
The influence of concomitant antidepressant medication on safety, tolerability and clinical...
67
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of The influence of concomitant antidepressant medication on safety, tolerability and clinical...


Editorial
Ethical aspects of publishingHans-Jurgen Moller ............................................................................................................................... 66
Review
Psychiatric symptoms associated with oculogyric crisis: A review of literature for thecharacterization of antipsychotic-induced episodes
Kazuhiko Abe ....................................................................................................................................... 70
Original Investigations
Effect of olanzapine treatment on platelet glutamine synthetase-like protein andglutamate dehydrogenase immunoreactivity in schizophrenia
Gulnur Sh. Burbaeva, Irina S. Boksha, Elena B. Tereshkina, Olga K. Savushkina,Marina S. Turishcheva, Lubov I. Starodubtseva, Oleg S. Brusov, Margarita A. Morozova .............. 75
The influence of concomitant antidepressant medication on safety, tolerability and clinicaleffectiveness of electroconvulsive therapy
Thomas C. Baghai, Alain Marcuse, Melanie Brosch, Cornelius Schule, Daniela Eser,Caroline Nothdurfter, Yvonne Steng, Ines Noack, Katrin Pietschmann, Hans-Jurgen Moller,Rainer Rupprecht .............................................................................................................................. 82
P300 differences exist between Tourette’s syndrome with and without attention deficiencyand hyperactivity disorder in children
Zhu Yan, Liu Po-Zi, Leung Kai-Man, Su Lin-Yan, Wu Da-Xing, Zhou Ming ...................................... 91
Summary of Original Research
Three-dimensional models of neurotransmitter transporters and their interactions withcocaine and S -citalopram
Aina Westrheim Ravna ......................................................................................................................... 99
Viewpoint
The problem of delusional ugliness: Is it really body dysmorphic disorder?Leonardo F. Fontenelle, Mauro V. Mendlowicz, Juliana Kalaf, Marcio Versiani .................................. 110
Case Reports/Case Series
Abdominal cutaneous nerve entrapment syndrome (ACNES) in a patient with apain syndrome previously assumed to be of psychiatric origin
Johannes Thome, Christian Egeler ........................................................................................................ 116
The treatment of recurring auditory hallucinations in schizophrenia with rTMSPaul B. Fitzgerald, Jessica Benitez, Jeff Z. Daskalakis, Anthony De Castella, Jayashri Kulkarni ......... 119
Aripiprazole in patients with Tourette syndromeEmanuel Bubl, Evgeniy Perlov, Ludger Tebartz Van Elst ..................................................................... 123
Announcement
‘‘2nd International Congress of Biological Psychiatry’’ ........................................................................ 125
Letter to the Editor
Addressing the limitations of the CATIE studySiegfried Kasper, Dietmar Winkler ....................................................................................................... 126
Instructions to Authors ....................................................................................................... 128
The World Journal of Biological PsychiatryVolume 7, No 2, 2006
Contents

EDITORIAL
Ethical aspects of publishing
Ethical aspects of publishing have become a matter
of greater interest over the past decade. Considera-
tion of the associated problems led to the formation
of respective societies and relevant congresses, for
example the International Committee of Medical
Journal Editors, The Vancouver Group (Walter and
Bloch 2001) and the Committee of Publication
Ethics in Great Britain (Smith 1997, 2003). Inter-
national congresses about publishing in biomedicine
with a focus on ethical problems took place as early
as 1989, 1993 and 1997 (Rennie 1990; Rennie and
Flanagin 1994, 1998).
Ethical problems affect various aspects of publish-
ing. Some of the most important ones will be
described briefly below, whereby only aspects related
to publishing itself will be discussed and not the
original basis for the publication, i.e. not the results
of scientific studies and the related problems con-
cerning study methods and execution.
The fact that the quality of a young scientist’s
research is nowadays measured by the number or
impact factor of his publications has resulted in a
strong pressure to publish for the individual and his
research group. But there are also other sources of
pressure to publish, e.g. the interest of the pharma-
ceutical companies to present the results of their
studies as widely and diversely as possible. As a
consequence, the results of a scientific investigation
or clinical pharmacological study are not published
in one paper, as would actually be meaningful and
necessary, but as many papers as possible are
published, for example by publication in various
languages. These publications are either real
duplicate publications, or some of them overlap
considerably with respect to the presented results.
Alternatively, new individual subsections are picked
out time and again that are often rather irrelevant
but are used to present repeatedly also the main
results of the corresponding scientific investigation.
Overall, duplication to a greater or lesser extent, or
fragmentation (‘salami tactic’), results in the pub-
lication of more papers than is actually necessary
and meaningful to present the results of a scientific
investigation. One of the results of this publication
technique is an undesirable increase in the flood of
publications, so that even the scientist active in the
respective area has trouble keeping an overview of
the available data. Furthermore, the joint origin of
the various publications, i.e. the actual data source,
is frequently not made apparent enough (often on
purpose!), so that the reader gains the impression
that results of several different studies are being
reported. This inflation of publications is often made
use of for studies with positive results. Besides the
purposeful or accepted deception of the readership,
this publication technique also causes difficulties for
meta-analyses, since it is often not clear to the meta-
analyst how many studies are being reported, thus
leading to a distortion of the data in the sense of a
positive bias. In the case of negative results, there is a
distortion in the other direction in the sense that
they are published only very briefly or not at all. The
technique of fragmented publication is often so
intransparent that it is sometimes not even noticed
by journal editors. For example, Walter and Bloch
(2001) pointed out that in the case of an antide-
pressant almost identical manuscripts were pub-
lished without the editors noticing.
A particularly serious case of ethical problems is
the publication of purposefully falsified data or of
data that are completely made up and do not have a
basis in the sense of an empirical study. Such cases
are almost certainly uncommon, but they are prob-
ably also rarely discovered and one must assume that
a certain number of cases go unnoticed. Several
years ago such a case was discovered in German
genetic research that caused a great stir, all the more
so because the research group had been given
generous support by the DFG (‘‘Deutsche For-
schungs Gesellschaft’’, German Research Associa-
tion) for years and because it had published in very
prestigious international journals. This case shows
that despite both the DFG’s complicated application
process and the careful and extensive review proce-
dures during the publication process at respected
journals, it can be difficult to recognise cleverly
performed falsifications. Editors, who do not usually
have access to the original data, hardly stand a
chance of recognising such falsifications, especially
when the published data appear to be plausible
enough.
Conflicts of interest can also represent a severe
dilemma, in particular the conflict between the
sponsor of a study, mostly the pharmaceutical
The World Journal of Biological Psychiatry, 2006; 7(2): 66�/69
ISSN 1562-2975 print/ISSN 1814-1412 online # 2006 Taylor & Francis
DOI: 10.1080/15622970600700108

industry, and the authors (Helmchen 2003; Miller
et al. 1999). The relationship between the author
and sponsor and the related financial interests
involved on both a personal and research-related
level can lead to a biased presentation, which is not a
falsification of data in the sense described above, but
perhaps a particularly positive depiction of the
results to the sponsor’s benefit, for example. This
can happen in such a way that is not at all evident to
the author, in face of the often very good personal
relationship to the sponsor, but which is almost
subconscious, normally at least unintentional. In
order to reveal this ‘‘conflict’’, most journals these
days require the author to describe any relevant
conflict of interest through a ‘‘disclosure’’ (Hender-
son et al. 2003), i.e. the author discloses the
source(s) of financial support and assistance with
the study, or by whom he was or is supported. He is
required to disclose everything that is potentially
related to the topic of the paper. This requirement
does not infer that the author has a conflict of
interest but serves the purpose to reveal to the reader
the author’s relationships in the matter at hand so
that the reader is specially sensitised for a possibly
biased presentation.
However, the focus on the sponsoring pharma-
ceutical industry, which mostly forms the basis of the
demand for disclosures, is possibly too narrow.
There are definitely other influencing factors which
are probably just as important, for example focussing
on a certain orientation of the overall subject area
(e.g. biological psychiatry versus psychotherapy),
rivalries between people, etc. (Horton 1997). How-
ever, such factors are much more elusive than the
rather ‘‘material relationships’’ with the sponsor of a
study or the sponsoring of a scientist’s research by
the pharmaceutical industry. Since for these reasons
a disclosure that is solely aimed at the sponsoring
by the pharmaceutical industry cannot fulfil the
idealistic demands placed on it, some colleagues
completely disapprove of the disclosure in the way
it is currently practised and brand it as a ‘‘new
McCarthyism’’ (Rothman 1993).
Insufficiently justified authorship is a further
problem in this respect. Under the pressure of the
need to publish and achieve as high an impact factor
as possible, it has increasingly become a habit that
many authors are mentioned on a publication who
may not be associated with, or who may have only a
limited association with the scientific investigation
or the publication. In this respect, unjustified
senior-authorships as a ‘‘favour’’ as well as unjusti-
fied reciprocal co-authorships of members of the
same research group or different research groups
from the same institution should be mentioned, all
of which serve to increase the personal impact factor
or the number of an individual’s publications.
Although the consideration of several publications
offers a chance to obtain clues as to which author has
perhaps just been named as a favour, it requires a lot
of work and the attributes of a detective. Admittedly,
it is sometimes difficult to decide who is entitled to
be named as a co-author of a publication (Helmchen
2001). Proposals of the Vancouver Group (Interna-
tional Committee of Medical Journal Editors 1997)
attempt to describe on different levels the respective
contribution that resulted in co-authorship, e.g.
contribution to study conception and design, con-
tribution to analysis or interpretation of data and
contribution to critical review of the work, etc.
However, on the other side these criteria are still
seen by some to be too soft. It has thus been
demanded that at the end of a paper the respective
contribution of each individual author/co-author is
described in detail.
Besides the naming of co-authors who have not
made a sufficient contribution to the scientific work
or the manuscript, there is the other problem that
scientists who have worked on a project are not
named in the publication for various personal or
institutional reasons. A further problem is the order
of the authors. One normally presumes that the
person who wrote the manuscript is also the first
author and therefore takes responsibility for the
content. Others also give the senior authors a special
role, as he is often head of the research group. The
fact that the order of names is not unimportant
becomes apparent when problems occur, e.g. in the
research falsification mentioned above: the senior
author claimed that he could not take any respon-
sibility for the details of the manuscript because he
had not been involved in the details of data collec-
tion.
Finally, a further bias in the publication process
should be mentioned that is not on the level of the
authors but on that of the editors: an editor and the
reviewers he chooses may follow a certain school of
thought of the subject area and promote this
direction preferentially by accordingly positive pub-
lications.
The editors of scientific journals thus have
an important and increasingly complex task,
which can hardly be accomplished ‘‘on the side’’.
Great demands are placed on the editor with
respect to fair ‘‘handling’’ of the manuscript
through to careful execution of the review process
under consideration of the special problems
mentioned above (Young 2003; Young and Joffe
2004).
An ethical problem of a completely different
dimension is the fact that only colleagues from the
rich industrial nations of the so-called western world
Editorial 67

have adequate access to scientific publications in
highly respected international journals. Colleagues
in economically poorer countries, the so-called
developing countries, often have no access to such
journals since they do not have the financial means
to buy their own copies and often also the university
libraries do not have enough money to buy these
journals. This problem is being increasingly dis-
cussed (Delucchi 2005). As far as I am aware, The
World Journal of Biological Psychiatry was the first
psychiatric journal to take this matter sufficiently
into account. With the help of an unrestricted grant
from the pharmaceutical industry (at that time from
Janssen Cilag and Organon), this journal was sent
free of charge to all members of the WFSBP, so that
also colleagues from the economically poor countries
could profit from the information it contained.
However, this procedure was also criticised at it
was feared that the sponsoring companies may find
an opportunity to influence the content to their
advantage. As Chief Editor of this journal I remain
fully convinced that this was not the case, although I
must admit that theoretically this could have been
the consequence. It is therefore definitely better if
sponsoring by one or two companies is replaced by a
‘‘pool sponsoring’’ by several companies as this
makes it much easier to avoid the possible develop-
ment of any bias.
The World Journal of Biological Psychiatry also
attempted to deal with another problem, although
perhaps in too ideological a way. For several
reasons it is often very difficult for scientists
from economically poor countries to publish the
results of their research. Often, the research is not
on an internationally competitive level, due among
other things to the poor financial setting or an
insufficient tradition or method of research. An-
other important reason is the fact that scientists in
economically poor countries do not have an
adequate command of English scientific language
and the English publication style. A publication is
therefore often rejected by the editor of a re-
spected international journal for reasons of lan-
guage and style, without it even being sent to
reviewers. One of the primary objectives of The
World Journal of Biological Psychiatry was to work
against this publication bias, which was to the
disadvantage of the colleagues from economically
poorer parts of the world. One such approach was
to increase the likelihood that manuscripts by
colleagues from such countries were accepted for
publication by assigning them to a member of the
Editorial Board or a specially chosen reviewer who
helped the author to improve their paper under
these aspects. Admittedly, this idealistic approach
could only be realised to a very limited extent. It
requires so much time and effort that voluntary
editors, members of the Editorial Board or re-
viewers can only be asked to perform such an
additional task in exceptional cases. Principally, it
would be desirable that a journal provides addi-
tional means for this task.
In the context of considerations about how to
make up-to-date knowledge available to all
interested parties in a fair way, the demand was
also expressed that a scientific journal should
realise ‘‘open access’’ in the internet right from
the start (Smith 2004; Suber 2002). However,
this contradicts the economical conditions in
which a journal is published and means must
thus be sought to achieve a balance of all interests
(Moller 2005).
Hans-Jurgen Moller
Chief Editor
The World Journal of Biological Psychiatry
Correspondence:
Professor Hans-Jurgen Moller
Department of Psychiatry
Ludwig-Maximilians-University
Nussbaumstr. 7, 80336 Munich
Germany
Tel: 49 89 5160 5501
Fax: 49 89 5160 5522
E-mail: [email protected]
References
Delucchi G. 2005. Regarding open access to scientific journals.
World J Biol Psychiatry 6:60.
Helmchen H. 2001. [Clinical research methods]. Fortschr Neurol
Psychiatrie 69:291�/299.
Helmchen H. 2003. [Psychiatrists and the pharmaceutical in-
dustry]. Nervenarzt 74:953�/964.
Henderson C, Howard L, Wilkinson G. 2003. Acknowledgement
of psychiatric research funding. Br J Psychiatry 183:273�/
275.
Horton R. 1997. Conflict of interest in clinical research: Oppro-
brium or obsession? Lancet 349:1112�/1113.
International Committee of Medical Journal Editors. 1997. Uni-
form requirements for manuscripts submitted to biomedical
journals. Ann Int Med 126:36�/47.
Miller F, Pickar D, Rosenstein D. 1999. Addressing ethical issues
in the Psychiatric Research Literature. Arch Gen Psychiatry
56:763�/764.
Moller HJ. 2005. Access to The World Journal of Biological
Psychiatry. World J Biol Psychiatry 5:118.
Rennie D. 1990. Editorial peer review in biomedical
publication: The first international congress. J Am Med Assoc
263:1317.
Rennie D, Flanagin A. 1994. The second international congress
on peer review in biomedical publication. J Am Med Assoc
272:91.
Rennie D, Flanagin A. 1998. Congress on biomedical peer review.
J Am Med Assoc 280:213.
68 Editorial

Rothman K. 1993. Conflict of interest: The new McCarthysim in
science. J Am Med Assoc 269:2782�/2784.
Smith R. 1997. Misconduct in research: Editors respond �/ The
Committee on Publication Ethics is formed. Br Med J
315:201�/202.
Smith R. 2003. Draft code of conduct for medical editors. Br Med
J 327:1010.
Smith R. 2004. Travelling but never arriving: reflections of a
retiring editor. Br Med J 329:242�/244.
Suber P. 2002. Open access to the scientific journal literature. J
Biol 1:1�/3.
Walter G, Bloch S. 2001. Publishing ethics in psychiatry. Aust NZ
J Psychiatry 35:28�/35.
Young S. 2003. Peer review of manuscripts: Theory and practice. J
Psychiatry Neurosci 28:327�/330.
Young S, Joffe R. 2004. Ethical conduct of journal editors. Rev
Psychiatr Neurosci 29:334�/336.
Editorial 69

REVIEW
Psychiatric symptoms associated with oculogyric crisis: A review ofliterature for the characterization of antipsychotic-induced episodes
KAZUHIKO ABE
Faculty of Health and Welfare, Seinan Jogakuin University, Kitakyushu, Japan
AbstractAntipsychotics have been found to induce recurrent psychotic episodes lasting minutes to hours, mostly accompanied byoculogyric crisis (OGC). To characterize this side effect, antipsychotic-induced and postencephalitic OGCs that werereported in the literature were compared to find out common characteristics of OGCs and their associated symptoms. Bothpostencephalitic and antipsychotic-induced OGCs were found to occur late in the day and at regular intervals, and wereassociated with autonomic symptoms such as profuse sweating, facial flushing, transitory hypertension and difficulty inmicturition. They were often associated also with transient psychiatric episodes: visual hallucinations and illusions, auditoryhallucinations, delusions, catatonic phenomena, obsessive thoughts and panic attacks. These (OGC) characteristics will beuseful in recognizing antipsychotic-induced psychiatric episodes. The associated psychiatric episodes were noted to recuroccasionally also without OGC in a few postencephalic cases, and during gradual dose reduction or after a switch to a novelor low-potency antipsychotic in drug-induced cases. These findings suggest that episodes with the OGC characteristics butwithout OGC per se, may be less severe reactions to antipsychotic medication than those with OGC, and may representmanifestations of subclinical OGC.
Key words: Oculogyric crisis, antipsychotics, epidemic encephalitis, psychiatric symptoms, autonomic symptoms
Introduction
Antipsychotics, especially high-potency conventional
antipsychotics, have been found to induce recurrent
psychotic episodes lasting minutes to hours (as
described below in detail), mostly accompanied by
oculogyric crisis (OGC). However, since the OGCs
in such cases can be brief or subtle (Rogers 1989;
Thornton and McKenna 1994) and easily missed,
the psychotic episodes could be mistaken for exacer-
bation of the psychosis (Benjamin 1999) and the
antipsychotic medication may be increased rather
than reduced, resulting in further aggravation of the
condition.
To prevent this outcome, antipsychotic-induced
episodes should be characterized, so that they are
easily recognized as such. Although there are not
many case reports where these episodes were de-
scribed in detail, some authors noted similarities
between the episodes in their cases and those which
had been described in association with postencepha-
litic OGC (Sachdev and Tang 1992; Thornton and
McKenna 1994; Bemjamin 1999). However, such
similarities have not been systematically investigated.
Most of the postencephalitic cases were published
in the 1920s and 1930s, and in many of them OGC-
associated psychiatric episodes were described in
detail. In this paper, such episodes are compared to
those associated with antipsychotic-induced OGC,
to find out characteristics common to both, which
will be useful in recognizing antipsychotic-related
transient psychiatric episodes.
Method
For symptoms associated with postencephalitic
OGC, literature was searched by Index Medicus
1925�/1939 with key words, epidemic encephalitis,
sequelae, oculogyric crisis (or Schauanfall, Blickk-
rampf or ocular manifestations), and 30 articles
published in psychiatric, neurological or medical
journals in English, German or French were found.
Of these, 18 described OGC-associated psychiatric
symptoms and are reviewed in this paper. Papers
Correspondence: Professor Kazuhiko Abe, MD, Seinan Jogakuin University, 1-3-5 Ibori, Kokura-Kitaku, Kitakyushu, 803-0835 Japan.
Tel./Fax: +81 940 35 2982. E-mail: [email protected]
The World Journal of Biological Psychiatry, 2006; 7(2): 70�/74
(Received 21 March 2005; accepted 16 June 2005)
ISSN 1562-2975 print/ISSN 1814-1412 online # 2006 Taylor & Francis
DOI: 10.1080/15622970500222443

cited by Jelliffe (1929) and Onuaguluchi (1961) on
the subject are also reviewed for this paper.
For symptoms associated with antipsychotic-in-
duced OGC, a computer search was performed
using the key words, oculogyric crisis, neuroleptic-
induced and psychiatric symptoms. Nine articles
were found. These and papers cited by Benjamin
(1999) and Owens (1999) on drug-induced OGC
and associated psychiatric episodes are reviewed for
this paper.
Results
Postencephalitic OGC
Encephalitis lethargica (epidemic encephalitis)
chiefly occurred in the period 1917�/1928, but its
causative agent was never determined, though pre-
sumably it was viral in nature. One of its common
sequelae was parkinsonism, often accompanied by
OGC (Yahr 1978).
The time of the day of occurrence and associated
autonomic symptoms. Postencephalitic OGC tended
to occur late in the day and at more or less regular
intervals (McCowan and Cook 1928; Jelliffe 1929;
Hall 1931; Odegard 1932), and were usually accom-
panied by autonomic symptoms such as profuse
sweating, facial flushing, salivation, transitory hy-
pertension and difficulty in micturition (Euziere et
al. 1933; Onuaguluchi 1961) and often preceded by
forward staring (Onuaguluchi 1961).
The associated psychiatric symptoms. The OGCs were
also often accompanied by a variety of psychiatric
symptoms (Jelliffe 1929). Visual hallucinations
(Pascheff 1926; Marinesco and Nicolesco 1932;
van Bogaert 1933) and illusions (Flach and Palisa
1936), tactile (Georgi 1926; Petit et al. 1926;
Schwab et al. 1951) and auditory hallucinations
(Brody and Freed 1941; Rosner 1942) and distor-
tion in body schema (van Bogaert 1933) were
reported.
Delusions (Brody and Freed 1941; Rosner 1942;
Gillespie 1944; Schwab et al. 1951), unresponsive-
ness bordering on catatonic stupor (Ewald 1925;
Jelliffe 1929; Leigh et al. 1987), hypomania (Pardee
1928), depression (Jelliffe 1929; Urechia 1949;
Schwab et al. 1951), ‘‘sham rage’’ (Oller 1946;
Urechia 1949), obsessive thoughts (Ewald 1925;
Odegard 1932; Euziere et al. 1933; Rosner 1942)
and severe anxiety (Jelliffe 1929; Schwab et al. 1951)
were also reported. These associated psychiatric
symptoms varied from patient to patient but varied
little among OGCs in the same patient, and were
usually absent between the OGCs (Rosner 1942).
On the other hand, the same transient episodes
which were associated with OGC, such as depressive
stupor (Ewald 1925), paroxysmal depression (Ur-
echia 1949; Schwab et al. 1951) or visual hallucina-
tions (Marinesco and Nicolesco 1932) were noted to
occur sometimes without OGC.
The temporal relationship between the OGC and
the psychiatric episode was described in a few cases:
the episode appeared first, became intense and
culminated in OGC or the latter appeared in the
middle of the former (van Bogaert 1933; Brody and
Freed 1941; Gillespie 1944; Urechia 1949; Leigh et
al. 1987).
The influence of treatment. As to the treatment and
prophylaxy of OGC, the administration of scopola-
mine decreased the frequency of OGC (Marinesco
and Nicolesco 1932; van Bogaert 1933). It was also
noted that scopolamine administered during OGC
abolished it, but the associated visual hallucinations
persisted (Marinesco and Nicolesco 1932). Other
anticholinergic drugs, such as hyosciamine (Brody
and Freed 1941; Schwab et al. 1951) and carami-
phen (Schwab et al. 1951), antihistamine, such as
diphenhydramine and pheniramine (Schwab et al.
1951), and amphetamine (Brody and Freed 1941;
Oller 1946), were used.
Anticholinergic combined with amphetamine was
found effective for OGC and its associated delu-
sional episodes (Brody and Freed 1941). In Oller’s
case (1946), recurrent episodes of sham rage asso-
ciated with postencephalitic dystonia lasting 6�/8
hours decreased in frequency from 10 to 5 times per
2 months while on scopolamine 0.8 mg t.i.d. After a
superaddition of amphetamine 10 mg b.i.d., the
dystonia decreased further in duration and fre-
quency and was no longer accompanied by sham
rage. This is the only case where psychiatric episodes
disappeared before OGC due to treatment.
Antipsychotic-induced OGC
The associated psychiatric symptoms. Many decades
later, antipsychotics, especially high-potency con-
ventional antipsychotics, were found to induce
recurrent OGC accompanied by transient episodes
of psychiatric symptoms. Simpson et al. (1976)
described two cases of transient psychotic episodes
in association with antipsychotic-induced dystonia:
one patient became catatonic and hallucinated
hearing threatening demands to kill himself, and
another, a mentally retarded boy, had episodes of
stereotyped activity in association with OGC, pro-
fuse sweating and drooling. The authors stated that
anticholinergic agent terminated such episodes in 10
cases including the afore-mentioned, and considered
Psychiatric symptoms associated with oculogyric crisis 71

that these episodes might be one manifestation of
dystonic reactions.
Psychiatric symptoms that have been reported to
accompany the antipsychotic-induced OGC in pa-
tients with schizophrenia are as follows: visual
hallucinations (Thornton and McKenna 1994; Ben-
jamin 1999; Abe 2004) and illusions (Higuchi et al.
1997; Benjamin 1999), auditory hallucinations
(Simpson et al. 1976; Chiu 1989; Rogers 1989;
Sachdev and Tang 1992; Thornton and McKenna
1994; Benjamin 1999), delusions (Benjamin 1999)
and catatonic phenomena (Simpson et al. 1976;
Thornton and McKenna 1994).
Similar episodes associated with OGC, have also
been reported in non-schizophrenic patients who
were on antipsychotic: visual and auditory halluci-
nations in a patient with epilepsy, and severe anxiety
in a patient with primary hyperparathyroidism
(Leigh et al. 1987), lilliputian micropsia in a patient
with bipolar disorder (Shiratsuchi et al. 1994) and
exacerbation of psychotic features in a patient with
depression (Benjamin 1999).
These psychiatric episodes disappeared after the
medication was changed to low-potency (Chiu 1989;
Thornton and McKenna 1994) or novel (Thornton
and McKenna 1994) antipsychotic or after the
addition of anticholinergic (Simpson et al. 1976;
Chiu 1989; Benjamin 1999). As in postencephalitic
cases, various psychiatric symptoms have been
reported as associated with OGC and they varied
from patient to patient and with no particular
symptom appearing predominant, but the same
symptom tended to reappear stereotypically in the
same patient (Benjamin 1999).
However, some authors maintain that a particular
feature of the episode (‘‘paroxysmal perceptual
alteration’’), a transient increase in the intensity of
visual (and often also auditory) perception that
recurs in the afternoon (most likely in the late
afternoon), is pathognomonic of antipsychotic-in-
duced episodes (Higuchi et al. 1997; Uchida et al.
2003a,b). However, its specificity for antipsychotic-
induced episodes has not yet been established.
Although this feature has been noted in some
antipsychotic-induced cases (Watanabe 1991; Abe
2004, Case 2) and in one postencephalitic case
(Flach and Palisa 1936), it has not been noted in
many other cases, so its prevalence may not be high.
The temporal relationship of OGC and the
psychiatric symptoms was described in detail in a
few cases: the OGC occurred in the middle of the
psychiatric episodes (Rogers 1989; Thornton and
McKenna 1994; Abe 2004) or the psychiatric
symptoms preceded the OGC (Leigh et al. 1987;
Sachdev and Tang 1992).
The time of day of occurrence of OGC. As to the time of
day for the occurrence, most were found to occur
between 14:00 and 20:00 h, suggesting a diurnal
variation (Tan et al. 1994). Over 80% of the episodes
of acute neuroleptic-induced dystonia were seen to
occur in the afternoon (Mazurek and Rosebush
1996). The associated psychiatric episodes had also
been noted to occur in the afternoon (Sachdev 1991;
Watanabe 1991).
The associated autonomic symptoms. The OGC-asso-
ciated episodes were accompanied by autonomic
symptoms such as profuse sweating, salivation, facial
flushing or pallor (Simpson et al. 1976; Watanabe
1991; Thornton and McKenna 1994) and transitory
hypertension (Abe 2004).
Psychiatric episodes unaccompanied by OGC. As in
postencephalitic cases, the transient psychotic symp-
toms which used to appear with OGC can some-
times occur alone. Rogers (1989) reported a case
with transient psychotic episodes most but not all of
which were accompanied by OGC. In response to
treatment, the OGC was usually the first to dis-
appear: having disappeared after the switch to
chlorpromazine (Rogers 1989), or it was barely
noticeable after switching to clozapine (Thornton
and McKenna 1994) while psychiatric episodes
recurred. Transient recurrent visual hallucinations
(Abe 2004; Case 1) were accompanied by OGC
before antipsychotic dose reduction, but afterwards
they continued to recur without OGC. These
recurrent hallucinations also disappeared after a
further dose reduction. The author suggested that
the episodes with OGC might represent more severe
reactions to antipsychotic than those without.
There are also reports of cases with a series of
periodic episodes without OGC, which were con-
sidered antipsychotic-induced: episodes of panic
attacks (Bachman and Modestin 1987; Argyle
1990; Mandalos and Szarek 1999; Higuchi et al.
1999), visual illusions (Higuchi et al. 1997; Uchida
2003), visual and auditory illusions (Higuchi et al.
1997; Abe 2004, Case 2) and catatonia-like unre-
sponsiveness accompanied by OGC-related auto-
nomic symptoms (Abe 2004, Case 3), respectively.
Discussion
Characteristics of OGC
Both postencephalitic and antipsychotic-induced
OGC tended to occur at regular intervals and late
in the day, and were accompanied by autonomic
symptoms such as profuse sweating, facial flushing,
salivation, transient hypertension and difficulty in
72 K. Abe

micturition. Both may be associated with similar
psychiatric symptoms: visual hallucinations or illu-
sions, auditory hallucinations, catatonic phenomena,
transitory delusions or obsessive ideas. The asso-
ciated symptoms varied from patient to patient, but
the same symptoms tended to appear repeatedly in
the same patient.
In postencephalitic cases the psychiatric symptom
grew in intensity and appeared to culminate in
OGC. Similarly, in antipsychotic-induced cases,
the OGCs were noted to occur in the middle of
the psychiatric episodes and to last for a shorter time
than the episodes. In some postencephalitic and
drug-induced cases, OGC-associated psychiatric
symptoms were noted to occur sometimes also as
single episodes unaccompanied by OGC, and in
some other cases the treatment abolished OGC
while the psychiatric episodes continued to recur
periodically as before.
Even in cases unaccompanied by OGC, antipsy-
chotic-induced episodes tended to show the afore-
mentioned OGC characteristics: periodic transient
recurrence of the same psychiatric symptom in the
afternoon accompanied by the autonomic symp-
toms. Taken together, these findings suggest that
the OGC characteristics are useful in recognizing
antipsychotic-induced psychiatric episodes, and that
the episodes without OGC may be less severe
reactions to antipsychotic medication than those
with OGC, and represent manifestations of subcli-
nical OGC.
If its specificity for antipsychotic-induced episodes
is established, ‘‘paroxysmal perceptual alteration’’,
which some authors consider pathognomonic, could
be useful in recognizing such episodes.
Clinical implications for antipsychotic-induced
psychiatric episodes
If episodes with the OGC characteristics are ob-
served in a patient on antipsychotic, an adverse
reaction to the drug is a possibility. Such a possibility
is more likely if one or more of the past episodes had
been accompanied by OGC. Another feature sup-
portive of the diagnosis is a significant decrease in
the frequency or duration of the episodes on anti-
psychotic dose reduction or switch to one of the
novel antipsychotics.
The treatment may be directed to the responsible
antipsychotic and its gradual reduction may be tried
in cases where this is practicable. The OGC may
disappear first, followed by the psychiatric episodes.
Alternative effective measures taken in previous
cases are a switch to a low-potency or novel
antipsychotic, or a superaddition of an anticholiner-
gic. Benzodiazepines have been found useful in the
treatment of OGC (Benjamin 1999; Owens 1999)
and may also be effective in OGC-associated epi-
sodes. However, they could obscure etiology and
cause dependence.
OGC and dopaminergia
The pathophysiology of dystonia is unknown at
present, but it is widely accepted that striatal
cholinergic hyperactivity underlies dystonia. As to
striatal dopaminergic activity, both its hypoactivity
and hyperactivity have been postulated. According
to Marsden and Jenner (1980), antipsychotic-in-
duced dystonias are due to a transient compensatory
dopamine excess. The psychotic episodes accompa-
nied by OGC have also been explained as transient
hyperdopaminergic states (Chiu 1989; Thornton
and McKenna 1994).
It is interesting to note that the side effects of
levodopa resemble the symptoms associated with
OGC (Abe 2002). Its psychiatric side effects include
illusions and hallucinations, which are predomi-
nantly visual but may also be auditory, olfactory or
tactile (Goodwin 1972; Moskovitz et al. 1978);
delusions, hypomania, confusion and panic attacks
(Goodwin 1972; Rondot et al. 1984). Its autonomic
effects are; palpitation and flushing, hypertension,
excessive sweating, urinary frequency and retention
(Parfitt 1999). Furthermore, levodopa can induce
OGC and peak-dose dystonia confined to the face
and neck (as in antipsychotic-induced dystonia) in
patients with Parkinson’s disease (Poewe et al.
1988).
This symptomatic resemblance between the levo-
dopa-induced and OGC-associated symptoms is
compatible with the hyperdopaminergic hypothesis
of OGC, but further studies are required to draw a
conclusion regarding the pathogenesis of OGC and
its associated symptoms.
Statement of interest
The author has no conflict of interest with any
commercial or other associations in connection with
the submitted article.
References
Abe K. 2002. Antipsychotic-induced recurrent transient psychia-
tric episodes, compared to amphetamine- and L-dopa-induced
symptoms. Presented at the XII World Congress of Psychiatry,
August 24�/29 2002, Yokohama, Japan.
Abe K. 2004. Antipsychotic medications can induce transient
psychotic episodes without oculogyric crises. J Nerv Ment Dis
192:164�/166.
Argyle N. 1990. Panic attacks in chronic schizophrenia. Br J
Psychiatry 157:430�/433.
Psychiatric symptoms associated with oculogyric crisis 73

