
Probabilistic prediction of protein–protein interactions from the protein sequences
12
Computers in Biology and Medicine 36 (2006) 1143 – 1154 www.intl.elsevierhealth.com/journals/cobm Probabilistic prediction of protein–protein interactions from the protein sequences Arunkumar Chinnasamy a , Ankush Mittal b, ∗ , Wing-Kin Sung c a Department of Computer Science, National University of Singapore, Singapore 117543 b Department of Electronics and Computer Engineering, Indian Institute ofTechnology, Roorkee, India 247667 c Department of Computer Science, National University of Singapore, Singapore 117543 Abstract Prediction of protein–protein interactions is very important for several bioinformatics tasks though it is not a straightforward problem. In this paper, employing only protein sequence information, a framework is presented to predict protein–protein interactions using a probabilistic-based tree augmented naïve (TAN) Bayesian network. Our framework also provides a confidence level for every predicted interaction, which is useful for further analysis by the biologists. The framework is applied to the yeast interaction datasets for predicting interactions and it is shown that our framework gives better performance than support vector machine (SVM). The framework is implemented as a webserver and is available for prediction. 2005 Elsevier Ltd. All rights reserved. Keywords: TAN Bayesian classifier; Protein–protein interaction; Protein feature extraction; Machine learning 1. Introduction Proteomics is the systematic study of the proteins in different cell types and different organisms. The main goal of proteomics is to predict the structures, interactions and functions of the proteins [1]. Human genome project alone results in difficult problem of understanding the structures, interactions and functions of approximately more than half a million human proteins encoded by some 30,000 genes. ∗ Corresponding author. E-mail addresses: [email protected] (A. Chinnasamy), [email protected] (A. Mittal), [email protected] (W.-K. Sung). 0010-4825/$ - see front matter 2005 Elsevier Ltd. All rights reserved. doi:10.1016/j.compbiomed.2005.09.005
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Probabilistic prediction of protein–protein interactions from the protein sequences

Computers in Biology and Medicine 36 (2006) 1143–1154www.intl.elsevierhealth.com/journals/cobm
Probabilistic prediction of protein–protein interactions from theprotein sequences
Arunkumar Chinnasamya, Ankush Mittalb,∗, Wing-Kin Sungc
aDepartment of Computer Science, National University of Singapore, Singapore 117543bDepartment of Electronics and Computer Engineering, Indian Institute of Technology, Roorkee, India 247667
cDepartment of Computer Science, National University of Singapore, Singapore 117543
Abstract
Prediction of protein–protein interactions is very important for several bioinformatics tasks though it is not astraightforward problem. In this paper, employing only protein sequence information, a framework is presented topredict protein–protein interactions using a probabilistic-based tree augmented naïve (TAN) Bayesian network. Ourframework also provides a confidence level for every predicted interaction, which is useful for further analysis bythe biologists. The framework is applied to the yeast interaction datasets for predicting interactions and it is shownthat our framework gives better performance than support vector machine (SVM). The framework is implementedas a webserver and is available for prediction.� 2005 Elsevier Ltd. All rights reserved.
Keywords: TAN Bayesian classifier; Protein–protein interaction; Protein feature extraction; Machine learning
1. Introduction
Proteomics is the systematic study of the proteins in different cell types and different organisms.The main goal of proteomics is to predict the structures, interactions and functions of the proteins [1].Human genome project alone results in difficult problem of understanding the structures, interactionsand functions of approximately more than half a million human proteins encoded by some 30,000 genes.
∗ Corresponding author.E-mail addresses: [email protected] (A. Chinnasamy), [email protected] (A. Mittal),
[email protected] (W.-K. Sung).
0010-4825/$ - see front matter � 2005 Elsevier Ltd. All rights reserved.doi:10.1016/j.compbiomed.2005.09.005

1144 A. Chinnasamy et al. / Computers in Biology and Medicine 36 (2006) 1143–1154
This task is extensive, complicated, time consuming, and requiring multi-methodological efforts frommulti-disciplinary researchers. These factors collectively led to naming the first decade of this millenniumas “Decade of Proteomics”, following the “Decade of Genomics”.
