
Phylogeny and biogeographic history of hake (genus Merluccius ), inferred from mitochondrial DNA...
12
J. Quinteiro Æ R. Vidal Æ M. Rey-Me´ ndez Phylogeny and biogeographic history of hake (genus Merluccius), inferred from mitochondrial DNA control-region sequences Received: 12 December 1998 / Accepted: 15 October 1999 Abstract Phylogenetic analyses of the left domain of the mitochondrial DNA control-region sequence have been used to examine the relationships among species of the genus Merluccius (Rafinesque, 1810), and to compare these with hypotheses based on morphological, meristic and allozyme characters. Analysis of aligned sequences revealed that transition bias was much lower than in mammalian mtDNA, and that nucleotide composition of control-region sequences was biased toward A and T. We have roughly calibrated a molecular clock for the genus, based on the rise of the Isthmus of Panama´, which is believed to have created a barrier to dispersal between marine species of the Atlantic and Pacific Oceans. Our mtDNA-based phylogeny was highly con- gruent with allozyme-based phylogenies, but poorly so with a previously described phylogeny based on mor- phology. Specifically, our phylogeny resolved two well- supported principal clades, one of American (west Atlantic and east Pacific) species and the other of Euro– African (east Atlantic) species. This suggests an evolu- tionary history during which the ancestral lineage of Merluccius was divided between two geographic regions, with subsequent dispersal and vicariant events resulting in the evolution and distribution of extant taxa. How- ever, the relationships between some taxa within the American clade could not be resolved. We suggest that this is consistent with an hypothesis of a rapid origin and radiation of these taxa. Introduction Species of the genus Merluccius (hake) are widely dis- tributed throughout cool waters (4 to 14 °C) of the main current system of the Atlantic, the Southern Ocean and the margin of the east Pacific (Soliman 1973). Although the taxonomy of this genus is complex, it is currently believed to comprise 12 species with highly conservative external morphology (Inada 1981; Cohen et al. 1990). Hake can perform long migrations, and physiological adaptations of the swimbladder and blood system fa- cilitate vertical mobility, enabling them to migrate from near the bottom during the day to mid-water or near- surface waters to feed at night (Olivar et al. 1988). Their eggs are pelagic, and the larvae remain in the water column for 2 mo. before descending to the bottom (Fahay 1974). In most areas, two hake species overlap for a considerable part of their geographical ranges. For example, the two species of Cape hake, M. capensis and M. paradoxus, overlap o southern Africa, the silver hake M. bilinearis and the oshore hake M. albidus o the eastern North American seaboard, the austral hake M. australis and the Argentine hake M. hubbsi o Pat- agonia, the Senegalese hake M. senegalensis and the Benguela hake M. polli o west Africa, and the austral hake M. australis and the Chilean hake M. gayi o southern Chile (Fig. 1) (Cohen et al. 1990; Howes 1991; Lo´pez Abella´n and Ariz Tellerı´a 1993). Our knowledge of the systematics and biogeography of Merluccius spp. relies mainly on the works of Inada (1981, 1989), Kabata and Ho (1981), Ferna´ndez (1985), Howes (1989), Cohen et al. (1990), and Ho (1990). Nevertheless, their interrelationships are poorly under- stood, and there is little agreement on the position of this group in relation to kindred taxa. Some investi- gators considered that Merluccius spp. should be in- cluded in the family Gadidae (see Inada 1989 for details), while others view the Merluccidae as a sepa- rate family occupying a relatively primitive position within the Gadidae (Marshall 1966; Cohen 1984; Marine Biology (2000) 136: 163–174 Ó Springer-Verlag 2000 Communicated by S.A. Poulet, Rosco J. Quinteiro Æ R. Vidal Æ M. Rey-Me´ndez (&) Universidade de Santiago de Compostela, Facultade de Bioloxı´a, Departamento de Bioquı´mica e Bioloxı´a Molecular, E-15706 Santiago de Compostela, Galicia, Spain Fax: 0034 (0)981 596904 e-mail: [email protected]
Transcript of Phylogeny and biogeographic history of hake (genus Merluccius ), inferred from mitochondrial DNA...

J. Quinteiro á R. Vidal á M. Rey-Me ndez
Phylogeny and biogeographic history of hake (genus Merluccius),inferred from mitochondrial DNA control-region sequences
Received: 12 December 1998 /Accepted: 15 October 1999
Abstract Phylogenetic analyses of the left domain of themitochondrial DNA control-region sequence have beenused to examine the relationships among species of thegenus Merluccius (Ra®nesque, 1810), and to comparethese with hypotheses based on morphological, meristicand allozyme characters. Analysis of aligned sequencesrevealed that transition bias was much lower than inmammalian mtDNA, and that nucleotide compositionof control-region sequences was biased toward A and T.We have roughly calibrated a molecular clock for thegenus, based on the rise of the Isthmus of Panama ,which is believed to have created a barrier to dispersalbetween marine species of the Atlantic and Paci®cOceans. Our mtDNA-based phylogeny was highly con-gruent with allozyme-based phylogenies, but poorly sowith a previously described phylogeny based on mor-phology. Speci®cally, our phylogeny resolved two well-supported principal clades, one of American (westAtlantic and east Paci®c) species and the other of Euro±African (east Atlantic) species. This suggests an evolu-tionary history during which the ancestral lineage ofMerluccius was divided between two geographic regions,with subsequent dispersal and vicariant events resultingin the evolution and distribution of extant taxa. How-ever, the relationships between some taxa within theAmerican clade could not be resolved. We suggest thatthis is consistent with an hypothesis of a rapid origin andradiation of these taxa.
Introduction
Species of the genus Merluccius (hake) are widely dis-tributed throughout cool waters (4 to 14 °C) of the maincurrent system of the Atlantic, the Southern Ocean andthe margin of the east Paci®c (Soliman 1973). Althoughthe taxonomy of this genus is complex, it is currentlybelieved to comprise 12 species with highly conservativeexternal morphology (Inada 1981; Cohen et al. 1990).Hake can perform long migrations, and physiologicaladaptations of the swimbladder and blood system fa-cilitate vertical mobility, enabling them to migrate fromnear the bottom during the day to mid-water or near-surface waters to feed at night (Olivar et al. 1988). Theireggs are pelagic, and the larvae remain in the watercolumn for �2 mo. before descending to the bottom(Fahay 1974). In most areas, two hake species overlapfor a considerable part of their geographical ranges. Forexample, the two species of Cape hake, M. capensis andM. paradoxus, overlap o� southern Africa, the silverhake M. bilinearis and the o�shore hake M. albidus o�the eastern North American seaboard, the austral hakeM. australis and the Argentine hake M. hubbsi o� Pat-agonia, the Senegalese hake M. senegalensis and theBenguela hake M. polli o� west Africa, and the australhake M. australis and the Chilean hake M. gayi o�southern Chile (Fig. 1) (Cohen et al. 1990; Howes 1991;Lo pez Abella n and Ariz Tellerõ a 1993).
Our knowledge of the systematics and biogeographyof Merluccius spp. relies mainly on the works of Inada(1981, 1989), Kabata and Ho (1981), Ferna ndez (1985),Howes (1989), Cohen et al. (1990), and Ho (1990).Nevertheless, their interrelationships are poorly under-stood, and there is little agreement on the position ofthis group in relation to kindred taxa. Some investi-gators considered that Merluccius spp. should be in-cluded in the family Gadidae (see Inada 1989 fordetails), while others view the Merluccidae as a sepa-rate family occupying a relatively primitive positionwithin the Gadidae (Marshall 1966; Cohen 1984;
Marine Biology (2000) 136: 163±174 Ó Springer-Verlag 2000
Communicated by S.A. Poulet, Rosco�
J. Quinteiro á R. Vidal á M. Rey-Me ndez (&)Universidade de Santiago de Compostela,Facultade de Bioloxõ a,Departamento de BioquõÂmica e Bioloxõ a Molecular,E-15706 Santiago de Compostela,Galicia, Spain
Fax: 0034 (0)981 596904e-mail: [email protected]

Okamura 1989). Markle (1989) ranked Merluccidae asa sister group to the Lotidae plus Gadidae families,while Howes recognized the derived character of thefamily Merluccidae (Howes 1990, 1991), and restrictedit to include only the genus Merluccius, ranking it as asister group of the Gadidae.
Paleontological information suggests that the genusarose in the middle Oligocene (Fedotov and Bannikov1989). There are two hypotheses concerning its originand dispersal: both agree that hake originated in theNorth Atlantic and entered the Paci®c through thethen-open Panamanian seaway, but di�er as to whetherM. hubbsi derived from an eastern South Paci®c stock(Szidat 1955, 1961; Inada 1981) or from a westernNorth Atlantic stock (Kabata and Ho 1981; Ho 1990).Likewise, morphological and biogeographic studies(Inada 1981; Ho 1990) suggest that hake from the Oldand the New World represent distinct clades. However,details of hake evolution in the eastern and westernAtlantic and the eastern Paci®c are still unclear, andtheories based on larval drift are unconvincing (Inada1981).