Bachmann KM, Modestin J. 1987. Neuroleptic-induced panic
attacks in a patient with delusional depression. J Nerv Ment Dis
175:373�/375.
Benjamin S. 1999. Oculogyric crisis. In: Joseph AB, Young RR,
editors. Movement disorders in neurology and neuropsychiatry.
Malden: Blackwell Science. pp 93�/103.
Bogaert L van. 1933. Sur les etats hallucinatoires au cours des
crises oculogyres de l’encephalite epidemique. Schweiz Arch
Neurol Psychiatrie 32:321�/334.
Brody MW, Freed H. 1941. Paranoid and compulsive symptoms
associated with oculogyric crises. Psychiatr Q 15:170�/176.
Chiu LPW. 1989. Transient recurrence of auditory hallucinations
during acute dystonia. Br J Psychiatry 155:110�/113.
Euziere J, Viallefont J, Vidal J, Bert P. 1933. Crises oculogyres a
direction variable avec troubles psychiques. Arch Soc Sci Med
Biol Montpellier 14:263�/266.
Ewald G. 1925. ‘Schauanfalle’ als postenzephalitische Storung.
(Zugleich ein Beitrag zur Frage psychischer Storungen bei
postenzephalitischen Zustanden). Monatsschr Psychiatr Neu-
rol 57:222�/253.
Flach A, Palisa C. 1936. Zur Psychopathologie des Zeiterlebens
im postencephalitischen Blickkrampf. Ztschr Neurol Psychia-
trie 154:599�/620.
Georgi F. Ungewohnliche postencephalitische Symptomenbilder
(zugleich ein Beitrag zur experimentellen Erzeugung sog.
Schauanfalle). Z Ges Neurol Psychiat 106: 602�/612.
Gillespie WH. 1944. A paranoid reaction associated with oculo-
gyric crises and parkinsonism. J Ment Sci 90:582�/587.
Goodwin FK. 1972. Behavioral effects of L-dopa in man. In:
Shader RI, editor. Psychiatric complication of medical drugs.
New York: Raven Press. pp 149�/174.
Hall AJ. 1931. Chronic epidemic encephalitis with special
reference to the ocular attacks. Br Med J ii:833�/837.
Higuchi H, Shimizu T, Hishikawa Y. 1997. Recurrent paroxysmal
episodes characterized by perceptual alteration in three schizo-
phrenic patients on neuroleptic medication. Psychiatry Clin
Neurosci 51:99�/101.
Higuchi H, Kamata M, Yoshimoto M, Shimizu T, Hishikawa Y.
1999. Panic attacks in patients with chronic schizophrenia: A
complication of long-term neuroleptic treatment. Psychiatry
Clin Neurosci 53:91�/94.
Jelliffe SE. 1929. Psychologic components in postencephalitic
oculogyric crises. Arch Neurol Psychiatry 21:491�/532.
Leigh RJ, Foley JM, Remler BF, Civil RH. 1987. Oculogyric
crisis: A syndrome of thought disorder and ocular deviation.
Ann Neurol 22:13�/17.
Mandalos GE, Szarek BL. 1999. New-onset panic attacks in a
patient treated with olanzapine. J Clin Psychopharmacol
19:191.
Marinesco G, Nicolesco M. 1932. Un cas de parkinsonism
accompagne de crises oculogyres et d’hallucinations colorees.
Rev Neurol 2:691�/693.
Marsden CD, Jenner P. 1980. The pathophysiology of extrapyr-
amidal side-effects of neuroleptic drugs. Psychol Med 10:55�/
72.
Mazurek MF, Rosebush PI. 1996. Circadian pattern of acute,
neuroleptic-induced dystonic reactions. Am J Psychiatry
153:708�/710.
McCowan PK, Cook LC. 1928. Oculogyric crises in chronic
epidemic encephalitis. Brain 51:285�/309.
Moskovitz C, Moses H, Klawans HL. 1978. Levodopa-induced
psychosis: A kindling phenomenon. Am J Psychiatry 135:669�/
675.
.Odegard O. 1932. A case of oculogyric fits in encephalitis
accompanied by obsessions and disturbance of ideation. Acta
Psychiatr Neurol 7:855�/865.
Oller CI. 1946. Paroxysmal autonomic crises in the postencepha-
litic state. Report of a case. Arch Neurol Psychiatry 55:388�/
396.
Onuaguluchi G. 1961. Crises in post-encephalitic Parkinsonism.
Brain 84:395�/415.
Owens DGC. 1999. A guide to the extrapyramidal side-effects of
antipsychotic drugs. Cambridge: Cambridge University Press.
pp 230�/234.
Pardee I. 1928. Paroxysmal oculogyric crises in parkinsonian
encephalitis. Am J Med Sci 175:683�/691.
Parfitt K. 1999. Martindale. The complete drug reference. 32nd
ed. London: The Pharmaceutical Press. pp 1137�/1141, 1447�/
1479.
Pascheff C. 1926. La coıncidence du tic ou syndrome de la
deviation periodique des yeux en haut avec des pseudo-
hallucinations visuelles dans l’encephalite lethargique epidemi-
que. Rev Gen Ophtalmol 40:333�/338.
Petit G, Bauer E, Chatagnon PE. 1926. Encephalite epidemique
fruste. Encephale 21:708�/709.
Poewe WH, Lees AJ, Stern GM. 1988. Dystonia in Parkinson’s
disease: Clinical and pharmacological features. Ann Neurol
23:73�/78.
Rogers DGC. 1989. Oculogyric crises and schizophrenia. Br J
Psychiatry 155:569�/570.
Rondot P, de Recondo J, Coignet A, Ziegler M. 1984. Mental
disorders in Parkinson’s desease after treatment with L-DOPA.
Adv Neurol 40:259�/269.
Rosner AA. 1942. Unit reaction states in oculogyric crises. Am J
Psychiatry 99:224�/228.
Sachdev P. 1991. Tardive oculogyric crisis and obsessional
thoughts. Br J Psychiatry 158:720�/721.
Sachdev P, Tang WM. 1992. Psychotic symptoms preceding
ocular deviation in a patient with tardive oculogyric crises. Aust
NZ J Psychiatry 26:666�/670.
Schwab RS, Fabing HD, Prichard JS. 1951. Psychiatric symptoms
and syndromes in Parkinson’s disease. Am J Psychiatry
107:901�/907.
Shiratsuchi T, Terao T, Ohga T. 1994. Paroxysmal perceptual
alteration during neuroleptic treatment in a woman with
bipolar disorder (in Japanese). Seishin-Igaku 36:1327.
Simpson GM, Varga E, Haher EJ. 1976. Psychotic exacerbations
produced by neuroleptics. Dis Nerv Syst 37:367�/369.
Tan C-H, Chiang P-C, Ng L-L, Chee K-T. 1994. Oculogyric
spasm in Asian psychiatric in-patients on maintenance medica-
tion. Br J Psychiatry 165:381�/383.
Thornton A, McKenna PJ. 1994. Acute dystonic reactions
complicated by psychotic phenomena. Br J Psychiatry
164:115�/118.
Uchida H, Suzuki T, Tanaka KF, Watanabe K, Yagi G, Kashima
H. 2003a. Recurrent episodes of perceptual alteration in
patients treated with antipsychotic agents. J Clin Psychophar-
macol 23:496�/499.
Uchida H, Suzuki T, Watanabe K, Yagi G, Kashima H. 2003b.
Antipsychotic-induced paroxysmal perceptual alteration. Am J
Psychiatry 160:2243�/2244.
Urechia CI. 1949. Les crises diencephaliques. Encephale 38:21�/
31.
Watanabe K. 1991. The syndromes of oculogyric crisis and
paroxysmal perceptual alteration in chronic schizophrenic
patients. A clinical inquiry into their pathophysiology (in
Japanese with English summary). Psychiatr Neurol 93:151�/
189.
Yahr MD. 1978. Encephalitis lethargica (von Economo’s disease,
epidemic encephalitis). In: Vinken PJ, Bruyn GW, editors.
Handbook of clinical neurology Vol. 34. Amsterdam: North-
Holland Publishing Company. pp 452�/457.
74 K. Abe

ORIGINAL INVESTIGATION
Effect of olanzapine treatment on platelet glutamine synthetase-likeprotein and glutamate dehydrogenase immunoreactivity inschizophrenia
GULNUR Sh. BURBAEVA, IRINA S. BOKSHA, ELENA B. TERESHKINA,
OLGA K. SAVUSHKINA, MARINA S. TURISHCHEVA, LUBOV I. STARODUBTSEVA,
OLEG S. BRUSOV & MARGARITA A. MOROZOVA
Mental Health Research Centre of Russian Academy of Medical Sciences, Moscow, Russia
AbstractAccording to contemporary views, the glutamatergic system is implicated in the pathogenesis of schizophrenia, and atypicalneuroleptics exert their effects (at least partially) through the glutamatergic system. Immunoreactive glutamate-metabolising enzymes, such as glutamine synthetase-like protein (GSLP) and two glutamate dehydrogenase isoenzymes(GDH), have been discovered in human platelets. The amount of GSLP in the platelets of 40 chronic patients withschizophrenia was found to be significantly higher than in 33 controls (consistent with our previous finding of increasedamounts of GSLP in the prefrontal cortex of chronic schizophrenia patients). Moreover, survival analysis of the group ofpatients treated with olanzapine for 28 weeks showed that the larger amount of GSLP measured in platelets beforetreatment, the shorter the treatment time needed to achieve a positive clinical response (defined a priori as ]/20% reductionin PANSS total score from the initial level before the treatment). Hence, GSLP level may serve as a predictor of thetreatment duration to achieve a positive outcome with olanzapine. Both GSLP and GDH were found significantly changedin the course of treatment; hence, treatment with olanzapine influences the amounts of glutamate-metabolising enzymes inthe platelets of chronic schizophrenia patients.
Key words: Schizophrenia, olanzapine, glutamate metabolising enzymes, platelets
Introduction
There are numerous lines of evidence implicating
impaired neurotransmitter systems in schizophrenia,
particularly failure of glutamatergic transmission was
proposed to be an important factor in schizophrenia
(Duncan et al. 1999; Krystal et al. 1999; Carlsson et
al. 2000; Tamminga and Frost 2001). The ‘gluta-
matergic hypothesis’ proposes that schizophrenia
may be caused by diminished activity of glutamater-
gic pathways, the major excitatory system in the
brain (Bartha et al. 1997; Krystal et al. 1999;
Tamminga and Frost 2001). Based primarily on
the fact of altered levels of glutamate neurotrans-
mitter, its receptors and transporters, the ‘glutama-
tergic hypothesis’ has been broadened into the field
of brain glutamate metabolism. In fact, significantly
changed levels of glutamate decarboxylase, glutami-
nase, glutamine synthetase (GS), glutamine synthe-
tase-like protein (GSLP, Boksha et al. 2000), and
glutamate dehydrogenase isoenzymes (GDH) have
been found in brains of patients with schizophrenia
compared to controls (Dracheva et al. 2002; Gluck
et al. 2002; McCullumsmith et al. 2002; Burbaeva et
al. 2003).
The search for a reliable peripheral marker linked
to alterations found in the central nervous system is
important for the prediction of development of
schizophrenia, which is essential for psychiatrists in
order to adopt a good strategy and for the optimisa-
tion of subsequent therapy in patients.
Platelets are known to be a rather satisfactory
model reflecting some processes that occur in
nervous tissue. Some components of the glutama-
tergic system have been found in platelets, such as
glutamate receptors and transporters (Berk et al.
Correspondence: Gulnur Sh. Burbaeva, MD, PhD, Mental Health Research Centre, Russian Academy of Medical Sciences, Laboratory of
Neurochemistry, Zagorodnoje Shosse 2-2, Moscow 117152, Russia. Tel: +7 95 952 9129. Fax: +7 95 952 8940. E-mail: burbaeva
@yahoo.com
The World Journal of Biological Psychiatry, 2006; 7(2): 75�/81
(Received 4 November 2004; accepted 18 April 2005)
ISSN 1562-2975 print/ISSN 1814-1412 online # 2006 Taylor & Francis
DOI: 10.1080/15622970510029957

2000; Ferrarese et al. 2001; Zoia et al. 2004), and
enzymatic activities of GDH and glutaminase have
been detected (Gluck et al. 2000). Moreover, defects
in glutamatergic systems, such as super sensitivity of
platelet glutamate receptors to glutamate, have been
revealed in platelets in schizophrenia patients (Berk
et al. 1999, 2000).
Recent evidence suggests that effects on glutama-
tergic transmission may contribute to the therapeutic
action of atypical antipsychotics (Goff et al. 2002).
In the present study, we tested a hypothesis that the
level of glutamate metabolising enzymes may be
altered in platelets of patients with schizophrenia
compared with controls, and that this level may be
influenced by treatment with the atypical neurolep-
tic, olanzapine, which is known to act as a modulator
of brain and serum glutamate concentrations (Goff
et al. 2002). Two enzymes involved in glutamate
metabolism, GSLP and GDH isoenzymes, have
been detected in extracts from human platelets.
The amounts of these proteins were determined in
platelets of patients before and during the course of
treatment with olanzapine.
Subjects
The control group consisted of 33 volunteers (11
men and 22 women, 19�/63 years old, healthy
persons with no history of mental or neurological
disorders), blood samples were taken once from each
person, and platelets were accumulated for the
control group during 2 years. Statistics for age are
summarised in Table I.
After receiving approval for the study from the
Ethics Committee, 40 patients (26 men and 14
women) with schizophrenia, paranoid type (DSM-
IV 295.30), were assigned to treatment with olanza-
pine. The main statistics for age, duration of illness,
and PANSS distributions are given in Table II.
Patients were included into the study if they met
the following inclusion criteria: (1) in-patients at
entry who satisfied Research Diagnostic Criteria
(RDC) (Spitzer et al. 1977) for chronic schizophre-
nia, paranoid, episodic type with no inter-episode
residual symptoms; (2) no less than two episodes of
schizophrenia in the previous 2 years; (3) men and
women 18�/60 years old; (4) a level of understanding
sufficient to communicate intelligently with the
investigator and nurse; (5) reliable and agreed to
cooperate with all tests and examinations required
by the protocol; (6) each patient (or a patient’s
authorised legal representative) understood the nat-
ure of the study and signed an informed consent
document; (7) an initial score (before treatment with
olanzapine) on the BPRS, extracted from the
PANNS, of at least 36; (8) an initial score (before
the treatment) on the CGI Severity of at least 3; (9)
concomitant medication (other permitted drug ther-
apy: benzodiazepines (diazepam, phenozepam) and
anticholinergics (trihexyphenidyl)).
Exclusion criteria were the following: (1) women
who were either pregnant or lactating; (2) uncor-
rected hypothyroidism or hyperthyroidism; (3)
myasthenia gravis; (4) narrow-angle glaucoma; (5)
chronic urinary retention and/or clinically significant
prostate hypertrophy; (6) one or more seizures
without clear and resolved etiology; (7) leucopenia;
(8) history of severe allergies or multiple adverse
drug reactions; (9) DSM-IV substance (alcohol or
other drugs) abuse or dependence within the pre-
vious 3 months; (10) judged clinically to be at
serious suicidal risk; (11) participation in clinical
trial of another investigation within 1 month prior to
study entry; (12) any other concomitant medication
with psychotropic activity, other than specified
above; (13) positive hepatitis surface antigen
(HbsAg) or positive IgM fraction of hepatitis core
antibody (anti-HBc(IgM)).
Medication
The study included an initial period, intended for
screening of patients and included the time require-
ment specified for washout from previous therapy
(9 days).
The treatment course consisted of an acute phase
(first 8 weeks) and a maintenance phase (following
20 weeks). The mean modal olanzapine dosage for
the total group of patients was 12.79/3.6 mg/day
(initiated at 5 mg/day, up to 20 mg/day during the
acute phase of the treatment, and finishing at 10
mg/day during the maintenance phase of the treat-
ment). About 15% of patients received benzodiaze-
pine drugs.
Methods
Isolation of platelets and method of extraction of proteins
from platelets
The method was elaborated for preparation of
platelets with optimum extraction of the target
Table I. Statistics for age and glutamine synthetase-like protein
(GSLP) and glutamate dehydrogenase (GDH) immunoreactivity
in control group.
Age
(years)
GSLP GDH
Median 26 75 82
Minimum 19 22 23
Maximum 63 176 214
GSLP and GDH immunoreactivity is expressed in relative units
(in percentages of the pooled control value).
76 G. Sh. Burbaeva et al.

proteins. Blood plasma prepared from 50 ml of
blood taken with citrate buffer, pH 5.7 (1:5, v/v),
was collected for centrifugation (10 000 rpm, 20
min, 58C). The pellet was washed with the citrate
buffer, re-suspended in 62.5 mM Tris�/HCl buffer,
pH 6.8, then frozen in liquid nitrogen and stored at
�/708C prior the analysis of proteins.
Platelet ‘lysate’ was obtained by the addition of
SDS to the platelets (after their thawing) to a final
concentration of 2%, then the samples were heated
(5 min, 958C), the protein concentration was
determined by the method of Lowry et al. (1951),
and 2-mercaptoethanol (to 5% final concentration)
was added to the samples before the electrophoresis.
The aliquots loaded on each lane of polyacrylamide
gel were matched in protein concentration. The
proteins were separated by electrophoresis (Laemmli
1970), with subsequent enzyme-coupled Western
immunoblotting, with chemiluminescent amplifica-
tion of the signal (ECL immunoblotting) for the
immunoreactivity determination.
Enzyme chemiluminescent amplified (ECLTM)
immunoblotting
Enzyme chemiluminescent amplified immunoblot-
ting was performed according to Towbin et al.
(1979) and the Amersham protocol using Hyper-
filmTM ECLTM nitrocellulose membranes, ECL re-
agents which were purchased from Amersham�/
Pharmacia Biotech (UK), and goat anti-rabbit IgG
antibody conjugate was purchased from Sigma
(USA).
GSLP immunoreactivity
GSLP immunoreactivity was evaluated using poly-
clonal antibodies described earlier and raised against
GSLP isolated from human brain (Boksha et al.
2000). Rabbit antiserum (at 1:9000 working dilu-
tion) stained the major protein band corresponding
to GSLP.
GDH immunoreactivity
GDH immunoreactivity was measured using poly-
clonal antibodies raised against two readily soluble
GDH isoenzymes isolated from human brain (Bur-
baeva et al. 2002). The rabbit antiserum (at 1:12 000
working dilution) stained two protein bands corre-
sponding to readily soluble GDH forms. Total
immunoreactivity for both GDH isoenzymes was
evaluated in the experiments as a measure of GDH
amount.
Quantitative analysis of films after ECL immuno-
blotting was performed using a Zeineh laser densit-
ometer and corresponding software (Biomed
Instruments, USA).
Immunoreactivity of GSLP and GDH is given in
relative units in Tables I and III. ‘Relative units’
mean percentages from a pooled control sample.
Statistical analysis
Complete statistical analysis was performed with
Statistica, Version 6.0 (nonparametric module,
Mann�/Whitney U-test, Wilcoxon matched pairs
test, and Spearman correlations were calculated for
comparisons of clinical and demography data with
biochemical measures).
The prognostic relevance of the biochemical tests
was analysed with the Survival Analysis (the Regres-
sion Model, Statistica Version 6). The ‘survival’ time
for each patient was specified as the time from the
start of the treatment right up to the clinical
response (in weeks). The ‘survival’ time distribution
was found to be exponential and proportional to the
values of a separate variable, or of a set of indepen-
Table II. Statistics for age, disease duration, and PANSS for schizophrenia group before and during the treatment with olanzapine.
Age
(years)
Disease duration
(years)
PANSS
(before treatment)
PANSS
(acute phase)
PANSS
(maintenance phase)
Median 31 0.5 76.5 51 49
Minimum 19 0.2 54 30 30
Maximum 56 43 127 102 97
Table III. Statistics for glutamine synthetase-like protein (GSLP) and glutamate dehydrogenase (GDH) immunoreactivity in schizophrenia
group before and during the treatment with olanzapine.
GSLP
(before treatment)
GSLP
(acute phase)
GSLP
(maintenance phase)
GDH
(before treatment)
GDH
(acute phase)
GDH
(maintenance phase)
Mediana 122 112 121.5 86.5 69 64.5
Minimum 12 7 19 7 7 7
Maximum 685 327 370 748 186 289
GSLP and GDH immunoreactivity are expressed in relative units (in percentages of the pooled control value).
Olanzapine treatment in schizophrenia 77

dent variables, before the treatment. The rate
parameter of the exponential distribution can then
be expressed as follows:
S(v)�exp(c�b1�v1�b2v2 � . . .�bmvm)
S(v) denotes the ‘survival’ times, c is a constant, bi
values are the regression parameters (b), and vi is an
independent variable.
Goodness-of-fit. Chi-squared goodness-of-fit was
computed as a function of the log-likelihood of the
model with all parameter estimated, and the log-
likelihood of the model in which all covariates were
forced to zero. If this chi-squared value was sig-
nificant, the null hypothesis was rejected and it was
assumed that the independent variables were sig-
nificantly related to ‘survival’ times.
The reliability of estimation of b coefficients was
determined from t values. The t value is the ratio of
the parameter estimates divided by their standard
errors. Note, the standard errors (and t values) are
computed from the second-order partial derivatives
of the log-likelihood function, resulting in asympto-
tic standard errors (and t values) for the parameters.
Note, if the regression parameter (b) is positive
for the independent variable, it means that the
higher the variable value, the longer the ‘survival’
time for the patient. Inverse relation will be obtained
if the regression parameter (b) is negative.
Results
Glutamine synthetase-like protein and glutamate
dehydrogenase isoenzymes in platelets of controls
Immunoreactive enzymes such as GSLP and two
isoenzymes of GDH were discovered in human
platelet extracts. Figure 1 shows the most typical
pattern for control cases. GSLP, left, and GDH
isoenzymes, right, display virtually the same mole-
cular masses (�/55 kDa, and �/58 and 56 kDa,
respectively) as described for the brain extracts
(Boksha et al. 2000; Burbaeva et al. 2003).
GSLP and GDH were detected in measurable
amounts in all subjects from the control group (33
persons). Relative amounts of the immunoreactive
proteins (GSLP, GDH) in each person from the
control group were evaluated using a calibration plot
drawn using a pooled control (reference) sample.
The immunoreactivity was expressed in relative
units (percentages from the sample pooled from
the five first controls accumulated).
GS and membrane-associated GDH immunor-
eactivities in platelets were below the detection
threshold by the same technique of immunostaining,
using either monoclonal antibody (Biogenesis, UK)
or polyclonal antibodies (Burbaeva et al. 2003),
respectively.
Distribution of GSLP and GDH immunoreactiv-
ity levels in subjects from the control group is
summarised in Table I (non-normal distribution
for both protein values was obtained).
A link between GSLP and GDH levels was found
in the control group (Spearman rank order correla-
tion R�/0.414; t�/2.404; P�/0.02), whereas no links
were found between any of these parameters and age
or gender in this group (P �/0.1).
Platelet GSLP and GDH in patients with schizophrenia
before treatment compared with control group
The staining patterns for subjects from control and
patient groups proved to be mainly similar for GSLP
as for GDH. GSLP and GDH were detected in
measurable amounts in all subjects from the patient
group. Relative amounts of the immunoreactive
GSLP and GDH in each subject from the patient
group were evaluated and expressed in the same way
as for controls. Table III summarises respective
distributions of GSLP and GDH amounts measured
before and during treatment (for the acute and
maintenance phases). A link between GSLP and
GDH levels was found before the treatment (Spear-
men R�/0.616; t�/4.823; P�/0.00002), whereas no
links between any of these parameters and age or
gender were found in the patient group (P �/0.1).
The absence of links between GSLP or GDH
levels and age or gender in both groups (controls and
patients) enabled a comparison of these groups with
respect to amounts of GSLP and GDH. Amount of
GSLP (immunoreactivity level) measured in patients
before treatment was found to be significantly higher
than in the control group (P�/0.003, Mann�/Whit-
ney U-test). A tendency to increased GDH in
Figure 1. Staining pattern of glutamine synthetase-like protein
(GSLP) and glutamate dehydrogenase (GDH) in control; 30 mg of
total protein (platelet extract) were applied on the gel followed by
ECL immunoblotting. GSLP (�/55 kDa band) was revealed by
rabbit antiserum (1:9000). Two isoenzymes of GDH (�/58 and
56 kDa bands) were stained by rabbit antiserum (1:12 000).
78 G. Sh. Burbaeva et al.

patients compared with controls was not significant
(P �/0.1).
Gamma correlation analysis of the patient group
before treatment (a search for a link between clinical
and biochemical �/ GSLP and GDH �/ parameters
from a correlation matrix) resulted in evaluation of
some links: GSLP was linked to unusual thought
content (g�/�/0.33, Z�/�/2.24, P�/0.025), and
GDH was linked to excitement level (g�/0.496,
Z�/ 3.41, P�/0.0006) and conceptual disorganisa-
tion (g�/0.283, Z�/2.21, P�/0.0269).
Platelet GSLP and GDH in patients during olanzapine
treatment
As a result of the treatment, the patients showed
significant improvement in the PANSS total, PANSS
positive and PANSS negative score changes through-
out the 8-week acute treatment phase, as well as
through the following 20-week treatment mainte-
nance period (P�/0.000001, Wilcoxon matched
pairs test). Dynamics of amounts of GSLP and
GDH as a result of the treatment was evaluated by
the same test: both parameters were found to be
significantly changed after the acute phase of treat-
ment (P�/0.038 and 0.012, respectively; Wilcoxon
matched pairs test). The link between amounts of
GSLP and GDH (found before the treatment)
remained rather tight in the acute and maintenance
phases of treatment (Spearman R�/0.553, t�/3.926,
P�/0.0004 and R�/0.405, t�/2.505, P�/0.02, re-
spectively).
As demonstrated in Table III, the data for
amounts of GSLP and GDH measured before
treatment seemed rather scattered, hence the data
were subjected to detailed analysis. The patient
subgroup with high initial GSLP level (higher than
the median value) was considered separately (20
persons). In this subgroup, the levels of GSLP were
found to be significantly changed after the acute and
maintenance phases of treatment (P�/0.007 and
0.004, respectively, Wilcoxon matched pairs test).
In this subgroup, the resulting decrease of GSLP
levels after the acute and maintenance phases of
treatment was significant (Mann-Whitney U-test:
P�/0.009 and 0.003, respectively), but still distin-
guished from the control group levels (Mann�/
Whitney U-test: P�/0.0001). The amount of GDH
in this subgroup was found to be significantly
changed as a result of treatment (P�/0.002 and
0.025 after acute and maintenance phases, respec-
tively, Wilcoxon matched pairs test), and indistin-
guishable from the control group (Mann�/Whitney
U-test: P �/0.1).
Search for predictor of positive clinical outcome of
olanzapine treatment
The data have been ordered by the results of clinical
tests. Clinical response was defined a priori as 20%
or greater reduction in PANSS total score from the
initial level before treatment. The positive clinical
response has been used as a categorised index of the
outcome in the subsequent search for the predictors
of outcome.
Evaluation of predictors of positive clinical out-
come was performed using the regression model of
survival analysis (see Methods).
The regression parameter (b) calculated for
GSLP in the range of maintenance treatment phase
is �/0.00276 (t�/�/2.453, constant c�/2.854, x2
(goodness-of-fit)�/4.795, P�/ 0.0285. The regres-
sion parameter (b) is negative. This means that
GSLP amounts have an inverse relation to ‘survival’
times of patients: the larger the amount of GSLP
measured in platelets before treatment, the less
treatment time needed to achieve a positive clinical
response. Thus, the time (weeks) to a positive
clinical response may be calculated according to
the following equation:
S�exp(2:854�0:00276�GSLP);
where S is number of weeks, and GSLP is amount of
GSLP measured before treatment.
So, amount of GSLP in platelets measured before
treatment has been found to be a possible predictor
of positive outcome in the olanzapine treatment.
Conclusion and discussion
In the present work, immunoreactivity of GSLP and
two readily soluble GDH isoenzymes have been
detected in human platelet lysates, and subunits of
these proteins possess virtually the same mobility on
electrophoresis as their brain forms (Figure 1),
whereas immunoreactivity of GS and membrane-
associated GDH isoenzyme are apparently below the
threshold of the detection technique (using antibo-
dies against brain forms of these proteins). Thus,
human platelets contain components of the gluta-
mate metabolising system, such as GSLP and two
GDH isoenzymes, along with previously found
glutaminase (Gluck et al. 2000), in addition to
glutamate transporters and receptors (Ferrarese et
al. 2001; Zoia et al. 2004).
Amount of GSLP in platelets of patients with
schizophrenia is significantly higher than in controls.
This is consistent with our findings of elevated
amounts of GSLP in the prefrontal cortex in brains
of patients with chronic schizophrenia (Burbaeva et
al. 2003).
Olanzapine treatment in schizophrenia 79

Our data indicate that amounts of GDH and
GSLP do not depend on age or gender in either
control persons or schizophrenic patients.
A rather tight link (high correlation coefficient)
has been observed between amounts of GSLP and
GDH measured in platelets of controls and patients
(before and after each phase of treatment). However,
the biochemical parameters were linked to different
clinical parameters (in the patient group before
treatment, amount of GSLP was linked to unusual
thought content, whereas amount of GDH reflected
excitement level and conceptual disorganisation).
The diversity of GSLP and GDH levels measured
in the patient group before treatment is obviously
larger than in the control group (such a high
variability is not limited to glutamatergic system
parameters, but can be found in nearly every
biological variable in schizophrenia). Maximum
amounts of GSLP and GDH are higher in the
patient group than in controls (4 and 3.5 times,
respectively), whereas minimum amounts of GSLP
and GDH are lower (2 and 3 times, respectively,
Tables I and III). This fact is explainable based on
the hypothesis of Krystal et al. (1999), suggesting a
higher variability of glutamatergic system activity in
the schizophrenic brain and/or disability of the
glutamatergic system tuning in patients with schizo-
phrenia.
In general, distributions of GSLP and GDH in the
patient group after the acute and maintenance
phases of treatment were not so widely scattered as
before treatment, wherein amplitude of GSLP tend
to diminish, still differing from the control group.
Hence, treatment with olanzapine affects the
amounts of both enzymes involved in glutamate
metabolism, and the observed phenomenon may
be described as: ‘the treatment makes the GSLP and
GDH levels smooth’ rather than ‘the treatment
normalises the amounts of GSLP and GDH’.
When patients with amount of GSLP higher than
the median value (before treatment) were considered
separately, a significant decrease was found in
amounts of both GSLP and GDH after treatment.
A remarkable positive link has been found be-
tween disease duration and amount of GDH mea-
sured either at the end of the acute or maintenance
phases of treatment (R�/0.412, t�/2.519, P�/0.02,
and R�/0.5295, t�/3.519, P�/0.001, respectively).
This link means that the longer the duration of
illness preceding the treatment course, the higher
level of GDH measured after treatment. Further
investigations are needed to understand whether this
phenomenon reflects the influence of the disease
itself, or neuroleptic treatment preceding the shift to
olanzapine.
To search for a reliable biochemical predictor of
treatment outcome, survival analysis was carried
out. This has enabled us to conclude that level of
GSLP for each individual patient with schizophrenia
may serve as a probable predictor of the treatment
duration needed to achieve a positive outcome of
treatment with olanzapine. The finding is worth
verifying in the future in other treatment trials.
To conclude, the immunoreactivity levels of glu-
tamate metabolising enzymes GSLP and GDH
found in human platelets are influenced by treat-
ment with olanzapine, wherein GSLP enables pre-
diction of the minimum duration of treatment
necessary to achieve a positive outcome. The change
of immunoreactivity levels of GSLP and GDH is
evidence for the effect of olanzapine treatment on
glutamate metabolism, which is consistent with the
finding mentioned above of modulation of serum
glutamate concentration by olanzapine (Goff et al.
2002).
Acknowledgements/Statement of interest
The study is supported by the Stanley Medical
Research Institute, US.
References
Bartha R, Williamson PC, Drost DJ, Malla A, Carr TJ, Cortese L,
Canaran G, Rylett RJ, Neufeld RWJ. 1997. Measurement of
glutamate and glutamine in the medial prefrontal cortex of
never-treated schizophrenic patients and healthy controls by
proton magnetic resonance spectroscopy. Arch Gen Psychiatry
54:959�/65.
Berk M, Plein H, Belsham B. 2000. The specificity of platelet
glutamate receptor supersensitivity in psychotic disorders. Life
Sci 66(25):2427�/32.
Berk M, Plein H, Czismadia T. 1999. Supersensitive platelet
glutamate receptors as a possible peripheral marker in schizo-
phrenia. Int Clin Psychopharmacol 14(2):119�/22.
Boksha IS, Tereshkina EB, Burbaeva GSh.. 2000. Glutamine
synthetase and glutamine synthetase-like protein from human
brain: Purification and comparative characterization. J Neuro-
chem 75:2574�/82.
Burbaeva GSh, Boksha IS, Turishcheva MS, Vorobyeva EA,
Savushkina OK, Tereshkina EB. 2003. Glutamine synthetase
and glutamate dehydrogenase in prefrontal cortex of patients
with schizophrenia. Prog Neuropsychopharmacol Biol Psychia-
try 27:675�/80.
Burbaeva GSh, Turishcheva MS, Vorobyeva EA, Savushkina OK,
Tereshkina EB, Boksha IS. 2002. Diversity of glutamate
dehydrogenase in human brain. Prog Neuropsychopharmacol
Biol Psychiatry 26(3):427�/35.
Carlsson A, Waters N, Waters S, Carlsson ML. 2000. Network
interactions in schizophrenia �/ Therapeutic implications. Brain
Res Rev 31(2/3):342�/9.
Dracheva S, Elhakem SL, Davis KL, Haroutunian V. 2002.
GAD65 and GAD67 protein expression in dorsolateral pre-
frontal cortex of elderly schizophrenia patients. Biol Psychiatry
51(Suppl):166.
Duncan GE, Zom S, Lieberman JA. 1999. Mechanisms of typical
and atypical antipsychotic drug action in relation to dopamine
80 G. Sh. Burbaeva et al.

