
Structural basis of Rnd1 binding to plexin Rho GTPase binding domains (RBDs)
Upload
independentCategory
view
7download
0
L E T T ERS
Rab35 GTPase and OCRL phosphatase remodel lipidsand F-actin for successful cytokinesisDaphné Dambournet1,2, Mickael Machicoane1,2, Laurent Chesneau1,2, Martin Sachse3, Murielle Rocancourt1,2,Ahmed El Marjou4,5, Etienne Formstecher6, Rémi Salomon7, Bruno Goud4,5 and Arnaud Echard1,2,8
Abscission is the least understood step of cytokinesis. Itconsists of the final cut of the intercellular bridge connectingthe sister cells at the end of mitosis, and is thought to involvemembrane trafficking as well as lipid and cytoskeletonremodelling1–6. We previously identified the Rab35 GTPase asa regulator of a fast recycling endocytic pathway that isessential for post-furrowing cytokinesis stages7. Here, we reportthat the phosphatidylinositol-4,5-bisphosphate (PtdIns(4,5)P2)5-phosphatase OCRL, which is mutated in Lowe syndromepatients8,9, is an effector of the Rab35 GTPase in cytokinesisabscission. GTP-bound (active) Rab35 directly interacts withOCRL and controls its localization at the intercellular bridge.Depletion of Rab35 or OCRL inhibits cytokinesis abscissionand is associated with local abnormal PtdIns(4,5)P2 andF-actin accumulation in the intercellular bridge. These divisiondefects are also found in cell lines derived from Lowe patientsand can be corrected by the addition of low doses of F-actindepolymerization drugs. Our data demonstrate thatPtdIns(4,5)P2 hydrolysis is important for normal cytokinesisabscission to locally remodel the F-actin cytoskeleton in theintercellular bridge. They also reveal an unexpected role for thephosphatase OCRL in cell division and shed new light on thepleiotropic phenotypes associated with Lowe disease.
After cytokinesis furrow ingression, sister cells are connected by anintercellular bridge that must be stabilized for several hours beforebeing cut by a poorly understood process called abscission1–3,5. Onthe basis of genome-wide RNA-mediated interference (RNAi) screens,remarkable progress has been made in the past decade in identifyingthe genes involved in cytokinesis10–12. These studies revealed anunexpected function of membrane trafficking, both exocytosis andendocytosis, in cytokinesis. However, the precise role of individualmembrane trafficking pathways in both intercellular bridge stabilityand cytokinesis abscission is still unknown. It has been suggested that
1Institut Pasteur, Membrane Traffic and Cell Division Lab. 25–28 rue du Dr Roux, 75724 Paris cedex 15, France. 2CNRS URA2582, France. 3Institut Pasteur,Imagopole, Plate-forme de microscopie ultrastructurale, 25–28 rue du Dr Roux, 75724 Paris cedex 15, France. 4Institut Curie, Molecular Mechanisms of IntracellularTransport Lab. 25 rue d’Ulm, 75005 Paris, France. 5CNRS UMR144, France. 6Hybrigenics SA, 3–5 impasse Reille, 75014 Paris, France. 7AP-HP Hôpital Necker,Service de Néphrologie Pédiatrique, Inserm U983, Paris, France.8Correspondence should be addressed to A.E. (e-mail: [email protected])
Received 29 November 2010; accepted 9 May 2011; published online 26 June 2011; DOI: 10.1038/ncb2279
cytokinesis abscission requires lipid and cytoskeleton remodelling,which could be controlled by membrane traffic3–6. To identify moreprecisely the transport routes involved and how they function incytokinesis, we previously conducted a systematic RNAi screen focusedon Rab genes, which encode key conserved GTPases that regulatemembrane trafficking in eukaryotic cells7. We found that Rab35regulates an endocytic pathway and is involved in the initial stabilityof the intercellular bridge, after furrow contraction, by controllingthe localization of SEPTIN2 (ref. 7). Interestingly, Rab35 depletionled to additional cytokinesis defects characterized by either delayedor inhibited abscission, indicating that Rab35 is essential for normalabscission, but the mechanism remained to be elucidated. Here wereport that Rab35 controls cytokinesis abscission through the functionof the phosphoinositide 5-phosphatase OCRL, the product of the generesponsible for a genetic disease, the oculocerebrorenal syndrome ofLowe8,13,14. Our data reveal that phosphatidylinositol-4,5-bisphosphate(PtdIns(4,5)P2) lipid hydrolysis is required for F-actin removal fromlate cytokinesis bridges, a limiting and important step for normalcytokinesis abscission.To investigate how Rab35 could control cytokinesis abscission, we
isolated potential Rab35 effectors using the GTPase-deficient mutantRab35Q67L as bait in a two-hybrid screen. Many hits (52 out of 227)encoded for the OCRL-b phosphatase, which has been shown tointeract in two-hybrid experiments with several other active RabGTPases, such as Rab1, Rab5 and Rab6 (refs 15,16). Full-lengthOCRL interacted specifically with the GTP-bound mutant Rab35(Rab35Q67L) but not with the GDP-bound, dominant-negative mutant(Rab35S22N; Fig. 1a). In addition, endogenous OCRL was pulled downfrom cell lysates by glutathione S-transferase (GST)–Rab35 loaded withGTP but not with GDP (Fig. 1b), and GFP–OCRL was preferentiallyco-immunoprecipitated with the GTP-bound Rab35 mutant (Fig. 1c).Finally, using purified recombinant proteins, the interaction betweenOCRL and Rab35 was found to be direct and to depend on Rab35binding to GTP (Fig. 1d,e). A solid-phase assay further showed that
NATURE CELL BIOLOGY VOLUME 13 | NUMBER 8 | AUGUST 2011 981
© 2011 Macmillan Publishers Limited. All rights reserved.

L E T T ERS
Myc–Rab35
Q67LWild typeS22N
IP anti-Myc WB anti-GFP
IP anti-Myc WB anti-Rab35
Input GFP–OCRL
Rab35 wild type
Rab35S22N
Rab11Q70L
Rab35Q67L
–His+His
1 893731540237119
RhoGAP like5-PasePH ASHpGAD–OCRL pGAD
alone
–His+His
6 × His–RabQ/L:
GST beads
GST–OCRL (540–893) beads
Rab 35
Bead input
WB
anti-6 × His
WB
anti-6 × His
Bead input
Input
6 × His–RabQ/L
Rab 1
Rab 5
Rab 6
Rab 11
Anti-OCRL
GST–Rab35
WB
Bead input
OCRL
Input
GST–Rab
GST
GST GST–Rab11
GDP GTP-γS GDP GTP-γS
c d
e
f
a b
GDP
Rab 35
Rab 6
Input 6 × His–Rab
GTP-γS6 × His–Rab35
6 × His–Rab6
6 × His–Rab11
Bead input
Bead input
WB
anti-6 × His
WB
anti-6 × His
GST–OCRL (540–893) beads
GST beads
GTP
-γS
GDP
GTP
-γS
GDP
GTP
-γS
GDPRab
11Rab 35
Rab 6
Rab 11
Ab
so
rban
ce 4
50
nm
6 × His–RabQ/L (ng)
0.1
0
0.2
0.3
0.4
6 × His–Rab11Q70L
6 × His–Rab1Q70L
6 × His–Rab5Q79L
6 × His–Rab6Q72L
6 × His–Rab35Q67L
500 100 150 200 250
Figure 1 OCRL directly interacts with GTP-bound Rab35. (a) TheSaccharomyces cerevisiae strain L40 was transformed with plasmidsencoding GAD fused to full-length OCRL-b (left) or GAD alone (control; right)to examine interactions with LexA fused to wild-type Rab35, Rab35Q67L
(GTP-bound mutant), Rab35S22N (GDP-bound mutant) and Rab11Q70L
(GTP-bound mutant). Growth on medium without His (− His) indicates aninteraction with the indicated proteins. Part of the ASH domain (amino acids540–676) was sufficient to interact specifically with GTP-bound Rab35in a yeast two-hybrid assay (data not shown). (b) Recombinant purifiedGST-tagged wild-type Rab35 or Rab11 proteins were loaded with eitherGDP or GTP-γS. After incubation with HeLa cell extracts, endogenous OCRLbound to either GST or GST–Rab beads was detected by western blot (WB)analysis using an anti-OCRL antibody. Input represents 5% load of thetotal cell extracts used in all conditions. (c) HeLa cells were co-transfectedwith plasmids encoding GFP–OCRL-b and either Myc-tagged wild-typeRab35, Rab35S22N or Rab35Q67L. Rab35 was immunoprecipitated (IP) withanti-Myc antibodies, and co-immunoprecipitated OCRL was revealed by
anti-GFP antibodies (middle). The amount of GFP–OCRL (1% lysate, top)and immunoprecipitated Rab35 (2%, bottom) is shown in all conditions. (d)Recombinant GST or GST–OCRL-b (amino acid 540–893) immobilized onglutathione beads was incubated with recombinant 6×His-tagged wild-typeRab35, Rab6 or Rab11 loaded with either GDP or GTP-γS (1% inputs areshown on the left). The amount of each Rab directly bound to GST (topwestern blot) or GST–OCRL-b (bottom western blot) was detected withanti-6×His antibodies. 1% bead inputs are shown below each western blot.(e) The same experiment as in d, except that recombinant 6×His-taggedGTP-bound mutant proteins (Q/L) of the indicated Rab were used. (f) 96-wellplates were coated with recombinant GST–OCRL-b (amino acids 540–893)and incubated with increasing amounts of recombinant 6×His-taggedGTP-bound Rab mutant proteins, as indicated (solid-phase assay). Afterwashes, Rab proteins bound to OCRL were detected (arbitrary units) usinganti-6×His antibodies and chromogenic substrate (mean±s.e.m., n = 3experiments). No binding to GST alone was detected (data not shown).Uncropped images of blots are shown in Supplementary Fig. S8a–d.
GTP–Rab35 bound to OCRL with a similar affinity as for Rab6(Fig. 1f), which was recently measured to be in the micromolarrange17. Of note, this binding was selective, as OCRL did not interact
with GTP-bound Rab11, another Rab involved in endocytosis andcytokinesis18 (Fig. 1a–f). We therefore conclude that OCRL directlyinteracts with Rab35, specifically when this GTPase is bound toGTP.
982 NATURE CELL BIOLOGY VOLUME 13 | NUMBER 8 | AUGUST 2011
© 2011 Macmillan Publishers Limited. All rights reserved.

L E T T ERS
OCRL 15Rab35 10
GM
13
0 O
CR
LO
CR
LG
M1
30
G
gg
G
GFP–Rab35mCherry–OCRL
6
42
3
87
5
1
Rab35 siRNA
Control siRNA
OC
RL
lab
elli
ng
at
the in
terc
ellu
lar
brid
ge
(perc
en
tag
e o
f co
ntr
ol)
∗∗∗
0
20
40
60
80
100
g
g
Rab
35 s
iRN
A
OCRL Aurora BOCRL
OCRL Aurora BOCRL
Co
ntr
ol siR
NA
gg
Control siRNA
Rab35
β-tubulin
Rab35 siRNA
OCRL Aurora BOCRL
a
d e
b c
Rab
35
S2
2N
Rab
35
Q6
7L
Co
ntr
ol
OC
RL
lab
elli
ng
at
the in
terc
ellu
lar
brid
ge
(perc
en
tag
e o
f co
ntr
ol)
0
20
40
60
80
100
120
140∗∗
∗∗∗
Rab35S22N Rab35Q67LControl
Figure 2 Rab35 activation controls the accumulation of an OCRL pool in thecytokinesis intercellular bridge. (a) HeLa cells in cytokinesis were stainedfor endogenous OCRL (green) and the Golgi marker GM130 (red). Right,higher-magnification images. Blue, DAPI (4,6-diamidino-2-phenylindole)staining. G, distal Golgi; g, proximal Golgi that is being reformed duringcytokinesis (see ref. 6). Arrow, intercellular bridge. Scale bar, 10 µm. (b)OCRL and Rab35 co-localize at the intercellular bridge. HeLa cells wereco-transfected with GFP-tagged wild-type Rab35 and mCherryFP–OCRL-b.Top left: OCRL channel. Top right: Rab35 channel. Bottom right: mergechannels (Rab35 in green and OCRL in red). These images correspond toa snapshot of a time-lapse movie acquired with a spinning-disc confocalmicroscope. Tracks (temporal projection) of individual OCRL vesiclestrafficking to (no. 1,2,4,5,7,8) or from (no. 3,6) the intercellular bridgeare indicated in the bottom left panel. Scale bar, 5 µm. (c) HeLa cellswere co-transfected with plasmids encoding GFP–OCRL-b and Myc-taggedwild-type Rab35. GFP-positive cells with late cytokinesis bridges (arrowhead,top panel, scale bar, 10 µm) were identified and fixed. Cells were thenprocessed for double immuno-electron microscopy localization of Rab35(10nm gold particles) and OCRL (15nm gold particles; middle panel).
