
Lam et al Forthcoming Relationship between road traffic noisescape and urban form in HK
Upload
independentCategory
view
2download
0
Toxicology in Vitro 24 (2010) 495–508
Contents lists available at ScienceDirect
Toxicology in Vitro
journal homepage: www.elsevier .com/locate / toxinvi t
Involvement of mitochondria mediated pathways in hepatoprotection conferred byFumaria parviflora Lam. extract against nimesulide induced apoptosis in vitro
Madhulika Tripathi a, Brijesh Kumar Singh a, Chetna Mishra a, Sheikh Raisuddin b, Poonam Kakkar a,*
a Herbal Research Section, Indian Institute of Toxicology Research (CSIR), P.O. Box-80, M.G. Marg, Lucknow 226001, Uttar Pradesh, Indiab Department of Medical Elementology and Toxicology, Hamdard University, New Delhi 110062, India
a r t i c l e i n f o
Article history:Received 25 May 2009Accepted 17 September 2009Available online 20 September 2009
Keywords:Primary hepatocytesNimesulideApoptosisReactive oxygen speciesFumaria parviflora Lam.Mitochondrial membrane potential
0887-2333/$ - see front matter � 2009 Elsevier Ltd. Adoi:10.1016/j.tiv.2009.09.011
Abbreviations: DWm, mitochondrial membrane poing factor; ApoAF-1, apoptosis activating factor-1; ABHA, butylated hydroxyanisole; BHT, butylated hydactivated DNase, CMF, chloromethylfluorescein; CMFcein diacetate; COX-II, cyclo-oxygenase-II; Cyt c, cytorescein; DCFH-DA, 20 ,70-dichlorofluorescein diacetateEDTA, ethylenediaminetetraacetic acid; EGTA, ethylenFood and Drug Administration; FITC, fluoresceinparviflora Lam.; GSH, reduced glutathione; HEPES, 4-neethanesulfonic acid; HPLC, high performance liquidradish peroxidase; JC-1, 50 ,50 ,60 ,60-tetrachloro-10 ,10 ,3carbocyanine iodide; LDH, lactate dehydrogenase; LPmalonyldialdehyde; MFI, mean fluorescence intensityability transition; MTT, 30[40 ,50-dimethylthiazol-2-ybromide; NBT, nitroblue tetrazolium; NSAIDs, nondrugs; ODS, octa dodcyl selanne; p-NA, p -nitro anilsaline; PVDF, polyvinylidene fluoride; ROS, reactivreverse phase high performance liquid chromatograptase; TBARS, thiobarbituric acid reactive subhydroperoxide.
* Corresponding author. Tel.: +91 0522 26213786,2628227.
E-mail address: [email protected] (P. Ka
a b s t r a c t
Nimesulide, a popular nonsteroidal anti-inflammatory drug, has been associated with serious hepatotox-icity. Reactive oxygen species (ROS) and mitochondrial perturbations have been implicated in druginduced hepatotoxicity, although their role in the pathway needs exploration. Study was undertakento elucidate the effect of Fumaria parviflora Lam. (Fp) on nimesulide induced cell death in primary rathepatocyte cultures. Fp extract treated cells showed increased viability as compared to nimesulidestressed cells as assessed by MTT assay. LDH leakage increased significantly at 500 lM nimesulide, andthe data suggested that apoptosis was the predominant mechanism responsible for cell death. Nimesu-lide induced apoptosis was further confirmed by DNA fragmentation and chromatin condensation.Nimesulide exposure increased intracellular ROS, translocation of Bax and Bcl2 followed by mitochon-drial depolarization and cytochrome c (Cyt c) release along with caspase-9/-3 activity confirming involve-ment of mitochondria in nimesulide induced apoptosis. Events like membrane depolarization ofmitochondria, expression of Bax, Bcl2, externalization of phosphatidyl serine are substantially reversedby the pre-treatment of Fp extract. Thus, the study indicates that Fp extract modulates critical events reg-ulating pro and anti-apoptotic proteins in mitochondria dependent apoptosis induced by nimesulide.
� 2009 Elsevier Ltd. All rights reserved.
1. Introduction than 100 million people worldwide take them on regular basis
Nonsteroidal anti-inflammatory drugs (NSAIDs) are amongstthe most commonly used analgesics. It is estimated that more
ll rights reserved.
tential; AIF, apoptosis induc-TP, adenosine triphosphate;
roxytoluene; CAD, caspases--DA, 50-chloromethylfluores-ochrome c; DCF, dichloroflu-; DMSO, dimethyl Sulfoxide;
e glycol tetraacetic acid; FDA,isothiocynate; Fp, Fumaria
(2-hydroxyethyl)-1-piperazi-chromatography; HRP,horse-0 ,30-tetraethylbenzamidazol-O, lipid peroxidation; MDA,
; MPT, mitochondrial perme-l]-20 , 50-diphenyltetrazolium-steroidal anti-inflammatoryine; PBS, phosphate bufferede oxygen species; RP-HPLC,hy; SOD, superoxide dismu-stance; t-BHP, tert-butyl
2627586x269; fax: +91 0522
kkar).
because of their analgesic, anti-inflammatory effectiveness andanti-pyretic properties. They do not cause sedation, mentalclouding, constipation or nausea (Berde and Sundel, 1998).Nimesulide is a preferential cyclo-oxygenase-II (COX-II) inhibitorand a non-carboxylic acid nonsteroidal anti-inflammatory drug. Itis chemically different from other NSAIDs because of the sulfo-nanilide moiety (Kulkarni, 2002). Nimesulide shows a good gas-trotolerability and low profile of gastrointestinal adverse effects.However, very few but serious cases of hepatic dysfunction andliver injury have been reported (Bolestreli, 2002). So far, themolecular mechanism underlying hepatotoxicity of nimesulidehas not been fully elucidated. The hepatic events that take placeare asymptomatic and reversible elevation of liver enzymes(Rainsford, 1998; Fosaluzza and Montagnani, 1989; Weiss et al.,1999), acute hepatitis associated with hepatocellular necrosis(Sbeit et al., 2001), cholestasis (Van Steenbergen et al., 1998)and some cases of fatal acute failure (Merlani et al., 2001) havebeen reported. A total of 900 drugs, toxins and herbs have beenreported to cause liver injury and among them drugs accountfor 20–40% of all instances of fulminant hepatic failure. Drugscontinue to be pulled from market with disturbing regularity be-cause of late discovery of hepatotoxicity. In the last few years,the US Food and Drug Administration (FDA) has withdrawn an

496 M. Tripathi et al. / Toxicology in
NSAID from the market for causing severe liver injury: Bromofe-nac which was introduced in 1997 as a short-term analgesic fororthopedic patients. This resulted into more than 50 cases of se-vere hepatic injury, and the drug was withdrawn in 1998 (Mehtaand Ozick, 2008). Formation of reactive oxygen species (ROS) inhepatocytes is reported to undergo drug induced hepatotoxicity.Consequently, this hepatic cell toxicity leads to a range of liverdiseases such as ischemia–reperfusion injury, fibrosis and liverfailure (Novo and Parola, 2008). Although molecular mechanismsresponsible for nimesulide-mediated liver injury is not fully elu-cidated yet, oxidative stress appears to play an important role init (Sheu et al., 2006). ROS is majorly generated in the mitochon-dria leading to serious damage to macromolecules of cellular sys-tem i.e. lipids, proteins, and DNA (Orrenius, 2007). Mitochondriainduced ROS plays an important role in the early stages of apop-tosis. Depolarized mitochondria induce ROS generation whichfurther results in an increase in the level of pro-apoptotic pro-teins in the cytosol (Emanuele et al., 2004). There are two princi-ple pathways for apoptosis: one requires the activation of cellsurface receptors such as Fas, while the other directly targetsmitochondria. Both the pathways subsequently activate proteo-lytic enzymes called effector caspase-3 that mediate rapid dis-mantling of cellular organelles and architecture (Cain et al.,2002). ROS generated during drug metabolism are detoxified bythe antioxidants present in the body leading to a state of equilib-rium. However, owing to inadequate antioxidant defense or over-production of ROS, the equilibrium tilts favoring an upsurge ofROS which further culminates in oxidative stress.
Antioxidants can offer protection against oxidative cellulardamage under disease/stress conditions. There are several syn-thetic antioxidants available such as butylated hydroxyanisole(BHA), butylated hydroxytoluene (BHT) but some toxicity is re-ported (Madhavi and Salunkhe, 1995). Plant derived antioxidantssuch as flavanoids and polyphenols are considered to be safe,hence considerable attempts are being made to identify plantswith antioxidant capacity. Fumaria parviflora Lam. (Fp), familyFumareaceae; is an annual creeper mainly found in the Indo–Pakistan subcontinent and Iran. In Iranian folk medicine, it is usedfor dermatological diseases, stimulation of liver function and gallbladder, as antiscabies, antiscorbite, antibronchite, diuretic,expectorant, anti-pyretic, diaphoretic, appetizer and antineoplas-tic. Its antinoceceptive effect has also been worked out (Heidariet al., 2004). Phytochemical analysis of Fp has indicated presenceof organic acids and isoquinoline alkaloids namely fumaric acid,protropine, cryptopine, sinactine, stylopine, dihydro-fumariline,per-fumidine, and dihydrosanguirine (Suau et al., 2002). Acetyl-cholinesterase and butyrylcholinesterase inhibitory activity of Fphas also been reported (Orhan et al., 2004). Significant oral anti-pyretic activity has been shown by hexane-chloroform andwater-soluble extracts of Fp in rabbits (Akhtar et al., 1984). Thepresent study was undertaken to investigate the role of mito-chondria and ROS/oxidative stress in nimesulide induced celldamage that ultimately leads to hepatotoxicity. In addition tothis, cytotoprotective and anti-apoptotic effect of 50% ethanolicextract of Fp was also investigated in nimesulide inducedhepatotoxicity.