and NMDA receptor hypofunction hypotheses of schizophre-
nia. Mol Psychiatry 4:418�/28.
Ferrarese C, Sala G, Riva R, Begni B, Zoia C, Tremolizzo L,
Galimberti G, Millul A, Bastone A, Mennini T, Balzarini C,
Frattola L, Beghi E. 2001. Decreased platelet glutamate uptake
in patients with amyotrophoc lateral sclerosis. Neurology
56:270�/2.
Gluck MR, Thomas RG, Sivak MA. 2000. Unaltered cytochrome
oxidase, glutamate dehydrogenase and glutaminase activities in
platelets from patients with sporadic amyotrophic lateral
sclerosis �/ A study of potential pathogenetic mechanisms in
neurodegenerative diseases. J Neural Transm 107(12):1437�/
47.
Gluck MR, Thomas RG, Davis KL, Haroutunian V. 2002.
Implications for altered glutamate and GABA metabolism in
the dorsolateral prefrontal cortex of aged schizophrenic pa-
tients. Am J Psychiatry 159:1165�/73.
Goff DC, Hennen J, Lyoo IK, Tsai G, Wald LL, Evins AE,
Yurgelun-Todd DA, Renshaw PF. 2002. Modulation of brain
and serum glutamatergic concentrations following a switch
from conventional neuroleptics to olanzapine. Biol Psychiatry
51(6):493�/7.
Krystal JH, Belger A, D’Souza DC, Anand A, Charney DS,
Aghajanian GK, Moghaddam B. 1999. Therapeutic implica-
tions of the hyperglutamatergic effects of NMDA antagonists.
Neuropsychopharmacol Suppl 21:143�/57.
Laemmli U. 1970. Cleavage of structural proteins during the
assembly of the head of bacteriophage T4. Nature 227:680�/5.
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. 1951. Protein
measurement with the Folin phenol reagent. J Biol Chem
193:265�/75.
McCullumsmith RE, Haroutunian V, Davis KL, Meador-Woo-
druff JH. 2002. Expression of glutaminase transcripts in the
thalamus of subjects with schizophrenia. Biol Psychiatry
51(Suppl):25.
Spitzer RL, Endicott J, Robins E. 1977. Research diagnostic
criteria for a selected group of functional disorders. 3rd ed.
New York: New York State Psychiatric Institute.
Tamminga CA, Frost DO. 2001. Changing concepts in
the neurochemistry of schizophrenia. Am J Psychiatry 158
(9):1365�/6.
Towbin H, Staehelin T, Gordon J. 1979. Electrophoretic transfer
of proteins from polyacrylamide gels to nitrocellulose sheets
procedure and some applications. Proc Natl Acad Sci USA
76(N9):4350�/4.
Zoia Ch, Cogliati T, Tagliabue E, Cavaletti G, Sala G, Galimberti
G, Rivolta I, Rossi V, Frattola L, Ferrarese C. 2004. Glutamate
transporters in platelets: EAAT1 decrease in aging and in
Alzheimer’s disease. Neurobiol Aging 25:149�/57.
Olanzapine treatment in schizophrenia 81

ORIGINAL INVESTIGATION
The influence of concomitant antidepressant medication on safety,tolerability and clinical effectiveness of electroconvulsive therapy
THOMAS C. BAGHAI, ALAIN MARCUSE, MELANIE BROSCH, CORNELIUS SCHULE,
DANIELA ESER, CAROLINE NOTHDURFTER, YVONNE STENG, INES NOACK,
KATRIN PIETSCHMANN, HANS-JURGEN MOLLER & RAINER RUPPRECHT
Department of Psychiatry and Psychotherapy, Ludwig-Maximilians-University, Munich, Germany
AbstractBackground: A major problem in the treatment of severe depression is the onset latency until clinical improvement. So far,electroconvulsive therapy (ECT) is the most effective somatic treatment of depression. This holds especially true fortreatment-refractory disturbances. However, not all patients respond to conventional unilateral ECT. In certain cases,subsequent clinical response can be achieved using bilateral or high-dose unilateral ECT. Also, a concomitantpharmacotherapy can be utilized to augment therapeutic effectiveness. Surprisingly, data in this field are widely lackingand only few studies showed advantages of an ECT/tricyclic antidepressant combination. Method: We retrospectivelyevaluated 5482 treatments in 455 patients to investigate possible therapeutic advantages in combination therapies versusECT monotherapy. Main outcome criteria were clinical effectiveness and tolerability. Moreover, treatment modalities andictal neurophysiological parameters that might influence treatment outcome were analysed. Results: A total of 18.2% of ourtreatments were ECT monotherapy, 8.87% were done with one antidepressant. Seizure duration was unaffected by the mostantidepressants. SSRI caused a lengthened seizure activity. Postictal suppression was lower in mirtazapine and higher inSSRI and SNRI treated patients. A significant enhancement of therapeutic effectiveness could be seen in the patient groupreceiving tricyclics, SSRI or mirtazapine. Serious adverse events were not recorded. Conclusion: Our study supports thehypothesis that mirtazapine can be used to enhance the therapeutic effectiveness of ECT. Controlled studies are necessaryto further investigate the possible advantages of ECT and pharmacotherapy combinations, especially the use of moderndually acting antidepressants which have proven their good effectiveness in treatment-resistant depression.
Key words: Electroconvulsive therapy, major depression, concomitant medication, antidepressant therapy
Introduction
ECT is still the most efficacious treatment option in
pharmacotherapy resistant depression
During recent years there has been considerable
progress in pharmacotherapeutic treatment options
for depressive disorders. Moreover, new treatment
principles are in progress to be established (Baghai
et al. 2006). Nevertheless, electroconvulsive ther-
apy is still one of the best tolerated and most
effective biological treatments of depressive disor-
ders. This holds especially true for severe treat-
ment-resistant major depressive disorder (ECT
Review Group 2003). Since the first publication
of a placebo-controlled double-blind study indicat-
ing the efficacy of ECT in the treatment of
depression (Greenblatt et al. 1964), a variety of
reports, which are summarized in a recent
review (ECT Review Group 2003), described the
excellent therapeutic effectiveness of this method.
Overall, a 20% better improvement in comparison
to tricyclic antidepressants (TCA) and a 45%
better improvement in comparison to monoami-
nooxidase inhibitors have been published in a
metaanalysis (Janicak et al. 1985). A better im-
provement in comparison to the selective serotonin
reuptake inhibitor (SSRI) paroxetine has been
described in a controlled study investigating the
HAMD-score reduction in a responder analysis
(Folkerts et al. 1997). In addition, a more rapid
improvement in comparison to pharmacotherapeu-
tic approaches has been published (Sackeim et al.
1993; Prudic et al. 1996; ECT Review Group
2003).
Correspondence: Thomas C. Baghai, MD, Department of Psychiatry and Psychotherapy, Ludwig-Maximilians-University,
Nussbaumstrasse 7, D-80336 Munich, Germany. Tel: +49 89 5160 5812. Fax: +49 89 5160 5391. E-mail: [email protected]
The World Journal of Biological Psychiatry, 2006; 7(2): 82�/90
(Received 3 March 2005; accepted 9 June 2005)
ISSN 1562-2975 print/ISSN 1814-1412 online # 2006 Taylor & Francis
DOI: 10.1080/15622970500213871
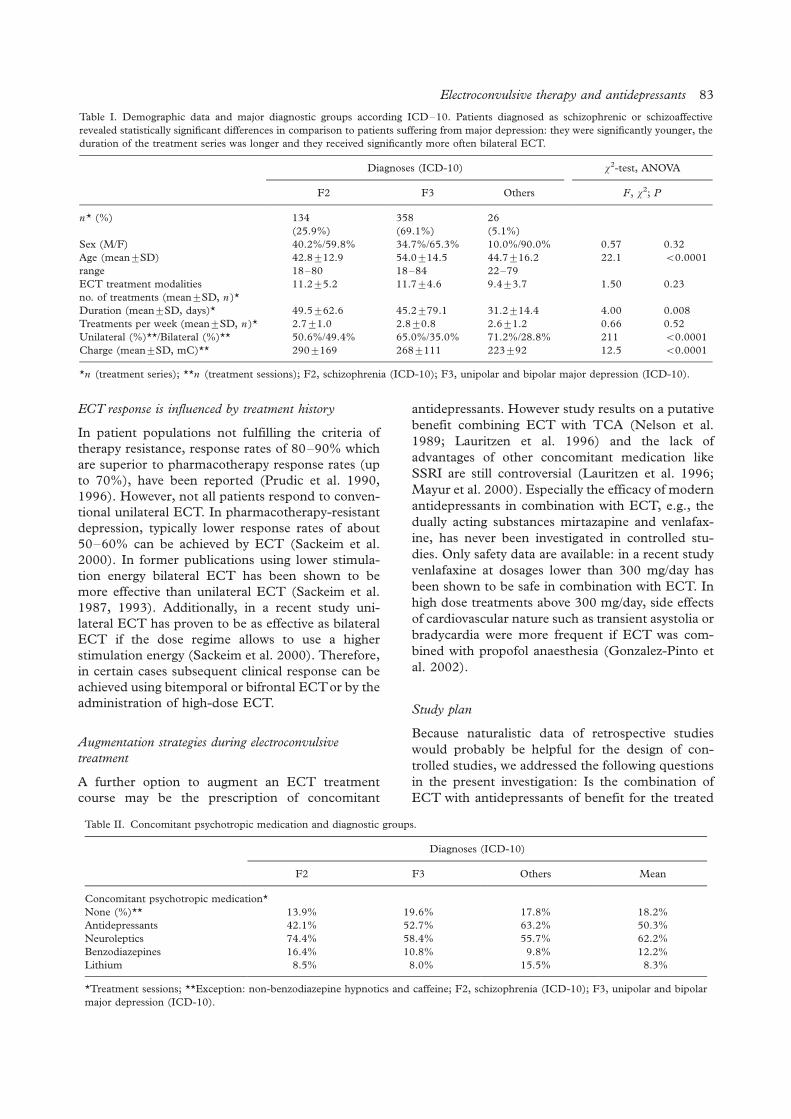
ECT response is influenced by treatment history
In patient populations not fulfilling the criteria of
therapy resistance, response rates of 80�/90% which
are superior to pharmacotherapy response rates (up
to 70%), have been reported (Prudic et al. 1990,
1996). However, not all patients respond to conven-
tional unilateral ECT. In pharmacotherapy-resistant
depression, typically lower response rates of about
50�/60% can be achieved by ECT (Sackeim et al.
2000). In former publications using lower stimula-
tion energy bilateral ECT has been shown to be
more effective than unilateral ECT (Sackeim et al.
1987, 1993). Additionally, in a recent study uni-
lateral ECT has proven to be as effective as bilateral
ECT if the dose regime allows to use a higher
stimulation energy (Sackeim et al. 2000). Therefore,
in certain cases subsequent clinical response can be
achieved using bitemporal or bifrontal ECTor by the
administration of high-dose ECT.
Augmentation strategies during electroconvulsive
treatment
A further option to augment an ECT treatment
course may be the prescription of concomitant
antidepressants. However study results on a putative
benefit combining ECT with TCA (Nelson et al.
1989; Lauritzen et al. 1996) and the lack of
advantages of other concomitant medication like
SSRI are still controversial (Lauritzen et al. 1996;
Mayur et al. 2000). Especially the efficacy of modern
antidepressants in combination with ECT, e.g., the
dually acting substances mirtazapine and venlafax-
ine, has never been investigated in controlled stu-
dies. Only safety data are available: in a recent study
venlafaxine at dosages lower than 300 mg/day has
been shown to be safe in combination with ECT. In
high dose treatments above 300 mg/day, side effects
of cardiovascular nature such as transient asystolia or
bradycardia were more frequent if ECT was com-
bined with propofol anaesthesia (Gonzalez-Pinto et
al. 2002).
Study plan
Because naturalistic data of retrospective studies
would probably be helpful for the design of con-
trolled studies, we addressed the following questions
in the present investigation: Is the combination of
ECT with antidepressants of benefit for the treated
Table I. Demographic data and major diagnostic groups according ICD�/10. Patients diagnosed as schizophrenic or schizoaffective
revealed statistically significant differences in comparison to patients suffering from major depression: they were significantly younger, the
duration of the treatment series was longer and they received significantly more often bilateral ECT.
Diagnoses (ICD-10) x2-test, ANOVA
F2 F3 Others F, x2; P
n* (%) 134 358 26
(25.9%) (69.1%) (5.1%)
Sex (M/F) 40.2%/59.8% 34.7%/65.3% 10.0%/90.0% 0.57 0.32
Age (mean9/SD) 42.89/12.9 54.09/14.5 44.79/16.2 22.1 B/0.0001
range 18�/80 18�/84 22�/79
ECT treatment modalities
no. of treatments (mean9/SD, n )*
11.29/5.2 11.79/4.6 9.49/3.7 1.50 0.23
Duration (mean9/SD, days)* 49.59/62.6 45.29/79.1 31.29/14.4 4.00 0.008
Treatments per week (mean9/SD, n )* 2.79/1.0 2.89/0.8 2.69/1.2 0.66 0.52
Unilateral (%)**/Bilateral (%)** 50.6%/49.4% 65.0%/35.0% 71.2%/28.8% 211 B/0.0001
Charge (mean9/SD, mC)** 2909/169 2689/111 2239/92 12.5 B/0.0001
*n (treatment series); **n (treatment sessions); F2, schizophrenia (ICD-10); F3, unipolar and bipolar major depression (ICD-10).
Table II. Concomitant psychotropic medication and diagnostic groups.
Diagnoses (ICD-10)
F2 F3 Others Mean
Concomitant psychotropic medication*
None (%)** 13.9% 19.6% 17.8% 18.2%
Antidepressants 42.1% 52.7% 63.2% 50.3%
Neuroleptics 74.4% 58.4% 55.7% 62.2%
Benzodiazepines 16.4% 10.8% 9.8% 12.2%
Lithium 8.5% 8.0% 15.5% 8.3%
*Treatment sessions; **Exception: non-benzodiazepine hypnotics and caffeine; F2, schizophrenia (ICD-10); F3, unipolar and bipolar
major depression (ICD-10).
Electroconvulsive therapy and antidepressants 83

patients regarding ictal electrophysiological para-
meters of potential prospective significance or the
actual clinical effectiveness? Is the combination
therapy of ECT and a concomitant antidepressant
pharmacotherapy as safe as an ECT monotherapy?
Is the incidence of cognitive or cardiovascular side
effects influenced by the concomitant medication?
Are there differences concerning the mentioned
variables between different classes of antidepres-
sants?
Materials and methods
Patients
We analysed data about 5482 ECT treatments in
455 psychiatric in-patients between the years 1995
and 2003 in a retrospective study. A total of
518 treatment series were recorded, because in 63
cases one patient receives more than one treatment
series during the observational time period. Patient
characteristics including major diagnostic groups
(ICD-10) (World Health Organization 1992),
demographic data and ECT treatment modalities
are given in Table I. We analysed data of patients
with treatment resistant paranoid hallucinatory schi-
zophrenia or schizoaffective disorder (F2), with
unipolar or bipolar major depression (F3) and rare
other diagnoses such as organic psychosis, person-
ality disorders and Gilles-de-la-tourette syndrome.
Before starting the treatment patients gave their
written informed consent for ECT procedures and
anaesthesia separately.
ECT treatments (Weiner et al. 1988)
ECT was performed using the Thymatron DGxTM
device during the years 1995 up to June 2000,
afterwards a Thymatron System-IVTM was used
from July 2000 up to 2003. The devices provide
stimulus delivery in form of constant current (900
mA) bi-directional pulse wave stimulation. The
voltage was up to 450 V, impulse width was 0.5�/1
ms (mean9/standard deviation (SD): 0.549/1.3 ms),
the frequency was 20�/70 Hz (51.99/17.7 Hz). The
length of a stimulus train was 0.14�/8 s (5.29/1.9 s).
Stimulus intensity was determined using the
modified age method in case of unilateral stimula-
tion and half-age method in case of bilateral stimula-
tion (Abrams 2002). Lower and upper charge limits
during the first stimulation were 30 and 60% device
adjustment representing a charge of 151 and 302
mC, respectively. Only in a few cases (B/5%) was
dose titration used before the initial treatment
session. In the case of dose titration, unilateral
treated patients received a first stimulus 2.5�/5-fold
over threshold, bilateral treated patients a stimulus
5% over threshold. Restimulation and dosage
elevation in 10% steps during the subsequent treat-
ments was recommended in case of too short EEG
(B/ 30 s) and EMG (B/ 25 s) seizure activity and a
too low postictal suppression index (B/80%). The
maximal charge was 504 mC during the years 1995�/
2001 and 1008 mC since January 2002. The mean
postictal suppression index was 88.09/11.9%, the
mean stimulation energy was 54.19/25.5% (272.79/
128.5 mC).
Electrode placement was primarily unilateral
according the d’Elia method (d’Elia G 1970; d’Elia
G et al. 1975). In the case of therapy resistance or
the knowledge of former inefficient unilateral ECT
bitemporal electrode placement was recommended.
Since electrode placement was not recorded during
clinical routine treatments, these data are lacking in
45.2% of treatments during the years 1995�/1999.
From 2000 on this information is available. 33.5% of
the investigated treatments were unilateral and
21.3% were bitemporal.
Seizure monitoring included one-lead EEG and
EMG monitoring up to June 2002 and two-lead
EEG, EMG and ECG monitoring from July 2002.
The one-lead EEG electrodes were placed left
frontopolar and over the ipsilateral mastoid (FP1�/
A1 according the international 10�/20 system). The
two-lead EEG electrodes were left and right fronto-
polar (FP1�/A1 and FP2�/A2) and over the ipsilateral
mastoid. EMG was conducted in 5�/10 cm over the
flexor carpi ulnaris muscle. The mean seizure
duration was 31.79/16.0 s (EEG) and 19.39/11.4 s
(EMG), respectively.
Anaesthesia
The administered anaesthetic agents were thiopental
(in 71.9% of treatments, mean dosage9/SD: 3649/
93 mg), propofol (20.0%, 1709/51 mg), methohex-
ital (5.5%, 1309/33 mg) and etomidate (2.6%, 329/
32 mg). Due to the clinical objective to achieve
adequate seizures, patients with higher seizure
threshold received more frequent methohexital
than other anaesthetics. During these treatments
most patients received no concomitant antidepres-
sant pharmacotherapy. For muscle relaxation, suc-
cinylcholine or pyridostigmine, together with
atracurium for premedication, were used.
Psychotropic medication
Information about the main treatment groups and
indication of concomitant medication is given in
Table I. A total of 813 treatments were done without
any psychiatric medication, 184 treatments were
done with concomitant non-benzodiazepine hypno-
84 T. C. Baghai et al.

tics. The analysis of all electrophysiological and
clinical parameters revealed no clinically relevant or
statistically significant differences between these two
groups. Due to this result and because no significant
psychotropic effects besides the sedating and sleep-
inducing properties were expected, we merged these
997 treatments in the group of patients without
psychotropic medication (noPT).
In the case of treatment with antidepressants,
42.1% of treatments were performed with concomi-
tant administration of tricyclic antidepressants
(TCA), 21.1% with tetracyclic antidepressants
(TTCA: 92.4% mirtazapine, 7.6% mianserine),
16.1% with selective serotonin reuptake inhibitors
(SSRI), 14.2% with venlafaxine (SNRI), 2.9% with
reboxetine, 2.7% with other antidepressants and
0.9% with monoaminooxidase inhibitors (predomi-
nantly moclobemide). In the case of treatment with
neuroleptics, 45.7% of the treatments were done
with concomitant administration of atypical sub-
stances, 34.3% with high potent classical neurolep-
tics, 12.3% with medium potency and 38.2% with
low potent neuroleptics (also combinations were
given).
Clinical assessments
We included clinical data and ECT treatment data
from the patient records, ECT treatment documen-
tation and ECT-device printouts in a relational
database. Both the diagnoses according ICD-10
(Sartorius 1983; ICD-10 Task Force 1987) and the
severity of disease according the clinical global
impression scale (CGI, Item 1) (National Institute
of Mental Health 1976) were assessed. CGI scores
were recorded in weekly intervals and after each
electroconvulsive treatment. Additional information
about therapeutic response and adverse effects such
as cognitive impairment were registered system-
atically (CGI, Items 3.1 and 3.2) after each ECT
according the clinicians judgments. Serious adverse
events, if applicable, should be recorded in free
form. Clinical, technical and electrophysiological
ECT data such as stimulation parameters, EEG
and EMG derivations including the length of con-
vulsions, postictal suppression, seizure energy and
seizure concordance were registered. Information
about all prescribed medication including psychia-
tric medication, medication prescribed for medical
conditions and substances used for anesthesia were
recorded in detail.
Electrophysiological measures
The minimal duration of generalized convulsions
measured using EEG and EMG techniques is a
necessary but not sufficient criterion for a probable
successful ECT treatment. A duration of at least 25 s
measured in the EEG and 20 s in the EMG is
recommended (Coffey et al. 1995). In most cases
computer-automated electroencephalographic and
electromyographic estimation of the seizure duration
is of sufficient quality (Swartz et al. 1994). But it is
also known that there is no significant correlation
between the therapeutic effectiveness of ECT and
the duration of convulsions (Abrams 1972, 2002;
Nobler et al. 1993). Especially during unilateral
ECT, a higher stimulation energy together with a
higher clinical efficacy (Sackeim et al. 2000) may
lead to shorter convulsions (Frey et al. 2001).
Therefore, other measures are of use to decide
whether a restimulation may be of use during an
individual treatment session. The postictal suppres-
sion index (PSI) shows how fast and complete the
EEG amplitude flattens directly after the convul-
sions. The index is calculated from the quotient of
the mean EEG amplitude during a 3-s derivation 0.5
s after the end of convulsions and the mean
amplitude of a 3-s passage during the convulsions.
Its unit ‘‘% suppression’’ is highly correlated to the
probability of clinical improvement (Suppes et al.
1996). Restimulation is recommended if the PSI is
under 80% (Weiner et al. 1991; Nobler et al. 1993).
The convulsion energy index (CEI) is the product of
the mean EEG amplitude and the duration of
convulsions as a measure of the intensity of the ictal
response after electrical stimulation (mV s) (Weiner
et al. 1991). The convulsion concordance index
(CCI) is a measure for the intracerebral general-
ization of the convulsions (Swartz et al. 1986). It is
calculated as follows: 100�EEG � EMG
EEG � EMG(EEG and
EMG represent the duration of the convulsions).
According to the Thymatron manufacturer So-
matics, restimulation should be considered if the
index is below 51%. Regular monitoring of EEG and
EMG quality and all treatment procedures was
provided by senior psychiatrists.
Statistical analyses
Statistical analyses were performed using SPSS for
Windows (Release 11.0.1, SPSS Inc., Chicago, IL
60606, USA). Mean differences in demographic and
clinical variables between the treatment groups were
compared using independent samples Student’s t-
tests and x2-tests. A one-way analysis of variance
(ANOVA procedure) was performed to detect sig-
nificant differences in electrophysiological variables
and clinical mean scores between the divergent
treatment groups. Bonferroni correction was used
Electroconvulsive therapy and antidepressants 85

in case of multiple testing. The level of significance
was set at 0.05.
Results
A total of 18.2% of the treatments was done without
any psychotropic medication. In 23.4% a psycho-
tropic monotherapy was prescribed. Comparing
these two groups, the influence of confounding
variables like pharmacokinetic and dynamic effects
combining several substances can be excluded.
Within the group of mono-pharmacotherapeutic
treatments with tetracyclic antidepressants (TTCA)
only 1.9% received mianserine and 98.1% mirtaza-
pine. During 30.4% of the treatments the patients
received two concomitant medications, in 18.3%
three medications and in 9.6% more than three and
up to six psychotropic acting agents were prescribed.
Due to the lack of randomization we could detect
differential applied stimulation energies and charges
in the different treatment groups (ANOVA
F(4,1154)�/13.2, P B/0.0001). Bonferroni cor-
rected post-hoc tests showed a significantly higher
applied charge in the group of treatments without
pharmacotherapy (mean9/SD: 2889/135 mC) in
comparison to treatments with concomitant TCA
(mean9/SD: 2199/103 mC, P B/0.0001), SSRI
(2289/95 mC, P�/0.001) or SNRI (2389/96 mC,
P�/0.039). The stimulation charge in the TTCA-
treated group showed a trend to higher values
(2529/76 mC, P�/0.26, n.s.) which was not statis-
tically significant.
As expected the ANOVA revealed a significant
differences in the duration of generalized convul-
sions (Figure 1; EEG: F(4,1076)�/6.27, P B/
0.0001; EMG: F(4,805)�/4.23, P�/0.002). Bonfer-
roni corrected post-hoc tests showed a significant
longer duration of convulsions in ECT/SSRI com-
bination therapies in comparison to treatments with
TCA (P�/0.012) or serotonin and noradrenalin
reuptake inhibitors (SNRI, venlafaxine) treatment
(P�/0.002) and in comparison to ECT monother-
apy (P�/0.001). TTCA (predominantly mirtaza-
pine) treatments resulted in significantly longer
convulsions in comparison to venlafaxine treatments
(P�/0.019).
The postictal suppression index showed signifi-
cant differences between the five treatment groups
(Figure 2; ANOVA F(4,837)�/5.59, P B/0.0001).
Bonferroni corrected post-hoc tests showed a sig-
nificantly lower PSI after ECT treatments under
TTCA in comparison to SSRI- (P�/0.023) or
SNRI-treated patients (P�/0.014). After treatments
without psychopharmacotherapy significantly lower
PSI than under SSRI (P�/0.015) or SNRI (P�/
0.015) were seen.
Both effectiveness and the rate of side effects were
affected by the concomitant medication. After the
first ECT treatment, the different treatment groups
showed no significant differences concerning the
severity of disease (CGI, Item 1: ANOVA
F(4,218)�/1.02, P�/0.39, n.s.), whereas it was
significantly influenced by the concomitant treat-
ment during the ECT treatment series (Figure 3;
Figure 1. Mean duration of generalized convulsions in EEG- and EMG-derivations. EEG: Significantly longer convulsions in ECT/SSRI
combination therapy in comparison to treatments with TCA, SNRI and treatments without concomitant medication. Significantly longer
convulsions during TTCA in comparison to venlafaxine treatment. EMG: Significant longer convulsions in SSRI- and TTCA-treated
patients in comparison to ECT treatments without psychotropic co-medication. no PT�/no psychotropic medication; exception: non-
benzodiazepine hypnotics in case of insomnia (n�/146 patients); TCA�/tricyclic antidepressants (n�/34 patients); TTCA�/tetracyclic
antidepressants (n�/14 patients); SSRI�/selective serotonin reuptake inhibitors (n�/10 patients); SNRI�/selective serotonin and
noradrenalin reuptake inhibitors (n�/14 patients).
86 T. C. Baghai et al.

ANOVA F(4,586)�/13.7, P B/0.0001). Bonferroni
corrected post-hoc tests showed a significantly
higher severity of disease during ECT treatments
without pharmacotherapy in comparison to treat-
ments combined with SSRI (P�/0.012) or TTCA
(P B/0.0001). TTCA-treated patients showed also
significant lower CGI Item 1 values in comparison to
TCA- (P B/0.0001) and SNRI (P�/0.002)-treated
patients. Similar results yielded CGI Item 3.1 (over-
all amelioration of symptoms): both treatments
without concomitant medication and SNRI-treated
patients showed higher mean scores and therefore
less improvement in comparison to TCA, TTCA
and SSRI treatments (Figure 3). The possibility to
apply a higher maximal charge since 2002 did not
influence the overall treatment results because high
dose treatments with charges between 504 and 1005
mC were applied infrequently.
The rate of cognitive and cardiovascular side
effects was significantly higher in SNRI-treated
patients in comparison to patients without psycho-
tropic medication. Considering transient cardiovas-
Figure 2. Postical suppression index: significantly higher indices in SSRI or SNRI combined ECT treatments in comparison to treatments
without psychopharmacological medication. Significantly higher convulsion energy index in treatments with TTCA in comparison to the
other treatment groups. No influence of concomitant treatment on convulsion concordance index. PSI�/postictal suppression index;
CCI�/convulsion concordance index; CEI�/convulsion energy index.
Figure 3. Clinical treatment efficacy (judged after each ECT treatment or in weekly intervals): significantly more benefit for the patients in
ECT treatments combined with tricyclics, SSRIs or TTCA. Significantly more cardiovascular or cognitive side effects in patients receiving
SNRI in comparison to patients without concomitant psychotropic medication. CGI Item 1: severity of disease (lower score indicates better
efficacy); CGI Item 3.1: efficacy (lower score indicates better efficacy); CGI Item 3.2: adverse effects (lower score indicates better
tolerability of the treatment).
Electroconvulsive therapy and antidepressants 87

cular side effects separately, the x2-test showed
two-fold higher rates than expected in the treatment
group receiving TTCA and less than half than
expected during the treatments with concomitant
application of TCA. These effects were statistically
significant (x2 (4,1151)�/20.9, P B/0.0001). There
were no reports about serious adverse events during
the observational time period. In addition there were
no reports about higher frequencies of therapeutic
interventions in case of side effects, e.g., high blood
pressure after ECT in one of the mentioned ther-
apeutic groups.
Discussion
Monotherapeutic antidepressant treatments using
TCA, SSRI or SNRI had significant impact on ictal
electrophysiological indices, the amelioration of
depressive symptoms and the incidence of side
effects.
As known from the literature, concomitant SSRI
treatment can lead to prolonged generalized seizures
(Gutierrez-Esteinou et al. 1989; Curran 1995)
independently from clinical outcome. The presented
investigation confirms this result and shows that also
TTCA, like mirtazapine, are able to lengthen seizure
duration. Patients receiving venlafaxine showed sig-
nificantly shorter seizures in comparison to SSRI
and TTCA treatments. The influence of confound-
ing variables, like concomitant benzodiazepine use,
was excluded, because only monotherapies with one
antidepressant were analysed. Because higher stimu-
lation energy can lead to shorter convulsions (Frey et
al. 2001), the lower applied energy in the SSRI-
treated group may contribute to these results.
Nevertheless, in neither SSRI- nor TTCA-treated
patients had seizures to be terminated using anaes-
thetics or benzodiazepines more often than in other
treatment groups. Also, a more frequent use of
caffeine in the indication to prolong seizures in one
of the treatment groups was not detected.
To evaluate the ECT treatment quality, other
electrophysiological parameters and indices of pro-
spective clinical significance were also used (Swartz
et al. 1986; Weiner et al. 1991; Nobler et al. 1993;
Suppes et al. 1996). In our study the PSI, which
indicates a positive correlation with clinical effec-
tiveness (Suppes et al. 1996), was significantly
enhanced in comparison to treatments without
psychotropic pharmacotherapy if SSRI or SNRI
treatment were prescribed at the same time. In
treatments with a concomitant TTCA the index
was significantly lower than in both mentioned
groups. On the contrary, these predominantly mir-
tazapine-treated patients showed a significantly
higher CEI in comparison to both the unmedicated
and all other medicated patients, which also indi-
cates a positive impact on therapeutic outcome
(Swartz et al. 1986; Weiner et al. 1991). Our
retrospective data show therefore that the CEI may
allow a better prediction of the treatment quality in
patients with concomitant antidepressant treatment
in comparison to both the PSI and the CCI, but due
to the retrospective characteristic of our analysis this
question cannot be answered finally. Postictal sup-
pression seems to be of limited usefulness to predict
the clinical effectiveness in our study, similar to
other prospective studies (Perera et al. 2004). In
addition, no data about the influence of venlafaxine
and mirtazapine on ictal electrophysiological mea-
sures during ECT have yet been published.
Therefore, we tried to investigate the interrelation
between the medication, the described findings and
clinical effectiveness. According the CGI Item 1
(severity of disease), patients receiving ECT and
TTCA had a significant lower score than patients
receiving ECT alone, patients receiving ECT and
tricyclics and patients receiving ECT and venlafax-
ine. SSRI-treated patients had also higher mean
scores than TTCA-treated patients, but this differ-
ence was not statistically significant after Bonferroni
correction. The difference between SSRI-treated
patients and the patients receiving ECT alone
achieved statistical significance. The mean ameliora-
tion of symptoms indicated by the mean CGI Item
3.1 score was also significantly more pronounced
during ECT treatments in combination with a TCA,
TTCA or SSRI therapy in comparison to ECT
alone. During treatments using ECTand venlafaxine
in combination a significantly weaker effect on CGI
Item 3.1 could be seen in comparison to TCA,
TTCA and SSRI combinations with ECT.
These results indicate that the PSI may not be the
best parameter to estimate the clinical effectiveness
of a single ECT treatment if ECT is not adminis-
tered as a monotherapy. This seems to be especially
true in ECT/mirtazapine combination therapy,
where the CEI may be better suitable to predict
clinical effectiveness. Another explanation of these
findings may be the lack of randomization in our
retrospective study of clinical routine treatments.
The patients were not randomized by chance to the
treatment groups and especially both dual acting
antidepressants are used very often in the case of
prior treatment-resistance. This is especially true in
patients receiving this medication in combination
with ECT. Some authors describe a similar effec-
tiveness in severe depression of tricyclics and SSRI
(Hirschfeld 1999), others report a better ameliora-
tion of depressive symptoms during TCA treatment
(Parker et al. 1999). Even if there are contradicting
publications, a favourable therapeutic outcome in
88 T. C. Baghai et al.