Despite their importance in the pelagic ecosystem(Pitcher and Alheit 1995), relatively little attention hasbeen devoted to the evolution of these organisms, anddispersal distances and the extent of consequent gene¯ow are currently unknown. With the exception of a fewstudies on allozyme polymorphism and restrictionfragment-length polymorphism (RFLP) of mitochon-
drial DNA (mtDNA) (Becker et al. 1988; Grant et al.1988; Rolda n 1995; Stepien and Rosenblatt 1996), thephylogenetic relationships among Merluccius spp. havenot been investigated at the DNA-sequence level.
An understanding of phylogenetic relationshipsamong species is necessary to a rigorous comparativeapproach in studies of adaptation (Felsenstein 1988),and can also illuminate biogeographic patterns (Avise1994). Studies on interspeci®c variation based on theanalysis of mtDNA sequences provide a powerful toolfor investigating the evolutionary history of ®shes(Moritz et al. 1987; Avise 1989). The usefulness ofmtDNA in evolutionary studies derives from its pre-dominantly maternal inheritance, high substitution rate,and apparent lack of recombination (reviewed by Wil-son et al. 1985). In addition, di�erent regions of themolecule and di�erent lineages of ®shes can evolve atdi�erent rates. Evolution of the control region can bethree to ®ve times faster than the remainder of the mi-tochondrial genome in some groups (Brown et al. 1986).However, several recent reports have challenged thegenerality of this evolutionary rate, especially in ®shes(Shedlock et al. 1992; Bernatchez and Danzmann 1993;Zhu et al. 1994).
In this study, we determined the sequence of the leftdomain of the control region of mtDNA in samplesfrom 11 of 12 currently recognized species of Merluccius.We then used tree-reconstruction techniques to investi-gate the evolutionary history of this genus on the basisof these sequences, and compared our conclusions withthose obtained previously by other techniques.
Fig. 1 Merluccius spp. Approximate distributions of major species
164

Materials and methods
Sampling
Nucleotide sequences of the left domain of the mtDNA controlregion of 26 individuals of 11 of the currently recognized species ofthe genus Merluccius (Ra®nesque, 1810) (Table 1) and two tenta-tive outgroup species, Macruronus magellanicus and M. novae-zelandiae (Quinteiro et al. unpublished data), were determined.Tissue from Merluccius angustimanus and the species recently de-scribed by Mathews (1985), M. hernandezi, was not available foranalysis. Tissue samples (muscle or heart) were collected and storedin 70% ethanol. The sampling strategy included an initial screeningto investigate intraspeci®c variation (Table 1), so that only non-identical haplotypes were included in the analysis.
Laboratory protocols
Total DNA was isolated by standard phenol/chloroform/isoamylalcohol procedure, and resuspended in 50 to 100 ll of sterile water(Kocher et al. 1989; Sambrook et al. 1989). Ampli®cations of con-trol-region sequences were carried out in 25 ll reaction volumescontaining Promega Bu�er A 1x, 1.5 mM MgCl2, 200 lM of each
dNTP, 0.1 lM of each primer and 0.5 or 1 ll of a 1/10 dilution ofstock total DNA. Two primers designed speci®cally for ®shes wereused: the light-strand primer L15998 (5¢-TACCCCAAACTCC-CAAAGCTA-3¢) complementary to tRNApro (Alvarado-Bremer1994); and a heavy-strand primer, B (5¢-ACGCTGGAAA-GAACGCCCGGCATGG-3¢), complementary to the central regionof the D-loop in gadoids (Lee et al. 1995). The ampli®cation proce-dure, performed in a Gene Amp PCR System 2400 (Perkin Elmer),was as follows: an initial denaturing step of 3 min at 94 °C, followedby 30 cycles, with each cycle comprised denaturing at 94 °C for 30 s,annealing at 50 to 54 °C for 40 s, followed by extension at 72 °C for1 min and a ®nal extension at 72 °C for 5 min. Standard precau-tions, including the use of negative controls, were taken to detectcontamination and related problems. The polymerase chain-reac-tion (PCR) mixture was digested for 30 min at 37 °C, with 1 ll ofshrimp alkaline phosphatase (2.0 U ll)1) and 1 ll of exonuclease I(10.0 U ll)1) contained in the PCR product pre-sequencing kit(USB, Cleveland, Ohio, USA). After enzyme inactivation, an ali-quot of 6 ll of the PCR mixture was used for cycle-sequencing withABI PRISMÒ DNA polymerase, FS (Perkin-Elmer Corporation,USA). Labelled extension products were gel-separated and detectedwith an automated DNA-sequencer (Model ABI PRISM 377, Per-kin-Elmer Corporation, USA). All samples were sequenced in theforward direction; problematic samples were subsequently se-quenced in the reverse direction to con®rm nucleotide assignments.
Table 1 Merluccius spp. Initialscreening of intraspeci®c varia-tion and sampling locations anddates of collection (MED Med-iterranean; NWA NW Atlantic;NEA NE Atlantic; NEP NEPaci®c; SWA SW Atlantic; SEASE Atlantic; SEP SE Paci®c)
Species abbreviations Common name Location Haplotypes
M. bilinearis Silver hake NWA, USA 3M. bil-1M. bil-2M. bil-3M. bil-4
M. australis Austral hake SEP, Chile 2M. aus-1M. aus-2M. aus-3
M. productus Paci®c hake NEP, USA 2M. pro-1M. pro-2
M. albidus O�shore hake NWA, USA 1M. alb-1M. alb-2
M. gayi Chilean hake SEP, Chile 1M. gay-1M. gay-2
M. hubbsi Argentine hake SWA, Argentina 1M. hub-1M. hub-2
M. capensis Shallow-water Cape hake SEA, South Africa 2M. cap-1M. cap-2
M. senegalensis Senegalese hake SEA, Senegal 2M. sen-1M. sen-2
M. merluccius European hake MED, NEA, Spain 4M. mer-1M. mer-2M. mer-3M. mer-4
M. polli Benguela hake SEA, Mauritania 2M. poll1-1M. polli-2
M. paradoxus Deep-water Cape hake SEA, South Africa 1M. pardx-1
165

Sequence analyses
Sequence alignments were performed using Clustal W (Thompsonet al. 1994), with subsequent checking and adjustment by eye. Thealigned sequences were then compared with published sequencesfor Gadiformes (Lee et al. 1995) to verify the boundaries of thetRNApro gene. Statistics on nucleotide composition were compiledusing the MEGA program (Kumar et al. 1993). Previous phylo-genetic analyses based on mtDNA have demonstrated that, as di-vergence time increases, many sites become saturated by multipletransitional substitutions, thus obscuring more ancient divergences(Brown et al. 1982). Transversions, however, appear to accumulatelinearly over time (Miyamoto and Boyle 1989; Irwin et al. 1991).Two approaches were used to investigate whether transition satu-ration occurred. (1) The number of transitions and transversionswere plotted against total sequence divergence (%) for all pairwisecomparisons. At saturation, the rate of increase in transitionsreaches a plateau, and transversions outnumber transitions. Se-quence divergences were estimated with the Tamura±Nei model(Tamura and Nei 1993), which corrects for nucleotide compositionbias and substitution bias. (2) We calculated the observed meanratio of transitions to transversions (TS:TV), and determinedwhether this ratio di�ered from that expected at phylogenetic sat-uration. The transition-to-transversion ratio expected at saturation(Rs:v) can be estimated from empirical-base frequencies (Holmquist1983): Rs:v � �PA � PG � PC � PT � : �PA � PG��PC � PT �where PA, PC,PT and PG are the base frequencies.
Phylogenetic analyses
Phylogenetic analyses were performed using two standard tree-re-construction methods, because no single approach has been shownto be consistently superior for ®nding the correct tree (Hillis andHuelsenbeck 1992; Hillis 1995).
First, a neighbor-joining (NJ) analysis without the assumptionof equal rates of evolution (Saitou and Nei 1987) was performedusing MEGA Version 1.01 (Kumar et al. 1993), considering bothtransitions and transversions, and the Tamura±Nei model of nu-cleotide substitution (Tamura and Nei 1993). In addition, we alsoperformed analyses considering transversions only to counteractthe potential e�ect of homoplasy. Missing data together with in-sertions and indels were handled with the complete-deletion option.The reliability of the NJ tree was evaluated by two 2000 bootstrapreplicates (Hedges 1992).
