
Quantum Chemical Modelling of Enzymatic and ... - DiVA Portal
100
Quantum Chemical Mo䘀䜀丩ਭㄊ⠀丩ਭㄊ⠀䬀倀 Enzymatic and Organometallic Reactions Ferran Planas Doctoral Thesis in Organic Chemistry at Stockholm University, Sweden 2021
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of Quantum Chemical Modelling of Enzymatic and ... - DiVA Portal

Quantum Chemical Modelling ofEnzymatic and OrganometallicReactions Ferran Planas
Ferran Planas Qu
antu
m C
hem
ical Modellin
g of Enzym
atic and O
rganom
etallic Reactions
Doctoral Thesis in Organic Chemistry at Stockholm University, Sweden 2021
Department of Organic Chemistry
ISBN 978-91-7911-454-1
Ferran Planas
In this thesis, density functional theory (DFT) is employed in the studyof two enzymes and two organometallic systems. First, the naturalreaction mechanism as well as the enantioselective formation ofhydroxyketones catalysed by two thiamine diphosphate (ThDP)-dependent enzymes, namely benzoylformate decarboxylase (BFDC) and2-succinyl-5-enolpyruvyl-6-hydroxy-3-cyclohexene-1-carboxylic-acid(SEPHCHC)- synthase (MenD) are investigated. For the two enzymes,the factors governing the enantioselectivity and, in the case of MenD,the regioselectivity, are described. Next, a Pummerer-like, C-Ccoupling reaction, is studied. The calculations unveil that the reactivespecies are open-cubane complexes, instead of the more common linearspecies. In the last study, a ruthenium-catalysed cyclopropanationreaction is investigated. The calculated free-energy profiles present avery complex scenario in which two cyclopropanation mechanisms,two side reactions and different possible catalysts need to beconsidered. A combined computational-experimental effort elucidateswhich is the active catalyst for the cyclopropanation reaction.


Quantum Chemical Modelling of Enzymatic andOrganometallic ReactionsFerran Planas
Academic dissertation for the Degree of Doctor of Philosophy in Organic Chemistry atStockholm University to be publicly defended on Friday 16 April 2021 at 14.00 in Magnélisalen,Kemiska övningslaboratoriet, Svante Arrhenius väg 16 B.
AbstractIn this thesis, density functional theory (DFT) is employed in the study of two enzymes and two organometallic systems.
First, the natural reaction mechanism, as well as the enantioselective formation of α-hydroxyketones catalysed bytwo thiamine diphosphate (ThDP)- dependent enzymes, namely benzoylformate decarboxylase (BFDC) and 2-succinyl-5-enolpyruvyl-6-hydroxy-3-cyclohexene-1-carboxylic-acid (SEPHCHC)- synthase (MenD), are investigated. To that end,different cluster models that account for the active sites of the enzymes are used. For BFDC, the calculated naturalreaction mechanism clarifies the roles of various active site residues and of the cofactor. Moreover, an unprecedentedtricyclic cofactor species is found to be kinetically relevant. The importance of this species is further explored in asecond study, in which the relative stabilities of the different ThDP-cofactor states are assessed in different enzymaticand non-enzymatic environments. In the last study of BFDC, the enantioselective carboligation mechanism between theenamine intermediate and two different acceptors, namely benzaldehyde or acetaldehyde, is studied. Moving into MenD,the calculated natural reaction mechanism gives insight into the formation tetrahedral post-decarboxylation intermediate,which has been extensively discussed in the literature. Moreover, the proton source in the keto-enol tautomerization inthe second part of the mechanism can also be elucidated. Finally, because MenD can perform 1,4- and 1,2-additions, thefactors governing the regioand enantioselectivity of two non-natural reactions are covered.
Next, a Pummerer-like, C-C coupling reaction, is studied, and the calculations show that an unstable open-cubanecomplex yields considerably lower barriers than the more typically suggested linear complexes. In the last study, aruthenium-catalysed cyclopropanation reaction is investigated. The calculated free-energy profiles indicate a very intricatescenario in which two cyclopropanation mechanisms and two side-reactions need to be considered. Importantly, one ofthese side-reactions, i.e. a migratory insertion of the carbene into the C-M bond of the ligand, results in the formationof a new catalyst, and a combined computational-experimental effort elucidates which is the active catalyst for thecyclopropanation reaction.
Keywords: DFT, enzyme, organometallic, mechanisms.
Stockholm 2021http://urn.kb.se/resolve?urn=urn:nbn:se:su:diva-190921
ISBN 978-91-7911-454-1ISBN 978-91-7911-455-8
Department of Organic Chemistry
Stockholm University, 106 91 Stockholm


QUANTUM CHEMICAL MODELLING OF ENZYMATIC ANDORGANOMETALLIC REACTIONS
Ferran Planas


Quantum Chemical Modellingof Enzymatic andOrganometallic Reactions
Ferran Planas

©Ferran Planas, Stockholm University 2021 ISBN print 978-91-7911-454-1ISBN PDF 978-91-7911-455-8 Printed in Sweden by Universitetsservice US-AB, Stockholm 2021

No has de parar delluitarDoncs has d'acabard'ensamblarEl teu cub de Rubik Som moviment - Aspencat


i
Abstract.
In this thesis, density functional theory (DFT) is employed in the study of two enzymes and two organometallic systems.
First, the natural reaction mechanism, as well as the enantioselective for-mation of α-hydroxyketones catalysed by two thiamine diphosphate (ThDP)- dependent enzymes, namely benzoylformate decarboxylase (BFDC) and 2-succinyl-5-enolpyruvyl-6-hydroxy-3-cyclohexene-1-carboxylic-acid (SEPHCHC)- synthase (MenD), are investigated. To that end, different clus-ter models that account for the active sites of the enzymes are used. For BFDC, the calculated natural reaction mechanism clarifies the roles of vari-ous active site residues and of the cofactor. Moreover, an unprecedented tri-cyclic cofactor species is found to be kinetically relevant. The importance of this species is further explored in a second study, in which the relative sta-bilities of the different ThDP-cofactor states are assessed in different enzy-matic and non-enzymatic environments. In the last study of BFDC, the en-antioselective carboligation mechanism between the enamine intermediate and two different acceptors, namely benzaldehyde or acetaldehyde, is studied. Moving into MenD, the calculated natural reaction mechanism gives insight into the formation tetrahedral post-decarboxylation intermediate, which has been extensively discussed in the literature. Moreover, the proton source in the keto-enol tautomerization in the second part of the mechanism can also be elucidated. Finally, because MenD can perform 1,4- and 1,2-additions, the factors governing the regio- and enantioselectivity of two non-natural reac-tions are covered.
Next, a Pummerer-like, C-C coupling reaction, is studied, and the calcula-tions show that an unstable open-cubane complex yields considerably lower barriers than the more typically suggested linear complexes. In the last study, a ruthenium-catalysed cyclopropanation reaction is investigated. The calculated free-energy profiles indicate a very intricate scenario in which two cyclopropanation mechanisms and two side-reactions need to be considered. Importantly, one of these side-reactions, i.e. a migratory insertion of the car-bene into the C-M bond of the ligand, results in the formation of a new cata-lyst, and a combined computational-experimental effort elucidates which is the active catalyst for the cyclopropanation reaction.

ii
Populärvetenskaplig sammanfattning.
Den moderna världen behöver renare och effektivare kemiska processer för att möta mänsklighetens ökande behov samtidigt som klimatet bevaras. Längs denna linje utvecklas modern organisk kemi med redesign och ska-pande av reaktioner för att möta dessa behov. Ett fält som har bidragit avsevärt till denna utveckling har varit användningen av katalysatorer, föreningar som ökar reaktionshastigheten och därmed möjliggör använd-ning av mildare förhållanden eller för utforskning av reaktioner som annars skulle vara omöjliga. Förutom katalys har också stökiometriska reaktioner baserade på billiga och icke-farliga material utvecklats därefter. I samtliga fall är detaljerad information önskvärd för att vägleda skapandet eller ut-vecklingen av nya kemiska reaktioner.
Följaktligen har utvecklingen av nya experimentella tekniker varit enorm, men studien av reaktionsintermediärer och andra kortlivade arter är fort-farande utmanande. Det är här beräkningstekniker har blivit grundläggande, eftersom deras användning ger tillgång till information som annars skulle vara ouppnåelig. Specifikt möjliggör kvantkemi atomistisk karakterisering av alla arter som är involverade i en reaktionsmekanism och för beräkning av deras inre energier. Viktigt är att detta ger detaljerad information om den relativa stabiliteten hos de olika arter som bildas i en reaktionsmekanism, som relaterar till den makroskopiska termodynamiken och kinetiken. Därför möjliggör användning av kvantkemiska beräkningar bland annat bestämn-ing av de faktorer som styr reaktionens termokemi, och således kan dessa metoder användas i den rationella utformningen av nya reaktioner.
I denna avhandling används kvantkemi för att studera reaktionsmekanis-mer för mycket olika system, allt från enzymer till små organometalliska komplex. När det gäller enzymerna studeras den naturliga reaktionen av två ThDP-beroende enzymer, såväl som den enantioselektiva karboligerings-reaktionen. De erhållna resultaten ger värdefull insikt i de relevanta resterna för enzymets aktivitet och avslöjar egenskaper hos de enzymer som var okända. De organometalliska komplexen, två system som mekanistisk un-dersökning visade sig vara en utmaning i tidigare experimentella studier, studeras i en kombinerad beräknings-experimentell insats. Tack vare detta tvärvetenskapliga synsätt kan ett antal öppna frågor som förblev svårfån-gade tidigare besvaras.

iii
List of publications and contributions of the author.
The following publications are included in the thesis:
I. A Theoretical Study of the Benzoylformate Decarboxylase
Reaction Mechanism. Ferran Planas, Xiang Sheng, Michael J. McLeish, Fahmi Himo. Front. Chem. 2018. 6: 205.
II. Computational characterization of enzyme-bound thiamin diphosphate reveals a surprisingly stable tricyclic state: im-plications for catalysis. Ferran Planas, Michael J. McLeish, Fahmi Himo. Beilstein J. Org. Chem. 2019, 15, 145-159.
III. Computational Study of Enantioselective Carboligation Catalyzed by Benzoylformate Decarboxylase. Ferran Planas, Michael J. McLeish, Fahmi Himo. ACS Catal. 2019, 9, 5657–5667.
IV. Biocatalytic Stetter Reaction: Computational Study of Mechanism and Origins of Regio- and Enantioselectivity of MenD Ferran Planas, Michael J. Mcleish, Fahmi Himo. Manuscript.
V. Computational and Experimental Study of Turbo-Organo-magnesium Amide Reagents: Cubane Aggregates as Reac-tive Intermediates in Pummerer Coupling. Ferran Planas, Stephanie V. Kohlhepp, Genping Huang, Abra-ham Mendoza, Fahmi Himo. Chem. Eur. J. 2021,27, 2767-2773.
VI. Cyclopropanation via Ruthenium-Derived Redox-Active Carbenes. Mechanistic Insights from Experiments and Com-putations. Ferran Planas, Matteo Costantini, Marc Montesinos-Magraner, Fahmi Himo, Abraham Mendoza. Manuscript.

iv
Permissions to reprint the following publications were obtained from their respective publishers:
I. F. Planas, X. Sheng, M. J. McLeish, F. Himo. Front. Chem. 2018. 6: 205. Copyright © 2018 Planas, Sheng, McLeish and Himo. Open ac-cess under creative commons attribution 4.0 license.
II. F. Planas, M. J. McLeish, F. Himo. Beilstein J. Org. Chem. 2019, 15, 145-159. Copyright © 2019 The authors. Open acess article under crea-tive commons attribution 4.0 license.
III. Reprinted with permission from F. Planas, M. J. McLeish, F. Himo. ACS Catal. 2019, 9, 5657–5667. Copyright © 2019 American Chemical Society.
IV. F. Planas, S. V. Kohlhepp, G. Huang, A. Mendoza, F. Himo. Chem. Eur. J. 2021, 27, 2767-2773. Copyright 2020 The authors. Chemistry – A European Journal published by Wiley-VCH GmbH. Open access under creative com-mons attribution 4.0 license.
The contribution by the author to the papers is as follows:
Papers I-IV: Performed all calculations, analysed the results, wrote a first
manuscript and participated in further corrections. Papers V and VI: Performed all calculations, analysed the results, wrote a
first manuscript and participated in further corrections of the computational part of the study.

v
Previous document based on this work.
This thesis is partly based on the author’s half-time report titled ”Quantum Chemical Modelling of Benzoylformate Decarboxylase”. The introduction (Chapter 1) and general theory (Chapter 2) have been updated to include the necessary background in organometallic chemistry, which was not included in the half-time report. Chapter 5 includes the three papers that were part of the half-time report (papers I-III), the introduction has been revised and up-dated, and an additional section on a project not included in the half-time has been written entirely for this thesis. Chapters 6, 7 and 8 are entirely new.

vi
Abbreviations.
QM Quantum mechanics DFT Density functional theory TS Transition State TST Transition State Theory ee enantiomeric excess HK Hohenberg-Kohn KS Kohn-Sham LDA Local-density approximation LSDA Local-spin density approximation GGA Generalized gradient approximation HF Hartree-Fock ThDP Thiamine diphosphate BFDC Benzoylformate decarboxylase SEPHCHC 2-succinyl-5-enolpyruvyl-6-hydroxy-3-cyclohex-
ene-1-carboxylic-acid SEPHCHC synthase MenD PDC Pyruvate decarboxylase BO But-3-en-2-one CA Cyclohex-1-ene-1-carbaldehyde DIPA Diisopropyl amide TMP 2,2,6,6-tetramethylpiperidine THF Tetrahydrofurane NHPI-DA N-hydroxyphthalimidyl diazoacetate AN Acetonitrile MI Migratory insertion DM Dimerization CP Cyclopropanation St Styrene Pr Propene

vii
Contents.
Abstract. .............................................................................................................. i
Populärvetenskaplig sammanfattning. ........................................................ ii
List of publications and contributions of the author. ............................. iii
Previous document based on this work. ..................................................... v
Abbreviations. ................................................................................................. vi
1. Introduction. ................................................................................................. 1
2. General theory. ............................................................................................. 3 2.1. Free energy profiles. ....................................................................................... 3 2.2. Transition state theory. ................................................................................. 4 2.3. Selectivity. ......................................................................................................... 4 2.4. Density functional theory. ............................................................................ 6
2.4.1. Dispersion. ................................................................................................ 8
3. Quantum chemical modelling of enzymatic reactions. ....................... 9 3.1. Michaelis-Menten kinetics. ........................................................................... 9 3.2. Quantum mechanical modelling of enzymes. ......................................... 10
3.2.1. Construction of the enzyme model. .................................................. 11 3.2.2. Limitations and considerations. ......................................................... 13 3.2.3. Modelling of asymmetric catalysis in enzymes. ............................ 13
4. Computational details. ............................................................................. 15 4.1. Additional remarks for the study of enzyme reaction mechanisms. . 15 4.2. Standard state correction. ........................................................................... 16
5. Quantum chemical modelling of ThDP dependent enzymes. (Papers I, II, III and IV) .............................................................................. 17
5.1. Benzoylformate Decarboxylase. ................................................................ 18 5.1.1. Active site model and computational details. ................................. 19 5.1.2. Catalytic mechanism of benzoylformate decarboxylase. (Paper I) .............................................................................................................................. 20 5.1.3. Computational characterization of enzyme-bound thiamine diphosphate. (Paper II) .................................................................................... 24 5.1.4. Computational study of the enantioselective carboligation catalysed by benzoylformate decarboxylase. (Paper III) ........................ 29
5.2. SEPHCHC synthase. .................................................................................... 33

viii
5.2.1. Active site models. ................................................................................ 34 5.2.2. Natural reaction mechanism. .............................................................. 35 5.2.3. Non-natural substrates: Analysis of the regioselectivity. ............ 39 5.2.4. Non-natural substrates: Enantioselectivity. ................................... 44
5.3. Conclusions. .................................................................................................... 44
6. Computational investigation of a Pummerer coupling in non-electrophilic media. (Paper V) .................................................................... 47
6.1. Results. ............................................................................................................ 48 6.2. Conclusions. .................................................................................................... 51
7. Cyclopropanation via ruthenium-derived redox-active carbenes. (Paper VI) ........................................................................................................ 53
7.1. Formation of the carbene intermediate. .................................................. 54 7.2. Reaction mechanisms of the side-reactions. ........................................... 55 7.3. Computational and experimental study of the cyclopropanation reaction. ................................................................................................................... 56 7.4. Analysis of the selectivity. .......................................................................... 60 7.5. Conclusions. .................................................................................................... 63
8. Concluding remarks. ................................................................................ 65
Acknowledgments. ........................................................................................ 67
References. ...................................................................................................... 68

1
1. Introduction.
Ever since the synthesis of urea in 1828,1 considered by many as the first organic synthesis, the interests that drive the development of organic chemi-stry have been constantly evolving. At first, the main goal was to increase molecular complexity as a way to mimic Nature, which resulted in an incre-dible growth of the chemical toolset during the first half of the 20th century.2 Over the past decades, the environmental concerns and the increasing needs of an ever-growing population have pushed organic chemistry towards the use of cleaner and simpler processes. In this evolution, the aspect that might have had the largest impact has been the use of catalysts, compounds that accelerate reaction rates without altering the thermodynamics of the re-action, nor being consumed themselves in the process.
Catalysts have been intimately related with organic synthesis since the beginning of the last century, when the topic merited two Nobel prices in 1909 and 1912. Their use allows for transformations that would otherwise require of extreme conditions or complicated procedures, reducing the waste and increasing the efficiency of reactions. A clear testament of the current importance of catalysis is that up to 90% of the industrial processes require of, at least, one catalyst.3
Catalysis is divided into three major branches: heterogeneous, homoge-neous, and biocatalysis. The first two are the most dominant forms in labor-atories and industry, and differ on whether one (homogeneous) or more than one (heterogeneous) phases are involved in the reaction. Biocatalysis is, in spite a considerable growth of its use in recent years, the least applied of the three branches, and is based on the use of biological systems, most commonly enzymes.
Despite the paramount role of catalysis in modern organic chemistry, stoi-chiometric reactions are, in some instances, an appealing alternative. For ex-ample, although there are several catalytic carbon-carbon cross-coupling procedures available, such as the ones based on nickel4 or paladium5 catalysts, stoichiometric C-C bond formation reactions are still being developed. Or-ganomagnesium, organolithium or organozinc compounds are a few exam-ples of reagents that, despite some associated procedure inconvenients, rep-resent a quite straightforward way to yield new C-C bonds, and offer a good variety in their scope.
Regardless of the nature of the chemical reaction (catalytic or non-cata-lytic), the development of new processes requires of increasingly precise

2
knowledge about the factors affecting the reactivity of a given system. Along this line, determination of reaction mechanisms is desirable, as thanks to them it is possible to rationalize certain experimental observables, such as the selectivity, kinetics or thermodynamics.
Modern experimental characterization techniques provide access to mech-anistic insight that was simply unimaginable in the past. However, experi-mental characterization of short-lived species remains challenging, and to determine structural features at atomistic level from an experimental ensem-ble is far from trivial.
Computational methodologies based on quantum mechanics (QM), on the other hand, allow for the calculation of the electronic structure of matter and the atomistic characterization of reactants, intermediates and products re-gardless of their kinetic and thermodynamic significance. With the use of computational chemistry, it is possible to draw a sequence of steps that shape a reaction mechanism, which in turn allows for the description of the factors that determine the reactivity or govern the selectivity. This information can then be used to develop or modify new catalysts in a more efficient manner.
The applicability of QM to organic systems has been boosted over the last decades thanks to the development of faster and more accurate computational methods, as well as the increasing computational power of modern super-computers. The calculation of reaction mechanisms is now a fundamental part in mechanistic investigations, and very diverse systems can be studied with QM methods. In this thesis, four systems as different as two enzymes, a metal-homogenous catalyst and Grignard reagents have been studied with density functional theory (DFT), which is a clear example of the wide ap-plicability of computational chemistry.
The present thesis is organised as follows. First, the fundamental theoret-ical concepts of the employed methodology and approximations are intro-duced in Chapters 2 to 4. Next, in Chapter 5, the reactivity and selectivity studies of two thiamine diphosphate (ThDP)-dependent enzymes, namely benzoylformate decarboxylase (BFDC) and SEPHCHC synthase (MenD), are discussed. In Chapter 6 the reaction mechanism of a non-catalytic, Pummerer-like, C-C coupling reaction is explained, and in Chapter 7, the re-sults on a combined experimental-computational study of a ruthenium-cata-lysed cyclopropanation are presented. Finally, the concluding remarks are presented in Chapter 8.