severe and treatment-resistant depression (Hirsch-
feld 1999; Thase et al. 2000) leads to an intensified
use of venlafaxine and mirtazapine. Of course a
higher degree of treatment-resistance, not only
during a pharmacotherapeutic approach but also
during an ECT treatment, leads to lower response
rates and higher estimations of the severity of
disease.
The rate of side effects was higher during ECT
treatments in combination with venlafaxine in com-
parison to unmedicated patients. It is known that a
higher rate of cognitive disturbances can be expected
in case of the use of higher stimulation charges
(Abrams et al. 1991; McCall et al. 2000). This effect
can be excluded to a large extend in our study,
because the treatments without concomitant phar-
macotherapy were done using the highest stimula-
tion charge. Otherwise, in spite of the published safe
use of this combination (Bernardo et al. 2000) in
daily dosages up to 150 mg (Gonzalez-Pinto et al.
2002), several reports about cardiovascular risks
(Agelink et al. 1998; Jha et al. 2002), especially
during high-dose venlafaxine therapy together with
the use of propofol anaesthesia (Gonzalez-Pinto et
al. 2002) and also prolonged seizures (Conway et al.
2001), are published for this combination therapy.
In our sample a total of 284 ECT treatments were
done with concomitant venlafaxine prescription. A
total of 195 (68.7%) of these prescriptions were in
high-dose range above 150 mg, 91 (32.0%) in the
highest doses above 300 mg. The mean dosage was
2559/120 mg/day. Comparably high dosages were
prescribed because of the high degree of treatment-
resistance in this patient group and may be the
explanation for the relatively lower clinical response
together with a higher incidence of side effects.
Nevertheless all observed side effects were only
transient and in most cases no specific treatment
was required.
It has to be mentioned that our analyses were
treatment session oriented and not patient oriented.
This includes the disadvantage that single treatment
series in singular patients who are treatment-resis-
tant in a special way and received more treatments
than other patients may influence the analysis more
than others. On the other hand, as an advantage,
changes in ECT and pharmacotherapy treatment
modalities within the ECT series do not influence
the results and have therefore not to be taken into
consideration.
Overall, our study shows an excellent tolerability
of ECT and ECT in combination with antidepres-
sant pharmacotherapy. The presented data give first
hints that it is not only tricyclic antidepressants that
lead to a therapeutic benefit in treatment-resistant
disorders. Also, other concomitant antidepressant
pharmacotherapies, during an ECT treatment
course like SSRI or the modern dually acting
antidepressant mirtazapine, may be able to enhance
treatment efficacy. Of course, this cannot be proven
in our study due to the naturalistic and retrospective
design. Our study provides data which justify and
demand further controlled evaluations using double-
blind and placebo-controlled conditions in a next
step to answer the question, if and which modern
antidepressants should be used in combination with
ECT to enhance responder rates, shorten the time
until clinical response and cause as fewer side effects
as possible, especially in the treatment of therapy-
resistant psychiatric disorders.
Acknowledgements/Statement of interest
The authors have no conflict of interest with any
commercial or other associations in connection with
the submitted article. The authors would like to
thank Mrs M. Ertl, Mrs S. Rauch and Mr K. Neuner
for patient nursing, ECT organization and database
management.
References
Abrams R. 1972. Recent clinical studies of ECT. Semin Psychia-
try 4:3�/12.
Abrams R. 2002. Stimulus titration and ECT dosing. J. ECT.
18:3�/9.
Abrams R, Swartz CM, Vedak C. 1991. Antidepressant effects of
high-dose right unilateral electroconvulsive therapy. Arch Gen
Psychiatry 48:746�/748.
Agelink MM, Zeit T, Klieser E. 1998. Prolonged bradycardia
complicates antidepressive treatment with venlafaxine and
ECT. Br J Psychiatry 173:441.
Baghai TC, Moller H-J, Rupprecht R. 2006. Recent progress in
pharmacological and non-pharmacological treatment options
of major depression. Curr Pharm Design 12:503�/515.
Bernardo M, Navarro V, Salva J, Arrufat FJ, Baeza I. 2000.
Seizure activity and safety in combined treatment with venla-
faxine and ECT: A pilot study. J ECT 16:38�/42.
Coffey CE, Lucke J, Weiner RD, Krystal AD, Aque M. 1995.
Seizure threshold in electroconvulsive therapy: I. Initial seizure
threshold. Biol. Psychiatry 37:713�/720.
Conway CR, Nelson LA. 2001. The combined use of bupropion,
lithium, and venlafaxine during ECT: A case of prolonged
seizure activity. J ECT 17:216�/218.
Curran S. 1995. Effect of paroxetine on seizure length during
electroconvulsive therapy. Acta Psychiatr Scand 92:239�/240.
D’Elia G. 1970. Unilateral electroconvulsive therapy. Acta Psy-
chiatr Scand Suppl 215:1�/98.
D’Elia G, Raotma H. 1975. Is unilateral ECT less effective than
bilateral ECT? Br J Psychiatry 126:83�/89.
ECT Review Group. 2003. Efficacy and safety of electroconvul-
sive therapy in depressive disorders: A systematic review and
meta-analysis. Lancet 361:799�/808.
Folkerts HW, Michael N, Tolle R, Schonauer K, Mucke S,
Schulze-Monking H. 1997. Electroconvulsive therapy vs.
paroxetine in treatment-resistant depression �/ a randomized
study. Acta Psychiatr Scand 96:334�/342.
Frey R, Heiden A, Scharfetter J, Schreinzer D, Blasbichler T,
Tauscher J, Felleiter P, Kasper S. 2001. Inverse relation
Electroconvulsive therapy and antidepressants 89

between stimulus intensity and seizure duration: Implications
for ECT procedure. J ECT 17:102�/108.
Gonzalez-Pinto A, Gutierrez M, Gonzalez N, Elizagarate E, Perez
De Heredia JL, Mico JA. 2002. Efficacy and safety of
venlafaxine-ECT combination in treatment-resistant depres-
sion. J Neuropsychiatry Clin Neurosci 14:206�/209.
Greenblatt M, Grosser GH, Wechsler HA. 1964. Differential
response of hospitalized depressed patients in somatic therapy.
Am J Psychiatry 935�/943.
Gutierrez-Esteinou R, Pope HG Jr. 1989. Does fluoxetine prolong
electrically induced seizures? Convuls Ther 5:344�/348.
Hirschfeld RM. 1999. Efficacy of SSRIs and newer antidepres-
sants in severe depression: comparison with TCAs. J Clin
Psychiatry 60:326�/335.
ICD-10 Task Force. 1987. Report of the ICD-10 Task Force.
American Medical Record Association. J Am Med Rec Assoc
58:51�/56.
Janicak PG, Davis JM, Gibbons RD, Ericksen S, Chang S,
Gallagher P. 1985. Efficacy of ECT: A meta-analysis. Am J
Psychiatry 142:297�/302.
Jha A, Tomar R. 2002. Interaction between ECT and venlafaxine.
Int J Geriatr Psychiatry 17:979�/980.
Lauritzen L, Odgaard K, Clemmesen L, Lunde M, Ohrstrom J,
Black C, Bech P. 1996. Relapse prevention by means of
paroxetine in ECT-treated patients with major depression: A
comparison with imipramine and placebo in medium-term
continuation therapy. Acta Psychiatr Scand 94:241�/251.
Mayur PM, Gangadhar BN, Subbakrishna DK, Janakiramaiah N.
2000. Discontinuation of antidepressant drugs during electro-
convulsive therapy: A controlled study. J Affect Disord 58:37�/
41.
McCall WV, Reboussin DM, Weiner RD, Sackeim HA. 2000.
Titrated moderately suprathreshold vs fixed high-dose right
unilateral electroconvulsive therapy: acute antidepressant and
cognitive effects. Arch Gen Psychiatry 57:438�/444.
National Institute of Mental Health. 1976. 028 CGI. Clinical
global impressions. In: Guy W, Bonato RR, editors. Manual for
the EDCEU Assessment Battery, 2. Rev. Chevy Chase, MD:
National Institute of Mental Health. pp 12-1�/12-6.
Nelson JP, Benjamin L. 1989. Efficacy and safety of combined ect
and tricyclic antidepressant drugs in the treatment of depressed
geriatric patients. Convuls Ther 5:321�/329.
Nobler MS, Sackeim HA, Solomou M, Luber B, Devanand DP,
Prudic J. 1993. EEG manifestations during ECT: Effects of
electrode placement and stimulus intensity. Biol. Psychiatry
34:321�/330.
Parker G, Mitchell P, Wilhelm K, Menkes D, Snowdon J,
Schweitzer I, Grounds D, Skerritt P, Roy K, Hadzi-Pavlovic
D. 1999. Are the newer antidepressant drugs as effective as
established physical treatments? Results from an Australasian
clinical panel review. Aust NZ J Psychiatry 33:874�/881.
Perera TD, Luber B, Nobler MS, Prudic J, Anderson C, Sackeim
HA. 2004. Seizure expression during electroconvulsive therapy:
Relationships with clinical outcome and cognitive side effects.
Neuropsychopharmacology 29:813�/825.
Prudic J, Sackeim HA, Devanand DP. 1990. Medication resis-
tance and clinical response to electroconvulsive therapy.
Psychiatry Res 31:287�/296.
Prudic J, Haskett RF, Mulsant B, Malone KM, Pettinati HM,
Stephens S, Greenberg R, Rifas SL, Sackeim HA. 1996.
Resistance to antidepressant medications and short-term clin-
ical response to ECT. Am J Psychiatry 153:985�/992.
Sackeim HA, Decina P, Kanzler M, Kerr B, Malitz S. 1987.
Effects of electrode placement on the efficacy of titrated, low-
dose ECT. Am J Psychiatry 144:1449�/1455.
Sackeim HA, Prudic J, Devanand DP, Kiersky JE, Fitzsimons L,
Moody BJ, McElhiney MC, Coleman EA, Settembrino JM.
1993. Effects of stimulus intensity and electrode placement on
the efficacy and cognitive effects of electroconvulsive therapy.
New Engl J Med 328:839�/846.
Sackeim HA, Prudic J, Devanand DP, Nobler MS, Lisanby SH,
Peyser S, Fitzsimons L, Moody BJ, Clark J. 2000. A prospec-
tive, randomized, double-blind comparison of bilateral and
right unilateral electroconvulsive therapy at different stimulus
intensities. Arch Gen Psychiatry 57:425�/434.
Sartorius N. 1983. The ICD-10 and depression. Acta Psychiatr
Scand Suppl 310:103�/105.
Suppes T, Webb A, Carmody T, Gordon E, Gutierrez-Esteinou R,
Hudson JI, Pope Jr HG. 1996. Is postictal electrical silence a
predictor of response to electroconvulsive therapy? J Affect
Disord 41:55�/58.
Swartz CM, Larson G. 1986. Generalization of the effects of
unilateral and bilateral ECT. Am J Psychiatry 143:1040�/1041.
Swartz CM, Abrams R, Rasmussen K, Pavel J, Zorumski CF,
Srinivasaraghavan J. 1994. Computer automated versus vi-
sually determined electroencephalographic and electromyo-
graphic seizure duration. Convuls Ther 10:165�/170.
Thase ME, Friedman ES, Howland RH. 2000. Venlafaxine and
treatment-resistant depression. Depress Anxiety 12(Suppl
1):55�/62.
Weiner RD, Weaver LA Jr, Sackeim HA. 1988. Reporting of
Technical Parameters in ECT Publications: Recommendations
for Authors. Convuls Ther 4:88�/91.
Weiner RD, Coffey CE, Krystal AD. 1991. The monitoring and
management of electrically induced seizures. Psychiatr Clin
North Am 14:845�/869.
World Health Organization. 1992. The ICD-10 Classification of
Mental and Behavioural Disorders: Clinical Descriptions and
Diagnostic Guidelines). Geneva: World Health Organization.
90 T. C. Baghai et al.

ORIGINAL INVESTIGATION
P300 differences exist between Tourette’s syndrome with and withoutattention deficiency and hyperactivity disorder in children
ZHU YAN, LIU PO-ZI, LEUNG KAI-MAN, SU LIN-YAN, WU DA-XING & ZHOU MING
Mental Health Department, The Second Affiliated Hospital (Yu Quan Hospital) of Medical School, Tsinghua University,
Beijing, China
AbstractObjective: To study the characteristics of P300 in Tourette’s syndrome (TS) with and without attention deficiency andhyperactivity disorder (ADHD). Method: Auditory evoked P300 were recorded in 19 TS only (TS�/ADHD) children, 15TS with ADHD (TS�/ADHD) children and 20 unaffected control subjects, and their waveforms, amplitudes, latencies andtopographies were compared at Fz, Cz, C3, C4 and Pz. Results: The TS�/ADHD group showed shorter latencies thancontrol subjects at all electrode sites (P B/0.05 or 0.01), and the TS�/ADHD group at CZ and PZ (P B/0.05); however, therewas no significant difference between control subjects and the TS�/ADHD group. The TS�/ADHD group showed smalleramplitudes than the control group at all electrode sites (P B/0.05), and the TS�/ADHD group at Cz (P B/0.05); however,there were no significant differences between control subjects and the TS�/ADHD group. There was no significantdifference in the prevalence of abnormal waveforms between the control, TS, TS�/ADHD and TS�/ADHD groups, butthere were significant differences in the variability of localization of P300 between the control and the TS group (P�/0.003),control and TS�/ADHD groups (P�/0.000), and the TS�/ADHD and TS�/ADHD groups (P�/0.039). P300 in the TS�/
ADHD group tended to spread out to the left and that of the TS�/ADHD group tended to spread out to the right.Conclusions: P300 differences exist between TS�/ADHD and TS�/ADHD in children. These suggested that establishmentdifferent development defects or delay of communications between different structures rather than a delay in maturation ofthe structures themselves may be involved in TS�/ADHD and TS�/ADHD children and ADHD symptoms in TS patientsare likely a trait rather than adventitious or acquired within the TS syndrome.
Key words: Tourette’s syndrome, attention deficiency and hyperactivity disorder, auditory evoked potentials, children
Introduction
Tourette’s syndrome (TS) is a chronic neuropsy-
chiatric disorder characterized by motor and phonic
tics and related somatosensory urges. Prevalence of
TS is higher than previously thought, and may be
present in up to 2% of the population (e.g., Faridi
and Suchowersky 2003). It is a heterogeneous group
of chronic neuropsychiatric diseases, and its aetiol-
ogy is likely multifactorial, including a variety of
biological and psychological factors. The complexity
and variability of these aetiological factors resulted in
various TS subtypes with differences in symptoma-
tology, chronicity and gender distribution, but more
importantly, different degrees of association with
other psychological disorders such as conduct pro-
blems, learning difficulties, emotional disorders and
especially attention deficit/hyperactivity disorder
(ADHD; e.g., Comings 1995; Walkup et al. 1995;
Leckman et al. 1997; Leckman 2002; Faridi and
Suchowersky 2003). Studies found that at least 40�/
50% of TS children had comorbid ADHD (TS�/
ADHD). This group of children was often impul-
sive, overly active and inattentive before develop-
ment of tics, with adverse effects on peer and family
relationships as well as the child’s ability to succeed
in school (e.g., Leckman et al. 1997; Faridi and
Suchowersky 2003). Moreover, treatment of TS�/
ADHD children is often complicated and contro-
versial. Psychostimulants, such as methylphenidate,
dexamphetamine, and related drugs are the most
effective agents for uncomplicated hyperkinetic dis-
order. But these drugs could be used with impunity
in some individuals with tics. Occasionally, they can
precipitate de novo tics or exacerbate pre-existing
Correspondence: Zhu Yan, MD, Mental Health Department, The Second Affiliated Hospital (Yu Quan Hospital) of Medical School of
Tsinghua University, 5 Shijinsan Road, Beijing 100049, China. Tel: +8610 010 86471980. Fax: +8610 010 88258319. E-mail:
The World Journal of Biological Psychiatry, 2006; 7(2): 91�/98
(Received 28 June 2005; accepted 25 November 2005)
ISSN 1562-2975 print/ISSN 1814-1412 online # 2006 Taylor & Francis
DOI: 10.1080/15622970500492723

tics in individuals with Tourette’s syndrome (e.g.,
Sandor 1995; Leckman et al. 1997; Scahill et al.
2000). The combination of TS�/ADHD is thus
highly important in child psychiatry. Better under-
standing of these disease subtypes may lead to better
understanding of their aetiology, better treatment
options and more accurate prognostication (e.g.,
Sandor 1995; Leckman et al. 1997; Scahill et al.
2000).
Differences between TS children with and without
ADHD were noted in recent clinical studies. ADHD
was found to cause greater impairment than the tics,
and the severity of ADHD was more predictive of
impaired social adjustment irrespective of the sever-
ity of the tic symptoms (e.g., Harris et al. 1995;
Sherman et al. 1998; Stephens and Sandor 1999;
Carter et al. 2000; Mostofsky et al. 2001; Brand
et al. 2002; Mahone et al. 2002; Sukhodolsky et al.
2003). Comorbid ADHD was found to be predictive
of increased morbidity and poor outcome (e.g.,
Leckman 2003).
Many clinical studies had observed these, but few
have investigated the neurobiological basis of this
difference. Drake et al. (1992) found that TS
patients with and without attention deficit disorder
(ADD) did not differ in brainstem auditory evoked
potential (BAEP) latencies, both groups were not
different from normal controls, and that AEP
latencies did not differ between TS patients and
controls. TS patients with ADD had longer N100
and N200 latencies than TS patients without ADD
(e.g., Drake et al. 1992). Oades et al. (1996) found
that both ADHD and TS groups showed very large
P2 components where the maxima were shifted
anteriorly. The differences in the later potentials
were of a topographical nature. Later indices of
stimulus processing (N2�/P3) showed a frontal
impairment in TS and a right hemispheric impair-
ment in ADHD patients (e.g., Oades et al. 1996).
Straube et al. (1997) found that GTS patients
showed a significant increased latency of antisac-
cades, highly impaired performance of sequences of
memory-guided saccades, and an isolated reduction
in the peak velocity of the antisaccades (e.g., Straube
et al. 1997).
There were also few studies on P300 wave in TS
patients. The P300 wave is a positive component of
the event-related potential (ERP) occurring approxi-
mately 300 ms after an infrequently presented target
stimulus (so-called ‘oddball’ paradigm). It is thought
to reflect underlying fundamental cognitive opera-
tions (e.g., Polich and Kok 1995). Some previous
studies of P300 in TS children showed prolonged
latencies and decreased amplitudes (e.g., Huang and
Liu 1999), while others showed no difference
between normal subjects and TS patients (e.g.,
Fowler and Mitchell 1997). The discordances may
be related to different methods in the recruitment of
subjects (TS is often associated with complications
that are both complex and variable), different
techniques in the recording of P300 and different
analytical methods being used.
We carefully selected 19 TS�/ADHD, 15 TS�/
ADHD and 20 control subjects and tried to show
that TS with and without ADHD are neurophysio-
logically heterogeneous by using P300 as an indi-
cator.
Method
Subjects
Thirty-four, 10�/14-year-old TS patients were
recruited through the Child and Adolescent Psy-
chiatric Health specialist clinic, Mental Health
Institute, Central South University between July
2000 and August 2004. The study was approved
by the Ethics Committee, South Central University
and Department of Health, China. Informed con-
sents for participation were obtained from parents of
all children. The diagnostic procedures started with
child and parent clinical interview. Board-certified
psychiatrists (Liu Po-zi and Su Lin-yan), each with
25 years of research and clinical experience treating
TS children, diagnosed each case on the basis of a
clinical interview with the parent, and completed the
Yale Global Tic Severity Scale (YGTSS). Diagnosis
of TS was based on DSM-IV diagnostic criteria, and
children with organic or somatic disorders, or had
taken haloperidol, tiapride and other medication in
the past month were excluded. Children who
evidenced transient tics or chronic motor or vocal
tics and comorbidities such as obsessive-compulsive
disorder, anxiety disorders, conduct problems,
learning difficulties, as well as bipolar and nonbipo-
lar mood disorders, were also excluded. All selected
TS children suffered severe tic symptoms by YGTSS
assessment. The 34 TS patients were further divided
into two groups, with or without ADHD, diagnosed
by the same board-certified psychiatrists, based on
DSM-IV criteria. The TS only (TS�/ADHD) group
included 19 children (17 boys and two girls)
between the ages of 10 and 14 years (mean�/
11.16, SD�/1.214), with average Ravens IQ score
of 105.46 (SD�/13.04). The average time since
diagnosis was 2.019/0.59 years. The TS with
ADHD (TS�/ADHD) group included 15 children
(11 boys and four girls) between the ages of 10 and
13 years (mean�/11.13, SD�/1.060, IQ�/99.25,
SD�/15.68), and average time of diagnosis being
2.089/1.03 years. Finally, a control group of 20
unaffected children (16 boys and four girls) between
92 Zhu Yan et al.

the ages of 10 and 13 years (mean�/11.45, SD�/
1.146, IQ�/119.25, SD�/12.64) were randomly
selected from local elementary and high schools.
They all received course credits or pecuniary
remuneration for participation. The three groups
were matched in age, sex ratio and IQ (F�/0.440,
x2�/1.493, F�/0.547, P �/0.05). The two study
groups were matched in average time from diagnosis
(Table I).
Measurement
A standard oddball paradigm (Stim-1 systems from
Neuroscan, USA) was used for P300 recording. The
experiments were carried out in a sound-proof room
with temperature controlled at about 258C. Audi-
tory stimuli were delivered through earphones.
Testing subjects, with eyes covered, were asked to
press a response key for target stimuli (20%) and to
ignore standard stimuli (80%). A total of 200 stimuli
were given, with interstimulus interval of 2.0 s.
Before formal recording, subjects were allowed to
get familiar with the procedure and to learn to use
the response key by left figure. Standard 32-elec-
trode cap electroencephalogram (EEG) was used
(ElectroCap International). The reference electrodes
were placed over the mastoid processes, and the
ground electrode on the forehead. The impedance
values were kept below 5 kV. Trials with overt eye or
muscular movements were rejected if the EOG
exceeded 65 mV above the eyes. Peak amplitude
and latency were recorded after the standard
(0.8 kHz), the high (2.0 kHz) and the low deviants
(1.4 kHz). The P300/P300-like peak was the largest
positive component at 240�/540 ms post stimuli
(Oades et al. 1995a,b). P300 amplitude (mv) was
defined as the voltage difference between a presti-
mulus baseline and the largest positive-going peak of
the ERP waveform within a latency window. Latency
(ms) was defined as the time from stimulus onset to
the point of maximum positive amplitude within the
latency window. EEG was amplified 500 times with
a band-pass filter set at 0.10�/40 Hz, sampled at
500 Hz and stored on a hard disk for off-line
analysis. P300 were averaged for each condition
separately. The averaging epoch was 1,000 ms, with
the first 200 ms before stimulus being the baseline.
Trials containing incorrect responses, EOG artifacts
or amplifier saturation were excluded from the
averages. Data were digitally filtered between 0 and
30 Hz. Grand averaged P300 by group average were
calculated using Neuroscan software. Graphs were
plotted after averaging mean amplitude values, and
colour topographic maps generated by superimpos-
ing the records using Neuroscan software. The
waveforms, amplitudes and latencies at FZ, PZ, CZ,
C3 and C4 were chosen and analysed by SPSS/PC�/
program. The mean latency and amplitude were
analysed in each different group by independent-
samples t-test. Waveform variables and topographic
distributions were analysed by 2�/2 table Pearson
Chi-squared test.
Results
P300 latency and amplitude
There was no significant difference in P300
latency between the TS group and control group
except at C4, where control group showed longer
latencies than TS group (P B/0.05). Similarly,
there was no significant difference in P300 latency
between the control group and the TS�/ADHD
group. When compared with the control group,
however, the TS�/ADHD group showed shorter
latencies at all electrode sites (P B/0.05 or 0.01).
Lastly, the TS�/ADHD group showed significantly
longer latencies at CZ and PZ (P B/0.05)
when compared with the TS�/ADHD group
(Tables II�/V).
There was significant difference in P300 ampli-
tude between the TS group and the control group.
TS group showed smaller amplitudes than the
control group at all electrode sites (P B/0.05 or
0.01). Similarly, there was significant difference in
P300 amplitude between the control group and the
TS�/ADHD groups at all electrode sites (P B/0.05 or
0.01). When compared with the control group and
the TS�/ADHD groups, however, the TS�/ADHD
group showed smaller amplitude only at PZ (P B/
0.05 or 0.01). Lastly, when compared with the
TS�/ADHD group, TS�/ADHD showed significant
smaller P300 amplitude only at CZ (P B/0.05)
(Tables II�/V).
Table I. Demographic and clinical characteristics by diagnostic group.
TS-only
(n�/19)
TS�/ADHD
(n�/15)
Controls
(n�/20) Statistical test P value
Mean age9/SD 11.169/1.214 11.139/1.060 11.459/1.146 F�/0.440 0.646
Gender (% male) 89.5 73.3 80.0 x2�/1.493 0.474
Mean IQ score9/SD 105.469/13.04 99.259/15.68 119.259/12.64 F�/0.547 0.594
Average time since diagnosis (years)9/SD 2.019/0.59 2.089/1.03 T�/0.267 0.792
P300 differences between TS�ADHD and TS+ADHD 93
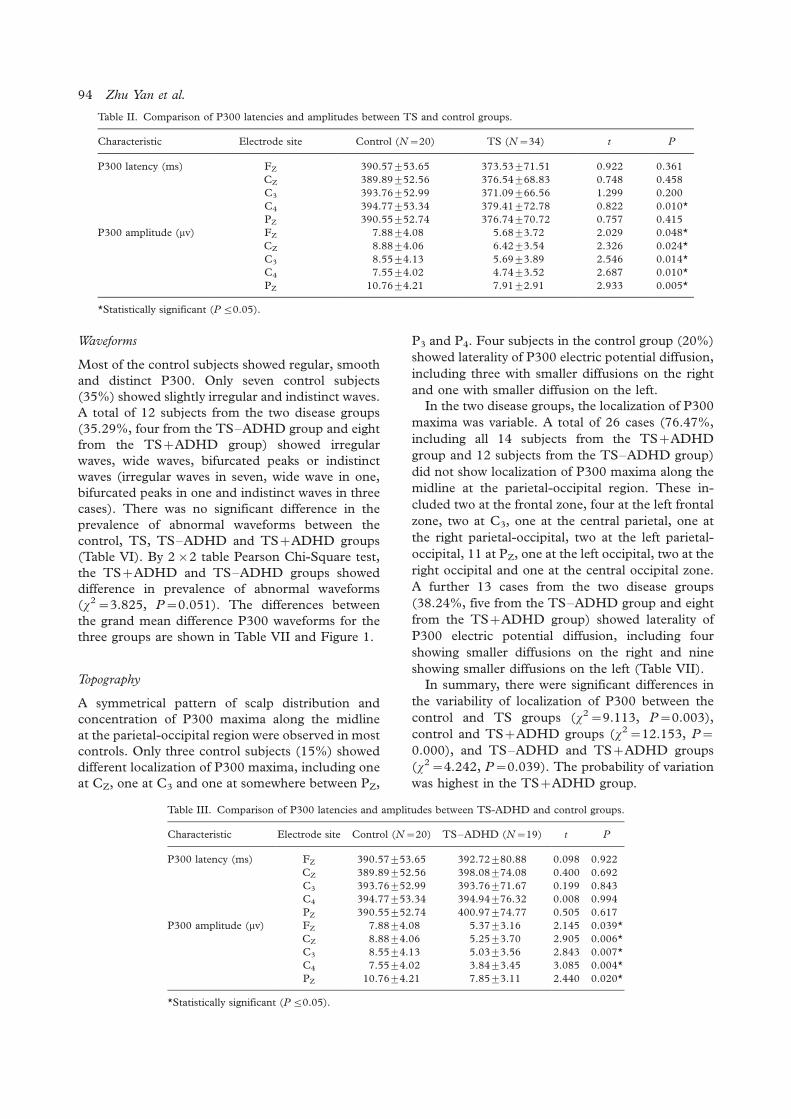
Waveforms
Most of the control subjects showed regular, smooth
and distinct P300. Only seven control subjects
(35%) showed slightly irregular and indistinct waves.
A total of 12 subjects from the two disease groups
(35.29%, four from the TS�/ADHD group and eight
from the TS�/ADHD group) showed irregular
waves, wide waves, bifurcated peaks or indistinct
waves (irregular waves in seven, wide wave in one,
bifurcated peaks in one and indistinct waves in three
cases). There was no significant difference in the
prevalence of abnormal waveforms between the
control, TS, TS�/ADHD and TS�/ADHD groups
(Table VI). By 2�/2 table Pearson Chi-Square test,
the TS�/ADHD and TS�/ADHD groups showed
difference in prevalence of abnormal waveforms
(x2�/3.825, P�/0.051). The differences between
the grand mean difference P300 waveforms for the
three groups are shown in Table VII and Figure 1.
Topography
A symmetrical pattern of scalp distribution and
concentration of P300 maxima along the midline
at the parietal-occipital region were observed in most
controls. Only three control subjects (15%) showed
different localization of P300 maxima, including one
at CZ, one at C3 and one at somewhere between PZ,
P3 and P4. Four subjects in the control group (20%)
showed laterality of P300 electric potential diffusion,
including three with smaller diffusions on the right
and one with smaller diffusion on the left.
In the two disease groups, the localization of P300
maxima was variable. A total of 26 cases (76.47%,
including all 14 subjects from the TS�/ADHD
group and 12 subjects from the TS�/ADHD group)
did not show localization of P300 maxima along the
midline at the parietal-occipital region. These in-
cluded two at the frontal zone, four at the left frontal
zone, two at C3, one at the central parietal, one at
the right parietal-occipital, two at the left parietal-
occipital, 11 at PZ, one at the left occipital, two at the
right occipital and one at the central occipital zone.
A further 13 cases from the two disease groups
(38.24%, five from the TS�/ADHD group and eight
from the TS�/ADHD group) showed laterality of
P300 electric potential diffusion, including four
showing smaller diffusions on the right and nine
showing smaller diffusions on the left (Table VII).
In summary, there were significant differences in
the variability of localization of P300 between the
control and TS groups (x2�/9.113, P�/0.003),
control and TS�/ADHD groups (x2�/12.153, P�/
0.000), and TS�/ADHD and TS�/ADHD groups
(x2�/4.242, P�/0.039). The probability of variation
was highest in the TS�/ADHD group.
Table II. Comparison of P300 latencies and amplitudes between TS and control groups.
Characteristic Electrode site Control (N�/20) TS (N�/34) t P
P300 latency (ms) FZ 390.579/53.65 373.539/71.51 0.922 0.361
CZ 389.899/52.56 376.549/68.83 0.748 0.458
C3 393.769/52.99 371.099/66.56 1.299 0.200
C4 394.779/53.34 379.419/72.78 0.822 0.010*
PZ 390.559/52.74 376.749/70.72 0.757 0.415
P300 amplitude (mv) FZ 7.889/4.08 5.689/3.72 2.029 0.048*
CZ 8.889/4.06 6.429/3.54 2.326 0.024*
C3 8.559/4.13 5.699/3.89 2.546 0.014*
C4 7.559/4.02 4.749/3.52 2.687 0.010*
PZ 10.769/4.21 7.919/2.91 2.933 0.005*
*Statistically significant (P 5/0.05).
Table III. Comparison of P300 latencies and amplitudes between TS-ADHD and control groups.
Characteristic Electrode site Control (N�/20) TS�/ADHD (N�/19) t P
P300 latency (ms) FZ 390.579/53.65 392.729/80.88 0.098 0.922
CZ 389.899/52.56 398.089/74.08 0.400 0.692
C3 393.769/52.99 393.769/71.67 0.199 0.843
C4 394.779/53.34 394.949/76.32 0.008 0.994
PZ 390.559/52.74 400.979/74.77 0.505 0.617
P300 amplitude (mv) FZ 7.889/4.08 5.379/3.16 2.145 0.039*
CZ 8.889/4.06 5.259/3.70 2.905 0.006*
C3 8.559/4.13 5.039/3.56 2.843 0.007*
C4 7.559/4.02 3.849/3.45 3.085 0.004*
PZ 10.769/4.21 7.859/3.11 2.440 0.020*
*Statistically significant (P 5/0.05).
94 Zhu Yan et al.