The regions indicated with arrows are presented at a higher magnification(bottom panels). Scale bar, 500nm. (d) Rab35 is required for properlocalization of OCRL at the intercellular bridge. HeLa cells were transfectedwith control or Rab35 -specific siRNA and stained for Aurora B kinase toidentify intercellular bridges (red) and for endogenous OCRL (green). Topleft: The western blot shows the efficiency of Rab35 RNAi (depletion by70%–85% of endogenous levels), using the β-tubulin signal as a loadingcontrol. Top right and bottom right panels: Higher-magnification images ofthe bridge regions for each siRNA. Bottom left: Quantification of the OCRLintensity at the intercellular bridge in control and Rab35-depleted cellsis presented (mean±s.e.m., n = 30 cells analysed per experiment, fourindependent experiments). g, proximal Golgi. Blue, DAPI staining. Scale bar,10 µm. (e) HeLa cells were transfected with either control (empty vector),active Rab35Q67L or dominant-negative Rab35S22N mutants. OCRL staining(right panels) and quantification in intercellular bridges (left panel) werecarried out as in d (mean±s.e.m., n =30 cells analysed per experiment,3–6 independent experiments). Blue, DAPI staining. Scale bar, 1 µm.** P <6×10−3 and *** P <3×10−5 (Student’s t -test). Uncropped imagesof blots are shown in Supplementary Fig. S8e.
We next examined the relative localization of Rab35 and OCRL inHeLa cells. The localization of OCRL during cell division is unknown,but in interphase OCRL is reported to be mainly localized at the
Golgi apparatus as well as at the plasma membrane, at clathrin-coated-pits and on internal endocytic vesicles15,19–21. The localizationof endogenous OCRL was confirmed at the Golgi in interphase cells
NATURE CELL BIOLOGY VOLUME 13 | NUMBER 8 | AUGUST 2011 983
© 2011 Macmillan Publishers Limited. All rights reserved.

L E T T ERS
(data not shown) and at Golgi complexes that reformed during celldivision (Fig. 2a, labelled as G and g). Interestingly, we identified a poolof OCRL that labelled both sides of the midbody in late cytokinesisbridges and did not correspond to Golgi structures (Fig. 2a). Thestaining was specific, as it disappeared in cells depleted for OCRL afterRNAi (Supplementary Fig. S1). OCRL-a and OCRL-b are two closelyrelated OCRL isoforms, and the OCRL-b isoform is the main variantexpressed in non-neuronal tissues22. High-resolution spinning-discconfocal time-lapse microscopy revealed that mCherryFP–OCRL-b (aswell as mCherryFP–OCRL-a, data not shown) was found at the plasmamembrane and on dynamic intracellular vesicles thatmoved to and, lessfrequently, from the intercellular bridge (vesicle tracks; Fig. 2b).Withinthe intercellular bridge, OCRL-b staining co-localized with GFP-fusedwild-type Rab35 (Fig. 2b). Double immuno-electron microscopylabelling confirmed that Rab35 is localized at the plasma membraneand on endosomes7,23–25, and revealed that at the intercellular bridgeRab35 was predominantly localized to the plasma membrane where itpartially co-localized with OCRL (Fig. 2c).We next investigated whether Rab35 controls the localization of
OCRL at the intercellular bridge during cytokinesis. All quantificationspresented hereafter were carried out on late cytokinesis bridges,characterized by thin Aurora B and α-tubulin stainings. As shownin Fig. 2d, Rab35 depletion by RNAi largely reduced the amount ofOCRL at the intercellular bridge. Consistent with Rab35 activationbeing important for OCRL localization, the level of OCRL staining wasincreased by 25% in cells overexpressing the Rab35Q67L mutant (GTP-bound), whereas it was decreased by twofold in cells overexpressingthe dominant-negative Rab35S22N mutant (Fig. 2e). Of note, OCRLlocalization at the Golgi was not reduced in Rab35S22N-expressing cellsor in Rab35-depleted cells (Fig. 2d). Interestingly, OCRL was reportedto interact with several other Rab GTPases15,16, but overexpression ofdominant-negative versions of each of them did not reduce OCRLstaining at the intercellular bridge during cytokinesis (SupplementaryFig. S2). In addition, the Rab11S22N mutant, which has been shownto perturb both cytokinesis and endocytosis/recycling18, did notinfluence OCRL localization during cell division (Supplementary Fig.S2). Together, these data indicate that Rab35 specifically contributesto OCRL localization at the intercellular bridge, and that Rab35activation is important for recruiting and/or targeting OCRL in thislocation during cytokinesis.At present, nothing is known about the potential function of OCRL
during cell division. Cytokinesis was analysed in HeLa cells followingOCRL RNAi, which led to an 85% decrease of endogenous OCRL at theprotein level (Supplementary Fig. S1). The number of cells connectedby acetyl-tubulin-positive intercellular bridges was increased from9.1%in control cells to 19.3% after OCRL depletion (n= 500 cells countedfor each of four independent experiments, P = 2× 10−4, Student’st -test). To directly determine whether OCRL is involved in cytokinesisabscission, we analysed cell divisions in control or OCRL-depletedcells over 48 h periods by time-lapse video microscopy (Fig. 3a). Inall cases, mitotic round-up, furrow contraction and formation of theintercellular bridge occurred normally (Fig. 3a). In OCRL-depletedcells, bridge instability was occasionally detected, giving rise to asmall proportion of binucleated cells (1.8%, compared with 0.9%in control cells, n= 500 cells counted for each of four independentexperiments, P = 4×10−2, Student’s t -test), as reported after Rab35
depletion7. However, abscission was either greatly delayed (meanabscission time was increased by twofold; Supplementary Fig. S3a) orcompletely blocked after OCRL depletion: 30% of the cells completedabscission more than 15 h after cytokinesis furrow ingression, andabscission was entirely blocked in ∼17% of the divisions (Fig. 3b).In these cases, each sister cell still connected by an intercellularbridge entered into a further round of mitosis. The bridge was eitherbroken at this stage (Supplementary Fig. S3b), or the cells remainedconnected as chains, indicating a complete inhibition of abscission(Fig. 3c and Supplementary Fig. S3c). Consistent with OCRL beingan effector of Rab35 in cell division, Rab35-depleted cells exhibitedabscission defects similar to those observed in OCRL-depleted cells(Fig. 3a,b and Supplementary Fig. S3a). Importantly, abscission defectsafter OCRL RNAi resulted specifically from OCRL depletion, as theywere completely suppressed by the expression of a short interferingRNA (siRNA)-resistant mRNA encoding either GFP-tagged OCRL-b(Fig. 3d and Supplementary Fig. S3d) or GFP-tagged OCRL-a (datanot shown). Interestingly, expression of an siRNA-resistant mRNAencoding GFP–OCRL-b (or GFP–OCRL-a, data not shown) with apoint mutation (H524R) known to abolish the phosphatase activity26
failed to rescue abscission defects in OCRL-depleted cells (Fig. 3d andSupplementary Fig. S3d). Together, these results reveal an unknownfunction of OCRL in cell division, and indicate that the phosphataseactivity of OCRL is required for normal cytokinesis abscission.Abscission defects observed after OCRL depletion in HeLa cells
were confirmed in renal cells that we established from a Lowe patientbearing a point-mutated version of OCRL (G421E; ref. 26). AlthoughOCRLG421E was still partially expressed in patient cells (Fig. 3e, inset),it had no significant in vitro PtdIns(4,5)P2 phosphatase activity(Fig. 3e). Importantly, mean abscission times were more than twofoldgreater in Lowe syndrome cells, when compared with the same cellstransfected with a plasmid encoding GFP–OCRL-b (or GFP–OCRL-a,data not shown) or when compared with renal cells that have beenestablished with the same protocol but from non-Lowe syndromedonors (Fig. 3f,g). Thus, these data indicate that the expression of amutated version of OCRL responsible for Lowe disease also causeddefects in cytokinesis abscission in patient cells.OCRL is a phosphoinositide 5-phosphatase whose preferred
substrate is PtdIns(4,5)P2 (refs 27,28). As its phosphatase activityseemed critical for OCRL function in cytokinesis (Fig. 3d–g), and givenOCRL localization during cell division (Fig. 2a–c), we investigatedPtdIns(4,5)P2 levels at the intercellular bridge, using an antibody thatspecifically detects this phosphoinositide29,30. We found a 50% increasein the level of the PtdIns(4,5)P2 signal within the intercellular bridgein OCRL-depleted HeLa cells, compared with control cells (Fig. 4a).A 40% increase was also observed at the intercellular bridge in Lowepatient renal cells (Fig. 4b). Of note, the global increase in the level ofPtdIns(4,5)P2 staining (outside the intercellular bridges) was much lessstriking (∼20%) than the one observed at the intercellular bridge, inboth HeLa cells depleted for OCRL and in patient cells (SupplementaryFig. S4a). This indicates that OCRL is critical during cytokinesis forcontrolling PtdIns(4,5)P2 levels locally, at the intercellular bridge.Consistent with Rab35 controlling the localization of the pool ofOCRL at the bridge (Fig. 2d), we found a 50% increase in thelevel of PtdIns(4,5)P2 at the intercellular bridge in Rab35-depletedHeLa cells (Fig. 4a). In contrast, no increase was observed after
984 NATURE CELL BIOLOGY VOLUME 13 | NUMBER 8 | AUGUST 2011
© 2011 Macmillan Publishers Limited. All rights reserved.

L E T T ERS
0
20
40
60
80
100
120
OCRL wild type
OCRLG421E OCRLH524R
Ptd
Ins(4
, 5)P
2 p
ho
sp
hata
se a
ctivity
(perc
enta
ge o
f th
e w
ild t
yp
e)
Anti- OCRL
Anti- β-tubulin
Normal cells
Lowe cells
+ GFP
Normal cells
+ GFP +GFP–OCRL wild type
Lowe cells
∗∗∗ ∗∗∗
0
2
4
6
8
10
12
Ab
scis
sio
n t
ime (h)
0
20
40
60
80
100
Time after furrow ingression (h)
61 11 16 21 26 31Cum
ula
tive p
erc
enta
ge o
f cells
th
at
co
mp
lete
d a
bscis
sio
n
a
c d
b
e f g
Normal cells + GFP
Lowe cells + GFP–OCRL wild type
Lowe cells + GFP
0
2
4
6
8
10
12
14A
bscis
sio
n t
ime (h
)
+ GFP
Control siRNA
OCRL siRNA
∗∗∗n.s.
