
Preparation of fibers with nanoscaled morphologies: Electrospinning of polymer blends
8
982 POLYMER ENGINEERING AND SCIENCE, JUNE 2001, Vol. 41, No. 6 INTRODUCTION T he manufacturing of polymer fibers characterized by small diameters in the range from several mi- crons down to a few tens of nanometers is of consider- able interest for various kinds of applications. Exam- ples are nanocomposites displaying reinforcing effects (1, 2), fiber mats composed of suitable polymers for applications in the area of tissue engineering, sensor technology or filtration (3, 4). Of special interest are fibers designed as templates for the preparation of functional nano- and mesotubes (5, 6). lt has been shown that electrospinning as a production method offers several advantages in comparison with extru- sion either from the melt or from solution (7–18). The electrospinning process involves the application of a strong electrostatic field to a capillary connected with a reservoir containing a polymer solution or melt. Under the influence of the electrostatic field, a pen- dant droplet of the polymer solution or melt at the capillary tip is deformed into a conical shape (Taylor cone) (19). If the voltage surpasses a threshold value, electrostatic forces overcome the surface tension, and a fine charged jet is ejected. The jet moves towards a counter electrode. Owing to its high viscosity and the presence of entanglements, it remains stable and does not transform into spherical droplets as expected for a liquid cylindrical thread (20). This results in the depo- sition of a thin polymer fiber on a substrate located on the counter electrode. It has been found that the de- position rate as well as the fiber diameter can be con- trolled within a broad range. By proper selection of the solvent and the polymer concentration as well as of the intrinsic conductivity, the viscosity and the sur- face tension of the solution, one obtains fibers with di- ameters down to a few tens of nanometers (7–18). For some applications it is desirable to control not only the diameter of the fibers and their internal mor- phology—and thus their intrinsic properties—but also their surface structure. Porous fibers or fibers with a large surface to volume ratio are of interest for filter applications or as templates for the formation of func- tional nanotubes (5, 6). Therefore, the scope of our research was to control the formation of the internal fiber structure, of intrinsic fiber properties and of the surface topology. The basic Preparation of Fibers With Nanoscaled Morphologies: Electrospinning of Polymer Blends MIKHAIL BOGNITZKI, THOMAS FRESE, MARTIN STEINHART, ANDREAS GREINER, and JOACHIM H. WENDORFF* Institute of Physical Chemistry and Material Science Center Philipps-University 35032 Marburg, Germany ANDREAS SCHAPER and MICHAEL HELLWIG Material Science Center Philipps-University 35032 Marburg, Germany The aim was to prepare fibers with diameters below the micrometer range char- acterized by specific bulk morphologies and surface topologies. Such materials are of interest for various applications including reinforcement, sensors or filtration as well as the formation of functional tubes by the use of fiber templates. We were able to manufacture highly structured submicrometer fibers by electrospinning from ternary solutions using polylactide (PLA) and polyvinylpyrrolidone (PVP) as polymer model components. Co-continuous phase morphologies resulted from phase sepa- ration processes taking place during fiber formation. In a subsequent step, specific surface topologies or fine pores were generated by selective removal of one of the components. *Corresponding author.
Transcript of Preparation of fibers with nanoscaled morphologies: Electrospinning of polymer blends

982 POLYMER ENGINEERING AND SCIENCE, JUNE 2001, Vol. 41, No. 6
INTRODUCTION
The manufacturing of polymer fibers characterizedby small diameters in the range from several mi-
crons down to a few tens of nanometers is of consider-able interest for various kinds of applications. Exam-ples are nanocomposites displaying reinforcing effects(1, 2), fiber mats composed of suitable polymers forapplications in the area of tissue engineering, sensortechnology or filtration (3, 4). Of special interest arefibers designed as templates for the preparation offunctional nano- and mesotubes (5, 6). lt has beenshown that electrospinning as a production methodoffers several advantages in comparison with extru-sion either from the melt or from solution (7–18).