Prediction of protein–protein interaction is a significant problem since it helps to understand the basisof cellular operations and other functions. It has been shown that proteins with similar functions are morelikely to interact [2]. If the function of one protein is known then the function of its binding partners islikely to be related. This helps to understand the functional roles of unannotated protein by knowing itsinteraction partners. Drug discovery is another area where protein–protein interaction prediction playsan important role.
The experimental techniques for finding protein–protein interactions such as yeast two-hybrid-basedmethods, mass spectroscopy protein chips and hybrid methods have several limitations which stimulatedthe research in computational way of predicting the interactions [3,4]. The study by Valencia and Pazos[5] describes several well-known computational methods of predicting protein–protein interaction. Animportant computation method based on phylogenetic profiles uses similarity of genes to predict theinteractions [6,7], where the similarity of genes is calculated based on presence or absence of genesin different species. Another method based on conservation of gene neighborhoods was employed inbacteria for prediction based on adjacency of genes in different species [8]. However, both methods needcomplete genomes for many species. Gene fusion method traces a single protein in other domains wherethe interacting proteins are same at some point [9]. However, this method is only applicable to proteinswith shared domains. Gomez et al. [10] proposed a solution based on domain interactions. They group theproteins based on conserved domains. Then, the interaction probability of a pair of proteins is estimatedby the known protein–protein domain interactions.
Our method predicts interactions based on protein sequences only. This work is similar to severalprevious works [11–13]. Bock and Gough [13] used the method of shuffling the sequences to get negativeexamples and the method of converting sequences of heterogeneous length to homogeneous features. Theyconstructed homogeneous features based on linear interpolation and then used SVM-based framework forprediction. Bandyopadhyay et al. [12] used multiple sequence alignment method for constructing featurevectors of homogeneous length. However, less number of negative examples was used in their experimentand conclusive results could not be obtained. Thus, there exists a lacuna for a framework that performsprotein–protein interactions given two protein sequences without making restrictive assumptions and withhigh accuracy. Our contribution lies in proposing such a framework.
This paper presents a framework with discretization of feature space and tree-augmented network(TAN) Bayesian classifier as foundation to address the problem. TAN Bayesian classifier is simple totrain, and yet it models the possibility of dependency relationship amongst the attributes that is notpermitted in naïve Bayesian models. There are several theoretical advantages of Bayesian network thatmakes it especially relevant to be employed in interaction prediction:
(a) Bayesian networks formalism allows the expert to input his or her model that is based on alreadyknown conclusions which may or may not be fully accurate. One can then refine the model with thestatistical data.
(b) Since the output of the Bayesian Networks is probabilities (i.e., likelihood), it facilitates furtheranalysis for the biologists.
(c) In making this part of a larger system based on several expert judges, it is easy to combine probabilitiesfrom each of the subsystem.

A. Chinnasamy et al. / Computers in Biology and Medicine 36 (2006) 1143–1154 1145
The rest of the paper is organized as follows. Section 2 presents a brief description of Bayesianclassifiers and TAN. In Section 3, the details of dataset and feature vector extraction are presented. Section4 describes our framework for prediction of protein–protein interaction followed by a discussion on theresults in Section 5. Section 6 briefly describes about our webserver for prediction. Finally, conclusions,contributions and scope for future work are presented.
2. Bayesian classifiers
Bayesian networks are directed cyclic graphs (DAG) which combine both statistical and graph theoryfor representing conditional independencies between a set of random variables in a probabilistic model[14]. A directed edge A → B indicates the causal relationship (A causing B) and thus Bayesian networksare quite intuitive. Optimal classifications can be achieved by reasoning about these probabilities alongwith observed data [14]. The Bayesian classifier was reported as effective classifier with high predictionaccuracy [16] as compared to other classifiers. In the study [17], the region where Bayesian classifiers areoptimal was provided as well as further evidence for the good performance. This classifier learns fromtraining data, the conditional probability of each attribute Ai given the class label C. Then classificationis done by applying Bayesian rule to compute the probability of C given the particular instance ofA1, A2, . . . , An and predicting the class with the highest probability.