2. Materials and method
Vitro 24 (2010) 495–508
2.1. Reagents
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide(MTT) was procured from Calbiochem (USA). DNA 100 bp ladder(O’GeneRuler, SM1143) was procured from Fermentas (EU),whereas agarose was obtained from GE Helthcare (USA). Goat poly-clonal IgG antibodies against cytochrome c (Cyt c), Bax, Bcl2 andCOX-2, mouse polyclonal antibody against b-Actin and horse-rad-ish peroxidase-conjugated rabbit-anti-goat and rabbit-anti-mousesecondary antibodies were obtained from Santa Cruz Biotechnology(Santa Cruz, CA). Chemiluminescent HRP Substrate-kit (Immobi-lonTM Western) was procured from Millipore (Bellarica, USA). CellTracker Green 50-Chloromethylflouroscein diacetate (CMF-DA)was procured from Molecular Probes (Eugene, Oregon, USA).5,50,6,60-Tetrachloro-1,10,3,30-tetraethylbenzamidazol carbocya-nine iodide (JC-1), 20,7-dichlorofluorescein diacetate (DCFH-DA),nimesulide (4-nitro-2-phenoxy methane sulfonanilide) and allother chemicals were purchased from Sigma (St. Louis, MO, USA)unless otherwise mentioned.
2.2. Animals
Animal handling in all experimental procedures was approvedby the Institutional Animal Ethics Committee, (ITRC/IAEC/20/2006). Sprague–Dawley male rats weighing 200 ± 20 g from IndianInstitute of Toxicology Research (IITR) animal colony were used forthe experiments. Rats were housed in an air conditioned room at25 ± 2 �C temperature with 60–70% humidity and a controlled12 h light/dark cycle. Rats were fed on standard pellet diet (Ashir-wad Pellet Diet, Mumbai, India) and water ad libitum. Chloroformwas used for euthanasia. Rats were fasted overnight before prepa-ration of primary hepatocytes.
2.3. Plant material and extract preparation
Fumaria parviflora Lam. was collected from Banthra centre ofNational Botanical Research Institute, Lucknow, India, authenti-cated by the taxonomy experts and a voucher specimen number-93991 against identified plant (i.e. Fumaria parviflora Lam.) was is-sued. Twenty gram powdered whole plant of Fp was suspended in100 ml of 50% ethanol and subjected to cold refluxing for 18 h. Theaqueous alcoholic extract obtained was filtered and the processwas repeated four times for maximum recovery. The filtrates werepooled and concentrated to 10 ml on rotory evaporator (BUCHIRotavapor R-114, Switzerland) followed by lyophilization in afreeze dryer (Heto Fd 3 drywinner). The powder obtained wasreconstituted into a working solution, 4 mg of lyophilized extractwas dissolved in 200 ll of DMSO followed by addition of ultrapuremilliQ (Direct Q5, Millipore, Bangalore, India) water to make up thevolume to 2.0 ml i.e. 2 mg/ml (10% DMSO). This reconstituted ex-tract was used for subsequent experiments.
2.4. Instrumentation and conditions for reverse phase HPLC
The reverse phase high performance liquid chromatography(RP-HPLC) system from Waters, Singapore, consisted of 515 HPLCpump, M-2998 photodiode array detector, pump control module-II, spherisorb ODS C-18 column (4.6 mm � 250 mm; 5 lm) foridentification of standards in 50% ethanolic Fp extract. Standardsand extract was monitored at 254 nm. The separation of fumaricacid was performed by isocratic elution with 0.1% acetic acid aque-ous mobile phase at a flow-rate of 0.3 ml/min and elution was ex-

M. Tripathi et al. / Toxicology in Vitro 24 (2010) 495–508 497
tracted at 226 nm. While separation of protopine was performedby isocratic elution with 0.01% acetic acid in methanol mobilephase at a flow-rate of 0.6 ml/min and extracted at 280 nm. Instru-ment was operated at ambient temperature. Chromatograms wereanalysed on Empower Pro V.2 software supplied with theinstrument.
2.5. Standard and sample preparations
Stock solution of standards i.e. fumaric acid and protopine(1 mg/ml) were prepared in milliQ water and methanol, respec-tively. Further dilution was done in respective solvents to obtain10 lg/ml solution of fumaric acid and 30 lg/ml concentration ofprotopine. 50% ethanolic Fp extract was dissolved in milliQ waterto give a final concentration of 50 mg/ml to analyse fumaric acid
and 10 mg/ml for detection of protopine. The sample was filteredby 0.22 lm syringe filter 20 ll sample from this extract was in-jected in HPLC for analysis.
2.6. Primary cell culture
Hepatocytes were isolated from liver of overnight fasted rataccording to the two step collagenase perfusion (Seglen, 1976).Cell viability was checked by trypan blue dye exclusion test with-in 1 h of cell isolation. Only preparations with cell viability great-er than 95% were used for subsequent experiments. Hepatocyteswere maintained in RPMI-1640 media supplemented with heat-inactivated 10% fetal bovine serum and 1% of 10,000 units Penicil-lin, 10 mg Streptomycin, 25 lg Amphotericin B, 1 mM sodiumpyruvate, 2 mM glutamine under an atmosphere of 5% CO2-95%air in an incubator (Thermo-forma) with controlled humidity at37 �C. The cells were seeded at a density of 1.0 � 104 cells/well(counted on hemocytometer) in 0.1% collagen pre-coated 96 wellplate, and used for drug exposure experiments after being cul-tured for 24 h.
2.7. Treatment of cells
Nimesulide was dissolved in 100% DMSO, filtered through0.22 lm filter and used for subsequent treatment as described inlinear diagram given below. The cells were pre-/post-incubatedwith drug as well as extract. In pre-treatment, cells were exposedto extract (30 min) prior to treatment with nimesulide (1 h),whereas in post-treatment, the cells were first exposed to nimesu-lide (500 lM) for 1 h and then extract was given for 30 min. Hepa-tocytes were also treated (control vehicle) with an equivalentvehicle concentration, i.e. 0.1% DMSO but no changes were ob-served (data not shown). For each set of experiment Silymarin(5 lg/10,000 cells) was used as a recovery control (a knownhepatoprotectent), whereas t-BHP (250 lM) was taken as positivecontrol for oxidative stress induced toxicity.
2.8. Quantitative analysis of viable cells
Mitochondrial metabolic activity in hepatocytes after followingthe treatment schedule was determined by colorimetric MTT as-say, as described by Mosmann (1983). After the treatment, culturemedium was carefully aspirated and MTT was added to each well.After 4 h incubation, 0.2 ml DMSO was added to dissolve the for-mazan crystal and incubated for 20 min. Absorbance was mea-sured at 530 nm using Spectramax PLUS 384 microplate reader(Soft max pro version 5.1; Molecular Devices, USA). The linear rela-tionship between absorbance and mitochondrial activity in cellwas taken into account. The data are expressed as a percentageof control viability measurement in untreated cells.
2.9. LDH activity based cytotoxicity assay
LDH (Lactate dehydrogenase) was measured using In vitro Tox-icology assay kit (Sigma, St. Louis, MO, USA). LDH activity was mea-sured in culture medium, dead floating cells and adherent viablecells. Floating cells were collected from the culture medium by

498 M. Tripathi et al. / Toxicology in Vitro 24 (2010) 495–508
centrifugation (240g) at 4 �C for 5 min and LDH content from thepellet was used as an index of apoptotic cell death (LDHp). TheLDH released in the culture medium (LDHe; extracellular LDH)was used as an index of necrotic cell death and the LDH presentin the adherent viable cells as intracellular LDH (LDHi). The per-centage of necrotic and apoptotic cell death was calculated as fol-lows (Yang et al., 2007):
% Necrosis : LDHe=ðLDHpþ LDHiþ LDHeÞ � 100% Apoptosis : LDHp=ðLDHpþ LDHiþ LDHeÞ � 100
2.10. Measurement of antioxidant status
2.10.1. SOD activitySOD activity was determined spectrophotometrically by inhibi-
tion of nicotinamide adenine dinucleotide (reduced)-phenazinemethosulfate-nitroblue tetrazolium reaction system, after adop-tion on microplate (Kakkar et al., 1984). Fifty percent inhibitionof formazan formation in 1 min is taken as one unit activity. Theactivity is reported as units/min/104 cells taking into account thenumber of cells in each well of microtiter plate.
2.10.2. MDA (malondialdehyde) determinationLipid peroxidation is quantified by measuring malondialdehyde
(MDA), a breakdown product formed from polyunsaturated fattyacids (PUFA) hydroperoxides. The concentration of thiobarbituricacid reactive substances (TBARS) was expressed as nmoles ofMDA formation in pre and post-treated hepatocytes using a freshlydiluted l,l,3,3-tetraethoxypropane for the standard calibrationcurve (Wallin et al., 1993). Trolox, an analog of Vit E was used aspositive control.
2.10.3. GSH contentGSH is a major intracellular antioxidant performing several
important biological functions. 50-Chloromethylfluorescein diace-tate (CMF-DA) was used to measure the total GSH spectrofluoro-metrically in a cellular system. The treated cells were incubatedwith the fluorescent dye (5 lg/ml) for 30 min in dark at 37 �Cand then read at Ex/Em-485/530 nm wavelength on spectrofluo-rometer (Synergy HT, Biotek, USA) (Okada et al., 1996).
2.11. Measurement of intracellular ROS generation
Intracellular ROS was detected by an oxidation-sensitive fluo-rescent probe 2070-dichlorofluorescein diacetate (DCFH-DA). Con-trol cells as well as cells with pre-/post-treatment wereincubated for 30 min with DCFH-DA (5 lg/ml) at 37 �C in dark.To evaluate ROS mediated oxidation of DCFH-DA to the fluorescentcompound DCF, samples were analyzed at an excitation wave-length of 480 nm and an emission wavelength of 525 nm by BD-LSR flow cytometer. Each determination is based on mean fluores-cence intensity of 10,000 events (Mohammad et al., 2001).
2.12. Preparation of sub-cellular fractions
Cytosolic and mitochondrial fractions were prepared as de-scribed by Zhang et al. (1999). Briefly, 7.5 � 106 cells were culturedin the flask and harvested, followed by single washing. These cellswere suspended in ice-cold RIPA buffer (20 mM HEPES-KOH, pH-7.5, 10 mM KCl, 1.5 mM MgCl2, 1.0 mM dithiothreitol, 1.0 mMEDTA, 1.0 mM EGTA, 1.0 mM phenylmethylsulfonyl fluoride) andsonicated for 10 s (Sartorius, Labsonic MTM). The lysate was centri-fuged (Sigma 3K18, Germany) at 800g for 4 min at 4 �C. Superna-tant was then centrifuged at 22,000g for 15 min at 4 �C. Resultingsupernatant was used as cytosolic fraction and pellet (mitochon-
dria) was re-suspended in cold RIPA buffer, incubated in ice for30 min followed by sonication for 10 s. The lysate was centrifugedat 15,000g for 15 min at 4 �C. The resultant supernatant was mito-chondrial fraction.