Second, the software package PHYLIP Version 3.5 (Felsenstein1993) was used to execute a maximum-likelihood (ML) analysis(Felsenstein 1981). Parameters speci®ed in the analysis includedone category of substitution, empirical base frequencies, and a 2.0:1value for the TS:TV ratio. This ratio, obtained from the sequencedata, constituted the mean TS:TV ratio for all pairwise compari-sons between ingroup taxa, estimated with the Tamura±Nei modelin the MEGA program, which corrects for transition bias (Wakeley1996) to give corrected ratios, R. Because the order of taxa in thedata set can a�ect the results (Ferris et al. 1995), we analyzed eachdata set ten times, using a randomized input order. Insertion/de-letion events are di�cult to take into account in the maximum-likelihood procedure, since sites with gaps in any of the sequencesunder consideration are excluded from the analysis (Goldman1997). The robustness of the ML tree was inferred from P valuescalculated for branch lengths.
In order to test whether the two tree topologies di�ered sig-ni®cantly, the Kishino and Hasegawa (1989) likelihood-ratio test,implemented in PHYLIP, was used: this test compares tree top-ologies on the basis of di�erences between their log-likelihoodvalues. We tested for the heterogeneity of rates of evolutionarychange along the di�erent lineages using the maximum-likelihoodmethod of Felsenstein (1993). The null hypothesis for this test isthat the rate of nucleotide substitutions is constant for all branchesof the tree.
Despite the fact that monophyly of the genus Merluccius hasbeen veri®ed by studies of both morphological and allozyme
characters (Ho 1990; Rolda n 1995), our choice of outgroup wasconstrained by the lack of available sequences and by the complextaxonomy of the genus (see ``Introduction''). Reported mtDNAsequences in GenBank for several members of the family Gadidae(Gadus morhua X99772; Melanogramus aegle®nus U59475; Micro-gadus tomcod U12058 and Pollachius virens U12069) are too di-vergent to provide informative comparisons. We therefore usedsequence data from the Patagonian grenadier and blue grenadier(Macruronus magellanicus and M. novaezelandiae: Quinteiro et al.unpublished data) to polarize the characters in some of our ana-lyses. Although our results are based on midpoint rooting, thetopologies of the trees remain unaltered when the Patagoniangrenadier and blue grenadier are included as an outgroup.
Results
After ampli®cations, a fragment of about 450 base pairs(bp) was obtained from each sampled individual ofMerluccius spp. Alignment gave a 412 bp sequence, withthe ®rst nucleotide located in Position 22 of the controlregion, since nucleotide assignments were uncertain forthe 5¢ end of some sequences.
Intraspeci®c sequence-divergence (range 0.3 to 0.8%)was negligible in comparison with observed interspeci®cdivergence, although pairwise comparisons using Me-rluccius merluccius did reveal somewhat higher values(range 0.5 to 1.8%: Table 2).
Patterns of nucleotide substitutionsand genetic divergence
A total of 121 variable sites and 107 phylogenetically in-formative sites were detected (Table 3). Base compositionwas strongly biased: adenine and thymine were repre-sented roughly equally, occurring at mean (�SE) per-centages of 31.8 � 0.28% and 31.7 � 0.16%,respectively, while cytosine occurred at 25.4 � 0.29% ofsites, and guanine at only 11.1 � 0.18% of sites. Thesebiases are similar to those found in the mtDNA control-region of other ®shes, including the left domain of thecontrol-region of other Gadiformes (Meyer et al. 1990;Bernatchez and Danzmann 1993; Brown et al. 1993; Zhuet al. 1994; Lee et al. 1995;McMillan and Palumbi 1997).
The ratio of transitions to transversions (TS:TV)(after correction for composition and transition bias) forpairwise interspeci®c comparisons ranged from 0.959 to7.146, with a mean of 2.0 � 0.76. The highest TS:TVratios were between species from the Paci®c Ocean,namely Merluccius productus and M. gayi (mean 7.1),the lowest between M. capensis and M. polli (mean 1.0).Among hake from the American and Euro-Africancoasts, the mean ratios were 3.0 � 1.39 and 2.0 � 1.04,respectively. TSs outnumbered TVs when closely relatedsequences were compared, but TVs were as common asTSs when some highly divergent sequences were com-pared, indicating possible saturation. However, a plot ofthe observed number of TSs and TVs against sequencedivergences revealed a fairly linear relationship, sug-gesting that saturation had not been reached (Fig. 2).
166

Furthermore, in no case did the TS:TV ratio (minimum0.959) approach the value expected at saturation (0.47;estimated following Holmquist 1983), and the meanTS:TV ratio (2.0) was signi®cantly higher than that ex-pected at saturation (Student t-test; t = 5.67, P <0.001).
Table 2 presents estimates of genetic distance be-tween all the individuals examined. Divergence rangedfrom as little as 2% between Merluccius gayi andM.productus to asmuch as 20%betweenM.bilinearis andM. senegalensis. Among the hake species of the westernAtlantic and the eastern Paci®c, the mean divergence(�SE) was 8.8 � 0.015%, while the mean divergenceamong species of the eastern Atlantic coast was slightlylower (6.0 � 0.012%). The mean divergence betweenspecies from di�erent continents was 14.0 � 0.021%.
Phylogenetic relationship
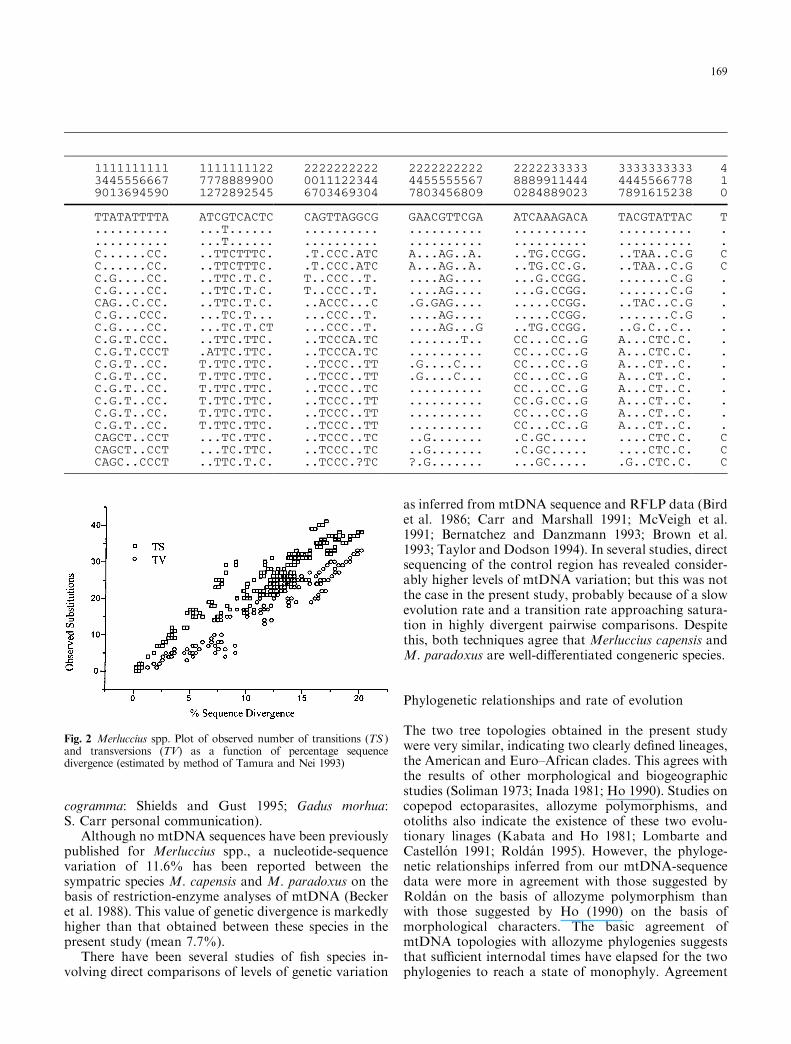
Gene tree-topologies obtained by the two methods (NJand ML) are shown in Fig. 3. The two trees are broadlysimilar, both showing two well-de®ned principal clades.The branching patterns obtained by the two methodswere largely congruent, although some branches exhib-ited low bootstrap support and con¯icting topology. Ingeneral, deeper nodes were well de®ned, whereas inter-mediate nodes had lower bootstrap values.