2.1. TAN Bayesian classifier
Structural relationship among the attributes is important for the Bayesian network classifier to constructthe relationship amongst various nodes. However, no clear structural relationship is known amongst theattributes and the class (though it seems plausible in the future). With less number of feature vectors andbiological knowledge, structure learning is impossible. Therefore, we chose TAN Bayesian classifier [16]rather than Bayesian network classifier as it seems more relevant to the problem considering the featurevector properties and relations. TAN allows dependency relationship amongst attributes in addition tothe class and the attributes. In other words, it has more generalized representation than naïve Bayesianclassifier as it allows a causal relationship between two attributes.
TAN Bayesian classifier learns a network in which each attribute has at most two parents. One parent isthe class while the optional parent can be any attribute. Attractive property of this classifier is that it learnsthe probabilities from the data in polynomial time. The inference is done on TAN Bayesian classifierfollowing a Bayesian rule as shown
P(Classk|A1, A2, . . . , An) = �P(Classk).
n∏
i=1
P(Ai |Classk, pa(Ai)), (1)
where � is normalization constant, assumed to be 0.5. P(Classk) is prior probability for class k. pa(Ai)
is the set of direct parents for node Ai . P(Ai |pa(Ai)) is estimated during the training phase of ouralgorithm.

1146 A. Chinnasamy et al. / Computers in Biology and Medicine 36 (2006) 1143–1154
3. Dataset and feature vectors extraction
To test the framework, we used the yeast interactions dataset which is collected from database ofinteracting proteins (DIP) [18]. At the time of doing the experiments, the yeast dataset contained 15,116interaction pairs, out of which 5522 interactions were validated by small-scale experiment, multipleexperiments and/or PVM validation. Hence, these 5522 interactions were extracted for the purpose ofexperiments. This composes the dataset of true positive interactions for the experiment. Negative dataset(protein pairs which do not interact with each other) is not available since there is no literature thatpublishes the negative dataset that are experimentally verified. In order to prepare the negative dataset,we reverse the sequence of one interacting protein in the interaction pairs. It has been shown that if asequence of one interacting partner is reversed or shuffled, then the probability of existence of interactionbetween these two is negligible [19]. Thus, the negative dataset is prepared by reversing the sequence ofthe right-side interacting partners. Final dataset contained 11,044 interactions, which is used for cross-validation experiments in our framework.
In order to perform empirical studies for the prediction, the amino acid sequence information ofheterogeneous length needs to be transformed into the feature vector information of homogeneous length.The feature vector representation should represent the sequence information responsible for the proteininteractions. From the past experiments it has been shown the hydrophobicity attribute is a good descriptorfor protein–protein interactions. Many studies confirm the role of hydrophobicity as a determinant ofprotein–protein interactions [20].
Feature vector extraction used in this work is similar to the one discussed in our earlier studies [22,23]for protein structure and fold prediction method except that we only consider the distributions. In ourearlier studies, feature vectors are constructed based on the following two steps:
(1) For each attribute, 20 amino acids are divided into three groups. For each protein sequence, everyamino acid is replaced by the index 1, 2, or 3 depending on its grouping. For example, proteinsequence KLLSHCLLVTLAAHLPAEFTPAV is replaced by 13322333323222322132232 based onthe attribute hydrophobicity division of amino acids.
(2) For each converted sequence computed in step 1, three descriptors “Composition” (C), “Transition”(T ), and “Distribution” (D), are calculated based on the definition given as follows.
Composition: Composition is calculated for each group based on the simple formula,
Ci = (ni/L)100, (2)
where Ci represents the percentage composition of each groupi , where ni represents total number ofgroupi residues in the sequences, and L represents the length of the sequence.
Transition: Transition (Ti,j ) is represented by the percentage frequency with which groupi is followedby groupj or groupj is followed by groupi , where i, j takes the values 1–3.