3
2. General theory.
The internal energy is one of the most important intrinsic properties that can be calculated with QM methods, as it determines the kinetic and thermo-dynamic significance of a chemical species relative to others. Importantly, the thermochemical properties of a reaction, such as reaction rates, can be deter-mined experimentally, and thus their comparison with molecular energies can be useful to assess a calculated reaction mechanism.
In this section, a theoretical framework to connect microscopic internal energies with experimental observables is briefly covered. First, the descrip-tion of free-energy profiles, an introduction to the transition state theory (TST) and the relationship between selectivity and calculated relative ener-gies are given. Next, the basics of density-functional theory and the details of the specific functional used in the present thesis are introduced.
2.1. Free energy profiles. Chemical reactions can be seen as the geometrical rearrangement of one or several molecules. The atoms that constitute these molecules can be arranged in a number of different geometrical distributions, and each of them is asso-ciated with a distinct energy. Therefore, a multi-dimensional energy lands-cape can be constructed in which all changes in atomic coordinates are asso-ciated with an energy change. In this energy landscape there are two kind of stationary points of chemical significance; minima and first-order saddle-points. Minima are points on the energy landscape in which all eigenvalues of the Hessian matrix (the second derivative of the energy with respect to all atomic positions) are positive, and relate to reactants, products and interme-diate species. First-order saddle-points are maxima on the free-energy lands-cape and have one negative eigenvalue of the Hessian matrix. The lowest in energy first-order saddle-point that connects two minima is defined as a tran-sition state (TS). A chemical pathway consists of the sequence of stationary points that connect reactants to products. In sake of clarity, free-energy landscapes can be simplified to free-energy profiles (FEP) with the use of the so called “reaction coordinate”, an abstract one-dimensional variable that re-presents the progress of the reaction. Importantly, the internal energy of a single molecule cannot be related to an experimental observable, but the re-lative energies of the different species can. That is, the energy necessary to

4
cross transition states relate to the kinetics of the reaction, whereas the rela-tive stability of reactants and products relate to the thermodynamics of the system.
2.2. Transition state theory. The main assumption in the transition state theory (TST)6 is that reactants and products are in a quasi-equilibrium with the transition state, a condition that is only true if the system is thermally equilibrated. The original formu-lation of the theory incorporates the transmission coefficient Κ, a factor that accounts for non-classical phenomena, such as recrossing and tunnelling. There is extensive research on the impact of these effects to the rate con-stant,7 and a more detailed discussion is beyond the scope of this thesis. Ho-wever, it must be noted that, in this thesis, the Κ value has been approxima-ted to 1 when invoking the TST.
𝑘 = 𝐾𝑘$𝑇ℎ𝑒(∆*‡/-. 2.1
In the equation above, kb is the Boltzmann constant, h the Plank constant, R the ideal gas constant, T the temperature and Κ the above-mentioned tran-simission coeafficient. The remaining term is the activation Gibbs free-en-ergy (∆𝐺‡). Therefore, the rate constant is expressed as a function of the ac-tivation free-energy, a value that can be obtained from a calculated reaction mechanism, and this theory allows for a direct comparison between macro-scopic kinetic data and microscopic internal energies.
2.3. Selectivity. Energy landscapes often entail different chemical paths, and therefore an initial combination of reactants can yield diverse products. When one pro-duct is predominantly formed over the rest, the reaction is selective. The se-lectivity can be determined in two ways: by kinetic or by thermodynamic control. When one product is favoured over the rest because its formation is associated with a lower barrier, the reaction is under kinetic control. Alter-natively, if the relative stability of the different products is what determines the selectivity, the reaction is under thermodynamic control.
A case of particular interest in organic chemistry are the reactions in which the two products than can form only differ in the spatial orientation of a newly formed chiral element, i.e. the two products are enantiomers. These
5
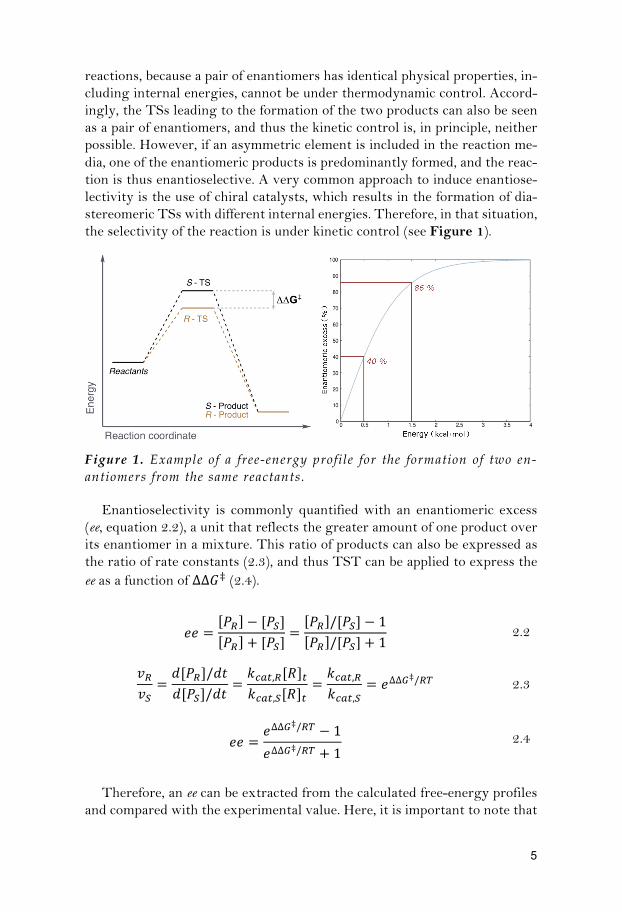
reactions, because a pair of enantiomers has identical physical properties, in-cluding internal energies, cannot be under thermodynamic control. Accord-ingly, the TSs leading to the formation of the two products can also be seen as a pair of enantiomers, and thus the kinetic control is, in principle, neither possible. However, if an asymmetric element is included in the reaction me-dia, one of the enantiomeric products is predominantly formed, and the reac-tion is thus enantioselective. A very common approach to induce enantiose-lectivity is the use of chiral catalysts, which results in the formation of dia-stereomeric TSs with different internal energies. Therefore, in that situation, the selectivity of the reaction is under kinetic control (see Figure 1).
Figure 1. Example of a free-energy profile for the formation of two en-antiomers from the same reactants.
Enantioselectivity is commonly quantified with an enantiomeric excess (ee, equation 2.2), a unit that reflects the greater amount of one product over its enantiomer in a mixture. This ratio of products can also be expressed as the ratio of rate constants (2.3), and thus TST can be applied to express the ee as a function of ∆∆𝐺‡ (2.4).
𝑒𝑒 =𝑃- − [𝑃3]𝑃- + [𝑃3]
=𝑃- /[𝑃3] − 1𝑃- /[𝑃3] + 1
2.2
𝑣-𝑣3
=𝑑[𝑃-]/𝑑𝑡𝑑[𝑃3]/𝑑𝑡
=𝑘:;<,-[𝑅]<𝑘:;<,3[𝑅]<
=𝑘:;<,-𝑘:;<,3
= 𝑒∆∆*‡/-. 2.3
𝑒𝑒 =𝑒∆∆*‡/-. − 1𝑒∆∆*‡/-. + 1
2.4
Therefore, an ee can be extracted from the calculated free-energy profiles
and compared with the experimental value. Here, it is important to note that

6
selectivity in general, and ee values in particular, are very sensitive to small energy differences. As an example, a change in energy of only 1 kcal/mol (from 0.5 kcal/mol to 1.5 kcal/mol) relates to a 46% better enatioselectivity (see Figure 1, right panel). Therefore, calculations on enantioselective reac-tions need to be highly accurate to reproduce the mentioned small energy differences, which makes of the modelling of these processes a very challeng-ing task. However, because in the calculation of enantioselective reactions the interest is in relative TS energies (∆∆𝐺‡) and the geometries of the TSs yielding the different enantiomeric products are very similar and represent the exact same chemical event, a good accuracy can be expected due to the cancellation of systematic errors. Indeed, many recent examples show that experimentally-observed selectivity trends can be successfully reproduced by calculations.8
2.4. Density functional theory. Nowadays, there is a wide variety of computational methods that can be used to study and characterize chemical compounds. When focusing on the reacti-vity, the description of the electronic structure of the system of interest is necessary, and the methods that account for it are based on quantum mechanics (QM). The reliability of these methods has been thoroughly as-sessed over the years,9 and they are now commonly used to determine the geometry of the species that form throughout a reaction mechanism, as well as their energies and other molecular properties. QM methods can be broadly divided in two branches. On one hand, there are the wave-function based methods which, as the name indicates, express the energy (and all molecular properties) as a function of the wave-function. These methods can be further divided in different classes depending on their treatment of the wave-function, but a detailed explanation is beyond the scope of this thesis. On the other hand, there are the density functional theory (DFT)-based methods, which express the energy of the system as a functional of the electron density, E[ρgs(r)]. These are the currently most used methods due to their good ba-lance between accuracy and cost,10 and have been shown to be a valid and reliable tool to study chemical reactions.11
DFT is a reformulation of quantum mechanics substantiated on the Ho-henberg-Kohn (HK) theorems.11 The first theorem states that the properties of the ground state of a many-electron non-degenerate system are described by a unique electron density.12 This in turn simplifies the many-body prob-lem, as the electron density only depends on the three-spatial coordinates, while the wave-function depends on the coordinates of all electrons and nu-clei. The second theorem states that the energy calculated from a guessed electron density, ρguess(r), different from the real one, ρgs(r), is an upper bound

7
to the true ground state energy (2.5). That is, the variational principle applies to DFT and, therefore, the electronic density of a given system is the one that minimizes the total energy.12
𝐸[𝜌ABCDD(𝑟)] ≥ 𝐸[𝜌AD(𝑟)] 2.5
The energy functional E can be divided into the kinetic energy (T), the nuclei-electron attraction (VNe) and the electron-electron (Vee) repulsion terms:
𝐸 𝜌(𝑟) = 𝑇 𝜌(𝑟) + 𝑉JC 𝜌 𝑟 + 𝑉CC 𝜌 𝑟 2.6
The electron-electron repulsion term (Vee) can then be divided into the Couloumb term (J) and the exchange-correlation energy (Exc), which con-tains the non-classical self-interaction, exchange and correlation effects.
𝐸 𝜌(𝑟) = 𝐸 𝜌(𝑟) + 𝑉JC 𝜌 𝑟 + 𝐽 𝜌 𝑟 + 𝐸L: 𝜌 𝑟 2.7
Kohn and Sham formulated an expression for the kinetic energy term that could be divided into two contributions: the kinetic energy contribution of a non-interacting system (KS system) with the same electron density as the real system, and a correction term defined as the difference between the real kinetic energy (T) and the kinetic energy of the non-interacting system (Tni). This latter term is included into the Exc term, arriving at the following ex-pression:
𝐸 𝜌(𝑟) = 𝑇MN 𝜌(𝑟) + 𝑉JC 𝜌 𝑟 + 𝐽 𝜌 𝑟 + 𝐸L: 𝜌 𝑟 2.8
Importantly, the VNe and J terms are known, and Tni can be calculated exactly by introducing the Kohn-Sham orbitals, single-particle orbitrals solely used for practicality. However, the Exc term remains elusive and DFT methods differ essentially on how they account for it. Two common approx-imations are the local density approximation (LDA) and the generalised gra-dient approximation (GGA), and both of them have been developed in vari-ous forms. A very popular GGA functional is B3LYP,13-15 which expresses the exchange term as a combination of LSDA (Local Spin Density Approxi-mation) exchange, pure Hartree-Fock (HF) exchange and an exchange term developed by Becke.15 The correlation, on the other hand, is expressed as a combination of LSDA correlation and a term developed by Lee, Yang and Parr.13 The total contribution of each of the terms is weighted by three em-pirical factors a, b and c, and the resulting expression for Exc is as follows:
𝐸L:OPQRS = 1 − 𝑎 𝐸LQ3UV + 𝑎𝐸LWX + 𝑏𝐸LO + 1 − 𝑐 𝐸:Q3UV + 𝑐𝐸:QRS 2.9

8
The accuracy of B3LYP has been extensively assessed against bench-marks,16 and a vast number of publications are the testimony of the applica-bility of this functional in the study of reaction mechanisms.
2.4.1. Dispersion. B3LYP has, of course, limitations and a well-known shortcoming is the in-adequate description of dispersion forces, interactions that arise from electron correlation and electron density fluctuations. These forces, although weak, can have an impact on the geometries and energies of the systems, an effect that can be significant in large systems such as enzymes.
In the last decade, several different approaches have been developed to account for dispersion interactions in DFT methods, and that has resulted in an increase of their accuracy. One solution has been to incorporate empirical corrections to the total energy,17 an approximation that increases accuracy without adding to the computational cost. Specifically, in the present thesis the atom-pairwise DFT-D3(BJ) protocol of Grimme has been used, which includes a two-body term:18
𝐸OPQRS(UP(O[) = 𝐸OPQRS − 𝐸\ND] 2.10
𝐸\ND] = 𝐸(^) 2.11
The two body term (E(2), truncated to the first two scaling factors) is the following:
𝐸(^) = −12
𝑠a𝐶aVO
𝑟VOa[𝑓\,a 𝑟VO ] +
VdO
𝑠e𝐶eVO
𝑟VOe[𝑓\,e 𝑟VO ] 2.12
Where the CnAB are the dispersion coefficients for atom pairs, sn (n=6,8) are the scaling factors that depend on the choice of the functional, and rAB are the internuclear distances. The fd,n term denotes the damping functions. In the B3LYP-D3(BJ) functional, the Becke-Johnsson (BJ) damping function is used instead of the standard zero-damping.15a

9
3. Quantum chemical modelling of enzymatic reactions.
The sources of enzyme proficiency are, to this date, not completely clear and despite decades of extensive experimental and computational work, these macromolecules still present open questions. The complexity of enzymes makes their computational study a more complicated task than similar stud-ies of smaller, and simpler, organic systems, and alternative methodologies are often needed. Moreover, the kinetic treatment of enzymatic reactions is also different than in other organic reactions. In this chapter, the Michaelis-Menten model, which is the most broadly used kinetics model for enzymatic reactions, is briefly described. Next, the computational methodology for the study of enzymatic catalytic mechanisms used in this thesis, its implications and considerations are explained.
3.1. Michaelis-Menten kinetics. Enzymatic reactions do not only consist of the chemical steps where bonds are broken and formed, but also encompass substrate binding and product release. Thus, a kinetic model that accounts for all these various steps is nec-essary. The Michaelis-Menten is one of the models that includes all the men-tioned events, and probably is the most widely used one. In this model, the reaction mechanism is divided into binding of substrate S to an enzyme E to form ES, and an irreversible chemical reaction to form the product P and regenerate the enzyme.19,20
3.1
In the reaction above, kcat is the catalytic rate constant, k1 is the substrate
binding rate constant, and k-1 is the substrate dissociation rate constant. The reaction rate can be expressed as a function of kcat and the enzyme-substrate complex [ES] concentration at any given time (3.2).
𝑣 =𝑑[𝑃]𝑑𝑡
= 𝑘:;<[𝐸𝑆] 3.2
E S ES+k1
k-1
kcatE P+

10
The rate expression can be reformulated as shown in equation 3.3 by as-suming a steady state regime and expressing [ES] in terms of [E]0.
𝑣 =𝑘:;< 𝑆 [𝐸]g
𝑆 + 𝑘:;< + 𝑘(h𝑘h
3.3
With the introduction of the so-called Michaelis constant (KM, equation 3.4), the reaction rate expression is simplified and the Michaelis-Menten equation is obtained (3.5).
𝐾i =𝑘:;< + 𝑘(h
𝑘h 3.4
𝑣 =𝑘:;< 𝑆 [𝐸]g𝐾i + [𝑆]
3.5
From the expression above, at high substrate concentration ([S]>>KM), the reaction will reach the maximum velocity Vmax, and the kcat value can then be obtained:
𝑉j;L = 𝑘:;<[𝐸]g 3.6
As a reminder, transition state theory (TST) allows for the comparison between reaction rate constants, such as kcat, and calculated free-energy bar-riers. However, it must be kept in mind that kcat also includes the product release from the active site, a step that is associated with significant energy inaccuracies when calculating a free-energy profile. In order to overcome this discrepancy, and as long as there is no evidence of the contrary, the product release is assumed not to be rate-limiting, and thus kcat can be directly com-pared to the free-energy profile of the calculated mechanism.
From expression 3.5, at low substrate concentration ([S]<<KM), a reac-tion rate with an apparent rate constant kcat/KM is obtained (3.7). The kcat/KM value is often used to mesure the catalytic efficiency of an enzyme, a concept that will be discussed below.
𝑣 =𝑘:;< 𝑆 [𝐸]
𝐾i 3.7
3.2. Quantum mechanical modelling of enzymes. There is always a trade-off between accuracy and time in QM-based studies, and the large size of enzymes compromises this balance, as the calculation of the whole molecule at QM level is unaffordable. Importantly, when describ-ing an enzymatic reaction, our interest resides on the residues conforming

11
the active site, which are expected to capture most of the relevant interac-tions. Thus, the remaining part of the enzyme body can be simplified and its effects, estimated. This can be done by different approaches21 and, probably, the most widely applied is the quantum mechanics/molecular mechanics (QM/MM) methodology. In this method, a small part of the enzyme, includ-ing the substrate and a selection of residues in its proximity, is treated at QM level, and the rest of the enzyme is studied with force field methods.22
Another methodology, which is the one adopted in the present thesis, is the quantum chemical cluster approach, in which a selection of residues con-stituting the active site (a cluster) is studied explicitly at QM level and the rest of the enzyme is modelled implicitly. This approach has been successfully applied in the study of the catalytic mechanisms of a wide range of enzymes.23 A brief explanation of the protocol used to design a cluster model is given in the following sections.
3.2.1. Construction of the enzyme model. The construction of an active site model in the cluster approach is schemati-cally summarized in Figure 2. The model is constructed on the basis of a crystallographic structure of the enzyme and consists of a careful selection of residues that form the active site. The chosen residues are extracted to-gether with the cofactor, if any, the substrate, and any crystallographic water molecule that could be relevant
Although there are some general guidelines to follow in order to design a model, the architecture of each enzyme is unique, and an individual analysis of each system is always necessary. Usually, the residues that interact with the substrate and/or the cofactor are included in the model. Moreover, if the properties of any of those residues is potentially altered by residues in the vicinity, these should also be included (for example, a glutamic acid with a hydrogen bond to a histidine). Finally, extra surrounding residues can be incorporated in order to avoid unrealistic movements of the residues or an incomplete representation of the environment.

12
Figure 2 . Representation of the construction of an active site model in the quantum-chemical cluster approach.
The selected residues are not necessarily incorporated in their entirety in the model, and those that are not sequential are usually truncated at their α-carbon, i.e. only the side chain is included to the model. The crystallographic arrangement of the active site, which in the real system is maintained due to the strain that the protein body imposes, is here conserved with a locking scheme in which the carbons at which the model is truncated are restrained. The locking scheme can be extended to a second carbon or hydrogen if un-realistic movements occur during the geometry optimisations. However, the number of locked atoms should always be kept to the minimum necessary in order to have the sufficient flexibility of the active site. Importantly, the use of a locking scheme is associated with substantial inaccuracies in the calcula-tion of the entropy term, and therefore only the zero-point energy (ZPE), a large-basis correction, and solvation corrections (see below) are included in the final energies.
The electrostatic environment is included using a homogenous polariza-ble medium with a dielectric constant ε of 4, the contribution of which is

13
added as a single-point correction to the final energy. Despite the somewhat arbitrary value of ε, the errors associated with this approximation are less significant with an increasing size of the model, as the included residues al-ready account for most of the electrostatics on the reactive species. Indeed, different studies have shown that solvation effects saturate rapidly with the size of the model.24
3.2.2. Limitations and considerations. The reasons behind enzymatic efficiency (kcat/KM) are still an open debate. En-zymatic reactions have been suggested to benefit from lower entropy penal-ties than the reaction in solution.25 Namely, in an enzyme, the cost associated with the loss of certain translational and rotational motions to form the TS would already be paid at substrate binding, and the protein environment would already be pre-organised to stabilize the transition state. However, the actual weight of each of these effects to enzymatic efficiency, in general, and to the reduction of the entropic penalty, in particular, is not clear.26
Along this line, to totally neglect the entropy term in the cluster approach might seem a somewhat controversial approximation. However, it must be noted that the calculated mechanisms are compared to the catalytic rate con-stant (kcat) with the use of the transition state theory, and not to enzyme effi-ciency (kcat/KM). Actually, it is known that the entropic effect on the chemical steps of an enzymatic reaction is very low26b,c and thus to neglect this term in the calculation of an enzymatic reaction mechanism is a reasonable ap-proximation.
3.2.3. Modelling of asymmetric catalysis in enzymes. Enzymes are, by nature, asymmetric molecules that can be used as chiral cat-alysts in enantioselective reactions. Therefore, in the computational study of enzymatic asymmetric reactions, a comparision between the experimentally measured and the computationally obtained ee, as shown in section 2.3, can be relevant. However, as mentioned in section 3.1, the rate expression of an enzymatic reaction also accounts for the processes of substrate binding and product release, and thus the expressions derived in section 2.3 might need to be revised for the case of an enzymatic enantioselective reaction.
In this thesis, two assumptions have been made in order to compare cal-culated energies with experimentally observed enzymatic selectivities. First, the different bindings of the substrate S to promote the formation of two enantiomers (ESS and ESR) are assumed to readily interconvert, and second, product release is assumed not to be rate-limiting. With these approxima-tions, the expressions presented in section 2.3 can also be used in an enzy-matic reaction, and the enantiomeric excess (ee) can thus be related to the

14
difference in activation free energies for the two enantiomers (∆∆𝐺‡) relative to a common zero (see Figure 3).
Figure 3 . Energy profile of an enantioselective enzymatic reaction .
To model an enantioselective reaction catalysed by an enzyme is especially difficult, as the vast conformational space might lead to inconsistencies, mainly related to the multiple-minima problem. These effects might not be relevant for the study of a general reaction mechanism, but can have signifi-cant implications when calculating the mechanism of an enantioselective re-action, which requires of an accuracy on the order of 1 kcal/mol on relative barriers. Despite the daunting and challenging task that the study of enanti-oselective mechanisms represents, the quantum chemical cluster approach has been successfully used to study enantioselectivity in quite a number of different enzymes.27
ΔΔG
E+S ESS
R - TS
S - TS
Ener
gy
Reaction coordinate
ESR
EPS
EPR E+P

15
4. Computational details.
All calculations in the present thesis were performed using the B3LYP-D3(BJ) functional13,14,18 as implemented in Gaussian 0928 software with the exception of the ruthenium-catalysed cyclopropanation in which Gaussian 16 was used.29 For the geometry optimisations, the 6-31G(d,p) basis set was used for all atoms with the exception of ruthenium in Chapter 7, for which the LANL2DZ pseudopotential.30
All final energies are corrected to the 6-311+G(2d,2p) basis set with sin-gle-point calculations (LANL2TZ31 for ruthenium in Chapter 7). The vibra-tional frequencies were calculated at the same level of theory as the geometry optimisation to confirm the nature of the structures and to calculate the ZPE correction in Chapter 5, and Gibbs free energy corrections in Chapters 6 and 7. Finally, the solvation energy was also added as a single-point correction with the SMD32 model for Chapters 5 and 6, and PCM33 for Chapter 7. In the enzyme models of Chapter 5, a dielectric constant ε of 4 is used, while in Chapters 6 and 7 the dielectric constant and the other necessary parameters of tetrahydrofurane and dichloromethane solvents were used, respectively, as these were the solvents used in the experimental works.
4.1. Additional remarks for the study of enzyme reaction mechanisms. As explained in section 3.2, entropy effects can be neglected in the chemical mechanisms of enzymes. However, in reactions where the molecularity changes an entropy correction must be added. In the modelling of BFDC and MenD, CO2 is released as a gas molecule and, therefore, its entropic contri-bution needs to be considered. In previous works, the entropy gain was esti-mated to be equal to the translational entropy for the free gas molecule and calculated to be 11.1 kcal/mol.34 This correction is therefore added to all steps after CO2 formation.

16
4.2. Standard state correction. Standard state corrections were added to account for the conversion from the 1 atm ideal gas to the 1 M standard state of the solutes in Chapters 6 and 7. Thus, the correction term -RT ln (1/24.5) = 1.9 kcal/mol was added to the energies of all complexes, except for the tetrahydrofurane solvent in Chapter 6 for which a term -RT ln(1/24.5 L/mol × 12.3 mol/L) = 3.4 is added, and for dinitrogen in Chapter 7, which is in the gaseous state and no correction was added.

