
Epidemic spreading over networks - A view from neighbourhoods

Propagation of Spreading DepressionInversely Correlates with Cortical Myelin
ContentDoron Merkler, MD,1 Florian Klinker, MD,2 Tanja Jurgens, MSc,1 Raoul Glaser, MD,2 Walter Paulus, MD,2
Bastian G. Brinkmann, MD,3 Michael W. Sereda, MD,2,3 Christine Stadelmann-Nessler, MD,1
Rubem C. A. Guedes, PhD,4 Wolfgang Bruck, MD,1,5 and David Liebetanz, MD2
Objective: Cortical myelin can be severely affected in patients with demyelinating disorders of the central nervous system.However, the functional implication of cortical demyelination remains elusive. In this study, we investigated whether corticalmyelin influences cortical spreading depression (CSD).Methods: CSD measurements were performed in rodent models of toxic and autoimmune induced cortical demyelination, inneuregulin-1 type I transgenic mice displaying cortical hypermyelination, and in glial fibrillary acidic protein–transgenic miceexhibiting pronounced astrogliosis.Results: Cortical demyelination, but not astrogliosis or inflammation per se, was associated with accelerated CSD. In contrast,hypermyelinated neuregulin-1 type I transgenic mice displayed a decelerated CSD propagation.Interpretation: Cortical myelin may be crucially involved in the stabilization and buffering of extracellular ion content that isdecisive for CSD propagation velocity and cortical excitability, respectively. Our data thus indicate that cortical involvement inhuman demyelinating diseases may lead to relevant alterations of cortical function.
Ann Neurol 2009;66:355–365
Cortical spreading depression (CSD) was first de-scribed as a slowly propagating wave of suppressedelectrical activity in the rabbit cortex.1 It is character-ized by a wave of depolarization of neurons and gliathat propagates in the gray matter of the central ner-vous system (CNS).2,3 CSD can be recorded in variousexperimental pathological conditions that affect the ce-rebral cortex including brain trauma, subarachnoidhemorrhage, and stroke.4–6 However, the exact causeand the pathophysiological correlate of CSD remainelusive, and only a few studies have identified CSD inhuman brains.7,8 Several studies suggest that increasedcortical excitability is associated with an acceleration ofCSD propagation.9,10 A recent in vivo imaging studyprovided evidence that CSD is linked to hypoxia and isaccompanied by transient morphological abnormalityin apical dendrites in the upper cortical layer.11 Theoccurrence of spontaneous CSD is commonly seen asan important pathophysiological hallmark for migraine,
but it has also been associated with epilepsy.8,12–14
Furthermore, a facilitated induction and a faster prop-agation of CSD were noted in a genetic animal modelof familial hemiplegic migraine type 1, corroboratingthe strong association of CSD with migraine aura andcortical hyperexcitability.15
Interestingly, an increased prevalence of epilep-sy16–19 is noted in multiple sclerosis (MS) patients. Be-cause it became clear in recent years that the cerebralcortex can become extensively involved during the dis-ease course of MS,20,21 the association of gray matterpathology with certain comorbidities has gained newattention. Accordingly, a recent neuroradiological studysuggested an association of cortical inflammation withepilepsy in MS patients.22 Nevertheless, the clinical im-plications of gray matter lesions in MS are still poorlyunderstood.23
The aim of this study is to determine the role ofcortical myelin in CSD propagation. We provide here
From the Departments of 1Neuropathology and 2Clinical Neuro-physiology, University Medical Center, Georg August University;3Research Group “Molecular Neurology,” Department of Neuroge-netics, Max Planck Institute for Experimental Medicine, Gottingen,Germany; 4Department of Nutrition, Universidade Federal de Per-nambuco, Recife, PE, Brazil; and 5Institut fur Multiple-Sklerose-Forschung, University Medical Center Gottingen and Gemeinnutz-ige Hertie-Stiftung, Gottingen, Germany.
Address correspondence to Doran Merkler, Department of Neuropa-thology, Robert-Koch-Str. 40, 37099 Gottingen, Germany. E-mail:[email protected]
Potential conflict of interest: Nothing to report.
D.M. and F.K. contributed equally to this work.
Additional Supporting Information may be found in the online ver-sion of this article.
Received Sep 23, 2008, and in revised form Apr 27, 2009. Acceptedfor publication May 1, 2009. Published online in Wiley Inter-Science (www.interscience.wiley.com). DOI: 10.1002/ana.21746
© 2009 American Neurological Association 355

several lines of evidence in favor of the hypothesis thatthe velocity of CSD propagation inversely correlateswith cortical myelin content. Furthermore, our datasuggest that cortical demyelination may be associatedwith accelerated CSD propagation, and thus may be apathological substrate for increased cortical excitabilityand proneness to cortical symptoms such as migraineand epilepsy in MS.
Materials and MethodsThe experiments were conducted in adult female Lewis rats(n � 41) obtained from Harlan (Horst, Netherlands) and inadult mice (n � 64) bred on a C57Bl/6 background. Theanimals were kept in groups on a 12:12 light/dark cycle un-der standard laboratory conditions with food and water adlibitum. All experiments were approved by the Governmentof Lower Saxony, Germany.
Cuprizone Model of Toxic Demyelination andRemyelinationCuprizone treatment was used as a model of toxic, nonin-flammatory cortical demyelination and remyelination.24 Cu-prizone experiments were begun with female C57Bl/6 mice(8 weeks; 19.1 � 0.19gm). They were either fed for 5 weekswith 0.2% (wt/wt) cuprizone (Sigma, St. Louis, MO) in theground breeder chow (n � 14 animals) or received chowwithout cuprizone admixture (control group, n � 11 ani-mals). We conducted two series of experiments. In the firstexperimental setup, CSD recordings (see later) were per-formed at the end of the 5 weeks of cuprizone treatment(demyelinating paradigm, n � 8 for cuprizone-treated ani-mals; n � 6 for age-matched control subjects). In the secondsetup, the cuprizone diet was discontinued after 5 weeks andanimals were maintained for further 6 weeks under normaldiet to allow spontaneous remyelination (remyelinating par-adigm). These animals were then subjected to CSD record-ing (n � 6 for cuprizone-treated animals; n � 5 for age-matched control animals). In a further set of animals (age,14 weeks; n � 8), CSD recording was performed after short-term cuprizone treatment for 3 days to study possible directmetabolic effects of cuprizone on CSD, independent ofevents related to demyelination. Untreated age-matched ani-mals served as control animals (n � 7 animals).
Neuregulin-1 Type I Transgenic Animals as a Modelof Cortical HypermyelinationCSD measurements were performed in a total of 16 adultmice, aged 4.5 months. One group consisted of 10neuregulin-1 (Nrg1) type I transgenic mice (6 female and 4male mice). The control group consisted of three female andthree male wild-type mice. The transgenic and wild-typemice were littermates. The animals were investigated in ran-domized order. All investigators were blinded to the geno-type of the animals.
Human Glial Fibrillary Acidic Protein TransgenicMice as Model of AstrogliosisTransgenic mice expressing the human glial fibrillary acidicprotein (hGFAP; TgN(GFAP)Mes1; referred to as Tg73.7)
gene were maintained on a C57Bl/6 background (�10 gen-erations backcrossed) as hemizygotes as described previous-ly.25 CSD measurements (see below) were performed onadult (5.5 months old; 22.3 � 0.87gm) hGFAP-transgenicmice (n � 5) or their wild-type littermates as controls (n �6). All investigators were blinded about the genotype of theanimals.
