
Plasminogen activators promote excitotoxicity-induced retinal damage
10
The FASEB Journal • Research Communication Plasminogen activators promote excitotoxicity-induced retinal damage Raghuveer S. Mali, Mei Cheng, and Shravan K. Chintala 1 Eye Research Institute of Oakland University, 409 Dodge Hall, Rochester, Michigan, USA ABSTRACT Increased levels of extracellular L-gluta- mate have been suggested to play a role in retinal damage in a number of blinding diseases such as glaucoma and diabetic retinopathy. Although glutamate can cause retinal damage in part by hyperstimulating its receptors (“excitotoxicity”), the downstream events that lead to retinal damage are poorly understood. In this study, we injected kainic acid (KA), a glutamate receptor agonist that specifically hyperstimulates non- NMDA-type receptors, into the vitreous humor of CD-1 mice and have investigated the role of plasminogen activators (PAs) [tissue plasminogen activator (tPA) and urokinase plasminogen activator (uPA)] in excitotoxic- ity-induced retinal damage. Injection of KA into the vitreous humor led to an up-regulation in tPA and an induction in uPA activity in the retina and this was associated with activation of zymogen plasminogen to active plasmin. Immunocytochemical analysis indicated that retinal ganglion cells (RGCs), constitutively ex- press tPA and release it into the extracellular space upon KA injection. Immunocytochemical analysis also indicated an increase in uPA in the nerve fiber layer after KA injection that was absent in the control retinas. These events were associated with apoptotic death of cells initially in the ganglion cell layer and subsequently in the inner and outer nuclear layer, associated with loss of RGCs and amacrine cells. These phenomena were inhibited when recombinant plasminogen activator in- hibitor (rPAI-1) or tPA-STOP were injected into the vitreous humor with KA, whereas a plasmin inhibitor, -2-antiplasmin, failed to attenuate KA-induced retinal damage. Taken together, these results suggest that inhibition of plasminogen activators might attenuate retinal damage in blinding retinal diseases in which hyperstimulation of glutamate receptors is implicated as a causative factor to retinal damage.—Mali, R. S., Cheng, M., Chintala, S. K. Plasminogen activators pro- mote excitotoxicity-induced retinal damage. FASEB J. 19, 1280 –1289 (2005) Key Words: retina ganglion cells excitotoxicity kainic acid tissue plasminogen activator urokinase plasminogen activa- tor and non-NMDA receptors Retinal damage is a major concern in blinding retinal diseases, including diabetic retinopathy and glaucoma (1–5). Although the mechanisms that underlie retinal damage in these diseases are poorly understood, previ- ous studies have suggested that excitatory amino acid (EAA) such as l-glutamate might play a role (6 –11). Accumulation of glutamate into the extracellular space activates two major classes of receptors: the ionotropic (N-methyl-d-aspartate [NMDA], -amino-3-hydroxy-5- methyl-4-isoxazolepropionic acid [AMPA]/kainic acid [KA]-type) and the G-protein-coupled metabotropic receptors (12–14). A number of cells including gan- glion cells and amacrine cells in the retina express these receptors (15, 16). Although activation of these receptors constitutes a physiological phenomenon, hy- perstimulation of NMDA and non-NMDA type iono- tropic receptors can lead to cell death. Indeed, previ- ous studies have reported that glutamate and its receptor agonists such as NMDA and KA hyperstimu- late glutamate receptors (“excitotoxicity”) and contrib- ute to retinal damage (10, 17–25). Elevated levels of glutamate and glutamate-mediated excitotoxicity have been implicated in ganglion cell loss in animals models for retinal ischemia (7, 26, 27), axonal injury (28), and glaucoma (6, 29). Furthermore, previous studies have reported the presence of elevated levels of glutamate in human glaucoma (6). In spite of these observations, the role of glutamate in glaucoma is controversial; the mechanisms that underlie glutamate-mediated excito- toxic-retinal damage are poorly understood and remain open to debate. Due to the lack of a clear understanding of the mechanisms involved in excitotoxic retinal damage, it is worthwhile to investigate and possibly draw some par- allels with other neurodegenerative diseases in which the role of excitotoxicity has been investigated and continues to be investigated. Recent investigations into the mechanisms of excitotoxicity in the central nervous system (CNS) have identified tissue plasminogen acti- vator (tPA) as one of the contributing factors to neu- ronal damage (30 –32). Plasminogen activators (PAs), urokinase plasminogen activator (uPA), and tPA are serine proteases that convert plasminogen, a physiolog- ical zymogen, into active plasmin, a trypsin-like endo- peptidase of broad substrate specificity (33). Endoge- nous plasminogen activator inhibitors (PAIs) control plasminogen activation by regulating the activity of 1 Correspondence: Eye Research Institute, 409 Dodge Hall, Oakland University, Rochester, MI, 48309, USA. E-mail: [email protected] doi: 10.1096/fj.04-3403com 1280 0892-6638/05/0019-1280 © FASEB
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Plasminogen activators promote excitotoxicity-induced retinal damage

The FASEB Journal • Research Communication
Plasminogen activators promote excitotoxicity-inducedretinal damage
Raghuveer S. Mali, Mei Cheng, and Shravan K. Chintala1
Eye Research Institute of Oakland University, 409 Dodge Hall, Rochester, Michigan, USA
ABSTRACT Increased levels of extracellular L-gluta-mate have been suggested to play a role in retinaldamage in a number of blinding diseases such asglaucoma and diabetic retinopathy. Although glutamatecan cause retinal damage in part by hyperstimulating itsreceptors (“excitotoxicity”), the downstream eventsthat lead to retinal damage are poorly understood. Inthis study, we injected kainic acid (KA), a glutamatereceptor agonist that specifically hyperstimulates non-NMDA-type receptors, into the vitreous humor of CD-1mice and have investigated the role of plasminogenactivators (PAs) [tissue plasminogen activator (tPA) andurokinase plasminogen activator (uPA)] in excitotoxic-ity-induced retinal damage. Injection of KA into thevitreous humor led to an up-regulation in tPA and aninduction in uPA activity in the retina and this wasassociated with activation of zymogen plasminogen toactive plasmin. Immunocytochemical analysis indicatedthat retinal ganglion cells (RGCs), constitutively ex-press tPA and release it into the extracellular spaceupon KA injection. Immunocytochemical analysis alsoindicated an increase in uPA in the nerve fiber layerafter KA injection that was absent in the control retinas.These events were associated with apoptotic death ofcells initially in the ganglion cell layer and subsequentlyin the inner and outer nuclear layer, associated with lossof RGCs and amacrine cells. These phenomena wereinhibited when recombinant plasminogen activator in-hibitor (rPAI-1) or tPA-STOP were injected into thevitreous humor with KA, whereas a plasmin inhibitor,�-2-antiplasmin, failed to attenuate KA-induced retinaldamage. Taken together, these results suggest thatinhibition of plasminogen activators might attenuateretinal damage in blinding retinal diseases in whichhyperstimulation of glutamate receptors is implicatedas a causative factor to retinal damage.—Mali, R. S.,Cheng, M., Chintala, S. K. Plasminogen activators pro-mote excitotoxicity-induced retinal damage. FASEB J.19, 1280–1289 (2005)
Key Words: retina � ganglion cells � excitotoxicity � kainic acid �tissue plasminogen activator � urokinase plasminogen activa-tor and non-NMDA receptors
Retinal damage is a major concern in blinding retinaldiseases, including diabetic retinopathy and glaucoma(1–5). Although the mechanisms that underlie retinaldamage in these diseases are poorly understood, previ-
ous studies have suggested that excitatory amino acid(EAA) such as l-glutamate might play a role (6–11).Accumulation of glutamate into the extracellular spaceactivates two major classes of receptors: the ionotropic(N-methyl-d-aspartate [NMDA], �-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid [AMPA]/kainic acid[KA]-type) and the G-protein-coupled metabotropicreceptors (12–14). A number of cells including gan-glion cells and amacrine cells in the retina expressthese receptors (15, 16). Although activation of thesereceptors constitutes a physiological phenomenon, hy-perstimulation of NMDA and non-NMDA type iono-tropic receptors can lead to cell death. Indeed, previ-ous studies have reported that glutamate and itsreceptor agonists such as NMDA and KA hyperstimu-late glutamate receptors (“excitotoxicity”) and contrib-ute to retinal damage (10, 17–25). Elevated levels ofglutamate and glutamate-mediated excitotoxicity havebeen implicated in ganglion cell loss in animals modelsfor retinal ischemia (7, 26, 27), axonal injury (28), andglaucoma (6, 29). Furthermore, previous studies havereported the presence of elevated levels of glutamate inhuman glaucoma (6). In spite of these observations, therole of glutamate in glaucoma is controversial; themechanisms that underlie glutamate-mediated excito-toxic-retinal damage are poorly understood and remainopen to debate.
Due to the lack of a clear understanding of themechanisms involved in excitotoxic retinal damage, it isworthwhile to investigate and possibly draw some par-allels with other neurodegenerative diseases in whichthe role of excitotoxicity has been investigated andcontinues to be investigated. Recent investigations intothe mechanisms of excitotoxicity in the central nervoussystem (CNS) have identified tissue plasminogen acti-vator (tPA) as one of the contributing factors to neu-ronal damage (30–32). Plasminogen activators (PAs),urokinase plasminogen activator (uPA), and tPA areserine proteases that convert plasminogen, a physiolog-ical zymogen, into active plasmin, a trypsin-like endo-peptidase of broad substrate specificity (33). Endoge-nous plasminogen activator inhibitors (PAIs) controlplasminogen activation by regulating the activity of
1 Correspondence: Eye Research Institute, 409 Dodge Hall,Oakland University, Rochester, MI, 48309, USA. E-mail:[email protected]
doi: 10.1096/fj.04-3403com
1280 0892-6638/05/0019-1280 © FASEB

plasminogen activators. In the CNS tPA plays a role inneuronal plasticity (34, 35) and neuronal regeneration(36–38), whereas uPA plays a role in astrocyte prolifer-ation and tissue remodeling (39, 40). Previous studieshave demonstrated the presence of tPA and uPA invarious eye tissues, including retinal neovascular mem-branes (41, 42), and in normal (43–46) and diabeticretinas (47). In a clinical setting, tPA has been used totreat stroke patients (48, 49) as well as patients withsubmacular hemorrhage (50–54). Although the func-tional roles of these proteases in the retina are unclear,mounting evidence indicates that tPA and uPA mightplay a degenerative role in the CNS (30–32, 55–60)and retina (42, 61–64).