17
5. Quantum chemical modelling of ThDP dependent enzymes. (Papers I, II, III and IV)
Thiamine diphosphate (ThDP)-dependent enzymes are a super-family of en-zymes the members of which catalyse the cleavage and formation of C-C, C-O, C-N and C-S bonds.35 It is known that the activity of all members of the super-family is based on the umpolung chemistry, i.e. the reversal of the po-larity of the carbonyl group, and follow very similar reaction mechanisms. Namely, the activation of the C2 by deprotonation (ylid formation), the addi-tion of the carbanion to the carbonyl of the substrate, the formation of the enamine and its subsequent reaction with an electrophile.
Depending on the substrate that the enzyme accepts to form the enamine, and its subsequent reactivity, ThDP-dependent enzymes are divided into ox-oreductases (OR), ketoacid dehydrogenases (K), transketolases (TK), and de-carboxylases (DC) (see Figure 4a).
Figure 4. a) Mechanism for formation of the enamine intermediate in ThDP-dependent enzymes and different reactivities of this species. b) Asymmetric synthesis of hydroxyketones.
One important feature of ThDP-dependent enzymes is their ability to cat-alyse enantioselective carboligation reactions (see Figure 4b).36 Different de-carboxylases and transketolases have been shown to catalyse the asymmetric synthesis of α-hydroxyketones from an activated donor (an aldehyde or an α-ketoacid) and an acceptor, most generally an aldehyde. Interestingly, with few exceptions, ThDP-dependent enzymes generally display R-selectivity.37 This has been suggested to be result of two main factors. First, the necessity of a parallel orientation of the carbonyl of the acceptor with the hydroxyl
N
NNS
H2N
R
H
1’
3’4’
2 N
NHNS
H2N
R NS
R
HO R2
RO
R2R
NS
R
HOR
C-H
C-C
C-N
C-O
C-S
R2
R1
O
H R1
O
O
OHor +R2
O
H
ThDP enzyme O
R1OH
R2
a.
b.
R-selectivity

18
group of the enamine (Breslow intermediate). Second, the differential binding of the substituent of the acceptor into two cavities,38 a large and exposed to the solvent one, which promotes the formation of the R-product, and a much smaller cavity conformed by hydrophobic and bulky residues, which pro-motes the S-enantiomer.38
In the articles presented in this chapter, the cluster approach was used to study two ThDP-dependent enzymes, namely benzoylformate decarboxylase (BFDC) and 2-succinyl-5-enolpyruvyl-6-hydroxy-3-cyclohexene-1-carbox-ylic-acid (SEPHCHC) synthase (MenD). For both enzymes, the natural re-action mechanism was explored, and different non-natural reactions were also studied in order to elucidate the factors governing the enantioselectivity for BFDC, and the enantio- and regioselectivity for MenD.
5.1. Benzoylformate decarboxylase. ThDP-dependent decarboxylases are known to preserve similar architecture, fold, and residue roles across all enzymes of the family. All these properties are nowadays well-established, mostly thanks to extensive experimental and computational work on pyruvate decarboxylase (PDC), which is considered to be the archetypal ThDP decarboxylase.39 Benzoylformate decarboxylase is, however, an exception to this, and although it conserves PDC’s three-di-mensional structure, its active site shows key differences.40,41 Namely, BFDC does not conserve two consecutive histidines, nor two ionisable (acid/base) residues that are present in PDC and are known to be involved in the cata-lytic mechanism.40 Instead, BFDC has two separate histidines (His70 and His281) and one Serine (Ser26) in the active site (see Figure 5).41
The specific roles of the three residues were assigned based on X-ray crys-tallography, site-directed mutagenesis and kinetics studies. His70 and His281 were described to have an acid/base role in different steps of the mechanism and Ser26 was pointed out to be relevant, although its role was less clear.42 However, a later mutagenesis study casted a shadow of doubt on these roles, as the mutation of the histidines to hydrophobic residues, which cannot perform acid/base activity, did not reduce significantly the reaction rate.43
Besides the natural reaction, and similarly to other decarboxylases, BFDC catalyses the formation of α-hydroxyketones from two aldehydes and, in-terestingly, the preferred selectivity of this reaction is known to be different than the common R-selectivity. That is, while the self-addition of benzalde-hyde results in the formation of (R)-benzoin with full enantiocontrol,44 the addition of acetaldehyde to benzaldehyde yields (S)-2-hydroxypropiophe-none, (S)-HPP, with an ee of ca 90%.45 This makes of BFDC the only ThDP-

19
dependant enzyme with natural S-selectivity, and the only one with dual-selectivity.
In this section, the results on an extensive computational study on BFDC using the cluster approach are presented. First, the natural reaction mecha-nism is explained, followed by the energetics of the different cofactor states in different environments, and finally the two enantioselective carboligation reactions are discussed.
Figure 5 . X-Ray structure of the BFDC active site. (PDB 1MCZ).41 The blue area represents the solvent exposed cavity and corresponds to the large cavity. The red area represents the small cavity formed by Leu461 and the cofactor.
5.1.1. Active site model and computational details. The three papers on BFDC are based on the same active site model, which was built on the basis of the crystal structure of the enzyme in complex with the inhibitor (R)-mandelate (PDB 1MCZ). The cofactor, without the diphos-phate group, was included in the model together with Ser26, His70 and His281, which are the three residues argued to be catalytically relevant. Moreover, Glu28 and Glu47 are included, as they form hydrogen bonds with His70 and the N1’ of the cofactor, respectively. The surrounding residues Asn26, Pro24, Asn27, Ala44, Leu45, Asn77, Leu110, Gly401, Gly402, Leu403, Tyr433, Tyr458, Ala460, and Phe464 were also included.

20
5.1.2. Catalytic mechanism of benzoylformate decarboxylase. (Paper I) In this section, the mechanism for the decarboxylation reaction of the natural substrate, benzoylformate, is presented. It must be noted that the N1’ and N4’ positions of the cofactor are ionisable, and that the cofactor can be in different states depending on the protonation state of these two positions. An extensive study on the energetics of all possible cofactor states is presented in the next section, and for the reaction mechanism the cofactor was assumed to be in its IP state at the starting enzyme-substrate complex (ES).
The energy profile for the calculated mechanism is shown in Figure 6 and the catalytic cycle in Figure 7. Selected geometries of the intermediates and TSs are shown in Figure 8. Starting from the ES complex, the energy of which is set to 0.0 kcal/mol, the first step is the activation of the cofactor via an intramolecular proton transfer from C2 to N4’. The barrier for this step (TS1) and the resulting ylid Int1 are calculated to be +8.6 and +3.9 kcal/mol, respectively. In addition to the activation, the IP state of the cofac-tor at ES can also undergo an intramolecular nucleophilic attack which yields an off-cycle species OC1. This species is calculated to be 5.4 kcal/mol lower in energy than ES and therefore raises the energy barrier for the ylid for-mation.
Figure 6 . Calculated energy profile for the decarboxylation of benzo-ylformate catalysed by BFDC. Red colour indicates the discussed alter-native paths. Energies are in kcal/mol.
Following the mechanism from Int1, the ylid attacks the carbonyl group of the substrate and forms a first tetrahedral alkoxide intermediate, Int2, which is stabilized by an oxyanion hole formed by His70 and the N4’ group of the cofactor. The barrier for this step, TS2, is calculated to be 15.2 kcal/mol relative to OC1. Int2 subsequently forms the enamine Int4, a transformation that can occur via two different paths. In one path (the Int2-Int3a-Int4), the alkoxide in Int2 is first protonated by His70 and the result-ing intermediate Int3a subsequently decarboxylates. In the other path (the
12.0
TS4a
CO2
8.6
3.9
9.8
13.5
-11.1
-3.8 1.5
-8.7
0.2
-2.6
4.8
-8.3
-1.7
-4.2
ES
TS1
Int1
Int2
Int4Int5
Int6
Int7
TS2
TS3b
TS5
TS7
TS8EP0.0
15.9
CO2
Int3b
18.9 17.4
-5.4OC1
-10.9OC2
15.2
7.0
Int4-CO2
Int3a-4.7

21
Int2-Int3b-Int4 path), Int2 first decarboxylates and the alkoxide is after-wards protonated by His70. The decarboxylation transition states TS3b and TS4a have calculated energies of 18.9 and 17.4 kcal/mol relative to OC1, respectively, and considering the small energy difference it is not possible to identify which of the two paths is operational. Importantly, in both cases Ser26 seems to be a relevant residue for the decarboxylation, as the hydrogen bond between its hydroxyl group and the carboxylate of the substrate needs to be broken in the two C-C bond cleavage TSs. Release of CO2, which ben-efits from a stabilization of ca 18 kcal/mol, ends up forming the enamine Int4, which is calculated to be the most stable intermediate with an energy of -11.1 kcal/mol relative to ES. Moreover, it is important to note that in the protonation of the alkoxide, His70 is only acting as a shuttle, and the net proton transfer is to Glu28.
Figure 7 . Detailed mechanism for BFDC obtained on the basis of the calculations.
NH
SN
O
H2N
N
NON
O
HH
O
OO
H
H
Ser26
Gly25
Glu28
His70
TS1
TS2
Int2 TS3a
Int3a
TS3b
Int5
TS5
TS4a
TS6
TS7
Int6
Int7
EP
CO2
O
HO
O
O
OC1
CO2
TS8
O
NH
SN
O
H2N
N
HNO
N
O
HH
O
OO
H
Ser26
Gly25
Glu28
His70
O
NH
SN
O
H2N
N
HNO
N
O
HH
OO
H
Ser26
Gly25
Glu28
His70
Int3b
NH
SN
O
H2N
N
NON
O
HH
OHO
H
Ser26
Gly25
Glu28
His70
NH
SN
O
HN
N
HNO
N
O
HH
OO
H
Ser26
Gly25
Glu28
His70
H
NH
SN
O
HN
N
NO
N
O
HH
OHO
H
Ser26
Gly25
Glu28
His70
H
NH
SN
O
H2N
N
HNO
N
O
HH
OO
Ser26
Gly25
Glu28
His70
H
NH
SNO
H2N
N
HNO
N
O
HH
OO
Ser26
Gly25
Glu28
His70
H
NH
SN
OHN
N
NON
O
HH
O
OO
H
H
Ser26
Gly25
Glu28
His70
O H
N
SN
OH2N
N
NON
O
HH
O
OO
H
H
Ser26
Gly25
Glu28
His70
OES
Int1
Int4
NH
SN
OHN
N
NON
O
HH
O
OO
H
H
Ser26
Gly25
Glu28
OH

22
Figure 8 . Selected optimized structures of intermediates and TSs along the reaction pathway. For clarity, only a small part of the model is shown. Distances are given in angstroms.
Following Int4, the next step is the intramolecular protonation of the enamine by N4’ to form a second tetrahedral intermediate Int5. Next, the benzylic alcohol is deprotonated to form the alkoxide Int6, and the N4’ group of the cofactor is protonated to recover the NH2 form and yield Int7. This latter step has a calculated barrier of 15.9 kcal/mol relative to the enamine (Int4→TS7 energy difference). Similarly to the previous steps, in these two proton transfers His70 is acting as a relay and the net transfer is to and from Glu28. In the final step of the mechanism the enzyme-product complex EP is formed upon a C-C bond cleavage which yields benzaldehyde and recovers the ylid state of the cofactor. Closing the catalytic cycle requires recovery of

23
the IP form of the cofactor, release of benzaldehyde and binding of a new molecule of benzoylformate.
Overall, according to the calculations, the rate-determining step would be the decarboxylation, with a barrier of 17.4 kcal/mol. This barrier is in good agreement with the reported kcat of ca 300 s-1,40 which can be converted to a barrier of ca 14 kcal/mol. However, as mentioned in section 3.1, the decar-boxylation step is subject to uncertainties due to entropic effects. This, to-gether with the close barriers for TS2 (15.2 kcal/mol) and TS7 (15.9 kcal/mol) make it difficult to confidently conclude which is the rate-deter-mining step. Nevertheless, it has been reported that decarboxylation (TS4a) is ca 30-fold faster than the nucleophilic attack (TS2) and the enamine pro-tonation/product release (TS7).40 These individual rate constants corre-spond to energy differences of ca 2 kcal/mol, which are in line with the cal-culated energy profile.
Thanks to the calculated mechanism, the roles of His281, His70 and Ser26 can be assigned. His281, which has been suggested to be responsible of enamine protonation,42b is only involved in the binding of the substrate, and the N4’ group of the cofactor is the one that protonates the enamine. In the present mechanism, His70 is involved in the Int2 → Int3a, Int3b → Int4, Int4 →Int6 and Int6 → Int7 proton transfer steps. However, in all cases the residue is acting as a shuttle and the net proton/donor acceptor is Glu28. His70 is also relevant for pre-decarboxylation intermediates, as it stabilizes the alkoxide in Int2 by forming an oxyanion hole together with the NH2 group. These two roles of His70 are consistent with the experimental results showing that mutation of this position to non-acid/base residues does not result in significant loss of activity, and that His70 was relevant for the for-mation of the first tetrahedral intermediate.43 Finally, the main role of Ser26 is in binding, and appears to be specifically relevant in the decarboxylation step.
As a final remark, it is important to note the significance of including the tricyclic off-cycle species of the cofactor in the calculated mechanism. As shown, this species is lower in energy than the ES complex and therefore raises the energies of all the subsequent steps until the decarboxylation. Spe-cifically, the barrier for the nucleophile attack would have been unrealisti-cally low without OC1 (9.8 kcal/mol instead of 15.2 kcal/mol) considering the evidences that show that it has a similar rate as decarboxylation. The relevant impact of this species prompted us to conduct a more extensive char-acterization of the cofactor and its states, which will be presented in the next section.

24
5.1.3. Computational characterization of enzyme-bound thiamine diphosphate. (Paper II) As described in the previous section, the cofactor ThDP can form different states, which essentially differ on the protonation state of N1’, N4’ and C2. Figure 9 shows all possible states that can form. Starting from the one com-monly described as the natural form of the cofactor, the AP state, protonation of the N1’ position results in the APH+ state. A subsequent deprotonation of C2 yields the ylid state YIH+. Similarly to the AP state, the ylid can also exist with a deprotonated N1’ (YI).
The relevance and role of each of these states has been extensively dis-cussed in the literature, and it is now very well known that the two ylid states are the pillars of ThDP catalysis,46 as these are the only states that can func-tionalize the substrate. Moreover, there has been great efforts to characterize all the states and stablish which is the predominant form in the active site.47
There are, however, two cofactor states that are largely ignored in the literature. These are two tricyclic forms of the cofactor (TC and TCH+) which form upon a nucleophile attack of N4’ at C2 (see Figure 9). These states were first described for the cofactor in solution,48 and later character-ized by X-ray crystallography in the enzymes phosphoketolase (PK)49 and acetolactate synthase (AHAS).50 Importantly, as shown in the previous chap-ter, these two states are not only a structural curiosity, but can also have a relevant impact on the kinetics of the reaction.
In this section, a detailed study on the different states of the cofactor ThDP in the active site of BFDC is presented. The aim of this work was to investigate the relative stability of each of the states and the effect of the enzyme environment. To that end, five models representing different enzy-matic and non-enzymatic environments were used.
Figure 9 . Common equilibriums involved in ThDP-dependent decarbox-ylases.
N
NNS
H2N
R
H N
NHNS
H2N
R
H
N
NHNS
HN
R
H
N
NHNS
H2N
R
NS
R
HO CO2
R
NS
R
HOR
N
NNS
H2N
R
AP APH+
IP
O
O
OR
CO2
N
NHN
NH
SR
H
TCH+
N
NN
NH
SR
H
TC
YIH+YI
C-C
C-O
C-H
PDC, BFDCPOX
AHAS
1’
3’4’
2
R = CH2CH2OPP
25
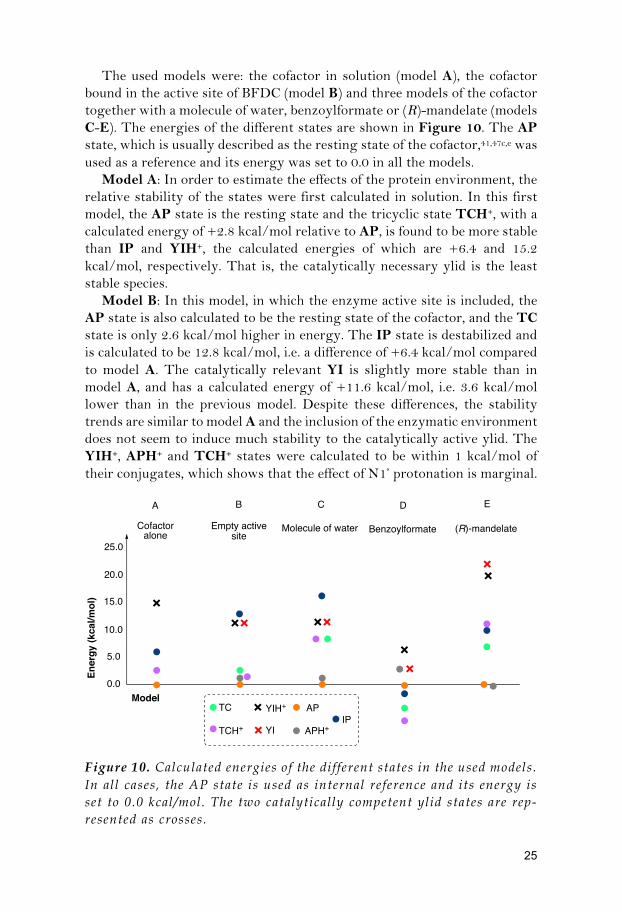
The used models were: the cofactor in solution (model A), the cofactor bound in the active site of BFDC (model B) and three models of the cofactor together with a molecule of water, benzoylformate or (R)-mandelate (models C-E). The energies of the different states are shown in Figure 10. The AP state, which is usually described as the resting state of the cofactor,41,47c,e was used as a reference and its energy was set to 0.0 in all the models.
Model A: In order to estimate the effects of the protein environment, the relative stability of the states were first calculated in solution. In this first model, the AP state is the resting state and the tricyclic state TCH+, with a calculated energy of +2.8 kcal/mol relative to AP, is found to be more stable than IP and YIH+, the calculated energies of which are +6.4 and 15.2 kcal/mol, respectively. That is, the catalytically necessary ylid is the least stable species.
Model B: In this model, in which the enzyme active site is included, the AP state is also calculated to be the resting state of the cofactor, and the TC state is only 2.6 kcal/mol higher in energy. The IP state is destabilized and is calculated to be 12.8 kcal/mol, i.e. a difference of +6.4 kcal/mol compared to model A. The catalytically relevant YI is slightly more stable than in model A, and has a calculated energy of +11.6 kcal/mol, i.e. 3.6 kcal/mol lower than in the previous model. Despite these differences, the stability trends are similar to model A and the inclusion of the enzymatic environment does not seem to induce much stability to the catalytically active ylid. The YIH+, APH+ and TCH+ states were calculated to be within 1 kcal/mol of their conjugates, which shows that the effect of N1’ protonation is marginal.
Figure 10. Calculated energies of the different states in the used models. In all cases, the AP state is used as internal reference and its energy is set to 0.0 kcal/mol. The two catalytically competent ylid states are rep-resented as crosses.
A B C D E
Cofactoralone
Empty activesite
Molecule of water Benzoylformate (R)-mandelate
0.0
5.0
10.0
15.0
20.0
25.0
TCH+IP
YIH+
APH+
TC
YI
AP
Ener
gy (k
cal/m
ol)
Model

26
Model C: The inclusion of a water molecule has a negative effect on the stability of the TC state, which is calculated to be +8.9 kcal/mol relative to the most stable state AP, i.e. a difference of +6.3 kcal/mol compared to model A. The IP state is also destabilized, and it is calculated to be +16.0 kcal/mol in this model, which represents an increase of 5.4 kcal/mol relative to model B and as much as 9.6 kcal/mol relative to model A. The YI state has a calcu-lated energy of 11.6 kcal/mol. Thus, it could be thought that inclusion of a molecule of water is, at least, not detrimental for YI which is calculated to be as stable as in model B and 4.1 kcal/mol lower in energy than in model A. However, formation of the ylid is in most cases described to be an intramo-lecular proton transfer starting at IP and, under this assumption, a raise in energy of IP hinders the ylid formation.
Model D: Binding of benzoylformate in the active site drastically changes the stabilities of the different states. In this model, the IP and YI states are calculated to be -0.9 and +6.0 kcal/mol relative to AP, respectively. There-fore, the three states are within ca 6 kcal/mol, which is in stark contrast to the previous models. Not only that, but in this model the most stable of the three states is IP. This indicates that the inclusion of the substrate facilitates the access to the catalytically active species IP and YI. However, the tricyclic states also benefit from the inclusion of the natural substrate and are calcu-lated to be the most stable species with calculated energies of -6.3 kcal/mol for TCH+ and -4.9 kcal/mol for TC. Therefore, the YI state is as disfavoured as in models B and C, as the energy difference between the most stable spe-cies (TCH+ in the present model and AP in the previous two) and the YI state is ca 10 kcal/mol.
Model E: Binding of (R)-mandelate results in the widest energy span be-tween states. In this model, the AP and APH+ states are the most stable states and the YI and YIH+ states are calculated to be as much as +23.2 and 20.2 kcal/mol, respectively. The ylid states are also substantially more un-stable than TCH+, TC and IP, which have calculated energies of +11.3, +7.9, and +10.2 kcal/mol relative to AP, respectively. Overall, the presence of (R)-mandelate stabilizes the AP state and makes the ylid states energetically un-feasible.
It is interesting to compare this model with the model including benzo-ylformate (model D), as both substrates have a carboxylic moiety, an oxygen at the α position and a phenyl ring substituent, but the obtained energies are drastically different. A comparison between the AP state for the two models shows that the (R)-mandelate has more interactions with the surrounding residues than benzoylformate (see Figure 11). Namely, the benzylic hy-droxyl of (R)-mandelate forms hydrogen bonds with the exocyclic NH2 and one with His70, while the carbonyl group of benzoylformate does not inter-act with any of the groups. When considering the ylid states, the negative

27
charge of the carbanion is facing the hydrogen of an sp3 carbon in the case of (R)-mandelate, but an electrophilic sp2 carbon in the case of benzoylformate.
Overall, the calculations on the different ThDP states show that the sta-bility of the different forms is highly dependent on the environment (see Fig-ure 10). For example, the AP-YI energy gap is 3.0 kcal/mol in model D and 23.2 kcal/mol in model E. However, it is interesting to see that, despite this high variability, the energy cost to form the ylid, i.e. the difference between the most stable state and the ylid, is rather similar in models B, C and D.
The calculated relative stabilities also suggest that the tricyclic states are more relevant than expected as these states are consistently more stable than the catalytically active ylid in all models. The model with the natural sub-strate is especially interesting, as the TCH+ state is calculated to be the most stable species overall.
The stability of the tricyclic states and the effect of the environment give important messages for the all studies concerning ThDP-dependent en-zymes. First, it points out that the largely ignored tricyclic species have an actual relevance in ThDP catalysis. That is, in the specific case of BFDC, the tricyclic states are adding a penalty of ca 5 kcal/mol to the ylid formation, which corresponds to a 1000-fold slower rate for this step. Furthermore, the results of this work alert that to directly start a computational study from the ylid state, which is commonly done, can result in an incomplete descrip-tion of the subsequent reaction mechanism.