The early evolutionary history of hake is character-ized by the divergence into two major basic lineages: oneAmerican clade comprising west Atlantic and east Pa-ci®c species, and the other Euro±African clade includingeast Atlantic species. The Euro±African clade comprisesspecies that are relatively homogeneous in size andecology. This clade was well-resolved, and bifurcatedinto two groups. The ®rst comprised Merluccius polli, ablackish species distributed along the west coast ofequatorial Africa, and M. paradoxus, which is distrib-uted along the continental shelf o� southern and south-western Africa. The second group comprisedM. capensisand its sister group M. senegalensis plus M. merluccius.The distribution of M. polli overlaps with that of M.senegalensis o� the coasts of Mauritania and Senegal,although the former inhabits deeper waters. Likewise, afurther two species with similar external morphology,M. capensis and M. paradoxus, overlap o� the south-western coast of Africa, but with the latter occurring indeeper water (Inada 1981). Despite such microallopatricdistributions (deep and shallow water), M. senegalensisand M. polli were separated by at least 37 nucleotidesubstitutions (sequence divergence: 10.2%), and M.capensis and M. paradoxus by at least 30 nucleotidesubstitutions (sequence divergence = 7.6%).
The American clade, containing all species occurringin the western Atlantic and eastern Paci®c, is more dif-®cult to interpret in view of the alternative topologiesobtained and the low bootstrap support for some nodesin the NJ tree. In the ML analysis, Merluccius hubbsiwas ranked as the sister taxon of the clade comprisingT
able
2Merlucciusspp.Genetic-distance
matrix
(Tamura±Nei
model)ofhakemtD
NA
control-regionsequences(Speciescodes
asin
Table
1)
Species
12
34
56
78
910
11
12
13
14
15
16
17
18
19
20
1:Mbil-1
2:Mbil-2
0.005
3:Mbil-3
0.008
0.003
4:Maus-1
0.171
0.164
0.167
5:Maus-2
0.164
0.158
0.161
0.005
6:Mpro-1
0.120
0.113
0.113
0.073
0.073
7:Mpro-2
0.122
0.116
0.116
0.075
0.075
0.003
8:Malb-1
0.144
0.139
0.138
0.090
0.091
0.053
0.055
9:Mgay-1
0.107
0.101
0.101
0.082
0.082
0.021
0.023
0.047
10:Mhub-1
0.123
0.116
0.116
0.078
0.078
0.034
0.036
0.053
0.034
11:Mcap-1
0.182
0.176
0.179
0.153
0.147
0.131
0.134
0.134
0.121
0.128
12:Mcap-2
0.185
0.178
0.182
0.156
0.150
0.134
0.137
0.137
0.124
0.131
0.008
13:Msen-1
0.198
0.191
0.195
0.168
0.162
0.139
0.142
0.136
0.139
0.140
0.067
0.069
14:Msen-2
0.201
0.195
0.198
0.171
0.165
0.142
0.139
0.139
0.142
0.143
0.069
0.072
0.003
15:Mmer-1
0.178
0.171
0.174
0.146
0.140
0.124
0.127
0.127
0.121
0.121
0.053
0.055
0.034
0.036
16:Mmer-2
0.178
0.172
0.175
0.143
0.136
0.118
0.121
0.25
0.121
0.115
0.050
0.053
0.031
0.034
0.013
17:Mmer-3
0.178
0.172
0.175
0.149
0.143
0.124
0.127
0.124
0.121
0.121
0.050
0.053
0.026
0.029
0.008
0.005
18:Mmer-4
0.191
0.185
0.188
0.156
0.156
0.137
0.141
0.138
0.134
0.134
0.058
0.061
0.045
0.048
0.016
0.018
0.018
19:Mpolli-1
0.172
0.166
0.169
0.146
0.140
0.130
0.133
0.140
0.127
0.133
0.075
0.072
0.100
0.103
0.086
0.077
0.083
0.098
20:Mpolli-2
0.169
0.163
0.166
0.149
0.143
0.127
0.130
0.137
0.124
0.130
0.075
0.072
0.100
0.103
0.086
0.077
0.083
0.098
0.003
21:Mpardx-1
0.169
0.162
0.165
0.153
0.146
0.115
0.118
0.125
0.112
0.124
0.078
0.075
0.109
0.112
0.100
0.092
0.098
0.107
0.034
0.031
167

M. australis, M. productus, M. albidus and M. gayi. Inthe NJ analysis, in contrast, M. australis and M. albidusdid not cluster as sister-species, and 55% of the pseu-doreplicates placed M. hubbsi as the sister taxon ofM. productus plus M. gayi. An analysis considering onlytranversions revealed basically the same topology,although with di�erent patterns for M. australis,M. albidus and M. hubbsi, for which bootstrap valueswere not signi®cantly improved. Despite this, the twoanalyses agree in at least two respects: (1) The primaryspeciation event gave rise toM. bilinearis, a silvery-whitespecies distributed along the North American Atlanticcoast (Inada 1981); (2) both analyses clustered M. prod-uctus (from the North Paci®c) and M. gayi (from thewest coast of South America) as sister-species, with arelatively high bootstrap support for this node (72%).Comparison of the NJ and ML trees by the log-likeli-hood test indicated that both are equally likely (NJln = )1689.8274, ML ln = )1678.5834; SD = 5.8281).
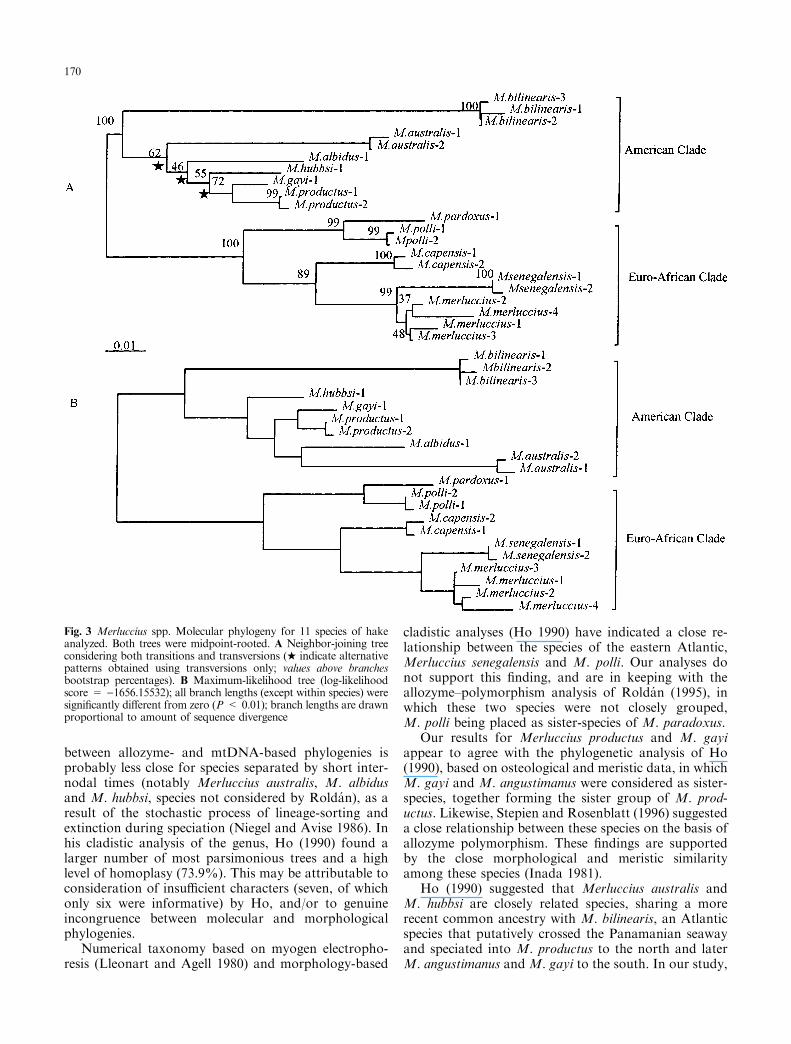
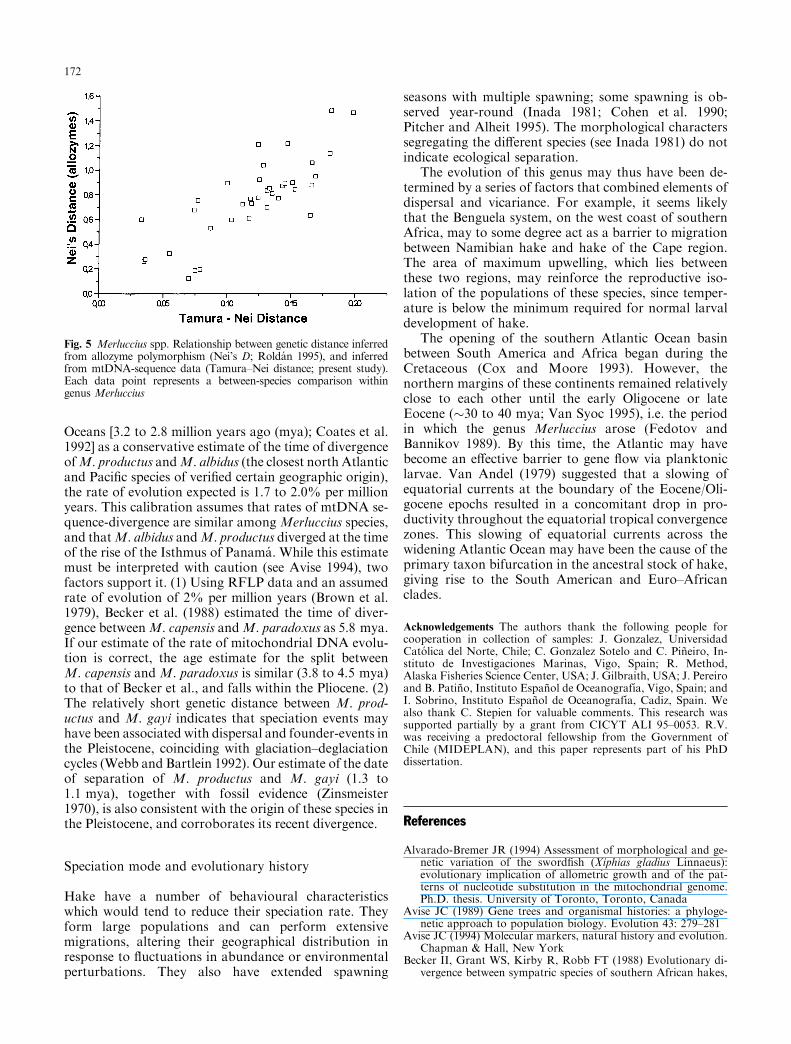
Phenograms based on genetic distance of allozymesin nine hake species (Rolda n 1995) were more similar tophylogenies found with mtDNA control-region se-quences than to phylogenies based on morphologicalcharacters (Ho 1990) (Fig. 4). There was a signi®cantcorrelation of allozymic genetic distances and sequence-divergence (Fig. 5; r = 0.8, Mantel's test: P < 0.001).