Previous studies from our laboratory have suggestedthat hyperstimulation of non-NMDA receptors (excito-toxicity) in the retina by kainic acid leads to an up-regulation of extracellular modulating proteases suchas matrix metalloproteinase-9 (MMP-9); this in turnplays a role in retinal damage (65). However, inhibitionof MMP activity by a synthetic inhibitor failed to offercomplete protection against KA-induced cell loss, indi-cating the possible role of other proteases such asplasminogen activators uPA and tPA in retinal damage.Therefore, we injected kainic acid (KA) into the vitre-ous humor of normal CD-1 mice (to hyperstimulatenon-NMDA-type glutamate receptors in the retina) andinvestigated the role of tPA and uPA in retinal damage.We provide some novel findings that hyperstimulationof non-NMDA-type receptors leads to an increase in tPAand uPA activity and protein levels in the retina. Weshow that the degenerative events associated with hy-perstimulation of non-NMDA type glutamate receptorscan be attenuated by injection of recombinant plasmin-ogen activator inhibitor-1 (rPAI-1) or tPA-STOP intothe vitreous humor along with KA. These results for thefirst time, to our knowledge, indicate that up-regulationof plasminogen activators (both tPA and uPA) play acausative role in retinal damage mediated by hyper-stimulation of glutamate receptors.
MATERIALS AND METHODS
Intravitreal injections
All the experiments on animals were performed under gen-eral anesthesia according to institutional protocol guidelinesand the Association for Research in Vision and Ophthalmol-ogy (ARVO) statement for the use of Animals in Ophthalmol-ogy and Vision Research. Normal adult CD-1 mice (6–8 wkold; Charles River Breeding Labs, Wilmington, MA, USA)were anesthetized by an intraperitoneal injection of 1.25%avertin (2,2,2-tribromoethanol in tert-amyl alcohol; 0.017mL/g body weight). Kainic acid was injected into the vitreoushumor as described previously (65). Unless otherwise indi-cated, intravitreal injections were performed in a final volumeof 2 �L. For control experiments, eyes (n�6 retinas each; 3independent experiments) were injected with 2 �L of 0.1Mphosphate-buffered saline (PBS, pH 7.4) alone and for treat-ment groups, eyes (n�6 retinas each; 3 independent experi-ments) were injected with 2 �L of 5, 10, and 20 mM
(corresponding to 10, 20, and 40 nmol) kainic acid (Sigma,St. Louis, MO, USA) prepared in PBS. For time coursestudies, 20 nmol KA was injected into the vitreous humor andproteins were extracted from 6 retinas each at each timepoint (4 independent experiments). In separate experiments(n�6 retinas each, 3 independent experiments), 2 �L of 10mM KA (corresponding to 20 nmol) plus rPAI-1 (correspond-ing to 0.25 and 5 �g; American Diagnostica, Stamford, CT,USA) were injected into the vitreous humor. In separateexperiments (n�6 retinas each; 3 independent experiments),1 �L of 20 mM KA (corresponding to 20 nmol) plus 1 �L ofa synthetic tPA inhibitor, tPA-STOP (2,7-bis-(4-amidonoben-zylidene)-cycloheptanone-1-one dihydrochloride salt (corre-sponding to 1 and 4 �M; American Diagnositica) or 1 �L ofKA (20 nmol) plus 1 �L of plasmin inhibitor �-2-antiplasmin(corresponding to 4 �g; ICN Biochemicals, Aurora, OH,USA) were injected into the vitreous humor.
Protein extraction
Six hours, 12 h, 1 day, and 2 days after intravitreal injectionanimals were anesthetized with an overdose of avertin, andtheir eyes were enucleated. Retinas were carefully removedand washed three times with PBS (pH 7.4) to remove anyvitreous humor that may have adhered to the retina. Three tofour retinas each were placed in Eppendorff tubes containing40 �L of extraction buffer (1% nonidet-P40, 20 mM Tris-HCl,150 mM NaCl, 1 mM Na3VO4, pH 7.4) and the tissues werehomogenized. Tissue homogenates were centrifuged at10,000 rpm for 5 min at 4°C and the supernatants werecollected. Protein concentration in supernatants was deter-mined using the Bio-Rad protein assay (Bio-Rad Laboratories,Hercules, CA, USA).
Zymography assays
Activities of plasminogen activators (both tPA and uPA) andplasmin were determined by substrate zymography accordingto methods described previously (66). Briefly, retinal extractscontaining equal amounts of protein (25 �g) were mixedwith SDS gel-loading buffer and loaded without reduction orheating onto 10% SDS polyacrylamide gels containing fibrin-ogen (5.5 mg/mL) and plasminogen (50 �g/mL) to deter-mine the activity of plasminogen activators (both tPA anduPA) and gels containing 0.2% �-casein to determine theactivity of plasmin. After electrophoresis, gels were washedthree times with 2.5% Triton-X 100 (15 min each time),placed in 0.1M glycine buffer (pH 8.0; for detection of tPAand uPA) or 0.1M Tris-buffer (pH 8.0; for detection ofplasmin), and incubated overnight at 37°C to allow proteol-ysis of the substrates in the gels. The gels were stained with0.1% Coomassie Brilliant Blue-R250, then destained with asolution containing 25% methanol and 10% acetic acid.Samples containing standard recombinant tPA or plasminwere coelectrophoresed for comparison. tPA activity in zymo-grams was confirmed by incubating the gels with rPAI-1 ortPA-STOP (data not shown) and by Western blot analysis (asdescribed below). A reduced molecular weight size standardwas included on all gels (data not shown; Life Technologies,Gaithersburg, MD, USA. The area cleared by tPA and uPA inthe zymograms was scanned by a flat-bed scanner, relativeprotease activity level was determined using image analysissoftware (Scion Corporation, Frederick, MD, USA), and theresults from four independent experiments were representedas mean arbitrary denistometric units �se. Statistical signifi-cance was analyzed by using a nonparametric Newman-Keulsanalog procedure (GB-Stat Software, Dynamic Microsystems,Silver Spring, MD, USA) and expressed as the mean �se.
1281PLASMINOGEN ACTIVATORS PROMOTE RETINAL DAMAGE
Immunohistochemistry
Eyes enucleated after KA injection were fixed with 4% para-formaldehyde for 1 h at room temperature and embedded inOCT compound (Sakura Finetek USA, Torrance, CA, USA).Traverse, 10 micron-thick cryostat sections were cut andplaced onto superfrost plus slides (Fisher Scientific, Pitts-burgh, PA, USA). Sections were incubated with antibodiesagainst uPA (1:100 dilution in PBS, Innovative Research, MI,USA), calretinin (amacrine cell marker, 1:100 dilution inPBS, Chemicon, CA, USA), and neurofilament light (gan-glion cell marker, 1:100 dilution in PBS, Santa Cruz Biotech-nology, Santa Cruz, CA, USA) to determine the tissue local-ization of these proteins. Sections were washed three times(15 min each) with Tris-HCl buffer (pH 7.4) and incubatedwith appropriate Alexa FluorR-568-conjugated secondary an-tibodies (1:200 dilution in PBS for NF-L and calretinin;Molecular Probes, Eugene, OR, USA) or Alexa FluorR-486-conjugated secondary antibodies (1:200 dilution in PBS foruPA) for 1 h at room temperature. Sections were washedagain with Tris-HCl buffer (three times, 15 min each) andmounted with a coverslip. Sections were observed under aNikon bright-field microscope equipped with epifluores-cence; digitized images were obtained using a SPOT digitalcamera. Images were converted into gray scale images usingAdobe Photoshop Software, versions 5.5 and 7.0 (Adobesystem Inc., Mountain View, CA, USA).