∗∗∗
+ GFP–OCRLH524R
+ GFP + GFP–OCRL
wild type
GFP
PLC-γ
Wild type H524R
OCRL siRNA + GFP–OCRL
OC
RL
siR
NA
2 12 22 32 42 52
Cum
ula
tive p
erc
enta
ge o
f cells
that
co
mp
lete
d a
bscis
sio
n
0
20
40
60
80
100
Time after furrow ingression (h)
Control siRNAOCRL siRNA Rab35 siRNA
Rab
35 s
iRN
AO
CR
L siR
NA
0 min 2 h 40 min 14 h 40 min 14 h 50 min
0 min 2 h 30 min 4 h 50 min 5 h
0 min 2 h 30 min 12 h 10 min 12 h 20 min
Co
ntr
ol siR
NA
Furrow ingression Early bridge Late bridgeAbscissioncompleted
Figure 3 Cytokinesis abscission defects in cells depleted for Rab35 orOCRL, and in Lowe syndrome patient cells. (a) Snapshots of time-lapsephase-contrast microscopy movies of HeLa cells transfected with eithercontrol, OCRL- or Rab35 -specific siRNAs. Insets, high-magnification imagesof the intercellular bridges (black arrowheads) before and after abscission.(b) Distribution of the abscission times in the cell populations describedin a, as indicated. The distributions after OCRL and Rab35 depletion aredifferent from the control, with P < 10−5 (Kolmogorov–Smirnov test). (c)Snapshot of OCRL-depleted cells in which cytokinesis abscission failedat two successive cell divisions (see also Supplementary Fig. S3c). Notethe presence of a chain of four cells connected by the old intercellularbridge (arrowhead) and by two new intercellular bridges (arrows). (d) Left,mean abscission times were measured on time-lapse movies in HeLa cellstransfected first with control or OCRL siRNAs, then with either GFP alone,wild-type GFP–OCRL-b or the GFP–OCRL-bH524R catalytically inactive mutant(mean±s.e.m., n=40–70 cells, three independent experiments). n.s.= notsignificant, *** P <8×10−5 (Student’s t -test). Right, western blot analysis
showing that the amounts of wild-type and mutant GFP–OCRL expressedare comparable (loading control, PLC-γ1). (e) The recombinant phosphatasedomain (amino acids 202–618) of either wild-type, G421E or H524R OCRLfused to GST was purified and the in vitro phosphatase activity towardsdC4-PtdIns(4,5)P2 was measured (mean±s.e.m., representative triplicateexperiment). Inset, amount of inactive OCRLG421E expressed in the Lowepatient cells analysed in this study (western blot loading control, β-tubulin).(f) Renal epithelial cells from a patient expressing the OCRLG421E mutantwere transfected with GFP alone or wild-type GFP–OCRL-b and abscissiontimes were measured as in b. For comparison, renal epithelial cells from adonor not mutated in OCRL (normal cells) were analysed after transfection ofGFP alone (mean±s.e.m., n=60–80 cells, three independent experiments).*** P < 2×10−3 (Student’s t -test). (g) Distribution of the abscissiontimes in the different cell populations described in f. Distributions in Lowecells transfected with GFP versus GFP–OCRL wild type are different, withP <10−5 (Kolmogorov–Smirnov test). Uncropped images of blots are shownin Supplementary Fig. S8g,h.
functional inhibition of Rab5 (Supplementary Fig. S4c,d), anotherendosomal Rab GTPase known to interact with OCRL (refs 15,21). Together, these results are consistent with the hypothesis thatOCRL is a downstream effector of Rab35 during cytokinesis and
that Rab35 specifically controls OCRL localization, and thus localPtdIns(4,5)P2 levels, at the intercellular bridge. PtdIns(4,5)P2 is akey signalling molecule that controls a number of cellular pathways,and in particular, promotes actin polymerization9,31. We formed a
NATURE CELL BIOLOGY VOLUME 13 | NUMBER 8 | AUGUST 2011 985
© 2011 Macmillan Publishers Limited. All rights reserved.
L E T T ERS
F-a
ctin
lab
elli
ng
at
the
inte
rcellu
lar
brid
ge
(perc
en
tag
e o
f co
ntr
ol)
Normal cells
∗∗∗
Lowe cells
0
50
100
150
200
F-a
ctin
A
uro
ra B
Lowe cellsNormal cells
∗∗∗ ∗∗
0
50
100
150
200
Control siRNA
OCRL siRNA
Rab35 siRNA
F-a
ctin
lab
elli
ng
at
the
inte
rcellu
lar
brid
ge
(perc
en
tag
e o
f co
ntr
ol)
Control siRNA OCRL siRNA Rab35 siRNA
F-a
ctin
A
uro
ra B
a b
c d
Ptd
Ins(4
, 5
)P2 la
belli
ng
at
the
inte
rcellu
lar
brid
ge
(perc
en
tag
e o
f co
ntr
ol)
Control siRNA
OCRL siRNA
Rab35 siRNA
∗∗∗∗∗∗
0
16014012010080604020
180
Control siRNA OCRL siRNA Rab35 siRNA
Ptd
Ins(4
, 5
)P2
α
-tu
bu
lin
Ptd
Ins(4
, 5
)P2 lab
elli
ng
at
the
inte
rcellu
lar
brid
ge
(perc
en
tag
e o
f co
ntr
ol)
160140120100806040200
Normal cells
Lowe cells
∗∗∗
Ptd
Ins(4
, 5
)P2
α
-tu
bu
lin
Normal cells Lowe cells
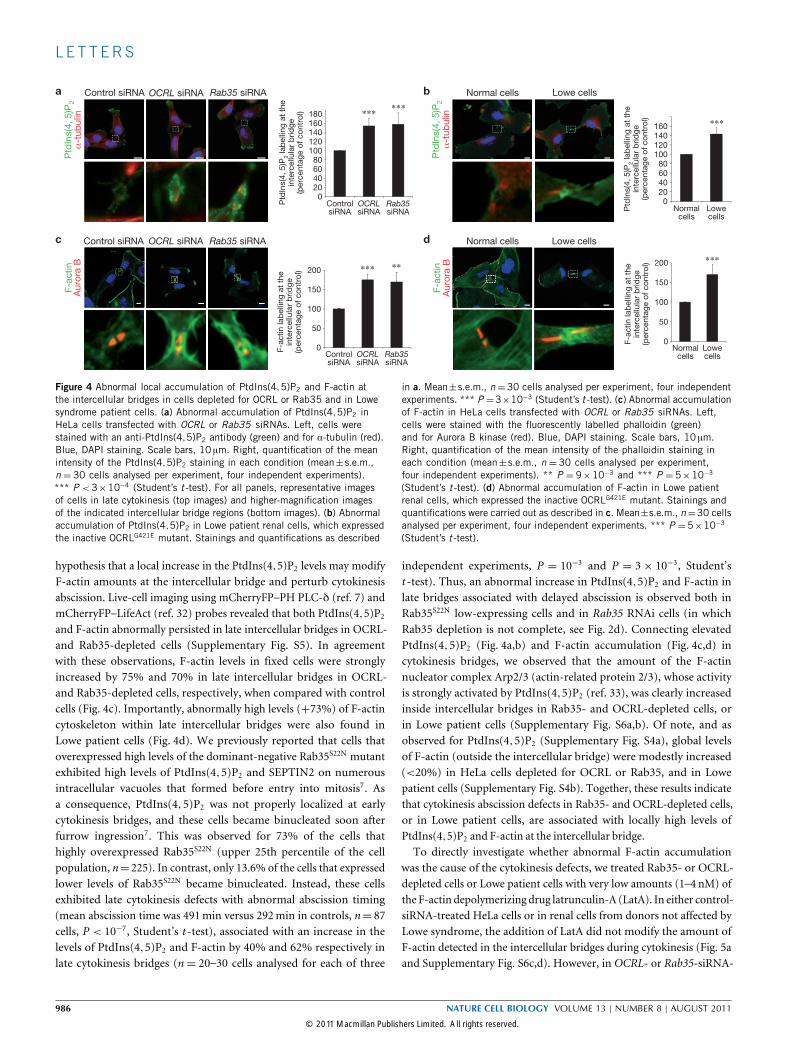
Figure 4 Abnormal local accumulation of PtdIns(4,5)P2 and F-actin atthe intercellular bridges in cells depleted for OCRL or Rab35 and in Lowesyndrome patient cells. (a) Abnormal accumulation of PtdIns(4,5)P2 inHeLa cells transfected with OCRL or Rab35 siRNAs. Left, cells werestained with an anti-PtdIns(4,5)P2 antibody (green) and for α-tubulin (red).Blue, DAPI staining. Scale bars, 10 µm. Right, quantification of the meanintensity of the PtdIns(4,5)P2 staining in each condition (mean±s.e.m.,n = 30 cells analysed per experiment, four independent experiments).*** P <3×10−4 (Student’s t -test). For all panels, representative imagesof cells in late cytokinesis (top images) and higher-magnification imagesof the indicated intercellular bridge regions (bottom images). (b) Abnormalaccumulation of PtdIns(4,5)P2 in Lowe patient renal cells, which expressedthe inactive OCRLG421E mutant. Stainings and quantifications as described
in a. Mean±s.e.m., n=30 cells analysed per experiment, four independentexperiments. *** P =3×10−3 (Student’s t -test). (c) Abnormal accumulationof F-actin in HeLa cells transfected with OCRL or Rab35 siRNAs. Left,cells were stained with the fluorescently labelled phalloidin (green)and for Aurora B kinase (red). Blue, DAPI staining. Scale bars, 10 µm.Right, quantification of the mean intensity of the phalloidin staining ineach condition (mean± s.e.m., n = 30 cells analysed per experiment,four independent experiments). ** P = 9×10−3 and *** P = 5×10−3
(Student’s t -test). (d) Abnormal accumulation of F-actin in Lowe patientrenal cells, which expressed the inactive OCRLG421E mutant. Stainings andquantifications were carried out as described in c. Mean±s.e.m., n=30 cellsanalysed per experiment, four independent experiments. *** P =5×10−3
(Student’s t -test).
hypothesis that a local increase in the PtdIns(4,5)P2 levels may modifyF-actin amounts at the intercellular bridge and perturb cytokinesisabscission. Live-cell imaging using mCherryFP–PH PLC-δ (ref. 7) andmCherryFP–LifeAct (ref. 32) probes revealed that both PtdIns(4,5)P2
and F-actin abnormally persisted in late intercellular bridges in OCRL-and Rab35-depleted cells (Supplementary Fig. S5). In agreementwith these observations, F-actin levels in fixed cells were stronglyincreased by 75% and 70% in late intercellular bridges in OCRL-and Rab35-depleted cells, respectively, when compared with controlcells (Fig. 4c). Importantly, abnormally high levels (+73%) of F-actincytoskeleton within late intercellular bridges were also found inLowe patient cells (Fig. 4d). We previously reported that cells thatoverexpressed high levels of the dominant-negative Rab35S22N mutantexhibited high levels of PtdIns(4,5)P2 and SEPTIN2 on numerousintracellular vacuoles that formed before entry into mitosis7. Asa consequence, PtdIns(4,5)P2 was not properly localized at earlycytokinesis bridges, and these cells became binucleated soon afterfurrow ingression7. This was observed for 73% of the cells thathighly overexpressed Rab35S22N (upper 25th percentile of the cellpopulation, n= 225). In contrast, only 13.6% of the cells that expressedlower levels of Rab35S22N became binucleated. Instead, these cellsexhibited late cytokinesis defects with abnormal abscission timing(mean abscission time was 491min versus 292min in controls, n= 87cells, P < 10−7, Student’s t -test), associated with an increase in thelevels of PtdIns(4,5)P2 and F-actin by 40% and 62% respectively inlate cytokinesis bridges (n= 20–30 cells analysed for each of three
independent experiments, P = 10−3 and P = 3× 10−3, Student’st -test). Thus, an abnormal increase in PtdIns(4,5)P2 and F-actin inlate bridges associated with delayed abscission is observed both inRab35S22N low-expressing cells and in Rab35 RNAi cells (in whichRab35 depletion is not complete, see Fig. 2d). Connecting elevatedPtdIns(4,5)P2 (Fig. 4a,b) and F-actin accumulation (Fig. 4c,d) incytokinesis bridges, we observed that the amount of the F-actinnucleator complex Arp2/3 (actin-related protein 2/3), whose activityis strongly activated by PtdIns(4,5)P2 (ref. 33), was clearly increasedinside intercellular bridges in Rab35- and OCRL-depleted cells, orin Lowe patient cells (Supplementary Fig. S6a,b). Of note, and asobserved for PtdIns(4,5)P2 (Supplementary Fig. S4a), global levelsof F-actin (outside the intercellular bridge) were modestly increased(<20%) in HeLa cells depleted for OCRL or Rab35, and in Lowepatient cells (Supplementary Fig. S4b). Together, these results indicatethat cytokinesis abscission defects in Rab35- and OCRL-depleted cells,or in Lowe patient cells, are associated with locally high levels ofPtdIns(4,5)P2 and F-actin at the intercellular bridge.To directly investigate whether abnormal F-actin accumulation
was the cause of the cytokinesis defects, we treated Rab35- or OCRL-depleted cells or Lowe patient cells with very low amounts (1–4 nM) ofthe F-actin depolymerizing drug latrunculin-A (LatA). In either control-siRNA-treated HeLa cells or in renal cells from donors not affected byLowe syndrome, the addition of LatA did not modify the amount ofF-actin detected in the intercellular bridges during cytokinesis (Fig. 5aand Supplementary Fig. S6c,d). However, in OCRL- or Rab35-siRNA-
986 NATURE CELL BIOLOGY VOLUME 13 | NUMBER 8 | AUGUST 2011
© 2011 Macmillan Publishers Limited. All rights reserved.