Targeted Cortical Experimental AutoimmuneEncephalomyelitis as a Model of Autoimmune-Induced Cortical Demyelination and RemyelinationA first set of experiments was performed on 32 adult femaleLewis rats (184.6 � 6gm). The animals were immunizedagainst recombinant myelin oligodendrocyte glycoprotein(rMOG; n � 27) corresponding to the N-terminal sequenceof rat MOG (amino acids 1–125) 18 days before intracere-bral stereotactic injection as described previously.26 In brief,the rats were anesthetized by isoflurane inhalation and in-jected subcutaneously at the base of the tail with a total vol-ume of 100�l rMOG (50�g MOG) emulsified in incom-plete Freund’s adjuvant (Sigma-Aldrich, Buchs, Switzerland).The remaining five animals were immunized with 100�l in-complete Freund’s adjuvant only. In a further control exper-iment, nine female Lewis rats (189 � 1.7gm) were immu-nized with 100�g guinea pig myelin basic protein (MBP)peptides 72 to 85 (MBP72-85) emulsified in completeFreund’s adjuvants 9 days before intracerebral stereotactic in-jection.
Sensitized rats were anesthetized by isoflurane inhalationand mounted in a stereotactic device. A fine hole was drilledthrough the skull giving access to the surface of the brain1mm caudal to the bregma, and 1 and 3mm lateral to thesagittal suture. A finely calibrated glass capillary was then ste-reotactically inserted, targeting the cortex (approximately1.5mm depth) at each location (Fig 1). The rats were theninjected with 1�l of a cytokine mixture composed of 250ngrecombinant rat tumor necrosis factor-� (R&D Systems,Abingdon, United Kingdom) and 150 units recombinant ratinterferon-� (PeproTech, London, United Kingdom) dis-solved in phosphate-buffered saline over a 3-minute period.A trace of monastral blue (Sigma-Aldrich) was added as amarker dye for better visibility on injection site. After injec-tion, the glass capillary was carefully withdrawn and the op-eration site was sealed by suture. Postsurgical recovery wasalways uneventful, with no overt clinical signs. CSD record-ings were performed on day 3 (n � 12), 8 (n � 8), or 14(n � 7) after stereotactical injection with rMOG-immunizedanimals and 3 days after stereotactical injection with MBP-immunized (n � 9) and incomplete Freund’s adjuvant–im-munized (n � 5) control animals. In all conditions, the non-injected contralateral cortex was used for intraindividualcontrol recordings.
Cortical Spreading Depression RecordingsMice were anesthetized with a mixture of urethane (1g/kg)and chloralose (0.4g/kg) (both from Sigma-Aldrich ChemieGmbH, Steinheim, Germany). They were positioned in astereotactic frame (TSE Systems, Bad Homburg, Germany).Body temperature was maintained at 36.8°C to 37.2°C.Three 2mm-diameter holes were drilled over the right hemi-
356 Annals of Neurology Vol 66 No 3 September 2009

sphere 2mm lateral of the sagittal suture, one 1mm rostral,and one 2.2mm and one 5mm posterior of the coronal su-ture. Two Ag-AgCl-Agar-Ringer electrodes were gentlyplaced onto the dura mater at the two rostral trepanationswithin a defined interelectrode distance (3.2mm) to recordCSD. A common reference electrode of the same type wasmounted on the nasal bone. The caudal trepanation wasused to elicit the CSD (see Fig 1).
The surgical procedure for the rats used in the focal ex-perimental autoimmune encephalomyelitis (fEAE) experi-ments was similar except that the burr holes were positionedbilaterally 1mm rostral and 2.2 and 5mm caudal of the
bregma, with a lateral distance of 3mm from the sagittal su-ture.
CSD was always elicited by applying a cotton ball (1–2mm diameter) soaked in a 2% KCl solution to the caudalburr hole for about 1 minute. CSD propagation was inves-tigated in the posterofrontal direction. The spontaneous cor-tical electrical activity (electrocorticogram) and the slow di-rect current (DC) potential change accompanying CSD wererecorded at 128Hz using a Neuroprax-DC-EEG amplifier(eldith GmbH, Ilmenau, Germany).
The time required for a CSD wave to pass the distancebetween the two epicortical recording electrodes was definedas �latency. CSD velocity (expressed in millimeters perminute [mm/min]) was calculated as the ratio of interelec-trode distance divided by �latency. Four CSDs were re-corded in each animal with an interval of 20 minutes be-tween 2 sessions. The first recording was not used forstatistical analysis because it showed a greater variation thanthe second to fourth recordings.9
In the fEAE experiments, CSD was elicited bilaterally onalternating sides four times each side. The side of the firstrecording was chosen randomly, and an interval of 20 min-utes was adhered to between the recordings.
Data Processing and EvaluationThe raw data were exported from the Neuroprax-EEG am-plifier in an ASCII format. Further processing and evaluationwas conducted using a Pentium 4 Notebook and MatlabR2006a software. The electroencephalographic data were fil-tered using a 60Hz low-pass filter. Subsequently, both con-trol and verum recordings were presented graphically in ran-domized order by a custom-designed Matlab program. Thetime points for the beginning of the slow potential change ofCSD were marked by the investigator in the leads of elec-trode 1 and 2 (see Fig 1), and the �latency was calculatedautomatically. The investigator was not aware of the condi-tions.
Histological and Immunohistochemical AnalysisAfter CSD measurements (cuprizone and targeted experi-mental autoimmune encephalomyelitis [EAE] model), theanimals were immediately perfused transcardially under deepanaesthesia with ketamine/xylazine with 4% paraformalde-hyde for light microscopic analysis.26,27
Fixed brains were embedded in paraffin, and coronal sec-tions (3�m) were stained with hematoxylin and eosin to ver-ify the absence of brain damage caused by surgery that couldinterfere with CSD measurements (eg, hemorrhage, traumaas a consequence of stereotactic injection). In adjacent serialsections, immunohistochemistry was performed using anti-bodies for macrophages/activated microglia (clone ED1; Se-rotec, Oxford, United Kingdom), MBP (A0623; DakoCyto-mation), and glial fibrillary acidic protein (GFAP;DakoCytomation, Hamburg, Germany). Bound antibodieswere visualized using an avidin-biotin technique with 3.3%diaminobenzidine as chromogen.
Density of myelinated fibers (expressed as myelin densityscore) in mice was determined by counting the intersectionsof myelin fibers with a stereological 25-point grid as de-scribed previously.28 No intersection would give a score of 0,
Fig 1. Experimental setup for measuring cortical spreadingdepression (CSD) latency in the targeted experimental autoim-mune encephalomyelitis (EAE) model. Two Ringer electrodeswere placed at defined distances on the dura caudally (record-ing electrode 1) and rostrally (recording electrode 2) in rela-tion to the cortical cytokine injection sites for EAE lesion in-duction, as well as on the corresponding site of thecontralateral hemisphere. A common reference electrode of thesame type was mounted on the nasal bones (reference elec-trode). CSD was elicited bilaterally on alternating sites fourtimes per side from the caudal trepanation by applying KClsolution. CSD propagation was measured in the posterofrontaldirection. The CSD �latency was defined as the time requiredfor one CSD wave to pass the distance between the two epi-cortical recording electrodes. CSD velocity was calculated asthe ratio of distance between the two recording electrodes di-vided by �latency. Four CSDs were recorded in each animalwith an interval of 20 minutes between two sessions. fEAE �experimental autoimmune encephalomyelitis.