Immunogold labeling of tPA
For tPA immunogold labeling (see Fig. 2), 10 micron-thickcryostat sections (prepared as described above) were incu-bated with a solution containing 10% BSA (in PBS, pH 7.4)and 0.3% Triton X-100 for 1 h at room temperature. Sectionswere washed three times with PBS (15 min each) andincubated overnight with polyclonal antibodies against tPA(1:100 dilution; Innovative Research, Southfield, MI, USA;antibody was diluted in a solution containing 1% BSA and0.3% Triton X-100 in PBS). The next morning sections were
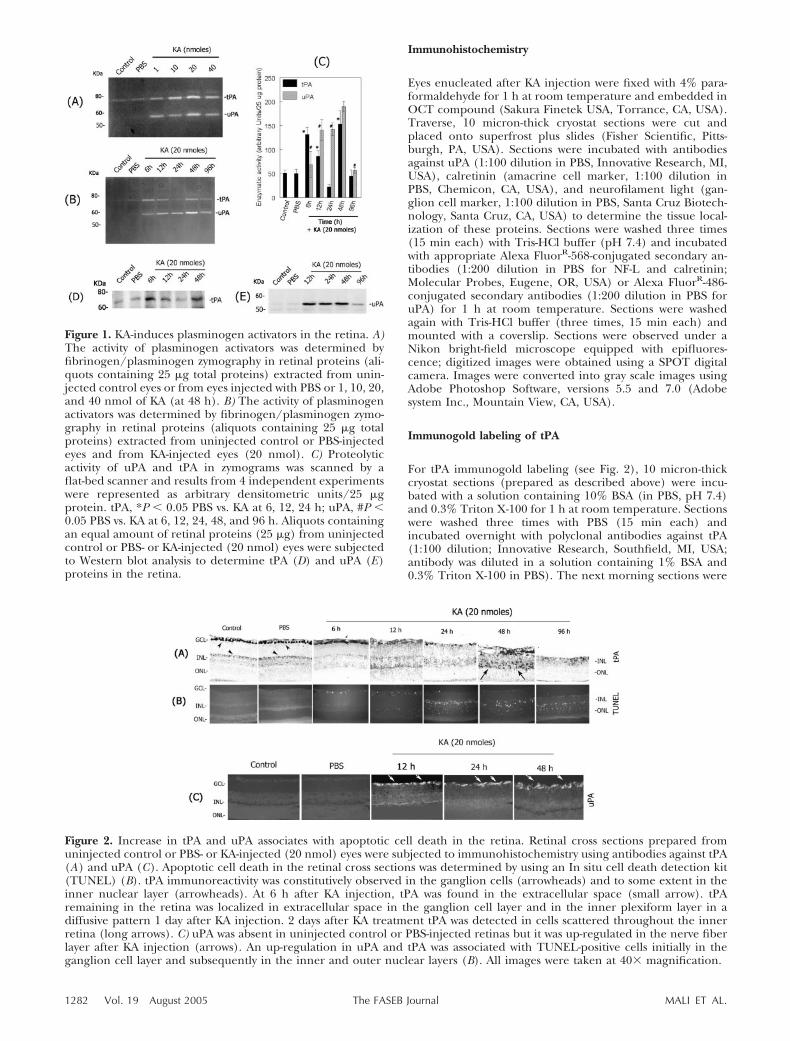
Figure 1. KA-induces plasminogen activators in the retina. A)The activity of plasminogen activators was determined byfibrinogen/plasminogen zymography in retinal proteins (ali-quots containing 25 �g total proteins) extracted from unin-jected control eyes or from eyes injected with PBS or 1, 10, 20,and 40 nmol of KA (at 48 h). B) The activity of plasminogenactivators was determined by fibrinogen/plasminogen zymo-graphy in retinal proteins (aliquots containing 25 �g totalproteins) extracted from uninjected control or PBS-injectedeyes and from KA-injected eyes (20 nmol). C) Proteolyticactivity of uPA and tPA in zymograms was scanned by aflat-bed scanner and results from 4 independent experimentswere represented as arbitrary densitometric units/25 �gprotein. tPA, *P � 0.05 PBS vs. KA at 6, 12, 24 h; uPA, #P �0.05 PBS vs. KA at 6, 12, 24, 48, and 96 h. Aliquots containingan equal amount of retinal proteins (25 �g) from uninjectedcontrol or PBS- or KA-injected (20 nmol) eyes were subjectedto Western blot analysis to determine tPA (D) and uPA (E)proteins in the retina.
Figure 2. Increase in tPA and uPA associates with apoptotic cell death in the retina. Retinal cross sections prepared fromuninjected control or PBS- or KA-injected (20 nmol) eyes were subjected to immunohistochemistry using antibodies against tPA(A) and uPA (C). Apoptotic cell death in the retinal cross sections was determined by using an In situ cell death detection kit(TUNEL) (B). tPA immunoreactivity was constitutively observed in the ganglion cells (arrowheads) and to some extent in theinner nuclear layer (arrowheads). At 6 h after KA injection, tPA was found in the extracellular space (small arrow). tPAremaining in the retina was localized in extracellular space in the ganglion cell layer and in the inner plexiform layer in adiffusive pattern 1 day after KA injection. 2 days after KA treatment tPA was detected in cells scattered throughout the innerretina (long arrows). C) uPA was absent in uninjected control or PBS-injected retinas but it was up-regulated in the nerve fiberlayer after KA injection (arrows). An up-regulation in uPA and tPA was associated with TUNEL-positive cells initially in theganglion cell layer and subsequently in the inner and outer nuclear layers (B). All images were taken at 40� magnification.
1282 Vol. 19 August 2005 MALI ET AL.The FASEB Journal

washed three times with PBS (30 min each) and incubatedwith goat anti-rabbit IgG conjugated to 5 nm gold particles(1:100 dilution; Accurate Chemical and Scientific Corpora-tion, Westbury, NY, USA) in 1% BSA containing 0.3% Triton-X-100) for 1 h at room temperature. Sections were washedfour times (20 min each) with PBS and four times (20 mineach) with distilled water and silver enhancement was per-formed on the sections according to manufacturer’s instruc-tions (Silver Enhancement Kit light Microscopy, AccurateChemical and Scientific Corporation, Westbury, NY, USA).Finally, sections were mounted using standard aqueousmounting solution and observed under a bright-field micro-scope. Digitized images were obtained using a SpotR digitalcamera attached to the microscope and compiled usingPhotoshop software (Adobe System Inc.).
Apoptosis assay
Apoptotic cell death in retinal cross sections was determinedusing a commercially available kit. Briefly, 10 micron-thickcryostat sections (n�8–10 sections for each treatment from 4independent experiments) prepared as described above andapoptotic cell death was detected by TdT-mediated dUTPnick-end labeling (TUNEL) assay, using an In situ cell deathdetection kit with fluorescein (Roche Biochemicals, Mann-heim, Germany) and the protocol provided by the manufac-turer. Tissue sections were examined using a Nikon micro-scope equipped with epifluorescence; digital images wereobtained with a SPOT digital camera and compiled usingAdobe Photoshop Software, versions 5.5 and 7.0 (AdobeSystem). The remaining number of TUNEL-positive cells inretinal cross sections was quantitated using image analysissoftware (Scion Corporation); results were represented astotal number of TUNEL-positive cells, mean �se. Statisticalsignificance was analyzed by using a nonparametric Newman-Keuls analog procedure (GB-Stat Software, Dynamic Micro-systems).
Western blot analysis
Aliquots containing an equal amount of retinal proteinextracts (25 �g) were mixed with gel loading buffer andseparated on 10% SDS-polyacrylamide gels. After electro-phoresis, the proteins were transferred onto nylon mem-branes and nonspecific binding was blocked with 10% nonfatdry milk in Tris-buffered saline containing 0.1% Tween-20(TBS-T). Membranes were then probed with antibodiesagainst tPA (1:1000 dilution; Innovative Research, Southfield,MI, USA), uPA (1:2000 dilution; Innovative Research), andplasminogen (1:2000; Innovative Research). After incubationwith the primary antibodies, membranes were washed withTBS-T and incubated with appropriate horse radish peroxi-dase (HRP) -conjugated secondary antibodies (1:4000 dilu-tion; Santa Cruz Biotechnology) for 1 h at room temperature.Finally, the proteins on the membranes were detected usingan ECL chemiluminescence kit (Amersham Pharmacia Bio-tech, Piscataway, NJ, USA) and exposing the membranes toX-ray film. Recombinant uPA, tPA, and plasminogen werecoelectrophoresed as positive standards (data not shown).
Retrograde labeling of retinal ganglion cells
Ganglion cells were retrogradely labeled as described previ-ously (65, 67). Briefly, 1.5 �L of a 5% solution of Aminostil-bamidine (Molecular Probes) in PBS was injected into thesuperior colliculi of anesthetized mice using a stereotaxicapparatus. KA was injected into the vitreous humor 1 wk afterAminostilbamidine application. Various times after KA injec-
tion, the animals were anesthetized and their eyes wereenucleated and fixed in 4% paraformaldehyde for 1 h atroom temperature. Retinas were detached from the eye cupsand rinsed with PBS (two times, 15 min each). After rinsing,retinas were overlaid on a glass slide and four small incisionswere made at the periphery to flatten the retina. The retinaswere mounted with coverslips using an aqueous mountingmedium (containing an anti-fading agent; GEL/MOUNT;Biomeda Corporation, Foster City, CA, USA). Alternatively,retinas were detached from the eyecups, embedded in OCTcompound and processed for preparation of retinal crosssections as described above. Ten micron-thick retinal crosssections were prepared and aminostilbamidine-positive gan-glion cells were observed under a fluorescence microscope(Nikon, Tokyo, Japan). Retinal ganglion cells located approx-imately the same distance from the optic disk were observedunder a fluorescence microscope (�155 sq. microns, 40�magnification) and photographed using a SpotR digital cam-era attached to the fluorescence microscope.
RESULTS
Kainic acid up-regulates tPA and uPA in the retinaand contributes to retinal damage
To determine whether hyperstimulation of non-NMDA-type glutamate receptors modulates tPA and uPA levelsin the retina, KA was injected into the vitreous humorin CD-1 mice and total retinal proteins were extracted.tPA and uPA activity in retinal protein extracts wasdetermined by fibrinogen/plasminogen zymographyassays. Zymography assays indicated a very low andconstitutive level of tPA activity in control or PBS-injected eyes (Fig. 1A). In contrast, KA injected eyesshowed a dose-dependent (Fig. 1A) and time-related(Fig. 1B) increase in tPA activity in the retina. tPAactivity was increased as early as 6 h after KA injection,returned to lower levels by 1 day, and increased again at2 days after KA injection (Fig. 1B). This type of tPAactivity profile was consistently observed in four inde-pendent experiments as shown by arbitrary densitomet-ric units (Fig. 1C). Western blot analysis of retinalprotein extracts from a similar experiment confirmedthe expression pattern of tPA in the retina (Fig. 1D).Although low levels of PAI-1 and PAI-2 were observedconstitutively in retinal proteins extracted from unin-jected control or PBS-injected eyes, Western blot anal-ysis indicated no significant change in PAI-1 and PAI-2proteins in KA injected eyes (at 6, 12, 24, 48, and 96 h)compared with uninjected control or PBS-injected eyes(data not shown). Zymography assays indicated theabsence of uPA activity in retinal proteins extractedfrom uninjected control retinas (Fig. 1A, B). In con-trast, intravitreal injection of KA led to a transientup-regulation in uPA activity in the retina. uPA levelswere induced as early as 6 h after KA injection, reacheda peak between 1 and 2 days, and returned to lowerlevels at day 4 (Fig. 1B, C).