28
Figure 11 . Optimized structures of four representative ThDP states for models D (including benzoylformate) and E (including (R)-mandelate). For clarity, only a small part of the model is shown.

29
5.1.4. Computational study of the enantioselective carboligation catalysed by benzoylformate decarboxylase. (Paper III) In this section the mechanisms for the self-addition of benzaldehyde to form benzoin and for the addition of benzaldehyde to acetaldehyde to yield 2-hyd-roxypropiophenone (HPP) are presented. Considering that the first half of the carboligation reaction (formation of the enamine from benzaldehyde) is common for all studied reactions, the study of the two carboligation reactions started from the binding of the acceptor.
For the two studied acceptors, different binding conformations were con-sidered. The full mechanism was calculated for all poses, and the most stable conformations for the different intermediates and TS were used to obtain the energy graphs. The calculated mechanisms consist of the same steps regard-less of the acceptor and are summarized in Figure 12. The first step involves the nucleophilic attack of the donor on the acceptor, which yields a first tet-rahedral intermediate Int1. This C-C bond formation results in an alkoxide, which in the next step is neutralized by an intramolecular proton transfer from the hydroxyl group of the enamine to form Int2. Finally, the C-C bond in cleaved, the enzyme-product complex EP is formed and the ylid form of the cofactor is formed.
Figure 12 . Mechanism for the carboligation reaction starting at the enamine.
For the self-addition of benzaldehyde, the energy graphs for the S- and R- products are shown in Figure 13. The most stable binding mode is found to be En(BA)-R1 and its energy is set to 0.0 kcal/mol. En(BA)-S1 is the most stable pro-S pose, i.e. a binding mode that eventually results in the formation of the S-product, with an energy of +1.1 kcal/mol. However, the first pro-ductive pro-S binding mode is En(BA)-S2, the energy of which is calculated to be +7.2 kcal/mol. The energy difference between the R- and S- paths in-creases even more upon C-C bond formation, with a calculated energy of 10.7 kcal/mol for TS1(BA)-R and as much as 25.1 kcal/mol for TS1(BA)-S. The subsequent intramolecular proton transfer is calculated to have low or no barrier for the R-path, and a barrier of 4.0 kcal/mol for the S-path. The final C-C bond cleavage step is the rate-determining step and has a calculated en-ergy of 18.3 and 27.6 for TS3(BA)-R and TS3(BA)-S, respectively.
Int1 Int2 EP
TS3TS2TS1
NH
SN
OH
H2N
OH
R1
NH
SN
OH
H2N
OHR
NH
SN
O
H2N
OHHR
NH
SN
OH2N
OHHR
En .

30
Figure 13 . Calculated potential energy graphs for the carboligation re-action of benzaldehyde. Energies in kcal/mol. The energy of the TS for the proton transfer step (Int1(BA)-R→ Int2(BA)-R) was lower than the preceding intermediate after adding the corrections,
Figure 14 . Optimized geometries of the two TS1 for the reaction with benzaldehyde as acceptor. For clarity, only a small part of the active site model is shown here.
5.8
18.3
10.7
0.0
25.1 22.926.9
9.4
18.0
27.6
8.6
9.5
9.3
En(BA)-R1
Int1(BA)-R
Int2(BA)-R
TS2(BA)-STS3(BA)-S
TS1(BA)-S
Int1(BA)-S
Int2(BA)-S
EP(BA)-R
EP(BA)-S
1.1En(BA)-S1
7.2En(BA)-S2
TS1(BA)-R
TS3(BA)-R

31
Although TS3 is the rate-determining and selectivity-determining step, the S-path is found to be unfeasible already at TS1(BA)-S, as it is calculated to be 14.4 kcal/mol higher in energy than TS1(BA)-R. A close analysis of the geometries of TS1(BA)-S and TS1(BA)-R shows the reasons of such a large difference (see Figure 14). In both transition states the nascent alkox-ide is stabilized by the formation of two hydrogen bonds with the OH group and the peptide bond of Ser26, with the difference that TS1(BA)-R benefits from an extra hydrogen bond with the hydroxyl group of the enamine. More-over, the Ser26 hydrogen bonds hinder the mobility of the acceptor and forces its substituent to point into the large cavity at TS1(BA)-R and into the small cavity at TS1(BA)-S. In the latter case, this causes clashes between the aromatic substituent and Leu461, Ala460 and the cofactor. Importantly, the calculated energy difference in favour of the R-product is in line with the experimental observation that only (R)-benzoin is formed.44a
Next, the reaction mechanisms for the addition of the enamine to acetal-dehyde were calculated. The potential energy graphs for HPP formation are shown in Figure 15, and as seen, these are quite different than those for ben-zoin formation.
The first step is calculated to have low or no barrier for the formation of both enantiomers and the resulting intermediates Int1(AA)R and Int1(AA)-S, with calculated energies of -3.1 and -4.8 kcal/mol, respectively, are found to be more stable than the starting binding modes. Similarly, the subsequent intramolecular proton transfer is calculated to have no significant barrier in the two paths.
Similarly to benzoin formation, the rate- and selectivity-determining step is the C-C bond cleavage, the energy of which is calculated to be 5.8 kcal/mol for TS3(AA)-S and 6.1 kcal/mol for TS3(AA)-R. This small energy differ-ence of 0.3 kcal/mol in favour of (S)-HPP indicates that the two paths are operational, and the major product would be the S-enantiomer. The observed S 90 % ee45b corresponds to an energy difference of ca 1.5 kcal/mol and thus the calculated energy difference is clearly underestimated. Nevertheless, the fact that the same trend is observed, i.e. two operational paths and the S-product being favoured, is considered to be satisfactory. Comparison of TS3(AA)-S and TS3(AA)-R (Figure 16), shows that the methyl group fits, almost indistinguishably, in both cavities, and that the hydroxyl group forms an identical hydrogen bond network in the two TSs. Considering the small size of the substituent, these similarities are not surprising but raise the ques-tion of what is inducing the selectivity in HPP formation. That is, the sizes of the cavities do not change depending on the substrate, and therefore the large cavity would always create less clashes than the small one. Conversely, the methyl substituient shows a preference for the small cavity, which could be a result of the favourable dispersion interactions between the methyl and the surroundings (mostly the cofactor and Leu461). These interactions are

32
highly dependent on a good fit of the substrate in the cavity. Indeed, the me-thyl substituent is the only one that shows selectivity over the S-product, and carboligation with the next aliphatic aldehyde, propanal, already selec-tively forms the R-product.
Figure 15 . Calculated potential energy graphs for the carboligation re-action of acetaldehyde. Energies in kcal/mol.
Figure 16 . Optimized geometries of the two TS3 for the reaction with acetaldehyde as acceptor. For clarity, only a small part of the active site model is shown here.
To summarize, in this chapter two enantioselective reactions have been studied, i.e. the formation of benzoin and HPP. The obtained potential energy graphs indicate that (R)-benzoin would be formed exclusively, and (S)-HPP would be the favoured product although its enantiomer would be also formed. Importantly, the obtained trends and the reversal of the enantiose-lectivity are in agreement with the experimental studies.
5.8
-4.8
-3.1
6.1
0.0
-5.1
0.2-3.2
-1.8
0.3
Int1(AA)-RInt2(AA)-R
TS3(AA)-R
Int1(AA)-S Int2(AA)-S
TS3(AA)-S EP(AA)-S
EP(AA)-R
En(AA)-R1
En(AA)-S1
4.7

33
5.2. SEPHCHC synthase. In addition to the 1,2-additions to form α-hydroxyketones that most of ThDP-dependant enzymes catalyse, a small group of ThDP-dependent en-zymes are also able to catalyse the conjugated addition of ketoacids to α,β-unsaturated carbonyl compounds. This reaction, named Stetter reaction, yields 1,4-dicarbonyl compounds,51,52 which are of high interest due to their high applicability but challenging preparation.
2-succinyl-5-enolpyruvyl-6-hydroxy-3-cyclohexene-1-carboxylic-acid (SEPHCHC) synthase, also named MenD, is a decarboxylase responsible of the conversion of isochorismate and 2-oxoglutarate (α-ketoglutarate) to SE-PHCHC as part of the menaquinone biosynthesis (see Figure 17).53 Its abil-ity to catalyse the formation of 1,4-dicarbonyl compounds as well as the C4 selective chain elongation of carboxylic substrates make MenD of potential interest in biocatalysis.54
Figure 17. Natural reaction catalysed by MenD.
Importantly, two reaction intermediates have been characterized by X-ray crystallography, providing privileged detail of the reaction mechanism. The first one is a tetrahedral post-decarboxylation intermediate, which was suggested to be either the carbanion form of the Breslow intermediate or its protonated form at the C2α position (see Figure 18).55 The second interme-diate is a donor-acceptor adduct corresponding to the second part of the mechanism (see Figure 18). This structure shed light on the role of Gln118,56 a highly conserved residue, which was described to be key for the stability of this crystallized intermediate.
Moreover, kinetics studies indicated that binding of ketoglutarate was rate-determining for the first half of the reaction, that the formation of the tetrahedral post-decarboxylation intermediate was irreversible in absence or in low concentrations of the acceptor, and that SEPHCHC formation or its release from the active site are rate-determining for the second half of the reaction.57 However, despite the excellent insight provided by these works, the specific reaction mechanism remained unknown.
In addition to the natural reaction, MenD has also been shown to catalyse the 1,2-addition to saturated acceptors,58 and both 1,2 and Stetter additions
OHO
O
OCO2
-OH
OO
OCO2
-
O
O
O
H
12
34O
O
O
O
O
ketoglutarate
isochorismate
SEPHCHCCO2

34
to α,β-unsaturated carbonyl compounds.59 Interestingly, the enzyme shows different regioselectivities depending on the acceptor, and while short-chain ketone Michael acceptors favour the formation of the Stetter product, alde-hydes selectively form the 1,2-product.58 Moreover, in the latter case, the reaction is enantioselective and MenD predominantly forms the R-prod-uct.60-62
In this section, the natural reaction catalysed by MenD and two reactions with non-natural acceptors are presented. The used acceptors were but-3-en-2-one (BO) and cyclohex-1-ene-1-carbaldehyde (CA), which show different selectivity and form the 1,4- and 1,2-product, respectively.58,59 These two re-actions were used to investigate the factors governing enantio- and regiose-lecticity.
Figure 18. Proposed mechanism and intermediates for the natural reac-tion of MenD. The two rendered images correspond to the two interme-diates characterized by X-Ray crystallography.
5.2.1. Active site models. The active site model of MenD used to study the first half of the reaction mechanism was built on the basis of the crystal structure containing the post-decarboxylation intermediate from E. coli (PDB 5EJ5). For the second half, a model based on the crystal structure of the donor-acceptor adduct from M. tuberculosis (PDB 5ESU) was used. Both models contain the same residues, and comprise the cofactor and the immediately surrounding Ala29, Pro30, Gly31, Ser32, Arg33, Glu55, Thr78, Ser79, Gly80, Arg107, Asn118, Asn117, Glu172, Arg395, Arg413, Ser416, Gly417, Ile418, Ile474, Phe475 and Leu478 (using the residue numbers of the E. coli variant). Position Ser416 corresponds to Ala402 in the M. tuberculosis variant. Therefore, this position

35
was manually mutated (Ala → Ser) in order to have the same sequence in the two models. In total, the model for the first half has 319 atoms, and the model for the second half, 345 atoms.
5.2.2. Natural reaction mechanism. In this section, the reaction mechanism for the synthesis of SEPHCHC from α-ketoglutarate and isochorismate catalysed by MenD is presented. The first part in the study of the reaction mechanism of MenD was an investigat-ion of the different states of the cofactor and their energies in the active site. Starting from the iminopyrimidine state (IP), formation of the tricyclic state TC state is associated with a barrier of 10.9 kcal/mol, and the transformation is calculated to be exothermic in 2.4 kcal/mol. Alternatively, IP can form the YI state, the energy of which is calculated to be -2.7 kcal/mol, and the cor-responding barrier 6.2 kcal/mol. Importantly, in the case of MenD the AP and APH+ states cannot form, as that would require of a proton source to donate a proton to N4’, and the active site does not have any sufficiently close.
Next, the reaction mechanism starting from the ylid was studied. The en-ergy of this species is set to 0.0 in the following discussion, and the calculated mechanism is shown in Figure 19. The associated energy profile is shown in Figure 20, and selected optimised geometries are shown in Figure 21. Start-ing from YI, the C2 carbanion adds to the carbonyl group of ketoglutarate with a calculated barrier of 10.5 kcal/mol (TS1) to form a first tetrahedral intermediate Int1, the energy of which is calculated to be -7.0 kcal/mol. This intermediate can either decarboxylate via TS2 or undergo an intramolecular proton transfer to neutralize the C2α-alkoxide. This latter transformation results in an off-cycle species (OC) that cannot decarboxylate and is calcu-lated to be 1.9 kcal/mol lower in energy than Int1. Thus, the presence of OC raises the decarboxylation barrier from 15.2 to 17.1 kcal/mol.
The decarboxylation from Int1 results in the formation of the enamine Int2, which is calculated to be -7.9 kcal/mol after CO2 release, relative to YI, and only 1 kcal/mol higher in energy than OC. At this stage, the enamine intermediate is suggested to form the tetrahedral post-decarboxylation in-termediate upon a proton transfer from the N4’H2 group to C2α, and thus to yield the imino form of the N4’ moiety.57 However, this transformation is calculated to be thermodynamically unfavourable by 7 kcal/mol. Alterna-tively, an estimation of the energy for protonation of C2α from the bulk, while keeping the N4’ group in the amino form, was found to be exothermic. The favourable formation of the post decarboxylation intermediate only if N4’ is in the amino form is in line with the calculated mechanisms of other ThDP-dependent enzymes.63

36
Figure 19. Detailed mechanism obtained on the basis of the calculations for the natural reaction catalysed by MenD.
Figure 20. Calculated energy profile for the natural reaction catalysed by MenD.
Int2
Int3
Int4Int5
EP
TS1
TS2
TS4
TS5
CO2O
OH
HO2C
H+
YIInt1
CO2
SEPHCHC
CO2
O
O2C
N
SN
O2C O
O2C
H2NHO
O
N
SN
O2CO
H2NHO
O
N
SN
O2COH
H2N
HO
O
HO2C
CO2
HOO
N
SN
O2COH
H2N
HO
O
HO2C
O O
N
SN
O2CO
H2N
HO
O
HO2C
CO2
H
N
SN
O2C
O
H2NHO
O
CO2H
CO2
H HOO
HOO
HOO
N
SN
O2COH
O2C
HNHO
O
N
SN
O2C
O
O2C
H2NHO
O
TS6
1st half-reaction
2nd half-reaction
OC
N
SN
O2COH
H2N
HO
O
HO2C
O O
HOO
N
SN
O2C
O
H2NHO
O
H
H+
0.0
10.5
-7.0 -7.9-8.9
8.2
0.0
5.82.5
8.3
-0.4
~7
~3
1st half-reaction 2nd half-reaction
CO2YI
TS1
Int1
TS2
Int2OC
Int3
TS4Int4
TS5
Int5
TS6*
EP*
17.1

37
Figure 21. Optimised geometries of a selection of intermediates and transition states of the second half of the reaction mechanism. For clar-ity, only few residues and polar hydrogens are included in the figure.

38
To calculate the second part of the reaction mechanism, a second active site model was used. In this second model the acceptor isochorismate is bound in the active site, and a proton is added to neutralize the C2α-O group to form Int3. From this intermediate, C2α adds to the β-carbon of the ac-ceptor to form the enolate intermediate Int4. The barrier for this step (TS4) and the resulting intermediate are calculated to be 5.8 kcal/mol and 2.5 kcal/mol, respectively. Next, the α-carbon of the acceptor moiety extracts the proton from the C2α-OH group with a calculated barrier of 8.3 kcal/mol relative to Int3, which results in the formation of Int5, the energy of which is calculated to be -0.4 kcal/mol. In the last step, Int5 undergoes a C-C bond cleavage to form the EP complex. Here it must be noted that the geometries corresponding to the transition state for this transformation (TS6) and the resulting EP complex could not be located, as optimisation of the two geom-etries always resulted in the formation of Int5. Therefore, the geometries and energies of these two stationary points were estimated from the highest-energy point along a linear transit of the C-C coordinate, and from a geome-try optimisation of the EP complex in which the forming carbonyl was con-strained to be planar.
In this section the reaction between α-ketoglutarate and isochorismate catalysed by MenD has been studied. As seen from the calculated energy profile (Figure 20), the decarboxylation (TS2) and proton transfer steps (TS5) are associated with the highest barriers for the first and second halves of the mechanism, with calculated barriers of 17.1 and 8.3 kcal/mol, respec-tively. Experimentally, the rate constant for the first half of the reaction, i.e. from substrate binding to decarboxylation, has been been determined to be 83.8 s-1,57 which corresponds to a barrier of ca 15 kcal/mol. This rate con-stant was assigned to the substrate binding event,57 and thus the associated barrier can only be used as an upper limit. The calculated barrier of 17.1 kcal/mol can be considered to be in good agreement with the experimental observations. The rate constant for the second half of the reaction, i.e. the steps from acceptor binding to product release, has been experimentally de-termined to be 0.3 s-1.57 This rate constant can be approximated to a barrier of ca 19 kcal/mol, and has been proposed to correspond to either the step of SEPHCHC formation in the active site or to product release. The considera-bly low calculated barriers for the second part of the reaction mechanism provide indirect evidence to support that product release, and not formation of SEPHCHC, is the rate-determining step. Moreover, the most stable spe-cies in the second part of the mechanism is the keto tautomer of the donor-acceptor adduct, which is in line with the fact that this intermediate has been crystallized.56
The calculations also revealed that formation of the post decarboxylation tetrahedral intermediate via the proposed C2α protonation by the N4’H2 group is not thermodynamically favourable, and instead C2α is protonated

39
by a proton from the bulk. Therefore, the calculated mechanism indicates that the crystallographically characterized post-decarboxylation intermedi-ate does not have the N4’ group in the imino form, but in the amino form.
5.2.3. Non-natural substrates: Analysis of the regioselectivity. In this section, the reaction mechanisms for the 1,4- and 1,2-additions of α-ketoglutarate to but-3-en-2-one (BO) and cyclohex-1-ene-1-carbaldehyde (CA) are covered. Because all steps leading to the formation of the enamine are independent of the acceptor, only the second part of the reaction mecha-nisms was studied. The reaction mechanisms are the same for the two accep-tors, and consist of the steps summarized in Figure 22.
For the two acceptors, different binding modes were first studied. As ex-plained in the introduction, the active site of ThDP-dependent enzymes can be divided into a small hydrophobic pocket, and a large, solvent-exposed, pocket. In the case of MenD, the small pocket is shaped by the thiazole ring of the cofactor, and the residues Phe475, Ile474 and Leu478. The most stable binding modes have all in common the parallel orientation of the hydroxyl of the enamine and the carbonyl of the acceptor, and differ on the conform-ation of the acceptor (cis or trans) and whether its large substituient (in) or the small substituient (out) is facing into the small pocket of the active site.
In this section, first, the 1,2- and 1,4-addition reactions for the two accep-tors will be discussed and compared in order to analyse the factors governing the regioselectivity. Next, the reaction with the aldehyde will be used to dis-cuss the enantioselectivity.

40
Figure 22. Possible reaction mechanisms between the enamine interme-diate and a α ,β-unsaturated acceptor. The coloured areas indicate the large (blue) and small (red) pockets.

41
Reaction mechanisms for but-3-en-2-one. Figure 23 shows the calculated energy profile for the 1,4-addition reaction and the 1,2-pathway associated with the lowest barriers. The two mecha-nisms diverge at the Int1→Int2 step, which corresponds to an enolate-keto tautomerization for the 1,4-pathway, and to an alcohol to alkoxide proton transfer for the 1,2-pathway. The resulting intermediates BO-Int2-14 and BO-Int2-12 are calculated to be -5.2 and +9.9 kcal/mol relative to En•BO-trans-out, respectively, i.e. a difference of 15.3 kcal/mol between paths. This energy difference slightly increases at the last step. That is, while the en-zyme-product complex for the 1,4 path (BO-EP-14) is calculated to be 0.1 kcal/mol, the energy of BO-EP-12 is as much as 18.5 kcal/mol. It must be noted that the transition state for this step (BO-TS3-12) could not be lo-cated, as all attempts resulted in the formation of BO-Int2-12. However, the energy difference of 18.4 kcal/mol between the two enzyme-product com-plexes is sufficient to conclude that the 1,2-path is non-operational. There-fore, the calculated mechanisms for the addition to the ketone indicate that the 1,4-product would be the only one to form, a result in line with the ob-served selectivity.58
Figure 23. Calculated energy profiles for the 1,2 and 1,4 additions of the enamine intermediate to but-3-en-2-one. * The transition state was lo-cated, but its energy after corrections was lower than the preceding in-termediate. ** The transition state could not be located.
Reaction mechanisms for cyclohex-1-ene-1-carbaldehyde. Figure 24 shows the calculated energy profiles for two the 1,2-additions (le-ading to the formation of the R- and S- products) and the 1,4-addition of the enamine to CA. As seen from the profile, the initial nucleophilie attack of the

42
enamine to the acceptor in the 1,4-addition (CA-TS1-14) is calculated to be higher in energy than the entire 1,2-addition pathways by, at least, ca 5 kcal/mol. Therefore, the 1,4-addition path can be concluded to be non-oper-ational, which is in line with the experimental regioselectivity.58
Figure 24. Calculated energy profiles for the 1,2 and 1,4 additions of the enamine intermediate to cyclohex-1-ene-1-carbaldehyde. * The transition state was not located, and the maximum in energy in the linear transit for the C-C coordinate is the same as EP. ** The transition state was not located, and the energy was estimated as the maximum in the linear transit along the C-C coordinate.
It is here instructive to compare the different reactivity of the two accep-tors, BO and CA, to analyse the factors governing the different regiose-lectivity of the two reactions. As seen from the two calculated 1,2-addition mechanisms (see Figures 23 and 24), the first step presents a barrier of only 1 kcal/mol and is exothermic for CA, while in the case of BO the same step is endothermic and is associated with a barrier of more than 10 kcal/mol. The reasons for this very different energetics is a combination of sterics and the inherently different reactivity of the two acceptors. That is, in the reac-tion of BO, the vinyl substituent points into the small cavity and clashes with the hydrophobic residues present in the pocket. In contrast, in the reaction of CA, the much smaller hydrogen is in the small pocket, therefore causing much smaller steric repulsion. In addition, the carbonyl of an aldehyde is a better electrophile than the carbonyl of a ketone, thus favouring the 1,2-ad-dition reaction in the former case.
The same factors can be invoked to understand the different profiles in the case of the1,4-addition. Namely, while in the case of BO the reaction starts with the vinyl substituent bound in the small pocket, in the reaction of CA the cyclohexenyl substituent is the one that binds into the small pocket.