Merkler et al: Spreading Depression and Myelin 357

whereas 100% intersections would give a maximal score of25. Intersections of 20 high-power fields per animal compris-ing cortical lamina IV to VI were counted, and the averageof intersections of all counted fields were calculated.
Demyelinated cortical area in the targeted EAE model wasmeasured as described previously.26 In brief, digital images oftissue sections were recorded through an Olympus light/flu-orescent microscope (Olympus Optical Company Ltd, Ham-burg, Germany) with a charge-coupled device camera (ColorView II; Soft Imaging System, Munster, Germany). The ex-tent of demyelination (in square millimeters) was then quan-tified by measuring the lesion area on overview photographs(40� magnification) of MBP-immunostained sections usingthe Analysis Software Color View II (Soft Imaging System).
Density of ED1 cells was determined within the injectedhemisphere in cortical layers I-II for targeted EAE. Densitiesof Mac3 and GFAP cells were determined in both hemi-spheres within layers IV-VI in mice. For this purpose, thenumbers of immunolabeled cells within 10 (rats) and 20(mice) optical fields at 400� magnification were quantifiedfor each animal and location using a counting grid.
For electron microscopy (Nrg1 type I animals), the ani-mals were perfused with 4% paraformaldehyde and 2.5%glutaraldehyde in 0.1M phosphate-buffered saline, and thecerebral cortex was removed, contrasted with osmium tetrox-ide, embedded in Epon, and ultrathin sections were made asdescribed previously.29
Statistical AnalysisData were analyzed using SPSS Version 11 for Windows.The graphs were generated using GraphPad Prism version4.00 for Windows (GraphPad Software, San Diego, CA).Statistical analysis was performed with paired t test if intra-individual corresponding controls were used, or unpaired ttest if two independent groups were compared. A p value 0.05 was considered significant. Data are presented asmeans � standard error of the means.
ResultsCortical Spreading Depression Propagation afterCuprizone-Induced Demyelination and SubsequentRemyelinationIn the first set of experiments, we tested whether alter-ation of cortical myelin as a result of cuprizone treat-ment24 affects CSD propagation velocity. For this pur-pose, we measured CSD propagation velocity in ademyelinating (5 weeks of cuprizone) or remyelinating(5 weeks of cuprizone followed by 6 weeks of normaldiet) experimental paradigm.
Immunohistochemical staining for MBP showedsubtotal cortical demyelination in animals exposed for5 weeks to a 0.2% (wt/wt) cuprizone diet as comparedwith age-matched control animals, which was reflectedby substantially reduced density of myelinated fibers(Figs 2A, C). Furthermore, cortical demyelination wasaccompanied by moderate reactive astrocytosis, as evi-denced by the detection of GFAP-positive, stellate-shaped, hypertrophic astrocytes within the demyeli-
nated cortex of cuprizone-fed animals (see Figs 2A, C).When measuring the propagation of CSD, we foundthat demyelinated animals showed a significant increasein CSD velocity to 4.17 � 0.34mm/min (mean �standard error of the mean; n � 8; p 0.05) as com-pared with age-matched control animals subjected to anormal diet with 3.22 � 0.17mm/min (n � 9) (seeFig 2B). To determine whether changes in CSD veloc-ity might be caused by a direct metabolic effect of cu-prizone, independent of the detectable alteration incortical myelin or astrocytic reaction, we gave a subsetof animals a cuprizone diet for only 3 days (short-termexperiment). Within this short time span, demyelina-tion or astrocytic reaction was not evident in histolog-ical sections (see Supplementary Fig 1). However, mildbut statistical significant increase in numbers of acti-vated microglia cells could already be noted after cu-prizone feeding for 3 days (see Supplementary Fig 1).The measured velocity of CSD in cuprizone-fed ani-mals in this short-term experiment was 3.91 �0.28mm/min (mean � standard error of the mean;n � 8; p � 0.47) and was not significantly different(p � 0.47) from age-matched control animals (3.65 �0.33mm/min; n � 7). These data indicate that acutemetabolic changes caused by cuprizone exposure, butunrelated to myelin damage or astrocytic activation, donot account for altered CSD propagation.
In the next step, we evaluated CSD velocity 6 weeksafter cuprizone termination, a time point when exten-sive restoration of myelin is expected.30 Quantitativeanalysis of MBP-immunostained sections demonstratedextensive cortical remyelination when compared withdemyelinated animals but still slightly reduced myelindensity as compared with age-matched control group(see Figs 2A, C). CSD velocity of previously cuprizone-exposed but remyelinated animals (3.57 � 0.21mm/min; n � 6; p � 0.51) was, however, unaltered ascompared with age-matched control animals (3.69 �0.08mm/min, n � 5; see Fig 2B).