To determine the cell types that synthesize tPA anduPA, immunohistochemical analysis was performed onretinal cross sections prepared at 6, 12, 24, 48, and 96 h
1283PLASMINOGEN ACTIVATORS PROMOTE RETINAL DAMAGE
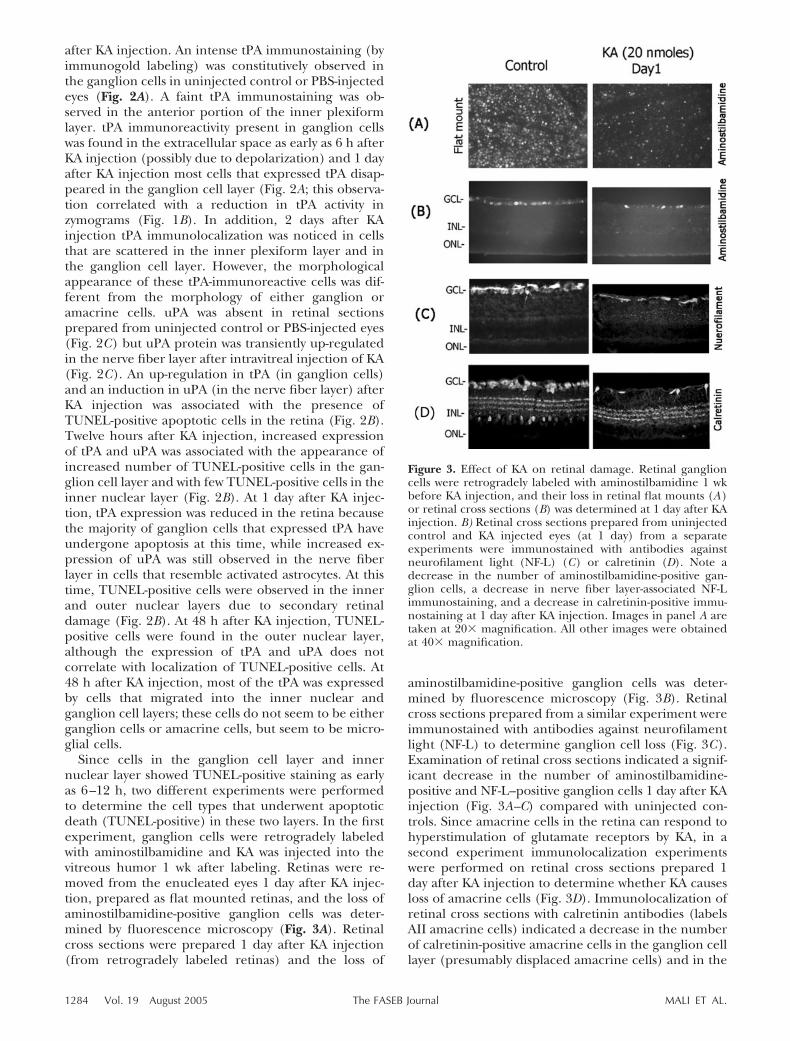
after KA injection. An intense tPA immunostaining (byimmunogold labeling) was constitutively observed inthe ganglion cells in uninjected control or PBS-injectedeyes (Fig. 2A). A faint tPA immunostaining was ob-served in the anterior portion of the inner plexiformlayer. tPA immunoreactivity present in ganglion cellswas found in the extracellular space as early as 6 h afterKA injection (possibly due to depolarization) and 1 dayafter KA injection most cells that expressed tPA disap-peared in the ganglion cell layer (Fig. 2A; this observa-tion correlated with a reduction in tPA activity inzymograms (Fig. 1B). In addition, 2 days after KAinjection tPA immunolocalization was noticed in cellsthat are scattered in the inner plexiform layer and inthe ganglion cell layer. However, the morphologicalappearance of these tPA-immunoreactive cells was dif-ferent from the morphology of either ganglion oramacrine cells. uPA was absent in retinal sectionsprepared from uninjected control or PBS-injected eyes(Fig. 2C) but uPA protein was transiently up-regulatedin the nerve fiber layer after intravitreal injection of KA(Fig. 2C). An up-regulation in tPA (in ganglion cells)and an induction in uPA (in the nerve fiber layer) afterKA injection was associated with the presence ofTUNEL-positive apoptotic cells in the retina (Fig. 2B).Twelve hours after KA injection, increased expressionof tPA and uPA was associated with the appearance ofincreased number of TUNEL-positive cells in the gan-glion cell layer and with few TUNEL-positive cells in theinner nuclear layer (Fig. 2B). At 1 day after KA injec-tion, tPA expression was reduced in the retina becausethe majority of ganglion cells that expressed tPA haveundergone apoptosis at this time, while increased ex-pression of uPA was still observed in the nerve fiberlayer in cells that resemble activated astrocytes. At thistime, TUNEL-positive cells were observed in the innerand outer nuclear layers due to secondary retinaldamage (Fig. 2B). At 48 h after KA injection, TUNEL-positive cells were found in the outer nuclear layer,although the expression of tPA and uPA does notcorrelate with localization of TUNEL-positive cells. At48 h after KA injection, most of the tPA was expressedby cells that migrated into the inner nuclear andganglion cell layers; these cells do not seem to be eitherganglion cells or amacrine cells, but seem to be micro-glial cells.
Since cells in the ganglion cell layer and innernuclear layer showed TUNEL-positive staining as earlyas 6–12 h, two different experiments were performedto determine the cell types that underwent apoptoticdeath (TUNEL-positive) in these two layers. In the firstexperiment, ganglion cells were retrogradely labeledwith aminostilbamidine and KA was injected into thevitreous humor 1 wk after labeling. Retinas were re-moved from the enucleated eyes 1 day after KA injec-tion, prepared as flat mounted retinas, and the loss ofaminostilbamidine-positive ganglion cells was deter-mined by fluorescence microscopy (Fig. 3A). Retinalcross sections were prepared 1 day after KA injection(from retrogradely labeled retinas) and the loss of
aminostilbamidine-positive ganglion cells was deter-mined by fluorescence microscopy (Fig. 3B). Retinalcross sections prepared from a similar experiment wereimmunostained with antibodies against neurofilamentlight (NF-L) to determine ganglion cell loss (Fig. 3C).Examination of retinal cross sections indicated a signif-icant decrease in the number of aminostilbamidine-positive and NF-L–positive ganglion cells 1 day after KAinjection (Fig. 3A–C) compared with uninjected con-trols. Since amacrine cells in the retina can respond tohyperstimulation of glutamate receptors by KA, in asecond experiment immunolocalization experimentswere performed on retinal cross sections prepared 1day after KA injection to determine whether KA causesloss of amacrine cells (Fig. 3D). Immunolocalization ofretinal cross sections with calretinin antibodies (labelsAII amacrine cells) indicated a decrease in the numberof calretinin-positive amacrine cells in the ganglion celllayer (presumably displaced amacrine cells) and in the
Figure 3. Effect of KA on retinal damage. Retinal ganglioncells were retrogradely labeled with aminostilbamidine 1 wkbefore KA injection, and their loss in retinal flat mounts (A)or retinal cross sections (B) was determined at 1 day after KAinjection. B) Retinal cross sections prepared from uninjectedcontrol and KA injected eyes (at 1 day) from a separateexperiments were immunostained with antibodies againstneurofilament light (NF-L) (C) or calretinin (D). Note adecrease in the number of aminostilbamidine-positive gan-glion cells, a decrease in nerve fiber layer-associated NF-Limmunostaining, and a decrease in calretinin-positive immu-nostaining at 1 day after KA injection. Images in panel A aretaken at 20� magnification. All other images were obtainedat 40� magnification.
1284 Vol. 19 August 2005 MALI ET AL.The FASEB Journal
inner nuclear layer (Fig. 3D). These results indicatethat intravitreal injection of KA leads to an up-regula-tion in tPA and uPA activity in the retina and causes lossof ganglion cells and amacrine cells by an apoptoticmechanism.