The topographic maps from individuals of each
group were superimposed to create a grand mean
group map by group average (Neuroscan). In the
control group, the P300 electric potential diffusion
contour lines formed concentric and symmetric
circles centred around the midline, and the P300
maxima were located along the midline at the
parietal-occipital region. In theTS�/ADHD and
TS�/ADHD groups, P300 spread out in a more
irregular fashion. In general, P300 in the TS�/
ADHD group tended to spread out to the left and
that of the TS�/ADHD group tended to spread out
to the right (Figures 2�/6).
Discussion
P300 evoked potential is a positive component that
appears approximately at 300 ms post-event. Also
known as cognitive potential, P300 is one of the
means to study the reception and cognitive proces-
sing of information by the human brain, an integral
part related to higher brain functions such as
attention, memory, judgment and abstract thinking.
The usual interpretation of P300 latency is that it is a
measure of stimulus classification speed. In this
context, it is important to note that P300 latency is
correlated negatively with mental function in normal
subjects, such that shorter latencies are related to
superior cognitive performance, a view that is con-
sistent with the observation that P300 latency
increases systematically as cognitive capability
decreases because of dementing illness. It is reason-
able to conclude that individual differences in P300
values obtained from a simple stimulus discrimina-
tion task provide a reliable indication of individual
variability in neuroelectric processing capability and
speed (Polich and Kok 1995; Oades et al. 1996).
Simply put, prolonged latency and decreased ampli-
tude in P300 reflect slowdown in information
processing and decreased energetic resources. Stu-
dies showed TS children had developmental delay
(e.g., Pennington and Ozonoff 1996; Palumbo et al.
1997; Bradshaw and Sheppard 2000). This refers to
a delay in the establishment of communications
between different structures rather than a delay in
maturation of the structures themselves (e.g., Polich
and Herbst 2000).
Our study showed that the TS�/ADHD children
had significantly shorted P300 latencies than the
control group, but the TS�/ADHD group did not
show this difference. The TS�/ADHD children also
had shorted P300 latencies than the TS�/ADHD at
CZ and PZ. The TS�/ADHD children showed
significantly decreased amplitudes than the control
group, but this was not seen in the TS�/ADHD
group. The TS�/ADHD children had smaller P300
Table IV. Comparison of P300 latencies and amplitudes between TS�/ADHD and control groups.
Characteristic Electrode site Control (N�/20) TS�/ADHD (N�/15) t P
P300 latency (ms) FZ 390.579/53.65 349.239/50.10 2.319 0.027*
CZ 389.899/52.56 349.259/51.80 2.227 0.029*
C3 393.769/52.99 347.449/52.60 2.567 0.015*
C4 394.779/53.34 359.749/65.23 1.748 0.090*
PZ 390.559/52.74 346.069/52.86 2.467 0.019*
P300 amplitude (mv) FZ 7.889/4.08 6.089/1.14 1.253 0.219
CZ 8.889/4.06 7.909/0.72 0.795 0.433
C3 8.559/4.13 6.549/1.10 1.411 0.168
C4 7.559/4.02 5.889/0.87 1.300 0.203
PZ 10.769/4.21 7.999/0.71 2.217 0.034*
*Statistically significant (P 5/0.05).
Table V. Comparison of P300 latencies and amplitudes between TS�/ADHD and TS-ADHD groups.
Characteristic Electrode site TS�/ADHD (N�/19) TS�/ADHD (N�/15) t P
P300 latency (ms) FZ 392.729/80.88 349.239/50.10 1.821 0.078
CZ 398.089/74.08 349.259/51.80 2.166 0.038*
C3 393.769/71.67 347.449/52.60 1.913 0.065
C4 394.949/76.32 359.749/65.23 1.422 0.165
PZ 400.979/74.77 346.069/52.86 2.406 0.022*
P300 amplitude (mv) FZ 5.379/3.16 6.089/1.14 0.546 0.589
CZ 5.259/3.70 7.909/0.72 2.305 0.028*
C3 5.039/3.56 6.549/1.10 1.125 0.269
C4 3.849/3.45 5.889/0.87 1.727 0.094
PZ 7.859/3.11 7.999/0.71 0.129 0.090
*Statistically significant (P 5/0.05).
P300 differences between TS�ADHD and TS+ADHD 95

amplitudes than the TS�/ADHD at Cz, These
results suggested that TS�/ADHD children had
faster information processing, and TS�/ADHD chil-
dren had decreased energetic resources according to
previous studies (e.g., Polich and Kok 1995). This
suggested that there may be functional differences
between the subgroups of these TS children.
Decreased P300 amplitude is often observed in
attention deficits (e.g., Posner and Petersen 1990;
Hansenne 2000). It was interesting that our study
found that TS�/ADHD children showed significantly
decreased P300 amplitude at CZ, C3, C4, PZ when
compared with controls. This results indicated there
maybe still have another unclear biology cause of
decreased amplitude, or suggested that attention
deficit was not the only cause of decreased amplitude
at least in TS children.
Cortical generators of P300 may include the
hippocampus, parahippocampal gyrus and the
amygdala, and different neurotransmitter substances
such as the cholinergic and the serotoninergic
systems and their radiations may also be involved.
Functional impairment in any of these structures
may affect P300 waveform (e.g., Posner and Peter-
sen 1990). Our study found no significant difference
in P300 waveforms between the TS and control
groups, TS�/ADHD and control groups, TS�/
ADHD and control groups and TS�/ADHD and
TS�/ADHD groups. This suggested that the TS
(including both TS�/ADHD and TS�/ADHD) only
suffer mild developmental defects or delays in
different brain structures. It is worth noting that
the TS�/ADHD group showed a tendency for more
alterations in P300 waveforms than the TS only
group, although this has not reached a statistically
significant level.
Developmental defects or functional impairment
in communications between different structures of
the brain may cause alteration in topographic
distribution of P300 potentials (e.g., Polich and
Herbst 2000). In this study, the control, TS�/
ADHD and TS�/ADHD groups showed differences
in P300 distribution in multiple regions. The P300
electric potential diffusion contour lines formed
concentric and symmetric circles centred around
the midline in the control group. However, the TS�/
ADHD and TS�/ADHD groups showed laterality of
Table VI. The probabilities of irregular P300 waveforms in control and TS groups.
Waveform Control (N�/20) TS (N�/34) TS�/ADHD (N�/19) TS�/ADHD (N�/15)
Irregular 7 12 4 8
Regular 13 22 15 7
% Irregular 35.0 35.3 21.1 53.3
Table VII. The probabilities of topography alterations of P300 in control and TS groups.
Topography Control (N�/20) TS (N�/34) TS�/ADHD (N�/19) TS�/ADHD (N�/15)
Irregular 7 26 12 14
Regular 13 8 7 1
% Irregular 35.0 76.5 63.5 100.0
Figure 1. The P300 waveforms from individuals of each group
were superimposed by group average at Fz.
Figure 2. The P300 waveforms from individuals of each group
were superimposed by group average at Pz.
96 Zhu Yan et al.
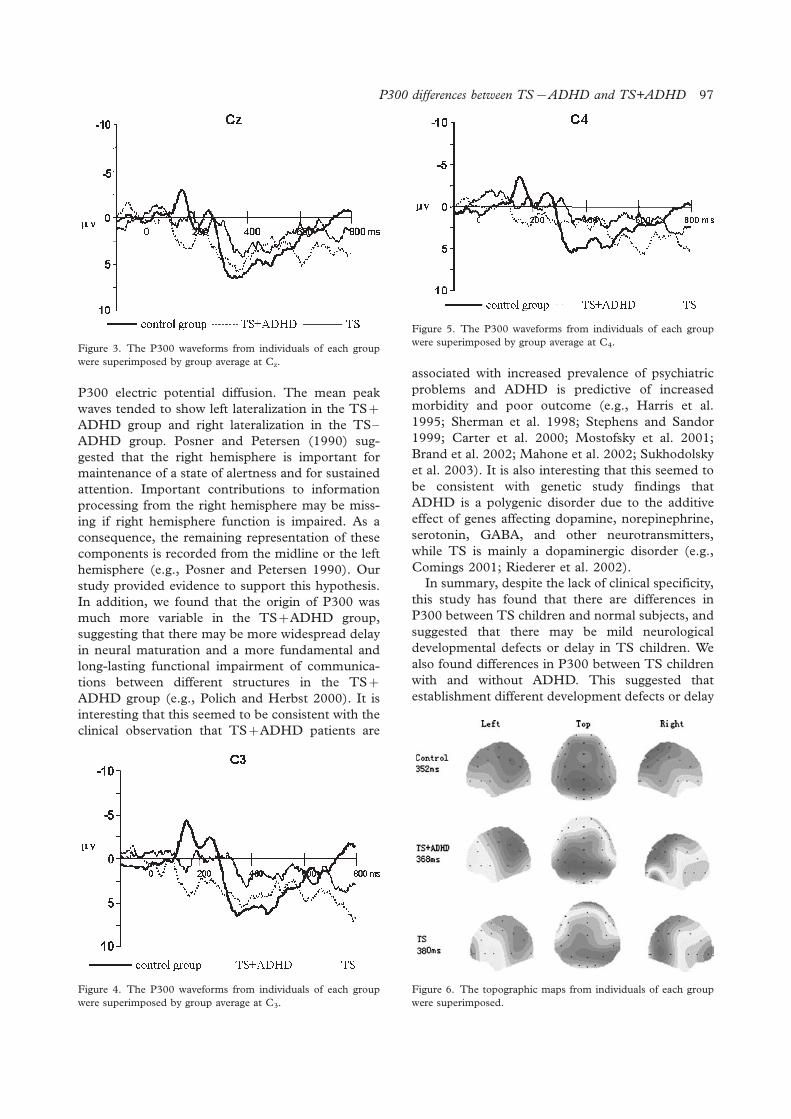
P300 electric potential diffusion. The mean peak
waves tended to show left lateralization in the TS�/
ADHD group and right lateralization in the TS�/
ADHD group. Posner and Petersen (1990) sug-
gested that the right hemisphere is important for
maintenance of a state of alertness and for sustained
attention. Important contributions to information
processing from the right hemisphere may be miss-
ing if right hemisphere function is impaired. As a
consequence, the remaining representation of these
components is recorded from the midline or the left
hemisphere (e.g., Posner and Petersen 1990). Our
study provided evidence to support this hypothesis.
In addition, we found that the origin of P300 was
much more variable in the TS�/ADHD group,
suggesting that there may be more widespread delay
in neural maturation and a more fundamental and
long-lasting functional impairment of communica-
tions between different structures in the TS�/
ADHD group (e.g., Polich and Herbst 2000). It is
interesting that this seemed to be consistent with the
clinical observation that TS�/ADHD patients are
associated with increased prevalence of psychiatric
problems and ADHD is predictive of increased
morbidity and poor outcome (e.g., Harris et al.
1995; Sherman et al. 1998; Stephens and Sandor
1999; Carter et al. 2000; Mostofsky et al. 2001;
Brand et al. 2002; Mahone et al. 2002; Sukhodolsky
et al. 2003). It is also interesting that this seemed to
be consistent with genetic study findings that
ADHD is a polygenic disorder due to the additive
effect of genes affecting dopamine, norepinephrine,
serotonin, GABA, and other neurotransmitters,
while TS is mainly a dopaminergic disorder (e.g.,
Comings 2001; Riederer et al. 2002).
In summary, despite the lack of clinical specificity,
this study has found that there are differences in
P300 between TS children and normal subjects, and
suggested that there may be mild neurological
developmental defects or delay in TS children. We
also found differences in P300 between TS children
with and without ADHD. This suggested that
establishment different development defects or delay
Figure 6. The topographic maps from individuals of each group
were superimposed.
Figure 5. The P300 waveforms from individuals of each group
were superimposed by group average at C4.Figure 3. The P300 waveforms from individuals of each group
were superimposed by group average at Cz.
Figure 4. The P300 waveforms from individuals of each group
were superimposed by group average at C3.
P300 differences between TS�ADHD and TS+ADHD 97

of communications between different structures,
rather than a delay in maturation of the structures
themselves, may be involved in TS�/ADHD and
TS�/ADHD children, and that ADHD symptoms in
TS patients are likely a trait rather than adventitious
or acquired within the TS syndrome. While altera-
tion of P300 amplitude or latency is indicative of
impaired cognitive efficiency in general, the changes
are non-specific and can be associated with a wide
range of brain disorders affect the fundamental
cognitive operations. More detailed studies of P300
neuropsychological origins are needed in future
studies.
Statement of interest
This study was supported by the National Natural
Science and Technology Fund of China (30300118).
References
Bradshaw JL, Sheppard DM. 2000. The neurodevelopmental
frontostriatal disorders: Evolutionary adaptiveness and anom-
alous lateralization. Brain Lang 73:297�/320.
Brand N, Geenen R, Oudenhoven M, Lindenborn B, van der Ree
A, Cohen-Kettenis P, Buitelaar JK. 2002. Brief report: Cogni-
tive functioning in children with Tourette’s syndrome with and
without comorbid ADHD. J Pediatr Psychol 27:203�/208.
Carter AS, O’Donnell DA, Schultz RT, Scahill L, Leckman JF,
Pauls DL. 2000. Social and emotional adjustment in children
affected with Gilles de la Tourette’s syndrome: Associations
with ADHD and family functioning. Attention Deficit Hyper-
activity Disorder. J Child Psychol Psychiatry 41:215�/223.
Comings DE. 2001. Clinical and molecular genetics of ADHD
and Tourette syndrome. Two related polygenic disorders. Ann
NY Acad Sci 931:50�/83.
Comings DE. 1995. Tourette’s syndrome: A behavioral spectrum
disorder. Adv Neurol 65:293�/303.
Drake ME Jr, Hietter SA, Padamadan H, Bogner JE, Andrews
JM, Weate S. 1992. Auditory evoked potentials in Gilles de la
Tourette syndrome. Clin Electroencephalogr 23:19�/23.
Faridi K, Suchowersky O. 2003. Gilles de la Tourette’s syndrome.
Can J Neurol Sci 30(Suppl 1):S64�/71.
Fowler B, Mitchell I. 1997. Biological determinants of P300: the
effects of a barbiturate on latency and amplitude. Biol Psychol
46:113�/124.
Harris EL, Schuerholz LJ, Singer HS, Reader MJ, Brown JE, Cox
C, Mohr J, Chase GA, Denckla MB. 1995. Executive function
in children with Tourette syndrome and/or attention deficit
hyperactivity disorder. J Int Neuropsychol Soc 1:511�/516.
Hansenne M. 2000. The p300 cognitive event-related potential. I.
Theoretical and psychobiologic perspectives. Neurophysiol
Clin 30:191�/210.
Huang xinhua, Liu chusheng. 1999. The relation of Auditory
evoked potentials and behaviors in Gilles de la Tourette
syndrome. Chin J Pract Pediatr 14:553�/554.
Leckman JF. 2003. Phenomenology of tics and natural history of
tic disorders. Brain Dev 25(Suppl 1):S24�/28.
Leckman JF, Peterson BS, George M. 1997. Pathogenesis of
Tourette’s syndrome. J Child Psychol Psychiatry 38:119�/142.
Leckman J F. 2002. Tourette’s syndrome. Lancet 360:1577�/
1586.
Mahone EM, Cirino PT, Cutting LE, Cerrone PM, Hagelthorn
KM, Hiemenz JR, Singer HS, Denckla MB. 2002. Validity of
the behavior rating inventory of executive function in children
with ADHD and/or Tourette syndrome. Arch Clin Neuropsy-
chol 17:643�/662.
Mostofsky SH, Lasker AG, Singer HS, Denckla MB, Zee DS.
2001. Oculomotor abnormalities in boys with tourette syn-
drome with and without ADHD. J Am Acad Child Adolesc
Psychiatry 40:1464�/1472.
Oades RD, Dittmann-Balcar A, Schepker R, Eggers C, Zerbin D.
1996. Auditory event-related potentials (ERPs) and mismatch
negativity (MMN) in healthy children and those with attention-
deficit or tourette/tic symptom. Biol Psychol 43:163�/185.
Palumbo D, Maughan A, Kurlan R. 1997. Hypothesis III.
Tourette syndrome is only one of several causes of a develop-
mental basal ganglia syndrome. Arch Neurol 54:475�/483.
Polich J, Kok A. 1995. Cognitive and biological determinants of
P300: An integrative review. Biol Psychol 41:103�/146.
Posner MI, Petersen SE. 1990. The attention system of the
human brain. Annu Rev Neurosci 13:25�/42.
Polich J, Herbst KL. 2000. P300 as a clinical assay: Rationale,
evaluation, and findings. Int J Psychophysiol 38:3�/19.
Pennington BF, Ozonoff S. 1996. Executive functions and
developmental psychopathology. J Child Psychol Psychiatry
37:51�/87.
Riederer F, Stamenkovic M, Schindler SD, Kasper S. 2002.
Tourette’s syndrome: A review. Nervenarzt 73:805�/819.
Sandor P. 1995. Clinical management of Tourette’s syndrome and
associated disorders. Can J Psychiatry 40:577�/583.
Scahill L, Chappell PB, King RA, Leckman JF. 2000. Pharma-
cologic treatment of tic disorders. Child Adolesc Psychiatr Clin
N Am 9:99�/117.
Sherman EM, Shepard L, Joschko M, Freeman RD. 1998.
Sustained attention and impulsivity in children with Tourette
syndrome: Comorbidity and confounds. J Clin Exp Neuropsy-
chol 20:644�/657.
Stephens RJ, Sandor P. 1999. Aggressive behaviour in children
with Tourette syndrome and comorbid attention-deficit hyper-
activity disorder and obsessive-compulsive disorder. Can J
Psychiatry 44:1036�/1042.
Sukhodolsky DG, Scahill L, Zhang H, Peterson BS, King RA,
Lombroso PJ, Katsovich L, Findley D, Leckman JF. 2003.
Disruptive behavior in children with Tourette’s syndrome:
Association with ADHD comorbidity, tic severity, and func-
tional impairment. J Am Acad Child Adolesc Psychiatry
42:98�/105.
Straube A, Mennicken JB, Riedel M, Eggert T, Muller N. 1997.
Saccades in Gilles de la Tourette’s syndrome. Mov Disord
12:536�/546.
Walkup JT, Scahill LD, Riddle MA. 1995. Disruptive behavior,
hyperactivity, and learning disabilities in children with Tour-
ette’s syndrome. Adv Neurol 65:259�/272.
98 Zhu Yan et al.

SUMMARY OF ORIGINAL RESEARCH
Three-dimensional models of neurotransmitter transporters and theirinteractions with cocaine and S-citalopram
AINA WESTRHEIM RAVNA
Department of Pharmacology, Institute of Medical Biology, University of Tromsø, Tromsø, Norway
AbstractDrugs that act on the human serotonin transporter (hSERT), human dopamine transporter (hDAT) and humannoradrenaline transporter (hNET) are important in antidepressant treatment and well known in drug abuse. Theinvestigation of their molecular mechanisms of action is very useful for designing new ligands with a therapeutic potential.The detailed three-dimensional molecular structure of any monoamine transporter is not known, but the three-dimensionalelectron density projection map of Escherichia coli Na�/H� antiporter (NhaA) has provided structural basis forconstructing models of such transporters using molecular modelling techniques. Three-dimensional models of thesedrug targets give insight into their structure, mechanisms and drug interactions. In these molecular modelling studies, anEscherichia coli NhaA model was first constructed based on its three-dimensional electron density projection map andexperimental studies on NhaA and the Escherichia coli lactose permease symporter (Lac permease). Then three-dimensionalmodels of the neurotransmitter transporters hDAT, hSERT and hNET were constructed based on the NhaA model andstudies of ligand binding to mutated dopamine transporter (DAT) and serotonin transporter (SERT). The structuralproperties of these neurotransmitter transporter models have been examined, and their interactions with cocaine and S -citalopram have been investigated.
Key words: Three-dimensional structure, molecular modelling, S-citalopram, cocaine, secondary transporter
Introduction
The science of pharmacology seeks to identify
interactions of drugs with various drug targets, and
to understand the biochemical and physiological
effects of drugs. The molecular interactions between
drugs and their targets in the human body can be
investigated using molecular modelling techniques.
The interactions seen in drug�/target complexes can
lead to understanding of the intermolecular forces
determining the specificity and the potency of the
drug. Such knowledge is important for structure-
aided drug design, where novel drugs are designed
based on information from three-dimensional mod-
els of drug target proteins.
Advances in crystallography, providing experi-
mental three-dimensional structures, have increased
relevant structural information for making molecular
models of drug targets. A model of a drug target with
an unknown three-dimensional structure may also
be constructed based on the X-ray structure of a
homologous protein by homology modelling.
Homology between two proteins indicates the pre-
sence of similar features because of a common
ancestor. Sequence similarity of the functional parts
of two proteins indicates that these two proteins have
homologous fold, but since their amino acid com-
position in the binding site area may differ from each
other, two homologous proteins may bind different
drugs. However, if no detailed structural informa-
tion about the drug target protein or homologous
proteins is available, information from ligand bind-
ing studies and indirect structural knowledge from
experimental studies may be used as a guideline for
making a molecular model of the drug target.
Most drug targets are membrane proteins. In
general, X-ray crystallography and nuclear magnetic
resonance (NMR) techniques are used for structural
determination of proteins at an atomic resolution,
but making crystals of membrane proteins is techni-
cally difficult, and molecular modelling may be used
as a step forward towards structural knowledge of
drug targets when no experimental structure is
available. However, very few three-dimensional
Correspondence: Dr Aina Westrheim Ravna, Department of Pharmacology, IMB, University of Tromsø, NO-9037 Tromsø, Norway.
Tel: +47 7764 4706. Fax: +47 7764 5310. E-mail: [email protected]
The World Journal of Biological Psychiatry, 2006; 7(2): 99�/109
(Received 21 March 2005; accepted 6 October 2005)
ISSN 1562-2975 print/ISSN 1814-1412 online # 2006 Taylor & Francis
DOI: 10.1080/15622970500402144

structures of membrane proteins have been experi-
mentally determined with a quality good enough for
traditional homology modelling.
When the transporter modelling studies described
in this paper (Ravna et al. 2001, 2003a,b) were
performed, only four high-resolution three-dimen-
sional structures of transporters had been reported
from X-ray crystallographic studies, namely the
Escherichia coli multidrug efflux transporter Acri-
flavine resistance protein B (AcrB) (Murakami et al.
2002), the Escherichia coli lipid flippase Lipid A
export ATP-binding/permease protein (MsbA)
(Chang and Roth 2001), the Escherichia coli vitamin
B12 transporter BtuCD (Locher et al. 2002), and
the calcium ATPase SERCA1a, which is localised in
skeletal muscle sarcoplasmatic reticulum
(Toyoshima et al. 2000). However, none of these
transporters were regarded as suitable templates for
modelling neurotransmitter transporters. When our
papers describing the neurotransmitter transporter
modelling were in press (Ravna et al. 2003a,b), a
breakthrough happened regarding secondary trans-
porter structures. X-ray structures of two different
secondary transporters were published; the Escher-
ichia coli lactose permease symporter (Lac per-
mease) (Abramson et al. 2003) and the Escherichia
coli glycerol-3-phosphate transporter (GlpT)
(Huang et al. 2003). When the molecular modelling
studies (Ravna et al. 2001, 2003a,b) described in
this article were performed, the most detailed
structure information about secondary transporter
protein structure was the three-dimensional electron
density projection map of the Escherichia coli
NhaA, determined by electron cryo-microscopy
(Williams 2000).
Secondary transporters
Secondary transporters are ion-coupled membrane
transport proteins that include a diverse group of
proteins, like the sodium:neurotransmitter sympor-
ter family (SNF) transporters for dopamine, nora-
drenaline and serotonin, glucose transporters, the
Na�/H� antiporter (NhaA) and the Lac permease.
Secondary transporter proteins carry small mole-
cules across cell membranes against a concentration
gradient using ion gradients as energy source, thus
facilitating solute accumulation and toxin removal.
Most secondary transporters are predicted to
have 12 transmembrane alpha-helices (TMHs)
with amino and carboxy terminals localised intra-
cellularly (Figure 1) (Reizer et al. 1994; Rothman
et al. 1996; Kaback and Wu 1997; Williams 2000).
In August 2003, the X-ray structures of the
major facilitator superfamily (MFS) transporters
Lac permease (Abramson et al. 2003) and GlpT
(Huang et al. 2003) were reported, giving a more
detailed insight into the three-dimensional structure
of secondary transporters. This has provided an
experimental basis for molecular modelling of sec-
ondary transporters, including the serotonin trans-
porter (SERT), the noradrenaline transporter
(NET), and the dopamine transporter (DAT), based
on the hypothesis that the 12TMH secondary
transporters might belong to a common folding
class.
NhaA
The NhaAs have a central role in maintaining the
circulation of H� and Na� of prokaryotic and
eukaryotic cells. The transporters contribute to the
pH and Na� homeostasis of cells, and in Escherichia
coli it regulates the internal pH at the alkaline pH
range (Padan et al. 1993; Padan and Schuldiner
1994; Padan and Schuldiner 1999).
The sub-cellular location of the Escherichia coli
NhaA is in the bacterial inner membrane. The
antiporter uses the inwardly directed H� electro-
chemical gradient to expel Na� from the cytoplasm
(Taglicht et al. 1991; Padan et al. 1999), with a
stoichiometry of two H� ions per Na� ion.
The activity (Vmax) of NhaA may vary by three
orders of magnitude in the pH range between 7 and
8.5 (Taglicht et al. 1991; Padan et al. 1999). A pH
shift from 7 to 8.5 induces a conformational change
in the protein structure increasing its activity (Pinner
et al. 1995; Padan et al. 1999). High pH sensitivity is
characteristic of transporters involved in pH regula-
tion (Padan et al. 1999).
NhaA crystallises as a dimer (Williams 2000), and
biochemical studies has shown that it also may exist
as an oligomer in the membrane, although it is
unclear whether dimerisation is required for NhaA
monomer function (Gerchman et al. 2001).
SNF and neurotransmitter modulation
The sequence identity within the SNF family is 40�/
90% (Reizer et al. 1994), and the family includes the
transporter molecules for the monoamines seroto-
nin, dopamine and noradrenaline. Monoamine
transporter function and expression is regulated by
Figure 1. Proposed topology of ion-coupled transporters: 12
TMHs.
100 A. Westrheim Ravna

several kinase and phosphatase pathways including
Protein kinase C (PKC) cascades (Jess et al. 2002;
Lin et al. 2003). Experimental studies have indicated
that SERT (Chang et al. 1998; Kilic and Rudnick
2000) and DAT (Hastrup et al. 2001; Torres et al.
2003) may exist as oligomers, but it is unclear
whether oligomerisation has a functional role.
The SNF proteins act as co-transporters of
sodium and chloride ions, and of transmitter mole-
cules (Barker and Blakely 1995) (Figure 2). The
energy for the inward movement of the transmitter
molecules is provided by the sodium gradient across
the cell membrane. Thus the action of the neuro-
transmitter is regulated.
SERT and antidepressant therapy
In the central nervous system (CNS) serotonergic
neurons occur in the pons and medulla (raphe
nuclei), projecting to the cortex, limbic system,
hypothalamus and spinal cord. The serotonergic
pathways in the brain are associated with mood
and emotion, sleep and wakefulness, sensory path-
ways (including nociception), hallucinations, body
temperature, feeding behaviour and vomiting
(Barnes and Sharp 1999).
According to the monoamine theory of depres-
sion, proposed in 1965, depression results from
functionally deficient transmission of serotonin and
noradrenaline in the CNS (Schildkraut 1965). This
theory was based on the observation that antide-
pressant drugs such as tricyclic antidepressants
(TCA) and monoamine oxidase inhibitors (MAOI)
facilitate monoaminergic transmission, but other
pharmacological evidence fails to support this the-
ory. Newer and more complex theories are currently
replacing this theory, and it has been suggested that
mood disorders are associated with impairments of
structural plasticity in the CNS (Manji and Duman
2001). However, the monoamine theory of depres-
sion currently remains a good basis for understand-
ing the action of antidepressants.
SERT is a membrane protein with 630 amino
acids (Ramamoorthy et al. 1993). The class of
antidepressant drugs termed selective serotonin
reuptake inhibitors (SSRIs) elevates the concentra-
tion of serotonin at the synapse by binding to SERT
thus inhibiting presynaptic reuptake of serotonin. In
addition to their antidepressant action, SSRIs are
also prescribed for treatment of obsessive-compul-
sive disorder, panic disorder, bulimia (Stahl 1998)
and pre-menstrual dysphoric disorder (Lin and
Thompson 2001). Inhibition of serotonin reuptake
is achieved rapidly in vitro , but improvement of
mood occurs after 2�/3 weeks of antidepressant
therapy. Long-term exposure of SERT to citalopram
results in down-regulation of the SERT protein at
the expression level (Horschitz et al. 2001).
Both therapeutic drugs, such as TCA, and drugs
of abuse, such as cocaine, bind to SERT and inhibit
serotonin reuptake. Ligand binding studies have
indicated more than one binding site at SERT, and
that cocaine and citalopram bind to the same site
and inhibit serotonin reuptake competitively. Parox-
etine seems to inhibit reuptake by allosteric modula-
tion (Akunne et al. 1992; Boja et al. 1994; Hyttel
1994; Sur et al. 1998; Barker et al. 1999).
DAT- and cocaine-induced reward
There are three main dopaminergic pathways in the
brain, the nigrostriatal pathway involved in motor
control, the mesolimbic/mesocortical pathway in-
volved in emotion- and drug-induced reward sys-
tems, and the tuberohypophyseal neurons involved
in regulation of secretions from pituitary gland. The
reward system, which is linked to drug abuse, starts
at the ventral tegmental area, follows the neuron to
the nucleus accumbens, and then on to the pre-
frontal cortex. The reward system gets activated
when a person receives positive reinforcement for
certain behaviours, which can be both natural
rewards and artificial rewards such as addictive
drugs (Bardo 1998).
DAT is an integral membrane protein with 620
amino acids (Giros et al. 1992). It terminates the
action of dopamine by reuptake of the neurotrans-
mitter into presynaptic neurons, and plays a key role
in neuronal dopamine recycling (Uhl et al. 1998).
When cocaine binds to DAT, the dopamine con-
centration is elevated, resulting in a ‘‘reward’’. The
rewarding properties of cocaine are largely due to its
inhibition of DAT on terminals of mesolimbic-
mesocortical dopaminergic neurons (Uhl et al.
1998), but SERT and NET inhibition also contri-
bute to cocaine reward and cocaine aversion (Hall
et al. 2002; Uhl et al. 2002).
NET and noradrenaline
The cell bodies of noradrenergic neurons are loca-
lised in the pons and medulla. Their axons project toFigure 2. Molecular mechanism of SERT. Transport of serotonin
mediated by inward directed sodium gradient.
Neurotransmitter transporters and cocaine and S-citalopram 101

the cerebral cortex, limbic system, hypothalamus,
cerebellum and the spinal cord. The noradrenergic
neurons are believed to participate in the reward
system, mood, arousal, blood pressure and neuroen-
docrine regulation. A functional deficiency in nora-
drenergic transmission may result in depression.
Drugs that act on the noradrenergic transmission
in the CNS include cocaine, amphetamines, anti-
depressants and some antihypertensive drugs (clo-
nidine, methyldopa).
Therapeutic drugs versus drugs of abuse
Although cocaine and SSRIs share similar molecular
mechanisms of action, cocaine is a highly addictive
drug and SSRIs are therapeutic drugs prescribed for
the treatment of depression. While the purpose of
investigating the molecular interactions of SSRIs
may be to design novel antidepressants with fewer
side effects than the existing ones, the investigation
of cocaine’s molecular interactions may aid to
develop blockers of the binding of cocaine to DAT
without inhibiting the reuptake of dopamine. Such
blockers might be effective in the treatment of
cocaine addiction.
SERT, DAT and NET binding sites
The ligand binding sites of SERT, DAT and NET
probably have a core area for cocaine binding in
common, since cocaine has similar affinities for all
three transporters. Binding studies of SSRIs show
that they are from 300 to 3500 times more selective
towards SERT over NET, and generally have low
affinity for DAT (Tatsumi et al. 1997). This in-
dicates differences in their binding areas that can be
investigated by comparing putative binding modes of
cocaine and S-citalopram in molecular modelling
studies. Both ligands block transmitter reuptake
competitively, and while cocaine is non-selective,
S-citalopram is a selective SERT inhibitor (Barker
and Blakely 1995).
Molecular modelling
Physical and chemical properties of molecules and
interactions between molecules are easier to under-
stand when the molecular structures are constructed
and visualised in three-dimensional models. A tool
for investigating psychotropic drug interactions with
their targets in the CNS is molecular modelling.
Molecular modelling is the generation, manipula-
tion and representation of three-dimensional mole-
cular structures and associated physical, chemical,
biological and pharmacological properties. Molecu-
lar models may be constructed in order to examine
three-dimensional structure and function of CNS
drug targets.
General tools for molecular modelling of mem-
brane proteins are amino acid sequence alignments
and predictions of start- and endpoints of TMHs,
interactive computer graphics and calculations. Ex-
perimental data are implemented in order to gen-
erate realistic molecular models. The modelling
approach may be based on a protein homologue
with a known three-dimensional structure, or, as in
the molecular modelling studies reviewed in this
paper (Ravna et al. 2001, 2003a,b), based on
structural analysis of low-resolution structural data
and site-directed mutagenesis studies.
Molecular modelling comprises both molecular
mechanics and quantum mechanics. Molecular
mechanics is used for calculations concerning
atomic nuclei, while quantum mechanics is used
for calculations concerning electronic system. In
molecular mechanics the atomic structure of a
molecule is considered to be a collection of atomic
masses that interact with each other via harmonic
forces. Molecular mechanics calculations are per-
formed by equations based on Newton’s Classical
Mechanics. The Born�/Oppenheimer approxima-
tion, which states that atomic nuclei move much
slower than electrons, so the vibrational and rota-
tional motions of a molecule can be separated from
the electronic motion, forms the basis of using
classical mechanics for these calculations.
Molecular mechanics calculations include energy
minimisation and molecular dynamics (MD) calcu-
lations. Energy minimisation is the calculation of the
lowest energy conformation of a molecule. The
reason for calculating the low-energy molecular
conformation is based on The Laws of Thermo-
dynamics, which state that molecules seek the lowest
potential energy spontaneously. MD is the simula-
tion of molecular motion during a short period of
time.
In the design of hypotheses regarding the patho-
physiology of affective disorders, molecular
mechanics calculations may be used in the construc-
tion of putative neurotransmitter transporter mod-
els. Such models may be helpful for understanding
the mechanism of molecular interactions between
psychotropic drugs and their CNS targets. In addi-
tion, genetic variants of neurotransmitter transpor-
ters can be modelled, and their possible structural
differences from wild type transporters can be
investigated.
Quantum mechanics enable the calculation of the
energy of an electronic system. Thus geometries
and structures of small molecules can be predicted,
but protein molecules are too big to be solved by
the Schrodinger equation (HC�/EC). Quantum
102 A. Westrheim Ravna

mechanics is useful for calculation of electrostatic
potentials (ESP) of small molecules.
Molecular modelling and structure-aided drug
design
When information about the three-dimensional
structure of a drug and drug target, and the drug�/
target complexation, is investigated using molecular
modelling techniques, it might be possible to under-
stand the intermolecular forces determining
the specificity and potency of their interaction.
The investigation of the drug�/target interaction at
the molecular level may also give insight into the
structural changes of both the drug and the drug
target for adopting an energetically favourable com-
plex. The obtained information may be used to
predict how a designed drug will fit into the target.
Eventually, new chemical compounds with fewer
side effects may be designed to act as drugs.
Transporter modelling as a working tool for
further studies
The models presented in this paper are mainly
considered as working tools for generating hypoth-
eses and designing further experimental studies
related to secondary transporter structure and func-
tion and their ligand interactions. Site-directed
mutagenesis studies and transporter modelling are
complementary to each other in an iterating process
towards a better understanding of the structure and
function of these proteins. As illustrated in Figure 3,
the construction of these transporter models, dock-
ing of ligand molecules into their putative binding
site and identifying amino acids in the model
interacting with the ligand will aid the selection of
amino acids for further site-directed mutagenesis
studies. If the observations of ligand binding affi-
nities made in the experimental observations are in
accordance with the effects proposed by the model-
ling study, the transporter model may be partly
correct. If not, an adjustment of the model must be
performed. Thus, experimental studies based on
assumptions made from the models may be useful
for further model refinements.
Molecular modelling and falsificationism
Falsificationism, a philosophy of science introduced
by Karl Popper, has the key idea that scientific
theories are falsifiable (Chalmers 2002). Theories
are construed as speculative and tentative conjec-
tures, and the science progresses by trial and
error. According to Popper, science starts with
problems, and, as solution to the problem, falsifiable
hypotheses are proposed by scientists. The hypoth-
eses are then criticised and tested. If the result does
not support the hypothesis, the hypothesis must be
adjusted or eliminated, and if the hypothesis is
validated, it must be subject to even more stringent
criticism and testing. Figure 4 shows the transporter
modelling process viewed from a falsificationistic
point of view. The scientific problem is the three-
dimensional structure of the transporters. A mole-
cular model of the transporter is constructed as a
hypothesis. Ligands are docked into the putative
binding site of the model, and logical predictions are
deducted from the hypothesis by suggesting amino
acids that may be involved ligand binding. Then the
hypothesis is tested experimentally by making single
point mutations in the transporters and testing
ligand binding affinities.
Construction of models
NhaA
A three-dimensional molecular model of the NhaA
was constructed (Figure 5a), using interactive mo-
lecular graphics and energy calculations (Ravna et al.
2001). The model was based on a three-dimensional
Figure 3. Transporter modelling procedure.
Neurotransmitter transporters and cocaine and S-citalopram 103

electron density projection map of the ion-coupled
membrane protein Escherichia coli NhaA (Williams
2000). Information from biophysical studies deter-
mining the assignment of the 12 TMHs and site-
directed mutagenesis studies of NhaA and Lac
permease were also important for model building
(Gerchman et al. 1993; Inoue et al. 1995; Kaback
and Wu 1997; Kuroda et al. 1997; Noumi et al.
1997; Olami et al. 1997; Gerchman et al. 1999;
Rimon et al. 1998). As indicated by the model
(Figure 5b), a pore was constituted by TMHs 1, 2,
3, 4, 5, 7, 8, 11 and 12. The pore may be the ion
translocation area, and it may be possible for ligands
to inhibit transport by binding to this pore area. The
loop between TMH4 and TMH5, located in the
cytoplasm, divided the pore area into two possible
translocation areas.
The modelling study suggested that the pH-
dependent activity of NhaA might be explained by
charge changes in the intracellular loop between
TMH8 and TMH9. When pH is increased, the
positively charged amino acids within the loop may
be neutralised thus leading to conformational
changes in the transporter protein. In the model,
conformational changes in this loop were seen after
180 ps of MD simulation. Such conformational
changes may alter the positions of TMHs 4, 5 and
11 relative to each other, such that a pore area of the
transporter protein is opened (Ravna et al. 2001).
DAT, SERT and NET
Three-dimensional models of DAT (Ravna et al.
2003b), SERT and NET (Ravna et al. 2003a) were
constructed based on the three-dimensional NhaA
model (Ravna et al. 2001) and site-directed muta-
genesis data on DAT and SERT (Kitayama et al.
1992; Kitayama et al. 1996; Chen et al. 1997; Barker
et al. 1998, 1999; Mitsuhata et al. 1998; Lee et al.
1999; Lin et al. 2000). The SERT model is shown in
Figure 6.
The S-citalopram and cocaine molecules were
docked into the putative ligand binding areas of the
SERT, NET and DAT models. The docking indi-
cated that the four most important residues for
ligand interaction were the conserved amino acids
Asp-98 (TMH1), Tyr-267 (TMH4) and Tyr-289
(TMH5) in SERT (Figure 7), which correspond to
Figure 4. Transporter modelling procedure and falsificationism.
Figure 5. Energy refined NhaA model. (A) Ca traces of the entire
structure, viewed in the membrane plane. The upper side is the
periplasmic side. (B) Ca traces of the TMHs, viewed from the
periplasmic side.
104 A. Westrheim Ravna