Felsenstein's maximum-likelihood test indicated thatthe rate of evolutionary change did not di�er signi®-cantly among the di�erent lineages (P < 0.05), sug-gesting that hake mtDNA control-region is evolving in aclock-like fashion. The optimal log-likelihood valueunder the constraint of a molecular clock was
)1668.1753 (vs )1656.1553 when this constraint wasrelaxed).
Discussion
Like many other ®sh groups, Merluccius spp. have verysimilar body forms and must be diagnosed by an arrayof subtle and statistically-determined meristic characters(Inada 1981, 1989). Nucleotide-sequence data from mi-tochondrial DNA, on the other hand, provide a largesource of potentially phylogenetic characters that areindependent of morphology, including problematicmeristic characters.
Rate of variation
As has been reported for other ®sh mtDNA control-regions (Meyer et al. 1990; Sturmbauer and Meyer 1992;Zhu et al. 1994), we found that transitions accumulatemore rapidly than transversions in hake mtDNA, whichhas a high A + T content (>60%). However, our re-sults indicate a low rate of nucleotide substitution and alow TS:TV ratio. There are indications from the litera-ture that the mitochondrial control-region may not be asvariable in some ®sh species as it is in mammals (Meyeret al. 1990; Bernatchez et al. 1992; Bernatchez andDanzmann 1993; Zhu et al. 1994; Turner 1997). Fur-thermore, a low rate of evolution has been reported forthe control region of other gadiforms (Theragra chal-
Table 3 Merluccius spp. Aligned sequences for the 11 species ofhake examined; only variable nucleotide positions are shown(? Ambiguities; dots indicate identity with ®rst sequence for
M. bilinearis, Mbil-1; species codes as in Table 1). Representativesequences of each species reported here have been submitted toGenBank: Accession Nos.: AF112245-AF112255
SpeciesCode No.
Variable nucleotide positions
1111 1111111111 11111111111122 2233344555 5566677778 8888890000 0001111111 2223333333
3456785746 8901534235 6702336890 2456751236 7890234789 1590134568
Mbil-1 AGCACAATCT TAAAATGATA TTTGGGGCCA ATTGAAAGAC AATAAAATCC ACTTTTTAGAMbil-2 .......... ......A... .......... .......... .......... ..........Mbil-3 .......... ..C...A... .......... .......... .......... ..........Maus-1 .......AGC .T..G.AG.T ...AATATTT ...TGG.... ......T..T TTCCCCCCATMaus-2 .......AGC ....G.AG.T ...AA.ATTT ...TGG.... ......T..T TTCCCCCCATMpro-1 ...G...AGC .GG.G.A..T ......AGTT ...TGG.... .........T T.C....TACMpro-2 ...G...AGC .GG.GAA..T ......AGTT ...TGG.... .........T T.C....TACMalb-1 .....G.AGC ..G.G.A..T C.....A..T ...TGG...T ........TT TTC....TACMgay-1 .......AGC .GG.G.A..T ......A.TT ...TGG...T .........T T.C....TACMhub-1 .....G..GC C.G.G.A... ......A.TT ...TGG.... .........T TTC....TACMcap-1 G....T..GC .G....AC.T C.AA..ATTT .ACTGGCAGT ..A.TG...T .TCG.A.TACMcap-2 G....T..GC .G....AC.T C.AA..ATTT .ACTGGCAGT ..A.TG...T .TCG.A.TACMsen-1 GCACTT..GC .G....ATCC C.ATA.ATTT .A.TGGT.GT G.A.T..CTT CTCG...TACMsen-2 GCACTT..GC .G...AATCC C.ATA.ATTT .A.TGGT.GT G.A.T..CTT CTCG...TACMmer-1 G....TG.GC ??.C..ATCC ..ATA.ATTT .A.TGGTA.T ..A.T..CTT CTCG...TACMmer-2 G....TG.GC G.....AT.C C.ATA.ATTT .A.TGGTA.T ..A.T..CTT CTCG...TACMmer-3 G....TG.GC G.....ATCC C.ATA.ATTT .A.TGGTA.T ..A.T..CTT CTCG...TACMmer-4 G....TG.GC ??....AC.? CAATATATTT GA.TGGTA.T .GAGT..CTT CTCG...TACMpolli-1 .....T.AGC .G....AT.C C.AA..ATT. .CCTGGTAGT ..A.T..A.T C.CC.C.TACMpolli-2 .....T.AGC .G....AT.C C.AA..ATT. .CCTGGTAGT ..A.T..A.T C.C..C.TACMpardx-1 ....TT.AGC .G....AC.. C.A...ATTT ..CTGGTAGT ..A.T..A.T C.C..A.TAC
168
cogramma: Shields and Gust 1995; Gadus morhua:S. Carr personal communication).
Although no mtDNA sequences have been previouslypublished for Merluccius spp., a nucleotide-sequencevariation of 11.6% has been reported between thesympatric species M. capensis and M. paradoxus on thebasis of restriction-enzyme analyses of mtDNA (Beckeret al. 1988). This value of genetic divergence is markedlyhigher than that obtained between these species in thepresent study (mean 7.7%).
There have been several studies of ®sh species in-volving direct comparisons of levels of genetic variation
as inferred from mtDNA sequence and RFLP data (Birdet al. 1986; Carr and Marshall 1991; McVeigh et al.1991; Bernatchez and Danzmann 1993; Brown et al.1993; Taylor and Dodson 1994). In several studies, directsequencing of the control region has revealed consider-ably higher levels of mtDNA variation; but this was notthe case in the present study, probably because of a slowevolution rate and a transition rate approaching satura-tion in highly divergent pairwise comparisons. Despitethis, both techniques agree that Merluccius capensis andM. paradoxus are well-di�erentiated congeneric species.