Distribution: Distribution descriptor Di,j is defined as the fractions of the entire sequence, where thefirst residue of a given group is located, and where 25%, 50%, 75%, and 100% of those are contained foreach of the three groups (j). Here i represents either 1%, 25%, 50%, 75% or 100%
The composition and transition features are omitted. Since the sequences are reversed, the differencebetween positive interactions and negative interactions is only the distribution of the groups. Compositionand transition features have the same values in both positive and negative interactions. Feature vectors

A. Chinnasamy et al. / Computers in Biology and Medicine 36 (2006) 1143–1154 1147
Table 1Division of amino acids based on hydrophobicity
Group 1 Group 2 Group 3
R, K, E, D, Q, N G,A, S, T, P,H,Y C, V, L, I, M, F, W
are constructed in two steps. In step 1, 20 amino acids are divided into three groups based on the attributehydrophobicity division of amino acids (Table 1). Hence, each protein sequence is replaced by a sequenceof ‘Group 1’, ‘Group 2’ and ‘Group 3’. Then, we calculate the fractions of the entire sequence, where thefirst residue of a given group is located, and where 25%, 50%, 75%, and 100% of groups are contained.Hence, each protein, sequence is replaced by attributes of size 15 and total dimensionality of training setis 31 (15 features of protein ‘A’ +15 features of protein ‘B’ +1 (class)).
3.1. Discretization
In our dataset, the feature vectors are of continuous nature. Though the Bayesian classifier supports bothcontinuous and discrete probability distributions [15], it was experimentally found that the continuousprobability distribution is not suitable for these datasets. In protein sequence analysis study, we didcomparative study between continuous and discrete methods. And the results for discrete method arearound 40% higher than the continuous one. The reason for poor continuous feature vector performanceis the nature of physiochemical properties. The data does not exist in the form of normal or other continuousprobability distributions and therefore normal assumption yield poor results. Therefore, we preprocesseddata by converting the continuous attribute data to discrete attribute data. There are several existingmethods that can be employed for discretization. One popular and simple approach is range discretization,which divides the interval (min(k) and max(k)) into n intervals of the same range.
However, this approach is not suitable for our problem. Some of the discretized partitions becomeover-populated while others remain empty leaving to poor discretization. In order to avoid this problem,we employ frequency-based discretization which partitions the attributes into intervals containing almostthe same number of instances. Several frequency based discretization methods were employed with ‘3’intervals, ‘4’ intervals, ‘5’ intervals, ‘7’ intervals and ‘10’ intervals. By experimental verification, themethod with ‘4’ intervals yielded better classification performance than other methods and therefore, itwas chosen.
4. Prediction of protein–protein interaction
An overview of a TAN Bayesian classifier for predicting protein–protein interaction is shown in Fig. 1.Experimentally found protein interactions obtained from Database of Interacting Proteins (DIP) are usedfor training the TAN Bayesian classifier. Interaction partners, ‘protein A’and ‘protein B’, are converted tofeature vectors based on hydrophobicity attribute. Discretization is performed to convert the continuousfeature vectors to discrete states. Finally, to predict if two proteins can interact, we pass their featurevector into the TAN Bayesian classifier and generate the prediction. The maximum probability of a class

1148 A. Chinnasamy et al. / Computers in Biology and Medicine 36 (2006) 1143–1154
O/P
Protein Sequence ‘A’
Protein Sequence ‘B’
HydrophobicityFeature Vectors
Extraction
HydrophobicityFeature Vectors
Extraction
TAN BayesianClassifier
Discretization And
Concatenation
Fig. 1. Overview of Bayesian network based protein–protein interaction prediction method.
PredictionClassA1
A2
An B1 B2
Bn
Fig. 2. Type I TAN Bayesian classifier structure.
PredictionClassA1
A2
AnB1 B2
Bn
Fig. 3. Type II TAN Bayesian classifier structure.
node in the classifier is given as the prediction result. We employed two different TAN Bayesian classifiersto predict the interactions. These two classifiers differ from each other by their network structures.