Asp-79, Tyr-252 and Tyr-274 in DAT, and Asp-75,
Tyr-249 and Tyr-271 in NET.
The nitrile group of S-citalopram interacted with
Tyr-95 in SERT. The docking of S-citalopram into
the binding areas of NET and DAT indicated that
the nitrile group of S-citalopram interacted with a
phenylalanine instead of a tyrosine. The amino acids
corresponding to Tyr-95 in SERT are Phe-76 in
DAT and Phe-72 in NET. Thus, it was hypothesised
that Tyr-95 may play a role in S-citalopram’s
selectivity to SERT (Ravna et al. 2003a). This may
explain why S-citalopram has a 3500 times higher
affinity to SERT than to NET (Tatsumi et al. 1997).
The role of Tyr-95 in the selective binding of
S-citalopram to SERT has also been demonstrated
by mutating this residue to a phenylalanine (Barker
et al. 1998).
Validity of three-dimensional secondary
transporter models
In the modelling of three-dimensional secondary
transporters reviewed in the present paper (Ravna et
al. 2001, 2003a,b), no suitable X-ray structure was
available as a template. Therefore, there are more
uncertainties than in traditional homology modelling
with available structural templates, and the results
must also be considered with that in mind.
The helical arrangement of the NhaA model was
based on the electron density projection map of the
Escherichia coli NhaA (Williams 2000). However,
the helical arrangement (Figure 5b) could not be
determined from the projection map, and was based
on site-directed mutagenesis studies on NhaA and
site-directed biophysical studies on Lac permease
(Ravna et al. 2001). The packing arrangement of
TMHs 7, 8, 9, 10 and 11 relative to each other was
derived from Lac permease studies (Kaback and Wu
1997). Furthermore, studies of Lac permease sug-
gest that TMH5 should be adjacent to TMH8
(Kaback and Wu 1997), while studies of NhaA
indicated that TMH4 should be adjacent to TMH11
(Rimon et al. 1998). TMH12 was placed adjacent to
TMH11 due to the short loop between these TMHs,
and the localisation chosen for TMH6 accounts for
the short loop between TMH6 and TMH7 on the
cytoplasmic side where these two TMHs are tilted
towards each other. TMHs 1, 2 and 3 were placed
based on site-directed mutagenesis studies on DAT
and SERT, assuming these TMHs may be a part of a
ligand binding site. In the model, TMH1 has limited
contact with the membrane in accordance with the
hydrophilic character of this segment. TMHs 1, 2, 3,
4, 5, 7, 8, 11 and 12 form a pore area (Figure 5b)
that may be separated into two translocation path-
ways by loop 4�/5 on the cytoplasmic side.
The DAT (Ravna et al. 2003b), SERT and NET
models (Ravna et al. 2003a) were based on the
TMH arrangement of the NhaA model. In these
models the positions of TMH2 and TMH3 were
interchanged compared with the NhaA model in
order to obtain the interactions of TMH3 with
cocaine and (�/)-2b-carbomethoxy-3b-(4-fluoro-
phenyl)tropane (CFT), as suggested by site-directed
mutagenesis data (Chen et al. 1997; Lee et al. 2000).
The positions of TMH2 and TMH3 were the most
uncertain parts of the helix arrangement of the
NhaA model.
There are several uncertainties in the TMH arra-
ngement suggested the models in these modelling
Figure 6. Energy refined SERT model (Ca traces) viewed in the
membrane plane. The upper side is the extracellular side.
Figure 7. S -Citalopram docked into SERT. Amino acids impor-
tant for ligand binding are displayed in the figure (Asp-98
(TMH1), Tyr-267 (TMH4) and Tyr-289 (TMH5)).
Neurotransmitter transporters and cocaine and S-citalopram 105

studies (Ravna et al. 2001, 2003a,b). It remains to
be firmly proven that the proteins within the group
of secondary transporters share a common folding
motif. Even though their functional mechanism
using an ion gradient as energy source for transloca-
tion of molecules across membranes is common
within the group, some of the transporters, like
NhaA, are antiporters, while others, like Lac per-
mease, DAT and SERT, are symporters. The type of
ions and substrates also differs among these trans-
porters. The low sequence identity within the
transmembrane regions between NhaA and the
SNF proteins SERT, DAT and NET, which is 8�/
11%, may argue against using NhaA as a template
for reliable comparative modelling. However, the use
of bacteriorhodopsin as a template in early molecular
models of G protein-coupled receptors (GPCRs)
(Dahl et al. 1991; Hibert et al. 1991), where the
sequence identity within the transmembrane regions
is 6�/11% (Hibert et al. 1991), was later confirmed
by the X-ray structure of the GPCR rhodopsin
(Palczewski et al. 2000) to be a reasonable approach
for the initial models of GPCRs. Despite the low
sequence identity, bacteriorhodopsin and GPCRs
share a similar overall three-dimensional folding
pattern with seven TMHs.
Several X-ray structures of 12 TMH transporters
were not regarded as suitable templates for DAT,
mainly due to differing functional mechanisms. The
X-ray structure of the Escherichia coli MsbA, which
belongs to the multidrug resistance ATP-binding
cassette (ABC) transporters (Chang and Roth
2001), was not considered as a suitable template
for the present modelling due to relative large
differences in functional mechanisms between the
SNF transporters and the MsbA. The crystal
structure of the bacterial multidrug efflux transpor-
ter AcrB (Murakami et al. 2002) was also considered
as not suitable as a template since the TMH
arrangement of AcrB does not fit with site-directed
mutagenesis data on DAT and SERT (Ravna et al.
2003b). These data suggest that TMH1 and TMH7
are involved in ligand binding, and this does not
seem possible with the AcrB helical arrangement.
AcrB is organised as a homotrimer, an organisation
that has not been suggested by experimental studies
for DAT and SERT. Furthermore, the AcrB protein
is part of an efflux system where it interacts with
other proteins as part of its mechanism, and has a
broad substrate specificity. The Escherichia coli
vitamin B12 transporter BtuCD (Locher et al.
2002) and the calcium ATPase SERCA1a
(Toyoshima et al. 2000) X-ray structures were not
considered as suitable templates due to the fact that
these transporters have 10 TMHs per domain.
Apparently, the NhaA electron density projection
map (Williams 2000) may have provided the best
available structural basis for molecular modelling of
SNF transporter proteins.
The TMH arrangements of the models (Ravna
et al. 2001, 2003a,b) were guided by site-directed
mutagenesis studies. The interpretation of site-
directed mutagenesis studies is, in general, not
unambiguous. The observed effects of mutations
on binding may be due to direct disruption of side
chain�/ligand interactions, or it may be due to other
structural changes in the protein affecting the bind-
ing site and the ligand binding indirectly. The
present models are based on indirect structural
information, and there are always possibilities of
incorrect interpretation of the data, both by mod-
ellers and those carrying out the experimental
studies. Description of experimental conditions
should be accurate in order to avoid misinterpreta-
tion of data. Clearly, the interpretation of literature
data may induce considerable levels of uncertainty of
the models.
Even though there is accordance between site-
directed mutagenesis data on cocaine binding to
mutated SERT and DAT and residues involved in
the binding site of the models, the overall architec-
tures of the models are uncertain. Focus was kept on
qualitative information derived from the models
rather than quantitative information such as ligand
binding energy. Lipid bilayer molecules and water
molecules were not included in the models due to
their relatively low level of accuracy.
Comparison with Lac permease X-ray
structures
In August 2003, the X-ray structures of the MFS
transporters Lac permease (Abramson et al. 2003)
and GlpT (Huang et al. 2003) were published. This
was a major contribution to knowledge of the
structure of secondary transporters, and it provided
a possibility to validate the hypothesis in these
modelling studies (Ravna et al. 2001, 2003a,b)
regarding transporter structure and function. As
shown in Figure 8, there is a striking resemblance
between the membrane spanning domains of the Lac
permease X-ray structure (Abramson et al. 2003)
and the NhaA model (Ravna et al. 2001; Dahl et al.
2004). This demonstrates that molecular modelling
studies such as those presented in this review may
give a reasonably good model of a membrane
protein, provided sufficient biochemical data are
available in addition to a low-resolution structure.
The main common features between the Lac
permease X-ray structure (Abramson et al. 2003)
and the NhaA model (Ravna et al. 2001) are the
architecture of TMHs 1�/6, and the putative binding
106 A. Westrheim Ravna

site consisting of TMHs 1, 2, 3, 4, 5, 7, 8 and 11.
The main discrepancies are the localisations of
TMH10 and TMH12. According to the Lac per-
mease X-ray structure, it seems that TMH10 is
probably involved in ligand binding, and that
TMH12 is not lining the translocation pathway.
Some differences between the two transporter struc-
tures should also be expected due to amino acid
sequence differences.
In spite of lacking a detailed template structure in
the molecular modelling studies presented here
(Ravna et al. 2001, 2003a,b), the similarities be-
tween the theoretical models and the X-ray struc-
tures indicate that these models can give a prediction
of ligand binding mechanisms. As a test for the
validity of our previous transporter models, the Lac
permease X-ray structure has been used as a
template to construct updated models of the human
serotonin transporter (hSERT), the human dopa-
mine transporter (hDAT) and the human noradre-
naline transporter (hNET) (Ravna et al., 2006).
However, it still remains to be proven whether
secondary transporters share a common architecture
Conclusions
Three-dimensional models of the neurotransmitter
transporters hDAT, hSERT and hNET (Ravna et al.
2003a,b) have been constructed based on the NhaA
model (Ravna et al. 2001) and studies of ligand
binding to mutated DAT and SERT. In lack of any
suitable X-ray structures when these molecular
modelling studies were performed, the NhaA elec-
tron density projection map (Williams 2000) was
used as a structural basis. The recent X-ray structure
of lac permease (Abramson et al. 2003) confirms the
hypothesis regarding secondary transporter ligand
binding and substrate translocation, supporting the
modelling approach used and the electron density
projection map as a basis for the modelling. This
indicates that molecular models based on low
resolution structural information may predict ligand
binding mechanisms and give new insight into the
structure and function of transporter proteins. The
neurotransmitter transporters hDAT, hSERT and
hNET models may represent useful tools in the
discovery more efficient new drugs with fewer side
effects for use in antidepressant therapy and in
treatment of cocaine addiction.
Acknowledgements
The author would like to thank Professor Svein
G. Dahl and Professor Ingebrigt Sylte for their
excellent guidance, encouragement and constructive
criticism.
Statement of interest
The author has no conflict of interest with any
commercial or other associations in connection with
the submitted article.
References
Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata
S. 2003. Structure and mechanism of the lactose permease of
Escherichia coli . Science 301:610�/615.
Akunne HC, de Costa BR, Jacobson AE, Rice KC, Rothman RB.
1992. [3H]Cocaine labels a binding site associated with the
serotonin transporter in guinea pig brain: allosteric modulation
by paroxetine. Neurochem Res 17:1275�/1283.
Bardo MT. 1998. Neuropharmacological mechanisms of drug
reward: beyond dopamine in the nucleus accumbens. Crit Rev
Neurobiol 12:37�/67.
Barker EL, Blakely RD. 1995. Norepinephrine and serotonin
transporters: molecular targets of antidepressant drugs. In:
Bloom FE, Kupfer DJ, editors. Psychopharmacology: The
fourth generation of progress. New York: Raven Press. pp
321�/334.
Barker EL, Moore KR, Rakhshan F, Blakely RD. 1999. Trans-
membrane domain I contributes to the permeation pathway for
serotonin and ions in the serotonin transporter. J Neurosci
19:4705�/4717.
Barker EL, Perlman MA, Adkins EM, Houlihan WJ, Pristupa ZB,
Niznik HB, Blakely RD. 1998. High affinity recognition of
serotonin transporter antagonists defined by species-scanning
mutagenesis. An aromatic residue in transmembrane domain I
dictates species-selective recognition of citalopram and mazin-
dol. J Biol Chem 273:19459�/19468.
Barnes NM, Sharp T. 1999. A review of central 5-HT receptors
and their function. Neuropharmacology 38:1083�/1152.
Boja JW, Vaughan R, Patel A, Shaya EK, Kuhar MJ. 1994. The
dopamine transporter. In: Niznik HB, editor. Dopamine
receptors and transporters. New York: Marcel Dekker. pp
611�/644.
Chalmers AF. 2002. What is this thing called science?. Bucking-
ham: Open University Press.
3
11
1
4
5
2
9
10
86
7
12
3
11
1
4
5
2
9
10
86
7
12
3
11
1
4
5
2
9
10
86
7
12
Figure 8. Proposed NhaA TMH localisation (Theoretical model)
(Ravna et al. 2001) (yellow filling) and Lac permease TMH
localisation (X-ray structure) (Abramson et al. 2003) (red lines),
viewed perpendicular to the membrane plane.
Neurotransmitter transporters and cocaine and S-citalopram 107

Chang AS, Starnes DM, Chang SM. 1998. Possible existence of
quaternary structure in the high-affinity serotonin transport
complex. Biochem Biophys Res Commun 249:416�/421.
Chang G, Roth CB. 2001. Structure of MsbA from E. coli : A
homolog of the multidrug resistance ATP binding cassette
(ABC) transporters. Science 293:1793�/1800.
Chen JG, Sachpatzidis A, Rudnick G. 1997. The third transmem-
brane domain of the serotonin transporter contains residues
associated with substrate and cocaine binding. J Biol Chem
272:28321�/28327.
Dahl SG, Edvardsen O, Sylte I. 1991. Molecular dynamics of
dopamine at the D2 receptor. Proc Natl Acad Sci USA
88:8111�/8115.
Dahl SG, Sylte I, Ravna AW. 2004. Structures and models of
transporter proteins. J Pharmacol Exp Ther 309:853�/860.
Gerchman Y, Olami Y, Rimon A, Taglicht D, Schuldiner S, Padan
E. 1993. Histidine-226 is part of the pH sensor of NhaA, a
Na�/H� antiporter in Escherichia coli . Proc Natl Acad Sci
USA 90:1212�/1216.
Gerchman Y, Rimon A, Padan E. 1999. A pH-dependent
conformational change of NhaA Na(�/)/H(�/) antiporter of
Escherichia coli involves loop VIII-IX, plays a role in the pH
response of the protein, and is maintained by the pure protein
in dodecyl maltoside. J Biol Chem 274:24617�/24624.
Gerchman Y, Rimon A, Venturi M, Padan E. 2001. Oligomeriza-
tion of NhaA, the Na�//H�/ antiporter of Escherichia coli in the
membrane and its functional and structural consequences.
Biochemistry 40:3403�/3412.
Giros B, el Mestikawy S, Godinot N, Zheng K, Han H, Yang-
Feng T, Caron MG. 1992. Cloning, pharmacological char-
acterization, and chromosome assignment of the human
dopamine transporter. Mol Pharmacol 42:383�/390.
Hall FS, Li XF, Sora I, Xu F, Caron M, Lesch KP, Murphy DL,
Uhl GR. 2002. Cocaine mechanisms: enhanced cocaine,
fluoxetine and nisoxetine place preferences following mono-
amine transporter deletions. Neuroscience 115:153�/161.
Hastrup H, Karlin A, Javitch JA. 2001. Symmetrical dimer of
the human dopamine transporter revealed by cross-linking
Cys-306 at the extracellular end of the sixth trans-
membrane segment. Proc Natl Acad Sci USA 98:10055�/
10060.
Hibert MF, Trumpp-Kallmeyer S, Bruinvels A, Hoflack J. 1991.
Three-dimensional models of neurotransmitter G-binding
protein-coupled receptors. Mol Pharmacol 40:8�/15.
Horschitz S, Hummerich R, Schloss P. 2001. Structure, function
and regulation of the 5-hydroxytryptamine (serotonin) trans-
porter. Biochem Soc Trans 29:728�/732.
Huang Y, Lemieux MJ, Song J, Auer M, Wang DN. 2003.
Structure and mechanism of the glycerol-3-phosphate trans-
porter from Escherichia coli . Science 301:616�/620.
Hyttel J. 1994. Pharmacological characterization of selective
serotonin reuptake inhibitors (SSRIs). Int Clin Psychopharma-
col 9(Suppl 1):19�/26.
Inoue H, Noumi T, Tsuchiya T, Kanazawa H. 1995. Essential
aspartic acid residues, Asp-133, Asp-163 and Asp-164, in the
transmembrane helices of a Na�/H� antiporter (NhaA) from
Escherichia coli . FEBS Lett 363:264�/268.
Jess U, El Far O, Kirsch J, Betz H. 2002. Interaction of the C-
terminal region of the rat serotonin transporter with Mac-
MARCKS modulates 5-HTuptake regulation by protein kinase
C. Biochem Biophys Res Commun 294:272�/279.
Kaback HR, Wu J. 1997. From membrane to molecule to the
third amino acid from the left with a membrane transport
protein. Q Rev Biophys 30:333�/364.
Kilic F, Rudnick G. 2000. Oligomerization of serotonin transpor-
ter and its functional consequences. Proc Natl Acad Sci USA
97:3106�/3111.
Kitayama S, Morita K, Dohi T, Wang JB, Davis SC, Uhl GR.
1996. Dissection of dopamine and cocaine binding sites on the
rat dopamine transporter expressed in COS cells. Ann NY
Acad Sci 801:388�/393.
Kitayama S, Shimada S, Xu H, Markham L, Donovan DM, Uhl
GR. 1992. Dopamine transporter site-directed mutations
differentially alter substrate transport and cocaine binding.
Proc Natl Acad Sci USA 89:7782�/7785.
Kuroda T, Shimamoto T, Mizushima T, Tsuchiya T. 1997.
Mutational analysis of amiloride sensitivity of the NhaA Na�/
H� antiporter from Vibrio parahaemolyticus . J Bacteriol
179:7600�/7602.
Lee SH, Chang MY, Lee KH, Park BS, Lee YS, Chin HR. 2000.
Importance of valine at position 152 for the substrate transport
and 2beta-carbomethoxy-3beta-(4-fluorophenyl)tropane bind-
ing of dopamine transporter. Mol Pharmacol 57:883�/889.
Lin J, Thompson DS. 2001. Treating premenstrual dysphoric
disorder using serotonin agents. J Womens Health Gend Based
Med 10:745�/750.
Lin Z, Wang W, Kopajtic T, Revay RS, Uhl GR. 1999. Dopamine
transporter: transmembrane phenylalanine mutations can se-
lectively influence dopamine uptake and cocaine analog recog-
nition. Mol Pharmacol 56:434�/447.
Lin Z, Zhang PW, Zhu X, Melgari JM, Huff R, Spieldoch RL, Uhl
GR. 2003. Phosphatidylinositol 3-kinase, protein kinase C, and
MEK1/2 kinase regulation of dopamine transporters (DAT)
require N-terminal DAT phosphoacceptor sites. J Biol Chem
278:20162�/20170.
Locher KP, Lee AT, Rees DC. 2002. The E. coli BtuCD
structure: a framework for ABC transporter architecture and
mechanism. Science 296:1091�/1098.
Manji HK, Duman RS. 2001. Impairments of neuroplasticity and
cellular resilience in severe mood disorders: implications for the
development of novel therapeutics. Psychopharmacol Bull
35:5�/49.
Mitsuhata C, Kitayama S, Morita K, Vandenbergh D, Uhl GR,
Dohi T. 1998. Tyrosine-533 of rat dopamine transporter:
involvement in interactions with 1-methyl-4-phenylpyridinium
and cocaine. Brain Res Mol Brain Res 56:84�/88.
Murakami S, Nakashima R, Yamashita E, Yamaguchi A. 2002.
Crystal structure of bacterial multidrug efflux transporter
AcrB. Nature 419:587�/593.
Noumi T, Inoue H, Sakurai T, Tsuchiya T, Kanazawa H. 1997.
Identification and characterization of functional residues in a
Na�/H� antiporter (NhaA) from Escherichia coli by random
mutagenesis. J Biochem (Tokyo) 121:661�/670.
Olami Y, Rimon A, Gerchman Y, Rothman A, Padan E. 1997.
Histidine 225, a residue of the NhaA-Na�/H� antiporter of
Escherichia coli is exposed and faces the cell exterior. J Biol
Chem 272:1761�/1768.
Padan E, Gerchman Y, Rimon A, Rothman A, Dover N, Carmel-
Harel O. 1999. The molecular mechanism of regulation of the
NhaA Na�/H� antiporter of Escherichia coli , a key transporter
in the adaptation to Na� and H�. Novartis Found Symp
221:183�/196.
Padan E, Schuldiner S. 1993. Na�/H� antiporters, molecular
devices that couple the Na� and H� circulation in cells. J
Bioenerg Biomembr 25:647�/669.
Padan E, Schuldiner S. 1994. Molecular physiology of the Na�/
H� antiporter in Escherichia coli . J Exp Biol 196:443�/456.
Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H,
Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE,
Yamamoto M, Miyano M. 2000. Crystal structure of rhodop-
sin: A G protein-coupled receptor. Science 289:739�/745.
Pinner E, Padan E, Schuldiner S. 1995. Amiloride and harmaline
are potent inhibitors of NhaB, a Na�/H� antiporter from
Escherichia coli . FEBS Lett 365:18�/22.
108 A. Westrheim Ravna

Ramamoorthy S, Bauman AL, Moore KR, Han H, Yang-Feng T,
Chang AS, Ganapathy V, Blakely RD. 1993. Antidepressant-
and cocaine-sensitive human serotonin transporter: Molecular
cloning, expression, and chromosomal localization. Proc Natl
Acad Sci USA 90:2542�/2546.
Ravna AW, Sylte I, Dahl SG. 2003a. Molecular mechanism of
citalopram and cocaine interactions with neurotransmitter
transporters. J Pharmacol Exp Ther 307:34�/41.
Ravna AW, Sylte I, Dahl SG. 2001. Molecular model of the
Echerichia coli Na�/H� antiporter NhaA. Receptors Channels
7:319�/328.
Ravna AW, Sylte I, Dahl SG. 2003b. Molecular model of the
neural dopamine transporter. J Comput Aided Mol Des
17:367�/382.
Ravna AW, Sylte I, Kristiansen K, Dahl SG. 2006. Putative drug
binding conformations of monoamine transporters. Bioorg
Med Chem 14:666�/675.
Reizer J, Reizer A, Saier MH Jr. 1994. A functional superfamily of
sodium/solute symporters. Biochim Biophys Acta 1197:133�/
166.
Rimon A, Gerchman Y, Kariv Z, Padan E. 1998. A point
mutation (G338S) and its suppressor mutations affect both
the pH response of the NhaA-Na�/H� antiporter as well as the
growth phenotype of Escherichia coli . J Biol Chem 273:26470�/
26476.
Rothman A, Padan E, Schuldiner S. 1996. Topological analysis of
NhaA, a Na�/H� antiporter from Escherichia coli . J Biol Chem
271:32288�/32292.
Schildkraut JJ. 1965. The catecholamine hypothesis of affective
disorders: A review of supporting evidence. Am J Psychiatry
122:509�/522.
Stahl SM. 1998. Mechanism of action of serotonin selective
reuptake inhibitors. Serotonin receptors and pathways mediate
therapeutic effects and side effects. J Affect Disord 51:215�/
235.
Sur C, Betz H, Schloss P. 1998. Distinct effects of imipramine on
5-hydroxytryptamine uptake mediated by the recombinant rat
serotonin transporter SERT1. J Neurochem 70:2545�/2553.
Taglicht D, Padan E, Schuldiner S. 1991. Overproduction and
purification of a functional Na�/H� antiporter coded by nhaA
(ant) from Escherichia coli . J Biol Chem 266:11289�/11294.
Tatsumi M, Groshan K, Blakely RD, Richelson E. 1997.
Pharmacological profile of antidepressants and related com-
pounds at human monoamine transporters. Eur J Pharmacol
340:249�/258.
Torres GE, Carneiro A, Seamans K, Fiorentini C, Sweeney A,
Yao WD, Caron MG. 2003. Oligomerization and trafficking of
the human dopamine transporter. Mutational analysis identi-
fies critical domains important for the functional expression of
the transporter. J Biol Chem 278:2731�/2739.
Toyoshima C, Nakasako M, Nomura H, Ogawa H. 2000. Crystal
structure of the calcium pump of sarcoplasmic reticulum at 2.6
A resolution. Nature 405:647�/655.
Uhl G, Lin Z, Metzger T, Dar DE. 1998. Dopamine transporter
mutants, small molecules, and approaches to cocaine antago-
nist/dopamine transporter disinhibitor development. Methods
Enzymol 296:456�/465.
Uhl GR, Hall FS, Sora I. 2002. Cocaine, reward, movement and
monoamine transporters. Mol Psychiatry 7:21�/26.
Williams KA. 2000. Three-dimensional structure of the ion-
coupled transport protein NhaA. Nature 403:112�/115.
Neurotransmitter transporters and cocaine and S-citalopram 109

VIEWPOINT
The problem of delusional ugliness: Is it really body dysmorphicdisorder?
LEONARDO F. FONTENELLE1, MAURO V. MENDLOWICZ2,3, JULIANA KALAF1 &
MARCIO VERSIANI1
1Anxiety and Depression Research Program, Institute of Psychiatry, Federal University of Rio de Janeiro (IPUB/UFRJ),
Rio de Janeiro, Brazil, 2Ethics Research Program, Institute of Psychiatry of the Federal University of Rio de Janeiro
(IPUB/UFRJ), Rio de Janeiro, Brazil, and 3Department of Psychiatry and Mental Health (MSM), Fluminense Federal
University, Brazil
Articles published in the Viewpoint section of this Journal may not meet the strict editorial and scientific standards that are
applied to major articles in The World Journal of Biological Psychiatry. In addition, the viewpoints expressed in these articles
do not necessarily represent those of the Editors or the Editorial Board.
AbstractCurrently, the DSM-IV allows individuals with dysmorphic delusions to be diagnosed with either delusional bodydysmorphic disorder (BDD) and delusional disorder, somatic type (DDST). However, given the growing acceptance of adimensional perspective in psychopathology, it is conceivable that future editions of the DSM may recommend theexclusion of the diagnosis of DDST whenever isolated dysmorphic delusions are present, arguing that the latter should beconsidered no more than a symptom of BDD. But is the concept of DDST condemned to extinction in favor of that ofdelusional BDD? While some studies suggest that non-delusional and delusional BDD/DDST may be indistinguishablefrom the clinical, neuroanatomical, and therapeutic perspectives, several facts support the utility of the DDST concept.Firstly, DDST is a wider construct than delusional BDD. Secondly, it is unclear whether DDST in general (as opposed todelusional BDD) belongs to the obsessive-compulsive spectrum. Thirdly, differential pharmacological response may not bean adequate criteria for blending non-delusional and delusional BDD/DDST. Fourthly, ‘‘delusional’’ BDD may not bedelusional at all. Finally, there is more about delusion than just an ‘‘extreme’’ conviction. Future studies are urgently neededin order to substantiate our judgement regarding the existence of diagnostic limits between delusional BDD and DDST withdysmorphic delusions.
Key words: Delusional disorder, body dysmorphic disorder, psychopathology
Introduction
It is beyond question that body dismorphic disorder
(BDD) is a severe mental disorder that can manifest
itself in the form of seriously life-threatening cases
that are associated with high levels of suicidality
(Phillips et al. 1993) and with severe (O’Sullivan
et al. 1999), sometimes bizarre (Fontenelle et al.
2002), self-injurious behaviors (Phillips et al. 1995).
Currently, the DSM-IV-TR (APA, 2000) allows
delusional BDD to be double-coded, i.e. individuals
with dysmorphic delusions may be diagnosed with
both BDD and delusional disorder, somatic type
(DDST). While double-coding is awkward, and has
the drawback of diagnosing the same syndrome as
two different disorders, it also reflects the possibility
that BDD’s delusional and non-delusional variants
may actually constitute the same disorder rather
than being distinct (Phillips 2004).
Given the increasing acceptance of a dimensional
perspective in psychopathology in general, it is
conceivable that future editions of the DSM may
recommend the exclusion of the diagnosis of DDST
whenever dysmorphic delusions are present, arguing
that the latter should be considered no more than a
symptom of BDD. But is there enough empirical
evidence to support such a bold diagnostic shift?
Correspondence: Dr Leonardo F. Fontenelle, Rua Lopes Trovao # 88, Apartment 1501, Bloco A, Icaraı, Niteroi, RJ, CEP: 24220-071,
Brazil. Tel: +55 21 2710 7857. Fax: +55 21 2710 5161. E-mail: [email protected]
The World Journal of Biological Psychiatry, 2006; 7(2): 110�/115
(Received 22 April 2005; accepted 18 July 2005)
ISSN 1562-2975 print/ISSN 1814-1412 online # 2006 Taylor & Francis
DOI: 10.1080/15622970500287420