2.13. Immunoblot analysis for COX-II expression, translocation of Bax,Bcl2 and Cyt c
The protein content corresponding to each treatment was quan-tified using Lowry’s method (Lowry et al., 1951). Samples wereincubated with Lammeli’s buffer for 5 min at 96 �C and immedi-ately kept in ice. Thirty microgram of protein sample from cyto-solic or mitochondrial fraction was separated on 12% SDS–polyacrylamide gel electrophoresis and electroblotted on PVDFmembrane (HybondTM –P Amersham biosciences, UK limited,NA). After blocking non-specific sites with 1 � blocking buffer,washing was performed using PBS containing 0.1% Tween-20.The membrane was then incubated for 1 h with goat polyclonalIgG antibodies of Bcl2, Cyt c, Bax and COX-II and mouse polyclonalIgG antibody of b-Actin (Santa Cruz Biotechnology, Inc.) in 1:500dilutions. This was followed by washing with PBS and incubationwith horse-radish peroxidase-conjugated Rabbit anti-goat IgG orgoat anti-mouse IgG secondary antibodies in 1:1000 dilutions for1 h at room temperature. Membrane was rewashed and the immu-noblot was revealed using ImmobilonTM Western ChemiliminescentHRP susbtrate kit. (Millipore, Corporation, MA, USA) b-actin wasused as internal standard. PageRulerTM Prestained Protein Ladder(5 ll), (SM-0671 from Fermentas, EU) was used to determinemolecular weight of the protein bands. Densitometry of the bandsobtained was done using NIH software Image J version 1.41 (USA).Expression of COX-II and level of Cyt c was assessed in cytosolicfraction whereas Bcl2 and Bax translocation from cytosol to mito-chondria was assessed in mitochondrial fraction.
2.14. Measurement of mitochondrial membrane potential (DWm)
JC-1 is a cationic fluorescent dye that selectively accumulateswithin mitochondrial matrix and forms red fluorescent J-aggre-gates under polarized condition, while under depolarized condi-tions it exists as green monomers. Cultured cells were incubatedwith 2.5 lg/ml of JC-1 fluoroprobe at 37 �C in dark for 30 min. Afterwashing the hepatocytes, DWm was assessed by comparing twofluorescence, i.e. red (Ex/Em-580/590 nm)/green (Ex/Em-510/527 nm) using flowcytometer (BD-LSR) and analysis was per-formed on Cell Quest software (Cossarizza et al., 1996).
2.15. Caspases-3 and -9 activity
Caspase-3 and caspase-9 activity was measured by colorimetricassay kits (Sigma–Aldrich, St. Louis, MO, USA and R&D Biosystems,Inc., MN, USA) according to the manufacturer’s protocol. In brief,isolated hepatocytes were pre and post-incubated with Fp andnimesulide following the treatment schedule. After incubation,the medium was discarded, adherent cells were harvested in PBSand sedimented by centrifugation at 600g for 3 min. The pelletswere then re-suspended in lysis buffer for 30 min on ice. The lysedcells were centrifuged at 10,000g for 3 min and to the supernatant,reaction mixture and buffer were added. The concentration of thep-nitro aniline released from the substrate was calculated fromthe absorbance value at 405 nm using a calibration curve.
2.16. DNA fragmentation analysis
DNA was isolated from control and treated hepatocytes. Afterincubation, cells were washed with PBS and lysed with lysis buffer(250 mM Sucrose, 5.0 mM Tris–HCl (pH 8.0), 1.0 mM EDTA, 20%
M. Tripathi et al. / Toxicology in Vitro 24 (2010) 495–508 499
SDS) and incubated overnight at 37 �C. Subsequently, RNase A wasadded for 1 h at 37 �C followed by addition of 8.0 M potassium ace-tate. The lysate was extracted twice with an equal volume of chlo-roform/isoamylalcohol (24:1) and centrifuged at 1000g for 5 min.The upper aqueous layer was separated, measured, twice the vol-ume of absolute ethanol was added to it and incubated at �20 �Cin order to precipitate the DNA. The pellet obtained after centrifu-gation at 14,000g for 15 min was air dried and then dissolved inTris–EDTA buffer. DNA quantification was done spectrophotomet-
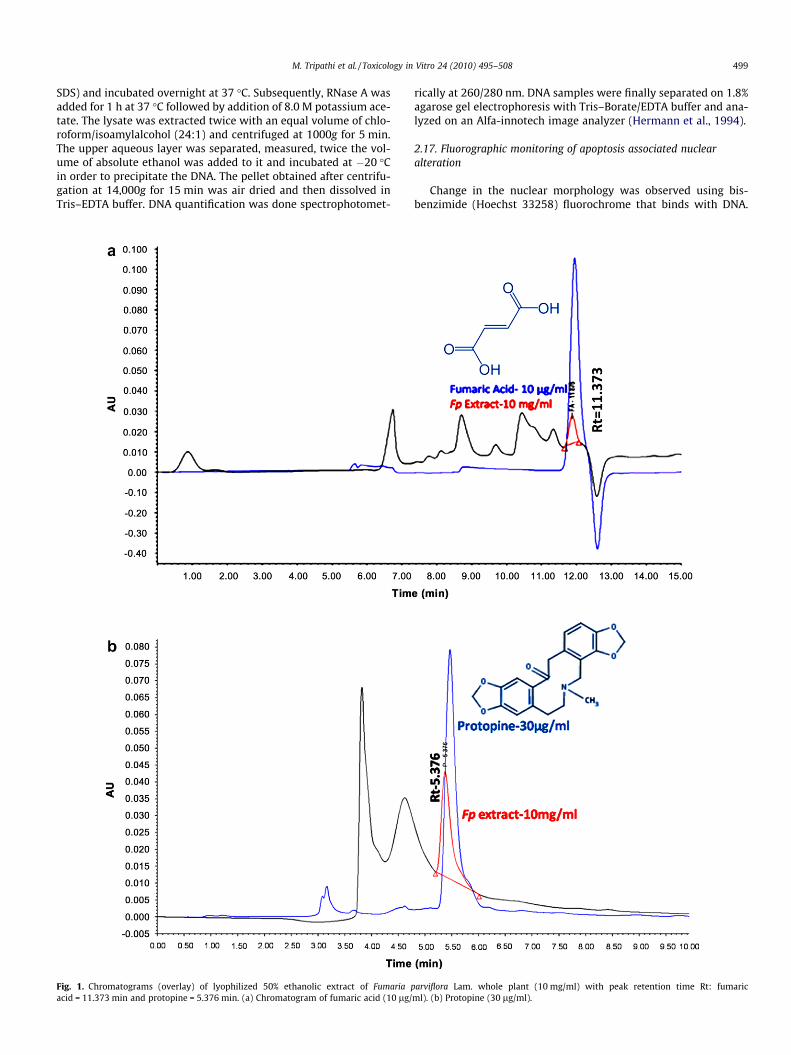
Fig. 1. Chromatograms (overlay) of lyophilized 50% ethanolic extract of Fumaria pacid = 11.373 min and protopine = 5.376 min. (a) Chromatogram of fumaric acid (10 lg/
rically at 260/280 nm. DNA samples were finally separated on 1.8%agarose gel electrophoresis with Tris–Borate/EDTA buffer and ana-lyzed on an Alfa-innotech image analyzer (Hermann et al., 1994).
2.17. Fluorographic monitoring of apoptosis associated nuclearalteration
Change in the nuclear morphology was observed using bis-benzimide (Hoechst 33258) fluorochrome that binds with DNA.
arviflora Lam. whole plant (10 mg/ml) with peak retention time Rt: fumaricml). (b) Protopine (30 lg/ml).
500 M. Tripathi et al. / Toxicology in Vitro 24 (2010) 495–508
Primary hepatocyte (treated and control) monolayers were fixed inice-cold methanol/acetic acid (3:1) for 5 min. Cells were stainedwith Hoechst 33258 (5 lg/ml) for 10 min, washed and mountedin a solution of 20 mM citric acid, 50 mM disodium orthophos-phate, and 50% glycerol (pH 5.5). Apoptosis associated nuclearalterations were examined at a wavelength of Ex/Em-350/460 nmusing Nikon microscope with fluorescence attachment (Baylyet al., 1994).