Kainic acid induces plasminogen activation
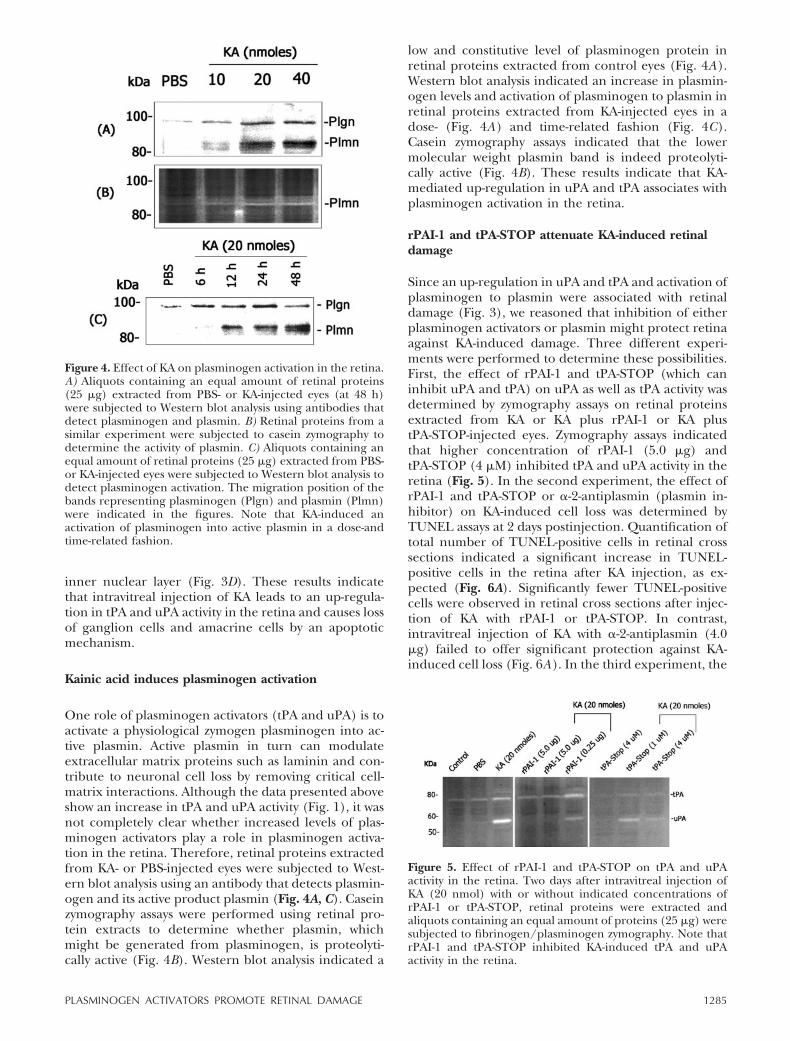
One role of plasminogen activators (tPA and uPA) is toactivate a physiological zymogen plasminogen into ac-tive plasmin. Active plasmin in turn can modulateextracellular matrix proteins such as laminin and con-tribute to neuronal cell loss by removing critical cell-matrix interactions. Although the data presented aboveshow an increase in tPA and uPA activity (Fig. 1), it wasnot completely clear whether increased levels of plas-minogen activators play a role in plasminogen activa-tion in the retina. Therefore, retinal proteins extractedfrom KA- or PBS-injected eyes were subjected to West-ern blot analysis using an antibody that detects plasmin-ogen and its active product plasmin (Fig. 4A, C). Caseinzymography assays were performed using retinal pro-tein extracts to determine whether plasmin, whichmight be generated from plasminogen, is proteolyti-cally active (Fig. 4B). Western blot analysis indicated a
low and constitutive level of plasminogen protein inretinal proteins extracted from control eyes (Fig. 4A).Western blot analysis indicated an increase in plasmin-ogen levels and activation of plasminogen to plasmin inretinal proteins extracted from KA-injected eyes in adose- (Fig. 4A) and time-related fashion (Fig. 4C).Casein zymography assays indicated that the lowermolecular weight plasmin band is indeed proteolyti-cally active (Fig. 4B). These results indicate that KA-mediated up-regulation in uPA and tPA associates withplasminogen activation in the retina.
rPAI-1 and tPA-STOP attenuate KA-induced retinaldamage
Since an up-regulation in uPA and tPA and activation ofplasminogen to plasmin were associated with retinaldamage (Fig. 3), we reasoned that inhibition of eitherplasminogen activators or plasmin might protect retinaagainst KA-induced damage. Three different experi-ments were performed to determine these possibilities.First, the effect of rPAI-1 and tPA-STOP (which caninhibit uPA and tPA) on uPA as well as tPA activity wasdetermined by zymography assays on retinal proteinsextracted from KA or KA plus rPAI-1 or KA plustPA-STOP-injected eyes. Zymography assays indicatedthat higher concentration of rPAI-1 (5.0 �g) andtPA-STOP (4 �M) inhibited tPA and uPA activity in theretina (Fig. 5). In the second experiment, the effect ofrPAI-1 and tPA-STOP or �-2-antiplasmin (plasmin in-hibitor) on KA-induced cell loss was determined byTUNEL assays at 2 days postinjection. Quantification oftotal number of TUNEL-positive cells in retinal crosssections indicated a significant increase in TUNEL-positive cells in the retina after KA injection, as ex-pected (Fig. 6A). Significantly fewer TUNEL-positivecells were observed in retinal cross sections after injec-tion of KA with rPAI-1 or tPA-STOP. In contrast,intravitreal injection of KA with �-2-antiplasmin (4.0�g) failed to offer significant protection against KA-induced cell loss (Fig. 6A). In the third experiment, the
Figure 5. Effect of rPAI-1 and tPA-STOP on tPA and uPAactivity in the retina. Two days after intravitreal injection ofKA (20 nmol) with or without indicated concentrations ofrPAI-1 or tPA-STOP, retinal proteins were extracted andaliquots containing an equal amount of proteins (25 �g) weresubjected to fibrinogen/plasminogen zymography. Note thatrPAI-1 and tPA-STOP inhibited KA-induced tPA and uPAactivity in the retina.
Figure 4. Effect of KA on plasminogen activation in the retina.A) Aliquots containing an equal amount of retinal proteins(25 �g) extracted from PBS- or KA-injected eyes (at 48 h)were subjected to Western blot analysis using antibodies thatdetect plasminogen and plasmin. B) Retinal proteins from asimilar experiment were subjected to casein zymography todetermine the activity of plasmin. C) Aliquots containing anequal amount of retinal proteins (25 �g) extracted from PBS-or KA-injected eyes were subjected to Western blot analysis todetect plasminogen activation. The migration position of thebands representing plasminogen (Plgn) and plasmin (Plmn)were indicated in the figures. Note that KA-induced anactivation of plasminogen into active plasmin in a dose-andtime-related fashion.
1285PLASMINOGEN ACTIVATORS PROMOTE RETINAL DAMAGE
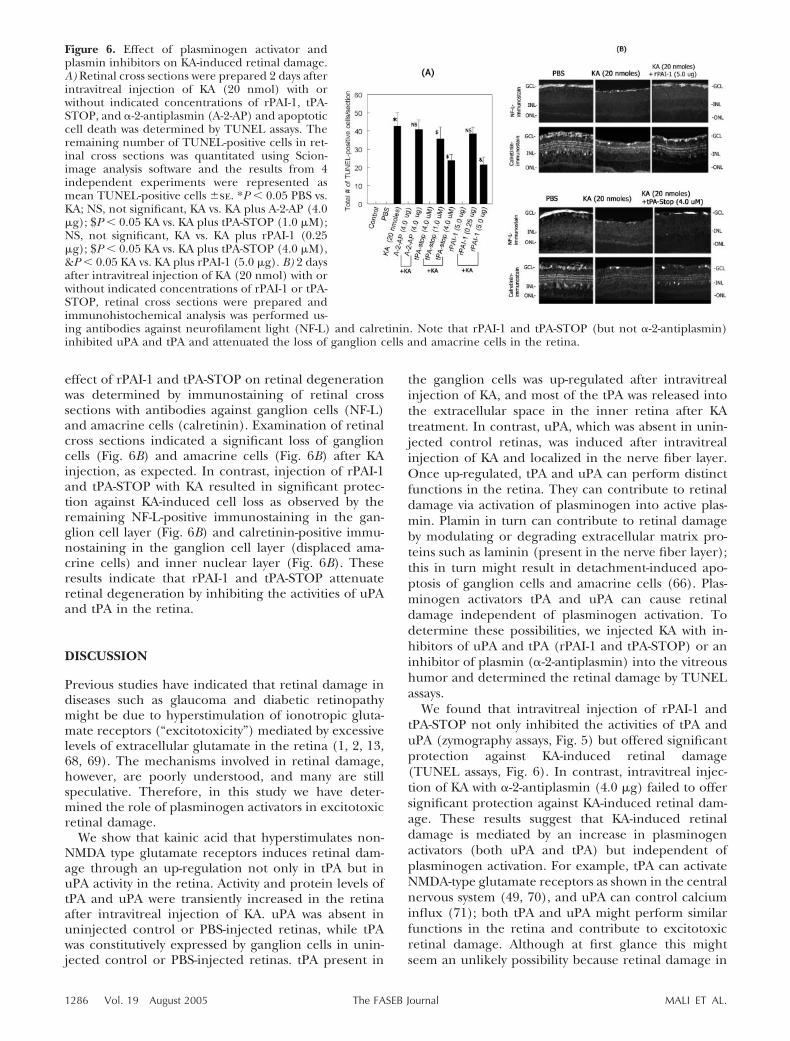
effect of rPAI-1 and tPA-STOP on retinal degenerationwas determined by immunostaining of retinal crosssections with antibodies against ganglion cells (NF-L)and amacrine cells (calretinin). Examination of retinalcross sections indicated a significant loss of ganglioncells (Fig. 6B) and amacrine cells (Fig. 6B) after KAinjection, as expected. In contrast, injection of rPAI-1and tPA-STOP with KA resulted in significant protec-tion against KA-induced cell loss as observed by theremaining NF-L-positive immunostaining in the gan-glion cell layer (Fig. 6B) and calretinin-positive immu-nostaining in the ganglion cell layer (displaced ama-crine cells) and inner nuclear layer (Fig. 6B). Theseresults indicate that rPAI-1 and tPA-STOP attenuateretinal degeneration by inhibiting the activities of uPAand tPA in the retina.
DISCUSSION
Previous studies have indicated that retinal damage indiseases such as glaucoma and diabetic retinopathymight be due to hyperstimulation of ionotropic gluta-mate receptors (“excitotoxicity”) mediated by excessivelevels of extracellular glutamate in the retina (1, 2, 13,68, 69). The mechanisms involved in retinal damage,however, are poorly understood, and many are stillspeculative. Therefore, in this study we have deter-mined the role of plasminogen activators in excitotoxicretinal damage.