Role of Reactive Astrogliosis for Cortical SpreadingDepression PropagationOur as well as previous results demonstrated thatcuprizone-induced demyelination is accompanied byreactive astrogliosis within the cerebral cortex.24,31,32
Therefore, we further investigated whether altered as-trocytic activation associated with astrocytic hypertro-phy and gliosis could account for the observed alteredCSD velocity during cuprizone-induced demyelination.To address this premise, we measured CSD velocity inmice transgenically overexpressing hGFAP in astro-cytes.25 Although no obvious alteration of cortical my-elinated fibers was detectable by immunohistochemicalanalysis for MBP in hGFAP mice, these animals exhib-ited large numbers of diffusely distributed reactive-appearing astrocytes within the cerebral cortex as visu-
358 Annals of Neurology Vol 66 No 3 September 2009
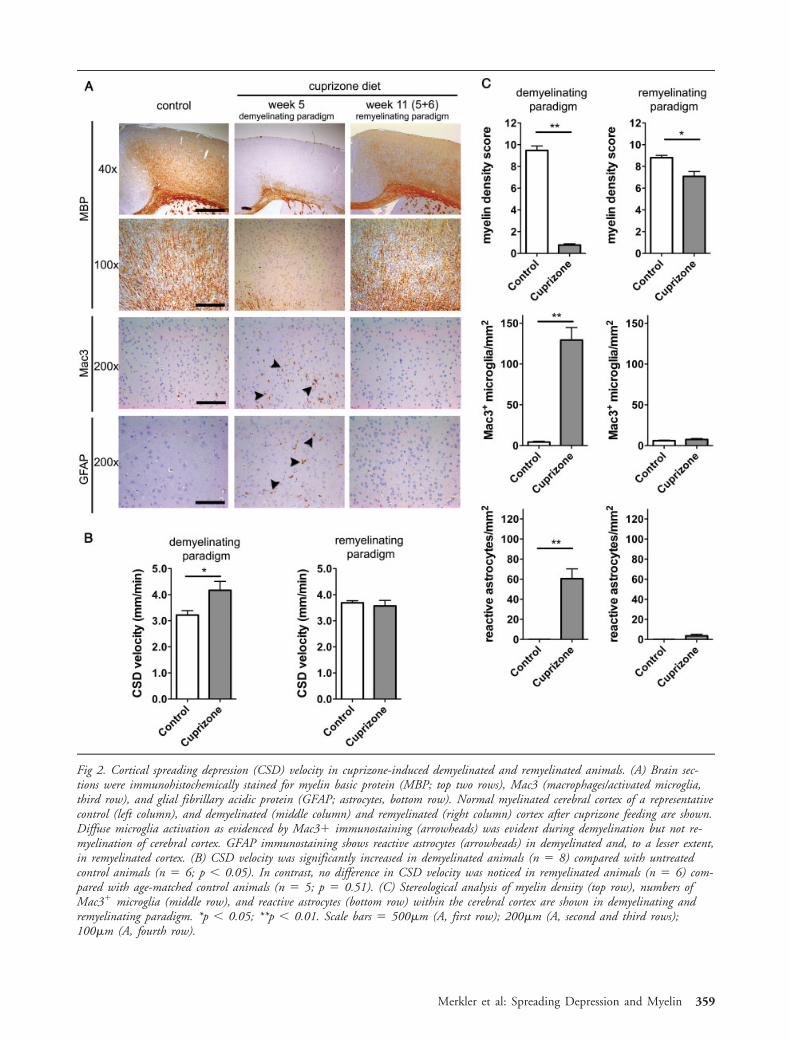
Fig 2. Cortical spreading depression (CSD) velocity in cuprizone-induced demyelinated and remyelinated animals. (A) Brain sec-tions were immunohistochemically stained for myelin basic protein (MBP; top two rows), Mac3 (macrophages/activated microglia,third row), and glial fibrillary acidic protein (GFAP; astrocytes, bottom row). Normal myelinated cerebral cortex of a representativecontrol (left column), and demyelinated (middle column) and remyelinated (right column) cortex after cuprizone feeding are shown.Diffuse microglia activation as evidenced by Mac3 immunostaining (arrowheads) was evident during demyelination but not re-myelination of cerebral cortex. GFAP immunostaining shows reactive astrocytes (arrowheads) in demyelinated and, to a lesser extent,in remyelinated cortex. (B) CSD velocity was significantly increased in demyelinated animals (n � 8) compared with untreatedcontrol animals (n � 6; p 0.05). In contrast, no difference in CSD velocity was noticed in remyelinated animals (n � 6) com-pared with age-matched control animals (n � 5; p � 0.51). (C) Stereological analysis of myelin density (top row), numbers ofMac3 microglia (middle row), and reactive astrocytes (bottom row) within the cerebral cortex are shown in demyelinating andremyelinating paradigm. *p 0.05; **p 0.01. Scale bars � 500�m (A, first row); 200�m (A, second and third rows);100�m (A, fourth row).
Merkler et al: Spreading Depression and Myelin 359
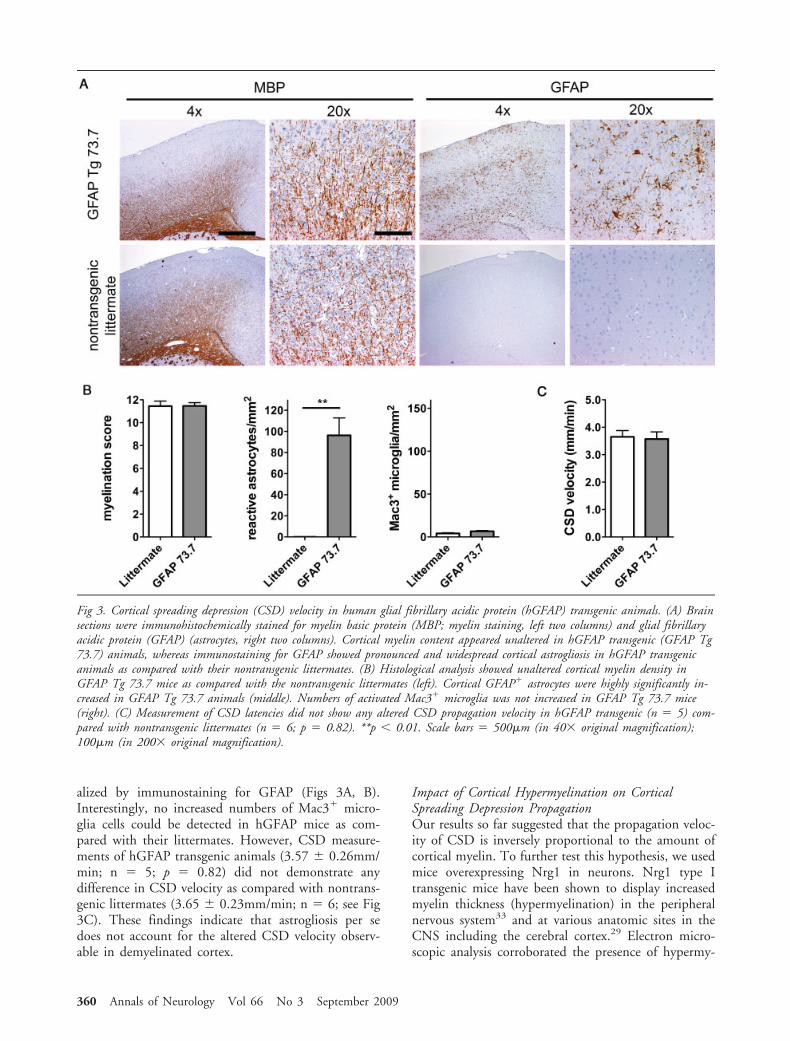
alized by immunostaining for GFAP (Figs 3A, B).Interestingly, no increased numbers of Mac3 micro-glia cells could be detected in hGFAP mice as com-pared with their littermates. However, CSD measure-ments of hGFAP transgenic animals (3.57 � 0.26mm/min; n � 5; p � 0.82) did not demonstrate anydifference in CSD velocity as compared with nontrans-genic littermates (3.65 � 0.23mm/min; n � 6; see Fig3C). These findings indicate that astrogliosis per sedoes not account for the altered CSD velocity observ-able in demyelinated cortex.
Impact of Cortical Hypermyelination on CorticalSpreading Depression PropagationOur results so far suggested that the propagation veloc-ity of CSD is inversely proportional to the amount ofcortical myelin. To further test this hypothesis, we usedmice overexpressing Nrg1 in neurons. Nrg1 type Itransgenic mice have been shown to display increasedmyelin thickness (hypermyelination) in the peripheralnervous system33 and at various anatomic sites in theCNS including the cerebral cortex.29 Electron micro-scopic analysis corroborated the presence of hypermy-
Fig 3. Cortical spreading depression (CSD) velocity in human glial fibrillary acidic protein (hGFAP) transgenic animals. (A) Brainsections were immunohistochemically stained for myelin basic protein (MBP; myelin staining, left two columns) and glial fibrillaryacidic protein (GFAP) (astrocytes, right two columns). Cortical myelin content appeared unaltered in hGFAP transgenic (GFAP Tg73.7) animals, whereas immunostaining for GFAP showed pronounced and widespread cortical astrogliosis in hGFAP transgenicanimals as compared with their nontransgenic littermates. (B) Histological analysis showed unaltered cortical myelin density inGFAP Tg 73.7 mice as compared with the nontransgenic littermates (left). Cortical GFAP astrocytes were highly significantly in-creased in GFAP Tg 73.7 animals (middle). Numbers of activated Mac3 microglia was not increased in GFAP Tg 73.7 mice(right). (C) Measurement of CSD latencies did not show any altered CSD propagation velocity in hGFAP transgenic (n � 5) com-pared with nontransgenic littermates (n � 6; p � 0.82). **p 0.01. Scale bars � 500�m (in 40� original magnification);100�m (in 200� original magnification).