The structure of the first TAN Bayesian classifier is similar to the classifier structure used in structureprediction problem [22,23] with only distribution features used. Let A1, A2, . . . , An and B1, B2, . . . , Bn
be the feature vectors for protein sequences ‘A’ and ‘B’, respectively. Then node Ai has parental rela-tionship with Ai+1 and Bi has parental relationship with Bi+1 where ‘i’ takes the values 1, 2, . . . , n − 1.The structure of second TAN Bayesian classifier is different from first TAN Bayesian classifier as in thesecond TAN classifier, node Ai has parental relationship with Bi where ‘i’ take values 1, 2, . . . , n. Thestructures of two TAN Bayesian classifiers are shown in Figs. 2 and 3.
A. Chinnasamy et al. / Computers in Biology and Medicine 36 (2006) 1143–1154 1149
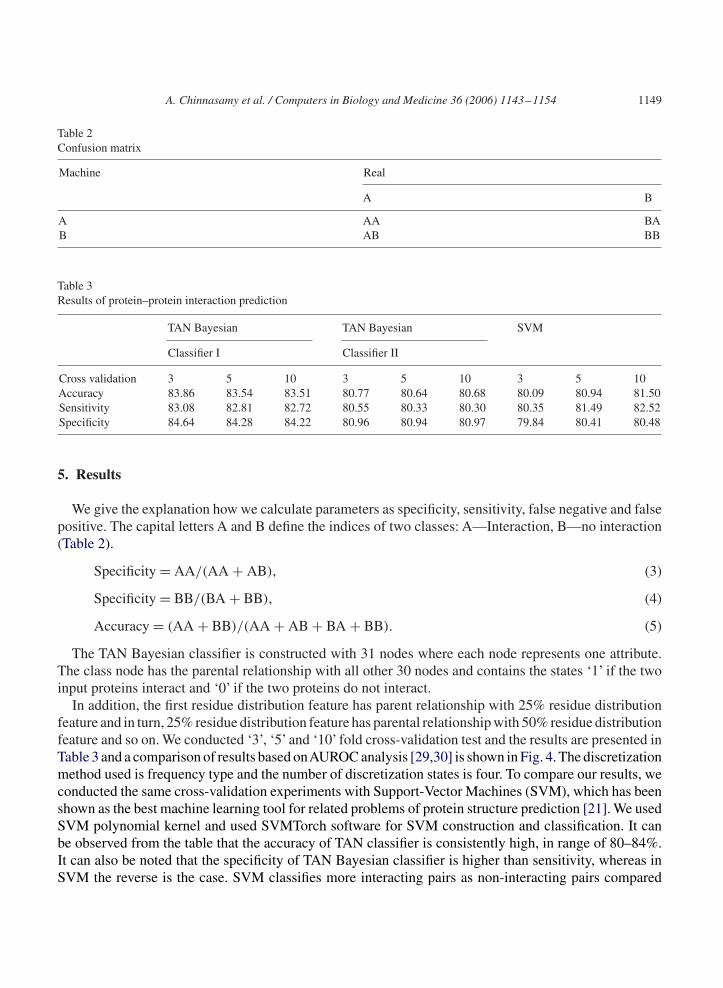
Table 2Confusion matrix
Machine Real
A B
A AA BAB AB BB
Table 3Results of protein–protein interaction prediction
TAN Bayesian TAN Bayesian SVM
Classifier I Classifier II
Cross validation 3 5 10 3 5 10 3 5 10Accuracy 83.86 83.54 83.51 80.77 80.64 80.68 80.09 80.94 81.50Sensitivity 83.08 82.81 82.72 80.55 80.33 80.30 80.35 81.49 82.52Specificity 84.64 84.28 84.22 80.96 80.94 80.97 79.84 80.41 80.48
5. Results
We give the explanation how we calculate parameters as specificity, sensitivity, false negative and falsepositive. The capital letters A and B define the indices of two classes: A—Interaction, B—no interaction(Table 2).
Specificity = AA/(AA + AB), (3)
Specificity = BB/(BA + BB), (4)
Accuracy = (AA + BB)/(AA + AB + BA + BB). (5)
The TAN Bayesian classifier is constructed with 31 nodes where each node represents one attribute.The class node has the parental relationship with all other 30 nodes and contains the states ‘1’ if the twoinput proteins interact and ‘0’ if the two proteins do not interact.