We show that kainic acid that hyperstimulates non-NMDA type glutamate receptors induces retinal dam-age through an up-regulation not only in tPA but inuPA activity in the retina. Activity and protein levels oftPA and uPA were transiently increased in the retinaafter intravitreal injection of KA. uPA was absent inuninjected control or PBS-injected retinas, while tPAwas constitutively expressed by ganglion cells in unin-jected control or PBS-injected retinas. tPA present in
the ganglion cells was up-regulated after intravitrealinjection of KA, and most of the tPA was released intothe extracellular space in the inner retina after KAtreatment. In contrast, uPA, which was absent in unin-jected control retinas, was induced after intravitrealinjection of KA and localized in the nerve fiber layer.Once up-regulated, tPA and uPA can perform distinctfunctions in the retina. They can contribute to retinaldamage via activation of plasminogen into active plas-min. Plamin in turn can contribute to retinal damageby modulating or degrading extracellular matrix pro-teins such as laminin (present in the nerve fiber layer);this in turn might result in detachment-induced apo-ptosis of ganglion cells and amacrine cells (66). Plas-minogen activators tPA and uPA can cause retinaldamage independent of plasminogen activation. Todetermine these possibilities, we injected KA with in-hibitors of uPA and tPA (rPAI-1 and tPA-STOP) or aninhibitor of plasmin (�-2-antiplasmin) into the vitreoushumor and determined the retinal damage by TUNELassays.
We found that intravitreal injection of rPAI-1 andtPA-STOP not only inhibited the activities of tPA anduPA (zymography assays, Fig. 5) but offered significantprotection against KA-induced retinal damage(TUNEL assays, Fig. 6). In contrast, intravitreal injec-tion of KA with �-2-antiplasmin (4.0 �g) failed to offersignificant protection against KA-induced retinal dam-age. These results suggest that KA-induced retinaldamage is mediated by an increase in plasminogenactivators (both uPA and tPA) but independent ofplasminogen activation. For example, tPA can activateNMDA-type glutamate receptors as shown in the centralnervous system (49, 70), and uPA can control calciuminflux (71); both tPA and uPA might perform similarfunctions in the retina and contribute to excitotoxicretinal damage. Although at first glance this mightseem an unlikely possibility because retinal damage in
Figure 6. Effect of plasminogen activator andplasmin inhibitors on KA-induced retinal damage.A) Retinal cross sections were prepared 2 days afterintravitreal injection of KA (20 nmol) with orwithout indicated concentrations of rPAI-1, tPA-STOP, and �-2-antiplasmin (A-2-AP) and apoptoticcell death was determined by TUNEL assays. Theremaining number of TUNEL-positive cells in ret-inal cross sections was quantitated using Scion-image analysis software and the results from 4independent experiments were represented asmean TUNEL-positive cells �se. *P � 0.05 PBS vs.KA; NS, not significant, KA vs. KA plus A-2-AP (4.0�g); $P � 0.05 KA vs. KA plus tPA-STOP (1.0 �M);NS, not significant, KA vs. KA plus rPAI-1 (0.25�g); $P � 0.05 KA vs. KA plus tPA-STOP (4.0 �M),&P � 0.05 KA vs. KA plus rPAI-1 (5.0 �g). B) 2 daysafter intravitreal injection of KA (20 nmol) with orwithout indicated concentrations of rPAI-1 or tPA-STOP, retinal cross sections were prepared andimmunohistochemical analysis was performed us-ing antibodies against neurofilament light (NF-L) and calretinin. Note that rPAI-1 and tPA-STOP (but not �-2-antiplasmin)inhibited uPA and tPA and attenuated the loss of ganglion cells and amacrine cells in the retina.
1286 Vol. 19 August 2005 MALI ET AL.The FASEB Journal

this study was determined after hyperstimulation ofnon-NMDA type receptors (by KA), ganglion cells andamacrine cells express both NMDA and non-NMDAtype glutamate receptors; tPA up-regulated in the ret-ina after KA injection can proteolytically processNMDA-type glutamate receptors and might contributedirectly to retinal damage. Although we found anincrease in uPA activity and protein levels and anassociation of uPA with plasminogen activation, little isknown about the additional roles of uPA in the retina atthis time. Studies aimed in this direction are necessaryto delineate additional roles of plasminogen activatorsin the retina. Although inhibition of plasmin activitycan attenuate detachment-induced apoptosis, in thepresence of increased amounts of excitotoxin such askainic acid and in the presence of increased levels ofuPA and tPA, inhibition of plasmin alone does notseem to confer protection against retinal damage.
In a previous study we observed that the optic nerveligation-induced retinal damage is dependent on plas-minogen activation; in this study, we found that KA-induced retinal damage is independent of plasminogenactivation. Although the exact mechanisms for thedifferential role of plasminogen activation in retinaldamage are not clear, there is one major differencebetween the animal model employed in this study(intravitreal injection of kainic acid) and the animalmodel used in our previous study (optic nerve ligation).In our earlier study, relatively higher plasminogenlevels were found in the retina after optic nerve ligationcompared with levels found in the retina after kainicacid injection into the vitreous humor. The increase inplasminogen levels found in the retina after optic nerveligation was due to the damage to the central retinalartery, compromise in blood retinal barrier (BRB,determined indirectly by albumin Western blot analy-sis) and leakage of plasminogen into the retina,whereas similar compromise in BRB was not observedafter intravitreal injection of kainic acid (data notshown). Clearly, plasminogen activation seems to play adifferential role in retinal damage depending on themodel system used.
We made an intriguing observation in this study withregards to the activity and protein profile of tPA duringtime course experiments. Constitutive levels of tPApresent in the uninjected control or PBS-injected eyeswere increased as early 6–12 h after KA injection,decreased by 24 h, and increased again at 48 h (Fig. 1).This observation can be explained by two possibilities.First, a reduction in tPA activity could simply be due tothe loss of ganglion cells that contributed to tPAproduction in the retina. This possibility was supportedby immunohistochemical analysis of tPA in Fig. 2. Datain Fig. 2 show that tPA present in the ganglion cells wasgradually released into the extracellular space startingat 6–12 h possibly due to membrane depolarization(72). At 24 h after KA injection, the majority of theganglion cells that expressed tPA were killed by KA,hence zymography and Western blot assays indicatedreduced levels of tPA in the retina. Second, tPA remain-
ing in the extracellular space at 24 h KA injection couldbe recycled by glial cells (48), degraded by additionalproteolytic mechanisms, or inhibited by protease inhib-itors such as neuroserpin. Although speculative at thistime, KA could activate metabotropic glutamate recep-tors (due to the release of endogenous l-glutamate bydying neurons), and these receptors in turn couldcounteract up-regulation of tPA mediated by non-NMDA receptors.
In this study we found that two different cell typessynthesize tPA in the retina. This was supported byimmunolocalization studies for tPA on retinal crosssections prepared 48 and 96 h after KA injection (Fig.2). The results presented in Fig. 2 indicate that 48 hafter KA injection, a different cell type showed tPA-positive staining in the retina. tPA-positive cells werefound throughout the inner retina in a scattered fash-ion; they were morphologically different from ganglioncells and morphology of tPA-positive cells resemblesthat of microglial cells. Although coimmunolocaliza-tion experiments could have been a better approach todetermine the origin of tPA in the retina, these exper-iments were unsuccessful due to technical difficultiesand nonavailability of suitable antibodies. The resultspresented above, however, indicate that two differentcell types, ganglion cells and microglial cells (73),contribute to the origin of tPA in the retina. Althoughthe exact reason why microglial cells synthesize tPAafter KA injection is not clear, they might synthesizetPA and use it to migrate into the inner retina toremove debris from dying cells (74–76), but they cancontribute to secondary retinal damage. It is also pos-sible that microglial cells can be migrated into theinner retina independent of tPA up-regulation (32, 73,77). Although spatial expression of plasminogen acti-vators in the retina (at 24 h to 96 h) does not seem tocorrelate with the localization of TUNEL-positive cells,the presence of TUNEL-positive cells in the inner andouter nuclear layers could be due to secondary eventsof damage subsequent to the primary events initiated inthe ganglion cell layer, which then continues to affectcells in remaining retinal layers. Secondary degenera-tion can be mediated by factors that could be releasedby degenerating ganglion and amacrine cells, includingendogenous glutamate and cytokines such as tumornecrosis factor-� and interleukin-1�.
In addition to up-regulation of tPA and uPA, hyper-stimulation of non-NMDA receptors by KA can result inup-regulation of other proteases such as matrix metal-loproteinases (MMPs), as previously reported (65).This seems quite plausible because both rPAI-1 andtPA-STOP failed to completely protect the KA-mediatedretinal damage in this study. Previous studies from thislaboratory have reported that inhibition of MMP activ-ity alone do not offer complete protection againstKA-mediated retinal damage (65). These results sug-gest that MMPs and plasminogen activators mightcollectively play a role in excitotoxic retinal damage. Atthis time, we cannot rule out the possible role of otherproteases that can contribute to retinal damage.
1287PLASMINOGEN ACTIVATORS PROMOTE RETINAL DAMAGE

Although there is some evidence in the CNS regard-ing the role of tPA in neuronal damage (30, 32), thenovelty of the observations made in this study is thatup-regulation of not only tPA but also uPA plays acausative role in retinal damage in response to intrav-itreal injection of KA, which hyperstimulates glutamatereceptors. As indicated above, in the absence of a clearunderstanding of the mechanisms involved in ischemiaor/and excitotoxic retinal damage (due to hyperstimu-lation of glutamate receptors), the results provided inthis study suggest that strategies aimed at reducing theactivity of plasminogen activators might offer protec-tion against retinal damage in blinding retinal diseasesin which glutamate receptor-mediated excitotoxicityhas been implicated as a causative factor (6, 8, 28, 68,69).