L E T T ERS
1 6 11 16 21 260
20
40
60
80
100
Cum
ula
tive p
erc
enta
ge o
f cells
th
at
co
mp
lete
d a
bscis
sio
n
Lowe cells + DMSO
Normal cells + DMSO
Lowe cells + LatA
Normal cells + LatA
Time after furrow ingression (h) Time after furrow ingression (h)
Cum
ula
tive p
erc
enta
ge o
f cells
th
at
co
mp
lete
d a
bscis
sio
n
0
20
40
60
80
100
2 7 12 17 22 27 32 37
Control + DMSOControl + LatAOCRL siRNA + DMSOOCRL siRNA + LatARab35 siRNA + DMSORab35 siRNA + LatA
∗∗n.s.
∗∗n.s.a b
c d e
Ab
scis
sio
n t
ime (h) 12
14
10
8
6
4
2
0
n.s.
∗∗
6
4
2
8
0
Ab
scis
sio
n t
ime (h)
Normal cells Lowe cells
DMSO
DMSO DMSO
DMSO DMSO DMSO DMSO DMSO DMSO DMSO
∗∗∗n.s.
Control OCRL Rab35siRNA:Normal cells Lowe cellsControl OCRL Rab35siRNA:
LatA
LatA LatA
LatA LatA LatA LatA LatA LatA LatA
∗∗∗n.s.
∗∗∗n.s.
F-a
ctin lab
elli
ng
at
the inte
rcellu
lar
brid
ge (p
erc
enta
ge o
f co
ntr
ol)
0
100
50
150
200
250
Figure 5 Abscission defects observed in cells depleted for OCRL or Rab35and in Lowe syndrome patient cells are suppressed by low doses of F-actindepolymerizing drugs. (a) Abnormal F-actin accumulation in HeLa cellsdepleted for OCRL or Rab35 (left) and in Lowe patient cells (right) issuppressed by LatA treatment. HeLa cells transfected with control, OCRLor Rab35 siRNAs were treated with either dimethylsulphoxide (DMSO) orDMSO+4nM LatA, as indicated. Renal cells from a donor not mutatedin OCRL (normal cells) and from the Lowe patient were treated witheither DMSO or DMSO+1nM LatA, as indicated. F-actin intensitiesat the intercellular bridges were quantified as described in Fig. 4c,d(mean± s.e.m., n = 90–150 cells, 3–5 independent experiments). (b)HeLa cells depleted for OCRL or Rab35 were treated with either DMSO orDMSO+LatA as described in a. Mean abscission times in each conditionwere measured by video microscopy (mean±s.e.m., n =60–77 cells, threeindependent experiments). (c) Normal and Lowe patient cells were treated
with either DMSO or DMSO+LatA as described in a and abscission timeswere measured as described in b (mean±s.e.m., n = 59–80 cells, threeindependent experiments). (d) Low doses of LatA are sufficient to restorenormal distributions of abscission times in HeLa cells depleted for OCRLor Rab35. The cells were treated with either DMSO or DMSO+LatA asdescribed in b. Distributions were plotted as described in Fig. 3b. (e) Lowdoses of LatA are sufficient to restore normal distributions of abscissiontimes in Lowe patient cells. Normal and Lowe patient cells were treatedwith either DMSO or DMSO+LatA as described in c. Distributions wereplotted as described in Fig. 3g. Whereas the distributions between red,blue and light green curves to their respective controls were different, withP <1.5×10−2–10−5 (Kolmogorov–Smirnov test), the distributions betweenblack, grey, cyan and yellow curves (d) or between black, grey and darkgreen curves (e) were not significantly different. n.s.= not significant. ***P <10−3 and ** P <2×10−2 (Student’s t -test).
treatedHeLa cells, or in Lowe syndrome renal cells, such LatA treatmentwas sufficient to restore normal amounts of F-actin at the intercel-lular bridges (Fig. 5a and Supplementary Fig. S6c,d). Remarkably,cytokinesis defects associated with OCRL depletion or with Rab35depletion were completely rescued by LatA treatment (Fig. 5b,d). Inaddition, normal abscission was also observed in Lowe patient cellstreated with LatA (Fig. 5c,e). These observations demonstrate thatabnormal F-actin levels are responsible for the defects in cytokinesisabscission observedwhenRab35 orOCRL functions are impaired.Our results indicate that Rab35- and OCRL-dependent remodelling
of lipids and F-actin is important for the last step of cell division.It has been suggested that hydrolysis of PtdIns(4,5)P2 is involvedin post-furrowing steps of cytokinesis34. Here, we demonstrate thatPtdIns(4,5)P2 hydrolysis is required for normal cytokinesis abscission,that the OCRL phosphatase is responsible for PtdIns(4,5)P2 hydrolysisduring cytokinesis, and that PtdIns(4,5)P2 hydrolysis is requiredfor preventing F-actin accumulation in late cytokinesis bridges, andconsequently for the physical separation of the daughter cells. Wepropose that the Rab35 GTPase locally regulates PtdIns(4,5)P2 levelsand thus cytokinesis abscission by controlling the localization of a
pool of the OCRL phosphatase at the intercellular bridge. In addition,impairing the Rab35–OCRL abscission pathway does not perturbthe localization of CHMP4B (component of the ESCRT abscissionpathway35–37) or PtdIns(3)P (component of the FYVE-CENT abscissionpathway38; Supplementary Fig. S7). Thus, several pathways mustcooperate to remodel lipids and cytoskeletons to allow cytokinesiscompletion. Our work also reveals an unexpected function in celldivision for OCRL, whose inactivation is responsible for Lowesyndrome and for 15% of cases of Dent disease. Lowe syndrome isa pleiotropic genetic disease characterized by reabsorption defects inrenal cells, congenital cataracts,mental retardation and aggressiveness13.In addition to endocytic recycling defects, abnormal cytokinesisabscission in specific cell types during development may also explainsome of the observed neurological and congenital defects. Finally, ithas previously been reported that short F-actin cytoplasmic filamentsare more frequent in Lowe syndrome patient fibroblasts in interphase,indicating abnormal F-actin dynamics in patient cells39. Strikingly,during cell division, cytokinesis defects in Lowe patient cells can berescued by the addition of F-actin depolymerizing agents at a dose thatdoes not perturb wild-type cells. This raises the possibility that other
NATURE CELL BIOLOGY VOLUME 13 | NUMBER 8 | AUGUST 2011 987
© 2011 Macmillan Publishers Limited. All rights reserved.

L E T T ERS
phenotypes associated with OCRL inactivation may also be correctedby reducing excessive F-actin levels and could represent a potentialtherapeutic lead, as recently suggested for other diseases40. �
METHODSMethods and any associated references are available in the onlineversion of the paper at http://www.nature.com/naturecellbiology
Note: Supplementary Information is available on the Nature Cell Biology website
ACKNOWLEDGEMENTSWe thank R. Woscholski (Imperial College, London, UK), O. Dorseuil (InstitutCochin, Paris, France), A. Gautreau (CNRS Lebs, Gif, France), A. Alcover (InstitutPasteur, Paris), S. Miserey-Lenkei, P. Benaroch and F. Nagano (Institut Curie, Paris,France) for providing reagents and plasmids; V. Chauvet, A. Gautreau, A. Houdusse,B. Payrastre, N. Vitale and R. Weil for helpful discussions; S. Miserey-Lenkei andE. Crowell for critical reading of the manuscript; F. Legendre (Hôpital Necker, Paris,France) for the establishment of patients cell lines; and J. Lunardi (CHU Grenoble,France) and G. Baujat for the pediatric network. We thank the ‘Association duSyndrome de Lowe’, patients and parents. We thank the Plate-Forme d’ImagerieDynamique (PFID) and Imagopole, Institut Pasteur, for microscopes and assistance.The authors thank V. Fraisier, L. Sengmanivong, J-B. Sibarita and J. Salamero forsupport in microscopy, and acknowledge the Nikon Imaging Centre at InstitutCurie-CNRS. This work has been supported by the Institut PASTEUR (G5 program),the Institut CURIE, the CNRS, the INSERM, the Agence Nationale pour laRecherche (grants ANR-Maladies Rares, GIS-Maladies Rares to B.G. and R.S., andANR 07-JCJC-0089 to A.E.) and the Schlumberger Foundation for Education andResearch—FSER (A.E.). D.D. and M.M. have been supported by the Ministère de laRecherche et de l’Enseignement Supérieur, and D.D. and L.C. have been supportedby the Association pour la Recherche sur le Cancer.
AUTHOR CONTRIBUTIONSD.D., M.M., L.C., M.S. and A.E. designed and analysed the experiments; D.D.,M.M., L.C., M.S., M.R., A.E.M., E.F. and A.E. did the experimental work; R.S. andB.G. provided reagents; D.D., M.M., L.C., M.S., B.G. and A.E. wrote or editedthe manuscript.
COMPETING FINANCIAL INTERESTSThe authors declare no competing financial interests.
Published online at http://www.nature.com/naturecellbiologyReprints and permissions information is available online at http://www.nature.com/reprints
1. Glotzer, M. The molecular requirements for cytokinesis. Science 307,1735–1739 (2005).
2. Eggert, U. S., Mitchison, T. J. & Field, C. M. Animal cytokinesis: from parts list tomechanisms. Annu. Rev. Biochem. 75, 543–566 (2006).
3. Barr, F. A. & Gruneberg, U. Cytokinesis: placing and making the final cut. Cell 131,847–860 (2007).
4. Echard, A. Membrane traffic and polarization of lipid domains during cytokinesis.Biochem. Soc. Trans. 36, 395–399 (2008).
5. Steigemann, P. & Gerlich, D. W. Cytokinetic abscission: cellular dynamics at themidbody. Trends Cell Biol. 19, 606–616 (2009).
6. Montagnac, G., Echard, A. & Chavrier, P. Endocytic traffic in animal cell cytokinesis.Curr. Opin. Cell Biol. 20, 454–461 (2008).
7. Kouranti, I., Sachse, M., Arouche, N., Goud, B. & Echard, A. Rab35 regulates anendocytic recycling pathway essential for the terminal steps of cytokinesis. Curr. Biol.16, 1719–1725 (2006).