Phylogenetic relationships and rate of evolution
The two tree topologies obtained in the present studywere very similar, indicating two clearly de®ned lineages,the American and Euro±African clades. This agrees withthe results of other morphological and biogeographicstudies (Soliman 1973; Inada 1981; Ho 1990). Studies oncopepod ectoparasites, allozyme polymorphisms, andotoliths also indicate the existence of these two evolu-tionary linages (Kabata and Ho 1981; Lombarte andCastello n 1991; Rolda n 1995). However, the phyloge-netic relationships inferred from our mtDNA-sequencedata were more in agreement with those suggested byRolda n on the basis of allozyme polymorphism thanwith those suggested by Ho (1990) on the basis ofmorphological characters. The basic agreement ofmtDNA topologies with allozyme phylogenies suggeststhat su�cient internodal times have elapsed for the twophylogenies to reach a state of monophyly. Agreement
1111111111 1111111122 2222222222 2222222222 2222233333 3333333333 43445556667 7778889900 0011122344 4455555567 8889911444 4445566778 19013694590 1272892545 6703469304 7803456809 0284889023 7891615238 0
TTATATTTTA ATCGTCACTC CAGTTAGGCG GAACGTTCGA ATCAAAGACA TACGTATTAC T.......... ...T...... .......... .......... .......... .......... ........... ...T...... .......... .......... .......... .......... .C......CC. ..TTCTTTC. .T.CCC.ATC A...AG..A. ..TG.CCGG. ..TAA..C.G CC......CC. ..TTCTTTC. .T.CCC.ATC A...AG..A. ..TG.CC.G. ..TAA..C.G CC.G....CC. ..TTC.T.C. T..CCC..T. ....AG.... ...G.CCGG. .......C.G .C.G....CC. ..TTC.T.C. T..CCC..T. ....AG.... ...G.CCGG. .......C.G .CAG..C.CC. ..TTC.T.C. ..ACCC...C .G.GAG.... .....CCGG. ..TAC..C.G .C.G...CCC. ...TC.T... ...CCC..T. ....AG.... .....CCGG. .......C.G .C.G....CC. ...TC.T.CT ...CCC..T. ....AG...G ..TG.CCGG. ..G.C..C.. .C.G.T.CCC. ..TTC.TTC. ..TCCCA.TC .......T.. CC...CC..G A...CTC.C. .C.G.T.CCCT .ATTC.TTC. ..TCCCA.TC .......... CC...CC..G A...CTC.C. .C.G.T..CC. T.TTC.TTC. ..TCCC..TT .G....C... CC...CC..G A...CT..C. .C.G.T..CC. T.TTC.TTC. ..TCCC..TT .G....C... CC...CC..G A...CT..C. .C.G.T..CC. T.TTC.TTC. ..TCCC..TC .......... CC...CC..G A...CT..C. .C.G.T..CC. T.TTC.TTC. ..TCCC..TT .......... CC.G.CC..G A...CT..C. .C.G.T..CC. T.TTC.TTC. ..TCCC..TT .......... CC...CC..G A...CT..C. .C.G.T..CC. T.TTC.TTC. ..TCCC..TT .......... CC...CC..G A...CT..C. .CAGCT..CCT ...TC.TTC. ..TCCC..TC ..G....... .C.GC..... ....CTC.C. CCAGCT..CCT ...TC.TTC. ..TCCC..TC ..G....... .C.GC..... ....CTC.C. CCAGC..CCCT ..TTC.T.C. ..TCCC.?TC ?.G....... ...GC..... .G..CTC.C. C
Fig. 2 Merluccius spp. Plot of observed number of transitions (TS )and transversions (TV) as a function of percentage sequencedivergence (estimated by method of Tamura and Nei 1993)
169
between allozyme- and mtDNA-based phylogenies isprobably less close for species separated by short inter-nodal times (notably Merluccius australis, M. albidusand M. hubbsi, species not considered by Rolda n), as aresult of the stochastic process of lineage-sorting andextinction during speciation (Niegel and Avise 1986). Inhis cladistic analysis of the genus, Ho (1990) found alarger number of most parsimonious trees and a highlevel of homoplasy (73.9%). This may be attributable toconsideration of insu�cient characters (seven, of whichonly six were informative) by Ho, and/or to genuineincongruence between molecular and morphologicalphylogenies.
Numerical taxonomy based on myogen electropho-resis (Lleonart and Agell 1980) and morphology-based
cladistic analyses (Ho 1990) have indicated a close re-lationship between the species of the eastern Atlantic,Merluccius senegalensis and M. polli. Our analyses donot support this ®nding, and are in keeping with theallozyme±polymorphism analysis of Rolda n (1995), inwhich these two species were not closely grouped,M. polli being placed as sister-species of M. paradoxus.
Our results for Merluccius productus and M. gayiappear to agree with the phylogenetic analysis of Ho(1990), based on osteological and meristic data, in whichM. gayi and M. angustimanus were considered as sister-species, together forming the sister group of M. prod-uctus. Likewise, Stepien and Rosenblatt (1996) suggesteda close relationship between these species on the basis ofallozyme polymorphism. These ®ndings are supportedby the close morphological and meristic similarityamong these species (Inada 1981).
Ho (1990) suggested that Merluccius australis andM. hubbsi are closely related species, sharing a morerecent common ancestry with M. bilinearis, an Atlanticspecies that putatively crossed the Panamanian seawayand speciated into M. productus to the north and laterM. angustimanus and M. gayi to the south. In our study,
Fig. 3 Merluccius spp. Molecular phylogeny for 11 species of hakeanalyzed. Both trees were midpoint-rooted. A Neighbor-joining treeconsidering both transitions and transversions (w indicate alternativepatterns obtained using transversions only; values above branchesbootstrap percentages). B Maximum-likelihood tree (log-likelihoodscore = )1656.15532); all branch lengths (except within species) weresigni®cantly di�erent from zero (P < 0.01); branch lengths are drawnproportional to amount of sequence divergence
170

the phylogenetic relationships among M. australis, M.albidus and M. hubbsi are uncertain, because of the al-ternative tree topologies, weak bootstrap support, andthe very short internode lengths compared with the longterminal branches. We suggest that the inability to re-solve clearly between these species may arise from theirrapid origin and radiation over a short time-span.
Pamilo and Nei (1988) argued that a tree constructedfrom DNA sequences of a single gene may di�er con-siderably from the species tree, if the time of divergencebetween species is short. This makes it di�cult to reachthe resolution required to clarify the phylogenetic rela-tionships between groups in fast cladogenetic lineages.As evolution proceeds along the terminal branches, it ismore likely to result in changes that are homoplasticwith the character states of the internal branches (Fel-
senstein 1978, 1988). Our control-region sequences ap-pear to resolve both basal and terminal but notintermediate relationships among hake. Thus, the lackof resolution at the intermediate level of divergence mayre¯ect rapid phyletic radiation.
The fossil record for hake is notoriously poor and in-complete, and it is di�cult to calibrate a rate of evolutionspeci®c to the genus Merluccius on the basis of our se-quence data. However, of particular interest is the puta-tive role played by the Isthmus of Panama in the evolutionof hake, since its closure created a barrier to dispersal. Weconsider that divergence between allopatric western At-lantic and eastern Paci®c species may provide a usefulbench mark by which to assess the time of divergence ofsplits in the genus Merluccius. Taking the earliestestimated time of separation of the tropical and Paci®c
Fig. 4 Merluccius spp. Summa-ry of current status of phyloge-netic relationships in hake.A Cladistic analysis of mor-phological and meristic data(adapted from Ho 1990);B UPGMA tree based on Nei'sgenetic distance (adapted fromRolda n 1995)
171
Oceans [3.2 to 2.8 million years ago (mya); Coates et al.1992] as a conservative estimate of the time of divergenceofM. productus andM. albidus (the closest north Atlanticand Paci®c species of veri®ed certain geographic origin),the rate of evolution expected is 1.7 to 2.0% per millionyears. This calibration assumes that rates of mtDNA se-quence-divergence are similar among Merluccius species,and thatM. albidus andM. productus diverged at the timeof the rise of the Isthmus of Panama . While this estimatemust be interpreted with caution (see Avise 1994), twofactors support it. (1) Using RFLP data and an assumedrate of evolution of 2% per million years (Brown et al.1979), Becker et al. (1988) estimated the time of diver-gence betweenM. capensis andM. paradoxus as 5.8 mya.If our estimate of the rate of mitochondrial DNA evolu-tion is correct, the age estimate for the split betweenM. capensis and M. paradoxus is similar (3.8 to 4.5 mya)to that of Becker et al., and falls within the Pliocene. (2)The relatively short genetic distance between M. prod-uctus and M. gayi indicates that speciation events mayhave been associated with dispersal and founder-events inthe Pleistocene, coinciding with glaciation±deglaciationcycles (Webb and Bartlein 1992). Our estimate of the dateof separation of M. productus and M. gayi (1.3 to1.1 mya), together with fossil evidence (Zinsmeister1970), is also consistent with the origin of these species inthe Pleistocene, and corroborates its recent divergence.
Speciation mode and evolutionary history
Hake have a number of behavioural characteristicswhich would tend to reduce their speciation rate. Theyform large populations and can perform extensivemigrations, altering their geographical distribution inresponse to ¯uctuations in abundance or environmentalperturbations. They also have extended spawning
seasons with multiple spawning; some spawning is ob-served year-round (Inada 1981; Cohen et al. 1990;Pitcher and Alheit 1995). The morphological characterssegregating the di�erent species (see Inada 1981) do notindicate ecological separation.