This work was supported by National Institutes projectgrant EY13643 (to S.K.C.) and Vision Research InfrastructureDevelopment Grant EY014803.
REFERENCES.
1. Osborne, N. N., Melena, J., Chidlow, G., and Wood, J. P. (2001)A hypothesis to explain ganglion cell death caused by vascularinsults at the optic nerve head: possible implication for thetreatment of glaucoma. Br. J. Ophthalmol. 85, 1252–1259
2. Osborne, N. N., Casson, R. J., Wood, J. P., Chidlow, G., Graham,M., and Melena, J. (2004) Retinal ischemia: mechanisms ofdamage and potential therapeutic strategies. Prog. Retin. Eye Res.23, 91–147
3. Flammer, J., Orgul, S., Costa, V. P., Orzalesi, N., Krieglstein,G. K., Serra, L. M., Renard, J. P., and Stefansson, E. (2002) Theimpact of ocular blood flow in glaucoma. Prog. Retin. Eye Res. 21,359–393
4. Selles-Navarro, I., Villegas-Perez, M. P., Salvador-Silva, M., Ruiz-Gomez, J. M., and Vidal-Sanz, M. (1996) Retinal ganglion celldeath after different transient periods of pressure-inducedischemia and survival intervals. A quantitative in vivo study.Invest. Ophthalmol. Vis. Sci. 37, 2002–2014
5. Osborne, N. N., and Larsen, A. K. (1996) Antigens associatedwith specific retinal cells are affected by ischaemia caused byraised intraocular pressure: effect of glutamate antagonists.Neurochem. Int. 29, 263–270
6. Dreyer, E. B., Zurakowski, D., Schumer, R. A., Podos, S. M., andLipton, S. A. (1996) Elevated glutamate levels in the vitreousbody of humans and monkeys with glaucoma. Arch. Ophthalmol.114, 299–305
7. Lipton, S. A. (2003) Possible role for memantine in protectingretinal ganglion cells from glaucomatous damage. Surv. Ophthal-mol. 48, Suppl. 1, S38–S46
8. Ambati, J., Chalam, K. V., Chawla, D. K., D'Angio, C. T., Guillet,E. G., Rose, S. J., Vanderlinde, R. E., and Ambati, B. K. (1997)Elevated gamma-aminobutyric acid, glutamate, and vascular endo-thelial growth factor levels in the vitreous of patients with prolifer-ative diabetic retinopathy. Arch. Ophthalmol. 115, 1161–1166
9. Barber, A. J. (2003) A new view of diabetic retinopathy: aneurodegenerative disease of the eye. Prog. Neuropsychopharma-col. Biol. Psychiatry 27, 283–290
10. Schwarcz, R., and Coyle, J. T. (1977) Kainic acid: neurotoxic effectsafter intraocular injection. Invest. Ophthalmol. Vis. Sci. 16, 141–148
11. Sucher, N. J., Lipton, S. A., and Dreyer, E. B. (1997) Molecularbasis of glutamate toxicity in retinal ganglion cells. Vision Res.37, 3483–3493
12. Nakanishi, S. (1992) Molecular diversity of glutamate receptorsand implications for brain function. Science 258, 597–603
13. Osborne, N. N., Ugarte, M., Chao, M., Chidlow, G., Bae, J. H.,Wood, J. P., and Nash, M. S. (1999) Neuroprotection in relationto retinal ischemia and relevance to glaucoma. Surv. Ophthalmol.43, Suppl. 1, S102–S128
14. Osborne, N. N., Larsen, A., and Barnett, N. L. (1995) Influenceof excitatory amino acids and ischemia on rat retinal cholineacetyltransferase-containing cells. Invest. Ophthalmol. Vis. Sci. 36,1692–1700
15. Brandstatter, J. H., Hartveit, E., Sassoe-Pognetto, M., andWassle, H. (1994) Expression of NMDA and high-affinity kai-nate receptor subunit mRNAs in the adult rat retina. Eur.J. Neurosci. 6, 1100–1112
16. Fletcher, E. L., and Kalloniatis, M. (1997) Localisation of aminoacid neurotransmitters during postnatal development of the ratretina. J. Comp. Neurol. 380, 449–471
17. Lucas, D. R., and Newhouse, J. P. (1957) The toxic effects sosodium l-glutamate on the inner layers of the retina. Arch.Ophthalmol. 58, 193–201
18. Olney, J. W. (1969) Glutamate-induced retinal degeneration inneonatal mice. Electron microscopy of the acutely evolvinglesion. J. Neuropathol. Exp. Neurol. 28, 455–474
19. Sisk, D. R., Kuwabara, T., and Kirsch, A. D. (1984) Behavioralrecovery in albino rats with glutamate-damaged retinas. Invest.Ophthalmol. Vis. Sci. 25, 1124–1128
20. Sisk, D. R., and Kuwabara, T. (1985) Histologic changes in theinner retina of albino rats following intravitreal injection ofmonosodium l-glutamate. Graefes Arch. Clin. Exp. Ophthalmol.223, 250–258
21. Morgan, I. G., and Ingham, C. A. (1981) Kainic acid affects bothplexiform layers of chicken retina. Neurosci. Lett. 21, 275–280
22. Izumi, Y., Benz, A. M., Kurby, C. O., Labruyere, J., Zorumski, C. F.,Price, M. T., and Olney, J. W. (1995) An ex vivo rat retinal preparationfor excitotoxicity studies. J. Neurosci. Methods 60, 219–225
23. Izumi, Y., Kirby-Sharkey, C. O., Benz, A. M., Labruyere, J., Price,M. T., Wozniak, D. F., Zorumski, C. F., and Olney, J. W. (1995)Age dependent sensitivity of the rat retina to the excitotoxicaction of N-methyl-d-aspartate. Neurobiol. Dis. 2, 139–144
24. Siliprandi, R., Canella, R., Carmignoto, G., Schiavo, N., Zanellato,A., Zanoni, R., and Vantini, G. (1992) N-methyl-d-aspartate-in-duced neurotoxicity in the adult rat retina. Vis. Neurosci. 8, 567–573
25. Manabe, S., and Lipton, S. A. (2003) Divergent NMDA signalsleading to proapoptotic and antiapoptotic pathways in the ratretina. Invest. Ophthalmol. Vis. Sci. 44, 385–392
26. Yoles, E., and Schwartz, M. (1998) Elevation of intraocularglutamate levels in rats with partial lesion of the optic nerve.Arch. Ophthalmol. 116, 906–910
27. Kim, T. W., Kang, K. B., Choung, H. K., Park, K. H., and Kim, D. M.(2000) Elevated glutamate levels in the vitreous body of an in vivomodel of optic nerve ischemia. Arch. Ophthalmol. 118, 533–536
28. Vorwerk, C. K., Zurakowski, D., McDermott, L. M., Mawrin, C., andDreyer, E. B. (2004) Effects of axonal injury on ganglion cellsurvival and glutamate homeostasis. Brain Res. Bull. 62, 485–490
29. Brooks, D. E., Garcia, G. A., Dreyer, E. B., Zurakowski, D., andFranco-Bourland, R. E. (1997) Vitreous body glutamate concen-tration in dogs with glaucoma. Am. J. Vet. Res. 58, 864–867
30. Chen, Z. L., and Strickland, S. (1997) Neuronal death in thehippocampus is promoted by plasmin-catalyzed degradation oflaminin. Cell 91, 917–925
31. Tsirka, S. E., Rogove, A. D., Bugge, T. H., Degen, J. L., andStrickland, S. (1997) An extracellular proteolytic cascade pro-motes neuronal degeneration in the mouse hippocampus.J. Neurosci. 17, 543–552
32. Tsirka, S. E., Rogove, A. D., and Strickland, S. (1996) Neuronalcell death and tPA. Nature (London) 384, 123–124
33. Dano, K., Andreasen, P. A., Grondahl-Hansen, J., Kristensen, P.,Nielsen, L. S., and Skriver, L. (1985) Plasminogen activators,tissue degradation, and cancer. Adv. Cancer Res. 44, 139–266
34. Seeds, N. W., Williams, B. L., and Bickford, P. C. (1995) Tissueplasminogen activator induction in Purkinje neurons aftercerebellar motor learning. Science 270, 1992–1994
35. Huang, Y. Y., Bach, M. E., Lipp, H. P., Zhuo, M., Wolfer, D. P.,Hawkins, R. D., Schoonjans, L., Kandel, E. R., Godfraind, J. M.,Mulligan, R., et al. (1996) Mice lacking the gene encoding tissue-type plasminogen activator show a selective interference withlate-phase long-term potentiation in both Schaffer collateral andmossy fiber pathways. Proc. Natl. Acad. Sci. USA 93, 8699–8704
36. Friedman, G. C., and Seeds, N. W. (1995) Tissue plasminogenactivator mRNA expression in granule neurons coincides withtheir migration in the developing cerebellum. J. Comp. Neurol.360, 658–670
1288 Vol. 19 August 2005 MALI ET AL.The FASEB Journal

37. Krystosek, A., and Seeds, N. W. (1981) Plasminogen activatorsecretion by granule neurons in cultures of developing cerebel-lum. Proc. Natl. Acad. Sci. USA 78, 7810–7814
38. Krystosek, A., and Seeds, N. W. (1981) Plasminogen activatorrelease at the neuronal growth cone. Science 213, 1532–1534
39. Moonen, G., Grau-Wagemans, M. P., and Selak, I. (1982)Plasminogen activator-plasmin system and neuronal migration.Nature (London) 298, 753–755
40. Nakajima, K., Reddington, M., Kohsaka, S., and Kreutzberg,G. W. (1996) Induction of urokinase-type plasminogen activatorin rat facial nucleus by axotomy of the facial nerve. J. Neurochem.66, 2500–2505