8. Lowe, M. Structure and function of the Lowe syndrome protein OCRL1. Traffic 6,711–719 (2005).
9. Vicinanza, M., D’Angelo, G., Di Campli, A. & De Matteis, M. A. Functionand dysfunction of the PI system in membrane trafficking. EMBO J. 27,2457–2470 (2008).
10. Echard, A., Hickson, G. R., Foley, E. & O’Farrell, P. H. Terminal cytokinesis eventsuncovered after an RNAi screen. Curr. Biol. 14, 1685–1693 (2004).
11. Eggert, U. S. et al. Parallel chemical genetic and genome-wide RNAi screens identifycytokinesis inhibitors and targets. PLoS Biol. 2, e379 (2004).
12. Skop, A. R., Liu, H., Yates, J., III, Meyer, B. J. & Heald, R. Dissection of themammalian midbody proteome reveals conserved cytokinesis mechanisms. Science305, 61–66 (2004).
13. Lowe, C. U., Terrey, M. & Mac, L. E. Organic-aciduria, decreased renal ammoniaproduction, hydrophthalmos, and mental retardation; a clinical entity. Am. J. Dis.Child 83, 164–184 (1952).
14. Attree, O. et al. The Lowe’s oculocerebrorenal syndrome gene encodes aprotein highly homologous to inositol polyphosphate-5-phosphatase. Nature 358,239–242 (1992).
15. Hyvola, N. et al. Membrane targeting and activation of the Lowe syndrome proteinOCRL1 by rab GTPases. EMBO J. 25, 3750–3761 (2006).
16. Fukuda, M., Kanno, E., Ishibashi, K. & Itoh, T. Large scale screening for novel rabeffectors reveals unexpected broad Rab binding specificity. Mol. Cell Proteomics 7,1031–1042 (2008).
17. Hou, X. et al. A structural basis for Lowe syndrome caused by mutations in theRab-binding domain of OCRL1. EMBO J. 30, 1659–1670 (2011).
18. Wilson, G. M. et al. The FIP3-Rab11 protein complex regulates recycling endosometargeting to the cleavage furrow during late cytokinesis. Mol. Biol. Cell 16,849–860 (2005).
19. Choudhury, R. et al. Lowe syndrome protein OCRL1 interacts with clathrin andregulates protein trafficking between endosomes and the trans-Golgi network. Mol.Biol. Cell 16, 3467–3479 (2005).
20. Faucherre, A. et al. Lowe syndrome protein Ocrl1 is translocated to membrane rufflesupon Rac GTPase activation: a new perspective on Lowe syndrome pathophysiology.Hum. Mol. Genet. 14, 1441–1448 (2005).
21. Erdmann, K. S. et al. A role of the Lowe syndrome protein OCRL in early steps ofthe endocytic pathway. Dev. Cell 13, 377–390 (2007).
22. Choudhury, R., Noakes, C. J., McKenzie, E., Kox, C. & Lowe, M. Differential clathrinbinding and subcellular localization of OCRL1 splice isoforms. J. Biol. Chem. 284,9965–9973 (2009).
23. Sato, M. et al. Regulation of endocytic recycling by C. elegans Rab35 and itsregulator RME-4, a coated-pit protein. EMBO J. 27, 1183–1196 (2008).
24. Zhang, J., Fonovic, M., Suyama, K., Bogyo, M. & Scott, M. P. Rab35 controlsactin bundling by recruiting fascin as an effector protein. Science 325,1250–1254 (2009).
25. Allaire, P. D. et al. The Connecdenn DENN domain: a GEF for Rab35 mediatingcargo-specific exit from early endosomes. Mol. Cell 37, 370–382 (2010).
26. Hichri, H. et al. From Lowe syndrome to Dent disease: correlations betweenmutations of the OCRL1 gene and clinical and biochemical phenotypes. Hum. Mutat.32, 379–388 (2011).
27. Zhang, X., Jefferson, A. B., Auethavekiat, V. & Majerus, P. W. The protein deficientin Lowe syndrome is a phosphatidylinositol-4,5-bisphosphate 5-phosphatase. Proc.Natl Acad. Sci. USA 92, 4853–4856 (1995).
28. Schmid, A. C., Wise, H. M., Mitchell, C. A., Nussbaum, R. & Woscholski, R. TypeII phosphoinositide 5-phosphatases have unique sensitivities towards fatty acidcomposition and head group phosphorylation. FEBS Lett. 576, 9–13 (2004).
29. Hammond, G. R., Schiavo, G. & Irvine, R. F. Immunocytochemical techniques revealmultiple, distinct cellular pools of PtdIns4P and PtdIns(4,5)P(2). Biochem. J. 422,23–35 (2009).
30. Begle, A., Tryoen-Toth, P., de Barry, J., Bader, M. F. & Vitale, N. ARF6 regulatesthe synthesis of fusogenic lipids for calcium-regulated exocytosis in neuroendocrinecells. J. Biol. Chem. 284, 4836–4845 (2009).
31. Di Paolo, G. & De Camilli, P. Phosphoinositides in cell regulation and membranedynamics. Nature 443, 651–657 (2006).
32. Miserey-Lenkei, S. et al. Rab and actomyosin-dependent fission of transport vesiclesat the Golgi complex. Nat. Cell Biol. 12, 645–654 (2010).
33. Ma, L., Cantley, L. C., Janmey, P. A. & Kirschner, M. W. Corequirement of specificphosphoinositides and small GTP-binding protein Cdc42 in inducing actin assemblyin Xenopus egg extracts. J. Cell Biol. 140, 1125–1136 (1998).
34. Wong, R. et al. PIP2 hydrolysis and calcium release are required for cytokinesis inDrosophila spermatocytes. Curr. Biol. 15, 1401–1406 (2005).
35. Carlton, J. G. & Martin-Serrano, J. Parallels between cytokinesis and retroviralbudding: a role for the ESCRT machinery. Science 316, 1908–1912 (2007).
36. Lee, H. H., Elia, N., Ghirlando, R., Lippincott-Schwartz, J. & Hurley, J. H. Midbodytargeting of the ESCRT machinery by a noncanonical coiled coil in CEP55. Science322, 576–580 (2008).
37. Guizetti, J. et al. Cortical constriction during abscission involves helices ofESCRT-III-dependent filaments. Science 331, 1616–1620 (2011).
38. Sagona, A. P. et al. PtdIns(3)P controls cytokinesis through KIF13A-mediatedrecruitment of FYVE-CENT to the midbody. Nat. Cell Biol. 12, 362–371 (2010).
39. Suchy, S. F. & Nussbaum, R. L. The deficiency of PIP2 5-phosphatasein Lowe syndrome affects actin polymerization. Am. J. Hum. Genet. 71,1420–1427 (2002).
40. Kim, J. et al. Functional genomic screen for modulators of ciliogenesis and ciliumlength. Nature 464, 1048–1051 (2010).
988 NATURE CELL BIOLOGY VOLUME 13 | NUMBER 8 | AUGUST 2011
© 2011 Macmillan Publishers Limited. All rights reserved.

DOI: 10.1038/ncb2279 METHODS
METHODSAntibodies, reagents and plasmids. The following antibodies were usedfor immunocytochemistry experiments: mouse anti-acetylated-tubulin antibody(611B1, Sigma-Aldrich); rabbit anti-MKLP2 (ref. 41); mouse anti-Aurora Bantibody (AIM-1, BD Bioscience); human anti-tubulin (F2C, gift from F. Perez,Institut Curie, Paris, France, ref. 42); mouse anti-PtdIns(4,5)P2 (2C11, SantaCruz Biotechnology); mouse anti-GM130 (BD Biosciences); mouse anti-p34-Arc(Upstate); rabbit anti-mouse IgG (Dako). Protein A gold was obtained from theCell Microscopy Center, UMC, Utrecht, The Netherlands. Rabbit antisera againstrecombinant full-length human OCRL-b tagged with hexahistidine (6×His) wereraised by Covalab and affinity-purified on GST recombinant full-length humanOCRL-b protein, as described in ref. 41. Fluorescently labelled secondary antibodieswere purchased from Jackson Laboratories. Phalloidin–rhodamine and LatA werepurchased from Molecular Probes. Anti-β-tubulin (Clone Tub 2.1, Sigma Aldrich),rabbit anti-Rab5 (BD Bioscience), anti-GFP (Roche), anti-PLC-γ1 (clone B-6-4,Millipore, gift from A. Alcover, Institut Pasteur, Paris), anti-6×His (Clone HIS-1,Sigma Aldrich) and secondary horseradish-peroxidase-coupled antibodies (JacksonLaboratories) were used in western blot experiments. Chromogenic TMB substratewas purchased from BD Biosciences. pEGFP–OCRL-b (gift from F. Nagano, InstitutCurie, Paris), pGEX–OCRL 5-phosphatase domain (gift from R. Woscholski,Imperial College, London, UK, ref. 28) and pEGFP–2×FYVE and pFlag–CHMP4B(gifts from P. Benaroch, Institut Curie, Paris) were used. Other plasmids weredescribed in refs 7,32. Standard molecular biology protocols were used to transferOCRL into pGAD and pmCherryFP. Mutants were obtained by QuickChange(Invitrogen).
Patient cell lines. The Lowe patient renal cell line has been established as previouslydescribed in ref. 43, after informed consent was obtained from the patient and hisparents, in accordance with French law. The patient was a male juvenile admittedto the Paediatric Nephrology Unit who fulfilled the following diagnosis criteria forLowe Syndrome: bilateral congenital cataracts, proximal tubulopathy and mentalretardation. OCRL was screened for mutations by single-strand conformationanalysis sequencing techniques as previously described44. The G421E mutationdisrupted the highly conserved amino-acid stretch WxGDxN(Y/F)R found in5-phosphatase.
Cell culture and transfection. HeLa cells were grown in DMEM medium(Gibco BRL) supplemented with 10% fetal bovine serum, 100Uml−1 peni-cillin/streptomycin and 2mM glutamine. Lowe cells and control cells were grownin DMEM/F12 (GIBCO BRL) supplemented with 10% fetal bovine serum, in-sulin transferrin selenium, 4 pgml−1 triiodothyronine, 36 ngml−1 dexamethasone,10 ngml−1 EGF, 100Uml−1 penicillin/streptomycin and 2mM glutamine at 33 ◦C.Cells were seeded onto six-well plates, 35-mm dishes (Iwaki) or 12-mm glasscoverslips and grown for 24 h before transfection. For expression of the constructsused in this study, HeLa cells or Lowe cells were transfected using either thecalcium phosphate precipitation method or FuGENE-6 (Roche) following themanufacturer’s instructions. For silencing experiments, HeLa cells were transfectedwith the corresponding siRNA using HiPerFect (Qiagen) following the manu-facturer’s instructions. Plasmids were transfected for 24 h (Figs 2b,c, 3d,f andSupplementary Figs S3d and S7) or 72 h (other figures). siRNAs were transfectedfor 72 h.
RNAi. The sequences of the siRNAs used in this study are as follows: humanRab35, 5′-GCUCACGAAGAACAGUAAA-3′ (ref. 7, Dharmacon); human OCRL,5′-GAAAGGAUCAGUGUCGAUA-3′ (from a SMARTpool, Dharmacon); humanRab5A, 5′-AAAGGAAUCAGUGUUGUAGUA-3′ (ref. 45); control luciferase,5′-CGUACGCGGAAUACUUCGA-3′ (ref. 7, Proligo-Sigma).
Yeast two-hybrid screen and experiments. A yeast two-hybrid screen withpLexA human Rab35Q67L as a bait was carried out by Hybrigenics SA, using a humanplacenta complementaryDNAGal4 activating domain (GAD)-fusion library. A totalof 1.08×108 diploids were tested. Subsequent yeast two-hybrid experiments werecarried out as described previously41.