360 Annals of Neurology Vol 66 No 3 September 2009

elination (of fibers smaller than 0.4�m), as well as anincreased number of myelinated axons within the neo-cortical layer II-III of the cerebral cortex of Nrg1 typeI transgenic mice (Fig 4A), whereas oligodendroglialprocesses and astroglial cell components appeared un-altered.29 Interestingly, and as postulated by our previ-ous results, Nrg1 type I transgenic mice exhibited dra-matically decreased CSD velocity (2.27 � 0.16mm/min; n � 10) compared with their nontransgeniclittermates (4.09 � 0.59mm/min; n � 6; p 0.01;see Fig 4B).
Role of Cortical Inflammation for Cortical SpreadingDepression Propagation in the Presence or Absence ofCortical DemyelinationAcute white matter MS lesions are further associatedwith cellular and humoral immune components anddisrupted blood–brain barrier.34 The cuprizone animalmodel, however, does not reflect these additional his-topathological hallmarks of MS lesions. Therefore, wetested whether inflammatory cortical demyelinationthat is associated with transient blood–brain barrierbreakdown causes altered CSD propagation. We there-fore applied the autoimmune animal model of unilat-erally targeted cortical EAE lesions.26
Three days after cytokine induction, the ipsilateralcortex exhibited widespread subpial demyelination withnumerous perivascular demyelinated spots and corticalinfiltrates of macrophages/microglia within the affectedhemisphere in rMOG-primed animals (Fig 5A; seeSupplementary Fig 2). Cortical demyelination wasmost extensive at day 3 after cytokine injection, butsubpial demyelination was still evident at day 8 aftercytokine injection (see Figs 5A, B). At day 15 afterlesion induction, cortical myelin was substantially re-
stored. Similarly, infiltrates of macrophages/microgliapeaked at day 3 after cytokine injection but markedlydeclined at days 8 and 15 after cytokine injection (seeFig 5C). At all investigated time points, cortical demy-elination and inflammatory activity were confined tothe ipsilateral cortex of stereotactic injection.
Together with these histological observations, CSDwas significantly accelerated within the affected ipsilat-eral hemisphere as compared with the control site atdays 3 (�v(EAE-control) � 0.39 � 0.15mm/min; n �12; p 0.05) and 8 (�v(EAE-control) � 0.51 �0.21mm/min; n � 8; p 0.05) after lesion induction(see Fig 5D). Two weeks after lesion induction, whencortical lesions already showed a substantial restorationof cortical myelin, CSD propagation appeared unal-tered as compared with the nonaffected contralateralhemisphere (�v(EAE-control) � 0.09 � 0.13mm/min;n � 7; p � 0.57; see Fig 5D).
To further test whether inflammation per se, in theabsence of demyelination, would be sufficient for theobserved CSD alteration, we performed experimentsinjecting cytokines into the cortex of animals previ-ously primed against guinea pig MBP72-85. MBP72-85
immunization causes a monophasic inflammatory CNSdisease in Lewis rats, reminiscent of acute disseminatedencephalomyelitis, which is characterized by denseperivascular T-cell and macrophage infiltrates, and isfurther accompanied by a blood–brain barrier break-down.35,36 But in contrast with rMOG-induced EAE,inflammatory lesions after MBP72-85 immunization arenot associated with extensive demyelination.37 Corticalcytokine injection of previously MBP72-85-primed ani-mals induced accumulation of ED1 macrophages/mi-croglia cells within the ipsilateral cerebral cortex withsimilar density as in rMOG-primed animals 3 days af-ter cytokine injection but did not result in widespreaddemyelination (see Figs 5E–G). Nevertheless, CSD ve-locity was unaltered in the inflamed hemisphere com-pared with the contralateral cortex (�v(EAE-control) ��0.05 � 0.21mm/min; n � 9; p � 0.77) 3 days aftercytokine injection (see Fig 5H). Similarly, 8 days aftercytokine injection, CSD was unchanged comparedwith the contralateral cortex (data not shown), but in aconsiderable proportion of the animals, histological ex-amination demonstrated widespread inflammatory in-filtrates that were not only restricted to the ipsilateralside of injection but also involved the contralateralbrain. Disseminated lesions involving also the spinalcord were furthermore accompanied by characteristicparaplegic clinical signs.38 Because we measured thedifference in CSD velocity between ipsilateral and con-tralateral (control) cortex in this experimental para-digm, we cannot exclude that this might have disguiseda difference in CSD velocity at this measurement timepoint. In contrast, results obtained 3 days after cyto-kine injection with MBP-immunized animals strongly
Fig 4. Cortical spreading depression (CSD) velocity in hyper-myelinated neuregulin-1 (Nrg1) overexpressing animals. (A)Representative electron microscopic picture of cerebral cortex ofnontransgenic (non tg) littermate and mice overexpressingNrg1 (Nrg1 type I tg). Of note, more myelinated axons withthicker myelin sheaths are present in the cerebral cortex ofNrg1 type I overexpressing animals. (B) CSD measurementsshowed strongly reduced CSD velocity in Nrg1 type I–overex-pressing animals (n � 10) compared with their wild-typelittermates (n � 6). **p 0.05. Scale bar � 1�m.