43
Moreover, the steric strain in the mechanism with BO is substantially re-duced upon the initial C-C bond formation between C2α and the terminal carbon of the vinyl group, as this transformation displaces the whole acceptor towards the large pocket. On the other hand, the cyclic nature of CA causes a large fraction of the ring susbtituent to stay in the small pocket across the whole mechanism. This results in a greater steric repulsion in the reaction with CA. Moreover, it is known that the enolate tautomer (Int1-14 in the mechanism) is relatively more favoured over the keto form (Int2-14) for an aldehyde, such as CA, than for a ketone, such as BO. The mentioned factors result in a favourable protonation of the α-carbon at Int1-14 for BO, but a kinetically and thermodynamically less favoured protonation for CA.
Figure 25. Considered binding modes for the addition of the enamine to cyclohex-1-ene-1-carbaldehyde

44
5.2.4. Non-natural substrates: Enantioselectivity. As seen from Figure 24, the 1,2-addition of the enamine to CA can result in the formation of two enantiomers of the product. The calculated mechanisms show that the energies associated with the pathway leading to the formation of the S-product are always higher than the R-pathway, in agreement with the observed exclusive formation of the latter enantiomer.58 The main factor for this difference is sterics. That is, formation of the R- and S-products need of the acceptor to bind with its Si or Re face towards the donor, respectively. This factor, along with the common parallel orientation of the hydroxyl of the donor and the carbonyl of the acceptor, results on the binding of the cyclohenxenyl substituent in the large pocket in the R-pathway, and in the small hydrophobic pocket in the S-patwhay, which results in the steric re-pulsion between the acceptor and Phe475 and Ile474. These intereactions are partly alleviated in the initial C-C bond formation and the subsequent proton transfer. However, in the last step the energy between paths is approximately 3 kcal/mol in favour of the R-pathawy, which corresponds to a fully selective reaction.
To summarize, in this section the regioselectivity and enantioselectivity of two reactions have been studied. As seen from the calculated energy pro-files, the sterics and the keto-enol equilibrium are found to play a relevant role in the regioselectivity, while the steric repulsion between the acceptor and the small pocket of the active site are what determine the enantioselec-tivity.
5.3. Conclusions. The mechanisms of the natural reactions has been studied for the ThDP-dependent enzymes benzoylformate decarboxylase (BFDC) and SEPHCHC synthase (MenD). All calculated mechanisms are in good agreement with the experimental observations. The experimental enantioselectivity trends are reproduced by the calculations, which is a further testament to the applica-bility of the cluster approach in the study of enantioselectivity in enzymes.
For BFDC, the calculated mechanism sheds light on the roles of His70, His281 and Ser26. The two latter appear to be involved only in binding, and His70 acts as a shuttle in several proton transfers where the donor/acceptor is Glu28. It is also shown that enamine protonation is an intramolecular pro-ton transfer from the exocyclic NH2 group and does not involve any residues in the active site. Moreover, the usually ignored tricyclic states of the cofac-tor are shown to be relevant for the kinetics of the reaction. Accordingly, the characterization of the different ThDP states is, to our knowledge, the most extensive computational study done on the issue. The calculations on BFDC show that the stabilities of the states are highly sensitive to the environment,

45
and supports the importance of the tricyclic states in ThDP-dependent en-zymes. Therefore, this work sets an important precedent, not only for future computational studies of ThDP-dependent enzymes, but also for the kinetic characterization of the different cofactor species in other ThDP-dependent enzymes.
For MenD, the calculated mechanism provides new insight into the reac-tion, and shows that the hydroxyl group of the enamine is the proton source in the formation of the product. Moreover, formation of the post-decarboxy-lation intermediate via a proton transfer from N4’ to C2α, as previously sug-gested,57 is found to be endothermic and the more likely scenario is that for-mation of these species happens via protonation from a proton from bulk sol-vent.
In addition to the natural reactions, different reactions with non-natural acceptors have also been studied for the two enzymes in order to establish the factors governing the enantio- and regioselectivity. In the case of MenD, the mechanisms of the 1,2- and 1,4-additions of the enamine to two α,β-un-saturated substrates have been studied. The calculated mechanisms show that the regioselectivity of the reaction is result of a combination of unfa-vourable sterics and the relative stability of the keto-enolate tautomarization in the 1,4-path, and the inherently different reactivity of the acceptor in the 1,2-addition.
When considering the enantioselectivity of the 1,2-additions, the neces-sary parallel orientation of the hydroxyl of the donor with the carbonyl of the acceptor and the steric clashed between the acceptor and the residues present in the small pocket of the active site are the factors governing the selectivity. Importantly, the reduction of the steric strain in the small pocket of the active site (via mutations or the use of smaller acceptors) is beneficial for both the 1,4- and the 1,2-S-selectivity.

46

47
6. Computational investigation of a Pummerer coupling in non-electrophilic media. (Paper V)
Organosulfur chemistry has been prominent in organic synthesis due to the rich chemistry of sulfur, and nowadays there is a plethora of synthesis methods based on the use of these compounds.64 Sulfoxides are stable and versatile building blocks65 that can be used, for example, as the electrophilic partner in Pummerer reactions and its different variations.64,66 However, in these reactions a strong electrophilic medium is necessary, which limits the scope of nucleophiles that can be used. Accordingly, the use of Grignard re-agents in Pummerer reactions, which are strong C-nucleophiles, has been rather scarce.67 Nevertheless, incorporation of these reagents is still very ap-pealing, as they display good chemoselectivities and are based on easily ac-cessible and inexpensive materials.
In 2017, Mendoza and co-workers reported a novel Pummerer-like, non-catalytic, C-C coupling reaction between a Grignard reagent and a sulfoxide in the presence of the turbo-Hauser base DIPAMgCl·LiCl (see Figure 26a).68 Importantly, this protocol allows for the C-H functionalization of a sulfoxide without the need for directing groups, accepts a broad range of nucleophiles, and side-products are marginally observed.
The mechanistic details of this reaction are, however, largely unknown. The formation of a sulfonium intermediate upon a β-oxide elimination from the sulfoxide-Mg complex has been proposed (see Figure 26a) based on chi-rality-transfer experiments.68 Moreover, the sensitivity of the experiments to the size of the amide and the presence of LiCl (the use of DIPAMgCl re-sulted in significantly worse yields, see Figure 26a) suggested that for-mation of turbo-organomagnesium amides, i.e. complexes between the Gri-gnard reagent and the turbo-Hauser base, was likely. However, the compli-cated nature of the solution equilibria of Grignard reagents and Hauser bases makes of the experimental characterization of turbo-organomagnesium am-ides a very challenging task. To date, there is only solid-state evidence of the structure of organomagnesium amides,69 i.e. without added LiCl, and their behaviour in solution is largely unknown (see Figure 26c).
To shed light on the mechanism of this Pummerer-like reaction, we per-formed a computational and experimental mechanistic investigation using methyl phenyl sulfoxide, DIPAMgCl·LiCl and the Grignard reagent i-

48
PrMgCl as model substrates. In this chapter, the results of the investigation are presented.
Figure 26 . a) Summary of the previous experimental results. b) Equilib-ria between the Grignard reagent and the turbo-Hauser base. c) Solid state structure of an organomagnesium amide. TMP=2,2,6,6-tetra-methylpiperidine. DIPA= diisopropylamide
6.1. Results. The first part of the computational investigation consisted of the calculation of several different complexes that can form from the starting materials. This study was divided into two parts: first, the complexes involving the base, the organomagnesium reagent, and the solvent, i.e. prior to the inclusion of the sulfoxide, were considered. Next, the two most stable complexes overall, which adopt a linear structure, and a cubane-like complex were used to ge-nerate new structures including a sulfoxide molecule. The resulting com-plexes are shown in Figure 27.
OS
’R MeDIPAMgCl
R-MgX
OS
’R MgS
’RS
’RR
“[MgXO]-“
proposed intermediate
DIPAMgClLiClR-MgX +
S
RNMg
NMg
S
R
(i-Pr)2
unknown structure in solution
DIPAMgClR-MgX +
(i-Pr)2
solid state structure
a. Studied reaction
c. Structure of an organoMg amide
b. Structure of a turbo-organoMg amide
THF, r.t. 94
+ base
LiCl
1 (%)
41
1
base
DIPAMgCl·LiClTMPMgCl· 14

49
Figure 27 . Structures of the three complexes used to study the reaction mechanism. Relative energies are indicated in kcal/mol.
Next, the three complexes were used as starting points to calculate the reaction mechanism, which was found to encompass the same steps regard-less of the considered initial species. Namely, a) an isomerization to form a σ-amide/µ-sulfoxide complex, b) an intramolecular proton transfer in which the amide deprotonates the α-carbon to form a sulfoxide enolate, and c) a nucleophilic addition of the isopropyl moiety to the sp2 carbon, which hap-pens concomitantly with the S-O bond cleavage.
Figure 28 shows the obtained free-energy profiles for the linear com-plexes. In both cases, the last step is calculated to be rate-determining with calculated overall barriers of 27.1 kcal/mol for complex A and 27.9 kcal/mol for complex B. These barriers are too high for the used experimental condi-tions (30 oC and 3 h reaction time), and thus the two linear complexes can be disregarded as active species. Moreover, the almost identical mechanisms and free-energy profiles for the two complexes suggest that other linear com-plexes are not likely to yield lower barriers.
Figure 28. Free-energy profiles for the reaction mechanism of complexes A and B.

50
Figure 29 shows the calculated free-energy profile for the reaction start-ing at the cubane complex C. Differently to the linear complexes, complex C undergoes two isomerization steps prior to the deprotonation. First, there is the formation of the µ-sulfoxide complex, and second the lithium ion binds to the oxygen to form Int2-C.
Figure 29. Free-energy profile for the reaction mechanism of complex C.
After the proton transfer via TS1-C, the energy of which is calculated to be 20.1 kcal/mol relative to complex A, the carbanionic species Int3-C is formed. In this intermediate, the α-carbon binds to the magnesium ion and displaces the oxygen of the sulfoxide (see Figure 30). Next, the sulfoxide enolate/carbanion Int4-C is formed upon a rearrangement that breaks the Mg-Cα interaction and results in the sp2 hybridized carbon. This intermedi-ate undergoes the final C-C bond formation concertedly with the cleavage of the S-O bond. The transition state for this step, TS2-C, is calculated to be 15.2 kcal/mol relative to Int3-C. In this last step, there is an increase of the strain due to the shortening of the Li-O distance from 1.99 Å at Int4-C to 1.82 at TS2-C (see Figure 30), which in turn results in the decoordination of one bridging chloride to adopt a terminal position.
Figure 30 : Selected optimised of the reaction mechanism of complex C. Distances in Å.
Int2-C13.2
TS1-C20.1
Int3-C-0.9
TS2-C14.3
C3.3 4.6
Int4-C
A0.0
Int1-C11.0
-63.0
MgN
MgCl
i-Pr
Cl
O
Cl
(i-Pr)2
Li
S
Ph
S Mg MgCl
i-Pr
Cl ClLi
S
OSPh
N (i-Pr)2
Mg MgCl
i-Pr
Cl ClLi
S
OSPh
N (i-Pr)2
Mg MgCl
i-Pr
Cl ClLi
S
OSPh
N (i-Pr)2
H
Mg MgCl
i-Pr
Cl ClLi
S
OS
Ph
NH(i-Pr)2
Mg MgCl
i-Pr
Cl ClLi
S
O
S Ph
NH(i-Pr)2
Mg MgCl
C
Cl ClLi
S
OSPh
NH(i-Pr)2
Mg MgCl
Cl ClLi
S
OSPh
NH(i-Pr)2
MgNMg
Cl
i-Pr
O
S
Cl
(i-Pr)2
SMe
Ph
Prod-C

51
Overall, the reaction mechanism for complex C has a total barrier of 20.1 kcal/mol (A→TS1-C), i.e. 7.0 kcal/mol less than the mechanism for the lin-ear complex A. Comparison of the calculated free-energy profiles for the two kinds of complexes show that the linear complexes, despite being somewhat more stable, are associated with barriers of 27-28 kcal/mol, while the open-cubane complexes have barriers calculated to be ca 7 kcal/mol lower. This is a consequence of the greater stabilization of the negative charge of the oxy-gen in the C-C bond formation/S-O bond cleavage step, which in turn results in a 12 kcal/mol lower in energy transition state for the cubane complex compared to complex A. Moreover, the sulfoxide enolate in complex C is more electrophilic than the same species in complex B (calculated charge of -0.82 vs -0.97 for the α-carbon), and also presents a longer S-O distance (1.82 Å vs 1.72 Å), two factors that presumably facilitate the nucleophilic addition.
An important difference between the calculated mechanisms for the two kind of complexes is the nature of the rate-determining steps. That is, while the proton transfer step is associated with the highest barrier for the cubane complexes, the C-C bond formation is the rate-determining step for the linear complexes. The fact that the proton transfer in the former case is irreversible implies that a primary kinetic isotope effect (KIE) should be observed.
Gratifyingly, the reaction with the deuterated methyl phenyl sulfoxide only reached 48% conversion in the same time that the non-deuterated coun-terpart achieved full conversion. Although an accurate KIE number could not be obtained due to the complicated kinetics of the system, the signifi-cantly lower conversion of the deuterated sulfoxide gives strong support to the calculated mechanism.
6.2. Conclusions. In this chapter, the reaction mechanism for a Pummerer-like, non-catalytic, C-C coupling reaction has been described. This study provided new insights into the structure of the reactive complexes and reaction intermediates that are otherwise very difficult to obtain due to the complexity of the aggregat-ion equilibria and complicated associated kinetics of the studied system. Accordingly, the calculations showed that a cubane complex, despite not being the most stable one, yield considerably lower barriers than the more typical, and stable, linear complexes. The lower barriers associated with the cubane topology could also explain the observed better performance of the coupling when LiCl is added. Moreover, the calculations showed that the re-action does not proceed via sulfonium intermediate, as previously suggested, but instead features an unusual coupling between two anionic carbons.

52

53
7. Cyclopropanation via ruthenium-derived redox-active carbenes. (Paper VI)
The cyclopropyl moiety has attracted interest in organic synthesis due to its prominent presence in biologically active molecules.70 This relevance has promoted the development of several different protocols for the synthesis of cyclopropane derivatives, which in most cases rely on the use of a transition-metal catalyst and a diazoacetate (two precursors to form a metal-carbene complex), and an olefin.71
Recently, Mendoza and co-workers reported a novel cyclopropanation re-action between the N-hydroxyphthalimidyl diazoacetate (NHPI-DA) and various olefins catalysed by a Ru-phenyloxazolidine (Ru-Pheox) complex.72 Importantly, the cyclopropanation reaction was remarkably selective over the diazoacetate dimerization, a very common side-reaction, even when poorly reactive aliphatic olefins were used. Moreover, the cyclopropane prod-ucts were formed with good enantio- and diastereoselectivities (see Figure 31). This cyclopropanation opened new endeavours in the derivatization of cyclopropanes, as the obtained N-hydroxyphthalimide esters are versatile building blocks that can be used as radical precursors or acyl donors in fur-ther functionalizations.
Mechanistic aspects of metal-catalysed cyclopropanation reactions have been extensively studied over the past decades, and it is now well established that cyclopropanation reactions can happen via an inner-sphere mechanism,73 which proceeds via metalacyclobutane intermediate formed upon a [2+2] cycloaddition between the carbene and an olefin, or via an outer-sphere mechanism,74 which consists of the insertion of an olefin into the carbene.
Figure 31. Summary of the previous experimental results and evidences.
H
FG
R
up to 99:1 e.r.
cyclopropanationcycle
dimerizationcycle
[(S)-Ru-Pheox]
[LnRu]CONHPI
H
EWGEWG
RN2 EWG
+ N2
N2
N2
redox-active
acyl donor
H
CONHPI
R
IPHNOC

54
In this chapter, the two reaction mechanisms for the cyclopropanation re-action as well as the possible side-reactions are described, and the chemo-, enantio- and diastereoselectivities of the reaction are analysed on the basis of the calculated free-energy profiles.
7.1. Formation of the carbene intermediate. First, the geometries of a number of complexes that can form from the star-ting materials were optimised, and their energies compared, in order to establish which is initial form of the catalyst. The ruthenium complex satura-ted with four acetonitriles (Ru-Pheox, Figure 32) was found to be the most stable species, and its energy was set to 0.0 and used as the reference in all calculated reaction mechanisms.
The first step for all studied reactions is the formation of the carbene in-termediate. Importantly, due to the lack of symmetry of the catalyst, four different carbenes can form at the four positions initially occupied by acetoni-triles. That is, two equatorial carbenes, in trans and cis to the oxazolidine (Eqtrans and Eqcis), and two apical carbenes in anti and syn to the phenyl sub-stituent of the pheox ligand (Apanti and Apsyn) with respect to the equatorial plane (see Figure 32). The barriers for the carbene formation are calculated to be 14.3, 15.1, 16.0 and 20.4 kcal/mol at the Apanti, Apsyn, Eqtrans and Eqcis positions, respectively. In all cases, the resulting carbene is calculated to be more stable than the Ru-Pheox complex by 5-20 kcal/mol. No interconver-sion path was found between carbenes, and considering the very similar car-bene formation barriers at Apanti, Apsyn and Eqtrans positions, these three car-benes must be considered to be likely to form, and their reactivity will be discussed individually. On the other hand, formation of the Eqtrans carbene is associated with a barrier 4.1 kcal/mol higher than Apanti carbene, and thus this position can be considered as non-operational.

55
Figure 32. Calculated energies (kcal/mol) for the carbene formation step at the various positions of the Ru-Pheox catalyst. AN = acetonitrile.
7.2. Reaction mechanisms of the side-reactions. As explained above, the dimerization of the diazoacetate is the most com-monly observed side-reaction in a metal-catalysed cyclopropanation with a carbene intermediate. Figure 33 shows the calculated free-energy profile for the dimerization of diazoacetate starting from Int2-Apanti, and the inserted table presents the calculated barrier at the other two positions. The mecha-nism starts with the exchange of the acetonitrile molecule at the Eqcis position for a second NHPI-DA molecule, which binds to the metal centre via one carbonyl of the NHPI moiety. The resulting intermediate Int3-DM-Apanti is calculated to be +0.8 kcal/mol, and subsequently undergoes the dimerization via TS-DM-Apanti, the energy of which is calculated to be 14.5 kcal/mol, both energies relative to Int2-Apanti. The dimerization reaction at Apsyn and Eqtrans positions is associated with calculated barriers of 14.8 and 20.4 kcal/mol re-lative to their respective carbene. Quite surprisingly, a second side-reaction was found to have a lower barrier than the diazoacetate dimerization. Na-mely, the carbene at Int2-Apanti can undergo a migratory insertion into the Ru-C bond of the ligand. The barrier for this transformation, TS-MI-Apanti, is calculated to be 8.2 kcal/mol relative to the carbene, i.e. 6.3 kcal/mol lower than the dimerization reaction (see ∆∆𝐺‡ik(Ui , Figure 33). Importantly, this transformation results in the formation of a much more stable ruthenium species Int3-MI-Apanti, the energy of which is calculated to be -40.7 kcal/mol relative to Int2-Apanti.
0.0
-20.8-20.5-19.5Int2
Carbene
-5.1
10.111.311.4
20.4
14.315.116.0
N2
N2
TS2
5.5
(Pheox)(AN)3RuCOONHPI
H
[(Pheox)Ru(CH3CN)4]
N2
(Pheox)(AN)3Ru COONHPI
H
(Pheox)(AN)3RuCOONHPI
H
N2
NHPI-DACH3CN
Int1
Apanti
ON
Ru
Apsyn
Eqtrans Eqcis
Ru-Pheox

56
Figure 33. Calculated free energy profile (kcal/mol) for the dimerization and migratory insertion reactions starting from the Int2-Apanti carbene. Inserted table shows the energies for reactions at the other positions, each relative to its own carbene intermediate.
The migratory insertion is favoured over the dimerization reaction also at the Apsyn position (∆∆𝐺‡ik(Ui=-6.4 kcal/mol), while the opposite is true at Eqtrans (∆∆𝐺‡ik(Ui=+11.4). Importantly, the migratory insertion results in the formation of a new chiral center, and two different diastereomers of the product can form. Indeed, Int3-MI-Apanti and Int3-MI-Apsyn are the two dia-stereomers of the migratory insertion product. Moreover, as shown in Paper VI, these two complexes can subsequently catalyse the dimerization and cy-clopropanation reactions. Therefore, a favourable migratory insertion could result in the coexistence of up to three catalysts (Ru-Pheox and the two Int3-MI complexes), a scenario that would have major implications in the overall selectivity of process.
7.3. Computational and experimental study of the cyclopropanation reaction. With the aim to elucidate which complex (Ru-Pheox or the migratory insertion products Int3-MI) catalyses the cyclopropanation reaction, two

57
different experiments were designed. It is here important to highlight that the very short reaction times (1-10 minutes) and the unstable nature of the intermediates makes not possible to isolate the organometallic species gene-rated during the reaction. However, if the migratory insertion is substanti-ally faster than the cyclopropanation and dimerization reactions, the use of stoichiometric to super-stoichiometric amounts of the catalyst relative to the NHPI-DA should result in an inhibition of the cyclopropanation reaction, as the migratory insertion would monopolize all NHPI-DA. Figure 34 shows the obtained yields and selectivities for the cyclopropanation reaction of p-methylstyrene at different [Ru-Pheox]0/[NHPI-DA]0 ratios. As shown, the excess of catalyst does not result in any noticeable change in the performance of the reaction, thus indicating that the migratory insertion is not the domi-nant reaction for the studied system.
Figure 34. Measured yields and enantiomeric ratios for different cata-lyst/diazoacetate ratios.
An alternative scenario to the one presented above would be that only a small amount of the Ru-Pheox undergoes the migratory insertion, and the resulting species is exceedingly more active in the cyclopropanation reaction than the Ru-Pheox. To assess this possibility, the efficiency and selectivity of the cyclopropanation reaction with a new diazocompound Ph4-NHPI-DA were also explored in an experiment in which cyclopropanation of p-me-thylstyrene with the new diazoacetate was followed by cyclopropanation of β-methylstyrene with the standard NHPI-DA. The goal of the experiment was the in situ generation of a migratory insertion complex featuring a much larger Ph4-NHPI moiety (Int3-MI-Ph4, see Figure 35a), and its subsequent use to catalyse the second cyclopropanation reaction. If the presented hy-pothesis is true, the use of Int3-MI-Ph4 would presumably result in a differ-ent selectivity due to the larger size of the intermediate than the reaction in standard conditions (see Figure 35b)
The results of the experiments show that the sequential addition of the two olefins and diazoacetates result in the same yield and selectivity as the reaction performed at standard conditions (see Figure 35c), indicating that migratory insertion is not likely to occur, or that in case it happens, its con-tribution to the cyclopropanation reaction is irrelevant. Considering these
Me H
CONHPI
NHPI-DA, 1.2 equiv(S)-RuPheox, 1 mol %
CH2Cl2, 0 °C40 min slow addition Me
48%, > 20:1 dr83:17 e.r.
H
CONHPI
β-methylstyrene, 0.1 mmolNHPI-DA, 0.12 mmol
CH2Cl2, 0 °C40 min slow addition
Me
47%, > 20:1 dr82:18 e.r.
H
COPhNHPI
PhNHPI-DA, 0.02 mmol(S)-RuPheox, 1 mol %
CH2Cl2, 0 °C, 5 min
0.03 mmol
> 95 %
Me
Me
In situ formation of bulky insertion product
Cyclopropanation with NHPI-DA
b:
c:
H
CONHPI
NHPI-DA(S)-RuPheox
CH2Cl2, 0 °C, 5 min
0.03 mmol
Me
Me
a:
Yield (%) e.r.
97 96:4
95 93:7
96 94:6
97 96:4
95 96:4
[Cat]0/[Diazo]0
0.01
0.5
1
2
5