The electrospinning process involves the applicationof a strong electrostatic field to a capillary connectedwith a reservoir containing a polymer solution or melt.Under the influence of the electrostatic field, a pen-dant droplet of the polymer solution or melt at thecapillary tip is deformed into a conical shape (Taylorcone) (19). If the voltage surpasses a threshold value,
electrostatic forces overcome the surface tension, anda fine charged jet is ejected. The jet moves towards acounter electrode. Owing to its high viscosity and thepresence of entanglements, it remains stable and doesnot transform into spherical droplets as expected for aliquid cylindrical thread (20). This results in the depo-sition of a thin polymer fiber on a substrate located onthe counter electrode. It has been found that the de-position rate as well as the fiber diameter can be con-trolled within a broad range. By proper selection ofthe solvent and the polymer concentration as well asof the intrinsic conductivity, the viscosity and the sur-face tension of the solution, one obtains fibers with di-ameters down to a few tens of nanometers (7–18).
For some applications it is desirable to control notonly the diameter of the fibers and their internal mor-phology—and thus their intrinsic properties—but alsotheir surface structure. Porous fibers or fibers with alarge surface to volume ratio are of interest for filterapplications or as templates for the formation of func-tional nanotubes (5, 6).
Therefore, the scope of our research was to controlthe formation of the internal fiber structure, of intrinsicfiber properties and of the surface topology. The basic
Preparation of Fibers With Nanoscaled Morphologies:Electrospinning of Polymer Blends
MIKHAIL BOGNITZKI, THOMAS FRESE, MARTIN STEINHART,ANDREAS GREINER, and JOACHIM H. WENDORFF*
Institute of Physical Chemistry and Material Science CenterPhilipps-University
35032 Marburg, Germany
ANDREAS SCHAPER and MICHAEL HELLWIG
Material Science CenterPhilipps-University
35032 Marburg, Germany
The aim was to prepare fibers with diameters below the micrometer range char-acterized by specific bulk morphologies and surface topologies. Such materials areof interest for various applications including reinforcement, sensors or filtration aswell as the formation of functional tubes by the use of fiber templates. We were ableto manufacture highly structured submicrometer fibers by electrospinning fromternary solutions using polylactide (PLA) and polyvinylpyrrolidone (PVP) as polymermodel components. Co-continuous phase morphologies resulted from phase sepa-ration processes taking place during fiber formation. In a subsequent step, specificsurface topologies or fine pores were generated by selective removal of one of thecomponents.
*Corresponding author.

concept was to couple the fiber formation during elec-trospinning with phase separation processes and tofreeze the generated phase structure in an early stage.This should result in the preservation of co-continu-ous or interpenetrating phase morphologies within thefibers. A subsequent step to obtain fibers with specificsurface topologies or pore structures was the selectiveremoval of one component. In this article, we describeour approach towards structured ultra-thin polymerfibers.
EXPERIMENTAL
We selected polylactide (PLA) and the amorphouspolymer polyvinylpyrrolidone (PVP) as model compo-nents. The chemical structures are shown in SchemeI. PLA was obtained as poly-L-lactide (PLLA) fromBoehringer and as poly-D,L-lactide (PDLLA) fromSigma-Aldrich. PLLA is an isotactic polymer composedof L-lactyl units and therefore partially crystalline.PDLLA is characterized by a statistical sequence of D-lactyl and L-lactyl units. Hence, this polymer is atac-tic and amorphous. The molecular weights of thepolymers, their glass transition temperatures and themelting temperature of PLLA are given in Table 1. As acommon solvent we selected dichloromethane thatevaporates rapidly at room temperature [boiling point:40°C (21)]. We prepared solutions containing eitherPVP and PLLA or PVP and PDLLA. The PVP/PLAweight ratios were varied as follows: 17/83, 50/50and 83/17. The total polymer concentration was inthe range from 2 wt% to 5 wt%. The compositions ofthe processed solutions are displayed by solid sym-bols in Fig. 1.
The electrospinning setup used here has been de-scribed previously (5, 6). The polymer solution, stored
within a reservoir, was pumped through a metal capil-lary using a peristaltic pump. The capillary was con-nected with a voltage supply. Its circular orifice had adiameter of 500 microns. A circular shaped counterelectrode with a diameter of 18 cm was located belowthe reservoir, so that a vertical arrangement of theelectrodes resulted. Fibers were collected on selectedsubstrates, e.g., glass slides, placed on the counterelectrode. The distance between the tip of the capillaryand the counter electrode was 20 cm, the applied volt-age was 35 kV in all cases described here.