Merkler et al: Spreading Depression and Myelin 361
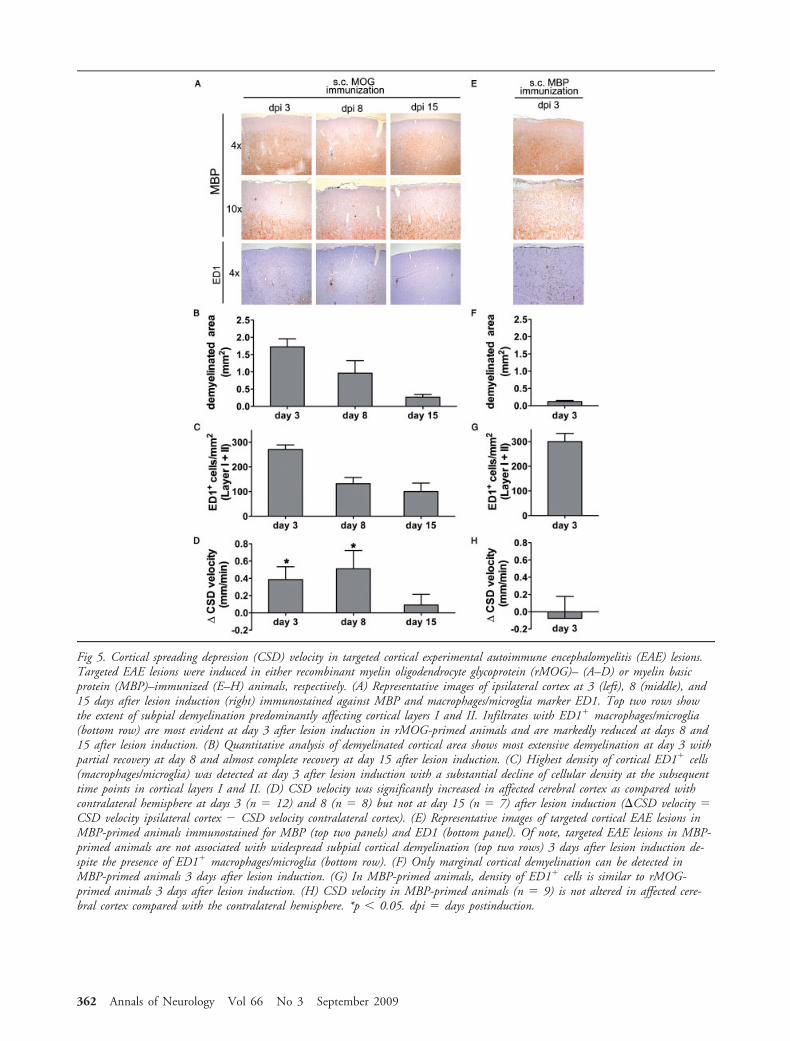
Fig 5. Cortical spreading depression (CSD) velocity in targeted cortical experimental autoimmune encephalomyelitis (EAE) lesions.Targeted EAE lesions were induced in either recombinant myelin oligodendrocyte glycoprotein (rMOG)– (A–D) or myelin basicprotein (MBP)–immunized (E–H) animals, respectively. (A) Representative images of ipsilateral cortex at 3 (left), 8 (middle), and15 days after lesion induction (right) immunostained against MBP and macrophages/microglia marker ED1. Top two rows showthe extent of subpial demyelination predominantly affecting cortical layers I and II. Infiltrates with ED1 macrophages/microglia(bottom row) are most evident at day 3 after lesion induction in rMOG-primed animals and are markedly reduced at days 8 and15 after lesion induction. (B) Quantitative analysis of demyelinated cortical area shows most extensive demyelination at day 3 withpartial recovery at day 8 and almost complete recovery at day 15 after lesion induction. (C) Highest density of cortical ED1 cells(macrophages/microglia) was detected at day 3 after lesion induction with a substantial decline of cellular density at the subsequenttime points in cortical layers I and II. (D) CSD velocity was significantly increased in affected cerebral cortex as compared withcontralateral hemisphere at days 3 (n � 12) and 8 (n � 8) but not at day 15 (n � 7) after lesion induction (�CSD velocity �CSD velocity ipsilateral cortex � CSD velocity contralateral cortex). (E) Representative images of targeted cortical EAE lesions inMBP-primed animals immunostained for MBP (top two panels) and ED1 (bottom panel). Of note, targeted EAE lesions in MBP-primed animals are not associated with widespread subpial cortical demyelination (top two rows) 3 days after lesion induction de-spite the presence of ED1 macrophages/microglia (bottom row). (F) Only marginal cortical demyelination can be detected inMBP-primed animals 3 days after lesion induction. (G) In MBP-primed animals, density of ED1 cells is similar to rMOG-primed animals 3 days after lesion induction. (H) CSD velocity in MBP-primed animals (n � 9) is not altered in affected cere-bral cortex compared with the contralateral hemisphere. *p 0.05. dpi � days postinduction.
362 Annals of Neurology Vol 66 No 3 September 2009

suggest that the acceleration of CSD does not correlatewith inflammation.
DiscussionOur data suggest that cortical myelin constitutes animportant determinant for the propagation of CSD.Hypermyelinated cerebral cortex was associated withslower CSD propagation speed, whereas cortical demy-elination, being caused by toxic or autoimmune pro-cesses, caused acceleration. In contrast, induction ofcortical astrogliosis or inflammation in the absence ofpronounced cortical demyelination was not associatedwith an alteration in CSD propagation. Furthermore,cuprizone-treated animals that were allowed to remy-elinate showed a restoration of CSD propagation ve-locity.
The pathophysiological hallmark of CSD consists ofa substantial redistribution of ions between intracellularand extracellular compartments that causes a transientdepolarization of neurons and glial cells.3 It is thereforesomewhat surprising that CSD propagation inverselycorrelated with cortical myelin, in contrast with thenerve conduction velocity, which is known to posi-tively correlate with the thickness of myelin sheets un-der physiological conditions. This apparently surprisingfinding indicates that the two processes, CSD andnerve impulse propagation, propagate by means of dis-tinct mechanisms, which is supported by the differentranges of propagation velocities, found in the twocases. Nevertheless, first indications that myelin mayaffect CSD derive from nutritional studies: Malnutri-tion of young rats was shown to cause dysmyelinationwithin the CNS.39 Interestingly, malnourished ratsearly in life displayed greater CSD velocities than well-nourished control animals.40 It was thus speculatedthat deficient myelination may account for this obser-vation, but other causalities such as altered cell-packingdensity, neuron-to-glia ratio, or synaptic neurotrans-mitters were also envisaged.
Several studies suggest that CSD is associated withslow Ca2 waves in astrocytes for which gap junctionsare essential.41,42 Interestingly, gene-expression analysisin hGFAP-transgenic mice demonstrated that astroglio-sis observed in these animals was associated with im-paired expression of various genes involved in calciumsignaling, including astroglial gap junctions proteinConnexin-26 and -30.43 Nonetheless, our measure-ments did not show any altered CSD propagation ve-locity in hGFAP-transgenic animals, indicating that thealtered expression of astroglial gap junction proteins inGFAP-transgenic astrocytes had limited impact, if any,on CSD velocity. In line with this observation, previ-ous reports showed that an inhibition of astrocyticCa2 waves does not depress CSD propagation.44 Itwas furthermore speculated that astrocytic Ca2 wavesare probably involved in the initiation of CSD and
contribute to the homeostatic failure observed duringCSD, but other factors such as K, glutamate, andpurinergic receptors are necessary for sustained propa-gation.45 This concept stands in some contrast withthe recent notion that astrocytic inactivation ofConnexin-43 was associated with an accelerated hip-pocampal CSD in mice.46 It might therefore be possi-ble that distinct mechanisms become differentially in-volved in CSD propagation, depending on theanatomic localization envisaged. Whether and how cor-tical myelin modulates CSD propagation has not beeninvestigated before. Our data, however, favor a directand active involvement of myelin components in thepropagation of CSD instead of an indirect (eg, neuro-toxic) effect as a consequence of demyelination. Thisassumption is based on the findings that CSD velocityis modulated dichotomously, namely, acceleration indemyelinated and deceleration in hypermyelinated an-imals. Furthermore, hypermyelination in Nrg1 trans-genic animals is not accompanied by increased oligoden-droglial cell density.29 It appears therefore reasonablethat the cytosolic compartment of compact myelin ap-pears to modulate CSD. Several studies have demon-strated the presence of connexins47 and AMPA (�-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid)/kainate receptors48,49 in mature myelin, suggesting aninvolvement in signal transduction and translocation ofions and molecules, respectively. Furthermore, it wasshown that CSD propagation could be blocked by glu-tamate antagonists.50 It is therefore conceivable thatsuch a blockade may at least partially also act via mye-linic N-methyl-D-aspartate receptors.51 Other studiessuggested a diffusion reaction process underlying CSDpropagation (for review, see Somjen3), a hypothesis thatwould also be consistent with the dichotomous associa-tion of myelin content and CSD velocity found in ourstudy.