58
Figure 35. a) Measured yield and enantiomeric ratio for the cyclopropa-nation of p-methylstyrene with Ph4-NHPI-DA and proposed intermedi-ate. b) Measured yield and enantiomeric ratio for the cyclopropanation of b-methylstyrene with NHPI-DA and proposed intermediate. c) Se-quential addition experiment.
results, the migratory insertion product can be ignored in the computational study of the cyclopropanation reaction, which will only be focused on the Ru-Pheox complex.
In the following paragraphs, the two possible cyclopropanation mecha-nisms, i.e. inner- and outer-sphere, will be presented. The full free-energy profiles starting from the Apanti, Apsyn and Eqtrans carbenes were calculated for styrene and propene as representative cases of aromatic and aliphatic olefins. For clarity, only the cyclopropanation of styrene at the Apanti position will be described, and the relevant energies for all other calculated mechanism will be presented in tables.
Figure 36 shows the calculated free energy profiles for the inner- and outer-sphere cyclopropanation mechanisms of styrene. The inner-sphere mechanism starts with a ligand exchange between an acetonitrile and an ole-fin at the Eqcis position to form Int3-IS-Apanti, the energy of which is calcu-lated to be 4.1 kcal/mol. This intermediate undergoes a formal [2+2] cy-cloaddition with a calculated barrier of 7.7 (TS4-IS-Apanti) kcal/mol and forms the metallacyclobutane intermediate Int4-IS-Apanti, the energy of
Me H
CONHPI
NHPI-DA, 1.2 equiv(S)-RuPheox, 1 mol %
CH2Cl2, 0 °C40 min slow addition Me
48%, > 20:1 dr83:17 e.r.
H
CONHPI
β-methylstyrene, 0.1 mmolNHPI-DA, 0.12 mmol
CH2Cl2, 0 °C40 min slow addition
Me
47%, > 20:1 dr82:18 e.r.
H
COPhNHPI
PhNHPI-DA, 0.02 mmol(S)-RuPheox, 1 mol %
CH2Cl2, 0 °C, 5 min
0.03 mmol
> 95 %
Me
Me
In situ formation of insertion product Int2-MI-Ph4
Cyclopropanation with NHPI-DA
b:
c:
H
COPhNHPI
PhNHPI-DA, 1.2 equiv(S)-RuPheox, 1 mol %
CH2Cl2, 0 °C40 min slow addition
86%, > 20:1 dr97:3 e.r.
Me
Me
RuNO
Ph(NCMe)4
PF6-
NO
OPh
Ph
PhPh
O
O
a:
RuNO
Ph(NCMe)4
PF6-
NO
OO
O
Cyclopropanation via Int3-MI?
Cyclopropanation via Int3-MI-Ph4?
Int3-MI
Int3-MI-Ph4
β-methylstyrene
p-methylstyrene

59
which is calculated to be +3.1 kcal/mol. This intermediate subsequently un-dergoes a reductive elimination (TS5-IS-Apanti) with an associated barrier of 10.9 kcal/mol to form the product. Alternatively, the cyclopropanation can happen via the outer-sphere mechanism, which consists of a direct insertion of an olefin molecule from the second shell into the carbene, and is associated with a barrier of 6.0 kcal/mol (TS-OS-Apanti), i.e. 4.9 kcal/mol less than the inner-sphere mechanism.
Figure 36. Calculated free energy profile for the inner-sphere (black) and outer-sphere (orange) cyclopropanation mechanisms of styrene at the Apanti position.
As seen from the calculated energies of the reductive elimination TS (which is always calculated to have the larger barrier among the two steps that entail the inner-sphere mechanism) and the outer-sphere transition states, the latter mechanism is, in all cases, favoured over the former (see Table 1). Moreover, cyclopropanation of styrene is almost always associated with lower barriers than propene, with the only exception of the inner-sphere mechanism at the Eqtrans position, in line with the better reactivity of the aro-matic olefin.
-20.5-16.4
-12.8R CH3CN
-17.4
-9.6
Carbene
TS4-IS
-32.4
R
TS5-IS
Metallacyclobutane
Product
Int2 Int3-IS Int4-IS
ΔGIS-st
ΔGOS-st
(Pheox)Ru
IPHNOOC H(Pheox)Ru
IPHNOOC H
R(Pheox)Ru
IPHNOOC H
R(Pheox)Ru
IPHNOOC H
R(Pheox)Ru
IPHNOOC H
-14.5
(Pheox)Ru
IHPHNOOC HR
TS-OS
-33.2
TS-OS
R
(AN)3(AN)2
(AN)2
(AN)2
(AN)2
(AN)3

60
Table 1. Calculated barriers for the outer-sphere mechanism and the reductive elimination transition state. All values are in kcal/mol and are calculated relative to the respective carbene intermediate.
Styrene Propene
∆𝐺‡l3(D< ∆𝐺‡k3(D< ∆𝐺‡l3(]m ∆𝐺‡k3(]m
Int2-Apanti 6.0 10.9 12.1 14.7
Int2-Apsyn 6.5 14.7 13.6 16.9
Int2-Eqtrans 13.1 21.7 15.3 20.3
7.4. Analysis of the selectivity. Up to this point, the two different side-reactions and the cyclopropanation reaction (via the inner- or outer-sphere mechanisms) have been described as independent reactions. However, it must be kept in mind that the presented reactions are the four possible outcomes to a single experimental procedure, and thus all calculated paths must be compared to each other. Figure 37 presents all the reaction than can happen starting from the Ru-Pheox com-plex. Importantly, and as shown in section 7.1 above, the barriers for the carbene formation at the Apanti, Apsyn and Eqtrans positions are calculated to be within ca 2 kcal/mol, and therefore the three carbenes need to be considered as likely to form. Next, each of these carbenes can react with a second NHPI-DA molecule to form the dimeric product (DM), with an olefin to cyclopro-panate (CP) or can undergo the migratory insertion (MI), a transformation that yields a second catalyst Int3-MI.
This appears to be a very convoluted scenario where two main factors can have an impact on the selectivity of the reaction: the relative formation of each carbene, and the interplay between the different mechanisms at each of the three positions. Therefore, comparison between the predicted selectivity from the calculated profiles at each position and the experimental outcome can be helpful to determine which of the three carbenes is the catalytically competent one for the cyclopropanation reaction. It is here instructive to pre-sent the enantio- and diastereoselectivity of the cyclopropane product that each position promotes, as it would help to distinguish the cyclopropane out-come obtained from the Apanti, Apsyn, and Eqtrans carbenes.

61
Figure 37 . Summary of the various mechanistic pathways presented in this chapter.
Experimentally, the cyclopropanation reaction favours the formation of the trans products, i.e. the cyclopropanes with the NHPI and the substituent of the olefin in an anti disposition. In addition, the major enantiomer has been determined to be the (R,R).72
The calculated energies of the most stable outer-sphere transition states leading to the formation of each diastereomer are listed in Table 2. For all the three carbene positions, the inner-sphere transition states are much higher (5-8 kcal/mol) than the outer-sphere ones, and can therefore be left out in the enantio- and diastereoselectivity analysis.
Table 2. Comparison of the calculated barriers (kcal/mol) of the vari-ous reactions that can take place at the carbene intermediate, Int2 for the styrene substrate. For each carbene, the lowest barrier is set to zero. DM = Dimerization. MI = Migratory insertion.
Cyclopropanation DM MI
TS-OS TS5-IS TS-DM TS-MI
R,R S,S S,R R,S
Int2-Apanti +3.9 0.0 +5.9 +5.1 +4.9 +8.5 +2.2
Int2-Apsyn 0.0 +1.8 +3.0 +4.2 +8.2 +8.3 +1.9
Int2-Eqtrans +1.1 +0.8 +5.1 0.0 +8.6 +7.3 +18.7
R
Dimerization(DM)
Cyclopropanation(CP)
Migratory Insertion
(MI)
N2
Int3-MI-Apanti
H
CONHPI
R
COONHPI COONHPI
H
N2
O
ONHPI
ApantiO
NRu
Apsyn
Eqtrans Eqcis
ONRu
Apsyn
Eqcis
ONRu
Apsyn
Eqtrans Eqcis
COONHPIHO
NRuEqtrans Eqcis
CP vs DM
MI vs CP vs DM
IPHNOOC H
IPHNOOCH
Apanti Apanti
H
N2
O
ONHPI
vsvsTS2
TS-DM
TS-MI
Int2-Eqtrans Int2-Apsyn Int2-Apanti
Inner-sphereTS5-IS
Outer-sphereTS-OSvs
ONRu
H COONHPI
(AN)4
+Int3-MI-Apsyn
Diastereomers

62
Starting from the Apanti position, which has the lowest barrier for the car-bene formation, the calculations predict the (S,S) product to be formed. The barriers leading to the other products are ca 4-6 kcal/mol higher in energy. On the other hand, starting from the other apical position Apsyn, the (R,R) product is correctly predicted, and the barriers leading to the other products are ca 2-4 kcal/mol higher. Finally, starting from the Eqsyn carbene, the cal-culation predict the (S,R) product to be dominant, with the (S,S) and (R,R) being only ca 1 kcal/mol higher in energy. Comparison of the calculated en-ergies for the four considered reactions (see Table 2) show that, at the three positions, the cyclopropanation is favoured over the other reactions by 2-7 kcal/mol, which is in good agreement with the experimentally determined selectivity (formation of the diazoacetate dimer product is not observed, and 99% of the NHPI-DA is converted into the cyclopropane product).72 The cal-culations on the enantio- and diastereoselectivity show that Apsyn is the likely operational carbene, as it is the only one that predicts the favourable for-mation of the experimentally observed major product (R,R). It is here im-portant to remember that formation of the carbene is independent of the na-ture of the olefin used in the reaction. Thus, the possible consequences of the favourable formation of Apsyn for the cyclopropanation of propene should also be evaluated.
Table 3 shows the relative energy barriers for the four mechanisms for the propene substrate. The calculated values show that at the Apsyn position, the migratory insertion is associated with a barrier 5.2 kcal/mol lower than the cyclopropanation reaction, indicating that the migratory insertion prod-uct would be the catalyst for the cyclopropanation of propene.
Table 3. Comparison of the calculated barriers (kcal/mol) of the var-ious reactions that can take place at the carbene intermediate, Int2 for the propene substrate. For each carbene, the lowest barrier is set to zero.
Cyclopropanation Dimerization Migratory Insertion
TS-OS TS5-IS TS-DM TS-MI
Int2-Apanti +3.9 +6.5 +6.3 0.0
Int2-Apsyn +5.2 +8.5 +6.4 0.0
Int2-Eqtrans 0.0 +5.0 +5.1 +16.5

63
Importantly, the experimental results presented in section 7.3, which showed that the migratory insertion product does not catalyse the cyclopro-panation reaction, are based on an aromatic olefin, and thus the same conclu-sion might not necessarily apply to aliphatic substrates. However, to have different catalysts operating depending on the olefin is considered to be highly unlikely, since the ee and dr trends observed for aliphatic and aromatic olefins are the same.72
An alternative scenario would be that operational carbene is the one for-ming at the Eqtrans position, as in this position the barrier for the migratory insertion is as much as 16.5 and 18.7 kcal/mol higher in energy than the cyclopropanation of propene and styrene, respectively. However, the pre-dicted selectivity in the case of propene (no formation of the dimer product) is at odds with the experimental one (22% formation of the dimeric product with a similar aliphatic olefin), and the enantioselectivity of the cyclopropa-nation of styrene is neither well predicted (see Table 2).
To summarize, the cyclopropanations of aromatic and aliphatic olefins with a NHPI-diazoacetate catalysed by the Ru-Pheox complex happens in a very intricate scenario. Starting from the Ru-Pheox catalyst, three different carbene intermediates can form (Apanti, Apsyn, and Eqtrans), and each of them can evolve via four different reactions: outer-sphere cyclopropanation, inner-sphere cyclopropanation, dimerization, and migratory insertion. Moreover, the outer-sphere and inner-sphere cyclopropanation result in different ste-reochemical outcomes. The interplay between all these options is very deli-cate and involves small energy differences between the different possible cal-culated paths. It is, therefore difficult to reconcile only one of the calculated reaction pathways with all experimental facts.
7.5. Conclusions. In this chapter, a combined experimental-computational study on the
cyclopropanation reaction between a diazoacetate and an olefin catalysed by the Ru-Pheox complex has been presented. The calculations revealed that, in addition to the common diazoacetate dimerization side-reaction, the car-bene intermediate can also undergo a migratory insertion to form a new ca-talyst. This reaction, although well precedented in the context of C-H functionalizations,75 has never been invoked in cyclopropanation reactions. In light of the seemingly favourable formation of the migratory insertion product, two sets of experiments were performed with the goal to elucidate the nature of the catalyst for the cyclopropanation reaction. The results sho-wed that the migratory insertion path was not likely to be operational in presence of an aromatic olefin. However, the migratory insertion path must

64
be considered as a potential competitive reaction, and formation of the mi-gratory insertion complex in the absence of olefins or in the presence of poorly reactive alkenes cannot be completely disregarded.
Among the two cyclopropanation reactions, the outer-sphere mechanism was found to be the operational one for the two olefins, and to favour the formation of the experimentally observed major product at the Apsyn position. However, two other positions presented similar barrier for the carbene for-mation, and thus the final observed selectivity is also affected by the relative formation of each carbene.
Comparison of the computationally predicted and experimentally deter-mined selectivities showed that a complete reconcilement between the two is not possible. Considering the very small differences in energy between some of the involved species, the calculations might not be able to capture all the small features that could allow for a better determination of the factors gov-erning the selectivity of the studied reaction. This unfortunate result is, on the other hand, an educational example of the true complexity behind certain, seemingly simple, systems, and of the thoroughness that is needed to study them.

65
8. Concluding remarks.
In this thesis, the reaction mechanism of the natural reaction of two ThDP-dependent enzymes, Benzoylformate decarboxylase (BFDC, Papers I, II and III) and SEPHCHC synthase (MenD, Paper IV) have been studied with the quantum chemical cluster approach. For BFDC, the calculated mechanism clarifies the roles of three residues present in the active site that were sug-gested to be relevant, although their specific roles were still unclear. Namely, Ser26 is found to be relevant for the decarboxylation step, His70 is found to be a proton shuttle between the ThDP-substrate adduct and Glu28, and His281 is found to be involved only in binding. Moreover, the calculations demonstrate that the cofactor is not only involved in formation of the ThDP-donor adduct to subsequently yield the enamine, but also is the proton donor at the key step of the enamine protonation. The calculations also indicate that a catalytically inactive state of the cofactor is kinetically relevant. Along this line, the stability of all possible cofactor states have been assessed in five dif-ferent models, showing that the relative abundance of each of them strongly depends on the environment.
For MenD, the calculated mechanism shows that the previously proposed protonation of the enamine intermediate by N4’ to form the post-decarboxy-lation tetrahedral intermediate is not energetically feasible, and instead pro-tonation by an external proton source is the most likely scenario. Moreover, for the second part of the reaction mechanism, the C2α-hydroxyl group is found to be the proton donor in the the enol-keto tautomerization of the ac-ceptor moiety.
Finally, the enantioselectivity of the addition of the enamine intermediate to a carbonyl acceptor has been studied for both BFDC and MenD. For BFDC, the parallel orientation of the hydroxyl moiety of the donor with the carbonyl of the acceptor is determined to be necessary in order the reaction to proceed. This, combined with the differential accommodation of the sub-stituent of the acceptor into the large or small pocket that conform the active site, is found to govern the enantioselectivity. The very same features are found to determine the enantiomeric outcome in the reaction catalysed by MenD. Importantly, the predicted selectivity trends are in both cases in agre-ement with the experimental evidences and, remarkably, the characteristic enantioselectivity reversal of BFDC is successfully reproduced. In the case of

66
MenD, the regioselectivity of the reaction has been also studied, as the en-zyme catalyses both the 1,2- and 1,4-additions to α,β-unsaturated acceptors. The two reaction mechanisms starting from the enamine have been studied for two different acceptors, and the regioselectivity is found to be governed by a combination of sterics and the relative stability of the keto-enol equili-bria.
Besides the enzymatic reactivity, in this thesis a non-catalytic, Pummerer-like, C-C coupling reaction (Paper V) and a ruthenium-catalysed cyclopropa-nation reaction (Paper VI) have also been presented. For the Pummerer-like coupling, the calculations give insight that would have been otherwise dif-ficult to elucidate, as an unstable open-cubane complex is found to yield considerably better barriers than the more typically suggested linear com-plexes. Importantly, the proposed mechanism by the calculations is suppor-ted by a positive kinetic-isotope effect (KIE).
Moving into the ruthenium-catalysed cyclopropanation, the reaction mechanism of two side-reactions as well as the two possible cyclopropanation mechanisms have been presented. The calculated free-energy profiles indica-ted that the migratory insertion of the carbene into the C-M bond of the ligand, although not previously suggested in the context of a cyclopropanat-ion, is a potential side-reaction. Importantly, this paths results in the form-ation of a new catalyst. Further experimental work contributed to determine that the migratory insertion product is most likely not the catalyst for the cyclopropanation mechanism. Finally, the selectivity analysis shows that the operational cyclopropanation mechanism is the outer-sphere one, and that the final stereochemical outcome is influenced by the carbene formation step.

67
Acknowledgments.
First and foremost, I would like to show my gratitude to my supervisor, Fahmi Himo, for giving me the chance to pursue a PhD in his group. These have been four fantastic years in which I have learnt a lot of chemistry, and perhaps even more about other life lessons. I would also like to thank the following people: Michael J. McLeish, for being such a great collaborator. Per Siegbahn and Margareta Blomberg, for sharing your expertise. Pher G. Andersson, for showing interest in my half-time report and my thesis. Abraham Mendoza, for sharing your knowledge during our unexpected, but eventually very long, collaboration. Also, all time spend on tennis courts. Xiang Sheng, for being such a great assistant in my first project, and such a great colleague these years. Henrik Daver, for teaching me so much about cluster management, and never, ever, loosing your patience in the process. All past and present members of the FH group that I have had the pleasure to work with. Bianca, Sara, Masoud, Oriana, Wen-jie, Joep, Mario, Li Man, Ma-ria, Akinobu, Jiji, Binh. Genping and Liao, for being such great hosts during my days in China. Marc and Miguel, for these years of friendship, for being great computational dummies and for proof-reading my thesis. Also, Amparo, Aitor, Victor, Alba, Pedro, Pablo, Alejandro, Elisa, Alberto, Samuel and other members of the Span-ish crew I might be forgetting, thanks for all dinners, board games and beers. Magda, for all shared sweets and your charming talent in complaining 24/7. All people from Desert Island and Kammakargatan, for making my arrival to Sweden much easier. All personnel and students at the department, thank you for creating a nice atmosphere. Åforsk and Knut and Alice Wallenbergs agencies for funding my trips to differ-ent conferences. Ramon Crehuet for convincing me that computational modelling of enzymes was an attractive field, and for introducing me to it. Anna Grandas, for her kind words whenever I needed support. Als meus pares i al meu germà. Gràcies per sempre donar-me suport, ànims i per cuidar en Nuk aquests anys. A la meva cosina Anna, per compartir les meves frustracions i per adoptar a la Nuba. A la meva àvia. Gràcies. M’hagués agradat dir-t’ho en persona.