RESULTS AND DISCUSSION
Ternary Phase Diagrams
Electrospinning of polymer solutions involves, to afirst approximation, rapid evaporation of the solventas the jet surface increases sharply within millisec-onds after its formation. This is due to an elongationof the jet caused by counteracting electrostatic andviscous forces (16, 18). Starting from a homogeneousternary solution, phase separation sets in as the sol-vent concentration is reduced so much that phaseboundaries are crossed (22, 23). Two phases charac-terized by different concentrations of the polymercomponents will thus be formed. A time scale wellbelow one second possibly down to the millisecondrange characterizes the phase separation during elec-trospinning. In fact, it has been reported that the de-position rate of the fibers is in the range of severalmeters per second (8, 12).
This has several consequences: The phase-separatedregions are not able to coarsen strongly prior to solidi-fication so that very fine phase morphologies could bepreserved. The solidification is controlled by the onsetof a glass transition process or by crystallization,
Preparation of Fibers With Nanoscaled Morphologies
POLYMER ENGINEERING AND SCIENCE, JUNE 2001, Vol. 41, No. 6 983
Table 1. Molecular Weights, Glass Transition Temperatures and Melting Temperatures of the Polymers Used.
Mw Mn Mw/Mn Tg [°C] Tm [°C]
PLLA 148,000 297,000 2.0 63 181PDLLA 54,000 89,000 1.6 52 —PVP 360,000 — — 180 —
Scheme 1. Structures of poly-vinylpyrrolidone PVP (a) and poly-lactide PLA (b).

which will happen as the concentration of the solventbecomes less than a critical value for a given tempera-ture. A second consequence is that the tendency to-wards a spinodal rather than a binodal phase separa-tion will be enhanced since nucleation phenomenarequire more time to set in than the initial growth ofunstable concentration fluctuations. We thus expecteda certain preference for co-continuous structuresrather than matrix-dispersed phase morphologies (24).
Phase diagrams for the ternary systems consideredhere are displayed in Fig. 1. Figure 1a shows thephase behavior of the system PVP/PDLLA/CH2Cl2. Itis obvious that high concentrations of the solvent arerequired in order to obtain homogeneous solutions.The compositions of the solutions used for electro-spinning (solid symbols) are rather adjacent to thephase boundary. Phase separation will thus takeplace in an early stage of the jet formation, i.e., bothgenerated phases will contain a rather high solvent
portion at this stage. Furthermore, the phase diagramreveals that the mutual miscibility of PDLLA and PVPis very low i.e. the two phases are predominantly com-posed of the pure polymer components and the sol-vent. The phase diagram becomes rather asymmetricalin case of solutions containing PLLA and PVP (Fig.1b). One reason for the different phase behavior ifPDLLA is replaced by PLLA is certainly the larger mol-ecular weight of PLLA. Yet, the conclusions with re-spect to the early onset of phase separation duringelectrospinning and the composition of the generatedcoexisting phases are similar for systems containingPLLA or PDLLA. Coarsening takes place on a limitedtime scale owing to the early solidification by theonset of a glass transition and/or by crystallization asthe solvent concentration decreases further duringthe electrospinning process.
To gain information on the structure formation dur-ing phase separation processes induced by rapidsolvent evaporation, we prepared thin films from thesolutions used for electrospinning by spincoating.Electrospinning as well as spincoating are character-ized by a structure formation process on a time scalefrom seconds down to milliseconds before solidifica-tion. The resulting film thicknesses were in the orderof one micron.
We observed that phase separation sets in indepen-dent of the PVP/PLA weight ratio in the original solu-tion i.e. phase separation cannot be suppressed de-spite the rapid evaporation of the solvent and thecorresponding rapid solidification. In general, co-con-tinuous phase morphologies were found with typicaldimensions well below one micron down to about 300nm (Fig. 2).
Topology of As-Spun Fibers
X-ray studies as well as calorimetric studies re-vealed that PVP and PDLLA fibers are amorphous asexpected whereas PLLA fibers are partially crystalline
Mikhail Bognitzki et al.