Is the concept of DDST condemned to extinction
in favor of that of delusional BDD? We will
now briefly discuss arguments for and against this
suggestion:
For
1. Non-delusional and delusional BDD/DDST are
indistinguishable from the clinical, neuroanatomical, and
prognostic standpoints
In one of the most often quoted and provocative
articles on this subject, Phillips et al. (1994)
compared 48 patients with non-delusional BDD
with 52 patients with delusional BDD [or, according
to some authors, DDST]. The two groups did not
differ significantly in terms of most variables exam-
ined, including demographics, phenomenology,
course, associated features, comorbidities pattern,
and treatment response. However, delusional sub-
jects had higher total scores in the modified
Y-BOCS, suggesting that the delusional variant of
BDD (or DDST) may be a more severe form of the
non-delusional BDD. In the same vein, recent
neuroimaging studies have identified dysfunctions
in the parietal cortices in both BDD in general
(Carey et al. 2004) and DDST (Wada et al. 1999;
Ota et al. 2003; Hayashi et al. 2004), thus suggesting
that these disorders may share a common neuroa-
natomical basis.
2. Non-delusional and delusional BDD/DDST may
respond preferentially to serotonergic antidepressants
Case series reports suggest that both non-delusional
and delusional BDD/DDST in adults (Phillips 1993,
1994; Torres et al. 1996; Wada et al. 1999; Phillips
et al. 2001; Hayashi et al. 2004) and in children
and adolescents (Sondheimer 1988; Albertini and
Phillips 1999) respond favorably to monotherapy
with serotonin reuptake inhibitors (SRI).
This impression was further reinforced by the
findings of two double blind, placebo-controlled
treatment studies using SRI in BDD. Hollander
et al. (1999) randomized 29 patients with BDD in a
16-week, double-blind, crossover design study com-
paring clomipramine and desipramine. Clomipra-
mine was superior to desipramine in the acute
treatment of BDD and, most strikingly, was equally
effective regardless of whether the patients had
insight or held their dysmorphic misperception
with delusional intensity. Likewise, Phillips and co-
workers (2002) randomized 64 patients with the
DSM-IV diagnosis of BDD or its delusional variant
(DDST) in a 12-week study in which fluoxetine was
found to be significantly more effective than placebo.
Once again, BDD symptoms of delusional patients
were found to be as likely as those of non-delusional
patients to respond to an SRI, namely the fluoxetine.
3. Non-delusional and delusional BDD/DDST do not
respond favorably to treatment with antipsychotics
While some early studies suggested that low-dose
pimozide might be a quite effective approach in the
treatment of DDST (Munro and Mok 1995), a
number of recent reports [including case series
(Phillips 1991, 1994), and a controlled trial (Phillips
2005)] point toward the opposite direction, i.e. that
antipsychotics in general (including pimozide) may
be of no help in the treatment of non-delusional or
delusional BDD/DDST (Phillips 2004). For exam-
ple, in a retrospective study by Phillips (1994), only
one out of 45 trials with antipsychotics was effective
for delusional BDD/DDST. Similarly, in a recent
8-week, placebo-controlled, double-blind, parallel-
group study of pimozide augmentation of fluoxetine
in the treatment of BDD, the antipsychotic was
found to be no more effective than placebo even in
the more highly delusional patients (Phillips 2005).
In fact, early studies employing pimozide may have
had some significant bias that need to be highlighted.
DDST is a heterogeneous construct with several
putative subtypes (Riding and Munro 1975). For
example, Modell et al. (1996) speculated that
affective changes may be more frequent among
patients with dysmorphic delusions while organic
brain diseases may be more common among indivi-
duals with delusions of infestation. This is of para-
mount importance, since the two controlled studies
that have posited the effectiveness of pimozide in the
treatment of DDST have included solely patients
with delusions of infestation (Hamann and Avnstorp
1982; Ungvari and Vladar 1986). Therefore, we
believe that the issue of the efficacy of antipsychotics
as a monotherapy to treat dysmorphic delusions
(delusional BDD/DDST) is, as of yet, far from clear.
Against
We have discussed some preliminary evidence sug-
gesting that delusional ugliness may be just another
facet of BDD. Nevertheless, we feel that it is still
unclear whether this syndrome might be best de-
scribed as BDD or as DDST. In other words, and as
previously stated, is there enough evidence to sup-
port the removal of the concept of DDST in favor of
that of delusional BDD? This is an important
question to be made since in the last few years the
literature on delusional BDD has been rising sig-
nificantly in comparison to that on DDST. We will
now list a number of reasons that we believe support
the utility of the construct of DDST:
Delusional ugliness 111

1. DDST is a wider construct than delusional BDD
The concept of DDST encompasses not only in-
dividuals with dysmorphic delusions but also some
patients suffering from delire des negations [Cotard
syndrome (Berrios and Luque 1995)] and those with
delusions of infestation [Ekbom syndrome (Baker
et al. 1995)], of body odour or halitosis [olfactory
reference syndrome (Pryse-Phillips 1971)], of ab-
normal dental conditions [phantom bite syndrome
(Marbach 1978)] and of infectious diseases (Wurtz,
1998). The recurrent reporting of such cases high-
lights the descriptive utility of the concept of DDST.
2. It is unclear whether DDST belongs to the obsessive-
compulsive spectrum
It has already been suggested that the diagnosis of
BDD (and of its delusional form) should be moved
from the somatoform disorders section of the DSM-
IV to a new section of the future DSM-V that would
be entitled ‘‘obsessive-compulsive spectrum disor-
der’’ (Phillips et al. 2003). Although there have been
previous attempts to incorporate some of these
delusional entities into the so-called obsessive-com-
pulsive spectrum [for example, the olfactory refer-
ence syndrome (Stein et al. 1998)], many of these
categories seem to fit rather forcibly into this
putative spectrum (Swerdlow 2000).
3. Differential profile of pharmacological response may
not be an adequate criteria for blending non-delusional
and delusional BDD/DDST
As noted above, one of the main arguments for the
blending of the delusional BDD/DDSTand the non-
delusional BDD concepts is that both disorders
respond to SRI but not to antipsychotics (Phillips
2004). Nevertheless, it seems counterintuitive to
suggest that delusional BDD/DDST does not re-
spond to the latter class of drugs if BDD is said to
belong to the OCD spectrum (Phillips et al. 1995)
and OCD itself responds to augmentation with
typical (McDougle et al. 1994) as well as atypical
antipsychotics (McDougle et al. 2000; Bystritsky et
al. 2004; Denys et al. 2004). Denys et al. (2004), for
instance, reported a trend towards a significant
reduction in delusionality (according to the BABS
scores) after an 8-week treatment with quetiapine of
patients with OCD. In an interesting report, Grant
(2001) described the efficacy of olanzapine in the
treatment of a patient with non-delusional BDD. We
believe, therefore, that future studies should further
investigate the potential utility of antipsychotics in
the treatment of non-delusional and of delusional
BDD/DDST.
4. Delusional BDD may not be delusional at all
Although several research groups (McElroy et al.
1993; Hollander et al. 1999; Phillips et al. 1994,
2002) could not find any significant clinical and
therapeutic difference between delusional and non-
delusional forms of BDD and concluded that the
concept of BDD might therefore incorporate that of
some forms of DDST, their studies could be
criticized on the basis that they have employed
controversial concepts of ‘‘delusion’’. In the studies
by McElroy et al. (1993) and Phillips et al. (1994),
for instance, subjects were considered delusional if
they were or had ever been completely (or ‘‘100%’’)
convinced for a significant period of time that their
views of their supposed defect were accurate and
undistorted. In their fluoxetine study, Phillips et al.
(2002) employed the Brown Assessment of Beliefs
Scale (BABS), which assesses both dimensionally
and categorically the appealing construct of delusion-
ality but is nevertheless restricted to the ‘‘past
week’’. However, as pointed out by Munro (1999),
delusional disorders are fundamentally longstanding
illnesses, not episodic ones.
The definition adopted by Phillips et al. (1994)
may have inadvertently led to the inclusion in their
study not only of patients with genuine DDST
[i.e. chronically or persistently delusional patients
(Opjordsmoen 1993)] but also of individuals with
BDD with an episodic or fluctuating ‘‘delusional’’
course. These inclusion criteria might have concei-
vably attenuated putative differences between BDD
and DDST.
We acknowledge that some authors have sug-
gested that delusions in general may be more fluid
over relatively short periods of time than it has been
admitted in many classical descriptions and con-
temporary formulations. For example, based on a
1-year follow-up study with 1,136 acutely hospita-
lized psychiatric patients, Appelbaum et al. (2004)
found that only 15.1% of patients were persistently
delusional at each follow-up appointment (every
10 weeks). This study, however, included mostly
patients with schizophrenia, mood, and schizoaffec-
tive disorders. Only 3.5% of the sample comprised
‘‘other psychotic disorders’’, a subgroup that may
have included individuals with delusional disorders
(such as delusional BDD/DDST).
Using obsessive-compulsive disorder (OCD) as a
reference point, Insel and Akiskal (1986) have
suggested that a shift from an obsession to a
‘‘delusion’’ may occur when a disturbed affective
process leads to the abandonment of resistance (the
internal struggle against an obsessional urge or idea)
and loss of insight. We believe that a similar under-
standing can be applied to BDD. For instance, it is
112 L. F. Fontenelle et al.

conceivable that somatic obsessions with dys-
morphic content may evolve into ‘‘understandable’’
secondary delusions or delusion-like ideas [in
Jaspers’ terms (1963)] in patients with non-delu-
sional BDD who develop either a major depressive
episode or a brief psychotic disorder whenever they
are submitted to a significant amount of stressful life
events (Munro, 1999).
In other words, some patients with ‘‘delusional’’
BDD may be in fact suffering from ‘‘delusional-like’’
BDD. The role of disturbed affective processes in
these ‘‘delusional’’ transformations might certainly
explain why the ‘‘delusional’’ patients with BDD
described by Phillips and co-workers (2004) re-
sponded preferentially to SRI rather than to anti-
psychotics. It could also account for the fact that, as
reported by McElroy et al. (1993), patients with
delusional BDD were more likely to be female, to
have experienced suicidal ideation, and to have
received the diagnosis of major depression as com-
pared to those with non-delusional BDD.
5. There is more about delusion than just conviction
Mullen (2003) have argued the identification of
delusions should not be based only on the presence
of extreme or absolute conviction. As seen above,
reports of non-psychotic beliefs (such as those found
in BDD) eventually evolving into a full-blown
delusional system and leading to the establishment
of a diagnosis of a delusional disorder (or any other
psychotic disorder) frequently rely on changes in the
levels of conviction expressed by the subject. This
emphasis on the level of conviction has been
considered naıve by some (Cutting, 1997), since it
ignores the other elements that comprise the delu-
sion construct and which are referred to in various
commonly accepted (modern or classical) defini-
tions of delusion, including that adopted by the
DSM-IV (Mullen, 2003).
Several different features of delusions were de-
scribed in a handful of factor-analytic studies, mostly
with patients with schizophrenia, schizoaffective,
and mood disorder (Kendler et al. 1983; Garety
and Hemsley, 1987; Appelbaum et al. 1999; Oulis et
al. 2000). Conviction seems to be just one of the
basic dimensions, which also include constructs such
as extension or pervasiveness (Kendler et al. 1983;
Appelbaum et al. 1999), bizarreness (Kendler et al.
1983; Oulis et al. 2000), pressure or preoccupation
(Kendler et al. 1983; Appelbaum et al. 1999),
delusional actions (Kendler et al. 1983; Garety and
Hemsley, 1987; Appelbaum et al. 1999; Oulis et al.
2000), negative affect or emotional impact (Kendler
et al. 1983; Garety and Hemsley, 1987; Appelbaum
et al. 1999; Oulis et al. 2000) and delusional
‘‘kinematics’’ [duration and stability of the delusion]
(Oulis et al. 2000). These multiple dimensions,
though, have loaded on a quite variable number of
factors, ranging from two (Kendler et al. 1983) to six
(Oulis et al. 2000).
The developed of the BABS, which also assesses
the multidimensional construct of delusionality and
may be more suitable to be employed in patients
with BDD and OCD, is a significant advance in the
field, although its results seem to load on a single
factor closely related to the level of conviction (Eisen
et al. 1998). In a recent study using the BABS, for
example, Eisen et al. (2004) found that patients with
BDD not only had significantly poorer global insight
than patients with OCD but also displayed signifi-
cantly stronger conviction that their central (dys-
morphic) beliefs were essentially correct, were more
certain that other people’s views were less accurate
than theirs, were more confident that other people
considered that the subjects’ beliefs were true, were
more reluctant to consider the possibility that their
belief could be false, and had less insight regarding
the psychiatric cause of their belief.
Conclusions
We must admit that in many cases it may not be
possible to fully disentangle ‘‘delusional’’ BDD from
DDST from a phenomenological and cross-sectional
point of view (de Leon et al. 1989). In fact, we
believe that the available evidence is not yet suffi-
cient to draw any definitive conclusion regarding the
nosological status of ‘‘delusional’’ BDD/DDST. We
agree with Phillips (2004) in that double-coding
dysmorphic delusions with both BDD and DDST is
awkward, and has the drawback of diagnosing the
same syndrome as two different disorders. However,
this coding also reflects the possibility that delusional
BDD and DDST may actually do not constitute the
same disorder.
Two types of studies are urgently needed in order
to substantiate our judgement regarding the limits
between delusional BDD and DDST with dys-
morphic delusions. Firstly, prospective, long-term
studies of BDD may help unveil how the level of
insight of patients with BDD fluctuates along the
days, weeks and years, and which factors lead to
such variations. Secondly, additional comparisons
between the demographic, clinical, biological, and
therapeutic characteristics of patients with BDD,
with their ‘‘episodic’’ lack of insight, and those of
patients with DDST, with their chronic dysmorphic
delusions, need to be performed in order to deline-
ate precise diagnostic boundaries between these two
disorders.
Delusional ugliness 113

Statement of interest
The authors have no conflict of interest with any
commercial or other associations in connection with
the submitted article.
References
Albertini RS, Phillips KA. 1999. Thirty-three cases of body
dysmorphic disorder in children and adolescents. J Am Acad
Child Adolesc Psychiatry 38:453�/459.
American Psychiatric Association. 2000. Diagnostic and statistical
manual of mental disorders. 4th ed. Text revision. Washington,
DC: American Psychiatric Press.
Appelbaum PS, Robbins PC, Roth LH. 1999. Dimensional
approach to delusions: Comparison across types and diagnoses.
Am J Psychiatry 156:1938�/1943.
Appelbaum PS, Robbins PC, Vesselinov R. 2004. Persistence and
stability of delusions over time. Compr Psychiatry 45:317�/324.
Baker PB, Cook BL, Winokur G. 1995. Delusional infestation.
The interface of delusions and hallucinations. Psychiatr Clin
North Am 18:345�/361.
Berrios GE, Luque R. 1995. Cotard’s syndrome: Analysis of 100
cases. Acta Psychiatr Scand 91:185�/188.
Bystritsky A, Ackerman DL, Rosen RM, Vapnik T, Gorbis E,
Maidment KM, et al. 2004. Augmentation of serotonin
reuptake inhibitors in refractory obsessive-compulsive disorder
using adjunctive olanzapine: A placebo-controlled trial. J Clin
Psychiatry 65:565�/568.
Carey P, Seedat S, Warwick J, van Heerden B, Stein DJ. 2004.
SPECT imaging of body dysmorphic disorder. J Neuropsy-
chiatry Clin Neurosci 16:357�/359.
Cutting J. 1997. Principles of psychopathology. Oxford: Oxford
University Press.
de Leon J, Bott A, Simpson GM. 1989. Dysmorphophobia: Body
dysmorphic disorder or delusional disorder, somatic subtype?
Compr Psychiatry 30:457�/472.
Denys D, de Geus F, van Megen HJ, Westenberg HG. 2004. A
double-blind, randomized, placebo-controlled trial of quetia-
pine addition in patients with obsessive-compulsive disorder
refractory to serotonin reuptake inhibitors. J Clin Psychiatry
65:1040�/1048.
Eisen JL, Phillips KA, Baer L, Beer DA, Atala KD, Rasmussen
SA. 1998. The Brown Assessment of Beliefs Scale: Reliability
and validity. Am J Psychiatry 155:102�/108.
Fontenelle LF, Mendlowicz MV, Mussi TC, Marques C, Versiani
M. 2002. The man with the purple nostrils: A case of
rhinotrichotillomania secondary to body dysmorphic disorder.
Acta Psychiatr Scand 106:464�/466.
Garety PA, Hemsley DR. 1987. Characteristics of delusional
experience. Eur Arch Psychiatry Neurol Sci 236:294�/298.
Grant JE. 2001. Successful treatment of nondelusional body
dysmorphic disorder with olanzapine: A case report. J Clin
Psychiatry 62:297�/298.
Hamann K, Avnstorp C. 1982. Delusions of infestation treated by
pimozide: A double-blind crossover clinical study. Acta Der-
matol Venereol 62:55�/58.
Hayashi H, Oshino S, Ishikawa J, Kawakatsu S, Otani K. 2004.
Paroxetine treatment of delusional disorder, somatic type. Hum
Psychopharmacol 19:351�/352.
Hollander E, Allen A, Kwon J, Aronowitz B, Schmeidler J, Wong
C, et al. 1999. Clomipramine vs desipramine crossover trial in
body dysmorphic disorder: Selective efficacy of a serotonin
reuptake inhibitor in imagined ugliness. Arch Gen Psychiatry
56:1033�/1039.
Insel TR, Akiskal HS. 1986. Obsessive-compulsive disorder with
psychotic features: A phenomenologic analysis. Am J Psychia-
try 143:1527�/1533.
Jaspers K. 1963. General Psychopathology. Manchester: Man-
chester University Press.
Kendler KS, Glazer WM, Morgenstern H. 1983. Dimensions of
delusional experience. Am J Psychiatry 140:466�/469.
Marbach JJ. 1978. Phantom bite syndrome. Am J Psychiatry
135:476�/479.
McDougle CJ, Goodman WK, Leckman JF, Lee NC, Heninger
GR, Price LH. 1994. Haloperidol addition in fluvoxamine-
refractory obsessive-compulsive disorder. A double-blind, pla-
cebo-controlled study in patients with and without tics. Arch
Gen Psychiatry 51:302�/308.
McDougle CJ, Epperson CN, Pelton GH, Wasylink S, Price LH.
2000. A double-blind, placebo-controlled study of risperidone
addition in serotonin reuptake inhibitor-refractory obsessive-
compulsive disorder. Arch Gen Psychiatry 57:794�/801.
McElroy SL, Phillips KA, Keck PE Jr, Hudson JI, Pope HG Jr.
1993. Body dysmorphic disorder: Does it have a psychotic
subtype? J Clin Psychiatry 54:389�/395.
Modell S, Kurtz G, Hoff P. 1996. Dysmorphophobia: Differen-
tial-diagnostic aspects. Psychopathology 29:126�/130.
Munro A. 1999. Delusional disorder: Paranoia and related
illnesses. Cambridge: Cambridge University Press.
Munro A, Mok H. 1995. An overview of treatment in paranoia/
delusional disorder. Can J Psychiatry 40:616�/622.
Mullen R. 2003. Delusions: The continuum versus category
debate. Aust NZ J Psychiatry 37:505�/511.
Opjordsmoen S. 1993. The duration criteria of delusional
disorder in modern classification. Psychopathology 26:85�/89.
O’Sullivan RL, Phillips KA, Keuthen NJ, Wilhelm S. 1999. Near-
fatal skin picking from delusional body dysmorphic disorder
responsive to fluvoxamine. Psychosomatics 40:79�/81.
Ota M, Mizukami K, Katano T, Sato S, Takeda T, Asada T. 2003.
A case of delusional disorder, somatic type with remarkable
improvement of clinical symptoms and single photon emission
computed tomograpy findings following modified electrocon-
vulsive therapy. Prog Neuropsychopharmacol Biol Psychiatry
27:881�/884.
Oulis P, Mamounas J, Hatzimanolis J, Christodoulou GN. 2000.
Clinical dimensions of delusional beliefs: A factor-analytic
study. Psychopathology. 33:81�/85.
Phillips KA. 1991. Body dysmorphic disorder: The distress of
imagined ugliness. Am J Psychiatry 148:1138�/1149.
Phillips KA. 2004. Psychosis in body dysmorphic disorder.
J Psychiatr Res 38:63�/72.
Phillips KA. 2005. Placebo-controlled study of pimozide aug-
mentation of fluoxetine in body dysmorphic disorder. Am J
Psychiatry 162:377�/379.
Phillips KA, Taub SL. 1995. Skin picking as a symptom of body
dysmorphic disorder. Psychopharmacol Bull 31:279�/288.
Phillips KA, McElroy SL, Keck PE Jr, Pope HG Jr, Hudson JI.
1993. Body dysmorphic disorder: 30 cases of imagined ugli-
ness. Am J Psychiatry 150:302�/308.
Phillips KA, McElroy SL, Keck PE Jr, Hudson JI, Pope HG Jr.
1994. A comparison of delusional and nondelusional body
dysmorphic disorder in 100 cases. Psychopharmacol Bull
30:179�/186.
Phillips KA, Albertini RS, Siniscalchi JM, Khan A, Robinson M.
2001. Effectiveness of pharmacotherapy for body dysmor-
phic disorder: A chart-review study. J Clin Psychiatry 62:
721�/727.
Phillips KA, Albertini RS, Rasmussen SA. 2002. A randomized
placebo-controlled trial of fluoxetine in body dysmorphic
disorder. Arch Gen Psychiatry 59:381�/388.
114 L. F. Fontenelle et al.

Phillips KA, Price LH, Greenberg BD, Rasmussen SA. 2003.
Should the DSM diagnostic grouping be changed? In: Phillips
KA, First MB, Pincus HA, editors. Advancing DSM: Dilem-
mas in psychiatric diagnosis. Washington, DC: American
Psychiatric Press. pp 57�/83.
Pryse-Phillips W. 1971. An olfactory reference syndrome. Acta
Psychiatr Scand 47:484�/509.
Riding J, Munro A. 1975. Pimozide in the treatment of mono-
symptomatic hypochondriacal psychosis. Acta Psychiatr Scand
52:23�/30.
Sondheimer A. 1988. Clomipramine treatment of delusional
disorder-somatic type. J Am Acad Child Adolesc Psychiatry
27:188�/192.
Stein DJ, Le Roux L, Bouwer C, Van Heerden B. 1998. Is
olfactory reference syndrome an obsessive-compulsive spec-
trum disorder? Two cases and a discussion. J Neuropsychiatry
Clin Neurosci 10:96�/99.
Swerdlow NR. Blurry Spectrum Disorders. 2000. In: Maj M,
Sartorius N, Okasha A, Zohar J, editors. Obsessive-Com-
pulsive Disorder. Chichester: John Wiley & Sons Ltd. pp
235�/238.
Torres AR, Tiosso AM, Gambarini-Zen AM. 1996. [Um caso de
delırio de infestacao parasitaria tratado com clomipramina].
Revista ABP-APAL 18:29�/34.
Ungvari G, Vladar K. 1986. Pimozide treatment for delusion of
infestation. Act Nerv Super (Praha) 28:103�/107.
Wada T, Kawakatsu S, Komatani A, Okuyama N, Otani K. 1999.
Possible association between delusional disorder, somatic type
and reduced regional cerebral blood flow. Prog Neuropsycho-
pharmacol Biol Psychiatry 23:353�/357.
Wurtz R. 1998. Psychiatric diseases presenting as infectious
diseases. Clin Infect Dis 26:924�/932.
Delusional ugliness 115

CASE REPORT
Abdominal cutaneous nerve entrapment syndrome (ACNES) in apatient with a pain syndrome previously assumed to be of psychiatricorigin
JOHANNES THOME1 & CHRISTIAN EGELER2
1Department of Psychiatry, School of Medicine, University of Wales Swansea, Swansea, UK, and 2Department of
Anaesthesia, Morriston Hospital, Swansea, UK
AbstractA 21-year-old female patient with chronic abdominal pain was referred to a psychiatric outpatient clinic aftergastroenterological and gynaecological pathogeneses had been excluded and a treatment with an antidepressant had hadno beneficial effects. The mental state examination, however, revealed no psychopathology whatsoever. The patient wasinjected with a local anaesthetic loco dolenti which resulted in immediate pain relief. She was diagnosed with abdominalcutaneous nerve entrapment syndrome (ACNES); no psychiatric diagnosis was given.
Key words: Depression, functional, liaison psychiatry, pain, psychosomatic
Introduction
Patients with pain syndromes represent an important
group in psychiatry (Merskey 1989; Pridmore et al.
2001). Often they are referred to mental health units
when no somatic explanation of the symptomatology
can be found. In these cases, it is important to
differentiate between ‘‘real’’ pain syndromes with
somatic background and functional or psychogenic
pain syndromes, even if extensive diagnostic steps
have already been taken (Steinmuller 1979).
Case report
The 21-year-old female patient suffered from
chronic pain for over 2 years. She had presented
herself in gastroenterology and gynaecology, but
no explanation for the pain syndrome could
be found despite extensive diagnostic measures
(X-ray, ultrasound, laparoscopy, barium enema).
A treatment with an antidepressant was suggested
(SSRI); however, the patient stopped taking this
medication after a continuous treatment over a
period of 3 weeks with 20 mg fluoxetine did not
affect her condition at all. She did not exhibit any
side effects either.
When referred to our psychiatric outpatient clinic,
the patient reported to feel frustrated that all
measures so far had failed to produce a diagnosis
and an effective treatment strategy. However, there
were no depressive or any other psychiatric symp-
toms present. Also, there was no sign of any form of
neurotic, stress-related or somatoform disorder,
personality disorder and none of the criteria for
somatoform pain disorders were met. There was
no history of organic mental disorder or use of
psychoactive substances.
The patient was actually able to cope with the
demands of daily life and handled stressful life events
quite well (separation from partner, life-threatening
disease of mother). There was no relation whatso-
ever between the course of these events and the
development and progression of the pain syndrome.
As a student in health sciences, she successfully
attended all teaching sessions and enjoyed social
contacts with her peers. There was no family history
of neuropsychiatric disorders.
Interestingly, the patient mentioned that she had
perused the internet with respect to her pain
syndrome and asked whether it could be associated
with a trapped nerve.
Correspondence: Johannes Thome, MD, PhD, Professor of Psychiatry, School of Medicine, University of Wales, Swansea, Singleton Park,
Swansea SA2 8PP, UK. Tel: +44 1792 602205. Fax: +44 1792 513430. E-mail: [email protected]
The World Journal of Biological Psychiatry, 2006; 7(2): 116�/118
(Received 23 January 2005; accepted 20 June 2005)
ISSN 1562-2975 print/ISSN 1814-1412 online # 2006 Taylor & Francis
DOI: 10.1080/15622970500222377

She reported that she had been troubled with
right iliac fossa pain for over 2 years, at times even
necessitating acute admission to hospital with
exacerbation of the pain. The pain was not
associated to her menstrual cycle or to her bowel
motions but could give her quite sharp spasmodic
pain, with a dull background ache. She could
pinpoint the source of the pain deep in the right
inguinal area. On deep palpation there was a
nodule of relatively hard tissue palpable. About
3 months after she had stopped taking the
antidepressant, she agreed to have this injected
with 10 ml 0.25% marcain and 20 mg depome-
drone. She had excellent immediate relief, lasting
well over 2 months. She was diagnosed with
ACNES. About 5 months after this excellent relief
from the trigger-point injection, she found the
pain recurring, but was still considerably better
than previously. An injection with 5 ml 0.25%
marcain and 20 mg depomedrone was repeated
which, again, gave her immediate relief. Since this
course is not untypical for ACNES, this diagnosis
was maintained.
Discussion
ACNES represents a common cause of abdo-
minal pain without other clinically significant
symptoms, often misdiagnosed as irritable bowel,
spastic colon, gastritis, psychoneurosis, depression,
anxiety, hysteria and malingering (Applegate
1973; Hershfield 1992; Suleiman and Johnston
2001).
Its pathophysiology is characterised by a fibrous
ring in the rectus muscle, through which a neuro-
vascular bundle travels. This bundle can move
freely, however compression against the ring
causes nerve ischemia and the acute or chronic
symptoms of ACNES (Applegate and Buck-
walter 1997).
Clinical presentation of ACNES symptoms can
vary, but usually acute or chronic abdominal pain
is described which may depend on physical activity
or certain movements. ACNES pain usually affects
only one side and is typically well localised. On
physical examination, it is normally possible to
palpate a tender spot and underneath a muscular
foramen at the very localisation indicated by the
patient (Applegate 2002).
The injection of a local anaesthetic will immedi-
ately relieve the ACNES-related pain and also
confirm the diagnosis (Applegate 2002; Doouss
and Boas 1975). Addition of steroids to the injec-
tate prolongs pain relief considerably and reduces
tissue tension within the nerve entrapment area.
Whilst repeat injections are often required, these
tend to produce longer pain relief until the pain
virtually disappears.
With respect to the therapy success, it is
important to discuss the possibility of a so-called
placebo effect. However, given the fact that
the patient fulfilled the classical criteria for
ACNES, including a palpable nodule of hard
tissue loco typico, and since psychosocial circum-
stances were clearly unrelated to the pain symp-
toms, it can be concluded that this patient was not
suffering from a psychogenic pain syndrome,
which then was ‘‘cured’’ by the placebo effect of
an injection.
There have been several reports in the literature
about cases in which it took a considerable amount
of time until the correct diagnosis of ACNES
was made, thereby unnecessarily prolonging the
patients’ suffering (Hershfield 1992; Peleg 1999;
Suleiman and Johnston 2001). When comprehensive
diagnostic batteries remain inconclusive, patients
suffering from pain syndromes are often referred to
psychiatrists and psychotherapists, suggesting a
psychogenic or functional disorder. However, it is
important to note that allegedly psychogenic pain
syndromes may well represent somatic conditions
such as ACNES.
Psychiatrists should, thus, be aware of this
syndrome in order to avoid an unnecessary prolon-
gation of their patients’ pain syndrome and to
direct them towards the proper therapy which
will result in immediate relief and is likely to
provide a cure.
Statement of interest
The authors have no conflict of interest with any
commercial or other associations in connection
with the submitted article.
References
Applegate WV. 1973. Abdominal cutaneous nerve entrapment
syndrome. Am Fam Physician 8:132�/133.
Applegate WV. 2002. Abdominal cutaneous nerve entrap-
ment syndrome (ACNES): A commonly overlooked cause of
abdominal pain. The Permanente Journal 6:http://xnet.kp.org/
permanentejournal/sum02/acnes.html. Accessed 22 October
2004.
Applegate WV, Buckwalter NR. 1997. Microanatomy of the
structures contributing to abdominal cutaneous nerve entrap-
ment syndrome. J Am Board Fam Pract 10:329�/332.
Doouss TW, Boas RA. 1975. The abdominal cutaneous nerve
entrapment syndrome. NZ Med J 81:473�/475.
Hershfield NB. 1992. The abdominal wall. A frequently
overlooked source of abdominal pain. J Clin Gastroenterol
14:199�/202.
Merskey H. 1989. Psychiatry and chronic pain. Can J Psychiatry
34:329�/336.
Abdominal cutaneous nerve entrapment syndrome 117

Peleg R. 1999. Abdominal wall pain caused by cutaneous nerve
entrapment in an adolescent girl taking oral contraceptive pills.
J Adolesc Health 24:45�/47.
Pridmore S, Oberoi G, Harris N. 2001. Psychiatry has much to
offer for chronic pain. Aust NZ J Psychiatry 35:145�/149.
Steinmuller R. 1979. The use and abuse of psychiatry in dealing
with pain patients. Psychiatr Q 51:184�/188.
Suleiman S, Johnston DE. 2001. The abdominal wall:
an overlooked source of pain. Am Fam Physician 64:431�/
438.
118 J. Thome & C. Egeler