Co-immunoprecipitation. HeLa cells were co-transfected with GFP–OCRL andeitherMyc-tagged wild-type Rab35, Rab35S22N or Rab35Q67L. Cells were lysed in lysisbuffer (20mM Tris at pH 7.4, 100mM KCl, 2mM MgCl2, 10% glycerol and 0.1%NP-40). Post-nuclear supernatants were incubated with Protein G Sepharose Beads(Protein G Sepharose 4 FastFlow, GE HealthCare) for 30min (preclarification).Supernatants were then incubated with monoclonal mouse anti-Myc antibody(9E10) and Protein G Sepharose beads for 1 h 30min. After the washes, beadswere lysed in Laemmli buffer and boiled at 95 ◦C for 5min. The amount ofco-immunoprecicipated OCRL in each condition was probed by western blotanalysis using mouse anti-GFP antibodies (1:1,000).
Bacterial expression and recombinant protein purifications. GST fused tothe wild-type, G421E or H524R OCRL 5-phosphatase domain (Val 202–Glu 618)was expressed in the BL21 pLysS strain of Escherichia coli after inductionwith 100 µM isopropyl-β-d-thiogalactopyranoside at 18 ◦C overnight. Cells werelysed in 50mM HEPES at pH 7.5, 3mM MgCl2, 2mM EGTA, 500mM NaCl,1% Triton X-100 and 1mM dithiothreitol. The GST fusion proteins wereaffinity-purified using glutathione Sepharose 4B (Pharmacia) and eluted with25mM HEPES at pH 7.5, 1mM EGTA, 500mM NaCl, 3mM MgCl2 and 20mMreduced glutathione. GST-fused wild-type Rab11A or Rab35, 6×His–Rab1AQ70L,6×His–Rab5AQ79L, 6×His–Rab6AQ72L or 6×His–Rab35Q67L, and wild-type6×His–Rab6A, 6×His–Rab11AQ72L or 6×His–Rab35 were purified as describedpreviously32. GST-fused OCRL-b (540–893) was expressed in the BL21 strain afterinduction with 500 µM isopropyl-β-d-thiogalactopyranoside at 20 ◦C overnight.Cells were lysed in PBS, 1mM dithiothreitol and 1mgml−1 lysozyme. Proteinsfused toGSTwere affinity-purified using glutathione Sepharose 4B (Pharmacia) andeluted with 25mMHEPES at pH 7.5, 1mMEGTA, 500mMNaCl, 3mMMgCl2 and20mM reduced glutathione.
GST-pulldown, solid-phase assays and western blot experiments. To investi-gate the interactions between GST–Rab35 and endogenous OCRL, HeLa cells weretrypsinized, washed twice in cold PBS and incubated on ice for 30min in lysis buffer(25mMTris at pH 7.5, 10mMMgCl2, 150mMNaCl and 0.1% Triton X-100). Cellswere centrifuged for 20min at 20,000g to remove cell fragments. The extracts wereprocessed for GST-pulldown as described previously32, except that the incubationbuffer used was 25mMTris at pH 7.5, 10mMMgCl2, 50mMNaCl and 0.1% TritonX-100. Western blotting experiments were carried out as described previously32
(anti-β-tubulin 1:1,000; anti-OCRL 1:200; anti-Rab35 1:1,000; anti-PLC-γ 1:10,000;anti-Rab5 1:500).
Direct interaction between 6×His–Rab wild-type or active mutant andGST–OCRL ASH–RhoGAP was carried out as previously described32. 6×His–Rab was incubated with GST or GST–OCRL(540–893) for 90min at 4 ◦C.6×His–Rab bound to GST or GST–OCRL was detected by western blot analysisusing anti-6×His antibodies (1:1,000). Solid-phase assays were carried out asdescribed previously15, except that 96-well plates (Nunc) were coated with 10 µg ofeither GST or GST–OCRL(450-893) and that 0–250 ng recombinant 6×His–RabGTPase-defective mutant proteins was used. Protein interaction was revealedby anti-6×His antibodies (1:100), using 100 µl TMD (BD Biosciences) as achromogenic substrate at room temperature for 10min. The reactions were stoppedby the addition of sulphuric acid. Absorbance was measured at 450 nm in aplate reader.
In vitro PtdIns(4,5)P2 phosphatase malachite green assay. The enzymaticactivity of the 5-phosphatase domain of wild-type and mutants of OCRL wasmeasured using the malachite green assay (protein tyrosine phosphatase assay,Sigma Aldrich) as previously described28. This assay was carried out using50 µMwater-soluble phosphoinositides dibutanoyl (diC4)-PtdIns(4,5)P2 (Echelon),in a 5min reaction at room temperature.
Immunofluorescence microscopy. HeLa and Lowe patient cells were grown oncoverslips and fixed in 4% paraformaldehyde for 10min at room temperature,except for OCRL and PtdIns(4,5)P2 stainings, and processed for immunoflu-orescence microscopy as previously described7. For OCRL labelling, cells werefixed in 1% paraformaldehyde and 0.1% Triton X-100 for 5min at 4 ◦C. Foranti-PtdIns(4,5)P2, immunofluorescence microscopy was carried out accordingvalidated protocols described in ref. 29. The following primary antibodies were used:anti-OCRL (1:100), anti-PtdIns(4,5)P2 (1:900), anti-GM130 (1:1,000), anti-AuroraB (1:250), anti-acetylated-tubulin (1:20,000), anti-Flag (1:200), anti-p34Arc (1:500)and anti-MKLP2 (1:2,000). Labelled phalloidin 1:2,000. For quantitative analysis,cells were examined under an inverted microscope (Leica DM-RXA2), equippedwith a ×60 1.4NA PL-APO objective lens for optical sectioning. Image stacks wereacquired usingMetamorph software (MDS) through a cooled CCD (charge-coupleddevice) camera (Photometrics Coolsnap HQ). Other images were acquired with anA1r inverted Ti E Nikon confocal microscope controlled by NIS element software,using a ×100 1.4NA PL-APO objective lens. The following lasers were used:405LD:max. 38mW,Multi-Ar (457/488/514): max. 65mW, 488DPSS:max. 75mW,561DPSS: max. 25mW, 543HeNemax: 1mW, 638LD: max. 10mW PMT.
Correlative light electron microscopy. Cells were seeded on MatTek glass-bottom dishes (MatTek) with a gridded coverslip coated with poly-lysine (Sigma).After co-transfection with GFP–OCRL-b and wild-type Myc-tagged Rab35, cellswere fixed with 2% formaldehyde+ 0.05% glutaraldehyde in 0.1M Soerensen’sphosphate buffer at pH 7.2. Transfected cells in late cytokinesis were selected by lightmicroscopy. Flat-embedding for Tokuyasu sectioning was carried out as describedelsewhere46. Selected areas were cut out with a piece of razor blade. To visualize
NATURE CELL BIOLOGY
© 2011 Macmillan Publishers Limited. All rights reserved.

METHODS DOI: 10.1038/ncb2279
cells of interest blocks were stained briefly with 1% toluidine blue in 2.3% sucrosesolution before freezing. Cryosections were cut with a nominal feed of 70 nmusing a UC6/FC6 (Leica Microsystems) and picked up with a 1:1 mixture of 2%methylcellulose and 2.3M sucrose. Double labelling using protein A gold was doneas previously described47 and was observed with a JEOL 1010 instrument operatedat 80 kV, equipped with a KeenView camera (Olympus, Soft Imaging Systems).
Time-lapse microscopy. For time-lapse phase-contrast microscopy, transfectedcells were plated on 35mm glass dishes (Iwaki) and put in an open chamber (LifeImaging) equilibrated in 5% CO2 and maintained at 37 ◦C. Time-lapse sequenceswere recorded at 10min for 60 h on a Nikon Eclipse Ti inverted microscope with a×20 0.45NAPlan Fluor ELWDobjective lens controlled byMetamorph 6.1 software(Universal Imaging). This microscope was equipped with a cooled CCD camera(HQ2; Roper Scientific).
For time-lapse fluorescent spinning-disc confocal microscopy, transfected cellswere plated on 35mm glass dishes (Iwaki) filled with culture medium. Time-lapseimaging was carried out at 37 ◦C using a spinning-disc microscope mounted on aninvertedmotorizedmicroscope (Nikon TE2000-U) through a×100 1.4NAPL-APOobjective lens. The apparatus is composed of a Yokogawa CSU-22 spinning-dischead, a Roper Scientific laser lounge, a Photometrics Coolsnap HQ2 CCD camerafor image acquisition and Metamorph software (MDS) to control the set-up.Acquisition parameters were 100ms exposure for the GFP channel and 100ms forthemCherry channel. The laser was set to 60% in each case. The images shown in thefigures correspond to the maximal intensity projection through the Z axis carriedout with Image J software (NIH Image). Tracking of the mCherry–OCRL vesicleswas carried out with the Manual tracking plugin on Image J software (NIH Image).
Quantitative analysis. For quantification of OCRL, PtdIns(4,5)P2 and F-actinlabelling at the intercellular bridge, 30 transfected cells were imaged for each
condition in at least three independent experiments. Images of control ortransfected/depleted cells were acquired using the same parameters, withoutautomatic scaling and gain adjustment, avoiding saturated pixels. In all cases, theintercellular bridge area was defined by Aurora B or tubulin labelling, and the meanintensity of bridge-associatedOCRL, PtdIns(4,5)P2 or F-actin fluorescence intensityin this area wasmeasured using Image J software (NIH Image). For quantification ofPtdIns(3)P or CHMP4B labelling at the intercellular bridge, 30 transfected cells withsiRNAs and pGFP–2×FYVE or pFlag–CHMP4B-encoding plasmids were imagedfor each condition in at least three independent experiments. Images of controlor depleted cells were acquired as described above. In all cases, the intercellularbridge area was defined by Aurora B and MKLP2 labelling, and the mean intensityof bridge-associated PtdIns(3)P or CHMP4B fluorescence intensity in this area wasmeasured using Image J software (NIH Image).41. Echard, A. et al. Interaction of a Golgi-associated kinesin-like protein with Rab6.
Science 279, 580–585 (1998).42. Nizak, C. et al. Recombinant antibodies against subcellular fractions used to track
endogenous Golgi protein dynamics in vivo. Traffic 4, 739–753 (2003).43. Racusen, L. C., Wilson, P. D., Hartz, P. A., Fivush, B. A. & Burrow, C. R.
Renal proximal tubular epithelium from patients with nephropathic cystinosis:immortalized cell lines as in vitro model systems. Kidney Int. 48, 536–543 (1995).
44. Monnier, N., Satre, V., Lerouge, E., Berthoin, F. & Lunardi, J. OCRL1 mutationanalysis in French Lowe syndrome patients: implications for molecular diagnosisstrategy and genetic counseling. Hum. Mutat. 16, 157–165 (2000).
45. Uzan-Gafsou, S. et al. Rab11A controls the biogenesis of Birbeck granules byregulating Langerin recycling and stability. Mol. Biol. Cell 18, 3169–3179 (2007).
46. Oorschot, V., de Wit, H., Annaert, W. G. & Klumperman, J. A novel flat-embeddingmethod to prepare ultrathin cryosections from cultured cells in their in situorientation. J. Histochem. Cytochem. 50, 1067–1080 (2002).
47. Slot, J. W. & Geuze, H. J. Cryosectioning and immunolabeling. Nat. Protoc. 2,2480–2491 (2007).
NATURE CELL BIOLOGY
© 2011 Macmillan Publishers Limited. All rights reserved.