2.18. Annexin V-FITC binding assay
The extrusion of phosphotidylserine on cell membrane surfacewas detected by FITC tagged annexin V stain using Annexin V-FITCApoptosis detection kit (APOAF; Sigma, St. Louis, MO, USA). Briefly,hepatocytes (Control, nimesulide and Fp pre-treatment, Fp post-treatment, Sliymarin pre and post-treatment) were aliquoted andto this aliquot Annexin V fluorescein isothiocynate (FITC) wasmixed and incubated for 15 min and the PI was added to distin-guish the necrotic cells. (a) Viable hepatocytes are negative forboth Annexin V and PI, (b) Early apoptotic hepatocytes were la-beled with Annexin V while were negative with PI, (c) Late apopto-tic cells were labeled with both AnnexinV and PI, (d) Necrotic cellsonly take up PI (Jie et al., 2003). Acquisition of stained cells was
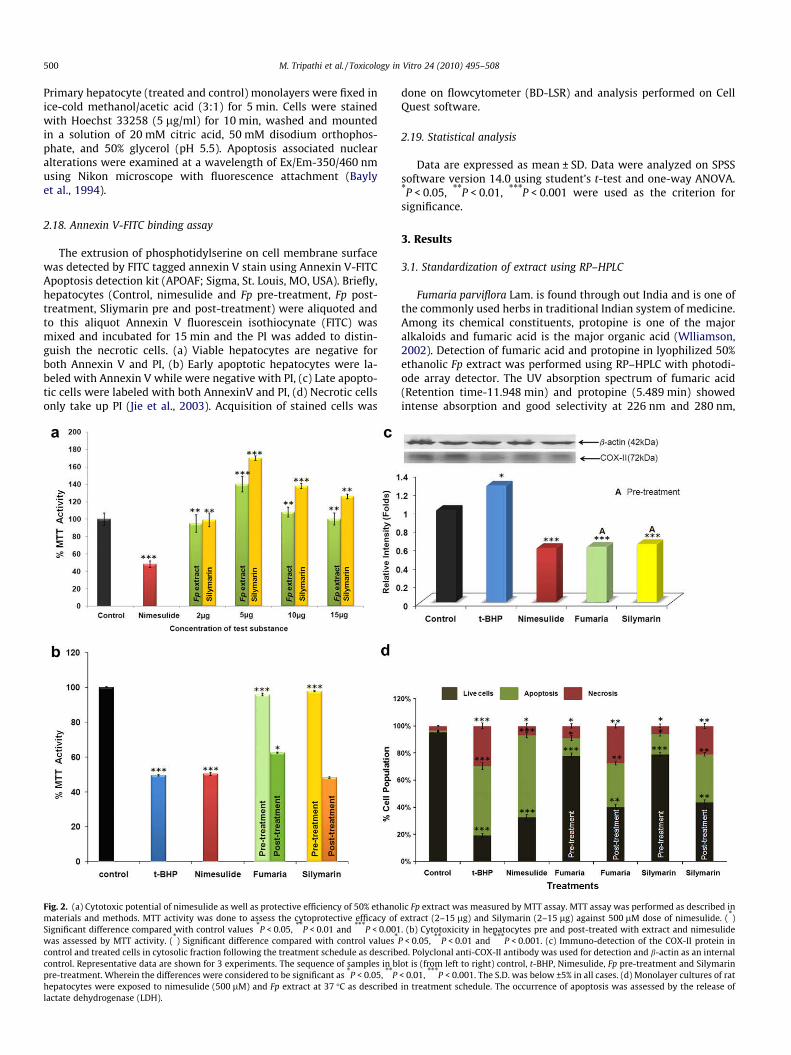
Fig. 2. (a) Cytotoxic potential of nimesulide as well as protective efficiency of 50% ethanomaterials and methods. MTT activity was done to assess the cytoprotective efficacy ofSignificant difference compared with control values *P < 0.05, **P < 0.01 and ***P < 0.001was assessed by MTT activity. (*) Significant difference compared with control values*
control and treated cells in cytosolic fraction following the treatment schedule as describcontrol. Representative data are shown for 3 experiments. The sequence of samples in blpre-treatment. Wherein the differences were considered to be significant as *P < 0.05, **Phepatocytes were exposed to nimesulide (500 lM) and Fp extract at 37 �C as describedlactate dehydrogenase (LDH).
done on flowcytometer (BD-LSR) and analysis performed on CellQuest software.
2.19. Statistical analysis
Data are expressed as mean ± SD. Data were analyzed on SPSSsoftware version 14.0 using student’s t-test and one-way ANOVA.*P < 0.05, **P < 0.01, ***P < 0.001 were used as the criterion forsignificance.
3. Results
3.1. Standardization of extract using RP–HPLC
Fumaria parviflora Lam. is found through out India and is one ofthe commonly used herbs in traditional Indian system of medicine.Among its chemical constituents, protopine is one of the majoralkaloids and fumaric acid is the major organic acid (Wlliamson,2002). Detection of fumaric acid and protopine in lyophilized 50%ethanolic Fp extract was performed using RP–HPLC with photodi-ode array detector. The UV absorption spectrum of fumaric acid(Retention time-11.948 min) and protopine (5.489 min) showedintense absorption and good selectivity at 226 nm and 280 nm,
lic Fp extract was measured by MTT assay. MTT assay was performed as described inextract (2–15 lg) and Silymarin (2–15 lg) against 500 lM dose of nimesulide. (*). (b) Cytotoxicity in hepatocytes pre and post-treated with extract and nimesulide
P < 0.05, **P < 0.01 and ***P < 0.001. (c) Immuno-detection of the COX-II protein ined. Polyclonal anti-COX-II antibody was used for detection and b-actin as an internalot is (from left to right) control, t-BHP, Nimesulide, Fp pre-treatment and Silymarin< 0.01, ***P < 0.001. The S.D. was below ±5% in all cases. (d) Monolayer cultures of ratin treatment schedule. The occurrence of apoptosis was assessed by the release of

Table 1Restoration of antioxidant status.
Treatment SOD activity(units/min/104
cells)
LPO (nM MDAformation/104
cells)
GSH content(CMF-fluorescence)
Control 13.9 ± 1.9 0.05 ± .001 90 ± 5.9t-BHP 7.0 ± 0.89*** 0.09 ± .004*** 34 ± 2.1***
Nimesulide 7.4 ± 0.71*** 0.10 ± .003*** 39 ± 1.8***
Fumaria pre-treatment
12.7 ± 1.2*** 0.041 ± .002*** 83 ± 3.8***
Fumariapost-treatment
7.0 ± 1.1 0.093 ± .004 39 ± 1.5
Silymarinpre-treatment
12.9 ± 1.5*** 0.043 ± .003*** 81 ± 2.6***
Silymarinpost-treatment
7.1 ± .98 0.081 ± .006 38 ± 1.1
SOD activity and lipid peroxidation (nM MDA formation) was quantified by col-ourimetric assay, whereas total glutathione content was quantified by spectroflu-orometry using CMF-DA fluorescent dye. Fumaria pre-treatment was found to beeffective over nimesulide induced oxidative injury. Value represents the mean ± SDof five independent experiments. t-BHP and nimesulide were compared to controlwhile Fumaria and Silymarin were compared to nimesulide. Results were consid-ered significant, ***P < 0.001.
M. Tripathi et al. / Toxicology in Vitro 24 (2010) 495–508 501
respectively. Fig. 1a shows overlay of HPLC chromatograms for fu-maric acid and Fp extract (Rt-11.876 min). Fig 1b is an overlay ofprotopine and Fp extract (Rt-5.376 min). This chemical fingerprintanalysis confirms the standardization and validation of the extractused in the study.
3.2. Cytoprotective effect of Fp extract on nimesulide toxicity
Varying concentrations of Fp extract (2–15 lg) were tested todetermine the effective concentration, at which no toxicity was ob-served. Fp (5 lg) extract enhanced the cell viability by 44%(P < 0.001) with respect to control. Silymarin (5 lg), a knownhepatoprotectent (Pradhan and Girish, 2006; Fraschni et al.,2002), used as a positive control, showed increase in viability by40% (P < 0.001). In order to evaluate the cytotoxic potential ofnimesulide, the hepatocytes were treated with varying concentra-tions of nimesulide (100–700 lM) and Inhibitory concentration(IC50) was determined by MTT cell viability assay. 500 lM ofnimesulide caused decrease in the viability by 52% (P < 0.001) withrespect to control whereas the IC50 for t-BHP, a known oxidativestress generator, was found to be 250 lM (48.29%; P < 0.01). To se-lect the dose of extract, cultured hepatocytes were treated with500 lM of nimesulide and varying concentration of Fp. 5 lg of ex-tract caused increase in viability by 91.8% P < 0.001 (Fig 2a) as com-pared to nimesulide stressed cells. The selected concentrations ofFp/Silymarin (5 lg), Nimesulide (500 lM) and t-BHP (250 lM)were used in the subsequent experiments. Cells pre-incubatedwith extract (Fp) showed significant protection and cell survivalwas found to increase by 45% (P < 0.001) as compared to nimesu-lide stressed cells. Cells pre-treated with silymarin also showedprotective effect and the viability was found to increase by 47%;P < 0.001 (Fig 2b). Primary hepatocytes post-incubated with ex-tract and silymarin did not show pronounced protection i.e. only12% with Fp and 2% with silymarin when compared to nimesulidetreated cells.
3.3. Effect of Fp pre-treatment on COX-II expression
Selective inhibition can make a difference in terms of side-ef-fects as nimesulide is a selective COX-II inhibitor over COX-I.Therefore, to see the effect of Fp extract on the pharmacologicalproperty of nimesulide, COX-II expression was detected in the cellstreated with nimesulide as well as cells pre-treated with Fp extract.For this purpose, an anti-COX-II antibody was taken and immuno-blot was performed. Immunoblot analysis (Fig 2c) revealed that Fpextract has no effect on nimesulide modulated COX-II expression.
3.4. Nimesulide induces cell death in hepatocytes by apoptosis which isprevented by Fp extract
Oxidative stress induced cell death may be accomplished bytwo distinct mechanisms, necrosis or apoptosis. Characterizationwas done to observe the mechanism involved in nimesulide in-duced hepatic cell death and to study the effectiveness of Fp ex-tract. The ratio of necrosis and apoptosis in primary hepatocyteswas analyzed using LDH activity-based assay. Intracellular LDH re-lease was evaluated as a result of the breakdown of plasma mem-brane of hepatocytes and alteration of its permeability. The cellswere exposed to nimesulide for 1 h and LDH activity in floatingcells and culture media was taken as the cell death indicator(either apoptosis or necrosis). As shown in Fig 2d. LDH leakage in-creased significantly in the presence of 500 lM nimesulide for 1 hand the number of apoptotic and necrotic cells were 60.62% and6.67%, respectively, suggesting that apoptosis as well as necrosiswas triggered by nimesulide but apoptosis was the predominantmechanism responsible for cell death. Cells pre-incubated with
Fp extract showed decrease in the number of apoptotic cells at5 lg i.e. 13.1%, whereas silymarin treatment also decreased thenumber of cells undergoing apoptosis. Post application of extractand silymarin did not show significant effect (40.16% and 43.75%,respectively). Results indicate that 5 lg of Fp extract showed sig-nificant decrease in apoptosis induced by nimesulide, suggestingits cytoprotective and anti-apoptotic efficacy.
3.5. Restoration of antioxidant capacity
3.5.1. SOD activityIn cultured primary hepatocytes, the SOD activity was found to
be 13.9 ± 1.9 units/min/1 � 104 cells. Cultured hepatocytes whentreated with selected dose of nimesulide (500 lM) and t-BHP(250 lM) showed decrease in SOD activity by 7.4 units/min/1 � 104 cells & 7 units/min/1� 104 cells (P < 0.001), respectively(Table 1). Cells that were pre-incubated with Fp extract (5 lg)showed 12.7 units/min/1 � 104 cells (P < 0.001) superoxide dismu-tase activity indicating levels comparable to untreated controlcells. The activity was significantly higher than the nimesulidestressed cells. Whereas cells post-incubated with extract did notshow changes in SOD activity as compared to nimesulide stressedcells. SOD activity was found to increase by 12.9 units/min/1 � 104 cells (P < 0.001) in hepatocytes pre-incubated with silyma-rin and the response was comparable to cells pre-treated with Fpextract. Post-treatment was not found to be effective.