CASE SERIES
The treatment of recurring auditory hallucinations in schizophreniawith rTMS
PAUL B. FITZGERALD1, JESSICA BENITEZ1, JEFF Z. DASKALAKIS2, ANTHONY DE
CASTELLA1 & JAYASHRI KULKARNI1
1Alfred Psychiatry Research Centre, The Alfred and Monash University Department of Psychological Medicine, Victoria,
Australia, and 2Centre for Addiction and Mental Health, Clarke Division, Toronto, Ontario, Canada
AbstractBackground: Auditory hallucinations are a common and disabling problem for many patients with schizophrenia and oftenfail to respond to optimal antipsychotic therapy. Repetitive transcranial magnetic stimulation (rTMS) has recently beentrialled as an alternative treatment option for these patients. These studies have generally been positive, but treatment hasonly been provided for short periods of time and little is known about the longer-term impact of TMS on the course ofhallucinations. Method: We describe two cases in which rTMS was provided to patients upon relapse of hallucinationsfollowing initial successful rTMS treatment in a clinical trial. Results: A repeat course of rTMS resulted in a markedimprovement in the symptoms experienced by these two patients. Conclusions: rTMS appears to have potential as a long-term treatment for patients with auditory hallucinations, but requires ongoing systematic investigation.
Key words: Hallucinations, schizophrenia, repetitive transcranial magnetic stimulation
Introduction
Persistent auditory hallucinations (AHs) are a
common problem in patients with schizophrenia
and contribute to ongoing disability and morbidity.
Repetitive transcranial magnetic stimulation (rTMS)
has recently been developed and trialled as a
potential new treatment for patients with treatment
resistant AHs. To date rTMS has been subjected to
several small randomised controlled trials. Initially
Hoffman and co-workers applied 1 Hz rTMS over
the left temporoparietal region in two pilot studies,
followed by a randomised sham controlled double-
blind trial (Hoffman et al. 2003), all with positive
results. The double-blind trial involved 24 patients
receiving stimulation over nine treatment sessions
(Hoffman et al. 2003). The active treatment group
experienced a significant improvement in hallucina-
tion severity compared to the sham group, and there
were no significant adverse events reported. There
was a persistence of therapeutic benefit in the
months following the study in a significant propor-
tion of the patients, although none received ongoing
rTMS treatment. Although one study failed to
replicate the findings of Hoffman’s pilot study design
(McIntosh et al. 2004), there have been several
positive replications of the clinical trial work. First,
Poulet and colleagues found a beneficial effect of
stimulation with therapeutic results obtained after
only 5 days of treatment (Poulet et al. 2005).
Second, a therapeutic benefit of 1 Hz rTMS over
both the left and right auditory region was found
with 20 minutes stimulation per day in a 10-day
treatment trial (Lee et al. 2005).
Despite these replications, little is known about
the long-term impact of rTMS on the clinical
outcome of patients with persistent auditory hallu-
cinations. In particular, it is unknown whether
rTMS will have beneficial clinical effects if halluci-
nations return after a successful period of treatment.
In this paper we describe the repeated treatment
with rTMS in two patients with persistent auditory
hallucinations. These cases suggest that TMS may
have an ongoing role to play in the treatment of
patients with hallucinations over time. Both patients
gave written informed consent and this study was
Correspondence: Associate Professor Paul B. Fitzgerald, Alfred Psychiatry Research Centre, First Floor, Old Baker Building, The Alfred,
Commercial Rd Melbourne, Victoria, 3004 Australia. Tel: +61 3 9276 6552. Fax: +61 3 9276 6556. E-mail: paul.fitzgerald
@med.monash.edu.au
The World Journal of Biological Psychiatry, 2006; 7(2): 119�/122
(Received 11 August 2005; accepted 2 November 2005)
ISSN 1562-2975 print/ISSN 1814-1412 online # 2006 Taylor & Francis
DOI: 10.1080/15622970500474705

approved by the human research committee of the
Alfred Hospital. Assessments of symptom severity in
both cases were made by a single trained rater using
the Hallucination Change Scale (HCS): this was
initially described by Hoffman et al. (2003) and
consists of a single rating from 0 (non-voices) to 20
(greatest severity) of hallucination severity. At base-
line, the rating is set at 10 with each patient
providing an individual description of the severity
of his/her voices. We also used the hallucinations
item of the Positive and Negative Syndrome Scale
(PANSS) (Kay et al. 1987).
Case report
Patient 1
The first patient, GC, was a 47-year-old male with a
7-year history of schizophrenia characterised by the
persistence of severe auditory hallucinations. He had
been treated over this time with multiple typical and
atypical antipsychotic medications at full therapeutic
doses. These had resulted in an improvement in his
general clinical state but had little impact on the
presence or severity of his hallucinations. At the time
of initial rTMS he had been receiving treatment with
a combination of risperidone (4 mg/day) and aripi-
prazole (30 mg/day) for over 6 months. He described
hearing voices that were negative, derogatory and
often commanding in nature including commands to
commit suicide. The voices were experienced every
day, for many hours at a time and inside his head.
GC was initially randomised to receive placebo
rTMS and this was provided for 10 days as part of
a double-blind sham controlled trial (Fitzgerald et
al. 2005). At the end of this time there was no
change in the severity of his AHs. Immediately after
this he was enrolled in an open active phase of
treatment and received 10 days of rTMS. This was
provided for 15 minutes per day, at 1 Hz, to the TP3
EEG site in the left temporoparietal cortex at 90% of
the resting motor threshold (Fitzgerald et al. 2005).
This resulted in a dramatic reduction of his AHs
from 10 to 0 on the HCS and from 5 to 1 on the
PANSS AH item. His total PANSS score decreased
from 87 at baseline to 52 at study end.
Seven months after rTMS treatment he was re-
referred by his treating psychiatrist due to a gradual
recurrence of his AHs over the previous month.
There had been no change in medication type or
dose since the end of the original rTMS treatment
course; he continued to receive 4 mg of risperidone
and 30 mg of aripiprazole per day. He was believed
by his treating psychiatrist to have remained com-
pliant with medication throughout this period
although this had not been confirmed with plasma
levels. The symptoms had returned to a similar level
of severity to that preceding his original treatment.
GC was provided repeat treatment with rTMS
under identical treatment conditions (same site,
intensity, frequency, number of pulses and dura-
tion). There was a very similar good clinical response
with the second period of treatment. At the end of
the 10 days he reported that he had not experienced
auditory hallucinations for the duration of the
second week of treatment and there was a significant
improvement in his overall level of functioning and
mental well-being. His score on the PANSS AH item
had reduced from 5 to 1 and his score on the HCS
from 10 to 0.
Patient 2
The second patient was an 18-year-old female with
childhood onset schizophrenia beginning at the age
of 9. Over the 9 years of her illness, LM, became
increasingly treatment resistant to the point where
she continued to experience persistent symptoms,
despite treatment with multiple antipsychotic med-
ication. Twelve months prior to referral for rTMS
she commenced treatment with clozapine, which
was progressively increased to 400 mg/day. This
resulted in a significant improvement in her general
positive symptoms and level of disorganisation, but
had little impact on her AHs. LM described multiple
persistent AHs which she experienced almost con-
tinuously. She described hearing several extremely
loud voices that made constant, derogatory and
abusive comments in the second and third person.
LM was also given active rTMS as part of a double-
blind randomised trial (10 sessions given on con-
secutive weekdays, 15 minutes of 1 Hz stimulation at
90% of the resting motor threshold, to the TP3 EEG
site in the left temporoparietal cortex) (Fitzgerald et
al. 2005). This resulted in a reduction in the severity
of the AHs from a rating of 10 to a rating of 3 (HCS)
(6 to 2 on the PANSS AH item). She remained on
400 mg of clozapine throughout treatment and the
follow-up period with medication supervised both
during and after TMS by family members. One
month after the end of treatment, she rated the
severity of the voices as 1 on the HCS. The
improvement in the AHs experienced by LM lasted
approximately 5 months during which time she
remained on an unchanged dose of clozapine. After
this time they gradually returned and increased in
intensity and frequency. Six months after initial
treatment with rTMS she received a further 10
sessions applied in an identical way. She again
experienced a significant improvement in her audi-
tory hallucinations. LM described a reduction in
AHs from being continuously present to occurring
120 P. B. Fitzgerald et al.

fleetingly once every 3�/4 days (HCS 10 to 3,
PANSS AH item 6 to 2). Following this second
trial, LM’s AHs were barely present for a period of 3
months, after which they increased to the previous
level. As a third treatment trial, rTMS was provided
in an identical way, but this time over 15 treatment
sessions (3 weeks). This resulted in a further
response with an almost complete remission of the
AHs which were only experienced approximately
once per day (HCS 10 to 2, PANSS AH item 6 to 2).
Four months after this period of treatment the
patient reported that her AHs remained at the
same minimally present level.
Discussion
Persistent auditory hallucinations are a serious
clinical problem that continue to cause significant
problems for patients with schizophrenia. Repetitive
TMS shows promise in the treatment of this difficult
clinical symptom of schizophrenia. Randomised
trials to date have suggested that rTMS has acute
efficacy in the relief of persistent auditory hallucina-
tions ( Hoffman et al. 2003; Lee et al. 2005; Poulet
et al. 2005). However, little is known with regard to
the long-term impact of rTMS on the clinical course
of patients with hallucinations. Importantly, halluci-
nations are likely to be a recurring and incapacitating
clinical symptom for some patients with schizophre-
nia and it is not clear that continual treatment with
antipsychotic medication results in a continued relief
of AHs after the end of a course of rTMS treatment.
Data on the use of rTMS treatment in the acute
treatment is gradually accumulating. Over the last
year, several positive replication studies (Chibbaro et
al. 2005; Hoffman et al. 2005; Lee et al. 2005;
Poulet et al. 2005) have supported the initial trials by
Hoffman et al. (2003, 2000). However, not all of the
published reports are strongly positive. In fact, the
trial in which the two patients in this report
participated failed to show a strong positive effect
of rTMS treatment (Fitzgerald et al. 2005). In that
study there was significantly greater reduction in the
loudness of AHs reported in patients with active as
compared to sham treatment, but no overall differ-
ence in HCS severity. It is possible that minor
differences in stimulation technique could have
explained the difference in response in this trial.
Perhaps more likely, however, is that even with a
successful treatment the conduct of multiple small
treatment studies in highly treatment non-responsive
populations is likely to produce some negative trial
results. It seems quite timely for the conduct of a
substantial multi-site trial of the efficacy of acute
rTMS treatment, very much as studies of this sort
are currently underway for rTMS treatment in
depression.
The two cases that we have described provide
early evidence that rTMS may have a role in the
treatment of persistent and recurring AHs. Although
the non-blinded treatment of the patients in these
cases introduces the possibility of the influence of
confounding factors, the dramatic nature of the
improvement in symptoms from previously severe
impairment suggest that a placebo effect alone is
unlikely. In addition, there did not seem to be any
other significant factors, such as changes in medica-
tion or site of treatment to account for these effects.
Further systematic evaluation of the effects of rTMS
treatment of AHs over time is clearly warranted.
This should include not just the evaluation of the
effects of rTMS in the treatment of acute episodes
Figure 1. Hallucination change scores (HCS) and Positive and Negative Symptom Scale (PANSS) Auditory Hallucination (AH) item
scores for each subject across the treatment phases.
rTMS and hallucinations 121

but also potentially its role in the maintenance
treatment of these disabling symptoms.
Acknowledgements/Statement of interest
The study was supported by a grant from The
Stanley Medical Research Institute and by Con-
stance and Stephen Lieber through a NARSAD
Young Investigator award (PF).
References
Chibbaro G, Daniele M, Alagona G, Di Pasquale C, Cannavo M,
Rapisarda V, Bella R, Pennisi G. 2005. Repetitive transcranial
magnetic stimulation in schizophrenic patients reporting audi-
tory hallucinations. Neurosci Lett 383:54�/57.
Fitzgerald PB, Benitez J, Brown T, Marston NAU, Daskalakis ZJ,
De Castella AR, Kulkarni J. 2005. A Double�/blind sham
controlled trial of repetitive transcranial magnetic stimulation
in the treatment of refractory auditory hallucinations. J Clin
Psychopharmacol 25:358�/362.
Hoffman RE, Boutros NN, Berman RM, Roessler E, Belger A,
Krystal JH, Charney DS. 1999. Transcranial magnetic stimula-
tion of left temporoparietal cortex in three patients reporting
hallucinated ‘‘voices’’. Biol Psychiatry 46:130�/132.
Hoffman RE, Boutros NN, Hu S, Berman RM, Krystal JH,
Charney DS. 2000. Transcranial magnetic stimulation and
auditory hallucinations in schizophrenia. Lancet 355:1073�/
1075.
Hoffman RE, Hawkins KA, Gueorguieva R, Boutros NN, Rachid
F, Carroll K, Krystal JH. 2003. Transcranial magnetic stimula-
tion of left temporoparietal cortex and medication-resistant
auditory hallucinations. Arch Gen Psychiatry 60:49�/56.
Hoffman RE, Gueorguieva R, Hawkins KA, Varanko M, Boutros
NN, Wu YT, Carroll K, Krystal JH. 2005. Temporoparietal
transcranial magnetic stimulation for auditory hallucinations:
Safety, efficacy and moderators in a fifty patient sample. Biol
Psychiatry 58:97�/104.
Kay SR, Fiszbein A, Opler LA. 1987. The positive and negative
syndrome scale (PANSS) for schizophrenia. Schizophr Bull
13:261�/276.
Lee SH, Kim W, Chung YC, Jung KH, Bahk WM, Jun TY, Kim
KS, George MS, Chae JH. 2005. A double blind study showing
that two weeks of daily repetitive TMS over the left or right
temporoparietal cortex reduces symptoms in patients with
schizophrenia who are having treatment-refractory auditory
hallucinations. Neurosci Lett 376:177�/181.
McIntosh AM, Semple D, Tasker K, Harrison LK, Owens DG,
Johnstone EC, Ebmeier KP. 2004. Transcranial magnetic
stimulation for auditory hallucinations in schizophrenia. Psy-
chiatry Res 127:9�/17.
Poulet E, Brunelin J, Bediou B, Bation R, Forgeard L, Dalery J,
d’Amato T, Saoud M. 2005. Slow transcranial magnetic
stimulation can rapidly reduce resistant auditory hallucinations
in schizophrenia. Biol Psychiatry 57:188�/191.
122 P. B. Fitzgerald et al.

CASE SERIES
Aripiprazole in patients with Tourette syndrome
EMANUEL BUBL, EVGENIY PERLOV & LUDGER TEBARTZ VAN ELST
Department of Psychiatry, Albert-Ludwigs-University, Freiburg, Germany
AbstractThe treatment of the Gilles de la Tourette syndrome (GTS) is often challenging. One reason for this is the highneuropsychiatric cormorbidity in terms of ADHD or obsessive-compulsive symptoms. Dopaminergic modulation e.g. withantidopaminergic medication is an important part of the medical therapy aimed at motor and vocal tics. We report recentexperiences with treatment with aripiprazole, a novel antipsychotic agent, which not only improved motor and vocal tics butalso ameliorated some behavioural symptoms of the GTS cluster. Furthermore, we discuss possible pharmacologicalmechanisms for the observed effects.
Key words: Gilles de la Tourette syndrome (GTS), aripiprazole, ADD, dopamine
Introduction
The Gilles de la Tourette syndrome (GTS) is a
complex neuropsychiatric disorder, which is char-
acterized by the occurrence of several vocal and
motor tics at the same time (Leckman 2002). Many
patients suffer from co-morbid obsessive-compulsive
disorder (OCD) or attention-deficit hyperactivity
disorder (ADHD) (Leckman 2002). Dopaminergic
modulation, e.g., with classic neuroleptics such as
haloperidol have been an important part of therapy
(Lucas 1967; Devinsky 1983). The treatment of
GTS is particularly difficult when it is associated
with co-morbid psychiatric symptoms. Treatment of
ADHD or OCD symptoms with psychostimulants or
SSRIs can deteriorate tics, whereas treatment of
motor and vocal tics with antidopaminergic medica-
tion sometimes worsens ADHD or OCD. In recent
years atypical neuroleptics such as risperidone have
been widely discussed (Bruun and Budman 1996).
We present a report of a women and a man with
Tourette syndrome successfully treated with aripi-
prazole.
Case reports
Mrs H, a 19-year-old caucasian female with GTS
complained to her psychiatrist about classic motor
and vocal tics of the facial and upper limb muscles
and echopraxia. Furthermore, the patient herself as
well as her partner and family described a marked
impulsivity and verbal aggression which led to
serious interpersonal problems. Apart from that
Mrs H did not suffer from any other medical or
neuropsychiatric disorder, e.g., obsessive-compul-
sive disorder. There was no history of birth compli-
cations, delayed development or streptococcal
infections and no family history of any neuro-
psychiatric disorder. Treatment with haloperidol
1.5 mg did improve the tics to a small extent.
However, there were still major interpersonal pro-
blems due to her impulsive and verbally aggressive
behaviour. Because of this sertraline was started up
to 100 mg with only minor positive effects. While
haloperidol, at the mentioned dosage, was well
tolerated, a therapeutic trial with aripiprazole
15 mg was started in order to minimise the risk of
tardive dyskinesia. Haloperidol was tapered at the
same time. This intervention led to a dramatic
improvement, not only of the motor and verbal tics
but also of the impulsiveness. Due to a decrease of
effectiveness after 4 weeks, the patient herself tried
to increase the dosage up to 40 mg a day. This
dosage proved to be effective and well tolerated, with
a sustained improvement of tics as well as impulsivity
for the follow-up period of over 1 year. Therefore,
the patient was left on this high dosage following
informed consent with respect to off-label treatment.
Correspondence: Ludger Tebartz Van Elst, MD, Department of Psychiatry, Albert-Ludwigs-University, Hauptstr. 5, D-79104 Freiburg,
Germany. Tel: +49 761 270 6501. Fax: +49 761 270 6619. E-mail: [email protected]
The World Journal of Biological Psychiatry, 2006; 7(2): 123�/125
(Received 28 July 2005; accepted 9 November 2005)
ISSN 1562-2975 print/ISSN 1814-1412 online # 2006 Taylor & Francis
DOI: 10.1080/15622970500474770

Mr H, a 21-year-old caucasian male, presented in
our clinic with classical vocal and motor tics
beginning in early childhood. Following normal
blood tests, EEG, and neuroimaging, GTS was
diagnosed. Birth history and early development
were normal. There was no family history of any
neuropsychiatric disorder. Psychiatric and neurolo-
gical examinations were essentially normal apart
from the tics. The patient did not show a history
of obsessive-compulsive symptoms. Given our
earlier experience and the good tolerability of
aripiprazole, we started with this substance at
15 mg in the morning. Again, there was a sustained
good response of vocal and motor tics without
significant side effects for the present follow-up
period of about 4 months.
Discussion
These results support similar findings in a number
of case series where aripiprazole was used with a
good efficacy and tolerability in patients with GTS
(Hounie et al. 2004; Dehning et al. 2005; Kastrup
et al. 2005; Murphy et al. 2005). In addition, in
our first case, not only the tics but also the
impulsivity, a common problem in GTS, improved
significantly. Atypical as well as typical neuroleptics
have been widely used in the treatment of GTS
and often show a good response, which is probably
due to modification of dopaminergic neurotrans-
mission. Dopaminergic alterations in the pathophy-
siology of GTS have been discussed earlier
(Devinsky 1983). Pharmacological as well as ima-
ging studies support this assumption (Shapiro et al.
1989; Sallee et al. 1997). Tics often worsen after
discontinuation of antidopaminergic drugs or ad-
ministration of psychostimulants (Mesulam 1986;
Albin et al. 2003).
One major problem in treating tics with typical
or present atypical neuroleptics is the side effect
profile of the different drugs. Whereas classical
neuroleptics are associated with an increased risk
for extrapyramidal symptoms, akathisia or tardive
dyskinesia, many second-generation drugs do have
specific side effects such as weight gain, sedation,
akathisia etc. (Weiden and Bruun 1987). Moreover,
the treatment of psychiatric comorbid conditions
often poses a problem in the medical treatment of
GTS. For example, treating ADHD symptoms is
often challenging, since psychostimulants might
deteriorate tics, as mentioned above. But even SSRIs
are associated with a worsening of tics in some cases
(Ruth et al. 2000).
Aripiprazole is a partial D2 antagonist as well as a
partial 5-HT1A agonist and a 5-HT2A antagonist.
Being a partial D2-agonist, aripiprazole exhibits
functional agonistic properties under hypodopami-
nergic conditions and functional antagonistic prop-
erties under hyperdopaminergic conditions. The
main anti-tic property of aripiprazole is probably
due to the resulting stabilizing effects on dopami-
nergic neurotransmission. However, the serotonergic
action might be of additional benefit in indirectly
attenuating mesolimbic and/or mesocortical dopa-
minergic pathways (Ichikawa et al. 2001). Possible
serotonergic effects on prefrontal serotonergic pro-
jection sites could be responsible for the positive
effects on OCD symptoms or impulsivity.
Given this background and our first positive
experiences, aripiprazole might well turn out to be
an efficacious and well-tolerated drug in the treat-
ment of GTS. It might not only improve tics but also
the common co-morbid (ADHD spectrum) symp-
toms like impulsivity and emotional instability.
Statement of interest
The authors have no conflict of interest with any
commercial or other associations in connection with
the submitted article.
References
Albin RL, Koeppe RA, Bohnen NI, Nichols TE, Meyer P,
Wernette K, Minoshima S, Kilbourn MR, Frey KA. 2003.
Increased ventral striatal monoaminergic innervation in Tour-
ette syndrome. Neurology 61:310�/315.
Bruun RD, Budman CL. 1996. Risperidone as a treatment for
Tourette’s syndrome. J Clin Psychiatry 57:29�/31.
Dehning S, Riedel M, Muller N. 2005. Aripiprazole in a patient
vulnerable to side effects. Am J Psychiatry 162:625.
Devinsky O. 1983. Neuroanatomy of Gilles de la Tourette’s
syndrome. Possible midbrain involvement. Arch Neurol
40:508�/514.
Hounie A, De Mathis A, Sampaio AS, Mercadante MT. 2004.
Aripiprazole and Tourette syndrome. Rev Bras Psiquiatr
26:213.
Ichikawa J, Ishii H, Bonaccorso S, Fowler WL, O’Laughlin IA,
Meltzer HY. 2001. 5-HT(2A) and D(2) receptor blockade
increases cortical DA release via 5-HT(1A) receptor activation:
A possible mechanism of atypical antipsychotic-induced cor-
tical dopamine release. J Neurochem 76:1521�/1531.
Kastrup A, Schlotter W, Plewnia C, Bartels M. 2005. Treatment
of tics in tourette syndrome with aripiprazole. J Clin Psycho-
pharmacol 25:94�/96.
Leckman JF. 2002. Tourette’s syndrome. Lancet 360:1577�/1586.
Lucas AR. 1967. Gilles de la Tourette’s disease in children:
Treatment with haloperidol. Am J Psychiatry 124:243�/245.
Mesulam MM. 1986. Cocaine and Tourette’s syndrome. New
Engl J Med 315:398.
Murphy TK, Bengtson MA, Soto O, Edge PJ, Sajid MW, Shapira
N, Yang M. 2005. Case series on the use of aripiprazole for
Tourette syndrome. Int J Neuropsychopharmacol 8:489�/490.
Ruth U, Mayer-Rosa J, Schlamp D, Freisleder FJ. 2000. Tour-
ette’s syndrome and antidepressant therapy: Exacerbation of
nervous tics with paroxetine. Z Kinder Jugendpsychiatr Psy-
chother 28:105�/108.
124 E. Bubl et al.

Sallee FR, Nesbitt L, Jackson C, Sine L, Sethuraman G. 1997.
Relative efficacy of haloperidol and pimozide in children and
adolescents with Tourette’s disorder. Am J Psychiatry
154:1057�/1062.
Shapiro E, Shapiro AK, Fulop G, Hubbard M, Mandeli J, Nordlie
J, Phillips RA. 1989. Controlled study of haloperidol, pimozide
and placebo for the treatment of Gilles de la Tourette’s
syndrome. Arch Gen Psychiatry 46:722�/730.
Weiden P, Bruun R. 1987. Worsening of Tourette’s disorder due
to neuroleptic-induced akathisia. Am J Psychiatry 144:504�/
505.
Aripiprazole in patients with Tourette syndrome 125

LETTER TO THE EDITOR
Addressing the limitations of the CATIE study
SIEGFRIED KASPER & DIETMAR WINKLER
Department of General Psychiatry, Medical University of Vienna, Austria
Letters published in this Journal do not necessarily reflect the opinions of the Editors or the Editorial Board.
The Clinical Antipsychotic Trials of Intervention
Effectiveness (CATIE) study by Lieberman et al.
(2005) is important because it represents one of the
largest comparative trials on the differential effec-
tiveness of atypical antipsychotics that has not been
sponsored directly by the pharmaceutical industry.
However, the study shows several severe methodo-
logical limitations, which we would like to discuss
point by point:
1. The treatment groups cannot be compared
easily because the equivalent dosage of anti-
psychotics was not equal: olanzapine, for ex-
ample, was dosed higher compared to the other
compounds. The maximum daily dosage al-
lowed for olanzapine during this study (30 mg/
day) also exceeded the maximum daily dosage
recommended by the company (20 mg/day).
2. The design of the study is biased since the
inclusion/exclusion criteria for the treatment
groups differed: patients with tardive dyskinesia
could enrol, but the randomization scheme
prevented the assignment to the perphenazine
group. This leaves the study with non-compar-
able groups since we know that patients with
tardive dyskinesia might be different in regard
to duration of illness, chronicity and even the
rate of response to treatment (Kasper et al.
2006). Given that the rate of patients with
tardive dyskinesia was as high as 19% in the
group of atypical antipsychotics (15% of all
subjects), this is a severe bias and limits
generalization of the results. Unfortunately
this point is not discussed in the article.
3. The study is not powered adequately to show
differences between the different groups. The
trial only had a statistical power of 76% for
comparisons involving perphenazine and of
58% for comparisons involving ziprasidone.
Therefore a type II error is not unlikely.
4. The study was carried out in different centres,
university hospitals, state hospitals and private
sites, as well as mixed sites, all with different
kinds of patient groups, which makes up for a
large heterogeneity of the study population.
5. It is not clear why the patients were included in
the study. It is only mentioned that they had to
suffer from schizophrenia and that only 28%
had an exacerbation of clinical symptomatology
within the previous 3 months.
6. It is not described if patients with known
treatment resistance to one of the drugs were
randomized to this very substance or excluded
by the protocol.
7. The dosing schedule is quite disconcerting: To
protect blinding, half the patients randomly
assigned to perphenazine, olanzapine, and ris-
peridone were assigned to twice-daily dosing and
half to once-daily dosing. A much more adequate
procedure would have been to assign all patients
to twice-daily dosing, because patients with once-
daily dosing were a priori known to be neither on
quetiapine nor on ziprasidone. Furthermore
differences in dosing are known to influence
compliance (Greenberg 1984) which might
have influenced the rate of discontinuation.
8. The rate of discontinuation of treatment for any
cause is the only primary outcome measure,
which makes this trial different from all the
other studies, but also impedes comparisons
with other studies.
9. The reader is left puzzled: why was quetiapine
associated with a higher rate of anticholinergic
Correspondence: Prof. Siegfried Kasper, Department of General Psychiatry, Medical University of Vienna, Wahringer Gurtel 18�/20,
A-1090 Vienna, Austria. Tel: �/43 1 40400 3568. Fax: �/43 1 40400 3099. E-mail: [email protected]
The World Journal of Biological Psychiatry, 2006; 7(2): 126�/127
ISSN 1562-2975 print/ISSN 1814-1412 online # 2006 Taylor & Francis
DOI: 10.1080/15622970600685424

effects than the other antipsychotics, given that
quetiapine patients used less anticholinergic
medication? This point is not mentioned in
the discussion.
10. It is brought up in the introduction that atypical
antipsychotics in general have weight gain and
alterations of glucose and lipid metabolism as
side effects. This is an uncritical statement. The
authors should note that the risk of metabolic
side effects is different between the substances.
For example, ziprasidone and aripiprazole do
not influence weight, lipids or glucose metabo-
lism (Lublin et al. 2005; Newcomer 2005).
Interestingly, their own study also documented
differences, e.g. olanzapine was associated with
greater weight gain and increases in measures of
glucose and lipid metabolism.
Altogether, it is our opinion that this study compares
non-comparable groups and leaves the reader
with results that could lead to wrong conclusions.
Not everybody is an expert with methodological
considerations of randomized controlled trials
and therefore might get the impression that in the
light of higher direct costs prescribing atypical
antipsychotics has no advantage over typical neuro-
leptics.
Statement of interest
Siegfried Kasper has received research grants, con-
sultancy fees and lecture fees from a number of
pharmaceutical companies in the area of CNS
development. Dr Dietmar Winkler has no conflict
of interest with any commercial or other associations
in connection with the submitted article..
References
Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck
RA, Perkins DO, Keefe RS, Davis SM, Davis CE, Lebowitz
BD, Severe J, Hsiao JK for the Clinical Antipsychotic Trials of
Intervention Effectiveness (CATIE) Investigators. 2005. Effec-
tiveness of antipsychotic drugs in patients with chronic schizo-
phrenia. New England Journal of Medicine 353:1209�/1223
Kasper S, Lowry AJ, Hodge A, Bitter I, Dossenbach M. 2006.
Tardive dyskinesia: Analysis of outpatients with schizophrenia
from Africa and the Middle East, Asia, Central and Eastern
Europe, and Latin America. Schizophrenia Research 81:139�/
143.
Greenberg RN. 1984. Overview of patient compliance with
medication dosing: A literature review. Clinical Therapy
6:592�/599.
Newcomer JW. 2005. Second-generation (atypical) antipsychotics
and metabolic effects: A comprehensive literature review. CNS
Drugs 19(Suppl 1):1�/93.
Lublin H, Eberhard J, Levander S. 2005. Current therapy issues
and unmet clinical needs in the treatment of schizophrenia: A
review of the new generation antipsychotics. International
Clinical Psychopharmacology 20:183�/198.
The limitations of the CATIE study 127

The World Journal of Biological Psychiatry
Instructions to Authors
The aim of The World Journal of Biological Psychiatry is to increase theworldwide communication of knowledge in clinical and basic research onbiological psychiatry. The composition of The World Journal of BiologicalPsychiatry , with its diverse categories that allow communication of a greatvariety of information, ensures that it is of interest to a wide range ofreaders. It offers the opportunity to educate (through critical reviewpapers), to publish original work and observations (original papers andcase reports), and to express personal opinions (Viewpoints/Letters to theEditor). The World Journal of Biological Psychiatry is thus an extremelyimportant medium in the field of biological psychiatry all over the world.
Manuscripts must be written in standard and grammatical English andshould present new results as well as be of scientific value. Contributionswill be considered for the following categories:
/� Editorial/� Reviews/Mini-reviews/� Original investigations/Summaries of original research/� Brief reports/� Viewpoints/� Case reports/Case series/� Letters to the editors/� Book reviews/� Invitations/Announcements
The World Journal of Biological Psychiatry’s Manuscript Central, web-based manuscript submission and handling system, is now available.Please submit all manuscripts online via Journal’s manuscriptCentral site that is accessible via the Journal’s home page:www.tandf.no/wjbp
Click on ‘‘online submission’’ which directs you to Manuscript Central’slog in page. Here either create an account or enter an existing account tolog in your ‘‘author centre’’ to upload manuscripts.
If you have difficulties in submitting your manuscript electronically, contactThe World Journal of Biological Psychiatry’s Editorial Office.
The receipt of the manuscript will be acknowledged by an e-mail whichincludes a manuscript ID number. Each manuscript will be assigned to atleast two reviewers. The manuscript ID number should be quoted in allcorrespondence with the Chief Editor and Editorial Office.
Submit manuscript text as a Word file and Figures as separate TIFF or EPSformat files. Use Times New Roman 12 point and double spacingthroughout the manuscript. Use a clear system of headings to divide upand clarify the text, with not more than three grades of headings. Thedesired position of figures and tables should be indicated in the manu-script.. Submission of a manuscript implies: that the work described has notbeen published before (except in the form of an abstract or as part of apublished lecture, review or thesis); that it is not under consideration forpublication anywhere else; that its publication has been approved by all co-authors, if any, as well as by responsible authorities �/ tacitly or explicitly �/
at the institute where the work has been carried out.
Manuscripts submitted for publication must contain a statement to theeffect that all human studies have been approved by the appropriate ethicscommittee and have therefore been performed in accordance with theethical standards laid down in the 1964 Declaration of Helsinki. It shouldalso be stated clearly in the text that all persons gave their informed consentprior to their inclusion in the study. Details that might disclose the identityof the subjects under study should be omitted.
Reports of animal experiments must state that the Principles of laboratoryanimal care (NIH publication No. 86-23, revised 1985) were followed, aswell as specific national laws (e.g., the current version of the German Lawon the Protection of Animals) where applicable.
The editors reserve the right to reject manuscripts that do not comply withthe above-mentioned requirements. The author will be held responsible forfalse statements or for failure to fulfill the above-mentioned requirements.
Manuscript specifications
1. The manuscript text file shall contain a title page. On the title page thefollowing shall be stated: The title of the article, which should beconcise but informative. A short title not exceeding 40 letters andspaces. The full names, affiliations and addresses of all the contribu-tors, and e-mail address, telephone number and fax number for thecorresponding author. The title page should also include details of thenumber of words, figures, and tables.
2. Each article must include a short Abstract of the essential results (notexceeding 200 words).
3. Key words. Up to five key words should be inserted below the abstract.Use terms from the Medical Subject Headings list from Index Medicus.
4. References. All references cited in the text are to be listed (double-spaced) at the end of the paper in alphabetical order under the name ofthe first author. If there are more than six authors, list the first sixauthors and use ‘et al.’ Articles ‘in press’ may be included, but muststate the journal that has accepted them. Personal communicationsshould be avoided. Journal titles should be abbreviated according toIndex Medicus.
Citations in the text should be by author and date in parentheses. Ifthere are two authors, both should be named (e.g., Agar and Douglas1955); if an article with more than two authors is cited, only the firstauthor’s name plus ‘et al.’ need be given (e.g., Komor et al. 1979); ifthere is more than one reference by the same author or team of authorsin the same year, then a, b, c, etc., should be added to the year, both inthe text and the list of references. When styling references, thefollowing examples should be followed:
Brain WR. 1958. The physiological basis of consciousness. Acritical review. Brain 81:426 �/455.
Kuhlenbeck H. 1954. The human diencephalon. A summary ofdevelopment, structure, function and pathology. Basel: Karger.
Teuber HL. 1964. The riddle of frontal lobe function in man. In:Warren T, Akert CH, editors. The frontal and granular cortex andbehavior. New York: McGraw-Hill. pp 252�/271.
5. Tables should be typed on separate sheets and numbered consecutivelywith Arabic numbers. All tables should include a brief descriptivecaption.
6. Figures. All illustrations, whether photographs, graphs or diagrams,should be numbered consecutively with Arabic numerals and sub-mitted on separate sheets. Each illustration should be referred to in thetext as ‘Figure 1’, ‘Figures 1�/4’, etc. The figures should have a shorttitle followed by a concise description. All abbreviations and symbolsappearing in the figure must be explained in the legend. Remarks suchas ‘for explanation (or details) see text’ should be avoided. Each figureshould have a legend containing sufficient information to render thefigure intelligible without reference to the text. If a figure has beenpublished previously, acknowledge the original source and submitwritten permission from the copyright holder to reproduce it. Ifphotographs of persons are used, either the subjects must not beidentifiable or their written permission to use the photographs mustaccompany the manuscript. Figures can be sent electronically asseparate files in EPS or TIFF format.
7. Copyright. It is a condition of publication that authors vest copyrightin their articles, including abstracts, in Taylor & Francis GroupLtd. This enables us to ensure full copyright protection and todisseminate the article, and the journal, to the widest possiblereadership in print and electronic formats as appropriate. Authorsmay, of course, use the material elsewhere after publication providingthat prior permission is obtained from Taylor & Francis GroupLtd. Authors are themselves responsible for obtaining permission toreproduce copyright material from other sources. To view the ‘Copy-right Transfer Frequently Asked Questions’ please visit http://www.tandf.co.ukljoumalslcopyright.asp
8. Proofs and offprints. Proofs will be sent as a PDF file to thecorresponding author’s e-mail address unless otherwise requested.The corrected proof must be returned to the publisher within 3 days.Authors will be liable to pay for excessive alterations in the proof.Offprints can be ordered by filling out the form accompanying theproofs. The corresponding authors will be supplied with a free copy ofthe relevant issue.
9. For further information about the journal, including links to the onlinesample copy and contents pages, visit the journal homepage:www.tandf.no/wjbp or contact the Editorial Office at the followingaddress:
Jacqueline Klesing, Editorial Assistant, Department of Psychiatry,Ludwig-Maximilians-University, Nussbaumstrasse 7, 80336 Munich,Germany. Tel: +49 89 5160 5531 Fax: +49 89 5160 5530. E-mail:[email protected]






