984 POLYMER ENGINEERING AND SCIENCE, JUNE 2001, Vol. 41, No. 6
(b)
Fig. 1. Ternary phase diagrams obtained by turbidity mea-surements at room temperature: a) PVP/PDLLA/CH2Cl2, b)PVP/PLLA/CH2Cl2. The solid symbols show the compositionsof the solutions used for electrospinning, the hollow squaresmark the phase boundary. The concentrations are given inwt%.
(a)
Fig. 2. Incident light micrograph of a film composed of PVPand PLLA at a weight ratio of 1:1 with a thickness of one mi-cron. The film was prepared by spincoating of a solution ofboth polymers in dichloromethane.
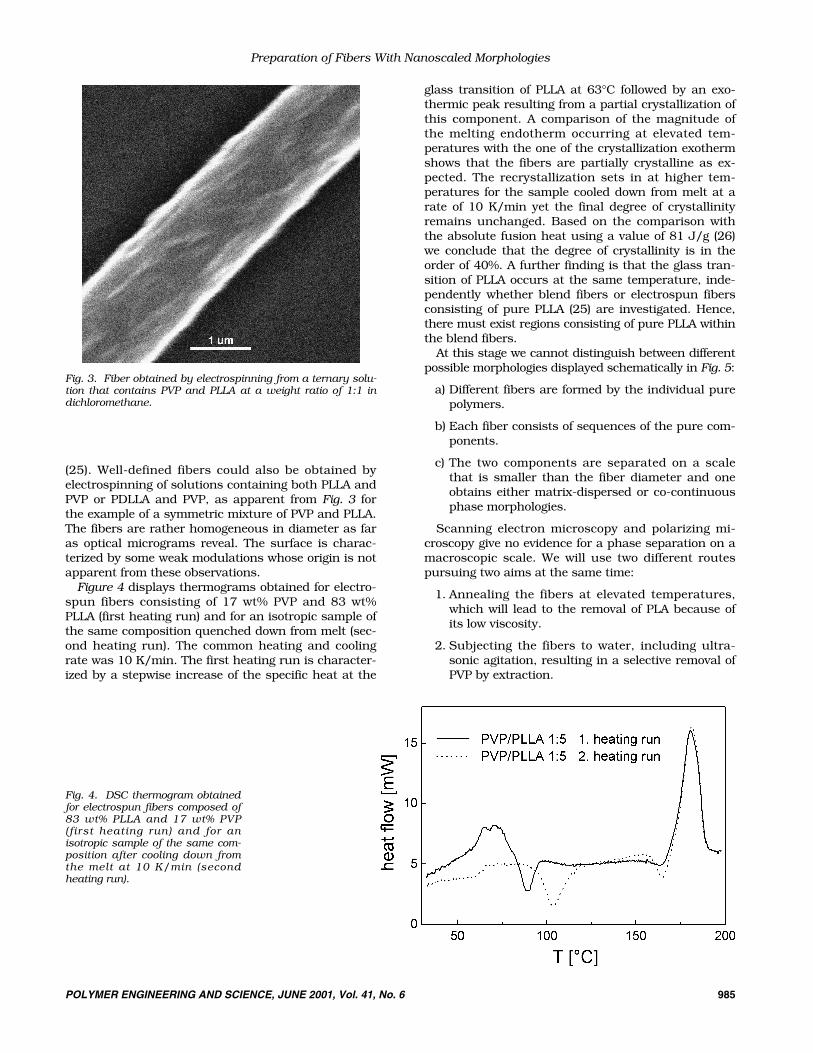
(25). Well-defined fibers could also be obtained byelectrospinning of solutions containing both PLLA andPVP or PDLLA and PVP, as apparent from Fig. 3 forthe example of a symmetric mixture of PVP and PLLA.The fibers are rather homogeneous in diameter as faras optical micrograms reveal. The surface is charac-terized by some weak modulations whose origin is notapparent from these observations.