S U P P L E M E N TA RY I N F O R M AT I O N
WWW.NATURE.COM/NATURECELLBIOLOGY 1
DOI: 10.1038/ncb2279
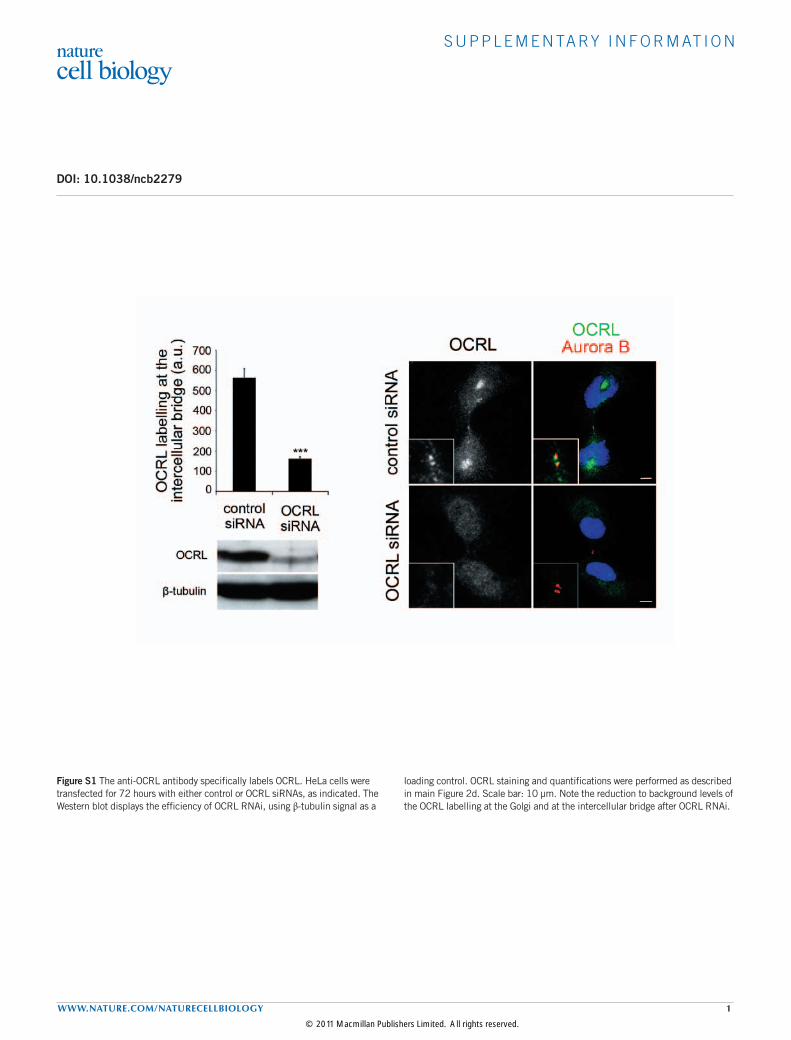
Figure S1 The anti-OCRL antibody specifically labels OCRL. HeLa cells were transfected for 72 hours with either control or OCRL siRNAs, as indicated. The Western blot displays the efficiency of OCRL RNAi, using β-tubulin signal as a
loading control. OCRL staining and quantifications were performed as described in main Figure 2d. Scale bar: 10 µm. Note the reduction to background levels of the OCRL labelling at the Golgi and at the intercellular bridge after OCRL RNAi.
© 2011 Macmillan Publishers Limited. All rights reserved.
S U P P L E M E N TA RY I N F O R M AT I O N
2 WWW.NATURE.COM/NATURECELLBIOLOGY
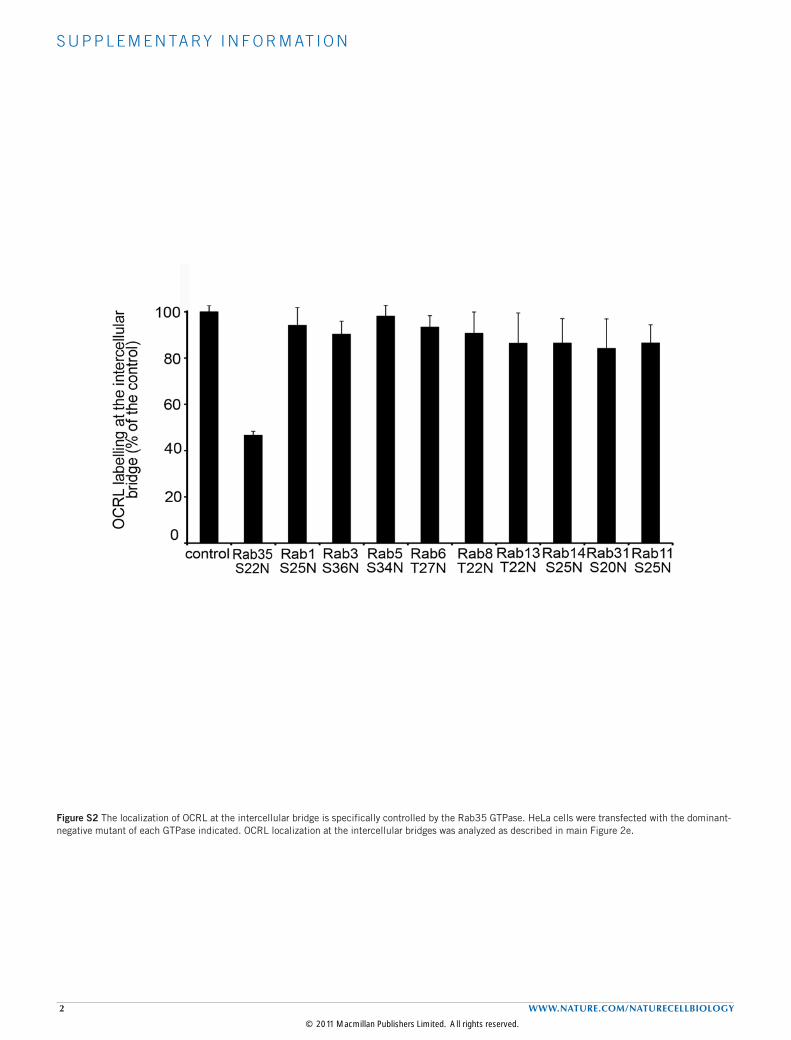
Figure S2 The localization of OCRL at the intercellular bridge is specifically controlled by the Rab35 GTPase. HeLa cells were transfected with the dominant-negative mutant of each GTPase indicated. OCRL localization at the intercellular bridges was analyzed as described in main Figure 2e.
© 2011 Macmillan Publishers Limited. All rights reserved.
S U P P L E M E N TA RY I N F O R M AT I O N
WWW.NATURE.COM/NATURECELLBIOLOGY 3
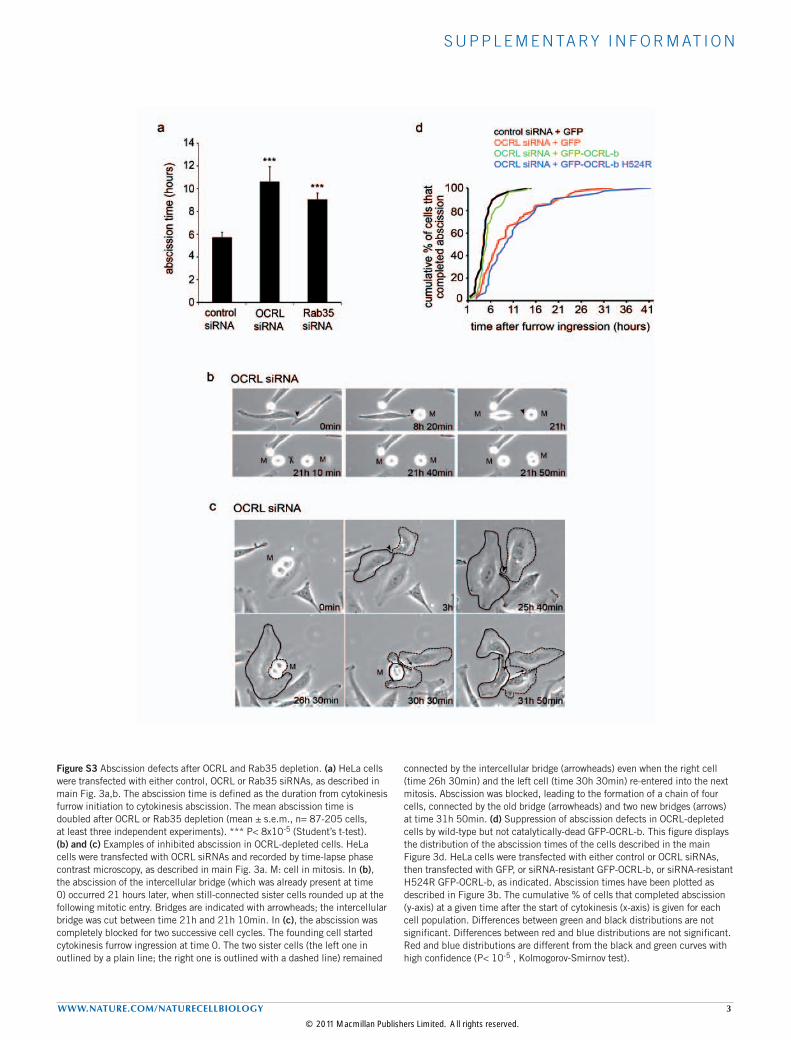
Figure S3 Abscission defects after OCRL and Rab35 depletion. (a) HeLa cells were transfected with either control, OCRL or Rab35 siRNAs, as described in main Fig. 3a,b. The abscission time is defined as the duration from cytokinesis furrow initiation to cytokinesis abscission. The mean abscission time is doubled after OCRL or Rab35 depletion (mean ± s.e.m., n= 87-205 cells, at least three independent experiments). *** P< 8x10-5 (Student’s t-test). (b) and (c) Examples of inhibited abscission in OCRL-depleted cells. HeLa cells were transfected with OCRL siRNAs and recorded by time-lapse phase contrast microscopy, as described in main Fig. 3a. M: cell in mitosis. In (b), the abscission of the intercellular bridge (which was already present at time 0) occurred 21 hours later, when still-connected sister cells rounded up at the following mitotic entry. Bridges are indicated with arrowheads; the intercellular bridge was cut between time 21h and 21h 10min. In (c), the abscission was completely blocked for two successive cell cycles. The founding cell started cytokinesis furrow ingression at time 0. The two sister cells (the left one in outlined by a plain line; the right one is outlined with a dashed line) remained
connected by the intercellular bridge (arrowheads) even when the right cell (time 26h 30min) and the left cell (time 30h 30min) re-entered into the next mitosis. Abscission was blocked, leading to the formation of a chain of four cells, connected by the old bridge (arrowheads) and two new bridges (arrows) at time 31h 50min. (d) Suppression of abscission defects in OCRL-depleted cells by wild-type but not catalytically-dead GFP-OCRL-b. This figure displays the distribution of the abscission times of the cells described in the main Figure 3d. HeLa cells were transfected with either control or OCRL siRNAs, then transfected with GFP, or siRNA-resistant GFP-OCRL-b, or siRNA-resistant H524R GFP-OCRL-b, as indicated. Abscission times have been plotted as described in Figure 3b. The cumulative % of cells that completed abscission (y-axis) at a given time after the start of cytokinesis (x-axis) is given for each cell population. Differences between green and black distributions are not significant. Differences between red and blue distributions are not significant. Red and blue distributions are different from the black and green curves with high confidence (P< 10-5 , Kolmogorov-Smirnov test).
© 2011 Macmillan Publishers Limited. All rights reserved.