In addition, the first residue distribution feature has parent relationship with 25% residue distributionfeature and in turn, 25% residue distribution feature has parental relationship with 50% residue distributionfeature and so on. We conducted ‘3’, ‘5’ and ‘10’ fold cross-validation test and the results are presented inTable 3 and a comparison of results based onAUROC analysis [29,30] is shown in Fig. 4. The discretizationmethod used is frequency type and the number of discretization states is four. To compare our results, weconducted the same cross-validation experiments with Support-Vector Machines (SVM), which has beenshown as the best machine learning tool for related problems of protein structure prediction [21]. We usedSVM polynomial kernel and used SVMTorch software for SVM construction and classification. It canbe observed from the table that the accuracy of TAN classifier is consistently high, in range of 80–84%.It can also be noted that the specificity of TAN Bayesian classifier is higher than sensitivity, whereas inSVM the reverse is the case. SVM classifies more interacting pairs as non-interacting pairs compared

1150 A. Chinnasamy et al. / Computers in Biology and Medicine 36 (2006) 1143–1154
0.79
0.8
0.85
0.84
0.83
0.82
0.81
0.165 0.17 0.175 0.18 0.185 0.19 0.195 0.2
TAN I
TAN II
SVM
False Positive Rate
Tru
e P
osit
ive
Rat
e
Fig. 4. Comparison of classifier results based on AUROC approach.
to TAN Bayesian classifier which gives the high specificity of TAN than SVM. The AUROC measuresthe predictive power of a rating model. A model that discriminates perfectly between defaulters and non-defaulters would receive an AUROC of 100% [29,30]. The area under the ROC-Curve (AUROC) servesas a measure for the discriminatory power. The AUROC for TAN I is greater than other two classifierswhich is clearly evident from Fig. 4 and shows superiority of TAN I compared to other two classifiers.
In SVM, only a fixed (usually small) number of training-set vectors determine the parameters of thedecision rule and, since no probability density is estimated, it becomes highly sensitive to noise. Witha finite training sample, a high-dimensional feature space is almost empty [27] and many separators inSVM tool may perform well on the training data, but only few would generalize well. It has been shownby [28] that both linear SVMs and nonlinear SVMs perform badly in the situation of many irrelevantfeatures.
The performance of classification tools also depends on the distribution of the data. For instance, SVMis a useful tool to classify populations characterized by abrupt decreases in the density functions. However,in the real world, we do not have data with sharp linear boundaries. In other words, the data classes donot have clear boundaries amongst them such that they can be separated by a linear plane.
In addition to that, SVM provides less insight into the structure of the data space and it is difficult tohandle data containing missing entries. SVMs often serve as black boxes in classification and it is verydifficult for humans to comprehend how the decision is made. On the other hand, in Bayesian network,the inference rules and network structure can be easily read-off.
In addition to a binary classification, it is desirable for a classifier to output a scalar value showing itsbelief in classification. The Bayesian network classifiers give their output as probabilities but this is noteasy to obtain from SVM. In addition to that, SVM also leads to a large number of kernels (in spite ofsparsity) as it solves multi-class problem as pairs of two classes.

A. Chinnasamy et al. / Computers in Biology and Medicine 36 (2006) 1143–1154 1151
Fig. 5. Screen shot of the input page of interaction prediction webserver.
6. Web server for protein–protein interaction prediction
The utility of our prediction method help biological researchers and technologists to deal with interac-tion prediction related problems such as in drug discovery. The interaction prediction webserver imple-menting our method is provided at the following address: http://www-appn.comp.nus.edu.sg/∼bioinfo/PPinteraction/. The server is trained with the dataset containing 11,044 protein sets. The inputs to theprediction server are protein sequences ‘A’ and ‘B’ whose interaction needs to be predicted. A screenshotof the input page is shown in Fig. 5. A screenshot of the result page is shown in Fig. 6. The output reportswhether the two proteins interact or not, and the associated probability value of the prediction.