The evolution of this genus may thus have been de-termined by a series of factors that combined elements ofdispersal and vicariance. For example, it seems likelythat the Benguela system, on the west coast of southernAfrica, may to some degree act as a barrier to migrationbetween Namibian hake and hake of the Cape region.The area of maximum upwelling, which lies betweenthese two regions, may reinforce the reproductive iso-lation of the populations of these species, since temper-ature is below the minimum required for normal larvaldevelopment of hake.
The opening of the southern Atlantic Ocean basinbetween South America and Africa began during theCretaceous (Cox and Moore 1993). However, thenorthern margins of these continents remained relativelyclose to each other until the early Oligocene or lateEocene (�30 to 40 mya; Van Syoc 1995), i.e. the periodin which the genus Merluccius arose (Fedotov andBannikov 1989). By this time, the Atlantic may havebecome an e�ective barrier to gene ¯ow via planktoniclarvae. Van Andel (1979) suggested that a slowing ofequatorial currents at the boundary of the Eocene/Oli-gocene epochs resulted in a concomitant drop in pro-ductivity throughout the equatorial tropical convergencezones. This slowing of equatorial currents across thewidening Atlantic Ocean may have been the cause of theprimary taxon bifurcation in the ancestral stock of hake,giving rise to the South American and Euro±Africanclades.
Acknowledgements The authors thank the following people forcooperation in collection of samples: J. Gonzalez, UniversidadCato lica del Norte, Chile; C. Gonzalez Sotelo and C. PinÄ eiro, In-stituto de Investigaciones Marinas, Vigo, Spain; R. Method,Alaska Fisheries Science Center, USA; J. Gilbraith, USA; J. Pereiroand B. PatinÄ o, Instituto EspanÄ ol de Oceanografõ a, Vigo, Spain; andI. Sobrino, Instituto EspanÄ ol de Oceanografõ a, Cadiz, Spain. Wealso thank C. Stepien for valuable comments. This research wassupported partially by a grant from CICYT ALI 95±0053. R.V.was receiving a predoctoral fellowship from the Government ofChile (MIDEPLAN), and this paper represents part of his PhDdissertation.
References
Alvarado-Bremer JR (1994) Assessment of morphological and ge-netic variation of the sword®sh (Xiphias gladius Linnaeus):evolutionary implication of allometric growth and of the pat-terns of nucleotide substitution in the mitochondrial genome.Ph.D. thesis. University of Toronto, Toronto, Canada
Avise JC (1989) Gene trees and organismal histories: a phyloge-netic approach to population biology. Evolution 43: 279±281
Avise JC (1994) Molecular markers, natural history and evolution.Chapman & Hall, New York
Becker II, Grant WS, Kirby R, Robb FT (1988) Evolutionary di-vergence between sympatric species of southern African hakes,
Fig. 5 Merluccius spp. Relationship between genetic distance inferredfrom allozyme polymorphism (Nei's D; Rolda n 1995), and inferredfrom mtDNA-sequence data (Tamura±Nei distance; present study).Each data point represents a between-species comparison withingenus Merluccius
172

Merluccius capensis and M. paradoxus. II. Restriction enzymeanalysis of mitochondrial DNA. Heredity, Lond 61: 21±30
Bernatchez L, Danzmann RG (1993) Congruence in control-regionsequence and restriction-site variation in mitochondrial DNAof brook charr (Salvelinus fontalinalis Mitchill). Molec BiolEvolut 10: 1002±1014
Bernatchez L, Gyuomard R, Bonhomme F (1992) Sequence vari-ation of the mitochondrial control region among geographicallyand morphologically remote European brown trout (Salmotrutta, L.) populations. Molec Ecol 1: 161±173
Bird P, Green JM, Davidson WS (1986) Analysis of mitochondrialDNA in allopatric anadromous and nonanadromous Atlanticsalmon, Salmo salar. Can J Zool 64: 118±120
Brown GG, Gadaleta G, Pepe G, Saccone C, Sbisa E (1986) Struc-tural conservation and variation in the D-loop containing regionof vertebrate mitochondrial DNA. J molec Biol 192: 503±511
Brown JR, Beckenbach AT, Smith MJ (1993) Interspeci®c DNAsequence variation of the mitochondrial control region of whitesturgeon (Acipenser transmontanus). Molec Biol Evolut 10: 326±341
Brown WM, George MJr, Wilson AC (1979) Rapid evolution ofanimal mitochondrial DNA. Proc natn Acad Sci USA 76:1967±1971
Brown WM, Prager EM, Wang A, Wilson AC (1982) Mitochon-drial DNA sequence of primates: tempo and mode of evolution.J molec Evolut 18: 239±255
Carr SM, Marshall HD (1991) Detection of intraspeci®c DNAsequence variation in the mitochondrial cytochrome b gene ofAtlantic cod (Gadus morhua) by the polymerase chain reaction.Can J Fish aquat Sciences 48: 48±52
Coates AG, Jackson JBC, Collins LS, Cronin TM, Dowsett HJ,Bybell LM, Jung P, Obando JA (1992) Closure of the Isthmusof Panama, the near-shore marine record of Costa Rica andwestern Panama. Bull geol Soc Am 104: 814±828
Cohen DM (1984) Gadiformes: overview. Spec Publs Am SocIchthyol Herpetol 1: 259±269
Cohen DM, Inada T, Iwamoto T, Scialabba N (1990) FAO speciescatalogue. Gadiform ®shes of the world (order Gadiformes).FAO Fish Synopsis 125 (10): 1±442
Cox CB, Moore PT (1993) Biogeography: an ecological and evo-lutionary approach. Blackwell Scienti®c Publications, London
Fahay MP (1974) Occurrence of silver hake, Merluccius bilinearis,eggs and larvae along the Middle Atlantic continental shelfduring 1966. Fish Bull US 72: 813±834
Fedotov VF, Bannikov AF (1989) On phylogenetic relationships offossil Gadidae. Sci Ser nat Hist Mus Los Ang Cty 32: 187±195
Felsenstein J (1978) Cases in which parsimony and compatibilitymethods will be positively misleading. Syst Zool 27: 401±410
Felsenstein J (1981) Evolutionary trees from DNA sequences: amaximum likelihood approach. J molec Evolut 17: 368±376
Felsenstein J (1988) Phylogenies from molecular sequences: infer-ence and reliability. A Rev Genet 22: 521±565
Felsenstein J (1993) PHYLIP (phylogeny inference package). Ver-sion 3.5c. (Distributed by the author)
Fernandez J (1985) A parasitological study of Merluccius australis(Hutton, 1872) (Pisces: Merluccidae): systematic, statistical andzoogeographic aspects. Boln Soc Biol Concepcio n, Chile 56: 31±41
Ferris JS, Kallersjo M, Kluge AC, Bult C (1995) Constructing asigni®cance test for incongruence. Syst Biol 44: 570±572
Grant WS, Becker II, Leslie RW (1988) Evolutionary divergencebetween sympatric species of southern African hakes, Mer-luccius capensis andM. paradoxus. I. Electrophoretic analysis ofproteins. Heredity, Lond 61: 13±20
Goldman N (1997) Phylogeny estimation. In: Bishop MJ, RawlingsCJ (eds) DNA and protein sequence analysis: a practical ap-proach. Oxford University Press, Oxford, pp 279±311
Hedges SM (1992) The number of replications needed for accurateestimation of the bootstrap P value in phylogenetic studies.Molec Biol Evolut 9: 366±369
Hillis DM (1995) Approaches for assessing phylogenetic accuracy.Syst Biol 44: 3±16
Hillis DM, Huelsenbeck JP (1992) Signal, noise and reliability inmolecular phylogenetic analyses. J Hered 83: 189±195
Ho J-S (1990) Phylogeny and biogeography of hakes (Merluccius:Teleostei): a cladistic analysis. Fish Bull US 88: 95±104
Holmquist R (1983) Transitions and transversions in evolutionarydescent: an approach to understanding. J molec Evolut 19: 134±144
Howes GJ (1989) Phylogeny relationships of macrouroid andgadoid ®shes based on cranial myology and arthrology. Sci Sernat Hist Mus Los Ang Cty 32: 113±128
Howes GJ (1990) The syncranial osteology of the southern eel-codfamily Muraenolepididae, with comments on its phylogeneticrelationships and on the biogeography of sub-Antarctic gadoid®shes. Zool J Linn Soc 100: 73±100
Howes GJ (1991) Biogeography of gadoid ®shes. J Biogeogr 18:595±622