S U P P L E M E N TA RY I N F O R M AT I O N
4 WWW.NATURE.COM/NATURECELLBIOLOGY
Figure S4 Modest global increase of PI(4,5)P2 and F-actin in cells depleted for OCRL or Rab35 and in Lowe syndrome patient cells. (a) The graphs display the global PI(4,5)P2 increase, outside the intercellular bridge, in OCRL- or Rab35-depleted HeLa cells, and in normal or Lowe patient cells described in Fig. 4a,b. mean± s.e.m., n=30 cells per experiment, three independent experiments. n.s.= not significant. ** P=2x10-2 (Student’s test). (b) The graphs display the global F-actin increase, outside the intercellular bridge, in OCRL- or Rab35-depleted HeLa cells, and in normal or Lowe patient cells described in Fig. 4c,d. mean± s.e.m., n=30 cells analyzed per experiment, 3-4 independent experiments. n.s.= not significant. ** P=2x10-2 *** P< 5x10-4 (Student’s test). (c) and (d) No
significant variation of PI(4,5)P2 in intercellular bridges after the functional inhibition of the Rab5 GTPase. (c) PI(4,5)P2 levels in intercellular bridges of HeLa cells transfected with plasmids encoding control or Rab5 dominant negative mutant (Rab5 S34N) were quantified as described in main Fig. 4a. mean ± s.e.m., n= 30 cells analyzed per experiment, 3 independent experiments. P= 0.3, Student’s t-test. (d) PI(4,5)P2 levels in intercellular bridges of HeLa cells transfected with control or Rab5 siRNAs were quantified as described in (c). mean ± s.e.m., n= 30 cells analyzed per experiment, 3 independent experiments. P= 0.4, Student’s t-test. n.s.= not significant. The Western blot displays the efficiency of Rab5 RNAi, using β-tubulin signal as a loading control.
© 2011 Macmillan Publishers Limited. All rights reserved.
S U P P L E M E N TA RY I N F O R M AT I O N
WWW.NATURE.COM/NATURECELLBIOLOGY 5
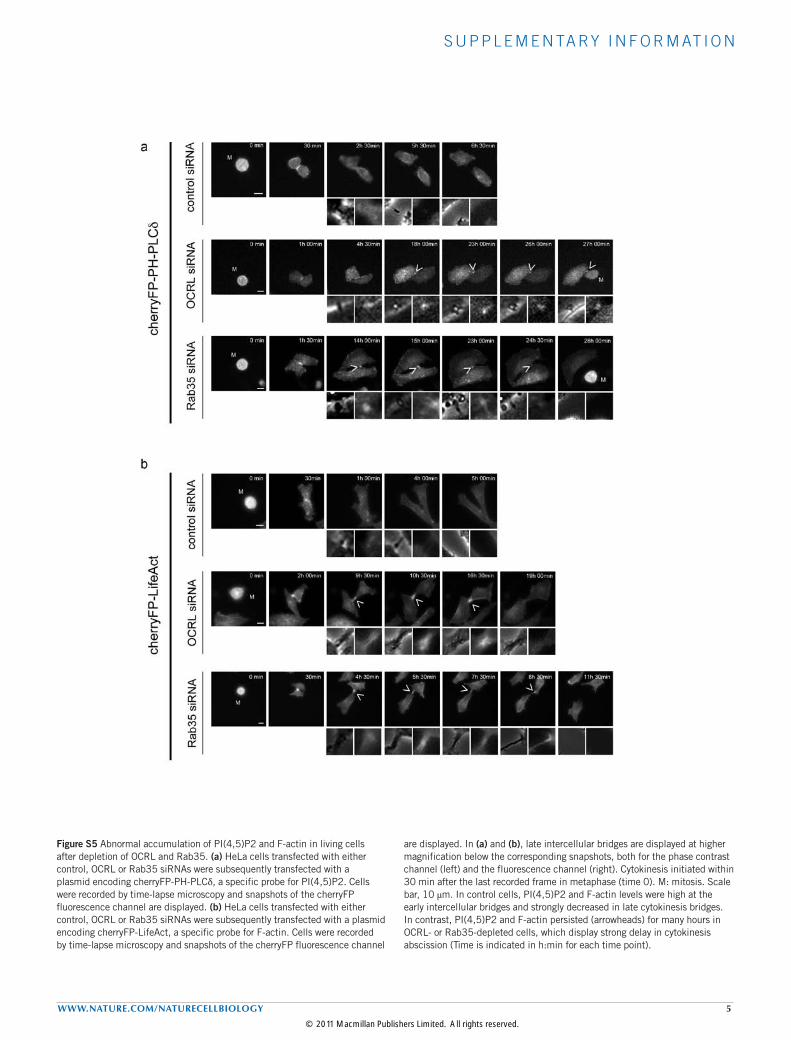
Figure S5 Abnormal accumulation of PI(4,5)P2 and F-actin in living cells after depletion of OCRL and Rab35. (a) HeLa cells transfected with either control, OCRL or Rab35 siRNAs were subsequently transfected with a plasmid encoding cherryFP-PH-PLCδ, a specific probe for PI(4,5)P2. Cells were recorded by time-lapse microscopy and snapshots of the cherryFP fluorescence channel are displayed. (b) HeLa cells transfected with either control, OCRL or Rab35 siRNAs were subsequently transfected with a plasmid encoding cherryFP-LifeAct, a specific probe for F-actin. Cells were recorded by time-lapse microscopy and snapshots of the cherryFP fluorescence channel
are displayed. In (a) and (b), late intercellular bridges are displayed at higher magnification below the corresponding snapshots, both for the phase contrast channel (left) and the fluorescence channel (right). Cytokinesis initiated within 30 min after the last recorded frame in metaphase (time 0). M: mitosis. Scale bar, 10 μm. In control cells, PI(4,5)P2 and F-actin levels were high at the early intercellular bridges and strongly decreased in late cytokinesis bridges. In contrast, PI(4,5)P2 and F-actin persisted (arrowheads) for many hours in OCRL- or Rab35-depleted cells, which display strong delay in cytokinesis abscission (Time is indicated in h:min for each time point).
© 2011 Macmillan Publishers Limited. All rights reserved.

S U P P L E M E N TA RY I N F O R M AT I O N
6 WWW.NATURE.COM/NATURECELLBIOLOGY
Figure S6 Strong increase of p34-Arc positive intercellular bridges in OCRL- or Rab35-depleted HeLa cells, and in Lowe patient cells. F-actin staining in cells depleted for OCRL or Rab35 and in Lowe syndrome patient cells, after treatment with low doses of Latrunculin-A. (a) Left panel: The p34-Arc component of the Arp2/3 F-actin nucleator complex (green) was stained in HeLa cells transfected with control, OCRL or Rab35 siRNAs. Areas of the intercellular bridges (red) delimited with dashed lines are displayed at higher magnification (bottom images). Right panel: Quantification of the number of intercellular bridges positive for p34-Arc2 is displayed. mean ± s.e.m., n= 30 cells analyzed per experiment, 3 independent experiments. *** P< 5x10-4 (Student’s t-test). (b) Same
stainings and quantifications as in (a) for normal and Lowe patient renal cells. mean ± s.e.m., n= 30 cells analyzed per experiment, 3 experiments. *** P= 5x10-4 (Student’s t-test). (c) HeLa cells transfected with control, OCRL or Rab35 siRNAs (left panels) or control and Lowe patient cells (right panels), as indicated, were treated with DMSO (control), as described in main Fig. 5a. Representative images of cells in cytokinesis (top images) and higher magnifications of the intercellular bridge regions (bottom images) are shown. Cells were stained with the fluorescently-labelled phalloidin (green) and for Aurora B kinase (red). Blue: DAPI staining. Scale bar, 10 μm. (d) Cells as indicated in (c) but treated with DMSO + Latrunculin-A, as described in main Fig. 5a.
© 2011 Macmillan Publishers Limited. All rights reserved.
S U P P L E M E N TA RY I N F O R M AT I O N
WWW.NATURE.COM/NATURECELLBIOLOGY 7
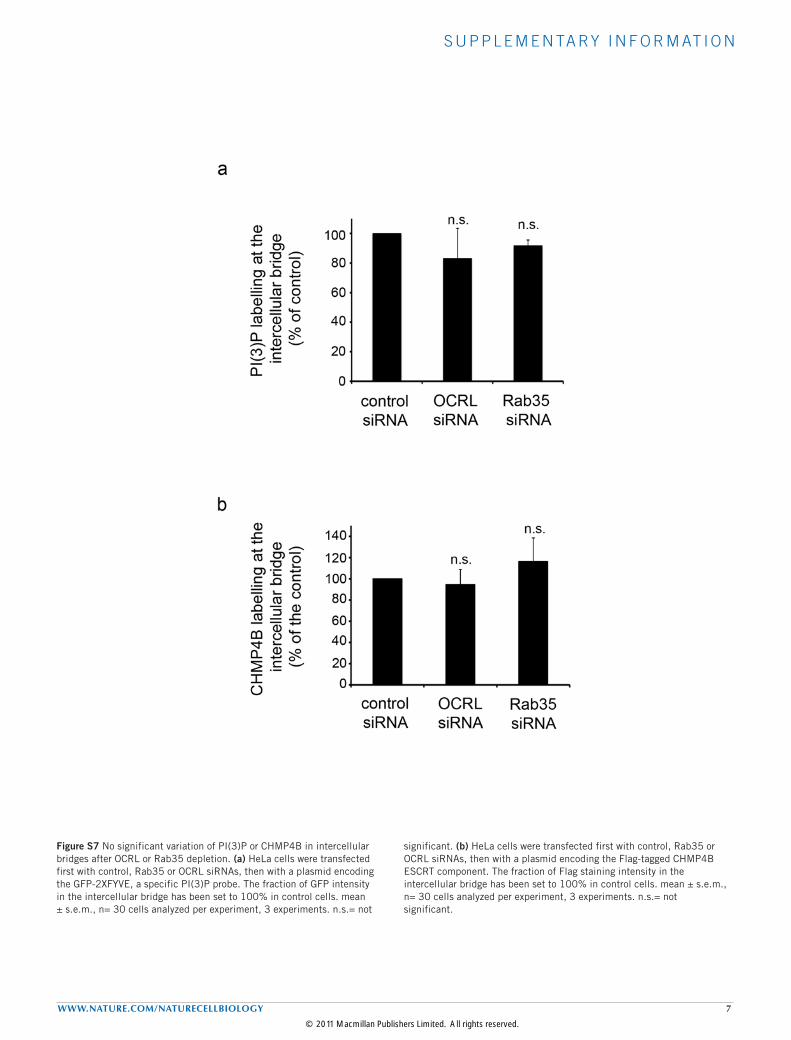
Figure S7 No significant variation of PI(3)P or CHMP4B in intercellular bridges after OCRL or Rab35 depletion. (a) HeLa cells were transfected first with control, Rab35 or OCRL siRNAs, then with a plasmid encoding the GFP-2XFYVE, a specific PI(3)P probe. The fraction of GFP intensity in the intercellular bridge has been set to 100% in control cells. mean ± s.e.m., n= 30 cells analyzed per experiment, 3 experiments. n.s.= not
significant. (b) HeLa cells were transfected first with control, Rab35 or OCRL siRNAs, then with a plasmid encoding the Flag-tagged CHMP4B ESCRT component. The fraction of Flag staining intensity in the intercellular bridge has been set to 100% in control cells. mean ± s.e.m., n= 30 cells analyzed per experiment, 3 experiments. n.s.= not significant.
© 2011 Macmillan Publishers Limited. All rights reserved.

S U P P L E M E N TA RY I N F O R M AT I O N
8 WWW.NATURE.COM/NATURECELLBIOLOGY
Figure S8 uncropped gel scans for all presented Western blots. (a) corresponds to Fig 1b; (b) corresponds to Fig. 1c; (c) corresponds to Fig. 1d; (d) corresponds to Fig. 1e; (e) corresponds to Fig. 2d; (f)
corresponds to Fig. S1; (g) corresponds to Fig. 3d; (h) corresponds to Fig. 3e; (i) corresponds to Fig. S4d. Relative molecular weights are indicated in kDa.
© 2011 Macmillan Publishers Limited. All rights reserved.
Copyright © 2022 FDOKUMEN


![[Cool] Gas Chromatography and Lipids](https://static.fdokumen.com/doc/165x107/6325a4b1852a7313b70e98e9/cool-gas-chromatography-and-lipids.jpg)

