3.5.2. Lipid peroxidation (LPO)Oxidative stress induced in the hepatocytes by free radical gen-
eration due to 500 lM of nimesulide and 250 lM of t-BHP caused0.1 nM MDA (P < 0.001) and 0.09 nM MDA (P < 0.001) formation/1 � 104 cells, respectively. On pre-treatment with 5 lg of Fp ex-tract, LPO reduced to 0.041 nM MDA/1 � 104 cells (P < 0.001),whereas in cells pre-incubated with silymarin (5 lg) the peroxida-tive decomposition of phospholipids reduced to 0.043 nM MDA/1 � 104 cells (P < 0.001) (Table 1). In cells post-incubated with5 lg of Fp extract and silymarin, the peroxidative decompositionof phospholipids reduced to 0.093 and 0.081 nM MDA/1 � 104 cells, indicating no significant change during post-incuba-tion in vitro.

502 M. Tripathi et al. / Toxicology in Vitro 24 (2010) 495–508
3.5.3. Intracellular levels of reduced glutathione (GSH)Glutathione is the key regulator of intracellular redox status.
The fluorescence intensity of CMF was captured using spectrofluo-rometer, which reflects the GSH content in the cellular system. Inour experiments, nimesulide enhanced oxidative stress by alteringglutathione levels. Spectrofluorometric estimation revealed thatthe cells stressed with nimesulide (500 lM) showed rapid reduc-tion in cellular GSH by 57% as compared to control. The cells pre-treated with Fp extract and silymarin were able to prevent GSHdepletion in cells by 2.13 and 2.07 folds, respectively when com-pared to nimesulide stressed cells, (Table 1). Post-treatment didnot show significant restoration in GSH content. Thus, the resultsreflect that extract (Fp) pre-treatment is effective in abrogatingoxidative damage and GSH depletion by nimesulide.
3.6. ROS generation
To further determine the status of ROS generation in nimesulidetreated cells, intracellular ROS level was measured using DCFH-DAfluoroprobe. Hepatocytes that were stressed with nimesulideshowed an increase in the ROS generation by 4.3 folds (P < 0.001)as compared to control. At the same time cells pre-treated withFp extract showed a significant decline in the ROS generation i.e.27% (P < 0.001) as compared to nimesulide stressed cells and thisresponse was comparable to silymarin treated cells i.e. 25%(P < 0.001, Recovery Control). Cells post-treated with Fp extractand silymarin showed inhibition in ROS generation by only 44%and 38% (P < 0.01), respectively. The data suggests that increasein ROS generation due to nimesulide causes hepatocyte toxicity,however pre-treatment with Fp extract inhibited the ROS genera-tion caused by nimesulide (Fig 3), thus rendering protection.
3.7. Possible Involvement of Mitochondria
3.7.1. Involvement of Bcl2 and BaxSince Bcl2 family plays a crucial role in cell apoptosis, western
blot analysis was carried out to assess the expression of the Bcl2family protein Bax and Bcl2. Bax, a pro-apoptotic Bcl2 family mem-
Fig. 3. Effect of nimesulide and Fp extract on ROS production in primary cultured hepatomean ± SD evaluated from three different sets of experiments. (*) significant difference co±5% in all cases.
ber resides in the cytosol and translocates to mitochondria uponinduction of apoptosis (Kakkar and Singh, 2007; Borner, 2003).After treatment of nimesulide and t-BHP the level of Bcl2 was de-ceased by 30% each. On pre-treatment with extract the level wasfound to be increased and the results were comparable to controlas evident from Fig 4a. Increased translocation of Bax from cytosolto mitochondria was evident from its enhanced level by 4.11 foldswith respect to control. The cells pre-treated with extract andsilymarin showed decrease in Bax by 3.39 and 3.38 folds, respec-tively (P < 0.001) when compared to nimesulide treated cells (Fig4b), confirming the protective role of Fp extract in vitro.
3.7.2. Mitochondrial membrane potentialMitochondria are vulnerable targets for various toxicants be-
cause of their important role in maintaining cellular integrity andfunctions. The functional alterations occur in mitochondria dueto changes in mitochondrial membrane potential (DWm). Disrup-tion of the mitochondrial membrane potential (i.e. depolarization)is one of the earliest indicators of cellular disturbance. Lowering ofDWm, causes dissipation of JC-1 aggregates into monomers andleads to shift from red to green fluorescence captured on flowcy-tometer. Control and Nimesulide treated hepatocytes showed18.81 % and 51.61% green fluorescence, respectively. This shiftwas also found in the cells treated with 250 lM of t-BHP(54.78%). In the cells pre-treated with Fp extract, there was com-plete recovery in nimesulide induced DWm and the mitochondriawith green fluorescence were found to be only 17.71% which iscomparable to control hepatocytes (i.e.18.81%). Pre-treatment withsilymarin, gave similar results i.e. 26.17% cells with green fluores-cence were observed. Fp extract alone did not affect DWm (datanot shown), confirming the protective effect of Fp extract pre-treatment (Fig. 4c and d). Results clearly indicate that nimesulideinduced cell death via mitochondrial permeability transition isinhibited by Fp extract.
3.7.3. Cytochorme c releaseRecently, Bax has been reported to induce Cyt c release and cas-
pase activation in vitro as well as in vivo (Kroemer et al., 1997; Pe-
cytes. DCFH-DA dye was used to assess the ROS production in cells. Data shown arempared with control values *P < 0.05, **P < 0.01 and ***P < 0.001. The S.D. was below

Fig. 4. (a) Bcl2 family proteins are characterized as cell death regulators. Immuno-detection of the Bcl2 protein was done with rabbit polyclonal anti-Bcl2 antibody inmitochondrial fraction and b-actin was used as an internal control. The sequence of samples in blot is (from left to right) control, t-BHP, Nimesulide, Fp pre-treatment andSilymarin pre-treatment. The S.D. was below ±5% in all cases. (b) Bax translocation from cytosol to mitochondria was assessed using immunoblot in the mitochondrialfraction from the stressed and pre-treated hepatocytes. Immuno-detection of the Bax protein was done with rabbit polyclonal anti-Bax antibody and b-actin was used as aninternal control. The sequence of samples in blot is (from left to right) control, t-BHP, Nimesulide, Fp pre-treatment and Silymarin pre-treatment. The S.D. was below ±5% in allcases. (c) Effect of pre and post-treatment of nimesulide and Fp on the mitochondrial membrane potential (DWm) of rat primary hepatocytes. Samples were taken at theindicated time points and assayed for depolarized mitochondria (i.e. mitochondria having low membrane potential). Changes in mitochondrial membrane potential (DWm)were observed using mitochondria specific fluorescent probe JC-1. Monomer green fluorescence increased as DWm dropped. In dotplot, Y-axis represents mitochondria withhigh membrane potential (Red Fluorescence) and X-axis represents mitochondria with low membrane potential (Green fluorescence). The S.D. was below ±5% in all cases. (d)Graph (100% scale) indicates percent mitochondrial population differentiated on membrane potential by flow cytometer. (e) Western blot analysis of Cyt c expression incontrol and treated cells in cytosolic fraction after the treatment schedule as described. Immuno-detection of the Cyt c protein was done with polyclonal anti-Cyt c antibodyand b-actin was used as an internal control. Representative data are shown for 3 experiments. The sequence of samples in blot is (from left to right) control, t-BHP,Nimesulide, Fp pre-treatment and Silymarin pre-treatment. The S.D. was below ±5% in all cases. (f) Effect of treatment on relative caspase-9 activity using spectrophotometeras assayed in whole cell lysate using R&D biosystem kit. The S.D. was below ±5% in all cases. (g) Effect of treatment on relative caspase-3 activity was assessed in whole celllysate using sigma kit Data shown are mean ± SD evaluated from three different sets of experiments. Treatment of t-BHP and nimesulide were compared to control while Fpand Silymarin treated cells were compared to nimesulide stressed cells. *P < 0.05, **P < 0.01, ***P < 0.001. The S.D. was below ±5% in all cases. (For interpretation of thereferences to colour in this figure legend, the reader is referred to the web version of this article.)
M. Tripathi et al. / Toxicology in Vitro 24 (2010) 495–508 503
tit et al., 1990). The release of Cyt c from the mitochondria into thecytosol is a key event in mitochondria dependent apoptosis (Lam-bert et al., 2004; Fleury et al., 2002). In the presence of sufficientATP, Cyt c binds to apoptosis activating factor-1 (ApoaF-1) andforms apoptosome that finally activates caspase-9 in the cytosol.Once the caspase-9 is activated, it in turn activates the executionercaspase-3. Fig 4e shows that there was a substantial release of Cyt cfrom mitochondria to cytosol on exposure to nimesulide i.e. 3.53folds increase in the Cyt c release. In contrast, cells pre-treatedwith extract showed very weak signals indicating that Cyt c releasewas prevented in the cells pre-treated with Fp extract. Silymarinpre-treated cells also showed decrease in Cyt c release by 2.96
folds. Post-treatment was not found to be effective (Data notshown).