41. Majka, S., McGuire, P., Colombo, S., and Das, A. (2001) Thebalance between proteinases and inhibitors in a murine model ofproliferative retinopathy. Invest. Ophthalmol. Vis. Sci. 42, 210–215
42. Das, A., McGuire, P. G., Eriqat, C., Ober, R. R., DeJuan, E., Jr.,Williams, G. A., McLamore, A., Biswas, J., and Johnson, D. W.(1999) Human diabetic neovascular membranes contain highlevels of urokinase and metalloproteinase enzymes. Invest. Oph-thalmol. Vis. Sci. 40, 809–813
43. Tripathi, R. C., Tripathi, B. J., and Park, J. K. (1990) Localiza-tion of urokinase-type plasminogen activator in human eyes: animmunocytochemical study. Exp. Eye Res. 51, 545–552
44. Tripathi, R. C., Park, J. K., Tripathi, B. J., and Millard, C. B.(1988) Tissue plasminogen activator in human aqueous humorand its possible therapeutic significance. Am. J. Ophthalmol. 106,719–722
45. Tripathi, B. J., Geanon, J. D., and Tripathi, R. C. (1987)Distribution of tissue plasminogen activator in human andmonkey eyes. An immunohistochemical study. Ophthalmology 94,1434–1438
46. Schacke, W., Beck, K. F., Pfeilschifter, J., Koch, F., and Hatten-bach, L. O. (2002) Modulation of tissue plasminogen activatorand plasminogen activator inhibitor-1 by transforming growthfactor-beta in human retinal glial cells. Invest. Ophthalmol. Vis.Sci. 43, 2799–2805
47. Lutty, G. A., Ikeda, K., Chandler, C., and McLeod, D. S. (1991)Immunolocalization of tissue plasminogen activator in the dia-betic and nondiabetic retina and choroid. Invest. Ophthalmol. Vis.Sci. 32, 237–245
48. Fernandez-Monreal, M., Lopez-Atalaya, J. P., Benchenane, K.,Leveille, F., Cacquevel, M., Plawinski, L., MacKenzie, E. T., Bu,G., Buisson, A., and Vivien, D. (2004) Is tissue-type plasminogenactivator a neuromodulator? Mol. Cell. Neurosci. 25, 594–601
49. Benchenane, K., Lopez-Atalaya, J. P., Fernandez-Monreal, M.,Touzani, O., and Vivien, D. (2004) Equivocal roles of tissue-typeplasminogen activator in stroke-induced injury. Trends Neurosci. 27,155–160
50. Johnson, M. W., Olsen, K. R., and Hernandez, E. (1992) Tissueplasminogen activator thrombolysis during surgical evacuation ofexperimental subretinal hemorrhage. Ophthalmology 99, 515–521
51. Lewis, H., Resnick, S. C., Flannery, J. G., and Straatsma, B. R.(1991) Tissue plasminogen activator treatment of experimentalsubretinal hemorrhage. Am. J. Ophthalmol. 111, 197–204
52. Lewis, H., and VanderBrug Medendorp, S. (1997) Tissue plas-minogen activator-assisted surgical excision of subfoveal choroi-dal neovascularization in age-related macular degeneration: arandomized, double-masked trial. Ophthalmology 104, 1847–1851(discussion 1852)
53. Hesse, L., Schmidt, J., and Kroll, P. (1999) Management ofacute submacular hemorrhage using recombinant tissue plas-minogen activator and gas. Graefes Arch. Clin. Exp. Ophthalmol.237, 273–277
54. Hesse, L., Schroeder, B., Heller, G., and Kroll, P. (2000)Quantitative effect of intravitreally injected tissue plasminogenactivator and gas on subretinal hemorrhage. Retina 20, 500–505
55. Wang, Y. F., Tsirka, S. E., Strickland, S., Stieg, P. E., Soriano,S. G., and Lipton, S. A. (1998) Tissue plasminogen activator(tPA) increases neuronal damage after focal cerebral ischemiain wild-type and tPA-deficient mice. Nat. Med. 4, 228–231
56. Traynelis, S. F., and Lipton, S. A. (2001) Is tissue plasminogenactivator a threat to neurons? Nat. Med. 7, 17–18
57. Masos, T., and Miskin, R. (1997) mRNAs encoding urokinase-type plasminogen activator and plasminogen activator inhibi-tor-1 are elevated in the mouse brain following kainate-medi-ated excitation. Mol. Brain Res. 47, 157–169
58. Tsirka, S. E. (1997) Clinical implications of the involvement oftPA in neuronal cell death. J. Mol. Med. 75, 341–347
59. Tsirka, S. E., Bugge, T. H., Degen, J. L., and Strickland, S.(1997) Neuronal death in the central nervous system demon-strates a non-fibrin substrate for plasmin. Proc. Natl. Acad. Sci.USA 94, 9779–9781
60. Tsirka, S. E., Gualandris, A., Amaral, D. G., and Strickland, S.(1995) Excitotoxin-induced neuronal degeneration and seizureare mediated by tissue plasminogen activator. Nature (London)377, 340–344
61. Irvine, W. D., Johnson, M. W., Hernandez, E., and Olsen, K. R.(1991) Retinal toxicity of human tissue plasminogen activator invitrectomized rabbit eyes. Arch. Ophthalmol. 109, 718–722
62. Hrach, C. J., Johnson, M. W., Hassan, A. S., Lei, B., Sieving, P. A.,and Elner, V. M. (2000) Retinal toxicity of commercial intravit-real tissue plasminogen activator solution in cat eyes. Arch.Ophthalmol. 118, 659–663
63. Chen, S. N., Ho, C. L., Kuo, Y. H., and Ho, J. D. (2001) Intravit-reous tissue plasminogen activator injection and pneumatic dis-placement in the management of submacular hemorrhage com-plicating scleral buckling procedures. Retina 21, 460–463
64. Esser, P., Heimann, K., Bartz-Schmidt, K. U., Walter, P., Krott,R., and Weller, M. (1997) Plasminogen in proliferative vitreo-retinal disorders. Br. J. Ophthalmol. 81, 590–594
65. Zhang, X., Cheng, M., and Chintala, S. K. (2004) Kainicacid-mediated upregulation of matrix metalloproteinase-9 pro-motes retinal degeneration. Invest. Ophthalmol. Vis. Sci. 45,2374–2383
66. Zhang, X., Chaudhry, A., and Chintala, S. K. (2003) Inhibitionof plasminogen activation protects against ganglion cell loss in amouse model of retinal damage. Mol. Vis. 9, 238–248
67. Chintala, S. K., Zhang, X., Austin, J. S., and Fini, M. E. (2002)Deficiency in matrix metalloproteinase gelatinase B (MMP-9)protects against retinal ganglion cell death after optic nerveligation. J. Biol. Chem. 277, 47461–47468
68. Vorwerk, C. K., Gorla, M. S., and Dreyer, E. B. (1999) Anexperimental basis for implicating excitotoxicity in glaucomatousoptic neuropathy. Surv. Ophthalmol. 43, Suppl. 1, S142–S150
69. Vorwerk, C. K., Lipton, S. A., Zurakowski, D., Hyman, B. T.,Sabel, B. A., and Dreyer, E. B. (1996) Chronic low-dose gluta-mate is toxic to retinal ganglion cells. Toxicity blocked bymemantine. Invest. Ophthalmol. Vis. Sci. 37, 1618–1624
70. Nicole, O., Docagne, F., Ali, C., Margaill, I., Carmeliet, P.,MacKenzie, E. T., Vivien, D., and Buisson, A. (2001) Theproteolytic activity of tissue-plasminogen activator enhancesNMDA receptor-mediated signaling. Nat. Med. 7, 59–64
71. Christow, S. P., Bychkov, R., Schroeder, C., Dietz, R., Haller, H.,Dumler, I., and Gulba, D. C. (1999) Urokinase activates calcium-dependent potassium channels in U937 cells via calcium releasefrom intracellular stores. Eur. J. Biochem. 265, 264–272
72. Gualandris, A., Jones, T. E., Strickland, S., and Tsirka, S. E. (1996)Membrane depolarization induces calcium-dependent secretion oftissue plasminogen activator. J. Neurosci. 16, 2220–2225
73. Rogove, A. D., Siao, C., Keyt, B., Strickland, S., and Tsirka, S. E.(1999) Activation of microglia reveals a non-proteolytic cytokinefunction for tissue plasminogen activator in the central nervoussystem. J. Cell Sci. 112, 4007–4016
74. Thanos, S. (1991) The relationship of microglial cells to dyingneurons during natural neuronal cell death and axotomy-induced degeneration of the rat retina. Eur. J. Neurosci. 3,1189–1207
75. Thanos, S., Mey, J., and Wild, M. (1993) Treatment of the adultretina with microglia-suppressing factors retards axotomy-in-duced neuronal degradation and enhances axonal regenerationin vivo and in vitro. J. Neurosci. 13, 455–466
76. Thanos, S., and Naskar, R. (2004) Correlation between retinalganglion cell death and chronically developing inherited glau-coma in a new rat mutant. Exp. Eye Res. 79, 119–129
77. Siao, C. J., and Tsirka, S. E. (2002) Tissue plasminogen activatormediates microglial activation via its finger domain throughannexin II. J. Neurosci. 22, 3352–3358
Received for publication November 11, 2004.Accepted for publication March 30, 2005.
1289PLASMINOGEN ACTIVATORS PROMOTE RETINAL DAMAGE




