Figure 4 displays thermograms obtained for electro-spun fibers consisting of 17 wt% PVP and 83 wt%PLLA (first heating run) and for an isotropic sample ofthe same composition quenched down from melt (sec-ond heating run). The common heating and coolingrate was 10 K/min. The first heating run is character-ized by a stepwise increase of the specific heat at the
glass transition of PLLA at 63°C followed by an exo-thermic peak resulting from a partial crystallization ofthis component. A comparison of the magnitude ofthe melting endotherm occurring at elevated tem-peratures with the one of the crystallization exothermshows that the fibers are partially crystalline as ex-pected. The recrystallization sets in at higher tem-peratures for the sample cooled down from melt at arate of 10 K/min yet the final degree of crystallinityremains unchanged. Based on the comparison withthe absolute fusion heat using a value of 81 J/g (26)we conclude that the degree of crystallinity is in theorder of 40%. A further finding is that the glass tran-sition of PLLA occurs at the same temperature, inde-pendently whether blend fibers or electrospun fibersconsisting of pure PLLA (25) are investigated. Hence,there must exist regions consisting of pure PLLA withinthe blend fibers.
At this stage we cannot distinguish between differentpossible morphologies displayed schematically in Fig. 5:
a) Different fibers are formed by the individual purepolymers.
b) Each fiber consists of sequences of the pure com-ponents.
c) The two components are separated on a scalethat is smaller than the fiber diameter and oneobtains either matrix-dispersed or co-continuousphase morphologies.
Scanning electron microscopy and polarizing mi-croscopy give no evidence for a phase separation on amacroscopic scale. We will use two different routespursuing two aims at the same time:
1. Annealing the fibers at elevated temperatures,which will lead to the removal of PLA because ofits low viscosity.
2. Subjecting the fibers to water, including ultra-sonic agitation, resulting in a selective removal ofPVP by extraction.
Preparation of Fibers With Nanoscaled Morphologies
POLYMER ENGINEERING AND SCIENCE, JUNE 2001, Vol. 41, No. 6 985
Fig. 4. DSC thermogram obtainedfor electrospun fibers composed of83 wt% PLLA and 17 wt% PVP(first heating run) and for anisotropic sample of the same com-position after cooling down fromthe melt at 10 K/min (secondheating run).
Fig. 3. Fiber obtained by electrospinning from a ternary solu-tion that contains PVP and PLLA at a weight ratio of 1:1 indichloromethane.

986 POLYMER ENGINEERING AND SCIENCE, JUNE 2001, Vol. 41, No. 6
Mikhail Bognitzki et al.
On the one hand, these procedures uncover the in-ternal phase structure of the fibers. On the otherhand, they result in fibers with interesting surfacetopologies as shown in Fig. 5.
Selective Removal of IndividualPolymer Compounds
We used water as solvent for the selective removalof PVP. First, we will discuss the amount of PVP thatcan be removed totally as a function of the composi-tion of the fibers. These data were obtained by a com-parison of the weight of dry fibers before and afterwater treatment. The results are shown in Fig. 6. Wefound that the amount of removable PVP relative tothe total amount is small for fibers composed predom-inantly of PLLA and approaches 100% if PVP is themajor component. This indicates that no macroscopicphase separation exists corresponding to the cases a)or b) depicted in Fig. 5 but rather that situation c) isobserved. Regions composed of PVP have to be locatedwithin the fibers in such a way that water cannot pene-trate them. The presence of amorphous PDLLA allowseasier extraction of PVP even in case of low PVP por-tions (Fig. 6). Very similar results were obtained forthe selective removal of PLA by heat treatment. An-nealing leads to a decomposition of PLA but also to aflow out of the blend fibers as apparent from the oc-currence of separated small droplets in the areas be-tween residual PVP fibers.
Selective Removal of One Component FromFibers of Symmetric Blends
Conclusions on the distribution of the two polymerswithin the fibers can be drawn from microscopical ob-servations of their shape and surface structure aftertreatment with water either in the absence or pres-ence of additional ultrasonic agitation. Continuousresidual fibers are obtained even if PVP is removed se-lectively from fibers containing PVP and PLA in equal
portions. The fibers become porous and display a reg-ular surface structure (Fig. 7). Surface undulations aswell as pores are observed with dimensions in theorder of 100 nm to 200 nm in case of PVP/PLLA mix-tures (Fig. 7a). In case of PVP/PDLLA mixtures thestructure patterns are much coarser, in the order of500 nm (Fig. 7b).