3.7.4. Caspases activationA family of cysteine proteases, termed caspases, play a central
part in both induction as well as execution of apoptosis. Caspase-9 and caspase-3 are the main effector and executor caspases in-volved in apoptosis and play a critical role in the disintegrationof the cells undergoing apoptosis. Analysis of caspase-9/-3 activa-tion was performed in primary cultured hepatocytes treated withnimesulide (500 lM) and Fp extract. Caspase-9 activity was foundto increase by 1.63 folds by nimesulide treatment. Cells that were
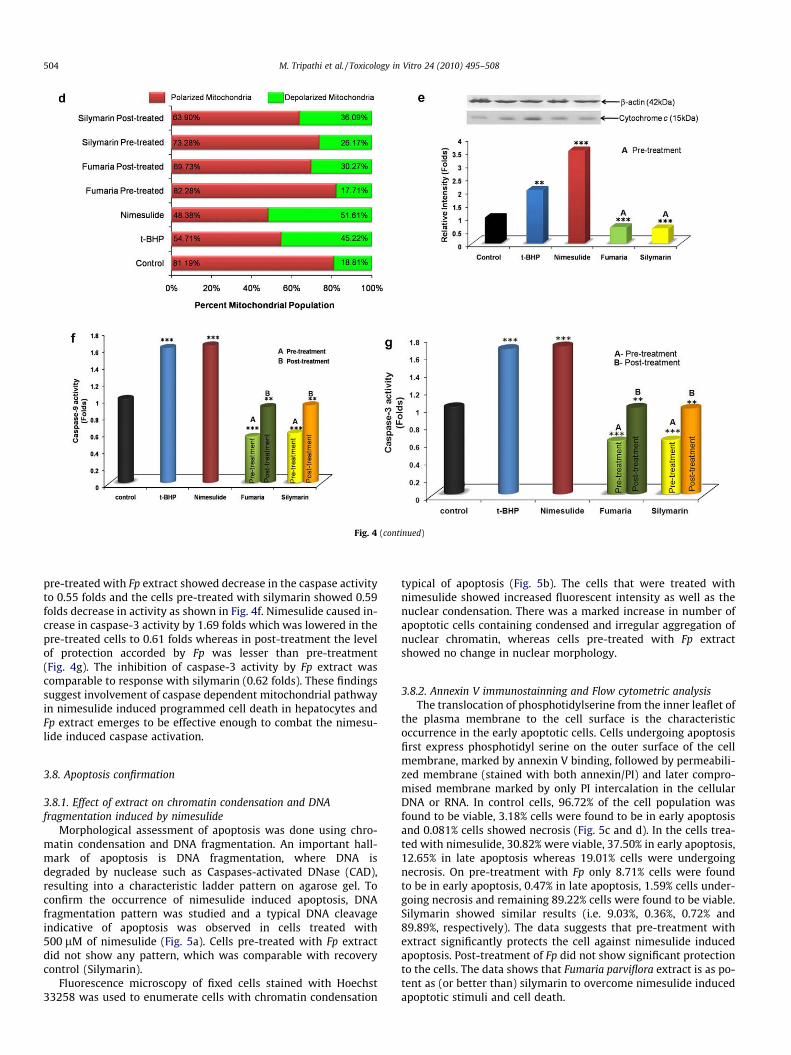
Fig. 4 (continued)
504 M. Tripathi et al. / Toxicology in Vitro 24 (2010) 495–508
pre-treated with Fp extract showed decrease in the caspase activityto 0.55 folds and the cells pre-treated with silymarin showed 0.59folds decrease in activity as shown in Fig. 4f. Nimesulide caused in-crease in caspase-3 activity by 1.69 folds which was lowered in thepre-treated cells to 0.61 folds whereas in post-treatment the levelof protection accorded by Fp was lesser than pre-treatment(Fig. 4g). The inhibition of caspase-3 activity by Fp extract wascomparable to response with silymarin (0.62 folds). These findingssuggest involvement of caspase dependent mitochondrial pathwayin nimesulide induced programmed cell death in hepatocytes andFp extract emerges to be effective enough to combat the nimesu-lide induced caspase activation.
3.8. Apoptosis confirmation
3.8.1. Effect of extract on chromatin condensation and DNAfragmentation induced by nimesulide
Morphological assessment of apoptosis was done using chro-matin condensation and DNA fragmentation. An important hall-mark of apoptosis is DNA fragmentation, where DNA isdegraded by nuclease such as Caspases-activated DNase (CAD),resulting into a characteristic ladder pattern on agarose gel. Toconfirm the occurrence of nimesulide induced apoptosis, DNAfragmentation pattern was studied and a typical DNA cleavageindicative of apoptosis was observed in cells treated with500 lM of nimesulide (Fig. 5a). Cells pre-treated with Fp extractdid not show any pattern, which was comparable with recoverycontrol (Silymarin).
Fluorescence microscopy of fixed cells stained with Hoechst33258 was used to enumerate cells with chromatin condensation
typical of apoptosis (Fig. 5b). The cells that were treated withnimesulide showed increased fluorescent intensity as well as thenuclear condensation. There was a marked increase in number ofapoptotic cells containing condensed and irregular aggregation ofnuclear chromatin, whereas cells pre-treated with Fp extractshowed no change in nuclear morphology.
3.8.2. Annexin V immunostainning and Flow cytometric analysisThe translocation of phosphotidylserine from the inner leaflet of
the plasma membrane to the cell surface is the characteristicoccurrence in the early apoptotic cells. Cells undergoing apoptosisfirst express phosphotidyl serine on the outer surface of the cellmembrane, marked by annexin V binding, followed by permeabili-zed membrane (stained with both annexin/PI) and later compro-mised membrane marked by only PI intercalation in the cellularDNA or RNA. In control cells, 96.72% of the cell population wasfound to be viable, 3.18% cells were found to be in early apoptosisand 0.081% cells showed necrosis (Fig. 5c and d). In the cells trea-ted with nimesulide, 30.82% were viable, 37.50% in early apoptosis,12.65% in late apoptosis whereas 19.01% cells were undergoingnecrosis. On pre-treatment with Fp only 8.71% cells were foundto be in early apoptosis, 0.47% in late apoptosis, 1.59% cells under-going necrosis and remaining 89.22% cells were found to be viable.Silymarin showed similar results (i.e. 9.03%, 0.36%, 0.72% and89.89%, respectively). The data suggests that pre-treatment withextract significantly protects the cell against nimesulide inducedapoptosis. Post-treatment of Fp did not show significant protectionto the cells. The data shows that Fumaria parviflora extract is as po-tent as (or better than) silymarin to overcome nimesulide inducedapoptotic stimuli and cell death.
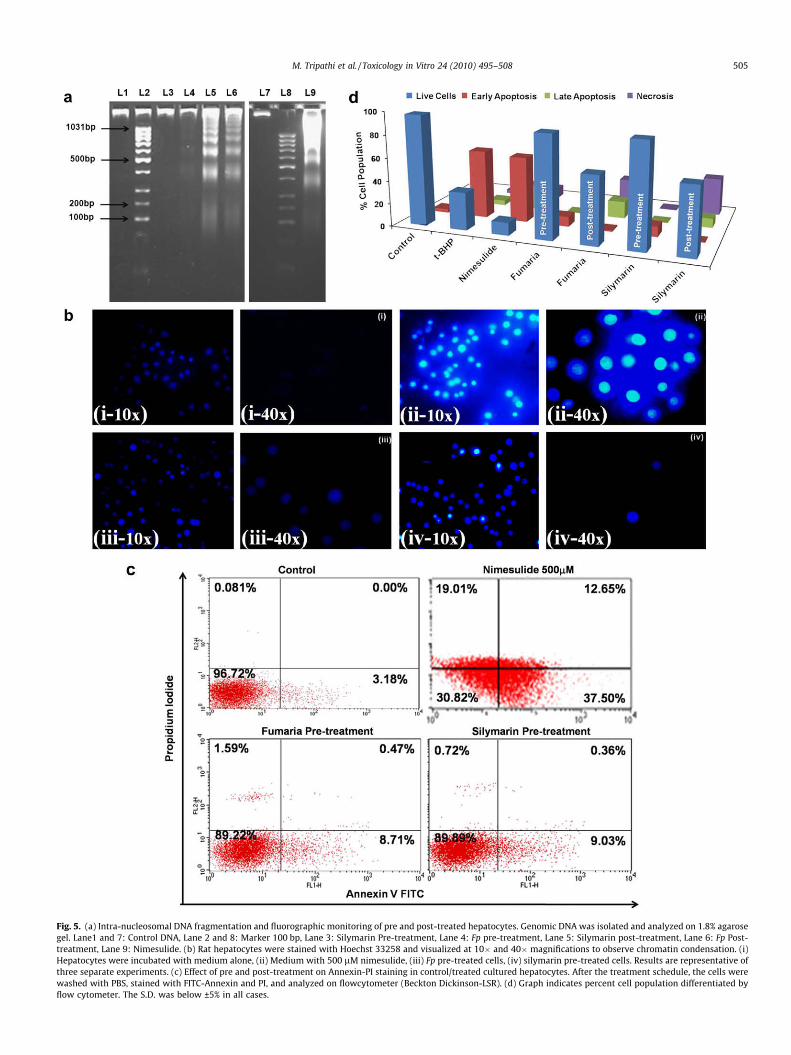
Fig. 5. (a) Intra-nucleosomal DNA fragmentation and fluorographic monitoring of pre and post-treated hepatocytes. Genomic DNA was isolated and analyzed on 1.8% agarosegel. Lane1 and 7: Control DNA, Lane 2 and 8: Marker 100 bp, Lane 3: Silymarin Pre-treatment, Lane 4: Fp pre-treatment, Lane 5: Silymarin post-treatment, Lane 6: Fp Post-treatment, Lane 9: Nimesulide. (b) Rat hepatocytes were stained with Hoechst 33258 and visualized at 10� and 40� magnifications to observe chromatin condensation. (i)Hepatocytes were incubated with medium alone, (ii) Medium with 500 lM nimesulide, (iii) Fp pre-treated cells, (iv) silymarin pre-treated cells. Results are representative ofthree separate experiments. (c) Effect of pre and post-treatment on Annexin-PI staining in control/treated cultured hepatocytes. After the treatment schedule, the cells werewashed with PBS, stained with FITC-Annexin and PI, and analyzed on flowcytometer (Beckton Dickinson-LSR). (d) Graph indicates percent cell population differentiated byflow cytometer. The S.D. was below ±5% in all cases.
M. Tripathi et al. / Toxicology in Vitro 24 (2010) 495–508 505

506 M. Tripathi et al. / Toxicology in Vitro 24 (2010) 495–508
4. Discussion
Toxicity is a well known reason for drug withdrawal, addition ofblack box warnings post approval, or delay in the development ofsafe therapeutics. More than 50% of cases of hepatotoxicity havebeen recognized to be a major reason for drug withdrawal basedon safety, especially effect on liver as it is the major site of metab-olism for most drugs (Atlante et al., 2003; Lasser et al., 2002)Nimesulide is a preferential COX-II inhibitor with high gastrotoler-ability but due to some reported cases of hepatotoxicity, its safetyis suspect. The adverse clinical effects of nimesulide are being stud-ied extensively. Ongoing animal studies and clinical reports, sug-gest that nimesulide causes impairment of kidney function also(Fung et al., 2002). The aim of this study was to investigate themolecular mechanism of nimesulide induced apoptosis in rathepatocytes and its possible amelioration by Fp extract withoutaffecting its native activity as COX-II inhibitor.