68
References.
[1] Wöhler, F. Ueber künstliche Bildung des Harnstoffs. Annalen der Physik und Chemie. 1828, 88 (2), 253–256.
[2] Nicolaou, K. C. The Emergence and Evolution of Organic Synthesis and Why It Is Important to Sustain It as an Advancing Art and Science for Its Own Sake. Isr. J. Chem. 2018, 58 (1–2), 104–113.
[3] Hagen, J. Industrial Catalysis: A Practical Approach. 3rd ed., Wiley-VCH. 2015.
[4] Tasker, S. Z.; Standley, E. A.; Jamison, T. F. Recent Advances in Homo-geneous Nickel Catalysis. Nature. 2014, 502, 299–309.
[5] de Vries, J. G. Palladium-Catalysed Coupling Reactions. Springer, 2012.
[6] Eyring, H. The Activated Complex in Chemical Reactions. J. Chem. Phys. 1935, 3 (2), 107–115.
[7] For selected examples, see: (a) Roca, M.; Moliner, V.; Tuñón, I.; Hynes, J. T. Coupling between Protein and Reaction Dynamics in Enzymatic Pro-cesses: Application of Grote-Hynes Theory to Catechol O-Methyltransfer-ase. J. Am. Chem. Soc. 2006, 128 (18), 6186–6193. (b) Ruiz-Pernía, J. J.; Tuñón, I.; Moliner, V.; Hynes, J. T.; Roca, M. Dynamic Effects on Reaction Rates in a Michael Addition Catalyzed by Chalcone Isomerase. Beyond the Frozen Environment Approach. J. Am. Chem. Soc. 2008, 130 (23), 7477–7488. (c) Roca, M.; Oliva, M.; Castillo, R.; Moliner, V.; Tuñón, I. Do Dynamic Ef-fects Play a Significant Role in Enzymatic Catalysis? A Theoretical Analysis of Formate Dehydrogenase. Chem. Eur. J. 2010, 16 (37), 11399–11411. (d) Boekelheide, N.; Salomón-Ferrer, R.; Miller, T. F. Dynamics and Dissipation in Enzyme Catalysis. Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (39), 16159–16163. (e) García-Meseguer, R.; Martí, S.; Ruiz-Pernía, J. J.; Moliner, V.; Tuñón, I. Studying the Role of Protein Dynamics in an SN2 Enzyme Reac-tion Using Free-Energy Surfaces and Solvent Coordinates. Nat. Chem. 2013, 5 (7), 566–571. (f) Ruiz-Pernía, J. J.; Behiry, E.; Luk, L. Y. P.; Loveridge, E. J.; Tuñón, I.; Moliner, V.; Allemann, R. K. Minimization of Dynamic Effects in the Evolution of Dihydrofolate Reductase. Chem. Sci. 2016, 7 (5), 3248–

69
3255. (g) Wu, J.; Gao, L. G.; Varga, Z.; Xu, X.; Ren, W.; Truhlar, D. G. Water Catalysis of the Reaction of Methanol with OH Radical in the Atmosphere Is Negligible. Angew. Chem. Int. Ed. 2020, 132 (27), 10918–10922.; For selected reviews and perspectives, see: (a) Pu, J.; Gao, J.; Truhlar, D. G. Multidimen-sional Tunneling, Recrossing, and the Transmission Coefficient for Enzy-matic Reactions. Chem. Rev. 2006, 106 (8), 3140–3169. (b) Meana-Pañeda, R.; Fernández-Ramos, A. Tunneling Transmission Coefficients: Toward More Accurate and Practical Implementations. In Kinetics and Dynamics (pp 481–500). Springer, 2010. (c) De La Lande, A.; Řezáč, J.; Lévy, B.; Sanders, B. C.; Salahub, D. R. Transmission Coefficients for Chemical Reactions with Mul-tiple States: Role of Quantum Decoherence. J. Am. Chem. Soc. 2011, 133 (11), 3883–3894. (d) Hay, S.; Scrutton, N. S. Good Vibrations in Enzyme-Cata-lysed Reactions. Nat. Chem. 2012, 4, 161–168. (e) Tuñón, I.; Laage, D.; Hynes, J. T. Are There Dynamical Effects in Enzyme Catalysis? Some Thoughts Concerning the Enzymatic Chemical Step. Arch. Biochem. Biophys. 2015, 582, 42–55. (f) Warshel, A.; Bora, R. P. Perspective: Defining and Quantifying the Role of Dynamics in Enzyme Catalysis. J. Chem. Phys. 2016, 144 (18), 180901.(g) Zinovjev, K.; Tuñón, I. Quantifying the Limits of Tran-sition State Theory in Enzymatic Catalysis. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (47), 12390–12395.
[8] (a) Lan, Y.; Liu, P.; Newman, S. G.; Lautens, M.; Houk, K. N. Theoretical Study of Pd(0)-Catalyzed Carbohalogenation of Alkenes: Mechanism and Or-igins of Reactivities and Selectivities in Alkyl Halide Reductive Elimination from Pd(Ii) Species. Chem. Sci. 2012, 3 (6), 1987–1995. (b) Wang, Y.; Guo, X.; Wu, B.; Wei, D.; Tang, M. Mechanistic and Stereoselectivity Study for the Reaction of Trifluoropyruvates with Arylpropenes Catalyzed by a Cati-onic Lewis Acid Rhodium Complex. RSC Adv. 2015, 5 (121), 100147–100158. (c) Wang, Q.; Lübcke, M.; Biosca, M.; Hedberg, M.; Eriksson, L.; Himo, F.; Szabó, K. J. Enantioselective Construction of Tertiary Fluoride Stereocenters by Organocatalytic Fluorocyclization. J. Am. Chem. Soc. 2020, 142 (47), 20048–20057. (d) Zhu, C.; Liu, J.; Mai, B. K.; Himo, F.; Bäckvall, J. E. Efficient Stereoselective Carbocyclization to Cis-1,4-Disubstituted Heter-ocycles Enabled by Dual Pd/Electron Transfer Mediator (ETM) Catalysis. J. Am. Chem. Soc. 2020, 142 (12), 5751–5759.
[9] (a) Mata, R. A.; Suhm, M. A. Benchmarking Quantum Chemical Meth-ods: Are We Heading in the Right Direction? Angew. Chem. Int. Ed. 2017, 56 (37), 11011–11018. (b) Sedlak, R.; Janowski, T.; Pitoňák, M.; Řezáč, J.; Pulay, P.; Hobza, P. Accuracy of Quantum Chemical Methods for Large Noncova-lent Complexes. J. Chem. Theory Comput. 2013, 9 (8), 3364–3374. (c) Hickey,

70
A. L.; Rowley, C. N. Benchmarking Quantum Chemical Methods for the Cal-culation of Molecular Dipole Moments and Polarizabilities. J. Phys. Chem. A 2014, 118 (20), 3678–3687. (d) Sirirak, J.; Lawan, N.; Van der Kamp, M. W.; Harvey, J. N.; Mulholland, A. J. Benchmarking Quantum Mechanical Meth-ods for Calculating Reaction Energies of Reactions Catalyzed by Enzymes. PeerJ. Phys. Chem. 2020, 2, e8.
[10] Haunschild, R.; Barth, A.; Marx, W. Evolution of DFT Studies in View of a Scientometric Perspective. J. Cheminform. 2016, 8 (1), 1-12.
[11] Cheng, G.-J.; Zhang, X.; Chung, L. W.; Xu, L.; Wu, Y.-D. Computa-tional Organic Chemistry: Bridging Theory and Experiment in Establishing the Mechanisms of Chemical Reactions. J. Am. Chem. Soc. 2015, 137 (5), 1706–1725.
[12] a) Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136 (3B), B864–B871. b) Kohn, W.; Sham, L. J. Self-Consistent Equa-tions Including Exchange and Correlation Effects. Phys. Rev. 1965, 140 (4A), A1133–A1138.
[13] Lee, C.; Yang, W.; Parr, R. G. Development of the Colle-Salvetti Cor-relation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B. 1988, 37 (2), 785–789.
[14] Becke, A. D. Density-functional Thermochemistry. III. The Role of Ex-act Exchange. J. Chem. Phys. 1993, 98 (7), 5648–5652.
[15] Becke, A. D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A. 1988, 38 (6), 3098–3100.
[16] a) Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32 (7), 1456–1465. (b) Mládek, A.; Krepl, M.; Svozil, D.; Čech, P.; Otyepka, M.; Banáš, P.; Zgarbová, M.; Jurečka, P.; Šponer, J. Benchmark Quantum-Chemical Calculations on a Complete Set of Rotameric Families of the DNA Sugar-Phosphate Backbone and Their Comparison with Modern Density Functional Theory. Phys. Chem. Chem. Phys. 2013, 15 (19), 7295–7310. (c) Steinmetz, M.; Grimme, S. Benchmark Study of the Performance of Density Functional Theory for Bond Activations with (Ni,Pd)-Based Transition-Metal Catalysts. ChemistryOpen. 2013, 2 (3), 115–124.
[17] Grimme, S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27 (15), 1787–1799.

71
[18] Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132 (15), 154104.
[19] Michaelis, L., and Menten, M. L. Die Kinetik der Invertinwirkung. Bi-ochem. Z. 1913, 49, 333−369.
[20] Johnson, K. A.; Goody, R. S. The Original Michaelis Constant: Trans-lation of the 1913 Michaelis–Menten Paper. Biochemistry. 2011, 50 (39), 8264–8269.
[21] (a) Cerqueira, N. M. F. S. A.; Fernandes, P. A.; Ramos, M. J. Protocol for Computational Enzymatic Reactivity Based on Geometry Optimisation. ChemPhysChem. 2018, 19 (6), 669–689. (b) Ahmadi, S.; Barrios Herrera, L.; Chehelamirani, M.; Hostaš, J.; Jalife, S.; Salahub, D. R. Multiscale Modeling of Enzymes: QM-Cluster, QM/MM, and QM/MM/MD: A Tutorial Re-view. Int. J. Quantum Chem. 2018, 118 (9), e25558.
[22] (a) Warshel, A.; Levitt, M. Theoretical Studies of Enzymic Reactions: Dielectric, Electrostatic and Steric Stabilization of the Carbonium Ion in the Reaction of Lysozyme. J. Mol. Biol. 1976, 103 (2), 227–249. (b) Warshel, A. Energetics of Enzyme Catalysis. Proc. Natl. Acad. Sci. U. S. A. 1978, 75 (11), 5250–5254. (c) Acevedo, O.; Jorgensen, W. L. Advances in Quantum and Mo-lecular Mechanical (QM/MM) Simulations for Organic and Enzymatic Re-actions. Acc. Chem. Res. 2010, 43 (1), 142–151. (d) Senn, H. M.; Thiel, W. QM/MM Methods for Biomolecular Systems. Angew. Chemie Int. Ed. 2009, 48 (7), 1198–1229. (e) van der Kamp, M. W.; Mulholland, A. J. Combined Quantum Mechanics/Molecular Mechanics (QM/MM) Methods in Compu-tational Enzymology. Biochemistry. 2013, 52 (16), 2708–2728.
[23] (a) Siegbahn, P. E. M.; Himo, F. Recent Developments of the Quantum Chemical Cluster Approach for Modeling Enzyme Reactions. JBIC J. Biol. Inorg. Chem. 2009, 14 (5), 643–651. (b) Siegbahn, P. E. M.; Himo, F. The Quantum Chemical Cluster Approach for Modeling Enzyme Reactions. WIREs Comput. Mol. Sci. 2011, 1 (3), 323–336. (c) Himo, F. Recent Trends in Quantum Chemical Modeling of Enzymatic Reactions. J. Am. Chem. Soc. 2017, 139 (20), 6780–6786. (d) Blomberg, M. R. A.; Borowski, T.; Himo, F.; Liao, R.-Z.; Siegbahn, P. E. M. Quantum Chemical Studies of Mechanisms for Metalloenzymes. Chem. Rev. 2014, 114 (7), 3601–3658.
[24] (a) Sevastik, R.; Himo, F. Quantum Chemical Modeling of Enzymatic Reactions: The Case of 4-Oxalocrotonate Tautomerase. Bioorg. Chem. 2007, 35 (6), 444–457. (b) Hopmann, K. H.; Himo, F. Quantum Chemical Modeling

72
of the Dehalogenation Reaction of Haloalcohol Dehalogenase. J. Chem. The-ory Comput. 2008, 4 (7), 1129–1137. (c) Georgieva, P.; Himo, F. Quantum Chemical Modeling of Enzymatic Reactions: The Case of Histone Lysine Methyltransferase. J. Comput. Chem. 2010, 31 (8), 1707-1714. (d) Liao, R.-Z.; Yu, J.-G.; Himo, F. Quantum Chemical Modeling of Enzymatic Reactions: The Case of Decarboxylation. J. Chem. Theory Comput. 2011, 7 (5), 1494–1501.
[25] (a) Jencks, W. P. Binding Energy, Specificity, and Enzymic Catalysis: The Circe Effect. Adv. Enzymol. Relat. Areas Mol. Biol. 2006, 43, 219− 410. (b) Jencks, W. P. From Chemistry to Biochemistry to Catalysis to Movement. Annu. Rev. Biochem. 1997, 66, 1−18.
[26] (a) Villà, J.; Štrajbl, M.; Glennon, T. M.; Sham, Y. Y.; Chu, Z. T.; Warshel, A. How Important Are Entropic Contributions to Enzyme Cataly-sis? Proc. Natl. Acad. Sci. U. S. A. 2000, 97 (22), 11899–11904. (b) Kazemi, M.; Himo, F.; Åqvist, J. Enzyme Catalysis by Entropy without Circe Effect. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (9), 2406–2411. (c) Mulholland, A. J. Dispelling the Effects of a Sorceress in Enzyme Catalysis. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (9), 2328–2330. (d) Åqvist, J.; Kazemi, M.; Isaksen, G. V.; Brandsdal, B. O. Entropy and Enzyme Catalysis. Acc. Chem. Res. 2017, 50 (2), 199–207.
[27] For selected examples, see: (a) Lind, M. E. S.; Himo, F. Quantum Chem-istry as a Tool in Asymmetric Biocatalysis: Limonene Epoxide Hydrolase Test Case. Angew. Chem. Int. Ed. 2013, 52 (17), 4563–4567. (b) Lind, M. E. S.; Himo, F. Quantum Chemical Modeling of Enantioconvergency in Soluble Epoxide Hydrolase. ACS Catal. 2016, 6 (12), 8145–8155. (c) Lind, M. E. S.; Himo, F. Theoretical Study of Reaction Mechanism and Stereoselectivity of Arylmalonate Decarboxylase. ACS Catal. 2014, 4 (11), 4153–4160. (d) Payer, S. E.; Sheng, X.; Pollak, H.; Wuensch, C.; Steinkellner, G.; Himo, F.; Glueck, S. M.; Faber, K. Exploring the Catalytic Promiscuity of Phenolic Acid De-carboxylases: Asymmetric, 1,6-Conjugate Addition of Nucleophiles Across 4-Hydroxystyrene. Adv. Synth. Catal. 2017, 359 (12), 2066-2075. (e) Sheng, X.; Himo, F. Theoretical Study of Enzyme Promiscuity: Mechanisms of Hy-dration and Carboxylation Activities of Phenolic Acid Decarboxylase. ACS Catal. 2017, 7 (3), 1733-1741. (f) Moa, S.; Himo, F. Quantum chemical study of mechanism and stereoselectivity of secondary alcohol dehydrogenase. J. Inorg. Biochem. 2017, 175, 259-266.; For a review, see: Sheng, X.; Kazemi, M.; Planas, F.; Himo, F. Modeling Enzymatic Enantioselectivity using Quantum Chemical Methodology. ACS Catal. 2020, 10, 6430-6449.

73
[28] Gaussian 09, Revision D.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Men-nucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers. K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian, Inc., Wallingford CT, 2013.
[29] Gaussian 16, Revision C.01,M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Iz-maylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian, Inc., Wallingford CT, 2019.
[30] (a) Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molec-ular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1984. 82 (1), 270-283. (b) Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985. 82 (1), 284-298
[31] A New Basis Set Exchange: An Open, Up-to-date Resource for the Mo-lecular Sciences Community. Benjamin P. Pritchard, Doaa Altarawy, Brett Didier, Tara D. Gibson, Theresa L. Windus. J. Chem. Inf. Model. 2019, 59 (11), 4814-4820.
[32] Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the

74
Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Ten-sions. J. Phys. Chem. B 2009, 113 (18), 6378–6396.
[33] Tomasi, J.; Persico, M. Molecular Interactions in Solution: An Over-view of Methods Based on Continuous Distributions of the Solvent. Chem. Rev. 1994, 94, 2027-2094.
[34] (a) Blomberg, M. R. A.; Siegbahn, P. E. M. Mechanism for N2O Gener-ation in Bacterial Nitric Oxide Reductase: A Quantum Chemical Study. Bio-chemistry 2012, 51 (25), 5173–5186. (b) Sheng, X.; Lind, M. E. S.; Himo, F. Theoretical Study of the Reaction Mechanism of Phenolic Acid Decarbox-ylase. FEBS J. 2015, 282 (24), 4703–4713. (c) Sheng, X.; Zhu, W.; Huddle-ston, J.; Xiang, D. F.; Raushel, F. M.; Richards, N. G. J.; Himo, F. A Combined Experimental-Theoretical Study of the LigW-Catalyzed Decarboxylation of 5-Carboxyvanillate in the Metabolic Pathway for Lignin Degradation. ACS Catal. 2017, 7 (8), 4968–4974.
[35] Bunik, V. I.; Tylicki, A.; Lukashev, N. V. Thiamin Diphosphate-De-pendent Enzymes: From Enzymology to Metabolic Regulation, Drug De-sign and Disease Models. FEBS J. 2013, 280 (24), 6412–6442.
[36] (a)Müller, M.; Gocke, D.; Pohl, M. Thiamin Diphosphate in Biological Chemistry: Exploitation of Diverse Thiamin Diphosphate-Dependent En-zymes for Asymmetric Chemoenzymatic Synthesis. FEBS J. 2009, 276 (11), 2894–2904. (b) Müller, M.; Sprenger, G. A.; Pohl, M. CC Bond Formation Using ThDP-Dependent Lyases. Curr. Opin. Chem Biol. 2013, 17 (2) 261–270.
[37]. For selected examples, see: (a) Demir, A. S.; Şeşenoglu, Ö.; Eren, E.; Hosrik, B.; Pohl, M.; Janzen, E.; Kolter, D.; Feldmann, R.; Dünkelmann, P.; Müller, M. Enantioselective Synthesis of α-Hydroxy Ketones via Benzalde-hyde Lyase-Catalyzed C-C Bond Formation Reaction. Adv. Synth. Catal. 2002, 344 (1), 96–103. (b) Engel, S.; Vyazmensky, M.; Geresh, S.; Barak, Z.; Chipman, D. M. Acetohydroxyacid Synthase: A New Enzyme for Chiral Syn-thesis OfR-Phenylacetylcarbinol. Biotechnol. Bioeng. 2003, 83 (7), 833–840. (c) Domínguez de María, P.; Pohl, M.; Gocke, D.; Gröger, H.; Trauthwein, H.; Stillger, T.; Walter, L.; Müller, M. Asymmetric Synthesis of Aliphatic 2-Hydroxy Ketones by Enzymatic Carboligation of Aldehydes. European J. Org. Chem. 2007, 2007 (18), 2940–2944. (d) Rother neé Gocke, D.; Kolter, G.; Gerhards, T.; Berthold, C. L.; Gauchenova, E.; Knoll, M.; Pleiss, J.; Müller, M.; Schneider, G.; Pohl, M. S-Selective Mixed Carboligation by Structure-Based Design of the Pyruvate Decarboxylase from Acetobacter Pasteurianus. ChemCatChem 2011, 3 (10), 1587–1596. (e) Beigi, M.; Loschonsky, S.;

75
Lehwald, P.; Brecht, V.; Andrade, S. L. A.; Leeper, F. J.; Hummel, W.; Müller, M. α-Hydroxy-β-Keto Acid Rearrangement-Decarboxylation: Impact on Thiamine Diphosphate-Dependent Enzymatic Transformations. Org. Bio-mol. Chem. 2013, 11 (2), 252–256. For a review, see: Hailes, H. C.; Rother, D.; Müller, M.; Westphal, R.; Ward, J. M.; Pleiss, J.; Vogel, C.; Pohl, M. Engi-neering Stereoselectivity of ThDP-Dependent Enzymes. FEBS J. 2013, 280 (24), 6374–6394.
[38] Knoll, M.; Müller, M.; Pleiss, J.; Pohl, M. Factors Mediating Activity, Selectivity, and Substrate Specificity for the Thiamin Diphosphate-Depend-ent Enzymes Benzaldehyde Lyase and Benzoylformate Decarboxylase. ChemBioChem. 2006, 7 (12), 1928–1934.
[39] (a) Schütz, A.; Sandalova, T.; Ricagno, S.; Hübner, G.; König, S.; Schnei-der, G. Crystal Structure of Thiamindiphosphate-Dependent Indolepyruvate Decarboxylase from Enterobacter Cloacae , an Enzyme Involved in the Bio-synthesis of the Plant Hormone Indole-3-Acetic Acid. Eur. J. Biochem. 2003, 270 (10), 2312–2321. (b) Berthold, C. L.; Gocke, D.; Wood, M. D.; Leeper, F. J.; Pohl, M.; Schneider, G.; IUCr. Structure of the Branched-Chain Keto Acid Decarboxylase (KdcA) from Lactococcus Lactis Provides Insights into the Structural Basis for the Chemoselective and Enantioselective Carboligation Reaction. Acta Crystallogr. Sect. D Biol. Crystallogr. 2007, 63 (12), 1217–1224. (c) Versées, W.; Spaepen, S.; Vanderleyden, J.; Steyaert, J. The Crystal Struc-ture of Phenylpyruvate Decarboxylase from Azospirillum Brasilense at 1.5 Å Resolution. FEBS J. 2007, 274 (9), 2363–2375.
[40] (a) Candy, J. M.; Duggleby, R. G. Structure and Properties of Pyruvate Decarboxylase and Site-Directed Mutagenesis of the Zymomonas Mobilis Enzyme. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1998, 1385 (2), 323–338. (b) Liu, M.; Sergienko, E. A.; Guo, F.; Wang, J.; Tittmann, K.; Hüb-ner, G.; Furey, W.; Jordan, F. Catalytic Acid−Base Groups in Yeast Pyruvate Decarboxylase. 1. Site-Directed Mutagenesis and Steady-State Kinetic Stud-ies on the Enzyme with the D28A, H114F, H115F, and E477Q Substitutions. Biochemistry 2001, 40 (25), 7355–7368. (c) Sergienko, E. A.; Jordan, F. Cata-lytic Acid−Base Groups in Yeast Pyruvate Decarboxylase. 2. Insights into the Specific Roles of D28 and E477 from the Rates and Stereospecificity of Formation of Carboligase Side Products. Biochemistry 2001, 40 (25), 7369–7381.
[41] Hasson, M. S.; Muscate, A.; McLeish, M. J.; Polovnikova, L. S.; Gerlt, J. A.; Kenyon, G. L.; Petsko, G. A.; Ringe, D. The Crystal Structure of Ben-

76
zoylformate Decarboxylase at 1.6 Å Resolution: Diversity of Catalytic Resi-dues in Thiamin Diphosphate-Dependent Enzymes. Biochemistry 1998, 37 (28), 9918–9930.
[42] (a) Sergienko, E. A.; Wang, J.; Polovnikova, L.; Hasson, M. S.; McLeish, M. J.; Kenyon, G. L.; Jordan, F. Spectroscopic Detection of Transient Thia-min Diphosphate-Bound Intermediates on Benzoylformate Decarboxylase. Biochemistry 2000, 39 (45), 13862–13869. (b) Polovnikova, E. S.; McLeish, M. J.; Sergienko, E. A.; Burgner, J. T.; Anderson, N. L.; Bera, A. K.; Jordan, F.; Kenyon, G. L.; Hasson, M. S. Structural and Kinetic Analysis of Catalysis by a Thiamin Diphosphate-Dependent Enzyme, Benzoylformate Decarbox-ylase. Biochemistry. 2003, 42 (7), 1820–1830.
[43] Yep, A.; Kenyon, G. L.; McLeish, M. J. Saturation Mutagenesis of Pu-tative Catalytic Residues of Benzoylformate Decarboxylase Provides a Chal-lenge to the Accepted Mechanism. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (15), 5733–5738.
[44] (a) Demir, A. S.; Dünnwald, T.; Iding, H.; Pohl, M.; Müller, M. Asym-metric Benzoin Reaction Catalyzed by Benzoylformate Decarboxylase. Tet-rahedron Asymmetry 1999, 10 (24), 4769–4774. (b) Iding, H.; Dünnwald, T.; Greiner, L.; Liese, A.; Müller, M.; Siegert, P.; Grötzinger, J.; Demir, A. S.; Pohl, M. Benzoylformate Decarboxylase from Pseudomonas Putida as Stable Catalyst for the Synthesis of Chiral 2-Hydroxy Ketones. Chem. Eur. J. 2000, 6 (8), 1483–1495. (c) Dünkelmann, P.; Kolter-Jung, D.; Nitsche, A.; Demir, A. S.; Siegert, P.; Lingen, B.; Baumann, M.; Pohl, M.; Müller, M. Develop-ment of a Donor-Acceptor Concept for Enzymatic Cross-Coupling Reactions of Aldehydes: The First Asymmetric Cross-Benzoin Condensation. J. Am. Chem. Soc. 2002, 124 (41), 12084–12085. (d) Lingen, B.; Grötzinger, J.; Kolter, D.; Kula, M.-R.; Pohl, M. Improving the Carboligase Activity of Ben-zoylformate Decarboxylase from Pseudomonas Putida by a Combination of Directed Evolution and Site-Directed Mutagenesis. Protein Eng. Des. Sel. 2002, 15 (7), 585–593.
[45] (a) Wilcocks, R.; Ward, O. P.; Dewdney, N. J.; Hong, Y.; Prosen, E. Acyloin Formation by Benzoylformate Decarboxylase from Pseudomonas Putida Whole Cells and Cell Extracts of Pseudomonas Putida Grown in a Medium Containing Ammonium Mandelate. Appl. Environ. Microbiol. 1992, 1699–1704. (b) Siegert, P.; McLeish, M. J.; Baumann, M.; Iding, H.; Kneen, M. M.; Kenyon, G. L.; Pohl, M. Exchanging the Substrate Specificities of Pyruvate Decarboxylase from Zymomonas Mobilis and Benzoylformate De-carboxylase from Pseudomonas Putida. Protein Eng. Des. Sel. 2005, 18 (7), 345–357.