Inada T (1981) Studies on the merluccid ®shes. Bull Far Seas FishRes Lab, Japan 18: 1±172
Inada T (1989) Current status of the systematics of gadiform ®shes.Sci Ser nat Hist Mus Los Ang Cty 32: 196±207
Irwin DM, Kocher TD, Wilson AC (1991) Evolution of the cyto-chrome b gene of mammals. J molec Evolut 32: 128±144
Kabata Z, Ho J-S (1981) The origin and dispersal of hake (genusMerluccius: Pisces: Teleostei) as indicated by its copepod par-asites. Oceanogr mar Biol A Rev 19: 381±404
Kishino H, Hasegawa M (1989) Evaluation of the maximum like-lihood estimate of the evolutionary tree topologies from DNAsequence data, and the branching order in Hominoidea. J molecEvolut 29: 170±179
Kocher TD, Thomas WK, Meyer A, Edwards SV, Paabo S,Villablanca FX, Wilson AC (1989) Dynamics of mitochon-drial DNA evolution in animals: ampli®cation and sequencingwith conserved primers. Proc natn Acad Sci USA 86: 6196±6200
Kumar S, Tamura K, Nei M (1993) MEGA: molecular evolu-tionary genetics analysis. Version 1.01. Pennsylvania StateUniversity, University Park
Lee W-J, Conroy JA, Howell WH, Kocher TD (1995) Structureand evolution of the teleost mitochondrial control-regions.J molec Evolut 41: 54±66
Lleonart J, Agell J (1980) Taxonomõ a nume rica del ge nero Mer-luccius (Ra®nesque, 1810) a partir de la electrofore sis delmio geno. Investigacio n pesq 44: 461±470
Lombarte A, Castello n A (1991) Interspeci®c and intraspeci®cotolith variability in the genus Merluccius as determined byimage analysis. Can J Zool 69: 2442±2449
Lo pez Abella n LJ, Ariz Tellerõ a J (1993) Aspectos generales de ladistribucio n y biologõ a de las especies del ge nero MerlucciusRa®nesque, 1810, en aguas de Senegal y Gambia (16° 00¢ N±12°25 N). Boln Inst esp Oceanogr 9: 101±121
Markle DF (1989) Aspects of character homology and phylogenyof the Gadiformes. Sci Ser nat Hist Mus Los Ang Cty 32: 59±98
Marshall NB (1966) The relationships of the anacanthine ®shesMacruronus, Lyconus and Steindachneria. Copeia 1966: 275±280
Mathews CP (1985) Meristic studies of the Gulf of Californiaspecies Merluccius, with a description of a new species. J natHist 19: 697±718
McMillan WO, Palumbi SR (1997) Rapid rate of control-regionevolution in Paci®c butter¯y®shes (Chaetodontidae). J molecEvolut 45: 473±484
McVeigh HP, Barlett SE, Davidson WS (1991) Polymerase chainreaction/direct sequence analysis of the cytochrome b gene inSalmo salar. Aquaculture, Amsterdam 95: 225±233
Meyer A, Kocher TD, Basasibwaki PA, Wilson AC (1990)Monophylectic origin of Lake Victoria cichlid ®shes suggestedby mitochondrial DNA sequences. Nature, Lond 347: 550±553
Miyamoto MM, Boyle SM (1989) The potencial importance ofmitochondrial DNA sequence data to eutherian mammal phy-logeny. In: Franholm R, Bremer K, Jornvall H (eds) Thehierarchy of life: molecules and morphology in phylogeneticanalysis. Elsevier Science, Amsterdam, pp 437±450
173

Moritz C, Dowling TE, Brown WM (1987) Evolution of animalmitochondrial DNA: relevance for population biology andsystematics. A Rev Ecol Syst 18: 269±292
Niegel JE, Avise JC (1986) Phylogenetic relationships of mito-chondrial DNA under various demographic models of specia-tion In: Nevo E, Karlin S (eds) Evolutionary processes andtheory. Academic Press, New York, pp 515±534
Okamura O (1989) Relationships of the suborder Macruroidei andrelated groups, with comments on Merlucciidae and Stein-dachneria. Sci Ser nat Hist Mus Los Ang Cty 32: 129±142
Olivar MP, Rubes P, Salat J (1988) Early life history and spawningof Merluccius capensis in the northern Benguela current. S Afr Jmar Sci 6: 245±254
Pamilo P, Nei M (1988) Relationship between gene trees and spe-cies trees. Molec Biol Evolut 5: 568±583
Pitcher TJ, Alheit J (1995) What makes a hake? A review of thecritical biological features that sustain global hake ®sheries. In:Alheit J, Pitcher TJ (eds) Hake, biology, ®sheries and markets.Chapman & Hall, London, pp 1±14
Rolda n MI (1995) Relaciones ®logene ticas en el ge nero Merlucciusy estructura ge netica poblacional en la merluza Argentina(Merluccius hubbsi) y la merluza Europea (Merluccius mer-luccius). Ph.D. thesis. University of Girona, Girona, Spain
Saitou N, Nei M (1987) The neighbour-joining method: a newmethod for reconstructing phylogenetic trees. Molec BiolEvolut 4: 406±425
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning. 2ndedn. Cold Spring Harbor Laboratory Press, Cold Spring Har-bor, New York
Shedlock AM, Parker JD, Crispin DA, Pietsch TW, Burmer GC(1992) Evolution of the salmonid mitochondrial control region.Molec Phylogenetics Evolut 1: 179±192
Shields GF, Gust JR (1995) Lack of geographic structure in mi-tochondrial DNA sequences of Bering Sea walleye pollock,Theragra chalcogramma. Molec mar Biol Biotechnol 4: 69±82
Soliman IA (1973) Variations of ®shes of genus Merluccius in At-lantic Ocean and Mediterranean Sea. Acta ichthyol Piscat3: 29±64
Stepien CA, Rosenblatt RH (1996) Genetic divergence in anti-tropical pelagic marine ®shes (Trachurus, Merluccius, andScomber) between north and south America. Copeia 3: 586±598
Sturmbauer C, Meyer A (1992) Genetic divergence, speciation andmorphological stasis in a lineage of African cichlid ®shes.Nature, Lond 359: 578±581
Szidat L (1955) La fauna de para sitos de ``Merluccius hubbsi'' comocaracter auxiliar para la solucio n de los problemas siste maticos
y zoogeogra ®cos del gene ro ``Merluccius'' L. Comun Inst nacInvest Cienc nat, B Aires 3: 1±54
Szidat L (1961) Versuch einer Zoogeographie des SudAtlantik mitHilfe von Leitparsiten der Meeres®sche. Parasit SchReihe 13: 1±98
Tamura K, Nei M (1993) Estimation of the number of nucleotidesubstitutions in the control region of mitochondrial DNA inhumans and chimpanzees. Molec Biol Evolut 10: 512±526
Taylor EB, Dodson JJ (1994) A molecular analysis of relationshipsand biogeography within a species complex of holartic ®sh(genus Osmerus). Molec Ecol 3: 235±248
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W:improving the sensitivity of progressive multiple sequencealignment through sequence weighting, positions-speci®c gappenalties and weight matrix choice. Nucleic Acids Res 22: 4673±4680
Turner TF (1997) Mitochondrial control region sequences andphylogenetic systematics of darters (Teleostei: Percidae).Copeia 1997 (2): 319±338
Van Andel TH (1979) An eclectic overview of plate tectonics,paleogeography, and paleoceanography. In: Gray J, BoucotAJ (eds) Proceedings of the Annual Biology Colloquiumand Selected Papers. Oregon State University, Corvallis, pp9±25
Van Syoc RJ (1995) Barnacle mitochondrial DNA: determininggenetic relationships among species of Pollicipes. In: SchramFR, Hoeg JT (eds) New frontiers in barnacle evolution. A.A.Balkema, Rotterdam, pp 269±296
Wakeley J (1996) The excess of transitions among nucleotide sub-stitutions: new methods of estimating transition bias underscoreits signi®cance. Trends Ecol Evolut 11: 158±163
Webb T III, Bartlein PJ (1992) Global changes during the last 3million years: climatic controls and biotic responses. A RevEcol Syst 23: 141±173
Wilson AC, Cann RL, Carr SM, George MJr, Gyllensten UB,Helm-Bychowski KM, Higuchi RG, Palumbi SR, Prager EM,Sage RD, Stoneking M (1985) Mitochondrial DNA and twoperspectives on evolutionary genetics. Biol J Linn Soc 26: 375±400
Zhu D, Jamieson BGM, Hugall A, Moritz C (1994) Sequenceevolution and phylogenetic signal in control-region and cyto-chrome b sequences of rainbow ®shes (Melanotaeniidae). MolecBiol Evolut 11: 672±683
Zinsmeister WJ (1970) A late Pliocene macrofossil fauna of New-port Beach, Orange County, California. Bull Sth Calif Acad Sci69: 121±125
174




















