for endoscopic evaluation ofnonsteroidal anti-inflammatory drug-induced mucosal damage in the stomach and the duodenum,Scand. J. Gastroenterology25, 443–448.
A. Anthony, C. Trasivoulou, A. P. Dhillon,et al. (1995). Pre-ulcerative villous contraction andmicrovascular occlusion induced by indomethacin in the rat jejunum,Aliment. Pharmacol. Ther.9, 605–613.
I. Bjarnason, J. Hayllar, A. J. Macpherson,et al. (1993). Side effects of nonsteroidal anti-inflammatorydrugs on the small and large intestine,Gastroenterology104, 1832–1847.
I. Bjarnason, A. J. M. Macpherson and D. Hollander (1995). Intestinal permeability: An overview,Gastroenterology108, 1566–1581.
I. Bjarnason, P. Smethurst, G. C. Fenn,et al. (1989). Misoprostol reduces indomethacin inducedchanges in human small intestinal permeability,Dig. Dis. Sci.34, 407–411.
R. Langenbach, S. G. Morham, H. F. Tiano,et al. (1995). Prostaglandin synthase 1 gene disruption inmice reduced arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration,Cell 83, 483–492.
T. Mahmud, S. S. Rafi, D. L. Scott,et al. (1996). Nonsteroidal antiinflammatory drugs and uncouplingof mitochondrial oxidative phosphorylation,Arth. Rheum.39, 1998–2003.
G. Nygard, A. Anthony, C. Piasecki,et al. (1994). Acute indomethacin-induced jejunal injury in therat: Early morphological and biochemical changes,Gastroenterology106, 567–575.

Gastrointestinal tolerability of nimesulide 187
G. Sigthorsson, M. Jacob, J. M. Wrigglesworth,et al. (1998). A comparison of indomethacinand nimesulide, a selective cyclooxygenase-2 inhibitor, on key pathophysiological steps in thepathogenesis of NSAID enteropathy in the rat,Scand. J. Gastroenterol.33, 728–735.
G. Sigthorsson, J. Tibble, J. Hayllar,et al. (1998). Intestinal permeability and inflammation in patientson NSAIDs,Gut.43, 506–511.
S. Somasundaram, J. Hayllar, S. Rafi,et al. (1995). The biochemical basis of NSAID-induced damageto the gastrointestinal tract: A review and a hypothesis,Scand. J. Gastroenterol.30, 289–299.
S. Somasundaram, S. Rafi, J. Hayllar,et al. (1997). Mitochondrial damage: A possible mechanism ofthe ‘topical’ phase of NSAID-induced injury to the rat intestine,Gut.41, 344–353.
J. Svensson and K. Samuelson (1983). Inhibition of platelet function by low dose acetylsalicylic acidin patients with cardiovascular disease,Thrombosis31, 499–503.
K. F. Swingle, G. G. I. Moore and T. J. Grant (1976). 4-Nitro-2-phenoxymethanesulfonanilide (R-805): a chemically novel anti-inflammatory agent,Arch. Int. Pharmacodyn.221, 132–139.
J. Tibble, G. Sigthorsson, R. Foster,et al. (1999). Faecal calprotectin: A simple method for thediagnosis of NSAID-induced enteropathy,Gut.45, 362–366.
O. Tofanetti, I. Casciarri, P. V. Cipolla,et al. (1989). Effect of nimesulide on cyclo-oxygenase activityin rat gastric mucosa and inflammatory exudate,Med. Sci. Res.17, 745–746.
G. P. Velo (1991). The anti-inflammatory, analgesic and antipyretic activity of nimesulide inexperimental models,Drug Investigation3 (Suppl. 3), 10–13.




