77
[46] Breslow, R. On the Mechanism of Thiamine Action. IV. Evidence from Studies on Model Systems. J. Am. Chem. Soc. 1958, 80 (14), 3719–3726.
[47] (a) Kochetov, G. A.; Usmanov, R. A.; Merzlov, V. P. Thiaminepyro-phosphate Induced Changes in the Optical Activity of Baker’s Yeast Trans-ketolase. FEBS Lett. 1970, 9 (5), 265–266. (b) Jordan, F.; Zhang, Z.; Ser-gienko, E. Spectroscopic Evidence for Participation of the 1ʹ,4ʹ-Imino Tauto-mer of Thiamin Diphosphate in Catalysis by Yeast Pyruvate Decarboxylase. Bioorg. Chem. 2002, 30 (3), 188–198. (c) Polovnikova, E. S.; McLeish, M. J.; Sergienko, E. A.; Burgner, J. T.; Anderson, N. L.; Bera, A. K.; Jordan, F.; Kenyon, G. L.; Hasson, M. S. Structural and Kinetic Analysis of Catalysis by a Thiamin Diphosphate-Dependent Enzyme, Benzoylformate Decarbox-ylase. Biochemistry. 2003, 42 (7), 1820–1830. (d) Nemeria, N.; Korotchkina, L.; McLeish, M. J.; Kenyon, G. L.; Patel, M. S.; Jordan, F. Elucidation of the Chemistry of Enzyme-Bound Thiamin Diphosphate Prior to Substrate Bind-ing: Defining Internal Equilibria among Tautomeric and Ionization States. Biochemistry. 2007, 46 (37), 10739–10744. (e) Brandt, G. S.; Kneen, M. M.; Chakraborty, S.; Baykal, A. T.; Nemeria, N.; Yep, A.; Ruby, D. I.; Petsko, G. A.; Kenyon, G. L.; McLeish, M. J.; et al. Snapshot of a Reaction Intermediate: Analysis of Benzoylformate Decarboxylase in Complex with a Benzo-ylphosphonate Inhibitor. Biochemistry. 2009, 48 (15), 3247–3257. (f) Patel, H.; Nemeria, N. S.; Andrews, F. H.; McLeish, M. J.; Jordan, F. Identification of Charge Transfer Transitions Related to Thiamin-Bound Intermediates on Enzymes Provides a Plethora of Signatures Useful in Mechanistic Studies. Biochemistry 2014, 53 (13), 2145–2152. (g) Balakrishnan, A.; Paramasivam, S.; Chakraborty, S.; Polenova, T.; Jordan, F. Solid-State Nuclear Magnetic Resonance Studies Delineate the Role of the Protein in Activation of Both Aromatic Rings of Thiamin. J. Am. Chem. Soc. 2012, 134 (1), 665–672.
[48] Washabaugh, M. W.; Yang, C. C.; Hollenbach, A. D.; Chen, P. Hydrol-ysis of Thiamin: Evidence for Rate-Limiting Breakdown of the Tricyclic Di-hydrothiachromine Intermediate in Neutral Aqueous Solution. Bioorg. Chem. 1993, 21 (2), 170–191.
[49] Suzuki, R.; Katayama, T.; Kim, B. J.; Wakagi, T.; Shoun, H.; Ashida, H.; Yamamoto, K.; Fushinobu, S. Crystal Structures of Phosphoketolase: Thia-mine Diphosphate-Dependent Dehydration Mechanism. J. Biol. Chem. 2010, 285 (44), 34279–34287.
[50] Pang, S. S.; Duggleby, R. G.; Schowen, R. L.; Guddat, L. W. The Crys-tal Structures of Klebsiella Pneumoniae Acetolactate Synthase with Enzyme-Bound Cofactor and with an Unusual Intermediate. J. Biol. Chem. 2004, 279 (3), 2242–2253.

78
[51] Williamson, N. R.; Simonsen, H. T.; Ahmed, R. A. A.; Goldet, G.; Slater, H.; Woodley, L.; Leeper, F. J.; Salmond, G. P. C. Biosynthesis of the Red Antibiotic, Prodigiosin, in Serratia: Identification of a Novel 2-Methyl-3-n-Amyl-Pyrroie (MAP) Assembly Pathway, Definition of the Terminal Con-densing Enzyme, and Implications for Undecylprodigiosin Biosynthesis in Streptomyces. Mol. Microbiol. 2005, 56 (4), 971–989.
[52] Kasparyan, E.; Richter, M.; Dresen, C.; Walter, L. S.; Fuchs, G.; Leeper, F. J.; Wacker, T.; Andrade, S. L. A.; Kolter, G.; Pohl, M.; Müller, M. Asym-metric Stetter Reactions Catalyzed by Thiamine Diphosphate-Dependent Enzymes. Appl. Microbiol. Biotechnol. 2014, 98 (23), 9681–9690.
[53] (a) Jiang, M.; Cao, Y.; Guo, Z. F.; Chen, M.; Chen, X.; Guo, Z. Mena-quinone Biosynthesis in Escherichia Coli: Identification of 2-Succinyl-5-Enolpyruvyl-6-Hydroxy-3-Cyclohexene-1-Carboxylate as a Novel Interme-diate and Re-Evaluation of MenD Activity. Biochemistry 2007, 46 (38), 10979–10989. (b) Dawson, A.; Fyfe, P. K.; Hunter, W. N. Specificity and Re-activity in Menaquinone Biosynthesis: The Structure of Escherichia Coli MenD (2-Succinyl-5-Enolpyruvyl-6-Hydroxy-3-Cyclohexadiene-1-Carbox-ylate Synthase). J. Mol. Biol. 2008, 384 (5), 1353–1368. (c) Dawson, A.; Chen, M.; Fyfe, P. K.; Guo, Z.; Hunter, W. N. Structure and Reactivity of Bacillus Subtilis MenD Catalyzing the First Committed Step in Menaquinone Bio-synthesis. J. Mol. Biol. 2010, 401 (2), 253–264.
[54] Aleku, G.A.; Nowicka, B.; Turner, N. J. Biocatalytic Potential of En-zymes Involved in the Biosynthesis of Isoprenoid Quinones. 2017, 10, 124-135.
[55] Fiedler, E.; Thorell, S.; Sandalova, T.; Golbik, R.; König, S.; Schneider, G. Snapshot of a Key Intermediate in Enzymatic Thiamin Catalysis: Crystal Structure of the α-Carbanion of (α,β-Dihydroxyethyl)-Thiamin Diphosphate in the Active Site of Transketolase from Saccharomyces Cerevisiae. Proc. Natl. Acad. Sci. U. S. A. 2002, 99 (2), 591–595.
[56] Jirgis, E. N. M.; Bashiri, G.; Bulloch, E. M. M.; Johnston, J. M.; Baker, E. N. Structural Views along the Mycobacterium Tuberculosis MenD Reac-tion Pathway Illuminate Key Aspects of Thiamin Diphosphate-Dependent Enzyme Mechanisms. Structure 2016, 24 (7), 1167–1177.
[57] Qin, M.; Song, H.; Dai, X.; Chan, C.; Chan, W.; Guo, Z. Single-Turnover Kinetics Reveal a Distinct Mode of Thiamine Diphosphate-Dependent Ca-talysis in VitaminK Biosynthesis. ChemBioChem 2018, 19 (14), 1514–1522.

79
[58] Kurutsch, A.; Richter, M.; Brecht, V.; Sprenger, G. A.; Müller, M. MenD as a Versatile Catalyst for Asymmetric Synthesis. J. Mol. Catal. B En-zym. 2009, 61 (1–2), 56–66.
[59] Beigi, M.; Waltzer, S.; Zarei, M.; Müller, M. New Stetter Reactions Catalyzed by Thiamine Diphosphate Dependent MenD from E. Coli. J. Bio-technol. 2014, 191, 64–68.
[60] Schapfl, M.; Baier, S.; Fries, A.; Ferlaino, S.; Waltzer, S.; Müller, M.; Sprenger, G. A. Extended Substrate Range of Thiamine Diphosphate-De-pendent MenD Enzyme by Coupling of Two C–C-Bonding Reactions. Appl. Microbiol. Biotechnol. 2018, 102 (19), 8359–8372.
[61] Westphal, R.; Waltzer, S.; Mackfeld, U.; Widmann, M.; Pleiss, J.; Beigi, M.; Müller, M.; Rother, D.; Pohl, M. (S)-Selective Mend Variants from Esch-erichia Coli Provide Access to New Functionalized Chiral α-Hydroxy Ke-tones. Chem. Commun. 2013, 49 (20), 2061–2063.
[62] Westphal, R.; Hahn, D.; Mackfeld, U.; Waltzer, S.; Beigi, M.; Widmann, M.; Vogel, C.; Pleiss, J.; Müller, M.; Rother, D.; Pohl, M. Tailoring the S-Selectivity of 2-Succinyl-5-Enolpyruvyl-6-Hydroxy-3- Cyclohexene-1-Car-boxylate Synthase (MenD) from Escherichia Coli. ChemCatChem 2013, 5 (12), 3587–3594.
[63] (a) Wang, J.; Dong, H.; Li, S.; He, H. Theoretical Study toward Under-standing the Catalytic Mechanism of Pyruvate Decarboxylase. J. Phys. Chem. B 2005, 109 (39), 18664–18672. (b) Hou, Q.; Gao, J.; Liu, Y.; Liu, C. A QM/MM Study on the Catalytic Mechanism of Pyruvate Decarboxylase. Theor. Chem. Acc. 2012, 131 (10), 1–9. (c) Sheng, X.; Liu, Y.; Zhang, R. A Theoretical Study of the Catalytic Mechanism of Oxalyl-CoA Decarbox-ylase, an Enzyme for Treating Urolithiasis. RSC Adv. 2014, 4 (67), 35777–35788. (d) Zhang, J.; Sheng, X.; Hou, Q.; Liu, Y. Theoretical Investigation on the Dissociation of (R)-Benzoin Catalyzed by Benzaldehyde Lyase. Int. J. Quantum Chem. 2014, 114 (6), 375–382. (e) Planas, F.; Sheng, X.; McLeish, M. J.; Himo, F. A Theoretical Study of the Benzoylformate Decarboxylase Reaction Mechanism. Front. Chem. 2018, 6, 205.
[64] Kaiser, D.; Klose, I.; Oost, R.; Neuhaus, J.; Maulide, N. Bond-Forming and -Breaking Reactions at Sulfur(IV): Sulfoxides, Sulfonium Salts, Sulfur Ylides, and Sulfinate Salts. Chem. Rev. 2019, 119 (14), 8701–8780.
[65] Pulis, A. P.; Procter, D. J. C−H Coupling Reactions Directed by Sulfox-ides: Teaching an Old Functional Group New Tricks. Angew. Chem. Int. Ed. 2016, 55 (34), 9842–9860.

80
[66] Smith, L. H. S.; Coote, S. C.; Sneddon, H. E.; Procter, D. J. Beyond the Pummerer Reaction: Recent Developments in Thionium Ion Chemistry. An-gew. Chem. Int. Ed. 2010, 49 (34), 5832–5844.
[67] (a) Kobayashi, K.; Horita, M.; Irisawa, S.; Matsunaga, A.; Morikawa, O.; Konishi, H. Magnesium Amide-Induced Pummerer-Type Reactions of Cy-clopropyl Sulfoxides. Synthesis of Cyclopropanone Dithioacetals. Bull. Chem. Soc. Jpn. 2002, 75 (6), 1367–1369. (b) Kobayashi, K.; Kawakita, M.; Yokota, K.; Mannami, T.; Yamamoto, K.; Morikawa, O.; Konishi, H. Reactions of Sul-foxides with Magnesium Amides. Transformation of Sulfoxides into Sulfides, Dithioacetals, and Vinyl Sulfides. Bull. Chem. Soc. Jpn. 1995, 68 (5), 1401–1407. (c) Kobayashi, K.; Yokota, K.; Akamatsu, H.; Morikawa, O.; Konishi, H. Reductive α-Substitution of Sulfoxides with Grignard Reagents Promoted by a Magnesium Amide. Bull. Chem. Soc. Jpn. 1996, 69 (2), 441–443.
[68] Colas, K.; Martín-Montero, R.; Mendoza, A. Intermolecular Pummerer Coupling with Carbon Nucleophiles in Non-Electrophilic Media. Angew. Chem. Int. Ed. 2017, 129 (50), 16258–16262.
[69] Henderson, K. W.; Mulvey, R. E.; Dorigo, A. E. Solvent Induced Dis-proportionation of Alkyl(Amido)Magnesium Species Containing a (2-Pyridyl)Amido Unit: Synthetic, Theoretical and NMR Spectroscopic Studies of Bis(Amido)Magnesium Compounds. J. Organomet. Chem. 1996, 518 (1–2), 139–146.
[70] (a) Wu, W.; Lin, Z.; Jiang, H. Recent Advances in the Synthesis of Cy-clopropanes. Organic and Biomolecular Chemistry. 2018, 41 (13), 7315–7329. (b) de Meijere, A.; Kozhushkov, S. I.; Schill, H. Three-Membered-Ring-Based Molecular Architectures. Chem. Rev. 2006, 41 (13), 4926–4996. (c) Chen, D. Y. K.; Pouwer, R. H.; Richard, J. A. Recent Advances in the Total Synthesis of Cyclopropane-Containing Natural Products. Chem. Soc. Rev. 2012, 41 (13), 4631–4642. (d) Ebner, C.; Carreira, E. M. Cyclopropanation Strategies in Re-cent Total Syntheses. Chem. Rev. 2017, 117 (18), 11651–11679. (e) Bartoli, G.; Bencivenni, G.; Dalpozzo, R. Asymmetric Cyclopropanation Reactions. Synthesis. 2014, 46 (8), 979–1029.
[71] (a) Donaldson, W. A. Synthesis of Cyclopropane Containing Natural Products. Tetrahedron. 2001, 57 (41), 8589–8627. (b) Pellissier, H. Recent Developments in Asymmetric Cyclopropanation. Tetrahedron. 2008, 64 (30–31), 7041–7095. (c) Nozaki, H.; Moriuti, S.; Takaya, H.; Noyori, R. Asymmet-ric Induction in Carbenoid Reaction by Means of a Dissymmetric Copper Chelate. Tetrahedron Lett. 1966, 7 (43), 5239– 5244. (d) Yao, X.; Qiu, M.; Lü, W.; Chen, H.; Zheng, Z. Substituted salen–Ru(II) Complexes as Catalysts in

81
the Asymmetric Cyclopropanation of Styrene by Ethyl Diazoacetate: The In-fluence of Substituents and Achiral Additives on Activity and Enantioselec-tivity. Tetrahedron: Asymmetry. 2001, 12 (2), 197–204. (e) Gregg, T. M.; Far-rugia, M. K.; Frost, J. R. Rhodium-Mediated Enantioselective Cyclopropana-tion of Allenes. Org. Lett. 2009, 11 (19), 4434–4436. (f) Zhu, S.; Ruppel, J. V.; Lu, H.; Wojtas, L.; Zhang, X. P. Cobalt-Catalyzed Asymmetric Cyclopropa-nation with Diazosulfones: Rigidification and Polarization of Ligand Chiral Environment via Hydrogen Bonding and Cyclization. J. Am. Chem. Soc. 2008, 130 (15), 5042– 5043.
[72] Montesinos-Magraner, M.; Costantini, M.; Ramírez-Contreras, R.; Mu-ratore, M. E.; Johansson, M. J.; Mendoza, A. General Cyclopropane Assem-bly by Enantioselective Transfer of a Redox-Active Carbene to Aliphatic Olefins. Angew. Chemie Int. Ed. 2019, 58 (18), 5930–5935.
[73] Early works on inner sphere cyclopropanation: (a) Bernardi, F.; Bot-toni, A.; Miscione, G. Pietro. DFT Study of the Olefin Metathesis Catalyzed by Ruthenium Complexes. Organometallics 2003, 22 (5), 940–947. (b) Ras-mussen, T.; Jensen, J. F.; Østergaard, N.; Tanner, D.; Ziegler, T.; Norrby, P. O. On the Mechanism of the Copper-Catalyzed Cyclopropanation Reaction. Chem. Eur. J. 2002, 8 (1), 177–184.
[74] For seminal works, see: (a) Fraile, J. M.; García, J. I.; Martínez-Merino, V.; Mayoral, J. A.; Salvatella, L. Theoretical (DFT) Insights into the Mecha-nism of Copper-Catalyzed Cyclopropanation Reactions. Implications for En-antioselective Catalysis. J. Am. Chem. Soc. 2001, 123 (31), 7616–7625. (b) Ta-gliatesta, P.; Pastorini, A. Remarkable Selectivity in the Cyclopropanation Reactions Catalysed by an Halogenated Iron Meso-Tetraphenylporphyrin. J. Mol. Catal. A Chem. 2003, 198, 57–61.; For selected recent works, see: (a) Fuller, J. T.; Harrison, D. J.; Leclerc, M. C.; Baker, R. T.; Ess, D. H.; Hughes, R. P. A New Stepwise Mechanism for Formation of a Metallacyclobutane via a Singlet Diradical Intermediate. Organometallics 2015, 34 (21), 5210–5213. (b) Dzik, W. I.; Xu, X.; Zhang, X. P.; Reek, J. N. H.; De Bruin, B. Carbene Radicals in CoII(Por)-Catalyzed Olefin Cyclopropanation. J. Am. Chem. Soc. 2010, 132 (31), 10891–10902. (c) Wei, Y.; Tinoco, A.; Steck, V.; Fasan, R.; Zhang, Y. Cyclopropanations via Heme Carbenes: Basic Mechanism and Ef-fects of Carbene Substituent, Protein Axial Ligand, and Porphyrin Substitu-tion. J. Am. Chem. Soc. 2018, 140 (5), 1649–1662. (d) Xue, Y. S.; Cai, Y. P.; Chen, Z. X. Mechanism and Stereoselectivity of the Rh(II)-Catalyzed Cyclo-propanation of Diazooxindole: A Density Functional Theory Study. RSC Adv. 2015, 5 (71), 57781–57791. (e) Nakagawa, Y.; Nakayama, N.; Goto, H.;

82
Fujisawa, I.; Chanthamath, S.; Shibatomi, K.; Iwasa, S. Computational Chem-ical Analysis of Ru(II)-Pheox–Catalyzed Highly Enantioselective Intramo-lecular Cyclopropanation Reactions. Chirality 2019, 31 (1), 52–61. (f) Wang, H.; Guptill, D. M.; Varela-Alvarez, A.; Musaev, D. G.; Davies, H. M. L. Rho-dium-Catalyzed Enantioselective Cyclopropanation of Electron-Deficient Alkenes. Chem. Sci. 2013, 4 (7), 2844–2850. (g) Torrent-Sucarrat, M.; Arras-tia, I.; Arrieta, A.; Cossío, F. P. Stereoselectivity, Different Oxidation States, and Multiple Spin States in the Cyclopropanation of Olefins Catalyzed by Fe-Porphyrin Complexes. ACS Catal. 2018, 8 (12), 11140–11153. (h) Casali, E.; Gallo, E.; Toma, L. An In-Depth Computational Study of Alkene Cyclopro-panation Catalyzed by Fe(Porphyrin)(OCH3) Complexes. The Environmen-tal Effects on the Energy Barriers. Inorg. Chem. 2020, 59 (16), 11329–11336. (i) Nakagawa, Y.; Imokawa, Y.; Fujisawa, I.; Nakayama, N.; Goto, H.; Chan-thamath, S.; Shibatomi, K.; Iwasa, S. Ligand Exchange Reaction on a Ru(II)-Pheox Complex as a Mechanistic Study of Catalytic Reactions. ACS Omega 2018, 3 (9), 11286–11289.
[75] (a) Borah, G.; Ramaiah, D.; Patel, P., Synthesis of Isoquinoline from Benzimidates via Ru(II)-Catalyzed C–H Alkylation/Annulations with Diazo Compounds. ChemistrySelect 2018, 3 (37), 10333-10337. (b) Kumar, G. S.; Khot, N. P.; Kapur, M., Oxazolinyl-Assisted Ru(II)-Catalyzed C−H Functionalization Based on Carbene Migratory Insertion: A One-Pot Three-Component Cascade Cyclization. Advanced Synthesis & Catalysis 2019, 361 (1), 73-78. (c) Li, Y.; Qi, Z.; Wang, H.; Yang, X.; Li, X., Ruthenium(II)-Catalyzed C−H Activation of Imidamides and Divergent Couplings with Diazo Compounds: Substrate-Controlled Synthesis of Indoles and 3H-Indoles. Angew. Chem. Int. Ed. 2016, 55 (39), 11877-11881 . (d) Su, L.; Yu, Z.; Ren, P.; Luo, Z.; Hou, W.; Xu, H., Ruthenium(II)-catalyzed synthesis of indazolone-fused cinnolines via C–H coupling with diazo compounds. Organic. Biomolecular Chemistry. 2018, 16 (39), 7236-7244. (e) Wang, C.; Maity, B.; Cavallo, L.; Rueping, M., Manganese Catalyzed Regioselective C–H Alkylation: Experiment and Computation. Organic Letters. 2018, 20 (10), 3105-3108.




