Furthermore, we removed PLA selectively by anneal-ing the as-spun fibers at temperatures well above theglass transition temperatures of the components andthe melting temperature of PLLA, i.e., at 200°C forabout 3 h. Annealing at elevated temperatures resultsin a selective removal of PLA partially because of de-composition and partially because of the flow of low-viscous PLA out of the fibers. This is obvious from an-nealing studies on pure PLA fibers. As expected forfluid threads in the absence of external constraints,the fibers become unstable and are transformed intosmall separated droplets owing to the growth of peri-odic fluctuations of the fiber diameter (27). In contrastto PLA fibers, no alteration of the shape of pure PVPfibers has been observed under these conditions.
Similar to the preservation of continuous residualPLA fibers in case of the selective removal of PVP, se-lective removal of PLA by annealing results in thepreservation of continuous residual PVP fibers. Theirwell-defined surface structure (Fig. 7c) resembles thesurface structure of the residual PLA fibers (Fig. 7a).Our tentative conclusion is that predominantly co-continuous phase structures are generated by phaseseparation during electrospinning and preserved byrapid solidification. The phase structure can be un-covered by the selective removal of one component.
Selective Removal of One Component FromFibers Containing Either PLA or PVP asthe Major Component
A significantly different result is obtained if eitherPLA or PVP is the major component. The fibers remain
Fig. 5. Schematic representation of possible fiber morphologies: a) PVP and PLA fibers consisting of the pure polymers, b) fibers com-posed of sequences of PLA and PVP, c) matrix-dispersed or co-continuous phase morphologies with dimensions smaller than the fiberdiameter.

POLYMER ENGINEERING AND SCIENCE, JUNE 2001, Vol. 41, No. 6 987
Preparation of Fibers With Nanoscaled Morphologies
Fig. 6. Relative amount of PVP thatcan be removed from electrospuncomposite fibers by water extractionas a function of the absolute PVPcontent.
Fig. 7. SEM micrographs of PVP/PLA 1:1 fibers: a) residualPLLA fiber after selective removal of PVP; b) residual PDLLAfiber after selective removal of PVP; and c) residual PVP fibersafter selective removal of PLLA.

988 POLYMER ENGINEERING AND SCIENCE, JUNE 2001, Vol. 41, No. 6
Mikhail Bognitzki et al.
compact without any alteration of the surface struc-ture if, for instance, PVP is the major component andone tries to remove the minor component PLA by an-nealing (Fig. 8a). This indicates the existence of a core-shell morphology with PVP forming the shell and PLAforming the core. If such fibers are subjected to water,we find a porous structure as in case of symmetricmixtures and a decrease in diameter (Fig. 8b). Similarresults are obtained if PLA is the major component.Water extraction does not yield porous structures incontrast to the thermal removal of PLA, as apparentfrom Fig. 8c.
We thus obtain highly structured fibers, which maybe of use for several applications described earlier.The co-continuous structures are elongated along thefiber axis. This indicates the occurrence of drawing
during structure formation. This is obvious from Figs.7 and 8. Previous studies on anisotropic patterns ofelectrospun fibers (28) are confirmed by these find-ings.
CONCLUSIONS
Electrospinning of polymer blends from solutionproduces ultra-thin fibers characterized by intrinsicstructure patterns on a 100 nm scale. The fibers dis-play an internal phase morphology that is evidentlycontrolled by rapid phase separation and rapid solidi-fication. By selective removal of one component,porous fibers and fibers with specific surface topolo-gies are accessible. Such fibers are of interest for abroad range of applications in areas such as medi-cine, sensor technology or filtration. Furthermore,
Fig. 8. SEM micrographs of a) PVP/PDLLA 5:1 fibers after an-nealing, b) a residual PDLLA fiber after selective removal ofPVP (original weight ratio PVP/PDLLA � 5:1) and c) a resid-ual PVP fiber after annealing (original weight ratio PVP/PDLLA � 1:5).