There are two major pathways which cause apoptosis i.e.extrinsic and intrinsic pathway. In intrinsic pathway, mitochondriaplay a key role in mediating apoptosis. Mitochondria are known tobe a major source of intracellular ROS generation and are particu-larly vulnerable to oxidative stress. Bcl2 family proteins play a ma-jor role in controlling mitochondrial pathway of apoptosis. It hasalso been reported that Bcl2 can prevent apoptosis by regulatingthe cellular redox potential and can act as an antioxidant byincreasing the glutathione pool and by redistributing GSH to vari-ous cellular compartments, thus preventing ROS production, GSHdepletion and cellular damage (Voehringer, 1999). It is suggestedthat GSH depletion is a frequent event during oxidative stress (Sch-attner and Sokolovskaya, 2000) and can also alter mitochondrialmembrane fluidity. ROS increase can be regarded both as causativeor downstream factor of the impairment of mitochondrial integrity
Fig. 6. Proposed mechanism of nimesulide induced hepatotoxicity. Nimesulide like NSAoxidant/antioxidant imbalance. Nimesulide, downregulates Bcl2, increases Bax expressiophenomenon in which mitochondrial membrane becomes non-selectively permeable focontinuous ROS formation occurs in mitochondria which further damage mitochondrialmitochondria to cytosol. In cytosol Cyt c, procaspase-9 along with dATP along with Apoafcaspase-9. This activated caspase-9 activates caspase-3 to execute apoptosis in hepatocytgeneration. ROS generation accelerates under mitochondrial dysfunction, resulting in celphosphotidylserines to surface of cell membrane. Under increased ROS and activated capoptotic signals. Fp extract was found to significantly modulate key steps of this mecha
and function, typical of the apoptotic process. It is reported thatseveral nitroaromatic drugs can readily produce superoxide anionradicals during their nitro-reductive metabolism (Rodrigo et al.,2002). In the present study, cells treated with nimesulide showedsignificant decrease in the glutathione content and increase in ROSconfirming that nimesulide causes oxidative stress in the hepato-cytes possibly via GSH depletion.
Apoptosis is mainly characterized by overall shrinkage in thevolume of the cells and its nucleus, the loss of adhesion to neigh-boring cells, phosphotidylserine externalization, blebbing, DNAfragmentation, changes in membrane structure (Susin et al.,1998). In our study the injury/toxicity in cultured hepatocytes in-duced by nimesulide was found to be due to apoptosis, since alarge number of cells were stained with annexin V (phosphotidylserine externalization). In addition nimesulide also caused nuclearmorphology change, as assessed by chromatin condensation usinghoechst stain and DNA fragmentation.
The relationship between mitochondrial pathway of apoptosisand ROS has been under study for many years (Herrera et al.,2001). Mitochondrial dysfunction, a hallmark of programmed celldeath (Apoptosis), produces ROS that influence numerous cell pro-cesses (Boelsterli, 2003). Earlier it has been documented that open-ing of high conductance MPT pore increases the permeability of themitochondrial inner membrane, resulting in disruption of ionichomeostasis, release of apoptogenic proteins such as Cyt c, and col-lapse of DWm, resulting in subsequent cell death occurs (Candeet al., 2004). Anti-apoptotic proteins like Bcl2 are localized mainlyin mitochondrial outer membranes where they block membranepermeabilization whereas pro-apoptotic members like Bax aretranslocated from the cytosol to mitochondrial outer membrane,facilitating the permeabilization of mitochondrial membrane andrelease of Cyt c (Pham et al., 2006; Ferri and Kroemer, 2001). This
IDs induce hepatocyte toxicity via Bax dependent mitochondrial pathway involvingn, in hepatocytes which induces mitochondria to undergo permeability transition, ar proteins less than 1500 Da, followed by DWm collapse. Due to DWm collapse, a
membrane. Under permeability transition, Cyt c and procaspase-9 are released fromform apoptosome. Apoptosome activates dimerization of procaspase-9 to activated
es. On the other hand, nimesulide in cytosol causes GSH depletion and increases ROSlular membrane lipid proxidation, giving early apoptosis signals by translocation ofaspase-3, chromatin condensation and DNA fragmentation occurs which gives latenism by which it prevents nimesulide induced hepatocyte cell death i.e. apoptosis.

M. Tripathi et al. / Toxicology in Vitro 24 (2010) 495–508 507
Cyt c using the energy provided by ATP, binds the apoptotic prote-ase activating factor (Apoaf), followed by aggregation of thesecomplexes to form a large heptameric complex also known as‘apoptosome’, which further binds to another protease caspase-9and further caspase cascade sets off. Our data, confirms theinvolvement of this pathway in treated cultured rat hepatocytes.Immunoblot analysis revealed significant decrease in the expres-sion of Bcl2 in nimesulide treated cells, increase in the levels ofBax translocated from cytosol to mitochondria, and mitochondrialdepolarization. Data also shows that nimesulide increases cytosolicCyt c, a critical step in the formation of apoptosome, and caspase-9activation which ultimately leads to the activation of caspase-3 asshown in Fig 6.
As evident from the data discussed above, we report here thatthe test drug nimesulide at a concentration of 500 lM was ableto cause cell death via apoptosis, remarkably involving the mito-chondria mediated pathway. Perhaps play a vital role in the path-ogenesis of the liver injury and oxidative stress can affect cellintegrity only when antioxidant mechanisms are no longer ableto cope with free radical generation. The administration of antiox-idant rich supplement along with the nimesulide can offer protec-tion against drug induced ROS mediated damage.
The second aspect of the study was to observe the cytoprotec-tive and antioxidative potential of Fp extract, to overcome nimesu-lide induced toxicity in cultured cells. Lyophilized 50% ethanolicextract was analysed to confirm the presence of fumaric acid andprotopine content in it (as major organic acid and alkaloid) withthe help of RP–HPLC. Pre-treatment of whole plant extract of Fpwas able to revert all the critical end points of nimesulide inducedapoptosis that further lead to cytotoxicity i.e. modulation in trans-location of anti/pro-apoptotic proteins from cytosol to mitochon-dria (i.e. increased Bcl2 and decreased Bax in mitochondrialfraction), MMP collapse, Cyt c release, phosphatidylserine external-ization, chromatin condensation, DNA fragmentation (Fig 6) ascompared to post-treatment. Effect of Fp extract on COX-II activityof nimesulide was also checked by using specific antibody for COX-II. Result reveals that Fp extract has no effect on the therapeuticefficiency of nimesulide (i.e. inhibition of COX-II). Thus it can besuggested that the extract does not suppresses the native propertyof drug and still protects hepatocytes from its toxicity caused bythe regular use/overdose of drug (nimesulide).
5. Conclusion
Nimesulide at 500 lM causes apoptosis in the primary culturedrat hepatocytes via mitochondria dependent pathway where ROS isthe main intermediate signaling molecule. Generation of ROS hasbeen shown to be an important apoptotic signal and indeed, induc-tion of ROS, depletion in the level of GSH and mitochondrial dys-function by nimesulide was clearly evident in the present study.The toxicity caused by nimesulide was abrogated by Fp extractwithout altering its therapeutic effect by modulating key steps ofapoptosis. On the basis of these findings, we have tried to draw amechanism involved in nimesulide induced hepatotoxicity. Wesuggest that oxidative damage synchronized with mitochondrialdamage and subsequent apoptosis is the main cause of nimesulideinduced cytotoxicity in hepatic cells. This study gives insight intothe possible use of herbal supplement which can reduce the oxida-tive stress generated by drug and is capable of reducing the hepa-totoxicity as it would be a boon for patients who are compelled totake high doses of nimesulide due to severe ailment.
Conflict of Interest statement
There is no conflict of interest.
Acknowledgment
Authors are grateful to Director, IITR, for his kind support to thiswork. The authors are thankful to Dr. Y. Shukla and Dr. A.B. Pant forproviding flow cytometer and fluorescence microscopy facilities.Financial support received under the Supra-Institutional Project(SIP-08) is acknowledged. Authors are also grateful to Council ofScientific and Industrial Research and Indian Council of MedicalResearch, New Delhi for fellowships.
Appendix A. Supplementary material
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.tiv.2009.09.011.
References
Akhtar, M.S., Khan, Q.M., Khaliq, T., 1984. Effects of Euphorbia prostrata and Fumariaparviflora in normoglycemic and alloxan-treated hyperglycaemic rabbits. PlantaMedica 50, 138–142.
Atlante, A., De Bari, L., Bobba, A., Marra, E., Calissano, P., Passarella, S., 2003.Cytochrome c, released from cerbellar granule cells undergoing apoptosis orexcytotoxic death, can generate protonmotive force and derive ATP synthesis inisolated mitochondria. Journal of Neurochemistry 86, 591–604.
Bayly, A.C., Roberts, A.R., Dive, C., 1994. Suppression of liver cell apoptosis in vitroby the non-genotoxic hepatocarcinogen and peroxisome proliferator nafenopin.Journal of Cell Biology 33, 191–198.
Berde, C., Sundel, R., 1998. COX-2 Inhibitors: A status report. Technical corner fromIASP newsletter. International Association for the study of Pain. p. 36.
Boelsterli, U.A., 2003. Idiosyncratic drug hepatotoxicity revisited: new insights frommechanistic toxicology. Toxicology Mechanisms and Methods 13, 3–20.
Bolestreli, U.A., 2002. Mechanism of NSAID-induced hepatotoxicity – focus onnimesulide. Drug Safety 25, 633–648.
Borner, C., 2003. The Bcl-2 protein family: sensors and checkpoints for life-or-deathdecisions. Molecular Immunology 39, 615–647.
Cain, K., Bratton, S.B., Cohen, G.M., 2002. The Apoaf-1 apoptosome; a large caspaseactivating complex. Biochimie 84, 203–214.
Cande, C., Vahsen, N., Metivier, D., Tourriere, H., Chebli, K., Garrido, C., 2004.Regulation of cytoplasmic stress granules by apoptosis-inducing factor. Journalof Cell Science 117, 4461–4468.