For MS, the most common demyelinating disease ofthe CNS, it has become clear in recent years that thecerebral cortex can become extensively demyelinatedespecially in chronic progressive MS.52 Magnetic reso-nance imaging studies suggested an association of cog-nitive impairments with cortical demyelination inMS.53,54 However, it is still a matter of debate to whatextent cortical demyelination contributes to functionaldeficits in human MS patients.23 Because in our tar-geted rMOG-induced EAE model demyelination ispredominantly restricted to the cerebral cortex, alter-ation of CSD propagation is most likely caused bychanges in the local environment of the affected cortex.Thus, cortical demyelination may not only precipitateCSD acceleration but also cause a general alteration incortical excitability, for example, by activity-related im-paired buffering of extracellular potassium concentra-tions and glutamate-mediated depolarization.9,10 Clin-ically, increased cortical excitability, as determined by
Merkler et al: Spreading Depression and Myelin 363

transcranial magnetic stimulation, was noted in MS pa-tients in the relapsing phase of the disease.55 Moreover,a recent magnetic resonance imaging study, applying adouble inversion recovery sequence, demonstrated anassociation of extensive cortical involvement in MSwith the occurrence of epilepsy.22
Nevertheless, although CSD is thought to be a hall-mark of migraine, to the best of our knowledge, noassociation between cortical involvement in MS andmigraine has been reported so far, and whether MSpatients have increased risk to suffer from migraine isstill a matter of debate. Some magnetic resonance im-aging studies suggested, however, a link between brain-stem lesions and migraine in MS.56,57 Although thismay hold true, one has to keep in mind that corticallesions are unlikely to be detected by conventional T2-weighted magnetic resonance images such as those ap-plied in most of these studies, and that cortical lesionsmay thus have been neglected.58 Furthermore, mi-graine is sometimes associated with T2 lesions on mag-netic resonance imaging.59 Future studies are thereforeneeded to unravel a potential association between cor-tical involvement in MS and probably migraine.
In summary, our study provides several lines of ex-perimental evidence that the extent of cortical myeli-nation inversely correlates with CSD propagation ve-locity. Thus, cortical demyelination, as can be noted inchronic MS and in other demyelinating diseases of theCNS,60 may represent a pathological substrate for al-tered CSD.
This study was supported by the SFB TRR43 (to D.M. and W.B.)and Walter and Ilse Rose Stiftung (T298/14375/2004) (to W.P.), aresearch fellowship from the Brazilian National Research CouncilCNPq (302565/2007-8, R.C.A.G.), FINEP/IBN-NET (01.06.0842-00), MCT-CNPq/MS-SCTIE-DECIT (17/2006), and the Del Mar-mol Foundation (M.W.S.).
We thank Prof A. Messing for kindly providing thehGFAP transgenic mice. We also thank M. Scheden-sack for expert technical assistance.
References1. Leao A. Spreading depression of activity in the cerebral cortex.
J Neurophysiol 1944:359–390.2. Sugaya E, Takato M, Noda Y. Neuronal and glial activity dur-
ing spreading depression in cerebral cortex of cat. J Neuro-physiol 1975;38:822–841.
3. Somjen GG. Mechanisms of spreading depression and hypoxicspreading depression-like depolarization. Physiol Rev 2001;81:1065–1096.
4. Hopwood SE, Parkin MC, Bezzina EL, et al. Transient changesin cortical glucose and lactate levels associated with peri-infarctdepolarisations, studied with rapid-sampling microdialysis.J Cereb Blood Flow Metab 2005;25:391–401.
5. Busch E, Gyngell ML, Eis M, et al. Potassium-induced corticalspreading depressions during focal cerebral ischemia in rats:contribution to lesion growth assessed by diffusion-weightedNMR and biochemical imaging. J Cereb Blood Flow Metab1996;16:1090–1099.
6. Petzold GC, Haack S, von Bohlen Und Halbach O, et al. Ni-tric oxide modulates spreading depolarization threshold in thehuman and rodent cortex. Stroke 2008;39:1292–1299.
7. Mayevsky A, Doron A, Manor T, et al. Cortical spreading de-pression recorded from the human brain using a multiparamet-ric monitoring system. Brain Res 1996;740:268–274.
8. Fabricius M, Fuhr S, Bhatia R, et al. Cortical spreading depres-sion and peri-infarct depolarization in acutely injured humancerebral cortex. Brain 2006;129:778–790.
9. Liebetanz D, Fregni F, Monte-Silva KK, et al. After-effects oftranscranial direct current stimulation (tDCS) on corticalspreading depression. Neurosci Lett 2006;398:85–90.
10. Fregni F, Liebetanz D, Monte-Silva KK, et al. Effects of trans-cranial direct current stimulation coupled with repetitive elec-trical stimulation on cortical spreading depression. Exp Neurol2007;204:462–466.
11. Takano T, Tian GF, Peng W, et al. Cortical spreading depres-sion causes and coincides with tissue hypoxia. Nat Neurosci2007;10:754–762.
12. Rogawski MA. Common pathophysiologic mechanisms in mi-graine and epilepsy. Arch Neurol 2008;65:709–714.
13. Margineanu DG, Klitgaard H. Brivaracetam inhibits spreadingdepression in rat neocortical slices in vitro. Seizure 2009;18:453–456.
14. Berger M, Speckmann EJ, Pape HC, Gorji A. Spreading de-pression enhances human neocortical excitability in vitro.Cephalalgia 2008;28:558–562.
15. van den Maagdenberg AM, Pietrobon D, Pizzorusso T, et al. ACacna1a knockin migraine mouse model with increased suscep-tibility to cortical spreading depression. Neuron 2004;41:701–710.
16. Spatt J, Chaix R, Mamoli B. Epileptic and non-epileptic sei-zures in multiple sclerosis. J Neurol 2001;248:2–9.
17. Ghezzi A, Montanini R, Basso PF, et al. Epilepsy in multiplesclerosis. Eur Neurol 1990;30:218–223.
18. Engelsen BA, Gronning M. Epileptic seizures in patients withmultiple sclerosis. Is the prognosis of epilepsy underestimated?Seizure 1997;6:377–382.
19. Moreau T, Sochurkova D, Lemesle M, et al. Epilepsy in pa-tients with multiple sclerosis: radiological-clinical correlations.Epilepsia 1998;39:893–896.
20. Kidd D, Barkhof F, McConnell R, et al. Cortical lesions inmultiple sclerosis. Brain 1999;122(pt 1):17–26.
21. Bo L, Vedeler CA, Nyland HI, et al. Subpial demyelination inthe cerebral cortex of multiple sclerosis patients. J NeuropatholExp Neurol 2003;62:723–732.
22. Calabrese M, De Stefano N, Atzori M, et al. Extensive corticalinflammation is associated with epilepsy in multiple sclerosis.J Neurol 2008;255:581–586.
23. Kutzelnigg A, Lassmann H. Cortical demyelination in multiplesclerosis: a substrate for cognitive deficits? J Neurol Sci 2006;245:123–126.
24. Skripuletz T, Lindner M, Kotsiari A, et al. Cortical demyelina-tion is prominent in the murine cuprizone model and is strain-dependent. Am J Pathol 2008;172:1053–1061.
25. Messing A, Head MW, Galles K, et al. Fatal encephalopathywith astrocyte inclusions in GFAP transgenic mice. Am J Pathol1998;152:391–398.