7. Conclusions and future work
In this paper, the problem of predicting the protein–protein interactions from the sequences only istackled. We addressed the problem of lack of negative dataset and validated our method with a largenumber of datasets. To help biologists, we designed a real time web server for interaction predictionproblem.
We applied feature vectors based on distribution of amino acids by considering hydrophobic index. Thetwo TAN Bayesian structures are based on biological knowledge and in agreement with the biologists.

1152 A. Chinnasamy et al. / Computers in Biology and Medicine 36 (2006) 1143–1154
Fig. 6. Screenshot of the output page of interaction prediction webserver.
In TAN II the hydrophobicity distribution of one protein depends on distribution on other protein. Bystructural analysis, it is found that the protein sequences are not necessarily linear in nature. But in TANI classifier, the node relationships 1%–25%; 25%–50%; 50%–75%; 75%–100% are more appropriate,because, certainly there are local linear relationship in the sequence. Compared to SVM, our modelsupports direct analysis of interaction by reading the probabilistic values. In addition, one can identifythe reverse relationship between hydrophobicity and interaction during docking studies.
We can extend this work for protein function prediction based on interactions. It has been accepted thatproteins are more likely to interact with other proteins which have the same functional class [2,25,26].By adding our framework of predicting interaction to these methods, the performance can be improved.
References
[1] Editorial. The promise of proteomics, Nature 402 (6763) (1999) 703.[2] M. Deng, K. Zhang, S. Mehta, T. Chen, F. Sun, Prediction of protein function using protein–protein interaction data, IEEE
Computer Society Bioinformatics Conference, 2002, pp. 197–206.[3] J.H. Lakey, E.M. Raggett, Measuring protein–protein interactions, Curr. Opin. Struct. Biol. 8 (1998) 119–123.[4] P. Legrain, J. Wojcik, J.M. Gauthier, Protein–protein interaction maps: a lead towards cellular functions, Trends Genet. 17
(2001) 346–352.[5] A. Valencia, F. Pazos, Computational methods for the prediction of protein interactions, Curr. Opin. Struct. Biol. 12 (2002)
368–373.[6] M. Pellegrini, E.M. Marcotte, M.J. Thompson, D. Eisenberg, T.O. Yeates, Assigning protein functions by comparative
genome analysis: protein phylogenetic profiles, Proc. Natl. Acad. Sci. USA 96 (1999) 4285–4288.[7] T. Gaasterland, M.A. Ragan, Microbial genescapes: phyletic and functional patterns of ORF distribution among prokaryotes,
Microb. Comp. Genomics 3 (1998) 199–217.[8] J. Tamames, G. Casari, C. Ouzounis,A.Valencia, Conserved clusters of functionally related genes in two bacterial genomes,
J. Mol. Evol. 44 (1997) 66–73.

A. Chinnasamy et al. / Computers in Biology and Medicine 36 (2006) 1143–1154 1153
[9] A.J. Enright, I. Iliopoulos, N.C. Kyrpides, C.A. Ouzounis, Protein interaction maps for complete genomes based on genefusion events, Nature 402 (1999) 86–90.
[10] S.M. Gomez,A. Rzhetsky,Towards the prediction of complete protein–protein interaction networks, Pac. Symp. Biocomput.7 (2002) 413–424.