Cossarizza, A., Ceccarelli, D., Masini, A., 1996. Functional heterogeneity of anisolated mitochondrial population revealed by cytofluorometric analysis at thesingle organelle level. Experimental Cell Research 22, 84–94.
Emanuele, S., D’Anneo, A., Bellavia, G., Vassallo, B., Lauricella, M., Blasio, A.D., Vento,R., Tesoriere, G., 2004. Sodium butyrate induces apoptosis in human hepatomacells by a mitochondria/caspase pathway, associated with degradation of beta-catenin, pRb and Bcl-XL. European Journal of Cancer 40, 1441–1452.
Ferri, K.F., Kroemer, G., 2001. Organelle-specific initiation of cell death pathways.Nature Cell Biology 3, E255–E263.
Fleury, C., Mignotte, B., Vayssière, J.L., 2002. Mitochondrial reactive oxygen speciesin cell death signaling. Biochimie 84, 131–141.
Fosaluzza, V., Montagnani, G., 1989. Efficacy and tolerability of nimesulide inelderly patients with osteoarthritis: double blind trial versus naproxen. Journalof International Medical Research 17, 295–303.
Fraschni, F., Demartini, G., Esposti, D., 2002. Pharmacology of silymarin. ClinicalDrug Investigation 22, 51–65.
Fung, M.F., Johnston, M.E., Eisenhauer, E.A., Elit, L., Hirte, H.W., Rosen, B., 2002.Chemotherapy for recurrent epithelial ovarian cancer previously treated withplatinum-a systematic review of the evidence from randomized trials. EuropeanJournal of Gynaecological Oncology 23, 104–110.
Heidari, R.M., Mandgary, A., Enayati, M., 2004. Antinociceptive effects and toxicityof Fumaria parviflora Lam. in mice and rats. DARU 12, 136–140.
Hermann, M., Lorenz, H.M., Vol, I.R., Grunke, M., Woith, W., Kalden, J.R., 1994. Arapid and simple method for the isolation of apoptotic DNA fragments. NucleicAcid Research 22, 5506–5507.
Herrera, J., Nava, M., Romero, F., Rodriguez-Iturbe, B., 2001. Melatonin preventsoxidative stress resulting from iron and erythropoietin administration.American Journal of Kidney Diseases 37, 750–757.
Jie, L., Takahito, Y., Hiroshi, S., Hiroyoshi, M., Dong-Sheng, S., Naoshi, M., Masao, Y.,Junji, M., Zaishun, J., Itara, Y., Narioki, T., 2003. Annexin vs. assay-proven anti-apoptotic effect of ascorbic acid 2-glucoside after cold ischemia/reperfusioninjury in rat liver transplantation. Acta Medica Okayama 57, 209–216.
Kakkar, P., Singh, B.K., 2007. Mitochondria: a hub of redox activities and cellulardistress control. Molecular Cellular Biochemistry 305, 235–253.
Kakkar, P., Das, B., Viswanathan, P.N., 1984. A modified spectrophotometric assay ofsuperoxide dismutase. Indian Journal of Biochemistry and Biophysics 21, 130–132.
Kroemer, G., Zamzanmi, N., Susin, S.A., 1997. Mitochondrial control of apoptosis.Immunology Today 18, 44–51.
Kulkarni, S.K., 2002. On the safety of nimesulide, a preferential COX-2 inhibitor.Current Science 83, 1442–1443.

508 M. Tripathi et al. / Toxicology in Vitro 24 (2010) 495–508
Lambert, C., Apel, K., Biesalski, H.K., Frank, J., 2004. 2-Methoxyestradiol inducescaspase-independent, mitochondria-centered apoptosis in DS-sarcoma cells.International Journal of Cancer 108, 493–501.
Lasser, K.E., Allen, P.D., Woolhandler, S.J., Himmelstein, D.U., Wolfe, S.M., Bor, D.H.,2002. Timing of new black box warnings and withdrawals for prescriptionmedications. Journal of the American Medical Association 287, 2215–2220.
Lowry, O.H., Rosenbourgh, N.J., Farr, A.L., Randall, R.J., 1951. Protein measurementwith folin phenol reagent. Journal of Biological Chemistry 193, 265–275.
Madhavi, D.L., Salunkhe, D.K., 1995. Toxicological aspects of food antioxidants. In:Madavi, D.L., Deshpande, S.S., Salunkhe, D.K. (Eds.), Food Antioxidants. Dekker,New York, p. 267.
Mehta, N., Ozick, L., 2008. Drug Induced Hepatotoxictiy. Found at <http://emedicine.medscape.com/article/169814-overview>.
Merlani, G., Fox, M., Oehen, H.P., Cathomas, G., Renner, E.L., Fattinger, K.,Schneemann, M., Kullak-Ublick, G.A., 2001. Fatal hepatotoxicity secondary tonimesulide. European Journal of Clinical Pharmacology 57, 321–326.
Mohammad, N.S., Kirsten, M.O., Torstein, L., 2001. Effect of extracellular Mg2+ onROS and Ca2+ accumulation during reoxygenation of rat cardiomyocytes.American Journal of Physiology Heart and Circulatory Physiology 280, 344–353.
Mosmann, T., 1983. Rapid colorimetric assay for cellular growth and survival:application to proliferation and cytotoxicity assays. Journal of ImmunologicalMethods 65, 55–63.
Novo, E., Parola, M., 2008. Redox mechanisms in hepatic chronic wound healing andfibrogenesis. Fibrogenesis and Tissue Repair 1, 5.
Okada, Y., Oyama, Y., Chikahisa, L., Satoh, M., Kanemaru, K., Sakai, H., 1996. Tri-n-butyltin-induced change in cellular level of glutathione in rat thymocytes: aflow cytometric study. Toxicology Letters 117, 123–128.
Orhan, I., Sener, B., Choudhary, M.I., Khalid, A., 2004. Acetylcholinesterase andbutyrylcholinesterase inhibitory activity of some Turkish medicinal plants.Journal of Ethnopharmacology 91, 57–60.
Orrenius, S., 2007. Reactive oxygen species in mitochondria-mediated cell death.Drug Metabolism Reviews 39, 443–455.
Petit, P.X., O’Conner, J.E., Grunwald, D., Brown, S.C., 1990. Analysis of the membranepotential of rat- and mouse-liver mitochondria by flow cytometry and possibleapplications. European Journal of Biochemistry 194, 389–397.
Pham, T.N.D., Marion, M., Denizeau, F., Jumarie, C., 2006. Cadmium-inducedapoptosis in rat hepatocytes does not necessarily involve caspase-dependentpathways. Toxicology In Vitro 20, 1331–1342.
Pradhan, S.C., Girish, C., 2006. Hepatoprotective herbal drug, silymarin fromexperimental pharmacology to clinical medicine. Indian Journal of MedicalResearch 124, 491–504.
Rainsford, K.D., 1998. An analysis from clinic- epidemiological data of the principleadverse events from the COX-2 selective NSAID, nimesulide, with particularreference to hepatic injury. Inflammopharmacology 6, 203–221.
Rodrigo, P., De Francisco, R., Perez-Pariente, J.M., Cadahia, V., Tojo, R., Rodriguez, M.,Lucena, M.I., 2002. Nimesulide induced severe haemolytic anemia and acuteliver failure leading to liver transplantation. Scandinavian Journal ofGastroenterology 37, 1341–1343.
Sbeit, W., Krivoy, N., Shiller, M., Farah, R., Cohen, H.I., Struminger, L., Reshef, R., 2001.Nimesulide induced acute hepatitis. Annals of Pharmacotherapy 35, 1049–1051.
Schattner, A., Sokolovskaya, J.C., 2000. Fatal hepatitis and renal failure duringtreatment with nimesulide. Journal of Internal Medicine 247, 153–155.
Seglen, P.O., 1976. Preparation of isolated rat liver cells. Methods in Cell Biology 13,29–83.
Sheu, S.S., Nauduri, D., Anders, M.W., 2006. Targeting antioxidants to mitochondria:a therapeutic direction. Biochimica et Biophysica Acta 1762, 256–265.
Suau, R., Cabezudo, B., Rico, R., Nájera, F., López-Romero, J.M., 2002. Directdetermination of alkaloid contents in Fumaria species by GC–MS.Phytochemical Analysis 13, 363–367.
Susin, S.A., Zamzami, N., Kroemer, G., 1998. Mitochondria as regulators of apoptosis:doubt no more. Biochimica et Biophysica Acta 1366, 151–165.
Van Steenbergen, W., Peeters, P., De Bondt, J., Staesse, D., Buscher, H., Laporta, T.,Desnet, T., Roskams, V., 1998. Nimesulide induced acute hepatitis: evidencefrom six cases. Journal of Hepatology 29, 135–141.
Voehringer, D.W., 1999. BCL-2 and glutathione: alterations in cellular redox state thatregulate apoptosis sensitivity. Free Radical Biology and Medicine 27, 945–950.
Wallin, B., Rosengren, B., Shertzer, H.G., Camejo, G., 1993. Lipoprotein oxidation andmeasurement of thiobarbituric acid reacting substances formation in a singlemicrotiter plate: its use for evaluation of antioxidants. Analytical Biochemistry208, 10–15.
Weiss, P., Mouallem, M., Bruck, R., Hassin, D., Tanay, A., Brickman, C.M., Farfel, Z.,Bar-Meir, S., 1999. Nimesulide induced acute hepatitis and acute liver failure.Israel Medical Association Journal 1, 89–91.
Wlliamson, E.M., 2002. Major Herbs of Ayurveda. Churchill Livingstone, New York.p. 151.
Yang, J., Wu, L.J., Tashino, S.I., Onodera, S., Ikejima, T., 2007. Critical roles of reactiveoxygen species in mitochondrial permeability transisiton in mediatingevodiamine-induced human melanoma A375–S2 cell apoptosis. Free RadicalResearch 41, 1099–1108.
Zhang, Q.H., Sheng, H.P., Brenanner, T.A., Loh, T.T., 1999. Redistribution ofcytochrome c release is not an essential requirement in C2-ceramide inducedapoptosis in HL-60 cells. Life Sciences 65, 1715–1723.
Copyright © 2022 FDOKUMEN




