26. Merkler D, Ernsting T, Kerschensteiner M, et al. A new focalEAE model of cortical demyelination: multiple sclerosis-like le-sions with rapid resolution of inflammation and extensive re-myelination. Brain 2006;129:1972–1983.
364 Annals of Neurology Vol 66 No 3 September 2009

27. Liebetanz D, Merkler D. Effects of commissural de- and remy-elination on motor skill behaviour in the cuprizone mousemodel of multiple sclerosis. Exp Neurol 2006;202:217–224.
28. Bruck W, Bitsch A, Kolenda H, et al. Inflammatory centralnervous system demyelination: correlation of magnetic reso-nance imaging findings with lesion pathology. Ann Neurol1997;42:783–793.
29. Brinkmann BG, Agarwal A, Sereda MW, et al. Neuregulin-1/ErbB signaling serves distinct functions in myelination of theperipheral and central nervous system. Neuron 2008;59:581–595.
30. Merkler D, Boretius S, Stadelmann C, et al. Multicontrast MRIof remyelination in the central nervous system. NMR Biomed2005;18:395–403.
31. Komoly S, Hudson LD, Webster HD, Bondy CA. Insulin-likegrowth factor I gene expression is induced in astrocytes duringexperimental demyelination. Proc Natl Acad Sci U S A 1992;89:1894–1898.
32. Hiremath MM, Saito Y, Knapp GW, et al. Microglial/macrophage accumulation during cuprizone-induced demyeli-nation in C57BL/6 mice. J Neuroimmunol 1998;92:38–49.
33. Michailov GV, Sereda MW, Brinkmann BG, et al. Axonalneuregulin-1 regulates myelin sheath thickness. Science 2004;304:700–703.
34. Hemmer B, Nessler S, Zhou D, et al. Immunopathogenesis andimmunotherapy of multiple sclerosis. Nat Clin Pract Neurol2006;2:201–211.
35. Tenembaum S, Chitnis T, Ness J, Hahn JS. Acute disseminatedencephalomyelitis. Neurology 2007;68:S23–S36.
36. Menge T, Hemmer B, Nessler S, et al. Acute disseminatedencephalomyelitis: an update. Arch Neurol 2005;62:1673–1680.
37. Zhou D, Srivastava R, Nessler S, et al. Identification of apathogenic antibody response to native myelin oligodendrocyteglycoprotein in multiple sclerosis. Proc Natl Acad Sci U S A2006;103:19057–19062.
38. Weissert R, Svenningsson A, Lobell A, et al. Molecular and ge-netic requirements for preferential recruitment of TCRBV8S2T cells in Lewis rat experimental autoimmune encephalomyelitis.J Immunol 1998;160:681–690.
39. Almeida MF, Silveira AC, Guedes RC, et al. Quantitative ul-trastructural evidence of myelin malformation in optic nerves ofrats submitted to a multideficient diet. Nutr Neurosci 2005;8:91–99.
40. Do Monte-Silva KK, Assis FL, Leal GM, Guedes RC.Nutrition-dependent influence of peripheral electrical stimula-tion during brain development on cortical spreading depressionin weaned rats. Nutr Neurosci 2007;10:187–194.
41. Chuquet J, Hollender L, Nimchinsky EA. High-resolution invivo imaging of the neurovascular unit during spreading depres-sion. J Neurosci 2007;27:4036–4044.
42. Nedergaard M, Cooper AJ, Goldman SA. Gap junctions arerequired for the propagation of spreading depression. J Neuro-biol 1995;28:433–444.
43. Hagemann TL, Gaeta SA, Smith MA, et al. Gene expressionanalysis in mice with elevated glial fibrillary acidic protein andRosenthal fibers reveals a stress response followed by glial acti-vation and neuronal dysfunction. Hum Mol Genet 2005;14:2443–2458.
44. Peters O, Schipke CG, Hashimoto Y, Kettenmann H. Differentmechanisms promote astrocyte Ca2 waves and spreading de-pression in the mouse neocortex. J Neurosci 2003;23:9888–9896.
45. Martins-Ferreira H, Nedergaard M, Nicholson C. Perspectiveson spreading depression. Brain Res 2000;32:215–234.
46. Theis M, Jauch R, Zhuo L, et al. Accelerated hippocampalspreading depression and enhanced locomotory activity in micewith astrocyte-directed inactivation of connexin43. J Neurosci2003;23:766–776.
47. Kamasawa N, Sik A, Morita M, et al. Connexin-47 andconnexin-32 in gap junctions of oligodendrocyte somata, mye-lin sheaths, paranodal loops and Schmidt-Lanterman incisures:implications for ionic homeostasis and potassium siphoning.Neuroscience 2005;136:65–86.
48. Brand-Schieber E, Werner P. AMPA/kainate receptors inmouse spinal cord cell-specific display of receptor subunits byoligodendrocytes and astrocytes and at the nodes of Ranvier.Glia 2003;42:12–24.
49. Li S, Stys PK. Mechanisms of ionotropic glutamate receptor-mediated excitotoxicity in isolated spinal cord white matter.J Neurosci 2000;20:1190–1198.
50. Lauritzen M, Rice ME, Okada Y, Nicholson C. Quisqualate,kainate and NMDA can initiate spreading depression in theturtle cerebellum. Brain Res 1988;475:317–327.
51. Micu I, Jiang Q, Coderre E, et al. NMDA receptors mediatecalcium accumulation in myelin during chemical ischaemia.Nature 2006;439:988–992.
52. Kutzelnigg A, Lucchinetti CF, Stadelmann C, et al. Corticaldemyelination and diffuse white matter injury in multiple scle-rosis. Brain 2005;128:2705–2712.
53. Rovaris M, Filippi M, Minicucci L, et al. Cortical/subcorticaldisease burden and cognitive impairment in patients with mul-tiple sclerosis. AJNR Am J Neuroradiol 2000;21:402–408.
54. Geurts JJ, Bo L, Pouwels PJ, et al. Cortical lesions in multiplesclerosis: combined postmortem MR imaging and histopathol-ogy. AJNR Am J Neuroradiol 2005;26:572–577.
55. Caramia MD, Palmieri MG, Desiato MT, et al. Brain excitabil-ity changes in the relapsing and remitting phases of multiplesclerosis: a study with transcranial magnetic stimulation. ClinNeurophysiol 2004;115:956–965.
56. Gee JR, Chang J, Dublin AB, Vijayan N. The association ofbrainstem lesions with migraine-like headache: an imagingstudy of multiple sclerosis. Headache 2005;45:670–677.
57. Tortorella P, Rocca MA, Colombo B, et al. Assessment of MRIabnormalities of the brainstem from patients with migraine andmultiple sclerosis. J Neurol Sci 2006;244:137–141.
58. Geurts JJ, Pouwels PJ, Uitdehaag BM, et al. Intracortical le-sions in multiple sclerosis: improved detection with 3D dou-ble inversion-recovery MR imaging. Radiology 2005;236:254 –260.
59. Swartz RH, Kern RZ. Migraine is associated with magnetic res-onance imaging white matter abnormalities: a meta-analysis.Arch Neurol 2004;61:1366–1368.
60. Moll NM, Rietsch AM, Ransohoff AJ, et al. Cortical demyeli-nation in PML and MS: Similarities and differences. Neurology2008;70:336–343.
Merkler et al: Spreading Depression and Myelin 365
Copyright © 2022 FDOKUMEN



