[11] J.R. Bock, D.A. Gough, Whole-proteome interaction mining, Bioinforrnatics 19 (1) (2003) 125–134.[12] R. Bandyopadhyay, K. Maatthews, D. Subramanian, X.X. Tan, Predicting protein-ligand interactions from primary
structure, Rice University, Department of Computer Science, Technical Report TR02-387, February 2002.[13] J.R. Bock, D.A. Gough, Predicting protein–protein interactions from primary structure, Bioinformatics 17-5 (2001)
455–460.[14] F.V. Jensen, Bayesian Networks and Decision Graphs, Springer, New York, 2001.[15] G.H. John, P. Langley, Estimating continuous distributions in Bayesian classifiers, in: Proceedings of the Eleventh
Conference on Uncertainty in Artificial Intelligence, 1995, pp. 338– 345.[16] N. Friedman, D. Geiger, M. Goldszmidt, Bayesian network classifiers, Mach. Learn. 29 (2–3) (1997) 131–163.[17] P. Domingos, M. Pazzani, On the optimality of the simple Bayesian classifier under zero–one loss, Mach. Learn. 29 (1997)
103–130.[18] I. Xenarios, L. Salwinski, X.J. Duan, P. Higney, S. Kim, D. Eisenberg, DIP: The database of interacting proteins. A research
tool for studying cellular networks of protein interactions, NAR 30 (2002) 303–305.[19] Y. Kandel, R. Matias, R. Unger, P.M. Winkler, Shuffling biological sequences, Discrete Appl. Math. 71 (1996) 171–185.[20] L. Young, R.L. Jernigan, D.G. Covell, A role for surface hydrophobicity in protein– protein recognition, Protein Sci. 3
(1994) 717–729.[21] H.Q. Ding, I. Dubchak, Multi-class protein fold recognition using support vector machines and neural networks,
Bioinformatics 4 (17) (2001) 349–358.[22] A. Chinnasamy, W.-K. Sung, A. Mittal, Protein structure and fold prediction using tree-augmented Bayesian classifier, Pac.
Symp. Biocomput. 6–10 (2004) 387–398.[23] A. Chinnasamy, W.-K. Sung, A. Mittal, Protein structure and fold prediction using tree-augmented Bayesian classifier, J.
Bioinformatics Comput. Biol. 3 (4) (2005) 803–820.[25] H. Hishigaki, K. Nakai, T. Ono, A. Tanigami, T. Takagi, Assessment of prediction accuracy of protein function from
protein–protein interaction data, Yeast 18 (2001) 523–531.[26] S. Letovsky, S. Kasif, Predicting protein function from protein/protein interaction data: a probabilistic approach,
Bioinformatics 19 (1) (2003) I197–I204.[27] P.S. Bradley, O.L. Mangasarian, Feature selection via concave minimization and SVMS, Proceedings of the International
Conference on Machine Learning, 1998, pp. 82–90.[28] J. Weston, S. Mukherjee, O. Chapelle, M. Pontil, V. Vapnik, T. Poggio, Feature selection for SVMs, Adv. Neural Inform.
Process. Syst. (2000) 668–674.[29] B. Engelmann, E. Hayden, D. Tasche, Testing rating accuracy, Risk 16 (2003) 82–86.[30] J. Sobehart, S. Keenan, Measuring default accurately, credit risk special report, Risk 14 (2001) 31–33.
Arunkumar Chinnasamy received his Bachelor of Engineering Degree in Information technology from Bharathiar University,India and Master Degree from National University of Singapore under the supervision of Dr. Ken Sung and Dr. Ankush Mittal.During his stay in National University of Singapore, he was involved in protein sequence analyses and snake genome analysisprojects. Currently, he is working at A∗STAR, Bioinformatics Institute, Singapore as Research Associate. His interests are datawarehousing and data mining of life science databases.
Ankush Mittal received the B. Tech. (Computer Science and Engineering) and M.S. Research degrees from the Indian Institute ofTechnology (IIT), Delhi in 1996 and 1998, respectively. He obtained his Ph.D. degree from Electrical and Computer Engineering,The National University of Singapore (NUS). From March 2001 for around two years, he was a faculty member in the Departmentof Computer Science, NUS. At present, he serves as an Assistant Professor in IIT Roorkee, India. His research interests includemachine learning, image processing, E-learning and bioinformatics.
Wing-Kin Sung received both B.Sc. and Ph.D. degree in the Department of Computer Science from the University of HongKong. Then, he worked as a Post-Doctoral Fellow inYale University and worked as a Senior Technology Officer in the University

1154 A. Chinnasamy et al. / Computers in Biology and Medicine 36 (2006) 1143–1154
of Hong Kong. Currently, he is an assistant professor in the Department of Computer Science, National University of Singapore,Singapore. He also works as a Senior Group Leader in the Department of Information and Mathematical Science, GenomeInstitute of Singapore, Singapore.




