POLYMER ENGINEERING AND SCIENCE, JUNE 2001, Vol. 41, No. 6 989
Preparation of Fibers With Nanoscaled Morphologies
porous PLA fibers can be used as templates for theformation of tubes (5, 6). PLA fiber templates can becoated with various materials like metals, metal ox-ides, glass, ceramics and even polymers by vapor de-position. In a subsequent step the PLA fiber templatesare removed by annealing at elevated temperatures.This is a versatile method to manufacture tubes withspecific surface topologies.
ACKNOWLEDGMENT
We would like to thank Uwe Zimmermann for thedetermination of the molecular weights of the polylac-tides.
REFERENCES1. M. M. Bergshoef and G. J. Vancso, Adv. Mater., 11,
1362 (1999).2. J.-S. Kim and D. H. Reneker, Polym. Comp., 20, 124
(1999).3. “The Nonwovens Industry Meets The Filtration Busi-
ness,” M. Jacobson, ed., Nonwovens Industry, May1991.
4. P. W. Gibson, H. L. Schreuder-Gibson, and D. Rivin,AIChE J., 45, 190 (1999).
5. M. Bognitzki, H. Hou, M Ishaque, T. Frese, M. Hellwig,C. Schwarte, A. Schaper, J. H. Wendorff, and A.Greiner, Polym. Prepr. (ACS, PMSE), 82, 115 (2000).
6. M. Bognitzki, H. Hou, M. Ishaque, T. Frese, M. Hellwig,C. Schwarte, A. Schaper, J. H. Wendorff, and A.Greiner, Adv. Mater., 12, 637 (2000).
7. A. Formhals, U. S. Patent 1,975,504 (1934).8. P. K. Baumgarten, J. Coll. Interf. Sci., 36, 71 (1971).9. L. Larrondo and R. S. Manley, J. Polym. Sci. Part B, 19,
909 (1981).
10. L. Larrondo and R. S. Manley, J. Polym. Sci. Part B, 19,921 (1981).
11. L. Larrondo and R. S. Manley, J. Polym. Sci. Part B, 19,933 (1981).
12. J. Doshi, PhD thesis, University of Akron (1994).13. M. M. Bergshoef, PhD thesis, Universiteit Twente (1999).14. D. H. Reneker and I. Chun, Nanotechnology (IOP Pub-
lishing), 7, 216 (1996). 15. R. Jaeger, M. M. Bergshoef, C. Martin i Batlle, H.
Schönherr, and G. J. Vancso, Macromol. Symp. (Hüthig& Wepf), 127, 141 (1998).
16. J. Doshi and D. H. Reneker, J. Electrostatics, 35, 151(1995).
17. H. Fong and D. H. Reneker, J. Polym. Sci. Part B, 37,3488 (1999).
18. D. H. Reneker, A. L. Yarin, H. Fong, and S. Koomb-hongse, J. Appl. Phys., 87, 4531 (2000).
19. G. I. Taylor, Proc. R. Soc. A, 313, 453 (1969).20. L. Rayleigh, Philos. Mag., 44, 184 (1882).21. Merck-Catalogue, Darmstadt (1999), p. 506.22. P. J. Flory, Principles of Polymer Chemistry, Cornell Uni-
versity Press, Ithaca, N.Y. (1953).23. H. Tompa, Polymer Solutions, Butterworth, London
(1956).24. T. Hashimoto, Material Science and Technology, Vol. 12,
p. 252, R. W. Cahn, P. Haasen, and E. J. Kramer, eds.,VCH-Verlag, Weinheim (1993).
25. M. Bognitzki, W. Czado, T. Frese, A. Schaper, M. Hell-wig, M. Steinhart, A. Greiner, and J. H. Wendorff, Adv.Mater., 13, 70 (2001).
26. E. W. Fischer, H. J. Sterzel, and G. Wegner, Coll. Polym.Sci., 251, 980 (1973).
27. S. Tomotika, Proc. R. Soc. A, 150, 322 (1935).28. R. Jaeger, H. Schönherr, and G. J. Vansco, Macromole-
cules, 29, 7634 (1996).




















